

Abstract
Abnormalities of one carbon, glutathione and sulfide metabolisms have recently emerged as novel pathomechanisms in diseases with mitochondrial dysfunction. However, the mechanisms underlying these abnormalities are not clear. Also, we recently showed that sulfide oxidation is impaired in Coenzyme Q10 (CoQ10) deficiency. This finding leads us to hypothesize that the therapeutic effects of CoQ10, frequently administered to patients with primary or secondary mitochondrial dysfunction, might be due to its function as cofactor for sulfide:quinone oxidoreductase (SQOR), the first enzyme in the sulfide oxidation pathway. Here, using biased and unbiased approaches, we show that supraphysiological levels of CoQ10 induces an increase in the expression of SQOR in skin fibroblasts from control subjects and patients with mutations in Complex I subunits genes or CoQ biosynthetic genes. This increase of SQOR induces the downregulation of the cystathionine β-synthase and cystathionine γ-lyase, two enzymes of the transsulfuration pathway, the subsequent downregulation of serine biosynthesis and the adaptation of other sulfide linked pathways, such as folate cycle, nucleotides metabolism and glutathione system. These metabolic changes are independent of the presence of sulfur aminoacids, are confirmed in mouse models, and are recapitulated by overexpression of SQOR, further proving that the metabolic effects of CoQ10 supplementation are mediated by the overexpression of SQOR. Our results contribute to a better understanding of how sulfide metabolism is integrated in one carbon metabolism and may explain some of the benefits of CoQ10 supplementation observed in mitochondrial diseases.
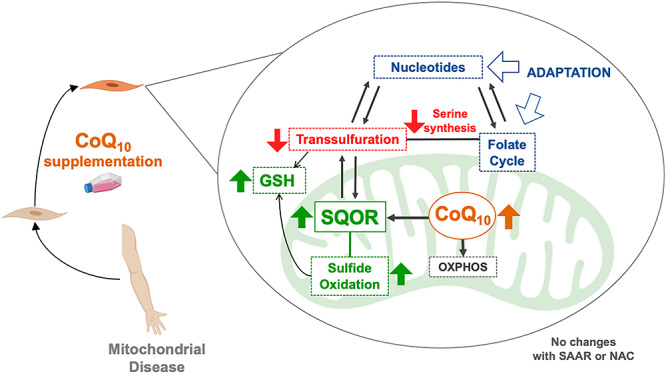
Graphical Abstract
Graphical Abstract.
Introduction
Mitochondria are the primary site of cellular energy production and can also have a broad range of other vital functions for the cells. Dysfunction of mitochondria is observed in common age-related disorders, including neurodegenerative diseases, metabolic syndrome, cardiomyopathies and sarcopenia (1). Mitochondrial dysfunction is likewise the primary characteristic of monogenic disorders that affect one of the genes that encode the estimated 1000 proteins identified in mitochondria (2). Among these, mutations in genes encoding essential proteins for the Oxidative Phosphorylation (OXPHOS) system account for the most common group of inborn errors of metabolism, the so-called mitochondrial diseases, which are clinically heterogeneous and can involve any organ or tissue. This clinical heterogeneity suggests that multiple pathogenic and adaptive mechanisms are involved in the clinical manifestations of the mitochondrial diseases.
Recently, the remodeling of folate cycle, and its link to transsulfuration pathway (the hydrogen sulfide biosynthetic pathway) and nucleotides metabolism, have been proposed as novel mechanisms contributing to the pathophysiological features of mitochondrial diseases, mainly those caused by deletions or depletion of the mitochondria DNA (mtDNA) (3–5). Also, alterations of hydrogen sulfide (H2S) metabolism are observed after pharmacological inhibition of complex I (CI), the first enzyme of the mitochondrial respiratory chain (6), and two recent studies have shown that Coenzyme Q (CoQ) deficiency severely decreases the levels of Sulfide Quinone Oxidoreductase (SQOR), the first enzyme in the mitochondrial H2S oxidation pathway. Thus, the decrease in SQOR leads to an impairment of H2S oxidation, leading to accumulation of H2S and depletion on the glutathione system (7,8). However, the regulation of these pathways and how they interplay with each other is unknown, and therapeutic interventions aimed to revert their alterations have failed (3).
The heterogenous combination of all these pathomechanisms makes it difficult to develop effective therapeutic interventions. Thus, the therapeutic options for mitochondrial disorders are mostly limited to palliative care (9,10), although promising therapeutic options are being developed for specific diseases (11,12). In general, CoQ10 supplementation is recommended for patients with mitochondrial disorders or other diseases with secondary mitochondrial dysfunction, and clinical improvements have been reported in some cases, but others do not show any positive response (13). The therapeutic efficacy of CoQ10 supplementation in the treatment of mitochondrial disorders might be explained by the development of secondary CoQ10 deficiency, which frequently occurs (13–16). Alternatively, we can postulate that supraphysiological levels of CoQ10 could induce an increase in the levels of SQOR, with consequent modifications in the up-stream transsulfuration pathway, which supplies H2S to the oxidation pathway.
Collectively, these data led us to hypothesize that (1) changes in the function of the mitochondrial respiratory chain or supraphysiological levels of CoQ10, might alter sulfide metabolism through the modification of the levels and activity of SQOR, which in turn may induce changes in other sulfide enzymes that are metabolically connected with other metabolic pathways, such as, serine biosynthesis, folate cycle or nucleotides metabolism; and (2) these pathways might be modulated by the sulfur aminoacids availability as a source of H2S. Thus, we have used biased and unbiased approaches to assess how metabolic pathways change in different models of OXPHOS disorders, how they regulate each other, and how they are modified by CoQ10 supplementation and/or sulfur aminoacids availability.
Results
CoQ10 supplementation modifies sulfide metabolism and the pathways associated to one carbon metabolism
We first analyzed the effect of CoQ10 supplementation in the transcriptomic profiles of skin fibroblasts from a control subject and a patient with Complex I deficiency due to a mutation in the NDUFS1 gene (Supplementary Material, Table S1; Data File S1). Both control and NDUFS1 mutant cells have similar levels of CoQ10 and supplementation with 100 μm CoQ10 significantly increases the cellular content of CoQ10 (Supplementary Material, Table S2), although the majority of these CoQ10 molecules would be located in the cell membrane and a low proportion would enter into the mitochondria (17,18). The increase of the cell content of CoQ10 modifies a variety of canonical pathways involved in cell metabolism, being the number of the altered pathways higher in the treated mutant cells than in the control treated cells (Fig. 1A and B).
Figure 1.
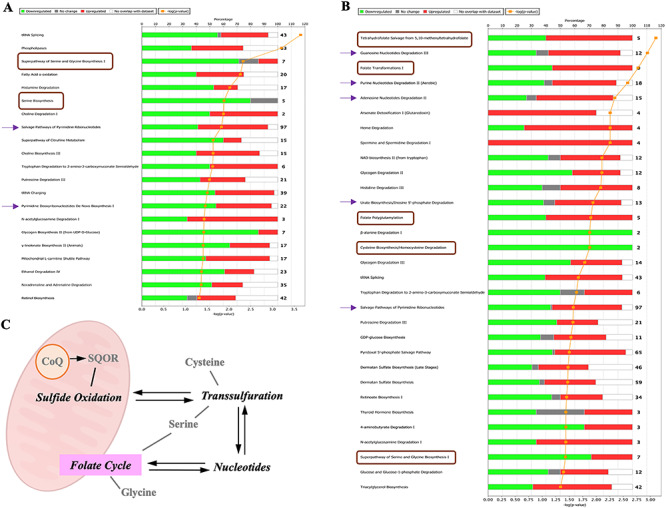
CoQ10 supplementation induces changes in pathways that are direct or indirectly related to sulfide metabolism. (A and B) Results of the pathway analysis showing the canonical pathways most enriched in the differentially expressed gene list of the CoQ10 treated cells dataset. The pathways related to sulfide metabolism are indicated by a brown square or a purple arrow. (A) Analysis of the control cells. (B) Analysis of the NDUFS1 mutant cells. (C) Pathways related to sulfide metabolism. SQOR, the first enzyme in the mitochondrial hydrogen sulfide oxidation pathway, is the target of CoQ10.
About 20–35% of the canonical pathways that CoQ10 modifies are related to sulfide metabolism or to pathways associated to one carbon metabolism. Specifically, CoQ10 supplementation in control cells affects the pathways of serine and glycine biosynthesis, the salvage pathway of pyrimidine biosynthesis and the pyrimidine deoxyribonucleotides de novo biosynthesis (Fig. 1A). CoQ10 supplementation in the NDUFS1 mutant cells modifies the tetrahydrofolate salvage from 5,10-methenyltetrahydrefolate, folate transformation I, folate polyglutamylation, cysteine biosynthesis/homocysteine degradation, the pathway of serine and glycine biosynthesis, the guanosine nucleotides degradation III, the purine nucleotides degradation II, the adenosine nucleotides degradation II, the urate biosynthesis/inosine 5′-phosphate degradation and the salvage pathways of pyrimidine ribonucleotides (Fig. 1B). Importantly, all these pathways are interconnected in the cell’s metabolism (Fig. 1C).
