

Purpose of review
The aim of this review was to compare and contrast the application of molecular epidemiology approaches for the improved management and understanding of the HIV versus SARS-CoV-2 epidemics.
Recent findings
Molecular biology approaches, including PCR and whole genome sequencing (WGS), have become powerful tools for epidemiological investigation. PCR approaches form the basis for many high-sensitivity diagnostic tests and can supplement traditional contact tracing and surveillance strategies to define risk networks and transmission patterns. WGS approaches can further define the causative agents of disease, trace the origins of the pathogen, and clarify routes of transmission. When coupled with clinical datasets, such as electronic medical record data, these approaches can investigate co-correlates of disease and pathogenesis. In the ongoing HIV epidemic, these approaches have been effectively deployed to identify treatment gaps, transmission clusters and risk factors, though significant barriers to rapid or real-time implementation remain critical to overcome. Likewise, these approaches have been successful in addressing some questions of SARS-CoV-2 transmission and pathogenesis, but the nature and rapid spread of the virus have posed additional challenges.
Summary
Overall, molecular epidemiology approaches offer unique advantages and challenges that complement traditional epidemiological tools for the improved understanding and management of epidemics.
Keywords: contact tracing, HIV, molecular epidemiology, SARS-CoV-2, whole genome sequencing
INTRODUCTION
The principal goal of epidemiology is to identify the causative and correlative factors that drive a disease to enable a rational basis for infection prevention and disease control. This includes addressing the basic questions of what is the causative agent, how is it spread, who is at risk, where is it prevalent, when is it a threat, and why does it cause disease? At the beginning of the HIV pandemic, these questions were addressed through the use of contact tracing, case finding, and well executed case–control studies, the basic tools of infectious disease epidemiologic investigation. With advances in molecular biology, most notably PCR and gene sequencing, new molecular-based approaches to perform epidemiological investigations were developed, oftentimes directly in response to the HIV epidemic itself. Today, molecular epidemiology is indispensable to the investigation of a new disease or disease outbreak. Indeed, these approaches were deployed very early in the SARS-like coronavirus 2 (SARS-CoV-2) epidemic to inform urgent outstanding questions of cause, origin, transmission, and risk. Despite these successes, several limitations to these approaches have been exposed by these two different epidemics, especially in regards to implementation. Here, we discuss some of the tools of molecular epidemiology, their use and limitations as applied to the HIV epidemic, and lessons we have learned so far in applying these approaches to SARS-CoV-2.
Box 1.

no caption available
Molecular diagnostics: antibody and PCR-based testing
One of the first challenges faced by clinicians, public health experts, and epidemiologists during a new epidemic is the need for accurate and sensitive diagnostic tools. Traditionally, diagnostic procedures were heavily dependent on clinical symptom tracking, history of exposure, and standard microbiology practices to isolate infectious agents. Although often sufficient for extracting population level trends, these tools alone are often complicated by variability in clinical presentation, incomplete medical histories, insufficient resources or inadequate protocols for microbial isolation, and high rates of overall uncertainty [1–3]. The development of molecular diagnostic tools for the detection of specific pathogens, or an immune response to specific pathogens, revolutionized our capacity to diagnose infectious diseases accurately within large populations. Although the sensitivity of these tests is still largely dependent on the quality and timing of sample collection relative to the infection time course, their specificity is generally high when properly controlled [1–3].
The first molecular diagnostic tool for HIV was an IgG antibody test developed in 1985, just 2 years after the isolation and discovery of the virus as the causative agent of AIDS [4–6]. This first-generation test was an ELISA that used HIV-1 infected cell lysates as the fixed antigen over which patient serum would be applied. Anti-HIV antibodies would stick to the HIV antigens for detection with IgG-specific secondary antibodies that could be quantified by a chemiluminescence readout. In order to rule out false-positive tests (due to pregnancy, autoimmune disease, and other undetermined reasons) and further differentiate HIV-1 from HIV-2, a subsequent validation of these results was required by immunoblotting or immunoflourescence [7,8]. This two-part testing algorithm (serology with secondary confirmation) would be refined in the second and third generations to improve the breadth of HIV subtypes that could be detected and to standardize the antigens used as bait for mass production. Although these algorithms had high sensitivity and specificity, their reliance on antibody detection dictated a significant lag time between exposure and diagnosis. In other words, due to the time it takes for the body to mount a specific antibody response detectable in the blood (i.e. the time to seroconversion), these tests were not be able to detect infection for 3–12 weeks following exposure [9–11]. To narrow this negative window, fourth-generation tests that incorporated direct antigen detection were developed, first becoming available in 1997 (Fig. 1a). These tests similarly relied on ELISA methodology, but for detection of both HIV p24 antigen as well as anti-HIV antibodies [9,12]. Fifth-generation tests that included separate readouts for antigens and antibodies were developed in 2015. These tests are usually effective at detecting HIV infection within 18–45 days following exposure. These later tests also allowed for more rapid and improved differentiation between HIV-1 and HIV-2 infection [13].
FIGURE 1.
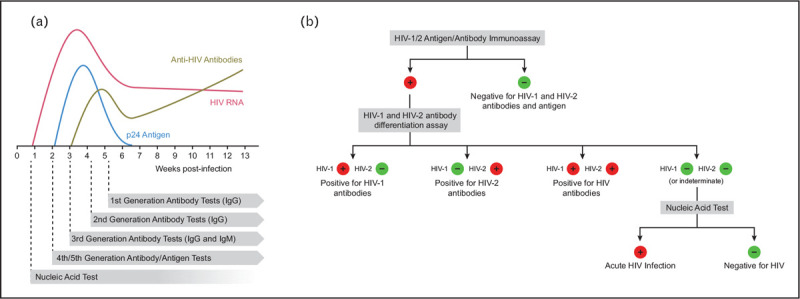
Evolution of HIV diagnostic algorithms. (a) This cartoon depicts the abundance of HIV diagnostic substrates in the serum of a figurative patient during the first several weeks following infection. Although first-generation HIV tests that relied on detection of anti-HIV IgG antibodies in patient serum had a large ‘negative window’ between infection and detection, each successive generation of tests reduced this gap. Nucleic acid tests are primarily used for medical management of disease, but are used for diagnostic/screening purposes in some algorithms. (b) The current HIV testing algorithm based on guidelines from the Centers for Disease Control.
Serological tests for diagnostic purposes are relatively cheap, have a low barrier to entry and can be readily adapted to rapid, at-home or point-of-care testing platforms [14,15]. These ‘rapid’ tests generally rely on immunochromatography wherein differences in antibody movement in the presence or absence of its antigen can be detected by laminar flow. Rapid tests generally have lower specificity than the traditional ELISA-based tests, and so require result confirmation, but enable outreach and testing to a much broader population than otherwise would be accessible [14,15]. There is currently one FDA-approved rapid self-test for HIV in the United States (OraQuick), which detects anti-HIV antibodies from an oral swab. Rapid, point-of-care tests are also available that use a single drop of blood from a fingertip (i.e. Alere Determine, among others) [14–16].
Although serological assays are the recommended diagnostic tests for HIV, PCR-based testing is also a helpful adjunct in certain diagnostic situations. Rather than detecting the virus-specific antibodies or viral proteins, these tests rely on detection of viral nucleic acids [17–19]. In these tests, viral RNA is extracted from blood samples, converted into complementary DNA (cDNA), and amplified with HIV-specific oligonucleotide primers. If done in a quantifiable way, these assays can also be used to determine viral load, which can be an important determinant of transmission and pathogenesis [20–24]. Although these tests are generally more expensive, more prone to false positivity than the current antibody testing algorithms, and can be subject to longer turnaround times, they can be used to detect HIV infection within 3–14 days following exposure, often prior to the appearance of quantifiable p24 antigen or anti-HIV antibodies in the blood [17–19]. PCR-based assays are primarily used as a diagnostic adjunct in cases wherein acute infection is suspected very soon after a high-risk exposure (with follow-up antibody algorithm confirmation) [18], for the detection of mother-to-child transmission (to differentiate infant infection given the passive transfer of antibody in utero) [25], and for surveillance of blood donations [26]. Following acute infection, some individuals may have viral set points below the limit of detection for PCR-based assays, so serological tests are often preferred at time points beyond the immediate post-exposure period. As such, PCR assays for plasma HIV RNA are primarily used for medical management and not generally recommended as a stand alone diagniostic.
The Centers for Disease Control and Prevention's (CDC's) current HIV testing algorithm calls for primary screening with a fourth or fifth-generation HIV-1/2 antigen/antibody immunoassay (Fig. 1b) [27]. A majority of these tests, such as the Food and Drug Administration (FDA) approved Abbott Architect HIV Ag/Ab test or the GS Combo Ag/Ab EIA test, have been extensively evaluated with reported sensitivity and specificity ranging from 99.7 to 100% [27–29]. Due to the high sensitivity, a follow-up test is not recommended, unless an exposure event is suspected within the test's negative window (roughly within the previous 2–4 weeks), in which case a PCR-based nucleic acid test is suggested. If the initial assay gives a positive result, a follow-up immunoassay to confirm diagnosis and differentiate between HIV-1 and HIV-2 is required. If a negative result is obtained in follow-up, a PCR-based nucleic acid test is again suggested to test for acute infection in the negative window. If a preliminary positive result is obtained from a rapid, point-of-care test, result confirmation using this algorithm in a certified clinical laboratory is required.
