

Abstract
Epidemiological studies suggest that individuals in the Mediterranean region with a loss-of-function, non-synonymous single nucleotide polymorphism (SNP; S188F), in glucose-6-phosphate dehydrogenase (G6pd) are less susceptible to vascular diseases. However, this association has not yet been experimentally proven. Here, we set out to determine whether the Mediterranean mutation confers protection from vascular diseases and to discover the underlying protective mechanism. We generated a rat model with the Mediterranean SNP (G6PDS188F) using CRISPR-Cas9 genome editing. In rats homozygous for the mutation, G6PD activity, but not expression, was reduced to 20% of wild-type (WT) littermates. Additionally, unbiased metabolomics analysis revealed that the pentose phosphate pathway (PPP) and other ancillary metabolic pathways connected to the PPP were reduced (P<0.05) in the arteries of G6PDS188F versus WT rats. Intriguingly, G6PDS188F mutants, as compared to WT rats, developed less large arterial stiffness and hypertension evoked by high fat diet and nitric oxide synthase inhibition with L-NG-nitroarginine methyl ester (L-NAME). Intravenous injection of a voltage-gated L-type Ca2+ channel agonist (methyl 2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]-1,4-dihydropyridine-3-carboxylate; Bay K8644) acutely increased blood pressure in WT but not in G6PDS188F rats. Finally, our results suggested that: 1) lower resting membrane potential of smooth muscle caused by increased expression of K+ channel proteins and 2) decreased voltage-gated Ca2+ channel activity in smooth muscle contributed to reduced hypertension and arterial stiffness evoked by L-NAME and high fat diet to G6PDS188F mutants as compared to WT rats. In summary, a mutation resulting in the replacement of a single amino acid (S188F) in G6PD, the rate-limiting enzyme in the PPP, ascribed properties to the vascular smooth muscle that shields the organism from risk factors associated with vascular diseases.
Keywords: NADPH, Pentose Phosphate Pathway, Glycolysis, Vascular, Hypertension, Stiffness
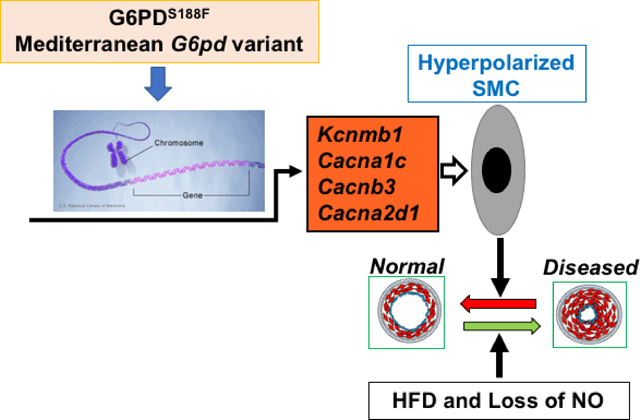
Graphical Abstract
Introduction
G6PD is the rate-limiting enzyme in the pentose phosphate pathway (PPP) – a fundamental component of cellular metabolism of glucose.1 The biochemical reactions that constitute the PPP are found in most life forms highlighting their essential role in metabolism. It is well established that G6PD and the PPP are a significant source of ribose sugar and NADPH, the reducing potential, in almost all cell types. Additionally, G6PD and the PPP are connected to ancillary metabolic pathways, mostly through the regulation of the levels of the reducing equivalent NADPH.2 Deficiency of G6PD is the most common enzymopathy in humans with over 400 million people worldwide suffering from different degrees of enzymopathy.3, 4 G6PD deficiency increases the susceptibility of red blood cells (RBCs) to oxidative hemolysis – a factor complicating anti-malaria treatment with pro-oxidant quinone drugs.5 However, although the PPP has been implicated in neurodegenerative disease,1, 6 cardiovascular disease,1, 7, 8 cancer,1, 9, 10 and aging,11 very little is known regarding the impact of G6PD deficiency on cardiovascular physiology and pathophysiology.
Epidemiological studies suggest that individuals in the Mediterranean region with a loss-of-function, non-synonymous single nucleotide polymorphism (SNP; S188F), in G6PD are less susceptible to vascular diseases.12–14 If this is true, then understanding the fundamental molecular basis for the cardiovascular protection afforded by the Mediterranean G6PD mutant will be of paramount importance in developing novel strategies that reduce the risk of coronary artery disease, peripheral vascular disease, and stroke – the leading causes of morbidity and mortality in the USA and worldwide.15
G6PD-derived NADPH is an essential cofactor for the generation of nitric oxide, the most potent vasodilator, by endothelial nitric oxide synthase.16 How individuals in the Mediterranean region with a G6PD deficiency are protected from vascular diseases is unknown, and there is no experimental proof of the association between the loss-of-function SNP (S188F) in G6PD and protection from vascular diseases. Therefore, to directly address whether the Mediterranean SNP is or is not protective in the cardiovascular system, we generated a rat carrying the Mediterranean SNP (G6PDS188F) using CRISPR-Cas9 gene editing and determined the effects of high fat diet and loss of nitric oxide on vascular function in G6PDS188F rats. We found that replacement of a single amino acid (S188F) in G6PD ascribed properties to the vascular smooth muscle that shielded the rats from some vascular diseases.