Further analysis of the indicated pathways shows that, in the sulfide metabolism, CoQ10 supplementation up-regulates SQOR (sulfide oxidation pathway) and down-regulates CSE and CBS (transsulfuration pathway), in both control and mutant cells (Fig. 2A and B and Supplementary Material, Fig. S1). In the serine biosynthesis, five key genes (PHGDH, PSAT1, PSDH, PSAT1P3 and PSAT1P4), are under-expressed after CoQ10 supplementation in both control and mutant cells (Fig. 2C and D and Supplementary Material, Fig. S1). An unbalance in the genes that encode mitochondrial proteins is observed in the folate cycle, in form of a decrease in the expression of SHMT2 and MTHFD2 and an increase in the expression of MTHFD2L after CoQ10 supplementation in both control and mutant cells (Fig. 2E and F and Supplementary Material, Fig. S1). Additionally, in the nucleotides’ metabolism, PRKCH, MAP2K8, MAP2K6, DMPK, AK4 and IMPDH2 are under-expressed, while EIF2AK2, CMPK2, APOBEC3G, APOBEC3F, APOBEC3B, AK5 and XDH are over-expressed after CoQ10 supplementation in both control and mutant cells (Fig. 2G and H and Supplementary Material, Fig. S2). The direction of the fold change for some of these genes after CoQ10 supplementation, e.g. SQOR, CSE, PSAT1P4, PHGDH, MTHFD1, PRKCH, EIF2AK2, DMK, CMPK2, APOBEC3G, APOBEC3F, APOBEC3B or AK4, is the opposite to the fold change direction in the mutant cells compared with control cells (Supplementary Material, Fig. S3). To check whether those changes in genes expression result in a metabolic adaptation, we also quantified the levels of serine and glycine, two metabolites involved in one carbon metabolism that are altered under mitochondrial dysfunction. Serine/glycine ratio slightly decreases in fibroblasts with NDUFS1 mutation, compared with control fibroblasts. Interestingly, serine/glycine ratio increases in control and mutant cells (Fig. 2I). Therefore, these data indicate that CoQ10 supplementation modifies sulfide metabolism and associated pathways, being all on them related to mitochondrial metabolism (Supplementary Material, Fig. S4).
Figure 2.
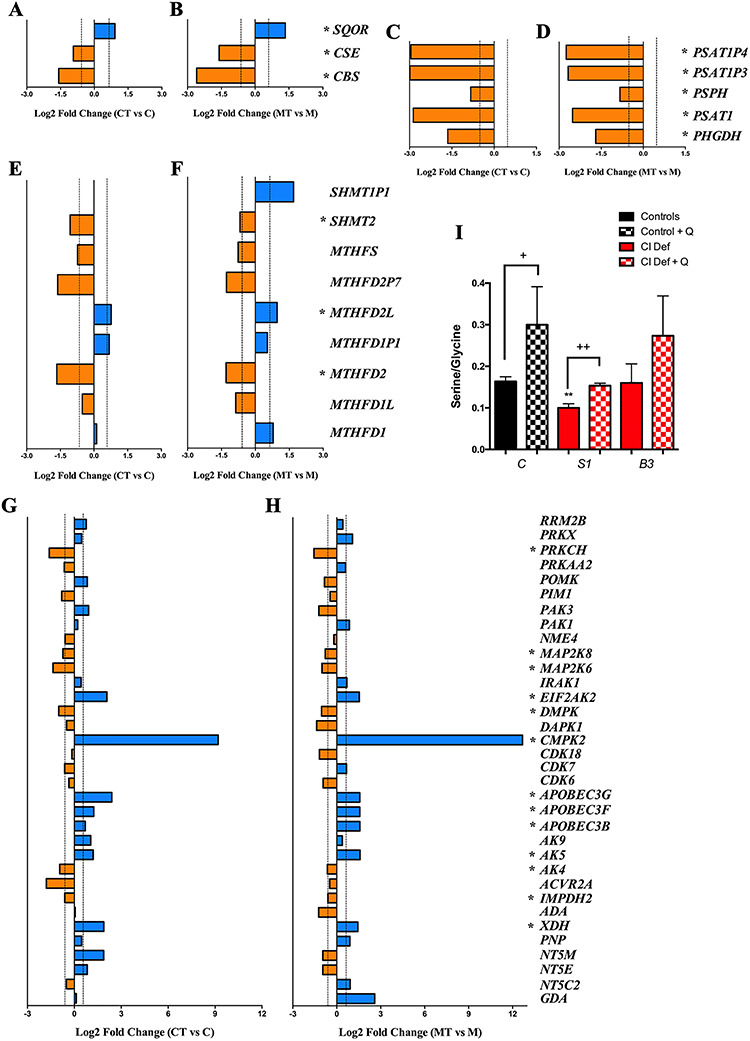
Changes in gene expression of pathways related to sulfide metabolism in response to CoQ10 supplementation. (A and B) Changes in the expression of genes related to cysteine biosynthesis and sulfide metabolism in control (A) and mutant (B) cells after CoQ10 supplementation. (C and D) Changes in the expression of genes related to serine/glycine biosynthesis in control (C) and mutant (D) cells after CoQ10 supplementation. (E and F) Changes in the expression of genes related to the folate cycle in control (E) and mutant (F) cells after CoQ10 supplementation. (G and H) Changes in the expression of genes related to nucleotides metabolism in control (G) and mutant (H) cells after CoQ10 supplementation. Bars in blue indicate genes overexpressed; bars in orange indicates genes underexpressed. Dotted lines indicate the threshold considered for overexpression (log2 FC = 0.585) or underexpression (log2 FC = −0.585). Asterisks indicates the genes that exceed the fold change threshold and have P-values less than 0.05 in the comparison of treated and untreated cells in both control and mutant cells. C, control; CT, control treated with CoQ10; M, NDUFS1 mutant; MT, NDUFS1 mutant treated with CoQ10. (I) Serine/Glycine ratio in control (black bars) and patients’ cells (red bars) with mutations in NDUFS1 and NDUFB3, and their response to CoQ10 supplementation (checkered bars). Data are expressed as mean ± SD. **P < 0.01; differences versus control. +P < 0.05; ++P < 0.01; control versus control + CoQ10 or Complex I deficiency versus Complex I deficiency + CoQ10 (t-test; n = 3 for each group; n = 3 for each group).
CoQ10 supplementation modifies sulfide metabolism gene expression in cells with mutations in different Complex I or CoQ-biosynthetic genes
Among all changes described above, the upregulation of SQOR is likely the primary effect of CoQ10 since that enzyme requires CoQ10 for its normal activity and CoQ10 deficiency causes a reduction in the levels of SQOR (7,8). Furthermore, SQOR needs the supply of hydrogen sulfide produced by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) in the transsulfuration pathway (Supplementary Material, Fig. S4). Thus, we extended the study by quantifying of the gene expression of enzymes of the hydrogen sulfide oxidation pathway, i.e. SQOR, thiosulfate sulfurtransferase (TST) and sulfite oxidase (SUOX), and the transsulfuration/hydrogen sulfide production pathways, i.e. CBS, CSE and 3-mercaptopyruvate sulfurtransferase (3MST), in additional cells that carry mutations in different Complex I subunits or CoQ-biosynthetic genes (Supplementary Material, Table S1).
Those cells have different residual levels of CI activity (Supplementary Material, Table S1), and we observed that the abnormalities in the levels of SQOR and CSE detected in the NDUFS1 mutant cells are not a common pattern in all Complex I deficiency cells. Specifically, SQOR mRNA levels are 10–20% significantly reduced in NDUFS1, NUUFS3 and NDUFS8 mutant cells compared with controls cells (Fig. 3A). TST mRNA levels are significantly increased in NDUFS3, NUUFS8, ND3, COQ4 and PDSS2 mutant cells compared with control cells (Fig. 3B). The levels of SOUX and CBS mRNA are similar in mutant and control cells (Fig. 3C and D). CSE mRNA levels are significantly increased in ND6, COQ4 and PDSS2 mutant cells compared with control cells (Fig. 3E). 3MST mRNA levels are only significantly increased in COQ4 mutant cells compared with control cells (Fig. 3B). Therefore, the changes on SQOR and CSE detected in the RNAseq analysis in the NDUFS1 cells are not a common pattern in all Complex I deficiency cells.
Figure 3.
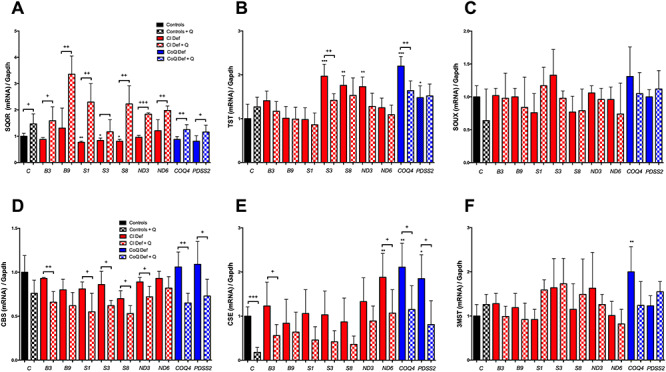
CoQ10 supplementation modifies the expression of key genes in the mitochondrial hydrogen sulfide oxidation pathway and the transsulfuration pathway. (A–C) Expression levels of genes involved in the mitochondrial hydrogen sulfide oxidation pathway. (D–F) Expression levels of genes involved in the transsulfuration pathway. Black bars indicate control cells; red bars indicate cells with mutation in Complex I genes; blue bars indicate cells with mutations in CoQ10 biosynthetic genes. Solid bars indicate untreated cells; checkered bars indicate CoQ10-treated cells. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; differences versus Control. +P < 0.05; ++P < 0.01; +++P < 0.001; control versus control + CoQ10 or Complex I deficiency versus Complex I deficiency + CoQ10 or CoQ10 deficiency versus CoQ10 deficiency + CoQ10 (t-test; n = 3 for each group; n = 3 for each group). C, control; B3, NDUFB3 mutant; B9, NDUFB9 mutant; S1, NDUFS1 mutant; S3, NDUFS3 mutant; S8, NDUFS8 mutant; ND3, ND3 mutant; ND6, ND6 mutant; COQ4, COQ4 mutant; PDSS2, PDSS2 mutant; CI Def, cells with Complex I deficiency; CoQ Def, cells with CoQ10 deficiency; Q, CoQ10.
CoQ10 supplementation significantly increases the cellular content of CoQ10 in all cell lines (Supplementary Material, Table S2). That increase in the cell content of CoQ10 induces a consistent increase of SQOR mRNA levels and decrease of CBS and CSE mRNA levels in all cells (Fig. 3A, D and E), although no generalized changes are detected on TST, SOUX or 3MST mRNA levels (Fig. 3B, C and F). Specifically, the increase of SQOR mRNA levels is statistically significant in controls cells and NDUFB3, NDUFB9, NDUFS1, NDUFS8, ND3, ND6, COQ4 and PDSS2 mutant cells (Fig. 3A); the decrease of CBS mRNA levels is statistically significant in NDUFB3, NDUFS1, NDUFS3, NDUFS8, ND3, COQ4 and PDSS2 mutant cells (Fig. 3D); and the decrease of CSE mRNA levels is statistically significant in control cells and NDUFB3, ND3, ND6, COQ4 and PDSS2 mutant cells (Fig. 3E). Also, a significant decrease in TST mRNA levels is detected only in NDUFS3 and COQ4 mutant cells after CoQ10 supplementation (Fig. 3B). Therefore, we found a remarkable inverse correlation between the mRNA levels of SQOR and the mRNA levels of CBS and CSE after CoQ10 supplementation.
