

Abstract
Epithelial ovarian carcinoma tissues express high levels of tumor necrosis factor-alpha (TNF-α) and other inflammatory cytokines. The underlying mechanism leading to the abnormal TNF-α expression in ovarian cancer remains poorly understood. In the current study, we demonstrated that lysophosphatidic acid (LPA), a lipid mediator present in ascites of ovarian cancer patients, induced expression of TNF-α mRNA and release of TNF-α protein in ovarian cancer cells. LPA also induced expression of interleukin-1β (IL-1β) mRNA but no significant increase in IL-1β protein was detected. LPA enhanced TNF-α mRNA through NF-κB-mediated transcriptional activation. Inactivation of ADAM17, a disintegrin and metalloproteinase, with a specific inhibitor TMI-1 or by shRNA knockdown prevented ovarian cancer cells from releasing TNF-α protein in response to LPA, indicating that LPA-mediated TNF-α production relies on both transcriptional upregulation of the TNF-α gene and the activity of ADAM17, the membrane-associated TNF-α-converting enzyme. Like many other biological responses to LPA, induction of TNF-α by LPA also depended on transactivation of epidermal growth factor receptor (EGFR). Interestingly, our results revealed that ADAM17 was also the shedding protease responsible for the transactivation of EGFR by LPA in ovarian cancer cells. To explore the biological outcomes of LPA-induced TNF-α, we examined the effects of a TNF-α neutralizing antibody and recombinant TNF-α soluble receptor on LPA-stimulated expression of pro-tumorigenic cytokines and chemokines overexpressed in ovarian cancer. Blockade of TNF-α signaling significantly reduced the production of IL-8, IL-6, and CXCL1, suggesting a hierarchy of mechanisms contributing to the robust expression of the inflammatory mediators in response to LPA in ovarian cancer cells. In contrast, TNF-α inhibition did not affect LPA-dependent cell proliferation. Taken together, our results establish that the bioactive lipid LPA drives expression of TNF-α to regulate an inflammatory network in ovarian cancer.
Keywords: LPA, TNF-α, ovarian cancer, cytokines, ADAM17
INTRODUCTION
Previous mRNA expression array studies from independent groups have shown that the proinflammatory cytokine TNF-α mRNA was overexpressed in epithelial ovarian carcinomas compared to normal ovarian tissues [1–3]. Most of these array studies of homogenized tumor tissues could not exclude a potential contribution from tumor-infiltrating immune cells such as macrophages, the primary cell type to express TNF-α. More critical analyses have used RNA in-situ hybridization technique to confirm the location of increased mRNA of TNF-α within ovarian tumor cells [4–6]. As further evidence for tumor cell-associated expression of TNF-α, primary ovarian tumor cells freshly isolated from ascites expressed TNF-α protein constitutively or exhibited TNF-α inducibility [1]. TNF-α and other inflammatory mediators were detected at elevated levels in the serum and ascites of ovarian cancer patients [7–10]. It is now well established that, in addition to host immune cells, ovarian tumor cells are a clinically relevant source of these elevated inflammatory cytokines in vivo.
Cancer cell-derived TNF-α plays pleiotropic roles in cancer development and progression [11–14]. It contributes to the chronic or sustained inflammation in the tumor milieu [15]. It is also implicated in tumor/stromal communication and tumor angiogenesis [14, 16]. Low endogenous levels of TNF-α acts as a mitogen towards cancer cells [13, 17, 18]. In contrast to these well-known biological actions of TNF-α, the mechanism driving TNF-α overexpression and the inflammatory phenotype in cancer remains poorly understood. Lysophosphatidic acid (LPA), a lysophospholipid mediator, is present in elevated concentrations in ascites of ovarian cancer patients [19–21]. LPA through its cognate G protein-coupled receptors (GPCRs) regulates many oncogenic processes in cancer cells [22–25]. We and others have demonstrated that LPA induced activation of various transcription factors, reprogramming expression of many target genes involved in cell proliferation, survival, migration and invasion, angiogenesis, glycolytic and lipogenic metabolism and inflammation [26–33]. The most prominent target genes of LPA identified by gene arrays were cytokines and chemokines such as interleukin-8 (IL-8), interleukin-6 (IL-6), and C-X-C motif chemokine ligand 1 (CXCL1) [28, 30, 34, 35]. LPA has been also shown to induce the lipid inflammatory molecules prostaglandins via upregulation of COX-2 in a subset of ovarian cancer cell lines [29, 31]. These prior observations highlight the potential of LPA as a promoter of the inflammatory phenotype of ovarian cancer. However, no study has connected LPA to cancer cell expression of TNF-α, the master regulator of systemic inflammation and expression of other cytokines and chemokines. One previous study found that the LPA1 receptor signaling was required for lipopolysaccharide (LPS)-induced TNF-α expression in rodent microglial cells [36]. In another report, LPA administration decreased serum TNF-α levels in both control mice and those treated with acetaminophen, hinting at inhibition of TNF-α production by LPA in liver cells [37].
In the present study, we showed that LPA potently induced TNF-α in ovarian cancer cells via NF-κB-dependent upregulation of TNF-α transcription and ADAM17-mediated processing and maturation of TNF-α protein. LPA induction of TNF-α protein required transactivation of EGFR as many other biological functions of LPA. Our analysis revealed that ADAM17 also mediated transactivation of EGFR by LPA. Using specific TNF-α-blocking agents, we showed that the induced TNF-α is functionally significant to contribute to LPA-induced production of IL-8, IL-6 and CXCL1 in ovarian cancer cells. Therefore, the tumor microenvironmental factor LPA drives expression of TNF-α to regulate the inflammatory phenotype in ovarian cancer.
MATERIALS AND METHODS
Reagents
LPA (1-oleoly, 18:1) was obtained in chloroform solvent from Avanti Polar Lipids, Inc. (Alabaster, AL). Prior to use, LPA was dried and dissolved in PBS containing 0.5% fatty acid-free bovine serum albumin (BSA) from Roche (Indianapolis, IN). The ELISA Max™ Deluxe Sets for TNF-α, IL-1β, IL-6 and IL-8 were purchased from BioLegend (San Diego, CA). The corresponding ELISA kits were prepared according to the manufacturer. The human CXCL1 DuoSet ELISA kit was from R&D Systems, Inc. (Minneapolis, MN). The TaqMan Universal PCR Master Mix and qPCR probes for TNF-α, IL-1β and GAPDH were obtained from ThermoFisher Scientific (Waltham, MA). The ADAM17 inhibitor TMI-1, NF-κB inhibitor (BAY11–7085), EGFR inhibitor AG1478, and anti-ADAM17 antibody (ABT94) were obtained from Sigma-Aldrich (St. Louis, MO). Pertussis toxin (PTX) was purchased from List biological labs (Campbell, CA). Anti-EGFR antibody and anti-phospho-EGFR (Y1068) antibody were from Cell Signaling Technology (Danvers, MA). Anti-IL6 antibody (SC-7920) and COX-2 antibody (SC-1745) were from Santa Cruz (Austin, TX). Anti-TNF-α neutralizing monoclonal antibody (TNF-α Ab) was from BioLegend. The soluble form of the TNF-α receptor (TNF-α SR) was obtained from Amgen (Thousand Oaks, CA). Plasmid DNAs were purified using the endo-free purification kit from Qiagen (Valencia, CA).
Cells
The sources of the ovarian cancer cell lines Caov-3, SKOV-3, and OVCAR-3 were described previously [38]. These cells were either from ATCC or from Dr. GB Mills and Dr. RC Bast, Jr. (MD Anderson Cancer Center). All cell lines used in this study were authenticated routinely by STR DNA fingerprinting at the Characterized Cell Line Core Facility of MD Anderson Cancer Center (Houston, TX). The cells were cultured in either RPMI medium 1640 (SKOV-3 and OVCAR-3) or high glucose DMEM (Caov-3) supplemented with 10% FBS and antibiotics. The cell lines were frozen at early passages and used for less than 1 month in continuous culture.
Gene knockdown
The shRNA target sequences for ADAM17 (5’-AGATCATCGCTTCTACAGA-3’) or control non-targeting sequence (5’-CCTAAGGTTAAGTCGCCCTCG-3’) were cloned into the shRNA lentiviral vector pGreenPuro (System Biosciences, Mountain View, CA). The recombinant lentivirus was produced in 293TN cells by co-transfection with lentiviral vectors and packaging plasmids using lipofectamine 2000 (Life Technologies). The culture supernatants were collected 2–2.5 days posttransfection and used for infection of ovarian cancer cells in culture. The transduced cells were selected with puromycin to obtain stably ADAM17 silenced cells.
The expression of NF-κB RelA/p65 was transiently silenced with SignalSilence® NF-κB p65 siRNA from Cell Signaling (#6261, Danvers, MA). The siRNAs for LPA1 (s4450), LPA2 (s17525) and LPA3 (s24110) were obtained from Thermo Fisher Scientific. Ovarian cancer cell lines in 6-well plates were transfected with 30 pmol gene-specific siRNA or control siRNA (#6568, Cell Signaling Technology) and 5 μl Lipofectamine RNAiMAX according to the manufacturer (Thermo Fisher Scientific). Two days posttransfection, the cells were starved for 24 hours before stimulation with LPA for the indicated durations.
Quantitative PCR
Total cellular RNA was extracted with Trizol (Thermo Fisher Scientific) from cells treated with or without LPA. Complementary DNA (cDNA) was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). The cDNA abundances of TNF-α, IL-1β, IL-8, IL-6, CXCL1, ADAM17, and GAPDH were quantified using gene-specific probes, the TaqMan Universal PCR Master Mix, and the BIO-RAD CFX Connect™ Real-Time PCR System. The results of TNF-α and IL-1β were normalized to the level of GAPDH and presented as LPA-induced fold changes relative to that of untreated control cells (defined as 1 fold).
ELISA
The concentrations of TNF-α, IL-1β, IL-6, IL-8, and CXCL1 in culture supernatants were determined using the above described ELISA kits according to the manufacturers’ instructions. The concentrations of these cytokines were calculated by comparing the absorbance of samples to standard curves and normalized to cell numbers. The results were presented as pg/5×105 cells/24 hours.
Immunoblotting analysis
Cells were lysed with a lysis buffer containing 40 mM HEPES pH 7.4, 150 mM NaCl, 1% Triton X-100, 10 mM NaF, 5 mM Na pyrophosphate, 1 mM β-glycerophosphate, 1 mM NaV and the protease/phosphatase inhibitor cocktail from Roche Diagnostics (Indianapolis, IN, USA). Protein concentrations were quantified with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). For analysis of ADAM17, a membrane-associated hydrophobic protein, lysate samples were solubilized in 1XSDS and incubated at 65 °C for 10 minutes instead of boiling to prevent heat-induced aggregation of the hydrophobic protein. The regular boiling procedure applied to analysis of other proteins. Equal amounts of proteins were resolved by SDS polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to immunoblot membrane (polyvinylidene difluoride) (BIO-RAD, Hercules, CA), and immunoblotted with antibodies following the protocols of the manufacturers. Immunocomplexes were visualized by an enhanced chemiluminescence detection kit (Thermo Fisher Scientific) using horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology).
Measurement of ADAM17 activity in live cells
ADAM17 activity in live cells were measured as previously described [39]. Briefly, cells in 24-well plates at around 60% confluence were serum-starved and stimulated in phenol red-free medium with 10 μM LPA for 30 minutes before addition of the quenched fluorogenic TACE substrate (Abz-LAQAVRSSSR-Dpa) (15 μM) (Sigma, #616407). The conversion of the substrate to its fluorogenic product was measured in a plate reader for 1 hour at emission 405 nm and extinction 320 nm. The activity was quantified by calculating the area under curve of the fluorescent signal over time, normalized to cell numbers and presented as fold changes relative to un-stimulated control cells (defined as 1 fold).
Statistics
All numerical data were presented as mean ± SD of triplicate assays, representative of three independent experiments. The statistical significances were analyzed using Student’s t-test unless otherwise stated, where p < 0.05 was considered statistically significant. In all figures, the statistical significances were indicated with n.s. (not significant) if p>0.05, * if p < 0.05 or ** if p < 0.01.
RESULTS
LPA induces expression of TNF-α
As we reported previously, the induction of cytokines and chemokines such as IL-8, IL-6, and CXCL1 is one of the most prominent effects of LPA towards cancer cells [28, 30, 34]. To explore LPA-TNF-α communications, we examined the effect of LPA on expression of TNF-α mRNA in ovarian cancer cell lines. Caov-3, SKOV-3, and OVCAR-3 cells were treated with LPA for different durations. LPA-induced TNF-α mRNA expression was assessed with RT-qPCR analysis of total cellular RNA from control and LPA-treated cells. As shown in Supplementary Fig. S1A, TNF-α mRNA expression was robustly induced by LPA in these cell lines. In Caov-3, LPA induction of TNF-α reached maximum at 2–4 hours of LPA treatment. More than 20 fold increase in TNF-α mRNA abundance was seen at 4 hours. In SKOV-3, the maximal induction was detected at 1 hour of treatment. The mRNA expression decreased thereafter but remained prominent (>10 fold) at 2 or 4 hours of LPA stimulation. Fig. 1A summarized the results of an independent experiment where the cells were treated with LPA for 2 hours in Caov-3 and OVCAR-3 or 1 hour in SKOV-3 cells. We then determined whether TNF-α mRNA upregulation led to TNF-α protein secretion as determined by ELISA. Consistently, TNF-α protein levels were significantly increased in culture supernatants of cells treated with 10 μM LPA for 24 hours (Fig. 1A, lower). The maximal response was observed when more than 10 μM LPA was used (Supplementary Fig. S1B).
Figure 1. LPA induces TNF-α mRNA expression and protein release.
