

Abstract
Humans share a core intestinal microbiome and yet human microbiome differs by genes, species, enterotypes (ecology), and gene count (microbial diversity). Achievement of microbiota metagenomic analysis has revealed that the microbiome gene count is a key stratifier of health in several immune disorders and clinical conditions. We review here the progress of the metagenomic pipeline analysis, and how this has allowed us to define the host–microbe symbiosis associated with a healthy status. The link between host–microbe symbiosis disruption, the so-called dysbiosis and chronic diseases or iatrogenic conditions is highlighted. Finally, opportunities to use microbiota modulation, with specific nutrients and/or live microbes, as a target for personalized nutrition and therapy for the maintenance, preservation, or restoration of host–microbe symbiosis are discussed.
Introduction
Homo sapiens are essentially symbiotic organisms. Humans are born virtually sterile and they meet the microbial world and develop a microbiota at the same time as they develop their immune system. A microbiota is defined as an “assemblage of microorganisms (all the bacteria, archaea, eukaryotes, and viruses) present in a defined environment” and is found in all multicellular organisms including plants. The synonymous term microbiome describes either the collective genomes of the microorganisms that reside in an environmental niche or the microorganisms themselves. The microbiota contributes to trophic functions, metabolism, barrier function, immune stimulation, and signalization to virtually all organs of the body.
The intestinal innate and adaptive immunity coordinate and interact with the symbionts contributing to intestinal homeostasis through establishment of a mutually beneficial relationship by tolerating symbiotic microbiota, and retaining the ability to exert proinflammatory response towards invasive pathogens. Maintenance of symbiosis is essential to health and well-being. Disruption of gut microbiota increases susceptibility to infection and sepsis through several mechanisms, including (a) allowing for expansion of pathogenic intestinal bacteria, (b) priming the immune system for a robust pro-inflammatory response, and (c) decreasing production of beneficial microbial products such as short-chain fatty acids.1 If there is disruption at the level of barrier immunity, there will be loss of immune homeostasis with an associated risk of immune or inflammatory disorders. Therefore, it is essential to understand symbiosis and develop the tools to monitor and fine-tune it to prevent or mitigate the risks, or even for use in microbiome-targeted therapeutics.
There are 100 trillion bacteria in the microbial ecosystem of the human body. Interactions with these microorganisms take place daily at the level of the skin, in the urogenital tract, mouth, pharynx, and respiratory system, and the digestive tract which contains by far, the largest density and diversity of microorganisms. Study of the human intestinal microbiota has been neglected for many years, while it is at the interface between ingested food and the gut epithelium and is in contact with the 1st pool of immune cells and the 2nd pool of neural cells of the body. It is now gaining recognition as a true organ that plays a major role in health and disease. By culture, it was possible to assess only about 30% of the dominant intestinal microbiota,2–4 a very limited perspective. Therefore, culture-independent tools were developed to enable an in-depth evaluation of the microbiota. With shotgun metagenomics, knowledge about the microbiome has been advancing quite rapidly. Each individual has 23,000 human genes and about 600,000 microbial genes. Thus, the human component contributes only to less than 4% of the total hologenome.5–7 The aim of this review is to offer an updated vision of the human intestinal microbiome, and describe the transition from symbiosis to dysbiosis and its implications in human disease.
Introduction to metagenomics
The metagenome is the name given to the combined genomes of all dominant microbes within the human intestinal tract. Knowledge is built up by the extraction of total DNA from human intestinal contents, mainly from stool samples but also from biopsies, and applying whole genome shotgun sequencing, assembling and annotating the genes and then according to the initial objectives of MetaHIT8 in Europe and the Human Microbiome Project (HMP)7 in the USA, to create a catalog of these microbial genes from the intestinal tract and move towards rapid gene counting methodologies. The reference gene catalog which has been constructed, highlights both the core metagenome and rare genes.5,9 Bioinformatic tools were designed to reconstruct genomes of microbial species present in complex metagenomic samples using a method, that clustered genes by co-abundance across series of metagenomic samples without the need for reference sequences.10 A collection of high-quality genomes is hence being constructed from essentially yet uncultured “metagenomic species”.
The construction of the reference gene catalog led to an initial description of 3.3 million gut genes from 124 European subjects in 2010 which was expanded to 10 million gut genes by 2014, corresponding to 1500 metagenomic species (MGS) from 1267 subjects from Europe, China and the USA.9 Constantly expanding, this catalog of genes is now a major asset for future metagenomic profiling. Owing to inter-individual variability, the number of genes increases with the number of subjects analysed. Yet more than 50% of the cohorts exhibited a small number of the essentially most common microorganisms—a core microbiome—which may be the most clinically relevant in relation to common diseases, and these were captured after the analysis of the first 100 individuals. Animal studies in 286 pigs11 and in 300 chickens are currently underway and it is evident that there is also a massive diversity of genes in these ecosystems, with 7.7 and 9.7 million genes, respectively, corresponding to 719 and 2300 MGS, respectively.
Although proportions of a species vary greatly between individuals, genes of a species will precisely co-vary in abundance in each individual. Just clustering genes based on co-variation will allow one to construct pools of genes that will correspond to genomes.10 In 2014, we described 741 large MetaGenomic Units (MGUs) of more than 700 genes and were able to demonstrate that they correspond to bacterial genomes of MGS, 85% of which are as yet unknown (have not been represented in culture collections). This work also revealed that over 6500 small MGUs correspond to phages, plasmids, or CRISPR elements that are most commonly associated with one single microbial genome.10
Quantitative metagenomics pipeline
The quantitative metagenomics pipeline starts with recommendations for stool sample collection (e.g., in clinical trials) and provision of collection kits, total DNA extraction, which is now in the process of being automated, and high throughput sequencing generating millions of short-sequence tags that may be used to directly map onto the reference gene catalog rather than to reconstruct genes.9 This mapping allows the counting of genes and generation of gene abundance profiles, which can be directly translated into MGS abundance. Gene profiles can also be mapped onto metabolic pathways.12 Finally, when these are integrated with human clinical data, with response to treatment or diets, the relevant microbial players can be identified and prediction models built and tested.10 A bar code appearance is produced which illustrates the occurrence of genes, where columns depict an individual and rows correspond to vital species. Therefore, it is imperative that the data generated in each of many large projects involved in human metagenome research be optimally comparable. From 2011 to 2015, the International Human Microbiome Standards (IHMS) project, (a consortium comprising partners and investigators from many countries), coordinated the development of standard operating procedures designed to optimize data quality and comparability in the human microbiome field.13,14 This consortium recommended a standardized DNA extraction method for human fecal samples, for which transferability across labs was established. Its adoption allows improvement of comparability of human gut microbiome studies and facilitates meta-analyses. Protocols implemented by 21 laboratories for DNA extraction were analysed which resulted in two procedures that were recognized as convenient for future work (www.microbiome-standards.org).
