

Abstract
Recombinant immunotoxins (RITs) are fusion proteins that join antibodies to protein toxins for targeted cell killing. RITs armed with Pseudomonas exotoxin A (PE) are undergoing clinical trials for the treatment of cancer. The current design of PE-based RITs joins an antibody fragment to the catalytic domain of PE using a polypeptide linker that is cleaved by the protease furin. Intracellular cleavage of native PE by furin is required for cytotoxicity, yet the PE cleavage site has been shown to be a poor furin substrate. Here we describe the rational design of more efficiently cleaved furin linkers in PE-based RITs, and experiments evaluating their effects on cleavage and cytotoxicity. We found that changes to the furin site could greatly influence both cleavage and cytotoxicity, but the two parameters were not directly correlated. Furthermore, the effects of alterations to the furin linker were not universal. Identical mutations in the anti-CD22 RIT HA22-LR often displayed different cytotoxicity from mutations in the anti-mesothelin RIT SS1-LR/GGS, underscoring the prominent role of the target site in their intoxication pathways. Combining several beneficial mutations in HA22-LR resulted in a variant (HA22-LR/FUR) with a remarkably enhanced cleavage rate and improved cytotoxicity against five B cell lines and similar or enhanced cytotoxicity in five out of six hairy cell leukemia patient samples. This result informs the design of protease-sensitive linkers and suggests that HA22-LR/FUR may be a candidate for further preclinical development.
Graphical Abstract
INTRODUCTION
Pseudomonas exotoxin A (PE) is a highly toxic 66 kDa protein secreted by strains of Pseudomonas aeruginosa. Native PE and its intoxication pathway have been extensively studied, giving us a broad understanding of its overall trafficking route and mechanism of action (reviewed in ref 1). The canonical view of the PE intoxication pathway begins with receptor-mediated endocytosis through the low-density lipoprotein-related protein receptor-1 or variant lipoprotein-related protein receptor-1b. The toxin subsequently traffics through the endolysosomal system to reach the Golgi, where it undergoes retrograde transport to the endoplasmic reticulum due to a C-terminal endoplasmic reticulum-retention signal. During the trafficking process disulfide bonds in PE are reduced and it is cleaved by the protease furin. Together, these actions separate the catalytically active fragment of PE from the cell-binding fragment. The catalytic fragment of PE translocates from the endoplasmic reticulum to the cytosol, where it encounters and ADP-ribosylates elongation factor 2, an essential component of protein synthesis. This modification inactivates elongation factor 2, halts translation, and eventually leads to cell death.
PE has been developed into a therapeutic agent for the treatment of cancers by engineering the toxin to target malignant cells. These agents, termed recombinant immunotoxins (RITs), combine a cytotoxic fragment of PE with an antigen-binding fragment from an antibody to a tumor-associated cell surface protein. The RIT moxetumomab pasudotox2 (previously known as HA223 or CAT-80154) contains the disulfide-stabilized variable fragment of an anti-CD22 antibody attached to a 38 kDa fragment of PE (PE38). Moxetumomab pasudotox targets the mature B cell-specific CD22 receptor for the treatment of B cell malignancies; clinical trials have shown it to be particularly effective at treating drug-resistant hairy cell leukemia (HCL).5
The RIT SS1P targets mesothelin, a surface protein found on mesotheliomas and many other solid tumors.6 SS1P contains the disulfide-stabilized variable fragment of an anti-mesothelin antibody attached to PE38. Clinical trials have shown modest improvements in patients with mesothelioma when used as a single agent,7,8 but dramatic tumor regression when combined with immunosuppressive drugs.9
PE-based RITs are thought to undergo an intoxication pathway similar to native PE, but utilize a different cell-surface target to mediate internalization (reviewed in ref 1). Cell- and target-specific factors, such as the number of target sites on the cell, the rate of target internalization, and the fate of the internalized target, all play roles in RIT cytotoxicity by affecting the uptake and trafficking of PE. Although these factors are target-dependent and difficult to adjust, the RITs themselves are amenable to improvements engineered to enhance their therapeutic characteristics. For example, the affinity of the RIT for its target can be increased in order to improve productive internalization. The Fv elements of both SS1P10 and HA2211 are affinity-enhanced variants of the original anti-mesothelin (SS) Fv and anti-CD22 (RFB4) Fv through the use of phage display selection. Additional attempts to improve PE-based RITs have included replacing the C-terminal ER-retention sequence of PE (REDL) with a more potent sequence (KDEL),12 generating a variant that is resistant to lysosomal proteases (PE-LR),13 improving the stability of RITs through engineering disulfide bonds,14-16 and decreasing the immunogenicity of RITs by identifying and removing B cell and T cell epitopes.17-20
Other steps in the PE intoxication pathway may also be targets for protein engineering. A prime example is the furin cleavage step, which separates the cell-binding from the catalytic fragments. The serine endoprotease furin, a membrane-bound proprotein convertase (reviewed in ref 21), traffics between the plasma membrane and the trans-Golgi network. At some point during this cycle, furin encounters and cleaves PE. In native PE, furin cleavage is essential for intoxication,22 but requires a further disulfide bond reduction to separate the cleaved fragments and generate an active toxin.23 The absolute requirement of furin cleavage in PE-based RITs has not been fully established. Recent work with PE-LR variant RITs indicates that furin cleavage is important, but is not essential for activity in some circumstances.13,24
Several studies have examined the polypeptide substrate preferences of furin and related proprotein convertases.25-29 When the PE furin cleavage site is compared to other furin substrate sequences, it meets the minimal amino acid requirements for furin cleavage (Arg at positions P4 and P1) but contains unfavorable residues at other positions. Analyses of specificity constants for various natural substrate sequences of furin indicated that the cleavage site of PE was a poor substrate for furin.26,27 One analysis in particular showed that the PE site sequence was the least efficiently cleaved out of 55 oligopeptide substrates tested, with a specificity constant greater than 1000-fold lower than the most efficiently cleaved furin substrate.26 These observations indicate that the furin site of PE is poorly optimized for cleavage efficiency, and suggests that mutations in the furin site may be able to dramatically enhance the cleavage rate. Furthermore, evidence suggests that enhancing furin cleavage may improve RIT cytotoxicity. Cells deficient in furin are resistant to PE intoxication, but can be made sensitive by transfection with furin cDNA30 or by pretreating PE-based RITs with furin.31
In this paper we describe experiments which examine the effect of mutations in the furin site on RIT cleavage, both in an in vitro assay and in cultured cells. The impact of these mutations on the cytotoxic activity of two RITs, the anti-CD22 HA22-LR and the anti-mesothelin SS1-LR/GGS, was also explored. We found that we could significantly and predictably alter furin cleavage through rational design of the PE furin cleavage site, but could not predict the effect of these mutations on RIT cytotoxicity. We conclude that the activity of HA22-LR, but not SS1-LR/GGS, may be enhanced through furin site mutations. These mutations may help to develop variants of HA22-LR that are more effective therapeutically, and are applicable to the design of bioconjugates utilizing a furin-cleavable polypeptide linker.