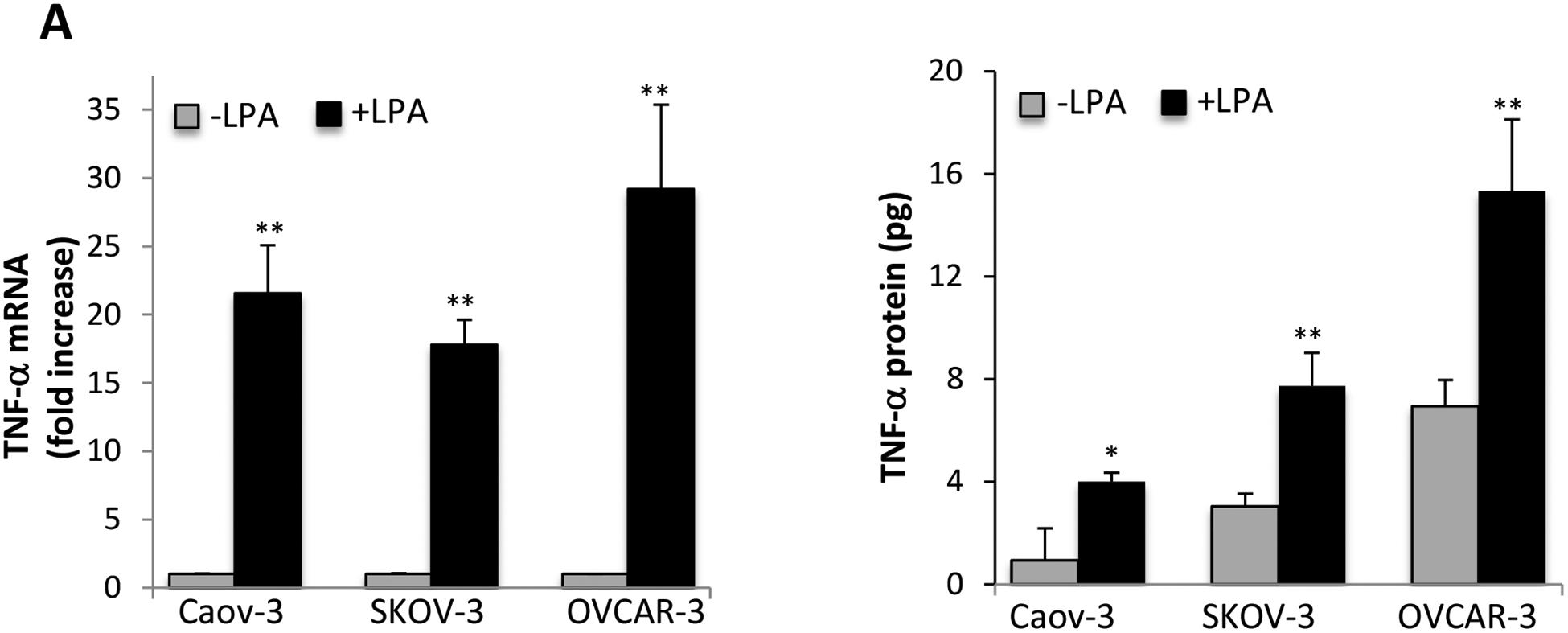
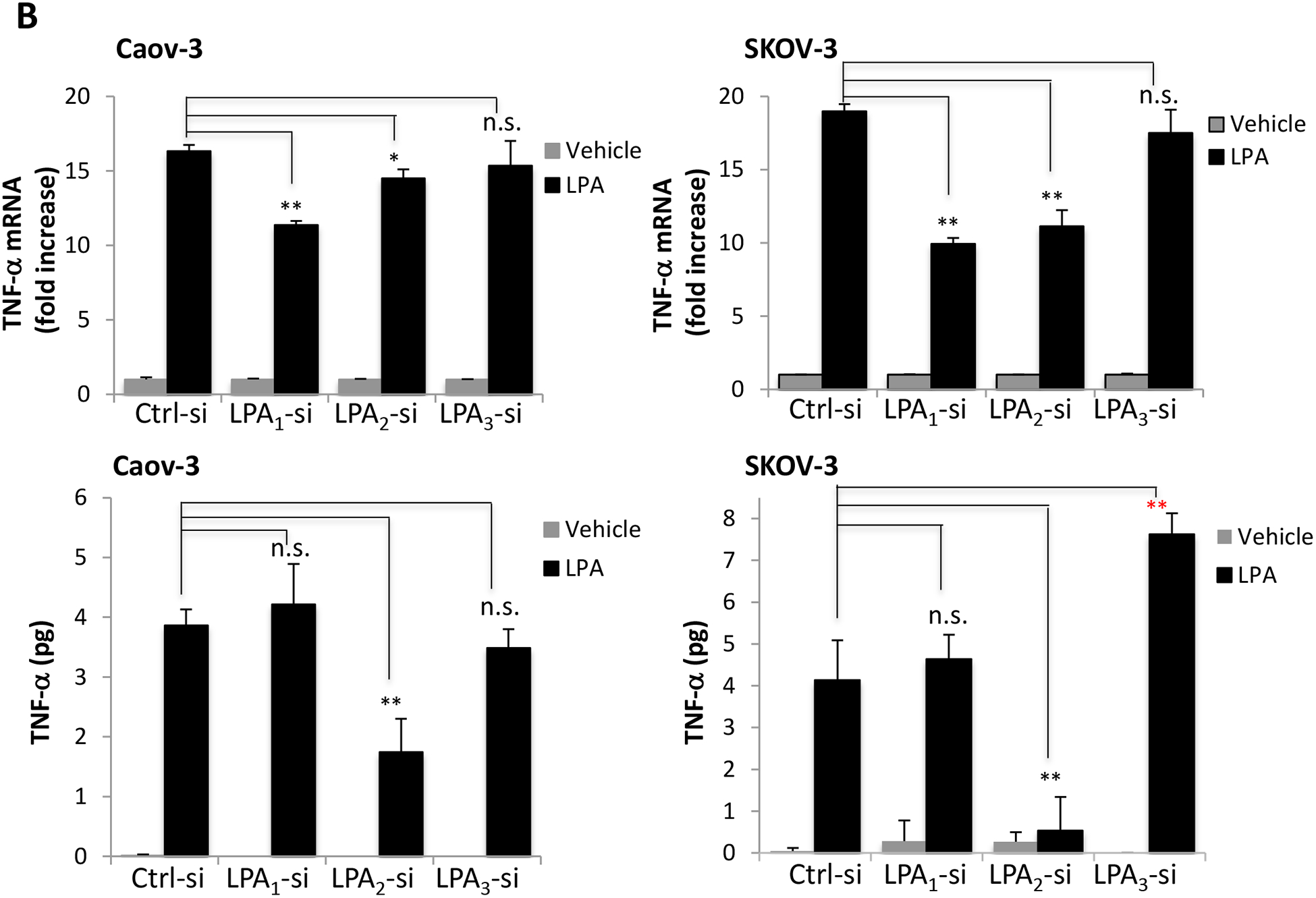
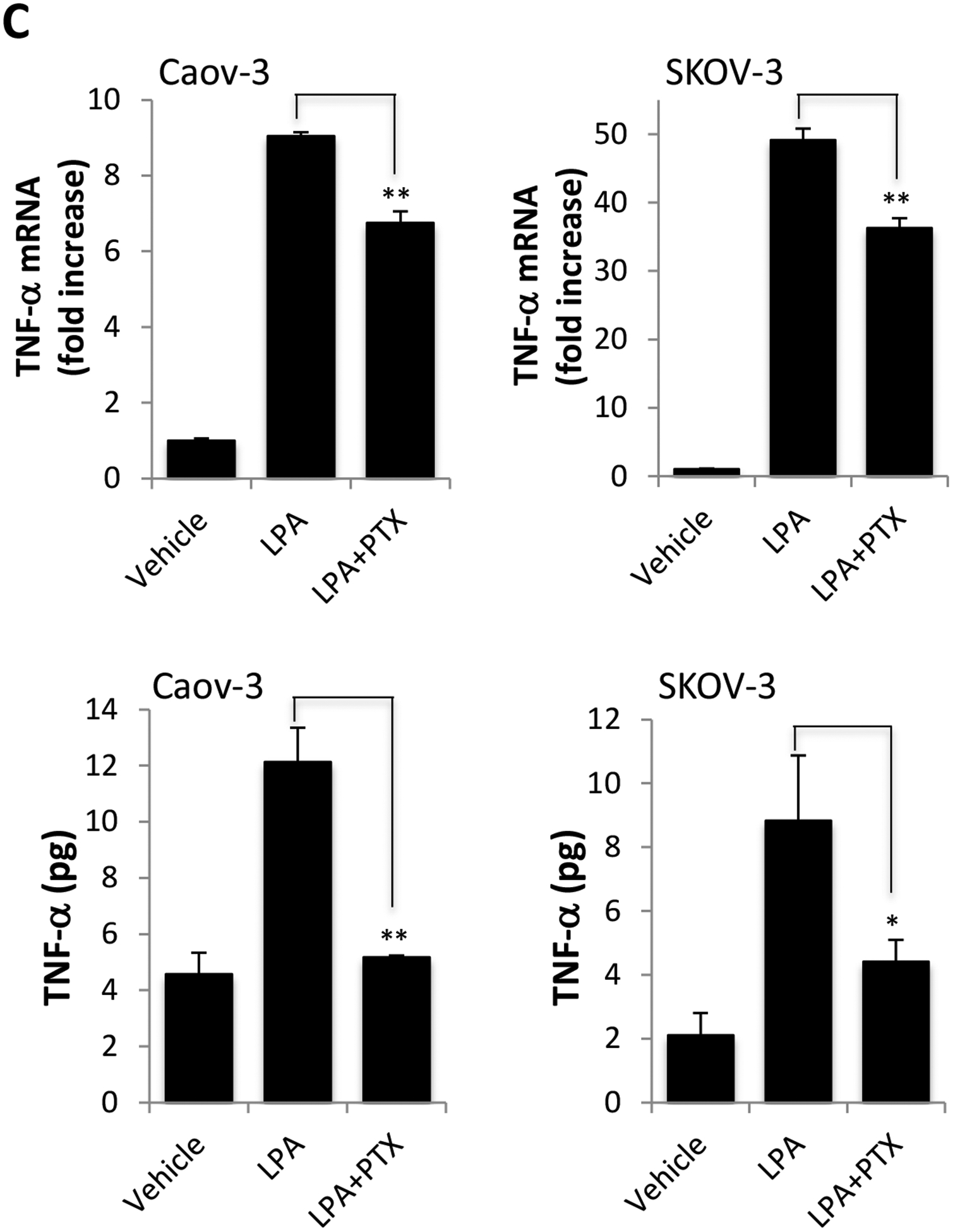
A. Ovarian cancer cell lines were serum starved for 24 hours and treated with 10 μM LPA in serum-free medium for 1 hour (SKOV-3) or 4 hours (Caov-3 and OVCAR-3) before total cellular RNA was isolated and analyzed by RT and quantitative PCR. The expression levels of TNF-α were normalized to GAPDH and presented as fold changes relative to untreated, control cells (upper). In the lower panel, Concentrations of secreted TNF-α protein in culture supernatants of control and LPA-treated cells in 12-well plates were determined by ELISA. The results were presented as pg/5×105 cells/24 hours. B. LPA1, LPA2 or LPA3 were knocked down by the corresponding subtype-specific siRNA to assess the effects of the Edg LPA receptors on LPA induced TNF-α mRNA (upper) and protein release (lower). C. Caov-3 and SKOV-3 cells were treated with LPA with or without PTX (50 ng/ml). PTX was added at least 3 hours before LPA. In both B and C, LPA-induced TNF-α mRNA and protein were assessed and presented as in A. In this and the following figures, the results were mean+SD of triplicates, representative of 3 independent experiments.
Since IL-1β is concomitantly induced along with TNF-α in most inflammatory conditions [40, 41], we examined the effects of LPA on IL-1β mRNA and protein release. Similarly, LPA stimulated multi-fold increases in IL-1β mRNA in ovarian cancer cell lines (Supplementary Fig. S2A). However, these cells expressed extremely low levels of IL-1β protein. Using ELISA, we did not detect any significant or consistent increase in IL-1β protein in culture supernatants of LPA-treated cells (Supplementary Fig. S2B). The inflammasome activation is the primary route leading to caspase 1-dependent processing/cleavage of IL-1β [42]. Immunoblotting analysis of caspase 1 status indicated that LPA induced early and slight cleavage of caspase 1 in SKOV-3 but not in Caov-3 or OVCAR-3 cells (Supplementary Fig. S2C). There was no corresponding decrease in pro-caspase 1, suggesting that the cleavage in SKOV-3 was modest in LPA-treated conditions. Furthermore, the weak cleavage of caspase 1 in SKOV-3 cells faded after 20 hours of LPA treatment. These results indicated that LPA was not capable of inducing a full-scale inflammasome activation or IL-1β production in ovarian cancer cells.
LPA induces TNF-α through LPA2 and the PTX-sensitive Gi protein
To identify LPA receptors that mediate TNF-α induction, we utilized gene specific siRNAs to knockdown each of the Edg LPA receptors LPA1, LPA2 and LPA3 (Supplementary Fig. S3). These LPA receptors are dysregulated in ovarian cancer [22, 23, 28]. In Caov-3 and SKOV-3 cells, knockdown of LPA1 or LPA2 partially reduced LPA induction of TNF-α mRNA (Fig. 1B). In contrast, LPA3 silencing did not inhibit the effect. We next examined the impact of the knockdown on LPA-induced TNF-α protein. As shown in Fig. 1B, ELISA analysis showed that only LPA2 knockdown significantly attenuated LPA-induced TNF-α protein production (by 55% in Caov-3 and 87% in SKOV-3). Despite its significant inhibition of TNF-α mRNA, LPA1 knockdown did not affect TNF-α protein production, suggesting that LPA-driven TNF-α release involves mechanisms beyond transcriptional regulation of TNF-α mRNA. Intriguingly, knockdown of LPA3 enhanced TNF-α protein level in SKOV-3 cells, implying that LPA3 acted as a negative receptor for this particular function of LPA. In Caov-3, knockdown of LPA3 did not affect LPA-driven TNF-α release. To further confirm the importance of LPA2 and the downstream G proteins in LPA-induced TNF-α, we examined the effect of PTX, an inhibitor of Gi protein. Treatment with PTX modestly decreased LPA-dependent TNF-α mRNA levels by less than 30% in Caov-3 and in SKOV-3 cells. However, PTX strongly attenuated LPA induction of TNF-α protein by 92% in Caov-3 and 66% in SKOV-3 (Fig. 1C). These results together establish that LPA2 and the Gi signaling cascade are involved in LPA regulation of TNF-α.