The modification of the expression of the sulfide oxidation genes after CoQ10 supplementation correlates with the changes in the levels of the sulfide oxidation proteins, and is independent of sulfur aminoacids availability
We assessed whether CoQ10 supplementation modified sulfide enzymes protein levels, as a result of the change in their gene expression. Moreover, since it has been reported that sulfide metabolism can be adapted to dietary restriction in sulfur aminoacids (SAAR) (19,20), we also evaluated whether the availability of sulfur aminoacids affects sulfide metabolism in CI and CoQ10 deficiency. Before CoQ10 supplementation, the levels of SQOR were significantly lower in two (out of three) of the cells with the lowest CI activity (NDUFB3 and NDUFS1) and in the CoQ10 deficiency cells, compared with controls, although the levels of SQOR were normal in the cells with 40–60% of residual CI activity, as well as in one (out of three) of the mutant cells with the lowest CI activity (NDUFB9) (Fig. 4A and B; Supplementary Material, Fig. S5A and B). The levels of TST and SUOX were similar in mutant and control cells, except for the increased levels of TST in NDUFS3 and COQ4 mutant cells (Fig. 4A, C and D; Supplementary Material, Fig. S5A, C and D). All these changes were similar in the cells cultured under SAAR (Fig. 4 and Supplementary Material, Fig. S5; quantitative data not shown). Therefore, there was a general correlation between mRNA and protein levels in the mutant cells, with some exceptions. Interestingly, the levels of SQOR are consistently increased in all cell types after CoQ10 supplementation compared with the untreated cells, being the differences statistically significant in controls, NDUFB3, NDUFS1, NDUFS8, ND3, COQ4 and PDSS2 mutant cells (Fig. 4A and B; Supplementary Material, Fig. S5A and B). Moreover, the levels of SQOR were rescued in NDUFB3, NDUFS1, COQ4 and PDSS2 mutant cells after CoQ10 supplementation (Fig. 4A and B; Supplementary Material, Fig. S5A and B). On the contrary, the levels of TST and SUOX did not change after CoQ10 supplementation, except for a partial decrease of TST levels in ND6 mutant cells after CoQ10 supplementation (Fig. 4A, C and D; Supplementary Material, Fig. S5A, C and D). All these changes were similar in the cells cultured under SAAR (Fig. 4 and Supplementary Material, Fig. S5; quantitative data not shown). Therefore, SQOR is the most affected enzyme in the H2S oxidation pathway under CI deficiency or low/high levels of CoQ10, and those changes are not affected by SAAR.
Figure 4.
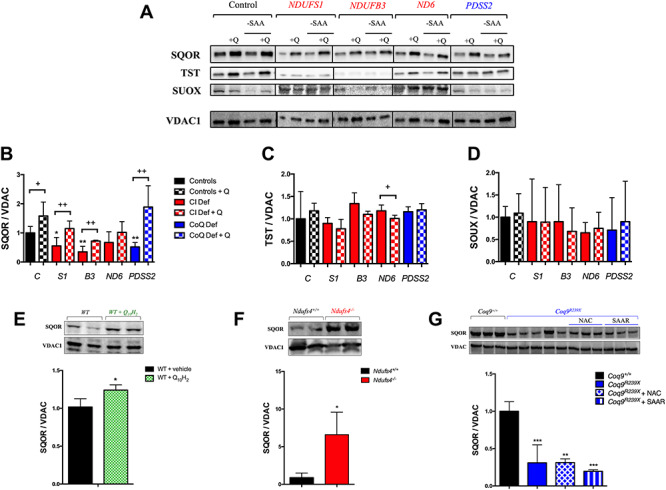
The levels of SQOR are modified under mitochondrial dysfunction or CoQ10 supplementation, independently of sulfur amino acids availability. (A–D) Levels of the proteins of the mitochondrial hydrogen sulfide oxidation pathway in human skin fibroblasts from control and patients with mutations in Complex I subunits or CoQ10 biosynthetic genes. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; differences versus Control. +P < 0.05; ++P < 0.01; +++P < 0.001; Control versus Control + CoQ10 or Complex I deficiency versus Complex I deficiency + CoQ10 or CoQ10 deficiency versus CoQ10 deficiency + CoQ10 (t-test; n = 3 for each group; n = 3 for each group). C, control; S1, NDUFS1 mutant; B3, NDUFB3 mutant; S3, NDUFS3 mutant; ND6, ND6 mutant; PDSS2, PDSS2 mutant; CI Def, cells with Complex I deficiency; CoQ Def, cells with CoQ10 deficiency; Q, CoQ10; -SAA, medium without sulfur aminoacids. The blot image (A) has been made from three different membranes, as follow: blot 1, control and PDSS2; blot 2, NDUFS1 and ND6; blot 3, NDUFB3. (E) Levels of SQOR in the liver of C57bl6j mice supplemented with ubiquinol-10 for 1 month. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; WT versus WT + CoQ10H2 (t-test; n = 4 for each group). (F) Levels of SQOR in the brain of Ndufs4−/− mice. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; Ndufs4+/+ versus Ndufs4−/− (t-test; n = 5 for each group). (G) Levels of SQOR in the kidneys of Coq9R239X with vehicle, NAC or under SAAR. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; differences versus Coq9+/+ (t-test; n = 3 for each group; n = 3–5 for each group).
To assess whether the effects of CoQ10 supplementation, CI deficiency or different availability of sulfur aminoacids on SQOR levels observed in vitro also occur in vivo, we measured SQOR levels in three different systems: (1) in the liver of C57BL/6 J mice after CoQ10H2 supplementation, because the liver is the tissue that accumulates more CoQ10 when it is orally administrated (21,22); (2) in the brain of Ndufs4−/− mice, the symptomatic tissue in this model of Leigh Syndrome due to CI deficiency (23) and (3) in the kidney of Coq9R239X mice under supplementation with N-acetylcysteine (NAC), a sulfur-containing aminoacid, or a diet with restriction in sulfur aminoacids (SAAR), since the kidney has a very active sulfide metabolism and it has reduced levels of SQOR in this model of CoQ deficiency (8,24). The levels of total CoQ (CoQ9 + CoQ10) increase in the liver of C57BL/6J mice after 1 month of water-soluble CoQ10H2 supplementation (Supplementary Material, Table S3). Consistent with this result, also the levels of SQOR increase in the liver of the same treated mice, compared with the untreated mice (Fig. 4E). The brain of Ndufs4−/− mice shows normal levels of CoQ9, as compared with those in Ndufs4+/+ mice (Supplementary Material, Table S3). However, the SQOR levels are significantly increased in mitochondria from the brain of Ndufs4−/− mice, compared with Ndufs4+/+ mice (Fig. 4F). The levels of CoQ9 and SQOR are significantly lower in the kidneys of Coq9R239X mice than in the kidneys of Coq9+/+, and they are not rescued by NAC or SAAR supplementation (Supplementary Material, Table S3 and Fig. 4G). These results indicate that CoQ10 is one of the factors that may regulate the levels of SQOR in vivo, independently of the sulfur-aminoacids availability. Moreover, SQOR is up-regulated, probably as a compensatory response, in the brain of the mouse model of Leigh Syndrome due to severe CI deficiency.
The abnormalities of SQOR levels in mitochondrial dysfunction are independent of mitochondrial supercomplexes formation
Since we have not identified a robust correlation between the SQOR levels and the residual CI activity, and because SQOR could interact with the mitochondrial supercomplexes though one of its components, i.e. CoQ, we evaluated the mitochondrial supercomplexes in control and mutant cells and mice. In the in vitro experiments, we used NDUFS8, NDUFS1 and NDUFB3 mutant cells, which have 54, 37 and 17% of residual CI activity, respectively, and a decrease in SQOR levels in the two latter cases. A decrease in the CI bound to supercomplexes is observed in the three mutant cells compared with control cells (Supplementary Material, Fig. S6A–C), while free CI is almost undetectable (Supplementary Material, Fig. S6A and D). Moreover, a decrease in the CIII bounds to supercomplexes and an increase of the free CIII are observed in the three mutant cells compared with control cells (Supplementary Material, Fig. S6E–H). Therefore, we did not observe a correlation between the disruption of the mitochondrial supercomplexes and the decrease in SQOR levels.
To test whether SQOR could be detected in association with some mitochondrial complex/supercomplex, we performed BNGE-Page with mitochondria from kidneys of wild-type, Ndufs4−/− and Coq9R239X mice and incubated the membranes with an anti-SQOR antibody. The pattern of bands is similar to those obtained for CIII (Supplementary Material, Fig. S6I). However, if we incubate the membrane with the anti-rabbit secondary antibody a similar pattern of bands is obtained (Supplementary Material, Fig. S6J), and this mainly corresponds to CIII (Supplementary Material, Fig. S6K), indicating some kind of cross-reaction between anti-rabbit antibody and some subunit of the CIII. Thus, we are not able to detect SQOR associated to any mitochondrial complex/supercomplex. Those results correlate with the data obtained by Van Strien and colleagues in a proteomic analysis in blue native gels run with samples of skin fibroblasts from control subjects (25). The representation of those data shows that the major proportion of SQOR is not bound to respiratory complexes or supercomplexes (Supplementary Material, Fig. S6L–N).
CoQ10 supplementation downregulates the transsulfuration pathway, independently of sulfur aminoacids availability
To evaluate whether the transsulfuration pathway may adapt to the changes of SQOR levels, we quantified the levels of CBS, CSE and 3MST in vitro in the same experimental groups used to study the H2S oxidation pathway. The levels of CBS are significantly increased in NDUFS3, COQ4 and PDSS2 mutant cells (Fig. 5A and B; Supplementary Material, Fig. S7A and B). The levels of CSE are increased in NDUFS1, NDUFS3, ND3 and COQ4 mutant cells (Fig. 5A and C; Supplementary Material, Fig. S7A and B). The levels of 3MST do not change in any mutant cells compared with control cells. Therefore, CBS and CSE are increased in certain cases of CI or CoQ deficiency.
