

Summary
Upon fertilization, terminally differentiated gametes are transformed to a totipotent zygote, which gives rise to an embryo. How parental epigenetic memories are inherited and reprogrammed to accommodate parental-to-zygotic transition remains a fundamental question in developmental biology, epigenetics, and stem cell biology. With the rapid advancement of ultra-sensitive or single-cell epigenome analysis methods, unusual principles of epigenetic reprogramming begin to be unveiled. Emerging data reveal that in many species, the parental epigenome undergoes dramatic reprogramming followed by subsequent re-establishment of the embryo epigenome, leading to epigenetic “rebooting.” Here, we discuss recent progress in understanding epigenetic reprogramming and their functions during mammalian early development. We also highlight the conserved and species-specific principles underlying diverse regulation of the epigenome in early embryos during evolution.
Keywords: epigenetics, epigenome, epigenetic reprogramming, mammalian development, early embryogenesis
Graphical Abstract
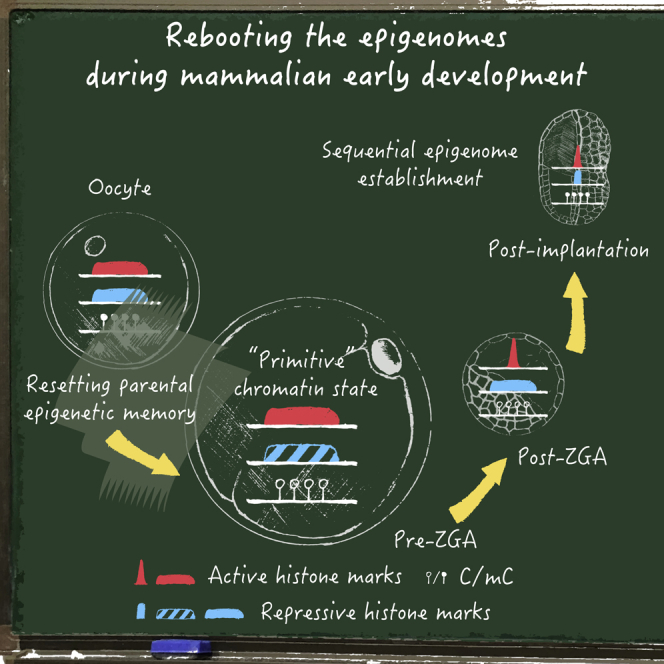
In this review article, Xie and Xia discuss the recent progress for understanding the dynamics and functions of epigenetic reprogramming in early mammalian development. Particularly, the authors summarize distinct fates and functions of the parental epigenomes during early mammalian development, and highlight the “primitive” chromatin state prior to zygotic genome activation that connects the parental epigenomes to the embryonic epigenome.
Main Text
Introduction
Epigenetic regulators, including DNA methylation, histone modifications, high-order chromatin structure, and regulatory RNAs, function as key players of spatiotemporal gene expression in development (Bird, 2002; Kouzarides, 2007). During mitosis, epigenetic modifications can be robustly propagated to daughter cells to facilitate the maintenance of the cell identity (Bird, 2002; Kouzarides, 2007). However, the epigenome undergoes a global reprogramming during early embryogenesis after the fusion of terminally differentiated oocyte and sperm (Burton and Torres-Padilla, 2014; Eckersley-Maslin et al., 2018; Xu and Xie, 2018). This gives rise to a totipotent zygote that subsequently develops into an embryo as the foundation of all future body plans (Chazaud and Yamanaka, 2016). Deficiency in epigenetic reprogramming results in severe developmental defects (Burton and Torres-Padilla, 2014; Eckersley-Maslin et al., 2018; Xu and Xie, 2018). How and why this transformation occurs have remained long-standing and challenging questions in the past, in part due to the scarcity of the research materials. With the rapid development of ultra-sensitive epigenome profiling techniques, accumulating studies are now starting to illuminate this “black box” at the molecular level. Here, we review the recent progress in the understanding of epigenetic reprogramming in early embryos and how it contributes to the mammalian early development, including: (1) non-canonical patterns and functions of the gametic epigenomes; (2) widespread resetting of parental epigenetic memory after fertilization; (3) the emergence of a “primitive” chromatin state prior to zygotic genome activation (ZGA); and (4) the establishment of embryonic epigenome during ZGA and implantation. Collectively, this results in a “rebooting” of the epigenomes during parental-to-embryonic transition. This review is primarily based on findings from mouse, and related data from human and other species are also discussed.
Gametic Epigenomes
Gametes are derived from primordial germ cells (PGCs), a process known for its developmental asymmetry between the two genders (Spiller et al., 2017). The epigenome also manifests distinct dynamics during sex-specific germ cell development. For instance, in both human and mouse, DNA methylation is globally removed during PGC development (Lee et al., 2014; Smith and Meissner, 2013). DNA remethylation initiates shortly thereafter and is completed before birth in male germ cells. By contrast, oocytes acquire DNA methylation during their growth after birth. Details of DNA methylation reprogramming have been well summarized elsewhere (Lee et al., 2014; Smith and Meissner, 2013; Wu and Zhang, 2014).
The Epigenome of Sperm
In mammals, one of the most characteristic features of mature sperm epigenome is that the majority of histones are replaced by protamine for DNA packaging and genome compaction (Rathke et al., 2014). Nevertheless, chemical modifications on the remaining histones largely retain canonical patterns shared by somatic cells (Hammoud et al., 2009, 2014). Despite the presumable compact chromatin, many promoters in mouse sperm are shown to be accessible and occupied by RNA polymerase II (Pol II) and mediator, while distal enhancers are bound by transcription regulators such as FOXA1, ERα, and AR, and enrich for histone variants H2A.Z and H3.3 (Jung et al., 2017, 2019). Intriguingly, compared with dynamic gene expression and histone modifications, DNA methylation appears to be rather stable during both the commitment of germline stem cells and subsequent gametogenesis (Hammoud et al., 2014). DNA methylation is even present at low-CG promoters that bear high Pol II and H3K4me3, which usually antagonize DNA methylation (Hammoud et al., 2014). The functions and the mechanisms of such stable DNA methylome remain unknown. It would be interesting to find out whether this is related to the downregulation of regulatory enzymes including de novo DNA methyltransferase and TET proteins (Wu and Zhang, 2014).
Besides the epigenetic marks, the proper execution of genome functions also requires appropriate folding of chromatin into three-dimensional (3D) structures (Bickmore, 2013; Gibcus and Dekker, 2013). In interphase cells, chromatin is often hierarchically organized into loops and Topologically Associating Domains (TADs) at the hundreds of kilobases scale, spatial compartments at the megabase level, and chromosome territories at the whole nucleus scale (Bickmore, 2013; Gibcus and Dekker, 2013). TADs are large insulating chromatin domains established by CTCF/cohesin-regulated loop extrusion (Rowley and Corces, 2018). Chromatin compartmentalization is believed to be driven by phase separation of chromatin with similar physical states (Falk et al., 2019). These high-order chromatin structures present an additional layer for transcriptional regulation in development and pathogenesis (Lupianez et al., 2016; Zheng and Xie, 2019). Interestingly, although tightly packaged by protamine, mouse and monkey sperm display clear conventional TADs and chromatin compartmentalization (Battulin et al., 2015; Du et al., 2017; Jung et al., 2017; Ke et al., 2017; Wang et al., 2019b). Unexpectedly, TADs are largely lost in human sperm (Chen et al., 2019a), showing species-specific regulation. The details of the sperm epigenome have been reviewed elsewhere (see, for example, Gold et al., 2018), and here we mainly focus on the oocyte epigenome.
The Unusual Epigenome of Oocyte
Of note, mammalian oocytes exhibit distinct epigenomes compared with those of sperm or somatic cells (Figures 1 and 2). DNA methylation, for example, is established in a transcription-dependent manner in mouse oocyte during the follicular growth (Sendzikaite and Kelsey, 2019). As a result, DNA methylation is mainly restricted to the transcribed gene bodies, leaving intergenic regions and non-transcribing genes hypomethylated, forming partially methylated domains (PMDs). Such transcription-dependent DNA methylation relies on the Pol II-SETD2-H3K36me3-DNMT3A/3L pathway (Bourc'his et al., 2001; Kaneda et al., 2004; Kobayashi et al., 2012; Shirane et al., 2013; Smallwood et al., 2011; Veselovska et al., 2015; Xu et al., 2019). Specifically, Pol II can recruit SETD2, the methyltransferase that deposits H3K36me3 at gene bodies, which further guides DNA methylation by recruiting DNMT3A/DNMT3B via the interaction with their PWWP domain (Dhayalan et al., 2010). Deficiency in Setd2 in oocytes results in loss of H3K36me3 and DNA methylation in transcribing regions (Xu et al., 2019). By contrast, NSD1, which broadly deposits H3K36me2 in euchromatic regions, but not SETD2, is responsible for the establishment of bulk DNA methylation in the male germline, leading to distinct DNA methylation patterns between gametes (Shirane et al., 2020). One crucial function of such transcription-dependent DNA methylation is to establish genomic imprints, which are gamete-specific methylation marks that control allele-specific gene expression once passed on to the fertilized embryos (Bartolomei and Ferguson-Smith, 2011). In several cases, oocyte exploits alternative upstream promoters of imprinted genes, which initiates transcriptions leading to the methylation of entire transcribing regions, including the original promoters (Stewart et al., 2016).
Figure 1.

Epigenetic Reprogramming during the Gamete-to-Embryo Transition in Mouse
In mouse, global genome silencing starts in late-stage gametes and lasts until ZGA in embryos. In oocyte, DNA methylation is established in a transcription-dependent manner. Sperm genome is methylated in both transcribing and non-transcribing regions. After fertilization, DNA methylation undergoes global erasure and re-establishment in post-implantation embryos. While sperm largely exhibits canonical patterns of histone modification, non-canonical patterns of histone modifications are found in oocytes and early embryos. Broad domains of non-canonical H3K4me3 (ncH3K4me3) and H3K27me3 are established in oocytes and can be briefly transmitted into embryos (except promoter H3K27me3). On the paternal allele, sperm H3K4me3 and H3K27me3 are replaced by broad domains of de novo H3K4me3 and H3K27me3 in zygotes after fertilization. While H3K4me3 is reprogrammed to a canonical pattern upon ZGA, H3K27me3 domains can be maintained until blastocyst and reset to a canonical pattern in post-implantation embryos. At promoters of developmental genes, the bivalency (H3K4me3/H3K27me3) is lost in pre-implantation embryos but reappears at E6.5 at unusually strong levels, forming “super-bivalency.” It becomes attenuated again at E7.5. H3K36me3, which marks transcribing gene bodies, is established in gametes, removed after fertilization, and re-established after ZGA in early embryos. H3K9me3 marks LTRs in gametes. After fertilization, H3K9me3 is reset to a transitionary state that persists to blastocyst. Blastocyst-specific H3K9me3 starts to emerge from the 4-cell stage. In post-implantation embryos, H3K9me3 also marks lineage-specific genes. Sperm exhibits canonical compartments A/B and TADs, while oocytes show compartments A/B, TADs, and PADs (a non-canonical chromatin 3D structure) in FGOs and metaphase-like structures in MII oocytes. These structures become globally weakened in early embryos and are then gradually reconsolidated as development proceeds. PADs also briefly reappear in 1-cell stage (weak but detectable) and 2-cell stage. LADs are lost in FGOs but are de novo established starting from the 1-cell embryos. LADs show unique features on the maternal allele in zygotes and on both alleles at the 2-cell stage. Canonical (somatic-like) patterns of epigenomes are marked in gray, while non-canonical (oocyte or early embryo-specific) patterns of epigenomes are marked in colors. FGO, full-grown oocyte; LAD, lamina-associated domain; PAD, Polycomb associating domain; TAD, topological associating domain; ZGA, zygotic genome activation.
Figure 2.

