

Abstract
The ITCH/AIP4 ubiquitin E3 ligase was discovered independently by two groups searching for atrophin-1 interacting proteins and studying the genetics of mouse coat color alteration, respectively. ITCH is classified as a NEDD4 family E3 ligase featured with the C-terminal HECT domain for E3 ligase function and WW domains for substrate recruiting. ITCH deficiency in the mouse causes severe multi-organ autoimmune disease. Its roles in maintaining a balanced immune response have been extensively characterized over the past two and a half decades. A wealth of reports demonstrate a multifaceted role of ITCH in human cancers. Given the versatility of ITCH in catalyzing both proteolytic and non-proteolytic ubiquitination of its over fifty substrates, ITCH’s role in malignancies is believed to be context-dependent. In this review, we summarize the downstream substrates of ITCH, the functions of ITCH in both tumor cells and the immune system, as well as the implications of such functions in human cancers. Moreover, we describe the upstream regulatory mechanisms of ITCH and the efforts have been made to target ITCH using small molecule inhibitors.
Keywords: ITCH, Ubiquitin E3 ligase, Cancer, Cancer therapy, Tumor microenvironment
1. Introduction
Ubiquitination is a post-translational modification in which the 76-amino acid ubiquitin molecule is covalently linked, usually to Lys residues, in target proteins [1]. Protein ubiquitination is a highly co-ordinated enzymatic reaction which requires the E1 ubiquitin-activating enzymes, the E2 ubiquitin-conjugating enzymes, and the E3 ubiquitin-ligating enzymes [2]. E1 activates the ubiquitin molecule in an ATP-dependent fashion which forms a thioester bond between the catalytic cysteine in E1 and the carboxyl terminus in ubiquitin; the activated ubiquitin is then conjugated to the E2 enzyme. Finally, the E3 ubiquitin ligases mediate the transfer of ubiquitin to the ε-amino group of the lysine residue on the substrate protein through an interaction involving both the substrate and the E2 enzyme [2].
The ubiquitin E3 ligase ITCH, also named atrophin-1 interacting protein 4 (AIP4), is a member of the NEDD4 HECT-type family E3 ligases [3]. A yeast two-hybrid screen of atrophin-1 interacting proteins led to the discovery of AIP4, along with other AIPs, including AIP2/WWP2 and AIP5/WWP1, all of which are NEDD4 family E3 ligases [4]. In the same year, an independent study on the genetics of the non-agouti-lethal 18H (a18H) mouse strain, which exhibits immunological disorders, identified a locus that is paracentrically inverted with the agouti locus. Given the autoimmune phenotypes found in the a18H strain, the gene was named Itchy [5]. In the past two and a half decades since then, the functions of ITCH in the immune system and malignancies have been extensively characterized. ITCH controls a wide spectrum of signaling pathways by promoting the poly-ubiquitination of its over 50 target proteins: c-JUN, c-FLIP, LATS1, p63, p73, TCR-ζ, BRAF, and many others (Table 1). The dynamic and context-dependent roles of ITCH in both immune cells and tumor cells are reflected by the variety of substrates that ITCH regulates. It is fascinating that ITCH not only catalyzes the canonical K48-linked poly-ubiquitination of its substrates for proteolysis but also contributes to the ubiquitin linkage diversity by assembling K63, K27, and K33-linked poly-ubiquitin chains. The roles of ITCH in the immune system have been well summarized elsewhere [6]. The purpose of this article is to review the involvement of ITCH, along with its substrates and upstream regulators, in cancer cells and their immune microenvironment. We expect that the summary of such knowledge will shed light on the future development of strategies to target ITCH in human cancers.
Table 1.
Summary of the identified ubiquitin substrates for ITCH.
| Substrate | Linkage | Role ITCH plays | Mechanism summary | Ref# |
|---|---|---|---|---|
| BCL10 | K48? | PKC or T-cell receptor (TCR)/CD28 signaling results in the downregulation of BCL10 protein levels by Itch and NEDD4, thereby attenuating NF-κB transcriptional activity. | ITCH, together with NEDD4, targets the BCL10 for proteolysis, downmodulating CD3/ CD28-induced activation of NF-κB. | [135] |
| BRAF | K27 | ITCH facilitates BRAF/MEK/ERK signaling pathway activation. | BRAF is ubiquitinated by the ITCH E3 ligase in response to JNK-mediated phosphorylation abolishes 14–3–3-mediated suppression of BRAF kinase activity, leading to sustained BRAF/MEK/ERK signaling. | [103] |
| c-FLIP | K48? | ITCH facilitates TNFα-induced cell death. | TNFα activates JNK to phosphorylate ITCH, which then ubiquitinates c-FLIP and induces its proteasomal degradation. | [104] |
| c-JUN | K48 | ITCH antagonizes AP-1 function upon JNK pathway activation. | Ubiqui tination of c-JUN and JUNB by ITCH results in their proteasomal degradation. | [71] |
| CXCR4, Hrs | Unknown | ITCH facilitates the sorting of CXCR4 to lysosomal degradation. | ITCH facilitates the lysosomal degradation of chemokine receptor CXCR4 by ubiquitination of the endosomal protein Hrs. | [91] |
| cytoplasmic misfolded proteins | Unknown | ITCH targets misfolded proteins to ensure the protein quality control (QC) system which protects cells against cellular toxicity. | ITCH mediates the degradation of thermally denatured misfolded luciferase proteins. | [174] |
| Deltex | K29 | ITCH may restrain NOTCH signaling. | ITCH promotes Deltex degradation through catalyzing K29-linked poly ubiqui tin chains on Deltex. | [81] |
| DVL | K48? | ITCH inhibits WNT pathway activation. | Phosphorylation of DVL leads to its proteolytic ubiquitination by the E3 ligase ITCH. | [76] |
| Endophilin A1 | Unknown | ITCH regulates ubiquitin-mediated endosomal sorting. | ITCH promotes endophilin A1 ubiquitination by the interaction of proline-rich domain of ITCH and SH3 domain of endophilin Al. | [90] |
| ERBB3 | K48? | ITCH engages a HER3 antibody to induce the proteasomal degradation of HER3 in cancer cells. | [175] | |
| ERBB4 | mono, K63 | ITCH facilitates the sorting and degradation of the ERBB4 CYT-1 isoform, which suppresses EGF-dependent transcriptional activation. | ITCH catalyzes mono-ubiquitination and K63 polyubiquitination of ERBB4 CYT-1, promoted its localization to intracellular vesicles for subsequent degradation. | [97,98,176] |
| FOXO1 | K48 | ITCH is essential for the differentiation of Tfh cells, germinal center responses and IgG responses to acute viral infection. | FOXOl is ubiquitinated by ITCH for proteasomal degradation. | [156] |
| GLI1 | K48 | ITCH suppresses Hedgehog pathway activation. | GLI1 is ubiquitinated by ITCH with the assistance from the adaptor protein Numb, which suppresses the Hedgehog signals. | [42] |
| GLI-Similar 3 | K48? | ITCH functions as a negative regulator of Glis3-mediated transcriptional activity. | ITCH targets GLIS3 transcription factor for ubiquitination and proteolysis. | [59] |
| HEF1/NEDD9 | K48 | ITCH may suppress TGF-β signaling. | TGF-β promotes ITCH, SMAD3 and HEF1 to form a complex to catalyze HEF1 ubiquitination and subsequent degradation. | [67] |
| histone H1.2 | K48, K63 | ITCH suppresses cellular DNA damage response. | ITCH-mediated polyubiqui tination of HI. 2 suppressed RNF8/RNF 168-dependent formation of 53BP 1 foci, which plays an important role in DNA damage response. | [31] |
| influenza A virus A1 protein | K48? | ITCH suppresses influenza A virus cell entrance. | ITCH interacts with viral Ml protein to facilitates its ubiquitination. | [177] |
| ITCH | K63 | K6 3-linked ITCH auto ubiqui tination may stabilize ITCH. | ITCH auto ubiquitination is an inter molecular reaction that promotes K6 3-linked, but not K48-linked polyubiqui tin chains. | [43] |
| ITSN1 | mono and K48? | ITCH regulates endocytosis. | ITSN1 is regulated differentially by ITCH depending on which of its isoform is modified. The major isoform ITSNl-s is monoubiquitinated, while the minor isoform, ITSNl-22a, undergoes a combination of mono- and oligo ubiqui tination. The mono ubiquitination stabilizes ITSNl-s, whereas the oligo ubiquitination of the minor isoform leads to its proteasomal degradation. | [92] |
| JUNB | K48 | ITCH antagonizes AP-1 function upon JNK pathway activation. | Ubiquitination of c-JUN and JUNB by ITCH results in their proteasomal degradation. | [70,71] |
| LATS1 | K48 | ITCH promotes YAP signaling in tumor cells. | ITCH-mediated degradation of LATS1 reduces YAP phosphorylation and leads to the accumulation of nuclear YAP, ITCH enhances transcriptional coactivation function of YAP to promote tumorigenicity. | [49] |
| LRP6 | Unknown | ITCH promotes LRP6-mediated WNT signaling function | ITCH interacts with and ubiquitinates wild type LRP6 but not the LDLR repeat mutants. | [77] |
| MAVS | K48 | ITCH suppresses antiviral innate immunity. | Expression of PCBP2 is induced after viral infection, which recruits ITCH to catalyze K48-linked polyubiquitination and degradation of MAVS. | [178] |
| MKK4/SEK1 | K48? | ITCH degrades MKK4 as a negative feedback regulation of the JNK/p38 pathways. | ITCH binds to MKK4 to promote the ubiquitination of MKK4 at K140 and K143, which leads to MKK4 degradation. | [105] |
| Notch | K29 | ITCH may restrain NOTCH signaling. | ITCH promotes the ubiquitination of NOTCH, and likely in a ligand-independent | [79,80] |
| Occludin | Unknown | ITCH regulates tight junction in epithelial cells | manner. ITCH promotes the degradation of tight junction-specific protein occludin. |
[179] |
| p45/NF-E2 | K63 | ITCH may regulate NF-E2 function during the development of hematopoietic cell lineages. | ITCH suppresses the transactivation activity of p45/NF-E2 via K63-linked polyubiquitination of p45/NF-E2. | [180] |
| p63 | K48? | ITCH may modulate keratinocyte function, where p63 plays a role. | ITCH binds, ubiquitinates, and promotes the degradation of p63. | [83] |
| p73 | K48? | ITCH suppresses DNA damage responses through p73. | ITCH binds to p73 and promotes its ubiquitination and proteasome-dependent degradation in basal conditions. In response to DNA damage, ITCH is rapidly degraded, reducing p73 turnover. TAp73 levels increase. | [87] |
| PI4KIIα | Unknown | ITCH has been shown to form a functional complex with the phosphatidylinositol (PI) 4-kinase type Ila (PI4KIIα) through which ITCH promotes the non-proteolytic ubiquitination of PI4KIIα. | [93] | |
| PLC-γ1 | K48? | Sustained Ca2+ and calcineurin signaling engage ITCH and NEDD4 to destabilizes PLC-γ1 and thereby inhibiting T cell anergy. | NEDD4 and ITCH promote PLC-γ1 ubiquitination and degradation. | [136] |
| PTCH1 | K48? | ITCH suppresses SHH-independent Hedgehog pathway activation. | C-terminal domain (CTD) of PTCH1 interacts with and is ubiquitinated on K1413 by ITCH. | [60] |
| RASSF5/NORE1 | K48? | ITCH suppresses RASSF5-mediated growth inhibition. | RASSF5 is poly-ubiquitinated by ITCH, a process that is inhibited by the acetylation of RASSF5, which suppress the binding between ITCH and RASSF5. | [181] |
| RIP 2 | K63 | ITCH inhibits NOD 2/RIP 2-induced NF-κB activation and suppresses inflammatory responses at mucosal surfaces. | The NOD2 signaling partner, RIP2, is directly K63-polyubiquitinated by ITCH. | [182] |
| ROR-γt | K48 | ITCH regulates IL-17-mediated colonic inflammation and carcinogenesis. | ITCH negatively controls Thl7 differentiation through ubiquitination and degradation of transcription factor RORγt, which is the regulator of Thl7 differentiation. | [153] |
| SHP-1 | K27 | Mice lacking ITCH and WWP2 showed signs of autoimmunity and lung inflammation due to biased differentiation toward the Th2 lineage and hypo-responsiveness after TCR stimulation. | ITCH and WWP2 form a complex to catalyze K27-linked ubiquitination of the phosphatase SHP-1, which disrupts the interaction between SHP-1 and the tyrosine kinase Lck, and therefore promoting TCR signaling. | [137] |
| SIRT6 | K48? | ITCH promotes hepatic lipid infiltration through reduced fatty acid oxidation. | ITCH ubiquitinates SIRT6, leading to its proteolysis. | [183] |
| Smad2 | Unknown | ITCH enhances TGF-β-induced transcription. | ITCH promotes the ubiquitination of SMAD2 and augments SMAD2 phosphorylation and activation. ITCH facilitates complex formation between TGF-β receptor and SMAD2 and enhances TGF-β-induced transcription. | [63] |
| Smad7 | K48? | ITCH facilitates TGF-β-induced transcription. | ITCH is an E3 ubiquitin ligase that specifically targets SMAD7 for ubiquitin-dependent degradation. | [64] |
| STAM-1 | Unknown | Depletion of ITCH and STAM-1 by siRNA caused significant inhibition of CXCR4-induced ERK-1/2 activation. | ITCH-mediated ubiquitination of STAM-1 in caveolae coordinates activation of ERK-1/2 signaling. | [91] |
| SUFU | K63 | ITCH suppresses Hedgehog pathway activation. | β-Arrestin 2 forms a complex with ITCH to promote K63-linked polyubiquitination of the Hedgehog pathway tumor suppressor SUFU, which inhibits the GLI-dependent transcription program. | [40] |
| TAB1 | K48 | ITCH suppresses skin inflammation in the mouse. | ITCH ubiquitinates TAB1 for proteolysis to suppress p38a activation. | [184] |
| TAK1 | K48 | ITCH suppresses TNF-mediated inflammatory signaling. | The ITCH-CYLD complex sequentially cleaved K63-linked ubiquitin chains and catalyzed K48-linked ubiquitination on the kinase TAK1 to terminate inflammatory signaling via tumor necrosis factor. | [69] |
| TCR-ζ | K33 | In mice deficient in the E3 ubiquitin ligases Cbl-b and Itch, T cell activation was augmented, accompanied by spontaneous auto immunity. | ITCH and Cbl-b promote TCR-ζ polyubiquitination via a K33-linkage, which affects its phosphorylation and association with the ζ chain-associated protein kinase Zap-70, to control T cell activation and auto immunity. | [134] |
| TIEG1 | K27 | ITCH facilitates Treg cell function and contributes to T cell anergy in the mouse. | ITCH catalyzes the K27-linked poly-ubiquitination of TIEG1, which facilitates its transcriptional activation to boost Foxp3 expression. | [65,66] |
| TRPV4 | K27 | ITCH suppresses TRPV4-mediated Ca2+ entry to the cells. | ITCH promotes the ubiquitination of the transient receptor potential (TRP) family protein TRPV4 to terminate TRPV4-mediated Ca2+ uptake. | [39,185] |
| TXNIP | Unknown | ITCH suppresses TXNIP-mediated cardiomyocyte apoptosis | TXNIP is ubiquitinated and degraded by ITCH in cardiomyocytes to reduce cardiotoxicity. | [186] |
| vFLIP | K48? | ITCH antagonizes Kaposi’s sarcoma herpesvirus vFLIP-mediated NFkB signaling and viral latency. | ITCH is involved in the ubiquitination and degradation of vFLIP, a process that is induced by KSHV RTA. | [187] |
| YAP | K48? | ITCH, with Amotl 30 engaged, suppresses YAP signaling. | Atoml30 repurposes ITCH from its previously described role in degrading LATS1 to the inhibition of YAP. | [188] |
2. ITCH is a HECT-type, NEDD4 family ubiquitin E3 ligase
The HECT family is a relatively small (28 known members) group of E3 ligases. They feature highly conserved C-terminal HECT domains of about 350 amino acids in length (see reviews in [7,8]). As opposed to RING-type E3 ligases, HECT-type E3 ligases are considered E2-E3 hybrids owing to their capability to receive activated ubiquitin from E2 conjugating enzymes, a process mainly controlled by the HECT domain [7]. The HECT domain consists of N-terminal and C-terminal lobes linked by a flexible linker region. The E2-binding site is found in the N-lobe, while the C-lobe encodes the active-site cysteine forming the thioester bond with the ubiquitin molecule [2,9–12]. The substrate specificity of HECT-type E3 ligases is believed to be determined by the domains located in the N-terminus. HECT E3s are grouped into three subfamilies based on these N-terminal regions: the NEDD4 E3 ligases (9 members), which contain tryptophan-tryptophan (WW) domains for substrate interaction, the HERC (HECT and RCC1-like) E3 ligases (6 members) harboring RCC1-like domains (RLDs), and a group of other HECT E3 ligases without either RLD or WW domains [8,13–16].