The type of diagnostic test that is most effective is highly dependent on the nature of the virus and the goal of the test. HIV establishes a chronic infection throughout the lifetime of an individual, so while early diagnosis is optimal, diagnostic tools that are effective at any stage of disease are useful for protecting patient health and for controlling future spread [20,21,30▪▪]. Viruses that do not establish chronic infections, however, afford only a small window in which diagnostic tests can be used to inform treatment and prevent spread. By the time serological responses are developed against SARS-CoV-2, for example, a majority of people have already cleared the virus and cannot transmit the infection. Thus, although serological tests that indicate prior exposure to SARS-CoV-2 may be informative for transmission models, such results are of limited benefit for determining if there is a current risk of further transmission [31,32▪]. Given this limitation, PCR-based testing has emerged as the primary diagnostic test for detecting SARS-CoV-2 infection in the clinic. There are a large variety of these tests available, most of which rely on amplification of a portion of the viral nucleocapsid (N) or RNA-dependent RNA polymerase (RdRp) genes [31,33▪,34].
When properly controlled, the specificity of PCR-based diagnostic tests can be near perfect in high prevalence symptomatic populations given their dependency on unique sequences of nucleic acids. The sensitivity of PCR-based testing, however, may be substantially lower dependent on the timing of the sample, sample source, and sample collection quality [34]. For example, the sensitivity and specificity of most nucleic acid tests for HIV are greater than 99.5%, but the sensitivity of these assays drop if the specimen is collected too early or late relative to acute infection, if the specimen used is from an oral swab or dried blood spot as opposed to serum, or if the specimen is processed outside a certified clinical laboratory [27,35]. Variation in the sensitivity of PCR-based SARS-CoV-2 testing is similarly dependent on timing, specimen collection and sample processing. To avoid these issues, best practices include amplification of a housekeeping gene within each specimen to validate sample quality, repeat testing after possible exposure to validate negative results (especially in cases shortly after exposure) and limitation of testing to certified clinical labortaories with trained personnel [36]. Even when using best practices, however, initial studies estimate that the sensitivity of SARS-CoV-2 PCR-based testing for COVID-19 pneumonia may be as low as 70% [37▪▪]. The relatively low sensitivity of these tests and the subsequently higher probabilities of false negatives emphasize the need for additional surveillance mechanisms (contact tracing, symptom monitoring and so on) as well as for careful adherence to public health guidelines (social distancing, wear masks in public spaces, wash hands regularly and so on). More studies are needed to determine the true sensitivity of the various tests currently being employed in clinical versus population-based settings and to determine how much sensitivity varies in symptomatic versus asymptomatic people [37▪▪].
Given the frequency and scale with which SARS-CoV-2 testing is needed for diagnosis, surveillance and monitoring, additional innovations in diagnostic testing strategies are required. The development of point-of-care testing strategies that either enable more reliable self-sampling (i.e. through oral rinsates), faster turn around times (i.e. rapid tests such as the Abbott ID NOW or Cepheid Xpert Xpress) or less reliance on specialized equipment are being explored [38–40]. Much like with the development of rapid HIV tests, these are not meant to serve as a stand-in for clinical testing, but rather as a means to broaden capacity and outreach, link potential pateints to clinical care sites, and inform potentially contagious individuals of steps to limit transmission. Beyond changing the test procedure itself, assorted pooling strategies are being explored to better enable bulk testing [41,42]. Orginally developed as a way to increase the throughput of blood borne pathogen testing in the blood supply, these strategies rely on pooling a given number of patient samples prior to testing [26,43]. Individual specimens that compose negative pools are presumed negative, while individual specimens that compose positive pools are then routed to individual testing. The number of samples in each pool is dictated by both the local positivity rate and the overall sensitivity of the assay. Many of these strategies and others are being developed with support from the National Institutes of Health Rapid Acceleration of Diagnostics (RADx) programme [40,44].
In sum, molecular diagnostics have become a vital tool in epidemic management to link people to care, monitor transmission, enact preventative measures and answer the most basic of epidemiological questions: who is being or has been infected? Although these tests may seem definitive, the sensitivity and specificity of the test, its implementation, and the exact nature of the readout dictate the strengths and weaknesses of each approach. As such, they are best leveraged in the context of complete clinical care and health surveillance that includes symptom monitoring, contact tracing, and risk communication. Improved study of the limitations of these tests in clinical practice and improved communication of these limitations to health providers and patients will go a long way towards optimal implementation of molecular diagnostics in the context of an ongoing epidemic.
Virus genome sequencing
Although molecular diagnostics have become vital tools for infectious disease surveillance, genetic sequencing approaches have likewise matured to become important tools for understanding disease cause and transmission. The genetic information obtained from viral sequences is a high-resolution source of information that can be used not only for studying biological properties of pathogenic viruses, but also for understanding critical elements of viral spread that inform and direct public health policies [45–48]. The extraordinary development of sequencing technologies and analysis methods even within the last decade have accelerated these trends in recent years. In addition, the establishment of publicly available sequence databases such as the Los Alamos HIV sequence database, HIV sequence compendium, GenBank, and GISAID have provided scientists with rapid access to global viral sequences for an increased analysis power and much faster response for monitoring epidemic trends.
The first HIV genomic sequences were described in a series of papers in 1985 [49,50]. Due to the relatively low abundance of viral DNA in patient tissues, these viruses were first passaged in tissue culture cells prior to molecular cloning and sequencing using dideoxynucleotide chain termination. Eventually, methods were developed to sequence clinical specimens directly [51]. It quickly became clear that HIV genetic diversity was not only remarkably high, but also critically important for defining the biological nature of the disease as well as the clinical efficacy of early antiviral drugs [52–54]. Indeed, as sequencing of clinical isolates became cheaper and easier, and the clinical significance of antiviral drug resistance became clear, sequencing of patient HIV plasma RNA became a standard of care in order to check for known antiviral resistance mutations and inform the use of appropriate therapeutics [55,56]. Later, these same results began to inform public health departments of potential risk networks (see Molecularly assisted Contact Tracing below). For the clinical purposes of drug resistance and coreceptor tropism prediction, high-throughput sequencing methods that target selected regions of the HIV genome (most commonly the HIV polymerase gene, pol) are effective [57–61], though more recent whole-genome sequencing methods have been developed for other applications such as global and within-host evolutionary analyses of HIV [62–64].
Experience with sequencing HIV and other viruses has resulted in three major approaches to viral whole-genome sequencing: metagenomic, PCR amplicon, or target enrichment (reviewed in [65]). Each sequencing approach begins with extraction of total nucleic acids from the clinical specimen. When sequencing RNA viruses such as HIV or SARS-CoV-2, a reverse transcription step is first undertaken to convert RNA to cDNA. In metagenomic sequencing approaches, total cDNA obtained from the specimen is used to generate a platform-specific sequencing library for shotgun sequencing. The resulting sequence reads are then filtered to remove sequences of human host origin prior to being assembled and/or aligned to a reference genome sequence. Metagenomic sequencing has the advantage of being the fastest and most direct of the sequencing methods, requiring the fewest intermediate steps and, by minimizing PCR cycles, presents fewer opportunities for the introduction of PCR bias into the results. Metagenomic sequencing has the greatest potential for novel viral pathogen discovery and was the method used, in conjunction with targeted PCR, to first identify the SARS-CoV-2 virus sequence from a patient with COVID-19 symptoms at the beginning of the pandemic [66▪▪]. These advantages are offset, however, by the higher cost and lower sensitivity of the metagenomic approach, as considerably deeper sequencing is often required to obtain sufficient sequence relative to the large proportion of contaminating host or commensal organism nucleic acids.
In PCR amplicon sequencing, the viral genome is amplified directly from the clinical specimen using PCR primers against the viral genome sequence to generate a library of tiled and overlapping amplicons. Sequence reads are aligned to a reference viral genome sequence to identify variants. PCR amplicon sequencing approaches were quickly developed and implemented for SARS-CoV-2, and have been crucial for several early discoveries [67,68▪]. This approach has the advantage of being able to selectively enrich the viral genome prior to sequencing, minimizing the amount of contaminating DNA or RNA being sequenced. This both decreases the cost of sequencing and increases the efficiency and throughput of clinical investigations as many isolates can be indexed and sequenced simultaneously. The amplification step also improves the sensitivity for detecting viruses in low abundance in the clinical specimen. Limitations of this approach include increased complexity of the library preparation and the possibility that viral variability could result in primer mismatches and incomplete sequencing. Relative to HIV, however, the low mutation rate of SARS-CoV-2 somewhat tempers these concerns [69,70].
Target enrichment, also known as pull-down, bait, or capture assays, involve DNA or RNA probes complementary to the viral sequence bound to a solid phase such as magnetic beads or others. After library preparation from the clinical specimen, viral genomes are allowed to hybridize to leader sequences on the bead-bound probes. The captured nucleic acids then undergo a limited number of PCR amplification cycles and sequencing. Multiple target enrichment probe sets were quickly developed for SARS-CoV-2 [71,72]. This approach can include multiple probes against the same virus such that a mutation in any one region does not disrupt sequencing or get replaced with primer or probe sequence. In addition, as there are fewer PCR amplification steps, there is less risk of introducing mutations into the genome sequence not found in the host.