Epigenetic Reprogramming during Gamete-to-Embryo Transition in Human
In human, the genome also undergoes global silencing from late-stage gametes, and human ZGA happens at around the 8-cell stage. The gametic patterns (such as transcription-dependent DNA methylation in oocytes) and reprogramming (global DNA demethylation after fertilization and remethylation in post-implantation) of DNA methylation in human largely resemble those in mouse. In human sperms and oocytes, H3K4me3 and H3K27me3 show canonical distributions. After fertilization, H3K27me3 is globally removed before ZGA, with the paternal allele likely showing a faster pace, and is re-established as early as the morula stage. Widespread de novo H3K4me3 transiently appears at CG-rich regions in pre-ZGA embryos, which is then reprogrammed to a canonical pattern upon ZGA. The H3K4me3/H3K27me3 bivalency at developmental gene promoters is lost during the majority of the pre-implantation development. Human sperm shows no TADs. Both TADs and compartments are weak after fertilization, which becomes consolidated after ZGA. Canonical (somatic-like) epigenomes are marked in gray, while non-canonical (oocyte or early embryo-specific) epigenomes are marked in colors.
In the past several years, the rapidly developing low-input chromatin profiling technologies revealed unusual patterns of histone modifications in mammalian oocytes and early embryos. For example, H3K4me3, a conserved hallmark of promoters and transcription initiation (Rivera and Ren, 2013), is enriched at active gene promoters in mouse primary and growing oocytes (Stewart et al., 2015; Zhang et al., 2016). However, large domains of H3K4me3 appear at promoters as well as many distal PMDs in late-stage growing oocytes, and become widespread in full-grown oocytes (FGOs) (Dahl et al., 2016; Zhang et al., 2016; Hanna et al., 2018). What triggers the H3K4me3 transition remains unknown. Interestingly, the emergence of such non-canonical H3K4me3 (ncH3K4me3) is correlated with the genome silencing during oogenesis (Dahl et al., 2016; Zhang et al., 2016). During later stages of follicle growth, oocytes transit from “non-surrounded nucleolus” to “surrounded nucleolus” (SN) state, as chromatin collapses around the nucleolus-like body (Zuccotti et al., 1995), coinciding with Pol II chromatin dissociation and transcription silencing (Abe et al., 2010). Intriguingly, deficiency of MLL2, which deposits ncH3K4me3 (Hanna et al., 2018), or overexpression of KDM5B (H3K4me3 demethylase) leads to defects of genome silencing in SN oocytes (Andreu-Vieyra et al., 2010; Zhang et al., 2016). MLL2-deficient oocytes also show compromised ovulation and post-fertilization lethality (Andreu-Vieyra et al., 2010). The mechanism underlying such repressive function of ncH3K4me3 remains unclear, but as H3K4me3 can recruit transcription regulators such as TAF3 (Lauberth et al., 2013; Vermeulen et al., 2007), it is postulated that ncH3K4me3 may function as “sponge” that sequesters transcriptional resources away from promoters (Zhang et al., 2016). On the other hand, CXXC finger protein 1 (CFP1) is a key DNA-binding component of the H3K4me3 methyltransferase SET1A and SET1B complex, which is required for H3K4me3 deposition at actively transcribed promoters but not poised promoters in mouse embryonic stem cells (mESCs) (Clouaire et al., 2012). Knocking out Cfp1 in oocytes leads to decreased H3K4me3 and global transcriptional activity, as well as defects in meiosis and development potential after fertilization (Sha et al., 2018; Yu et al., 2017). It is possible that Cfp1 may be required for the canonical H3K4me3 in oocytes, which may have distinct functions compared with ncH3K4me3. H3K27me3, a repressive histone mark deposited to silent developmental genes by Polycomb repressive complex 2 (PRC2) (Bernstein et al., 2006; Margueron and Reinberg, 2011), also shows unusual distributions in mouse oocytes. In early-growing oocytes, widespread broad H3K27me3 domains can be found in most non-transcribed regions, which then retreat to a subset of PMDs in FGOs (Zheng et al., 2016). Another repressive mark, H3K9me2, is typically associated with nuclear lamina-associated domains (LADs) (Peric-Hupkes et al., 2010). However, H3K9me2 is also mainly enriched at PMDs in mouse oocytes (Au Yeung et al., 2019). Maternal depletion of G9a, the writer of H3K9me2, leads to a decrease of H3K9me2 and reorganization of heterochromatin in oocytes. Embryos derived from G9a knocked-out oocytes show abnormal chromosome segregation and frequent developmental arrest (Au Yeung et al., 2019).
The unique oocyte epigenomes are likely linked to the prolonged meiotic arrest at the diplotene stage, which presumably provides sufficient time for various types of epigenetic writers to deposit corresponding marks. These marks in turn are maintained by their delicate crosstalks. For example, both H3K4me3 and H3K27me3 show strong anticorrelation with DNA methylation in oocytes (Dahl et al., 2016; Zhang et al., 2016; Zheng et al., 2016). Ablation of KDM1A or KDM1B in oocytes results in increased H3K4me3 and decreased DNA methylation at a subset of promoters (Stewart et al., 2015). While abolishing DNA methylation alone in mouse oocytes has moderate impacts on H3K4me3 in gene bodies (Hanna et al., 2018), Setd2 knockout leads to the loss of H3K36me3 and DNA methylation as well as the invasion of H3K4me3 or H3K27me3 into former H3K36me3 territories (Xu et al., 2019). These regions encompass imprinting control regions, where ectopic H3K4me3, instead of DNA methylation, arises in Setd2 mutant oocytes. The invasion of H3K27me3 into gene bodies is also correlated with gene repression (Xu et al., 2019). Consequently, the embryos derived by Setd2-null oocytes cannot develop beyond 1-cell stage. Unexpectedly, human oocytes largely exhibit canonical H3K4me3 and H3K27me3 patterns like those in somatic cells (Xia et al., 2019), indicating that distinct mechanisms regulate the oocyte epigenome between mouse and human (Figure 2).
Non-conventional 3D chromatin structures also exist in mouse oocyte. While early-growing mouse oocytes show typical TADs and compartments, self-interacting compartment-like domains (Polycomb associating domain, or PAD) emerge in late-stage mouse oocytes, which align almost perfectly with broad H3K27me3 domains and are independent of TADs and conventional A/B compartments (Du et al., 2019; Flyamer et al., 2017). Deficiency of PRC1, rather than PRC2, leads to disruption of PADs and gene derepression in oocytes. PRC2, however, appears to be important for the brief re-establishment of PADs on the maternal allele of early embryos (Du et al., 2019). Why PADs uniquely appear in oocytes remains unclear, but their appearance coincides with their loss of LADs, which reflects nuclear lamina-chromatin association (Borsos et al., 2019). As LADs are typically repressive and closely related to compartments B (Bickmore, 2013; Gibcus and Dekker, 2013), the absence of lamina association perhaps provides an opportunity for other chromatin organization, such as PADs, to become prominent. Finally, metaphase II (MII) oocytes, which are arrested in the metaphase, lack TADs, PADs, and compartments A/B, as chromatin adopts a metaphase-like state (Du et al., 2017; Ke et al., 2017) presumably mediated by condensin (Gibcus et al., 2018).
Resetting Parental Epigenetic Marks after Fertilization
Given the distinct epigenetic landscapes in gametes, one important purpose of epigenetic reprogramming in early development is to equalize the epigenetic landscapes between alleles after fertilization (Figures 1 and 2). In mouse, DNA methylation is globally removed after fertilization from both alleles, mediated by active and passive mechanisms, except for imprinting control regions (ICRs) and certain repeats such as IAPs (Amouroux et al., 2016; Smith and Meissner, 2013; Wu and Zhang, 2014). Similar reprogramming of DNA methylation also occurs in human early embryos (Guo et al., 2014; Smith et al., 2014; Zhu et al., 2018). Hence, these results reveal both inheritance and erasure of epigenetic information between generations (Eckersley-Maslin et al., 2018; Wu and Zhang, 2014; Xu and Xie, 2018).
Recent studies also shed light on the reprogramming modes of histones and their chemical modifications. After fertilization, one of the earliest reprogramming events is the protamine-histone-exchange, a process that requires the splicing kinase SRPK-1-mediated protamine phosphorylation (Gou et al., 2020) and histone variant H3.3 incorporation by its chaperone HIRA (Inoue and Zhang, 2014; Lin et al., 2014). An accompanying recurrent theme is that many paternal histone modifications appear to be quickly depleted after fertilization, often prior to DNA replication in zygotes (Burton and Torres-Padilla, 2014; Xu and Xie, 2018). On the maternal allele, the reprogramming entails both substantial inheritance and erasure. For example, mouse oocyte H3K4me3 is inherited to embryos until the late-2-cell stage, when it is converted to the canonical promoter H3K4me3 pattern upon ZGA (Dahl et al., 2016; Zhang et al., 2016). The removal of ncH3K4me3 domains does not require DNA replication but does require ZGA, presumably because its two demethylases, KDM5A and KDM5B, are highly induced at this stage (Dahl et al., 2016; Zhang et al., 2016). Knockdown both demethylases results in compromised ZGA and developmental failure to reach blastocysts (Dahl et al., 2016). The reprogramming of maternal H3K27me3 in mouse embryos is more complex. H3K27me3 at the promoters of developmental genes is removed shortly after fertilization and is not restored (on both alleles) until around the blastocyst stage (Zheng et al., 2016). Nevertheless, these developmental genes are generally not expressed in the pre-implantation embryos, suggesting additional mechanisms to ensure their silencing or the lack of activators. The resetting of H3K27me3 is likely important for erasing previous gene repression memory, as the persisting somatic H3K27me3 was suggested to possibly serve as an epigenetic barrier for somatic cell nuclear transfer (SCNT) embryo development (Yang et al., 2018). On the contrary, distal H3K27me3 domains from mouse oocytes persist to blastocyst before being lost in post-implantation embryos (Zheng et al., 2016). During their brief presence in pre-implantation embryos, some of the H3K27me3 domains can function as an imprinting mechanism to regulate allelic expression (Inoue et al., 2017, 2018). For example, an H3K27me3 domain represses the maternal copy of Xist, a master regulator of X chromosome inactivation (XCI) (Loda and Heard, 2019), leading to paternal XCI in mouse early embryo (Harris et al., 2019; Inoue et al., 2018). Loss of maternal Embryonic Ectoderm Development (EED), a component of PRC2, results in defects of paternal XCI and male-biased lethality (Harris et al., 2019; Inoue et al., 2018). Such imprinting is lost in embryonic lineages after implantation but is maintained in extraembryonic tissues by DNA methylation instead, which is considered as a more stable epigenetic mark (Chen et al., 2019b). The oocyte H3K27me3-mediated imprinting is apparently missing in SCNT embryos, and artificially restoring the monoallelic expression states of four H3K27me3 imprinted genes can significantly improve mouse SCNT efficiency (Wang et al., 2020). Of note, H3K27me3 undergoes a global depletion in human early embryos before ZGA (with the paternal allele likely showing a faster kinetics) (van de Werken et al., 2014; Xia et al., 2019; Zhang et al., 2012), indicating that H3K27me3-mediated imprinting may be absent in humans. Consistently, XIST is biallelically expressed, and both X chromosomes undergo “transcription dampening” in human early embryos (Petropoulos et al., 2016). Notably, a number of DNA methylation-independent allelically expressed genes were identified in human morula (Zhang et al., 2019). It remains to be determined whether these candidate imprinted genes are also present in general population and whether they could be mediated by other mechanisms.
