

Espresso efficiently distinguishes true mutations from background noise common in deep sequencing data.
Abstract
Sensitive mutation detection by next-generation sequencing is critical for early cancer detection, monitoring minimal/measurable residual disease (MRD), and guiding precision oncology. Nevertheless, because of artifacts introduced during library preparation and sequencing, the detection of low-frequency variants at high specificity is problematic. Here, we present Espresso, an error suppression method that considers local sequence features to accurately detect single-nucleotide variants (SNVs). Compared to other advanced error suppression techniques, Espresso consistently demonstrated lower numbers of false-positive mutation calls and greater sensitivity. We demonstrated Espresso’s superior performance in detecting MRD in the peripheral blood of patients with acute myeloid leukemia (AML) throughout their treatment course. Furthermore, we showed that accurate mutation calling in a small number of informative genomic loci might provide a cost-efficient strategy for pragmatic risk prediction of AML development in healthy individuals. More broadly, we aim for Espresso to aid with accurate mutation detection in many other research and clinical settings.
INTRODUCTION
The process of single-nucleotide variant (SNV) accumulation is an important universal element of cancer initiation and progression. While the genetic landscape of the most common malignancies has been broadly described (1–3), accurate identification of driver mutations in specimens with low cancer DNA purity continues to be of great importance yet presents substantial challenges. Hybrid-capture–based next-generation sequencing (NGS) is one of the most common techniques being used for circulating tumor DNA profiling (4, 5), detection of therapy-resistant clones (6, 7) and preleukemia (8, 9), and monitoring disease burden during therapy (10). Nevertheless, in all of these settings, the relevant genomic alterations typically exist at low relative abundance.
Several different methods have been developed in recent years to address the barrier of identifying the minute fraction of DNA molecules harboring an alteration against the high background of NGS-associated errors. Among the various methods, state-of-the-art techniques for error suppression typically can be categorized into two groups: (i) those that incorporate unique molecular identifiers (UMIs) to suppress library amplification errors by the assembly of consensus sequences (11–13) and (ii) those that use probabilistic models to estimate background sequencing noise. The latter group can be further segregated into those that generate models that estimate error rates by the analysis of data from a single sample (i.e., single sample/tumor-only mode) (14–16), data from a single control sample (16–18), or data from multiple control samples (e.g., cohort of healthy controls) (19–21). In the case of paired patient’s tumor and matched normal sample, Bayesian statistics models are commonly used to identify tumor-specific somatic variants that are distinguishable from the background and the germline variants detected within the matched normal sample (22, 23). Some techniques rely on a ploidy assumption to calculate genotype probabilities (24), while others have adapted statistical models to analyze allele frequencies directly (16), thus allowing the identification of rare subclones in existing, complex cancer genomes. Since a single control sample cannot fully account for the stochastic nature of NGS errors, other algorithms have been developed to generate site-specific error estimations using a larger cohort of controls (19–21). This approach could be problematic as proper control samples are not always available. When control samples are completely lacking, stringent preprocessing steps can be applied to prioritize high-confidence mutations, for instance, thresholds on base quality scores, supporting read counts, and variant allele frequencies.
Despite advances enabled by the diverse approaches mentioned above, each is associated with inherent disadvantages that can lead to increased assay complexity, elevated sequencing costs, and/or suboptimal exchange between sensitivity and specificity (fig. S1, table S1, and Supplementary Note). To overcome these limitations, we characterized the contextual patterns of high-frequency errors observed during targeted hybrid-capture NGS in >1000 samples, divided across multiple technically diverse and clinically relevant human cohorts. On the basis of these patterns, we developed Espresso, a novel UMI-independent method that optimizes the suppression of artifacts from deep NGS for accurate SNV mutation calling.
RESULTS
Evaluation of error abundance and rates in multiple NGS datasets
To demonstrate the challenges associated with low–variant allele fraction (VAF) mutation calling from hybrid-capture–targeted NGS, we interrogated multiple benchmarking datasets that differ by their library preparation techniques, captured genomic loci, number of samples, and sequencing depths (Fig. 1A, table S2, and Materials and Methods). Briefly, these datasets include the following: (i) CB: a human cord blood dataset; (ii) CL: a cell line dilution series using genomic DNA from the acute myeloid leukemia (AML) cell line MOLM13 and the colon cancer cell line SW48; (iii and iv) pre-AML1 and pre-AML2: peripheral blood DNA from two separate cohorts, each composed of pre-AML cases (that is, blood was drawn before clinical diagnosis of AML) and age- and sex-matched controls (9); and (v) AML-MRD: a cohort composed of peripheral blood DNA samples obtained from patients with AML during the course of treatment.
Fig. 1. Sequencing depths and error abundance in the investigated datasets.
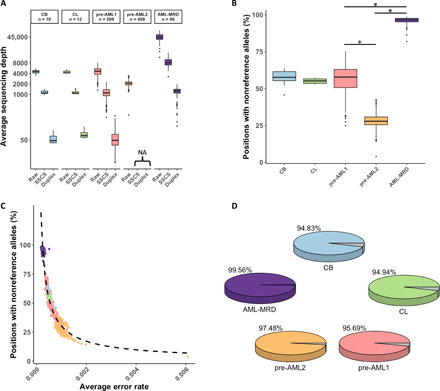
(A) Raw, SSCS, and duplex average sequencing depths across all the samples included in this study. Different colors represent different datasets, and these are consistent across all of the figure panels. (B) Sample-wide error abundance in the diverse NGS cohorts. The fraction of genomic positions being observed with at least one nonreference allele supporting read in each sample is indicated. Error burden is significantly different among the investigated datasets (Mann-Whitney test: P < 1.2 × 10−53 for the indicated comparisons). (C) Inverse correlation between the abundance of genomic positions with nonreference allele and their corresponding allele frequencies is demonstrated (Spearman’s rank order correlation: r = −0.95; *P < 2.2 × 10−16). Each dot represents a single sample. (D) Panel-wide error abundance in the diverse NGS cohorts as determined by the inclusion of positions with a minimum of one nonreference supporting read in at least one sample. NA, not applicable.
Three different target panels were used to sequence these cohorts, resulting in 83,000 to 1.2 million interrogated bases (table S2). Investigating these genomic loci revealed that the percentage of positions with nonreference alleles per sample varied widely among the different datasets and, in some cases, among samples within a particular dataset (Fig. 1B). Samples with a lower percentage of positions with nonreference alleles displayed higher average error rates (Fig. 1C). Furthermore, almost all genomic positions sequenced harbored a nonreference allele in at least one sample in each dataset (Fig. 1D). Overall, these observations reveal the magnitude of the challenge presented by potential false-positive variants produced by hybrid-capture NGS. Since such a large number of technical artifacts may mask clinically relevant variants, we conducted an unbiased exploration of multiple strategies aiming to specifically suppress NGS errors while maintaining high sensitivity in identifying real mutations.
Error rates and sequencing depth vary according to different sequence contexts
To evaluate the contextual dependencies of errors in the datasets described above, we investigated how error rates differ with respect to the substitution type, and its 5′ and 3′ one-base flanking genomic sequence. We found that error rates are highly heterogeneous across the 192 distinct trinucleotide sequence contexts (Fig. 2A, top, and fig. S2) and are highly variable between samples within the same experimental cohort (Fig. 2A, bottom). High error rates were frequently observed at C>A and C>T substitutions (Fig. 2, A and B, and fig. S2). C>T error rates were particularly high when they occurred at a CpG context (Fig. 2, A and C, and fig. S2). Initiated by spontaneous deamination of 5-methylcytosine, real mutations in this context accumulate during aging (25), are frequent in germline cells (Supplementary Note), and are also highly prevalent in cancer genomes (26), emphasizing the importance of evaluating error rates in relation to their associated genomic contexts.
Fig. 2. Error rates significantly differ between trinucleotide sequence contexts.
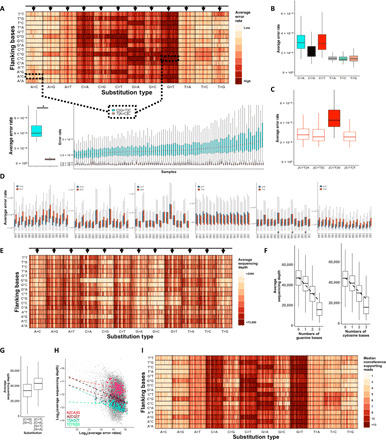
(A) Nonreference average error rates at the 192 distinct trinucleotide contexts are shown using the AML-MRD dataset. Vertical lines in each box represent individual samples. Samples’ order is kept among distinct contexts. Arrows represent a group of samples with high error rates across multiple contexts. The bottom panels exemplified variation among contextual error rates (*Wilcoxon signed-rank test: P < 1.8 × 10−17) and samples (Mann-Whitney test, samples with the highest and lowest error rates. C[G>T]C: P < 7.7 × 10−41, T[A>C]C: P < 3.6 × 10−6). (B). C>T and C>A substitutions are more frequent (Wilcoxon signed-rank test, P < 1.4 × 10−252 for all the comparisons with the other substitution types). (C) High error rates at CpG sites (Wilcoxon signed-rank test, P < 1.1 × 10−64 for all comparisons). (D) Error rates vary between error contexts and their reciprocals (Wilcoxon rank sum test, P < 0.05; #significance was not reached). (E) Average sequencing depths. Arrows represent a group of samples with low sequencing depths across multiple contexts. (F) Reduced sequencing depth at contexts that include reference cytosine and an increasing number of guanine (Pearson correlation: r = −0.35; P = 2.3 × 10−264) and at contexts that include reference guanine with an increasing number of cytosine (r = −0.29; P = 8.6 × 10−179). (G) Low sequencing depth at contexts with C>G or G>C base substitutions (Wilcoxon signed-rank test: P = 1.7 × 10−217). (H) Inverse correlation between depth and error rates (black dashed line, log-log scaled Pearson correlation: r = −0.27; P = 9.7 × 10−308). Correlation strengths differ among different error contexts (colored dashed lines). (I) The number of nonreference supporting reads at the 192 distinct trinucleotide contexts is shown. The samples’ order is identical across (A), (E), and (I).