Figure 5.
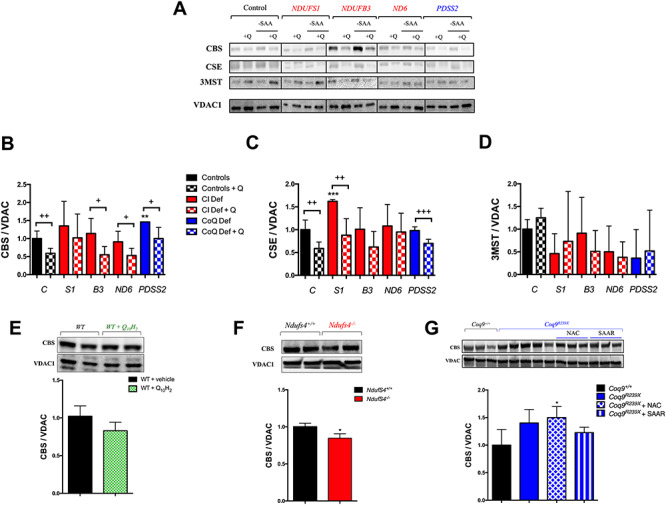
CoQ10 regulates the enzymes of transsulfuration pathway independently of sulfur amino acids availability. (A–D) Levels of the proteins of the transsulfuration pathway in human skin fibroblasts from control and patients with mutations in Complex I subunits or CoQ10 biosynthetic genes. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; differences versus Control. +P < 0.05; ++P < 0.01; +++P < 0.001; Control versus Control + CoQ10 or Complex I deficiency versus Complex I deficiency + CoQ10 or CoQ10 deficiency versus CoQ10 deficiency + CoQ10 (t-test; n = 3 for each group; n = 3 for each group). C, control; S1, NDUFS1 mutant; B3, NDUFB3 mutant; S3, NDUFS3 mutant; ND6, ND6 mutant; PDSS2, PDSS2 mutant; CI Def, cells with Complex I deficiency; CoQ Def, cells with CoQ10 deficiency; Q, CoQ10; -SAA, medium without sulfur aminoacids. The blot image (A) has been made from three different membranes, as follow: blot 1, control and PDSS2; blot 2, NDUFS1 and ND6; blot 3, NDUFB3. (E) Levels of CBS in the liver of C57bl6j mice supplemented with ubiquinol-10 for 1 month. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; WT versus WT + CoQ10H2 (t-test; n = 4 for each group). (F) Levels of CBS in the brain of Ndufs4−/− mice. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; Ndufs4+/+ versus Ndufs4−/− (t-test; n = 5 for each group). (G) Levels of SQOR in the kidneys of Coq9R239X with vehicle, NAC or under SAAR. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; differences versus Coq9+/+ (t-test; n = 3 for each group; n = 3–5 for each group).
After supplementation with CoQ10, the levels of CBS and CSE consistently decrease in all cell lines, compared with untreated cells (Fig. 5A–C; Supplementary Material, Fig. S7A–C), although the decrease is statistically significantly only in controls and NDUFB3, NDUFS3, ND3, ND6, COQ4 and PDSS2 mutant cells for CBS (Fig. 5B and Supplementary Material, Fig. S7B); and controls and NDUFB9, NDUFS1 and PDSS2 mutant cells for CSE (Fig. 5C and Supplementary Material, Fig. S7C). On the contrary, the levels of 3MST are not modified after CoQ10 supplementation (Fig. 5A and D; Supplementary Material, Fig. S7A and D). All these changes are similar in the cells cultured under SAAR (Fig. 5A and Supplementary Material, Fig. S7A; quantitative data not shown). Therefore, there is also an indirect correlation between the protein levels of SQOR and the protein levels of CBS and CSE after CoQ10 supplementation. The effect of CoQ10 over SQOR and CBS is also confirmed with a lower concentration of CoQ10 (Supplementary Material, Fig. S8).
In vivo, the levels of CBS are slightly decreased in the liver of C57BL/6 J mice treated with CoQ10H2 and in the brain of Ndufs4−/− mice, compared with the respective control animals (Fig. 5E and F). In the kidneys of Coq9R239X mice, the levels of CBS slightly increased compared with Coq9+/+ mice and this change was maintained under supplementation with NAC or SAAR (Fig. 5G). Therefore, the inverse correlation between SQOR and CBS also occurs in vivo.
CoQ10 indirectly regulates the transsulfuration pathway through SQOR and/or other transcriptional regulatory pathways, and independently of oxidative stress
To assess whether CoQ10 regulates the enzymes of the transsulfuration pathway directly or through the modulation of SQOR, we overexpressed SQOR in control cells and NDUFB3 and NDUFS1 mutant, and measured mRNA and protein levels of SQOR, CBS and CSE. The transduction with SQOR-LV produces significant increases of SQOR mRNA and protein levels in control and mutant cell, compared with the matched non-transduced cells (Fig. 6A, D and E). In parallel, the mRNA and protein levels of CBS and CSE are significantly decreased in the cells transduced with the SQOR-LV, compared with the matched non-transduced cells (Fig. 6B, C, D, F and G), suggesting that the effect of CoQ10 over CBS and CSE is an indirect action through the modulation of SQOR levels. We also assessed the effects of SQOR overexpression on serine biosynthesis, folate cycle and nucleotides metabolism, and observed that SQOR overexpression reproduces the gene expression profile changes caused by CoQ10 supplementation (Fig. 6H–K). Specifically, the expression of PSAT1 slightly decreases in both control and NDUFS1 SQOR-transduced cells (Fig. 6H); and the expression of MTHFFD2, CMPK2 and EIF2AK2 significantly increases in both control and NDUFS1 SQOR-transduced cells (Fig. 6I–K).
Figure 6.
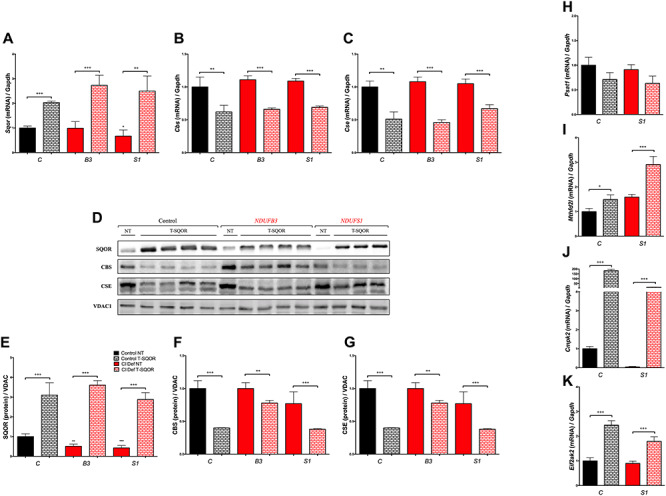
CoQ10 indirectly regulates the transsulfuration pathway through SQOR. (A–C) Effect of genetic overexpression of SQOR in the expression levels of the genes SQOR, CBS and CSE. (D–G) Effect of genetic overexpression of SQOR in the levels of the proteins SQOR, CBS and CSE. (H–K) Effect of genetic overexpression of SQOR in the expression levels of the genes PSAT1, MTHFD2L, CMPK2 and EIF2AK2. C, control; B3, NDUFB3 mutant; S1, NDUFS1 mutant; NT, non-transduced; T-SQOR, lentiviral vector containing SQOR cDNA. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; differences versus Control NT. +P < 0.05; ++P < 0.01; +++P < 0.001; Control NT versus Control T-SQOR or Complex I deficiency NT versus Complex I deficiency T-SQOR or CoQ10 deficiency NT versus CoQ10 deficiency T-SQOR (t-test; n = 3 for each group; n = 3–4 for each group).
Nevertheless, we also investigated whether the effects of CoQ10 on the changes of the expression of the identified genes could be attributed to a direct interaction of COQ10 with pathways involved in transcriptional regulation or cell signaling. Transcriptomics analysis reveals that CoQ10 partially modifies the STAT3 (Supplementary Material, Fig. S9A, B, E, F) and the HIF1α signaling (Supplementary Material, Fig. S9A, C, E, G) pathways in both control (Supplementary Material, Fig. S9A–C) and NDUFS1 mutant (Supplementary Material, Fig. S9E–G) cells. Moreover, the sumoylation pathway is altered in control cells treated with CoQ10 (Supplementary Material, Fig. S9A and D), and the NRF2 response in the NDUFS1 mutant cells treated with CoQ10 (Supplementary Material, Fig. S9A and D). Therefore, we cannot exclude that CoQ10 supplementation effects on the transsulfuration pathway are partially mediated by its effects on other transcriptional regulators.
To know whether the mechanism of action of CoQ10 over sulfide metabolism may be mediated by oxidative stress, we also tested the effects of other antioxidants, i.e. idebenone (18), NAC or melatonin (aMT) (26), in the levels of SQOR. Idebenone induces a profound cell death at the same dose than CoQ10, as reported in other studies (27). The treatments with NAC or melatonin do not modify the levels of SQOR in control cells and mutated in NDUFS1, NDUFB3 and PDSS2 (Supplementary Material, Fig. S10), suggesting that oxidative stress is not involve in the regulation of SQOR in vitro.