RESULTS AND DISCUSSION
Furin Cleavage Site.
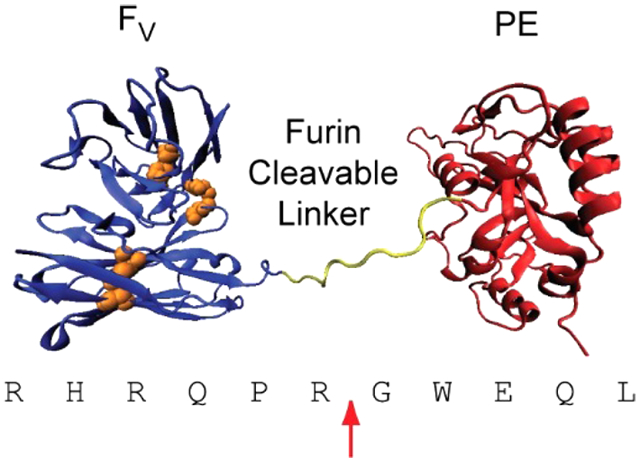
Furin recognizes a minimal sequence of R-X-X-R and hydrolyzes the peptide bond immediately C-terminal to the recognition sequence. In native PE, furin cleaves the peptide bond between Arg279 and Gly280. The current design of RITs has been engineered for resistance to lysosomal degradation through large deletions of protease-sensitive sites.13 These RITs have been given the LR (lysosome degradation resistant) designation. Although a significant fraction of native PE has been removed or replaced, LR RITs retain the furin cleavage site of PE as a cleavable linker between the antibody and toxin domains (Figure 1).
Figure 1. HA22-LR.

A. A hypothetical cartoon model structure of the PE-based RIT HA22-LR is shown. The Fv is colored blue, with cystines represented in orange. The catalytic fragment of PE is colored red, and the furin cleavage site is yellow. Residues 607–613 of native PE are disordered in the crystal structure and are not shown here. B. The furin cleavage site of HA22-LR is shown. The position and identity of amino acid residues at the native furin cleavage site of PE are shown. Residue numbering is based on the 613-residue native PE. Protease substrate position is based on standard Schechter and Berger nomenclature. Point mutations at the PE furin cleavage site, hypothesized to create a more efficient substrate for furin hydrolysis, are indicated.
Although furin cleavage is essential to native PE, RITs derived from PE have been extensively modified and may not behave identically to the native toxin. To confirm that furin cleavage is significant to the intoxication pathway of HA22-LR, we evaluated the cytotoxicity of HA22-LR against CA46 cells in the presence and absence of proprotein convertase inhibitor (PPCI), a potent inhibitor of furin.32 PPCI is well tolerated and can be incubated with the CA46 Burkitt’s lymphoma cell line at concentrations up to 10 μM without toxicity (data not shown). Higher concentrations were not evaluated, but have been previously shown to have minimal toxicity up to 50 μM.32 Cytotoxicity assays showed that the EC50 of HA22-LR against CA46 cells increased by approximately 4-fold in the presence of PPCI, from 0.28 ng/mL to 1.22 ng/mL (Figure S1). This suggests that furin cleavage is important, but not essential, for HA22-LR intoxication, and agrees with previous experiments showing that derivatives of HA22 (moxetumomab pasudotox) can function at a reduced level of activity without the furin site.13
We have previously identified the furin cleavage site in native PE as residues 274–284, corresponding to furin substrate positions P6–P5′.13 We analyzed the relative occurrence of residues at these 11 positions from 132 proteins with known furin cleavage sites (www.nuolan.net/substrates.html29) and compared the overall residue preferences to the residues in native PE. The PE site mirrors the most commonly occurring residues at positions P6, P4, and P1, but contains slightly less common residues at positions P3, P4′, and P5′, and much less common residues at positions P5, P2, P1′, P2′, and P3′ (Table S1). This information, along with furin cleavage preference data from past studies25-28 suggested potential point mutations that could enhance the cleavage efficiency of the PE furin site.
We selected one point mutation to attempt at each of ten positions in the furin cleavage site of HA22-LR (Figure 1). Positions near the site of hydrolysis were more likely to have a direct effect on cleavage efficiency, and were altered to conform to three major parameters: (1) commonly occurring residues at their respective sites (Table S1), (2) peptide cleavage efficiency,27 and (3) molecular modeling of the furin–substrate interaction.28 These three approaches made specific predictions concerning residue preference that were typically in close agreement for residues near the site of hydrolysis. Altered sites include P5 (H275R), P3 (Q277R), P2 (P278K), P1′ (G280S), P2′ (W281L), and P3′ (E283D). Positions P4 and P1 were not changed because they constitute the minimal furin recognition sequence and are the most commonly occurring residues at their respective sites.
Positions P6, P4′, and P5′ were farther from the site of hydrolysis and less likely to have a significant impact on cleavage efficiency. Position P6 is located at the N-terminal edge of the substrate in the core of the furin–substrate complex, and was predicted to favor positively charged residues by molecular modeling.28 An analysis of the most efficiently cleaved furin peptide substrates indicated that His occurred most frequently at P6.27 Although Arg was the most commonly occurring residue overall at P6, and was the existing residue in native PE, we chose to alter P6 to His in order to evaluate its impact on cleavage and toxicity. Furthermore, the presence of His at P6 limits the future possibility of creating a new cleavage site prior to the native site by introducing Arg at both P6 and P3. Positions P4′ and P5′ were predicted to favor small polar residues.28 An analysis of peptide cleavage efficiency indicated that the most efficiently cleaved substrates were most likely to contain Leu or Thr at position P4′ (P5′ was not evaluated).27 We thus altered positions P4′ (Q283T) and P5′ (L284S) to smaller polar residues. As a control, we also prepared a variant that cannot be cleaved by furin, in which the P1 Arg is changed to Gly (R279G). A detailed list of all variants is shown in Table S2. We prepared all mutants as described. No notable differences in yield, purity, or stability were observed during the purification.