Methods
Detail methods, experimental protocols, and additional results are available in online Supplement.
Experimental protocols using animals had approval (A3362–01) from the New York Medical College and Medical College of Wisconsin Institutional Animal Care and Use Committees and experiments were performed in blinded fashion. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
CRISPR-Cas9 genome editing:
A rat model harboring a non-synonymous (S188F) substitution in the coding region of the rat G6pd gene, close to the G6PD catalytic domain, was developed using a 3-component CRISPR editing approach17 in fertilized Sprague Dawley (Crl:SD, Charles River Laboratories) rat embryos.
CRISPOR18 was used to design a CRISPR RNA (crRNA) located in close proximity to the SNP variant. The sequence of this crRNA (CUGGUCCUCACGAAACAGAG) was submitted for synthesis (Synthego, Redwood City, CA) and used with a transactivating crRNA (tracrRNA) and a single-strand oligonucleotide (Ultramer, IDT: 5’- GAACCGCATCATAGTGGAGAAGCCCTTCGGGAGAGACCTGCAGAGCTCCAATCAACTGTCGAACCACATCTTTTCTCTGTTTCGTGAGGACCAGATCTACCGCATTGACCACTACCTGGGCAAAGAGATGGTCCAGAACCTCATGG −3’) serving as a homology-directed repair template carrying the SNP variant (bold underlined sequence, includes the SNP and a second mutation to disrupt the protospacer adjacent motif). Ribonucleoprotein complex (crRNA/tracrRNA/Cas9; 50 ng/ul) and ssODN (50 ng/ul) were co-injected by pronuclear injection into Crl:SD embryos. Genotyping rat pups was performed by PCR and Sanger sequencing. Founders were bred for germline transmission of the SNP allele. Predicted off-targeting events were determined with the CRISPOR tool;18 two predicted noncoding off-target sites were found to be > 4 Mb away from the SNP suggesting probable segregation upon breeding. These G6PDS188F mutants and their wild type (WT) age-matched littermates were used in this study.
Echocardiography, hemodynamics measurements, force measurements, membrane potential and intracellular Ca2+ levels, metabolomics, G6PD activity, nitrite measurements, histology, qPCR, and Western blotting, were performed as described previously19–27 and detail protocols are given in the online supplement.
Statistical Analysis.
Graphs and statistical analyses were prepared with GraphPad Prism 5.0 (GraphPad Software, Inc, La Jolla, CA) and Metaboanalyst 3.0. Values are presented as the mean ± SEM of the number of samples (n) from different animals. Two-way ANOVA with post-hoc Bonferroni correction was used to compare multiple groups, and unpaired Student’s t-test was used to compare two groups. Values of p < 0.05 were considered significant.
Results
Activity of PPP and other ancillary pathways was reduced in CRISPR-Cas9-edited G6PDS188F rats:
Sanger sequencing data demonstrate two nucleotide substitutions in G6pd (Fig. 1A). We substituted two nucleotides, preserving the S188F nonsynonymous substitution to simplify genotyping and destroy the PAM site (CCT). Although we did not perform whole genome sequencing for off-targets, the Cutting Frequency Determination specificity score of the crRNA was high (84) with only two predicted off-target sites on the X-chromosome and both noncoding sites are more than 4 Mb away from the on-target site based on the CRISPOR tool.18 Thus, we deem relevant off-targeting to be very low, if at all. There were no detectable differences in the baseline morphological (structure) and functional (physiological) phenotype between G6PDS188F and wild-type (WT) rats. Moreover, there was no difference in body weight and hematocrit between six-month-old G6PDS188F and WT rats. However, while the expression of G6PD was not different, activity was reduced by 80% in liver of G6PDS188F as compared to WT rats (Fig. 1B). G6PD is the rate limiting enzyme in the PPP that reduces six carbon glucose to five carbon ribose sugars and produces NADPH, a major reducing potential that maintains a ratio of reduced glutathione (GSH)-to-oxidized glutathione (GSSG), in the cell (Fig. 1C). The PPP integrates with other pathways in the cell (Supplement figure S1) Metabolomics analyses revealed that ribose phosphate (and isobaric pentose phosphate metabolites of the PPP) are decreased, and G6P and GSSG-to-GSH ratio are increased, in arteries of G6PDS188F as compared to WT rats (Fig. 1D, E). In addition, metabolites of the TCA cycle and ancillary metabolic pathways (polyamine pathway and one-carbon metabolism) that are involved in NADPH homeostasis were significantly lower in arteries of G6PDS188F rat (Fig. 2).