The levels of SQOR and CBS correlate with the levels of total GSH in vivo
We previously reported that the glutathione system is decreased in specific tissues of animals with CoQ deficiency, a fact that we attributed to the disruption of the sulfide metabolism (7,8). To confirm that premise, we evaluated the glutathione system in the kidneys of Coq9R239X treated with NAC or SAAR, as well as in the brain of Ndufs4−/− mice. The kidneys of Coq9R239X, which have decreased levels of SQOR and increased levels of CBS, show decreased levels of total glutathione, and that decrease is preserved after the supplementation with NAC or SAAR (Fig. 7A). Moreover, the GSSG/GSH ratio is reduced in untreated Coq9R239X mice, as well as in Coq9R239X mice treated with NAC or SAAR, compared with Coq9+/+ mice (Fig. 7B). The levels of the glutathione enzymes, GPx and GRd, are mildly decreased in the three experimental groups of Coq9R239X mice, compared with Coq9+/+ mice, although the decrease is statistically significant only in the case of GPx in untreated Coq9R239X mice (Fig. 7C). The brain of Ndufs4−/− mice, which has increased levels of SQOR and decreased levels of CBS, shows increased levels of total glutathione and GSSG/GSH ratio, compared with Ndufs4+/+ mice (Fig. 7D and E), but no differences were found in the levels of glutathione enzymes. Globally, these data confirm that sulfide metabolism influences the levels of glutathione, independently of the sulfur aminoacids availability.
Figure 7.
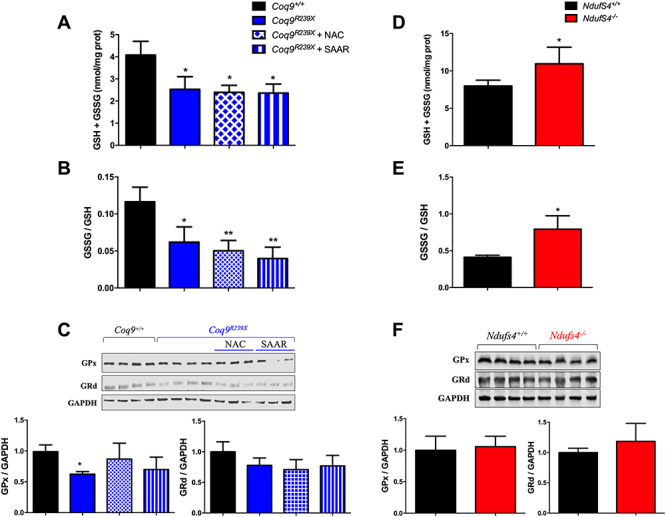
The changes in sulfide metabolism correlate with the alterations of the glutathione system in vivo. (A–C) Total glutathione (A), GSSG/GSH ratio (B) and glutathione enzymes levels (C) in Coq9R239X with regular chow, a diet supplemented with N-acetylcysteine (NAC) or a diet with restricted content of sulfur aminoacids (SAAR). Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; differences versus Coq9+/+ (t-test; n = 3 for each group; n = 3–4 for each group). (D–F) Total glutathione (D), GSSG/GSH ratio (E) and glutathione enzymes levels (F) in Ndufs4−/−. Data are expressed as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; differences versus Ndufs4+/+ (t-test; n = 4 for each group).
Discussion
Mitochondrial dysfunction causes heterogeneous clinical presentations, due to the variable involvement of multiple metabolic pathways related to the mitochondrial metabolism, e.g. one carbon metabolism or fatty acids oxidation (3–5,14). Therefore, finding therapeutic approaches for those dysfunctions is challenging. Oral CoQ10 supplementation is a common recommended therapeutic approach in patients with mitochondrial disorders and other diseases with secondary mitochondrial involvement. However, its rationale use is not well understood, particularly in patient with normal CoQ10 levels. Here, we show that supraphysiological levels of CoQ10 induce the overexpression of SQOR, the first enzyme of the mitochondrial hydrogen sulfide oxidation pathway (sulfide catabolization), leading to downregulation of CBS and CSE. These two latter enzymes belong to the transsulfuration pathway (sulfide biosynthesis), which is metabolically connected to serine biosynthesis, the folate cycle and the nucleotides metabolism (3,28). Thus, supraphysiological levels of CoQ10 result in a modulation of these pathways, restoring the function that is altered by mitochondrial dysfunction (3–5,14).
We previously reported that CoQ deficiency causes a severe reduction in the levels of SQOR, leading to a disruption of the sulfide metabolism and a decrease in the glutathione levels (7,8). In the current study, we show that CoQ deficiency also affects the transsulfuration pathway, independently of the availability of sulfur aminoacids. Additionally, we show that specific molecular defects in Complex I subunits cause disruption of the sulfide metabolism. However, those alterations do not correlate with the residual Complex I activity and are not due to an interaction of the SQOR with the mitochondrial supercomplexes. These findings suggest that the variable response of the sulfide metabolism to complex I deficiency could influence the heterogenous disease phenotype (29), in part because of the effects of that response on the metabolism of glutathione, which requires the balanced metabolism of cysteine and glycine (28,30).
Importantly, supplementation with CoQ10 in both controls and Complex I deficiency cells causes a consistent upregulation of SQOR and downregulation of the transsulfuration pathway, independently of the availability of sulfur aminoacids. This modulation of the sulfide metabolism causes a therapeutic adaptation of metabolic pathways that are closely connected to the transsulfuration pathway, i.e. the serine biosynthesis, the folate cycle and the nucleotides metabolism. These metabolic pathways are unbalanced in a variety of models of mitochondrial diseases (3–5,14), probably due to the activation of serine catabolism (31). Specifically, CBS, CSE, PSAT1, SHMT2 and PHGDH are upregulated after 24 h of pharmacological inhibition of Complex I in cell culture (6); Mthfd2 is induced in the skeletal muscle, heart and brown adipose tissue of the Deletor mice, resulting in a remodeling of the one carbon metabolism (3,5,32); Mthfd2 and Shmt2 are induced in the heart of five different mouse models of mitochondrial diseases due to disruption of key genes that regulate mtDNA gene expression, mtDNA replication or translation (14); CBS, CSE, PHGDH, PSPH and PSAT1 are induced after pharmacologically induced mtDNA depletion in vitro, leading to an increase in serine biosynthesis and transsulfuration (4); or MTHFD1L, MTHFD2, PSAT1, PHGDH and SHMT2 are upregulated in the muscle of patients with mitochondrial myopathy due to mtDNA single or multiple deletion(s) (5). Importantly, our in vitro analyses show that supraphysiological levels of CoQ10 can rescue all these abnormalities, inducing the downregulation of CBS, CSE, PHGDH, PSPH, PSAT1, MTHFD1L and MTHFD2, among others, in both control and Complex I deficiency cells. Furthermore, the serine/glycine ratio, which is decreased in mouse models of secondary CoQ deficiency (14), increases after CoQ10 supplementation, most likely due to a compensation in the biosynthesis and use of serine, together with a decrease of glycine production in the folate cycle. These changes in the transsulfuration, serine biosynthesis and folate cycle correlate with the increase in SQOR and an alteration in the nucleotide metabolism. However, these effects are not likely due to an stimulation of the OXPHOS system, where CoQ plays a central role in the Q junction (33), since CoQ10 is able to stimulate the mitochondrial complexes activities in cells with CoQ10 deficiency but its effect in cells with normal levels of CoQ10 is limited (34). Therefore, high doses of CoQ10 supplementation may provide therapeutic benefits in patients with mitochondrial disorders and alterations in sulfide metabolism, serine biosynthesis, folate cycle or nucleotide metabolism. Moreover, the effect of CoQ10 on the upregulation of SQOR and downregulation of CBS and CSE could reduce the clinical consequences of the accumulation of hydrogen sulfide and reduction in glutathione levels in patients with ethylmalonic encephalopathy due to mutations in ETHE1 (30,35) or in patients with Leigh Syndrome due to mutations in SQOR (36). Furthermore, the effects of CoQ10 in sulfide metabolism could benefit patients with colon cancer, thyroid carcinomas or Chron’s disease, since the increase of transsulfuration pathway has been identified as a key pathogenic mechanism in both cases (37–39). However, an important limitation of the exogenous CoQ10 supplementation is the low absorption and bioavailability of this molecule. CoQ10, orally or intraperitoneally administrated, mainly localizes in the spleen and the liver, with a low proportion localized in the heart, muscle and kidneys, and a very low proportion in the brain (17,21). Therefore, effective strategies to increase the bioavailability of CoQ10 are necessary to enhance its therapeutic potential.
The current study also contributes to elucidate the mechanisms of how CoQ10 is able to induces metabolic changes at the transcriptional level. Our data demonstrate that the overexpression of SQOR alone, could lead to the downregulation of the transsulfuration pathway and to the adaptation of the other metabolic pathways. Most likely, all these changes are mediated by the detection of the levels of the SQOR products, i.e. thiosulfate (SSO32−) or GSH persulfide (GSS−), as it happens with the CstR in bacteria (40). However, homologous sensors/regulators have not been identified in mammals yet (41). Nevertheless, the supplementation with thiosulfate in HUVEC cells induces a decrease in CSE levels (42). Additionally, the results of our transcriptome analysis suggest that some other mechanisms could mediate the adaptative response to the supraphysiological levels of CoQ10: (1) repression of STAT3 pathway (43), and the subsequent reduction of the binding of STAT3 into the CSE promoter, which reduces CSE expression (44); (2) activation of the Sumoylation that could downregulate CBS (45,46) and/or (3) alteration of HIF1α signaling that could downregulate CBS (47).
Although further studies are needed to elucidate the detailed mechanisms of the transcriptional regulation mediated by CoQ10, we demonstrate that supraphysiological levels of CoQ10 induce SQOR and downregulates the transsulfuration pathway, leading to a downregulation of serine biosynthesis and an adaptation of the linked pathways of folate cycle and nucleotides metabolism. All these metabolic changes could be mediated by the CoQ10–SQOR interaction, and they could explain the therapeutic benefits of CoQ10 supplementation in some patients with mitochondrial disorders. They, therefore, are very likely relevant for the treatment of mitochondrial dysfunction in other common diseases.