The repressive mark H3K9me3 has been shown to be a major epigenetic barrier for SCNT in mouse (Liu et al., 2016a; Matoba et al., 2014) and human (Chung et al., 2015). In mouse, unlike H3K4me3 and H3K27me3, H3K9me3 appears to undergo global resetting on both alleles after fertilization (Wang et al., 2018). The de novo H3K9me3 deposited in 1-cell embryos shows distinct distributions between the two parental alleles, with the paternal, but not the maternal, H3K9me3 being deposited by SUV39H2 (Burton et al., 2020; Wang et al., 2018). The allele asymmetry lasts as late as the blastocyst stage (Wang et al., 2018). “Late-stage embryo-specific” H3K9me3 can be found as early as the 4-cell stage, which is then progressively enhanced during pre-implantation development. These domains enrich for long terminal repeat (LTR) retrotransposons and HP1 binding sites in mESCs, presumably rendering an epigenetic relay from decreasing DNA methylation to increasing H3K9me3 at LTRs. For instance, MERVL, a class of retrotransposon highly expressed at 2-cell stage, starts to acquire H3K9me3 from the 4-cell stage, coinciding with its rapid downregulation (Wang et al., 2018). H3K9me3 exerts repression on LTRs in part through CHAF1A, a DNA replication-coupled histone deposition chaperone (Wang et al., 2018).
Notably, the loss of parental histone modifications appears to be more extensive in early embryos of lower organisms such as zebrafish and frog, where cell cycles are much faster than those in mammals and rapid cell division presumably dilutes parental histone marks (Akkers et al., 2009; Murphy et al., 2018; Vastenhouw et al., 2010; Zhang et al., 2018a). However, several studies also reported that oocyte-transmitted histone modifications can function in early fly embryos (Samata et al., 2020; Zenk et al., 2017). Overall, these fascinating findings beg the question as to why different parental marks adapt different reprogramming modes, as some are inherited while others are lost in early embryos. Growing evidence indicates that the different reprogramming modes of parental epigenetic marks during parental-to-zygotic transition may be closely related to their functions (Figure 3).
Figure 3.

Distinct Fates of the Parental Epigenomes
1. “Epigenome for gametes.” An example comes from oocyte H3K36me3, which is deposited in transcribed regions and plays critical functions in imprinting establishment and oocyte development. Oocyte H3K36me3 subsequently becomes possible “transcription fossils” when genome is silenced, and is removed after fertilization. 2. “Epigenome for the next generation.” Both DNA methylation- and maternal H3K27me3-mediated imprints are established in gametes and function in embryos. DNA methylation-mediated imprints can be faithfully maintained throughout development except in PGCs. The oocyte H3K27me3-mediated imprint can be inherited to pre-implantation embryos and is lost in post-implantation embryos, but is relayed by DNA methylation in extraembryonic lineages to maintain imprinting. 3. “Epigenome for both oocyte and early embryos.” Oocyte H3K4me3 is an example that plays roles in both mouse oocytes and embryos. In mouse FGOs, H3K4me3 promotes global genome silencing. In early embryos, H3K4me3 is required for establishing paternal LADs. 4. “Passenger marks?” Oocyte DNA methylation outside of ICRs is largely dispensable for oocyte development, raising the possibility that they may act as “passenger marks.” After fertilization, the majority of these DNA methylation marks undergoes global removal. 5. “Pre-erasure of parental epigenetic memory before fertilization.” The DNA methylation of enhancers often correlates with the loss of enhancer activities. While oocyte enhancers are progressively methylated after fertilization, sperm enhancers in zebrafish gain DNA methylation prior to fertilization, raising a possibility that the parental epigenetic memory (sperm enhancer) is pre-erased before fertilization. PGC, primordial germ cells.
Epigenetic Marks Required for Gametogenesis
Firstly, it is reasonable to believe that a substantial part of the epigenomes in gametes is established to primarily accommodate the needs of gametogenesis. For instance, protamine can facilitate sperm genome compaction and motility, which is then swiftly removed after fertilization (Balhorn, 2007). In zebrafish, active histone marks in sperm occupy genes that are involved in meiosis and spermatogenesis (Zhang et al., 2018a). The motif analysis at H3K27ac-marked enhancers revealed predominantly RFX factors, the major transcription factor (TF) for ciliogenesis (Zhang et al., 2018a). In mouse, H3K36me3, a mark of active transcription elongation, is inherited from transcribing FGOs to silenced MII oocytes, before it is rapidly lost in early embryos (Xu et al., 2019). Therefore, H3K36me3 in MII oocytes may reflect epigenetic “fossils” for previous transcription activities. As these functions become obsolete in early embryos, these epigenetic marks are removed soon after fertilization.
Epigenetic Marks Required for the Next Generation
DNA methylation-dependent imprints are classic examples of epigenetic marks that are established in gametes but play major roles in the next generation (Bartolomei and Ferguson-Smith, 2011). Similarly, H3K27me3 marked imprints may also belong to this category, as maternal deficiency of EED leads to defective silencing of maternal Xist and H3K27me3-dependent imprinted genes, and compromised development (Inoue et al., 2018).
Epigenetic Marks Required for Both Oocyte and Embryo Functions
There are a few examples of epigenetic marks that may have functions both in oocytes and early embryos. For example, H3K4me3 appears to be required for genome silencing in mouse oocytes (Andreu-Vieyra et al., 2010; Zhang et al., 2016). Interestingly, H3K4me3 is also required for establishing LADs on the paternal allele in mouse zygotes (Borsos et al., 2019). How this mysterious event occurs remains unknown.
“Passenger Marks”?
Notably, oocyte DNA methylation, besides imprints, appears to be largely dispensable for oocyte development as well as pre-implantation development (Hata et al., 2002; Kaneda et al., 2010). This result raises an interesting question as to whether some epigenetic marks may be “passenger marks,” analogous to the “passenger mutations” in cancer (Pon and Marra, 2015). One certainly cannot fully exclude the possibility that these marks also play critical roles in regulating certain genes beyond known imprinted genes in later development. For example, it was shown that normal development of trophoblast requires silencing of Scml2 by oocyte-derived DNA methylation (Branco et al., 2016). Nevertheless, the seemingly dispensable role of DNA methylation in oocytes is perhaps not surprising, given oocytes can stay genome-wide hypomethylated for an extended period in mammals (Sendzikaite and Kelsey, 2019). A similar observation was made for oocyte H3K27me3, as maternal Eed-null oocytes can also be fertilized and only show male-biased sublethality, presumably due to lack of H3K27me3-controlled imprinting (Inoue et al., 2018). One possibility for the emergence of such pervasive but non-essential epigenetic marks is perhaps promiscuous deposition during the extended cell-cycle arrest in oocytes. In fact, mounting evidence has revealed multiple mechanisms developed by oocytes to avoid excessive accumulation of repressive marks. For instance, despite being dispensable for oocyte methylome establishment, DNMT1 is highly abundant in mouse oocytes (Hirasawa et al., 2008). Its activity, however, is strictly limited by STELLA, an essential maternal factor that excludes UHRF1, a key co-factor of DNMT1, from nucleus (Li et al., 2018b). Stella-deficient oocytes show global DNA hypermethylation caused by the unleashed de novo activities of DNMT1. Notably, this contrasts with a previous finding showing that STELLA protects maternal DNA methylation (Nakamura et al., 2012). In addition, H3K36me3 can restrict H3K27me3 in oocytes (as well as H3K4me3, which also plays a repressive role in oocytes (Andreu-Vieyra et al., 2010; Zhang et al., 2016)) (Xu et al., 2019). Recently, it was shown the H3K9me3 demethylase KDM4A restricts H3K9me3 to safeguard the mouse oocyte epigenome (Sankar et al., 2020). Such excessive repressive marks can cause transcription and development defects in early embryos if not timely removed (Li et al., 2018b).
Pre-erasing Parental Epigenetic Memory Prior to Fertilization
Intriguingly, some parental epigenetic memories appear to be even pre-erased prior to fertilization. For instance, active enhancers are usually hypomethylated and the gain of DNA methylation often indicates the loss of enhancer functions (Rivera and Ren, 2013; Song et al., 2019). In zebrafish, oocyte enhancers become progressively methylated after fertilization. However, those in sperm are already hypermethylated before fertilization, which correlates with sperm-specific early silencing of tet genes (Zhang et al., 2018a). Such reprogramming in part contributes to the previous observation that the methylome on the maternal allele is reprogrammed after zebrafish fertilization to match that of the paternal allele (Jiang et al., 2013; Potok et al., 2013). It was proposed that the sperm methylome already adopts an “embryonic” pattern prior to fertilization (Zhang et al., 2018a). Further studies are needed to determine whether such pre-erasure of parental epigenetic memories facilitates subsequent embryo development. Notably, a recent study showed that human sperm lacks TADs due to the downregulation of CTCF (Chen et al., 2019a). Given that TADs are also highly relaxed after fertilization (Chen et al., 2019a), these data raise a question as to whether this is also another way of pre-erasing parental epigenetic memory. In summary, the above results reveal fine regulation of both inheritance and reprogramming of gametic epigenetic memories during parental-to-zygotic transition to accommodate the embryo development.
A Primitive Chromatin State in Pre-ZGA Embryos
Prior to ZGA, emerging data have unveiled unique epigenomes in this transcriptionally “dark” period, which is distinct from those in both gametes and post-ZGA embryos. These data collectively point to a “primitive” chromatin state that shows immature heterochromatin, widespread open chromatin and active marks, as well as a relaxed 3D genome, as described below. This likely reflects both the silenced state and the parental-to-zygotic transitionary nature of this period. Note that pre-ZGA here refers to the period prior to major ZGA, when thousands of genes are activated around a similar time. A small subset of genes is usually expressed ahead of major ZGA, termed “minor ZGA.” Here, we still consider minor ZGA as part of the pre-ZGA period.
Missing or Immature Heterochromatin
It was reported that mouse early embryos feature a highly relaxed chromatin state characterized by dispersed chromatin structure under electronic microscopy (Ahmed et al., 2010), increased histone mobility (Boskovic et al., 2014), and loss of heterochromatin foci (Probst and Almouzni, 2008). Expression of exogenous gene does not require enhancers in 1-cell mouse embryos but does so at the 2-cell stage when ZGA occurs, supporting permissive chromatin in 1-cell embryos but more repressed chromatin at the 2-cell stage (Schultz, 2002). In line with the notion that heterochromatin is extensively remodeled after fertilization, many repressive epigenetic marks, including DNA methylation, H3K27me3, and H3K9me3, are either missing or in the “immature state” in pre-implantation embryos (Burton et al., 2020; Lee et al., 2014; Liu et al., 2016b; Smith and Meissner, 2013; Wang et al., 2018; Xia et al., 2019; Zheng et al., 2016). For example, SUV39H1 is not expressed in 1-cell mouse embryos, coinciding with the absence of constitutive heterochromatin including chromocenters (Burton et al., 2020). Interestingly, de novo H3K9me3 can occur in 1-cell embryos and is mediated by SUV39H2 on the paternal pronuclei. However, such de novo H3K9me3 is non-repressive but instead bookmarks promoters for future compaction. Forced expression of SUV39H1 induced chromocenter formation at the 2-cell stage and compromised pre-implantation development, suggesting that the precocious restoration of constitutive heterochromatin is detrimental to embryo development (Burton et al., 2020). Besides, by using DamID, LADs are shown to be de novo established after fertilization but exhibit atypical genome distribution on the maternal allele in zygotes and on both alleles at the 2-cell stage (Borsos et al., 2019). Intriguingly, the paternal LADs in zygotes are dependent on H3K4me3 but not H3K9me2/me3, echoing the immature or non-canonical heterochromatin organization. The compromised heterochromatin is also suggested to underlie the activation of several key minor ZGA genes, such as Zscan4, which plays crucial roles in telomere lengthening and genome integrity quality check (Ko, 2016). Many transposable elements are highly active in early embryos (Peaston et al., 2004). For example, MERVL, a class of retrotransposable elements, often shows promiscuous transcription that proceeds far downstream beyond itself during mouse minor ZGA (Abe et al., 2015; Macfarlan et al., 2012; Peaston et al., 2004). Such activities of transposable elements also profoundly shape the chromatin states and regulate early development. For example, the transcription of MERVL is associated with broad peaks of assay for transposase-accessible chromatin sequencing (ATAC-seq) (Wu et al., 2016) and 3D chromatin boundary (Kruse et al., 2019). In addition, LINE-1 is activated in mouse early embryos, with the expression peaking at the 2-cell stage. Interference of both activation and silencing of LINE-1 leads to pre-implantation developmental defects in a transcript-independent way, indicating it primarily functions in cis (Jachowicz et al., 2017). Another study reported that LINE-1 RNA can recruit Nucleolin/KAP1 to repress 2-cell specific genes and MERVL through downregulating Dux. Inhibition of LINE-1 in embryos impairs ZGA and exits from the 2-cell stage (Percharde et al., 2018). In addition, ATAC-seq peaks are strongly enriched for SINE elements from 2- to 8-cell stages in mouse embryos (Lu et al., 2016; Wu et al., 2016). In human, widespread accessible chromatin is enriched by transposable elements including ERVK/HERV-K and SVA (Gao et al., 2018; Li et al., 2018a; Wu et al., 2018). Expression of HERVK was shown to trigger immune responses and was proposed to protect human embryos from viral infection (Grow et al., 2015), suggesting that repeat activation plays critical functions in mammalian early embryos rather than simply being by-products of epigenetic silencing deficiency.