Furin Cleavage.
We evaluated furin cleavage for each variant of HA22-LR (Figure 2A) using an in vitro time course of purified RITs incubated with the soluble catalytic domain of recombinant human furin. Samples at various time points were removed and analyzed by SDS-PAGE, followed by densitometry of the resulting bands. A ratio of the band density between cleaved and the whole RIT was plotted against time and fit to a four-parameter logistic model. Figure S2 provides examples of SDS-PAGE gels and curve fitting. From the fitted data, we determined the time required for furin to cleave half of the available RIT and compared each mutant to the value determined for HA22-LR. As expected, the only mutation to exhibit decreased cleavage efficiency was R279G, for which no cleavage was observed over a 24 h period. Two mutations, R274H and Q283T, were indistinguishable from HA22-LR. The mutations L284S (1.2-fold), E282D (1.6-fold), and Q277R (1.9-fold) each showed a slight enhancement in cleavage. Larger enhancements were observed for the mutations H275R (2.6-fold), G280S (7.8-fold), and P278K (12-fold). The largest increase in the cleavage rate for a point mutation was observed for W281L, which showed a 29-fold increase from HA22-LR.
Figure 2.
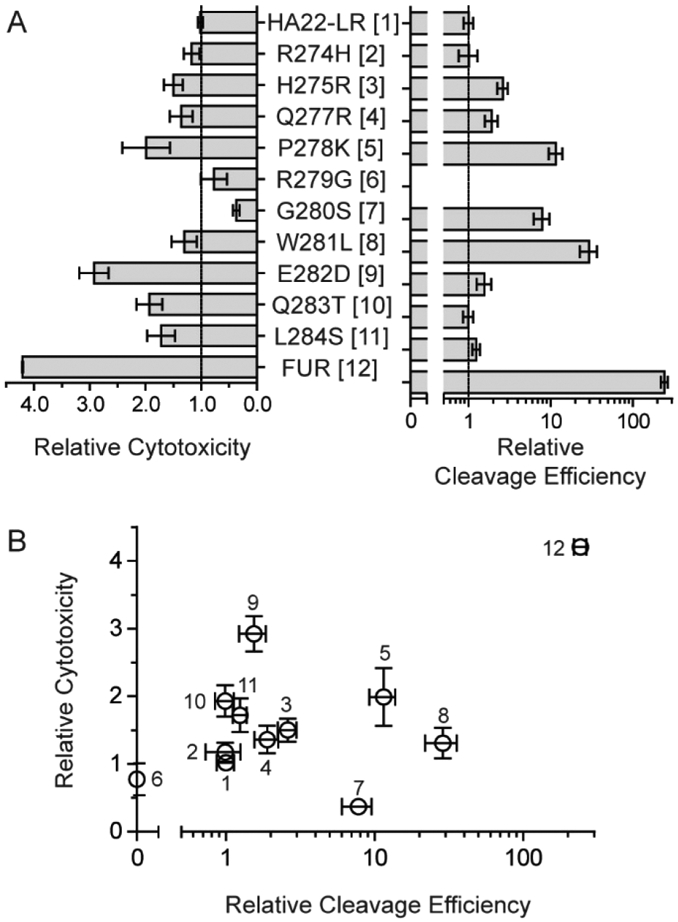
Properties of furin site mutants. A. Individual point mutations and the FUR multipoint combination mutant at the furin cleavage site of HA22-LR were compared using assays for both cytotoxicity and furin cleavage efficiency. Cytotoxicity is compared here as mean EC50 values of the mutants relative to the HA22-LR. Values are averaged from at least three different assays against the CA46 Burkitt’s lymphoma cell line. Error bars denote standard error of the mean values. The cleavage efficiency is compared here as the cleavage rate of mutants relative to HA22-LR using an in vitro assay followed by SDS-PAGE and band densitometry. Example data for individual RITs is presented in Figure S1. B. A correlation plot comparing relative cytotoxicity with relative cleavage efficiency. Individual proteins are numbered as indicated in panel A.
Cytotoxicity Assays.
Having demonstrated that we could enhance the cleavage rate of HA22-LR through mutations in the furin site, we next assessed the cytotoxicity of these variants on the CA46 cell line (Figure 2A). The resulting EC50 values from these experiments (n ≥ 3) were averaged and compared to HA22-LR. Two mutations, R279G (0.8-fold) and G280S (0.4-fold), showed decreased activity. The remainder of the point mutations typically showed modest enhancements in activity, ranging from 1.2-fold (R274H) to 2.0-fold (P278K). The only mutation to show a >2-fold increase in activity was E282D, which was nearly 3-fold more active than HA22-LR.
It is interesting to note that HA22-LR does not require furin cleavage for cytotoxicity, as shown by the activity of the R279G mutant. This mutation prevents furin cleavage of the RIT, and although it diminishes the cytotoxicity of HA22-LR slightly it does not abolish its cytotoxic activity. This indicates that HA22-LR is able to bypass furin cleavage during intoxication, and contrasts dramatically with the behavior of the anti-mesothelin RIT SS1-LR, which is inactivated by the R279G mutation.24 This stark difference may be due to the different targets of the two RITs, which likely play major roles in the internalization, intracellular trafficking, and cytotoxicity of the molecules. Research is ongoing to explore how the target of a RIT influences its intracellular trafficking itinerary and eventual cytotoxicity.
Correlation Plot.
We next compared the cytotoxicity and cleavage efficiency of each mutant in a correlation plot (Figure 2B). Using Spearman’s rank correlation test, the general trend of the plot has a positive slope (ρ(11) = 0.145), but is not statistically significant (P = 0.673). Thus, although the tendency is for greater cleavage efficiency to result in a more active protein, the two variables cannot be said to have a direct relationship. Certain mutations, in particular, are badly out of line with a direct correlation. For instance, the mutation G280S enhances cleavage efficiency by 7.8-fold, but diminishes cytotoxicity by 2.7-fold. Similarly, the W281L mutation shows a large cleavage enhancement of 29-fold, but has a minimal effect on cytotoxicity of 1.3-fold.
