

Abstract
Clinical ophthalmologists consider each retinal disease as a completely unique entity. However, various retinal diseases, such as uveitis, age-related macular degeneration, diabetic retinopathy, and primary open-angle glaucoma, share a number of common pathogenetic pathways. Whether a retinal disease initiates from direct injury to the blood-retinal barrier (BRB) or a defect/injury to retinal neurons or glia that impairs the BRB secondarily, the BRB is a pivotal point in determining the prognosis as self-limiting and recovering, or developing and progressing to a clinical phenotype. The present review summarizes our current knowledge on the physiology and cellular and molecular pathology of the BRB, which underlies its pivotal role in the initiation and development of common retinal diseases.
Keywords: Blood-retinal barrier, Retinal inflammatory diseases, Age-related macular degeneration, Diabetic retinopathy, Primary open-angle glaucoma, Neuroinflammation
Introduction
The blood-retinal barrier (BRB) is the most critical component underlying retinal homeostasis. There are two sub-types of the BRB. The main structure of the inner BRB (iBRB) is a microvascular network that nourishes the inner retinal layers. The outer BRB (oBRB) lies between the choriocapillaris and the neurosensory retina and is maintained by tight junctions (TJs) between the retinal pigment epithelial (RPE) cells. Notably, the BRB is unique in maintaining the retina as an immune-privileged site by separating the retina from systemic immune and inflammatory components.[1] Physiological neuronal or glial injury/death within the retina during aging is generally self-limited when the BRB remains structurally and functionally intact. Minor injury to the BRB may occur during some of these physiological processes or very early disease stage. Timely repair of BRB injury with complete recovery of its structure and function results in no clinical consequences. Otherwise, immune and inflammatory components from the systemic circulation enter the retina through the impaired BRB and initiate a self-exacerbating vicious cycle of neuroimmune responses that lead to the development of clinical disease phenotypes.
BRB impairment is detected in numerous retinal diseases, such as glaucoma,[2] diabetic retinopathy (DR),[3] and age-related macular degeneration (AMD)[4], and retinal inflammatory diseases, such as uveitis,[5,6] central serous chorioretinopathy,[7] and retinal vein occlusion.[8] Each of these retinal diseases is a completely independent and unique clinical entity. However, various retinal diseases share a few common pathogenetic pathways. Whether a retinal disease initiates from direct injury to the BRB or defect/injury to retinal neurons or glia that leads to BRB impairment secondarily, the BRB is a pivotal point in determining the fate or prognosis of the pathological outcomes as (1) self-limiting and recovering or (2) developing and progressing to a clinical disease phenotype. Retinal exudates (eg, water, proteins, lipids, and cells) and the neuroinflammation that recruits systemic immune components subsequent to BRB impairment are underlying all retinal diseases. Therefore, the investigation of cellular and molecular changes in the BRB is of paramount priority in our investigation of the pathogenesis and early detection of retinal diseases.
Structure of the BRB
The iBRB is formed and maintained via the interaction of vascular endothelial cells with neurons, pericytes, and glial cells. This interacting complex is referred to as the neurovascular unit (NVU) [Figure 1], which is relatively similar in structure and function to the blood-brain barrier (BBB).[9] Therefore, it is relatively understandable that any injury originating in neurons or glia may extend to the impairment of the iBRB.
Figure 1.
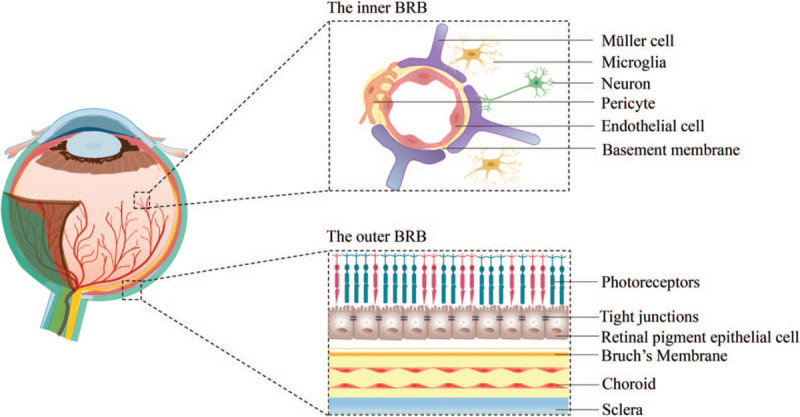
Structure of the blood-retinal barrier (BRB). The inner BRB comprises vascular endothelial cells, pericytes, glial cells, and neurons. The outer BRB is formed by interactions of the choroid, Bruch's membrane, and retinal pigment epithelium.
The iBRB is a highly selective regulator of molecules that enter the retina. This feature relies on two distinct and interdependent transport systems, transcellular transport (transcytosis) and paracellular transport.[9] A low rate of transcytosis through the iBRB may result from the low-level expression of receptors, transporters, and caveolae mediators within the endothelial cells.[10] Junctional complexes, specifically TJs, restrict paracellular transport between the adjacent vascular endothelial cells. TJs comprise more than 40 proteins, of which claudins, occludins, tricellulin (transmembrane proteins), and zonula occludens (ZO, intracellular proteins) are classic markers.[9,11] Increased paracellular or transcellular permeability indicates dysfunction of the BRB, and the abnormal expression of these classic marker proteins may represent the earliest molecular sign of BRB injury.
The oBRB is formed by a single layer of RPE cells that interact with fenestrated choriocapillaris and Bruch's membrane [Figure 1].[12] TJs interconnect neighboring RPE cells and control the paracellular transport of fluids and molecules between the choriocapillaris and the neurosensory retina. The oBRB is a major site for retinal immune surveillance. RPE cells produce several immunomodulatory mediators, including pro-inflammatory and inhibitory factors.[1]
BRB Impairment is a Hallmark of Retinal Dysfunction and Diseases
Retinal immune privilege is essential for the maintenance of retinal homeostasis and prevention of retinal diseases. The BRB insulates the retina from circulating immune cells within the capillaries, and it is the first-tier defense strategy to maintain the retina as an immune-privileged site.[13] Under certain circumstances, the BRB may be impaired, which results in the initiation and development of retinal diseases [Figure 2]. BRB impairment may be caused by direct injury or as a secondary response following defect/injury to retinal neurons or glia. External infections and trauma may directly damage the BRB.[5,14] These two extreme conditions cause circulating components, including exogenous antigens and leukocytes, to enter the retina and activate the innate immune response, which may further initiate adaptive immune responses to pathogen or self-antigen exposure, and cause excessive retinal inflammatory injury. Pathology related to the advanced stage of systemic diseases, such as diabetes and hypertension, also impairs the iBRB via direct injury to the retinal microvascular endothelial cells,[15,16] which leads to retinal inflammation.
