

Genome-wide RNA sequencing revealed a broader regulatory role for the TyrR transcription factor in the ecologically versatile bacterium Enterobacter ludwigii beyond that of aromatic amino acid synthesis and transport that constitute the role of the TyrR regulon of E. coli. In E. ludwigii, a plant symbiont and human gut commensal, the TyrR regulon is expanded to include genes that are beneficial for plant interactions and response to stresses. Identification of the genes regulated by TyrR provides insight into the mechanisms by which the bacterium adapts to its environment.
KEYWORDS: Cpx envelop stress response, Enterobacter ludwigii, RNA-Seq, TyrR
ABSTRACT
The TyrR transcription factor controls the expression of genes for the uptake and biosynthesis of aromatic amino acids in Escherichia coli. In the plant-associated and clinically significant proteobacterium Enterobacter ludwigii UW5, the TyrR orthologue was previously shown to regulate genes that encode enzymes for synthesis of the plant hormone indole-3-acetic acid and for gluconeogenesis, indicating a broader function for the transcription factor. This study aimed to delineate the TyrR regulon of E. ludwigii by comparing the transcriptomes of the wild type and a tyrR deletion strain. In E. ludwigii, TyrR positively or negatively regulates the expression of over 150 genes. TyrR downregulated expression of envelope stress response regulators CpxR and CpxP through interaction with a DNA binding site in the intergenic region between divergently transcribed cpxP and cpxR. Repression of cpxP was alleviated by tyrosine. Methyltransferase gene dmpM, which is possibly involved in antibiotic synthesis, was strongly activated in the presence of tyrosine and phenylalanine by TyrR binding to its promoter region. TyrR also regulated expression of genes for aromatic catabolism and anaerobic respiration. Our findings suggest that the E. ludwigii TyrR regulon has diverged from that of E. coli to include genes for survival in the diverse environments that this bacterium inhabits and illustrate the expansion and plasticity of transcription factor regulons.
IMPORTANCE Genome-wide RNA sequencing revealed a broader regulatory role for the TyrR transcription factor in the ecologically versatile bacterium Enterobacter ludwigii beyond that of aromatic amino acid synthesis and transport that constitute the role of the TyrR regulon of E. coli. In E. ludwigii, a plant symbiont and human gut commensal, the TyrR regulon is expanded to include genes that are beneficial for plant interactions and response to stresses. Identification of the genes regulated by TyrR provides insight into the mechanisms by which the bacterium adapts to its environment.
INTRODUCTION
Expression of genes for aromatic amino acid biosynthesis and uptake in Escherichia coli is regulated by the transcription factor TyrR, which represses or activates nine separate transcriptional units encompassing 12 genes (1, 2). TyrR controls the expression of genes encoding enzymes of the shikimate pathway for synthesis of chorismate (aroF, aroG, and aroL); for production of the aromatic amino acids phenylalanine and tyrosine (tyrA and tyrB) and folic acid (folA), which are derived from chorismate; genes encoding several aromatic amino acid transporters (aroP, mtr, and tyrP); and TyrR itself (tyrR) (1, 2). To regulate expression of these genes, TyrR binds to DNA at sites related to the consensus sequence TGTAAA-N6-TTTACA that overlap or are adjacent to sigma-70 promoters (3–5). The degree of agreement with the consensus sequence affects the affinity of TyrR binding to the sequence (3, 5). Generally, strong TyrR boxes have at least 10 nucleotide matches to the consensus, a high degree of sequence symmetry, and an AT-rich spacer sequence, while weak boxes have fewer than 10 matching nucleotides and less symmetry (6, 7). A key feature of functional TyrR boxes is the invariable G-N14-C bases; changes to either nucleotide result in a loss of TyrR binding. Each of the three aromatic amino acids, namely, tryptophan, phenylalanine, and tyrosine, functions as cofactors for TyrR, modulating its regulatory action such that each regulon member is uniquely controlled. The aromatic amino acids bind TyrR at two sites, one ATP-independent binding site for tryptophan and phenylalanine and a second ATP-dependent tyrosine binding site (1, 5). TyrR exists as a dimer in the absence of cofactors (8), but in the presence of tyrosine and ATP it undergoes further polymerization to form a hexamer, which may then interact simultaneously with multiple TyrR boxes (9, 10). Phenylalanine can facilitate the cooperative binding of TyrR dimers across multiple boxes through dimer-dimer interactions (11, 12).
TyrR functions as both a transcriptional repressor and activator, in some cases exhibiting both capacities at the same promoter. Promoters repressed by TyrR in the presence of tyrosine contain at least two adjacent TyrR boxes, one strong and one weak (7). The weak box is typically closer to, or overlapping with, the core promoter (−35 or −10 elements), and cofactor-mediated binding at this low-affinity site requires an adjacent strong box. Repression may involve the exclusion of RNA polymerase from the promoter, as is the case for tyrP, or the inhibition of subsequent steps of transcription, such as open complex formation or promoter escape, as for tyrB (13–16). TyrR activation of transcription occurs through direct interactions with the RNA polymerase alpha subunit carboxy-terminal domain, which recruits RNA polymerase to the promoter or otherwise stabilizes its interaction with the DNA (1). This requires TyrR to be positioned upstream of the −35 promoter element and on the same face of DNA to allow direct contact between the N-terminal domain of TyrR and RNA polymerase (17, 18). A further requirement for TyrR-mediated gene activation is an imperfect promoter, as seen with the GC-rich −10 elements of the tyrP, mtr, and aroPP3 genes (1, 19). In all instances of TyrR regulation, the location of the TyrR boxes and the spacing between them and the promoter are crucial determinants for the mode of transcriptional control exhibited by TyrR, which is further modulated by the oligomerization status of the protein influenced by the presence of aromatic amino acids (12, 14).
The TyrR regulon has been extensively studied in E. coli; however, in other gammaproteobacteria, additional TyrR-regulated genes have been identified that encode functions accessory to or beyond aromatic amino acid biosynthesis and uptake. The tpl gene of Citrobacter freundii and Erwinia herbicola, encoding tyrosine phenol lyase for tyrosine catabolism, is upregulated by cooperative TyrR binding across three boxes in conjunction with DNA-bending protein integration host factor (IHF) and cAMP receptor protein (20–22). In the entomopathogenic bacterium Photorhabdus luminescens, expression of the phenylalanine-ammonium lyase gene sltA is controlled by TyrR and is required for production of a stilbene antibiotic (23). The acid-inducible genes aniC and hyaB of Salmonella enterica subsp. enterica serovar Typhimurium, encoding an arginine:agmatine symporter and hydrogenlyase subunit, respectively, are activated by TyrR with tyrosine under acidic growth conditions (24). The Pseudomonas TyrR homologue, PhhR, controls genes for metabolism of aromatic compounds and amino acid catabolism, in addition to those for aromatic amino acid biosynthesis (25–27). Rodionov et al. inferred from reconstruction of transcriptional networks that TyrR has undergone regulon expansion in the Shewanella genus, controlling degradation pathways for phenylalanine, tyrosine, branched-chain amino acids, proline, and oligopeptides, in addition to the glyoxylate cycle for carbohydrate synthesis (28). Furthermore, genomic SELEX and bioinformatic analysis suggest that there are more TyrR regulon members in E. coli; however, direct regulation of the predicted TyrR-controlled genes is yet to be demonstrated (1, 2). The differences in TyrR-regulated genes among these bacteria may reflect adaptations to different habitats.
Enterobacter ludwigii, a member of the E. cloacae complex, is a plant growth-promoting rhizobacterium, as well as an enteric commensal and opportunistic human pathogen (29, 30). E. ludwigii UW5 (renamed from Enterobacter cloacae UW5 based on genomic average nucleotide identity) synthesizes high levels of the plant growth hormone indole-3-acetic acid (IAA) when tryptophan is supplied exogenously (29). In this bacterium, IAA is produced from tryptophan via indole-3-pyruvate, which is converted to indole-3-acetaldehyde by indolepyruvate decarboxylase, encoded by ipdC. The expression of ipdC is wholly dependent on TyrR and is upregulated by aromatic amino acids, especially phenylalanine (29, 31). Transcriptional activation of ipdC occurs by TyrR binding to a promoter-proximal strong box centered 30 bp upstream from the −35 element. A second TyrR box further upstream overlaps the −10 element of the divergently transcribed akr gene promoter and represses its expression (31). This second weak box has atypical spacing of the left and right palindromic arms, which are separated by 2 or 8 base pairs rather than by the canonical 6, yet binding by TyrR has been demonstrated in vitro (31). The akr homologue in E. coli encodes an l-glyceraldehyde-3-phosphate reductase, which along with l-glyceraldehyde dehydrogenase generates dihydroxyacetone phosphate for gluconeogenesis (32). The novel finding that, in E. ludwigii, TyrR controls expression of enzymes for tryptophan catabolism and gluconeogenesis indicates that its function is more diverse than that of E. coli TyrR. In this study, we aimed to delineate the TyrR regulon in E. ludwigii UW5 by comparing the genome-wide transcriptional profiles in the wild-type and a tyrR mutant strain. We found that TyrR controls an expanded regulon in E. ludwigii, significantly altering expression of genes and operons for anaerobic respiration and aromatic catabolism and directly activating transcription of a methyltransferase gene. Moreover, TyrR mediates changes in the Cpx stress envelope response pathway by repressing transcription of the cpx operon.
RESULTS
RNA sequencing reveals a large number of TyrR-regulated genes.
The scope of gene regulation by TyrR was assessed by comparing the global transcription profiles of wild-type E. ludwigii and a markerless tyrR deletion mutant by RNA sequencing. Four independent biological replicates of each strain were grown in Luria-Bertani (LB) medium to the mid-log phase for purification of RNA and subsequent sequencing. Each of the aromatic amino acids that are cofactors for TyrR is present in LB medium at concentrations (≥1 mM) that are typically used for gene expression studies with TyrR (33). Extracted RNA was of high quality, with an average RNA integrity number (RIN) of 7.95 (maximum RIN of 10 indicates fully intact RNA) (34), and optical density at 260/280 nm (OD260/280) ratios of 1.99. On average, each sample library yielded 16.5 million reads and 3.3 billion bases, providing a good level of coverage for coding sequences and other genomic features. Deletion of tyrR resulted in 155 genes (3.47% of all genes) showing differential gene expression compared to the wild-type strain (|log2 fold change [log2FC]| > 2; false-discovery rate [FDR] threshold-adjusted P value [Padj] < 0.001) (Fig. 1; see also Tables S4 and S5 in the supplemental material). Transcription was lower for 75 of these genes in the tyrR mutant (Table S4), of which the most downregulated was dmpM (log2FC = −5.36), encoding a predicted S-adenosylmethionine (SAM) O-methyltransferase. Eighty genes were derepressed (Table S5), with the highest fold increase observed for yebE (log2FC = 6.16), which encodes a predicted inner membrane protein of unknown function. The reliability of the transcriptome sequencing (RNA-Seq) data was supported by the relative expression levels of the ipdC locus (see Fig. S1A in the supplemental material), which is known to require TyrR for activation (29, 31). The RNA-Seq data confirm previous findings, showing strong downregulation of ipdC (log2FC = −4.28) and weak, but significant, upregulation of akr (log2FC = 1.11; Padj < 0.001) in the tyrR mutant strain. Consistent with reduced ipdC expression, production of the phytohormone IAA in the tyrR mutant was abolished (Fig. S1B).
FIG 1.