These observations indicate that the relationship between furin cleavage and cytotoxicity is more complex than a simple linear function, and must involve competing priorities with different residue preferences. Several possible explanations for this complexity can be postulated. It is possible that enhanced furin cleavage may be detrimental if the RIT is cleaved by extracellular furin prior to internalization. If this happens the cytotoxic domain of PE-LR RITs like HA22-LR would be released from the cell-binding domain, effectively inactivating the RIT by separating the domains prior to internalization. Therefore, enhancing furin cleavage to the point where extracellular cleavage becomes significant may decrease productive internalization. To our knowledge, there has been no demonstration of extracellular RIT cleavage by furin, but the presence of furin at the cell surface and in the extracellular space suggests that it is possible. Although data suggests that cell surface levels of furin are low, anthrax toxin protective antigen must be activated by furin cleavage prior to internalization.33 This indicates that a sufficient level of furin may exist outside of cells to inactive RITs based on PE-LR. Native PE and PE38-based RITs are not susceptible to this potential problem because the cell-binding and cytotoxic domains are joined by a disulfide bond in addition to the furin cleavable linker.
Another possible explanation is that the residue composition of the furin site is important for reasons other than cleavage, and it cannot be altered without some negative impact. Perhaps the newly revealed N-terminus of the furin-cleaved cytotoxic domain is involved in intracellular routing or toxin stability. This possibility is supported by the behavior of the first three N-terminal residues of the cytotoxic domain after furin cleavage, positions G280, W281, and E282. The mutation G280S shows an unexpected negative correlation between furin cleavage (7.8-fold) and cytotoxicity (0.4-fold), and the mutation W281L shows a poor positive correlation (29-fold vs 1.3-fold). The E282D mutant demonstrates a small increase in furin cleavage (1.6-fold) and a larger increase in cytotoxicity (2.9-fold). These three mutations show no clear relationship between cleavage and cytotoxicity, and the observed changes in cytotoxicity are likely to be the result of steps other than furin cleavage during the intoxication process. We are continuing to explore the intoxication pathway of RITs to identify additional contributing factors.
Combination Mutant.
Although furin cleavage did not directly correlate with cytotoxicity, the majority of the mutations had a positive influence on the activity of HA22-LR. To evaluate the possibility that a greater enhancement in cytotoxicity was attainable, we chose to combine all point mutations that did not diminish activity into a single mutant. This combination mutant, called HA22-LR/FUR, contained 8 individual mutations at the furin cleavage site: R274H, H275R, Q277R, P278K, W281L, E282D, Q283T, and L284S. The mutations R279G and G280S were not included because they exhibited reduced cytotoxicity. HA22-LR/FUR showed a remarkable enhancement in cleavage efficiency of 242-fold, and an appreciable enhancement in cytotoxicity of 4.2-fold on CA46 cells (Figure 2A).
To confirm that furin cleavage was enhanced in living cells, we evaluated a time course of CA46 cells treated with HA22-LR (Figure 3A), HA22-LR R279G (Figure 3B), and HA22-LR/FUR (Figure 3C). Lysates from treated cells were examined for full length and cleaved RITs by Western blot. Bands representative of full-length, reduced, and furin-cleaved RIT are indicated. Each band was evaluated by densitometry, and the cleaved fraction of total protein was plotted against time for each of the three RITs (Figure 3D). As expected, the HA22-LR R279G mutant showed no observable cleavage, HA22-LR showed a small fraction of cleaved protein, and HA22-LR/FUR showed a greatly enhanced fraction of cleaved protein. This supports the validity of the in vitro furin cleavage data for HA22-LR/FUR, and is consistent with our hypothesis that rationally designed furin site mutations can significantly enhance the rate of furin cleavage. In addition, the R279G mutation eliminates observable cleavage of HA22-LR, and indicates that other proteases cannot compensate for lack of furin cleavage.
Figure 3.
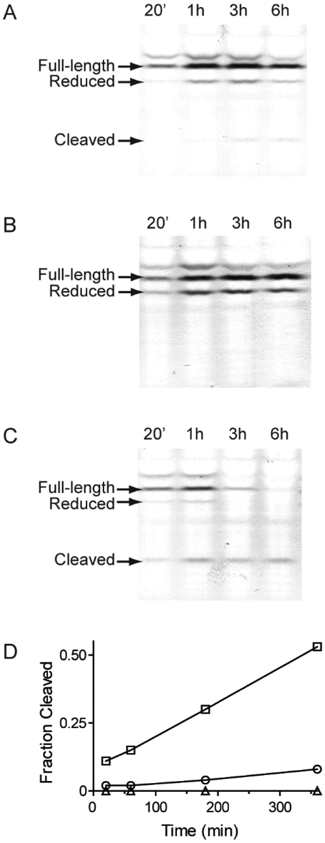
Internalization and cleavage of HA22-LR/FUR. We evaluated whole cell lysates from CA46 cells incubated for various time intervals with HA22-LR (A), HA22-LR R279G (B), or HA22-LR/FUR (C) by nonreducing Western blot for PE. Full-length, reduced, and furin-cleaved fragments are indicated. The intensity of each band was quantified and the ratio between the furin-cleaved band intensity and the total intensity of all bands at each time point was plotted over time (D). HA22-LR (open circles), HA22-LR R279G (open triangles), and HA22-LR/FUR (open squares) are shown.
Further analysis of the cytotoxicity of HA22-LR/FUR on a selection of CD22-positive B cell lines demonstrated that enhanced cytotoxicity was retained across all lines tested (Figure 4). The magnitude of the enhancement, however, was inconsistent. The greatest change was observed in the Daudi cell line, which showed a 7.2-fold increase in cytotoxicity relative to HA22-LR. The 4.3-fold enhancement seen in the Ramos line was similar to the 4.2-fold enhancement observed in CA46. The KOPN-8 (1.9-fold) and Raji (1.5-fold) cell lines showed smaller enhancements. These observations are consistent with our initial hypothesis that enhancing furin cleavage can increase RIT cytotoxicity, but the differences between cell lines supports the idea of a greater underlying complexity.