The NEDD4 family E3 ligases are among the most characterized HECT domain proteins; the members include NEDD4 (NEDD4–1), NEDD4L (NEDD4–2), ITCH (AIP4), WWP1 (AIP5), WWP2 (AIP2), NEDL1 (HECW1), NEDL2 (HECW2), SMURF1 and SMURF2. All of these proteins share a very similar domain composition: an N-terminal C2 domain, two to four WW domains, and the catalytic HECT domain at the C-terminus [13,15,16]. The C2 domain of NEDD4 family E3s binds to phospholipids and can direct NEDD4 proteins to plasma membranes, endosomes and multivesicular bodies (MVBs) [17,18]. WW domains found in the central part of NEDD4 family E3 ligases mainly serve to interact with substrate proteins owing to their intrinsic protein-protein interacting capability [19]. The most characterized NEDD4-WW interacting motifs include the PPxY motif and other proline-rich motifs [8]. The NEDD4 E3s tend to adopt an autoinhibitory conformation in basal status through direct interaction between the N-terminal C2 or WW domain and the C-terminal HECT domain [20–22]. Such interactions may help to prevent substrate or E2 access and function as a switch for fine-tuning NEDD4 E3 activity [3].
The human ITCH gene encodes a 903 amino acid polypeptide with a typical NEDD4 family protein domain structure with four WW domains to interact with its substrates (Fig. 1A) [23]. The ITCH proteins are highly conserved among humans and other mammals, for example, there is 91 % identity between human and mouse ITCH proteins. Like other HECT type E3 ligases, ITCH catalyzes substrate ubiquitination in a two-step reaction. The first step is the ubiquitin transfer from E2 to the catalytic cysteine in the HECT domain, which is often referred to as the trans-thioesterification reaction. The carboxyl terminus of the charged ubiquitin is then nucleophilically attacked by the primary amino group of the substrate protein, which forms a peptide bond between ubiquitin and the substrate protein [7]. HECT type E3 ligases catalyze substrate ubiquitination in a processive fashion and, the poly-ubiquitin chain linkage is considered to be determined by the HECT E3s, rather than the E2s [7]. However, some of the NEDD4 family E3s, including ITCH, exhibit a remarkable versatility in promoting different types of polyubiquitin linkages. As shown in Table 1, among the approximately 50 known ITCH ubiquitin substrates, ITCH has been shown to promote substrate poly-ubiquitination using K48, K63, K27, K29, and K33 linkages. Therefore, how ITCH and its accompanied E2 dictate linkage selection mechanistically is worthy of future investigation. A previous study uncovered a unique K48-K63 branched poly-ubiquitin chain assembled by HECT E3 HUWE1 and RING E3 TRAF6 [24]. Such branched ubiquitin chains facilitate NF-κB activation while preventing K63 deubiquitinase CYLD-mediated negative feedback regulation hence sustaining NF-κB signaling in cells. Given the flexibility of ITCH in promoting both K63 and K48-linked ubiquitination, it will be intriguing to assess if ITCH alone could catalyze a branched linkage in vitro and in cells.
Fig. 1.
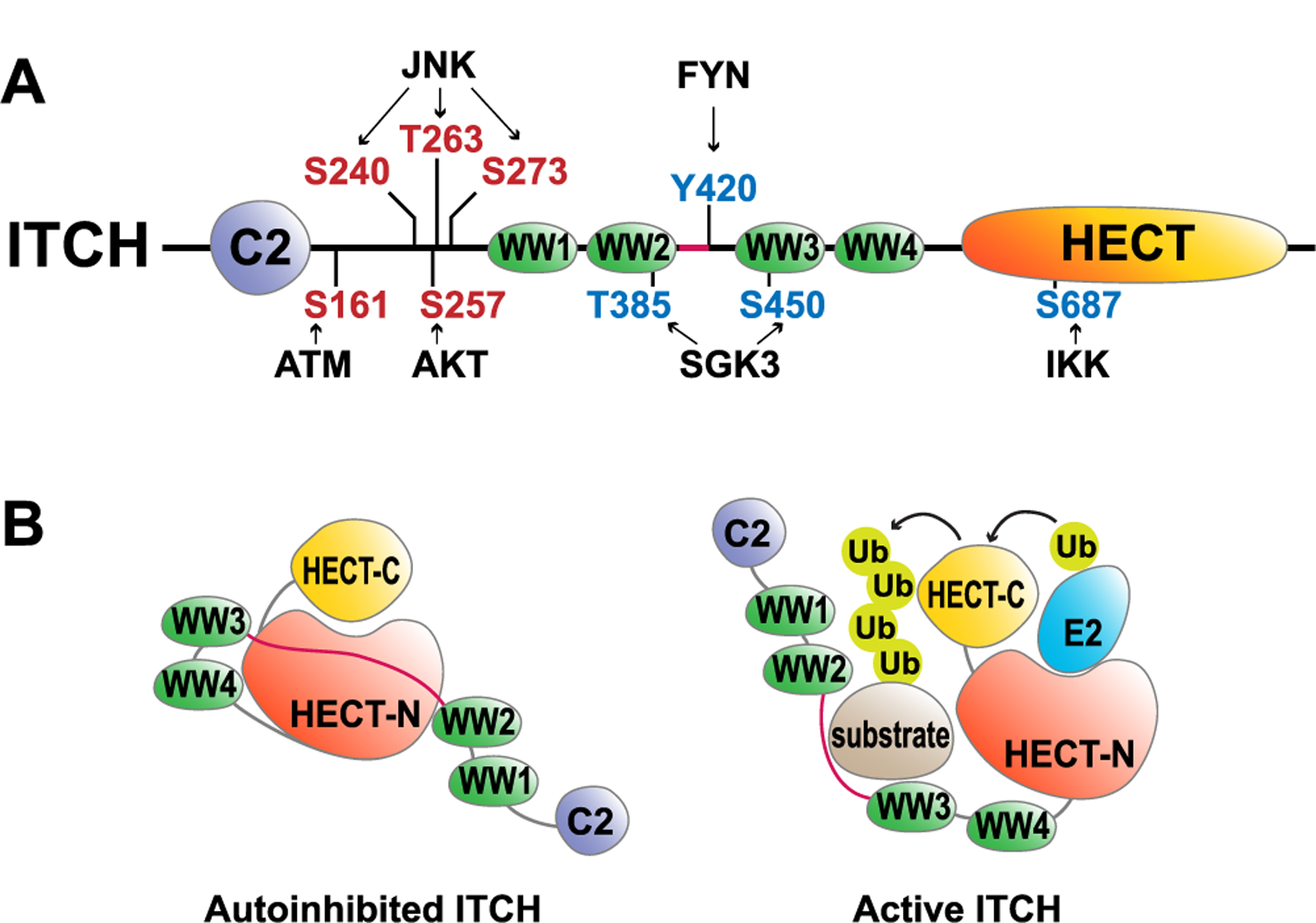
Schematic illustration of ITCH activation and regulatory mechanisms.
A) The domain structure of the human ITCH protein. The red residues indicate activation phosphorylation sites, while the blue residues indicate inhibitory phosphorylation sites. B) Inactive and active conformations of the ITCH protein based on the structural studies [27,28].
3. Regulation of ITCH
Previous studies unveiled that a subset of NEDD4 family E3 ligases including SMURF1, SMURF2, NEDD4, and WWP2 adopt an autoinhibitory conformation in which the C2 domain binds the HECT domain to prevent E2 and substrate access. Truncation mutants of these E3s without their N-terminal C2 domain exhibit an increased activity [20–22]. It is intriguing that ITCH with the C2 domain deleted (ΔC2-ITCH) displayed a similar activity as WT-ITCH, yet it is still less active compared to its HECT domain in vitro [20,25], suggesting other autonomous regulatory mechanisms. Indeed, Riling and colleagues found that WW2 and WW3 domains directly interact with the HECT domain to restrain ITCH E3 ligase activity, which could be relieved through the competitive binding of NDFIP1 (NEDD4 family-interacting protein 1) and NDFIP2 proteins [25,26]Table 2 More recently, the crystal structure of the inactive ΔC2-ITCH was resolved, and it shows consistency with previous biochemical data; it was found that a small linker region (aa 351–381) between WW2 and WW3 domains of ITCH was, in fact, bound to the HECT domain of ITCH in the autoinhibited state [27] (Fig. 1B). This model was also supported by a study focused on the autoinhibitory mechanism of WWP2 [28].
Table 2.
Summary of the identified regulations for ITCH.
| Upstream regulator | Mechanism summary | Ref# |
|---|---|---|
| AKT | AKT-mediated S257 phosphorylation of ITCH activates its function. | [31] |
| ATM | ATM positively modulates ITCH E3-ubiquitin ligase activity by phosphorylating ITCH on S161. | [30] |
| FYN | Tyrosine phosphorylation of ITCH at Y420 by FYN inhibits ITCH activity. | [32] |
| Herpes simplex virus UL56 | UL56 interacts with Itch, independent of additional viral proteins, and mediates degradation of Itch. | [189] |
| IKKα, IKKβ | ITCH is phosphorylated on S687 by IKKs, which suppresses ITCH activity | [190] |
| JNK | JNK phosphorylates ITCH at S240, S263, and T273 to promote ITCH-mediated JUNB ubiquitination and subsequent degradation | [29] |
| LRAD3 | LRAD3 contains two PPxY and functions similarly as NDFIPs to alter ITCH inhibitory conformation | [37] |
| miR-106b | hsa-miR-106b, which itself is down regulated in metastatic pancreatic cancer, directly interacts and inhibit ITCH expression | [191,192] |
| N4BP1 | N4BP1 association with the second WW domain (WW2) of ITCH interferes with E3 binding to its substrates, hence suppresses Itch activity | [175] |
| NDFIP1, NDFIP2 | WW domains 2 and 3 of Itch bind to the HECT domain, mediating autoinhibition. NDFIPs bind multiple WW domains through its PY motifs and relieve this autoinhibition, leaving other WW domains available to recruit substrate. | [25,26,27,36] |
| NF-κB p65 | Overexpression of NF-κB p65 increased ITCH expression, and RANKL promoted the binding of p65 onto the NF-κB binding sites in the ITCH promoter. | [193] |
| SGK3/CISK | SGK3 phosphorylates ITCH at T385 and S450 to suppress ITCH acitivity. | [33] |
| Spartin | Spartin acts as an adaptor protein that activates and recruits ITCH to lipid droplets and by this means regulates the level of ubiquitination of adipophilin and potentially other lipid-associated proteins. | [194] |
| USP9X | Functions as the DUB to antagonize the proteolytic ITCH autoubiquitination | [44] |
| YOD1 | Functions as the DUB to antagonize the proteolytic ITCH autoubiquitination | [45] |
In addition to NDFIP1-controlled activation of ITCH, an early study from the Karin group revealed that in T cells, upon anti-CD3 and anti-CD28 stimulation, the MEKK1/JNK kinase cascade phosphorylates ITCH at S240, S263, and T273 to promote ITCH-mediated JUNB ubiquitination and subsequent degradation [29]. Similarly, phosphorylation events that occur at the linker region between C2 and WW domains, which includes ATM-mediated S161 [30] and AKT-mediated S257 [31] phosphorylation, facilitates ITCH activation (Fig. 1A). These phosphorylation events may serve to alter the suppressive conformation of the ITCH N-terminal region or to recruit p-S/T interacting proteins to disrupt the WW-HECT interaction. On the other hand, FYN-mediated Y420 [32] and SGK3-mediated T385 and S450 [33] phosphorylation, which are found within the WW2-WW3 region (Fig. 1A), inhibits ITCH activity presumably through disrupting WW-substrate interactions. Such a notion was supported by an NMR study in which it was found that threonine phosphorylation in the WW3 domain of ITCH blocks its interaction with PPxY-containing ligands [34]. In addition to the phosphorylation at the N-terminus, IKK-mediated phosphorylation in the HECT domain at S687 has also been demonstrated to inhibit the E2-HECT interaction [35] Table 2.
Several ITCH-associated proteins have been identified to modulate ITCH activity. Binding to NDFIP proteins through the WW domains of ITCH allows the access of PPxY motif-containing substrates [26]. Indeed, NDFIP1 and NDFIP2 were found to enhance ITCH-mediated ubiquitination of JUNB and endophilin [25,27,36]. A very similar mechanism was observed for LRAD3, which contains two PPxY motifs within its intracellular domain and interacts with the WW domains of ITCH and NEDD4 to facilitate their auto-ubiquitination and degradation [37]. β-Arrestin 2, on the other hand, is required for ITCH-dependent endosomal sorting of the chemokine receptor CXCR4 [38], while its homolog, β-Arrestin 1, functions as an adaptor for ITCH by promoting the ubiquitination of transient receptor potential ion channel protein TRPV4 and thereby negatively regulating intracellular calcium signaling [39]. β-Arrestin 2 also forms a complex with ITCH to promote K63-linked polyubiquitination of the Hedgehog pathway tumor suppressor SUFU, which inhibits the GLI-dependent transcription program [40]. Likewise, the adaptor protein Numb facilitates ITCH-mediated degradation of Gli1, which also helps to terminate Hedgehog pathway activation [41,42] Table 2.
ITCH catalyzes its own ubiquitination through either the K48 or the K63 linkage [43]. It has been shown that the deubiquitinases USP9X [44] and YOD1 [45] antagonize the proteolytic K48-linked auto-ubiquitination of ITCH in vitro and in cells. On the other hand, K63-linked autoubiquitination of ITCH prevents proteolytic K48-linked autoubiquitination and stabilizes the ITCH protein [43]. Also, ITCH negatively regulates the deubiquitinase A20 and hence suppresses A20-mediated inactivation of the RIPK1 during NF-κB activation [46].
4. Cancer cell autonomous roles of ITCH in tumorigenesis
ITCH was initially identified as a key enzyme in maintaining a balanced immune response and is closely associated with autoimmune disease [47]. Its roles in malignancies have also been unveiled through the discovery of its approximately 50 ubiquitin substrates (Table 1). The ITCH gene is not frequently mutated, deleted or amplified in most human cancers (www.cbioportal.org). However, overexpression of ITCH has been observed in several human cancers including anaplastic thyroid carcinoma [48], breast cancer, ovarian cancer, and sarcomas [49]. Like other NEDD4 family genes, its regulation may primarily occur at the transcriptional and post-translational levels. ITCH demonstrates distinct roles through tagging different substrates for ubiquitination and hence modulating different signaling pathways. Therefore, it is not surprising that ITCH exhibits both oncogenic and tumor suppressor functions in different types of human cancers. ITCH is often activated when it receives stimulatory signals from upstream kinases or binding partners (Fig. 1A); thus, it may act primarily as a caretaker gene to fine-tune the downstream signaling pathways (Fig. 2).
