

Abstract
Radical reactions hold a number of inherent advantages in organic synthesis that may potentially impact the planning and practice for construction of organic molecules. However, the control of enantioselectivity in radical processes remains one of the longstanding challenges. While significant advances have recently been achieved in intramolecular radical reactions, the governing of asymmetric induction in intermolecular radical reactions still poses challenging issues. We herein report a catalytic approach that is highly effective for controlling enantioselectivity as well as reactivity of the intermolecular radical C–H amination of carboxylic acid esters with organic azides via Co(II)-based metalloradical catalysis (MRC). The key to the success lies in the catalyst development to maximize noncovalent attractive interactions through fine-tuning of the remote substituents of the D2-symmetric chiral amidoporphyrin ligand. This noncovalent interaction strategy presents a solution that may be generally applicable in controlling reactivity and enantioselectivity in intermolecular radical reactions. The Co(II)-catalyzed intermolecular C–H amination, which operates under mild conditions with the C–H substrate as the limiting reagent, exhibits a broad substrate scope with high chemoselectivity, providing effective access to valuable chiral amino acid derivatives with high enantioselectivities. Systematic mechanistic studies shed light into the working details of the underlying stepwise radical pathway for the Co(II)-based C–H amination.
Graphical Abstract
INTRODUCTION
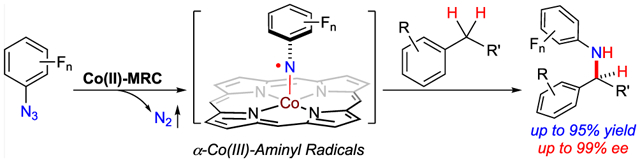
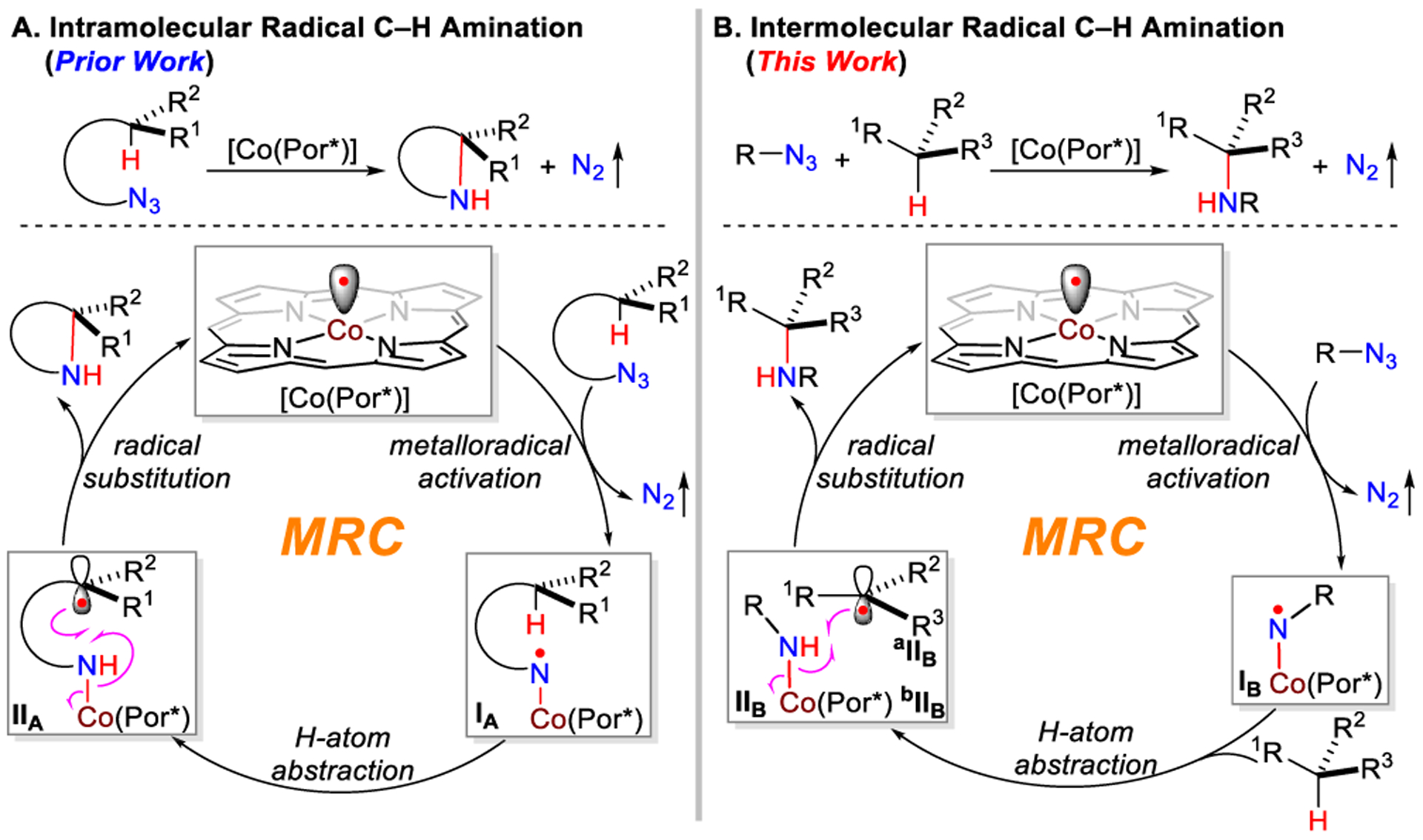
Nitrogen-centered radicals have been frequently exploited as reactive intermediates for the development of new methods in organic synthesis.1 Among different applications, free aminyl radicals have been demonstrated with the potential for the direct functionalization of C(sp3)–H bonds to form valuable nitrogen-containing compounds.2 Despite significant progress on this potentially powerful synthetic methodology, the control of enantioselectivity in regard to radical C–H amination remains a formidable task and largely unaddressed.3 Among considerable efforts in surmounting this challenge,4 metalloradical catalysis (MRC) represents a conceptually new approach that utilizes metalloradical complexes as a new kind of catalyst for the generation of metal-supported organic radicals as key intermediates to regulate subsequent radical reactions.5–7 To this end, Co(II) complexes of porphyrins, a family of stable 15e-metalloradicals, have been shown to homolytically activate organic azides to generate α-Co(III)-aminyl radicals, a new type of aminyl radical supported by metal complexes.8 With the employment of D2-symmetric chiral amidoporphyrins as the supporting ligands, we recently achieved enantioselective intramolecular radical C–H amination for stereoselective construction of chiral N-heterocycles.9 These catalytic reactions proceed through a stepwise radical pathway that involves metalloradical activation of organic azides to generate α-Co(III)-aminyl radical intermediates IA, followed by intramolecular H atom abstraction (HAA) to form ω-Co(III)-alkyl radical intermediates IIA, and subsequent intramolecular radical substitution (RS) resulting in the formation and release of the aminated product (Scheme 1A). In principle, the analogous stepwise radical mechanism should also operate for the intermolecular version of the radical C–H amination, involving the corresponding intermediates IB and IIB (Scheme 1B). However, there are additional challenges inherently associated with the control of reactivity and enantioselectivity in the intermolecular radical process. In the absence of the linkage between the N-centered radical and C–H bond (as in the case of intermediate IA), intermolecular H atom abstraction from the C–H bond of the substrate by α-Co(III)-aminyl radical intermediate IB could be complicated by issues with regioselectivity and chemoselectivity. Furthermore, without the covalent linkage in the resulting ∞-Co(III)-alkyl radical intermediate IIB, the C-centered radical aIIB would be virtually “free” to escape from the Co(III)-amido complex bIIB, which would terminate the desired catalytic cycle. Consequently, it could lead to side reactions and radical chain processes, resulting in further loss of reactivity and selectivity. Moreover, considering that the concentrations of both aIIB and bIIB are equally low (not higher than the catalyst concentration), the last step of intermolecular radical substitution would be intrinsically difficult as a bimolecular second-order reaction. Besides the issue of reactivity, the control of enantioselectivity in intermolecular radical substitution is a topic virtually unexplored. With “free” interconversion between two prochiral faces of typical C-centered radicals, we wondered what factors could be exploited to govern the asymmetric induction of C–N bond formation via radical substitution between aIIB and bIIB. Encouraged by our recent success in the development of enantioselective radical processes for intramolecular C–H amination,9 we envisioned that the key to addressing these issues would be the catalyst development to maximize noncovalent attractive interactions through fine-tuning of the environments of D2-symmetric chiral amidoporphyrin ligand. If achieved, intermolecular radical C–H amination via Co(II)-based MRC could potentially provide a generally applicable strategy for stereoselective synthesis of chiral amines directly from omnipresent C–H bonds with a wide range of organic azides.