Volcano plot displaying differentially expressed genes in E. ludwigii wild-type and tyrR mutant strains. The green dots indicate genes whose transcripts increased by >2 log2FC (false discovery rate [FDR]-adjusted P < 0.001) in the tyrR mutant compared to the wild-type strain (n = 75); the red dots indicate genes whose transcripts were decreased by >2 log2FC (FDR-adjusted P value < 0.001) (n = 80). Genes whose expression changed by |log2FC| of >3 are labeled. The y axis corresponds to the negative base 10 logarithm of the adjusted P values, and the x axis displays the log2 fold change (log2FC) value. The horizontal and vertical dotted lines indicate the threshold criteria of a Benjamini-Hochberg corrected P value of 0.001 and a |log2FC| value of 2, respectively.
Orthologs of known E. coli TyrR-regulated genes are similarly regulated in E. ludwigii.
TyrR regulon genes previously characterized in E. coli were similarly regulated by TyrR in E. ludwigii. In E. coli, the TyrR regulon was experimentally shown to consist of nine transcriptional units (1), and orthologs for each are present in E. ludwigii. In the E. ludwigii tyrR mutant, strong derepression of transcription was observed for aroF-tyrA, tyrP, aroP, and aroG (Table 1 and Table S5), consistent with repression by TyrR in the presence of either phenylalanine or tyrosine in E. coli (1). The aroL gene, which is also repressed by TyrR in E. coli (34), displayed increased levels of transcription in the E. ludwigii tyrR mutant (log2FC = 1.78; Padj < 0.001); however, the increase was below the 4-fold threshold for differential expression in this study. The folA gene, which is activated by TyrR in E. coli in the presence of tyrosine, was not appreciably regulated by TyrR in E. ludwigii. An examination of the sequence upstream of folA revealed that the two TyrR boxes in the E. coli folA promoter region are absent in E. ludwigii, while the IHF binding site and sigma-70 promoter are conserved (see Fig. S2 in the supplemental material). Therefore, the folA gene is not likely to be a TyrR regulon member in E. ludwigii. The mtr and tyrB genes, which are weakly regulated by TyrR in E. coli (35), were not differentially expressed by more than 4-fold in the E. ludwigii tyrR mutant (Table 1). Together, our results are in good agreement with previous measurements of TyrR-regulated genes, with regulon members displaying up- or downregulation consistent with expectations for a tyrR mutant.
TABLE 1.
TyrR regulation of aromatic amino acid transport and biosynthesis genes is similar in E. ludwigii and E. coli
| E. ludwigii locus tag | Gene | Product | Expression (log2FC)a
|
|
|---|---|---|---|---|
| E. ludwigii | E. colib | |||
| ABT55_19500 | aroF-tyrA | DAHP synthase, tyrosine repressible | 6.04 | 4.06 |
| ABT55_06645 | aroL | Shikimate kinase II | 1.78 | 1.18 |
| ABT55_16305 | tyrP | Tyrosine transporter | 4.31 | 1.74 |
| ABT55_22355 | mtr | High-affinity tryptophan transporter | −1.49 | −0.58 |
| ABT55_05725 | aroP | General aromatic amino acid transporter | 5.09 | 0.64 |
| ABT55_03500 | tyrB | Aminotransferase | −0.21 | 0.32 |
| ABT55_08825 | aroG | DAHP synthase, phenylalanine repressible | 2.75 | 0.79 |
| ABT55_05420 | folA | Dihydrofolate reductase | 1.50 | −1.30c |
RNA sequencing results were validated by quantitative RT-PCR.
To validate the RNA-Seq data and investigate the dynamics of TyrR-regulated gene expression, several genes with a range of expression levels were analyzed by quantitative reverse transcriptase PCR (RT-PCR). Genes were chosen that increased or decreased in expression in the tyrR mutant, and a known TyrR-regulated gene, aroG, was included as a control. The quantitative RT-PCR measurements of expression are in strong agreement with the values determined by RNA sequencing (Fig. 2). Two of the genes, dmpM and yagU, were consistently expressed at lower levels in the tyrR mutant compared to the wild-type strain (Fig. 2). Genes proP, eco, and aroG were highly expressed in the tyrR mutant as measured by both methods. Expression of cpxP was upregulated in the quantitative RT-PCR data (21.8-fold), but to a lesser extent than in the RNA-Seq results (65.3-fold). While the cpxR gene was not differentially expressed according to the conservative criteria used in this study, both methods indicated a consistent 3.5-fold increase in expression in a tyrR mutant background. Overall, there was a strong, significant correlation between expression levels measured by RNA-Seq and quantitative RT-PCR (Pearson’s r = 0.975; P = 1.82 × 10−4) (see Fig. S3 in the supplemental material).
FIG 2.

Verification of RNA sequencing results by quantitative reverse transcriptase PCR (RT-PCR). Differential expression of genes that were downregulated (A) or derepressed (B) in the tyrR mutant as measured by RNA-Seq (white bars) and quantitative RT-PCR (gray bars). Relative expression was determined using the threshold cycle (ΔΔCT) method for quantitative RT-PCR by comparing against the respective wild-type transcripts. The results are normalized against recA expression levels. Data represent the average from at least three biological replicates; error bars represent the standard error of the mean.
The E. ludwigii TyrR regulon has an expanded functional role.
Initial assessment of the RNA-Seq data revealed many genes with significant differential expression that were seemingly unrelated to aromatic amino acid metabolism, including genes encoding membrane-related functions such as transport, anaerobic respiration, and chemotaxis. To understand the broader function of TyrR, gene ontology (GO) terms were assigned to the E. ludwigii genome, and an enrichment analysis was performed with GOSeq using the Wallenius noncentral hypergeometric distribution for both the up- and downregulated genes in the tyrR mutant to determine which terms, if any, were significantly more frequent than the genome-wide background for those categories (36). GO terms that are enriched in the downregulated and derepressed genes for each of the three main functional classes (37) are clustered together based on semantic similarity. For all differentially expressed genes, enriched GO terms were primarily identified in the biological processes and molecular functions classifications, with fewer GO terms related to cellular components (see Fig. S4 in the supplemental material).
Analysis of significantly enriched GO terms among genes downregulated in the tyrR mutant, and therefore normally activated directly or indirectly by TyrR, included those that function in anaerobic respiration, the glyceraldehyde-3-phosphate dehydrogenase complex, formate oxidation, chemotaxis, and transmembrane signaling (Table 2). The expression of neighboring genes was often similarly upregulated, although in some cases at levels below the 4-fold cutoff, suggesting that the genes may be components of polycistronic operons (see Table S6 in the supplemental material). Operons that were downregulated included the arginine fermentation pathway operon arcABC, formate dehydrogenase operon fdnGHI, anaerobic glycerol-3-phosphate dehydrogenase operon glpABC, iron permease operon efeUOB, and threonine dehydratase operon tdcABC (Table S4). Expression of some respiratory operons was lower, including that of cytochrome d ubiquinol oxidase operon cydABXybgE and dimethyl sulfoxide (DMSO) reductase operon dmsAB. Of the respiratory nitrate reductase A operon narGHJI, only the narH gene was more than 4-fold lower in the tyrR mutant (log2FC = −2.29), while the other genes were slightly downregulated (log2FC = −1.26 to −1.70; Padj < 0.001). The fdhF gene, encoding formate dehydrogenase H, was also downregulated, as were two proteins of a hydrogenase complex. Operons for fimbria, flagella, and chemotaxis systems were also expressed at lower levels in the tyrR mutant (Table S4).
TABLE 2.
Significantly enriched GO terms among the genes up and downregulated in the tyrR mutant
| Expression | Gene ontology term(s)a | Process |
|---|---|---|
| Up | GO:0006805, GO:0071466, GO:0042178, GO:0009410 | Xenobiotic metabolism |
| GO:0042537, GO:0010124, GO:0019439, GO:1901361 | Aromatic catabolic processes | |
| GO:0072329, GO:0016054, GO:0046395 | Organic carboxylic acid breakdown | |
| Down | GO:0009061, GO:0009055 | Anaerobic respiration |
| GO:0009331, GO:0046168 | Glyceraldehyde-3-phosphate dehydrogenase | |
| GO:0015944 | Formate oxidation | |
| GO:0006935 | Chemotaxis | |
| GO:0004888 | Transmembrane signaling |
GOSeq called terms significantly enriched at a Padj value of <0.01, applying the Benjamini-Hochberg correction method.
Significantly enriched GO terms among the genes whose expression was enhanced in the tyrR mutant, and therefore normally downregulated by TyrR, included those that function in xenobiotic metabolism, aromatic catabolism, and catabolism of carboxylic acids (Table 2). Genes for central carbon metabolism, transporters, and several transcription factors were upregulated in the mutant (Table S5). Upregulated operons in the tyrR mutant included lldPRD, for conversion of l-lactaldehyde to pyruvate; paaCDEFGHI, for phenylacetate catabolism; tctCBA, encoding a tripartite citrate transporter, and its associated two-component response regulator tctDE; and acs-act, encoding an acetate/glycolate symporter and acetyl coenzyme A (acetyl-CoA) synthetase (Table S5). A 14-gene operon predicted to encode an aromatic dioxygenase and degradation pathway was also expressed at higher levels. Taken together, these results show that TyrR impacts the expression of diverse cellular processes beyond those related to aromatic amino acids.
TyrR directly activates expression of a predicted SAM-dependent O-methyltransferase.
The strongest downregulated gene in the tyrR mutant background was dmpM, which was predicted to encode an SAM-dependent O-methyltransferase (see Table S7 in the supplemental material), suggesting that TyrR is required for its expression. While originally annotated as a methyltransferase involved in the biosynthesis of the tyrosine-derived antibiotic puromycin, a BLAST search of the Streptomyces alboniger puromycin biosynthesis genes against the E. ludwigii genome indicated that E. ludwigii lacks homologous genes to produce this antibiotic. Despite sharing a conserved structural fold, SAM-dependent methyltransferases share little sequence identity and methylate a wide range of substances, making it difficult to assign physiological function to these enzymes (38). According to the structural classifications of SAM-dependent methyltransferases, the predicted DmpM polypeptide shares structural features with plant small molecule O-methyltransferases based on the presence of an extended N terminus comprised of a putative dimerization domain and an additional alpha helix inserted between beta sheet 5 and alpha helix D and the absence of alpha helix C in the main structural fold (38). The plant methyltransferases are involved in the secondary metabolism of a number of small molecules, including that of phenylpropanoids derived from phenylalanine and tyrosine. A search of the Protein Data Bank using HHpred with the DmpM amino acid sequence as a query returned several top hits for bacterial methyltransferases involved in antibiotic production and plant O-methyltransferases (39–41) (Table S7).
In wild-type E. ludwigii, expression of dmpM was significantly enhanced in the presence of tyrosine, and to a lesser extent phenylalanine, increasing 102.7- and 6.9-fold, respectively, compared to in M9 medium without aromatic amino acids (Fig. 3A). Expression in tryptophan was not significantly different from that in M9 minimal medium alone. The dmpM transcript levels were 100-fold lower in the tyrR mutant relative to those in the wild-type strain in the presence of tyrosine (Fig. 3B). These findings indicate that expression of dmpM is dependent on TyrR primarily in conjunction with tyrosine.
FIG 3.
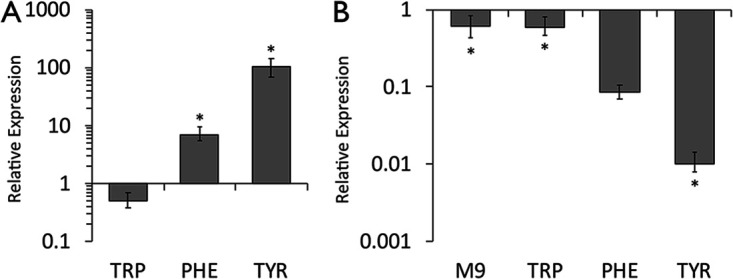
Activation of dmpM expression requires aromatic amino acids and TyrR. Relative expression of dmpM was determined in the E. ludwigii wild-type strain (A) and the isogenic tyrR mutant (B) grown in M9 minimal medium supplemented with or without 1 mM each aromatic amino acid by quantitative RT-PCR. Relative expression was determined using the ΔΔCT method by comparing to the respective wild-type transcripts in M9 minimal medium alone (A) or to the wild-type transcripts under each growth condition (B). Expression was normalized to recA expression levels. Data represent the average from at least three biological replicates; error bars represent the standard error of the mean. The asterisks indicate significant differences (P < 0.05) from the control treatment.