Several platforms for high throughput whole genome sequencing of viruses using these library preparation approaches are available. These include the short-read sequencing technologies such as Illumina's MiniSeq, MiSeq or NextSeq platforms with maximum read lengths of 300 bp and long-read platforms such as the Pacific Biosciences (PacBio) Sequel II or Oxford Nanopore MinION or GridION systems, which can each produce reads averaging several thousand bases in length. These platforms differ also in read accuracy with read sequences from short read platforms having error rates often below 1% compared with the approximately 5–15% error rate of the PacBio and Nanopore platforms [73]. The higher error rates of sequences produced on long-read platforms can complicate the resolution of HIV quasispecies in clinical specimens, which can result from prolonged chronic infection in the setting of high rates of both mutation and replication of the virus [74]. Hence, short read sequencing is currently more favoured for high throughput targeted or whole genome sequencing of HIV [75,76]. In contrast, the low mutation rate and limited infection duration of SARS-CoV-2 allow for either short or long-read sequencing platforms to be used to elucidate the sole or dominant variant in clinical specimens.
Phylogenetic and phylodynamic methods
Most viruses, especially RNA viruses, accumulate variability at very high rates due to their elevated mutation rates, high progeny production, and short replication cycles [77–81]. This variability is affected by transmission patterns, host population structure and selective processes operating on the viral population, such as immune responses or antiviral therapies [74,82–85]. Due to the effect of these factors on the viral genetic variability, we can study the viral sequences and their variability patterns to infer the different processes the viral populations have gone through. Although the high mutation rates of other viruses such as HIV (inter-host substitution rate of ∼5x10−3 substitutions/site/year [70]) or influenza virus (∼4x10−3 substitutions/site/year [86]) allow for robust variability analysis based on targeted sequencing of selected genes or genome regions, the relatively low mutation rate of SARS-CoV-2 (estimated substitution rate of ∼8x10−4 substitutions/site/year [69,87]) mandates complete viral genome sequencing to enable variability analyses.
Generally, the first step in variation analysis is to reconstruct a phylogenetic tree with a representative set of sequences by applying nucleotide substitution models that best fit the data (i.e. the model that best explains the observed rates at which each nucleotide is substituted by another) [88]. The most commonly used methods for viral phylogenetic tree reconstruction are Maximum Likelihood [89–91] and Bayesian approaches [92,93]. The inclusion of specimens from different time points allows for evolutionary rate calculation by including a temporal parameter in the tree reconstruction and assuming a time-dependent accumulation of mutations (i.e. a molecular clock) [94,95]. The inferred phylogeny will depict the genetic relationships between the viral sequences along the reconstructed branches of the tree (Fig. 2a). Using these phylogenies, analytical methods can be used to surmise epidemiological processes using both traditional phylogenetic and newly developed phylodynamic methods.
FIGURE 2.
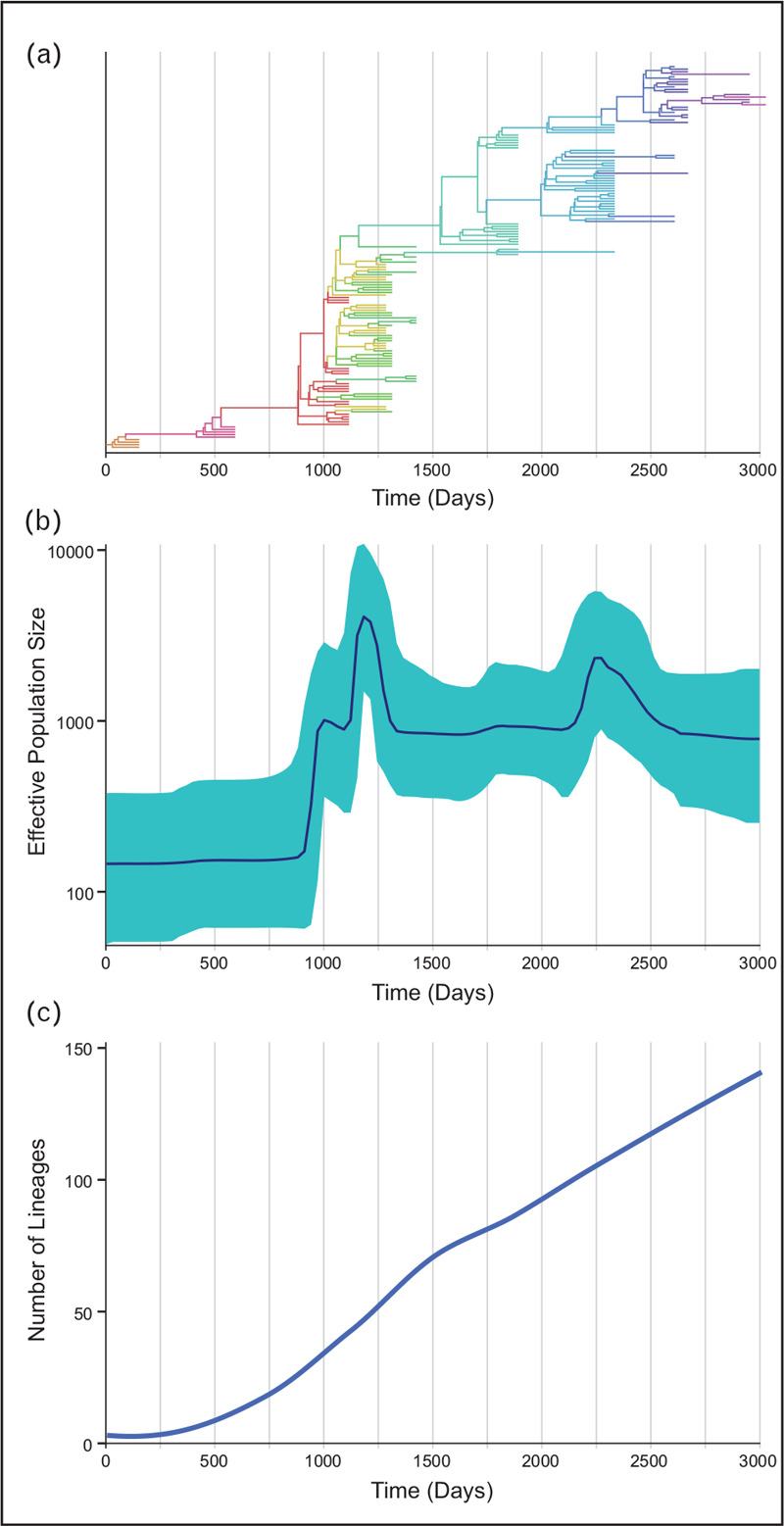
Phylodynamic methods for measuring epidemiological parameters. (a) A hypothetical, temporal phylodynamic tree with geographical information (indicated by colors) where the x-axis represents the time of sample isolation. The combination of genetic distance and spatiotemporal metadata allows for the visualization of outbreaks over time and for estimation of viral evolutionary rates, selective pressures, diversification patterns and migration events, among other parameters. (b) These same phylogenies can be used to estimate viral population sizes of the outbreak at different times. (c) The degree of genetic diversification over time, as shown by the number of lineages over time, in relation to population size changes and other inferred evolutionary dynamics, can help estimate epidemiological parameters like spread or transmission.
Traditional phylogenetic methods use molecular evolution and population genetics to infer characteristics of the viral populations being analysed, such as the most likely path of viral evolution and the origin of different viral lineages by statistical reconstruction of the most-probable ancestors of the sampled sequences along the phylogenetic tree [96,97]; the selective processes operating in the population by comparison of the rates of synonymous and nonsynonymous mutations [98–100]; and the viral population structure by measurement of the degree of compartmentalization [101,102] or neutrality [103] throughout the tree. Newer phylodynamic methods merge classic epidemiological models, such as the Susceptible-Infected-Recovered (SIR) model, with traditional phylogenetic methods to more directly study viral epidemics [74,83,104,105]. These methods allow the extraction of epidemiological parameters from the genetic information including viral origins, transmission networks, viral population size changes and risk factors (Fig. 2b,c). Moreover, these models allow the inclusion of geographical information in the analyses to study spatiotemporal dynamics of the viral populations using phylogeography [106]. Phylodynamic approaches have already been successfully applied in the study of multiple viral pathogens, including HIV-1 transmission networks [107▪,108–110,111▪▪] and epidemiological studies [105,112–114], influenza pandemics [115–117], Ebolavirus outbreaks [118–121] and hepatitis C virus studies [122,123], among others. More recently, phylodynamic analyses are being widely utilized to understand the new SARS-CoV-2 pandemic and inform public health measures [69,87,124,125].
Although traditional epidemiological studies based on surveys, detailed contact tracing, and mathematical modeling are instrumental for studying epidemics, they require careful examination of confounding factors, are limited by missing or misclassified clinical care data, and can be prone to sampling issues [126]. Likewise, molecular-based methods for estimating key epidemiological parameters face a number of limitations due to evolutionary complexities, recombination, sampling bias and incorrect rooting of the phylogenies (i.e. incorrect assumption of the most recent common ancestor) [127]. The lower mutation rate of SARS-CoV-2, in particular, necessitates a careful adaptation of some of the phylogenetic assumptions previously optimized for studying RNA viruses with higher mutation rates. However, when used properly, these methods can provide confident and precise estimates of epidemiological parameters with reasonable predictive power that is difficult to achieve through traditional approaches alone [47,104,126,128].