Widespread Deposition of Transcription-Independent De Novo Active Marks
Interestingly, despite the suspension of the transcription program, widespread de novo deposition of active histone marks can be found prior to ZGA, with distributions different from those in both gametes and post-ZGA embryos. In mouse zygote, while the maternal allele inherits H3K4me3 from oocytes, very broad and weak domains of H3K4me3 briefly appear in gene-rich regions on the paternal allele (Zhang et al., 2016). In human embryos, widespread de novo H3K4me3 transiently appear at CG-rich promoters and distal regions in pre-ZGA embryos, including the promoters of developmental genes and putative poised enhancers (Xia et al., 2019). Such H3K4me3 is absent in oocytes and does not predict future expression but is closely correlated with CpG density (Xia et al., 2019), raising a possibility that it may rely on DNA sequence feature as a “default” state (Bird, 2002; Wachter et al., 2014). These H3K4me3 marked regions resolve into either active or inactive states upon ZGA. This finding is also reminiscent of zebrafish early embryos, where de novo H3K27ac is deposited to promoters genome-wide prior to ZGA (Sato et al., 2019; Zhang et al., 2018a). Mechanistically, the widespread emergence of transcription-independent active histone marks may be ascribed to the unusually permissive chromatin and the lack of opposing repressive epigenetic marks at this stage. Although not directly correlated with transcription, it is tempting to speculate that these active histone marks in pre-ZGA embryos may promote chromatin openness and future gene activation. Consistently, knocking down maternal acetyltransferases in zebrafish results in defective ZGA and embryonic lethality (Zhang et al., 2018a). When inhibiting transcription or H3K27ac readers, the signals of elongating Pol II but not H3K27ac is blocked (Sato et al., 2019), further supporting that histone acetylation is upstream of transcription in this case.
Widespread Transcription-Independent Accessible Chromatin
The unique chromatin state of embryos at early stages is also manifested in accessible chromatin analysis. In both human and mouse, ATAC-seq and DNase sequencing (DNase-seq) can both well capture open chromatin in later-stage embryos (after the 8-cell stage). However, successes vary in earlier embryos for different methods (Gao et al., 2018; Guo et al., 2017; Inoue et al., 2017; Li et al., 2018a; Lu et al., 2016; Wu et al., 2016, 2018). The difficulty in detecting open chromatin in early-stage embryos may again hint at their unique chromatin states. For example, the permissive nature of early embryo chromatin may lead to overdigestion of DNase I or Tn5 transposase and subsequent loss of open chromatin peaks, as reported for high concentration of MNase in fly S2 cells (Mieczkowski et al., 2016). Nevertheless, the DNase-seq and ATAC-seq experiments at early stages that are indeed successfully provide a valuable glimpse of the unique chromatin states prior to ZGA. For example, a reanalysis of the dataset from Inoue et al. (2017) revealed distinct landscapes of accessible chromatin in 1-cell mouse embryos between the two parental alleles. Such chromatin accessibility may be shaped by a combination of both cis factors (such as inherited epigenetic marks) and trans factors (such as TFs). For example, the open chromatin detected by DNase-seq at maternal allele of mouse zygotes resembles that of oocytes (Wu et al., 2018), indicating transmission of epigenetic marks that maintain such chromatin states. On the other hand, accessible chromatin on both alleles, despite distinct landscapes, enrich for similar motifs of oocyte TFs, suggesting that these regions also share trans-acting factors (Wu et al., 2018). A large fraction of these distal accessible regions is quickly closed in a ZGA-dependent manner in both human and mouse (Wu et al., 2016, 2018), which possibly reflects the transition of maternal-to-embryonic transcription regulators that lead to the resetting and convergence of the maternal and paternal epigenetic landscapes (Lu et al., 2016; Wu et al., 2016). Along the same lines, in 1-cell SCNT mouse embryos, accessible chromatin shows a rapid donor-to-embryo transition that correlates with the switch of TF programs (Djekidel et al., 2018), again supporting the potent activities of trans-acting factors in sculpting the chromatin landscapes of early embryos.
A Hyper-Relaxed Higher-Order Chromatin after Fertilization
The permissiveness of chromatin in post-fertilization embryos is also reflected in their 3D chromatin organization. Hi-C analyses revealed highly weakened TAD and compartmentalization in 1-cell mouse embryos (Collombet et al., 2020; Du et al., 2017; Gassler et al., 2017; Ke et al., 2017). These structures are gradually consolidated as development proceeds, and reach a more mature state around the 8-cell stage. Such relaxed chromatin in early embryos is also conserved among fly, human (Chen et al., 2019a; Hug et al., 2017), and fish (Nakamura et al., 2018; Kaaij et al., 2018). Similar relaxed chromatin organization was also found in SCNT mouse embryos, although at a slightly late stage (2-cell) (Chen et al., 2020; Zhang et al., 2020). This extraordinary conservation during evolution raises a possibility that such relaxed chromatin states may very likely have important functions. Interestingly, our recent study revealed that cohesin, the core architectural protein required for TADs (Rowley and Corces, 2018), can repress minor ZGA genes such as Zscan4 in mESCs and differentiated neurons, and its removal facilitates minor ZGA and promotes SCNT development (Zhang et al., 2020). It remains to be determined whether this is also true in fertilized mouse embryos. In human embryos, the relaxed TADs are attributed to the low expression of CTCF (Chen et al., 2019a).
Establishing Embryonic Epigenomes in Post-ZGA Embryos
ZGA is a milestone event in early development as it initiates the maternal-to-zygotic transition. In mouse, minor and major ZGA occur in middle-to-late zygote and late 2-cell embryos, respectively; by contrast, ZGA occurs around the 8-cell stage in human (Eckersley-Maslin et al., 2018; Schulz and Harrison, 2019). How ZGA is initiated in mammals remains largely unclear. However, several candidate regulators were proposed. DUX (in mouse) and its homolog DUX4 (in human) were suggested to play roles for ZGA (De Iaco et al., 2017; Hendrickson et al., 2017; Whiddon et al., 2017). However, despite the sublethality, the majority of Dux knockout embryos still develop into adulthood with minor defects in ZGA (Chen and Zhang, 2019; Guo et al., 2019), suggesting the existence of additional ZGA regulators. Interestingly, the timely degradation of DUX beyond the 2-cell stage is required for proper embryo development (Guo et al., 2019). Furthermore, it was shown that DPPA2/4 can induce Dux expression and 2-cell-like transcriptional program in mESCs (De Iaco et al., 2019; Eckersley-Maslin et al., 2019; Yan et al., 2019), and likely function upstream of DUX. Other ZGA contributory factors include NFYA in mouse and OCT4 in human, which were shown to be required for the proper activation of hundreds of ZGA genes (Gao et al., 2018; Lu et al., 2016). More details about the mechanism regulating ZGA are reviewed elsewhere (Schulz and Harrison, 2019).
A major wave of establishment of the embryonic epigenome, mainly for active epigenetic marks, is often found at ZGA. For instance in both human and mouse, canonical H3K4me3 is established at the active promoters upon ZGA (Dahl et al., 2016; Liu et al., 2016b; Xia et al., 2019; Zhang et al., 2016). Around the same time, accessible chromatin and H3K27ac emerge at active promoters and putative enhancers (Gao et al., 2018; Li et al., 2018a; Lu et al., 2016; Wu et al., 2016, 2018; Xia et al., 2019). By contrast, pre-ZGA specific broad H3K4me3 and accessible chromatin are lost. Such transition strictly requires ZGA in both human and mouse (Wu et al., 2016, 2018; Xia et al., 2019; Zhang et al., 2016). It is possible that the onset of transcription may recruit epigenetic marks at active genes, which further facilitates and stabilizes transcription, leading to positive-feedback loops. This coincides with the depletion of many previously deposited active marks from inactive regions. In the case of transition from ncH3K4me3 to canonical H3K4me3 in mouse embryos, the H3K4me3 demethylases Kdm5a/5b are highly induced in ZGA, which potentially can remove ncH3K4me3 in non-transcribing regions. Consistently, knockdown of KDM5A/B results in substantial retention of H3K4me3 as shown by immunostaining (Dahl et al., 2016; Liu et al., 2016b), although this requires confirmation by chromatin immunoprecipitation sequencing (ChIP-seq) data. The dependence of active mark establishment on transcription is, however, not a universal rule. For instance, a significant fraction of de novo H3K4me3 and H3K27ac upon ZGA is independent of transcription in zebrafish embryos (Zhang et al., 2018a).
By contrast, the establishment of canonical repressive epigenetic marks for the embryonic epigenomes undergoes a slower pace. DNA methylation restoration starts around implantation in both mouse and human embryos (Smith et al., 2017; Zhang et al., 2018b; Zhou et al., 2019). In mouse, promoter H3K27me3 is not restored at developmental genes until around implantation (Zheng et al., 2016). In human, promoter H3K27me3 is not detected upon ZGA at the 8-cell stage but is readily found in morula, coinciding with the re-expression of PRC2 components (Xia et al., 2019; Zhang et al., 2019). In mouse, H3K9me3 adopts a transitionary state after fertilization, and blastocyst-specific H3K9me3 begins to emerge as early as the 4-cell stage (Wang et al., 2018). Lineage-specific H3K9me3 at gene promoters can be observed at embryonic day 6.5 (E6.5) to E7.5 after implantation. Therefore, while active marks of embryonic patterns are readily found immediately after ZGA (“leading establishment”), repressive marks experience a longer transitionary period and, thus, delayed establishment toward embryonic epigenomes (“lagging establishment”) (Figure 4). The immature repressive chromatin environment in early-stage embryos may be functionally linked to the emergence of totipotency and pluripotency. For instance, the immature state of repressive marks is partially recaptured in naive ESCs but is lost in primed ESCs (Hackett and Surani, 2014). Deletion of LSD/KDM1A leads to an increase of H3K4me2/me3 and H3K27ac, and decrease of H3K9me/me2, as well as expanded fate potential in mESCs (Macfarlan et al., 2011). Loss of SETDB1, the methyltransferase of H3K9me3, initiates a 2-cell-like transition program by inducing the expression of DUX in mESCs (Wu et al., 2020). Conversely, the restoration of repressive marks may restrict cell fate among distinct lineages. For instance, in post-implantation embryos, embryonic and extraembryonic lineages showed distinct methylome landscapes, as extraembryonic cells show lower global DNA methylation but higher DNA methylation at developmental gene promoters (Smith et al., 2017; Zhang et al., 2018b). Such differential patterning was suggested to be mediated by WNT and FGF (fibroblast growth factor) signaling (Smith et al., 2017). In addition, Nicetto et al. (2019) found that H3K9me3 marks near tens of thousands of coding genes in early, uncommitted cells at the germ layer stage. Upon differentiation, H3K9me3-marked heterochromatin undergoes rearrangement and reduction to pave the way for expression of cell-type-specific genes. It was proposed that H3K9me3 restricts lineage regulators at this stage before they are activated in a tissue-specific manner, and deficiency of H3K9me3 methyltransferases results in the failure of lineage commitment during organogenesis (Nicetto et al., 2019). Interestingly, such delayed establishment also applies to bivalent marks H3K4me3 and H3K27me3 at inactive developmental genes. The H3K4me3/H3K27me3 bivalency that bears both active and repressive marks is proposed to poise the gene for activation when deposited into the same promoters (Bernstein et al., 2006). The bivalency is absent in pre-implantation mouse embryos but strongly emerges in E6.5 epiblast (“super-bivalency”) (Xiang et al., 2019). It then becomes markedly reduced in fate-committed lineages at E7.5. Loss of bivalent H3K4me3 through knocking out MLL2/KMT2B is associated with impaired activation of developmental genes and subsequent embryonic lethality (Glaser et al., 2009; Xiang et al., 2019). More recently, DPPA2 and DPPA4 have been shown to function as epigenetic priming factors to facilitate bivalency establishment and counteract with DNA methylation in developmental genes (Eckersley-Maslin et al., 2020; Gretarsson and Hackett, 2020).