Fig. 2.
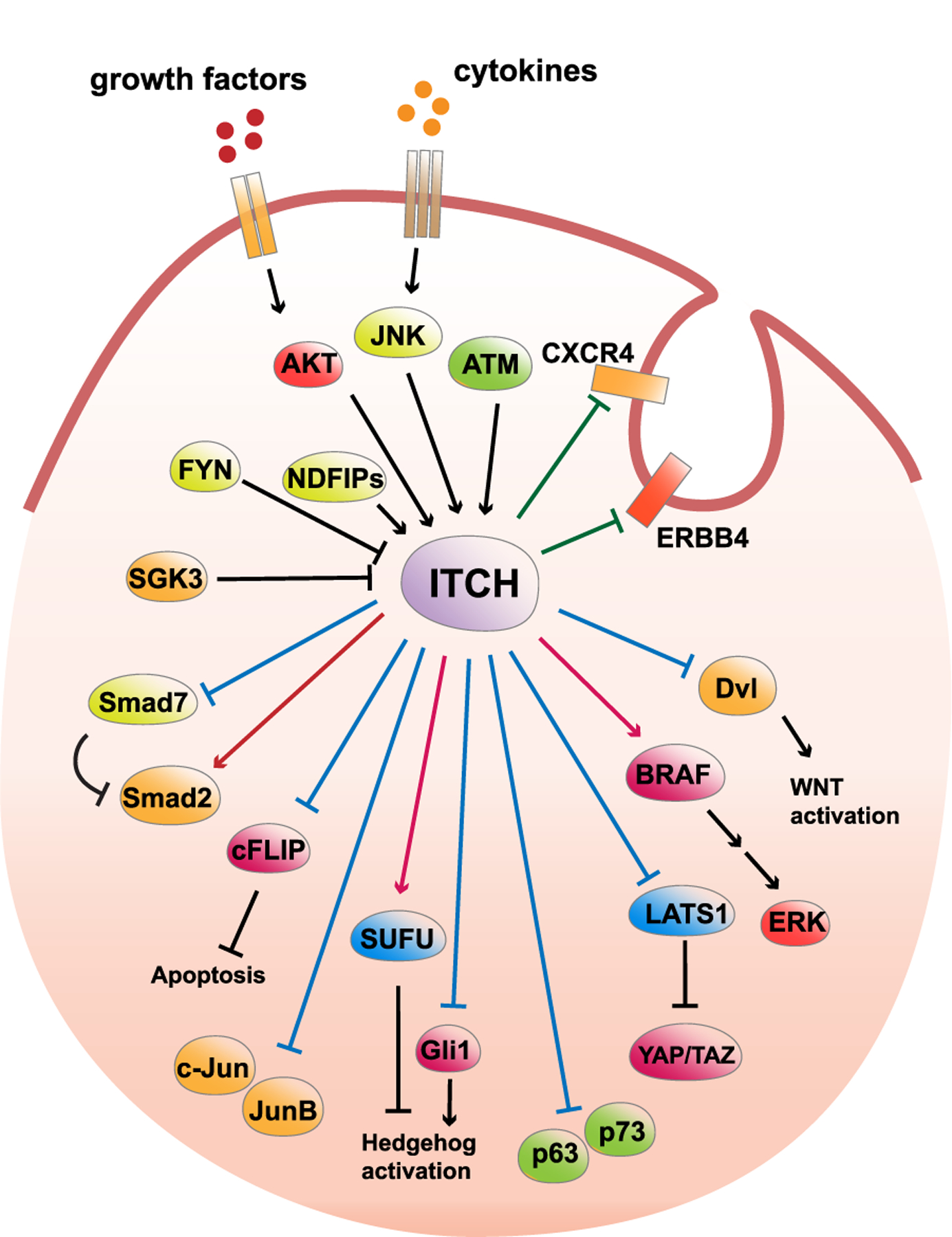
ITCH targets different signaling pathways in tumor cells.
The arrowhead from ITCH indicates positive regulation of the substrate protein, while the blunt head from ITCH indicates negative regulation of the substrate protein. Blue line indicates a K48-linked polyubiquitination of the substrate, red line indicates a non-proteolytic linkage, while green line indicates the target protein is subjected to endocytic degradation after modified by ITCH. The arrowhead towards ITCH indicate a positive regulation of ITCH E3 ligase activity, while the blunt head towards ITCH indicates negative regulation of ITCH E3 ligase activity.
4.1. ITCH in Hippo pathway regulation
The Hippo signaling pathway is a signaling relay carried out by a few key kinases and transcription factors which control cell proliferation, differentiation, and death [50]. The key components of the Hippo pathway consist of LATS1 and LATS2 kinases as well as YAP and TAZ transcriptional coactivators. Phosphorylation of YAP/TAZ by LATS1/2 leads to their cytoplasmic retention, degradation, and subsequently reduces their target gene expression [51]. LATS1 functions as a tumor suppressor by inducing G2-M arrest and promoting apoptosis [52,53]. Hyperactivation of YAP and TAZ is frequently found in human cancers to maintain cancer stem cell properties and to promote epithelial-mesenchymal transition (EMT) [54]. ITCH has been identified as an upstream E3 ligase for LATS1 to promote its proteolysis [49,55]. ITCH silencing in breast cancer cell lines MCF7 and MDA-MB-231 suppressed YAP-mediated cell growth [49,55], while the ectopic expression of ITCH transformed MCF10A cells in vitro [49]. A recent study uncovered that the deubiquitinase YOD1 stabilizes ITCH and enhances ITCH-mediated activation of YAP/TAZ transcription in liver cancer cells [45]. In contrast to the oncogenic roles of ITCH in destabilizing the tumor suppressor LATS1, Lim and colleagues discovered that ITCH also targets a YAP/TAZ transcription coactivator WBP2 for proteolysis in breast cancer cells, which positions ITCH as a tumor suppressor in breast cancer cells [56]. Notably, ITCH-controlled WBP2 degradation is blocked by WNT and EGFR activation through the disruption of ITCH/WBP2 interaction [56]. These findings suggest that ITCH could function as a potentiometer to precisely control the signal flux in response to different upstream inputs in tumor cells.
4.2. ITCH in Hedgehog pathway regulation
Like the Hippo pathway, the Hedgehog pathway was initially discovered from genetics studies using the fruit fly Drosophila melanogaster [57]. The Hedgehog pathway is composed of a chain of molecular events transmitted from the Hedgehog ligands via their receptors Patched (PTCH1) and GPCR-like protein Smoothened (SMO) to the GLI transcription factors. SUFU is a negative regulator for this signaling cascade [57]. The Hedgehog pathway drives the transcription of a panel of oncogenes including MYCN, CCND1, CCND2, BCL2, and many others [58]. ITCH seems to modulate the Hedgehog pathway at all the signaling transduction stages. GLI1 stability was found to be negatively regulated by ITCH with the assistance from the adaptor protein NUMB [42], indicating that ITCH opposes Hedgehog pathway activation. Similarly, ITCH targets GLI-similar 3 (GLIS3) transcription factors for proteolysis in pancreatic β cells [59]. Further, ITCH forms a complex with β-Arrestin 2 to catalyze K63-linked ubiquitination of SUFU, which increases the association of SUFU with GLI3 and facilitates the conversion of GLI3 into a repressor, thereby inhibiting the GLI-mediated Hedgehog pathway activation [40]. These findings together support a tumor suppressor role of ITCH through antagonizing the canonical GLI-dependent Hedgehog pathway.
In contrast to such a role, PTCH1 was found ubiquitinated at K1413 by ITCH, the ubiquitination provoked PTCH1 proteolysis, subsequent GLI activation, and cell survival [60]. On the other hand, ITCH-mediated activation of MAPK and TGF-β pathways facilitated PTCH1/SMO-independent activation of GLI transcription factors [61] and likely antagonized the proteolytic inhibition of the canonical Hedgehog signaling as described above.
4.3. ITCH in TGF-β pathway regulation
The TGF-β signaling pathway regulates cell proliferation, differentiation, apoptosis, and migration. Upon the binding of TGF-β to its receptor, SMAD2, SMAD3, and SMAD4 proteins are translocated from the cytoplasm to the nucleus for transcription initiation, whereas SMAD7 functioned as an inhibitory molecule to antagonize this circuit [62]. ITCH-dependent regulation of TGF-β signaling was uncovered by the Liu group through the identification of ITCH as the upstream E3 ligase for SMAD2. Interestingly, SMAD2 poly-ubiquitination by ITCH appeared to be non-proteolytic, which led to the increased activating phosphorylation of SMAD2 [63]. In contrast, ITCH suppressed TGF-β pathway activation by enhancing the association of SMAD7 with the activated TGFβ-RI [64]. In addition to modulating the canonical TGF-β pathway proteins, ITCH catalyzed the K27-linked poly-ubiquitination of TIEG1 which facilitated its transcriptional activation to boost FOXP3 expression [65,66]. ITCH was also found to regulate HEF1 stability by forming a complex with SMAD3 and HEF1 in a TGF-β-independent manner [67].
In addition to the canonical SMAD-dependent pathway, TGF-β also triggers JNK activation through TAK1 and MKK4, which are MAP3K and MAP2K for the JNK kinase [68]. JNK-dependent phosphorylation and subsequent activation of ITCH may further amplify the canonical SMAD-mediated transcription program. Intriguingly, ITCH also targets TAK1 for proteolytic ubiquitination in T cells [69], suggesting a negative feedback regulation of this noncanonical TGF-β cascade. Given the context-dependent role of TGF-β pathway in tumorigenesis, it is reasonable to postulate that ITCH could function differentially in different cellular contexts to either promote or suppress tumor cell proliferation and metastasis.
4.4. ITCH in AP-1 transcription factor regulation
The AP-1 transcription factors c-JUN and JUNB are the founding members of the ITCH ubiquitin substrates (Table 1) [70,71]. Upon TCR-mediated JNK activation in T cells, E3 ligase activity of ITCH is stimulated by JNK-mediated phosphorylation (Fig. 1A), which drives the ubiquitination of JUN proteins [29]. Paradoxically, JNK itself is the activating kinase for JUN family transcription factors. JNK phosphorylates c-JUN at its N-terminus to stimulate JUN transcriptional activity by promoting either an interaction with basal transcriptional machinery or coactivators [72]. In such a scenario, ITCH seems to provide negative feedback regulation on AP-1 activation. This is particularly important because AP-1 transcription factors are double-edged swords in tumor cells; they facilitate proliferation but at the same time provoke apoptosis [73]. The timely extinction of the JUN signal is vital to properly utilizing this oncogenic pathway; therefore, it is not surprising that JUN proteins are subjected to proteolytic regulation by ITCH, SCFFBW7, and CUL4COP1 E3 ligases [72]. However, it remains largely unknown if these JUN-upstream E3s work cooperatively or competitively to control JUN protein stability in tumor cells.
4.5. ITCH in WNT and NOTCH signaling pathways
Both WNT and NOTCH signaling pathways play indispensable roles in embryonic development and tumorigenesis [74,75]. ITCH ubiquitinates the phosphorylated form of Dishevelled (DVL), a key molecule in mediating WNT signal transduction, for proteolysis, such that ITCH antagonizes WNT activation [76]. However, a recent study unveiled that ITCH facilitates the ubiquitin-dependent endocytosis of the WNT receptor LRP6 and promotes canonical WNT activation [77]. These results suggest that ITCH could have both positive and negative impacts on the canonical WNT signals. DVL directly regulates small GTPases RHOA, RAC and CDC42 to enhance cell mobility [78]. Conversely, the destabilization of DVL by ITCH indicates that ITCH may have a negative impact on the non-canonical WNT pathway in tumor cells.
An early study in 2000 reported that ITCH promotes NOTCH poly-ubiquitination [79]. The poly-ubiquitin chain assembled on NOTCH was later characterized as the K29-linkage: K29-linked ubiquitination of NOTCH facilitates its lysosomal degradation in the absence of the ligand [80]. Intriguingly, ITCH also facilitates the K29-linked ubiquitination of another NOTCH signaling component, Deltex, for lysosomal degradation [81]. The association of ITCH with lysosomal regulation of WNT and NOTCH signaling modules is further supported by the findings linking ITCH with ubiquitin-dependent sorting and degradation as described below.
4.6. ITCH regulates p63/p73 transcription factors
The proteins p63 and p73 are p53 homolog transcription factors that play important roles in multiple biological processes including skin morphogenesis, regeneration, tumorigenesis, and response to chemotherapy [82]. In the epidermis, ITCH mediates the degradation of p63 to regulate epidermal keratinocyte differentiation [83]. On the other hand, p73 is upregulated in response to DNA damage, cell cycle arrest and apoptosis [84–86]. When cells are not under stressful conditions, the basal level of p73 is kept at a low level through ITCH-mediated ubiquitination and proteasome-dependent degradation. In response to DNA damage, ITCH is rapidly degraded which stabilizes p73 and leads to growth arrest and apoptosis [87]. Given the tumor suppressor roles of both p63 and p73 in human cancers, it is conceivable that ITCH may exhibit oncogenic functionality to accelerate p63 and p73 turnover in skin or lung cancers, where p63 and p73 display a profound tumor suppressor role.
4.7. ITCH regulates endosome and lysosome functions
In addition to proteasome-dependent proteolysis, ubiquitin-dependent sorting of integral membrane proteins for degradation in lysosomes is an important mechanism to terminate extracellular signals or to recycle molecules back to the plasma membrane [88]. Cancer cells have frequently been observed to escape from endocytic degradation through receptor tyrosine kinase (RTK) mutants [89]. ITCH has been demonstrated to participate in endocytic sorting by regulating several key endocytic regulators. ITCH was found localized within the endosomal system where it bound to endophilin A1 and promoted its endocytic degradation [90]. Similarly, ITCH mediates agonist-dependent ubiquitination of the chemokine receptor CXCR4 and the HGF-regulated tyrosine kinase substrate Hrs, the latter plays a central role in endocytic sorting [91]. PI3-kinase-dependent activation of the cytokine-independent survival kinase CISK/GSK3 antagonizes ITCH-mediated CXCR4 degradation and therefore sustains CXCR4-mediated oncogenic signals [33]. The endocytic scaffolding protein intersectin1 (ITSN1), on the other hand, is regulated differently by ITCH depending on the isoform that is modified. The major isoform ITSN1-s is monoubiquitinated, while the minor isoform, ITSN1–22a, undergoes a combination of mono- and oligoubiquitination. The monoubiquitination of ITSN1-s promotes its stabilization, whereas the oligoubiquitination of the minor isoform leads to its proteasomal degradation [92]. ITCH has also been shown to form a functional complex with the phosphatidylinositol (PI) 4-kinase type IIα (PI4KIIα) through which ITCH promotes the non-proteolytic ubiquitination of PI4KIIα, and as a result, facilitates the internalization of the WNT-activated frizzled 4 (Fz4) receptor [93].
4.8. ITCH in DNA damage response
Dysregulation of the DNA damage response (DDR) pathway is crucial for tumor cells to cope with genomic instability-triggered cell death pathways [94,95]. Poly-ubiquitinated histone H1 serves as an important signaling intermediate to stimulate RNF8/RNF168-mediated DDR at DNA damage sites for 53BP1 foci formation following ionizing radiation (IR) [96]. ITCH-mediated polyubiquitination of H1.2 suppresses RNF8/RNF168-dependent formation of the 53BP1 foci, which plays an important role in DNA damage response. AKT-mediated activation of the ITCH-H1.2 axis may confer triple-negative breast cancer (TNBC) cells to counteract the replication stress and to facilitate tumor cell survival in response to DNA damage [31].