Figure 4.
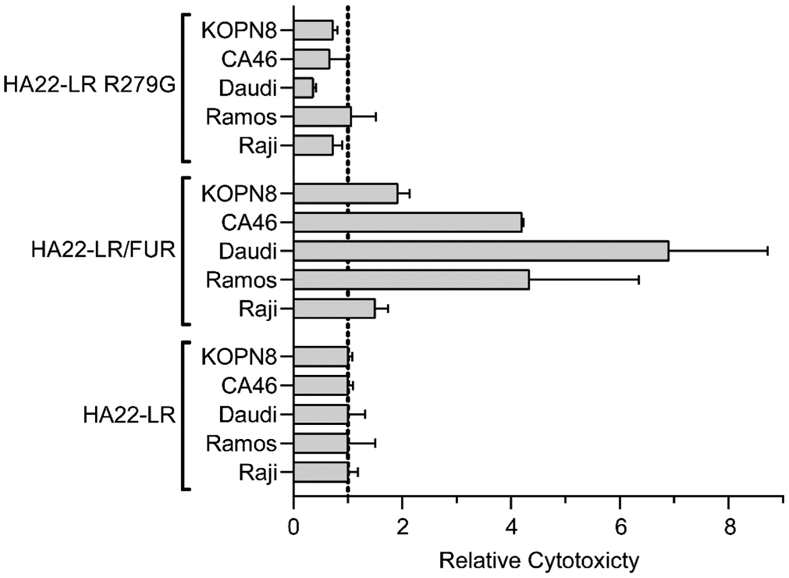
Relative cytotoxicity of HA22-LR/FUR We evaluated HA22-LR, HA22-LR R279G, and HA22-LR/FUR for cytotoxicity on several CD22-positive cell lines (KOPN8, CA46, Daudi, Ramos, and Raji). The relative mean EC50 values with standard error (ng/mL) from at least three separate experiments are presented here, normalized to HA22-LR.
In addition to HA22-LR/FUR, we also evaluated the activity of HA22-LR R279G on these cell lines. We found a generally consistent trend of diminished activity except for the Ramos cell line, which showed similar activity to HA22-LR (Figure 4). As discussed above, these results agree that furin cleavage is not an absolute requirement for intoxication, although in all cases the enhanced cleavage of HA22-LR/FUR consistently leads to enhanced cytotoxicity.
The level of furin expression in these cell lines may influence RIT cytotoxicity. We assessed furin expression by Western blot of lysates from the CA46, Raji, Ramos, and Daudi cell lines (Figure 5). The level of furin detected was visually similar in all cell lines. We confirmed this conclusion using band densitometry. Relative to the actin loading control, CA46 showed the highest levels of furin expression. The remaining cell lines showed slightly reduced expression levels compared to CA46, ranging from Raji (72% of CA46 levels) to Daudi (90% of CA46 levels). Although these differences are relatively small, a comparison of furin expression to the cytotoxicity results shows that the two parameters do not correlate.
Figure 5.

Furin expression levels. We evaluated lysates of the cell lines CA46, Raji, Ramos, and Daudi for furin by Western blot. Lysates were probed with antibodies for furin (A) and actin (B). The intensity of each band was determined and the ratio of the band densities for each cell line was normalized to CA46 to estimate relative expression levels (C).
Cytotoxicity toward HCL Patient-Derived Cells.
To determine whether the combination mutant would be effective against cells obtained directly from patients, we analyzed cells from six patients with HCL. In these assays, we compared the parent molecule, moxetumomab pasudotox (HA22), to variants HA22-LR, HA22-LR/FUR, and HA22-LR R279G. The resulting data from trials using both an ATP-depletion viability assay and a protein synthesis inhibition assay are summarized in Table 1. High levels of HA22-LR/FUR activity were observed for all patient cell populations tested, but the relative activity was variable. Relative to moxetumomab pasudotox, HA22-LR/ FUR exhibited enhanced activity in HH01 and RD27, similar activity in BL02, C117, and HH20, and reduced activity against C186. Relative to HA22-LR, HA22-LR/FUR exhibited similar or enhanced activity against all patient cells except for C186. Comparing the ATP-depletion assay, statistically significant differences (p < 0.001 in a two-tailed unpaired t test) were observed between HA22-LR and HA22-LR/FUR in patient cells C117, HH20, HH01, and C186. Interestingly, HA22-LR R279G was consistently much less active than the other RITs tested, frequently showing reductions in activity of 100-fold or greater. This result indicates that furin cleavage may be essential in HCL patient cells, suggesting an intrinsic difference in the intoxication pathway between these cells and the cell lines we evaluated.
Table 1.
Cytotoxicity toward HCL Patient Cells
| EC50 ± SD (ng/mL) | |||||
|---|---|---|---|---|---|
| patient | Moxetumomab pasudotox (HA22) | HA22-LR | HA22-LR/FUR | HA22-LR R279G | assay type |
| BL02 | 0.130 ± 0.003 | 0.150 ± 0.006 | 0.147 ± 0.003 | <100 | ATP |
| 0.368 ± 0.118 | 0.519 ± 0.135 | 0.402 ± 0.078 | 417 ± 100 | Protein synthesis | |
| C117*** | 0.140 ± 0.004 | 0.229 ± 0.007 | 0.128 ± 0.003 | >1000 | ATP |
| 0.142 ± 0.018 | 0.153 ± 0.026 | 0.132 ± 0.001 | 468 ± 46.3 | Protein synthesis | |
| HH20*** | <0.1 | 0.232 ± 0.017 | 0.068 ± 0.023 | <100 | ATP |
| <0.1 | <0.1 | 0.094 ± 0.035 | <100 | Protein synthesis | |
| RD27 | 0.784 ± 0.028 | 0.368 ± 0.040 | 0.352 ± 0.012 | >1000 | ATP |
| 0.532 ± 0.186 | 0.315 ± 0.092 | 0.237 ± 0.014 | 512 ± 352 | Protein synthesis | |
| HH01*** | 0.352 ± 0.002 | 0.449 ± 0.013 | 0.306 ± 0.005 | >1000 | ATP |
| 0.347 ± 0.016 | 0.441 ± 0.103 | 0.239 ± 0.039 | >1000 | Protein synthesis | |
| C186*** | 0.416 ± 0.019 | 0.629 ± 0.018 | 0.997 ± 0.031 | >1000 | ATP |
| 0.456 ± 0.035 | 0.780 ± 0.161 | 0.872 ± 0.115 | >1000 | Protein synthesis | |
p < 0.001 from an unpaired two-tailed t test comparing the ATP-depletion assay results of HA22-LR and HA22-LR/FUR.