While contextual error patterns were generally similar between their complementary counterparts, they did not always mirror each other perfectly within any particular sample (fig. S3A). Small yet statistically significant asymmetric error rates were consistently observed among the majority of error contexts in each of the cohorts (Fig. 2D and fig. S3B). For instance, we measured asymmetric error rates involving G>T/C>A, in line with prior observations (27). Error rate asymmetries were markedly directional and consistently elevated in specific contexts as compared with their matched reciprocals in all of the investigated datasets. As an example, each of the 16 trinucleotide contexts containing A>T substitutions demonstrated elevated error rates as compared with their corresponding reciprocal contexts containing T>A substitutions. Together, these results indicate that 192, rather than 96, contextual error types would need to be considered to accurately model error rates.
Next, we investigated how sequencing depth may influence error frequencies. As with error rates, sequencing depth differed between distinct contextual error types (Fig. 2E). We noticed a marked inverse correlation between sequencing depth and guanine or cytosine content within specific trinucleotide contexts, a possible reflection of the systemic under-coverage in GC-rich regions reported in NGS (Fig. 2F) (28, 29). Sequencing depth was also lower within trinucleotide contexts that included C>G and G>C substitutions as compared with those that included nucleotide substitutions that reduce GC content (Fig. 2G). These data illustrate how sequencing depth can be influenced by both the trinucleotide context and the nonreference allele.
Overall, a modest, statistically significant inverse correlation was observed between sequencing depth and error rates (Fig. 2H). Correlation strengths were not equal among distinct contextual error types. Further supporting this trend, individual samples with lower average sequencing depth displayed high error rates in multiple contextual error types (see arrows in Fig. 2, A and E). In contrast to the error rates, the absolute number of nonreference supporting reads at the distinct contextual error types showed reduced inter-sample differences in those samples; however, the differences between distinct contextual errors were preserved (Fig. 2I). Collectively, the results obtained here suggest that integration of intra-sample contextual error modeling of nonreference supporting reads at each of the 192 contexts may be a promising strategy for accurate suppression of errors produced by hybrid-capture NGS.
Using intra-sample contextual error modeling for reduction of false-positive calls
As described above, errors varied across samples yet were highly stereotypical according to sequence context and sequencing depth. We reasoned that intra-sample contextual error patterns could be leveraged for in silico error suppression. Such an approach could have several inherent advantages over existing error suppression methods that rely on UMIs, apply thresholds based on intra-sample–wide error rates, or use control samples to train error rate models. Therefore, we devised a computational approach, called Espresso, to model within a sample of interest the nonreference allele counts at each of the 192 distinct contextual error types. Espresso incorporates three distinct features that make it robust to different sequencing datasets (Supplementary Note): (i) pragmatic pre-filters that prepare the dataset for error modeling (fig. S4), (ii) automatic selection of the most appropriate probabilistic distribution for error modeling at a particular contextual error type (fig. S5), and (iii) utilization of nonreference supporting reads as opposed to VAF for error modeling (fig. S6). Unlike applying fixed and arbitrary cutoffs (e.g., minimum VAF, coverage, and number of supporting reads), nonreference alleles would not be indiscriminately eliminated by such an approach; rather, mutations would only be called if they reached statistical significance when compared to their corresponding error distributions (Fig. 3, A to E, and Materials and Methods).
Fig. 3. Integration of intra-sample contextual error modeling for error suppression.
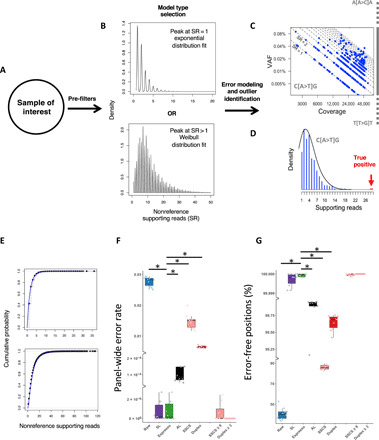
Flowchart illustrating the error modeling technique that is implemented by Espresso. (A) Following the summarization of the sequencing data to include the dominant alleles at each investigated genomic position, their corresponding read counts, and the average mapping read qualities in each sample of interest, a set of filters is being applied, aiming to deplete potential somatic SNVs and common polymorphism from being included in the error models. (B) On the basis of the distribution of the nonreference supporting reads in the enriched error list, Espresso selects between either the exponential or the Weibull probabilistic approaches. (C) The nonreference supporting read (SR) counts in each sample are being grouped based on the genomic sequence context to generate 192 context-specific distribution models. (D) The models are being reapplied to the entire sample’s data for outlier identification. True positives are being determined if they reach statistical significance when compared to their corresponding error distribution. (E) The cumulative distribution function graph displays the empirical data (black dots) and the theoretical data (blue line) generated by the 192 models in all the samples included in the CB dataset (top, exponential models) and the AML-MRD dataset (bottom, Weibull models). (F) Panel-wide error rates defined as the number of nonreference alleles supporting reads following error suppression, divided by all the reads from the same category (i.e., raw, SSCS, and duplex reads) across the entire 1,264,830-bp panel and (G) percentage of error-free positions in the 10 cord blood samples are illustrated. For error suppression, a cutoff P value ≤ 0.05 (Bonferroni-adjusted) was used. SSCS and duplex cutoffs are ≥1 nonreference supporting read unless indicated otherwise. * indicates Wilcoxon signed-rank test: P < 0.002.
To evaluate the performance of Espresso, we first applied it to the CB dataset. We reasoned that CB would have a minimal burden of somatic mutations, allowing for a more precise estimation of true error rates. We also tested in parallel other common error suppression techniques for unbiased comparative performance assessment (Materials and Methods). The techniques selected for comparison were representative of the spectrum of previously published tools. Specifically, we used two UMI-based methods, namely, single-strand consensus sequences (SSCSs) and duplex sequences (12), and two statistical methods for error correction that model background error distributions differently. Among the two statistical methods used, one relies on a training cohort to estimate error rates at the allele level (termed AL here) (20), and the other estimates error rates at the sample level (termed SL here) (14) without consideration for distinct sequence contexts.
Panel-wide error rates were highly similar among the 10 CB samples but varied significantly among the different error suppression methods (Fig. 3F). As compared with the various statistical approaches (i.e., SL, AL, and Espresso), the UMI-based methods demonstrated inferior error suppression capabilities. A minimum of nine nonreference supporting SSCS reads or three nonreference supporting duplex reads were required to achieve panel-wide error rates comparable to that of SL and Espresso in the CB dataset. We observed similar relative performance among the methods to maximize the number of error-free positions across the entire target panel (Fig. 3G). Considering the highest panel-wide error rate obtained by Espresso (2.74 × 10−6) and the lowest of the panel-wide error rate observed without error suppression (0.025) across the CB samples, Espresso achieved an error rate reduction of more than 9000-fold.
Analytical assessment of mutation detection accuracy
To evaluate the sensitivity and specificity exchange delivered by Espresso, we analyzed the sequencing data from the CL dataset, which consisted of a dilution series using two cancer cell lines, MOLM13 and SW48. For sensitivity measurements, we assessed the ability of the different methods to detect 119 MOLM13-specific germline variants at the different dilutions (table S3). To evaluate specificity, we assessed the miscalling of 186 AML-related somatic hotspot mutations that are covered by the target panel but are absent from both cell lines (table S3). Espresso outperformed all the other methods in distinguishing between true and false variants (Fig. 4A). In contrast, duplex sequencing achieved the smallest area under the receiver operator curve (AUC), highlighting the low diagnostic accuracy of this method and, consequently, its limited clinical utility in detecting variants across large hybrid-capture panels.
Fig. 4. Statistical measures of performance and constraints of contextual error modeling.
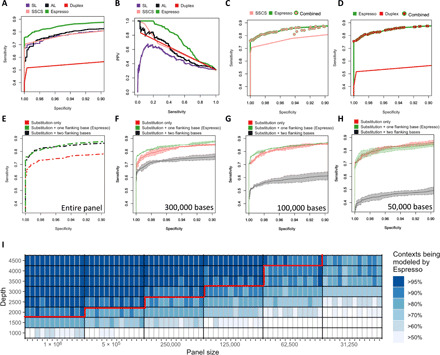
(A) Espresso demonstrates improved sensitivity versus specificity and (B) preferable precision-recall trade-offs as compared with the various indicated methods. The ability of each method to differentiate between 119 positive alleles and 186 negative control variants in a set of serially diluted cell line DNA samples was tested. (C and D) No substantial benefit of using UMIs to augment Espresso’s performance could be determined. Sensitivities and specificities were measured at all the possible combinations of the unique P values outputted by Espresso and the unique numbers of SSCS or duplex nonreference supporting reads that were observed in the dataset. The maximum sensitivities at each calculated value of specificity are illustrated. (E to H) Sensitivity versus specificity trade-offs derived by the reduced and extended contextual error modeling approaches are illustrated in comparison with Espresso. Ninety-five percent confidence intervals (shaded colors) and average values were derived by three random subsets of the data for each one of the indicated in silico decreased panel sizes. (I) Heatmap illustrating the percentage of contextual models that can be generated by Espresso when data are being restricted by either panel size reduction or sequencing depth reduction, or both. Data removal was controlled for both the reference and nonreference supporting reads, thus keeping the variant allele frequencies of the nonreference alleles similar to those in the original samples. The red line illustrates such combinations, of which 90% or more of the distinct contextual models could have been generated in every sample in the CL dataset. With datasets that fall below this line, the 12-model contextual error modeling approach can be used in addition to Espresso.