Figure 1:
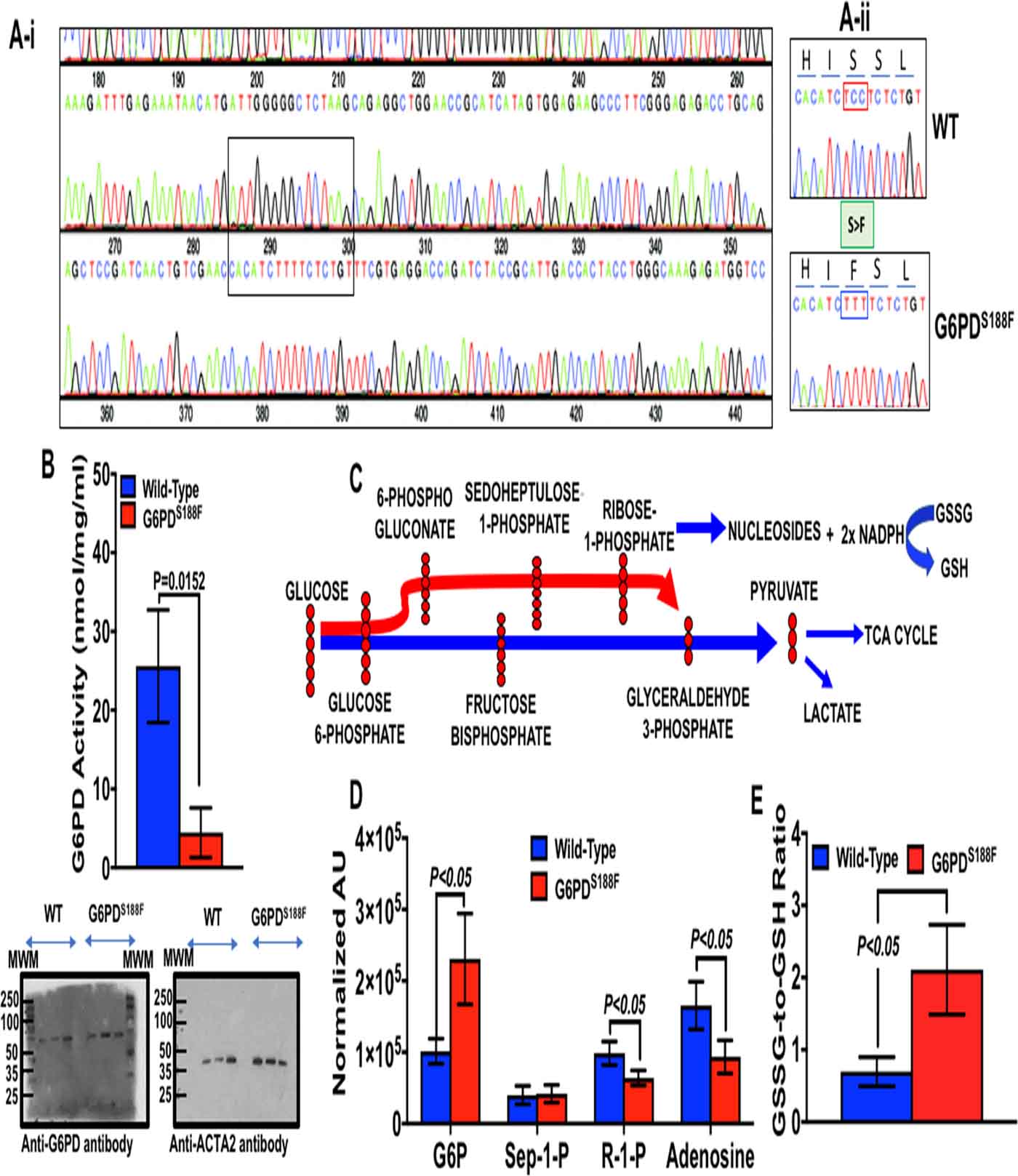
Genotyping, G6PD activity and expression, and glucose metabolism through the pentose phosphate pathway. In (A-i) Sanger sequence rats was performed to determine mutation in G6PD between 285 and 300 bp (TCT; shown in square box) by CRISPR-cas9 editing. (A-ii) Sanger sequence demonstrating C>T in G6pd resulting S188F switch in G6PD of the mutant (G6PDS188F) compared to wild-type (WT) rats. Red box is PAM and green box is S>F substitution. (B) G6PD activity (Top) and expression (Bottom) in liver of WT and G6PDS188F rats. (C) A schematic illustrating metabolism six-carbon (glucose) to five-carbon (ribose sugar), nucleoside, and synthesis of NADPH in the PPP. (D) metabolomic analysis showing increased glucose 6-phosphate and reduction in the PPP metabolite ribose 1-phosphate (R-1-P) and adenosine in arteries of G6PDS188F as compared WT rats. (E) The PPP-derived NADPH redox regulates oxidized (GSSG)-to-reduced (GSH) glutathione ratio in most cells and GSSG-to-GSH ratio is increased in arteries G6PDS188F than WT rats. N=8 to 9 rats in WT and G6PDS188F group.