Figure 4.

Establishment of the Embryonic Epigenome during Mammalian Early Development
A schematic model showing sequential establishment of embryonic epigenome in mammalian early development. In pre-ZGA embryos, the epigenome exists as a primitive, transitionary state, featured with widespread, transcription-independent active histone marks and permissive chromatin, as well as missing or immature repressive marks. After ZGA, the active marks of embryonic epigenomes are quickly established (“leading establishment”), while the repressive marks of embryonic epigenomes undergo a slower pace of establishment (“lagging establishment”) at later stages.
In human, epigenetic regulation during gastrulation and early lineage specification have remained poorly understood until recently. Interestingly, in human inner cell mass, H3K27me3 preferentially marks EPI/PE (epiblast/primitive endoderm) genes rather than TE (trophectoderm) genes (Xia et al., 2019), indicating that it may play roles in restricting EPI/PE segregations. A recent study employed the in vitro culture system to support the human embryo development until day 14, and investigated the transcriptome and DNA methylation using single-cell multi-omics profiling (Zhou et al., 2019). The general dynamics of DNA methylation follow a similar reprogramming process both in human and mouse, suggesting its conserved regulation. Of note, recent in vitro culture systems can support the development of non-human primate embryo beyond implantation (Ma et al., 2019; Niu et al., 2019). It would be interesting to study how the embryonic epigenome is established in primate embryos using these systems.
Perspectives
Deciphering how and why epigenetic reprogramming occurs during parental-to-zygotic transition is crucial toward understanding how life starts. In the past decade, ultra-sensitive chromatin analysis technologies have provided an unprecedented view of the dramatic epigenetic transitions during mammalian early development. Not surprisingly, these emerging data generated more intriguing questions than answers. Several challenges need to be met for further investigations. First, ultra-sensitive epigenetic profiling technology is still a major bottleneck for studying chromatin reprogramming in early development. In particular, mapping the binding of TFs and chromatin regulators in early embryos remains a daunting task thus far. Improved ChIP-based and chromatin immunocleavage-based genome-wide methods (e.g., itChIP-seq, ACT-seq, ChIL-seq, CUT&RUN, CUT&Tag, CoBATCH) (Ai et al., 2019; Carter et al., 2019; Harada et al., 2019; Kaya-Okur et al., 2019; Skene and Henikoff, 2017; Wang et al., 2019a) offered promising approaches, but optimizations are still needed for relatively low-abundance proteins (as in the cases of many TFs). These methods also heavily rely on sensitive and specific antibodies. Besides, delineating cell heterogeneity in early development requires the adaptation of current single-cell epigenomics and multi-omics technologies to low-input versions. In addition, live imaging and CRISPR-based lineage tracing (McKenna et al., 2016) would be critical toolsets for recording the continuous development in a highly refined spatiotemporal manner. Second, there are still major knowledge gaps between global epigenetic transformation and their biological functions in developmental events. For example, the functions of DNA demethylation after fertilization remain elusive, despite proposals suggesting that it may facilitate pluripotency (Lee et al., 2014; Smith and Meissner, 2013; Wu and Zhang, 2014) or the evolution of placenta through the exaptation of retrotransposable elements (Kaneko-Ishino and Ishino, 2013). How the epigenomes work in concert with TFs and developmental cues in development remains to be addressed. Deciphering these functions again can be greatly accelerated with future cutting-edge technologies. For example, easy-to-use genetic manipulation of interesting genes in early embryos remains a major demand in practice, especially for oocyte-supplied maternal factors, which are often challenging to be efficiently removed. Besides, epigenome editing (Holtzman and Gersbach, 2018) may greatly facilitate decoding the functions of epigenetic marks at specific loci. Third, increasing data have uncovered the species-specific epigenome reprogramming, which is in line with the “hourglass” model suggesting that the animal development is more conserved during gastrulation but less so at earlier and later stages (Irie and Kuratani, 2014). Alternative animal models are needed to study human-specific features that are not present in mouse. Lastly, emerging new in vitro models, such as embryoid, blastoid, or gastruloid (Shahbazi et al., 2019), present valuable opportunities to study important biology events, particularly in developmental windows that are often inaccessible for research in vivo. Additionally, they can be subjected to genetic screening and biochemical assays that are not feasible for early embryos. With the advent of these cutting-edge technologies, we envision that exciting findings will be uncovered in the forthcoming years in the fields of early development and stem cells, ultimately paving the way to regenerative medicine in the future.
Author Contributions
W. Xia and W. Xie researched data for the article, and discussed and wrote the manuscript.
Acknowledgments
We thank the laboratory members for the comments and help during the preparation of the manuscript, and H. Zheng for the original idea of the Graphical Abstract. The review included selected studies due to the space limitation, and we sincerely apologize to all the authors whose work was not cited.
Funding: This work was supported by the National Key R&D Program of China (grant no. 2019YFA0508901 to W. Xie), the National Natural Science Foundation of China (grant nos. 31988101, 31830047, and 31725018 to W. Xie), the THU-PKU Center for Life Sciences (W. Xie), and the Beijing Municipal Commission of Science and Technology (grant no. Z181100001318006 to W. Xie). W. Xie is an HHMI International Research Scholar.
References
- Abe K., Inoue A., Suzuki M.G., Aoki F. Global gene silencing is caused by the dissociation of RNA polymerase II from DNA in mouse oocytes. J. Reprod. Dev. 2010;56:502–507. doi: 10.1262/jrd.10-068a. [DOI] [PubMed] [Google Scholar]
- Abe K., Yamamoto R., Franke V., Cao M., Suzuki Y., Suzuki M.G., Vlahovicek K., Svoboda P., Schultz R.M., Aoki F. The first murine zygotic transcription is promiscuous and uncoupled from splicing and 3′ processing. EMBO J. 2015;34:1523–1537. doi: 10.15252/embj.201490648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed K., Dehghani H., Rugg-Gunn P., Fussner E., Rossant J., Bazett-Jones D.P. Global chromatin architecture reflects pluripotency and lineage commitment in the early mouse embryo. PLoS One. 2010;5:e10531. doi: 10.1371/journal.pone.0010531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai S., Xiong H., Li C.C., Luo Y., Shi Q., Liu Y., Yu X., Li C., He A. Profiling chromatin states using single-cell itChIP-seq. Nat. Cell Biol. 2019;21:1164–1172. doi: 10.1038/s41556-019-0383-5. [DOI] [PubMed] [Google Scholar]
- Akkers R.C., van Heeringen S.J., Jacobi U.G., Janssen-Megens E.M., Francoijs K.J., Stunnenberg H.G., Veenstra G.J. A hierarchy of H3K4me3 and H3K27me3 acquisition in spatial gene regulation in Xenopus embryos. Dev. Cell. 2009;17:425–434. doi: 10.1016/j.devcel.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amouroux R., Nashun B., Shirane K., Nakagawa S., Hill P.W., D'Souza Z., Nakayama M., Matsuda M., Turp A., Ndjetehe E. De novo DNA methylation drives 5hmC accumulation in mouse zygotes. Nat. Cell Biol. 2016;18:225–233. doi: 10.1038/ncb3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu-Vieyra C.V., Chen R., Agno J.E., Glaser S., Anastassiadis K., Stewart A.F., Matzuk M.M. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. 2010;8:e1000453. doi: 10.1371/journal.pbio.1000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au Yeung W.K., Brind'Amour J., Hatano Y., Yamagata K., Feil R., Lorincz M.C., Tachibana M., Shinkai Y., Sasaki H. Histone H3K9 methyltransferase G9a in oocytes is essential for preimplantation development but dispensable for CG methylation protection. Cell Rep. 2019;27:282–293.e4. doi: 10.1016/j.celrep.2019.03.002. [DOI] [PubMed] [Google Scholar]
- Balhorn R. The protamine family of sperm nuclear proteins. Genome Biol. 2007;8:227. doi: 10.1186/gb-2007-8-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomei M.S., Ferguson-Smith A.C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011;3:a002592. doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battulin N., Fishman V.S., Mazur A.M., Pomaznoy M., Khabarova A.A., Afonnikov D.A., Prokhortchouk E.B., Serov O.L. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 2015;16:77. doi: 10.1186/s13059-015-0642-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bickmore W.A. The spatial organization of the human genome. Annu. Rev. Genomics Hum. Genet. 2013;14:67–84. doi: 10.1146/annurev-genom-091212-153515. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Borsos M., Perricone S.M., Schauer T., Pontabry J., de Luca K.L., de Vries S.S., Ruiz-Morales E.R., Torres-Padilla M.E., Kind J. Genome-lamina interactions are established de novo in the early mouse embryo. Nature. 2019;569:729–733. doi: 10.1038/s41586-019-1233-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boskovic A., Eid A., Pontabry J., Ishiuchi T., Spiegelhalter C., Raghu Ram E.V., Meshorer E., Torres-Padilla M.E. Higher chromatin mobility supports totipotency and precedes pluripotency in vivo. Genes Dev. 2014;28:1042–1047. doi: 10.1101/gad.238881.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourc'his D., Xu G.L., Lin C.S., Bollman B., Bestor T.H. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- Branco M.R., King M., Perez-Garcia V., Bogutz A.B., Caley M., Fineberg E., Lefebvre L., Cook S.J., Dean W., Hemberger M. Maternal DNA methylation regulates early trophoblast development. Dev. Cell. 2016;36:152–163. doi: 10.1016/j.devcel.2015.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton A., Brochard V., Galan C., Ruiz-Morales E.R., Rovira Q., Rodriguez-Terrones D., Kruse K., Le Gras S., Udayakumar V.S., Chin H.G. Heterochromatin establishment during early mammalian development is regulated by pericentromeric RNA and characterized by non-repressive H3K9me3. Nat. Cell Biol. 2020;22:767–778. doi: 10.1038/s41556-020-0536-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton A., Torres-Padilla M.E. Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nat. Rev. Mol. Cell Biol. 2014;15:723–734. doi: 10.1038/nrm3885. [DOI] [PubMed] [Google Scholar]
- Carter B., Ku W.L., Kang J.Y., Hu G., Perrie J., Tang Q., Zhao K. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq) Nat. Commun. 2019;10:3747. doi: 10.1038/s41467-019-11559-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazaud C., Yamanaka Y. Lineage specification in the mouse preimplantation embryo. Development. 2016;143:1063–1074. doi: 10.1242/dev.128314. [DOI] [PubMed] [Google Scholar]