Two TyrR boxes were predicted in the nucleotide sequence of the dmpM promoter region (Fig. 4). Box 1 is centered 32 bp upstream from the predicted −35 sequence and matches the consensus sequence at 9/12 bp with an A/T rich spacer. The second predicted TyrR box is centered 63 bp upstream from the −35 sequence and also matches the consensus at 9/12 bp with an A/T rich spacer sequence. Thus, these binding sites are on the same face of the DNA, three and six helical turns upstream of the −35 site, respectively. Their location relative to the predicted RNA polymerase binding site, and to each other, strongly suggests that they may function as TyrR binding sites for the positive regulation of dmpM.
FIG 4.

Predicted regulatory elements in the dmpM promoter region. Two predicted TyrR binding sites are boxed, and point mutations made to each site are indicated below the sequence. The consensus sequence for the TyrR binding site is shown above the boxes. The dmpM start codon and putative −35 and −10 elements of a sigma-70 promoter are indicated in bold text. The primer binding sites used to generate probes for electrophoretic mobility shift assays (EMSAs) are marked with arrows.
DNase I footprinting assays using a 302-bp DNA probe containing the dmpM promoter region showed that TyrR protected two 20-bp regions spanning each of the predicted TyrR boxes from DNase I digestion in the presence and absence of tyrosine (Fig. 5; see also Fig. S5 in the supplemental material). Control DNase I footprinting assays, with bovine serum albumin (BSA) instead of TyrR, failed to protect the same, or any other region, of the DNA probe (see Fig. S6 in the supplemental material). Consistent with this, electrophoretic mobility shift assays (EMSAs) using a 6-carboxyfluorescein (FAM)-labeled DNA probe spanning predicted TyrR boxes 1 and 2 revealed a shift in the migration of the wild-type dmpM DNA probe incubated with purified TyrR and a decrease in the amount of free DNA with increasing protein concentration (Fig. 6). The amount of protein required to shift half of the wild-type DNA was between 0.44 μM and 0.87 μM, and at 2.19 μM almost all of the DNA probe was shifted. The migration of TyrR-bound DNA probe was further retarded as the concentration of protein increased, which may indicate multiple dimers of bound TyrR. There was no noticeable effect on the migration of the TyrR-DNA complexes when EMSAs were performed in the presence of aromatic amino acids (data not shown). Incubation of TyrR with a dmpM probe that contained key base pair substitutions in either TyrR box 1 or 2 impeded migration of the probe to a lesser extent compared to that under incubation with the wild-type probe, indicating that TyrR binds to each of the boxes (Fig. 6). The lack of a defined shifted band when the box 2 mutant probe was incubated with TyrR suggests that binding at box 2 increases binding of TyrR at box 1. A probe with mutations in both TyrR boxes failed to bind TyrR, and shifted DNA was abolished (Fig. 6). Therefore, the results of the footprinting assays and EMSAs support the direct interaction of TyrR with both of the TyrR boxes to promote expression of dmpM.
FIG 5.

DNase I footprinting assays indicate that TyrR binds to two sites upstream of the dmpM promoter. A carboxyfluorescein (FAM)-labeled PCR fragment containing the dmpM promoter region was incubated with increasing amounts (0, 0.18, 0.37, 0.75, 1.5, and 3.0 μg) of TyrR in the absence (left) or presence (right) of tyrosine prior to digestion with DNase I. Black bars highlight regions corresponding to predicted TyrR boxes 1 and 2 (underlined) that are protected from DNase I digestion as determined by capillary electrophoresis. Numbers shown on the upper trace and in the DNA sequence correspond to the nucleotide position in the PCR fragment.
FIG 6.

Sequence-specific binding of TyrR to the dmpM promoter. Electrophoretic mobility shift assays (EMSAs) were performed with 100 ng of 6-FAM-labeled DNA fragments spanning the wild-type dmpM promoter region or the same region containing point mutations in predicted TyrR boxes 1 and/or 2 (Fig. 4), and increasing concentrations (0.44, 0.87, 2.19, and 4.39 μM) of purified TyrR. The location of free unbound DNA (F) is indicated.
TyrR represses expression of envelope stress response regulators CpxP and CpxAR.
An interesting observation in the RNA-Seq data was the identification of several differentially expressed genes that are also members of the regulon of the two-component regulator CpxAR that controls the envelope stress response in E. coli. For example, the highest upregulated gene in the tyrR mutant, yebE, encoding an inner membrane protein of unknown function, is positively regulated by CpxR in E. coli (42). Expression of several other CpxR-activated genes was increased in the tyrR mutant, including ytfK, degP, aroG, yccA, ycfS, yceJ, spy, htpX, acrD, and yqaE (Table S5) (42–49). CpxR-repressed genes efeU and glpABC were highly repressed (log2FC = −2.91 to −4.98) in the tyrR mutant (Table S4). Genes controlled by CpxR in S. Typhimurium, including ybbA, ybiJ, mdtJ, eco, deoA, fimA, and dcuB, were among the differentially expressed genes (50). The cpxP and cpxA genes, which encode the periplasmic chaperone CpxP and transmembrane histidine kinase CpxA that together with the CpxR response regulator mediate the Cpx stress response, were significantly upregulated in the tyrR mutant (cpxP, log2FC = 6.03; cpxA, log2FC = 2.15) (Table S5), indicating that TyrR normally represses their expression. Expression of the cpxR gene was also higher in the mutant (log2FC = 1.81; Padj < 0.001) (Table S6), although it did not meet the conservative criteria for differential expression in this study. The divergently transcribed cpxP and cpxRA operons in E. coli, which share 84.4% nucleotide sequence identity with those of E. ludwigii, are activated by CpxR binding at three CpxR boxes in the cpxR-cpxP intergenic region (51, 52). The DNA-binding motifs recognized by CpxR and TyrR share sequence similarity, and in the E. coli aroG promoter the binding sites for these transcription factors overlap (42, 53). The enrichment of CpxR-regulated genes in the RNA-Seq data, including cpxP and cpxA, and the presence of orthologous CpxR boxes and promoter elements in the E. ludwigii cpxR-cpxP intergenic region (Fig. 7; see also Fig. S7 in the supplemental material) suggested that TyrR may repress the cpxP-RA operons by coopting the CpxR binding sites as weak TyrR boxes.
FIG 7.

Predicted regulatory elements in the intergenic region of divergently transcribed cpxR and cpxP. The locations of CpxR binding sites are boxed, and point mutations made to predicted TyrR binding sites (shaded) overlapping each are indicated below the sequence. The consensus sequence for the TyrR binding site is shown above the predicted sites. The cpxR and cpxP start codons are in bold, transcription start sites (+1) are in bold italics, and the −35 and −10 elements of the sigma-70 promoters are underlined. The primer binding sites used to generate probes for EMSAs are marked with arrows. The locations of promoter elements and CpxR binding sites are inferred from orthologous sequences in the E. coli cpx promoters, which share a high degree of sequence conservation (see Fig. S7 in the supplemental material).
The possibility that TyrR interacts with sequences in the E. ludwigii cpxP and cpxR promoter regions was examined by quantitative RT-PCR in the presence and absence of aromatic amino acids and by DNase I footprinting assays and EMSAs to assess specific protein-DNA interactions. Transcription from the cpxP promoter in wild-type E. ludwigii was significantly downregulated by tryptophan and phenylalanine (1.5- and 1.9-fold, respectively) compared to that in M9 minimal medium alone (Fig. 8A), while tyrosine induced a 24.9-fold increase in expression relative to that in M9 medium (Fig. 8A). In contrast, wild-type expression of cpxR was not different when E. ludwigii was cultured in minimal medium with or without any of the aromatic amino acids (Fig. 8A). Therefore, the downregulation of cpxP by tryptophan and phenylalanine, and upregulation by tyrosine in the wild-type strain, is not due to altered levels of CpxR. Relative to expression in the wild-type background, transcription of cpxP was significantly greater in the tyrR mutant than that in M9 medium alone (25.5-fold) or in medium supplemented with tryptophan (45.5-fold) or phenylalanine (50.1-fold) (Fig. 8B). Transcription of cpxP in tyrosine was not significantly different in the tyrR mutant relative to that in the wild-type strain. Expression of cpxR was consistently higher in the tyrR mutant strain under all conditions (3.9- to 4.8-fold increase), with aromatic amino acids having no effect (Fig. 8B). These results support the conclusion that transcription of cpxR and cpxP is repressed by TyrR and that cpxP is derepressed by tyrosine.
FIG 8.
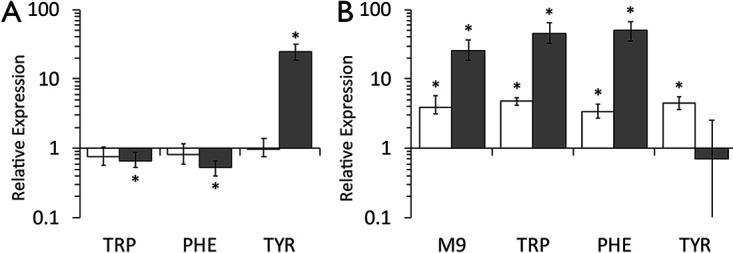
TyrR downregulates expression of the cpx operons. Relative expression of cpxR (white bars) and cpxP (gray bars) was determined in the E. ludwigii wild-type strain (A) and the isogenic tyrR mutant (B) grown in M9 minimal medium with or without 1 mM each aromatic amino acid by quantitative RT-PCR. Relative expression was determined using the ΔΔCT method by comparing to the respective wild-type transcripts in M9 minimal medium alone (A) or to the wild-type transcripts under each growth condition (B). Expression was normalized to expression levels of the recA housekeeping gene. Data represents the average of at least three biological replicates; error bars represent the standard error of the mean. The asterisks indicate significant differences (P < 0.05) from the control treatment.
While canonical TyrR boxes were not apparent in the cpxP or cpxR promoter regions, we proposed that TyrR may recognize and bind to one or more of the three CpxR DNA binding sites (Fig. 7 and Fig. S7). This hypothesis was predicated on the observations that (i) TyrR has previously been shown to bind a noncanonical site in E. ludwigii (31), (ii) TyrR may bind sequences lacking the conserved G-N14-C, although with lower affinity (54), and (iii) both TyrR and CpxR have been experimentally demonstrated to bind the same DNA sequence in the aroG promoter (42, 53), although no study has looked at the concurrent binding of both proteins. DNase I footprinting assays, using a 238-bp probe containing the cpxR-cpxP intergenic region, revealed that TyrR protected a 20-bp region encompassing a predicted TyrR binding site from DNase I digestion in the presence and absence of tyrosine (Fig. 9; see also Fig. S8 in the supplemental material). Protection of the region increased with increasing concentrations of TyrR. Control DNase I footprinting assays with BSA instead of TyrR failed to protect the same, or any other, region of the DNA probe (see Fig. S9 in the supplemental material). Binding of TyrR to the cpxR-cpxP intergenic region is supported by EMSAs. Incubation of a 6-FAM-labeled DNA probe corresponding to the wild-type cpxR-cpxP intergenic region with increasing concentrations of purified TyrR resulted in a clear retardation in the migration of the probe (Fig. 10). In the presence of 2.19 and 4.39 μM TyrR, 54% and 49% of the probes, respectively, had shifted (see Table S8 in the supplemental material). Point mutations were introduced into each of the CpxR boxes at nucleotides predicted to play similar roles to the highly conserved G and C in a TyrR box (1) (Fig. 7). Mutations in CpxR boxes 2 and 3 had no discernible effect on the ability of TyrR to bind to the DNA, as probes containing these sequences exhibited strong shifts in migration in the presence of 4.39 μM TyrR, to a position similar to the wild-type sequence. Mutations to CpxR box 1 resulted in a marked reduction in the proportion of shifted DNA probe to 19% (Fig. 10 and Table S8). Among the three predicted CpxR boxes, box 1 has the closest agreement to the TyrR consensus sequence with 8/12 bp matches, and its proximity to the −35 elements of both the cpxR and cpxP promoters may enable a bound TyrR dimer to interfere with RNA polymerase binding. In all EMSA reactions, TyrR was preincubated with a 10-fold excess of nonspecific competitor DNA to ensure sequence specific DNA binding. Inclusion of aromatic amino acids in the binding reactions did not affect TyrR-DNA migration (data not shown). These findings were reproducible in multiple experiments (data not shown), indicating that CpxR box 1 contributes to TyrR binding to the cpxR-cpxP intergenic region. Together, the results show that TyrR represses expression of the cpxP-cpxRA operons through direct binding of the promoter regions at a noncanonical site that overlaps a predicted CpxR binding site and thereby alters expression of the CpxAR regulon.