Figure 2:
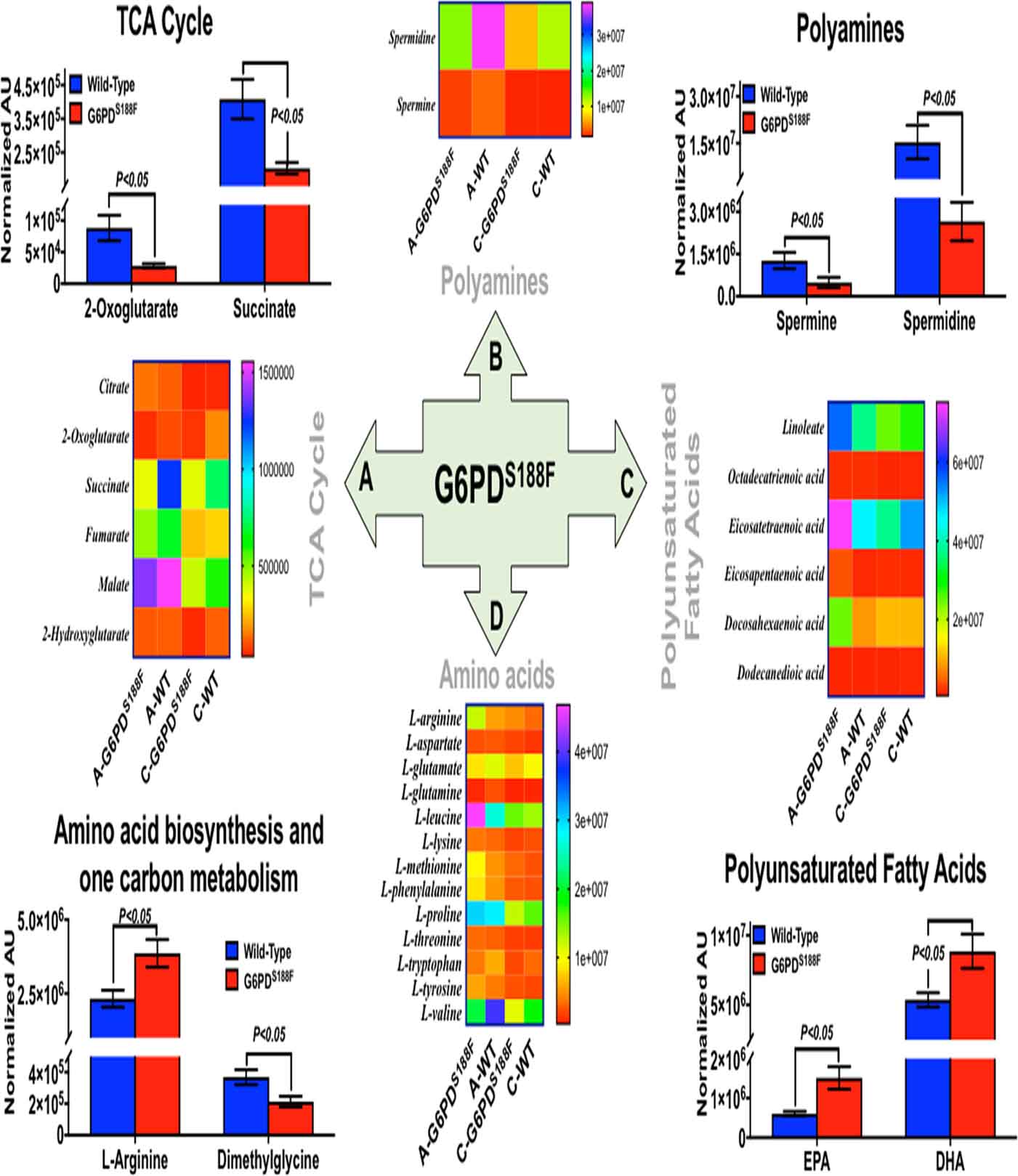
Metabolism in arteries of WT and G6PDS188F rats. Metabolic variations in arteries of WT and G6PDS188F rats were quantitatively determined by metabolomic analysis. Selected metabolites of: Tricarboxylic acid (TCA) cycle; Polyamines; Polyunsaturated Fatty Acids; and Amino acids and one carbon metabolism, in aorta and carotid artery combined, are significantly altered in G6PDS188F versus WT rats. Heat map illustrating levels of individual metabolites of four metabolic pathways quantitated in aorta (A-G6PDS188F and A-WT) and in carotid artery (C-G6PDS188F and C-WT). N=8 to 9 rats in WT and G6PDS188F group.
High fat diet elicited less severe hypertension and arterial stiffness in G6PDS188F mutants compared to WT rats:
Metabolic syndrome is on the rise worldwide.28 It is caused by high fat/calorie diet and is a major risk factor for life-threating coronary artery disease and stroke.15, 28 High fat diet (HFD) and hormonal changes increase expression and activity of G6PD.29 Therefore, our goal was to determine whether the HFD-mediated increase in risk factors for vascular diseases, large artery stiffness and hypertension, are amplified or reduced in G6PDS188F as compared to WT rats. We fed WT and G6PDS188F rats with HFD (containing 20% protein, 60% fat, and 20% carbohydrate; 60% fat of total calories; D12492, Research Diets Inc., USA) for four months. Both WT and G6PDS188F rats gained identical weight on HFD (Supplement figure S2). We performed echocardiography and catherization on these rats and found that carotid peak velocity and pulse wave velocity were elevated more (21.1% versus 14.3%) in WT than G6PDS188F rats (Fig. 3A, B) as determined from the blood flow in carotid and transit time between Doppler flow signals in the carotid and iliac arteries of WT and G6PDS188F rats on normal chow or HFD (Fig. 3C). Further, systolic and diastolic blood pressure (Fig. 3D–E) and arterial elastance (Fig. 3F) were increased in HFD fed WT but not in HFD fed G6PDS188F rats. There was no major remodeling (medial hypertrophy or neo-intimal formation) of carotid artery and aorta isolated from WT and G6PD S188F rats fed with normal chow or HFD (Supplement figure S3).