Scheme 1.
Intra- and Intermolecular Pathways for Radical C–H Amination via Co(II)-Based Metalloradical Catalysis
Development of catalytic systems for direct functionalization of ubiquitous C(sp3)–H bonds with nitrogen sources represents a highly attractive approach for general synthesis of valuable amines.10 Despite intensive research efforts, development of enantioselective catalytic systems for the synthesis of chiral amines by intermolecular C–H amination is still in its infancy. Notable examples include asymmetric amination systems by chiral catalysts based on metal complexes of rhodium,11 ruthenium,12 and manganese,13,12b which typically proceed via concerted C–H insertion involving electrophilic metallonitrene intermediates. Enantioselective intermolecular C–H amination has also been demonstrated with engineered iron–heme enzymes14 and rhodium-based catalyst under photoredox conditions.15 While they represent significant advances, these catalytic systems experienced limitations such as the need for excess C–H substrates, limited substrate scope, or low level of enantiocontrol. Evidently, there persists an unmet need of general and efficient catalytic systems for highly enantioselective intermolecular C–H amination. In comparison to ionic reactions, radical reactions are inherently more reactive and less sensitive to the electronic requirements of substrates, characteristics that may lead to the development of more effective catalytic systems with a broad substrate scope, including the amination of challenging electron-deficient C–H bonds. We herein report the first catalytic radical system via Co(II)-MRC for enantioselective intermolecular C–H amination. Specifically, we describe the development of Co(II)-based metalloradical system that can activate fluoroaryl azides16 for enantioselective amination of α-C–H bonds in carboxylic acid esters, a class of electron-deficient C–H bonds. This new Co(II)-catalyzed C–H amination provides a straightforward method for stereoselective synthesis of chiral α-amino acid derivatives directly from widely available carboxylic esters. We hope to show the importance of catalyst development in achieving effective control of both reactivity and enantioselectivity in this intermolecular radical process.
RESULTS AND DISCUSSION
Catalyst Development.
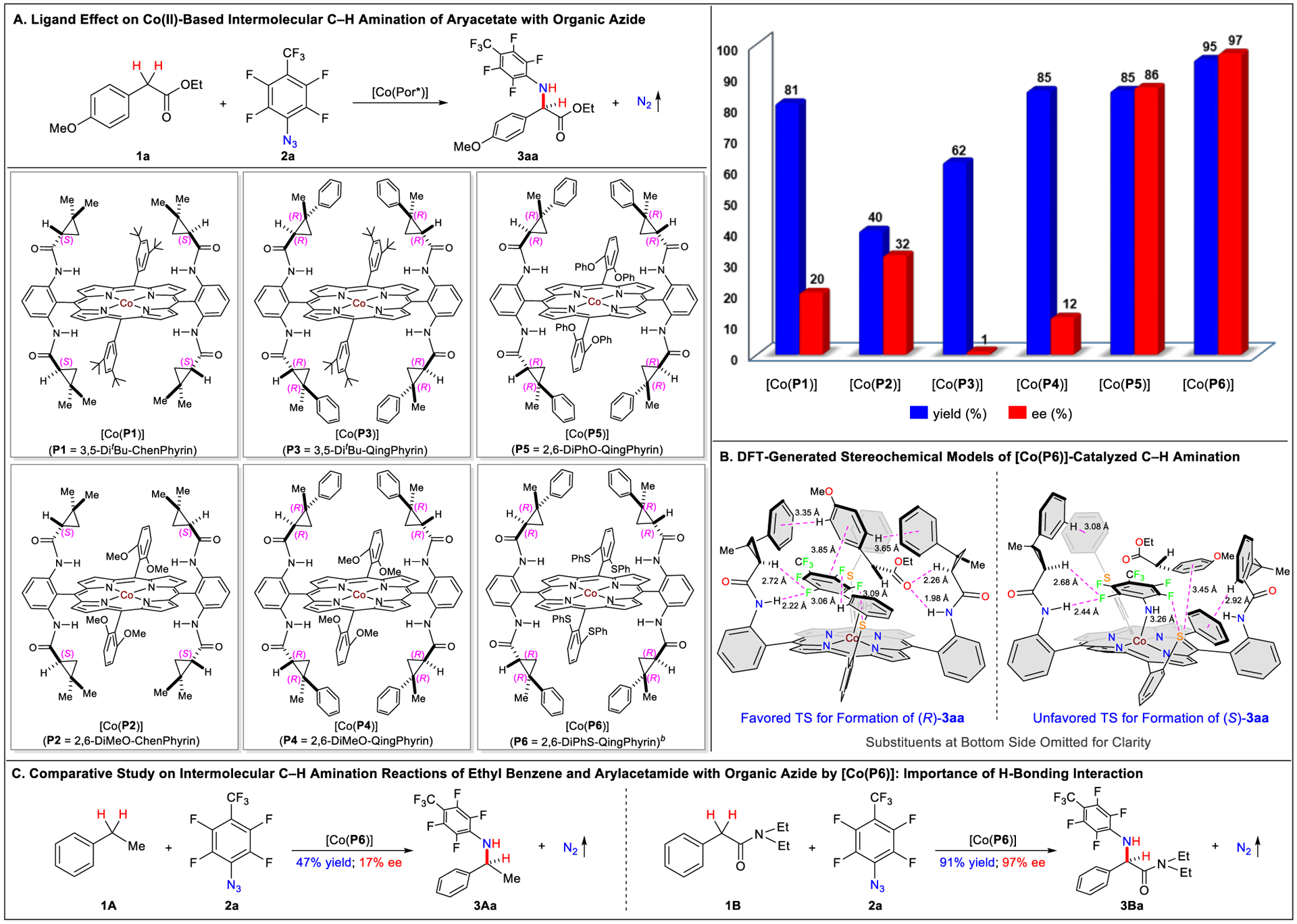
To assess the feasibility of the proposed intermolecular radical process, we first examined the α-C(sp3)–H amination reaction of ethyl (4-methoxyphenyl)-acetate (1a) with 4-trifluoromethyl-2,3,5,6-tetrafluorophenyl azide (2a) by Co(II) complexes of porphyrins (Scheme 2A and Table S1). With the use of the first-generation metalloradical catalyst [Co(P1)] (P1 = 3,5-DitBu-ChenPhyrin),17 it was gratifying to find that the C–H amination reaction could afford the desired α-aryl α-amino acid ester 3aa in high yield (81%) with low but significant enantioselectivity (20% ee). Replacing the catalyst by the analogous [Co(P2)] (P2 = 2,6-DiMeO-ChenPhyrin), which has methoxy groups at the 2,6-positions instead of tert-butyl groups at the 3,5-positions, resulted in some improvement in enantioselectivity (32% ee) but led to considerable decrease in reactivity (40% yield). Encouraged by these initial results, we then systematically investigated the ligand effect on the reactivity and selectivity of the Co(II)-catalyzed reaction. When second-generation metalloradical catalyst [Co(P3)] (P3 = 3,5-DitBu-QingPhyrin),18 which bears cyclopropanecarboxyamides with two contiguous stereocenters, was employed, the reaction occurred in a better reactivity (62% yield) but with almost no enantioselectivity. However, switching the catalyst [Co(P3)] to its analogous [Co(P4)] (P4 = 2,6-DiMeO-QingPhyrin) gave rise to even higher yield (85%) and resumed asymmetric induction at a low but significant level (12% ee), indicating once again the significant influence of the nonchiral substituents in the ligand on the catalytic reaction. To our delight, the use of [Co(P5)] (P5 = 2,6-DiPhO-QingPhyrin), in which the methoxy groups were replaced with phenoxy groups at the 2,6-positions, led to a significant increase in enantioselectivity (86% ee) without affecting the high reactivity (85% yield). Further catalyst development by substituting the