FIG 9.

DNase I footprinting assays indicate that TyrR binds to the cpxR-cpxP intergenic region. A FAM-labeled PCR fragment containing the cpxR-cpxP intergenic region was incubated with increasing amounts (0, 0.75, 1.5, 3.0, and 6.0 μg) of TyrR in the absence (left) or presence (right) of tyrosine prior to digestion with DNase I. The black bar highlights the region overlapping CpxR box 1 (Fig. 7) that is protected from DNase I digestion as determined by capillary electrophoresis. Predicted TyrR binding sites that overlap CpxR boxes 2 and 3 were not protected from DNase I digestion (see Fig. S8 in the supplemental material). Numbers shown on the upper trace and in the DNA sequence correspond to the nucleotide position in the PCR fragment; predicted TyrR binding sites are underlined.
FIG 10.

Sequence-specific binding of TyrR to the cpxR-cpxP promoter region. Electrophoretic mobility shift assays (EMSAs) were performed with 100 ng of 6-FAM-labeled DNA fragments spanning the wild-type cpxR-cpxP intergenic region or the same region containing point mutations in predicted TyrR binding sites that overlap with CpxR box 1, 2, or 3 (Fig. 7) and with increasing concentrations (0 to 4.39 μM) of purified TyrR. F, location of free unbound DNA.
DISCUSSION
The mechanism by which TyrR exerts control over gene expression is complex, and its ability to either activate or repress transcription of a target gene is dependent on a number of interconnected factors, including the number, location, and affinity of DNA binding sites, concentration of available TyrR protein in the cell, relative promoter strength of target genes, DNA conformation, nucleoid binding proteins, and the presence of aromatic amino acid cofactors (1, 55, 56). Thus, TyrR is a versatile transcription factor that integrates multiple signals to exert appropriate transcriptional control over each member of its regulon in response to physiological and environmental factors. While the regulon has been extensively studied in E. coli, there is growing evidence that it is more complex in other gammaproteobacteria that produce TyrR (23, 24, 28, 57), possibly reflecting lifestyle differences among these bacteria. In this study, the results of high-throughput RNA sequencing revealed that the TyrR regulon has undergone expansion in E. ludwigii, which is not unexpected for a bacterium that inhabits the diverse environments of the human intestinal tract and the plant rhizosphere.
The TyrR regulon overlaps the Cpx stress response regulon.
A surprising finding of this study was the overlap between the TyrR and CpxR regulons and the responsiveness of cpxP expression to aromatic amino acids. The cpxRA and cpxP operons encode a two-component signal transduction pathway, comprising sensor kinase CpxA, cytoplasmic response regulator CpxR, and periplasmic accessory protein CpxP. The E. coli proteins, which are well studied, share 96%, 95%, and 78% amino acid sequence identity, respectively, with those in E. ludwigii. CpxA autophosphorylates in response to diverse signals such as alkaline pH, high osmolarity, altered membrane lipid composition, misfolded proteins, copper, detergents, indole, EDTA, hydrophobic surfaces, or surface attachment via the outer membrane lipoprotein NlpE (52, 58–64). CpxA transphosphorylates CpxR, which controls a regulon that encompasses genes for the alleviation of envelope stress, type III secretion, motility, chemotaxis, adherence, biofilm formation, virulence, cell wall integrity, and responses to antibiotics (64–70). The primary function of CpxP is thought to be fine-tuning the Cpx response through inhibition of CpxA (58, 71, 72). It binds misfolded periplasmic proteins which titrate it away from CpxA, thereby increasing CpxA autophosphorylation and activation of CpxR (72, 73). Transcription of the cpxP gene is strongly upregulated by phosphorylated CpxR, providing a rapid negative-feedback mechanism to prevent constitutive activation of the Cpx response (74, 75).
Downregulation of the divergently transcribed cpxP and cpxRA operons by TyrR depends, at least in part, on the interaction of TyrR with a noncanonical binding site situated immediately upstream of the −35 elements of both the cpxR and cpxP promoters and overlapping a predicted CpxR binding site. The interaction between TyrR and the cpx promoters is predicted to be weak based on low sequence identity to the TyrR consensus binding site, lack of the conserved G-N14-C in the binding site, and the presence of a single TyrR box. Consistent with other DNase I footprinting studies with TyrR (53), the region of DNA protected by the protein extends two base pairs beyond the 18-bp motif required for DNA binding. This coverage would prevent both RNA polymerase and CpxR from binding to the cpx promoters, thereby repressing expression. In addition to loss of direct promoter interaction and hence derepression in the tyrR mutant, dysregulation of one or more operons due to loss of TyrR may also indirectly increase expression of the cpx operons.
Repression of cpxP is alleviated by tyrosine, which is unusual among TyrR-regulated genes, and DNase I footprinting assays suggest that this is not due to decreased affinity of TyrR for the cpxP promoter in the presence of tyrosine. Tyrosine, with ATP, induces hexamerization of TyrR and normally increases its DNA binding affinity and binding to promoter regions with multiple boxes (1). However, the presence of aromatic amino acid cofactors that alter the oligomerization status of TyrR and its relative affinity for a DNA binding site also influences the concentration of available TyrR. In the presence of tyrosine or phenylalanine, TyrR hexamers and dimers, respectively, are recruited to promoters with multiple TyrR boxes reducing available TyrR in the cell. Tyrosine-induced hexamerization may titrate TyrR away from the cpx promoters, which are predicted to be weakly bound, to other TyrR-regulated promoters that have a greater affinity for the protein, thereby derepressing cpxP expression. Consistent with this, cpxP expression levels are similar in the presence of tyrosine and in the absence of TyrR.
In addition to repression of the cpxP and cpxRA operons, TyrR may directly modulate expression of other genes of the Cpx regulon. In E. coli MG1655, CpxR activates expression of aroG, which encodes the phenylalanine-sensitive 3-deoxy-d-arabinoheptulosonate 7-phosphate (DAHP) synthase that catalyzes the initial step in synthesis of the aromatic amino acid precursor; aroG is also repressed by TyrR (42). CpxR also weakly activates expression of aroK, encoding shikimate kinase 1 of the chorismate biosynthetic pathway (42, 76). TyrR represses aroL, encoding shikimate kinase II, which is the dominant isoenzyme (77), and aroF, encoding a tyrosine-sensitive DAHP synthase (1). A current model suggests that CpxR upregulates the chorismate pathway to increase production of aromatic metabolites such as indole that enhances CpxR-mediated resistance to antimicrobial compounds (78). This may be a fine-tuning mechanism to provide aromatic metabolites when intracellular pools of chorismate are low due to TyrR-mediated repression of the chorismate biosynthetic pathway.
TyrR activates expression of a predicted SAM-dependent O-methyltransferase.
Despite the initial annotation of TyrR-activated dmpM as a SAM-dependent methyltransferase gene involved in synthesis of the antibiotic puromycin, the function of the encoded protein is unknown. SAM-dependent methyltransferases catalyze the transfer of a methyl group from SAM to a wide variety of molecular targets and are classified based on structure, substrate specificity, and the target atom for methylation (38). While they share a common structural fold, there is little conservation at the level of sequence identity, making sequence-based identification difficult. The strong upregulation of dmpM in the presence of tyrosine, and to a lesser extent phenylalanine, suggests that the encoded protein may utilize these amino acids or a metabolic derivative as the substrates. Puromycin is synthesized from tyrosine; however, E. ludwigii UW5 lacks other genes required for synthesis of this antibiotic, which seem to be restricted to Streptomyces alboniger (79). Several other bacterial SAM-methyltransferases involved in antibiotic production share a low level of sequence identity and predicted structural elements with DmpM.
The activation of dmpM transcription by TyrR fits known modes of upregulation. Activation by TyrR requires binding upstream of the −35 sequence of a promoter such that it interacts with the carboxyl-terminal domain of the alpha subunit of RNA polymerase (1). The two TyrR boxes in the dmpM promoter region are centered 32 and 63 bp upstream from the −35 element of a predicted sigma-70 promoter, placing them 3 and 6 full turns of the DNA helix, respectively, from the −35 sequence. Mutation of the E. coli tyrP promoter region to alter the spacing between the strong TyrR box and −35 sequence revealed that 31 or 32 bp between centers was optimal for transcription in the presence of phenylalanine and tyrosine (80). Therefore, the TyrR box upstream of dmpM is positioned to activate high levels of expression, as was confirmed by quantitative RT-PCR in the presence of phenylalanine and especially tyrosine. The strong (>100-fold) increase in dmpM expression in response to tyrosine suggests that this gene is upregulated by the hexameric form of TyrR.
The TyrR regulon includes genes for aromatic catabolism and anaerobic respiration.
A number of aromatic catabolic operons were repressed by TyrR in E. ludwigii, which is in contrast with the role of the orthologous regulator PhhR in Pseudomonas that is primarily responsible for activating aromatic catabolism (27). The 14-gene paa locus encoding the phenylacetate degradation pathway shares genome synteny with that of E. coli and is arranged in three transcriptional units comprised of paaZ, paaABCDEFGHIJK, and paaXY (see Fig. S10A in the supplemental material); in E. coli, expression of each operon is driven by a PaaX-controlled promoter (81). Only the main operon, paaCDEFGHI, was more than 4-fold upregulated in the tyrR mutant (see Fig. S10B and Table S5 in the supplemental material). Expression of the PaaX repressor was not significantly different between the strains (log2FC = 0.1) and there were no apparent TyrR boxes in the paaA promoter (Fig. S10C); therefore, derepression of the operon in the tyrR mutant is likely due to inhibition of PaaX by phenylacetyl-CoA, the first intermediate of the pathway (82). One or more peripheral pathways for phenylalanine catabolism may be upregulated in the tyrR mutant, yielding phenylacetate, which is then ligated with CoA by PaaK (not significantly altered in the mutant strain). These results suggest that phenylacetic acid catabolism is normally downregulated by TyrR, although it does not appear to be due to direct interaction of TyrR with the paaA promoter region or increased expression of repressor PaaX.
A second cluster of eight genes (ABT55_21185 to ABT55_21220), possibly comprising a single transcriptional unit involved in aromatic catabolism, was also significantly upregulated in the tyrR mutant, indicating that this locus is normally repressed by TyrR. The predicted functions of the genes in the cluster include an aromatic ring hydroxylating dioxygenase, several hydrolytic enzymes, a regulatory gene, and a transporter. Aromatic ring hydroxylating dioxygenases have relaxed substrate specificity and may oxidize a number of similar molecules, making assignment of function difficult without biochemical investigation (83). A putative TyrR binding site is located upstream of this operon and directly overlaps the −10 element of a predicted sigma-70 promoter.