Figure 3:
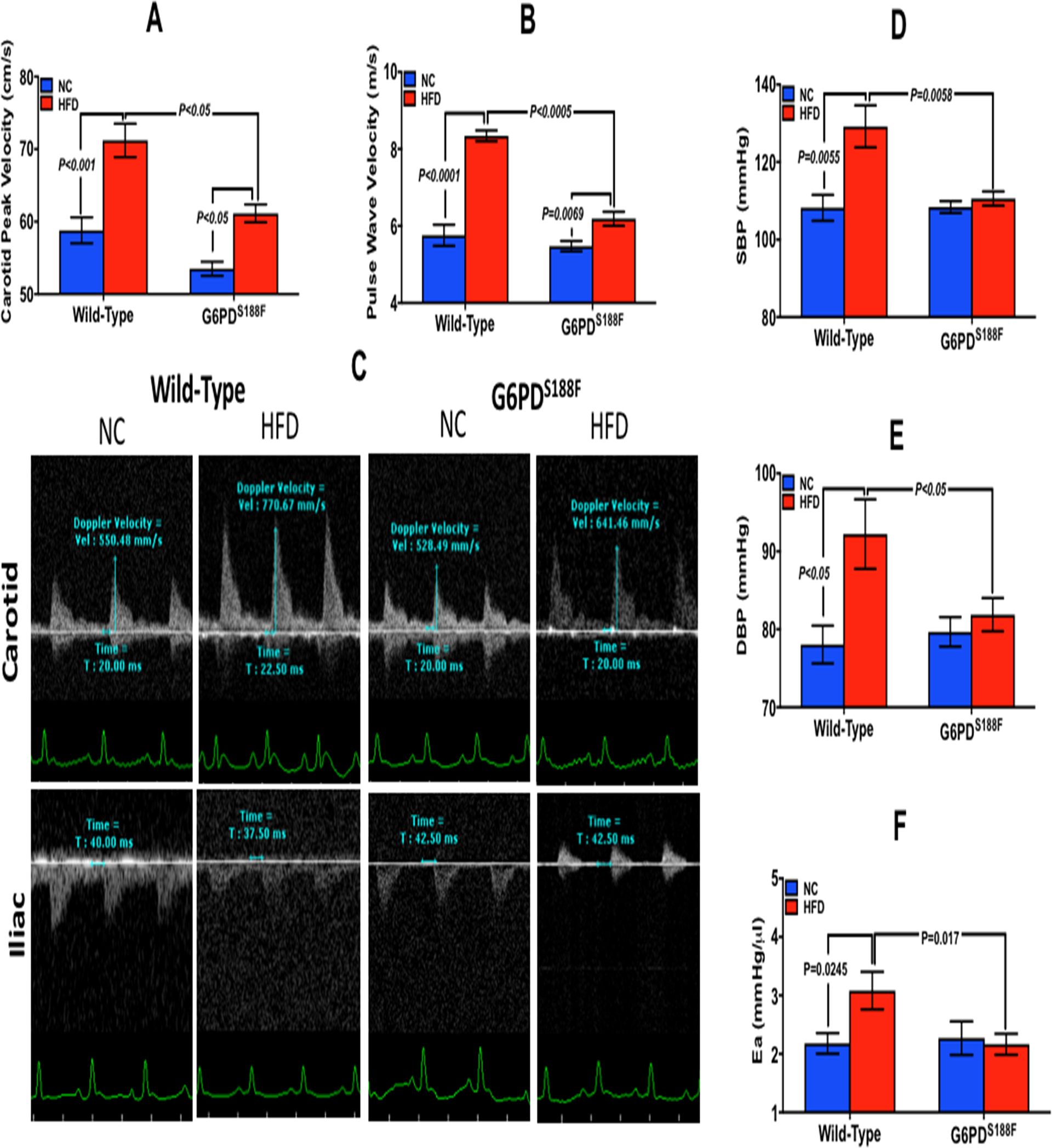
Effect of HFD on blood pressure, large artery stiffness, and common carotid artery stenosis in WT and G6PDS188F rats. (A) Common carotid artery peak velocity in G6PDS188F and WT rats on normal chow (NC) and high fat diet (HFD). (B) Pulse wave velocity (PWV), determined by the distance between carotid and iliac artery divided by time, increased less in G6PDS188F than in WT rats. (C) A representative echocardiography experiment showing the blood flow in carotid and iliac artery in WT and G6PDS188F rats on NC and HFD. (D-F) HFD-elicited systolic and diastolic blood pressure and arterial elastance (Ea) increased more in WT than G6PDS188F rats. N=5 rats in WT and G6PDS188F group.