LPA stimulates TNF-α transcription in an NF-κB-dependent manner
The NF-κB transcription factor plays a central role in the regulation of TNF-α in response to various stimuli such as LPS [43, 44]. It has been reported that LPA induced NF-κB activation in different cancer cells [28, 45]. The NF-κB inhibitor BAY11–7085 is an irreversible inhibitor of IκBα phosphorylation, preventing its release from the NF-κB complex and the subsequent NF-κB activation and nuclear import [46]. We applied this inhibitor to assess the potential involvement of NF-κB in LPA-driven TNF-α transcription. As shown in Fig. 2A, LPA-induced increases in TNF-α mRNA were strongly inhibited by BAY11–7085 at 10 μM. Higher concentrations such as 25 μM completely eliminated LPA induction of TNF-α mRNA.
Figure 2. LPA stimulates TNF-α transcription via the NF-κB transcription factor.
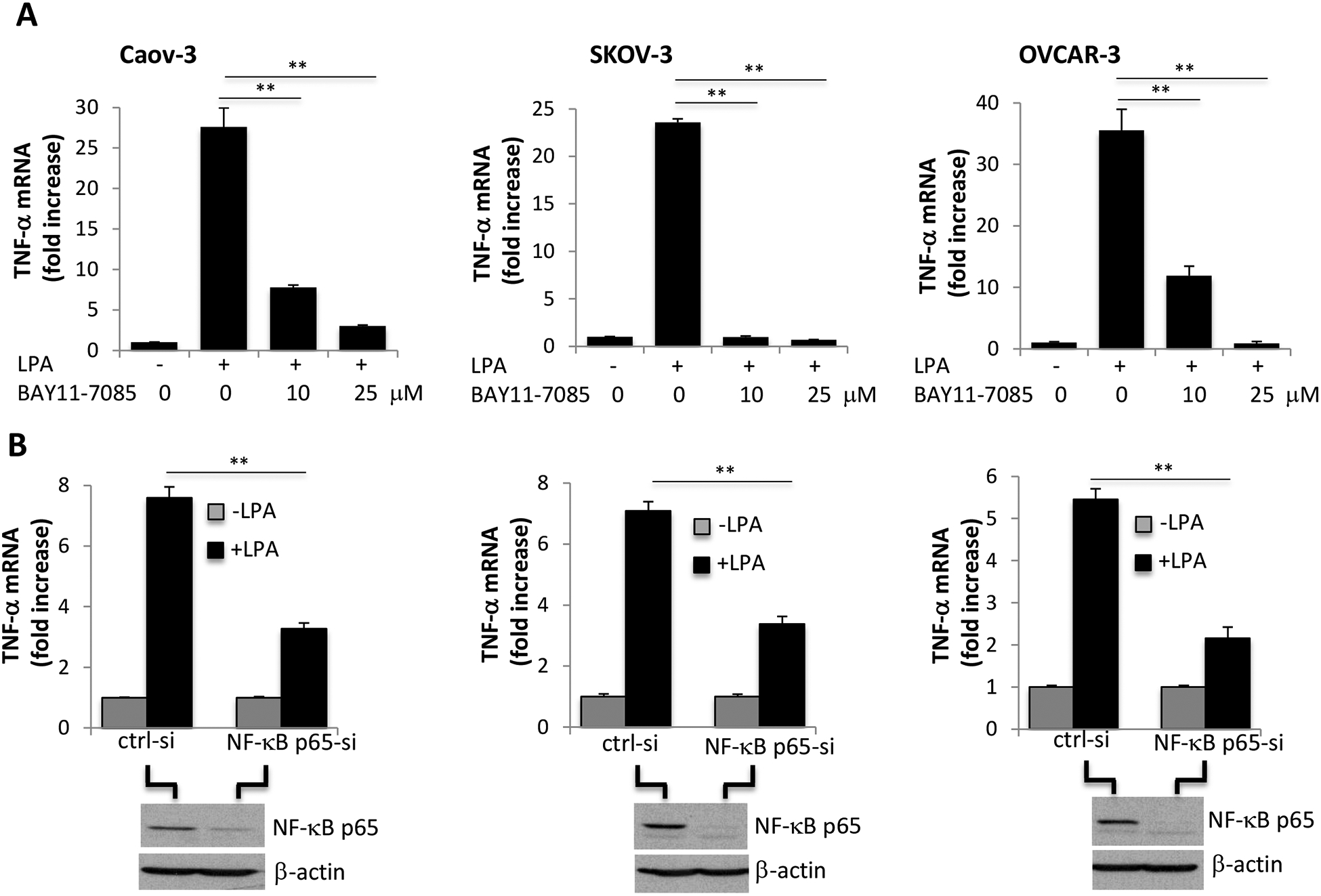
A. Caov-3, SKOV-3 and OVCAR-3 cells were treated with LPA as in Fig. 1A in the presence of the NF-κB inhibitor BAY11–7085. BAY11–7085 was added 30 minutes before LPA treatment. The effects of the indicated concentrations BAY11–7085 on LPA-induced TNF-α mRNA abundances were analyzed by RT-qPCR and presented as described in Fig. 1A. B. The RelA/p65 gene of the canonical NF-κB pathway was transiently silenced by siRNA to assess the effect of NF-κB p65 on LPA-induced TNF-α mRNA. The NF-κB p65 knockdown efficiency was confirmed by immunoblotting analysis.
To further confirm the crucial role of NF-κB in transcriptional activation of TNF-α by LPA, we silenced expression of RelA/p65 of the classical NF-κB pathway. In all ovarian cancer cell lines tested, the NF-κB p65 siRNA significantly inhibited LPA induction of TNF-α mRNA compared to the control, non-targeting siRNA (Fig. 2B). Of note, the transiently transfected cells did not respond to LPA as potently as non-transfected cells likely owing to the transfection-associated cell stress. Nevertheless, the molecular approach clearly showed that the classical NF-κB cascade was essential for LPA induction of TNF-α transcription.
LPA-driven TNF-α production relies on the TNF-α-converting enzyme ADAM17
The human TNF-α protein is expressed as a 233 amino acid-long, 27-kDa precursor. It is proteolytically cleaved to a 157 amino acid long, 17-kDa mature protein by ADAM17, the TNF-α-converting enzyme (TACE) [47, 48]. LPA treatment induced expression of ADAM17 in Caov-3 and SKOV-3 but not in OVCAR-3 (Fig. 3A). The LPA-induced increase in ADAM17 protein in Caov-3 and SKOV-3 cells was independent of the NF-κB transcription factor as LPA did not upregulate ADAM17 mRNA in SKOV-3 or only slightly increased ADAM17 mRNA in Caov-3 (~1.5 fold), which was insensitive to the NF-κB inhibitor BAY11–7085 (Supplementary Fig. S4). To determine if LPA induced ADAM17 activation, we measured the activity of ADAM17 in live cells using a fluorogenic TACE substrate. As shown in Fig. 3B, treatment with LPA increased the cleavage of the ADAM17 substrate in all three cell lines, which was prevented by the ADAM17 inhibitor, TMI-1 [49], indicating involvement of additional mechanism(s) more than the increase in protein abundance for ADAM17 activation.
Figure 3. LPA-driven TNF-α release relies on the activity of ADAM17.
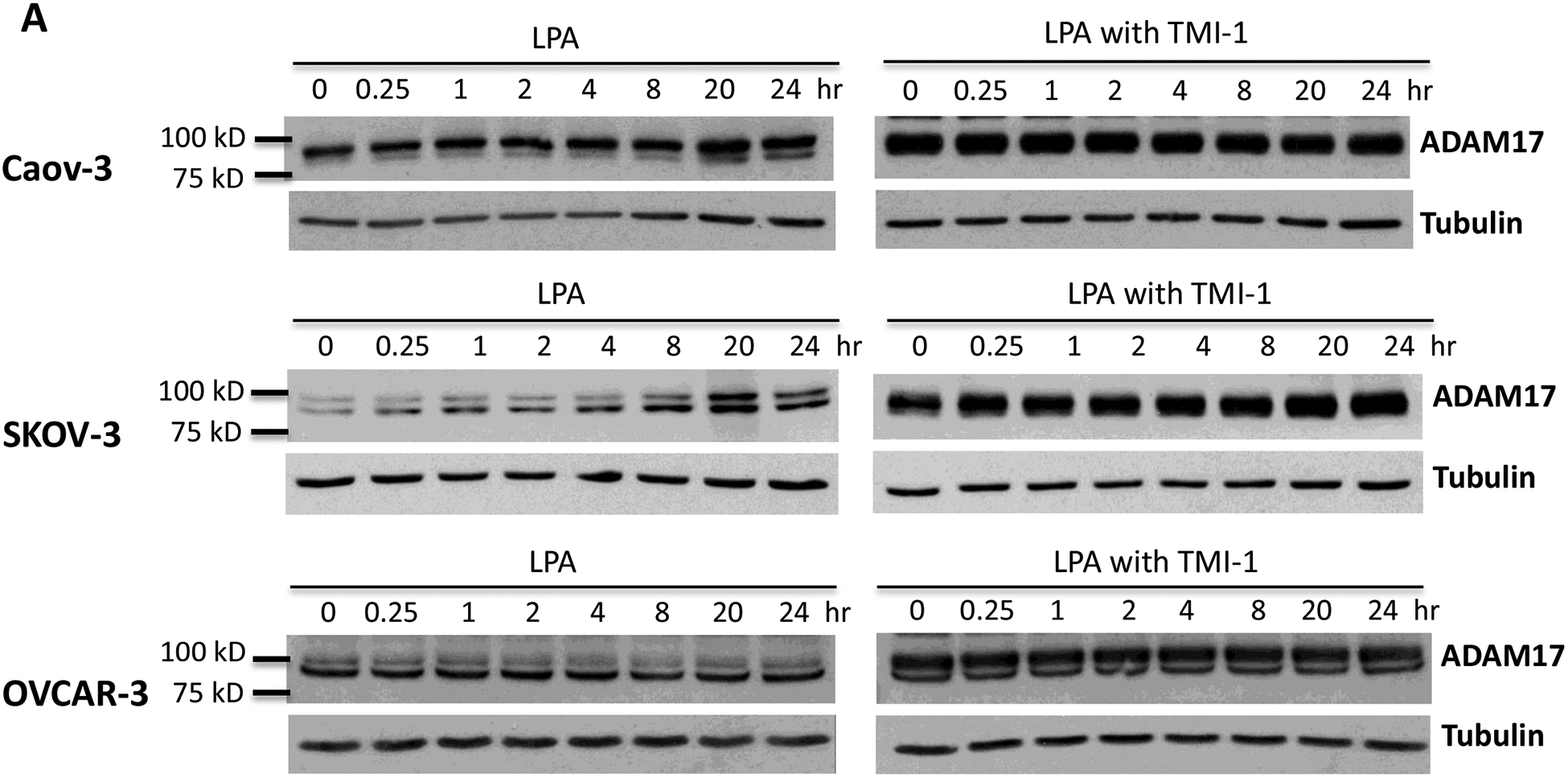
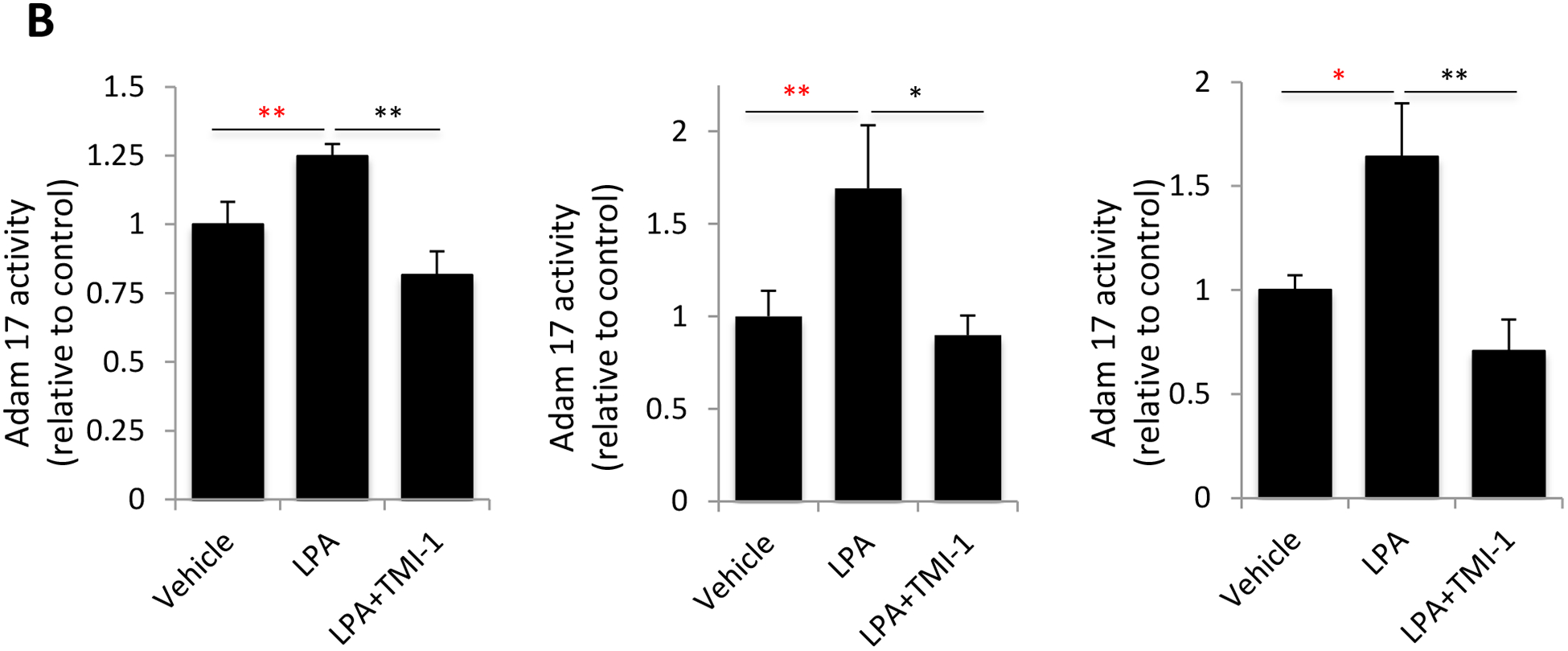
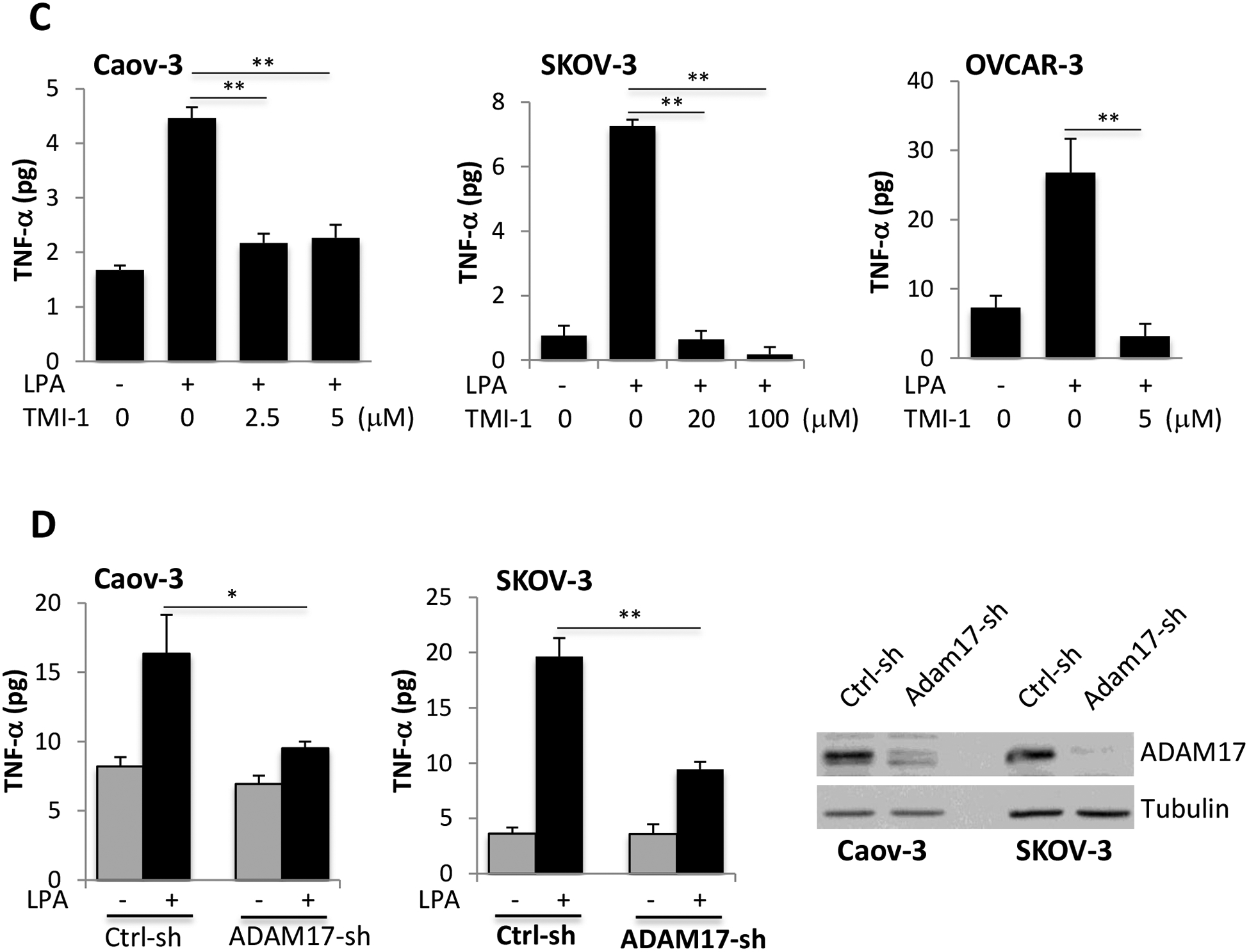
A. Caov-3, SKOV-3 and OVCAR-3 cells were treated with 10 μM LPA for the indicated periods of time with or without TMI-1. TMI-1 was added at 2.5, 20 and 5 μM to Caov-3, SKOV-3 and OVCAR-3, respectively, 1 hour before LPA treatment. Expression of ADAM17 was assessed by immunoblotting analysis. B. Ovarian cancer cell lines in 24-well plated were serum-starved and treated with LPA (10 μM) with or without MTI-1 (5 μM for Caov-3 or OVCAR-3 and 20 μM for SKOV-3) for 30 minutes before addition of the quenched fluorogenic substrate for measurement at 37 °C for 1 hour as detailed in Materials and Methods. The results were presented as fold changes in activity relative to the un-stimulated control cells (defined as 1 fold). C. The ovarian cancer cell lines in 12-well plates were stimulated with LPA in the presence of indicated concentrations of TMI-1. TNF-α levels in culture supernatants were quantified with ELISA and presented as in Fig. 1A. D. ADAM17 was silenced in Caov-3 and SKOV-3 cells by lentivirally-mediated shRNA to determine the effect of ADAM17 deficiency on LPA-induced TNF-α release. ADAM17 knockdown efficiency was confirmed by immunoblotting analysis.