In the tyrR mutant, significant dysregulation of genes for anaerobic metabolism was observed, which included reduced expression of respiratory nitrate reduction pathways and several dehydrogenases and terminal reductases that are components of anaerobic electron transport chains. Expression of both the membrane-bound respiratory nitrate reductase complex A (NarGHJI) for initial reduction of nitrate to nitrite and the cytoplasmic NirBD NADH-nitrite reductase for detoxificaton of nitrite and production of ammonium (84, 85) was downregulated in the E. ludwigii tyrR mutant. Nitrate reduction draws electrons from the membrane quinol pool, coupling reduction of nitrate to generation of the proton motive force (86). The nitrate-inducible formate dehydrogenase complex (fdnIHG), and pyruvate formate lyase (pflAB) were also downregulated in the mutant, suggesting that TyrR directly or indirectly upregulates both the quinone-reducing formate dehydrogenase and the quinol-oxidizing nitrate reductase that form a proton translocating redox loop. Formate, which is oxidized by formate dehydrogenase, is produced from anaerobic catabolism of organic substrates by pyruvate formate lyase (87).
Nitrate reductase may also accept electrons from both anaerobic (glpABC) and aerobic (glpD) glycerol-3-phosphate dehydrogenase, both of which were downregulated in the mutant. The nitric oxide tolerant cytochrome bd-I terminal oxidase encoded by cydAB-cydXybgE, along with Hcp/Hcr, is part of the Gram-negative response to bactericidal nitric oxide and enhances tolerance to nitrosative stress (87, 88); genes encoding these proteins were expressed at lower levels in the tyrR mutant. Other terminal reductases that were downregulated in the tyrR mutant included fumarate and DMSO reductases. The membrane potential generated by the activity of these protein complexes may drive uptake of amino acids (89, 90). Although significant changes in the expression of the respiratory complexes were observed in the tyrR mutant, it is not known whether this is due to loss of TyrR interaction with the promoter or a consequence of changes in the physiological state of the mutant brought on by loss of TyrR. Many of these pathways are regulated by specific transcription factors or are more broadly impacted by global transcription factors.
Conclusions.
In addition to confirming TyrR regulation of aromatic amino acid biosynthetic and transport genes in E. ludwigii, transcriptome analysis indicates that TyrR exerts broader control over the physiology and ecology of this bacterium, including the envelope stress response, nutrient acquisition, and energy metabolism. The results demonstrate that the TyrR regulon is more expansive in Enterobacteriaceae species than previously thought. However, the specific role of TyrR in many of these processes has yet to be investigated and may be direct through interaction with gene promoters or indirect through intricate regulatory networks with other transcription factors, such as CpxR. Nonetheless, the work presented here provides further insight into the complexity of TyrR regulatory interactions and its role beyond aromatic amino acid biosynthesis and uptake in the rhizosphere and enteric commensal bacterium E. ludwigii.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. Bacteria were routinely cultured in Luria-Bertani (LB) broth with shaking at 250 rpm and incubated at 37°C for E. coli or at 30°C for E. ludwigii. To measure TyrR-responsive gene expression, E. ludwigii was grown in M9 minimal medium supplemented with 1 mM l-tryptophan, l-phenylalanine, or l-tyrosine. Antibiotics were used at the following concentrations: 100 μg/ml ampicillin, 25 μg/ml kanamycin, 25 μg/ml gentamicin, or 25 μg/ml chloramphenicol.
Construction of a tyrR deletion mutant.
The entire tyrR coding sequence, up to and including the start and stop codons, was removed from the E. ludwigii chromosome. The tyrR sequence was initially replaced via homologous recombination with a kanamycin resistance gene flanked by flippase (Flp)-mediated Flp recognition target (FRT) sites that, following selection, was subsequently excised by Flp to avoid introduction of an antibiotic resistance gene (91). The upstream and downstream regions of the genome flanking the tyrR sequence were amplified from wild-type E. ludwigii genomic DNA using the primer pairs TUF-PstI and TUR-XbaI and TDF-XbaI and TDR-SacI, respectively (see Table S2 in the supplemental material). Purified PCR products were used as the templates for splicing by overlap extension PCR with primers TUF-PstI and TDR-SacI to yield a 1.5-kb fragment which was subsequently ligated into pGEM-T Easy (Promega), generating plasmid pTUD. Plasmid pTUD was transformed into E. coli JM109, and transformants were selected on ampicillin. The FRT-flanked kanamycin (Km) resistance gene was amplified from plasmid pKD4 using primers PKDP1 and PKDP2 and ligated into XbaI-digested pTUD, resulting in plasmid pTUD-Km, which was transformed into E. coli JM109. The 3-kb TUD-Km cassette was excised from pTUD-Km via NotI digestion and ligated with similarly digested pJQ200SK, creating pJQTUD-Km. pJQTUD-Km was transformed into E. coli S17-1λpir and subsequently introduced into wild-type E. ludwigii by conjugation. Single recombinants were identified by gentamicin and kanamycin resistance, sucrose sensitivity (conferred by sacB on pJQ200SK), and PCR amplification of the kanamycin resistance gene with primers PKDP1 and PKDP2. Subsequently, the pJQ200SK backbone was selected against by growing single recombinants overnight in LB broth with 10% sucrose, and plating on LB kanamycin with 10% sucrose. Double recombinants were screened for gentamicin sensitivity and kanamycin resistance, and verified by PCR amplification with primers TUF and TUR, and TDF and TDR, which anneal outside the region used for homologous recombination. The kanamycin resistance gene was removed by transforming calcium chloride-competent cells with plasmid pCP20 encoding Flp recombinase and selecting on chloramphenicol-containing medium. Positive transformants were grown overnight in LB broth at 37°C to induce expression of the Flp recombinase and inhibit pCP20 replication. Loss of the kanamycin resistance gene was verified by kanamycin sensitivity, PCR amplification with primers TUF and TDR, and sequencing of the amplified product. Verified tyrR mutants (E. ludwigii J224) were stored at −80°C.
IAA quantification.
Because IAA production in E. ludwigii is wholly dependent on TyrR-mediated expression of the ipdC gene (29), colorimetric quantification of IAA was used to validate the loss of TyrR activity. E. ludwigii wild-type and tyrR mutant strains were grown overnight in LB broth, pelleted, and washed twice with 1× M9 salts (Difco), before inoculation 1:100 into M9 minimal medium or M9 medium supplemented with 1 mM tryptophan. IAA production was quantified by the method of Gordon and Weber (92) after 24 h of incubation at 30°C. Culture turbidity at OD600 was recorded before pelleting 1 ml of cells and transferring 40 μl of culture supernatant to a 96-well plate containing 160 μl Salkowski’s reagent (36% [vol/vol] H2SO4, 10 mM FeCl3). Reaction mixtures were incubated at room temperature for 20 min before quantification of absorbance at OD535 on a SpectraMax M5 plate reader (Molecular Devices). Total IAA was determined by extrapolating from a standard curve of known concentration and normalized to culture turbidity.
RNA isolation and sequencing.
Overnight cultures from single colonies of wild-type and tyrR mutant E. ludwigii were inoculated 1:100 into 3 ml fresh LB in quadruplicate and incubated at 30°C and 250 rpm for ∼2.5 h until reaching the mid-logarithmic phase of growth (OD600 = 0.6 to 0.9). A total of 0.5 ml of cell suspension were then transferred to a 2 ml tube containing 1 ml RNAprotect bacteria reagent (Qiagen, Hilden, Germany), vortexed, and incubated for 5 min at room temperature before pelleting by centrifugation. Cell pellets were stored at −20°C until further use. Frozen cell pellets were thawed on ice and resuspended with 15 mg/ml lysozyme and 15 mg/ml proteinase K in 200 μl TE buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA) to lyse cells for 10 min at 4°C with periodic vortexing. Total RNA was then extracted using the RNeasy minikit (Qiagen) according to the manufacturer’s instructions. RNA quality and quantity were measured on a SpectraMax M5 plate reader and visualized by agarose gel electrophoresis to ensure sample integrity. Contaminating genomic DNA was removed by treating 5 μg total RNA in a 50 μl reaction mixture containing 5 units amplification grade DNase I (Invitrogen, CA) and 5 μl 10× DNase I digestion buffer and incubating at 37°C for 60 min. The reaction was terminated with 1 mM (final concentration) EDTA and heating at 70°C for 10 min. The DNase-treated RNA was purified using the Qiagen MinElute cleanup kit (Qiagen) and eluted into 30 μl of RNase-free water. DNA removal was verified by performing quantitative real-time PCR with the ipdC gene as the template on each DNase-treated RNA sample using primers ICRTIF and ICRTIR (see Table S2 in the supplemental material). Samples that yielded no amplicons or crossing point (Cp) values greater than 30 were considered acceptable for library preparation. The final quality and quantity of RNA samples were measured on an Agilent Bioanalyzer-RNA 6000 Pico chip (Agilent Technologies, CA). Construction of RNA-Seq libraries and sequencing were performed by the Genome Quebec Innovation Centre (Montreal, Quebec, Canada). rRNA was removed using the Ribo-Zero rRNA removal kit for Gram-negative bacteria (Illumina). The resulting enriched mRNA was used to construct a 100-bp paired-end stranded Illumina library using the TruSeq RNA library prep kit v2 (Illumina) following the manufacturer’s instructions. The libraries were individually barcoded and sequenced together on a single Illumina HiSeq 2000 lane.
RNA-Seq data analysis.
Sequence data were inspected for overall quality using FastQC v0.11.2 (S. Andrews; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Low-quality reads and contaminating Illumina TruSeq adapter sequences were removed using Trimmomatic v0.3 (93) with a sliding window size of 4 bp, cutting sequences when the average Phred score dropped below 15. Surviving paired reads were mapped to the E. ludwigii UW5 reference genome (GenBank accession number CP011798) (94) using Burrows-Wheeler Aligner (BWA) (v0.7.9-r789) (95) as single fragments with a maximum mismatch of 1%. Read alignments were sorted, indexed, and converted to BAM format with SAMtools v0.1.19-44428cd (96). Sample read alignments were visualized using the Integrative Genomics Viewer v2.3.32 (97) to ensure correct mapping of reads to genomic features. The number of reads that mapped to each genomic feature was calculated using the default parameters of HTSeq-count v0.6.1.p1 (98). Genes with significantly different levels of expression between wild-type and tyrR mutant strains were called using DESeq2 v1.12.4 (99). Genes were considered to be differentially expressed when there was at least a 4-fold change (i.e., |log2FC| > 2) in expression with a false-discovery rate (FDR) threshold-adjusted P value (Padj) of ≤0.001 as determined by the Benjamini-Hochberg correction method (99).
Pathway enrichment analysis.
Gene ontology (GO) terms were mapped to the annotated E. ludwigii UW5 genome using Blast2GO (37, 100, 101). R version 3.4.3 (102) was used for statistical analysis and plotting of data. The Bioconductor package GOseq 1.30.0 (36) was employed to identify over- and underrepresented GO terms present in the differentially expressed data set applying the Wallenius distribution approximation, and significantly enriched terms were determined by applying an FDR cutoff of 0.05 using the Benjamini-Hochberg adjustment. REVIGO was used to visualize the distribution of GO terms according to the major GO classifications (biological processes, cellular components, and molecular function) (103). Coverage plots for mapped RNA-Seq data were generated with the Bioconductor package Gviz 1.22.3 (104).
Quantitative reverse transcriptase PCR.