Inhibition of nitric oxide synthase elicited less increase in hypertension and arterial elastance in G6PDS188F than WT rats:
Nitric oxide (NO) is a potent vasodilator, and loss of endothelial-derived NO has been implicated in the pathogenesis of metabolic syndrome-associated vascular diseases. NO is synthesized by NO synthase, which requires NADPH co-factor as an electron donor. Therefore, since G6PD activity is reduced by G6PDS188F mutation, we measured NO levels and determined whether inhibition of NO synthase with L-NAME exacerbates hypertension and arterial stiffness in G6PDS188F versus WT rats. Nitrite levels were showed slight reducing trend (P=0.312) in aorta of G6PDS188F (934±135 μM/mg protein) as compared to WT (1049±179 μM/mg protein) rats. L-NAME (1 mg/ml in drinking water) increased systolic and diastolic blood pressure (Fig. 4A–B) and elastance (Fig. 4C) more in WT than G6PDS188F rats. Furthermore, isometric tension studies in carotid arterial rings revealed that KCl (Fig. 4D–F)- and phenylephrine (Supplement figure S4A–F)-induced contraction curves shifted to the left and maximum contraction was increased (Supplement figure S4B) in WT but not in G6PDS188F carotids. Acetylcholine-induced relaxation was reduced in WT (Supplement figure S4E) but not in G6PDS188F (Supplement figure S4F) rats. There was no major remodeling (medial hypertrophy or neo-intimal formation) of carotid artery and aorta from WT and G6PD S188F rats treated with L-NAME (Supplement figure S5).
Figure 4:
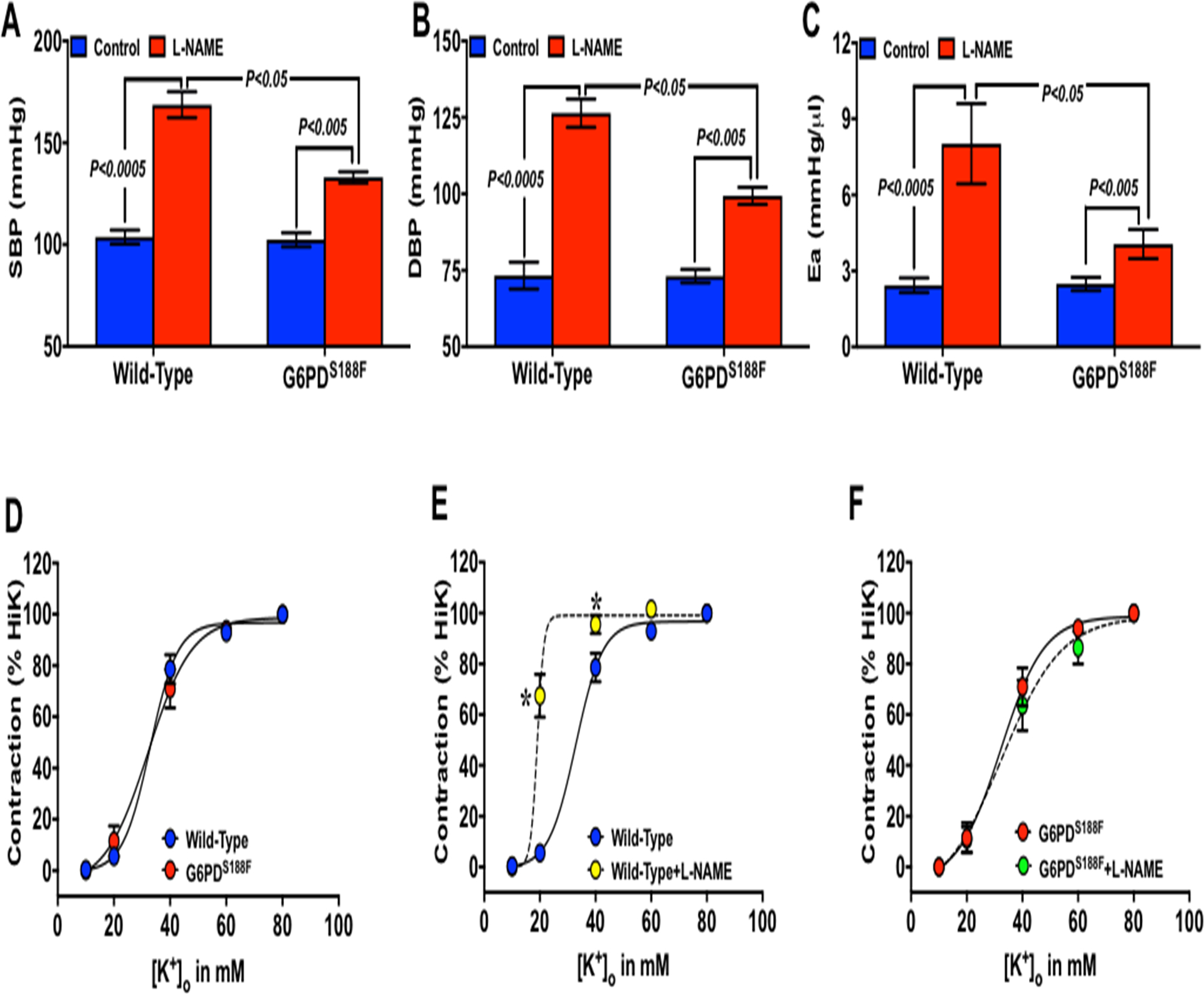
Effect of L-NAME on blood pressure, arterial elastance, and carotid artery function in WT and G6PDS188F rats. (A) Systolic and (B) diastolic pressure, and (C) arterial elastance in WT and G6PDS188F rats treated with vehicle control and L-NAME (1 mg/ml in drinking water) for 5 days is shown. (D-F) KCl-induced contraction of common carotid arteries isolated from WT and G6PDS188F rats treated with vehicle control and L-NAME. N=8 to 11 rats in WT and G6PDS188F group.