To determine whether ADAM17 was involved in LPA-mediated TNF-α shedding, we treated ovarian cancer cell lines with TMI-1. Intriguingly, TMI-1 treatment led to a quick and sustained elevation of ADAM17 protein likely as a compensatory response to TMI-1 targeting of the enzymatic activity (Fig. 3A). The strong induction of ADAM17 by TMI-1 was more obvious when lysates from vehicle- and TMI-1-treated cells were run on the same gel (Supplementary Fig. S5). Consistent with the effect on its target protein, TMI-1 effectively prevented LPA-induced accumulation of TNF-α present in culture supernatants no matter whether it was added 1 hour before LPA (Fig. 3C) or simultaneously with LPA (Supplementary Fig. S6).
To further confirm the specific role of ADAM17 in LPA-dependent production of TNF-α, ADAM17 expression was silenced with lentivirally-mediated ADAM17-shRNA in Caov-3 and SKOV-3 cells. In line with the effect of TMI-1, the downregulation of ADAM17 significantly decreased TNF-α levels in culture supernatants of LPA-treated cells (Fig. 3D).
LPA transactivation of EGFR is required for TNF-α protein production
Many biological functions of LPA depend on a permissive signal from transactivation of EGFR although the mechanism underlying such a requirement is not clear [50]. We next examined whether LPA-induced TNF-α also depended on the trans activity of EGFR. When cellular EGFR was repressed by the EGFR inhibitor AG1478 [51], LPA induction of TNF-α protein, but not TNF-α mRNA, was dramatically reduced as shown in Fig. 4. In fact, the abundances of LPA-induced TNF-α mRNA were modestly but significantly increased in the presence of AG1478 for an unknown mechanism (Fig. 4). The results suggested a specific role for EGFR transactivation in post-transcriptional regulation of TNF-α protein. The lack of an inhibitory effect on TNF-α mRNA was consistent with the primary role of NF-κB in transcriptional activation of TNF-α as we previously showed that LPA-induced NF-κB activity was independent of EGFR transactivation [52].
Figure 4. EGFR transactivation is required for LPA induction of TNF-α protein but not TNF-α mRNA expression.
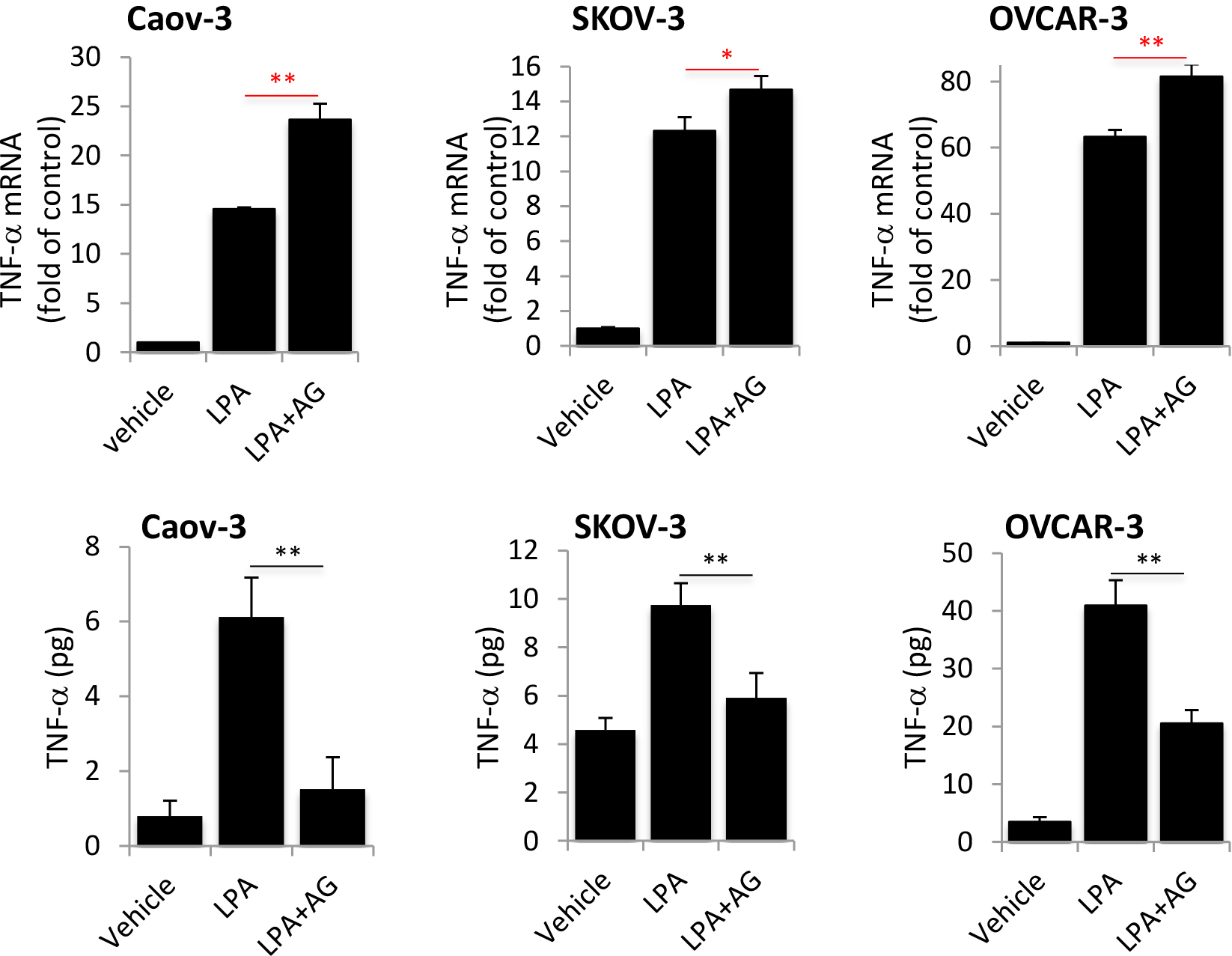
A. Caov-3, SKOV-3 and OVCAR-3 cells were treated with LPA as in Fig. 1A in the presence or absence of the EGFR inhibitor AG1478 (1 μM). TNF-α mRNA was analyzed by RT-qPCR to determine the effects of EGFR blocking on LPA-induced TNF-α mRNA expression. B. The effects of AG1478 on LPA-induced TNF-α protein present in culture supernatants were assessed and presented as in Fig. 1A.
ADAM17 mediates LPA transactivation of EGFR
In addition to processing TNF-α, ADAM17 has been also implicated in ectodomain shedding of other cytokines and growth factors including the EGFR ligands amphiregulin, heparin-binding EGF (HB-EGF) and transforming growth factor α (TGF-α) [53, 54]. We thus examined the possibility that ADAM17 might play a dual role in the cleavage of both TNF-α and EGFR ligands in LPA-treated cells. As shown in Fig. 5A, LPA induced immediate and sustained tyrosine phosphorylation of EGFR at Y1068 (as surrogate of EGFR activation) in ovarian cancer cell lines. The phosphorylation was highly sensitive to the ADAM17 inhibitor TMI-1 (Fig. 5A). As further molecular evidence for EGFR transactivation in an ADAM17-dependent manner, lentivirally-mediated knockdown of ADAM17 also attenuated the EGFR tyrosine phosphorylation in response to LPA in Caov-3 and SKOV-3 cells (Fig. 5B). It should be pointed out that effect of LPA on EGFR phosphorylation appeared to be biphasic with an early response at 15 minutes and a delayed one peaking between 8–20 hours. The late response was less sensitive to TMI-1 or shRNA knockdown. This could be due to incomplete inactivation of ADAM17 or adaptive activation of another metalloprotease. Taken together, the results indicated that in LPA-treated conditions, ADAM17 exerted dual effects on EGFR transactivation and TNF-α processing, which converge to turn on TNF-α protein yield.