Figure 2.
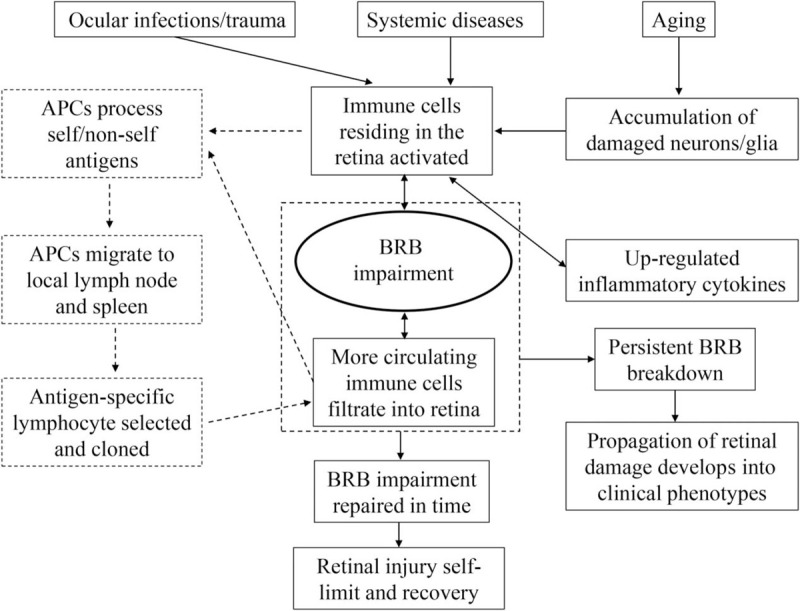
Blood-retinal barrier impairment initiates and propagates retinal inflammation. APC: Antigen-presenting cell; BRB: Blood-retinal barrier.
Aging is an important and independent risk factor for BRB breakdown.[13,17] During the aging process, the retina undergoes some pathophysiological changes primarily due to increased oxidative stress.[18] The number of retinal neurons, including rods, cones, and RPE cells, decreases significantly due to aging-related cell apoptosis. Normally, these apoptotic cells are cleared by resident phagocytes within the retina without pathological immune consequences. Microglia and other types of innate immune cells residing in the retina sense stress and initiate a mild inflammatory response (para-inflammation), which is essential for maintaining retinal homeostasis. However, if accumulated oxidative stress products or apoptotic cells are too many to be cleared in a timely manner, then classic inflammatory pathology may occur.
Various neuroimmune regulatory signals from neurons regulated microglial activation.[19,20] Abnormal neuron death with aging may initiate microglial activation and lead to the excessive secretion of pro-inflammatory cytokines, which may further exacerbate neuroinflammation and neuron loss. This vicious cycle may undermine vascular endothelial cells, followed by the release of more pro-inflammatory cytokines. Once the iBRB is impaired from massive endothelium loss, circulating leukocytes infiltrate into the retina and reach a stage of inflammatory pathology that is beyond a self-limiting recovery. Aging is also associated with an increased interaction between leukocytes and endothelia and causes impairment to the integrity of TJs.[21] Acute inflammation may breach the BRB within a significantly short period, but BRB impairment occurs insidiously over a notably long period during aging. When para-inflammation develops into chronic and consistent inflammation, age-related retinal diseases, such as glaucoma and AMD, may occur.[17,22]
BRB Breakdown: The Initiation and Development of Retinal Diseases
Clinically, ophthalmologists diagnose and treat each retinal disease according to the anomaly that manifests in the ocular fundus. Clinical signs, such as retinal hard exudation, edema, and hemorrhage, reflect BRB impairment of different severities. Basic research on retinal diseases, specifically uveitis, AMD, and DR, demonstrated that the presence of BRB alternations at an early disease stage, which is critical for all inflammatory expansion, promoted neuron injury. There is no way for circulating immune and inflammatory cells to enter the retina in the absence of BRB impairment. Therefore, no exacerbation of initial retinal injury and no clinical manifestations develop.
Circulating immune T cells were involved in the prolonged phase of retinal ganglion cell (RGC) degeneration in a transient high intraocular pressure (IOP) glaucoma mouse model.[23] Microglia are essential for T-cell infiltration,[6] and theoretically, BRB impairment is a prerequisite for T-cell entry into the retina. It is important to examine and clarify the time course and association between microglial activation, BRB impairment, and immuno-inflammatory cell infiltration in primary open-angle glaucoma (POAG). However, studies examining the presence of a BRB alteration in a special type of POAG, normal-tension glaucoma, have not been conducted yet. Therefore, knowledge of other retinal diseases may help us gain insights for further clinical and basic research on different sub-types of POAG.
Infectious uveitis and autoimmune uveitis
Uveitis is a complex group of diseases with multiple etiologies, including infection, autoimmunity, and various physical and chemical injuries. Uveitis is categorized by anatomical location and includes several sub-types: anterior uveitis, intermediate uveitis, posterior uveitis, and panuveitis. BRB impairment is commonly observed in posterior uveitis due to infection or autoimmunity.