Human intestinal tract metagenomics
When looking at a large number of subjects, only a small number of species were highly conserved. In a cohort of 124 subjects, only 57 species were seen in 90% of them, whereas only 18 species were present in nearly all subjects and belonging to the major phyla that are observed in the human gut microbiota.5 Thus, less than 20 out of an average of 300 dominant species are present per individual indicating that there is massive inter-individual variation. Stability over time for a given individual’s metagenome under healthy conditions is such that it represents an equivalent of a personal fingerprint. Reproducible patterns of variation in the microbiota have been observed in the adult human gut and have been separated into clusters termed “enterotypes”.15 Enterotypes could be used to stratify human gut microbiomes in order to improve the understanding of complex biological problems in human health and wellbeing. The number, or even existence of different community types, has since been a topic of considerable debate after the publication of the original approach. In 2018, Costea et al.16 revisited the enterotype concept by performing a refined meta-analysis and proposed a modified concept of enterotypes, with the goal of reconciling divergent viewpoints. They showed that human microbiomes differ at the level of ecological arrangements with three preferred patterns or enterotypes (comprising the genera Bacteroides, Ruminococcus, and Prevotella). These genera are not fixed populations because there is flexibility or plasticity in the ecological distribution between them which occurs with for instance, dietary changes, or antibiotic treatments, where a person can switch from one enterotype to another one.17 After the first description of these enterotypes, Wu et al.,18 investigated the association of dietary and environmental variables with the gut microbiota and showed that long-term dietary habits had an impact on the gut microbiome, and revealed that the Bacteroides enterotype is dominant among individuals with a “Western” fast-food type diet (rich in animal fat and proteins), whereas Prevotella is highly associated with a high-fiber diet (complex carbohydrates), dominated by fruits and vegetables. Thereafter, a connection was made between enterotypes and gene count or “richness”. The distribution of the human population with the number of individuals as a function of the number of genes in the human metagenome (<200,000 to >800,000) did not follow a normal bell-shaped curve but a curve shouldered to the left pointing at individuals with a low richness (diversity) of genes, and a higher main peak to the right denoting the high gene richness microbiomes. Among the three enterotypes, the Bacteroides enterotype was dominant on the left-hand side (in low richness individuals) while Prevotella and Ruminococcus were on the right-hand side (high richness individuals). The study showed that 68 species were specifically linked to gene richness and later, that low gene richness was a key stratifier in chronic conditions, associated with worst phenotypes, non-response to dietary change and with high-risk morbidities especially metabolic disorders.19
Furthermore, low gene richness of the dominant metagenome has also been coined as a key stratifier related to health, as individuals with a reduced microbial gene richness exhibit more metabolic dysfunction and low-grade inflammation if they are overweight or obese.20 They are non-responders to calorie restriction,20 more severe and possibly faster progressor patients in extreme obesity (Clément, pers. communication) and liver cirrhosis,21 and poorer responders to immune checkpoint blockers in cancer immunotherapy.22
Finally, metagenomics has also been used to describe the microbiota in several chronic disease conditions such as type 2 diabetes.23 Through quantitative metagenomic sequencing and analysis, it has been shown that several gut bacterial species can be linked to metabolic risk markers in obesity,19 or to liver cirrhosis.21
Chronic diseases incidence and environmental changes: a moving paradigm
Despite considerable medical progress resulting in a marked decline in the incidence of infectious diseases, chronic disease incidence has been rising for over 60 years. Autoimmune and autoinflammatory disorders such as inflammatory bowel disease (e.g., Crohn’s disease, ulcerative colitis), multiple sclerosis, type 1 diabetes, and allergic conditions such as asthma, are increasing. On the increase also, are metabolic conditions such as type 2 diabetes and obesity, and other disorders like cirrhosis; some cancers and neurological conditions. It is predicted that 1 in 4 individuals will suffer from one of these pathologies by 2025. Prevention is hence an urgent need and perhaps human longevity is at stake, as it is starting to decline in ‘developed’ countries. A stark example is that of autism incidence which affects 1 in 59 births in the USA and 1 in 150 births in France and in the UK according to the last census.
It is legitimate to wonder which of the significant changes made over this period could have affected the human microbiota and may explain the increased incidence of these chronic diseases, which is obviously too sharp to possibly correspond to a drift in human genetics. In the course of just a few generations, there are at least three alterations that may have played a role. First, birth modes have changed drastically. Based on the data from 121 countries, between 1990 and 2014, the global average C-section rate increased by 12.4% (from 6.7% to 19.1%) with an average annual rate of increase of 4.4% and reach up to 40.5% in some countries.24 This increase rate of C-section prevents the natural vertical transmission of microbes from mother to child and microbiota differs between C-section born and vaginally delivered infants over the first year of life, showing enrichment of Bifidobacterium spp., and reduction of Enterococcus and Klebsiella spp. in vaginally delivered infants, possibly contributing to an increase susceptibility to infection in C-section born children.25 Prophylactic antibiotic treatment at birth is also quite common given the recommendation to use them for C-section26 and in case of Streptococcus agalactiae maternal carriage or operative vaginal delivery.27 Second, levels of physical activity have decreased. Worldwide, the prevalence of insufficient physical activity was 27.5% (95% uncertainty interval 25.0–32.2) in 2016, with highest incidence in high-income western countries (42.3%, 39.1–45.4).28 Furthermore, prevalence of insufficient physical activity was even higher in adolescent aged 11–17 years, being 81.0% (95% uncertainty interval 77.8–87.7), with no impact of the country income group.29 Third, Industrial Period (nineteenth century) have fundamentally altered nutrition in Western countries. Nowadays, dairy products, cereals, refined sugars, refined vegetable oils, and alcohol make up 72.1% of the total daily energy consumed by all people in the United States of America.30 Therefore, the average dietary intake of fiber in the typical United States diet is 17.33 g/day,31 well below the recommended 25–30 g/day.32 Finally, the gut microbiota can be adversely affected by exposure to xenobiotics, among which are drugs but also environmental pollutants and food additives.33 An example is dietary emulsifiers, so commonly ingested in processed foods, which can induce intestinal inflammation.34 Importantly, antibiotic consumption continues to increase worldwide (antibiotic consumption rate increased 39% between 2000 and 2016) including in low-income and middle-income countries and projections of global antibiotic consumption in 2030, assuming no policy changes, are up to 200% higher than the 42 billion defined daily doses estimated in 2015.35
The common thread in chronic conditions
An altered microbiota disrupts the host–microbe symbiosis which can lead to an altered gut permeability (also known as intestinal barrier dysfunction or “leaky gut” syndrome, despite this term is no longer favored) and an inflammatory state, resulting in oxidative stress and promotion of systemic immune responses. In a “healthy” condition and symbiosis, we observe a rich and diverse microbiota, integrity of the mucosal barrier, immune tolerance, and a balanced redox state.36 When this symbiosis is disrupted, whether by alterations due to nutrition, antibiotics, xenobiotics, radio/chemotherapy, or immuno-biologics, a low richness microbiota is consistently observed with loss of symbionts (including species of the core metagenome) and an increase in pathobionts (essentially Gram-negative proinflammatory microbes). This results in increased intestinal permeability and low-grade to overt inflammation develops, yielding oxidative stress promoting further aggravation and alteration of the gut microbiota.37,38 All the ingredients of a vicious circle are therefore present. This supports the concept of a potential critical transition from a symbiotic state to one of dysbiosis, which is highly consistent with concepts of population ecology and more globally complex system dynamics (Fig. 1).39,40 Importantly, intestinal barrier dysfunction is commonly observed in aging vertebrates and invertebrates.41–45 A current hypothesis is that decreasing intestinal barrier function with aging can cause increased microbial translocation into the systemic blood circulation, that subsequently causes systemic inflammation (inflammaging) and significant clinical outcomes (e.g., metabolic syndrome, decreased physical function, and mortality).