Figure 5. ADAM17 is also the metalloproteinase mediating EGFR transactivation in LPA-treated cells.
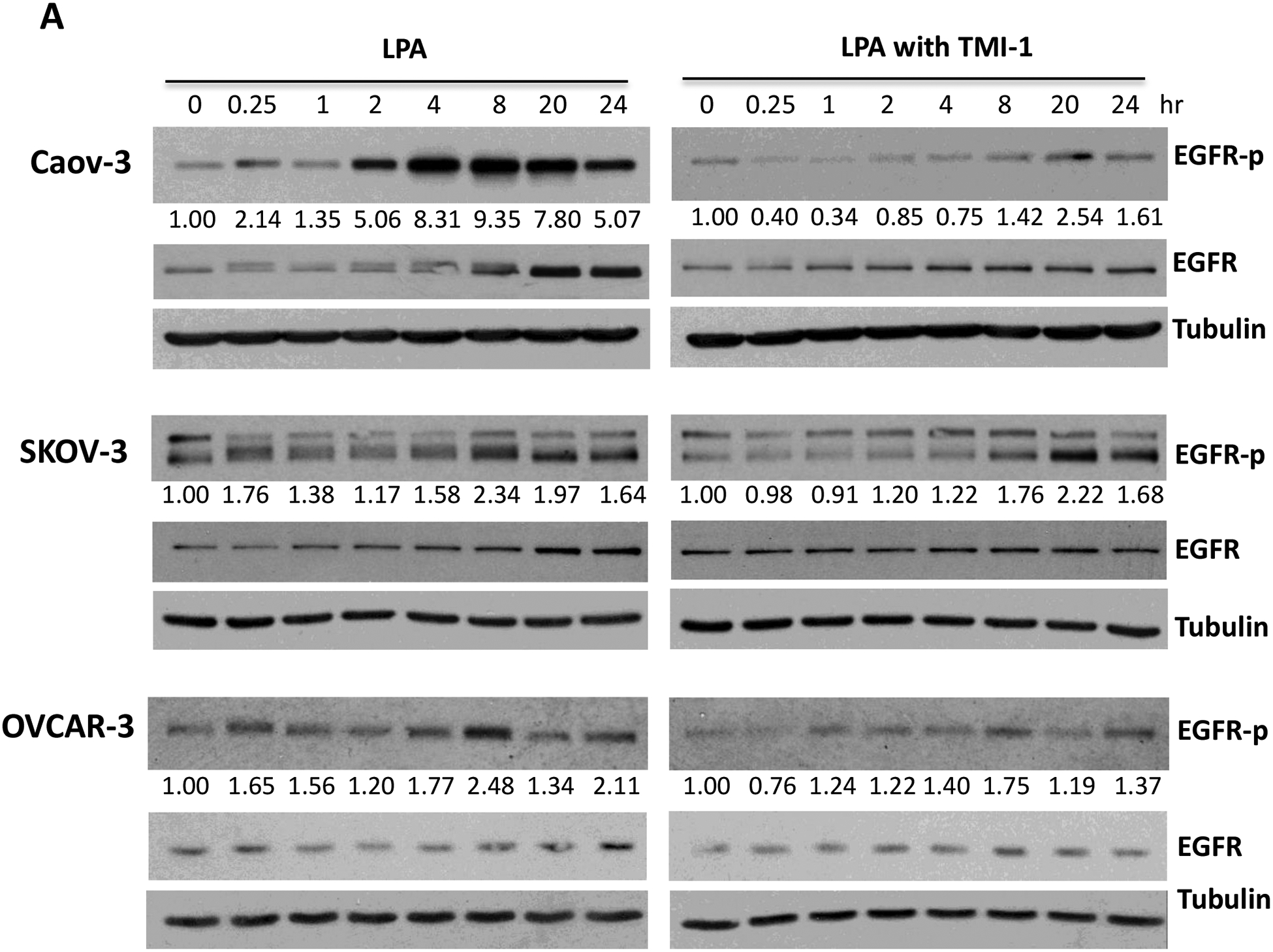
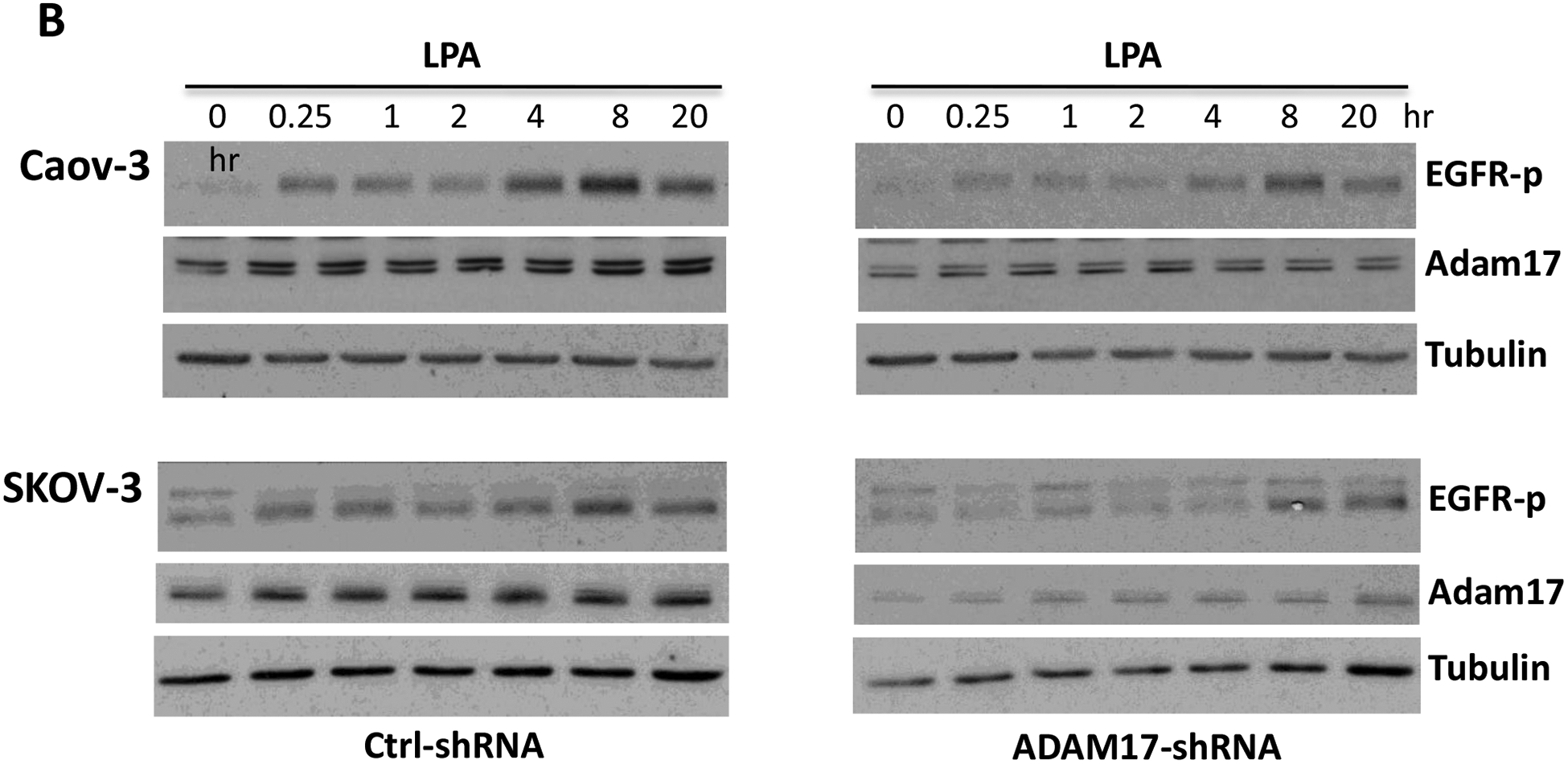
A. Caov-3, SKOV-3 and OVCAR-3 cells were treated with 10 μM LPA for the indicated periods of time in the presence or absence of the ADAM17 inhibitor TMI-1 (5 μM for Caov-3 or OVCAR-3, and 20 μM for SKOV-3) as described in Fig. 3C. EGFR tyrosine phosphorylation at Y1068, total EGFR and tubulin were examined by immunoblotting. Levels of phospho-EGFR (EGFR-p) were quantified by densitometry, normalized to tubulin and presented as fold changes relative to control cells. B. The effects of ADAM17-sh RNA knockdown on the LPA-induced EGFR phosphorylation were assessed as in A.
TNF-α reinforces LPA-elicited IL-8, IL-6 and CXCL1 production
TNF-α plays a pivotal role in orchestrating the production of a variety of pro-inflammatory cytokines including IL-8, IL-6 and IL-1 [55]. Therefore, TNF-α is considered to be a master regulator of pro-inflammatory cytokine networks. We and others have reported that LPA induced expression of IL-8, IL-6, and CXCL1 in ovarian and other types of cancer cells [28, 30, 34, 56]. We next assessed whether LPA-induced TNF-α provided a boost to the effects of LPA on the production of these cytokines and chemokines in ovarian cancer cell lines. We took advantage of two molecular tools to block TNF-α activity: TNF-α neutralizing antibody (TNF-α Ab) and recombinant TNF-α soluble receptor (TNF-α SR). Both approaches have been approved by the FDA for treatment of arthritis and other TNF-α-driven medical conditions [57]. Incubation of Caov-3, SKOV-3, and OVCAR-3 cells with one of the TNF-α-blocking agents decreased cellular production of IL-8 (Fig. 6A), IL-6 (Fig. 6B) and CXCL1 (Fig. 6D).
Figure 6. LPA-induced TNF-α is functionally sufficient to reinforce IL-8, IL-6 and CXCL1 production.