A significant number of studies demonstrated that infectious agents, including viruses,[24,25] parasites,[5,26] and bacteria,[27,28] caused posterior uveitis and damaged the BRB, specifically the oBRB. An in vitro oBRB model using a human RPE cell line showed that exposure to human immunodeficiency virus 1 increased the permeability of RPE monolayers due to the decreased expression of several TJ proteins, including ZO-1, occludin, and claudin, rather than influencing cellular viability. Disruption of intercellular TJs caused oBRB breakdown and further facilitated additional bacterial entry into the retina.[24] The most common parasitic infection that causes posterior uveitis is Toxoplasma gondii.[29] Monocytes are carriers of tachyzoites, and infected monocytes cross the choroidal vessels to the oBRB. The infiltrated monocytes disrupt the TJs, which leads to increased permeability of the oBRB.[5] Data from an in vivo mouse model injected with endogenous Staphylococcus aureus (S. aureus) and an in vitro RPE monolayer model inoculated with S. aureus demonstrated that the bacteria may alter TJs of the oBRB and directly cause endophthalmitis via the impaired barrier.[27] Other bacteria, such as Candida albicans, increase adhesion with retinal vascular endothelial cells and access the neural retina via transcytosis.[30,31] Notably, previous studies based on in vitro oBRB models neglect the roles of Bruch's membrane and choroid. Viral-induced alterations of the oBRB may be more complicated in humans than the simplified process in an in vitro experiment setting. Therefore, three-dimensional models integrating RPE, Bruch's membrane, and choroid may be necessary to provide a better research methodology.
Autoimmune uveitis is an important cause of blindness. Several studies showed that the development of autoimmune uveitis was closely associated with BRB breakdown.[32,33] The BRB provides the retina an immune-privileged environment. However, exposure of autoantigens inside the eye, such as retinal S-antigen/arrestin and interphotoreceptor retinoid-binding protein (IRBP),[34] may trigger an uncontrolled, dysregulated immune response under certain conditions. A murine model of experimental autoimmune uveitis (EAU) induced by systemic immunization with IRBP and additional adjuvants showed that autoreactive CD4+ T cells in local lymph nodes and spleen were cloned and polarized into pathogenic T helper (Th) 1 and Th17 cells. Although the dominant antigen-presenting cell (APC) population in the retina during the early stages of EAU is unclear, activation of microglia is indispensable for the infiltration of circulating immune cells into the retina through the impaired BRB, which initiates inflammation in EAU.[6] After immune cells transmigrate out of the retinal blood vessels, antigen-specific T cells interact with local APCs and trigger the subsequent wide-scale inflammatory response. Pro-inflammatory cytokines, such as tumor necrosis factor (TNF) and interleukin-1β (IL-1β), produced by activated T cells and the continuous infiltration of leukocytes further damage endothelial TJs and the iBRB.[35] Cell surface proteins of RPEs that are important for cell adhesion, transportation, and cell communication, including synaptotagmin 1, basigin, and collectrin, respectively, were significantly decreased on RPEs harvested from horse models with EAU.[36] These data support the idea that the oBRB may also be altered during early autoimmune uveitis.
In summary, infectious uveitis may directly alter the oBRB to initiate and expand inflammatory damage to the retina. Although EAU shows that a systemic immune response induces BRB disruption, the initial factor for BRB disruption in human autoimmune uveitis is unclear. BRB impairment is an essential first step in the pathogenesis of inflammatory retinal lesions in either scenario.
AMD
AMD is a macular disorder with a progressive loss of central vision and irreversible visual impairment. AMD is classified clinically into a dry form, or non-exudative AMD, and a wet form, or exudative AMD. Dry AMD features drusen and pathological extracellular deposit accumulation between the RPE and the inner Bruch's membrane.[37] Aging and genetic, environmental, and metabolic factors contribute to the development of AMD, but its pathogenesis remains largely unknown.
The distinctive characteristic of wet AMD is the formation of choroidal neovascularization. Neovascularization is the consequence of the increased production of angiogenic factors from RPE to compensate for the hypoxic status via dysfunctional choriocapillaris. Neovascularizations penetrate the oBRB and reach the neurosensory retina. Alternatively, neovascularization may occur after breakdown of the oBRB and the subsequent recruitment of circulating macrophages, which secrete more vascular endothelial growth factor (VEGF). Wet AMD may be diagnosed using fundus fluorescein angiography/indocyanine green angiography to identify the leakage of neovascularization or optical coherence tomography to detect the accumulation of subretinal and/or intraretinal fluid.
The lack of positive findings on these imaging techniques in dry AMD indicates a preserved intact oBRB. However, several studies showed sub-clinical leakage in dry AMD via analysis of abundant plasma proteins in postmortem AMD retinas from donors.[38,39] Albumin, fibrinogen, immunoglobulin G, and complement components should not be able to cross the BRB in theory, and the fact that these macromolecular substances were significantly higher in dry AMD retinas than controls indicates the presence of BRB impairment in the early stage of dry AMD.[39] AMD is accompanied by neuroinflammation. One possible mechanism is that the disruption of the retinal immune privilege, subsequent to impairment of the BRB, leads to an excessive activation of inflammation and immunological cascade with subsequent neuronal injury.[40]
DR
DR is a common complication of diabetes, and it remains a major cause of visual impairment and blindness in working-age people.[41] DR is traditionally considered as a microvascular disease with signs of microaneurysms, hemorrhages, vascular distortion, hard exudates, cotton wool spots, intraretinal microvascular abnormalities, and neovascularization,[42] some of which are the consequences of BRB impairment.