O atoms at the 2,6-positions of the nonchiral substituents in [Co(P5)] with S atoms led to the development of [Co(P6)] (P6 = 2,6-DiPhS-QingPhyrin; see the Supporting Information for X-ray structure), which proved to be an even more effective catalyst, producing α-amino acid ester 3aa in excellent yield (95%) with exceptional enantioselectivity (97% ee). The difference in performance between [Co(P6)] and [Co(P5)] demonstrates that even a ligand modification as subtle as heteroatom substitution of O atoms by S atoms can give rise to remarkable improvements in both reactivity and enantioselectivity, manifesting the effectiveness of catalyst development in controlling the radical process. As depicted in the proposed stereochemical model on the basis of DFT calculations (Scheme 2B), the effectiveness of [Co(P6)] in controlling reactivity and enantioselectivity may be attributed to the cooperative interplay of several noncovalent interactions among the two substrates and the catalyst, including multiple H-bonding and π-stacking interactions as well as van der Waals forces (see Supporting Information for details). Together, these attractive weak forces hold the two reacting substrates within the catalyst’s pocket in proximity and orient them in proper conformations to facilitate the stereoselective C–N bond formation. According to the DFT-optimized model, [Co(P6)] catalyzes the preferred formation of product 3aa as (R)-enantiomer over (S)-enantiomer, an outcome that is consistent with the experimental observation (see Table 1). As comparison, intermolecular C–H amination reactions were conducted under the same optimized conditions for substrates ethyl-benzene (1A) and N,N-diethyl-2-phenylacetamide (1B), which have no and similar H-bonding ability to 1a, respectively (Scheme 2C). While the reaction of 1B afforded the corresponding α-aryl α-amino acid amide 3Ba in 91% yield with 97% ee, amination of 1A produced the desired α-aminoethylbenzene 3Aa in 47% yield with 17% ee. Together, these results clearly revealed the importance of H-bonding interaction as suggested by the DFT model.
Scheme 2.
Ligand Effect on Co(II)-Based Asymmetric System for Intermolecular C–H Amination with Organic Azidesa
aCarried out with 1 (0.10 mmol) and 2a (0.15 mmol) in the presence of 4 Å molecular sieves by [Co(Por*)] (4 mol %) in trifluorotoluene (0.5 mL) at 40 °C for 48 h. Isolated yields. Enantiomeric excess was determined by chiral HPLC. bStructure of P6 was determined by X-ray crystallography.
Table 1.
Enantioselective Intermolecular Radical Amination of C(sp3)–H Bonds with Fluoroaryl Azides via Co(II)-MRCa
|
Carried out with 1 (0.10 mmol) and 2 (0.15 mmol) in the presence of 4 Å molecular sieves by [Co(P6)] (4 mol %) in trifluorotoluene (0.5 mL) at 40 °C for 48 h. Isolated yields. Enantiomeric excess was determined by chiral HPLC.
Performed on 3.0 mmol scale.
Absolute configuration determined by X-ray crystallography.
In benzene (0.5 mL).
Allylic regioisomers were observed as minor products.
With 1 (0.10 mmol) and 2a (0.12 mmol).
Substrate Scope.
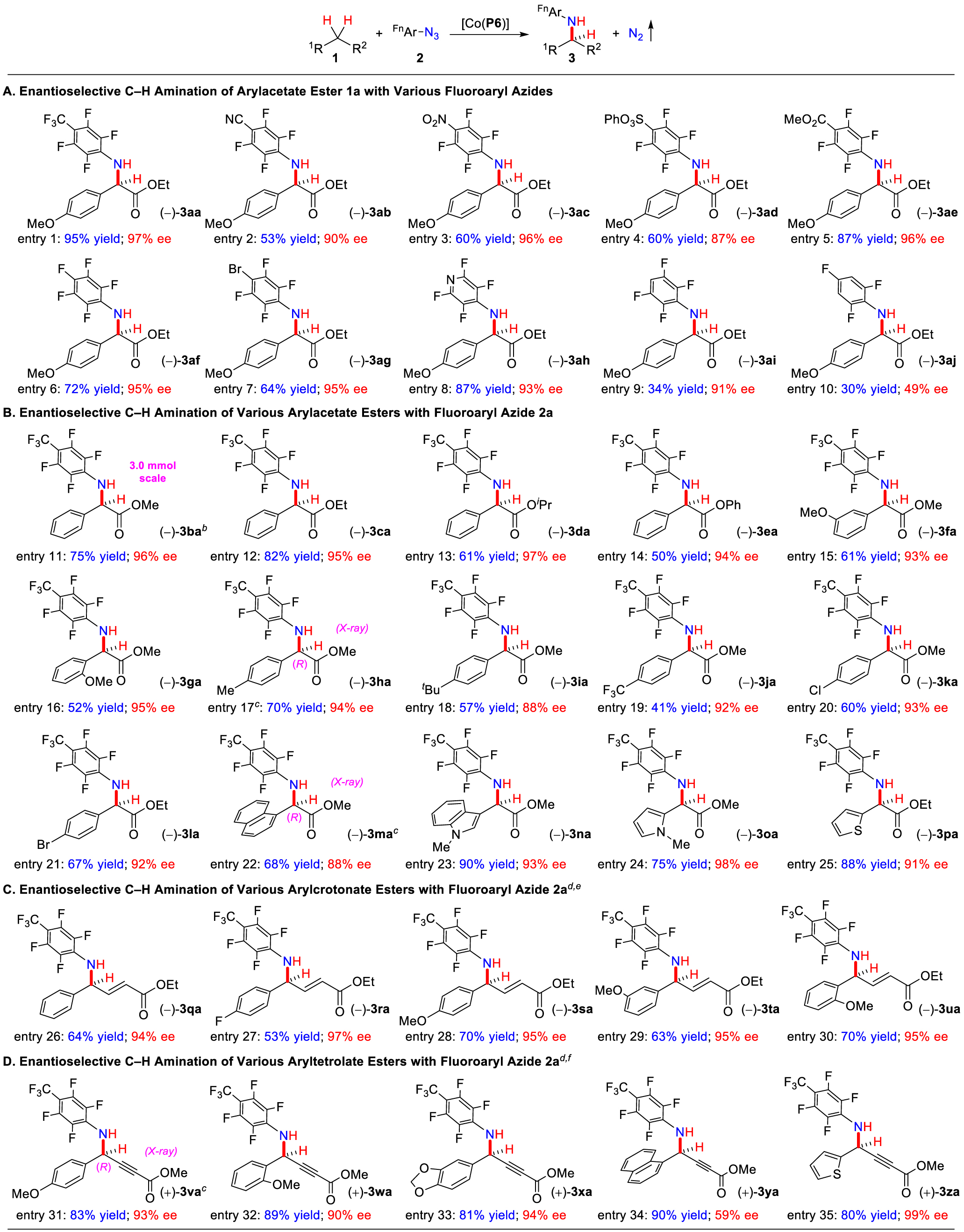
Using the optimized catalyst [Co(P6)], we then investigated the scope of fluoroaryl azides 2 for Co(II)-catalyzed C–H amination using arylacetate ester 1a as the standard substrate (Table 1A). Similar to 4-trifluoromethyl-2,3,5,6-tetrafluorophenyl azide (2a), analogues bearing other para-substituents such as −CN (2b), −NO2 (2c), −SO3Ph (2d), and −CO2Me (2e) could also be used as effective nitrogen sources for the C–H amination, generating the corresponding α-aryl α-amino acid esters 3ab–3ae in good to excellent yields with high enantioselectivities (Table 1; entries 1–5). In addition, both pentafluorophenyl azide (2f) and 4-bromotetrafluorophenyl azide (2g) were suitable aminating reagents for the catalytic process, leading to productive formation of N-fluoroaryl α-amino acid esters 3af and 3ag with excellent enantioselectivities (Table 1; entries 6 and 7). Interestingly, 4-tetrafluoropyridinyl azide (2h) could serve as a competent nitrogen precursor for highly enantioselective C–H amination of 1a to form the amino acid derivative 3ah without complications from the pyridine unit (Table 1; entry 8). Additionally, the Co(II)-based system could use 