These types of molecular surveillance methods have already been implemented to help understand and track HIV spread in both research and public health settings. Phylogenetics were used in a landmark HIV prevention clinical trial to determine if participants on antiretroviral therapy (ART) were the source of new infections [129,130] and by the Centers for Disease Control (CDC) to help stop an outbreak of HIV among needle sharing partners [131]. Phylodynamic approaches have also enabled identification of gaps in current HIV prevention, care and treatment programmes that can improve detection, monitoring and control of chronic, local HIV subepidemics [132,133]. In this case, molecular surveillance by sampling a large statewide repository of HIV-1 sequence data identified steady onward propagation over years of certain sequences, uncovering previously unrecognized characteristics of those local subepidemics [132]. Another report assessed sources of transmission among recently infected MSM in the Netherlands, finding that the majority of recent infections could have been prevented with a public health approach informed by phylogenetic analyses [134]. Likewise, these methods have been used to describe the likely origin of the SARS-CoV-2 outbreaks around the world, identify newly emergent variants of the virus associated with higher viral loads in patient airways and define the potential for re-infection.
Molecularly assisted contact tracing
Although retrospective genetic variation analyses, such as those highlighted above, have been unquestionably valuable, real-time use of molecular surveillance may be more relevant for public health control of both HIV and SARS-CoV-2 spread. Unfortunately, the application of genome sequencing approaches and their analysis is both time and cost intensive, while public health agencies have been, and are now, resource-constrained. They generally do not have the time, analytical expertise or necessary resources to learn and apply the computationally intensive phylogenetic and phylodynamic methods above, which therefore now seem to have greater potential for real-time viral surveillance in the research setting than in public health organizations. Towards this end, another analytic methodology, HIV TRAnsmission Cluster Engine (HIVTRACE), has enabled frontline public health personnel to perform near real-time HIV molecular surveillance [111▪▪]. This method uses pairwise comparisons of genetic distances between sequences, without constructing phylogenetic trees, to identify ‘molecular clusters’ of HIV sequences. HIVTRACE has been deployed (with security measures) in recent years by the CDC to many state and city health departments across the USA [111▪▪]. This has been enabled by laws in a majority of states that mandate diagnostic laboratories to report full nucleotide sequences of a short, subgenomic region of HIV-1 (the pol gene, most commonly). These sequences are determined from plasma virion RNA as part of routine HIV care to guide selection of antiretrovirals to which an individual patient's virus is not resistant due to viral pol gene mutations. The CDC has used HIV pol sequence comparisons on a national level to bring large intra and inter-state molecular clusters to attention of relevant local jurisdictions for investigations aimed at interdicting further spread.
At state and city health departments, research is ongoing to determine how HIVTRACE analyses may further augment HIV contact tracing. Large-scale contact tracing for sexually transmitted diseases (STDs) began during World War II, and local public health agencies evolved this effort in response to the HIV epidemic into what is now called ‘partner elicitation services’. This involves a public health worker interviewing a person newly diagnosed with HIV and/or recently entering HIV care to identify all their sexual and drug-use partners (within a certain time frame). The health department then reaches out to these reported contacts privately and confidentially to determine what, if any, services would benefit them and help curtail further HIV transmission. This includes HIV testing for those who are not diagnosed with HIV. Linkage to care is arranged with the goal of enabling ART-mediated viral suppression to prevent further spread for those newly testing positive. Named partners who test negative for HIV are considered for pre-exposure prophylaxis (PrEP) if they meet indications based on ongoing behavioral risk for HIV acquisition. Viremia suppression and possibly engagement (or re-engagement) to care are services provided to named partners who are already known to have HIV.
In many communities with high HIV incidences, timely access to partner services can be challenging. Ongoing research leveraging HIVTRACE is seeking to determine best practices for molecular surveillance with the goal of increasing the efficiency of partner services while maximally interdicting further HIV spread. The concept is that molecular clusters may include some individuals with related sequences who were not named as partners by the index case. In addition, the ‘riskiest’ molecular clusters might be prioritized, so that partner services workers can prioritize identifying the larger ‘transmission clusters’ that may be more problematic for public health. HIV-infected persons who lack sequences in the database because they were either not diagnosed or did not have HIV pol genotyping results reported to the health department would be added to relevant clusters by contact tracing. This, in turn, could facilitate further public health outreach to the larger ‘risk network’ that includes both HIV-infected persons (with and without HIV sequences) as well as uninfected persons who are at-risk via reported contact with those infected person(s) (Fig. 3).
FIGURE 3.
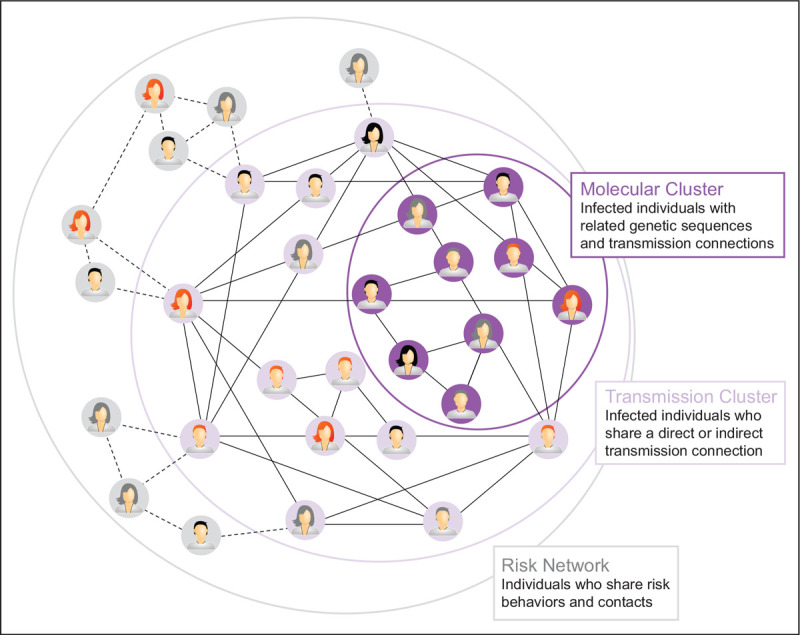
Interpretation of molecular clusters in an epidemiological outbreak. Molecular clusters identified by viral sequencing and phylogenetics represent a subset of individuals within a transmission cluster who are linked by direct or indirect transmission events. These individuals are a further subset of people in an overall risk network who may or may not have already been exposed to a disease. Determining how representative a molecular cluster is of a risk network depends on a combination of molecular surveillance, contact tracing, and community engagement.
Research applying a phylodynamic-like approach used HIVTRACE in combination with annual numbers of new diagnoses to determine that the molecular clusters with the most rapid and recent growth in the prior few years are at greatest risk for future cluster growth [110,135,136]. To define a growing cluster, the CDC now uses a tight genetic distance threshold (0.5%), a short time window for querying surveillance sequence databases (3 years), and any number above a small number of new infections (five new diagnoses) joining the molecular cluster within the most recent 12-month period [110]. However, more research is needed to define the optimal metric to monitor growing clusters and guide efforts to control further spread [137]. This includes developing more effective means to identify larger HIV transmission and risk networks from initial molecular clusters, for example, through iterative name elicitation or by using social network strategies [138–140].
How well these emerging lessons in HIV molecular surveillance might apply to SARS-CoV-2 is not yet clear. Differences in droplet/aerosol/contact route of transmission versus sexual transmission, as well as the shorter duration of infectivity of SARS-CoV-2, may make elicitation of names of potential contacts more difficult for those diagnosed with, or exposed to, SARS-CoV-2 than HIV. This could increase the potential benefits of adding molecular surveillance to public health epidemiological investigations. That being said, decreased SARS-CoV-2 genetic diversity, relative to HIV, may or may not be countered by using full genome sequences for clustering, complicating the interpretation of molecular clusters. Furthermore, the scale and pace of SARS-CoV-2 spread outstrips that of HIV, raising the possibility that sequence cluster assessments may be much more challenging to apply to COVID-19 control.
One critical lesson from these efforts for HIV is that molecular surveillance may complement, but not replace, current contact tracing that relies on elicitation of names of close contacts. It is important to note that there is far from complete concordance between partners named by an index case in a partner services interview and those persons with HIV sequences that molecularly cluster with that same index case. One report found that 48% of named partners had genetically closely related viruses, and that persons with genetically similar HIV sequences comprised 53% of named partners [141]. Persons diagnosed with HIV (female and male) who reported high-risk heterosexual contact were more likely to name at least one partner with a genetically similar virus than those reporting their HIV risk was via more stigmatized/unlawful behaviors, such as injection drug use or sex between men [141]. It is speculated that the latter HIV risk behaviours involve anonymity of partners, more partners or unwillingness to disclose names for multiple reasons; stigma and marginalization associated with those behavioral risks for HIV acquisition remain pervasive. Although only a subset of those undergoing contact tracing for SARS-CoV-2 exposure may face such repercussions for reporting (i.e. potentially based on immigration status, race, or adverse consequences enforced by educational or professional institutions), this highlights the importance of community engagement for successful control of viral spread by contact tracing. Indeed, the extensive literature on many persisting ethical issues raised by HIV molecular surveillance that still require work should inform the cautions and safeguards appropriate for research on potentially advancing molecular surveillance of SARS-CoV-2 [133,142–148].