Blood pressure elicited by L-type Ca2+ channel agonist but not by α1-adrenergic agonist was decreased in G6PDS188F as compared to WT rats:
Since KCl (Fig. 4)- and phenylephrine (Supplement figure S4)-induced contraction of carotid artery significantly increased in L-NAME-treated WT but not G6PDS188F rats, we sought to determine whether L-type Ca2+ channel- and α1-adrenergic receptor-mediated pathways of vasoconstriction were suppressed in G6PDS188F rats. To do so, we intravenously injected methyl 2.6-dimethyl-5-nitro-4-[2-(trfluoromethyl)phenyl]-1,4-dihydropyridine-3-carboxylate (Bay K8644; 1 μM), a selective L-type Ca2+ channel agonist,30 and phenylephrine (0.01, 0.100 and 1.0 μM), a specific α1-agonist that constricts smooth muscle in L-type Ca2+ channel-independent but predominantly by Rho kinase-dependent manner,31 in WT and G6PDS188F rats and determined blood pressure over a period of 30 minutes. Interestingly, systolic and diastolic blood pressure elicited by Bay K8644 (Fig. 5A, B) but not by phenylephrine (1 μM; Supplement figure S6) was significantly reduced in G6PDS188F as compared to WT rats. A lower concentration of phenylephrine did not increase blood pressure.
Figure 5:
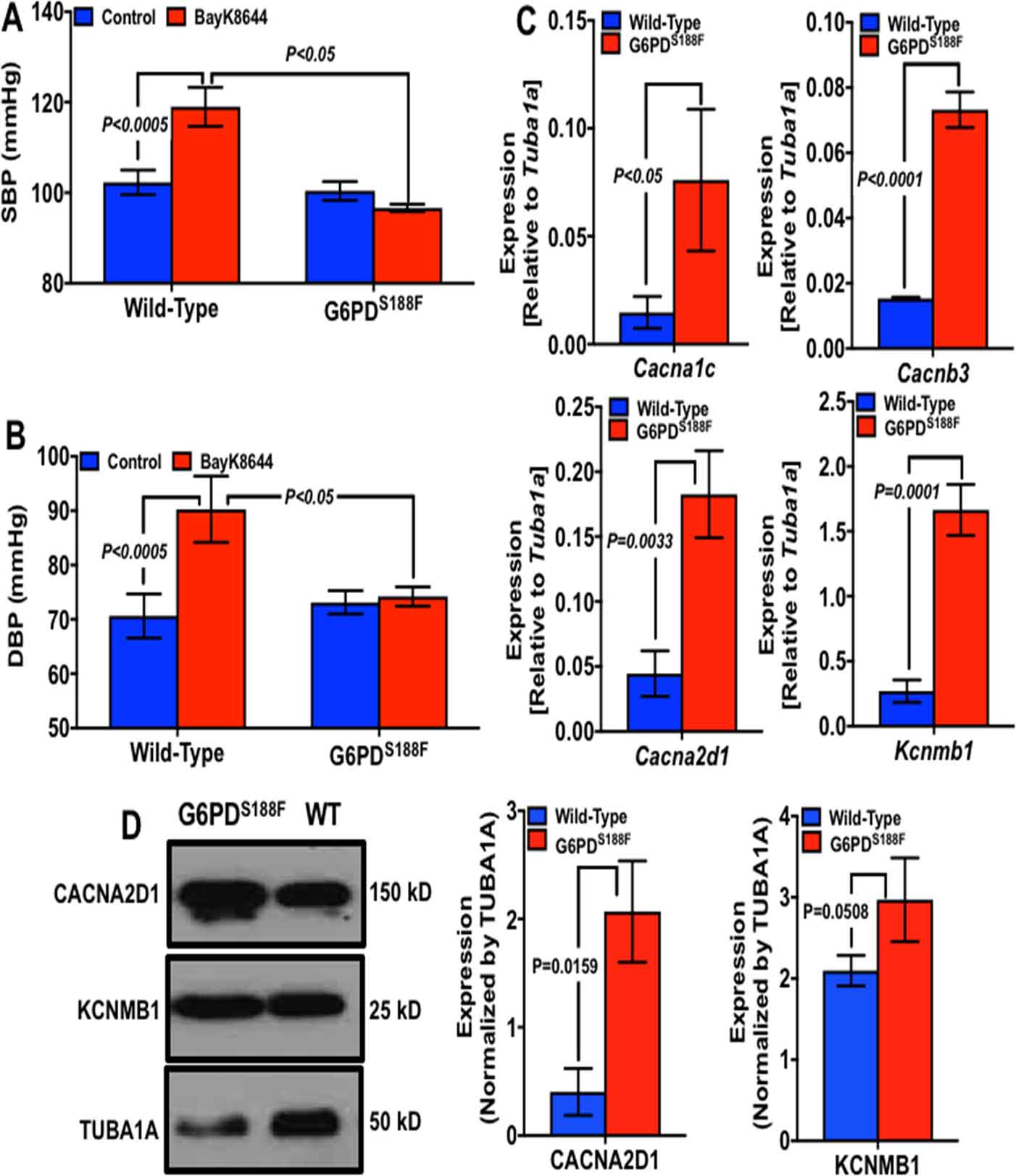
Effect of BayK8644 and phenylephrine on blood pressure and ion channel gene expression in aorta of WT and G6PDS188F rats. Systolic and diastolic blood pressure was recorded before (Control) and 10 minutes after intravenously injecting (A, B) BayK8644 (1 μM) to WT (N=7) and G6PDS188F (N=5) rats. Expression levels of: (C) alpha-, beta- and delta-subunits of L-type Ca2+ channels, and beta-subunit of BKCa channel gene in aorta of WT and G6PDS188F rats is shown. (D) Expression of delta-subunit of L-type Ca2+ (left and middle panel) and BKCa (middle and right panel) channel protein in aorta of WT and G6PDS188F rats is shown. N=5 in each group.