Emerging evidence suggests that several factors, such as hypoxia-ischemia, oxidative stress, and inflammation,[3,43–45] together are responsible for the breakdown of the iBRB and the oBRB during the pathogenesis of DR. Chronic hyperglycemia triggers metabolic abnormalities. Biochemical metabolic pathways including the polyol and hexamine pathways, accumulation of advanced glycation end products, activation of protein kinase C, and tissue renin-angiotensin system have been implicated in DR.[3,42,46] The overall effects of these disturbed metabolic pathways increase the production of reactive oxygen species (ROS). Under normal conditions, the antioxidant system clears the ROS to prevent cellular damage. However, an imbalance between oxidants and antioxidants was identified in DR.[47] The accumulation of ROS exacerbates mitochondrial injury and facilitates the production of ROS from the mitochondria.[48] This vicious cycle further leads to oxidative stress, which affects the metabolic status of multiple retinal cell types and results in damage to retinal vascular endothelia. The overproduction of ROS also stimulates inflammation via the activation of signal transduction pathways, including nuclear factor kappa light chain enhancer of activated B cells and mitogen-activated protein kinase cascades. DR murine models showed that oxidative stress and inflammation were augmented and coordinately promoted BRB breakdown.[49] This result is consistent with the fact that the expression of several inflammatory cytokines and growth factors is unregulated during the development of DR. TNF-α and IL-1β increase the expression of intercellular adhesion molecule-1(ICAM-1) on retinal endothelial cells and leukocytes, which recruits leukocytes to the vascular membrane.[50] The collagenase matrix metalloproteinase 9 degrades the extracellular matrix and releases netrin-1 fragments, which augments the vascular permeability.[51] Unregulated VEGF binds to cell-surface tyrosine kinase receptors, which decreases the level of intercellular TJ proteins and contributes to neovascularization.[52] The overall effects of these molecules facilitate leukostasis, the non-perfusion of the retinal vessels, retinal hypoxia-ischemia, and endothelial cell death, which work together and lead to a common end, that is, BRB impairment.
Endothelial cell damage received more attention in the disruption of the BRB, but the roles of other injured cells are also important. Traditionally, vascular pericyte dropout is one of the earliest hallmarks of DR, responsible for the early leakage of the iBRB.[53,54] A recent study demonstrated that pericyte dropout was not a direct causative factor of vascular leakage. Early pericyte loss did not increase BRB permeability. Nevertheless, pericyte dropout remains an essential factor that destabilizes retinal vascular endothelial cells and contributes to iBRB breakdown.[55] In patients with diabetic macular edema, a loss of astrocytes is observed in the thinned nerve fiber layer, which causes impairment of iBRB integrity and increased vascular leakage.[56] Activation of microglia accompanied by increased inflammatory cytokines and an excessive inflammatory response leads to vascular impairment and neuronal death.[57] Because the core structure of the iBRB is NVU, the dysfunction of pericytes and glial cells induced by metabolic disorder in diabetes accelerates iBRB breakdown. Photoreceptors release inflammatory cytokines, such as IL-1α, IL-1β, IL-6, IL-12, and TNF-α, in diabetic mice, which directly increase the permeability of the iBRB partially via a change in claudin expression.[58] Hyperglycemia also alters the products of RPEs and compromises TJs between RPEs, which affect the function of the oBRB.[59] Notably, retinal neurodegeneration is identified in DR patients, which may be the consequence of diabetic ischemia and microvascular damage.[60] However, retinal neurodegeneration may occur before clinically detectable vasculopathy. We hypothesize that neurodegeneration is consequent to minor impairments to the BRB. Continuous microvascular leakage may induce a chronic neural immune-inflammatory response, followed by the loss of neurons. Future studies may develop a novel technique to tackle these questions via examination of specific BRB damage-related molecular targets. The current research data allow us to conclude that a series of pathophysiological changes in diabetes cause BRB impairment, which progresses into clinically detectable retinopathy.
POAG
Glaucoma is a complex and multifactorial neurodegenerative disease that is characterized by a loss of RGCs and their axons with typical changes in the optic nerve head, which results in a progressive loss of visual function.[61] POAG is the most common form of glaucoma with no definitive etiology. Elevated IOP, aging, and abnormal blood pressure (hypertension or hypotension) are important risk factors,[62] but the pathophysiology of POAG is not well understood.
Because RGCs and their axons exist in a specific retinal microenvironment, and an intact BRB is essential for retinal homeostasis, dysfunction of the BRB may be an initial factor that contributes to the progressive loss of RGCs and their axons. Clinically, some POAG patients present with typical flame-shaped hemorrhage near the edge of a defective nerve fiber layer and atrophy of choriocapillaris and the RPE, which indicate a BRB impairment that may exacerbate neuronal injury. However, few basic research directly addressed the role of BRB breakdown in POAG. Retinal histology showed abnormal RPE cells, disruption of RPE continuity, increased permeability of retinal blood vessels, leukocyte infiltration, and an accumulation of CD3-positive T lymphocytes around some vessels in the inner retinal layers of the canine globes with primary glaucoma,[2] which are signs of BRB damage. A transient high IOP glaucoma mouse model demonstrated that T cells engaged in progressive ganglion cell death.[23] BRB impairment is a prerequisite for T-cell infiltration into the retina, and we must examine and clarify the time course and association between glial activation, BRB impairment, and immuno-inflammatory cell infiltration in POAG. One study used an acute short-term ocular hypertension (OHT) mouse model and showed that the tightness of the retinal vascular endothelium was unaltered during the entire course of experiment despite a time-dependent RGC loss, narrowing of the capillary plexus, microglial activation, and decrease of microvascular pericyte coverage.[63] This finding is largely consistent with the concept that a sudden pericyte dropout does not significantly disturb the BRB.[55] Although the OHT model does not investigate the presence of BRB breakdown, glaucomatous neurodegeneration in humans is generally a long-term course, and the aging population is a primary patient group. Taken together, BRB dysfunction may occur in the very early stages of POAG. However, this area remains a virgin territory for subsequent study.
The retina is an extension of the central nervous system (CNS), and the iBRB resembles the BBB. Aging is a strong risk factor for the development of CNS degeneration and POAG. Therefore, it is reasonable to hypothesize that some insights would be gained from knowledge on the pathogenetic mechanisms of CNS degenerative diseases.
Recent evidence of BBB impairment was identified in naturally aged mice and humans. Dysfunction in the NVU and consequent albumin extravasation triggered the transforming growth factor-β signaling cascade in astrocytes, which caused an abnormal function of neurons.[64] Circulating immune cell migration through an impaired BBB together with glial activation contributes to the progression of Alzheimer's disease (AD).[65] Similarly, we hypothesize that iBRB dysfunction and secondary neuro-inflammatory damage occur in POAG patients despite the absence of significantly elevated IOP. Vascular endothelial loss and deterioration of the iBRB were reported during the natural course of human aging.[66] However, age-related barrier (eg, the BBB and BRB) breakdown alone may not be adequate to cause neurodegenerative diseases, including AD and POAG.