Electronic medical record data
Although the approaches above focus on the who, what, where and when of epidemiological investigation, understanding how infectious diseases are spread and why certain people may be more at risk usually depend on the cultivation and maintenance of clinical care data that track patient demographics, spatiotemporal variables, symptoms, behaviours, medical history, exposure, timing, etc. In conjunction with molecular information, including diagnostics, viral load, viral sequence and even patient genotypic information, these datasets can yield an epidemiological goldmine of information that shed light on transmission dynamics, pathogenesis, risk factors, and treatment outcomes. Traditionally, these data had to be manually curated from medical records and case files, which was not only time consuming, but exceptionally laborious. Today, much of this information can be extracted from electronic medical record (EMR) systems, though several challenges with these types of data persist.
The AIDS pandemic began in an era without EMR systems and was first described by the CDC through standard epidemiological practices, including contact tracing, case finding and well executed case--control studies. This was done well before any laboratory had identified or sequenced HIV and this sentinel work defined routes of transmission and high-risk populations that continue to hold true today [149]. As EMR systems became popular and an essential part of medical care throughout the early to mid-2000s, HIV researchers have harnessed the power of EMRs to study real-world shifts in disease as individuals with HIV are aging and care has moved from in-hospital treatment of opportunistic infections to outpatient chronic management [150–153]. Early in the COVID-19 pandemic, EMR systems helped investigators quickly describe the clinical syndrome of hospitalized patients, define risk for poor outcomes and assess therapeutic interventions [154,155,156▪▪,157–159]. As we study the epidemiology of long-term complications of COVID-19 and of milder disease managed in the outpatient setting, we can expect to face several of the challenges we are currently facing in the use of EMR data to inform HIV research.
These global pandemics have highlighted the need for rapid, reliable and comprehensive clinical information to help inform the evolving epidemiology of old and new health problems. EMR systems can fill this role, but we must remember that they were originally developed not to serve as an epidemiological research database, but rather for documentation of clinical care, managing day to day care of patients and for billing payers of medical services. Generally, public health and epidemiological based research have been an afterthought with EMR generated data analysed in a retrospective fashion. As such, EMR datasets are often complicated by missing information, inconsistencies, subjectivity and mixtures of longitudinal and static information. Thus, although EMR-based researchers often have the power of ‘Big Data’, they are ultimately at the mercy of clinical care providers for the type and timing of data generated. EMR research requires advanced statistical methods and careful consideration of clinical data management and standardization across data systems. At times, this requires clinician case review of individual EMRs to verify information, understand the completeness of electronic data capture and minimize misclassification of critical disease outcomes [160–162].
The Centers for AIDS Research Network of Integrated Clinical Systems (CNICS; https://sites.uab.edu/cnics/) has been notably successful in harnessing the power of multi-site EMR data for HIV research and should serve as a model for how we move COVID-19 EMR-based research into the next phase of epidemiological and translational research [163–165]. This group consists of eight HIV clinical care sites throughout the USA with diverse demographics and geography. Each site is responsible for curating clinical data on HIV patients in care from the EMR and periodically submitting to the data management centre (DMC) at the University of Washington where quality checks, reconciliation and data standardization is a critical component of creating useful and rich research datasets [163,166▪]. Each site locally collects and stores biological samples (plasma and cells) annually on a subset of participants and CNICS has a centralized process to quickly identify research samples paired with clinical data. One of the unique strengths of CNICS is their approach of performing patient-reported outcome (PRO) assessments within the context of routine clinical care for in-depth longitudinal evaluations of substance/alcohol abuse, tobacco consumption, mental health, sexual behaviour and neurocognitive performance [167]. Finally, CNICS has an organized and efficient process to quickly perform research feasibility assessments, review research concept proposals, obtain letters for grant applications, give expert feedback and mentorship to investigators, and provide large research datasets. In addition to collaborative research across the network, CNICS sites have utilized their data to understand local HIV epidemics and identify potential participants for other observational or interventional HIV clinical studies.
Future COVID-19 research on the evolution of viral genetics, long-term outcomes, reinfection, immune dysfunction, immunity and risk for end organ disease will require a similar approach with a central process for standardization and verification of EMR data, robust biological sample collection, and thoughtful participant centered evaluations. A much larger national network of clinical care sites each contributing COVID-19 (and comparator) clinical data from EMRs for research analyses has rapidly been developed (https://ncats.nih.gov/n3c), providing the potential for even more powerful data analyses for COVID-19 given these lessons can be learned and applied.
As shown throughout this review, our understanding of disease diagnosis, pathogenesis and relevant outcomes will change with advancements in research and technology. At the same time, EMR systems must be flexible and evolve to play a bigger better role in providing high-quality information for public health surveillance and epidemiology research. In this way, we hope that lessons learned from years of HIV/AIDS EMR-based research can lead the path forward for COVID-19.
CONCLUSION
In the past few decades, major advances in molecular biology have revolutionized the ways in which we study and understand human health and disease. New technologies in serology, PCR, gene sequencing and computational modelling have revealed new methods to understand and identify the causative and correlative factors that drive disease. Molecular epidemiology approaches are providing new means for diagnosis, for tracking transmission and for understanding pathogenesis. These techniques, often developed in response to and optimized in conjunction with the ongoing HIV epidemic, are now being applied to the SARS-CoV-2 epidemic in force. Although the advantages of these approaches are real, they also face several limitations and challenges, particularly in regards to implementation, that must be overcome by further research to reach their full potential. In any case, these approaches have been found to complement and enhance, but not supplant, traditional means of epidemiological study. Moving forward, we should take this opportunity to advance and employ new, blended methods of traditional and molecular epidemiology to improve both our understanding and management of current and future epidemics.
Acknowledgements
None.
Financial support and sponsorship
This work was supported by the Gilead Sciences Research Scholars Program in HIV (JFH), NIH grant K22 AI136691 (JFH), NIH grant R01 AI150998 (JFH), the NIH-supported Third Coast CFAR P30 AI117943 (RLR, JFH, CJA, RTD), a supplement from the NIH-sponsored HARC Center P50 GM082250 (JFH), NIH grant R01 AI 136056 (RTD), the NIH National Center for Advancing Translational Sciences grant UL1 TR001422 (RTD), a CTSA supplement to NCATS UL1 TR002389 (JFH), the generous support of the Dixon Family Foundation (JFH, EAO), a supplement to the Northwestern University Cancer Center P30 CA060553 (JFH), as well as through a generous contribution from the Walder Foundation (JFH, EAO). The funding sources had no role in the study design, data collection, analysis, interpretation or writing of the report.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Naber SP. Molecular pathology–diagnosis of infectious disease. N Engl J Med 1994; 331:1212–1215. [DOI] [PubMed] [Google Scholar]
- 2.Jungkind D. Tech. Sight. Molecular testing for infectious disease. Science 2001; 294:1553–1555. [DOI] [PubMed] [Google Scholar]
- 3.Yang S, Rothman RE. PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings. Lancet Infect Dis 2004; 4:337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallo RC, Sarin PS, Gelmann EP, et al. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science 1983; 220:865–867. [DOI] [PubMed] [Google Scholar]
- 5.Barre-Sinoussi F, Chermann JC, Rey F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983; 220:868–871. [DOI] [PubMed] [Google Scholar]
- 6.Centers for Disease, C. Public Health Service guidelines for counseling and antibody testing to prevent HIV infection and AIDS. MMWR Morb Mortal Wkly Rep 1987; 36:509–515. [PubMed] [Google Scholar]
- 7.Centers for Disease, C. Interpretation and use of the western blot assay for serodiagnosis of human immunodeficiency virus type 1 infections. MMWR Suppl 1989; 38:1–7. [PubMed] [Google Scholar]
- 8.Centers for Disease, C. Update: serologic testing for antibody to human immunodeficiency virus. MMWR Morb Mortal Wkly Rep 1988; 36:833–840. 845. [PubMed] [Google Scholar]
- 9.Alexander TS. Human immunodeficiency virus diagnostic testing: 30 years of evolution. Clin Vaccine Immunol 2016; 23:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Branson BM. HIV testing updates and challenges: when regulatory caution and public health imperatives collide. Curr HIV/AIDS Rep 2015; 12:117–126. [DOI] [PubMed] [Google Scholar]
- 11.Delaney KP, Hanson DL, Masciotra S, et al. Time until emergence of HIV test reactivity following infection with HIV-1: implications for interpreting test results and retesting after exposure. Clin Infect Dis 2017; 64:53–59. [DOI] [PubMed] [Google Scholar]
- 12.Bentsen C, McLaughlin L, Mitchell E, et al. Performance evaluation of the Bio-Rad Laboratories GS HIV Combo Ag/Ab EIA, a 4th generation HIV assay for the simultaneous detection of HIV p24 antigen and antibodies to HIV-1 (groups M and O) and HIV-2 in human serum or plasma. J Clin Virol 2011; 52: Suppl 1: S57–S61. [DOI] [PubMed] [Google Scholar]
- 13.Salmona M, Delarue S, Delaugerre C, et al. Clinical evaluation of BioPlex 2200 HIV Ag-Ab, an automated screening method providing discrete detection of HIV-1 p24 antigen, HIV-1 antibody, and HIV-2 antibody. J Clin Microbiol 2014; 52:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franco-Paredes C, Tellez I, del Rio C. Rapid HIV testing: a review of the literature and implications for the clinician. Curr HIV/AIDS Rep 2006; 3:169–175. [DOI] [PubMed] [Google Scholar]
- 15.Myers JE, El-Sadr WM, Zerbe A, Branson BM. Rapid HIV self-testing: long in coming but opportunities beckon. AIDS 2013; 27:1687–1695. [DOI] [PubMed] [Google Scholar]