- Chen Z., Zhang Y. Loss of DUX causes minor defects in zygotic genome activation and is compatible with mouse development. Nat. Genet. 2019;51:947–951. doi: 10.1038/s41588-019-0418-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Ke Y., Wu K., Zhao H., Sun Y., Gao L., Liu Z., Zhang J., Tao W., Hou Z. Key role for CTCF in establishing chromatin structure in human embryos. Nature. 2019;576:306–310. doi: 10.1038/s41586-019-1812-0. [DOI] [PubMed] [Google Scholar]
- Chen Z., Yin Q., Inoue A., Zhang C., Zhang Y. Allelic H3K27me3 to allelic DNA methylation switch maintains noncanonical imprinting in extraembryonic cells. Sci. Adv. 2019;5:eaay7246. doi: 10.1126/sciadv.aay7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Zhu Q., Li C., Kou X., Zhao Y., Li Y., Xu R., Yang L., Yang L., Gu L. Chromatin architecture reorganization in murine somatic cell nuclear transfer embryos. Nat. Commun. 2020;11:1813. doi: 10.1038/s41467-020-15607-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y.G., Matoba S., Liu Y., Eum J.H., Lu F., Jiang W., Lee J.E., Sepilian V., Cha K.Y., Lee D.R. Histone demethylase expression enhances human somatic cell nuclear transfer efficiency and promotes derivation of pluripotent stem cells. Cell Stem Cell. 2015;17:758–766. doi: 10.1016/j.stem.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Clouaire T., Webb S., Skene P., Illingworth R., Kerr A., Andrews R., Lee J.H., Skalnik D., Bird A. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26:1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombet S., Ranisavljevic N., Nagano T., Varnai C., Shisode T., Leung W., Piolot T., Galupa R., Borensztein M., Servant N. Parental-to-embryo switch of chromosome organization in early embryogenesis. Nature. 2020;580:142–146. doi: 10.1038/s41586-020-2125-z. [DOI] [PubMed] [Google Scholar]
- Dahl J.A., Jung I., Aanes H., Greggains G.D., Manaf A., Lerdrup M., Li G., Kuan S., Li B., Lee A.Y. Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature. 2016;537:548–552. doi: 10.1038/nature19360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Iaco A., Planet E., Coluccio A., Verp S., Duc J., Trono D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 2017;49:941–945. doi: 10.1038/ng.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Iaco A., Coudray A., Duc J., Trono D. DPPA2 and DPPA4 are necessary to establish a 2C-like state in mouse embryonic stem cells. EMBO Rep. 2019;20:e47382. doi: 10.15252/embr.201847382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A., Rajavelu A., Rathert P., Tamas R., Jurkowska R.Z., Ragozin S., Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djekidel M.N., Inoue A., Matoba S., Suzuki T., Zhang C., Lu F., Jiang L., Zhang Y. Reprogramming of chromatin accessibility in somatic cell nuclear transfer is DNA replication independent. Cell Rep. 2018;23:1939–1947. doi: 10.1016/j.celrep.2018.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z., Zheng H., Huang B., Ma R., Wu J., Zhang X., He J., Xiang Y., Wang Q., Li Y. Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature. 2017;547:232–235. doi: 10.1038/nature23263. [DOI] [PubMed] [Google Scholar]
- Du Z., Zheng H., Kawamura Y.K., Zhang K., Gassler J., Powell S., Xu Q., Lin Z., Xu K., Zhou Q. Polycomb group proteins regulate chromatin architecture in mouse oocytes and early embryos. Mol. Cell. 2019;77:825–839.e7. doi: 10.1016/j.molcel.2019.11.011. [DOI] [PubMed] [Google Scholar]
- Eckersley-Maslin M.A., Alda-Catalinas C., Reik W. Dynamics of the epigenetic landscape during the maternal-to-zygotic transition. Nat. Rev. Mol. Cell Biol. 2018;19:436–450. doi: 10.1038/s41580-018-0008-z. [DOI] [PubMed] [Google Scholar]
- Eckersley-Maslin M., Alda-Catalinas C., Blotenburg M., Kreibich E., Krueger C., Reik W. Dppa2 and Dppa4 directly regulate the Dux-driven zygotic transcriptional program. Genes Dev. 2019;33:194–208. doi: 10.1101/gad.321174.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckersley-Maslin M.A., Parry A., Blotenburg M., Krueger C., Ito Y., Franklin V.N.R., Narita M., D'Santos C.S., Reik W. Epigenetic priming by Dppa2 and 4 in pluripotency facilitates multi-lineage commitment. Nat. Struct. Mol. Biol. 2020;27:696–705. doi: 10.1038/s41594-020-0443-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk M., Feodorova Y., Naumova N., Imakaev M., Lajoie B.R., Leonhardt H., Joffe B., Dekker J., Fudenberg G., Solovei I. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature. 2019;570:395–399. doi: 10.1038/s41586-019-1275-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyamer I.M., Gassler J., Imakaev M., Brandao H.B., Ulianov S.V., Abdennur N., Razin S.V., Mirny L.A., Tachibana-Konwalski K. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature. 2017;544:110–114. doi: 10.1038/nature21711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L., Wu K., Liu Z., Yao X., Yuan S., Tao W., Yi L., Yu G., Hou Z., Fan D. Chromatin accessibility landscape in human early embryos and its association with evolution. Cell. 2018;173:248–259.e15. doi: 10.1016/j.cell.2018.02.028. [DOI] [PubMed] [Google Scholar]
- Gassler J., Brandao H.B., Imakaev M., Flyamer I.M., Ladstatter S., Bickmore W.A., Peters J.M., Mirny L.A., Tachibana K. A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. EMBO J. 2017;36:3600–3618. doi: 10.15252/embj.201798083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibcus J.H., Dekker J. The hierarchy of the 3D genome. Mol. Cell. 2013;49:773–782. doi: 10.1016/j.molcel.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibcus J.H., Samejima K., Goloborodko A., Samejima I., Naumova N., Nuebler J., Kanemaki M.T., Xie L., Paulson J.R., Earnshaw W.C. A pathway for mitotic chromosome formation. Science. 2018;359:eaao6135. doi: 10.1126/science.aao6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser S., Lubitz S., Loveland K.L., Ohbo K., Robb L., Schwenk F., Seibler J., Roellig D., Kranz A., Anastassiadis K. The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenetics Chromatin. 2009;2:5. doi: 10.1186/1756-8935-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold H.B., Jung Y.H., Corces V.G. Not just heads and tails: the complexity of the sperm epigenome. J. Biol. Chem. 2018;293:13815–13820. doi: 10.1074/jbc.R117.001561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou L.T., Lim D.H., Ma W., Aubol B.E., Hao Y., Wang X., Zhao J., Liang Z., Shao C., Zhang X. Initiation of parental genome reprogramming in fertilized oocyte by splicing kinase SRPK1-catalyzed protamine phosphorylation. Cell. 2020;180:1212–1227.e14. doi: 10.1016/j.cell.2020.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gretarsson K.H., Hackett J.A. Dppa2 and Dppa4 counteract de novo methylation to establish a permissive epigenome for development. Nat. Struct. Mol. Biol. 2020;27:706–716. doi: 10.1038/s41594-020-0445-1. [DOI] [PubMed] [Google Scholar]
- Grow E.J., Flynn R.A., Chavez S.L., Bayless N.L., Wossidlo M., Wesche D.J., Martin L., Ware C.B., Blish C.A., Chang H.Y. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature. 2015;522:221–225. doi: 10.1038/nature14308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H., Zhu P., Yan L., Li R., Hu B., Lian Y., Yan J., Ren X., Lin S., Li J. The DNA methylation landscape of human early embryos. Nature. 2014;511:606–610. doi: 10.1038/nature13544. [DOI] [PubMed] [Google Scholar]
- Guo F., Li L., Li J., Wu X., Hu B., Zhu P., Wen L., Tang F. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017;27:967–988. doi: 10.1038/cr.2017.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M., Zhang Y., Zhou J., Bi Y., Xu J., Xu C., Kou X., Zhao Y., Li Y., Tu Z. Precise temporal regulation of Dux is important for embryo development. Cell Res. 2019;29:956–959. doi: 10.1038/s41422-019-0238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett J.A., Surani M.A. Regulatory principles of pluripotency: from the ground state up. Cell Stem Cell. 2014;15:416–430. doi: 10.1016/j.stem.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Hammoud S.S., Nix D.A., Zhang H., Purwar J., Carrell D.T., Cairns B.R. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460:473–478. doi: 10.1038/nature08162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud S.S., Low D.H., Yi C., Carrell D.T., Guccione E., Cairns B.R. Chromatin and transcription transitions of mammalian adult germline stem cells and spermatogenesis. Cell Stem Cell. 2014;15:239–253. doi: 10.1016/j.stem.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Hanna C.W., Taudt A., Huang J., Gahurova L., Kranz A., Andrews S., Dean W., Stewart A.F., Colome-Tatche M., Kelsey G. MLL2 conveys transcription-independent H3K4 trimethylation in oocytes. Nat. Struct. Mol. Biol. 2018;25:73–82. doi: 10.1038/s41594-017-0013-5. [DOI] [PubMed] [Google Scholar]
- Harada A., Maehara K., Handa T., Arimura Y., Nogami J., Hayashi-Takanaka Y., Shirahige K., Kurumizaka H., Kimura H., Ohkawa Y. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat. Cell Biol. 2019;21:287–296. doi: 10.1038/s41556-018-0248-3. [DOI] [PubMed] [Google Scholar]
- Harris C., Cloutier M., Trotter M., Hinten M., Gayen S., Du Z., Xie W., Kalantry S. Conversion of random X-inactivation to imprinted X-inactivation by maternal PRC2. Elife. 2019;8:e44258. doi: 10.7554/eLife.44258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata K., Okano M., Lei H., Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–1993. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- Hendrickson P.G., Dorais J.A., Grow E.J., Whiddon J.L., Lim J.W., Wike C.L., Weaver B.D., Pflueger C., Emery B.R., Wilcox A.L. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 2017;49:925–934. doi: 10.1038/ng.3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa R., Chiba H., Kaneda M., Tajima S., Li E., Jaenisch R., Sasaki H. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008;22:1607–1616. doi: 10.1101/gad.1667008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman L., Gersbach C.A. Editing the epigenome: reshaping the genomic landscape. Annu. Rev. Genomics Hum. Genet. 2018;19:43–71. doi: 10.1146/annurev-genom-083117-021632. [DOI] [PubMed] [Google Scholar]
- Hug C.B., Grimaldi A.G., Kruse K., Vaquerizas J.M. Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell. 2017;169:216–228.e19. doi: 10.1016/j.cell.2017.03.024. [DOI] [PubMed] [Google Scholar]
- Inoue A., Zhang Y. Nucleosome assembly is required for nuclear pore complex assembly in mouse zygotes. Nat. Struct. Mol. Biol. 2014;21:609–616. doi: 10.1038/nsmb.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A., Jiang L., Lu F., Suzuki T., Zhang Y. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature. 2017;547:419–424. doi: 10.1038/nature23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A., Chen Z., Yin Q., Zhang Y. Maternal Eed knockout causes loss of H3K27me3 imprinting and random X inactivation in the extraembryonic cells. Genes Dev. 2018;32:1525–1536. doi: 10.1101/gad.318675.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie N., Kuratani S. The developmental hourglass model: a predictor of the basic body plan? Development. 2014;141:4649–4655. doi: 10.1242/dev.107318. [DOI] [PubMed] [Google Scholar]
- Jachowicz J.W., Bing X., Pontabry J., Boskovic A., Rando O.J., Torres-Padilla M.E. LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat. Genet. 2017;49:1502–1510. doi: 10.1038/ng.3945. [DOI] [PubMed] [Google Scholar]