To quantify target mRNAs by PCR, cultures of E. ludwigii wild-type and tyrR mutant strains were grown (in triplicate) in LB broth or in M9 minimal medium supplemented with 1 mM aromatic amino acid. RNA was purified, quantified, and treated with DNase I to remove genomic DNA as described above for RNA-Seq sample preparation. cDNA was generated by mixing 100 ng DNase I-treated RNA, 20 pmol of gene-specific reverse primer (Table S2) and 10 nmol deoxynucleoside triphosphates (dNTPs) in a final volume of 13 μl. The reaction mixture was incubated at 65°C for 5 min before it was placed on ice for 1 min. To each cDNA reaction mixture, 4 μl first-strand synthesis buffer (Invitrogen), 1 μl 0.1 M dithiothreitol, 1 μl RNaseOUT (Invitrogen), and 1 μl SuperScript III reverse transcriptase (RT) (Invitrogen) were added, and the mixture was incubated at 55°C for 1 h. Controls without reverse transcriptase were prepared in identical reaction mixtures. cDNA synthesis was stopped by heating at 70°C for 10 min, and samples were stored at −20°C.
Duplicate real-time quantitative PCRs were performed with each cDNA in 25 μl reaction mixtures containing the following components: 200 nM each forward and reverse primer (Table S2), 12.5 μl Platinum Taq SYBR green mastermix (Invitrogen), and 4 μl cDNA corresponding to 100 ng total RNA. Quantitative RT-PCR was performed on a Rotor-Gene 6000 thermocycler (Corbett Life Science, Sydney, Australia) with the following cycling conditions: initial denaturation at 98°C for 2 min, followed by 40 cycles of denaturation at 98°C for 10 s, primer annealing at 51°C (aroG and proP), 57°C (recA, cpxP, and cpxR), or 61°C (yagU, eco, and dmpM) for 15 s, and elongation at 72°C for 20 s with acquisition of the fluorescent signal. A melting curve analysis was performed at the completion of each run by raising temperature from 60°C to 95°C in 1°C increments every 5 s with acquisition of fluorescent signal. The raw data were exported into LinRegPCR to determine individual reaction efficiencies and calculate corresponding Cp values (105). These values were imported into the Relative Expression Software Tool (REST) to calculate relative expression ratios between treatments, normalized with reference gene recA (106), using the threshold cycle (ΔΔCT) method (107, 108).
Purification of His6-TyrR.
Purification of His6-TyrR was performed as previously described (31). Briefly, overnight cultures of E. coli A118 were subcultured 1:100 into 50 ml fresh LB medium and grown at 37°C and 250 rpm for 4 h until reaching an OD600 value of 0.4. Expression of His6-TyrR from plasmid pQEtyrR was induced with 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG), and cells were incubated for a further 4 h before harvesting by centrifugation. Cell pellets were stored at −20°C until further use. Cell pellets were thawed on ice and resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole [pH 8.0]) with 1 mg/ml lysozyme (Sigma-Aldrich, ON, Canada) for 30 min. Cells were fully lysed while on ice with six 10-s bursts at 26 W using a Sonic Dismembrator model 100 (Fisher Scientific, ON, Canada). His6-TyrR was purified using Ni-nitrilotriacetic acid (NTA) resin (Qiagen) according to the manufacturer’s protocol for protein purification under native conditions, with the following exceptions: wash buffer 1, 50 mM NaH2PO4, 300 mM NaCl, and 20 mM imidazole (pH 8.0); wash buffer 2, 50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, and 10% glycerol (pH 8.0). Purified protein was eluted four times with 0.5 ml elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, and 8% glycerol [pH 8.0]) and stored in aliquots at −80°C.
Electrophoretic mobility shift assays.
Wild-type cpx and dmpM promoters, and cpx promoters with specific point mutations in predicted TyrR boxes (see Table S3 in the supplemental material) were synthesized by Integrated DNA Technologies (IDT, Coralville, IA) and provided on sequence-verified plasmids (Table S1). Site-directed mutagenesis was used to introduce mutations in each of the predicted TyrR boxes in the dmpM promoter (Table S3) creating pUCdmpM-mut1 and pUCdmpM-mut2 (Table S1). Triplicate PCRs were performed in 50-μl volumes containing 125 μmol each forward and reverse primer (box 1, SDM1-F and SDM1-R; box 2, SDM2-F and SDM2-R) (Table S2), 0.5 μl Phusion Taq polymerase (New England Biolabs [NEB], ON, Canada), 1 μl dNTPs, and 25 ng pUCdmpM as the template with the following cycling conditions: 95°C for 2 min, 12 cycles of at 95°C for 30 s, annealing at 60°C for 60 s, and elongation at 72°C for 90 s. PCR products were pooled and digested with 3 μl DpnI (NEB) at 37°C for 2 h. The digested DNA was PCR purified (Qiagen), eluted into 10 μl double-distilled water (ddH2O), and transformed into E. coli JM109. The TyrR box 1 and 2 double mutant (pUCdmpM-mut3) was created by repeating the mutagenesis PCR using primers SDM2-F2 and SDM2-R with pUCdmpM-mut1 as the template. All plasmid sequences were verified by sequencing. To generate 6-carboxyfluoroscein (FAM) end-labeled probes for EMSAs, plasmids carrying the wild-type and mutant promoter sequences were used as the templates for PCR with primers cpx-F1 and cpx-R-FAM or dmpM-F1 and dmpM-R-FAM (Table S2) to amplify the cpxR-cpxP intergenic region and dmpM promoter region, respectively. Labeled PCR products were purified using the Qiagen PCR purification kit.
Protein-DNA binding reactions were performed as described previously (31). Briefly, His6-TyrR was added to 5 μl 5× binding buffer (50 mM Tris-Cl [pH 7.5], 250 mM NaCl, 5 mM MgCl2, 20% glycerol, 2.5 mM dithiothreitol, and 2.5 mM EDTA), 1 μg poly(dI-dC) (nonspecific competitor), and 1 mM aromatic amino acid (with 0.1 mM ATP when tyrosine was added), and was incubated at room temperature for 5 min. Afterwards, 100 ng 6-FAM-labeled DNA was added to the reaction and incubated for 30 min at room temperature. Reactions were stopped with 5 μl loading buffer (0.25× Tris-borate-EDTA [TBE], 60% glycerol, and 0.001% bromophenol blue) and loaded onto prerun 6% native polyacrylamide gels containing 1 mM aromatic amino acid (with 0.1 mM ATP when tyrosine was added). Protein-DNA complexes were resolved at 100 V at 4°C for ∼120 min and visualized on a ChemiDoc MP imager (Bio-Rad, CA). Band intensity was quantified using Image Lab software v6.0.1 (Bio-Rad).
DNase I footprinting assays.
FAM end-labeled probes for DNase I footprinting assays were generated by PCR using primers dmpM-F-FAM and dmpM-R for the dmpM promoter region and primers cpx-F-FAM and cpx-R for the cpxR-cpxP intergenic region (Table S2), and pUCdmpM or pIDTcpx, respectively, as the templates (Table S1). Labeled PCR products (302 bp for dmpM and 238 bp for cpxR-cpxP) were purified from polyacrylamide gels using the Qiagen PCR purification kit. Probes (100 ng) were incubated with various concentrations of purified His6-TyrR (0 to 3 μg for dmpM and 0 to 6 μg for cpxR-cpxP) or bovine serum albumin (BSA, 0 to 30 μg) in binding buffer (10 mM Tris-Cl [pH 7.5], 50 mM NaCl, 1 mM MgCl2, 4% glycerol, 0.5 mM dithiothreitol, 0.5 mM EDTA, and 1 μg poly[d(I-C)]), with or without 1 mM tyrosine, for 30 min at room temperature. DNase I (0.02 U; NEB), 2.5 μl 10× DNase buffer, and 1.5 μl ddH2O were added, the mixture was incubated at room temperature for 2 min, and then the reaction was stopped with 1 μl 0.5 M EDTA and heating at 75°C for 10 min. Reaction mixtures were brought to a total volume of 100 μl with elution buffer (EB, Qiagen) and mixed with an equal volume of phenol-chloroform-isoamyl alcohol (25:24:1 vol/vol) with 1 min of vortexing. Samples were centrifuged for 5 min at 13,000 × g and the aqueous phase aspirated to a new tube. Each sample was mixed with 1 μg glycogen (NEB), 1:10 volume 3 M sodium acetate, and 4 volumes of 100% ethanol. DNA was precipitated overnight at −80°C. Samples were centrifuged for 30 min at 13,000 × g and the supernatant was discarded. The DNA pellet was washed twice with ice-cold 80% ethanol, centrifuged for 10 min at 13,000 × g, and ethanol discarded. After air drying the pellet to remove residual ethanol, DNA was resuspended in 10 μl EB buffer overnight at 4°C. Two microliters of each sample was mixed with GeneScan 500 LIZ size standard (Applied Biosystems) and resolved by capillary electrophoresis at the Genome Quebec Innovation Centre on an ABI-3730XL DNA Analyzer using Peak Scanner software (Applied Biosystems). The peaks representing DNA fragment sizes were analyzed using Geneious Prime 2020 software with the microsatellite plug-in (Biomatters Ltd., Auckland, New Zealand) and aligned with peaks from a dideoxynucleotide sequencing reaction (Thermo Sequenase Dye Primer Manual cycle sequencing kit, Thermo Fisher Scientific) with a FAM-labeled primer for each promoter region.
Data availability.