Anti-Mesothelin RIT.
Thus far, our experiments have been confined to the anti-CD22 RIT HA22-LR. To explore whether furin site mutations might be applicable to other PE-based RITs, we tested the same point mutations on the anti-mesothelin RIT SS1-LR/GGS.24 SS1-LR/GGS is constructed similarly to HA22-LR, but contains an anti-mesothlin Fv in place of the anti-CD22 Fv in HA22-LR and a Gly-Gly-Ser linker following the furin cleavage site. We initially evaluated the cytotoxicity of furin site mutants on the activity of SS1-LR/GGS against the mesothelin-positive cervical carcinoma cell line KB31 (Figure 6C). Our results showed that no mutation enhanced the activity of the RIT, and several mutations, most notably P278K, showed a significant decrease in cytotoxicity. These results were remarkably different from our observations of HA22-LR. To confirm these results, we further tested lung adenocarcinoma lines L55 (Figure 6B) and NCI-H322M (Figure 6B). The NCI-H322M line showed a cytotoxicity profile nearly identical to that of KB31. The L55 line showed a similar profile, but did show one mutation, E282D, with enhanced activity of 1.7-fold.
Figure 6.
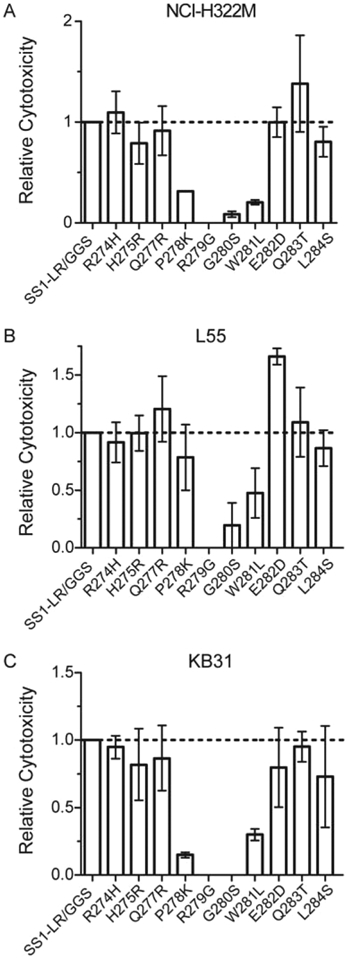
SS1-LR/GGS furin site mutants. We evaluated individual point mutations in the furin cleavage site of SS1-LR/GGS for cytotoxicity against three mesothelin-positive cell lines: KB31, L55, and NCI-H322M. Mean EC50 values (ng/mL) from at least three separate experiments are presented here relative to SS1-LR/GGS.
These results indicate that SS1-LR/GGS is not freely amenable to changes in the furin cleavage site, and highlight the importance of the RIT target to its cytotoxic behavior. Although the activity of HA22-LR on CD22-positive cell lines can be improved by mutations that enhance furin cleavage, SS1-LR/GGS fails to show a similar effect on mesothelin-positive lines. By extension, the data suggest a fundamental difference in the intoxication pathway between anti-CD22 and anti-mesothelin RITs. We speculate that the cause of this difference must lie in the initial stages of receptor-mediated internalization and intracellular trafficking of the RIT. Research into the intoxication pathway of another PE-based RIT, LMB2, has shown that differences in the intracellular route can significantly impact the cytotoxicity.34 The targets themselves are quite distinct. CD22 is a large transmembrane glycoprotein that shows constitutive endocytic recycling,35 while mesothelin is a glyocophosphatidylinositol (GPI)-linked protein6 and thus is internalized and sorted independently of cytosolic interactions.36 These differences between targets may significantly impact RIT intoxication. Research is under way to explore the differences between RIT target receptors, but further development of furin site mutations in the anti-mesothelin RIT SS1-LR/GGS is not warranted.
CONCLUSIONS
In the experiments presented here, we explored the hypotheses that (1) we could enhance the furin cleavage efficiency of PE-based RITs through rationally designed mutations in the furin-cleavable linker and (2) these mutations could enhance RIT cytotoxicity by increasing the rate of a restrictive step during intoxication. The data demonstrate that we can greatly enhance the efficiency of furin cleavage by rational design of the target site, but cannot predict a priori the cytotoxic effects of such mutations. We conclude that the role of furin in the intoxication pathway of PE-based RITs is more complex than a simple cleavage/activation, and more research is needed to determine the role of furin in the intracellular trafficking of PE and its derivatives. Mutations in the furin linker of the anti-CD22 RIT HA22-LR, however, are candidates for additional preclinical development. Future directions include in vivo mouse studies of the HA22-LR furin-site variants to evaluate antitumor activity, nonspecific toxicity, and pharmacokinetics. Furthermore, our data informs the design of furin-cleavable linkers for all bioconjugates and suggests that cleavage parameters may require optimization.
EXPERIMENTAL PROCEDURES
Proteins.
HA22-LR, SS1-LR, and all derivative mutants were expressed in Escherichia coli BL21(λDE3) from vectors expressing either the heavy chain polypeptide of the Fv (VH) joined to PE or the light chain polypeptide of the Fv (VL). The two polypeptides were subsequently refolded and purified as described.37 All RITs were prepared by the authors in the Laboratory of Molecular Biology, National Cancer Institute (NCI; Bethesda, MD). Mutations were generated either by Quikchange site-directed mutagenesis (Stratagene, La Jolla, CA) with primers from Lofstrand Laboratories Limited (Gaithersburg, MD), or by direct gene synthesis (GenScript USA Inc., Piscataway, NJ).
Cell Lines.
Several CD22-positive human-derived cell lines were used in this study: CA46, Raji, Ramos, Daudi, and KOPN-8. Mesothelin-positive cell lines KB31, L55, and NCI-H322 M were also used. CD22-positive lines were cultured in RPMI-1640 base medium supplemented with 1 mM sodium pyruvate, and mesothelin-positive lines were cultured in DMEM base medium. All lines were cultured at 37 °C with 5% CO2 in medium supplemented with 10% FBS, 2 mM L-glutamine, 100 U penicillin, and 100 μg streptomycin (Life Technologies).