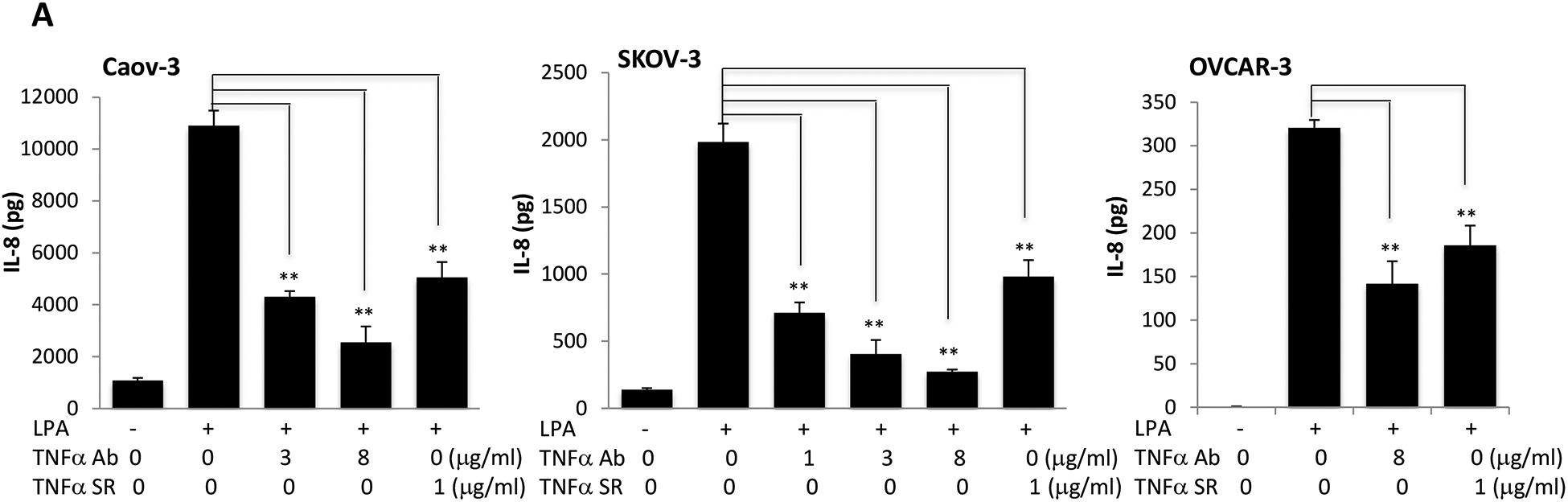
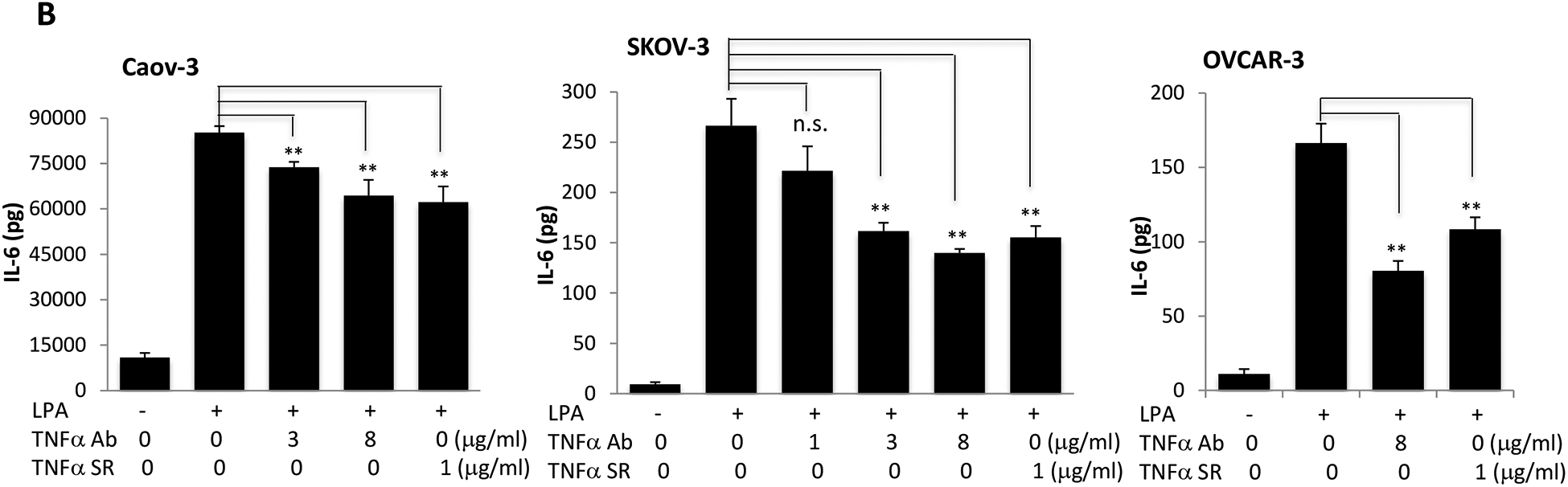
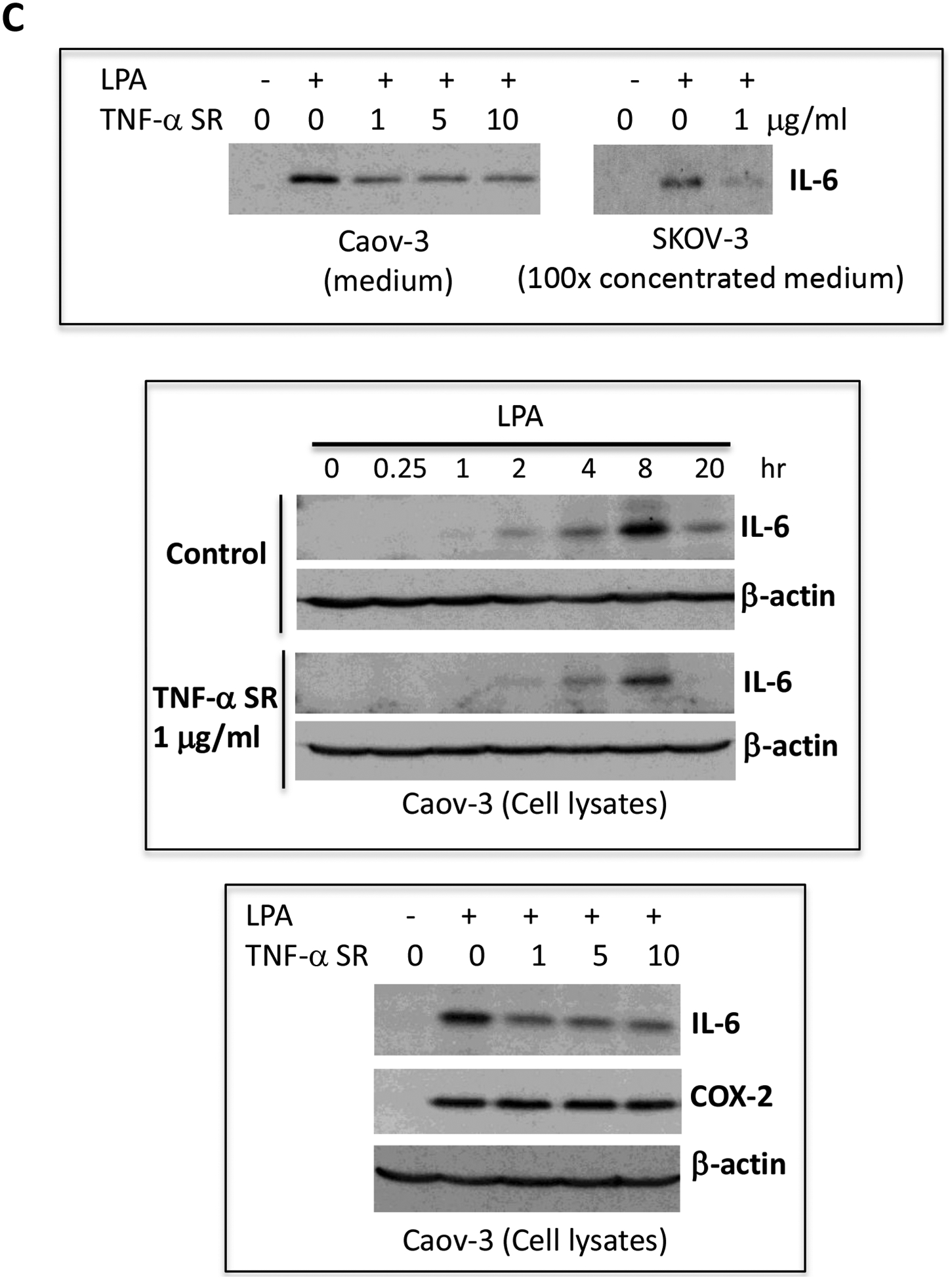
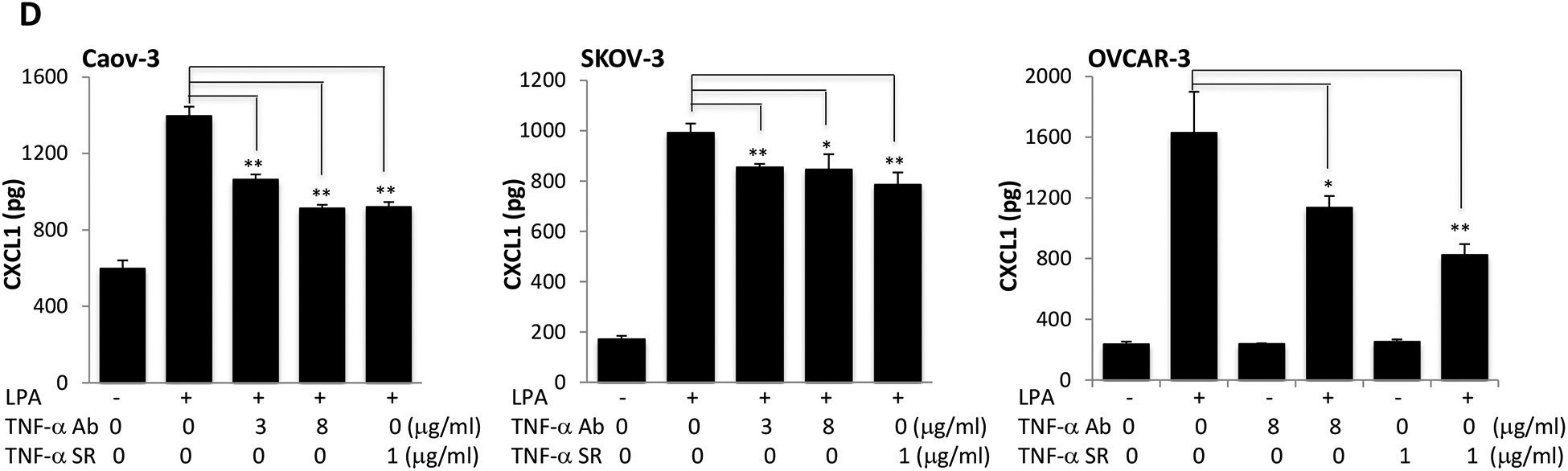
Caov-3, SKOV-3 and OVCAR-3 cells were treated for 24 hours with LPA as in Fig. 1 in the presence of vehicle, TNF-α Ab or TNF-α SR at the indicated concentrations. These TNF-α blocking agents were added simultaneously with LPA. Concentrations of IL-8 (A), IL-6 (B), CXCL1 (D) in culture supernatants were measured with ELISA and the results presented in the same way as TNF-α described in Fig. 1A. C. IL-6 protein in culture supernatants of Caov-3 cells or in 100X concentrated supernatants of SKOV-3 cells were assessed by immunoblotting analysis (upper). The time-dependent induction of intracellular IL-6 in the presence or absence of TNF-α SR was examined by immunoblotting analysis of cellular lysates (middle). The specificity of TNF-α SR inhibition of IL-6 was verified by lack of an inhibitory effect on cellular COX-2, another LPA-induced gene (lower).
In general, the inhibitory effect of TNF-α Ab and TNF-α SR on IL-6 production was less dramatic than that on IL-8, particularly in Caov-3 cells. This might be related to the overproduction of IL-6 in Caov-3 cells. The LPA-induced IL-6 protein in Caov-3 was 100–1,000 fold higher than in other cell lines. It was readily detectable in unconcentrated culture supernatants or cell lysates with direct immunoblotting (Fig. 6C). We confirmed the inhibitory effect of TNF-α SR on LPA-induced IL-6 protein present in both cell lysates and culture supernatants (Fig. 6C). These results together revealed a functional role of induced TNF-α in boosting LPA-mediated production of IL-8, IL-6 and CXCL1. In contrast to IL-8, IL-6 and CXCL1, TNF-α SR did not interfere with LPA-induced expression of COX-2 in Caov-3 cells (Fig. 6C).
TNF-α is not involved in LPA-driven cell proliferation
TNF-α inhibits or stimulates cell proliferation depending on doses or the context of the target cells. Low concentrations of TNF-α have been shown to promote proliferation while high doses could trigger apoptotic cell death in ovarian cancer cells [18, 58]. We examined the possibility that LPA-induced TNF-α might promote or inhibit LPA-dependent cell proliferation. Ovarian cancer cell lines were treated with LPA for over 30 hours in the presence or absence of the TNF-α Ab or TNF-α SR at doses that effectively repressed LPA-induced IL-8, IL-6 and CXCL1 production. As shown in Supplementary Fig. S7, LPA treatment increased cell numbers in Caov-3 and SKOV-3 but not in OVCAR-3 cells. The proliferative effect of LPA in these cells was not influenced by the presence of TNF-α Ab or TNF-α SR.
DISCUSSION
Epidemiological and clinical studies support a role of chronic inflammation in tumor initiation and progression [59, 60]. The concept is particularly relevant to ovarian oncogenesis. Ovulation, a known risk factor of ovarian cancer, is an inflammatory process involving localized elevations in cytokines and pro-inflammatory eicosanoids, wound healing and tissue repairing [61]. Elevated serum levels of TNF-α, IL-6 and C-reactive protein (CRP) are predictive of higher risk for development of ovarian cancer [62–67]. Many cytokines and chemokines including TNF-α, IL-6, IL-8, and CXCL1 are significantly increased in serum, ascites or tumor samples of ovarian cancer patients [7–10, 30]. Biological studies have shown that both ovarian tumor cells and tumor-infiltrating immune cells contribute to elevated levels of TNF-α and other inflammatory mediators in ovarian tumors [1, 2, 6].
As a primary pro-inflammatory factor, TNF-α acts as an endogenous promoter of a variety of oncogenic processes such as cellular transformation, survival, proliferation, invasion, angiogenesis, and metastasis [1, 11, 12, 14–17]. However, the molecular switch to turn on the TNF-α gene in tumor cells remains poorly defined. Few previous investigations have explored the communications between LPA and TNF-α, two extracellular mediators co-present in blood and malignant effusions. The present study is the first to show that LPA is a potent driver of the TNF-α gene transcription and protein release in ovarian tumor cells. Our study reveals a multitude of mechanisms underlying LPA-driven TNF-α and the biological significance of TNF-α induction as summarized in Fig. 7. LPA induces transcriptional upregulation of the TNF-α gene via NF-κB and activation of ADAM17 likely through LPA2. ADAM17 mediates the ectodomain shedding of TNF-α protein as well as EGF-related ligands to transactivate EGFR.
Figure 7.
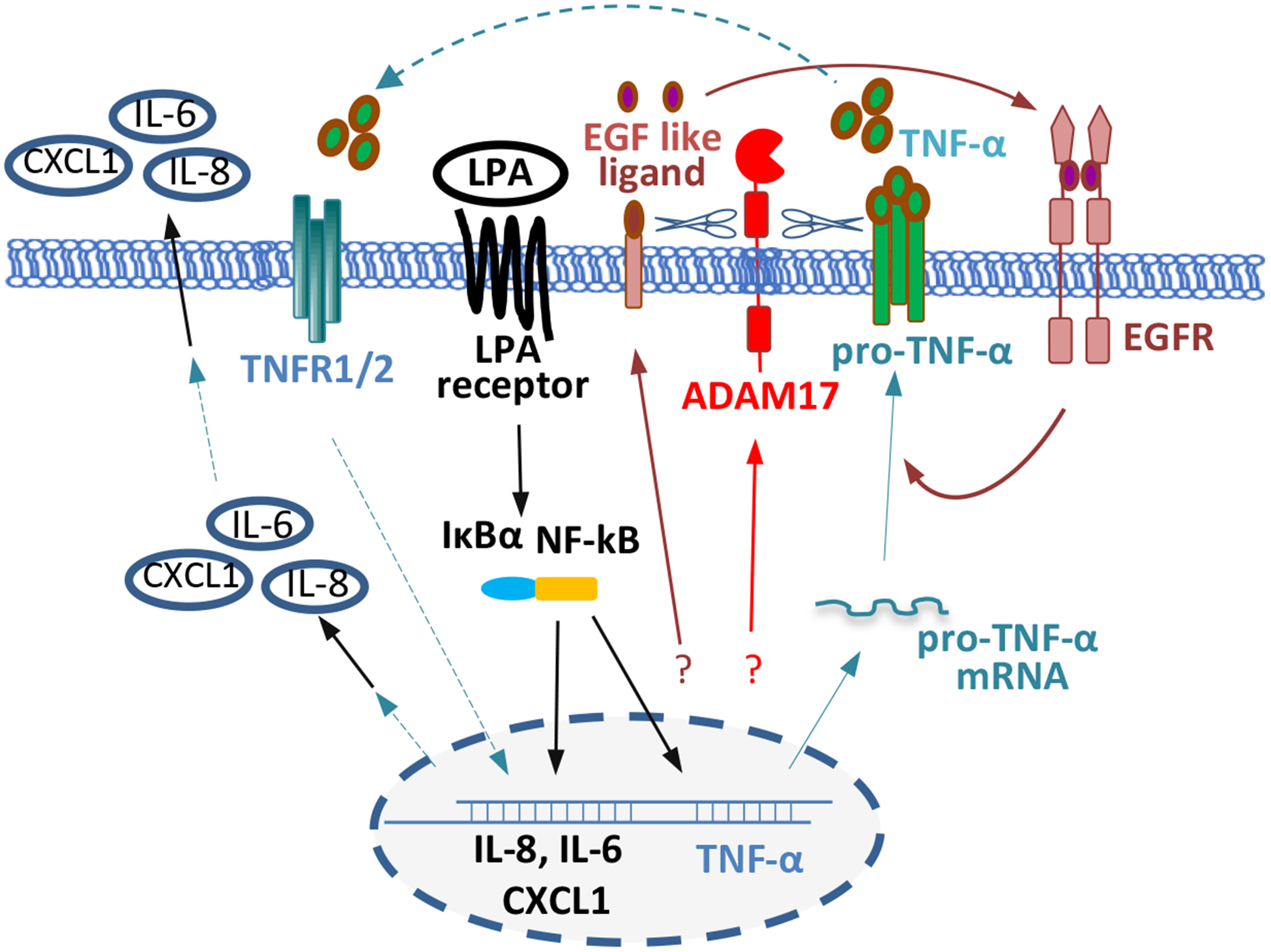
Summary of the mechanistic players to induce TNF-α and the connection of TNF-α to other pro-inflammatory mediators in LPA-treated ovarian cancer cells.