Materials and Methods
Human skin fibroblasts culture and treatments
Skin fibroblasts were provided by Holger Prokisch from the University of Munhen. Control and mutant skin fibroblasts were obtained from patients with mutations in different subunits of the mitochondrial Complex I or CoQ biosynthetic genes (Supplementary Material, Table S1). Fibroblasts were cultured at 37°C and 5% CO2 in high glucose DMEM-GlutaMAX medium supplemented 1% MEM non-essential amino acids, and 1% antibiotics/antimycotic. Fibroblasts were treated with 100 μm CoQ10, a dose commonly used in cell culture studies (34,48), during 7 days. A pilot study with 20 μm CoQ10, 100 μm idebenone, 100 μm NAC or 100 μm aMT for 7 days was also performed. CoQ10 was provided by Tishcon (USA) in a water-soluble formulation that contains gamma cyclodextrin. A control group with vehicle at the same dose was studied. CoQ10, idebenone or vehicle were dissolved in FBS prior to addition to culture medium (10% FBS). NAC was dissolved in phosphate buffered saline (PBS). Melatonin stock solution was prepared in 15% propylene glycol, giving a final concentration of propylene glycol in cell culture of 0.75%. Sulfur aminoacids restriction was carried out by incubating for up to 24 h in DMEM lacking Met and Cys (Gibco) with 10% dialyzed FBS.
Mice use and experimental treatment
Mice were housed in the Animal Facility of the University of Granada under a SPF zone with lights on at 7:00 a.m. and off at 7:00 p.m. Mice had unlimited access to water and rodent chow. All experiments were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the University of Granada (procedure number 9-CEEA-OH-2013) and in accordance with the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (CETS # 123), directive 2010/63/EU on the protection of animals used for scientific purposes and the Spanish law (R.D. 53/2013). Animals were randomly assigned in experimental groups. Data were randomly collected and processed as well.
Control C57Bl6 mice were treated with ubiquinol-10 (Q10H2), which has better bioavailability in vivo, in the drinking water at a dose of 240 mg/kg bw/day (21). Ubiquinol-10 was provided by Kaneka (Japan) in a water-soluble formulation that contains gamma cyclodextrin. A control group with vehicle at the same dose was studied. The treatment started at 1 month of age, and the mice were euthanized for the experimental assays at 2 months of age. Livers were extracted and frozen at −80°C until assayed for CoQ10 or protein quantification. Ndufs4+/+ and Ndufs4−/− mice (MGI: 3793713), a model of Leigh syndrome due to Complex I deficiency (23), were euthanized at 4–5 weeks of age. Brains were extracted and frozen at –80°C until assayed for CoQ10 or protein quantification. To quantify SQOR in the brain, mitochondria were freshly isolated, as previously described (8). Coq9R239X mice (MGI: 5473628) were treated with L-NAC added to drinking water at a concentration of 1%. Separately, mice were treated with sulfur amino acid restriction (SAAR) diet containing 0.15% Methionine and 0% Cystine (49), compared with control diet containing 0.6% Methionine and 0.42% Cystine.
Quantification of CoQ10 levels in cells and mouse tissues and measurement of Complex I activity
CoQ9 and CoQ10 levels were determined via reversed phase HPLC coupled to electrochemical (EC) detection (18,50). The results were expressed in ng CoQ/mg prot. Complex I activity was determined as previously described (51), normalized by citrate synthase activity, and represented as the percentage of the control values.
Serine and glycine quantification
Serine and glycine were quantified as described (52). Cell pellets were stored at −80°C and extracted immediately before analysis in ice-cold lysis/extraction buffer (methanol:acetonitrile:water 5:3:2 v/v). Samples were concentrated in a speedvac system and the resulting dry residues were resuspended in water. Standards of serine and glycine were dissolved in water at a concentration of 10, 100 and 10 000 ng/ml. Samples were separated through a 5 min isocratic elution on a Kinetex XB-C18 column (150 × 2.1 mm i.d., 1.7 μm particle size—Phenomenex, Torrance, CA, USA) at 250 μl/min (mobile phase: 5% acetonitrile, 95% 18 mΩ H2O, 0.1% formic acid; column temperature: 25°C). The UHPLC system was coupled online with a Q Exactive Focus mass spectrometer (Thermo, San Jose, CA, USA), scanning in Full MS mode (2 μscans) at 70 000 resolution from 60 to 200 m/z. Calibration was performed before each analysis using a positive calibration mix (Piercenet—Thermo Fisher, Rockford, IL, USA). Data were analyzed using Tracefinder 4.1 software (Thermo, San Jose, CA, USA). Analyte retention times were confirmed by comparison with external standard retention times.
Transcriptome analysis by RNA-Seq
The RNeasy Lipid Tissue Mini Kit (Qiagen) was used to extract total RNAs from the brainstem of five animals in each experimental group. The RNAs were precipitated and their quality and quantity assessed using Agilent Bioanalyzer 2100 and an RNA 6000 chip (Agilent Technologies). The cDNA libraries were then constructed using the TruSeq RNA Sample Prep Kit v2 (Illumina, Inc.) and their quality checked using Agilent Bioanalyzer 2100 and DNA 1000 chip (Agilent Technologies). The libraries were Paired End sequenced in a HiSeq 4000 system (Illumina, Inc.). We aimed for 4–5 Giga Bases outcome per sample. The quality of the resulting sequencing reads was assessed using FastQC. The Homo sapiens hg38 fasta and gtf files of the reference genome were downloaded from the Ensembl database and indexed using the bwtsw option of BWA (53). BWA, combined with xa2multi.pl and SAMtools (54), was also used for aligning the sequencing reads against the reference genome, and HTSeq was used for counting the number of reads aligned to each genomic locus (55). The alignments and counting were carried out in our local server following the protocols as described (56).
After elimination of the genomic loci that aligned to <5 reads in <5 samples and normalization of the read counts by library size, the differential gene expression was detected using the Generalized Linear Model (glmLRT option) statistic in EdgeR. We used a 0.05 P-level threshold. Annotation of the differentially expressed genes was obtained from H. sapiens hg38 genome.
The genes that passed the inclusion criteria were then subjected to gene classification, using a database of hand-curated literature on specific protein–protein interactions and regulatory networks (Ingenuity Pathway Analysis (IPA); Ingenuity Systems, Redwood City, CA, USA). The canonical pathways involved in the metabolism and transcriptional regulation where the significantly changed transcripts are involved were detected by Ingenuity Pathway Analysis were evaluated and P-values < 0.05 were considered significant. Further, specific functional networks, based on published knowledge on protein–protein interactions and regulatory networks, were constructed by Ingenuity Pathway Analysis (57).
Gene expression analyses
Total cellular RNA from frozen cell pellets were extracted and electrophoresed in 1.5% agarose gels in order to check the RNA integrity. RNA from skin fibroblasts was extracted using Real Total RNA Spin Plus Kit (Real). Total RNA was quantified by optical density at 260/280 nm and was used to generate cDNA with High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Amplification of human genes SQOR (Hs01126963_m1), CBS (Hs01598251_m1), CSE (Hs00542284_m1), TST (Hs00361812_m1), SUOX (Hs00166578_m1), PSAT1 (Hs00795278_mH), MTHFD2L (Hs01017321_m1), CMPK2 (Hs01013364_m1), EIF2AK2 (Hs00169345_m) and the human GADPH probe, as a standard loading control (Hs99999905_m1), was performed using quantitative real-time PCR, specific Taqman probes (from Applied Biosystems) and the standard curve method.
Sample preparation and western blot analysis in tissues and fibroblasts
For western blot analyses, a glass-Teflon homogenizer was used to homogenize mouse liver, brain and kidney samples at 1100 rpm in T-PER® buffer (Thermo Scientific) with protease inhibitor cocktail (Pierce). Homogenates were sonicated and centrifuged at 1000g for 5 min at 4°C, and the resultant supernatants were used for western blot analysis. For western blot analyses in cerebral mitochondria, the pellets containing the mitochondrial fraction were re-suspended in RIPA buffer with protease inhibitor cocktail (8). Mitochondrial isolation was performed as previously described (8). For western blot analyses in skin fibroblasts, the pellets containing the cells were re-suspended in RIPA buffer with protease inhibitor cocktail. About 30 μg of protein from the sample extracts was electrophoresed in 12% Mini-PROTEAN TGX™ precast gels (Bio-Rad) using the electrophoresis system mini-PROTEAN Tetra Cell (Bio-Rad). Proteins were transferred onto PVDF 0.45-μm membranes using a Trans-blot Cell (Bio-Rad) and probed with target antibodies. Protein–antibody interactions were detected using peroxidase-conjugated horse anti-mouse, anti-rabbit or anti-goat IgG antibodies and Amersham ECL™ Prime Western Blotting Detection Reagent (GE Healthcare, Buckinghamshire, UK). Band quantification was carried out using an Image Station 2000R (Kodak, Spain) and a Kodak 1D 3.6 software. Protein band intensity was normalized to VDAC1 (mitochondrial proteins), and the data were expressed in terms of percent relative to wild-type mice or control cells (8,58).
The following primary antibodies were used: anti-SQRDL (Proteintech, 17256-1-AP), anti-CBS (Proteintech, 14787-1-AP), anti-CSE (Proteintech, 12217-1-AP), anti-TST (Proteintech, 16311-1-AP), anti-ETHE1 (Sigma, HPA029029), anti-SUOX (Proteintech, 15075-1-AP), anti-3-MPST (Sigma, HPA001240), anti-GPx (Abcam, ab125066), anti-GRd (Santa Cruz Biotechnology, sc-32886) and anti-VDAC1 (Abcam, ab14734).
Evaluation of supercomplex formation by BNGE
Blue native gel electrophoresis (BNGE) was performed on crude mitochondrial fractions from mice kidneys or human fibroblasts. Mitochondrial isolation from kidneys was performed as previously described (59). One aliquot of the crude mitochondrial fraction was used for protein determination. The remaining samples were then centrifuged at 13 000g for 3 min at 4°C. The mitochondrial pellets were suspended in an appropriate volume of medium C (1 M aminocaproic acid, 50 mm Bis-Tris–HCl [pH 7.0]) to create a protein concentration of 10 mg/ml, and the membrane proteins were solubilized in digitonin (4 g/g) and incubated for 10 min in ice. After centrifugation for 30 min at 13 000g (4°C), the supernatants were collected and 3 μl of 5% Brilliant Blue G dye, prepared in 1 M aminocaproic acid, was added.