The integration of all known knowledge on the pathogenesis of POAG, other retinal diseases, and CNS neurodegenerative diseases allows us to introduce some theoretical hypotheses for further discussion and investigations: (1) significantly or relatively elevated IOP may cause RGC loss and retinal vascular injury due to ischemia-reperfusion; (2) massive physiological loss of RGCs and their axons during the aging process may be responsible for the excessive activation of retina-resident microglia and other innate immune cells, which leads to the production of pro-inflammatory cytokines, upregulation of vascular permeability, and impairment of the integrity of the iBRB; and (3) the antigens from CNS, the retina or circulating system may activate the immune system and cause circulating immune cells to infiltrate into the retina and damage the BRB, which disrupts retinal immune privilege and induces gradual RGC loss. Because the BRB plays a pivotal role in the initiation and amplification of neuroinflammation within the retina, it is definitively worthwhile to investigate whether BRB impairment is present in early stage of POAG. BRB breakdown, due to elevated IOP or other unknown etiologies, may provide us with a novel window to clearly understand the pathogenesis of POAG.
Barriers We Should Investigate and Understand in More Details
Although previous studies demonstrated the presence of BRB impairment in various retinal diseases, their exact mechanisms are not completely clarified. There are still several questions that need to be answered: (1) Is impairment of BRB the cause or consequence of retinal inflammation? (2) Is BRB impairment an early manifestation or an advanced injury in retinal diseases? (3) As an immune-privileged site, what triggers autoimmune retinal inflammation? (4) What is the exact mechanism underlying progressive RGC loss in glaucoma, even if IOP is effectively controlled or constantly in the normal range? (5) What exact role do glia play in iBRB breakdown? We will need comprehensive clinical and basic research to answer these questions. The latest developments in neuroscience and immunological investigations on CNS neurodegeneration will help shed light on retinal exploration.
Some recent studies demonstrated that the gastrointestinal microbiome modulated the development of CNS[67] and immune system,[68] and it played an important role in immune-mediated diseases, such as CNS degeneration[69,70] and some retinal diseases.[23,71] Some important barriers exist in physiological conditions, including the BBB, BRB, intestinal epithelial barrier (IEB), and gut-vascular barrier in the human body,[72] which protect the brain, retina, and intestine from harmful gut microbial cometabolites, thereby preventing or limiting the development of pathological immune responses. Intact microbiota contribute to the maturing process of the BBB and regulate the morphology and function of microglia,[73,74] and germ-free (GF) mice exhibit a leaky BBB, which indicates the essential role of intact microbiota.[73] Short-chain fatty acids produced by microbiota help maintain the integrity of the IEB.[75] Therefore, perturbed components of the microbiota may be involved in abnormal barrier permeability, which triggers systemic inflammatory processes via the translocation of gut-derived immune stimuli, such as microbe-associated molecular patterns and damage-associated molecular patterns. The BBB and BRB are two important bridges for these stimuli to enter the brain or the eye. Several recent studies showed that microbial dysbiosis increased barrier permeability and systemic inflammation, which promoted AD pathology via a neuroimmune-specific response.[76,77] A microbe-induced neuroimmune response was also found in glaucoma. A recent comprehensive study using a transient high IOP glaucoma mouse model in immunodeficient mice and GF mice demonstrated that T-cell responses partially mediated glaucomatous neurodegeneration. Exposure to commensal microflora-derived heat shock proteins (HSPs) sensitizes T cells, and memory T cells infiltrate into the retina and may be activated by host HSPs via molecular mimicry.[23] Although BRB breakdown was not investigated in this study, the permeability of the BRB must have been pre-compromised to allow T-cell migration into the retina. Similar findings were demonstrated in a novel mouse model of spontaneous uveitis, which showed that retina-specific T cells were activated by gut commensal microbiota antigen via autoreactive T-cell receptor.[78] Taken together, we can reasonably hypothesize that intestinal microorganisms are closely associated with several retinal diseases via the pivotal gate of the BRB. The role of gastrointestinal microbes in glaucoma is a nascent field, but existent data on the microbiome and CNS may provide us with valuable insights.
Conclusion and Perspective
The BRB plays a significant role in maintaining retinal homeostasis, and BRB impairment is pivotal in the initiation and development of several retinal diseases. An impaired BRB may serve as a gateway for macromolecular substances, such as leukocytes and microbes, to migrate from the blood circulation into the retina, which results in disruption of retinal immune privilege and expansion of retinal inflammation. Alterations of the BRB were identified in various retinal diseases, including inflammatory retinal diseases, AMD, DR, and POAG. However, the importance of the BRB as a common pivotal point in retinal diseases has been relatively underestimated, specifically in POAG. Based on data from other retinal diseases, CNS degeneration, and related glaucomatous research, BRB impairment in POAG is likely an essential step in pathogenesis and a prerequisite to exacerbated neuroinflammation and the progressive loss of RGCs and their axons.
Funding
This study was supported by grants from the Major Project of National Natural Science Foundation of China (NSFC)-Guangdong Province Joint Fund (No. 3030902113080), the Science and Technology Planning Project of Guangdong Province (No. 303090100502050-18), and the Guangzhou Science and Technology Plan Project (Nos. 201803040020 and 201903010065).
Conflicts of interest
None.