2,3,5,6-tetrafluorophenyl azide (2i) for the amination reaction, producing the desired product 3ai with excellent enantioselectivity, albeit in low yield (Table 1; entry 9). When 2,4,6-trifluorophenyl azide (2j) was used, however, the corresponding amination product 3aj was obtained in low yield with only moderate enantioselectivity (Table 1; entry 10). It is worth noting that the use of 2,4,5-trifluorophenyl azide afforded only a trace amount of the corresponding C–H amination product while no product was observed with the use of 3,4,5-trifluorophenyl azide under the same conditions, suggesting the importance of 2,6-difluoro substituents on the aryl azides for effective intermolecular C–H amination. In addition to rendering the Co(III)-aminyl radical electrophilic, both of the ortho-fluoro atoms may play important roles in facilitating the cooperative interplay of the multiple noncovalent interactions (Scheme 2B).
Subsequently, a wide range of arylacetate esters 1 were examined as C–H substrates for Co(II)-catalyzed amination by [Co(P6)] using azide 2a as a representative nitrogen source (Table 1B). Similar to ethyl ester (1a), various esters of phenylacetate, such as methyl (1b), ethyl (1c), isopropyl (1d), and phenyl (1e) esters, could be productively aminated to form the corresponding α-amino acid esters 3ba–3ea with excellent enantioselectivities (Table 1; entries 11–14). It is worth mentioning that the amination process for the synthesis of α-amino acid derivatives could be scaled up, as demonstrated by the synthesis of optically active compound 3ba on a 3.0 mmol scale in 75% yield with 96% ee. In addition to para-OMe-substituted 1a, arylacetate derivatives bearing substituents with varied electronic properties at different positions on the aromatic ring, such as meta-OMe (1f), ortho-OMe (1g), para-Me (1h), para-tBu (1i), para-CF3 (1j), para-Cl (1k), and para-Br (1l) groups, could all act as adequate C–H substrates for the Co(II)-based enantioselective amination, allowing for the convenient access to highly enantioenriched α-amino acid derivatives 3fa–3la bearing various functionalized α-aryl units (Table 1; entries 15–21). The C–H amination could also be applied to arylacetates with extended aromatic and heteroaromatic systems as shown in high-yielding formation of α-amino acid derivatives with α-naphthyl (3ma), α-indolyl (3na), α-pyrrolyl (3oa), and α-thiophenyl (3pa) groups with excellent enantioselectivities (Table 1; entries 22–25). The absolute configurations of the newly generated stereogenic centers in 3ha and 3ma were both established as (R) by X-ray crystallography.
Furthermore, the [Co(P6)]-based catalytic system could be expanded to the enantioselective C–H amination of both arylcrotonate esters (Table 1C) and aryltetrolate esters (Table 1D). For example, the allylic C–H bonds of ethyl phenylcrotonate (1q) could be effectively aminated by [Co(P6)] with azide 2a, producing the γ-aryl γ-amino acid ester 3qa in good yield with high enantioselectivity (Table 1; entry 26). The Co(II)-based system proved to be similarly effective for highly enantioselective amination of allylic C–H bonds in arylcrotonate esters (1r–1u) bearing aryl substituents (Table 1; entries 27–30). In all the cases, the corresponding allylic regioisomer γ-aryl α-amino acid esters were also generated but as the minor products. Likewise, [Co(P6)] was capable of catalyzing enantioselective amination of the propargylic C–H bonds in aryltetrolate esters as exemplified by efficient reactions of tetrolate derivatives 1v–1z containing disparate aryl groups with azide 2a, delivering the functionalized γ-aryl γ-amino acid derivatives 3va–3za in high yields with good enantioselectivities (Table 1; entries 31–35). The absolute configuration of the major enantiomer of 3va was established as (R) by X-ray crystallography, which is the same as 3ha and 3ma. Notably, the C–H amination process catalyzed by [Co(P6)] exhibited chemoselectivity as the normally more reactive C=C and C≡C bonds were unaffected. It is also worth noting that the [Co(P6)]-catalyzed amination displayed high regioselectivity at the γ-position over the α-position, two possible reactive sites that are associated with both the allylic and propargylic radical intermediates.
Mechanistic Studies.