Approximately, 2 × 106 cells were used for mitochondrial isolation in human skin fibroblasts. Cell pellets were resuspended in PBS, then digitonin was added at a concentration of 8 mg/ml and the mixture incubated for 10 min in ice for solubilizing cell membranes. The samples were centrifuged for 5 min at 10 000g (4°C) and resuspended in PBS, then centrifuged for a second time at the same conditions. The pellets were resuspended in mitochondrial cell buffer (1.5 M aminocaproic acid, 50 mm Bis-Tris–HCl [pH 7.0]). Then, the mitochondrial membrane proteins were solubilized with digitonin at a final concentration of 1% and incubated for 5 min in ice. Samples were centrifuged for 30 min at 18 000g (4°C). The supernatants were combined with 5% Brilliant Blue G dye, prepared in 1 M aminocaproic acid.
Mitochondrial proteins were then loaded and run on a 4–16% gradient native gel (Thermo Scientific, BN1002BOX) as previously described (60). After electrophoresis, the complexes were electroblotted onto PVDF membranes and sequentially tested with specific antibodies against CI, anti-NDUFA9 (Abcam, ab14713), CIII, anti-ubiquinol-cytochrome c reductase core protein I (Abcam, ab110252), anti-SQRDL (Proteintech, 17256-1-AP), anti-Vdac1 (Abcam, ab14734) and anti-rabbit (Thermofisher, 31460).
Lentiviral vectors constructs, vector production and cell transduction
The CSQORWP LV plasmid was constructed by standard cloning techniques using PstI/BamHI restriction enzymes to replace the eGFP in the CEWP backbone by the SQOR cDNA (obtained by gene synthesis from Genscript).
Vector production was performed by fast growing 293T cells plated on amine-coated petri dishes (Sarsted, Newton, NC) in order to achieve 80–90% confluence for transfection. 293T cells were cotransfected with the vector (CSQORWP), the packaging (pCMVΔR8.91) and the envelope (pMD2.G) plasmids using LipoD293 (SignaGen, Gainthersburg, MD, USA). Viral supernatants were collected 48 h after transfection and the particles were frozen or concentrated by ultrafiltration at 2000 g and 4°C, using 100 kDa centrifugal filter devices (Amicon Ultra-15, Millipore, Billerica, MA). LV particles were used to transduce skin fibroblasts by adding different volumes to the cell cultures in order to achieve the desired multiplicities of infection. The medium was changed after 5 h of incubation.
Glutathione measurement
Glutathione measurements were performed in total homogenate of mouse tissues. Tissues were homogenized in sodium phosphate buffer (A) (100 mm sodium phosphate, 5 mm EDTA-Na2, pH 8.0) and deproteinized using ice-cold TCA 40% and centrifuged at 20 000g for 15 min. For GSH measurement, the supernatant was incubated with a solution of (A) and orthophthalaldehyde/ethanol (B) (1 mg/ml) for 15 min at room temperature. The fluorescence of the samples was measured at 340 nm excitation and 420 nm emission wavelengths in a spectrofluorometer plate reader (Bio-Tek Instruments Inc., Winooski, VT, USA). For GSSG measurement, the supernatant was preincubated with N-ethylmaleimide solution (5 mg/ml) for 40 min then alkalinized with 0.1 N NaOH (C). Aliquots of these mixtures were incubated with (B) and (C) for 15 min at room temperature. Finally, the fluorescence was measured as before. GSH and GSSG concentrations were calculated according to standard curves that we previously prepared, and the levels of GSH and GSSG were expressed in nmol/mg protein (8).
Statistical analysis
All statistical analyses were performed using the Prism 6 scientific software. Data are expressed as the mean ± SD of 37 experiments per group. An unpaired Student’s t-test was used to compare the differences between CoQ10-treated and untreated cells with the same genotype, as well as to compare the differences between mutated and control cells/mice. A P-value of 0.05 or less was considered to be statistically significant. The statistical test used for the transcriptomics analyses is described in its respective section.
Supplementary Material
Acknowledgements
P.G.-G., A.H.-G. and C.M. led the study, developed most of the experiments, conducted the IPA analyses, analyzed the results, designed the figures, and wrote the manuscript. E.B.-C. contributed to the mitochondrial assays and developed the experiments with the LVSQOR. M.B. designed and analyzed the RNA-Seq and contributed to the discussion. M.Z. performed some western blot experiments and contributed in the conception of the idea. U.B.A. contributed with some of the experiments in mouse tissues. SSH developed the LV-SQOR. H.P. provided the skin fibroblasts from patients. F.M. designed LV-SQOR.
G.E. contributed to the discussion. C.M.Q. contributed in the conception of the idea and edited the manuscript. L.C.L. conceived the idea for the project, supervised the experiments, and edited the manuscript. All authors critically reviewed the manuscript. Some results shown in this article will constitute a section of the P.G.-G. and A.H.-G. doctoral theses at the University of Granada.
Conflict of Interest statement. The other authors have declared that no conflict of interest exists.
Data Availability
RNA-Seq data were generated as described above. The files have been uploaded to the SRA repository of the National Center for Biotechnology Information. Accession: PRJNA636102; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA636102/.
Funding
This work was supported by grants from Ministerio de Ciencia e Innovación, Spain, and the ERDF (RTI2018-093503-B-100); the Muscular Dystrophy Association (MDA-602322); the University of Granada (grant reference ‘UNETE’, UCE-PP2017-06) (L.C.L.) and the National Institute of Health (NIH, United States) P01 HD080642-01 (C.M.Q.). A.H.-G. and P.G.-G. are ‘FPU fellows’ from the Ministerio de Universidades, Spain. E.B.-C. was supported by the Junta de Andalucía. U.B.A. was supported by the Erasmus+ Program.
References
- 1. Nunnari J. and Suomalainen A. (2012) Mitochondria: in sickness and in health. Cell, 148, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. et al. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell, 134, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nikkanen J., Forsstrom S., Euro L., Paetau I., Kohnz R.A., Wang L., Chilov D., Viinamaki J., Roivainen A., Marjamaki P. et al. (2016) Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab., 23, 635–648. [DOI] [PubMed] [Google Scholar]
- 4. Bao X.R., Ong S.E., Goldberger O., Peng J., Sharma R., Thompson D.A., Vafai S.B., Cox A.G., Marutani E., Ichinose F. et al. (2016) Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife, 5, e10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Forsstrom S., Jackson C.B., Carroll C.J., Kuronen M., Pirinen E., Pradhan S., Marmyleva A., Auranen M., Kleine I.M., Khan N.A. et al. (2019) Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab., 30, 1040–1054 e1047. [DOI] [PubMed] [Google Scholar]
- 6. Krug A.K., Gutbier S., Zhao L., Poltl D., Kullmann C., Ivanova V., Forster S., Jagtap S., Meiser J., Leparc G. et al. (2014) Transcriptional and metabolic adaptation of human neurons to the mitochondrial toxicant MPP(+). Cell Death Dis., 5, e1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ziosi M., Di Meo I., Kleiner G., Gao X.H., Barca E., Sanchez-Quintero M.J., Tadesse S., Jiang H., Qiao C., Rodenburg R.J. et al. (2017) Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol. Med., 9, 96–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luna-Sanchez M., Hidalgo-Gutierrez A., Hildebrandt T.M., Chaves-Serrano J., Barriocanal-Casado E., Santos-Fandila A., Romero M., Sayed R.K., Duarte J., Prokisch H. et al. (2017) CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol. Med., 9, 78–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DiMauro S., Schon E.A., Carelli V. and Hirano M. (2013) The clinical maze of mitochondrial neurology. Nat. Rev. Neurol., 9, 429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang W., Karamanlidis G. and Tian R. (2016) Novel targets for mitochondrial medicine. Sci. Transl. Med., 8, 326rv323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garone C. and Viscomi C. (2018) Towards a therapy for mitochondrial disease: an update. Biochem. Soc. Trans., 46, 1247–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hidalgo-Gutierrez A., Barriocanal-Casado E., Bakkali M., Diaz-Casado M.E., Sanchez-Maldonado L., Romero M., Sayed R.K., Prehn C., Escames G., Duarte J. et al. (2019) Beta-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9 (R239X) mice. EMBO Mol. Med., 11, e9466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hargreaves I.P. (2014) Coenzyme Q10 as a therapy for mitochondrial disease. Int. J. Biochem. Cell Biol., 49, 105–111. [DOI] [PubMed] [Google Scholar]
- 14. Kuhl I., Miranda M., Atanassov I., Kuznetsova I., Hinze Y., Mourier A., Filipovska A. and Larsson N.G. (2017) Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife, 6, e30952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yubero D., Montero R., Martin M.A., Montoya J., Ribes A., Grazina M., Trevisson E., Rodriguez-Aguilera J.C., Hargreaves I.P., Salviati L. et al. (2016) Secondary coenzyme Q10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion, 30, 51–58. [DOI] [PubMed] [Google Scholar]
- 16. Fazakerley D.J., Chaudhuri R., Yang P., Maghzal G.J., Thomas K.C., Krycer J.R., Humphrey S.J., Parker B.L., Fisher-Wellman K.H., Meoli C.C. et al. (2018) Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife, 7, e32111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bentinger M., Dallner G., Chojnacki T. and Swiezewska E. (2003) Distribution and breakdown of labeled coenzyme Q10 in rat. Free Radic. Biol. Med., 34, 563–575. [DOI] [PubMed] [Google Scholar]
- 18. Lopez L.C., Quinzii C.M., Area E., Naini A., Rahman S., Schuelke M., Salviati L., DiMauro S. and Hirano M. (2010) Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: time- and compound-dependent effects. PLoS One, 5, e11897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kabil H., Kabil O., Banerjee R., Harshman L.G. and Pletcher S.D. (2011) Increased transsulfuration mediates longevity and dietary restriction in Drosophila. Proc. Natl. Acad. Sci. USA, 108, 16831–16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hine C. and Mitchell J.R. (2015) Calorie restriction and methionine restriction in control of endogenous hydrogen sulfide production by the transsulfuration pathway. Exp. Gerontol., 68, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garcia-Corzo L., Luna-Sanchez M., Doerrier C., Ortiz F., Escames G., Acuna-Castroviejo D. and Lopez L.C. (2014) Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim. Biophys. Acta, 1842, 893–901. [DOI] [PubMed] [Google Scholar]