The use of hybrid-capture NGS panels allows for the detection of mutations at thousands of genomic positions. However, their use also creates unique challenges for true variant identification across so many bases. In addition to high sensitivity and specificity, positive predictive value (PPV) must be prioritized to maximize utility. We assessed PPV in conjunction with sensitivity (i.e., precision-recall analysis). We focused on variants with expected VAF ≤ 0.2%, since accurate variant detection below this threshold is clinically important yet has proven to be a great challenge for existing hybrid-capture NGS platforms (5, 30). Espresso provided a sensitivity of 19.9%, thus achieving the highest number of true-positive, low-VAF alleles at 100% PPV among the tested methods (Fig. 4B). This corresponds to a 6.8-fold improvement as compared to AL, which was the next best-performing method to detect low-VAF alleles without scarifying PPV. Notably, SL performed far worse in this analysis than the other methods due to a high number of false-positive calls across various sensitivity thresholds. This result highlights the limited power of noncontextual, sample-level error modeling in detecting mutations with very low read support despite its ability to achieve an extremely high level of error suppression (Fig. 3, F and G). Further supporting this, we compared the false-positive and true-positive calls obtained by Espresso with that of Mutect2 (16) at “tumor-only mode.” Once more, Espresso demonstrated superior results (table S4).
Previously, the suppression of errors through statistical error modeling was shown to be enhanced by combination with UMI-based approaches (20). However, integrating UMI information with Espresso did not confer significant performance improvements (Fig. 4, C and D), suggesting that accurate detection of low-frequency variants can be achieved with Espresso alone. Collectively, the comparative analysis using the CL dataset indicates that the bioinformatic strategy applied here outperformed other methods in the reliable distinction of low-frequency errors from real SNVs.
Impact of panel size and sequencing depth on contextual error modeling
To characterize pragmatic constraints of our method, we compared Espresso with alternative sequence context-based error models. Specifically, we included (i) a simplified 12-model design that accounts only for the 12 possible distinct substitution types without consideration of flanking bases and (ii) an expanded 3072-model design that accounts for the substitution type and for two additional 3′ and 5′ flanking bases. We evaluated the impact of panel size (i.e., number of interrogated bases) and sequencing depth on the performance of Espresso and the alternative sequence context-based models using the CL dataset.
This comparative analysis exposed critical factors affecting the performance of the alternative models. On the one hand, the performance of the 3072-model approach suffered with reduced panel size (Fig. 4, E to H, and fig. S7A). This is an expected observation that is attributed to the reduction in the number of nonreference alleles being used to populate a relatively high number of models, thus resulting in either model generation failure or an inadequate estimation of the background error noise. In contrast, performance of the 12-model approach was less dependent on panel size since the relatively small number of models was easily populated with nonreference alleles (Fig. 4, E to H, and fig. S7B); however, Espresso consistently outperformed the 12-model approach, presumably because the 12 models were insufficient to account for errors arising within distinct sequence contexts. Moreover, the 12-model approach performed poorly on the largest panel size, possibly as a result of model overfitting from high-VAF errors that escape the initial filtering steps (Materials and Methods). The performance of Espresso was relatively consistent across a broad range of panel sizes from ~1 Mb down to ~50 kb (Fig. 4, E to H, and fig. S7C).
Next, we serially downsampled the CL dataset to simulate various practical scenarios of panel sizes (1 Mb to 32.5 kb) and sequencing depths (4500× to 1000×). At each simulated panel-depth combination, we determined the percentage of trinucleotide contexts that could be modeled directly by Espresso (Fig. 4I). Notably, low represented nonreference alleles that cannot be modeled directly by Espresso would still be analyzed automatically by alternative techniques that are included in the software package (see “Data and materials availability”). Overall, these results illustrate the performance dependencies of Espresso and related sequence context–based models to assist with their implementation in a wide range of sequencing settings.
Detection and monitoring of minimal residual disease
Having demonstrated Espresso’s high analytical performance in the CB and CL datasets, we next sought to evaluate its clinical utility. The presence of persistent AML clones that carry genetic abnormalities during or after treatment has been shown to carry crucial prognostic information (31). Therefore, we assembled a cohort of 42 patients with AML (AML-MRD; table S5) whose mutations were previously determined at diagnosis (table S3). Forty of the 42 patients had serial samples analyzed by ultra-deep hybrid-capture NGS at two time points during therapy; for the other two patients, single follow-up samples were available.
Since minimal/measurable residual disease (MRD) monitoring may guide clinical decisions (32–34), in addition to true positives, both false positives and false negatives could have tremendous implications for patient care. We therefore evaluated F1 scores, which represent the harmonic mean of PPV and sensitivity. For comparative performance evaluation, mutations reported at diagnosis were considered as true positives if they were detected in the follow-up samples of the same patient or as false positives if they were detected in other patients. We first applied a cutoff of α ≤ 0.05 (Bonferroni-adjusted) for the probabilistic methods SL, AL, and Espresso and a heuristic threshold of ≥1 nonreference supporting reads for the UMI-based methods SSCS and duplex. Tested on the subset of samples obtained at either the first time point (T1, closer to diagnosis) or the second time point (T2, further into treatment), Espresso delivered the highest F1 scores (0.71 at T1 and 0.74 at T2) followed by AL and duplex (Fig. 5A). We next applied the optimized SSCS and duplex cutoffs used in the CB analysis (i.e., ≥9 and ≥3 nonreference supporting reads, respectively). Although F1 scores improved with these parameters, they still fell short due to an increased number of false positives for SSCS ≥9 and an increased number of false negatives for duplex ≥3 in both the T1 and the T2 data subsets as compared with Espresso (Fig. 5B).
Fig. 5. Measurements of residual disease.
(A) Espresso provides a preferred balance between precision (PPV) and recall (sensitivity), as determined by the inspection of 78 SNVs reported across 35 of 42 patients at the time of AML diagnosis. Mutations were called in the patients’ sample at 21 different iterations. In each iteration, 6 random patients of the 42 were excluded. Median F1 scores and 1 SD are shown for the various methods tested at two time points during the course of treatment (T1 and T2, Wilcoxon signed-rank test: P ≤ 6.4 × 10–5 for all the comparisons with Espresso). (B) The variation in the mutations being called by Espresso (α ≤ 0.05, Bonferroni-adjusted), SSCS (≥9 nonreference supporting reads), and duplex (≥3 nonreference supporting reads) is illustrated. Red color indicates called mutations, while blue color indicates that mutations were not detected. FP, false positives; FN, false negatives. (C) Sensitivity versus specificity as determined by the different tested methods. (D) Enrichment of clones, carriers of TP53, and DNMT3A mutations is observed in patients with AML following therapy. The y axis represents the number of mutations detected, classified by the affected genes.
Despite the technical differences between the CL and AML-MRD datasets, Espresso once again produced the most preferred balance between sensitivity and specificity (Fig. 5C). We compared Espresso with additional algorithms and saw consistent outcomes. Espresso outperformed Mutect2 (16) in both the tumor-only mode and the “panel of normals” mode when samples obtained from 14 healthy adults were used (table S4). Espresso also outperformed deepSNV (18), a statistical algorithm that was developed specifically for the accurate detection of SNVs from deep targeted sequencing experiments. The comparison with deepSNV extrapolates beyond the probabilistic approaches being used and illustrates the benefits of other features implemented in our bioinformatic pipeline for the reduction of false-positive calls (fig. S8).
Having established Espresso as the preferred methodology to maximize the accuracy of SNV detection from peripheral blood, we next sought to implement it for the characterization of clonal dynamics in patients with AML. Since the competitive balance among different hematopoietic clones is likely to change during multiple rounds of chemotherapy, we hypothesized that Espresso would enable the identification of resistant clones that were not reported at diagnosis. We therefore extended our analysis to include an additional 147 highly recurrent AML SNVs that are covered by the AML-MRD hybrid-capture panel (table S3). Across all the samples, Espresso identified 92 mutations (α ≤ 0.05, Bonferroni-adjusted) with the lowest being reported at VAF = 0.0135% (table S6 and fig. S9). These correspond to 59 distinct mutations, out of which 47 (~80%) were present in at least two samples of the same patient (that is, reported at diagnosis and detected in at least one additional time point by Espresso or detected in the two follow-up samples by Espresso). Such a high percentage of validated mutations is an indicator of Espresso’s reliable mutation calling. Among these, Espresso has enabled the detection of 22 new putative driver SNVs not reported at diagnosis in 15 patients, including in 3 of the 7 patients (~43%) with no SNVs in the diagnostic report (table S6). Further supporting the validity of the mutations called by Espresso, most of these newly identified mutations were in genes that commonly contribute to positive clonal selection following cytotoxic chemotherapy (35–37), including TP53 and DNMT3A (Fig. 5D).