Comprehensive studies were carried out to gain insight into the underlying stepwise radical mechanism of the Co(II)-catalyzed intermolecular C–H amination (Scheme 3). To directly detect the α-Co(III)-aminyl radical intermediate I, the isotropic X-band electron paramagnetic resonance (EPR) spectrum was recorded at room temperature for the reaction mixture of [Co(P1)] with azide 2h in benzene without C–H substrate (Scheme 3A). The spectrum displays notable signals that are characteristic of α-Co(III)-aminyl radicals.9,8a The observed isotropic g-value of ~2.00 is consistent with the generation of organic radical I[Co(P1)]/2h upon spin translocation from the Co(II) center to the N atom during the process of metalloradical activation. Consistent with the spin delocalization in α-arylaminyl radical intermediate I[Co(P1)]/2h, the observed signals were broad and could be fittingly simulated by involving its three resonance forms on the basis of couplings by 59Co (I = 7/2), 14N (I = 1), and 19F (I = 1/2): 82% of N-centered radical at α-position NαI[Co(P1)]/2h (g = 2.04824; A(Co) = 117.9 MHz; A(N) = 139.5 MHz; A(F) = 0 MHz), 8% of C-centered radical at γ-position CγI[Co(P1)]/2h (g = 2.01921; A(Co) = 0 MHz; A(N) = 69.2 MHz; A(F) = 130.0 MHz), and 10% of N-centered radical at ε-position NεI[Co(P1)]/2h (g = 2.08007; A(Co) = 0 MHz; A(N) = 95.9 MHz; A(F) = 0 MHz). Furthermore, intermediate I[Co(P1)]/2h could be detected by high-resolution mass spectrometry (HRMS) with ESI ionization. The obtained spectrum evidently exhibited a signal corresponding to [I + [Co(P1)]/2h]+ (m/z = 1503.6881), resulting from the neutral α-Co(III)-aminyl radical I[Co(P1)]/2h by the loss of one electron. Both the experimentally determined exact mass and isotope distribution pattern matched well with those calculated from the formula of [(P1)Co(NC5NF4)+)] (m/z = 1503.6879; see Supporting Information for details). Correspondingly, α-Co(III)-aminyl radical intermediate I[Co(P6)]/2a, generated from the reaction mixture of [Co(P6)] with azide 2a, could also be detected by EPR with much stronger signals, which seems consistent with the higher activity of [Co(P6)] compared to [Co(P1)] (Scheme 3A). Similarly, the broad EPR signals of I[Co(P6)]/2a could be fitted nicely with three resonance structures: 88% of N-centered radical at α-position NαI[Co(P6)]/2a (g = 2.01362; A(Co) = 116.7 MHz; A(N) = 124.9 MHz; A(F) = 0 MHz), 4% of C-centered radical at γ-position CγI[Co(P6)]/2a (g = 2.02094; A(Co) = 0 MHz; A(N) = 110.3 MHz; A(F) = 81.0 MHz), and 8% of C-centered radical at ε-position CεI[Co(P6)]/2a (g = 2.07664; A(Co) = 0 MHz; A(N) = 0 MHz; A(F) = 0 MHz).
Scheme 3.
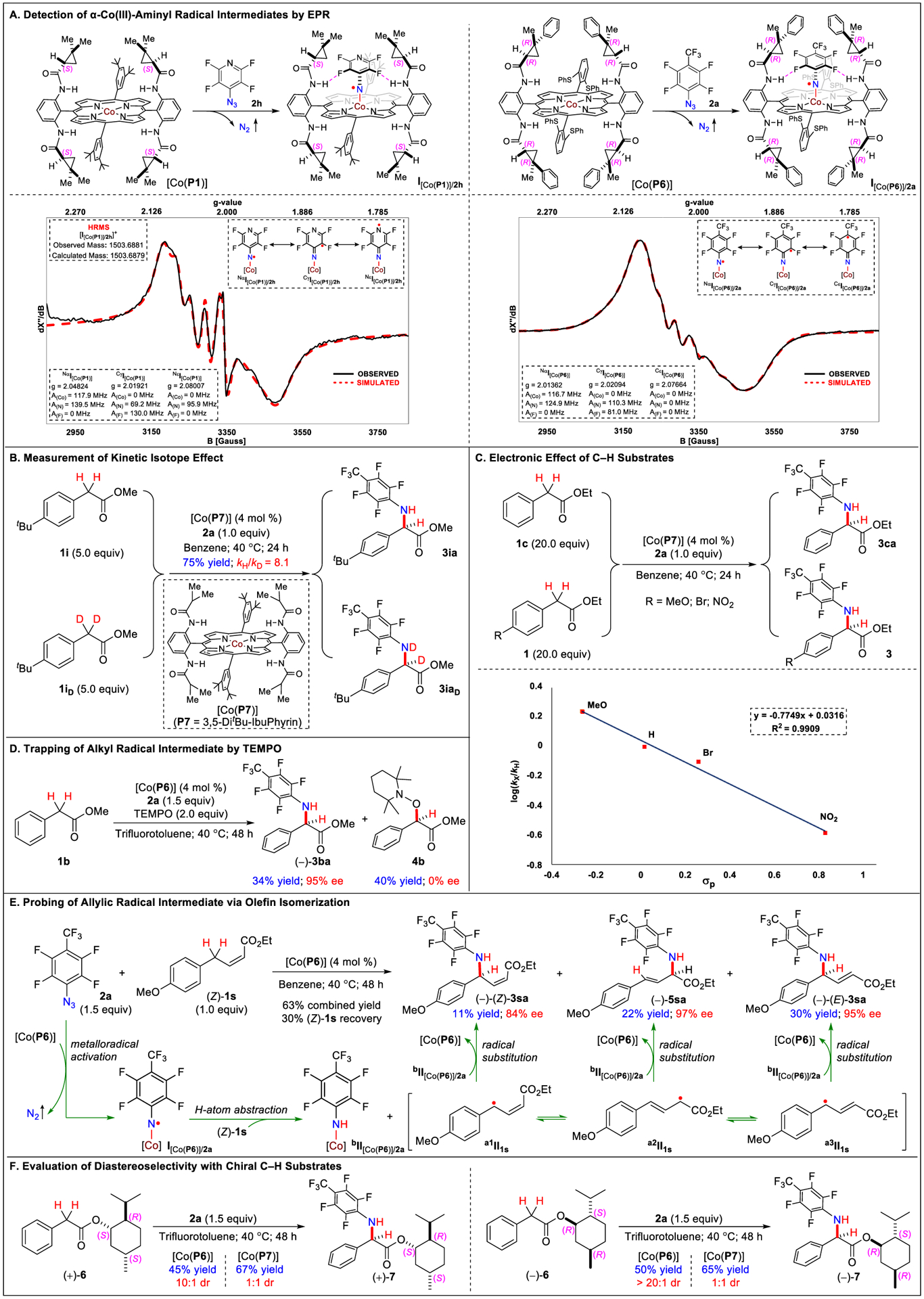
Mechanistic Studies on Co(II)-Catalyzed Intermolecular C–H Amination with Organic Azides
To determine the kinetic isotope effect (KIE), a direct competition experiment between the reactions of arylacetate ester 1i and its bisdeuterated analogue 1iD with azide 2a was conducted using achiral catalyst [Co(P7)] (P7 = 3,5-DitBu-IbuPhyrin) (Scheme 3B).19 A mixture of amination products 3ia and 3iaD was formed in a 75% combined yield. Analysis of the product mixture by 1H NMR provided an intermolecular KIE value (kH/kD) of 8.1. This high degree of primary KIE is consistent with the proposed step of C–H bond cleavage via intermolecular H atom abstraction by α-Co(III)-aminyl radical intermediate I[Co(P7)]/2a.9 To assess the potential electronic effect of the Co(II)-based radical process, competition reactions were performed for intermolecular C–H amination between ethyl phenylacetate ester (1c) and its para-substituted arylacetate analogs having wide-ranging electronic properties with azide 2a by [Co(P7)] (Scheme 3C). The results revealed a strong linear correlation between the log(kX/kH) and the Hammett constants (σp) of the para-substituents with a negative slope of −0.77. The Hammett plot signifies the electrophilic nature of the key radical intermediate I[Co(P7)/2a, which is likely a result of the strong electron-withdrawing effect of the fluoroaryl group. To directly trap ∞-Co(III)-alkyl radical intermediate aII generated from intermolecular H atom abstraction, the amination reaction of arylacetate ester 1b with azide 2a was conducted in the presence of TEMPO (2.0 equiv) using chiral catalyst [Co(P6)] (Scheme 3D). Remarkably, amination product 3ba was still formed with the similarly high enantioselectivity (95% ee) in spite of excess TEMPO, although in a much lower yield (34%) than the reaction without TEMPO. Concurrently, the reaction also produced compound 4b in 40% yield without any asymmetric induction, which was evidently formed from trapping of the “free” alkyl radical intermediate aII1b by TEMPO outside the chiral environment of the Co(III)-amido complex bII[Co(P6)]. Further TEMPO trapping experiments in different solvents indicated that solvent viscosity had some effect but was not a major factor to affect the outcome (Table S2).