It is well known that LPA elicits transactivation of EGFR and many biological functions of LPA depend on transactivated EGFR [54, 68–73] (also see Supplementary Fig. S8). We herein demonstrated that LPA-induced TNF-α production also required EGFR transactivation, which seemed to promote post-transcriptional generation of TNF-α protein. In contrast, EGFR trans activity was not required for induction of TNF-α mRNA that was mediated by NF-κB. We have previously shown that LPA-induced NF-κB lies downstream of Gq, independent of EFGR trans activity [31]. However, EGFR trans activity was required for Gi-elicited cellular response. The differential requirements suggest that a Gi-linked signal is necessary for TNF-α protein release. This speculation is also consistent with the observation that PTX blocked TNF-α protein production (Figure 2C).
The crosstalk between EGFR and agonists of GPCRs has been a subject of extensive research. While some investigations suggest a ligand-independent route involving activation of cytoplasmic tyrosine kinases such as Src [68, 74], more studies implicate the release of EGFR ligands to activate EGFR through an auto- or paracrine loop [54, 69–73, 75]. Diverse EGFR ligands such as amphiregulin, HB-EGF, and TNF-α have been reported to be released upon GPCR activation [54, 72]. Several members of the metalloprotease family including ADAM10, ADAM15, and ADAM17 are potentially involved in the cleavage of these EGFR ligands, depending on the cellular context and nature of GPCR agonists [54].
In LPA-treated squamous cell carcinoma cells, Gschwind et al showed clearly that ADAM17 mediated the shedding of amphiregulin to transactivate EGFR [70]. In ovarian cancer cells, LPA induced amphiregulin mRNA and protein secretion through the Hippo-YAP signaling pathway [73]. In another analysis in ovarian cancer cells, LPA was capable of inducing ectodomain shedding of ectopically transfected pro-HB-EGF by an unidentified metalloprotease [71]. A follow-up study demonstrated correlative overexpression of ADAM17 protein and HB-EGF in ovarian carcinoma tissues but not in normal ovarian epithelium [76]. Although these observations implied an intermediate role of endogenously expressed ADAM17 in shedding of HB-EGF, a direct link has not been established in ovarian cancer cells. In the present study, we used both pharmacological and molecular approaches to probing the role of ADAM17 in the process. TMI-1 potently and selectively inhibited ADAM17 as reflected by the compensatory induction of its target ADAM17. LPA-induced EGFR transactivation was highly sensitive to pretreatment of ovarian cancer cells with TMI-1. Similarly, silencing of ADAM17 with shRNA attenuated EGFR trans activity. These results altogether provide definitive evidence for an essential role of ADAM17 in LPA-mediated EGFR transactivation in ovarian cancer cells.
Due to the central role of TNF-α in the regulation of other pro-inflammatory cytokines, we asked whether LPA-induced TNF-α might reach biologically relevant levels to boost LPA-initiated production of other cytokines downstream of TNF-α. If so, LPA-induced TNF-α could amplify its pro-inflammatory activity by upregulating a cytokine and chemokine network. We and others have previously shown that LPA induced expression of IL-8, IL-6 and CXCL1 in ovarian cancer cells [28, 30, 34, 56]. These effects of LPA were apparently initiated by LPA per se as LPA induced early transcriptional activation of these genes before possible accumulation of TNF-α [28, 30, 34, 56]. However, when induced TNF-α reached a physiologically functional level, it could provide a second wave of stimulation to boost production of these inflammatory mediators. Indeed, our analyses using two specific TNF-α-blocking agents decreased the contents of IL-8, IL-6, and CXCL1 in culture supernatants of LPA-treated cells, confirming a significant contribution from the induced TNF-α to the robust and sustained levels of IL-8, IL-6 and CXCL1 in ovarian cancer cells. The results were also consistent with the prior observation of very high levels of these cytokines present in conditioned medium of LPA-treated cells [28, 30, 56, 77].
In sum, we have shown a novel function of LPA to induce TNF-α in ovarian cancer cells. The process relies on both NF-κB-mediated transcriptional activation of TNF-α gene and ADAM17-dependent shedding of TNF-α protein. We demonstrate that ADAM17 is also responsible for LPA-induced EGFR transactivation, which is involved in the post-transcriptional regulation of TNF-α. Finally, LPA-induced TNF-α is functionally significant as it boosts expression of other cytokines and chemokines to amplify the pro-inflammatory activity in ovarian cancer cells and potentially in the tumor milieu.
Supplementary Material
ACKNOWLEDGMENTS
The work was supported in part by NIH/NCI R01 CA229812, Astar Biotech Research Award, Massey Cancer Center Pilot Research Award, and the NIH/NCI grant P30 CA16059 to MCC of VCU School of Medicine. The authors have no conflict of interest to declare.
Abbreviations:
- LPA
lysophosphatidic acid
- TNF-α
tumor necrosis factor alpha
- GPCR
G protein-coupled receptor
- IL-1β
interleukin-1β
- IL-8
interleukin-8
- IL-6
interleukin-6
- CXCL1
C-X-C motif chemokine ligand 1
- EGF
epidermal growth factor
- EGFR
EGF receptor
- NF-κB
nuclear factor kappa B
Footnotes
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this article.
REFERENCES CITED
- [1].Szlosarek PW, Grimshaw MJ, Kulbe H, Wilson JL, Wilbanks GD, Burke F, Balkwill FR, Expression and regulation of tumor necrosis factor alpha in normal and malignant ovarian epithelium, Mol Cancer Ther, 5 (2006) 382–390. [DOI] [PubMed] [Google Scholar]
- [2].Kulbe H, Chakravarty P, Leinster DA, Charles KA, Kwong J, Thompson RG, Coward JI, Schioppa T, Robinson SC, Gallagher WM, Galletta L, Australian G Ovarian Cancer Study, M.A. Salako, J.F. Smyth, T. Hagemann, D.J. Brennan, D.D. Bowtell, F.R. Balkwill, A dynamic inflammatory cytokine network in the human ovarian cancer microenvironment, Cancer Res, 72 (2012) 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Piura B, Medina L, Rabinovich A, Dyomin V, Levy RS, Huleihel M, Distinct expression and localization of TNF system in ovarian carcinoma tissues: possible involvement of TNF-alpha in morphological changes of ovarian cancerous cells, Anticancer Res, 34 (2014) 745–752. [PubMed] [Google Scholar]
- [4].Naylor MS, Stamp GW, Foulkes WD, Eccles D, Balkwill FR, Tumor necrosis factor and its receptors in human ovarian cancer. Potential role in disease progression, J Clin Invest, 91 (1993) 2194–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vitonis AF, Titus-Ernstoff L, Cramer DW, Assessing ovarian cancer risk when considering elective oophorectomy at the time of hysterectomy, Obstet Gynecol, 117 (2011) 1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gupta M, Babic A, Beck AH, Terry K, TNF-alpha expression, risk factors, and inflammatory exposures in ovarian cancer: evidence for an inflammatory pathway of ovarian carcinogenesis?, Hum Pathol, 54 (2016) 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Moradi MM, Carson LF, Weinberg B, Haney AF, Twiggs LB, Ramakrishnan S, Serum and ascitic fluid levels of interleukin-1, interleukin-6, and tumor necrosis factor-alpha in patients with ovarian epithelial cancer, Cancer, 72 (1993) 2433–2440. [DOI] [PubMed] [Google Scholar]
- [8].Gadducci A, Ferdeghini M, Castellani C, Annicchiarico C, Gagetti O, Prontera C, Bianchi R, Facchini V, Serum levels of tumor necrosis factor (TNF), soluble receptors for TNF (55- and 75-kDa sTNFr), and soluble CD14 (sCD14) in epithelial ovarian cancer, Gynecol Oncol, 58 (1995) 184–188. [DOI] [PubMed] [Google Scholar]
- [9].Sipak-Szmigiel O, Wlodarski P, Ronin-Walknowska E, Niedzielski A, Karakiewicz B, Sluczanowska-Glabowska S, Laszczynska M, Malinowski W, Serum and peritoneal fluid concentrations of soluble human leukocyte antigen, tumor necrosis factor alpha and interleukin 10 in patients with selected ovarian pathologies, J Ovarian Res, 10 (2017) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kampan NC, Madondo MT, McNally OM, Stephens AN, Quinn MA, Plebanski M, Interleukin 6 Present in Inflammatory Ascites from Advanced Epithelial Ovarian Cancer Patients Promotes Tumor Necrosis Factor Receptor 2-Expressing Regulatory T Cells, Front Immunol, 8 (2017) 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, Holdsworth H, Turner L, Rollins B, Pasparakis M, Kollias G, Balkwill F, Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis, Nat Med, 5 (1999) 828–831. [DOI] [PubMed] [Google Scholar]
- [12].Balkwill F, TNF-alpha in promotion and progression of cancer, Cancer Metastasis Rev, 25 (2006) 409–416. [DOI] [PubMed] [Google Scholar]
- [13].Szlosarek P, Charles KA, Balkwill FR, Tumour necrosis factor-alpha as a tumour promoter, Eur J Cancer, 42 (2006) 745–750. [DOI] [PubMed] [Google Scholar]
- [14].Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, Gould D, Ayhan A, Balkwill F, The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells, Cancer Res, 67 (2007) 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sethi G, Sung B, Aggarwal BB, TNF: a master switch for inflammation to cancer, Front Biosci, 13 (2008) 5094–5107. [DOI] [PubMed] [Google Scholar]
- [16].Baluk P, Yao LC, Feng J, Romano T, Jung SS, Schreiter JL, Yan L, Shealy DJ, McDonald DM, TNF-alpha drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice, J Clin Invest, 119 (2009) 2954–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hagemann T, Robinson SC, Schulz M, Trumper L, Balkwill FR, Binder C, Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-alpha dependent up-regulation of matrix metalloproteases, Carcinogenesis, 25 (2004) 1543–1549. [DOI] [PubMed] [Google Scholar]
- [18].Wu S, Boyer CM, Whitaker RS, Berchuck A, Wiener JR, Weinberg JB, Bast RC Jr., Tumor necrosis factor alpha as an autocrine and paracrine growth factor for ovarian cancer: monokine induction of tumor cell proliferation and tumor necrosis factor alpha expression, Cancer Res, 53 (1993) 1939–1944. [PubMed] [Google Scholar]
- [19].Xu Y, Gaudette DC, Boynton JD, Frankel A, Fang XJ, Sharma A, Hurteau J, Casey G, Goodbody A, Mellors A, et al. , Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients, Clin Cancer Res, 1 (1995) 1223–1232. [PubMed] [Google Scholar]
- [20].Westermann AM, Havik E, Postma FR, Beijnen JH, Dalesio O, Moolenaar WH, Rodenhuis S, Malignant effusions contain lysophosphatidic acid (LPA)-like activity, Ann Oncol, 9 (1998) 437–442. [DOI] [PubMed] [Google Scholar]
- [21].Xiao YJ, Schwartz B, Washington M, Kennedy A, Webster K, Belinson J, Xu Y, Electrospray ionization mass spectrometry analysis of lysophospholipids in human ascitic fluids: comparison of the lysophospholipid contents in malignant vs nonmalignant ascitic fluids, Anal Biochem, 290 (2001) 302–313. [DOI] [PubMed] [Google Scholar]
- [22].Fang X, Schummer M, Mao M, Yu S, Tabassam FH, Swaby R, Hasegawa Y, Tanyi JL, LaPushin R, Eder A, Jaffe R, Erickson J, Mills GB, Lysophosphatidic acid is a bioactive mediator in ovarian cancer, Biochim Biophys Acta, 1582 (2002) 257–264. [DOI] [PubMed] [Google Scholar]
- [23].Mills GB, Moolenaar WH, The emerging role of lysophosphatidic acid in cancer, Nat Rev Cancer, 3 (2003) 582–591. [DOI] [PubMed] [Google Scholar]
- [24].Lee SC, Fujiwara Y, Tigyi GJ, Uncovering unique roles of LPA receptors in the tumor microenvironment, Receptors Clin Investig, 2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yun CC, Lysophosphatidic Acid and Autotaxin-associated Effects on the Initiation and Progression of Colorectal Cancer, Cancers (Basel), 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pustilnik TB, Estrella V, Wiener JR, Mao M, Eder A, Watt MA, Bast RC Jr., Mills GB, Lysophosphatidic acid induces urokinase secretion by ovarian cancer cells, Clin Cancer Res, 5 (1999) 3704–3710. [PubMed] [Google Scholar]
- [27].Hu YL, Tee MK, Goetzl EJ, Auersperg N, Mills GB, Ferrara N, Jaffe RB, Lysophosphatidic acid induction of vascular endothelial growth factor expression in human ovarian cancer cells, J Natl Cancer Inst, 93 (2001) 762–768. [DOI] [PubMed] [Google Scholar]