- 16.Spielberg F, Levine RO, Weaver M. Self-testing for HIV: a new option for HIV prevention? Lancet Infect Dis 2004; 4:640–646. [DOI] [PubMed] [Google Scholar]
- 17.Loussert-Ajaka I, Descamps D, Simon F, et al. Genetic diversity and HIV detection by polymerase chain reaction. Lancet 1995; 346:912–913. [DOI] [PubMed] [Google Scholar]
- 18.Busch MP, Lee LL, Satten GA, et al. Time course of detection of viral and serologic markers preceding human immunodeficiency virus type 1 seroconversion: implications for screening of blood and tissue donors. Transfusion 1995; 35:91–97. [DOI] [PubMed] [Google Scholar]
- 19.Abravaya K, Esping C, Hoenle R, et al. Performance of a multiplex qualitative PCR LCx assay for detection of human immunodeficiency virus type 1 (HIV-1) group M subtypes, group O, and HIV-2. J Clin Microbiol 2000; 38:716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mellors JW, Muñoz A, Giorgi JV, et al. Plasma viral load and CD4+ lymphocytes as prognostic markers of HIV-1 infection. Ann Intern Med 1997; 126:946–954. [DOI] [PubMed] [Google Scholar]
- 21.O’Brien WA, Hartigan PM, Martin D, et al. Changes in plasma HIV-1 RNA and CD4+ lymphocyte counts and the risk of progression to AIDS. Veterans Affairs Cooperative Study Group on AIDS. N Engl J Med 1996; 334:426–431. [DOI] [PubMed] [Google Scholar]
- 22.Michael NL, Vahey M, Burke DS, Redfield RR. Viral DNA and mRNA expression correlate with the stage of human immunodeficiency virus (HIV) type 1 infection in humans: evidence for viral replication in all stages of HIV disease. J Virol 1992; 66:310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark SJ, Saag MS, Decker WD, et al. High titers of cytopathic virus in plasma of patients with symptomatic primary HIV-1 infection. N Engl J Med 1991; 324:954–960. [DOI] [PubMed] [Google Scholar]
- 24.Schnittman SM, Greenhouse JJ, Psallidopoulos MC, et al. Increasing viral burden in CD4+ T cells from patients with human immunodeficiency virus (HIV) infection reflects rapidly progressive immunosuppression and clinical disease. Ann Intern Med 1990; 113:438–443. [DOI] [PubMed] [Google Scholar]
- 25.Cassol S, Butcher A, Kinard S, et al. Rapid screening for early detection of mother-to-child transmission of human immunodeficiency virus type 1. J Clin Microbiol 1994; 32:2641–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roth WK, Weber M, Seifried E. Feasibility and efficacy of routine PCR screening of blood donations for hepatitis C virus, hepatitis B virus, and HIV-1 in a blood-bank setting. Lancet 1999; 353:359–363. [DOI] [PubMed] [Google Scholar]
- 27.Centers for Disease Control and Prevention. Laboratory testing for the diagnosis of HIV infection. 2018. Retrieved October 7, 2020 from: https://stacks.cdc.gov/view/cdc/23447. [Google Scholar]
- 28.Nasrullah M, Wesolowski LG, Meyer WA, et al. Performance of a fourth-generation HIV screening assay and an alternative HIV diagnostic testing algorithm. AIDS 2013; 27:731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ly TD, Ebel A, Faucher V, et al. Could the new HIV combined p24 antigen and antibody assays replace p24 antigen specific assays? J Virol Methods 2007; 143:86–94. [DOI] [PubMed] [Google Scholar]
- 30▪▪.Rodger AJ, Cambiano V, Bruun T, et al. Risk of HIV transmission through condomless sex in serodifferent gay couples with the HIV-positive partner taking suppressive antiretroviral therapy (PARTNER): final results of a multicentre, prospective, observational study. Lancet 2019; 393:2428–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]; This final report from the PARTNER study strongly supports that people with HIV with an undetectable viral load cannot transmit the virus sexually, that is that Undetectable = Untransmittable.
- 31.Cheng MP, Papenburg J, Desjardins M, et al. Diagnostic testing for severe acute respiratory syndrome-related coronavirus 2: a narrative review. Ann Intern Med 2020; 172:726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32▪.Amanat F, Stadlbauer D, Strohmeier S, et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat Med 2020; 26:1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reported one of the first serology tests for SARS-CoV-2 infection.
- 33▪.Corman VM, Landt O, Kaiser M, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 2020; 25:2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reported one of the first diagnostic PCR tests for SARS-CoV-2 infection.
- 34.Mathuria JP, Yadav R, Rajkumar Laboratory diagnosis of SARS-CoV-2: a review of current methods. J Infect Public Health 2020; 13:901–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pant Pai N, Balram B, Shivkumar S, et al. Head-to-head comparison of accuracy of a rapid point-of-care HIV test with oral versus whole-blood specimens: a systematic review and meta-analysis. Lancet Infect Dis 2012; 12:373–380. [DOI] [PubMed] [Google Scholar]
- 36.Centers for Disease Control and Prevention. Coronavirus Disease 2019 (COVID-19) diagnostic testing. https://www.cdc.gov/coronavirus/2019-ncov/lab/testing.html 2020. [Google Scholar]
- 37▪▪.Woloshin S, Patel N, Kesselheim AS. False negative tests for SARS-CoV-2 infection: challenges and implications. N Engl J Med 2020; 383:e38. [DOI] [PubMed] [Google Scholar]; This perspective provides a careful discussion of false negative testing for SARS-CoV-2 and the implications of these results for testing strategy, clinical care and epidemiology.
- 38.Gibani MM, Toumazou C, Sohbati M, et al. Assessing a novel, lab-free, point-of-care test for SARS-CoV-2 (CovidNudge): a diagnostic accuracy study. Lancet Microbe 2020; doi: 10.1016/S2666-5247(20)30121-X. [Online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Czumbel LM, Kiss S, Farkas N, et al. Saliva as a candidate for COVID-19 diagnostic testing: a meta-analysis. Front Med (Lausanne) 2020; 7:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tromberg BJ, Schwetz TA, Pérez-Stable EJ, et al. Rapid scaling up of Covid-19 diagnostic testing in the United States: the NIH RADx Initiative. N Engl J Med 2020; 383:1071–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yelin I, Aharony N, Shaer Tamar E, et al. Evaluation of COVID-19 RT-qPCR test in multi-sample pools. Clin Infect Dis 2020; doi: 10.1093/cid/ciaa531. [Online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hogan CA, Sahoo MK, Pinsky BA. Sample pooling as a strategy to detect community transmission of SARS-CoV-2. JAMA 2020; 323:1967–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vargo J, Smith K, Knott C, et al. Clinical specificity and sensitivity of a blood screening assay for detection of HIV-1 and HCV RNA. Transfusion 2002; 42:876–885. [DOI] [PubMed] [Google Scholar]
- 44.National Institutes of Health. Rapid Acceleration of Diagnostics (RADx). https://www.nih.gov/research-training/medical-research-initiatives/radx. 2020. [Google Scholar]
- 45.Grad YH, Lipsitch M. Epidemiologic data and pathogen genome sequences: a powerful synergy for public health. Genome Biol 2014; 15:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Artika IM, Wiyatno A, Ma’roef CN. Pathogenic viruses: molecular detection and characterization. Infect Genet Evol 2020; 81:104215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wohl S, Schaffner SF, Sabeti PC. Genomic analysis of viral outbreaks. Annu Rev Virol 2016; 3:173–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gwinn M, MacCannell DR, Khabbaz RF. Integrating advanced molecular technologies into public health. J Clin Microbiol 2017; 55:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wain-Hobson S, Sonigo P, Danos O, et al. Nucleotide sequence of the AIDS virus, LAV. Cell 1985; 40:9–17. [DOI] [PubMed] [Google Scholar]
- 50.Ratner L, Haseltine W, Patarca R, et al. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 1985; 313:277–284. [DOI] [PubMed] [Google Scholar]
- 51.Li Y, Hui H, Burgess CJ, et al. Complete nucleotide sequence, genome organization, and biological properties of human immunodeficiency virus type 1 in vivo: evidence for limited defectiveness and complementation. J Virol 1992; 66:6587–6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Larder BA, Darby G, Richman DD. HIV with reduced sensitivity to zidovudine (AZT) isolated during prolonged therapy. Science 1989; 243:1731–1734. [DOI] [PubMed] [Google Scholar]
- 53.Larder BA, Kemp SD. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT). Science 1989; 246:1155–1158. [DOI] [PubMed] [Google Scholar]
- 54.Saag MS, Hahn BH, Gibbons J, et al. Extensive variation of human immunodeficiency virus type-1 in vivo. Nature 1988; 334:440–444. [DOI] [PubMed] [Google Scholar]
- 55.D’Aquila RT, Johnson VA, Welles SL, et al. Zidovudine resistance and HIV-1 disease progression during antiretroviral therapy. AIDS Clinical Trials Group Protocol 116B/117 Team and the Virology Committee Resistance Working Group. Ann Intern Med 1995; 122:401–408. [DOI] [PubMed] [Google Scholar]
- 56.Japour AJ, Welles S, D’Aquila RT, et al. Prevalence and clinical significance of zidovudine resistance mutations in human immunodeficiency virus isolated from patients after long-term zidovudine treatment. AIDS Clinical Trials Group 116B/117 Study Team and the Virology Committee Resistance Working Group. J Infect Dis 1995; 171:1172–1179. [DOI] [PubMed] [Google Scholar]
- 57.Callegaro A, Di Filippo E, Astuti N, et al. Early clinical response and presence of viral resistant minority variants: a proof of concept study. J Int AIDS Soc 2014; 17:19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dudley DM, Chin EN, Bimber BN, et al. Low-cost ultra-wide genotyping using Roche/454 pyrosequencing for surveillance of HIV drug resistance. PLoS One 2012; 7:e36494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ekici H, Rao SD, Sönnerborg A, et al. Cost-efficient HIV-1 drug resistance surveillance using multiplexed high-throughput amplicon sequencing: implications for use in low- and middle-income countries. J Antimicrob Chemother 2014; 69:3349–3355. [DOI] [PubMed] [Google Scholar]
- 60.Archer J, Weber J, Henry K, et al. Use of four next-generation sequencing platforms to determine HIV-1 coreceptor tropism. PLoS One 2012; 7:e49602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swenson LC, Daumer M, Paredes R. Next-generation sequencing to assess HIV tropism. Curr Opin HIV AIDS 2012; 7:478–485. [DOI] [PubMed] [Google Scholar]
- 62.Henn MR, Boutwell CL, Charlebois P, et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog 2012; 8:e1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Malboeuf CM, Yang X, Charlebois P, et al. Complete viral RNA genome sequencing of ultra-low copy samples by sequence-independent amplification. Nucleic Acids Res 2013; 41:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luk KC, Berg MG, Naccache SN, et al. Utility of metagenomic next-generation sequencing for characterization of HIV and human pegivirus diversity. PLoS One 2015; 10:e0141723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Houldcroft CJ, Beale MA, Breuer J. Clinical and biological insights from viral genome sequencing. Nat Rev Microbiol 2017; 15:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66▪▪.Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020; 579:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the discovery of SARS-CoV-2 as the caustive agent of COVID-19 via metagenomic sequencing of clinical samples.