4.9. ITCH in other signaling pathways
ITCH has been found to regulate the RTK-MAPK signaling module. Meijer et al. reported that ITCH catalyzes mono-ubiquitination and K63-linked poly-ubiquitination of ERBB4 isoform CYT-1, which promotes the endocytic degradation of ERBB4 CYT-1. ITCH-dependent degradation of ERBB4 suppresses EGF-dependent oncogenic programs [97,98].
Downstream of the RTK, BRAF plays an indispensable role in activating the MEK/ERK pathway to drive tumorigenesis in melanoma [99–102]. Our group reported that, in response to proinflammatory cytokines, BRAF is subjected to K27-linked poly-ubiquitination in melanoma cells by ITCH. K27-linked ubiquitination of BRAF recruits PP2A to antagonize the S365 phosphorylation and disrupt the inhibitory interaction with 14–3-3, leading to sustained BRAF activation and subsequent elevation of the MEK/ERK signaling [103].
ITCH has been demonstrated to ubiquitinate the anti-apoptotic protein c-FLIP for degradation [104]. The ubiquitination and degradation of c-FLIP are partly responsible for the cytotoxic activity of TNFα, which activates ITCH via JNK-mediated phosphorylation [29]. On the other hand, ITCH was found to destabilize MKK4, an upstream MAP2K that activates the JNK/p38 kinases, providing a form of feedback regulation to the MEKK1-MKK4-JNK signaling axis [105].
5. Immunological functions of ITCH and their implications in tumorigenesis
The most remarkable phenotype found in the Itchy mice (Itcha18H/a18H) is the presence of multi-organ inflammation and autoimmune disease [6]. Therefore, it is not surprising that ITCH is involved in the control of many aspects of lymphocytic function and host immune defense, which includes T-cell activation and anergy, T-helper cell differentiation, innate immunity, B cell development, and viral infection. Several ITCH substrates described above in the discussion of tumorigenesis are also central players or modulators of these processes. Though most immunological functions of ITCH were identified through studying the peripheral immune system of the Itchy mice, such mechanisms may shed light on the potential roles that ITCH plays in the tumor immune microenvironment (Fig. 3).
Fig. 3.
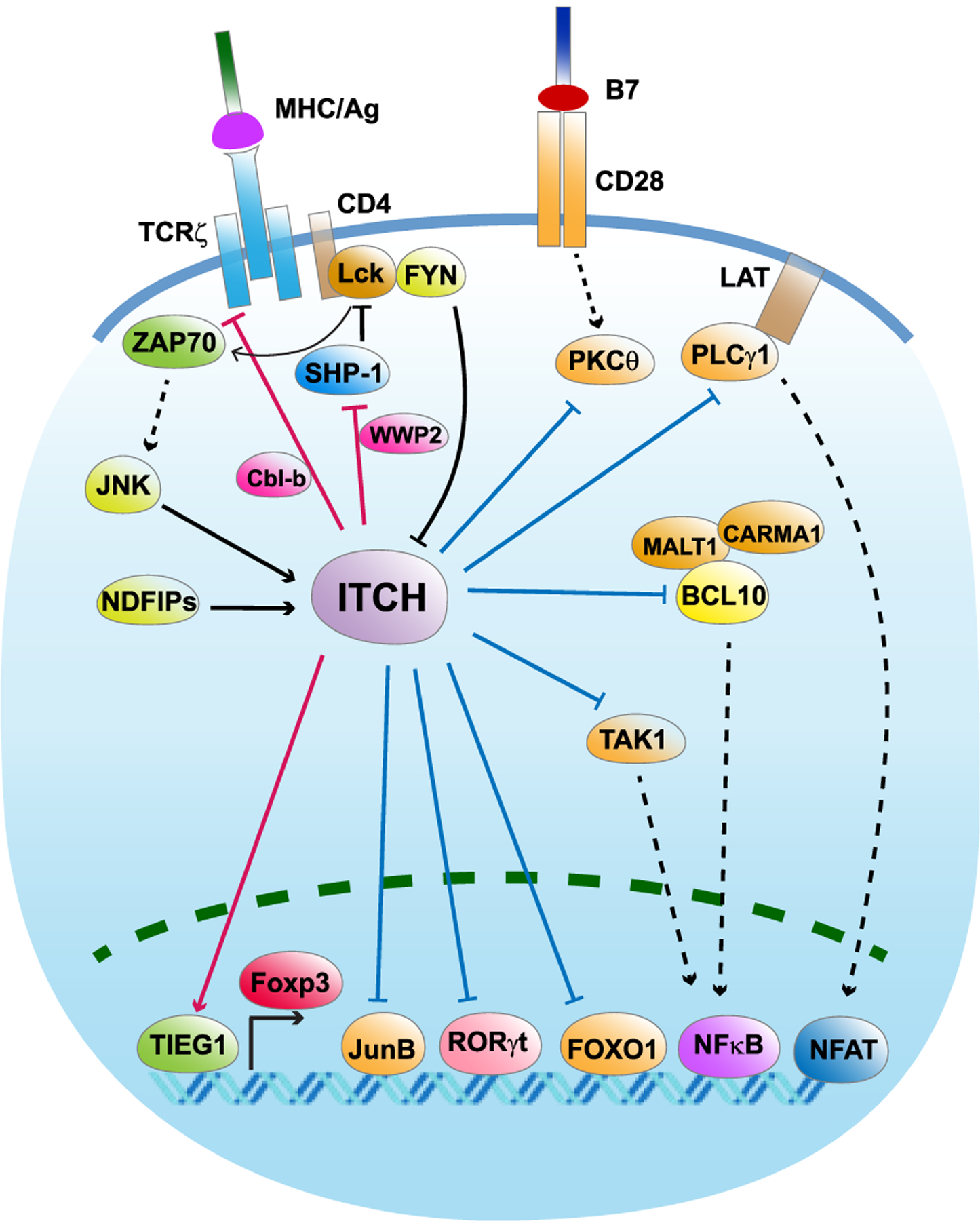
ITCH plays multifaceted roles in T cells.
The arrowhead from ITCH indicates positive regulation of the substrate protein, while the blunt head from ITCH indicates negative regulation of the substrate protein. Blue line indicates a K48-linked polyubiquitination of the substrate, while red line indicates a non-proteolytic linkage. The arrowhead towards ITCH indicates a positive regulation of ITCH E3 ligase activity, while the blunt head towards ITCH indicates negative regulation of ITCH E3 ligase activity.
5.1. The tumor immune microenvironment
Tumors are not isolated from the host tissues, but rather they are surrounded by the tumor microenvironment (TME), which contains blood vessels, extracellular matrix (ECM), signaling molecules and tumor infiltrated immune cells [106–111]. Immune cells present in the TME mainly include T lymphocytes, B lymphocytes, macrophages, dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), natural killer (NK) cells, and natural killer T (NKT) cells [112–115]. The communication between tumor cells and tumor infiltrated immune cells occurs at every stage of tumor development. The functions of the immune cells in recognizing, eliminating and promoting tumor cell development have been investigated for decades.
It is generally accepted that a high number of interferon (IFN)-γ secreting CD8+ T cells and CD4+ T helper 1 cells (Th1), tumor-associated M1 macrophages, NK cells, type I NKT cells and mature DCs probably correlates with good prognosis in cancer patients [116–122]. One the other hand, a subset of immune cells like Tregs, MDSCs, M2 macrophages, and immature DCs facilitates tumor progression by antagonizing antitumor immunity [113,123–125]. Although Th2 and Th17 cells are initially characterized as tumor-promoting lymphocytes, several groups reported that they play rather contradictory roles in different tumor settings [115,124,126–128]. Such context-dependent roles are also observed for B lymphocytes [106,129–131]. The double-sided roles of these cells in tumorigenesis are likely due to cytokines and other molecules they produced. These molecules exhibit distinct functions in different TME through regulating a wide variety of signaling pathways not only in tumor cells but also in immune and stromal cells [113,125,132]. One of such examples is the signaling networks ITCH influences in the immune system and its potential impact on the tumor immune microenvironment.
5.2. ITCH in T cell activation
The T cell antigen receptor (TCR) is the starting point of T cell activation upon its interaction with antigenic ligands, which are short peptide fragments bound to MHC class I or class II molecules [133]. ITCH has been shown to directly modulate TCR-ζ function [134]. ITCH also works together with Cbl-b to promote K33-linked ubiquitination of TCR-ζ, which inhibits TCR-ζ’s binding to ZAP70 and suppresses the phosphorylation and subsequent activation of ZAP70 [134]. This finding supports a model in which ITCH cooperates with Cbl-b to inhibit TCR activation. Further, ITCH has been demonstrated to regulate Ca2+-induced T cell anergy through the ubiquitination and degradation of PKCθ and PLC-γ1, both of which are vital for calcium signal-mediated T cell activation [135,136]. However, in a recent study, CD4+ T cells with both Itch and Wwp2 deleted exhibit a compromised TCR activation and a bias toward Th2 cells [137]. ITCH and WWP2 form a complex to catalyze K27-linked ubiquitination of the phosphatase SHP-1, which disrupts the interaction between SHP-1 and the tyrosine kinase Lck; this leads to the promotion of TCR signaling [137]. During TCR activation, Lck phosphorylates TCR-ζ and ZAP70 to facilitate the activation of downstream NF-κB and MAPK signals. SHP-1 functions as a negative regulatory element to terminate TCR signaling by depho-sphorylating Lck. These observations indicate that the balance between WWP2 and Cbl-b functions could play an important role in directing ITCH’s role in TCR signaling (Fig. 3).
5.3. ITCH in Th2 cell differentiation
T helper cells are activated after the naïve CD4+ Th cells recognize and interact with an antigen-MHC class II molecule complex. The activated Th cells secrete cytokines to facilitate the activation of B cells, cytotoxic T cells, macrophages, and various other cell types that participate in the immune response. Different sets of cytokines produced by activated Th cells dictate different immune responses [138]. There is mounting evidence suggesting the central, while complicated, roles of T helper cells in immune response and tumor immunity, which is largely owing to the complexity of T helper cell subtypes including Th1, Th2, Th9, Th17, and T-follicular helper (Tfh) cells [139].
Compared to Th1 cells, which produce IFN-γ and IL-2 as their signature cytokines, Th2 cells mainly secrete cytokines such as IL-4, IL-5, and IL-13; thus, Th2 cells are essential for host defense. Itch−/− T cells exhibit an elevated production of the Th2 cytokines IL-4 and IL-5 without affecting IFN-γ production, suggesting that ITCH negatively regulates Th2 polarization [70]. Such a phenotype was due to ITCH-mediated proteolytic ubiquitination of the JUNB transcription factor [70]. Upon TCR stimulation, JNK phosphorylates ITCH to facilitate its activation, leading to the ubiquitination and degradation of JUNB which in turn suppressed IL-4 production and Th2 differentiation [140]. Consistent with ITCH’s role in restraining Th2 differentiation, genetic ablation of Ndfip1 in the mouse phenocopied the biased differentiation of Th2 cells observed in Itcha18H/a18H mice [36]. Th2 cells have a dual role in tumor immunity due to the context-dependent functions of Th2 cytokines, a review on this topic could be found at [141].
Th2 cells are generally considered as tumor-promoting through secreting immunosuppressive cytokines such as IL-10 [141]. However, other Th2 cytokines may exhibit context-dependent functions in different tumor microenvironments and therefore provides a rather controversial role for Th2 cells in tumor immunity [141]. At least, it has been reported that in coordination with other cell types, such as tumor-infiltrating eosinophils, Th2 cells exhibit anti-tumor activity [142,143].
5.4. ITCH in Treg cells
The induction of regulatory T cells (Tregs) from naïve CD4+ T cells is driven by TGF-β and IL-2 signals, which stimulates the expression of the Foxp3 transcription factor [144]. There are two types of Treg cells identified, some are generated in the thymus early in life, thus named thymic Treg cells (tTreg or nTreg); others are generated in various tissues of the body throughout life, and are named peripheral Treg cells (pTreg or iTreg) [144]. pTreg, not tTreg, cells are found decreased in Itch deficient mice, suggesting a positive role of ITCH in promoting pTreg differentiation [65,66]. Mechanistically, IL-6 activates the tyrosine kinase Tyk2 to phosphorylate TIEG1 at Y179, which promotes ITCH-mediated K27-linked polyubiquitination of TIEG1. TIEG1 acts downstream of TGF-β to promote Foxp3 expression and pTreg cell differentiation [65,66]. In an effort to specifically investigate the roles ITCH plays involving Treg cells, Treg cell-specific Itch knockout mice were generated (Itchfl/fl;Foxp3-Cre) [145]. Surprisingly, the absence of Itch in tTregs does not affect Foxp3 expression or the overall tTreg development, suggesting a secondary effect of IL-4 production rather than an intrinsic role of ITCH in pTreg cells.
Treg cells suppress the proliferation and function of a variety of anti-tumor immune cell populations by producing IL-10, TGF-β and cytotoxic T-lymphocyte antigen 4 (CTLA4) [146]. Clinical studies show that tumor-infiltrating Tregs usually correlate with poor prognosis in a variety of cancers including lung cancer [147] and pancreatic cancer [148]. However, in certain types of cancers such as head and neck cancer [149] and bladder cancer [150], tumor-infiltrating Tregs seem to predict better prognosis.
5.5. ITCH in Th17 cells
Th17 cells are characterized by the production of IL-17 and IL-22 cytokines [151]. IL-17 cytokines are double-edged agents and, depending on different types of cancer, it can be either tumor-suppressive or tumorigenic [152]. ITCH negatively regulates Th17 differentiation through the ubiquitination and degradation of the RORγt transcription factor, a lineage marker for the Th17 cell population [153]. It has been consistently shown that NDFIP1, which activates the catalytic activity of ITCH, also promotes RORγt degradation and reduces IL-17 production in Th17 cells [154]. Accumulation of IL-17 in the colonic mucosa has been attributed to spontaneous colonic inflammation and colorectal carcinogenesis, suggesting a tumor suppressor role of ITCH in inflammation-induced colon cancer [153].
5.6. ITCH in Tfh and B lymphocytes
One of the major functions of CD4+ T cells is to assist B lymphocyte development and antibody production. T follicular helper (Tfh) cells are a specialized subset of CD4+ T cells required for germinal center reactions which promote B cell proliferation, somatic hypermutation, and class-switch recombination [155]. ITCH facilitates Tfh cell differentiation by targeting FOXO1, a negative regulator of Tfh differentiation, for proteolysis [156]. In addition to Tfh cells, FOXO1 has been shown to possess an important role in suppressing the activated Treg (aTreg) cell population [157]. Therefore, it is worth further investigating the role of ITCH in controlling aTreg cells, especially in tumor-infiltrating lymphocytes. Notably, although Tfh cells exhibit a dual role in the tumor immune microenvironment [158], CXCL13-associated Tfh and B cell infiltration has been demonstrated as a favorable prognostic marker in colon cancer patients [159].
B cells are responsible for humoral immunity through antigen presentation to T cells and secretion of antigen-specific antibodies, while dysregulated development and activation of B cells are associated with immunodeficiency and autoimmunity [160]. B cells are also subjected to ITCH-dependent regulations. There was an increased germinal center B cell number found in Itcha18H/a18H mice, and Itch deficient B cells which exhibited increased proliferation and glycolytic capacity largely due to mTORC1 activation [161]. However, the mechanism by which ITCH modulates B cell proliferation remains unexplored. In an independent study, conditional knockout of Itch in mouse B cells (Itchfl/fl;Cd19-Cre) resulted in fewer pro-B cells in the bone marrow, more small resting IgM-IgD-B cells in the periphery, and lower B-cell numbers in the lymph nodes [162].