Footnotes
How to cite this article: Yang X, Yu XW, Zhang DD, Fan ZG. Blood-retinal barrier as a converging pivot in understanding the initiation and development of retinal diseases. Chin Med J 2020;133:2586–2594. doi: 10.1097/CM9.0000000000001015
References
- 1.Shechter R, London A, Schwartz M. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat Rev Immunol 2013; 13:206–218. doi: 10.1038/nri3391. [DOI] [PubMed] [Google Scholar]
- 2.Mangan BG, Al-Yahya K, Chen CT, Gionfriddo JR, Powell CC, Dubielzig RR, et al. Retinal pigment epithelial damage, breakdown of the blood-retinal barrier, and retinal inflammation in dogs with primary glaucoma. Vet Ophthalmol 2007; 10: Suppl 1: 117–124. doi: 10.1111/j.1463-5224.2007.00585.x. [DOI] [PubMed] [Google Scholar]
- 3.Mahajan N, Arora P, Sandhir R. Perturbed biochemical pathways and associated oxidative stress lead to vascular dysfunctions in diabetic retinopathy. Oxid Med Cell Longev 2019; 2019:8458472.doi: 10.1155/2019/8458472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molins B, Méndez AP, Llorenç V, Zarranz-Ventura J, Mesquida M, Adan A, et al. C-reactive protein isoforms differentially affect outer blood-retinal barrier integrity and function. Am J Physiol Cell Physiol 2017; 312:C244–C253. doi: 10.1152/ajpcell.00057.2016. [DOI] [PubMed] [Google Scholar]
- 5.Song HB, Jun HO, Kim JH, Lee YH, Choi MH, Kim JH. Disruption of outer blood-retinal barrier by Toxoplasma gondii-infected monocytes is mediated by paracrinely activated FAK signaling. PLoS One 2017; 12:e0175159.doi: 10.1371/journal.pone.0175159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okunuki Y, Mukai R, Nakao T, Tabor SJ, Butovsky O, Dana R, et al. Retinal microglia initiate neuroinflammation in ocular autoimmunity. Proc Natl Acad Sci U S A 2019; 116:9989–9998. doi: 10.1073/pnas.1820387116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahlers C, Geitzenauer W, Stock G, Golbaz I, Schmidt-Erfurth U, Prünte C. Alterations of intraretinal layers in acute central serous chorioretinopathy. Acta Ophthalmol 2009; 87:511–516. doi: 10.1111/j.1755-3768.2008.01468.x. [DOI] [PubMed] [Google Scholar]
- 8.Hayreh SS, Zimmerman MB. Fundus changes in central retinal vein occlusion. Retina 2015; 35:29–42. doi: 10.1097/IAE.0000000000000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Díaz-Coránguez M, Ramos C, Antonetti DA. The inner blood-retinal barrier: cellular basis and development. Vision Res 2017; 139:123–137. doi: 10.1016/j.visres.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu X, Fliesler SJ, Zhao YY, Stallcup WB, Cohen AW, Elliott MH. Loss of caveolin-1 causes blood-retinal barrier breakdown, venous enlargement, and mural cell alteration. Am J Pathol 2014; 184:541–555. doi: 10.1016/j.ajpath.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erickson KK, Sundstrom JM, Antonetti DA. Vascular permeability in ocular disease and the role of tight junctions. Angiogenesis 2007; 10:103–117. doi: 10.1007/s10456-007-9067-z. [DOI] [PubMed] [Google Scholar]
- 12.Fields MA, Del Priore LV, Adelman RA, Rizzolo LJ. Interactions of the choroid, Bruch's membrane, retinal pigment epithelium, and neurosensory retina collaborate to form the outer blood-retinal-barrier. Prog Retin Eye Res 2020; 76:100803.doi: 10.1016/j.preteyeres.2019.100803. [DOI] [PubMed] [Google Scholar]
- 13.Chen M, Luo C, Zhao J, Devarajan G, Xu H. Immune regulation in the aging retina. Prog Retin Eye Res 2019; 69:159–172. doi: 10.1016/j.preteyeres.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romaniuk VM. Ocular trauma and other catastrophes. Emerg Med Clin North Am 2013; 31:399–411. doi: 10.1016/j.emc.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Flammer J, Konieczka K, Bruno RM, Virdis A, Flammer AJ, Taddei S. The eye and the heart. Eur Heart J 2013; 34:1270–1278. doi: 10.1093/eurheartj/eht023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Opdenakker G, Abu El-Asrar A. Metalloproteinases mediate diabetes-induced retinal neuropathy and vasculopathy. Cell Mol Life Sci 2019; 76:3157–3166. doi: 10.1007/s00018-019-03177-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res 2009; 28:348–368. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Robinson JV, James AL. Some observations on the effects produced in white mice following the injection of certain suspensions of corroding bacilli. Br J Exp Pathol 1975; 56:14–16. [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Li F, Yang Y, Chen J, Hu X. SIRP/CD47 signaling in neurological disorders. Brain Res 2015; 1623:74–80. doi: 10.1016/j.brainres.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mecha M, Carrillo-Salinas FJ, Feliú A, Mestre L, Guaza C. Microglia activation states and cannabinoid system: therapeutic implications. Pharmacol Ther 2016; 166:40–55. doi: 10.1016/j.pharmthera.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 21.Suematsu M, Suzuki H, Delano FA, Schmid-Schönbein GW. The inflammatory aspect of the microcirculation in hypertension: oxidative stress, leukocytes/endothelial interaction, apoptosis. Microcirculation 2002; 9:259–276. doi: 10.1038/sj.mn.7800141. [DOI] [PubMed] [Google Scholar]
- 22.Perez VL, Caspi RR. Immune mechanisms in inflammatory and degenerative eye disease. Trends Immunol 2015; 36:354–363. doi: 10.1016/j.it.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, Cho KS, Vu THK, Shen CH, Kaur M, Chen G, et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat Commun 2018; 9:3209.doi: 10.1038/s41467-018-05681-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tan S, Duan H, Xun T, Ci W, Qiu J, Yu F, et al. HIV-1 impairs human retinal pigment epithelial barrier function: possible association with the pathogenesis of HIV-associated retinopathy. Lab Invest 2014; 94:777–787. doi: 10.1038/labinvest.2014.72. [DOI] [PubMed] [Google Scholar]