- Jiang L., Zhang J., Wang J.J., Wang L., Zhang L., Li G., Yang X., Ma X., Sun X., Cai J. Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell. 2013;153:773–784. doi: 10.1016/j.cell.2013.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y.H., Sauria M.E.G., Lyu X., Cheema M.S., Ausio J., Taylor J., Corces V.G. Chromatin states in mouse sperm correlate with embryonic and adult regulatory landscapes. Cell Rep. 2017;18:1366–1382. doi: 10.1016/j.celrep.2017.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y.H., Kremsky I., Gold H.B., Rowley M.J., Punyawai K., Buonanotte A., Lyu X., Bixler B.J., Chan A.W.S., Corces V.G. Maintenance of CTCF- and transcription factor-mediated interactions from the gametes to the early mouse embryo. Mol. Cell. 2019;75:154–171.e5. doi: 10.1016/j.molcel.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaaij L.J.T., van der Weide R.H., Ketting R.F., de Wit E. Systemic loss and gain of chromatin architecture throughout zebrafish development. Cell Rep. 2018;24:1–10.e4. doi: 10.1016/j.celrep.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M., Okano M., Hata K., Sado T., Tsujimoto N., Li E., Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- Kaneda M., Hirasawa R., Chiba H., Okano M., Li E., Sasaki H. Genetic evidence for Dnmt3a-dependent imprinting during oocyte growth obtained by conditional knockout with Zp3-Cre and complete exclusion of Dnmt3b by chimera formation. Genes Cells. 2010;15:169–179. doi: 10.1111/j.1365-2443.2009.01374.x. [DOI] [PubMed] [Google Scholar]
- Kaneko-Ishino T., Ishino F. The evolution of the placenta and viviparity is related to LTR retrotransposon-derived genes in mammals. J. Mamm. Ova Res. 2013;30:16–23. [Google Scholar]
- Kaya-Okur H.S., Wu S.J., Codomo C.A., Pledger E.S., Bryson T.D., Henikoff J.G., Ahmad K., Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019;10:1930. doi: 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y., Xu Y., Chen X., Feng S., Liu Z., Sun Y., Yao X., Li F., Zhu W., Gao L. 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell. 2017;170:367–381.e20. doi: 10.1016/j.cell.2017.06.029. [DOI] [PubMed] [Google Scholar]
- Ko M.S. Zygotic genome activation revisited: looking through the expression and function of Zscan4. Curr. Top. Dev. Biol. 2016;120:103–124. doi: 10.1016/bs.ctdb.2016.04.004. [DOI] [PubMed] [Google Scholar]
- Kobayashi H., Sakurai T., Imai M., Takahashi N., Fukuda A., Yayoi O., Sato S., Nakabayashi K., Hata K., Sotomaru Y. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kruse K., Diaz N., Enriquez-Gasca R., Gaume X., Torres-Padilla M.-E., Vaquerizas J.M. Transposable elements drive reorganisation of 3D chromatin during early embryogenesis. bioRxiv. 2019 doi: 10.1101/523712. [DOI] [Google Scholar]
- Lauberth S.M., Nakayama T., Wu X., Ferris A.L., Tang Z., Hughes S.H., Roeder R.G. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.J., Hore T.A., Reik W. Reprogramming the methylome: erasing memory and creating diversity. Cell Stem Cell. 2014;14:710–719. doi: 10.1016/j.stem.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Guo F., Gao Y., Ren Y., Yuan P., Yan L., Li R., Lian Y., Li J., Hu B. Single-cell multi-omics sequencing of human early embryos. Nat. Cell Biol. 2018;20:847–858. doi: 10.1038/s41556-018-0123-2. [DOI] [PubMed] [Google Scholar]
- Li Y., Zhang Z., Chen J., Liu W., Lai W., Liu B., Li X., Liu L., Xu S., Dong Q. Stella safeguards the oocyte methylome by preventing de novo methylation mediated by DNMT1. Nature. 2018;564:136–140. doi: 10.1038/s41586-018-0751-5. [DOI] [PubMed] [Google Scholar]
- Lin C.J., Koh F.M., Wong P., Conti M., Ramalho-Santos M. Hira-mediated H3.3 incorporation is required for DNA replication and ribosomal RNA transcription in the mouse zygote. Dev. Cell. 2014;30:268–279. doi: 10.1016/j.devcel.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Liu X., Wang C., Gao Y., Gao R., Kou X., Zhao Y., Li J., Wu Y., Xiu W. Identification of key factors conquering developmental arrest of somatic cell cloned embryos by combining embryo biopsy and single-cell sequencing. Cell Discov. 2016;2:16010. doi: 10.1038/celldisc.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Wang C., Liu W., Li J., Li C., Kou X., Chen J., Zhao Y., Gao H., Wang H. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature. 2016;537:558–562. doi: 10.1038/nature19362. [DOI] [PubMed] [Google Scholar]
- Loda A., Heard E. Xist RNA in action: past, present, and future. PLoS Genet. 2019;15:e1008333. doi: 10.1371/journal.pgen.1008333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F., Liu Y., Inoue A., Suzuki T., Zhao K., Zhang Y. Establishing chromatin regulatory landscape during mouse preimplantation development. Cell. 2016;165:1375–1388. doi: 10.1016/j.cell.2016.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupianez D.G., Spielmann M., Mundlos S. Breaking TADs: how alterations of chromatin domains result in disease. Trends Genet. 2016;32:225–237. doi: 10.1016/j.tig.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Ma H., Zhai J., Wan H., Jiang X., Wang X., Wang L., Xiang Y., He X., Zhao Z.A., Zhao B. In vitro culture of cynomolgus monkey embryos beyond early gastrulation. Science. 2019;366:eaax7890. doi: 10.1126/science.aax7890. [DOI] [PubMed] [Google Scholar]
- Macfarlan T.S., Gifford W.D., Agarwal S., Driscoll S., Lettieri K., Wang J., Andrews S.E., Franco L., Rosenfeld M.G., Ren B. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev. 2011;25:594–607. doi: 10.1101/gad.2008511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan T.S., Gifford W.D., Driscoll S., Lettieri K., Rowe H.M., Bonanomi D., Firth A., Singer O., Trono D., Pfaff S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature. 2012;487:57–63. doi: 10.1038/nature11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R., Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S., Liu Y., Lu F., Iwabuchi K.A., Shen L., Inoue A., Zhang Y. Embryonic development following somatic cell nuclear transfer impeded by persisting histone methylation. Cell. 2014;159:884–895. doi: 10.1016/j.cell.2014.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., Findlay G.M., Gagnon J.A., Horwitz M.S., Schier A.F., Shendure J. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science. 2016;353:aaf7907. doi: 10.1126/science.aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski J., Cook A., Bowman S.K., Mueller B., Alver B.H., Kundu S., Deaton A.M., Urban J.A., Larschan E., Park P.J. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat. Commun. 2016;7:11485. doi: 10.1038/ncomms11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy P.J., Wu S.F., James C.R., Wike C.L., Cairns B.R. Placeholder nucleosomes underlie germline-to-embryo DNA methylation reprogramming. Cell. 2018;172:993–1006.e13. doi: 10.1016/j.cell.2018.01.022. [DOI] [PubMed] [Google Scholar]
- Nakamura T., Liu Y.J., Nakashima H., Umehara H., Inoue K., Matoba S., Tachibana M., Ogura A., Shinkai Y., Nakano T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–419. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- Nakamura R., Motai Y., Kumagai M., Nishiyama H., Durand N.C., Kondo K., Kondo T., Tsukahara T., Shimada A., Lieberman Aiden E. CTCF looping is established during gastrulation in medaka embryos. bioRxiv. 2018 doi: 10.1101/454082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicetto D., Donahue G., Jain T., Peng T., Sidoli S., Sheng L., Montavon T., Becker J.S., Grindheim J.M., Blahnik K. H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science. 2019;363:294–297. doi: 10.1126/science.aau0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y., Sun N., Li C., Lei Y., Huang Z., Wu J., Si C., Dai X., Liu C., Wei J. Dissecting primate early post-implantation development using long-term in vitro embryo culture. Science. 2019;366:eaaw5754. doi: 10.1126/science.aaw5754. [DOI] [PubMed] [Google Scholar]
- Peaston A.E., Evsikov A.V., Graber J.H., de Vries W.N., Holbrook A.E., Solter D., Knowles B.B. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev. Cell. 2004;7:597–606. doi: 10.1016/j.devcel.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Percharde M., Lin C.J., Yin Y., Guan J., Peixoto G.A., Bulut-Karslioglu A., Biechele S., Huang B., Shen X., Ramalho-Santos M. A LINE1-nucleolin partnership regulates early development and ESC identity. Cell. 2018;174:391–405.e19. doi: 10.1016/j.cell.2018.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric-Hupkes D., Meuleman W., Pagie L., Bruggeman S.W., Solovei I., Brugman W., Graf S., Flicek P., Kerkhoven R.M., van Lohuizen M. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos S., Edsgard D., Reinius B., Deng Q., Panula S.P., Codeluppi S., Plaza Reyes A., Linnarsson S., Sandberg R., Lanner F. Single-cell RNA-seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell. 2016;165:1012–1026. doi: 10.1016/j.cell.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pon J.R., Marra M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015;10:25–50. doi: 10.1146/annurev-pathol-012414-040312. [DOI] [PubMed] [Google Scholar]
- Potok M.E., Nix D.A., Parnell T.J., Cairns B.R. Reprogramming the maternal zebrafish genome after fertilization to match the paternal methylation pattern. Cell. 2013;153:759–772. doi: 10.1016/j.cell.2013.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst A.V., Almouzni G. Pericentric heterochromatin: dynamic organization during early development in mammals. Differentiation. 2008;76:15–23. doi: 10.1111/j.1432-0436.2007.00220.x. [DOI] [PubMed] [Google Scholar]
- Rathke C., Baarends W.M., Awe S., Renkawitz-Pohl R. Chromatin dynamics during spermiogenesis. Biochim. Biophys. Acta. 2014;1839:155–168. doi: 10.1016/j.bbagrm.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Rivera C.M., Ren B. Mapping human epigenomes. Cell. 2013;155:39–55. doi: 10.1016/j.cell.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley M.J., Corces V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018;19:789–800. doi: 10.1038/s41576-018-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samata M., Alexiadis A., Richard G., Georgiev P., Nuebler J., Kulkarni T., Renschler G., Basilicata M.F., Zenk F.L., Shvedunova M. Intergenerationally maintained histone H4 lysine 16 acetylation is instructive for future gene activation. Cell. 2020;182:127–144.e23. doi: 10.1016/j.cell.2020.05.026. [DOI] [PubMed] [Google Scholar]
- Sankar A., Lerdrup M., Manaf A., Johansen J.V., Gonzalez J.M., Borup R., Blanshard R., Klungland A., Hansen K., Andersen C.Y. KDM4A regulates the maternal-to-zygotic transition by protecting broad H3K4me3 domains from H3K9me3 invasion in oocytes. Nat. Cell Biol. 2020;22:380–388. doi: 10.1038/s41556-020-0494-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y., Hilbert L., Oda H., Wan Y., Heddleston J.M., Chew T.L., Zaburdaev V., Keller P., Lionnet T., Vastenhouw N. Histone H3K27 acetylation precedes active transcription during zebrafish zygotic genome activation as revealed by live-cell analysis. Development. 2019;146:dev179127. doi: 10.1242/dev.179127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz R.M. The molecular foundations of the maternal to zygotic transition in the preimplantation embryo. Hum. Reprod Update. 2002;8:323–331. doi: 10.1093/humupd/8.4.323. [DOI] [PubMed] [Google Scholar]