Fig. 1. Critical transition in chronic immune diseases (concept).
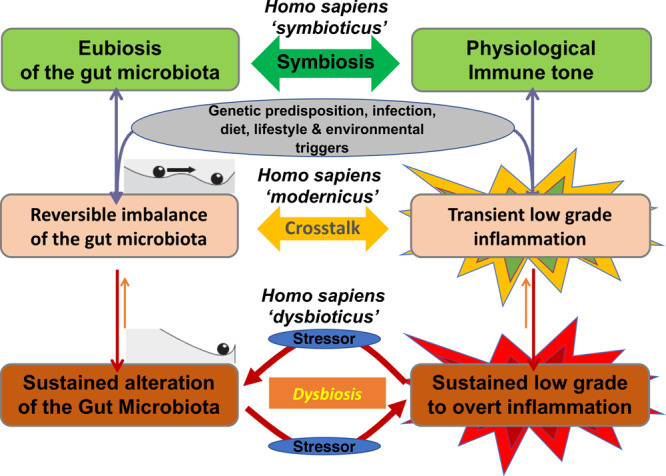
At baseline, in humans, eubiosis of the gut microbiota is associated with physiological immune tone (symbiosis—Homo sapiens “symbioticus”). Thereafter, genetic predisposition, infection, and modern changes in antibiotic use, diet, lifestyle, and environmental triggers lead to reversible imbalance of the gut microbiota and transient low-grade inflammation with a reciprocal impact on each other (crosstalk—Homo sapiens “modernicus”). Finally, exposure to stress signals above the system’s robustness can lead to a sustained alteration of the gut microbiota and inflammation (dysbiosis—Homo sapiens “dysbioticus”).
This latter hypothesis challenges the reassuring vision of the possibility to return from disease to health, and suggests the vicious circle could promote critical transitions and establishment of durably altered symbiosis that might be a common feature of most patients suffering from non-transmissible chronic diseases. This would have major implications in terms of potential innovations since diagnosis, prediction, prevention, and even therapy could address the four triggers that durably alter symbiosis following critical transition, namely low richness microbiota, intestinal permeability, inflammation, and oxidative stress.
The important message relating to critical transition and alternative stable states (based on the findings of Thom46 and Scheffer47) is that as long as the stress conditions applying to an ecosystem are reasonable, a shift can occur, but within the boundaries of ecological robustness, hence allowing resilience and return to the stable healthy state.48 With greater stress, at a tipping point, the ecosystem will bounce into a state of equilibrium that is typical of pre-disease or disease, a state that even if the stressor is removed, will not revert to the healthy state. Only if the stress conditions are dramatically lowered, can the ecosystem be returned to the normal condition.48
Microbiome-based innovations
At present, there is a great interest in innovative health outcomes of the research and discoveries of the gut microbiome. Our work published in 2017,49 identified four main areas of application: (i) stratification and monitoring; (ii) target for modulation; (iii) source of novel biologics and targets; and (iv) development of microbiota-derived drugs.
Several studies in different diseases conditions suggest a predictive value of microbiome profiling and richness. The microbiome of patients receiving an allogeneic hematopoietic stem cell transplant (allo-HSCT) varied significantly between patients. Patients with a high diversity microbiome (Inverse Simpson Index50 > 4) had a probability of transplant-related death rate of around 11% compared with a rate of about 50% in patients with a low diversity (Inverse Simpson Index < 2). The authors concluded that the diversity of the intestinal microbiota at engraftment is an independent predictor of mortality at 3 years in allo-HSCT recipients, indicating that the intestinal microbiota may be an important factor in the success or failure of allo-HSCT.51 This finding was confirmed in a large multicentre study including 1362 patients from United States of America, Germany, and Japan, where higher diversity of intestinal microbiota at the time of neutrophil engraftment was associated with lower mortality.52 Furthermore, it was shown that early use of broad spectrum antibiotics (i.e., before allo-HSCT) was associated with a decrease in fecal abundance of commensal Clostridiales and an increased risk of transplant-related mortality.53
Similarly, it has been shown that administration of antibiotics to cancer patients significantly shortens the overall survival (OS). In a study of patients with NSCLC (n = 140), renal cell cancer (n = 67), and urothelial carcinoma (n = 42), median OS was 20.6 months versus 11.5 months (P < 0.001) for no antibiotic versus antibiotic given in the two months that precede or the month following initiation of immunotherapy, respectively.22
Other work has previously shown that Gram-positive symbionts will act as adjuvants in anticancer therapy. Working with mouse models led to the hypothesis that gut permeability induced by chemotherapy treatment would result in dysbiosis and allow translocation of bacteria into secondary lymphoid organs, thereby priming a Th1/Th17 response that in tumor beds will favor the efficacy of the treatment. Antibiotic use actually knocked down the adjuvant microbes able to promote the efficacy of anticancer therapy. Transfer of Th17 cells partially restored the antitumor efficacy of cyclophosphamide.54 Disruption of the microbiota was also shown to impair the response of subcutaneous tumors to CpG-oligonucleotide immunotherapy and platinum chemotherapy.55
Furthermore, intestinal microbiota richness was found to be associated with a better response to anti PD-1 immunotherapies, namely in non-small cell lung cancer (NSCLC), renal cell carcinoma, urothelial carcinoma,22 and melanoma.56 In the former, the MGS count was higher in patients with long (>6 months) progression-free survival (PFS) than in those with short (<6 months) PFS. This was similar for gene count.22
The above-mentioned study in 112 melanoma patients documenting the modulation of the anticancer response to anti-PD-1 by the intestinal microbiota, found a significantly longer PFS in patients with a high abundance of Faecalibacterium species, after 600 days, compared with those with a low abundance (P = 0.03)56. Conversely, there was a shorter PFS in patients with a high abundance of bacteria of the order Bacteroidales, compared with those patients with a low abundance (P = 0.05). This order contains the dominant Gram-negative bacteria in the human gut. The strongest predictors of response to anti PD-1 on Cox proportional-hazard analysis were Faecalibacterium abundance (hazard ratio (HR) = 2.95; 95% CI = 1.31–7.29, P = 0.03), and prior immunotherapy (HR = 2.87; 95% CI = 1.10–7.89, P = 0.03).56
Similarly, stool samples metagenomics analysis of patient with non-small cell lung cancer, renal cell carcinoma, and urothelial carcinoma at diagnosis revealed correlations between clinical responses to immune checkpoint inhibitors and the relative abundance of Akkermansia muciniphila.22 Oral supplementation with A. muciniphila after FMT with nonresponder feces restored the efficacy of PD-1 blockade in an interleukin-12-dependent manner by increasing the recruitment of CCR9+ CXCR3+ CD4+ T lymphocytes into mouse tumor beds.