To further probe the existence of the ∞-Co(III)-alkyl radical intermediate, the (Z)-isomer of ethyl 4-methoxyphenylcrotonate (Z)-1s was employed as the C–H substrate for the catalytic amination with azide 2a using [Co(P6)] as the catalyst (Scheme 3E). Besides the C–H amination product (Z)-3sa and unreacted (Z)-1s with unchanged configuration, two other products, 5sa and (E)-3sa, were also generated, indicating the existence of three allylic radical isomers (a1II1s, a2II1s, and a3II1s). Interestingly, all three amination products (Z)-3sa, 5sa, and (E)-3sa were formed with high enantioselectivities, suggesting that the allylic radical intermediates were not “free” when reacting inside the chiral environment of the Co(III)-amido complex bII[Co(P6)]/2a. To evaluate diastereoselectivity of the Co(II)-catalyzed intermolecular C–H amination, (+)-menthyl phenylacetate (+)-6 and (–)-menthyl phenylacetate (−)-6 were utilized as chiral C–H substrates for amination with azide 2a using both [Co(P6)] and [Co(P7)] (Scheme 3F). While achiral catalyst [Co(P7)] gave almost no control of diastereoselectivity in both reactions with (+)-6 and (−)-6, chiral catalyst [Co(P6)] enabled the stereoselective formation of amination products (+)-7 and (−)-7, respectively, with excellent diastereoselectivities. These results indicate that the Co(II)-based catalytic system can effectively control the stereochemistry of intermolecular C–H amination over the substrate. The differences in the ratio of the two diastereomers most likely reflect a matched–mismatched effect of chirality between the ligand and the substrate.
CONCLUSIONS
In summary, we have demonstrated, for the first time, a highly enantioselective system for intermolecular radical C–H amination via Co(II)-based metalloradical catalysis (MRC). The Co(II)-catalyzed amination, which operates under mild conditions with C–H substrate as the limiting reagent, exhibits a broad substrate scope and high chemoselectivity, providing effective access to valuable chiral amino acid derivatives with high enantioselectivities. The key to the success of controlling both reactivity and enantioselectivity of this intermolecular radical process is the development of a Co(II)-based metalloradical catalyst through fine-tuning of the remote substituents of the D2-symmetric chiral amidoporphyrin ligand to maximize cooperative noncovalent attractive interactions.
This new enantioselective intermolecular C–H amination process, which is fundamentally different from traditional metallonitrene insertion process, has been shown to proceed through a stepwise radical pathway, involving sequential steps of (i) metalloradical activation (MRA) of organic azides, (ii) intermolecular hydrogen atom abstraction (HAA) from C–H substrates, and (iii) intermolecular radical substitution (RS) for C–N bond formation, with effective control of both reactivity and enantioselectivity. We anticipate that this radical approach for intermolecular C–H amination, revealed by Co(II)-based MRC, will become generally applicable. It is our hope that this work will spur the development of new catalytic radical systems for direct functionalization of omnipresent C(sp3)–H bonds to form valuable nitrogen-containing compounds with potential control of chemo-, regio-, and stereoselectivity.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support by NIH (Grant R01-GM132471) and in part by NSF (Grant CHE-1900375). We thank James Zhang (Johns Hopkins University) for helpful discussions with valuable suggestions.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10415.
Experimental details and analytical data for all new compounds (PDF)
Crystallographic information (ZIP)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c10415
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Xiong T; Zhang Q New Amination Strategies Based on Nitrogen-Centered Radical Chemistry. Chem. Soc. Rev 2016, 45, 3069–3087. [DOI] [PubMed] [Google Scholar]; (b) Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]
- (2).(a) Stateman LM; Nakafuku KM; Nagib DA Remote C–H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 2018, 50, 1569–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yi H; Zhang G; Wang H; Huang Z; Wang J; Singh AK; Lei A Recent Advances in Radical C–H Activation/Radical Cross-Coupling. Chem. Rev 2017, 117, 9016–9085. [DOI] [PubMed] [Google Scholar]
- (3).For selected reviews, see the following: [Google Scholar]; (a) Lu Q; Glorius F Radical Enantioselective C(sp3)–H Functionalization. Angew. Chem., Int. Ed 2017, 56, 49–51. [DOI] [PubMed] [Google Scholar]; (b) Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]; (c) Sibi MP; Manyem S; Zimmerman J Enantioselective Radical Processes. Chem. Rev 2003, 103, 3263–3296. [DOI] [PubMed] [Google Scholar]
- (4).For selected examples on approaches to controlling radical reactivity and enantioselectivity, see the following: [Google Scholar]; (a) Nakafuku KM; Zhang Z; Wappes EA; Stateman LM; Chen AD; Nagib DA Enantioselective Radical C–H Amination for the Synthesis of β-Amino Alcohols. Nat. Chem 2020, 12, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang X; Wang B; Wang Y; Jiang G; Feng J; Zhao H Photoenzymatic Enantioselective Intermolecular Radical Hydroalkylation. Nature 2020, 584, 69–74. [DOI] [PubMed] [Google Scholar]; (c) Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK Photoexcitation of Flavoenzymes Enables a Stereoselective Radical Cyclization. Science 2019, 364, 1166. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Proctor RSJ; Davis HJ; Phipps RJ Catalytic Enantioselective Minisci-Type Addition to Heteroarenes. Science 2018, 360, 419. [DOI] [PubMed] [Google Scholar]; (e) Kern N; Plesniak MP; McDouall JJW; Procter DJ Enantioselective Cyclizations and Cyclization Cascades of Samarium Ketyl Radicals. Nat. Chem 2017, 9, 1198–1204. [DOI] [PubMed] [Google Scholar]; (f) Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric Copper-Catalyzed C–N Cross-Couplings Induced by Visible Light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective Cyanation of Benzylic C–H bonds via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Lin J-S; Dong X-Y; Li T-T; Jiang N-C; Tan B; Liu X-Y A Dual-Catalytic Strategy To Direct Asymmetric Radical Amino-trifluoromethylation of Alkenes. J. Am. Chem. Soc 2016, 138, 9357–9360. [DOI] [PubMed] [Google Scholar]; (i) Brill ZG; Grover HK; Maimone TJ Enantioselective Synthesis of an Ophiobolin Sesterterpene via a Programmed Radical Cascade. Science 2016, 352, 1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Du J; Skubi KL; Schultz DM; Yoon TP A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Hashimoto T; Kawamata Y; Maruoka K An Organic Thiyl Radical Catalyst for Enantioselective Cyclization. Nat. Chem 2014, 6, 702–705. [DOI] [PubMed] [Google Scholar]; (l) Bergonzini G; Schindler CS; Wallentin C-J; Jacobsen EN; Stephenson CR Photoredox Activation and Anion Binding Catalysis in the Dual Catalytic Enantioselective Synthesis of β-amino Esters. Chem. Sci 2014, 5, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Huo H; Shen X; Wang C; Zhang L; Röse P; Chen L-A; Harms K; Marsch M; Hilt G; Meggers E Asymmetric Photoredox Transition-metal Catalysis Activated by Visible Light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]; (n) Arceo E; Jurberg ID;Álvarez-Fernández A; Melchiorre P Photo-chemical Activity of a Key Donor-Acceptor Complex Can Drive Stereoselective Catalytic α-Alkylation of Aldehydes. Nat. Chem 2013, 5, 750–756. [DOI] [PubMed] [Google Scholar]; (o) Rono LJ; Yayla HG; Wang DY; Armstrong MF; Knowles RR Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J. Am. Chem. Soc 2013, 135, 17735–17738. [DOI] [PubMed] [Google Scholar]; (p) Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Bauer A; Westkämper F; Grimme S; Bach T Catalytic Enantioselective Eeactions Driven by Photoinduced Electron Transfer. Nature 2005, 436, 1139–1140. [DOI] [PubMed] [Google Scholar]
- (5).For selected reviews and highlights on Co(II)-based MRC, see the following: [Google Scholar]; (a) Singh R; Mukherjee A Metalloporphyrin Catalyzed C–H Amination. ACS Catal 2019, 9, 3604–3617. [Google Scholar]; (b) Huang H-M; Garduño-Castro MH; Morrill C; Procter DJ Catalytic Cascade Reactions by Radical Relay. Chem. Soc. Rev 2019, 48, 4626–4638. [DOI] [PubMed] [Google Scholar]; (c) Demarteau J; Debuigne A; Detrembleur C Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries. Chem. Rev 2019, 119, 6906–6955. [DOI] [PubMed] [Google Scholar]; (d) Pellissier H; Clavier H Enantioselective Cobalt-Catalyzed Transformations. Chem. Rev 2014, 114, 2775–2823. [DOI] [PubMed] [Google Scholar]; (e) Che C-M; Lo VK-Y; Zhou C-Y; Huang J-S Selective Functionalisation of Saturated C–H Bonds with Metalloporphyrin Catalysts. Chem. Soc. Rev 2011, 40, 1950–1975. [DOI] [PubMed] [Google Scholar]; (f) Lu H; Zhang XP Catalytic C–H Functionalization by Metalloporphyrins: Recent Developments and Future Directions. Chem. Soc. Rev 2011, 40, 1899–1909. [DOI] [PubMed] [Google Scholar]; (g) Driver TG Recent Advances in Transition Metal-Catalyzed N-Atom Transfer Reactions of Azides. Org. Biomol. Chem 2010, 8, 3831–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Doyle MP Exceptional Selectivity in Cyclopropanation Reactions Catalyzed by Chiral Cobalt(II)-Porphyrin Catalysts. Angew. Chem., Int. Ed 2009, 48, 850–852. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Fantauzzi S; Caselli A; Gallo E Nitrene Transfer Reactions Mediated by Metallo-Porphyrin Complexes. Dalton Trans. 2009, 5434–5443. [DOI] [PubMed] [Google Scholar]
- (6).For selected examples of Ti(III)-based radical processes, see the following: [Google Scholar]; (a) Ye K-Y; McCallum T; Lin S Bimetallic Radical Redox-Relay Catalysis for the Isomerization of Epoxides to Allylic Alcohols. J. Am. Chem. Soc 2019, 141, 9548–9554. [DOI] [PubMed] [Google Scholar]; (b) Yao C; Dahmen T; Gansäuer A; Norton J Anti-Markovnikov Alcohols via Epoxide Hydrogenation through Cooperative Catalysis. Science 2019, 364, 764–767. [DOI] [PubMed] [Google Scholar]; (c) Hao W; Wu X; Sun JZ; Siu JC; MacMillan SN; Lin S Radical Redox-Relay Catalysis: Formal [3+2] Cycloaddition of N-Acylaziridines and Alkenes. J. Am. Chem. Soc 2017, 139, 12141–12144. [DOI] [PubMed] [Google Scholar]; (d) Gansaüer A; Hildebrandt S; Vogelsang E; Flowers RA II. Tuning the Redox Properties of the Titanocene(III)/(IV)-Couple for Atom-Economical Catalysis in Single Electron Steps. Dalton Trans. 2016, 45, 448–452. [DOI] [PubMed] [Google Scholar]; (e) Gansäuer A; Hildebrandt S; Michelmann A; Dahmen T; von Laufenberg D; Kube C; Fianu GD; Flowers RA II Cationic Titanocene(III) Complexes for Catalysis in Single-Electron Steps. Angew. Chem., Int. Ed 2015, 54, 7003–7006. [DOI] [PubMed] [Google Scholar]; (f) Gansäuer A; Fleckhaus A; Lafont MA; Okkel A; Kotsis K; Anoop A; Neese F Catalysis via Homolytic Substitutions with C–O and Ti–O Bonds: Oxidative Additions and Reductive Eliminations in Single Electron Steps. J. Am. Chem. Soc 2009, 131, 16989–16999. [DOI] [PubMed] [Google Scholar]; (g) Gansäuer A; Fan C-A; Keller F; Keil J Titanocene-Catalyzed Regiodivergent Epoxide Openings. J. Am. Chem. Soc 2007, 129, 3484–3485. [DOI] [PubMed] [Google Scholar]; (h) Gansäuer A; Rinker B; Pierobon M; Grimme S; Gerenkamp M; Mück-Lichtenfeld C A Radical Tandem Reaction with Homolytic Cleavage of a Ti-O Bond. Angew. Chem., Int. Ed 2003, 42, 3687–3690. [DOI] [PubMed] [Google Scholar]; (i) RajanBabu TV; Nugent WA Selective Generation of Free Radicals from Epoxides Using a Transition-Metal Radical. A Powerful New Tool for Organic Synthesis. J. Am. Chem. Soc 1994, 116, 986–997. [Google Scholar]; (j) Nugent WA; RajanBabu TV Transition-Metal-Centered Radicals in Organic Synthesis. Titanium(III)-Induced Cyclization of Epoxy Olefins. J. Am. Chem. Soc 1988, 110, 8561–8562. [Google Scholar]
- (7).For selected examples of catalytic radical processes involving metalloradical intermediates, see the following: [Google Scholar]; (a) Kapat A; Sperger T; Guven S; Schoenebeck F E-Olefins through Intramolecular Radical Relocation. Science 2019, 363, 391. [DOI] [PubMed] [Google Scholar]; (b) Kuo JL; Hartung J; Han A; Norton JR Direct Generation of Oxygen-Stabilized Radicals by H• Transfer from Transition Metal Hydrides. J. Am. Chem. Soc 2015, 137, 1036–1039. [DOI] [PubMed] [Google Scholar]; (c) Estes DP; Norton JR; Jockusch S; Sattler W Mechanisms by Which Alkynes React with CpCr(CO)3H. Application to Radical Cyclization. J. Am. Chem. Soc 2012, 134, 15512–15518. [DOI] [PubMed] [Google Scholar]; (d) Li G; Han A; Pulling ME; Estes DP; Norton JR Evidence for Formation of a Co–H Bond from (H2O)2Co(dmgBF2)2 under H2: Application to Radical Cyclizations. J. Am. Chem. Soc 2012, 134, 14662–14665. [DOI] [PubMed] [Google Scholar]; (e) Smith DM; Pulling ME; Norton JR Tin-Free and Catalytic Radical Cyclizations. J. Am. Chem. Soc 2007, 129, 770–771. [DOI] [PubMed] [Google Scholar]