- [28].Fang X, Yu S, Bast RC, Liu S, Xu HJ, Hu SX, LaPushin R, Claret FX, Aggarwal BB, Lu Y, Mills GB, Mechanisms for lysophosphatidic acid-induced cytokine production in ovarian cancer cells, J Biol Chem, 279 (2004) 9653–9661. [DOI] [PubMed] [Google Scholar]
- [29].Symowicz J, Adley BP, Woo MM, Auersperg N, Hudson LG, Stack MS, Cyclooxygenase-2 functions as a downstream mediator of lysophosphatidic acid to promote aggressive behavior in ovarian carcinoma cells, Cancer Res, 65 (2005) 2234–2242. [DOI] [PubMed] [Google Scholar]
- [30].Lee Z, Swaby RF, Liang Y, Yu S, Liu S, Lu KH, Bast RC Jr., Mills GB, Fang X, Lysophosphatidic acid is a major regulator of growth-regulated oncogene alpha in ovarian cancer, Cancer Res, 66 (2006) 2740–2748. [DOI] [PubMed] [Google Scholar]
- [31].Oyesanya RA, Lee ZP, Wu J, Chen J, Song Y, Mukherjee A, Dent P, Kordula T, Zhou H, Fang X, Transcriptional and post-transcriptional mechanisms for lysophosphatidic acid-induced cyclooxygenase-2 expression in ovarian cancer cells, FASEB J, 22 (2008) 2639–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Song Y, Wu J, Oyesanya RA, Lee Z, Mukherjee A, Fang X, Sp-1 and c-Myc mediate lysophosphatidic acid-induced expression of vascular endothelial growth factor in ovarian cancer cells via a hypoxia-inducible factor-1-independent mechanism, Clin Cancer Res, 15 (2009) 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mukherjee A, Ma Y, Yuan F, Gong Y, Fang Z, Mohamed EM, Berrios E, Shao H, Fang X, Lysophosphatidic Acid Up-Regulates Hexokinase II and Glycolysis to Promote Proliferation of Ovarian Cancer Cells, Neoplasia, 17 (2015) 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schwartz BM, Hong G, Morrison BH, Wu W, Baudhuin LM, Xiao YJ, Mok SC, Xu Y, Lysophospholipids increase interleukin-8 expression in ovarian cancer cells, Gynecol Oncol, 81 (2001) 291–300. [DOI] [PubMed] [Google Scholar]
- [35].Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, Zhang Y, Mazumdar A, Hilsenbeck SG, Mills GB, Brown PH, Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8, Cancer Res, 73 (2013) 3470–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kwon JH, Gaire BP, Park SJ, Shin DY, Choi JW, Identifying lysophosphatidic acid receptor subtype 1 (LPA1) as a novel factor to modulate microglial activation and their TNF-alpha production by activating ERK1/2, Biochim Biophys Acta Mol Cell Biol Lipids, 1863 (2018) 1237–1245. [DOI] [PubMed] [Google Scholar]
- [37].Bae GH, Lee SK, Kim HS, Lee M, Lee HY, Bae YS, Lysophosphatidic acid protects against acetaminophen-induced acute liver injury, Exp Mol Med, 49 (2017) e407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ma Y, Wang W, Idowu MO, Oh U, Wang XY, Temkin SM, Fang X, Ovarian cancer relies on glucose transporter 1 to fuel glycolysis and growth: Anti-tumor activity of BAY-876, Cancers (Basel), 11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cabron AS, El azzouzi K, Boss M, Arnold P, Schwarz J, Rosas M, Dobert JP, Pavlenko E, Schumacher N, Renne T, Taylor PR, Linder S, Rose-John S, Zunke F. Structural and functional analyses of the shedding protease ADAM17 in HoxB8-immortalized macrophages and dendritic-like cells. J Immunol, 201 (2018) 3106–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Luger TA, Schwarz T, Evidence for an epidermal cytokine network, J Invest Dermatol, 95 (1990) 100S–104S. [DOI] [PubMed] [Google Scholar]
- [41].Barnes PJ, The cytokine network in asthma and chronic obstructive pulmonary disease, J Clin Invest, 118 (2008) 3546–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schroder K, Tschopp J, The inflammasomes, Cell, 140 (2010) 821–832. [DOI] [PubMed] [Google Scholar]
- [43].Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV, Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages, J Exp Med, 171 (1990) 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu H, Sidiropoulos P, Song G, Pagliari LJ, Birrer MJ, Stein B, Anrather J, Pope RM, TNF-alpha gene expression in macrophages: regulation by NF-kappa B is independent of c-Jun or C/EBP beta, J Immunol, 164 (2000) 4277–4285. [DOI] [PubMed] [Google Scholar]
- [45].Li H, Ye X, Mahanivong C, Bian D, Chun J, Huang S, Signaling mechanisms responsible for lysophosphatidic acid-induced urokinase plasminogen activator expression in ovarian cancer cells, J Biol Chem, 280 (2005) 10564–10571. [DOI] [PubMed] [Google Scholar]
- [46].Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME, Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo, J Biol Chem, 272 (1997) 21096–21103. [DOI] [PubMed] [Google Scholar]
- [47].Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, Kohr WJ, Aggarwal BB, Goeddel DV, Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin, Nature, 312 (1984) 724–729. [DOI] [PubMed] [Google Scholar]
- [48].Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP, A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells, Nature, 385 (1997) 729–733. [DOI] [PubMed] [Google Scholar]
- [49].Zhang Y, Xu J, Levin J, Hegen M, Li G, Robertshaw H, Brennan F, Cummons T, Clarke D, Vansell N, Nickerson-Nutter C, Barone D, Mohler K, Black R, Skotnicki J, Gibbons J, Feldmann M, Frost P, Larsen G, Lin LL, Identification and characterization of 4-[[4-(2-butynyloxy)phenyl]sulfonyl]-N-hydroxy-2,2-dimethyl-(3S)thiomorpholinecar boxamide (TMI-1), a novel dual tumor necrosis factor-alpha-converting enzyme/matrix metalloprotease inhibitor for the treatment of rheumatoid arthritis, J Pharmacol Exp Ther, 309 (2004) 348–355. [DOI] [PubMed] [Google Scholar]
- [50].Wang Z, Transactivation of Epidermal Growth Factor Receptor by G Protein-Coupled Receptors: Recent Progress, Challenges and Future Research, Int J Mol Sci, 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Levitzki A, Gazit A, Tyrosine kinase inhibition: an approach to drug development, Science, 267 (1995) 1782–1788. [DOI] [PubMed] [Google Scholar]
- [52].Oyesanya RA, Greenbaum S, Dang D, Lee Z, Mukherjee A, Wu J, Dent P, Fang X, Differential requirement of the epidermal growth factor receptor for G protein-mediated activation of transcription factors by lysophosphatidic acid, Mol Cancer, 9 (2010) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, Peschon JJ, Castner BJ, Gerhart MJ, Paxton RJ, Black RA, Lee DC, Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability, J Biol Chem, 277 (2002) 12838–12845. [DOI] [PubMed] [Google Scholar]
- [54].Schafer B, Gschwind A, Ullrich A, Multiple G-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion, Oncogene, 23 (2004) 991–999. [DOI] [PubMed] [Google Scholar]
- [55].Kany S, Vollrath JT, Relja B, Cytokines in Inflammatory Disease, Int J Mol Sci, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chou CH, Wei LH, Kuo ML, Huang YJ, Lai KP, Chen CA, Hsieh CY, Up-regulation of interleukin-6 in human ovarian cancer cell via a Gi/PI3K-Akt/NF-kappaB pathway by lysophosphatidic acid, an ovarian cancer-activating factor, Carcinogenesis, 26 (2005) 45–52. [DOI] [PubMed] [Google Scholar]
- [57].Steeland S, Libert C, Vandenbroucke RE, A New Venue of TNF Targeting, Int J Mol Sci, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Li B, Vincent A, Cates J, Brantley-Sieders DM, Polk DB, Young PP, Low levels of tumor necrosis factor alpha increase tumor growth by inducing an endothelial phenotype of monocytes recruited to the tumor site, Cancer Res, 69 (2009) 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ness RB, Cottreau C, Possible role of ovarian epithelial inflammation in ovarian cancer, J Natl Cancer Inst, 91 (1999) 1459–1467. [DOI] [PubMed] [Google Scholar]
- [60].Shalapour S, Karin M, Immunity, inflammation, and cancer: an eternal fight between good and evil, J Clin Invest, 125 (2015) 3347–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Espey LL, Current status of the hypothesis that mammalian ovulation is comparable to an inflammatory reaction, Biol Reprod, 50 (1994) 233–238. [DOI] [PubMed] [Google Scholar]
- [62].McSorley MA, Alberg AJ, Allen DS, Allen NE, Brinton LA, Dorgan JF, Pollak M, Tao Y, Helzlsouer KJ, C-reactive protein concentrations and subsequent ovarian cancer risk, Obstet Gynecol, 109 (2007) 933–941. [DOI] [PubMed] [Google Scholar]
- [63].Lundin E, Dossus L, Clendenen T, Krogh V, Grankvist K, Wulff M, Sieri S, Arslan AA, Lenner P, Berrino F, Hallmans G, Zeleniuch-Jacquotte A, Toniolo P, Lukanova A, C-reactive protein and ovarian cancer: a prospective study nested in three cohorts (Sweden, USA, Italy), Cancer Causes Control, 20 (2009) 1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Clendenen TV, Koenig KL, Arslan AA, Lukanova A, Berrino F, Gu Y, Hallmans G, Idahl A, Krogh V, Lokshin AE, Lundin E, Muti P, Marrangoni A, Nolen BM, Ohlson N, Shore RE, Sieri S, Zeleniuch-Jacquotte A, Factors associated with inflammation markers, a cross-sectional analysis, Cytokine, 56 (2011) 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Toriola AT, Grankvist K, Agborsangaya CB, Lukanova A, Lehtinen M, Surcel HM, Changes in pre-diagnostic serum C-reactive protein concentrations and ovarian cancer risk: a longitudinal study, Ann Oncol, 22 (2011) 1916–1921. [DOI] [PubMed] [Google Scholar]
- [66].Poole EM, Lee IM, Ridker PM, Buring JE, Hankinson SE, Tworoger SS, A prospective study of circulating C-reactive protein, interleukin-6, and tumor necrosis factor alpha receptor 2 levels and risk of ovarian cancer, Am J Epidemiol, 178 (2013) 1256–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Trabert B, Pinto L, Hartge P, Kemp T, Black A, Sherman ME, Brinton LA, Pfeiffer RM, Shiels MS, Chaturvedi AK, Hildesheim A, Wentzensen N, Pre-diagnostic serum levels of inflammation markers and risk of ovarian cancer in the prostate, lung, colorectal and ovarian cancer (PLCO) screening trial, Gynecol Oncol, 135 (2014) 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chen Q, Olashaw N, Wu J, Participation of reactive oxygen species in the lysophosphatidic acid-stimulated mitogen-activated protein kinase kinase activation pathway, J Biol Chem, 270 (1995) 28499–28502. [DOI] [PubMed] [Google Scholar]
- [69].Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A, EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF, Nature, 402 (1999) 884–888. [DOI] [PubMed] [Google Scholar]
- [70].Gschwind A, Hart S, Fischer OM, Ullrich A, TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells, EMBO J, 22 (2003) 2411–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Miyamoto S, Hirata M, Yamazaki A, Kageyama T, Hasuwa H, Mizushima H, Tanaka Y, Yagi H, Sonoda K, Kai M, Kanoh H, Nakano H, Mekada E, Heparin-binding EGF-like growth factor is a promising target for ovarian cancer therapy, Cancer Res, 64 (2004) 5720–5727. [DOI] [PubMed] [Google Scholar]
- [72].Rodland KD, Bollinger N, Ippolito D, Opresko LK, Coffey RJ, Zangar R, Wiley HS, Multiple mechanisms are responsible for transactivation of the epidermal growth factor receptor in mammary epithelial cells, J Biol Chem, 283 (2008) 31477–31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cai H, Xu Y, The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells, Cell Commun Signal, 11 (2013) 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Shah BH, Catt KJ, Calcium-independent activation of extracellularly regulated kinases 1 and 2 by angiotensin II in hepatic C9 cells: roles of protein kinase Cdelta, Src/proline-rich tyrosine kinase 2, and epidermal growth receptor trans-activation, Mol Pharmacol, 61 (2002) 343–351. [DOI] [PubMed] [Google Scholar]
- [75].Pierce KL, Tohgo A, Ahn S, Field ME, Luttrell LM, Lefkowitz RJ, Epidermal growth factor (EGF) receptor-dependent ERK activation by G protein-coupled receptors: a co-culture system for identifying intermediates upstream and downstream of heparin-binding EGF shedding, J Biol Chem, 276 (2001) 23155–23160. [DOI] [PubMed] [Google Scholar]
- [76].Tanaka Y, Miyamoto S, Suzuki SO, Oki E, Yagi H, Sonoda K, Yamazaki A, Mizushima H, Maehara Y, Mekada E, Nakano H, Clinical significance of heparin-binding epidermal growth factor-like growth factor and a disintegrin and metalloprotease 17 expression in human ovarian cancer, Clin Cancer Res, 11 (2005) 4783–4792. [DOI] [PubMed] [Google Scholar]
- [77].Boucharaba A, Serre CM, Gres S, Saulnier-Blache JS, Bordet JC, Guglielmi J, Clezardin P, Peyruchaud O, Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer, J Clin Invest, 114 (2004) 1714–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.