These mouse model studies and the recent finding that primary resistance to immune checkpoint inhibitors can be attributed to abnormal gut microbiome composition,22 suggest that the gut microbiota helps shape the anticancer immune response.
Overall, this suggests that microbiota-based biomarkers will be useful for personalized medicine, patients being stratified according to their microbiota composition to receive, or not, a bacterial therapy. Nevertheless, so far, microbiome-based stratification remains a statistical notion based on the comparison between groups of patients, and it remains unclear whether such stratification can be done at the individual level. Only well-designed clinical trials with a stratification based on validated microbiome biomarkers will provide a definitive answer to that.
Bacterial therapy
Many species of bacteria have now been described in relation to their protective bioactivity and many are part of the metagenomic core of the microbiota. Some of these microbes synthesize specific bioactive molecules that are important in host–microbe symbiosis. Bacteroides fragilis has an immunomodulatory capsular polysaccharide A (PSA), which is a potent activator of CD4+ T cells and thus is important in the maintenance of immunological homeostasis.57 Several investigators have now started phase 1 and 2 clinical trials whereby a bioactive microbe is administered in capsules to patients in an attempt to treat or mitigate risks in specific disease conditions. One such example is the use of F. prausnitzii as a monostrain approach in clinical studies for the treatment of IBS. Another example of a bioactive commensal is Akkermansia muciniphila, which is inversely associated with diabetes, obesity, cardiometabolic diseases, and low-grade inflammation.58 Murine models of obesity and/or type 2 diabetes were found to be characterized by a lower abundance of A. muciniphila, and this microbe was strongly correlated with lower cardiometabolic risk factors.59 The purified, bioactive outer membrane protein (Amuc_1l00) of A. muciniphila is now being tested based on a large body of evidence, which established the first proof-of-concept study using Akkermansia in humans.58,60 Nevertheless, some studies suggest that A. muciniphila may have a detrimental role, in particular in graft-versus-host disease after allo-HSCT,61 or in intestinal Salmonella enterica Typhimurium-induced gut inflammation.62 Similarly, Bacteroides fragilis is a well-known opportunistic pathogen in particular in immunosuppressed patients.63,64 Overall, this raises the question of a potential detrimental role of bacterial therapy depending of the pathology and host context.
Many other bioactive commensals are currently being developed as next-generation probiotics such as Roseburia intestinalis or Blautia hydrogenotrophica. Similarly, commensal Hafnia alvei strain reduces food intake and fat mass in obese mice and appear as a new potential probiotic for appetite and body weight management.65
Finally, we must raise an important issue that is the capacity to establish an exogenous strain in a colonized intestine. In fact, a systematic review reported no effect of probiotics on fecal microbiome composition in six out of seven analysed studies.66 Furthermore, it was established that humans featured person-specific, region-specific, and strain-specific mucosal colonization patterns, hallmarked by predictive baseline host and microbiome features.67 In addition, early evidence arises that the baseline microbiome could actually dictate the permissivity of a microbiome to an administered probiotic or live biotherapeutic possibly explaining inter-individual variation in clinical responses.68 Overall, these data suggest that randomized clinical trial are mandatory to evaluate safety and efficacy of probiotics, and that personalized probiotic approaches may be necessary to establish exogeneous strain in a colonized intestine.
Fecal microbiota transfer (FMT)
FMT is a recognized treatment option for recurrent Clostridioides difficile infection. In 2013, van Nood et al.69 showed that infusion of donor feces was significantly more effective than the standard of care (vancomycin) for the treatment of recurrent C. difficile infection in 16 patients. One round of FMT cured approximately 80% of patients and two or more rounds cured over 90%, whereas the use of vancomycin was able to eradicate the pathogen in only 30% of 13 patients. These impressive results led to the discontinuation of the study at the interim analysis as it was non-ethical to pursue with such a large difference in efficacy in favor of FMT versus vancomycin. FMT is now recommended as standard treatment for second recurrence C. difficile infection70,71 and is performed routinely to cure these patients. This approach is being assessed experimentally in many other conditions with intestinal microbiota dysbiosis. Therefore, use of FMT has been reported in gastrointestinal conditions but also in extra-intestinal disorders such as inflammatory bowel diseases, irritable bowel syndrome (IBS), liver diseases, metabolic diseases, neurological and psychiatric conditions, and cancer.
Four randomized clinical trials evaluated FMT in patients with mild or moderate ulcerative colitis.72–75 While, FMT was associated with a significant improvement of the primary endpoint (endoscopic response or endoscopic remission) in two of the trials,74,75 the remaining two trials were terminated after an interim analysis showing no significant difference between FMT and placebo.72,73 Finally, a pooled-analysis of the four trials suggest a benefit of FMT over placebo on endoscopic remission (39/140, 28%, versus 13/137, 9%, p < 0.01).76 For Crohn’s disease, only small non-randomized studies are available with mixed results. Furthermore, while a meta-analysis in 71 patients reported a pooled remission rate of 52%77, this result was driven by one large cohort study78. Overall, FMT is still an experimental approach in inflammatory bowel disease, and should be performed within clinical trials to identify factors predictive of response in terms of disease characteristics, donors, or timing of FMT.