- 67.Quick J, Grubaugh ND, Pullan ST, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 2017; 12:1261–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68▪.Fauver JR, Petrone ME, Hodcroft EB, et al. Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell 2020; 181:990–996. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is one of several examples of the use of phylogenetics for tracing the spread and origin of SARS-CoV-2 early in the pandemic.
- 69.Boni MF, Lemey P, Jiang X, et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol 2020; 5:1408–1417. [DOI] [PubMed] [Google Scholar]
- 70.Lemey P, Rambaut A, Pybus OG. HIV evolutionary dynamics within and among hosts. AIDS Rev 2006; 8:125–140. [PubMed] [Google Scholar]
- 71.Xiao M, Liu X, Ji J, et al. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med 2020; 12:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wen S, Sun C, Zheng H, et al. High-coverage SARS-CoV-2 genome sequences acquired by target capture sequencing. J Med Virol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rang FJ, Kloosterman WP, de Ridder J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol 2018; 19:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grenfell BT, Pybus OG, Gog JR, et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004; 303:327–332. [DOI] [PubMed] [Google Scholar]
- 75.Brumme CJ, Poon AFY. Promises and pitfalls of Illumina sequencing for HIV resistance genotyping. Virus Res 2017; 239:97–105. [DOI] [PubMed] [Google Scholar]
- 76.Wymant C, Blanquart F, Golubchik T, et al. Easy and accurate reconstruction of whole HIV genomes from short-read sequence data with shiver. Virus Evol 2018; 4:vey007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Domingo E, Perales C. Viral quasispecies. PLoS Genet 2019; 15:e1008271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sanjuan R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci 2016; 73:4433–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Duffy S. Why are RNA virus mutation rates so damn high? PLoS Biol 2018; 16:e3000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanjuan R, Nebot MR, Chirico N, et al. Viral mutation rates. J Virol 2010; 84:9733–9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Peck KM, Lauring AS. Complexities of viral mutation rates. J Virol 2018; 92: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moya A, Holmes EC, Gonzalez-Candelas F. The population genetics and evolutionary epidemiology of RNA viruses. Nat Rev Microbiol 2004; 2:279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Volz EM, Koelle K, Bedford T. Viral phylodynamics. PLoS Comput Biol 2013; 9:e1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pybus OG, Rambaut A. Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet 2009; 10:540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bartha I, Carlson JM, Brumme CJ, et al. A genome-to-genome analysis of associations between human genetic variation, HIV-1 sequence diversity, and viral control. Elife 2013; 2:e01123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bedford T, Riley S, Barr IG, et al. Global circulation patterns of seasonal influenza viruses vary with antigenic drift. Nature 2015; 523:217–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gonzalez-Reiche AS, Hernandez MM, Sullivan MJ, et al. Introductions and early spread of SARS-CoV-2 in the New York City area. Science 2020; 369:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 2012; 9:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 1981; 17:368–376. [DOI] [PubMed] [Google Scholar]
- 90.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 2003; 52:696–704. [DOI] [PubMed] [Google Scholar]
- 91.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006; 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- 92.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001; 17:754–755. [DOI] [PubMed] [Google Scholar]
- 93.Bouckaert R, Vaughan TG, Barido-Sottani J, et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol 2019; 15:e1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Drummond AJ, Ho SY, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol 2006; 4:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Firth C, Kitchen A, Shapiro B, et al. Using time-structured data to estimate evolutionary rates of double-stranded DNA viruses. Mol Biol Evol 2010; 27:2038–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang Z, Kumar S, Nei M. A new method of inference of ancestral nucleotide and amino acid sequences. Genetics 1995; 141:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Joy JB, Liang RH, McCloskey RM, et al. Ancestral reconstruction. PLoS Comput Biol 2016; 12:e1004763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Suzuki Y. New methods for detecting positive selection at single amino acid sites. J Mol Evol 2004; 59:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith MD, Wertheim JO, Weaver S, et al. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol 2015; 32:1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kosakovsky Pond SL, Frost SD. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 2005; 22:1208–1222. [DOI] [PubMed] [Google Scholar]
- 101.Zarate S, Pond SL, Shapshak P, Frost SD. Comparative study of methods for detecting sequence compartmentalization in human immunodeficiency virus type 1. J Virol 2007; 81:6643–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting F(ST). Nat Rev Genet 2009; 10:639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bhatt S, Katzourakis A, Pybus OG. Detecting natural selection in RNA virus populations using sequence summary statistics. Infect Genet Evol 2010; 10:421–430. [DOI] [PubMed] [Google Scholar]
- 104.Kuhnert D, Wu CH, Drummond AJ. Phylogenetic and epidemic modeling of rapidly evolving infectious diseases. Infect Genet Evol 2011; 11:1825–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Volz EM, Kosakovsky Pond SL, Ward MJ, et al. Phylodynamics of infectious disease epidemics. Genetics 2009; 183:1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Comput Biol 2009; 5:e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107▪.Ratmann O, Grabowski MK, Hall M, et al. Inferring HIV-1 transmission networks and sources of epidemic spread in Africa with deep-sequence phylogenetic analysis. Nat Commun 2019; 10:1411. [DOI] [PMC free article] [PubMed] [Google Scholar]; This manuscript describes the use of HIV sequencing and phylogenetics to inform and provide directinality to tranmission networks.
- 108.Dennis AM, Volz E, Frost ASMSDW, et al. HIV-1 transmission clustering and phylodynamics highlight the important role of young men who have sex with men. AIDS Res Hum Retroviruses 2018; 34:879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Leitner T. Phylogenetics in HIV transmission: taking within-host diversity into account. Curr Opin HIV AIDS 2019; 14:181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Oster AM, France AM, Panneer N, et al. Identifying clusters of recent and rapid HIV transmission through analysis of molecular surveillance data 2018; 79:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111▪▪.Kosakovsky Pond SL, Weaver S, Leigh Brown AJ, Wertheim JO. HIV-TRACE (TRAnsmission Cluster Engine): a tool for large scale molecular epidemiology of HIV-1 and other rapidly evolving pathogens. Mol Biol Evol 2018; 35:1812–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents a computational approach to identify molecular clusters of transmission in near real-time, a tool that is being investigated to complement tranditional contact tracing in community settings.