Together, our results demonstrate substantial advantages of Espresso over other methods for SNV detection from peripheral blood of patients with AML during the course of therapy. Encouraged by a recent consensus document release from the European LeukemiaNet MRD Working Party (38), many studies are now underway to evaluate the prognostic and predictive significance of clonal dynamics in AML and the proposed role of MRD detection as a surrogate endpoint for clinical trials (39). Implementation of Espresso in these contexts has the potential for significant clinical utility.
Targeting informative genomic loci for improved practicality of pre-AML genomic screens
Age-related clonal hematopoiesis (ARCH) is a common phenomenon evident by the presence of somatic mutations in hematopoietic stem cells of otherwise healthy individuals that cause a clonal expansion of the stem cells and their progeny (40). Recently, our group reported several hundred ARCH-associated mutations spread across 27 genes with various contributions to the risk of AML transformation (9). Our study provided a proof of concept for risk prediction of AML. Nevertheless, large population screens using broad sequencing panels remain socioeconomically unattractive because of high costs, the relatively low incidence of AML, and the relatively high incidence of ARCH in the general population.
To address these challenges, we reasoned that interrogating a small number of highly recurrent AML mutations would be a more tractable approach than broad hybrid-capture sequencing. This approach could theoretically result in improved segregation between pre-AML and controls while reducing sequencing costs. The success of this approach relies on the accurate identification of preleukemic mutations in asymptomatic individuals.
We first compiled datasets that would allow comparisons among the distinct methods used in our previous analyses. For this reason, we focused initially on the pre-AML1 dataset, which contains UMIs in the sequencing reads, and the CB dataset, which could be used as a training set for error rate estimation at the AL. Putative driver SNVs (that is, mutations in coding sequences other than synonymous SNVs and mutations at splice sites) identified by each method at the recurrently mutated genomic loci were used to derive random forest classifiers that were trained and tested on their corresponding method’s mutation calls (table S7). For the probabilistic methods, α ≤ 0.05 (Bonferroni-adjusted) was used, and for the UMI-dependent methods, we applied either a threshold of one supporting consensus read or SSCS ≥ 9 and duplex ≥ 3. The Espresso-derived classifier exhibited the highest level of performance for discriminating pre-AML from controls (AUC: 0.74) and reported the highest sensitivity (46.8%) at 100% specificity (Fig. 6A). A reduction in specificity down to 96.3 or 93.7% was needed to achieve the same sensitivity with the SL-derived and SSCS-derived classifiers, respectively. The SSCS-derived model also underperformed the Espresso-derived classifier when the SSCS ≥ 1 cutoff was applied (AUC: 0.66, Fig. 6A, dashed line). The duplex ≥ 3 derived classifier had the poorest performance (AUC: 0.42), owing to poor duplex consensus efficiency (fig. S1B), low duplex coverage (Fig. 1A), and subsequent dropout of mutations not meeting the required cutoff. On the contrary, with a threshold of one supporting duplex read, a large number of putatively false-positive SNVs were called, resulting in poor classification accuracy (AUC: 0.65, Fig. 6A, dashed line). The AL-derived classifier also performed poorly due to a high number of false-positive SNVs (AUC: 0.62).
Fig. 6. AML transformation risk prediction using recurrently mutated loci.
Classification performance evaluation of pre-AML and control, mutated samples. (A) Each classifier was trained and tested on the mutations that were obtained from the classifier’s corresponding method. (B) Comparison between the Espresso and the SL-derived classifiers. In this iteration, each classifier was trained using its corresponding method’s mutation calls and was tested in its accuracy to classify pre-AML cases and controls, including mutated samples identified by the other method as well. (C) Comparative performance validation between the Espresso and the SL-derived classifiers to differentiate between pre-AML and control samples obtained from an additional validation dataset (8). Information regarding the study participant’s age, specific mutations, and their VAFs was obtained directly from the main text. (D) Performance estimation using the validation dataset and simulated controls. (E) Precision-recall trade-offs are calculated at the individual level (that is, serial samples are accounted for single individuals and individuals without any mutations are also included in the performance measurements). The red dot indicates AML’s incidence rate. This is equivalent to a situation where no screen is being conducted at all [PPV = incidence rate = 0.006% (44), SN = 100%]. The green dot indicates the model performance using an additional published dataset consisting of 11,262 individuals when the model was set to achieve 100% specificity in the training set. Horizontal color bars represent PPV ranges determined for screening mammography for breast cancer (54) and fecal immunochemical test for advanced adenomas and colorectal cancer (CRC) (55). Comparison with the genetic risk model performance shows the extent to which sensitivity must be compromised to achieve PPV comparable with these widely applied early detection tests.
There is a low cumulative risk of ARCH progression to hematologic neoplasms (41). For this reason, the implementation of a population-based pre-AML genomic screening test would need to achieve exceedingly high specificity and low false-positive rate. We therefore prioritized the Espresso- and SL-derived classifiers for subsequent performance evaluation. Additional mutations that were found by Espresso and SL in the pre-AML2 dataset were included in the analysis (table S7). Each classifier was trained on the mutations found by its corresponding method in both the datasets (pre-AML1 and pre-AML2) and tested on the data that include all the mutations detected by either of the two methods. The Espresso-derived classifier once more provided a better overall sensitivity-specificity balance and a greater sensitivity at 100% specificity (Fig. 6B). Similar trends were observed when both the classifiers were applied to an external validation set consisting of mutations called in 188 pre-AMLs and 181 controls (8), with the Espresso-derived classifier again displaying higher discriminatory accuracy (Fig. 6C). Together, the superior classifier performance using mutations called by Espresso illustrates that accurate mutation calling is imperative when designing genetic risk prediction models.
To estimate how well the winning classifier would perform as a population-wide screening test, we spiked the validation set into >4 million in silico simulated controls (prevalence ~0.005%; Materials and Methods). Despite the small genomic footprint (table S8), the Espresso-derived classifier resulted in accurate identification of the mutated pre-AML samples (AUC: 0.84; Fig. 6D). As an example, when the model was tuned to minimize false-positive calls based on the pre-AML1/pre-AML2 merged training dataset, a sensitivity of 29.3% and a specificity of 99.8% were obtained. Precision-recall analysis revealed the extent to which the Espresso-derived classifier may enrich for individuals at high risk of developing AML as compared with current practice (no screening, i.e., AML incidence rate) (Fig. 6E). Sensitivity was 4.8% at 100% PPV; this small subset detected with no false positives was enriched for highly penetrant SRSF2/IDH2 double-positive individuals with the highest risk for AML development (table S9). Last, we estimated the model performance in an additional published cohort of 11,262 individuals (42). In this cohort, when the model was tuned to minimize false positives within the training dataset, a sensitivity of 14.3% and a PPV of 4.8% were obtained (Fig. 6E and table S9).
DISCUSSION
In this study, we described the rationale, technical performance characteristics, and potential clinical utility for Espresso, a novel method to improve hybrid-capture sequencing–based SNV detection. Unlike many other NGS error suppression methods, including the representative published UMI-based and probabilistic model–based approaches tested here, Espresso does not rely on UMIs or a training set of controls for error rate estimations; therefore, Espresso improves practicality by reducing library preparation complexity, assay costs, and analysis time. We observed additional notable advantages of Espresso over alternative methods, and these were consistent across diverse datasets. Specifically, Espresso produced superior error suppression and an improved trade-off between sensitivity and specificity for detection of low-VAF alleles.
These advantages of Espresso were the result of several key features. First, Espresso applies a set of pre-filters to prepare the data for error modeling. Second, Espresso automatically selects between two statistical models to estimate the number of alternative supporting reads rather than the VAFs; thus, in addition to selecting the more appropriate error distribution model, it better accounts for error rate bias resulting from variation in sequencing depth within hybrid-capture NGS datasets. Third, Espresso markedly reduces false-positive calls by considering only the dominant nonreference allele at each interrogated genomic position. Fourth, Espresso leverages a large number of errors that share the same trinucleotide sequence context within the investigated sample; thus, it reduces the potential for misrepresentation of real error rates by relatively small control cohorts.
To explore its potential use in clinical settings, we tested the performance of Espresso to detect SNVs in serial peripheral blood samples from 42 patients with AML who achieved clinical remission. Consistent with the performance in the other investigated datasets, Espresso outperformed all the other tested methods in this setting. Using Espresso, we found resistant subclones enriched for TP53 and DNMT3A mutations that were genetically distinct from the AML clones present at diagnosis. In the future, more extensive cohort studies are needed to determine whether the selection and enrichment of such clones following induction therapy may affect patient outcomes in a nonautonomous fashion, similar to the observations in solid malignancies (43). Furthermore, combining accurate detection of persistent mutations together with other independent prognostic markers will be necessary to build clinically relevant models for accurate determination of the risk of relapse.