For IBS, a randomized clinical trial recently reported a significant decrease in IBS severity score at 3 months with FMT compared to placebo [65% (36/55) versus 43% (12/28), P = 0.049].79 Nevertheless, the difference was no longer statistically significant at 12 months. Future clinical trials will have to identify the subset of patients that benefit of FMT, and the frequency of FMT repetition in those patients to have a sustained response.
FMT for hepatic encephalopathy is also under intensive investigation based on the promising results of a first clinical trial that randomized standard of care alone or combined with FMT in 20 patients with liver cirrhosis and recurrent hepatic encephalopathy80. FMT appeared to be safe with fewer severe adverse events (2 versus 8, P = 0.02) and effective with no new episode of hepatic encephalopathy in the FMT group versus 6 in the standard of care alone group (P = 0.03). A second placebo-controlled randomized phase 1 study confirmed the safety of FMT in hepatic encephalopathy but failed to find an impact on hepatic encephalopathy episodes81, and larger studies are expected to decipher the exact role of FMT in those patients.
FMT was also evaluated outside of the gastro-intestinal setting. For example, two small randomized clinical trials showed that FMT from thin donors increased glucose clearance in obese Caucasian males with metabolic syndrome.82,83 However, no impact on clinical parameters was observed raising the question of the relevance of this approach in those patients. More promising results were reported in children ages 7–16 years with autism spectrum disorder and associated gastrointestinal symptoms (constipation or diarrhea).84 This open label study reported a decrease in both gastrointestinal and neuropsychiatric symptoms. Furthermore, a follow-up study, 2 years after treatment was completed, confirmed that most improvements in GI symptoms were maintained, and that autism-related symptoms improved even more after the end of treatment.85 Several studies are ongoing to confirm this promising result (NCT03408886, NCT03829878).
Regarding the use of FMT to eradicate multidrug-resistant bacteria (MDRB), small prospective studies reported mixed results with a complete decolonization achieved in 33–75% of patients.86–90 Therefore, randomized clinical studies are ongoing to evaluate the real impact of FMT for MDRB eradication (NCT04188743, NCT04146337). Interestingly, several studies were performed in highly immunosuppressed patients with hematologic malignancies.89,90 Bilinski et al. reported complete eradication of MDRB in 15 out of 20 patients with hematologic malignancies.89 Of note, eight patients received FMT after allo-HSCT. Similarly, we achieved successful MDRB eradication after FMT in 6 out of 10 allo-HCST patients, with no side effects.90 We also conducted a proof of concept study (Phase 1b/2a) evaluating autologuous FMT in 25 acute myeloid leukemia (AML) patients.91 Intensive chemotherapy of AML and subsequent use of large spectrum antibiotics are known to negatively impact gut microbiota composition, generating a profound state of dysbiosis accompanied by an inflammatory burst and to induce MDRB acquisition.92 Stool were screened, conditioned and frozen at diagnosis and before patients received any induction chemotherapy and antibiotic treatment. Around 1 month of post-diagnosis, after aplasia completion, patients were administered the autologous FMT, and 10 days later received consolidation chemotherapy. There was a marked disruption of the microbiota induced by intensive chemotherapy and antibiotics but, after auto FMT, the microbiome was restored to 90% of its initial richness, overall microbial diversity and Simpson diversity index. Furthermore, in this trial, auto-FMT showed an excellent safety profile and there was on average a reduction of 43% of genes coding for anti-microbial resistance.91 Furthermore, two studies recently reported that either autologous or third-party fecal microbiota transplantation was safe and effective for gut microbiota reconstitution after allo-HSCT.93,94 However, the ability of such strategies to reverse the deleterious effect of an impaired gut microbiota during the allo-HSCT course and thus impact a patient’s outcome, remains to be demonstrated.56
Several case reports also suggested that FMT is effective for the treatment of steroid-refractory gastro-intestinal acute graft-versus-host disease (aGVHD), a life-threatening complication after allo-HSCT. A small cohort study confirms this finding with a complete response in 10 out of 15 patients (67%) with steroid-refractory or steroid-dependant aGVHD 28 days after FMT and no safety event.95 This response was accompanied by an increase in gut microbial α-diversity, a partial engraftment of donor bacterial species, and increased abundance of butyrate-producing bacteria, including Clostridiales and Blautia species. We also reported the compassionate use of MaaT013, a standardized, pooled-donor, high-richness microbiota biotherapeutic, in 29 patients with steroid-refractory or steroid-dependent aGVHD96. The product was well tolerated and at day 28, overall response and complete remission rate were 59 and 31%, respectively. Therefore, FMT appear to be a promising strategy and several studies are ongoing to evaluate FMT for aGVHD management (NCT03812705, NCGT03492502, NCT03359980, NCT03720392, and NCT03678493).
Overall, outcomes of FMT are mixed depending on the indication. Therefore, well-designed randomized clinical trials are mandatory to evaluate the efficacy of FMT in the broad range of indications, and to identify patients who will benefit from FMT. Furthermore, growing use of FMT, including in highly immunosuppressed patients, raise the question of its safety. Recently, bacteraemia with extended-spectrum beta-lactamase (ESBL)-producing Escherichia coli was reported in two patients after they had undergone FMT in two independent clinical trials; both cases were linked to the same stool donor by means of genomic sequencing,97 one of the patients died. This highlights the importance of rigorous donors’ selection. A European consensus conference provide clear guidelines that must be followed for patients screening.98 Furthermore, in some countries health authorities provide mandatory recommendations for FMT donor screening. Nevertheless, despite a thorough screening it remains difficult to completely exclude the risk of transmission of new pathogens. The ongoing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic highlights this. While SARS-CoV-2 primarily infect the respiratory tracts, secondary evidence of gastrointestinal carriage of the virus lead to the prohibition of FMT donor screening, a quarantine of stool graft already collected and consequently an interruption of all FMT procedures in most countries. Nevertheless, reorganization of FMT services has allowed FMT resumption thanks to additional guidelines regarding donor selection, including now nasopharyngeal swab and reverse transcription polymerase chain reaction (RT-PCR) assays, serology, and validated molecular stool testing.99,100
Conclusions
Humans share a core intestinal microbiome and yet they differ by genes, species, enterotypes (ecology), and gene count (microbial diversity). The microbiome gene count is a key stratifier of health in several immune disorders and clinical conditions. Dysbiosis is an altered state of host–microbe crosstalk with auto-aggravating signals from both sides that may induce circular causalities and thereby durably altered symbioses. Microbiota modulation should be considered as a target for personalized nutrition and therapy. Specific nutrients and live microbes can be considered strategic bioactives for the maintenance, preservation or restoration of host–microbe symbiosis (Fig. 2). A major way of leveraging prevention in chronic diseases and iatrogenic conditions will hence be related to the management and the monitoring of intestinal ecology.