- 112.Wertheim JO, Leigh Brown AJ, Hepler NL, et al. The global transmission network of HIV-1. J Infect Dis 2014; 209:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rodrigo AG, Shpaer EG, Delwart EL, et al. Coalescent estimates of HIV-1 generation time in vivo. Proc Natl Acad Sci U S A 1999; 96:2187–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gifford RJ, de Oliveira T, Rambaut A, et al. Phylogenetic surveillance of viral genetic diversity and the evolving molecular epidemiology of human immunodeficiency virus type 1. J Virol 2007; 81:13050–13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bedford T, Cobey S, Beerli P, Pascual M. Global migration dynamics underlie evolution and persistence of human influenza A (H3N2). PLoS Pathog 2010; 6:e1000918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Koelle K, Cobey S, Grenfell B, Pascual M. Epochal evolution shapes the phylodynamics of interpandemic influenza A (H3N2) in humans. Science 2006; 314:1898–1903. [DOI] [PubMed] [Google Scholar]
- 117.Fraser C, Donnelly CA, Cauchemez S, et al. Pandemic potential of a strain of influenza A (H1N1): early findings. Science 2009; 324:1557–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Alizon S, Lion S, Murall CL, Abbate JL. Quantifying the epidemic spread of Ebola virus (EBOV) in Sierra Leone using phylodynamics. Virulence 2014; 5:825–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carroll MW, Matthews DA, Hiscox JA, et al. Temporal and spatial analysis of the 2014-2015 Ebola virus outbreak in West Africa. Nature 2015; 524:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dudas G, Rambaut A. Phylogenetic analysis of Guinea 2014 EBOV ebolavirus outbreak. PLoS Curr 2014; 6: doi: 10.1371/ecurrents.outbreaks.84eefe5ce43ec9dc0bf0670f7b8b417d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gire SK, Goba A, Andersen KG, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014; 345:1369–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Magiorkinis G, Magiorkinis E, Paraskevis D, et al. The global spread of hepatitis C virus 1a and 1b: a phylodynamic and phylogeographic analysis. PLoS Med 2009; 6:e1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pybus OG, Charleston MA, Gupta S, et al. The epidemic behavior of the hepatitis C virus. Science 2001; 292:2323–2325. [DOI] [PubMed] [Google Scholar]
- 124.Lu J, et al. Genomic epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell 2020; 181:997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lorenzo-Redondo R, Nam HH, Roberts SC, et al. A unique clade of SARS-CoV-2 viruses is associated with lower viral loads in patient upper airways. medRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rife BD, Mavian C, Chen X, et al. Phylodynamic applications in 21(st) century global infectious disease research. Glob Health Res Policy 2017; 2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Frost SD, Pybus OG, Gog JR, et al. Eight challenges in phylodynamic inference. Epidemics 2015; 10:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pybus OG, Fraser C, Rambaut A. Evolutionary epidemiology: preparing for an age of genomic plenty. Philos Trans R Soc Lond B Biol Sci 2013; 368:20120193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cohen MS, Chen YQ, McCauley M, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med 2011; 365:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ping LH, Jabara CB, Rodrigo AG, et al. HIV-1 transmission during early antiretroviral therapy: evaluation of two HIV-1 transmission events in the HPTN 052 prevention study. PLoS One 2013; 8:e71557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Peters PJ, Pontones P, Hoover KW, et al. HIV infection linked to injection use of oxymorphone in Indiana, 2014–2015. N Engl J Med 2016; 375:229–239. [DOI] [PubMed] [Google Scholar]
- 132.Dennis AM, Hué S, Billock R, et al. Human immunodeficiency virus type 1 phylodynamics to detect and characterize active transmission clusters in North Carolina. J Infect Dis 2020; 221:1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.France AM, Oster AM. The promise and complexities of detecting and monitoring HIV transmission clusters. J Infect Dis 2020; 221:1223–1225. [DOI] [PubMed] [Google Scholar]
- 134.Ratmann O, van Sighem A, Bezemer D, et al. Sources of HIV infection among men having sex with men and implications for prevention. Sci Transl Med 2016; 8:320ra322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wertheim JO, Murrell B, Mehta SR, et al. Growth of HIV-1 molecular transmission clusters in New York City. J Infect Dis 2018; 218:1943–1953. doi:10.1093/infdis/jiy431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wertheim JO, Panneer N, France AM, et al. Incident infection in high-priority HIV molecular transmission clusters in the United States. AIDS 2020; 34:1187–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wertheim JO, Chato C, Poon AFY. Comparative analysis of HIV sequences in real time for public health. Curr Opin HIV AIDS 2019; 14:213–220. [DOI] [PubMed] [Google Scholar]
- 138.Morgan E, Skaathun B, Schneider JA. Sexual, social, and genetic network overlap: a socio-molecular approach toward public health intervention of HIV. Am J Public Health 2018; 108:1528–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pagkas-Bather J, Young LE, Chen YT, Schneider JA. Social network interventions for HIV transmission elimination. Curr HIV/AIDS Rep 2020; 17:450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kimbrough LW, Fisher HE, Jones KT, et al. Accessing social networks with high rates of undiagnosed HIV infection: the social networks demonstration project. Am J Public Health 2009; 99:1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wertheim JO, Kosakovsky Pond SL, Forgione LA, et al. Social and genetic networks of HIV-1 transmission in New York City. PLoS Pathog 2017; 13:e1006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.McClelland A, Guta A, Gagnon M. The rise of molecular HIV surveillance: implications on consent and criminalization. Crit Public Health 2020; 30:487–493. [Google Scholar]
- 143.Gilbert M, Swenson L, Unger D, et al. Need for robust and inclusive public health ethics review of the monitoring of HIV phylogenetic clusters for HIV prevention. Lancet HIV 2016; 3:e461. [DOI] [PubMed] [Google Scholar]
- 144.Schairer C, Mehta SR, Vinterbo SA, et al. Perceptions of molecular epidemiology studies of HIV among stakeholders. J Public Health Res 2017; 6:992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Centers for Disease Control and Prevention. Data security and confidentiality guidelines. 2020. Retrieved October 7, 2020 from: https://www.cdc.gov/nchhstp/programintegration/docs/PCSIDataSecurityGuidelines.pdf. [Google Scholar]
- 146.National Alliance of State and Territorial AIDS Directors. HIV data privacy and confidentiality: legal and ethical considerations for health department data sharing. 2018. Retrieved October 7, 2020 from: https://www.nastad.org/resource/hiv-data-privacyandconfidentiality. [Google Scholar]
- 147.Dawson L, Benbow N, Fletcher FE, et al. Addressing ethical challenges in US-based HIV phylogenetic research. J Infect Dis 2020; doi: 10.1093/infdis/jiaa107. [Online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Evans D, Benbow N. Project Inform and Northwestern University. 2018; doi: 10.18131/G3MT7B. [Google Scholar]
- 149.Jaffe HW. Lessons from the early HIV/AIDS epidemic. AIDS 2018; 32:1719–1721. [DOI] [PubMed] [Google Scholar]
- 150.Todd CS, Mills SJ, Innes AL. Electronic health, telemedicine, and new paradigms for training and care. Curr Opin HIV AIDS 2017; 12:475–487. [DOI] [PubMed] [Google Scholar]
- 151.Castelnuovo B, Kiragga A, Afayo V, et al. Implementation of provider-based electronic medical records and improvement of the quality of data in a large HIV program in Sub-Saharan Africa. PLoS One 2012; 7:e51631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Greenberg AE, Hays H, Castel AD, et al. Development of a large urban longitudinal HIV clinical cohort using a web-based platform to merge electronically and manually abstracted data from disparate medical record systems: technical challenges and innovative solutions. J Am Med Inform Assoc 2016; 23:635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Herwehe J, Wilbright W, Abrams A, et al. Implementation of an innovative, integrated electronic medical record (EMR) and public health information exchange for HIV/AIDS. J Am Med Inform Assoc 2012; 19:448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kuno T, Takahashi M, Obata R, Maeda T. Cardiovascular comorbidities, cardiac injury, and prognosis of COVID-19 in New York City. Am Heart J 2020; 226:24–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020; 395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156▪▪.Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 2020; 395:1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is one of the first reports to describe clinical risk factors for severe COVID-19 using emergency medical record data.
- 157.Petrilli CM, Jones SA, Yang J, et al. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ 2020; 369:m1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pryor R, Atkinson C, Cooper K, et al. The electronic medical record and COVID-19: is it up to the challenge? Am J Infect Control 2020; 48:966–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chhiba KD, Patel GB, Vu THT, et al. Prevalence and characterization of asthma in hospitalized and nonhospitalized patients with COVID-19. J Allergy Clin Immunol 2020; 146:307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Coorevits P, Sundgren M, Klein GO, et al. Electronic health records: new opportunities for clinical research. J Intern Med 2013; 274:547–560. [DOI] [PubMed] [Google Scholar]
- 161.Cowie MR, Blomster JI, Curtis LH, et al. Electronic health records to facilitate clinical research. Clin Res Cardiol 2017; 106:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Floris-Moore M, Edmonds A, Napravnik S, Adimora AA. Computerized adjudication of coronary heart disease events using the electronic medical record in HIV clinical research: possibilities and challenges ahead. AIDS Res Hum Retroviruses 2020; 36:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kitahata MM, Rodriguez B, Haubrich R, et al. Cohort profile: the Centers for AIDS Research Network of Integrated Clinical Systems. Int J Epidemiol 2008; 37:948–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Aldous JL, Pond SK, Poon A, et al. Characterizing HIV transmission networks across the United States. Clin Infect Dis 2012; 55:1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mugavero MJ, Napravnik S, Cole SR, et al. Viremia copy-years predicts mortality among treatment-naive HIV-infected patients initiating antiretroviral therapy. Clin Infect Dis 2011; 53:927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166▪.Hood JE, Bradley H, Hughes JP, et al. Reconciling the evaluation of co-morbidities among HIV care patients in two large data systems: the Medical Monitoring Project and CFAR Network of Integrated Clinical Systems. AIDS Care 2018; 30:1551–1559. [DOI] [PubMed] [Google Scholar]; A recent example from the multicentre CNICS collaborative on the challenges and rewards of integrating emergency medical record data.
- 167.Rudolph JE, Cole SR, Edwards JK, et al. At-risk alcohol use among HIV-positive patients and the completion of patient-reported outcomes. AIDS Behav 2018; 22:1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]