Our results emphasize the importance of accurate mutation detection for the derivation of classification models in the setting of early detection of AML. Using Espresso, we derived a risk prediction model that is focused on a minimal yet highly informative set of genomic loci that are recurrently mutated in patients with AML. With only 1594 genomic bases being interrogated, our results imply that up to 29.3% of de novo AML cases can be predicted years in advance with a specificity of 99.8%. Although sensitivity may greatly suffer with elevated PPV, considering the incidence rates of AML in the general population (~6:100,000) (44), our approach would still provide meaningful patient enrichment. Modest sensitivity may be acceptable when screening the general population as long as specificity and PPV remain high. Further prospective validation studies are required to assess the feasibility, utility, and cost-effectiveness of this targeted approach. Our findings should also be extended to incorporate additional predictive biomarkers. As AML is a blood-borne disease, we envision that epigenetic and metabolomic perturbations within leukocytes may further improve prediction accuracy, thus making AML predictions more clinically useful. Our results indicate that certain biomarker-enriched populations may be at an exceedingly high risk of developing AML. In time, novel therapeutic developments and targeted therapies against blood cells with high-risk mutations may provide the minimal side effects necessary to deliver a favorable risk-benefit ratio that justifies the initiation of early intervention clinical studies.
In summary, we have described, benchmarked, and validated a new practical NGS error suppression technique. We have demonstrated the superiority of Espresso in detecting somatic SNVs as compared with existing state-of-the-art approaches and defined its limitations with respect to sequencing depths and hybrid-capture panel sizes. We used Espresso to derive new biological insights, augmenting our understanding of the genetic mutations that define high-risk malignant transformation and therapy resistance clones in patients with AML. We envision that Espresso will prove useful in guiding clinical decisions and scientific research alike.
MATERIALS AND METHODS
Cohorts description
CB dataset: This dataset is composed of 10 human umbilical cord blood genomic DNA samples obtained from Trillium Hospital (Mississauga, Ontario, Canada) with informed consent in accordance with guidelines approved by the University Health Network Research Ethics Board. Cord blood was processed 24 to 48 hours after delivery. Mononuclear cells were enriched using Ficoll-Paque followed by red blood lysis by ammonium chloride and CD34+ selection before DNA extraction. CL dataset: MOLM13 cell line DNA was mixed with SW48 cell line DNA at relative concentrations of 100, 5, 1, 0.2, 0.04, and 0% and was sequenced in duplicate. Pre-AML1 and pre-AML2 datasets: Detailed information regarding these cohorts is described elsewhere (9). Briefly, the pre-AML1 dataset contains peripheral blood genomic DNA samples obtained from a total of 509 individuals upon enrollment into the European Prospective Investigation into Cancer and Nutrition (EPIC) study (45) between 1993 and 1998. Together, 414 control individuals who did not develop any hematological disorders during the extended follow-up period and 95 individuals who developed AML were included in this study. The pre-AML2 dataset contains peripheral blood genomic DNA samples obtained from individuals enrolled in the EPIC-Norfolk longitudinal cohort study between 1994 and 2010. Samples were available from 37 patients with AML and 262 age- and sex-matched controls without a history of cancer or any hematological conditions. Samples taken at multiple time points were available for a fraction of the participants in this cohort. Notably, samples from eight pre-AML patients in the pre-AML2 cohort were separately sequenced in the pre-AML1 dataset (by independent investigators using a different methodology). To avoid statistical misrepresentation of AML predictions, we removed those samples from the pre-AML2 dataset before the derivation of the described genetic risk models. AML-MRD dataset: This dataset is composed of peripheral blood genomic DNA from 42 patients with AML treated at the Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, Canada. All 42 patients achieved morphologic leukemia-free state (MLFS) on chemotherapy. Complete count recovery occurred when absolute neutrophil count recovered to ≥1 × 109/liter and platelet count recovered to ≥100 × 109/liter up to 7 days following the bone marrow assessment that confirmed MLFS status. All patients were deidentified with patient IDs. Their demographic and clinical features were captured (table S5). All the samples in this study, including healthy individuals and patients with cancer, were collected with informed consent for research use and were approved by Institutional Review Boards in accordance with the Declaration of Helsinki. Protocols were approved by the following ethics committees: (i) International Agency for Research on Cancer Ethics Committee approval #14-31, (ii) East of England—Cambridgeshire and Hertfordshire Research Ethics Committee reference number 98CN01, and (iii) University Health Network Research Ethics Board # 01-0573.24.
NGS libraries construction and sequencing
Library construction and sequencing were done as previously described (9). Briefly, for each sample in the CB, CL, and pre-AML1 datasets, 100 ng of genomic DNA was sheared to 250–base pair (bp) fragments before library construction (KAPA HyperPrep Kit KK8504, Kapa Biosystems) with a Covaris E220 instrument using the recommended settings. After end repair and A-tailing, ligation of UMI-containing adaptors was performed with 100-fold molar excess. Agencourt AMPure XP beads (Beckman Coulter) were used for library cleanup following eight cycles of fragment amplification with 0.5 μM Illumina universal and indexing primers. Targeted hybrid-capture was carried out on pools of three indexed libraries. Five microliters of Cot-I DNA (1 mg ml−1; Invitrogen) and 1 nmol each of xGen Universal Blocking Oligo, TS-p5, and xGen Universal Blocking Oligo, TS-p7 (8 nucleotides) were added to each pool of adaptor-ligated DNA. The mixture was dried using a SpeedVac and then was resuspended in 1.1 μl of water, 3.4 μl of NimbleGen hybridization component A, and 8.5 μl of NimbleGen 2× hybridization buffer. The mixture was heat-denatured at 95°C for 10 min following the addition of 4 μl of xGen Lockdown Probes (3 pmol; xGen AML Cancer Panel v.1.0). Hybridization was conducted at 47°C for 72 hours. Washing and recovery of the captured DNA were initiated with 100 μl of clean streptavidin beads that were added to each capture. Following separation of the libraries and the supernatant using a magnet, 200 μl of 1× Stringent Wash Buffer was added, and the reaction was incubated for 5 min at 65°C. The supernatant containing unbound DNA was removed before repeating the high stringency wash for the second time. The bound DNA was then washed one time with 200 μl of each of the following: 1× Wash Buffer, 1× Wash Buffer II, and 1× Wash Buffer III. The washed DNA on beads was resuspended in 40 μl of nuclease-free water, and this volume was divided into two polymerase chain reaction (PCR) tubes that were subjected to 10 cycles of post-capture amplification (Kapa Biosystems, recommended conditions). Libraries were spiked with 2% PhiX before sequencing. The procedure used for the pre-AML2 dataset is described elsewhere (referred to as the validation cohort) (9). For each sample in the AML-MRD dataset, peripheral blood samples were collected during remission in PAXgene Blood DNA Tubes (PreAnalytiX, Hombrechtikon, Switzerland). DNA was extracted according to the manufacturer’s instructions. Illumina-compatible libraries were constructed from 100 ng of sheared genomic DNA using the Covaris M220 sonicator (Covaris, Woburn, MA, USA) and the KAPA HyperPrep Kit (#KK8504, Kapa Biosystems, Wilmington, MA, USA). Following end repair and A-tailing, adapter ligation was performed for 16 hours at 4°C using 100-fold molar excess of adapters. Agencourt AMPure XP beads (Beckman Coulter) were used for library cleanup, and ligated fragments were amplified by PCR for 6 cycles using 0.5 μM universal and indexed primers. Following hybrid-capture at 47°C for 72 hours, the captured DNA fragments were enriched with 12 cycles of PCR. Paired-end 2 × 125-bp sequencing was performed on an Illumina HiSeq 2500 instrument with eight libraries multiplexed into each lane.
Bioinformatics pipeline and consensus reads assembly
Paired-end sequencing data from the Illumina platform were converted to FASTQ format. When included, the unique molecular barcode information at each read of the pair was trimmed and was added to the read header. The Burrows-Wheeler aligner (BWA-mem) (46) was used for the alignment of the processed FASTQ files to the reference hg19 genome. To eliminate the chance of ambiguous short indel alignment on neighboring SNV miscalls, we removed reads with indels. We further cleaned the data from short and hard clipped reads and any nonunique read alignments. We found that, together, these preprocessing steps can improve SNV detection (fig. S8). Consensus read assembly into read families was done in a similar way to previous reports (47, 48). Specifically, reads that share the same molecular barcode sequence, the genomic position of where each read of the pair maps to the reference, and the CIGAR string were grouped. Families that consisted of at least two reads were used to generate SSCS, and a consensus base was called when there was full agreement. When a consensus base was called, it was assigned with the maximum base quality score observed in its corresponding precollapsed reads. Similarly, when two SSCSs with corresponding UMIs on the reciprocal strand were observed, duplex reads were generated. After converting the raw-, SSCS-, and duplex-containing sam files into coordinate-sorted bam files, we used samtools (49) version 1.2 and Varscan2 (14) version 2.2.8 to summarize the data. The following parameters were used: (i) mpileup parameters: -s -x -BQ0 -q1 -d100000 and (ii) pileup2cns parameters: --min-coverage 10 --min-reads2 1 --min-avg-qual 30 --min-var-freq 0.0001 --p-value 1 --strand-filter 0. These are rather permissive parameters allowing the output of all the dominant alleles in each one of the investigated genomic positions. To allow unbiased performance comparisons, we used this format as an input for all the probabilistic methods (SL, AL, and Espresso) and the UMI-based methods (SSCS and duplex).