The E. ludwigii UW5 genome used in this study was previously sequenced and annotated (94) and is available from the NCBI GenBank database under the accession number CP011798. The raw RNA sequence data are available in the Gene Expression Omnibus database under the accession number GSE122440.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a grant to C.L.P. and by a scholarship to T.J.D.C. from the Natural Sciences and Engineering Research Council of Canada.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Pittard AJ, Camakaris H, Yang J. 2005. The TyrR regulon. Mol Microbiol 55:16–26. doi: 10.1111/j.1365-2958.2004.04385.x. [DOI] [PubMed] [Google Scholar]
- 2.Yang J, Ogawa Y, Camakaris H, Shimada T, Ishihama A, Pittard AJ. 2007. folA, a new member of the TyrR regulon in Escherichia coli K-12. J Bacteriol 189:6080–6084. doi: 10.1128/JB.00482-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garner CC, Herrmann KM. 1985. Operator mutations of the Escherichia coli aroF gene. J Biol Chem 260:3820–3825. [PubMed] [Google Scholar]
- 4.Cornish EC, Argyropoulos VP, Pittard AJ, Davidson BE. 1986. Structure of the Escherichia coli K12 regulatory gene tyrR: nucleotide sequence and sites of initiation of transcription and translation. J Biol Chem 261:403–410. [PubMed] [Google Scholar]
- 5.Pittard AJ, Davidson BE. 1991. TyrR protein of Escherichia coli and its role as repressor and activator. Mol Microbiol 5:1585–1592. doi: 10.1111/j.1365-2958.1991.tb01904.x. [DOI] [PubMed] [Google Scholar]
- 6.Hwang JS, Yang J, Pittard AJ. 1999. Specific contacts between residues in the DNA-binding domain of the TyrR protein and bases in the operator of the tyrP gene of Escherichia coli. J Bacteriol 181:2338–2345. doi: 10.1128/JB.181.8.2338-2345.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pittard AJ. 1996. The various strategies within the TyrR regulon of Escherichia coli to modulate gene expression. Genes Cells 1:717–725. doi: 10.1111/j.1365-2443.1996.tb00012.x. [DOI] [PubMed] [Google Scholar]
- 8.Wilson TJ, Maroudas P, Howlett GJ, Davidson BE. 1994. Ligand-induced self-association of the Escherichia coli regulatory protein TyrR. J Mol Biol 238:309–318. doi: 10.1006/jmbi.1994.1294. [DOI] [PubMed] [Google Scholar]
- 9.Argaet VP, Wilson TJ, Davidson BE. 1994. Purification of the Escherichia coli regulatory protein TyrR and analysis of its interactions with ATP, tyrosine, phenylalanine, and tryptophan. J Biol Chem 269:5171–5178. [PubMed] [Google Scholar]
- 10.Cui J, Somerville RL. 1993. A mutational analysis of the structural basis for transcriptional activation and monomer-monomer interaction in the TyrR system of Escherichia coli K-12. J Bacteriol 175:1777–1784. doi: 10.1128/jb.175.6.1777-1784.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cobbett CS, Delbridge ML. 1987. Regulatory mutants of the aroF-tyrA operon of Escherichia coli K-12. J Bacteriol 169:2500–2506. doi: 10.1128/jb.169.6.2500-2506.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cobbett CS. 1988. Repression of the aroF promoter by the TyrR repressor in Escherichia coli K-12: role of the “upstream” operator site. Mol Microbiol 2:377–383. doi: 10.1111/j.1365-2958.1988.tb00042.x. [DOI] [PubMed] [Google Scholar]
- 13.Yang J, Pittard AJ. 1987. Molecular analysis of the regulatory region of the Escherichia coli K-12 tyrB gene. J Bacteriol 169:4710–4715. doi: 10.1128/jb.169.10.4710-4715.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chye ML, Pittard AJ. 1987. Transcription control of the aroP gene in Escherichia coli K-12: analysis of operator mutants. J Bacteriol 169:386–393. doi: 10.1128/jb.169.1.386-393.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeFeyter RC, Davidson BE, Pittard AJ. 1986. Nucleotide sequence of the transcription unit containing the aroL and aroM genes from Escherichia coli K-12. J Bacteriol 165:233–239. doi: 10.1128/jb.165.1.233-239.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, Camakaris H, Pittard J. 2002. Molecular analysis of tyrosine-and phenylalanine-mediated repression of the tyrB promoter by the TyrR protein of Escherichia coli. Mol Microbiol 45:1407–1419. doi: 10.1046/j.1365-2958.2002.03108.x. [DOI] [PubMed] [Google Scholar]
- 17.Dixon MP, Pau RN, Howlett GJ, Dunstan DE, Sawyer WH, Davidson BE. 2002. The central domain of Escherichia coli TyrR is responsible for hexamerization associated with tyrosine-mediated repression of gene expression. J Biol Chem 277:23186–23192. doi: 10.1074/jbc.M112184200. [DOI] [PubMed] [Google Scholar]
- 18.Lawley B, Fujita N, Ishihama A, Pittard AJ. 1995. The TyrR protein of Escherichia coli is a class I transcription activator. J Bacteriol 177:238–241. doi: 10.1128/jb.177.1.238-241.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, Hwang JS, Camakaris H, Irawaty W, Ishihama A, Pittard AJ. 2004. Mode of action of the TyrR protein: repression and activation of the tyrP promoter of Escherichia coli. Mol Microbiol 52:243–256. doi: 10.1111/j.1365-2958.2003.03965.x. [DOI] [PubMed] [Google Scholar]
- 20.Bai Q, Somerville RL. 1998. Integration host factor and cyclic AMP receptor protein are required for TyrR-mediated activation of tpl in Citrobacter freundii. J Bacteriol 180:6173–6186. doi: 10.1128/JB.180.23.6173-6186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith HQ, Somerville RL. 1997. The tpl promoter of Citrobacter freundii is activated by the TyrR protein. J Bacteriol 179:5914–5921. doi: 10.1128/jb.179.18.5914-5921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katayama T, Suzuki H, Yamamoto K, Kumagai H. 1999. Transcriptional regulation of tyrosine phenol-lyase gene mediated through TyrR and cAMP receptor protein. Biosci Biotechnol Biochem 63:1823–1827. doi: 10.1271/bbb.63.1823. [DOI] [PubMed] [Google Scholar]
- 23.Lango-Scholey L, Brachmann AO, Bode HB, Clarke DJ. 2013. The expression of stlA in Photorhabdus luminescens is controlled by nutrient limitation. PLoS One 8:e82152. doi: 10.1371/journal.pone.0082152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park KR, Giard JC, Eom JH, Bearson S, Foster JW. 1999. Cyclic AMP receptor protein and TyrR are required for acid pH and anaerobic induction of hyaB and aniC in Salmonella typhimurium. J Bacteriol 181:689–694. doi: 10.1128/JB.181.2.689-694.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song J, Jensen RA. 1996. PhhR, a divergently transcribed activator of the phenylalanine hydroxylase gene cluster of Pseudomonas aeruginosa. Mol Microbiol 22:497–507. doi: 10.1046/j.1365-2958.1996.00131.x. [DOI] [PubMed] [Google Scholar]
- 26.Herrera MC, Ramos J-L. 2007. Catabolism of phenylalanine by Pseudomonas putida: the NtrC-family PhhR regulator binds to two sites upstream from the phhA gene and stimulates transcription with sigma70. J Mol Biol 366:1374–1386. doi: 10.1016/j.jmb.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 27.Herrera MC, Duque E, Rodríguez-Herva JJ, Fernández-Escamilla AM, Ramos JL. 2010. Identification and characterization of the PhhR regulon in Pseudomonas putida. Environ Microbiol 12:1427–1438. doi: 10.1111/j.1462-2920.2009.02124.x. [DOI] [PubMed] [Google Scholar]
- 28.Rodionov DA, Novichkov PS, Stavrovskaya ED, Rodionova IA, Li X, Kazanov MD, Ravcheev DA, Gerasimova AV, Kazakov AE, Kovaleva GY, Permina EA, Laikova ON, Overbeek R, Romine MF, Fredrickson JK, Arkin AP, Dubchak I, Osterman AL, Gelfand MS. 2011. Comparative genomic reconstruction of transcriptional networks controlling central metabolism in the Shewanella genus. BMC Genomics 12:S3. doi: 10.1186/1471-2164-12-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryu RJ, Patten CL. 2008. Aromatic amino acid-dependent expression of indole-3-pyruvate decarboxylase is regulated by TyrR in Enterobacter cloacae UW5. J Bacteriol 190:7200–7208. doi: 10.1128/JB.00804-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li G, Hu Z, Zeng P, Zhu B, Wu L. 2015. Whole genome sequence of Enterobacter ludgwigii type strain EN-199, isolated from clinical specimens. FEMS Microbiol Lett 362:fnv033. [DOI] [PubMed] [Google Scholar]
- 31.Coulson TJD, Patten CL. 2015. The TyrR transcription factor regulates the divergent akr-ipdC operons of Enterobacter cloacae UW5. PLoS One 10:e0121241. doi: 10.1371/journal.pone.0121241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desai KK, Miller BG. 2008. A metabolic bypass of the triosephosphate isomerase reaction. Biochemistry 47:7983–7985. doi: 10.1021/bi801054v. [DOI] [PubMed] [Google Scholar]
- 33.Sezonov G, Joseleau-Petit D, D'Ari R. 2007. Escherichia coli physiology in Luria-Bertani broth. J Bacteriol 189:8746–8749. doi: 10.1128/JB.01368-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. 2006. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwok T, Yang J, Pittard AJ, Wilson TJ, Davidson BE. 1995. Analysis of an Escherichia coli mutant TyrR protein with impaired capacity for tyrosine‐mediated repression, but still able to activate at σ70 promoters. Mol Microbiol 17:471–481. doi: 10.1111/j.1365-2958.1995.mmi_17030471.x. [DOI] [PubMed] [Google Scholar]
- 36.Young MD, Wakefield MJ, Smyth GK, Oshlack A. 2010. Gene ontology analysis for RNA-Seq: accounting for selection bias. Genome Biol 11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene Ontology: tool for the unification of biology. Nat Genet 25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin JL, McMillan FM. 2002. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol 12:783–793. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 39.Zimmermann L, Stephens A, Nam S-Z, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, Alva V. 2018. A completely reimplemented MPI Bioinformatics Toolkit with a new HHpred server at its core. J Mol Biol 430:2237–2243. doi: 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 40.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 41.Soding J. 2005. Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 42.Price NL, Raivio TL. 2009. Characterization of the Cpx regulon in Escherichia coli strain MC4100. J Bacteriol 191:1798–1815. doi: 10.1128/JB.00798-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raivio TL, Leblanc SKD, Price NL. 2013. The Escherichia coli Cpx envelope stress response regulates genes of diverse function that impact antibiotic resistance and membrane integrity. J Bacteriol 195:2755–2767. doi: 10.1128/JB.00105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Danese PN, Snyder WB, Cosma CL, Davis LJ, Silhavy TJ. 1995. The Cpx two-component signal transduction pathway of Escherichia coli regulates transcription of the gene specifying the stress-inducible periplasmic protease, DegP. Genes Dev 9:387–398. doi: 10.1101/gad.9.4.387. [DOI] [PubMed] [Google Scholar]
- 45.Pogliano J, Lynch AS, Belin D, Lin EC, Beckwith J. 1997. Regulation of Escherichia coli cell envelope proteins involved in protein folding and degradation by the Cpx two-component system. Genes Dev 11:1169–1182. doi: 10.1101/gad.11.9.1169. [DOI] [PubMed] [Google Scholar]
- 46.Bury-Moné S, Nomane Y, Reymond N, Barbet R, Jacquet E, Imbeaud S, Jacq A, Bouloc P. 2009. Global analysis of extracytoplasmic stress signaling in Escherichia coli. PLoS Genet 5:e1000651. doi: 10.1371/journal.pgen.1000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimohata N, Chiba S, Saikawa N, Ito K, Akiyama Y. 2002. The Cpx stress response system of Escherichia coli senses plasma membrane proteins and controls HtpX, a membrane protease with a cytosolic active site. Genes Cells 7:653–662. doi: 10.1046/j.1365-2443.2002.00554.x. [DOI] [PubMed] [Google Scholar]
- 48.Hirakawa H, Nishino K, Hirata T, Yamaguchi A. 2003. Comprehensive studies of drug resistance mediated by overexpression of response regulators of two-component signal transduction systems in Escherichia coli. J Bacteriol 185:1851–1856. doi: 10.1128/jb.185.6.1851-1856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao J, Woodhall MR, Alvarez J, Cartron ML, Andrews SC. 2007. EfeUOB (YcdNOB) is a tripartite, acid-induced and CpxAR-regulated, low-pH Fe 2+ transporter that is cryptic in Escherichia coli K-12 but functional in E. coli O157:H7. Mol Microbiol 65:857–875. doi: 10.1111/j.1365-2958.2007.05802.x. [DOI] [PubMed] [Google Scholar]
- 50.Wells HC. 2015. Further characterisation of the envelope stress responses of Salmonella Typhimurium. PhD thesis. University of East Anglia, Norwich, England. [Google Scholar]