In Vitro Furin Cleavage Assay.
Furin (100 U/mL; New England Biolabs, Inc., Ipswich, MA) was incubated at 37 °C with individual RITs (0.25 mg/mL) in furin cleavage buffer (100 mM MES, 5 mM CaCl2, 50 mM NaCl, pH 5.5). At various time points, samples were removed, diluted 2-fold in 2× SDS tris-glycine sample buffer, heated to 85 °C for 2 min, and analyzed by nonreducing SDS-PAGE using Novex 4–20% acrylamide tris-glycine 1.5 mm 15-well gels (Life Technologies). The gels were stained with SimplyBlue Safestain (Invitrogen Corporation) and scanned into TIF files. The densities of the full-length and cleaved bands were quantified with ImageJ software.38 The ratio of the cleaved band to the full-length band was plotted against time and fit to a two-exponent association function using GraphPad PRISM software.
Cytotoxicity Assays.
Viability of cell lines treated with immunotoxins was measured using the Cell Counting Kit-8 WST-8 assay (Dojindo Molecular Technologies, Inc.). Cells (2000 cells/well) were plated in 96-well plates, left overnight to adhere, and incubated with varying concentrations of RITs for 72 h at a final volume of 0.2 mL. For assays using PPCI (EMD Millipore, Billerica, MA), PPCI was added to the culture media immediately prior to plating the cells and included with the RITs to maintain the appropriate concentration. At the end of the incubation period, 10 μL of the CCK-8 reagent was added to each well and the plates were incubated at 37 °C until the wells with the maximum absorbance at 450 nm reached values of ~1 OD. Values were normalized between controls of cycloheximide (10 μg/mL) and buffer (Dulbecco’s phosphate buffered saline without Ca and Mg (D-PBS) containing 0.2% human serum albumin), then fit to a sigmoidal equation with variable slopes for the plateau, baseline, and Hill slope using GraphPad PRISM software. The equation was subsequently used to interpolate the concentration of RIT, which reduced cell viability to the 50% level (EC50). Cells from patients with HCL were similarly evaluated using a protein synthesis inhibition assay36 and an ATP depletion assay39 as previously described.
RIT Internalization Assay.
CA46 suspension cells (2 × 106/well) were plated in a 6-well plate with 2 mL medium containing 0.1 μg/mL of the RIT to be evaluated. Cells were incubated at 37 °C for various time intervals, after which the cells were harvested and washed consecutively with 1 mL cold D-PBS, 1 mL cold stripping buffer (1 mg/mL BSA in 0.2 M glycine, pH 2.5), and 1 mL cold D-PBS. Cells were subsequently lysed with 100 μL RIPA buffer (150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% Na deoxycholate, 0.1% SDS, 50 mM Tris-Cl, pH 8.0) containing a protease inhibitor cocktail (Sigma, St. Louis, MO). Samples were analyzed by nonreducing Western blot using a rabbit anti-PE38 polyclonal antibody.
Furin Western Blots.
RIPA buffer lysates of the cell lines CA46, Raji, Ramos, and Daudi were evaluated for furin expression by Western blot. Blots were probed with rabbit anti-furin polyclonal antibody H-220 (Santa Cruz Biotechnology Inc., Dallas, TX) and loading control rabbit anti-actin polyclonal antibody (Thermo Fisher Scientific Inc., Waltham, MA). Bands were detected using an alkaline phosphatase-conjugated secondary antibody and the NBT/BCIP chromogenic substrate (Life Technologies).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and in part by the Fisher College of Science and Mathematics, Towson University.
ABBREVIATIONS
- D-PBS
Dulbecco’s phosphate buffered saline without Ca and Mg
- HCL
hairy cell leukemia
- PE
Pseudomonas exotoxin A
- PE38
a 38 kDa fragment of PE
- PPCI
proprotein convertase inhibitor
Footnotes
Supporting Information
Amino acid preferences at furin cleavage sites, amino acid sequences of proteins used in the experiments described in this paper, cytotoxicity of HA22-LR on CA46 cells in the presence and absence of PPCI furin inhibitor, and example analyses comparing furin cleavage and cytotoxicity between variant proteins described in this paper. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.5b00190.
The authors declare no competing financial interest.