5.7. ITCH in the regulation of inflammatory cytokines
Chronic inflammation is a risk factor for developing several types of human cancers. Epidemiology studies revealed a 14 % increase in prostate cancer risk due to prostatitis, a 25 % increase in colorectal cancer risk due to ulcerative colitis, and a 10–20-fold increase in the risk of pancreatic cancer for patients who have experienced pancreatitis [163]. Since multi-organ inflammation has been found in Itcha18H/a18H mice, ITCH may exhibit anti-tumorigenic roles in these types of cancers. Using a syngeneic Lewis lung carcinoma model, Ahmed and colleagues observed increased tumor development in Itcha18H/a18H and Cyld−/−mice [69]. The mechanistic study showed that ITCH is complexed with CYLD to promote the transition of K63-linkage to K48-linkage via sequentially cleaving the K63-linked ubiquitin chain followed by catalyzing K48- linked ubiquitination on the TAK1 protein. Such ubiquitin linkage switches triggers the proteasomal degradation of TAK1 and an attenuation of the inflammatory response [69]. Moreover, ITCH targets the scaffolding protein BCL10 for proteolysis [135]. BCL10 is a component of the lymphocyte-specific CARMA1/CARD11–BCL10–MALT1 (CBM-1) signalosome that stimulates the MALT1 protease to activate canonical NF-κB and JNK signaling [164].
6. Targeting ITCH in human cancers
Targeting ubiquitin enzymes is an attractive therapeutic approach for the treatment of multiple cancers. One of the best examples is the discovery of the FDA approved proteasome inhibitor Bortezomib and Carfilzomib for the treatment of multiple myeloma [165]. Compared to efforts against the proteasome or the E1/E2 enzymes, targeting ubiquitin E3 ligases appears to be a more specific approach given the fact that E3s dictate the substrate specificity in the ubiquitin–proteasome system. To date, only three compounds target the ubiquitin E3 ligase cereblon. Namely, the teratogenic compounds thalidomide, and its analogs lenalidomide and pomalidomide, have been approved by the FDA for this use [166]. Compared to RING family E3s, only limited efforts have been devoted to targeting HECT type E3 ligases. A pan-HECT inhibitor heclin was discovered from a screen that targets the HECT domain catalytic cysteine for oxidation, thereby inhibiting HECT E3 activity [167]. In another study, indole-3-carbinol (I3C) derivatives were found to bind the HECT domain of NEDD4 and suppress its E3 ligase activity [168]. Notably, I3C has recently been demonstrated to also inhibit WWP1 activity and restore PTEN tumor suppressor activity in a MYC-driven prostate cancer mouse model [169]. High throughput screening approaches (using ITCH autoubiquitination as a marker ITCH activity) identified clomipramine, which is an antidepressant drug, as a putative ITCH inhibitor that suppressed the growth of breast, prostate and bladder cancer cell lines. Mechanistically, clomipramine inhibits ITCH autoubiquitination by interfering with ubiquitin transthiolation activity in an irreversible manner [170]. Due to the highly conserved mechanisms of ubiquitin transfer and substrate recruitment among NEDD4 family E3s, there is uncertainty surrounding the specificity of these NEDD4 E3 inhibitors [171]. Therefore, future in-depth investigations on both the specificity and in vivo preclinical studies are urgently needed.
7. Conclusion and perspectives
Since its discovery in 1995, ITCH’s roles in maintaining a balanced immune response have been extensively investigated [172]. Our knowledge of the immunological functions of ITCH are mainly obtained from the studies of the non-lethal a18H agouti strain (Itcha18H/a18H) and the conditional knockout Itchfl/fl strain. Despite the autoimmune disease found in Itcha18H/a18H mice, no obvious malignancy was reported. The lack of malignancy in Itcha18H/a18H mice may be due to their short life span, which is typically 6–8 months on the C57BL/6 J background [70]. Given the accelerated tumor formation of colon cancer and lung cancer cells in Itcha18H/a18H mice [69,153], it will be of great interest to examine if loss of Itch facilitates colon cancer and lung cancer development in genetically engineered mouse models (GEMMs). Most of the evidence accumulated to date on the role of ITCH in tumorigenesis were derived from in vitro or ex vivo models. It is therefore likely that the absence of an intact tumor microenvironment might not fully reflect all of the pathophysiological changes that ITCH could influence at both the cancer cell-intrinsic and the systemic levels. In addition to germline deletions, specifically targeting Itch expression in conditional GEMMs may provide an important opportunity to examine the role of Itch in tumorigenesis, especially in aged mice. For example, an Itchfl/fl;KrasLSL−G12D/+;p53fl/fl lung cancer GEMM would be valuable to assess the roles Itch plays in KrasG12D-driven lung adenocarcinoma. More importantly, further introducing the Cd4-Cre allele will provide a set of genetically defined models to fully deduce the roles of Itch in the lung epithelium and the T helper cells.
Genetic alterations in the ITCH gene occur infrequently in the most common types of human cancer. Somatic mutations of ITCH were found in 3.4 % of HER2+ breast cancer and 7.9 % of triple negative breast cancer cases [56]. Among these mutations, the E855 K mutant of ITCH exhibits a loss of E3 ligase activity, and attenuation of ITCH function has been shown to promote WNT pathway activation and facilitate breast cancer cell survival [56]. In contrast to the low frequency of somatic ITCH mutations observed, there is good evidence that post-translational modifications modulate ITCH function in response to different upstream signals (Fig. 1A). Our group has recently identified a cytokine-stimulated maintenance of ERK activation by JNK/ITCH-mediated BRAF non-proteolytic ubiquitination in BRAFWT melanoma cells [103]. This provides a mechanistic explanation of the pro-survival roles of the inflammatory cytokines in melanoma cells [173]. DNA damage activates the ATM kinase, which promotes the activation of ITCH [30], and ITCH in turn destabilizes the p63 [83] and p73 [87] tumor suppressors to alleviate the apoptotic stress. This signaling axis may contribute to the oncogenic functions of ITCH in skin cancers. The adaptor proteins NDFIP1 and NDFIP2 facilitate ITCH activation by relieving the intramolecular interaction between the WW and HECT domains. Expression levels of these scaffolding proteins may impact ITCH activity in certain types of cancers. Of note, NDFIP1 has been found overexpressed in the TCGA-PAAD and TCGA-THYM datasets, indicating a potential hyper-activation of ITCH in pancreatic adenocarcinoma and thymoma tumors. Looking forward, the development of cancer type-specific ITCH animal models and the design of specific ITCH inhibitors will be important for us to better understand and to specifically target this important signaling molecule in human cancers.
Acknowledgments
This work was supported in part by the National Institutes of Health (NIH) grant to L.W. (CA183914) and the Melanoma Research Alliance.
Abbreviations:
- AIP4
Atrophin-interacting protein 4
- AP-1
activator protein-1
- ATM
ataxia telangiectasia mutated
- aTreg
activated Treg
- BCL10
B-cell lymphoma/leukemia 10
- c-FLIP
cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein
- CBL-b
Casitas B lymphoma-b
- CBM-1
CARMA1/CARD11-BCL10-MALT1
- CISK
cytokine-independent survival kinase
- COP1
constitutive photomorphogenesis 1
- CTD
C-terminal domain
- CTLA4
cytotoxic T-lymphocyte antigen 4
- CUL4
cullin 4
- CXCR4
C-X-C chemokine receptor type 4
- CYLD
cylindromatosis
- DDR
DNA damage response
- DTX
deltex
- DUB
deubi-quitinating enzyme
- E1
E1 activating enzyme
- E2
E2 conjugating enzyme
- E3
E3 ubiquitin ligase
- ECM
extracellular matrix
- EGFR
epidermal growth factor receptor
- EMT
epithelial-mesenchymal transition
- FBW7
F-box and WD repeat domain-containing 7
- FDA
food and drug administration
- FOXO1
forkhead box protein O1
- Fz4
frizzled 4
- GEMM
genetically engineered mouse model
- GLIs3
Gli-similar 3
- GPCR
G protein-coupled receptor
- HECT
homologous to E6AP C terminus
- HECW1
HECT, C2 and WW domain containing E3 ubiquitin protein ligase 1
- HECW2
HECT, C2 and WW domain containing E3 ubiquitin protein ligase 2
- HEF1
human enhancer of filamentation 1
- HER2
human epidermal growth factor receptor 2
- HERC
HECT and RCC1-like
- Hrs
HGF-regulated tyrosine kinase substrate
- HUWE1
HECT, UBA and WWE domain containing 1
- I3C
indole-3-carbinol
- IFN
interferon
- IKK
I-kappa B kinase complex
- IL
interleukin
- IR
ionizing radiation
- ITSN1
intersectin1
- JNK
c-JUN N-terminal kinase
- KSHV
Kaposi’s sarcoma-associated herpesvirus
- LATS1
large tumor suppressor kinase 1
- LDL
low-density lipoprotein
- LRAD3
the low-density-lipoprotein receptor class A domain containing 3
- LRP6
low-density lipoprotein related protein 6
- MAP2K
mitogen-activated protein kinase kinase
- MAPK
mitogen-activated protein kinase
- MDSC
myeloid suppressive cell
- MDSCs
myeloid-derived suppressor cells
- MEKK1
mitogen-activated protein kinase kinase kinase 1
- MHC
major histocompatibility complex
- MKK4
mitogen-activated protein kinase kinase 4
- mTORC1
mechanistic target of rapamycin complex 1
- MVBs
multivesicular bodies
- N4BP1
NEDD4-binding partner 1
- NDFIP1
NEDD4 family-interacting protein 1
- NDFIP2
NEDD4 family-interacting protein 2
- NDFIPs
NEDD4 family-interacting proteins
- NEDD4
neural precursor cell-expressed developmentally downregulated gene 4
- NEDD4L
NEDD4 like E3 ubiquitin protein ligase
- NEDL1
NEDD4-like ubiquitin protein ligase-1
- NEDL2
NEDD4-like ubiquitin protein ligase-2
- NF-E2
nuclear factor ery-throid- derived 2
- NF-κB
nuclear factor-kappa B
- NK
natural killer cell
- NKT
natural killer T cell
- NMR
nuclear magnetic resonance
- PAAD
pancreatic adenocarcinoma
- PCBP2
poly(rC) binding protein 2
- PI4KIIα
phosphatidylinositol (PI) 4-kinase type Iiα
- PKC
protein kinase C
- PLC-γ1
phospholipase C-γ1
- PTCH1
patched-1 protein
- pTreg or iTreg
peripheral Treg cells
- RANKL
receptor activator of NF-κB ligand
- RING
really interesting new gene
- RIP2
receptor interacting serine/threonine protein kinase 2
- RLDs
RCC1-like domains
- RORγt
retinoic acid-related orphan receptor gamma t
- RTA
transcription activator
- RTK
receptor tyrosine kinase
- SCF
SKP1-CUL1-F-box protein
- SGK3
serum and glucorticoid regulated kinase family member 3
- SHH
sonic Hedgehog
- SHP-1
Src homology 2 domain-containing protein tyrosine phosphatase 1
- SMAD2
mothers against decapentaplegic homolog 2
- SMAD3
mothers against decapentaplegic homolog 3
- SMAD7
mothers against decapentaplegic homolog 7
- SMURF1
Smad ubiquitination regulatory factor 1
- SMURF2
Smad ubiquitination regulatory factor 2
- STAM-1
signal-transducing adaptor molecule-1
- SUFU
suppressor of fused homolog
- TAK1
transforming growth factor-β activated kinase-1
- TAM
tumor associated macrophage
- TAp73
transcriptionally active p73
- TCGA
the cancer genome atlas
- TCR
T-cell receptor
- Tfh cells
T follicular helper cells
- TGF-β
transforming growth factor-β
- TGFβ-RI
transforming growth factor-β type I receptor
- Th cells
T helper cells
- THYM
thymoma
- TIEG1
TGF-β-inducible early gene 1 product
- TME
tumor microenvironment
- TNBC
triple-negative breast cancer
- TRAF6
Tumor necrosis factor receptor-associated factor 6
- Treg
regulatory T cell
- TRP
transient receptor potential
- TRPV4
transient receptor potential cation channel subfamily V member 4
- tTreg or nTreg
thymic Treg cells
- TXNIP
thioredoxin-interacting protein
- USP9X
ubiquitin specific peptidase 9, X-linked
- vFLIP
viral FLICE inhibitory protein
- WWP1
WW domain-containing protein 1
- WWP2
WW domain-containing protein 2
- YAP
Yes-associated protein
- ZAP70
zeta-chain-associated protein kinase 70
Footnotes
Declaration of Competing Interest
The authors declare no conflict of interest.