Probabilistic models for error correction
With Espresso, we deployed a novel approach to model errors based on their association with either one of the 192 contextual contexts (Fig. 3, A to E). These correspond to 12 base substitution types, four alternative 5′ bases, and four alternative 3′ bases. To mitigate the impact of outliers and real mutations on overfitting, a set of filters is applied to exclude specific variants from the contextual error models (Supplementary Note and fig. S4). These include the removal of alleles (i) that are observed as germline variants in the general population (50, 51) with minor allele frequency ≥ 0.1%, (ii) with VAF/error rates ≥5%, (iii) that have MapQual<59 and MapQual!=0 [for additional information, please refer to the manual of Varscan2 (14)], (iv) that describe recurrent cancer mutations, and (v) that disproportionally persist across multiple samples in the dataset (see the “Flagged alleles” section; Materials and Methods). Notably, to prevent performance comparison bias, we used these filters together with all the probabilistic methods (SL, AL, and Espresso) and the UMI-based methods (SSCS and duplex) tested.
To determine the more appropriate distribution type for error modeling, Espresso first investigates the overall distribution of nonreference supporting reads in a context-independent manner, in the sample’s filtered, error-enriched list. On the basis of the observed peak occurrence, either exponential or Weibull distribution models are selected to generate all the contextual models. If the peak corresponds to a single nonreference supporting read, exponential distribution will be used to represent the data; otherwise, if this value is larger than 1, Weibull distribution will be used. Either the “pexp” or “pweibull” R functions are then being used together with the modeled parameters from the fitdistrplus package (either rate or shape and scale) to determine how high any nonreference allele of interest is being represented above its corresponding contextual background. A Bonferroni-corrected P value ≤ 0.05 was used to determine whether any nonreference allele received significantly more supported reads.
For comparative performance analysis, error rate models at the AL were constructed as previously described (20). Briefly, if the total number of nonzero allele frequencies seen in the training set used for error modeling was ≤5, we used Gaussian distribution; otherwise, we fit a Weibull distribution to the allele frequencies observed in the training set. Specifically, the “pnorm” or pweibull R functions were used together with the modeled parameters (either mean and SD or shape and scale) to estimate the likelihood that any allele frequency value of interest is above the corresponding modeled distribution derived for the same interrogated position in the corresponding training set. The yielded P values were adjusted by incorporating the fraction of nonzero allele frequencies into the final models [for additional information, please refer to iDES (20)]. Training datasets were constructed as follows: (i) The pre-AML1 dataset was used for the CB analysis (Fig. 3) and the CL analysis (Fig. 4). (ii) A training set composed of peripheral blood genomic DNA samples from 14 healthy individuals was sequenced and used in the analysis of the AML-MRD data (Fig. 5). (iii) The CB dataset was used as a training set for the derivation of the AL-based model for AML risk prediction (Fig. 6). To evaluate allele mutated status at the SL, we used Varscan2 (14) that computes statistical significance in single samples by Fisher’s exact test.
Flagged alleles
While parameters such as specific genomic context, the presence of a repetitive region, and low base or read mapping quality may explain the basis of some errors, these do not always capture artifacts that may persist across multiple samples. We therefore derived a statistical approach to flag recurrently specious alleles. To flag potentially low-frequency artifactual alleles that escaped conventional filtering, we iterated between the 99 and 99.9% nonreference allele frequency quantiles in the entire investigated cohort in increments of 0.1% (user-defined parameters). The 10 derived VAF values were used consecutively to apply Fisher’s exact tests, determining whether errors with VAF above the quantile-derived cutoff distribute proportionately among all the observed nonreference alleles in the dataset or being clustered in a low number of alleles across many samples in an unbalanced fashion. Then, if included, we removed recurrent Catalogue of Somatic Mutations in Cancer (COSMIC) (52) mutations (that is, SNVs with classification other than synonymous with at least three case reports of hematopoietic and lymphoid tissues; COSMIC version 80) to derive a final list of dataset-specific flagged alleles to be excluded from contextual error modeling.
AML risk prediction
To derive with a list of mutations that are highly associated with leukemic transformation for AML risk prediction model derivation, we interrogated the COSMIC database (52) and ranked variants according to their evidence for functional relevance in AML. All the SNVs with classification other than synonymous with at least 10 case reports of “hematopoietic and lymphoid tissues” were considered hotspot variants. For the future implementation of our findings, we reasoned that any hybrid-capture probe design and short sequencing reads would efficiently encompass at least several genomic bases surrounding these hotspots. Therefore, we extended the variant calls to capture mutations with a putative deleterious effect that are within five–amino acid distance surrounding each hotspot variant. Genomic loci that were found to be mutated in the training cohort (pre-AML1 and pre-AML2) were used for the final model derivation (table S8). Notably, we discarded genomic loci with mutations in KIT, KRAS, and PHF6 as these were found solely in the training set’s controls. Such enrichment surely does not correlate with real-life evidence and can bias classification. We then used a random forest algorithm via the R package randomForest. Mutations were grouped by genes, and their VAFs were used to train the model together with the age of the individuals at sampling and the number of the mutations that they carry. If more than one mutation was detected in the same gene, the highest VAF was used. The number of features used for each one of the 5000 generated trees was two.
Simulated controls
To simulate a large population screen, we used the mutations detected by Espresso in the controls from the pre-AML1 and pre-AML2 (termed merged dataset here). We first calculated the frequency of controls that carry at least one mutation at the following age groups: 20 to 49, 50 to 64, 65 to 74, and >75 years old. For these age groups, we obtained the incidence rates of AML through the Surveillance, Epidemiology, and End Results Program (53). By assuming similar age distribution for the validation cohort (8) and the individuals interrogated in the merged dataset and knowing the number of pre-AML cases interrogated in the validation cohort (n = 188), we were able to estimate the number of simulated controls needed to mimic real incidence rates for each age group. Overall, 4,033,904 controls were simulated.
The frequency of ARCH and the number of mutations that each individual carries within each control age group from the merged dataset helped us to estimate how many of the simulated individuals are expected to carry mutations in the relevant genomic loci (table S8). Overall, 5.05, 7.69, 10.70, and 19.09% of the individuals within the age range of 20 to 49, for 50 to 64, for 65 to 74, and ≥75 years, respectively, were simulated to have ARCH. A total of 285,629 individuals (~7%) were simulated to carry one mutation, 934 with two mutations (~0.02%), and 156 with three mutations (~0.004%). We next assigned the specific mutations to the simulated individuals based on their association with each age group. For example, for the 149,423 simulated mutated controls with a simulated age of 50 to 64, we populated a list of 149,423 specific mutations that were detected in control individuals in the same age group or in younger age groups in the merged dataset. We also allowed 10% of the mutations detected in the merged dataset in one age group older to be randomly included. Last, we aimed to assign VAF to the simulated mutations. We observed that the VAF of the detected mutations in the merged dataset did not significantly correlate with age [R(Pearson) = 0.20; P = 0.07] and that a lognormal distribution accurately captures the VAF distribution among all the detected mutations. We therefore used the “rlnorm” R function to simulate VAFs. This resulted with a median VAF of 1.45% and a mean VAF of 2.45% for the simulated controls; 37.46% of the simulated VAFs received a value of VAF ≥ 2%. As intended, these values are highly comparable with those of the mutations found in the merge dataset’s controls (table S7).