- 25.Singh PK, Guest JM, Kanwar M, Boss J, Gao N, Juzych MS, et al. Zika virus infects cells lining the blood-retinal barrier and causes chorioretinal atrophy in mouse eyes. JCI Insight 2017; 2:e92340.doi: 10.1172/jci.insight.92340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furtado JM, Bharadwaj AS, Chipps TJ, Pan Y, Ashander LM, Smith JR. Toxoplasma gondii tachyzoites cross retinal endothelium assisted by intercellular adhesion molecule-1 in vitro. Immunol Cell Biol 2012; 90:912–915. doi: 10.1038/icb.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coburn PS, Wiskur BJ, Astley RA, Callegan MC. Blood-retinal barrier compromise and endogenous Staphylococcus aureus endophthalmitis. Invest Ophthalmol Vis Sci 2015; 56:7303–7311. doi: 10.1167/iovs.15-17488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mander KA, Finnie JW. Loss of endothelial barrier antigen immunoreactivity in rat retinal microvessels is correlated with clostridium perfringens type D epsilon toxin-induced damage to the blood-retinal barrier. J Comp Pathol 2018; 158:51–55. doi: 10.1016/j.jcpa.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Maenz M, Schlüter D, Liesenfeld O, Schares G, Gross U, Pleyer U. Ocular toxoplasmosis past, present and new aspects of an old disease. Prog Retin Eye Res 2014; 39:77–106. doi: 10.1016/j.preteyeres.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Fardini Y, Wang X, Témoin S, Nithianantham S, Lee D, Shoham M, et al. Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol 2011; 82:1468–1480. doi: 10.1111/j.1365-2958.2011.07905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jong AY, Stins MF, Huang SH, Chen SH, Kim KS. Traversal of Candida albicans across human blood-brain barrier in vitro. Infect Immun 2001; 69:4536–4544. doi: 10.1128/IAI.69.7.4536-4544.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee RW, Nicholson LB, Sen HN, Chan CC, Wei L, Nussenblatt RB, et al. Autoimmune and autoinflammatory mechanisms in uveitis. Semin Immunopathol 2014; 36:581–594. doi: 10.1007/s00281-014-0433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forrester JV, Kuffova L, Dick AD. Autoimmunity, autoinflammation, and infection in uveitis. Am J Ophthalmol 2018; 189:77–85. doi: 10.1016/j.ajo.2018.02.019. [DOI] [PubMed] [Google Scholar]
- 34.Diedrichs-Möhring M, Kaufmann U, Wildner G. The immunopathogenesis of chronic and relapsing autoimmune uveitis - lessons from experimental rat models. Prog Retin Eye Res 2018; 65:107–126. doi: 10.1016/j.preteyeres.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 35.Bamforth SD, Lightman S, Greenwood J. The effect of TNF-alpha and IL-6 on the permeability of the rat blood-retinal barrier in vivo. Acta Neuropathol 1996; 91:624–632. doi: 10.1007/s004010050476. [DOI] [PubMed] [Google Scholar]
- 36.Uhl PB, Szober CM, Amann B, Alge-Priglinger C, Ueffing M, Hauck SM, et al. In situ cell surface proteomics reveals differentially expressed membrane proteins in retinal pigment epithelial cells during autoimmune uveitis. J Proteomics 2014; 109:50–62. doi: 10.1016/j.jprot.2014.06.020. [DOI] [PubMed] [Google Scholar]
- 37.Mitchell P, Liew G, Gopinath B, Wong TY. Age-related macular degeneration. Lancet 2018; 392:1147–1159. doi: 10.1016/s0140-6736(18)31550-2. [DOI] [PubMed] [Google Scholar]
- 38.Mullins RF, Schoo DP, Sohn EH, Flamme-Wiese MJ, Workamelahu G, Johnston RM, et al. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol 2014; 184:3142–3153. doi: 10.1016/j.ajpath.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schultz H, Song Y, Baumann BH, Kapphahn RJ, Montezuma SR, Ferrington DA, et al. Increased serum proteins in non-exudative AMD retinas. Exp Eye Res 2019; 186:107686.doi: 10.1016/j.exer.2019.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buschini E, Piras A, Nuzzi R, Vercelli A. Age related macular degeneration and drusen: neuroinflammation in the retina. Prog Neurobiol 2011; 95:14–25. doi: 10.1016/j.pneurobio.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 41.Klein R, Lee KE, Gangnon RE, Klein BE. The 25-year incidence of visual impairment in type 1 diabetes mellitus the Wisconsin epidemiologic study of diabetic retinopathy. Ophthalmology 2010; 117:63–70. doi: 10.1016/j.ophtha.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet 2010; 376:124–136. doi: 10.1016/S0140-6736(09)62124-3. [DOI] [PubMed] [Google Scholar]
- 43.Klaassen I, Van Noorden CJ, Schlingemann RO. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res 2013; 34:19–48. doi: 10.1016/j.preteyeres.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Frey T, Antonetti DA. Alterations to the blood-retinal barrier in diabetes: cytokines and reactive oxygen species. Antioxid Redox Signal 2011; 15:1271–1284. doi: 10.1089/ars.2011.3906. [DOI] [PubMed] [Google Scholar]
- 45.Kusuhara S, Fukushima Y, Ogura S, Inoue N, Uemura A. Pathophysiology of diabetic retinopathy: the old and the new. Diabetes Metab J 2018; 42:364–376. doi: 10.4093/dmj.2018.0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kowluru RA, Mishra M. Therapeutic targets for altering mitochondrial dysfunction associated with diabetic retinopathy. Expert Opin Ther Targets 2018; 22:233–245. doi: 10.1080/14728222.2018.1439921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tien T, Zhang J, Muto T, Kim D, Sarthy VP, Roy S. High glucose induces mitochondrial dysfunction in retinal müller cells: implications for diabetic retinopathy. Invest Ophthalmol Vis Sci 2017; 58:2915–2921. doi: 10.1167/iovs.16-21355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kowluru RA, Mishra M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim Biophys Acta 2015; 1852:2474–2483. doi: 10.1016/j.bbadis.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 49.Imai A, Toriyama Y, Iesato Y, Hirabayashi K, Sakurai T, Kamiyoshi A, et al. Adrenomedullin suppresses vascular endothelial growth factor-induced vascular hyperpermeability and inflammation in retinopathy. Am J Pathol 2017; 187:999–1015. doi: 10.1016/j.ajpath.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 50.Noda K, Nakao S, Zandi S, Sun D, Hayes KC, Hafezi-Moghadam A. Retinopathy in a novel model of metabolic syndrome and type 2 diabetes: new insight on the inflammatory paradigm. FASEB J 2014; 28:2038–2046. doi: 10.1096/fj.12-215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miloudi K, Binet F, Wilson A, Cerani A, Oubaha M, Menard C, et al. Truncated netrin-1 contributes to pathological vascular permeability in diabetic retinopathy. J Clin Invest 2016; 126:3006–3022. doi: 10.1172/JCI84767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lally DR, Shah CP, Heier JS. Vascular endothelial growth factor and diabetic macular edema. Surv Ophthalmol 2016; 61:759–768. doi: 10.1016/j.survophthal.