REFERENCES
- (1).Weldon JE, and Pastan I (2011) A guide to taming a toxin -recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 278, 4683–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kreitman RJ, and Pastan I (2011) Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res 17, 6398–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Decker T, Oelsner M, Kreitman RJ, Salvatore G, Wang QC, Pastan I, Peschel C, and Licht T (2004) Induction of caspase-dependent programmed cell death in B-cell chronic lymphocytic leukemia by anti-CD22 immunotoxins. Blood 103, 2718–2726. [DOI] [PubMed] [Google Scholar]
- (4).Alderson RF, Kreitman RJ, Chen T, Yeung P, Herbst R, Fox JA, and Pastan I (2009) CAT-8015: a second-generation Pseudomonas exotoxin A-based immunotherapy targeting CD22-expressing hematologic malignancies. Clin. Cancer Res 15, 832–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, Fitzgerald DJ, Lechleider R, and Pastan I (2012) Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J. Clin. Oncol 30, 1822–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hassan R, Bera T, and Pastan I (2004) Mesothelin: a new target for immunotherapy. Clin. Cancer Res 10 (12 Pt 1), 3937–3942. [DOI] [PubMed] [Google Scholar]
- (7).Kreitman RJ, Hassan R, Fitzgerald DJ, and Pastan I (2009) Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin. Cancer Res 15, 5274–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, and Pastan I (2007) Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res 13, 5144–5149. [DOI] [PubMed] [Google Scholar]
- (9).Hassan R, Miller AC, Sharon E, Thomas A, Reynolds JC, Ling A, Kreitman RJ, Miettinen MM, Steinberg SM, Fowler DH, et al. (2013) Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci. Transl. Med 5, 208ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chowdhury PS, and Pastan I (1999) Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat. Biotechnol 17, 568–572. [DOI] [PubMed] [Google Scholar]
- (11).Salvatore G, Beers R, Margulies I, Kreitman RJ, and Pastan I (2002) Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin. Cancer Res 8, 995–1002. [PubMed] [Google Scholar]
- (12).Seetharam S, Chaudhary VK, FitzGerald D, and Pastan I (1991) Increased cytotoxic activity of Pseudomonas exotoxin and two chimeric toxins ending in KDEL. J. Biol. Chem 266, 17376–17381. [PubMed] [Google Scholar]
- (13).Weldon JE, Xiang L, Chertov O, Margulies I, Kreitman RJ, FitzGerald DJ, and Pastan I (2009) A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 113, 3792–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Liu W, Onda M, Kim C, Xiang L, Weldon JE, Lee B, and Pastan I (2012) A recombinant immunotoxin engineered for increased stability by adding a disulfide bond has decreased immunogenicity. Protein Eng. Des. Sel 25, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Brinkmann U, Reiter Y, Jung SH, Lee B, and Pastan I (1993) A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc. Natl. Acad. Sci. U.S.A 90, 7538–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Reiter Y, Brinkmann U, Webber KO, Jung SH, Lee B, and Pastan I (1994) Engineering interchain disulfide bonds into conserved framework regions of Fv fragments: improved biochemical characteristics of recombinant immunotoxins containing disulfide-stabilized Fv. Protein Eng. 7, 697–704. [DOI] [PubMed] [Google Scholar]
- (17).Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ, and Pastan I (2011) Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc. Natl. Acad. Sci. U.S.A 108, 5742–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Liu W, Onda M, Lee B, Kreitman RJ, Hassan R, Xiang L, and Pastan I (2012) Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc. Natl. Acad. Sci. U.S.A 109, 11782–11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mazor R, Vassall AN, Eberle JA, Beers R, Weldon JE, Venzon DJ, Tsang KY, Benhar I, and Pastan I (2012) Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc. Natl. Acad. Sci. U.S.A 109, E3597–E3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Mazor R, Eberle JA, Hu X, Vassall AN, Onda M, Beers R, Lee EC, Kreitman RJ, Lee B, Baker D, et al. (2014) Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc. Natl. Acad. Sci. U.S.A 111, 8571–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Thomas G (2002) Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol 3, 753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ogata M, Chaudhary VK, Pastan I, and FitzGerald DJ (1990) Processing of Pseudomonas exotoxin by a cellular protease results in the generation of a 37,000-Da toxin fragment that is translocated to the cytosol. J. Biol. Chem 265, 20678–20685. [PubMed] [Google Scholar]
- (23).McKee ML, and FitzGerald DJ (1999) Reduction of furin-nicked Pseudomonas exotoxin A: an unfolding story. Biochemistry 38, 16507–16513. [DOI] [PubMed] [Google Scholar]
- (24).Weldon JE, Xiang L, Zhang J, Beers R, Walker DA, Onda M, Hassan R, and Pastan I (2013) A recombinant immunotoxin against the tumor-associated antigen mesothelin reengineered for high activity, low off-target toxicity, and reduced antigenicity. Mol. Cancer Ther 12, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Duckert P, Brunak S, and Blom N (2004) Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel 17, 107–112. [DOI] [PubMed] [Google Scholar]
- (26).Remacle AG, Shiryaev SA, Oh ES, Cieplak P, Srinivasan A, Wei G, Liddington RC, Ratnikov BI, Parent A, Desjardins R, et al. (2008) Substrate cleavage analysis of furin and related proprotein convertases. A comparative study. J. Biol. Chem 283, 20897–20906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Izidoro MA, Gouvea IE, Santos JA, Assis DM, Oliveira V, Judice WA, Juliano MA, Lindberg I, and Juliano L (2009) A study of human furin specificity using synthetic peptides derived from natural substrates, and effects of potassium ions. Arch. Biochem. Biophys 487, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tian S (2009) A 20 residues motif delineates the furin cleavage site and its physical properties may influence viral fusion. Biochem. Insights 2, 9–20. [Google Scholar]
- (29).Tian S, Huang Q, Fang Y, and Wu J (2011) FurinDB: A database of 20-residue Furin cleavage site motifs, substrates and their associated drugs. Int. J. Mol. Sci. 12, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Moehring JM, Inocencio NM, Robertson BJ, and Moehring TJ (1993) Expression of mouse furin in a Chinese hamster cell resistant to Pseudomonas exotoxin A and viruses complements the genetic lesion. J. Biol. Chem 268, 2590–2594. [PubMed] [Google Scholar]
- (31).Chiron MF, Fryling CM, and FitzGerald D (1997) Furin-mediated cleavage of Pseudomonas exotoxin-derived chimeric toxins. J. Biol. Chem 272, 31707–31711. [DOI] [PubMed] [Google Scholar]
- (32).Becker GL, Lu Y, Hardes K, Strehlow B, Levesque C, Lindberg I, Sandvig K, Bakowsky U, Day R, Garten W, et al. (2012) Highly potent inhibitors of proprotein convertase furin as potential drugs for treatment of infectious diseases. J. Biol. Chem 287, 21992–22003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gawlik K, Remacle AG, Shiryaev SA, Golubkov VS, Ouyang M, Wang Y, and Strongin AY (2010) A femtomol range FRET biosensor reports exceedingly low levels of cell surface furin: implications for the processing of anthrax protective antigen. PLoS One 5, e11305. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (34).Tortorella LL, Pipalia NH, Mukherjee S, Pastan I, Fitzgerald D, and Maxfield FR (2012) Efficiency of immunotoxin cytotoxicity is modulated by the intracellular itinerary. PLoS One 7, e47320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).O’Reilly MK, Tian H, and Paulson JC (2011) CD22 is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B cells. J. Immunol 186, 1554–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Fujita M, and Kinoshita T (2012) GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim. Biophys. Acta 1821, 1050–1058. [DOI] [PubMed] [Google Scholar]
- (37).Pastan I, Beers R, and Bera TK (2004) Recombinant immunotoxins in the treatment of cancer. Methods Mol. Biol 248, 503–518. [DOI] [PubMed] [Google Scholar]
- (38).Schneider CA, Rasband WS, and Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bera TK, Onda M, Kreitman RJ, and Pastan I (2014) An improved recombinant Fab-immunotoxin targeting CD22 expressing malignancies. Leuk. Res 38, 1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.