Supplementary Material
Acknowledgments
We acknowledge the support from the Princess Margaret Cancer Foundation. We thank the Genome Technologies team at the Ontario Institute for Cancer Research as well as the Princess Margaret Genomics Centre for technical expertise with sequencing, data management, and QC analysis. We acknowledge the dedicated work of G. Vassiliou from the Wellcome Trust Sanger Institute for supervising sample acquisition from the EPIC-Norfolk longitudinal cohort and for the management of the sequencing data, its creation, and its sharing. Funding: S.A. received support from the Benjamin Pearl fellowship from the McEwen Centre for Regenerative Medicine and is funded by the Ontario Institute for Cancer Research. A.G.X.Z. was supported by the CIHR Vanier Scholarship. S.V.B., S.M.C., and T.J.P. were supported by the Gattuso-Slaight Personalized Cancer Medicine Fund at the Princess Margaret Cancer Centre. J.E.D. was supported by funds from the Princess Margaret Cancer Centre Foundation, Ontario Institute for Cancer Research, with funding from the Province of Ontario, Canadian Institutes for Health Research, Canadian Cancer Society Research Institute, Terry Fox Foundation, Genome Canada through the Ontario Genomics Institute, and the Canada Research Chair. Author contributions: S.A. developed the concept and the code, led and supervised the analysis of the data, and wrote the manuscript. A.G.X.Z. created the R package and contributed to the development of the analytical pipeline. I.N.-M. contributed to the development of the R package and the correspondence with the reviewers. S.W.K.N. provided support for genetic predictive model derivation. T.T.W. and T.J.P. provided bioinformatics support and contributed to the analysis of the data. M.D.M., T.M., and S.M.C. enabled sample acquisition and clinical data curation and provided clinical expertise. L.I.S. and P.A. contributed to the study design and/or in obtaining data and materials. J.E.D. and S.V.B. supervised all aspects of the study and wrote the manuscript. All authors commented on the manuscript at all stages. Competing interests: S.V.B. is co-inventor on patents, distinct from the subject matter of this manuscript, relating to circulating tumor DNA analysis, one of which has been licensed to Roche Molecular Diagnostics. S.V.B. provides consultation for DNAMx Inc. and has received research funding from Nektar Therapeutics. T.J.P. provides consultation for Merck, Chrysalis Biomedical Advisors, the Canadian Pension Plan Investment Board, and Illumina (compensated) and receives research support (institutional) from Roche/Genentech. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All the sequencing files used in this manuscript can be downloaded from the European Genome-phenome Archive (EGA) or the NCBI Sequence Read Archive under the following accession numbers: (i) pre-AML1, EGAD00001003583; (ii) pre-AML2, EGAD00001003703; (iii) CB, EGAD00001005261; (iv) AML-MRD, EGAD00001005270; and (v) CL, SRP141184. Software and supplemental code are available on GitHub at www.github.com/abelson-lab/Espresso and www.github.com/abelson-lab/Espresso_paper, respectively. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabe3722/DC1
REFERENCES AND NOTES
- 1.Cancer Genome Atlas Research Network, Weinstein J. N., Collisson E. A., Mills G. B., Shaw K. R. M., Ozenberger B. A., Ellrott K., Shmulevich I., Sander C., Stuart J. M., The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The International Cancer Genome Consortium , International network of cancer genome projects. Nature 464, 993–998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey M. H., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl M. C., Kim J., Reardon B., Ng P. K. S., Jeong K. J., Cao S., Wang Z., Gao J., Gao Q., Wang F., Liu E. M., Mularoni L., Rubio-Perez C., Nagarajan N., Cortés-Ciriano I., Zhou D. C., Liang W.-W., Hess J. M., Yellapantula V. D., Tamborero D., Gonzalez-Perez A., Suphavilai C., Ko J. Y., Khurana E., Park P. J., Van Allen E. M., Liang H.; MC3 Working Group; Cancer Genome Atlas Research Network, Lawrence M. S., Godzik A., Lopez-Bigas N., Stuart J., Wheeler D., Getz G., Chen K., Lazar A. J., Mills G. B., Karchin R., Ding L., Comprehensive characterization of cancer driver genes and mutations. Cell 173, 1034–1035.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shu Y., Wu X., Tong X., Wang X., Chang Z., Mao Y., Chen X., Sun J., Wang Z., Hong Z., Zhu L., Zhu C., Chen J., Liang Y., Shao H., Shao Y. W., Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci. Rep. 7, 583 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark T. A., Chung J. H., Kennedy M., Hughes J. D., Chennagiri N., Lieber D. S., Fendler B., Young L., Zhao M., Coyne M., Breese V., Young G., Donahue A., Pavlick D., Tsiros A., Brennan T., Zhong S., Mughal T., Bailey M., He J., Roels S., Frampton G. M., Spoerke J. M., Gendreau S., Lackner M., Schleifman E., Peters E., Ross J. S., Ali S. M., Miller V. A., Gregg J. P., Stephens P. J., Welsh A., Otto G. A., Lipson D., Analytical validation of a hybrid capture–based next-generation sequencing clinical assay for genomic profiling of cell-free circulating tumor DNA. J. Mol. Diagn. 20, 686–702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phallen J., Sausen M., Adleff V., Leal A., Hruban C., White J., Anagnostou V., Fiksel J., Cristiano S., Papp E., Speir S., Reinert T., Orntoft M.-B. W., Woodward B. D., Murphy D., Parpart-Li S., Riley D., Nesselbush M., Sengamalay N., Georgiadis A., Li Q. K., Madsen M. R., Mortensen F. V., Huiskens J., Punt C., van Grieken N., Fijneman R., Meijer G., Husain H., Scharpf R. B., Diaz L. A. Jr., Jones S., Angiuoli S., Ørntoft T., Nielsen H. J., Andersen C. L., Velculescu V. E., Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 9, eaan2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paweletz C. P., Sacher A. G., Raymond C. K., Alden R. S., O’Connell A., Mach S. L., Kuang Y., Gandhi L., Kirschmeier P., English J. M., Lim L. P., Jänne P. A., Oxnard G. R., Bias-corrected targeted next-generation sequencing for rapid, multiplexed detection of actionable alterations in cell-free DNA from advanced lung cancer patients. Cancer Res. 22, 915–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desai P., Mencia-Trinchant N., Savenkov O., Simon M. S., Cheang G., Lee S., Samuel M., Ritchie E. K., Guzman M. L., Ballman K. V., Roboz G. J., Hassane D. C., Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 24, 1015–1023 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abelson S., Collord G., Stanley W., Ng K., Weissbrod O., Cohen N. M., Niemeyer E., Barda N., Zuzarte P. C., Heisler L., Sundaravadanam Y., Luben R., Hayat S., Wang T. T., Zhao Z., Cirlan I., Pugh T. J., Soave D., Ng K., Latimer C., Hardy C., Raine K., Jones D., Hoult D., Britten A., Mcpherson J. D., Johansson M., Mbabaali F., Eagles J., Miller J. K., Pasternack D., Timms L., Krzyzanowski P., Awadalla P., Costa R., Segal E., Bratman S. V., Beer P., Behjati S., Martincorena I., Wang J. C. Y., Bowles K. M., Quirós J. R., Karakatsani A., La Vecchia C., Trichopoulou A., Salamanca-Fernández E., Huerta J. M., Barricarte A., Travis R. C., Tumino R., Masala G., Boeing H., Panico S., Kaaks R., Krämer A., Sieri S., Riboli E., Vineis P., Foll M., McKay J., Polidoro S., Sala N., Khaw K. T., Vermeulen R., Campbell P. J., Papaemmanuil E., Minden M. D., Tanay A., Balicer R. D., Wareham N. J., Gerstung M., Dick J. E., Brennan P., Vassiliou G. S., Shlush L. I., Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559, 400–404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman A. M., Bratman S. V., To J., Wynne J. F., Eclov N. C. W., Modlin L. A., Liu C. L., Neal J. W., Wakelee H. A., Merritt R. E., Shrager J. B., Loo B. W. Jr., Alizadeh A. A., Diehn M., An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 20, 548–554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kinde I., Wu J., Papadopoulos N., Kinzler K. W., Vogelstein B., Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. U.S.A. 108, 9530–9535 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmitt M. W., Hiatt J. B., Loeb L. A., Kennedy S. R., Fox E. J., Salk J. J., Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. 109, 14508–14513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim C. S., Mohan S., Ayub M., Rothwell D. G., Dive C., Brady G., Miller C., In silico error correction improves cfDNA mutation calling. Bioinformatics 35, 2380–2385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koboldt D. C., Zhang Q., Larson D. E., Shen D., McLellan M. D., Lin L., Miller C. A., Mardis E. R., Ding L., Wilson R. K., VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kockan C., Hach F., Sarrafi I., Bell R. H., McConeghy B., Beja K., Haegert A., Wyatt A. W., Volik S. V., Chi K. N., Collins C. C., Sahinalp S. C., SiNVICT: Ultra-sensitive detection of single nucleotide variants and indels in circulating tumour DNA. Bioinformatics 33, 26–34 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Cibulskis K., Lawrence M. S., Carter S. L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E. S., Getz G., Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saunders C. T., Wong W. S. W., Swamy S., Becq J., Murray L. J., Cheetham R. K., Strelka: Accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics 28, 1811–1817 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Gerstung M., Beisel C., Rechsteiner M., Wild P., Schraml P., Moch H., Beerenwinkel N., Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat. Commun. 