Fig. 2. Restoring symbiosis—from Homo dysbioticus to Homo symbioticus.
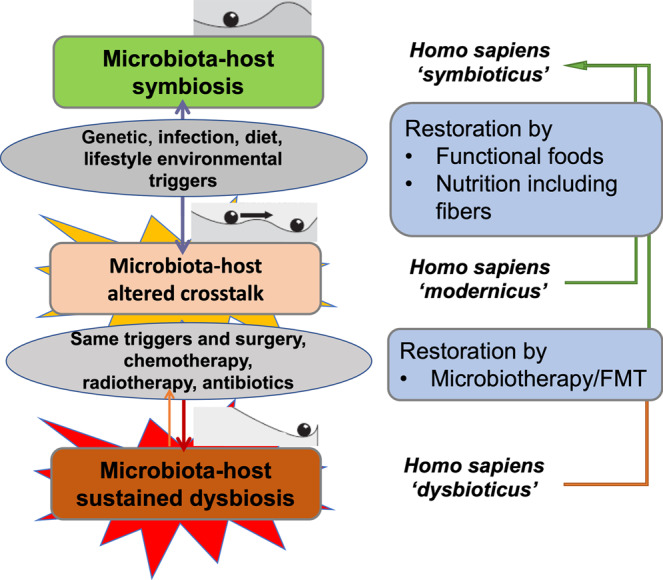
For restoration of the microbiota-host altered crosstalk, modification of nutrition can restore symbiosis. However, in case of microbiota-host sustained dysbiosis, restoration of symbiosis is more difficult and requires therapeutic intervention with microbiotherapy or fecal microbiota transplantation.
Acknowledgements
This review was permitted in part by funding to Joel Doré from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant agreement ERC-2017-AdG No. 788191—Homo.symbiosus).
Author contributions
All authors contributed substantially to the conception, writing, critical review, and final approval of the manuscript.
Competing interests
M.M. and J.D. received consultancy honoraria from MaaT Pharma outside the scope of this work. F.M. and B.G. did not disclose any relevant conflict of interest in relation to this work.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Florent Malard, Email: florent.malard@inserm.fr.
Mohamad Mohty, Email: mohamad.mohty@inserm.fr.
References
- 1.Adelman MW, et al. The gut microbiome’s role in the development, maintenance, and outcomes of sepsis. Crit. Care. 2020;24:278. doi: 10.1186/s13054-020-02989-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayashi H, Sakamoto M, Benno Y. Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol. Immunol. 2002;46:535–548. doi: 10.1111/j.1348-0421.2002.tb02731.x. [DOI] [PubMed] [Google Scholar]
- 3.Tannock GW, et al. Analysis of the fecal microflora of human subjects consuming a probiotic product containing Lactobacillus rhamnosus DR20. Appl. Environ. Microbiol. 2000;66:2578–DR2588. doi: 10.1128/AEM.66.6.2578-2588.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suau A, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl. Environ. Microbiol. 1999;65:4799–4807. doi: 10.1128/AEM.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grice EA, Segre JA. The human microbiome: our second genome. Annu. Rev. Genomics Hum. Genet. 2012;13:151–170. doi: 10.1146/annurev-genom-090711-163814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehrlich, S. D. in Metagenomics of the Human Body. (ed. Nelson, K. E.) 307–316 (Springer, New York, 2011).
- 9.Li J, et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014;32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen HB, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014;32:822–828. doi: 10.1038/nbt.2939. [DOI] [PubMed] [Google Scholar]
- 11.Xiao L, et al. A reference gene catalogue of the pig gut microbiome. Nat. Microbiol. 2016;1:16161. doi: 10.1038/nmicrobiol.2016.161. [DOI] [PubMed] [Google Scholar]
- 12.Vandeputte D, et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551:507–511. doi: 10.1038/nature24460. [DOI] [PubMed] [Google Scholar]
- 13.Costea PI, et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017;35:1069–1076. doi: 10.1038/nbt.3960. [DOI] [PubMed] [Google Scholar]
- 14.Cardona S, et al. Storage conditions of intestinal microbiota matter in metagenomic analysis. BMC Microbiol. 2012;12:158. doi: 10.1186/1471-2180-12-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costea PI, et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018;3:8–16. doi: 10.1038/s41564-017-0072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knights D, et al. Rethinking “enterotypes”. Cell Host Microbe. 2014;16:433–437. doi: 10.1016/j.chom.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu GD, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Chatelier E, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 20.Cotillard A, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585–588. doi: 10.1038/nature12480. [DOI] [PubMed] [Google Scholar]
- 21.Solé, C. et al. Alterations in gut microbiome in cirrhosis as assessed by quantitative metagenomics: relationship with acute-on-chronic liver failure and prognosis. Gastroenterology (2020). (Online ahead of print). [DOI] [PubMed]
- 22.Routy B, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 23.Qin J, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 24.Betrán AP, et al. The increasing trend in caesarean section rates: global, regional and national estimates: 1990–2014. PLoS ONE. 2016;11:e0148343. doi: 10.1371/journal.pone.0148343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reyman M, et al. Impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nat. Commun. 2019;10:4997. doi: 10.1038/s41467-019-13014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smaill, F. M. & Grivell, R. M. Antibiotic prophylaxis versus no prophylaxis for preventing infection after cesarean section. Cochrane Datab. Syst. Rev.10, Cd007482 (2014). [DOI] [PMC free article] [PubMed]
- 27.Knight M, et al. Prophylactic antibiotics in the prevention of infection after operative vaginal delivery (ANODE): a multicentre randomised controlled trial. Lancet. 2019;393:2395–2403. doi: 10.1016/S0140-6736(19)30773-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guthold R, Stevens GA, Riley LM, Bull FC. Worldwide trends in insufficient physical activity from 2001 to 2016: a pooled analysis of 358 population-based surveys with 1.9 million participants. Lancet Glob. Health. 2018;6:e1077–e1086. doi: 10.1016/S2214-109X(18)30357-7. [DOI] [PubMed] [Google Scholar]
- 29.Guthold R, Stevens GA, Riley LM, Bull FC. Global trends in insufficient physical activity among adolescents: a pooled analysis of 298 population-based surveys with 1.6 million participants. Lancet Child Adolesc. Health. 2020;4:23–35. doi: 10.1016/S2352-4642(19)30323-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cordain L, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am. J. Clin. Nutr. 2005;81:341–354. doi: 10.1093/ajcn.81.2.341. [DOI] [PubMed] [Google Scholar]
- 31.Han, S., Wu, L., Wang, W., Li, N. & Wu, X. Trends in dietary nutrients by demographic characteristics and BMI among US Adults, 2003–2016. Nutrients11, 2617 (2019). [DOI] [PMC free article] [PubMed]
- 32.Krauss RM, et al. AHA dietary guidelines: revision 2000: a statement for healthcare professionals from the Nutrition Committee of the American Heart Association. Circulation. 2000;102:2284–2299. doi: 10.1161/01.CIR.102.18.2284. [DOI] [PubMed] [Google Scholar]