- (8).For detailed studies on the radical mechanism of [Co(Por)]-catalyzed C–H amination, including EPR observation of α-Co(III)-aminyl radical intermediates (also known as Co(III)-nitrene radicals), see the following: [Google Scholar]; (a) Goswami M; Lyaskovskyy V; Domingos SR; Buma WJ; Woutersen S; Troeppner O; Ivanović-Burmazović I; Lu H; Cui X; Zhang XP; Reijerse EJ; DeBeer S; van Schooneveld MM; Pfaff FF; Ray K; de Bruin B Characterization of Porphyrin-Co(III)-’Nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc 2015, 137, 5468–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hopmann KH; Ghosh A Mechanism of Cobalt-Porphyrin-Catalyzed Aziridination. ACS Catal. 2011, 1, 597–600. [Google Scholar]; (c) Olivos Suarez AI; Jiang H; Zhang XP; de Bruin B The Radical Mechanism of Cobalt(II) Porphyrin-Catalyzed Olefin Aziridination and the Importance of Cooperative H-Bonding. Dalton Trans. 2011, 40, 5697–5705. [DOI] [PubMed] [Google Scholar]; (d) Lyaskovskyy V; Suarez AIO; Lu H; Jiang H; Zhang XP; de Bruin B Mechanism of Cobalt(II) Porphyrin-Catalyzed C–H Amination with Organic Azides: Radical Nature and H-Atom Abstraction Ability of the Key Cobalt(III)-Nitrene Intermediates. J. Am. Chem. Soc 2011, 133, 12264–12273. [DOI] [PubMed] [Google Scholar]
- (9).(a) Lang K; Torker S; Wojtas L; Zhang XP Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H Amination via Enantiodifferentiative H-Atom Abstraction and Stereo-retentive Radical Substitution. J. Am. Chem. Soc 2019, 141, 12388–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu Y; Lang K; Li C; Gill JB; Kim I; Lu H; Fields KB; Marshall M; Cheng Q; Cui X; Wojtas L; Zhang XP Enantioselective Radical Construction of 5-Membered Cyclic Sulfonamides by Metalloradical C–H Amination. J. Am. Chem. Soc 2019, 141, 18160–18169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li C; Lang K; Lu H; Hu Y; Cui X; Wojtas L; Zhang XP Catalytic Radical Process for Enantioselective Amination of C(sp3)–H Bonds. Angew. Chem., Int. Ed 2018, 57, 16837–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For selected reviews on direct C–H amination, see the following: [Google Scholar]; (a) Hazelard D; Nocquet P-A; Compain P Catalytic C–H Amination at Its Limits: Challenges and Solutions. Org. Chem. Front 2017, 4, 2500–2521. [Google Scholar]; (b) Park Y; Kim Y; Chang S Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]; (c) Ramirez TA; Zhao B; Shi Y Recent Advances in Transition Metal-Catalyzed sp3 C–H Amination Adjacent to Double Bonds and Carbonyl Groups. Chem. Soc. Rev 2012, 41, 931–942. [DOI] [PubMed] [Google Scholar]; (d) Collet F; Lescot C; Dauban P Catalytic C–H Amination: The Stereoselectivity Issue. Chem. Soc. Rev 2011, 40, 1926–1936. [DOI] [PubMed] [Google Scholar]
- (11).(a) Nasrallah A; Boquet V; Hecker A; Retailleau P; Darses B; Dauban P Catalytic Enantioselective Intermolecular Benzylic C(sp3)–H Amination. Angew. Chem., Int. Ed 2019, 58, 8192–8196. [DOI] [PubMed] [Google Scholar]; (b) Höke T; Herdtweck E; Bach T Hydrogen-Bond Mediated Regio-and Enantioselectivity in a C–H Amination Reaction Catalysed by a Supramolecular Rh(II) Complex. Chem. Commun 2013, 49, 8009–8011. [DOI] [PubMed] [Google Scholar]; (c) Reddy RP; Davies HM Dirhodium Tetracarboxylates Derived from Adamantylglycine as Chiral Catalysts for Enantioselective C–H Aminations. Org. Lett 2006, 8, 5013–5016. [DOI] [PubMed] [Google Scholar]; (d) Yamawaki M; Tsutsui H; Kitagaki S; Anada M; Hashimoto S Dirhodium(II) tetrakis[N-tetrachlorophthaloyl-(S)-tert-leucinate]: a New Chiral Rh(II) Catalyst for Enantioselective Amidation of C–H bonds. Tetrahedron Lett. 2002, 43, 9561–9564. [Google Scholar]; (e) Nägeli I; Baud C; Bernardinelli G; Jacquier Y; Moraon M; Müllet P Rhodium-(II)-Catalyzed CH Insertions with {[(4-Nitrophenyl)sulfonyl]imino}-phenyl-λ3-iodane. Helv. Chim. Acta 1997, 80, 1087–1105. [Google Scholar]
- (12).(a) Nishioka Y; Uchida T; Katsuki T Enantio-and Regioselective Intermolecular Benzylic and Allylic C–H Bond Amination. Angew. Chem., Int. Ed 2013, 52, 1739–1742. [DOI] [PubMed] [Google Scholar]; (b) Zhou X-G; Yu X-Q; Huang J-S; Che C-M Asymmetric Amidation of Saturated C–H Bonds Catalysed by Chiral Ruthenium and Manganese Porphyrins. Chem. Commun 1999, 2377–2378. [Google Scholar]
- (13).Kohmura Y; Katsuki T Mn (salen)-catalyzed Enantioselective C–H Amination. Tetrahedron Lett. 2001, 42, 3339–3342. [Google Scholar]
- (14).(a) Jia Z-J; Gao S; Arnold FH Enzymatic Primary Amination of Benzylic and Allylic C(sp3)–H Bonds. J. Am. Chem. Soc 2020, 142, 10279–10283. [DOI] [PubMed] [Google Scholar]; (b) Prier CK; Zhang RK; Buller AR; Brinkmann-Chen S; Arnold FH Enantioselective, Intermolecular Benzylic C–H Amination Catalysed by an Engineered Iron-Haem Enzyme. Nat. Chem 2017, 9, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Huang X; Webster RD; Harms K; Meggers E Asymmetric Catalysis with Organic Azides and Diazo Compounds Initiated by Photoinduced Electron Transfer. J. Am. Chem. Soc 2016, 138, 12636–12642. [DOI] [PubMed] [Google Scholar]
- (16).Jin L-M; Lu H; Cui Y; Lizardi CL; Arzua TN; Wojtas L; Cui X; Zhang XP Selective Radical Amination of Aldehydic C(sp2)–H Bonds with Fluoroaryl Azides via Co(II)-Based Metalloradical Catalysis: Synthesis of N-Fluoroaryl Amides from Aldehydes under Neutral and Nonoxidative Conditions. Chem. Sci 2014, 5, 2422–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen Y; Fields KB; Zhang XP Bromoporphyrins as Versatile Synthons for Modular Construction of Chiral Porphyrins: Cobalt-Catalyzed Highly Enantioselective and Diastereoselective Cyclopropanation. J. Am. Chem. Soc 2004, 126, 14718–14719. [DOI] [PubMed] [Google Scholar]
- (18).Xu X; Lu H; Ruppel JV; Cui X; Lopez de Mesa S; Wojtas L; Zhang XP Highly Asymmetric Intramolecular Cyclopropanation of Acceptor-Substituted Diazoacetates by Co(II)-Based Metalloradical Catalysis: Iterative Approach for Development of New-Generation Catalysts. J. Am. Chem. Soc 2011, 133, 15292–15295. [DOI] [PubMed] [Google Scholar]
- (19).Ruppel JV; Jones JE; Huff CA; Kamble RM; Chen Y; Zhang XP A Highly Effective Cobalt Catalyst for Olefin Aziridination with Azides: Hydrogen Bonding Guided Catalyst Design. Org. Lett 2008, 10, 1995–1998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.