3, 811 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Shiraishi Y., Sato Y., Chiba K., Okuno Y., Nagata Y., Yoshida K., Shiba N., Hayashi Y., Kume H., Homma Y., Sanada M., Ogawa S., Miyano S., An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 41, e89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman A. M., Lovejoy A. F., Klass D. M., Kurtz D. M., Chabon J. J., Scherer F., Stehr H., Liu C. L., Bratman S. V., Say C., Zhou L., Carter J. N., West R. B., Sledge G. W. Jr., Shrager J. B., Loo B. W. Jr., Neal J. W., Wakelee H. A., Diehn M., Alizadeh A. A., Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 34, 547–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerstung M., Papaemmanuil E., Campbell P. J., Subclonal variant calling with multiple samples and prior knowledge. Bioinformatics 30, 1198–1204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larson D. E., Harris C. C., Chen K., Koboldt D. C., Abbott T. E., Dooling D. J., Ley T. J., Mardis E. R., Wilson R. K., Ding L., SomaticSniper: Identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moriyama T., Imoto S., Hayashi S., Shiraishi Y., Miyano S., Yamaguchi R., A Bayesian model integration for mutation calling through data partitioning. Bioinformatics 35, 4247–4254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alexandrov L. B., Nik-Zainal S., Wedge D. C., Aparicio S. A. J. R., Behjati S., Biankin A. V., Bignell G. R., Bolli N., Borg A., Børresen-Dale A. L., Boyault S., Burkhardt B., Butler A. P., Caldas C., Davies H. R., Desmedt C., Eils R., Eyfjörd J. E., Foekens J. A., Greaves M., Hosoda F., Hutter B., Ilicic T., Imbeaud S., Imielinsk M., Jäger N., Jones D. T. W., Jonas D., Knappskog S., Koo M., Lakhani S. R., López-Otín C., Martin S., Munshi N. C., Nakamura H., Northcott P. A., Pajic M., Papaemmanuil E., Paradiso A., Pearson J. V., Puente X. S., Raine K., Ramakrishna M., Richardson A. L., Richter J., Rosenstiel P., Schlesner M., Schumacher T. N., Span P. N., Teague J. W., Totoki Y., Tutt A. N. J., Valdés-Mas R., van Buuren M. M., van ‘t Veer L., Vincent-Salomon A., Waddell N., Yates L. R.; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain, Zucman-Rossi J., Futreal P. A., McDermott U., Lichter P., Meyerson M., Grimmond S. M., Siebert R., Campo E., Shibata T., Pfister S. M., Campbell P. J., Stratton M. R., Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poulos R. C., Olivier J., Wong J. W. H., The interaction between cytosine methylation and processes of DNA replication and repair shape the mutational landscape of cancer genomes. Nucleic Acids Res. 45, 7786–7795 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costello M., Pugh T. J., Fennell T. J., Stewart C., Lichtenstein L., Meldrim J. C., Fostel J. L., Friedrich D. C., Perrin D., Dionne D., Kim S., Gabriel S. B., Lander E. S., Fisher S., Getz G., Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aird D., Ross M. G., Chen W.-S., Danielsson M., Fennell T., Russ C., Jaffe D. B., Nusbaum C., Gnirke A., Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 12, R18 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y., Speed T. P., Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 40, e72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Odegaard J. I., Vincent J. J., Mortimer S., Vowles J. V., Ulrich B. C., Banks K. C., Fairclough S. R., Zill O. A., Sikora M., Mokhtari R., Abdueva D., Nagy R. J., Lee C. E., Kiedrowski L. A., Paweletz C. P., Eltoukhy H., Lanman R. B., Chudova D. I., Talasaz A. A., Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin. Cancer Res. 24, 3539–3549 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Jongen-Lavrencic M., Grob T., Hanekamp D., Kavelaars F. G., al Hinai A., Zeilemaker A., Erpelinck-Verschueren C. A. J., Gradowska P. L., Meijer R., Cloos J., Biemond B. J., Graux C., van Marwijk Kooy M., Manz M. G., Pabst T., Passweg J. R., Havelange V., Ossenkoppele G. J., Sanders M. A., Schuurhuis G. J., Löwenberg B., Valk P. J. M., Molecular minimal residual disease in acute myeloid leukemia. N. Engl. J. Med. 378, 1189–1199 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Zhu H.-H., Zhang X.-H., Qin Y.-Z., Liu D.-H., Jiang H., Chen H., Jiang Q., Xu L.-P., Lu J., Han W., Bao L., Wang Y., Chen Y.-H., Wang J.-Z., Wang F.-R., Lai Y.-Y., Chai J.-Y., Wang L.-R., Liu Y.-R., Liu K.-Y., Jiang B., MRD-directed risk stratification treatment may improve outcomes of t(8;21) AML in the first complete remission: Results from the AML05 multicenter trial. Blood 121, 4056–4062 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Platzbecker U., Middeke J. M., Sockel K., Herbst R., Wolf D., Baldus C. D., Oelschlägel U., Mütherig A., Fransecky L., Noppeney R., Bug G., Götze K. S., Krämer A., Bochtler T., Stelljes M., Groth C., Schubert A., Mende M., Stölzel F., Borkmann C., Kubasch A. S., von Bonin M., Serve H., Hänel M., Dührsen U., Schetelig J., Röllig C., Kramer M., Ehninger G., Bornhäuser M., Thiede C., Measurable residual disease-guided treatment with azacitidine to prevent haematological relapse in patients with myelodysplastic syndrome and acute myeloid leukaemia (RELAZA2): An open-label, multicentre, phase 2 trial. Lancet Oncol. 19, 1668–1679 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Balsat M., Renneville A., Thomas X., de Botton S., Caillot D., Marceau A., Lemasle E., Marolleau J.-P., Nibourel O., Berthon C., Raffoux E., Pigneux A., Rodriguez C., Vey N., Cayuela J.-M., Hayette S., Braun T., Coudé M. M., Terre C., Celli-Lebras K., Dombret H., Preudhomme C., Boissel N., Postinduction minimal residual disease predicts outcome and benefit from allogeneic stem cell transplantation in acute myeloid leukemia with NPM1 mutation: A study by the acute leukemia french association group. J. Clin. Oncol. 35, 185–193 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Wong T. N., Ramsingh G., Young A. L., Miller C. A., Touma W., Welch J. S., Lamprecht T. L., Shen D., Hundal J., Fulton R. S., Heath S., Baty J. D., Klco J. M., Ding L., Mardis E. R., Westervelt P., Dipersio J. F., Walter M. J., Graubert T. A., Ley T. J., Druley T. E., Link D. C., Wilson R. K., Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 518, 552–555 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong T. N., Miller C. A., Jotte M. R. M., Bagegni N., Baty J. D., Schmidt A. P., Cashen A. F., Duncavage E. J., Helton N. M., Fiala M., Fulton R. S., Heath S. E., Janke M., Luber K., Westervelt P., Vij R., DiPersio J. F., Welch J. S., Graubert T. A., Walter M. J., Ley T. J., Link D. C., Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat. Commun. 9, 455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guryanova O. A., Shank K., Spitzer B., Luciani L., Koche R. P., Garrett-Bakelman F. E., Ganzel C., Durham B. H., Mohanty A., Hoermann G., Rivera S. A., Chramiec A. G., Pronier E., Bastian L., Keller M. D., Tovbin D., Loizou E., Weinstein A. R., Gonzalez A. R., Lieu Y. K., Rowe J. M., Pastore F., McKenney A. S., Krivtsov A. V., Sperr W. R., Cross J. R., Mason C. E., Tallman M. S., Arcila M. E., Abdel-Wahab O., Armstrong S. A., Kubicek S., Staber P. B., Gönen M., Paietta E. M., Melnick A. M., Nimer S. D., Mukherjee S., Levine R. L., DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat. Med. 22, 1488–1495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuurhuis G. J., Heuser M., Freeman S., Béné M.-C., Buccisano F., Cloos J., Grimwade D., Haferlach T., Hills R. K., Hourigan C. S., Jorgensen J. L., Kern W., Lacombe F., Maurillo L., Preudhomme C., van der Reijden B. A., Thiede C., Venditti A., Vyas P., Wood B. L., Walter R. B., Döhner K., Roboz G. J., Ossenkoppele G. J., Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 131, 1275–1291 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buccisano F., Maurillo L., Del Principe M. I., Di Veroli A., De Bellis E., Biagi A., Zizzari A., Rossi V., Rapisarda V., Amadori S., Voso M. T., Lo-Coco F., Arcese W., Venditti A., Minimal residual disease as a biomarker for outcome prediction and therapy optimization in acute myeloid leukemia. Expert Rev. Hematol. 11, 307–313 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Shlush L. I., Age-related clonal hematopoiesis. Blood 131, 496–504 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Steensma D. P., Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2, 3404–3410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zink F., Stacey S. N., Norddahl G. L., Frigge M. L., Magnusson O. T., Jonsdottir I., Thorgeirsson T. E., Sigurdsson A., Gudjonsson S. A., Gudmundsson J., Jonasson J. G., Tryggvadottir L., Jonsson T., Helgason A., Gylfason A., Sulem P., Rafnar T., Thorsteinsdottir U., Gudbjartsson D. F., Masson G., Kong A., Stefansson K., Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coombs C. C., Zehir A., Devlin S. M., Kishtagari A., Syed A., Jonsson P., Hyman D. M., Solit D. B., Robson M. E., Baselga J., Arcila M. E., Ladanyi M., Tallman M. S., Levine R. L., Berger M. F., Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 21, 374–382.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hao T., Li-Talley M., Buck A., Chen W., An emerging trend of rapid increase of leukemia but not all cancers in the aging population in the United States. Sci. Rep. 9, 12070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riboli E., Hunt K. J., Slimani N., Ferrari P., Norat T., Fahey M., Charrondière U. R., Hémon B., Casagrande C., Vignat J., Overvad K., Tjønneland A., Clavel-Chapelon F., Thiébaut A., Wahrendorf J., Boeing H., Trichopoulos D., Trichopoulou A., Vineis P., Palli D., Bueno-De-Mesquita H. B., Peeters P. H. M., Lund E., Engeset D., González C. A., Barricarte A., Berglund G., Hallmans G., Day N. E., Key T. J., Kaaks R., Saracci R., European prospective investigation into cancer and nutrition (EPIC): Study populations and data collection. Public Health Nutr. 5, 1113–1124 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Li H., Durbin R., Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kennedy S. R., Schmitt M. W., Fox E. J., Kohrn B. F., Salk J. J., Ahn E. H., Prindle M. J., Kuong K. J., Shen J.-C., Risques R.-A., Loeb L. A., Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 9, 2586–2606 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang T. T., Abelson S., Zou J., Li T., Zhao Z., Dick J. E., Shlush L. I., Pugh T. J., Bratman S. V., High efficiency error suppression for accurate detection of low-frequency variants. Nucleic Acids Res. 47, e87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.The 1000 Genomes Project Consortium , A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glusman G., Caballero J., Mauldin D. E., Hood L., Roach J. C., Kaviar: An accessible system for testing SNV novelty. Bioinformatics 27, 3216–3217 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Forbes S. A., Beare D., Boutselakis H., Bamford S., Bindal N., Tate J., Cole C. G., Ward S., Dawson E., Ponting L., Stefancsik R., Harsha B., YinKok C., Jia M., Jubb H., Sondka Z., Thompson S., De T., Campbell P. J., COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.SEER Cancer Stat Facts—National Cancer Institute (2017); https://seer.cancer.gov/statfacts/.
- 54.Venkatesan A., Kerlikowske K., Chu P., Smith-Bindman R., Sickles E. A., Positive predictive value of specific mammographic findings according to reader and patient variables. Radiology 250, 648–657 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Navarro M., Nicolas A., Ferrandez A., Lanas A., Colorectal cancer population screening programs worldwide in 2016: An update. World J. Gastroenterol. 23, 3632–3642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabe3722/DC1