References
- [1].Rape M, Ubiquitylation at the crossroads of development and disease, Nat. Rev. Mol. Cell Biol 19 (2018) 59–70, 10.1038/nrm.2017.83. [DOI] [PubMed] [Google Scholar]
- [2].Hershko A, Ciechanover A, The ubiquitin system, Annu. Rev. Biochem 67 (1998) 425–479, 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- [3].Rotin D, Kumar S, Physiological functions of the HECT family of ubiquitin ligases, Nat. Rev. Mol. Cell Biol 10 (2009) 398–409, 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- [4].Wood JD, Yuan J, Margolis RL, Colomer V, Duan K, Kushi J, Kaminsky Z, Kleiderlein JJ, Sharp AH, Ross CA, Atrophin-1, the DRPLA gene product, interacts with two families of WW domain-containing proteins, Mol. Cell. Neurosci 11 (1998) 149–160, 10.1006/mcne.1998.0677. [DOI] [PubMed] [Google Scholar]
- [5].Perry WL, Hustad CM, Swing DA, O’Sullivan TN, Jenkins NA, Copeland NG, The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice, Nat. Genet 18 (1998) 143–146, 10.1038/ng0298-143. [DOI] [PubMed] [Google Scholar]
- [6].Aki D, Zhang W, Liu Y-C, The E3 ligase Itch in immune regulation and beyond, Immunol. Rev 266 (2015) 6–26, 10.1111/imr.12301. [DOI] [PubMed] [Google Scholar]
- [7].Lorenz S, Structural mechanisms of HECT-type ubiquitin ligases, Biol. Chem 399 (2018) 127–145, 10.1515/hsz-2017-0184. [DOI] [PubMed] [Google Scholar]
- [8].Scheffner M, Kumar S, Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects, Biochim. Biophys. Acta 1843 (2014) 61–74, 10.1016/j.bbamcr.2013.03.024. [DOI] [PubMed] [Google Scholar]
- [9].Pickart CM, Eddins MJ, Ubiquitin: structures, functions, mechanisms, Biochim. Biophys. Acta 1695 (2004) 55–72, 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- [10].Bernassola F, Karin M, Ciechanover A, Melino G, The HECT family of E3 ubiquitin ligases: multiple players in cancer development, Cancer Cell 14 (2008) 10–21, 10.1016/j.ccr.2008.06.001. [DOI] [PubMed] [Google Scholar]
- [11].Fajner V, Maspero E, Polo S, Targeting HECT-type E3 ligases - insights from catalysis, regulation and inhibitors, FEBS Lett. 591 (2017) 2636–2647, 10.1002/1873-3468.12775. [DOI] [PubMed] [Google Scholar]
- [12].Morreale FE, Walden H, Types of ubiquitin ligases, Cell 165 (2016) 248–248.e1, 10.1016/j.cell.2016.03.003. [DOI] [PubMed] [Google Scholar]
- [13].Ingham RJ, Gish G, Pawson T, The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture, Oncogene 23 (2004) 1972–1984, 10.1038/sj.onc.1207436. [DOI] [PubMed] [Google Scholar]
- [14].Mari S, Ruetalo N, Maspero E, Stoffregen MC, Pasqualato S, Polo S, Wiesner S, Structural and functional framework for the autoinhibition of Nedd4-family ubiquitin ligases, Structure 22 (2014) 1639–1649, 10.1016/j.str.2014.09.006. [DOI] [PubMed] [Google Scholar]
- [15].Ye X, Wang L, Shang B, Wang Z, Wei W, NEDD4: a promising target for cancer therapy, Curr. Cancer Drug Targets 14 (2014) 549–556, 10.2174/1568009614666140725092430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen C, Matesic LE, The Nedd4-like family of E3 ubiquitin ligases and cancer, Cancer Metastasis Rev. 26 (2007) 587–604, 10.1007/s10555-007-9091-x. [DOI] [PubMed] [Google Scholar]
- [17].Dunn R, Klos DA, Adler AS, Hicke L, The C2 domain of the Rsp5 ubiquitin ligase binds membrane phosphoinositides and directs ubiquitination of endosomal cargo, J. Cell Biol 165 (2004) 135–144, 10.1083/jcb.200309026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Plant PJ, Lafont F, Lecat S, Verkade P, Simons K, Rotin D, Apical membrane targeting of Nedd4 is mediated by an association of its C2 domain with annexin XIIIb, J. Cell Biol 149 (2000) 1473–1484, 10.1083/jcb.149.7.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Staub O, Rotin D, WW domains, Structure 4 (1996) 495–499, 10.1016/S0969-2126(96)00054-8. [DOI] [PubMed] [Google Scholar]
- [20].Wiesner S, Ogunjimi AA, Wang H-R, Rotin D, Sicheri F, Wrana JL, Forman-Kay JD, Autoinhibition of the HECT-type ubiquitin ligase Smurf2 through its C2 domain, Cell 130 (2007) 651–662, 10.1016/j.cell.2007.06.050. [DOI] [PubMed] [Google Scholar]
- [21].Wan L, Zou W, Gao D, Inuzuka H, Fukushima H, Berg AH, Drapp R, Shaik S, Hu D, Lester C, Eguren M, Malumbres M, Glimcher LH, Wei W, Cdh1 regulates osteoblast function through an APC/C-independent modulation of Smurf1, Mol. Cell 44 (2011) 721–733, 10.1016/j.molcel.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang Z, Liu Z, Chen X, Li J, Yao W, Huang S, Gu A, Lei Q-Y, Mao Y, Wen W, A multi-lock inhibitory mechanism for fine-tuning enzyme activities of the HECT family E3 ligases, Nat. Commun 10 (2019) 3162, 10.1038/s41467-019-11224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Weber J, Polo S, Maspero E, HECT E3 ligases: a tale with multiple facets, Front. Physiol 10 (2019) 370, 10.3389/fphys.2019.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K, The K48-K63 branched ubiquitin chain regulates NF-κB signaling, Mol. Cell 64 (2016) 251–266, 10.1016/j.molcel.2016.09.014. [DOI] [PubMed] [Google Scholar]
- [25].Riling C, Kamadurai H, Kumar S, O’Leary CE, Wu K-P, Manion EE, Ying M, Schulman BA, Oliver PM, Itch WW domains inhibit its E3 ubiquitin ligase activity by blocking E2-E3 ligase trans-thiolation, J. Biol. Chem 290 (2015) 23875–23887, 10.1074/jbc.M115.649269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mund T, Pelham HRB, Control of the activity of WW-HECT domain E3 ubiquitin ligases by NDFIP proteins, EMBO Rep. 10 (2009) 501–507, 10.1038/embor.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhu K, Shan Z, Chen X, Cai Y, Cui L, Yao W, Wang Z, Shi P, Tian C, Lou J, Xie Y, Wen W, Allosteric auto-inhibition and activation of the Nedd4 family E3 ligase Itch, EMBO Rep. 18 (2017) 1618–1630, 10.15252/embr.201744454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen Z, Jiang H, Xu W, Li X, Dempsey DR, Zhang X, Devreotes P, Wolberger C, Amzel LM, Gabelli SB, Cole PA, A tunable brake for hect ubiquitin ligases, Mol. Cell 66 (2017) 342–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gallagher E, Gao M, Liu Y-C, Karin M, Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change, Proc. Natl. Acad. Sci. U.S.A 103 (2006) 1717–1722, 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Santini S, Stagni V, Giambruno R, Fianco G, Di Benedetto A, Mottolese M, Pellegrini M, Barilà D, ATM kinase activity modulates ITCH E3-ubiquitin ligase activity, Oncogene 33 (2014) 1113–1123, 10.1038/onc.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chang L, Shen L, Zhou H, Gao J, Pan H, Zheng L, Armstrong B, Peng Y, Peng G, Zhou BP, Rosen ST, Shen B, ITCH nuclear translocation and H1.2 polyubiquitination negatively regulate the DNA damage response, Nucleic Acids Res. 47 (2019) 824–842, 10.1093/nar/gky1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang C, Zhou W, Jeon M-S, Demydenko D, Harada Y, Zhou H, Liu Y-C, Negative regulation of the E3 ubiquitin ligase itch via Fyn-mediated tyrosine phosphorylation, Mol. Cell 21 (2006) 135–141, 10.1016/j.molcel.2005.11.014. [DOI] [PubMed] [Google Scholar]
- [33].Slagsvold T, Marchese A, Brech A, Stenmark H, CISK attenuates degradation of the chemokine receptor CXCR4 via the ubiquitin ligase AIP4, EMBO J. 25 (2006) 3738–3749, 10.1038/sj.emboj.7601267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Morales B, Ramirez-Espain X, Shaw AZ, Martin-Malpartida P, Yraola F, Sánchez-Tilló E, Farrera C, Celada A, Royo M, Macias MJ, NMR structural studies of the ItchWW3 domain reveal that phosphorylation at T30 inhibits the interaction with PPxY-containing ligands, Structure 15 (2007) 473–483, 10.1016/j.str.2007.03.005. [DOI] [PubMed] [Google Scholar]
- [35].Perez JM, Chen Y, Xiao TS, Abbott DW, Phosphorylation of the E3 ubiquitin protein ligase ITCH diminishes binding to its cognate E2 ubiquitin ligase, J. Biol. Chem 293 (2018) 1100–1105, 10.1074/jbc.RA117.000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Oliver PM, Cao X, Worthen GS, Shi P, Briones N, MacLeod M, White J, Kirby P, Kappler J, Marrack P, Yang B, Ndfip1 protein promotes the function of itch ubiquitin ligase to prevent T cell activation and T helper 2 cell-mediated inflammation, Immunity 25 (2006) 929–940, 10.1016/j.immuni.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Noyes NC, Hampton B, Migliorini M, Strickland DK, Regulation of itch and Nedd4 E3 ligase activity and degradation by LRAD3, Biochemistry 55 (2016) 1204–1213, 10.1021/acs.biochem.5b01218. [DOI] [PubMed] [Google Scholar]
- [38].Bhandari D, Trejo J, Benovic JL, Marchese A, Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4, J. Biol. Chem 282 (2007) 36971–36979, 10.1074/jbc.M705085200. [DOI] [PubMed] [Google Scholar]
- [39].Shukla AK, Kim J, Ahn S, Xiao K, Shenoy SK, Liedtke W, Lefkowitz RJ, Arresting a transient receptor potential (TRP) channel: beta-arrestin 1 mediates ubiquitination and functional down-regulation of TRPV4, J. Biol. Chem 285 (2010) 30115–30125, 10.1074/jbc.M110.141549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Infante P, Faedda R, Bernardi F, Bufalieri F, Lospinoso Severini L, Alfonsi R, Mazzà D, Siler M, Coni S, Po A, Petroni M, Ferretti E, Mori M, De Smaele E, Canettieri G, Capalbo C, Maroder M, Screpanti I, Kool M, Pfister SM, Guardavaccaro D, Gulino A, Di Marcotullio L, Itch/β-arrestin2-dependent non-proteolytic ubiquitylation of SuFu controls Hedgehog signalling and medullo-blastoma tumorigenesis, Nat. Commun 9 (2018) 976, 10.1038/s41467-018-03339-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Di Marcotullio L, Ferretti E, Greco A, De Smaele E, Po A, Sico MA, Alimandi M, Giannini G, Maroder M, Screpanti I, Gulino A, Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination, Nat. Cell Biol 8 (2006) 1415–1423, 10.1038/ncb1510. [DOI] [PubMed] [Google Scholar]
- [42].Di Marcotullio L, Greco A, Mazzà D, Canettieri G, Pietrosanti L, Infante P, Coni S, Moretti M, De Smaele E, Ferretti E, Screpanti I, Gulino A, Numb activates the E3 ligase Itch to control Gli1 function through a novel degradation signal, Oncogene 30 (2011) 65–76, 10.1038/onc.2010.394. [DOI] [PubMed] [Google Scholar]
- [43].Scialpi F, Malatesta M, Peschiaroli A, Rossi M, Melino G, Bernassola F, Itch self-polyubiquitylation occurs through lysine-63 linkages, Biochem. Pharmacol 76 (2008) 1515–1521, 10.1016/j.bcp.2008.07.028. [DOI] [PubMed] [Google Scholar]
- [44].Mouchantaf R, Azakir BA, McPherson PS, Millard SM, Wood SA, Angers A, The ubiquitin ligase itch is auto-ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X, J. Biol. Chem 281 (2006) 38738–38747, 10.1074/jbc.M605959200. [DOI] [PubMed] [Google Scholar]
- [45].Kim Y, Kim W, Song Y, Kim J-R, Cho K, Moon H, Ro SW, Seo E, Ryu Y-M, Myung S-J, Jho E-H, Deubiquitinase YOD1 potentiates YAP/TAZ activities through enhancing ITCH stability, Proc. Natl. Acad. Sci 114 (2017) 4691–4696, 10.1073/pnas.1620306114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shembade N, Harhaj NS, Parvatiyar K, Copeland NG, Jenkins NA, Matesic LE, Harhaj EW, The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20, Nat. Immunol 9 (2008) 254–262, 10.1038/ni1563. [DOI] [PubMed] [Google Scholar]
- [47].Lohr NJ, Molleston JP, Strauss KA, Torres-Martinez W, Sherman EA, Squires RH, Rider NL, Chikwava KR, Cummings OW, Morton DH, Puffenberger EG, Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease, Am. J. Hum. Genet 86 (2010) 447–453, 10.1016/j.ajhg.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ishihara T, Tsuda H, Hotta A, Kozaki K-I, Yoshida A, Noh JY, Ito K, Imoto I, Inazawa J, ITCH is a putative target for a novel 20q11.22 amplification detected in anaplastic thyroid carcinoma cells by array-based comparative genomic hybridization, Cancer Sci. 99 (2008) 1940–1949, 10.1111/j.1349-7006.2008.00900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Salah Z, Melino G, Aqeilan RI, Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity, Cancer Res. 71 (2011) 2010–2020, 10.1158/0008-5472.CAN-10-3516. [DOI] [PubMed] [Google Scholar]
- [50].Yu FX, Guan KL, The Hippo pathway: regulators and regulations, Genes Dev. 27 (2013) 355–371, 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hao Y, Chun A, Cheung K, Rashidi B, Yang X, Tumor suppressor LATS1 is a negative regulator of oncogene YAP, J. Biol. Chem 283 (2008) 5496–5509, 10.1074/jbc.M709037200. [DOI] [PubMed] [Google Scholar]
- [52].Zhao B, Li L, Lei Q, Guan K-L, The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version, Genes Dev. 24 (2010) 862–874, 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pan D, The Hippo signaling pathway in development and cancer, Dev. Cell 19 (2010) 491–505, 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Moroishi T, Hansen CG, Guan KL, The emerging roles of YAP and TAZ in cancer, Nat. Rev. Cancer 15 (2015) 73–79, 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ho KC, Zhou Z, She Y-M, Chun A, Cyr TD, Yang X, Itch E3 ubiquitin ligase regulates large tumor suppressor 1 stability, Proc. Natl. Acad. Sci 108 (2011) 4870–4875, 10.1073/pnas.1101273108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lim SK, Lu SY, Kang SA, Tan HJ, Li Z, Wee ZNA, Guan JS, Chichili VPR, Sivaraman J, Putti T, Thike AA, Tan PH, Sudol M, Virshup DM, Chan SW, Hong W, Lim YP, Wnt signaling promotes breast cancer by blocking ITCH-mediated degradation of YAP/TAZ transcriptional coactivator WBP2, Cancer Res. 76 (2016) 6278–6289, 10.1158/0008-5472.CAN-15-3537. [DOI] [PubMed] [Google Scholar]
- [57].Pak E, Segal RA, Hedgehog signal transduction: key players, oncogenic drivers, and cancer therapy, Dev. Cell 38 (2016) 333–344, 10.1016/j.devcel.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Riobo-Del Galdo NA, Lara Montero Á, Wertheimer EV, Role of Hedgehog signaling in breast Cancer: pathogenesis and therapeutics, Cells 8 (2019), 10.3390/cells8040375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].ZeRuth GT, Williams JG, Cole YC, Jetten AM, HECT E3 ubiquitin ligase itch functions as a novel negative regulator of gli-similar 3 (Glis3) transcriptional activity, PLoS One 10 (2015) e0131303, 10.1371/journal.pone.0131303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chen XL, Chinchilla P, Fombonne J, Ho L, Guix C, Keen JH, Mehlen P, Riobo NA, Patched-1 proapoptotic activity is downregulated by modification of K1413 by the E3 ubiquitin-protein ligase Itchy homolog, Mol. Cell. Biol 34 (2014) 3855–3866, 10.1128/MCB.00960-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].D’Amico D, Canettieri G, Translating Hedgehog in cancer: controlling protein synthesis, Trends Mol. Med 22 (2016) 851–862, 10.1016/j.molmed.2016.08.003. [DOI] [PubMed] [Google Scholar]
- [62].Wrighton KH, Lin X, Feng XH, Phospho-control of TGF-β superfamily signaling, Cell Res. 19 (2009) 8–20, 10.1038/cr.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bai Y, Yang C, Hu K, Elly C, Liu Y-C, Itch E3 ligase-mediated regulation of TGF-beta signaling by modulating smad2 phosphorylation, Mol. Cell 15 (2004) 825–831, 10.1016/j.molcel.2004.07.021. [DOI] [PubMed] [Google Scholar]
- [64].Lallemand F, Seo SR, Ferrand N, Pessah M, L’Hoste S, Rawadi G, Roman-Roman S, Camonis J, Atfi A, AIP4 restricts transforming growth factor-beta signaling through a ubiquitination-independent mechanism, J. Biol. Chem 280 (2005) 27645–27653, 10.1074/jbc.M500188200. [DOI] [PubMed] [Google Scholar]