- 22. Kleiner G., Barca E., Ziosi M., Emmanuele V., Xu Y., Hidalgo-Gutierrez A., Qiao C., Tadesse S., Area-Gomez E., Lopez L.C. et al. (2018) CoQ10 supplementation rescues nephrotic syndrome through normalization of H2S oxidation pathway. Biochim. Biophys. Acta Mol. Basis Dis., 1864, 3708–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quintana A., Kruse S.E., Kapur R.P., Sanz E. and Palmiter R.D. (2010) Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. USA, 107, 10996–11001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karmin O. and Siow Y.L. (2018) Metabolic imbalance of homocysteine and hydrogen sulfide in kidney disease. Curr. Med. Chem., 25, 367–377. [DOI] [PubMed] [Google Scholar]
- 25. Van Strien J., Guerrero-Castillo S., Chatzispyrou I.A., Houtkooper R.H., Brandt U. and Huynen M.A. (2019) COmplexome profiling ALignment (COPAL) reveals remodeling of mitochondrial protein complexes in Barth syndrome. Bioinformatics, 35, 3083–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Acuna C.D., Lopez L.C., Escames G., Lopez A., Garcia J.A. and Reiter R.J. (2011) Melatonin-mitochondria interplay in health and disease. Curr. Top. Med. Chem., 11, 221–240. [DOI] [PubMed] [Google Scholar]
- 27. Varricchio C., Beirne K., Heard C., Newland B., Rozanowska M., Brancale A. and Votruba M. (2020) The ying and yang of idebenone: not too little, not too much—cell death in NQO1 deficient cells and the mouse retina. Free Radic. Biol. Med., 152, 551–560. [DOI] [PubMed] [Google Scholar]
- 28. Ducker G.S. and Rabinowitz J.D. (2017) One-carbon metabolism in health and disease. Cell Metab., 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fassone E. and Rahman S. (2012) Complex I deficiency: clinical features, biochemistry and molecular genetics. J. Med. Genet., 49, 578–590. [DOI] [PubMed] [Google Scholar]
- 30. Sahebekhtiari N., Fernandez-Guerra P., Nochi Z., Carlsen J., Bross P. and Palmfeldt J. (2019) Deficiency of the mitochondrial sulfide regulator ETHE1 disturbs cell growth, glutathione level and causes proteome alterations outside mitochondria. Biochim. Biophys. Acta Mol. Basis Dis., 1865, 126–135. [DOI] [PubMed] [Google Scholar]
- 31. Yang L., Garcia Canaveras J.C., Chen Z., Wang L., Liang L., Jang C., Mayr J.A., Zhang Z., Ghergurovich J.M., Zhan L. et al. (2020) Serine catabolism feeds NADH when respiration is impaired. Cell Metab., 31, 809–821 e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tyynismaa H., Carroll C.J., Raimundo N., Ahola-Erkkila S., Wenz T., Ruhanen H., Guse K., Hemminki A., Peltola-Mjosund K.E., Tulkki V. et al. (2010) Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet., 19, 3948–3958. [DOI] [PubMed] [Google Scholar]
- 33. Lemieux H., Blier P.U. and Gnaiger E. (2017) Remodeling pathway control of mitochondrial respiratory capacity by temperature in mouse heart: electron flow through the Q-junction in permeabilized fibers. Sci. Rep., 7, 2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rodriguez-Hernandez A., Cordero M.D., Salviati L., Artuch R., Pineda M., Briones P., Izquierdo L.G., Cotan D., Navas P. and Sanchez-Alcazar J.A. (2009) Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy, 5, 19–31. [DOI] [PubMed] [Google Scholar]
- 35. Tiranti V., Viscomi C., Hildebrandt T., Di Meo I., Mineri R., Tiveron C., Levitt M.D., Prelle A., Fagiolari G., Rimoldi M. et al. (2009) Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med., 15, 200–205. [DOI] [PubMed] [Google Scholar]
- 36. Friederich M.W., Elias A.F., Kuster A., Laugwitz L., Larson A.A., Landry A.P., Ellwood-Digel L., Mirsky D.M., Dimmock D., Haven J. et al. (2020) Pathogenic variants in SQOR encoding sulfide:quinone oxidoreductase are a potentially treatable cause of Leigh disease. J. Inherit. Metab. Dis., 43, 1024–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mottawea W., Chiang C.K., Muhlbauer M., Starr A.E., Butcher J., Abujamel T., Deeke S.A., Brandel A., Zhou H., Shokralla S. et al. (2016) Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn's disease. Nat. Commun., 7, 13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Phillips C.M., Zatarain J.R., Nicholls M.E., Porter C., Widen S.G., Thanki K., Johnson P., Jawad M.U., Moyer M.P., Randall J.W. et al. (2017) Upregulation of cystathionine-beta-synthase in colonic epithelia reprograms metabolism and promotes carcinogenesis. Cancer Res., 77, 5741–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Turbat-Herrera E.A., Kilpatrick M.J., Chen J., Meram A.T., Cotelingam J., Ghali G., Kevil C.G., Coppola D. and Shackelford R.E. (2018) Cystathione beta-synthase is increased in thyroid malignancies. Anticancer Res., 38, 6085–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luebke J.L., Shen J., Bruce K.E., Kehl-Fie T.E., Peng H., Skaar E.P. and Giedroc D.P. (2014) The CsoR-like sulfurtransferase repressor (CstR) is a persulfide sensor in Staphylococcus aureus. Mol. Microbiol., 94, 1343–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shimizu T. and Masuda S. (2020) Persulphide-responsive transcriptional regulation and metabolism in bacteria. J. Biochem., 167, 125–132. [DOI] [PubMed] [Google Scholar]
- 42. Leskova A., Pardue S., Glawe J.D., Kevil C.G. and Shen X. (2017) Role of thiosulfate in hydrogen sulfide-dependent redox signaling in endothelial cells. Am. J. Physiol. Heart Circ. Physiol., 313, H256–H264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee S.Y., Lee S.H., Yang E.J., Kim J.K., Kim E.K., Jung K., Jung H., Lee K., Lee H.H., Lee B.I. et al. (2017) Coenzyme Q10 inhibits Th17 and STAT3 Signaling pathways to ameliorate colitis in mice. J. Med. Food, 20, 821–829. [DOI] [PubMed] [Google Scholar]
- 44. You J., Shi X., Liang H., Ye J., Wang L., Han H., Fang H., Kang W. and Wang T. (2017) Cystathionine- gamma-lyase promotes process of breast cancer in association with STAT3 signaling pathway. Oncotarget, 8, 65677–65686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kabil O., Zhou Y. and Banerjee R. (2006) Human cystathionine beta-synthase is a target for sumoylation. Biochemistry (Mosc), 45, 13528–13536. [DOI] [PubMed] [Google Scholar]
- 46. Agrawal N. and Banerjee R. (2008) Human polycomb 2 protein is a SUMO E3 ligase and alleviates substrate-induced inhibition of cystathionine beta-synthase sumoylation. PLoS One, 3, e4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Paul B.D. and Snyder S.H. (2015) H2S: a novel gasotransmitter that signals by sulfhydration. Trends Biochem. Sci., 40, 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De la Mata M., Garrido-Maraver J., Cotan D., Cordero M.D., Oropesa-Avila M., Izquierdo L.G., De Miguel M., Lorite J.B., Infante E.R., Ybot P. et al. (2012) Recovery of MERRF fibroblasts and cybrids pathophysiology by coenzyme Q10. Neurotherapeutics, 9, 446–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miller R.A., Buehner G., Chang Y., Harper J.M., Sigler R. and Smith-Wheelock M. (2005) Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell, 4, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Garcia-Corzo L., Luna-Sanchez M., Doerrier C., Garcia J.A., Guaras A., Acin-Perez R., Bullejos-Peregrin J., Lopez A., Escames G., Enriquez J.A. et al. (2013) Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet., 22, 1233–1248. [DOI] [PubMed] [Google Scholar]
- 51. Haack T.B., Madignier F., Herzer M., Lamantea E., Danhauser K., Invernizzi F., Koch J., Freitag M., Drost R., Hillier I. et al. (2012) Mutation screening of 75 candidate genes in 152 complex I deficiency cases identifies pathogenic variants in 16 genes including NDUFB9. J. Med. Genet., 49, 83–89. [DOI] [PubMed] [Google Scholar]
- 52. Nemkov T., D'Alessandro A. and Hansen K.C. (2015) Three-minute method for amino acid analysis by UHPLC and high-resolution quadrupole orbitrap mass spectrometry. Amino Acids, 47, 2345–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Linden D.R., Furne J., Stoltz G.J., Abdel-Rehim M.S., Levitt M.D. and Szurszewski J.H. (2012) Sulphide quinone reductase contributes to hydrogen sulphide metabolism in murine peripheral tissues but not in the CNS. Br. J. Pharmacol., 165, 2178–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and Genome Project Data Processing, S (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mishanina T.V., Libiad M. and Banerjee R. (2015) Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol., 11, 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bakkali M. and Martin-Blazquez R. (2018) RNA-Seq reveals large quantitative differences between the transcriptomes of outbreak and non-outbreak locusts. Sci. Rep., 8, 9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Raimundo N., Vanharanta S., Aaltonen L.A., Hovatta I. and Suomalainen A. (2009) Downregulation of SRF-FOS-JUNB pathway in fumarate hydratase deficiency and in uterine leiomyomas. Oncogene, 28, 1261–1273. [DOI] [PubMed] [Google Scholar]
- 58. Luna-Sanchez M., Diaz-Casado E., Barca E., Tejada M.A., Montilla-Garcia A., Cobos E.J., Escames G., Acuna-Castroviejo D., Quinzii C.M. and Lopez L.C. (2015) The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med., 7, 670–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fernandez-Vizarra E., Lopez-Perez M.J. and Enriquez J.A. (2002) Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods, 26, 292–297. [DOI] [PubMed] [Google Scholar]
- 60. Acin-Perez R., Fernandez-Silva P., Peleato M.L., Perez-Martos A. and Enriquez J.A. (2008) Respiratory active mitochondrial supercomplexes. Mol. Cell, 32, 529–539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data were generated as described above. The files have been uploaded to the SRA repository of the National Center for Biotechnology Information. Accession: PRJNA636102; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA636102/.