- 33.Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50. doi: 10.1016/j.cell.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chassaing B, et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. 2015;519:92–96. doi: 10.1038/nature14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klein EY, et al. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl Acad. Sci. USA. 2018;115:E3463–e3470. doi: 10.1073/pnas.1717295115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van de Guchte M, Blottiere HM, Dore J. Humans as holobionts: implications for prevention and therapy. Microbiome. 2018;6:81. doi: 10.1186/s40168-018-0466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bäumler AJ, Sperandio V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature. 2016;535:85–93. doi: 10.1038/nature18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Litvak Y, Bäumler AJ. Microbiota-Nourishing immunity: a guide to understanding our microbial self. Immunity. 2019;51:214–224. doi: 10.1016/j.immuni.2019.08.003. [DOI] [PubMed] [Google Scholar]
- 39.Scheffer M, Carpenter S, Foley JA, Folke C, Walker B. Catastrophic shifts in ecosystems. Nature. 2001;413:591–596. doi: 10.1038/35098000. [DOI] [PubMed] [Google Scholar]
- 40.Kefi S, et al. Early warning signals of ecological transitions: methods for spatial patterns. PLoS ONE. 2014;9:e92097. doi: 10.1371/journal.pone.0092097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rera M, Clark RI, Walker DW. Intestinal barrier dysfunction links metabolic and inflammatory markers of aging to death in Drosophila. Proc. Natl Acad. Sci. USA. 2012;109:21528–21533. doi: 10.1073/pnas.1215849110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh S, et al. Elevated muscle TLR4 expression and metabolic endotoxemia in human aging. J. Gerontol. Ser. A. 2015;70:232–246. doi: 10.1093/gerona/glu067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kavanagh K, et al. Microbial translocation and skeletal muscle in young and old vervet monkeys. Age. 2016;38:58. doi: 10.1007/s11357-016-9924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson QN, et al. Greater microbial translocation and vulnerability to metabolic disease in healthy aged female monkeys. Sci. Rep. 2018;8:11373. doi: 10.1038/s41598-018-29473-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kavanagh K, et al. Biomarkers of leaky gut are related to inflammation and reduced physical function in older adults with cardiometabolic disease and mobility limitations. Geroscience. 2019;41:923–933. doi: 10.1007/s11357-019-00112-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thom, R. Paraboles et catastrophes. Entretiens sur les mathématiques, la science et la philosophie (Flammarion, Paris, 1983).
- 47.Scheffer, M. Critical Transitions in Nature and Society (Princeton University Press, New Jersey, 2009).
- 48.Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336:1255–1262. doi: 10.1126/science.1224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dore J, Multon MC, Behier JM. The human gut microbiome as source of innovation for health: which physiological and therapeutic outcomes could we expect? Therapie. 2017;72:21–38. doi: 10.1016/j.therap.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 50.Simpson EH. Measurement of diversity. Nature. 1949;163:688–688. doi: 10.1038/163688a0. [DOI] [Google Scholar]
- 51.Taur Y, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood. 2014;124:1174–1182. doi: 10.1182/blood-2014-02-554725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peled JU, et al. Microbiota as predictor of mortality in allogeneic hematopoietic-cell transplantation. N. Engl. J. Med. 2020;382:822–834. doi: 10.1056/NEJMoa1900623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weber D, et al. Microbiota disruption induced by early use of broad-spectrum antibiotics is an independent risk factor of outcome after allogeneic stem cell transplantation. Biol. Blood Marrow Transplant. 2017;23:845–852. doi: 10.1016/j.bbmt.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viaud S, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–976. doi: 10.1126/science.1240537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iida N, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–970. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gopalakrishnan V, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 58.Cani PD, de Vos WM. Next-generation beneficial microbes: the case of Akkermansia muciniphila. Front. Microbiol. 2017;8:1765. doi: 10.3389/fmicb.2017.01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Everard A, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl Acad. Sci. USA. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plovier H, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017;23:107–113. doi: 10.1038/nm.4236. [DOI] [PubMed] [Google Scholar]
- 61.Shono Y, et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci. Transl. Med. 2016;8:339ra371. doi: 10.1126/scitranslmed.aaf2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ganesh BP, Klopfleisch R, Loh G, Blaut M. Commensal Akkermansia muciniphila exacerbates gut inflammation in Salmonella typhimurium-infected gnotobiotic mice. PLoS ONE. 2013;8:e74963. doi: 10.1371/journal.pone.0074963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murphy EC, Mörgelin M, Cooney JC, Frick IM. Interaction of Bacteroides fragilis and Bacteroides thetaiotaomicron with the kallikrein-kinin system. Microbiology. 2011;157:2094–2105. doi: 10.1099/mic.0.046862-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murthy, H. S. et al. Baseline gut microbiota composition is associated with major infections early after hematopoietic cell transplantation. Biol. Blood Marrow Transpl. 26, 2001-2010 (2020). [DOI] [PubMed]
- 65.Legrand R, et al. Commensal Hafnia alvei strain reduces food intake and fat mass in obese mice-a new potential probiotic for appetite and body weight management. Int. J. Obes. 2020;44:1041–1051. doi: 10.1038/s41366-019-0515-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kristensen NB, et al. Alterations in fecal microbiota composition by probiotic supplementation in healthy adults: a systematic review of randomized controlled trials. Genome Med. 2016;8:52. doi: 10.1186/s13073-016-0300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zmora N, et al. Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell. 2018;174:1388–1405. doi: 10.1016/j.cell.2018.08.041. [DOI] [PubMed] [Google Scholar]
- 68.Zhang C, et al. Ecological robustness of the gut microbiota in response to ingestion of transient food-borne microbes. ISME J. 2016;10:2235–2245. doi: 10.1038/ismej.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Nood E, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- 70.Debast SB, Bauer MP, Kuijper EJ. European society of clinical microbiology and infectious diseases: update of the treatment guidance document for Clostridium difficile infection. Clin. Microbiol. Infect. 2014;20:1–26. doi: 10.1111/1469-0691.12418. [DOI] [PubMed] [Google Scholar]
- 71.McDonald LC, et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the infectious diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA) Clin. Infect. Dis. 2018;66:987–994. doi: 10.1093/cid/ciy149. [DOI] [PubMed] [Google Scholar]