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 53.Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci 2011; 52:1156–1163. doi: 10.1167/iovs.10-6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hammes HP, Feng Y, Pfister F, Brownlee M. Diabetic retinopathy: targeting vasoregression. Diabetes 2011; 60:9–16. doi: 10.2337/db10-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park DY, Lee J, Kim J, Kim K, Hong S, Han S, et al. Plastic roles of pericytes in the blood-retinal barrier. Nat Commun 2017; 8:15296.doi: 10.1038/ncomms15296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haj Najeeb B, Simader C, Deak G, Vass C, Gamper J, Montuoro A, et al. The distribution of leakage on fluorescein angiography in diabetic macular edema: a new approach to its etiology. Invest Ophthalmol Vis Sci 2017; 58:3986–3990. doi: 10.1167/iovs.17-21510. [DOI] [PubMed] [Google Scholar]
- 57.Altmann C, Schmidt M. The role of microglia in diabetic retinopathy: inflammation, microvasculature defects and neurodegeneration. Int J Mol Sci 2018; 19:110.doi: 10.3390/ijms19010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tonade D, Liu H, Palczewski K, Kern TS. Photoreceptor cells produce inflammatory products that contribute to retinal vascular permeability in a mouse model of diabetes. Diabetologia 2017; 60:2111–2120. doi: 10.1007/s00125-017-4381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Willermain F, Scifo L, Weber C, Caspers L, Perret J, Delporte C. Potential interplay between hyperosmolarity and inflammation on retinal pigmented epithelium in pathogenesis of diabetic retinopathy. Int J Mol Sci 2018; 19:1056.doi: 10.3390/ijms19041056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barber AJ. Diabetic retinopathy: recent advances towards understanding neurodegeneration and vision loss. Sci China Life Sci 2015; 58:541–549. doi: 10.1007/s11427-015-4856-x. [DOI] [PubMed] [Google Scholar]
- 61.Liu SA, Zhao ZN, Sun NN, Han Y, Chen J, Fan ZG. Transitions of the understanding and definition of primary glaucoma. Chin Med J 2018; 131:2852–2859. doi: 10.4103/0366-6999.246069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weinreb RN, Leung CK, Crowston JG, Medeiros FA, Friedman DS, Wiggs JL, et al. Primary open-angle glaucoma. Nat Rev Dis Primers 2016; 2:16067.doi: 10.1038/nrdp.2016.67. [DOI] [PubMed] [Google Scholar]
- 63.Trost A, Motloch K, Bruckner D, Schroedl F, Bogner B, Kaser-Eichberger A, et al. Time-dependent retinal ganglion cell loss, microglial activation and blood-retina-barrier tightness in an acute model of ocular hypertension. Exp Eye Res 2015; 136:59–71. doi: 10.1016/j.exer.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 64.Senatorov VV, Jr, Friedman AR, Milikovsky DZ, Ofer J, Saar-Ashkenazy R, Charbash A, et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci Transl Med 2019; 11:eaaw8283.doi: 10.1126/scitranslmed.aaw8283. [DOI] [PubMed] [Google Scholar]
- 65.Mietelska-Porowska A, Wojda U. T lymphocytes and inflammatory mediators in the interplay between brain and blood in Alzheimer's disease: potential pools of new biomarkers. J Immunol Res 2017; 2017:4626540.doi: 10.1155/2017/4626540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.López-Luppo M, Catita J, Ramos D, Navarro M, Carretero A, Mendes-Jorge L, et al. Cellular senescence is associated with human retinal microaneurysm formation during aging. Invest Ophthalmol Vis Sci 2017; 58:2832–2842. doi: 10.1167/iovs.16-20312. [DOI] [PubMed] [Google Scholar]
- 67.Sampson TR, Mazmanian SK. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 2015; 17:565–576. doi: 10.1016/j.chom.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature 2016; 535:65–74. doi: 10.1038/nature18847. [DOI] [PubMed] [Google Scholar]
- 69.Spielman LJ, Gibson DL, Klegeris A. Unhealthy gut, unhealthy brain: the role of the intestinal microbiota in neurodegenerative diseases. Neurochem Int 2018; 120:149–163. doi: 10.1016/j.neuint.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 70.Wang HX, Wang YP. Gut microbiota-brain axis. Chin Med J 2016; 129:2373–2380. doi: 10.4103/0366-6999.190667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rinninella E, Mele MC, Merendino N, Cintoni M, Anselmi G, Caporossi A, et al. The role of diet, micronutrients and the gut microbiota in age-related macular degeneration: new perspectives from the gut retina axis. Nutrients 2018; 10:1677.doi: 10.3390/nu10111677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spadoni I, Fornasa G, Rescigno M. Organ-specific protection mediated by cooperation between vascular and epithelial barriers. Nat Rev Immunol 2017; 17:761–773. doi: 10.1038/nri.2017.100. [DOI] [PubMed] [Google Scholar]
- 73.Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med 2014; 6:263ra158.doi: 10.1126/scitranslmed.3009759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci 2015; 18:965–977. doi: 10.1038/nn.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016; 7:189–200. doi: 10.1080/19490976.2015.1134082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen CH, Lin CL, Kao CH. Irritable bowel syndrome is associated with an increased risk of dementia: a nationwide population-based study. PLoS One 2016; 11:e0144589.doi: 10.1371/journal.pone.0144589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen YG. Research progress in the pathogenesis of Alzheimer's disease. Chin Med J 2018; 131:1618–1624. doi: 10.4103/0366-6999.235112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Horai R, Zárate-Bladés CR, Dillenburg-Pilla P, Chen J, Kielczewski JL, Silver PB, et al. Microbiota-dependent activation of an autoreactive T cell receptor provokes autoimmunity in an immunologically privileged site. Immunity 2015; 43:343–353. doi: 10.1016/j.immuni.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]