- 51.Danese PN, Silhavy TJ. 1998. CpxP, a stress-combative member of the Cpx regulon. J Bacteriol 180:831–839. doi: 10.1128/JB.180.4.831-839.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Wulf P, Kwon O, Lin EC. 1999. The CpxRA signal transduction system of Escherichia coli: growth-related autoactivation and control of unanticipated target operons. J Bacteriol 181:6772–6778. doi: 10.1128/JB.181.21.6772-6778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baseggio N, Davies WD, Davidson BE. 1990. Identification of the promoter, operator, and 5′ and 3′ ends of the mRNA of the Escherichia coli K-12 gene aroG. J Bacteriol 172:2547–2557. doi: 10.1128/jb.172.5.2547-2557.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andrews AE, Lawley B, Pittard AJ. 1991. Mutational analysis of repression and activation of the tyrP gene in Escherichia coli. J Bacteriol 173:5068–5078. doi: 10.1128/jb.173.16.5068-5078.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Camakaris H, Pittard AJ. 1982. Autoregulation of the tyrR gene. J Bacteriol 150:70–75. doi: 10.1128/JB.150.1.70-75.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pittard AJ, Yang J. 2008. Biosynthesis of the aromatic amino acids. EcoSal Plus 3. doi: 10.1128/ecosalplus.3.6.1.8. [DOI] [PubMed] [Google Scholar]
- 57.Deng Z, Liu Z, He J, Wang J, Yan Y, Wang X, Cui Y, Bi Y, Du Z, Song Y, Yang R, Han Y. 2015. TyrR, the regulator of aromatic amino acid metabolism, is required for mice infection of Yersinia pestis. Front Microbiol 6:110. doi: 10.3389/fmicb.2015.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fleischer R, Heermann R, Jung K, Hunke S. 2007. Purification, reconstitution, and characterization of the CpxRAP envelope stress system of Escherichia coli. J Biol Chem 282:8583–8593. doi: 10.1074/jbc.M605785200. [DOI] [PubMed] [Google Scholar]
- 59.Mileykovskaya E, Dowhan W. 1997. The Cpx two-component signal transduction pathway is activated in Escherichia coli mutant strains lacking phosphatidylethanolamine. J Bacteriol 179:1029–1034. doi: 10.1128/jb.179.4.1029-1034.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keller R, Ariöz C, Hansmeier N, Stenberg-Bruzell F, Burstedt M, Vikström D, Kelly A, Wieslander Å, Daley DO, Hunke S. 2015. The Escherichia coli envelope stress sensor CpxA responds to changes in lipid bilayer properties. Biochemistry 54:3670–3676. doi: 10.1021/acs.biochem.5b00242. [DOI] [PubMed] [Google Scholar]
- 61.Jones CH, Danese PN, Pinkner JS, Silhavy TJ, Hultgren SJ. 1997. The chaperone-assisted membrane release and folding pathway is sensed by two signal transduction systems. EMBO J 16:6394–6406. doi: 10.1093/emboj/16.21.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirakawa H, Inazumi Y, Masaki T, Hirata T, Yamaguchi A. 2005. Indole induces the expression of multidrug exporter genes in Escherichia coli. Mol Microbiol 55:1113–1126. doi: 10.1111/j.1365-2958.2004.04449.x. [DOI] [PubMed] [Google Scholar]
- 63.Hunke S, Betton J-M. 2003. Temperature effect on inclusion body formation and stress response in the periplasm of Escherichia coli. Mol Microbiol 50:1579–1589. doi: 10.1046/j.1365-2958.2003.03785.x. [DOI] [PubMed] [Google Scholar]
- 64.Otto K, Silhavy TJ. 2002. Surface sensing and adhesion of Escherichia coli controlled by the Cpx-signaling pathway. Proc Natl Acad Sci U S A 99:2287–2292. doi: 10.1073/pnas.042521699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Surmann K, Ćudić E, Hammer E, Hunke S. 2016. Molecular and proteome analyses highlight the importance of the Cpx envelope stress system for acid stress and cell wall stability in Escherichia coli. MicrobiologyOpen 5:582–596. doi: 10.1002/mbo3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Liu F, Peng W, Yan K, Zhao H, Liu T, Cheng H, Chang P, Yuan F, Chen H, Bei W. 2018. The CpxA/CpxR two-component system affects biofilm formation and virulence in Actinobacillus pleuropneumoniae. Front Cell Infect Microbiol 8:72. doi: 10.3389/fcimb.2018.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gangaiah D, Raterman EL, Wu H, Fortney KR, Gao H, Liu Y, Jerse AE, Spinola SM. 2017. Both MisR (CpxR) and MisS (CpxA) are required for Neisseria gonorrhoeae infection in a murine model of lower genital tract infection. Infect Immun 85:e00307-17. doi: 10.1128/IAI.00307-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Delhaye A, Collet J-F, Laloux G. 2016. Fine-tuning of the Cpx envelope stress response is required for cell wall homeostasis in Escherichia coli. mBio 7:e00047-16. doi: 10.1128/mBio.00047-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tian Z-X, Yi X-X, Cho A, O’Gara F, Wang Y-P. 2016. CpxR activates MexAB-OprM efflux pump expression and enhances antibiotic resistance in both laboratory and clinical nalB-type isolates of Pseudomonas aeruginosa. PLoS Pathog 12:e1005932-22. doi: 10.1371/journal.ppat.1005932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ćudić E, Surmann K, Panasia G, Hammer E, Hunke S. 2017. The role of the two-component systems Cpx and Arc in protein alterations upon gentamicin treatment in Escherichia coli. BMC Microbiol 17:197. doi: 10.1186/s12866-017-1100-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raivio TL, Popkin DL, Silhavy TJ. 1999. The Cpx envelope stress response is controlled by amplification and feedback inhibition. J Bacteriol 181:5263–5272. doi: 10.1128/JB.181.17.5263-5272.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tschauner K, Hörnschemeyer P, Müller VS, Hunke S. 2014. Dynamic interaction between the CpxA sensor kinase and the periplasmic accessory protein CpxP mediates signal recognition in E. coli. PLoS One 9:e107383-12. doi: 10.1371/journal.pone.0107383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Raivio TL, Laird MW, Joly JC, Silhavy TJ. 2000. Tethering of CpxP to the inner membrane prevents spheroplast induction of the Cpx envelope stress response. Mol Microbiol 37:1186–1197. doi: 10.1046/j.1365-2958.2000.02074.x. [DOI] [PubMed] [Google Scholar]
- 74.DiGiuseppe PA, Silhavy TJ. 2003. Signal detection and target gene induction by the CpxRA two-component system. J Bacteriol 185:2432–2440. doi: 10.1128/jb.185.8.2432-2440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beloin C, Valle J, Latour-Lambert P, Faure P, Kzreminski M, Balestrino D, Haagensen JAJ, Molin S, Prensier G, Arbeille B, Ghigo J-M. 2004. Global impact of mature biofilm lifestyle on Escherichia coli K-12 gene expression. Mol Microbiol 51:659–674. doi: 10.1046/j.1365-2958.2003.03865.x. [DOI] [PubMed] [Google Scholar]
- 76.De Wulf P, McGuire AM, Liu X, Lin ECC. 2002. Genome-wide profiling of promoter recognition by the two-component response regulator CpxR-P in Escherichia coli. J Biol Chem 277:26652–26661. doi: 10.1074/jbc.M203487200. [DOI] [PubMed] [Google Scholar]
- 77.DeFeyter RC, Pittard AJ. 1986. Purification and properties of shikimate kinase II from Escherichia coli K-12. J Bacteriol 165:331–333. doi: 10.1128/jb.165.1.331-333.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Weatherspoon-Griffin N, Yang D, Kong W, Hua Z, Shi Y. 2014. The CpxR/CpxA two-component regulatory system up-regulates the multidrug resistance cascade to facilitate Escherichia coli resistance to a model antimicrobial peptide. J Biol Chem 289:32571–32582. doi: 10.1074/jbc.M114.565762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lacalle RA, Tercero JA, Vara J, Jimenez A. 1993. Identification of the gene encoding an N-acetylpuromycin N-acetylhydrolase in the puromycin biosynthetic gene cluster from Streptomyces alboniger. J Bacteriol 175:7474–7478. doi: 10.1128/jb.175.22.7474-7478.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Andrews AE, Dickson B, Lawley B, Cobbett C, Pittard AJ. 1991. Importance of the position of TYR R boxes for repression and activation of the tyrP and aroF genes in Escherichia coli. J Bacteriol 173:5079–5085. doi: 10.1128/jb.173.16.5079-5085.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Teufel R, Mascaraque V, Ismail W, Voss M, Perera J, Eisenreich W, Haehnel W, Fuchs G. 2010. Bacterial phenylalanine and phenylacetate catabolic pathway revealed. Proc Natl Acad Sci U S A 107:14390–14395. doi: 10.1073/pnas.1005399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ferrández A, García JL, Díaz E. 2000. Transcriptional regulation of the divergent paa catabolic operons for phenylacetic acid degradation in Escherichia coli. J Biol Chem 275:12214–12222. doi: 10.1074/jbc.275.16.12214. [DOI] [PubMed] [Google Scholar]
- 83.Parales RE, Resnick SM. 2006. Aromatic ring hydroxylating dioxygenases, p 287–340. In Romos J-L, Levesque RC (ed), Pseudomonas, vol. 4 Springer, Boston, MA. [Google Scholar]
- 84.Moreno-Vivián C, Cabello P, Martínez-Luque M, Blasco R, Castillo F. 1999. Prokaryotic nitrate reduction: molecular properties and functional distinction among bacterial nitrate reductases. J Bacteriol 181:6573–6584. doi: 10.1128/JB.181.21.6573-6584.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vine CE, Purewal SK, Cole JA. 2011. NsrR-dependent method for detecting nitric oxide accumulation in the Escherichia coli cytoplasm and enzymes involved in NO production. FEMS Microbiol Lett 325:108–114. doi: 10.1111/j.1574-6968.2011.02385.x. [DOI] [PubMed] [Google Scholar]
- 86.Simon J, van Spanning RJM, Richardson DJ. 2008. The organisation of proton motive and non-proton motive redox loops in prokaryotic respiratory systems. Biochim Biophys Acta 1777:1480–1490. doi: 10.1016/j.bbabio.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 87.Shepherd M, Achard MES, Idris A, Totsika M, Phan M-D, Peters KM, Sarkar S, Ribeiro CA, Holyoake LV, Ladakis D, Ulett GC, Sweet MJ, Poole RK, McEwan AG, Schembri MA. 2016. The cytochrome bd-I respiratory oxidase augments survival of multidrug-resistant Escherichia coli during infection. Sci Rep 6:35285. doi: 10.1038/srep35285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Borisov VB, Gennis RB, Hemp J, Verkhovsky MI. 2011. The cytochrome bd respiratory oxygen reductases. Biochim Biophys Acta 1807:1398–1413. doi: 10.1016/j.bbabio.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miki K, Lin EC. 1975. Electron transport chain from glycerol 3-phosphate to nitrate in Escherichia coli. J Bacteriol 124:1288–1294. doi: 10.1128/JB.124.3.1288-1294.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Garland PB, Downie JA, Haddock BA. 1975. Proton translocation and the respiratory nitrate reductase of Escherichia coli. Biochem J 152:547–559. doi: 10.1042/bj1520547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gordon SA, Weber RP. 1951. Colorimetric estimation of indolacetic acid. Plant Physiol 26:192–195. doi: 10.1104/pp.26.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coulson TJD, Patten CL. 2015. Complete genome sequence of Enterobacter cloacae UW5, a rhizobacterium capable of high levels of indole-3-acetic acid production. Genome Announc 3:e00843-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. 2008. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.The Gene Ontology Consortium. 2017. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 45:D331–D338. doi: 10.1093/nar/gkw1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- 103.Supek F, Bošnjak M, Škunca N, Šmuc T. 2011. REVIGO summarizes and visualizes long lists of Gene Ontology terms. PLoS One 6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hahne F, Ivanek R. 2016. Visualizing genomic data using Gviz and Bioconductor, p 335–351. In Mathé E, Davis S (ed), Statistical genomics: methods and protocols. Springer New York, New York, NY. [DOI] [PubMed] [Google Scholar]
- 105.Ramakers C, Ruijter JM, Deprez RHL, Moorman AFM. 2003. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339:62–66. doi: 10.1016/S0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- 106.English MM, Coulson TJD, Horsman SR, Patten CL. 2010. Overexpression of hns in the plant growth-promoting bacterium Enterobacter cloacae UW5 increases root colonization. J Appl Microbiol 108:2180–2190. doi: 10.1111/j.1365-2672.2009.04620.x. [DOI] [PubMed] [Google Scholar]
- 107.Pfaffl MW, Horgan GW, Dempfle L. 2002. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The E. ludwigii UW5 genome used in this study was previously sequenced and annotated (94) and is available from the NCBI GenBank database under the accession number CP011798. The raw RNA sequence data are available in the Gene Expression Omnibus database under the accession number GSE122440.