- [65].Venuprasad K, Huang H, Harada Y, Elly C, Subramaniam M, Spelsberg T, Su J, Liu Y-C, The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1, Nat. Immunol 9 (2008) 245–253, 10.1038/ni1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Peng D-J, Zeng M, Muromoto R, Matsuda T, Shimoda K, Subramaniam M, Spelsberg TC, Wei W-Z, Venuprasad K, Noncanonical K27-linked poly-ubiquitination of TIEG1 regulates Foxp3 expression and tumor growth, J. Immunol 186 (2011) 5638–5647, 10.4049/jimmunol.1003801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Feng L, Guedes S, Wang T, Atrophin-1-interacting protein 4/human Itch is a ubiquitin E3 ligase for human enhancer of filamentation 1 in transforming growth factor-beta signaling pathways, J. Biol. Chem 279 (2004) 29681–29690, 10.1074/jbc.M403221200. [DOI] [PubMed] [Google Scholar]
- [68].Frey RS, Mulder KM, Involvement of extracellular signal-regulated kinase 2 and stress-activated protein kinase/Jun N-terminal kinase activation by transforming growth factor beta in the negative growth control of breast cancer cells, Cancer Res. 57 (1997) 628–633. [PubMed] [Google Scholar]
- [69].Ahmed N, Zeng M, Sinha I, Polin L, Wei W-Z, Rathinam C, Flavell R, Massoumi R, Venuprasad K, The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation, Nat. Immunol 12 (2011) 1176–1183, 10.1038/ni.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Fang D, Elly C, Gao B, Fang N, Altman Y, Joazeiro C, Hunter T, Copeland N, Jenkins N, Liu Y-C, Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation, Nat. Immunol 3 (2002) 281–287, 10.1038/ni763. [DOI] [PubMed] [Google Scholar]
- [71].Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu Y-C, Karin M, Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch, Science 306 (2004) 271–275, 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- [72].Lopez-Bergami P, Lau E, Ronai Z, Emerging roles of ATF2 and the dynamic AP1 network in cancer, Nat. Rev. Cancer 10 (2010) 65–76, 10.1038/nrc2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Eferl R, Wagner EF, AP-1: a double-edged sword in tumorigenesis, Nat. Rev. Cancer 3 (2003) 859–868, 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- [74].Anastas JN, Moon RT, WNT signalling pathways as therapeutic targets in cancer, Nat. Rev. Cancer 13 (2013) 11–26, 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- [75].Bray SJ, Notch signalling in context, Nat. Rev. Mol. Cell Biol 17 (2016) 722–735, 10.1038/nrm.2016.94. [DOI] [PubMed] [Google Scholar]
- [76].Wei W, Li M, Wang J, Nie F, Li L, The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein, Mol. Cell. Biol 32 (2012) 3903–3912, 10.1128/MCB.00251-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Vijayakumar S, Liu G, Wen H-C, Abu Y, Chong R, Nastri H, Bornstein GG, Pan Z-Q, Aaronson SA, Extracellular LDLR repeats modulate Wnt signaling activity by promoting LRP6 receptor endocytosis mediated by the Itch E3 ubiquitin ligase, Genes Cancer 8 (2017) 613–627, 10.18632/genesandcancer.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Clevers H, Nusse R, Wnt/β-catenin signaling and disease, Cell. 149 (2012) 1192–1205, 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- [79].Qiu L, Joazeiro C, Fang N, Wang HY, Elly C, Altman Y, Fang D, Hunter T, Liu YC, Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase, J. Biol. Chem 275 (2000) 35734–35737, 10.1074/jbc.M007300200. [DOI] [PubMed] [Google Scholar]
- [80].Chastagner P, Israël A, Brou C, AIP4/Itch regulates Notch receptor degradation in the absence of ligand, PLoS One 3 (2008) e2735, 10.1371/journal.pone.0002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Chastagner P, Israël A, Brou C, Itch/AIP4 mediates Deltex degradation through the formation of K29-linked polyubiquitin chains, EMBO Rep. 7 (2006) 1147–1153, 10.1038/sj.embor.7400822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Botchkarev VA, Flores ER, p53/p63/p73 in the epidermis in health and disease, Cold Spring Harb. Perspect. Med 4 (2014), 10.1101/cshperspect.a015248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA, Croce CM, Melino G, The E3 ubiquitin ligase Itch controls the protein stability of p63, Proc. Natl. Acad. Sci. U.S.A 103 (2006) 12753–12758, 10.1073/pnas.0603449103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Agami R, Blandino G, Oren M, Shaul Y, Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis, Nature 399 (1999) 809–813, 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- [85].Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Levrero M, Wang JY, The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage, Nature 399 (1999) 806–809, 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- [86].Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang YY, Lu H, Kharbanda S, Weichselbaum R, Kufe D, p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage, Nature 399 (1999) 814–817, 10.1038/21704. [DOI] [PubMed] [Google Scholar]
- [87].Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu Y-C, Vousden KH, Cesareni G, Melino G, The ubiquitin-protein ligase Itch regulates p73 stability, EMBO J. 24 (2005) 836–848, 10.1038/sj.emboj.7600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Cullen PJ, Steinberg F, To degrade or not to degrade: mechanisms and significance of endocytic recycling, Nat. Rev. Mol. Cell Biol 19 (2018) 679–696, 10.1038/s41580-018-0053-7. [DOI] [PubMed] [Google Scholar]
- [89].Mellman I, Yarden Y, Endocytosis and cancer, Cold Spring Harb. Perspect. Biol 5 (2013) a016949, 10.1101/cshperspect.a016949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Angers A, Ramjaun AR, McPherson PS, The HECT domain ligase itch ubiquitinates endophilin and localizes to the trans-Golgi network and endosomal system, J. Biol. Chem 279 (2004) 11471–11479, 10.1074/jbc.M309934200. [DOI] [PubMed] [Google Scholar]
- [91].Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL, The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4, Dev. Cell 5 (2003) 709–722, 10.1016/s1534-5807(03)00321-6. [DOI] [PubMed] [Google Scholar]
- [92].Dergai O, Dergai M, Rynditch A, Ubiquitin-ligase AIP4 controls differential ubiquitination and stability of isoforms of the scaffold protein ITSN1, FEBS Lett. (2018), 10.1002/1873-3468.13118. [DOI] [PubMed] [Google Scholar]
- [93].Mössinger J, Wieffer M, Krause E, Freund C, Gerth F, Krauss M, Haucke V, Phosphatidylinositol 4-kinase IIα function at endosomes is regulated by the ubiquitin ligase Itch, EMBO Rep. 13 (2012) 1087–1094, 10.1038/embor.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Corcoran NM, Clarkson MJ, Stuchbery R, Hovens CM, Molecular Pathways: Targeting DNA Repair Pathway Defects Enriched in Metastasis, Clin. Cancer Res 22 (2016) 3132–3137, 10.1158/1078-0432.CCR-15-1050. [DOI] [PubMed] [Google Scholar]
- [95].Roos WP, Thomas AD, Kaina B, DNA damage and the balance between survival and death in cancer biology, Nat. Rev. Cancer 16 (2016) 20–33, 10.1038/nrc.2015.2. [DOI] [PubMed] [Google Scholar]
- [96].Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker-Jensen S, Mailand N, Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage, Nature 527 (2015) 389–393, 10.1038/nature15401. [DOI] [PubMed] [Google Scholar]
- [97].Sundvall M, Korhonen A, Paatero I, Gaudio E, Melino G, Croce CM, Aqeilan RI, Elenius K, Isoform-specific monoubiquitination, endocytosis, and degradation of alternatively spliced ErbB4 isoforms, Proc. Natl. Acad. Sci. U.S.A 105 (2008) 4162–4167, 10.1073/pnas.0708333105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Meijer IMJ, van Rotterdam W, van Zoelen EJJ, van Leeuwen JEM, Cbl and Itch binding sites in ERBB4 CYT-1 and CYT-2 mediate K48- and K63-poly-ubiquitination, respectively, Cell. Signal 25 (2013) 470–478, 10.1016/j.cellsig.2012.11.008. [DOI] [PubMed] [Google Scholar]
- [99].Wellbrock C, Karasarides M, Marais R, The RAF proteins take centre stage, Nat. Rev. Mol. Cell Biol 5 (2004) 875–885, 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- [100].Dhillon AS, Hagan S, Rath O, Kolch W, MAP kinase signalling pathways in cancer, Oncogene 26 (2007) 3279–3290, 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- [101].Wan L, Chen M, Cao J, Dai X, Yin Q, Zhang J, Song S-J, Lu Y, Liu J, Inuzuka H, Katon JM, Berry K, Fung J, Ng C, Liu P, Song MS, Xue L, Bronson RT, Kirschner MW, Cui R, Pandolfi PP, Wei W, The APC/C E3 ligase complex activator FZR1 restricts BRAF oncogenic function, Cancer Discov. 7 (2017) 424–441, 10.1158/2159-8290.CD-16-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Chen M, Wan L, Zhang J, Zhang J, Mendez L, Clohessy JG, Berry K, Victor J, Yin Q, Zhu Y, Wei W, Pandolfi PP, Deregulated PP1α phosphatase activity towards MAPK activation is antagonized by a tumor suppressive failsafe mechanism, Nat. Commun 9 (2018) 159, 10.1038/s41467-017-02272-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Yin Q, Han T, Fang B, Zhang G, Zhang C, Roberts ER, Izumi V, Zheng M, Jiang S, Yin X, Kim M, Cai J, Haura EB, Koomen JM, Smalley KSM, Wan L, K27-linked ubiquitination of BRAF by ITCH engages cytokine response to maintain MEK-ERK signaling, Nat. Commun 10 (2019) 1870, 10.1038/s41467-019-09844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Chang L, Kamata H, Solinas G, Luo J-L, Maeda S, Venuprasad K, Liu Y-C, Karin M, The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover, Cell 124 (2006) 601–613, 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- [105].Ahn Y-H, Kurie JM, MKK4/SEK1 is negatively regulated through a feedback loop involving the E3 ubiquitin ligase itch, J. Biol. Chem 284 (2009) 29399–29404, 10.1074/jbc.M109.044958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Balkwill FR, Capasso M, Hagemann T, The tumor microenvironment at a glance, J. Cell. Sci 125 (2012) 5591–5596, 10.1242/jcs.116392. [DOI] [PubMed] [Google Scholar]
- [107].Gajewski TF, Schreiber H, Fu Y-X, Innate and adaptive immune cells in the tumor microenvironment, Nat. Immunol 14 (2013) 1014–1022, 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA, Chronic Inflammation and Cytokines in the Tumor Microenvironment, J. Immunol. Res 2014 (2014) 1–19, 10.1155/2014/149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Li H, Fan X, Houghton J, Tumor microenvironment: The role of the tumor stroma in cancer, J. Cell. Biochem 101 (2007) 805–815, 10.1002/jcb.21159. [DOI] [PubMed] [Google Scholar]
- [110].Hanahan D, Coussens LM, Accessories to the crime: functions of cells recruited to the tumor microenvironment, Cancer Cell 21 (2012) 309–322, 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- [111].Hui L, Chen Y, Tumor microenvironment: sanctuary of the devil, Cancer Lett. 368 (2015) 7–13, 10.1016/j.canlet.2015.07.039. [DOI] [PubMed] [Google Scholar]
- [112].Whiteside TL, The tumor microenvironment and its role in promoting tumor growth, Oncogene 27 (2008) 5904–5912, 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Chanmee T, Ontong P, Konno K, Itano N, Tumor-associated macrophages as major players in the tumor microenvironment, Cancers 6 (2014) 1670–1690, 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Polyak K, Haviv I, Campbell IG, Co-evolution of tumor cells and their microenvironment, Trends Genet. 25 (2009) 30–38, 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- [115].Joyce JA, Fearon DT, T cell exclusion, immune privilege, and the tumor microenvironment, Science. 348 (2015) 74–80, 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- [116].Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S, Functions of natural killer cells, Nat. Immunol 9 (2008) 503–510, 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- [117].Baginska J, Viry E, Paggetti J, Medves S, Berchem G, Moussay E, Janji B, The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity, Front. Immunol 4 (2013) 490, 10.3389/fimmu.2013.00490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Taniguchi M, Seino K-I, Nakayama T, The NKT cell system: bridging innate and acquired immunity, Nat. Immunol 4 (2003) 1164–1165, 10.1038/ni1203-1164. [DOI] [PubMed] [Google Scholar]
- [119].Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, Kawano T, Pelikan SB, Crowe NY, Godfrey DI, Differential tumor surveillance by natural killer (NK) and NKT cells, J. Exp. Med 191 (2000) 661–668, 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Hadrup S, Donia M, Thor Straten P, Effector CD4 and CD8 T cells and their role in the tumor microenvironment, Cancer Microenviron. 6 (2013) 123–133, 10.1007/s12307-012-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Toes REM, Ossendorp F, Offringa R, Melief CJM, CD4 T Cells and Their Role in Antitumor Immune Responses, J. Exp. Med 189 (1999) 753–756, 10.1084/jem.189.5.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, Rizzuto GA, Lazarus JJ, Pamer EG, Houghton AN, Merghoub T, Wolchok JD, Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment, Cancer Res. 72 (2012) 876–886, 10.1158/0008-5472.CAN-11-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Ruffell B, Affara NI, Coussens LM, Differential macrophage programming in the tumor microenvironment, Trends Immunol. 33 (2012) 119–126, 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Gajewski TF, Meng Y, Harlin H, Immune suppression in the tumor microenvironment, J. Immunother 29 (2006) 233–240, 10.1097/01.cji.0000199193.29048.56. [DOI] [PubMed] [Google Scholar]
- [125].Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK, Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression, Semin. Cancer Biol 22 (2012) 275–281, 10.1016/j.semcancer.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Crespo J, Sun H, Welling TH, Tian Z, Zou W, T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment, Curr. Opin. Immunol 25 (2013) 214–221, 10.1016/j.coi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Jiang Y, Li Y, Zhu B, T-cell exhaustion in the tumor microenvironment, Cell Death Dis. 6 (2015) e1792, 10.1038/cddis.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kryczek I, Wei S, Zou L, Altuwaijri S, Szeliga W, Kolls J, Chang A, Zou W, Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment, J. Immunol 178 (2007) 6730–6733, 10.4049/jimmunol.178.11.6730. [DOI] [PubMed] [Google Scholar]
- [129].Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE, Coussens LM, DeClerck YA, Tumor microenvironment complexity: emerging roles in cancer therapy, Cancer Res. 72 (2012) 2473–2480, 10.1158/0008-5472.CAN-12-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Qin Z, Richter G, Schüler T, Ibe S, Cao X, Blankenstein T, B cells inhibit induction of T cell-dependent tumor immunity, Nat. Med 4 (1998) 627–630, 10.1038/nm0598-627. [DOI] [PubMed] [Google Scholar]
- [131].Inoue S, Leitner WW, Golding B, Scott D, Inhibitory effects of B cells on anti-tumor immunity, Cancer Res. 66 (2006) 7741–7747, 10.1158/0008-5472.CAN-05-3766. [DOI] [PubMed] [Google Scholar]