- 72.Rossen NG, et al. Findings from a randomized controlled trial of fecal transplantation for patients with ulcerative colitis. Gastroenterology. 2015;149:110–118. doi: 10.1053/j.gastro.2015.03.045. [DOI] [PubMed] [Google Scholar]
- 73.Moayyedi P, et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology. 2015;149:102–109. doi: 10.1053/j.gastro.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 74.Paramsothy S, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet. 2017;389:1218–1228. doi: 10.1016/S0140-6736(17)30182-4. [DOI] [PubMed] [Google Scholar]
- 75.Costello SP, et al. Effect of Fecal microbiota transplantation on 8-week remission in patients with ulcerative colitis: a randomized clinical trial. JAMA. 2019;321:156–164. doi: 10.1001/jama.2018.20046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Costello SP, et al. Systematic review with meta-analysis: faecal microbiota transplantation for the induction of remission for active ulcerative colitis. Aliment. Pharmacol. Ther. 2017;46:213–224. doi: 10.1111/apt.14173. [DOI] [PubMed] [Google Scholar]
- 77.Paramsothy S, et al. Faecal microbiota transplantation for inflammatory bowel disease: a systematic review and meta-analysis. J. Crohns Colitis. 2017;11:1180–1199. doi: 10.1093/ecco-jcc/jjx063. [DOI] [PubMed] [Google Scholar]
- 78.Cui B, et al. Fecal microbiota transplantation through mid-gut for refractory Crohn’s disease: safety, feasibility, and efficacy trial results. J. Gastroenterol. Hepatol. 2015;30:51–58. doi: 10.1111/jgh.12727. [DOI] [PubMed] [Google Scholar]
- 79.Johnsen PH, et al. Faecal microbiota transplantation versus placebo for moderate-to-severe irritable bowel syndrome: a double-blind, randomised, placebo-controlled, parallel-group, single-centre trial. Lancet Gastroenterol. Hepatol. 2018;3:17–24. doi: 10.1016/S2468-1253(17)30338-2. [DOI] [PubMed] [Google Scholar]
- 80.Bajaj JS, et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: a randomized clinical trial. Hepatology. 2017;66:1727–1738. doi: 10.1002/hep.29306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bajaj JS, et al. Fecal microbial transplant capsules are safe in hepatic encephalopathy: a phase 1, randomized, placebo-controlled trial. Hepatology. 2019;70:1690–1703. doi: 10.1002/hep.30690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vrieze A, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–916. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 83.Kootte RS, et al. Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab. 2017;26:611–619. doi: 10.1016/j.cmet.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 84.Kang DW, et al. Microbiota transfer therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome. 2017;5:10. doi: 10.1186/s40168-016-0225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kang DW, et al. Long-term benefit of microbiota transfer therapy on autism symptoms and gut microbiota. Sci. Rep. 2019;9:5821. doi: 10.1038/s41598-019-42183-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davido B, et al. Is faecal microbiota transplantation an option to eradicate highly drug-resistant enteric bacteria carriage? J. Hosp. Infect. 2017;95:433–437. doi: 10.1016/j.jhin.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 87.Dinh A, et al. Clearance of carbapenem-resistant enterobacteriaceae vs. vancomycin-resistant enterococci carriage after faecal microbiota transplant: a prospective comparative study. J. Hosp. Infect. 2018;99:481–486. doi: 10.1016/j.jhin.2018.02.018. [DOI] [PubMed] [Google Scholar]
- 88.Singh R, et al. Fecal microbiota transplantation against intestinal colonization by extended spectrum beta-lactamase producing enterobacteriaceae: a proof of principle study. BMC Res Notes. 2018;11:190. doi: 10.1186/s13104-018-3293-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bilinski J, et al. Fecal microbiota transplantation in patients with blood disorders inhibits gut colonization with antibiotic-resistant bacteria: results of a prospective, single-center Study. Clin. Infect. Dis. 2017;65:364–370. doi: 10.1093/cid/cix252. [DOI] [PubMed] [Google Scholar]
- 90.Battipaglia G, et al. Fecal microbiota transplantation before or after allogeneic hematopoietic transplantation in patients with hematologic malignancies carrying multidrug-resistance bacteria. Haematologica. 2019;104:1682–1688. doi: 10.3324/haematol.2018.198549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mohty M, et al. The odyssee study: prevention of dysbiosis complications with autologous fecal microbiota transfer (FMT) in acute myeloid leukemia (AML) patients undergoing intensive treatment: results of a prospective multicenter trial. Blood. 2018;132:1444. doi: 10.1182/blood-2018-99-112825. [DOI] [Google Scholar]
- 92.Galloway-Pena JR, et al. The role of the gastrointestinal microbiome in infectious complications during induction chemotherapy for acute myeloid leukemia. Cancer. 2016;122:2186–2196. doi: 10.1002/cncr.30039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Taur, Y. et al. Reconstitution of the gut microbiota of antibiotic-treated patients by autologous fecal microbiota transplant. Sci. Transl. Med.10, 460 (2018). [DOI] [PMC free article] [PubMed]
- 94.DeFilipp Z, et al. Third-party fecal microbiota transplantation following allo-HCT reconstitutes microbiome diversity. Blood Adv. 2018;2:745–753. doi: 10.1182/bloodadvances.2018017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Lier, Y. F. et al. Donor fecal microbiota transplantation ameliorates intestinal graft-versus-host disease in allogeneic hematopoietic cell transplant recipients. Sci. Transl. Med.12, 556 (2020). [DOI] [PubMed]
- 96.Malard, F. et al. Successful and Safe Treatment of Intestinal GvHD with Pooled-Donor Full Ecosystem Microbiota Biotherapeutics: results with a 29 patient-cohort of a compassionate use/expanded access treatment program. Blood 136, (Supplement 1) 15 (2020).
- 97.DeFilipp Z, et al. Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N. Engl. J. Med. 2019;381:2043–2050. doi: 10.1056/NEJMoa1910437. [DOI] [PubMed] [Google Scholar]
- 98.Cammarota G, et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut. 2017;66:569–580. doi: 10.1136/gutjnl-2016-313017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ianiro G, et al. Reorganisation of faecal microbiota transplant services during the COVID-19 pandemic. Gut. 2020;69:1555–1563. doi: 10.1136/gutjnl-2020-321829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ng SC, Chan FKL, Chan PKS. Screening FMT donors during the COVID-19 pandemic: a protocol for stool SARS-CoV-2 viral quantification. Lancet Gastroenterol. Hepatol. 2020;5:642–643. doi: 10.1016/S2468-1253(20)30124-2. [DOI] [PMC free article] [PubMed] [Google Scholar]