- Schulz K.N., Harrison M.M. Mechanisms regulating zygotic genome activation. Nat. Rev. Genet. 2019;20:221–234. doi: 10.1038/s41576-018-0087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendzikaite G., Kelsey G. The role and mechanisms of DNA methylation in the oocyte. Essays Biochem. 2019;63:691–705. doi: 10.1042/EBC20190043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha Q.Q., Dai X.X., Jiang J.C., Yu C., Jiang Y., Liu J., Ou X.H., Zhang S.Y., Fan H.Y. CFP1 coordinates histone H3 lysine-4 trimethylation and meiotic cell cycle progression in mouse oocytes. Nat. Commun. 2018;9:3477. doi: 10.1038/s41467-018-05930-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazi M.N., Siggia E.D., Zernicka-Goetz M. Self-organization of stem cells into embryos: a window on early mammalian development. Science. 2019;364:948–951. doi: 10.1126/science.aax0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirane K., Miura F., Ito T., Lorincz M.C. NSD1-deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb-associated silencing. Nat. Genet. 2020 doi: 10.1038/s41588-020-0689-z. [DOI] [PubMed] [Google Scholar]
- Shirane K., Toh H., Kobayashi H., Miura F., Chiba H., Ito T., Kono T., Sasaki H. Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet. 2013;9:e1003439. doi: 10.1371/journal.pgen.1003439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene P.J., Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. 2017;6:e21856. doi: 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood S.A., Tomizawa S., Krueger F., Ruf N., Carli N., Segonds-Pichon A., Sato S., Hata K., Andrews S.R., Kelsey G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 2011;43:811–814. doi: 10.1038/ng.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z.D., Chan M.M., Humm K.C., Karnik R., Mekhoubad S., Regev A., Eggan K., Meissner A. DNA methylation dynamics of the human preimplantation embryo. Nature. 2014;511:611–615. doi: 10.1038/nature13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z.D., Meissner A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- Smith Z.D., Shi J., Gu H., Donaghey J., Clement K., Cacchiarelli D., Gnirke A., Michor F., Meissner A. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature. 2017;549:543–547. doi: 10.1038/nature23891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y., van den Berg P.R., Markoulaki S., Soldner F., Dall'Agnese A., Henninger J.E., Drotar J., Rosenau N., Cohen M.A., Young R.A. Dynamic enhancer DNA methylation as basis for transcriptional and cellular heterogeneity of ESCs. Mol. Cell. 2019;75:905–920.e6. doi: 10.1016/j.molcel.2019.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller C., Koopman P., Bowles J. Sex determination in the mammalian germline. Annu. Rev. Genet. 2017;51:265–285. doi: 10.1146/annurev-genet-120215-035449. [DOI] [PubMed] [Google Scholar]
- Stewart K.R., Veselovska L., Kelsey G. Establishment and functions of DNA methylation in the germline. Epigenomics. 2016;8:1399–1413. doi: 10.2217/epi-2016-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart K.R., Veselovska L., Kim J., Huang J., Saadeh H., Tomizawa S., Smallwood S.A., Chen T., Kelsey G. Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev. 2015;29:2449–2462. doi: 10.1101/gad.271353.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Werken C., van der Heijden G.W., Eleveld C., Teeuwssen M., Albert M., Baarends W.M., Laven J.S., Peters A.H., Baart E.B. Paternal heterochromatin formation in human embryos is H3K9/HP1 directed and primed by sperm-derived histone modifications. Nat. Commun. 2014;5:5868. doi: 10.1038/ncomms6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw N.L., Zhang Y., Woods I.G., Imam F., Regev A., Liu X.S., Rinn J., Schier A.F. Chromatin signature of embryonic pluripotency is established during genome activation. Nature. 2010;464:922–926. doi: 10.1038/nature08866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M., Mulder K.W., Denissov S., Pijnappel W.W., van Schaik F.M., Varier R.A., Baltissen M.P., Stunnenberg H.G., Mann M., Timmers H.T. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Veselovska L., Smallwood S.A., Saadeh H., Stewart K.R., Krueger F., Maupetit-Mehouas S., Arnaud P., Tomizawa S., Andrews S., Kelsey G. Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol. 2015;16:209. doi: 10.1186/s13059-015-0769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter E., Quante T., Merusi C., Arczewska A., Stewart F., Webb S., Bird A. Synthetic CpG islands reveal DNA sequence determinants of chromatin structure. eLife. 2014;3:e03397. doi: 10.7554/eLife.03397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Liu X., Gao Y., Yang L., Li C., Liu W., Chen C., Kou X., Zhao Y., Chen J. Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nat. Cell Biol. 2018;20:620–631. doi: 10.1038/s41556-018-0093-4. [DOI] [PubMed] [Google Scholar]
- Wang Q., Xiong H., Ai S., Yu X., Liu Y., Zhang J., He A. CoBATCH for high-throughput single-cell epigenomic profiling. Mol. Cell. 2019;76:206–216.e7. doi: 10.1016/j.molcel.2019.07.015. [DOI] [PubMed] [Google Scholar]
- Wang Y., Wang H., Zhang Y., Du Z., Si W., Fan S., Qin D., Wang M., Duan Y., Li L. Reprogramming of meiotic chromatin architecture during spermatogenesis. Mol. Cell. 2019;73:547–561.e6. doi: 10.1016/j.molcel.2018.11.019. [DOI] [PubMed] [Google Scholar]
- Wang L.-Y., Li Z.-K., Wang L.-B., Liu C., Sun X.-H., Feng G.-H., Wang J.-Q., Li Y.-F., Qiao L.-Y., Nie H. Overcoming intrinsic H3K27me3 imprinting barriers improves post-implantation development after somatic cell nuclear transfer. Cell Stem Cell. 2020;27:315–325.e5. doi: 10.1016/j.stem.2020.05.014. [DOI] [PubMed] [Google Scholar]
- Whiddon J.L., Langford A.T., Wong C.J., Zhong J.W., Tapscott S.J. Conservation and innovation in the DUX4-family gene network. Nat. Genet. 2017;49:935–940. doi: 10.1038/ng.3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45–68. doi: 10.1016/j.cell.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Huang B., Chen H., Yin Q., Liu Y., Xiang Y., Zhang B., Liu B., Wang Q., Xia W. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature. 2016;534:652–657. doi: 10.1038/nature18606. [DOI] [PubMed] [Google Scholar]
- Wu J., Xu J., Liu B., Yao G., Wang P., Lin Z., Huang B., Wang X., Li T., Shi S. Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature. 2018;557:256–260. doi: 10.1038/s41586-018-0080-8. [DOI] [PubMed] [Google Scholar]
- Wu K., Liu H., Wang Y., He J., Xu S., Chen Y., Kuang J., Liu J., Guo L., Li D. SETDB1-Mediated cell fate transition between 2C-like and pluripotent states. Cell Rep. 2020;30:25–36.e6. doi: 10.1016/j.celrep.2019.12.010. [DOI] [PubMed] [Google Scholar]
- Xia W., Xu J., Yu G., Yao G., Xu K., Ma X., Zhang N., Liu B., Li T., Lin Z. Resetting histone modifications during human parental-to-zygotic transition. Science. 2019;365:353–360. doi: 10.1126/science.aaw5118. [DOI] [PubMed] [Google Scholar]
- Xiang Y., Zhang Y., Xu Q., Zhou C., Liu B., Du Z., Zhang K., Zhang B., Wang X., Gayen S. Epigenomic analysis of gastrulation identifies a unique chromatin state for primed pluripotency. Nat. Genet. 2019;52:95–105. doi: 10.1038/s41588-019-0545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Xiang Y., Wang Q., Wang L., Brind'Amour J., Bogutz A.B., Zhang Y., Zhang B., Yu G., Xia W. SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat. Genet. 2019;51:844–856. doi: 10.1038/s41588-019-0398-7. [DOI] [PubMed] [Google Scholar]
- Xu Q., Xie W. Epigenome in early mammalian development: inheritance, reprogramming and establishment. Trends Cell Biol. 2018;28:237–253. doi: 10.1016/j.tcb.2017.10.008. [DOI] [PubMed] [Google Scholar]
- Yan Y.L., Zhang C., Hao J., Wang X.L., Ming J., Mi L., Na J., Hu X., Wang Y. DPPA2/4 and SUMO E3 ligase PIAS4 opposingly regulate zygotic transcriptional program. PLoS Biol. 2019;17:e3000324. doi: 10.1371/journal.pbio.3000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Song L., Liu X., Bai L., Li G. KDM6A and KDM6B play contrasting roles in nuclear transfer embryos revealed by MERVL reporter system. EMBO Rep. 2018;19:e46240. doi: 10.15252/embr.201846240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C., Fan X., Sha Q.Q., Wang H.H., Li B.T., Dai X.X., Shen L., Liu J., Wang L., Liu K. CFP1 regulates histone H3K4 trimethylation and developmental potential in mouse oocytes. Cell Rep. 2017;20:1161–1172. doi: 10.1016/j.celrep.2017.07.011. [DOI] [PubMed] [Google Scholar]
- Zenk F., Loeser E., Schiavo R., Kilpert F., Bogdanovic O., Iovino N. Germ line-inherited H3K27me3 restricts enhancer function during maternal-to-zygotic transition. Science. 2017;357:212–216. doi: 10.1126/science.aam5339. [DOI] [PubMed] [Google Scholar]
- Zhang A., Xu B., Sun Y., Lu X., Gu R., Wu L., Feng Y., Xu C. Dynamic changes of histone H3 trimethylated at positions K4 and K27 in human oocytes and preimplantation embryos. Fertil. Steril. 2012;98:1009–1016. doi: 10.1016/j.fertnstert.2012.06.034. [DOI] [PubMed] [Google Scholar]
- Zhang B., Zheng H., Huang B., Li W., Xiang Y., Peng X., Ming J., Wu X., Zhang Y., Xu Q. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature. 2016;537:553–557. doi: 10.1038/nature19361. [DOI] [PubMed] [Google Scholar]
- Zhang B., Wu X., Zhang W., Shen W., Sun Q., Liu K., Zhang Y., Wang Q., Li Y., Meng A. Widespread enhancer dememorization and promoter priming during parental-to-zygotic transition. Mol. Cell. 2018;72:673–686.e6. doi: 10.1016/j.molcel.2018.10.017. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Xiang Y., Yin Q., Du Z., Peng X., Wang Q., Fidalgo M., Xia W., Li Y., Zhao Z.A. Dynamic epigenomic landscapes during early lineage specification in mouse embryos. Nat. Genet. 2018;50:96–105. doi: 10.1038/s41588-017-0003-x. [DOI] [PubMed] [Google Scholar]
- Zhang W., Chen Z., Yin Q., Zhang D., Racowsky C., Zhang Y. Maternal-biased H3K27me3 correlates with paternal-specific gene expression in the human morula. Genes Dev. 2019;33:382–387. doi: 10.1101/gad.323105.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Wu D.Y., Zheng H., Wang Y., Sun Q.R., Liu X., Wang L.Y., Xiong W.J., Wang Q., Rhodes J.D.P. Analysis of genome architecture during SCNT reveals a role of cohesin in impeding minor ZGA. Mol. Cell. 2020;79:234–250.e9. doi: 10.1016/j.molcel.2020.06.001. [DOI] [PubMed] [Google Scholar]
- Zheng H., Huang B., Zhang B., Xiang Y., Du Z., Xu Q., Li Y., Wang Q., Ma J., Peng X. Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol. Cell. 2016;63:1066–1079. doi: 10.1016/j.molcel.2016.08.032. [DOI] [PubMed] [Google Scholar]
- Zheng H., Xie W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019 doi: 10.1038/s41580-019-0132-4. [DOI] [PubMed] [Google Scholar]
- Zhou F., Wang R., Yuan P., Ren Y., Mao Y., Li R., Lian Y., Li J., Wen L., Yan L. Reconstituting the transcriptome and DNA methylome landscapes of human implantation. Nature. 2019;572:660–664. doi: 10.1038/s41586-019-1500-0. [DOI] [PubMed] [Google Scholar]
- Zhu P., Guo H., Ren Y., Hou Y., Dong J., Li R., Lian Y., Fan X., Hu B., Gao Y. Single-cell DNA methylome sequencing of human preimplantation embryos. Nat. Genet. 2018;50:12–19. doi: 10.1038/s41588-017-0007-6. [DOI] [PubMed] [Google Scholar]
- Zuccotti M., Piccinelli A., Giorgi Rossi P., Garagna S., Redi C.A. Chromatin organization during mouse oocyte growth. Mol. Reprod. Dev. 1995;41:479–485. doi: 10.1002/mrd.1080410410. [DOI] [PubMed] [Google Scholar]