- [132].Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S, Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response, J. Immunol 179 (2007) 977–983, 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- [133].Courtney AH, Lo W-L, Weiss A, TCR Signaling: Mechanisms of Initiation and Propagation, Trends Biochem. Sci 43 (2018) 108–123, 10.1016/j.tibs.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Huang H, Jeon M-S, Liao L, Yang C, Elly C, Yates JR, Liu Y-C, K33-linked polyubiquitination of T cell receptor-zeta regulates proteolysis-independent T cell signaling, Immunity 33 (2010) 60–70, 10.1016/j.immuni.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Scharschmidt E, Wegener E, Heissmeyer V, Rao A, Krappmann D, Degradation of Bcl10 induced by T-cell activation negatively regulates NF-kappa B signaling, Mol. Cell. Biol 24 (2004) 3860–3873, 10.1128/MCB.24.9.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Heissmeyer V, Macián F, Im S-H, Varma R, Feske S, Venuprasad K, Gu H, Liu Y-C, Dustin ML, Rao A, Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins, Nat. Immunol 5 (2004) 255–265, 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- [137].Aki D, Li H, Zhang W, Zheng M, Elly C, Lee JH, Zou W, Liu Y-C, The E3 ligases Itch and WWP2 cooperate to limit TH2 differentiation by enhancing signaling through the TCR, Nat. Immunol (2018), 10.1038/s41590-018-0137-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Seder RA, Ahmed R, Similarities and differences in CD4+ and CD8+ effector and memory T cell generation, Nat. Immunol 4 (2003) 835–842, 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- [139].Zhu J, Paul WE, Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors, Immunol. Rev 238 (2010) 247–262, 10.1111/j.1600-065X.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu Y-C, Karin M, Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch, Science 306 (2004) 271–275, 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- [141].Ellyard JI, Simson L, Parish CR, Th2-mediated anti-tumour immunity: friend or foe? Tissue Antigens 70 (2007) 1–11, 10.1111/j.1399-0039.2007.00869.x. [DOI] [PubMed] [Google Scholar]
- [142].Tepper RI, Coffman RL, Leder P, An eosinophil-dependent mechanism for the antitumor effect of interleukin-4, Science. 257 (1992) 548–551, 10.1126/science.1636093. [DOI] [PubMed] [Google Scholar]
- [143].Pericle F, Giovarelli M, Colombo MP, Ferrari G, Musiani P, Modesti A, Cavallo F, Di Pierro F, Novelli F, Forni G, An efficient Th2-type memory follows CD8+ lymphocyte-driven and eosinophil-mediated rejection of a spontaneous mouse mammary adenocarcinoma engineered to release IL-4, J. Immunol 153 (1994) 5659–5673. [PubMed] [Google Scholar]
- [144].Togashi Y, Shitara K, Nishikawa H, Regulatory T cells in cancer immunosuppression — implications for anticancer therapy, Nat. Rev. Clin. Oncol (2019), 10.1038/s41571-019-0175-7. [DOI] [PubMed] [Google Scholar]
- [145].Jin H, Park Y, Elly C, Liu Y-C, Itch expression by Treg cells controls Th2 inflammatory responses, J. Clin. Invest 123 (2013) 4923–4934, 10.1172/JCI69355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Tanaka A, Sakaguchi S, Regulatory T cells in cancer immunotherapy, Cell Res. 27 (2017) 109–118, 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi M-B, Harpole DH, Patz EF, Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients, Cancer 107 (2006) 2866–2872, 10.1002/cncr.22282. [DOI] [PubMed] [Google Scholar]
- [148].Hiraoka N, Onozato K, Kosuge T, Hirohashi S, Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions, Clin. Cancer Res 12 (2006) 5423–5434, 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- [149].Badoual C, Hans S, Rodriguez J, Peyrard S, Klein C, Agueznay NEH, Mosseri V, Laccourreye O, Bruneval P, Fridman WH, Brasnu DF, Tartour E, Prognostic value of tumor-infiltrating CD4+ T-cell subpopulations in head and neck cancers, Clin. Cancer Res 12 (2006) 465–472, 10.1158/1078-0432.CCR-05-1886. [DOI] [PubMed] [Google Scholar]
- [150].Winerdal ME, Marits P, Winerdal M, Hasan M, Rosenblatt R, Tolf A, Selling K, Sherif A, Winqvist O, FOXP3 and survival in urinary bladder cancer, BJU Int 108 (2011) 1672–1678, 10.1111/j.1464-410X.2010.10020.x. [DOI] [PubMed] [Google Scholar]
- [151].Zhu J, Yamane H, Paul WE, Differentiation of effector CD4 T cell populations (*), Annu. Rev. Immunol 28 (2010) 445–489, 10.1146/annurevimmunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Asadzadeh Z, Mohammadi H, Safarzadeh E, Hemmatzadeh M, Mahdian-Shakib A, Jadidi-Niaragh F, Azizi G, Baradaran B, The paradox of Th17 cell functions in tumor immunity, Cell. Immunol 322 (2017) 15–25, 10.1016/j.cellimm.2017.10.015. [DOI] [PubMed] [Google Scholar]
- [153].Kathania M, Khare P, Zeng M, Cantarel B, Zhang H, Ueno H, Venuprasad K, Itch inhibits IL-17-mediated colon inflammation and tumorigenesis by ROR-γt ubiquitination, Nat. Immunol 17 (2016) 997–1004, 10.1038/ni.3488. [DOI] [PubMed] [Google Scholar]
- [154].Layman AA, Sprout S, Phillips D, Oliver PM, Ndfip1 restricts Th17 cell potency by limiting lineage stability and proinflammatory cytokine production, Sci. Rep 7 (2017) 39649, 10.1038/srep39649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Crotty S, T follicular helper cell biology: a decade of discovery and diseases, Immunity 50 (2019) 1132–1148, 10.1016/j.immuni.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Xiao N, Eto D, Elly C, Peng G, Crotty S, Liu Y-C, The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells, Nat. Immunol 15 (2014) 657–666, 10.1038/ni.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Luo CT, Liao W, Dadi S, Toure A, Li MO, Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity, Nature 529 (2016) 532–536, 10.1038/nature16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Ma CS, Deenick EK, Human T follicular helper (Tfh) cells and disease, Immunol. Cell Biol 92 (2014) 64–71, 10.1038/icb.2013.55. [DOI] [PubMed] [Google Scholar]
- [159].Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, Bruneval P, Fridman WH, Becker C, Pagès F, Speicher MR, Trajanoski Z, Galon J, Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer, Immunity 39 (2013) 782–795, 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- [160].Largeot A, Pagano G, Gonder S, Moussay E, Paggetti J, The B-side of Cancer immunity: the underrated tune, Cells 8 (2019), 10.3390/cells8050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Moser EK, Roof J, Dybas JM, Spruce LA, Seeholzer SH, Cancro MP, Oliver PM, The E3 ubiquitin ligase Itch restricts antigen-driven B cell responses, J. Exp. Med 216 (2019) 2170–2183, 10.1084/jem.20181953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Liu X, Zhang Y, Wei Y, Wang Z, Zhu G, Fang Y, Zhai B, Xu R, Han G, Chen G, Xiao H, Hou C, Shen B, Li Y, Ma N, Wang R, The E3 ubiquitin ligase Itch is required for B-cell development, Sci. Rep 9 (2019) 421, 10.1038/s41598-018-36844-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Rayburn ER, Ezell SJ, Zhang R, Anti-inflammatory agents for cancer therapy, Mol. Cell. Pharmacol 1 (2009) 29–43, 10.4255/mcpharmacol.09.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Gehring T, Seeholzer T, Krappmann D, BCL10 - bridging CARDs to immune activation, Front. Immunol 9 (2018) 1539, 10.3389/fimmu.2018.01539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].van Beurden-Tan CHY, Franken MG, Blommestein HM, Uyl-de Groot CA, Sonneveld P, systematic literature review and network meta-analysis of treatment outcomes in relapsed and/or refractory multiple myeloma, J. Clin. Oncol 35 (2017) 1312–1319, 10.1200/JCO.2016.71.1663. [DOI] [PubMed] [Google Scholar]
- [166].Chanan-Khan AA, Swaika A, Paulus A, Kumar SK, Mikhael JR, Rajkumar SV, Dispenzieri A, Lacy MQ, Pomalidomide: the new immunomodulatory agent for the treatment of multiple myeloma, Blood Cancer J. 3 (2013) e143, 10.1038/bcj.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Mund T, Lewis MJ, Maslen S, Pelham HR, Peptide and small molecule inhibitors of HECT-type ubiquitin ligases, Proc. Natl. Acad. Sci. U.S.A 111 (2014) 16736–16741, 10.1073/pnas.1412152111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Quirit JG, Lavrenov SN, Poindexter K, Xu J, Kyauk C, Durkin KA, Aronchik I, Tomasiak T, Solomatin YA, Preobrazhenskaya MN, Firestone GL, Indole-3-carbinol (I3C) analogues are potent small molecule inhibitors of NEDD4–1 ubiquitin ligase activity that disrupt proliferation of human melanoma cells, Biochem. Pharmacol 127 (2017) 13–27, 10.1016/j.bcp.2016.12.007. [DOI] [PubMed] [Google Scholar]
- [169].Lee Y-R, Chen M, Lee JD, Zhang J, Lin S-Y, Fu T-M, Chen H, Ishikawa T, Chiang S-Y, Katon J, Zhang Y, Shulga YV, Bester AC, Fung J, Monteleone E, Wan L, Shen C, Hsu C-H, Papa A, Clohessy JG, Teruya-Feldstein J, Jain S, Wu H, Matesic L, Chen R-H, Wei W, Pandolfi PP, Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway, Science 364 (2019), 10.1126/science.aau0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Rossi M, Rotblat B, Ansell K, Amelio I, Caraglia M, Misso G, Bernassola F, Cavasotto CN, Knight RA, Ciechanover A, Melino G, High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy, Cell Death Dis. 5 (2014) e1203, 10.1038/cddis.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Fajner V, Maspero E, Polo S, Targeting HECT-type E3 ligases - insights from catalysis, regulation and inhibitors, FEBS Lett. 591 (2017) 2636–2647, 10.1002/1873-3468.12775. [DOI] [PubMed] [Google Scholar]
- [172].Hustad CM, Perry WL, Siracusa LD, Rasberry C, Cobb L, Cattanach BM, Kovatch R, Copeland NG, Jenkins NA, Molecular genetic characterization of six recessive viable alleles of the mouse agouti locus, Genetics. 140 (1995) 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Smith MP, Sanchez-Laorden B, O’Brien K, Brunton H, Ferguson J, Young H, Dhomen N, Flaherty KT, Frederick DT, Cooper ZA, Wargo JA, Marais R, Wellbrock C, O’brien K, Brunton H, Ferguson J, Young H, Dhomen N, Flaherty KT, Frederick DT, Cooper ZA, Wargo JA, Marais R, Wellbrock C, The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFa, Cancer Discov. 4 (2014) 1214–1229, 10.1158/2159-8290.CD-13-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Chhangani D, Upadhyay A, Amanullah A, Joshi V, Mishra A, Ubiquitin ligase ITCH recruitment suppresses the aggregation and cellular toxicity of cytoplasmic misfolded proteins, Sci. Rep 4 (2014) 5077, 10.1038/srep05077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Le Clorennec C, Lazrek Y, Dubreuil O, Larbouret C, Poul M-A, Mondon P, Melino G, Pèlegrin A, Chardès T, The anti-HER3 (ErbB3) therapeutic antibody 9F7-F11 induces HER3 ubiquitination and degradation in tumors through JNK1/2-dependent ITCH/AIP4 activation, Oncotarget. 7 (2016) 37013–37029, 10.18632/oncotarget.9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Omerovic J, Santangelo L, Puggioni EM-R, Marrocco J, Dall’Armi C, Palumbo C, Belleudi F, Di Marcotullio L, Frati L, Torrisi M-R, Cesareni G, Gulino A, Alimandi M, The E3 ligase Aip4/Itch ubiquitinates and targets ErbB-4 for degradation, FASEB J. 21 (2007) 2849–2862, 10.1096/fj.06-7925com. [DOI] [PubMed] [Google Scholar]
- [177].Mahesutihan M, Zheng W, Cui L, Li Y, Jiao P, Yang W, Liu W, Li J, Fan W, Yang L, Liu W, Sun L, CypA regulates AIP4-Mediated M1 ubiquitination of influenza a virus, Virol. Sin 33 (2018) 440–448, 10.1007/s12250-018-0058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, Jiang Z, PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4, Nat. Immunol 10 (2009) 1300–1308, 10.1038/ni.1815. [DOI] [PubMed] [Google Scholar]
- [179].Traweger A, Fang D, Liu Y-C, Stelzhammer W, Krizbai IA, Fresser F, Bauer H-C, Bauer H, The tight junction-specific protein occludin is a functional target of the E3 ubiquitin-protein ligase itch, J. Biol. Chem 277 (2002) 10201–10208, 10.1074/jbc.M111384200. [DOI] [PubMed] [Google Scholar]
- [180].Lee T-L, Shyu Y-C, Hsu T-Y, Shen C-KJ, Itch regulates p45/NF-E2 in vivo by Lys63-linked ubiquitination, Biochem. Biophys. Res. Commun 375 (2008) 326–330, 10.1016/j.bbrc.2008.07.164. [DOI] [PubMed] [Google Scholar]
- [181].Suryaraja R, Anitha M, Anbarasu K, Kumari G, Mahalingam S, The E3 ubiquitin ligase Itch regulates tumor suppressor protein RASSF5/NORE1 stability in an acetylation-dependent manner, Cell Death Dis. 4 (2013) e565, 10.1038/cddis.2013.91e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Tao M, Scacheri PC, Marinis JM, Harhaj EW, Matesic LE, Abbott DW, ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways, Curr. Biol 19 (2009) 1255–1263, 10.1016/j.cub.2009.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Stöhr R, Mavilio M, Marino A, Casagrande V, Kappel B, Möllmann J, Menghini R, Melino G, Federici M, ITCH modulates SIRT6 and SREBP2 to influence lipid metabolism and atherosclerosis in ApoE null mice, Sci. Rep 5 (2015) 9023, 10.1038/srep09023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Theivanthiran B, Kathania M, Zeng M, Anguiano E, Basrur V, Vandergriff T, Pascual V, Wei W-Z, Massoumi R, Venuprasad K, The E3 ubiquitin ligase Itch inhibits p38α signaling and skin inflammation through the ubiquitylation of Tab1, Sci. Signal 8 (2015) ra22, 10.1126/scisignal.2005903. [DOI] [PubMed] [Google Scholar]
- [185].Wegierski T, Hill K, Schaefer M, Walz G, The HECT ubiquitin ligase AIP4 regulates the cell surface expression of select TRP channels, EMBO J. 25 (2006) 5659–5669, 10.1038/sj.emboj.7601429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Otaki Y, Takahashi H, Watanabe T, Funayama A, Netsu S, Honda Y, Narumi T, Kadowaki S, Hasegawa H, Honda S, Arimoto T, Shishido T, Miyamoto T, Kamata H, Nakajima O, Kubota I, HECT-type ubiquitin E3 ligase ITCH interacts with thioredoxin-interacting protein and ameliorates reactive oxygen species-induced cardiotoxicity, J. Am. Heart Assoc 5 (2016), 10.1161/JAHA.115.002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Chmura JC, Herold K, Ruffin A, Atuobi T, Fabiyi Y, Mitchell AE, Choi YB, Ehrlich ES, The Itch ubiquitin ligase is required for KSHV RTA induced vFLIP degradation, Virology 501 (2017) 119–126, 10.1016/j.virol.2016.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Adler JJ, Heller BL, Bringman LR, Ranahan WP, Cocklin RR, Goebl MG, Oh M, Lim H-S, Ingham RJ, Wells CD, Amot130 adapts atrophin-1 interacting protein 4 to inhibit yes-associated protein signaling and cell growth, J. Biol. Chem 288 (2013) 15181–15193, 10.1074/jbc.M112.446534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Ushijima Y, Luo C, Kamakura M, Goshima F, Kimura H, Nishiyama Y, Herpes simplex virus UL56 interacts with and regulates the Nedd4-family ubiquitin ligase Itch, Virol. J 7 (2010) 179, 10.1186/1743-422X-7-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Perez JM, Chirieleison SM, Abbott DW, An IκB kinase-regulated feedforward circuit prolongs inflammation, Cell Rep. 12 (2015) 537–544, 10.1016/j.celrep.2015.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Luo Z-L, Luo H-J, Fang C, Cheng L, Huang Z, Dai R, Li K, Tian F-Z, Wang T, Tang L-J, Negative correlation of ITCH E3 ubiquitin ligase and miRNA-106b dictates metastatic progression in pancreatic cancer, Oncotarget 7 (2016) 1477–1485, 10.18632/oncotarget.6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Sampath D, Calin GA, Puduvalli VK, Gopisetty G, Taccioli C, Liu C-G, Ewald B, Liu C, Keating MJ, Plunkett W, Specific activation of microRNA106b enables the p73 apoptotic response in chronic lymphocytic leukemia by targeting the ubiquitin ligase Itch for degradation, Blood 113 (2009) 3744–3753, 10.1182/blood-2008-09-178707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Zhang H, Wu C, Matesic LE, Li X, Wang Z, Boyce BF, Xing L, Ubiquitin E3 ligase Itch negatively regulates osteoclast formation by promoting deubiquitination of tumor necrosis factor (TNF) receptor-associated factor 6, J. Biol. Chem 288 (2013) 22359–22368, 10.1074/jbc.M112.442459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Hooper C, Puttamadappa SS, Loring Z, Shekhtman A, Bakowska JC, Spartin activates atrophin-1-interacting protein 4 (AIP4) E3 ubiquitin ligase and promotes ubiquitination of adipophilin on lipid droplets, BMC Biol. 8 (2010) 72, 10.1186/1741-7007-8-72. [DOI] [PMC free article] [PubMed] [Google Scholar]