Ca2+ and K+ channel genes and proteins are increased in arteries of G6PDS188F rats:
Bay K8644 binds to the α-subunit of L-type Ca2+ channel32 and increases the open channel probability,33 which augments Ca2+ influx into SMCs to evoke contraction.34 This way Bay K8644 constricts arteries and raises blood pressure. Therefore, since Bay K8644 elicited less increase of blood pressure in G6PDS188F than WT rats , we considered whether L-type Ca2+ channel proteins are reduced in vascular smooth muscle of G6PDS188F rats. On the contrary, we found the expression of genes that encode Cacna1c, α-subunit of L-type Ca2+ channel, Cacnb3, β-subunit of L-type Ca2+ channel, Cacna2d1, δ-subunit of L-type Ca2+ channel, and Kcnmb1, β-subunit of big conductance K+ channel, were significantly increased in aorta of G6PDS188F as compared to WT rats (Fig. 5C). Furthermore, we found that CACNA2D1 and KCNMB1 protein expression were higher in aorta of G6PDS188F as compared to WT rats (Fig. 5D). Additionally, we determined the expression of these genes in aorta of HFD-fed WT and G6PDS188F rats (Supplement table S1). Intriguingly, HFD feeding, as compared to normal chow feeding, increased Cacna1c by 20-fold, Cacnb1 by 10-fold, Cacnb3 by 27-fold, and Cacna2d1 by 2.3-fold in WT rats, but not in G6PDS188F rats. Instead, HFD feeding significantly increased (3.1-fold) Kcnma1, α-subunit of big conductance K+ channel, in G6PDS188F but not in WT rats.
Membrane potential is less and Ca2+-influx-induced contraction is attenuated in arteries of G6PDS188F than in WT rats:
Since we found that Cacna1c was paradoxically increased in arteries of G6PDS188F rats, in which Bay K8644-elicited less increase of blood pressure than WT rats, we postulated that Bay K8644 binding to the open channels, which ensues with the depolarization of the membrane potential, would be reduced, perhaps due to less depolarized membrane potential in the smooth muscle cells of G6PDS188F versus WT rats. Therefore, we determined membrane potential by a slow-response membrane potential indicator, DiBAC4(3), in aorta of G6PDS188F and WT rats at resting condition and in response to KCl. Fluorescence of DiBAC4(3) increases with depolarization of the membrane potential.20 Our results showed that fluorescence of DiBAC4(3) was higher in the resting and KCl (20 and 40 mM)-treated arteries of WT versus G6PDS188F rats (Fig. 6A, B). Additionally, since nitrendipine, a structural analog of Bay K8644, decreases Ca2+ channel activity in SMCs with more depolarized membrane potentials,35 we determined whether nitrendipine elicited less relaxation of arteries isolated from G6PDS188F and WT rats pre-contracted with KCl (30 mM). As expected, nitrendipine-elicited less relaxation of aorta from G6PDS188F than WT rats (Fig. 6C). Consequences of lower depolarized membrane potential are: 1) reduced opening of the L-type Ca2+ channels; 2) less Ca2+ influx into the SMCs; and 3) attenuated contraction of arteries. Therefore, we measured intracellular Ca2+ ([Ca2+]i) levels in resting and KCl (30 mM)-treated SMCs by fluorescence imaging. These results showed that [Ca2+]i is higher in resting and KCl-treated SMCs from WT as compared to G6PDS188F rats (Fig. 6D). Next, we determined the Ca2+ influx-elicited isometric force generation (contraction) of aorta and carotid artery isolated from G6PDS188F and WT rats pre-treated with KCl (30 M) or Bay K8644 (1 μM). We found that KCl (Fig. 6E, F)- and Bay K8644 (Supplement figure S7)-elicited less contraction of aorta and carotid artery from G6PDS188F rats as compared to WT rats. Taken together, these findings suggest that G6PDS188F-induced hyperpolarization of membrane potential reduced Ca2+ influx and Ca2+-mediated SMC contraction.
Figure 6:
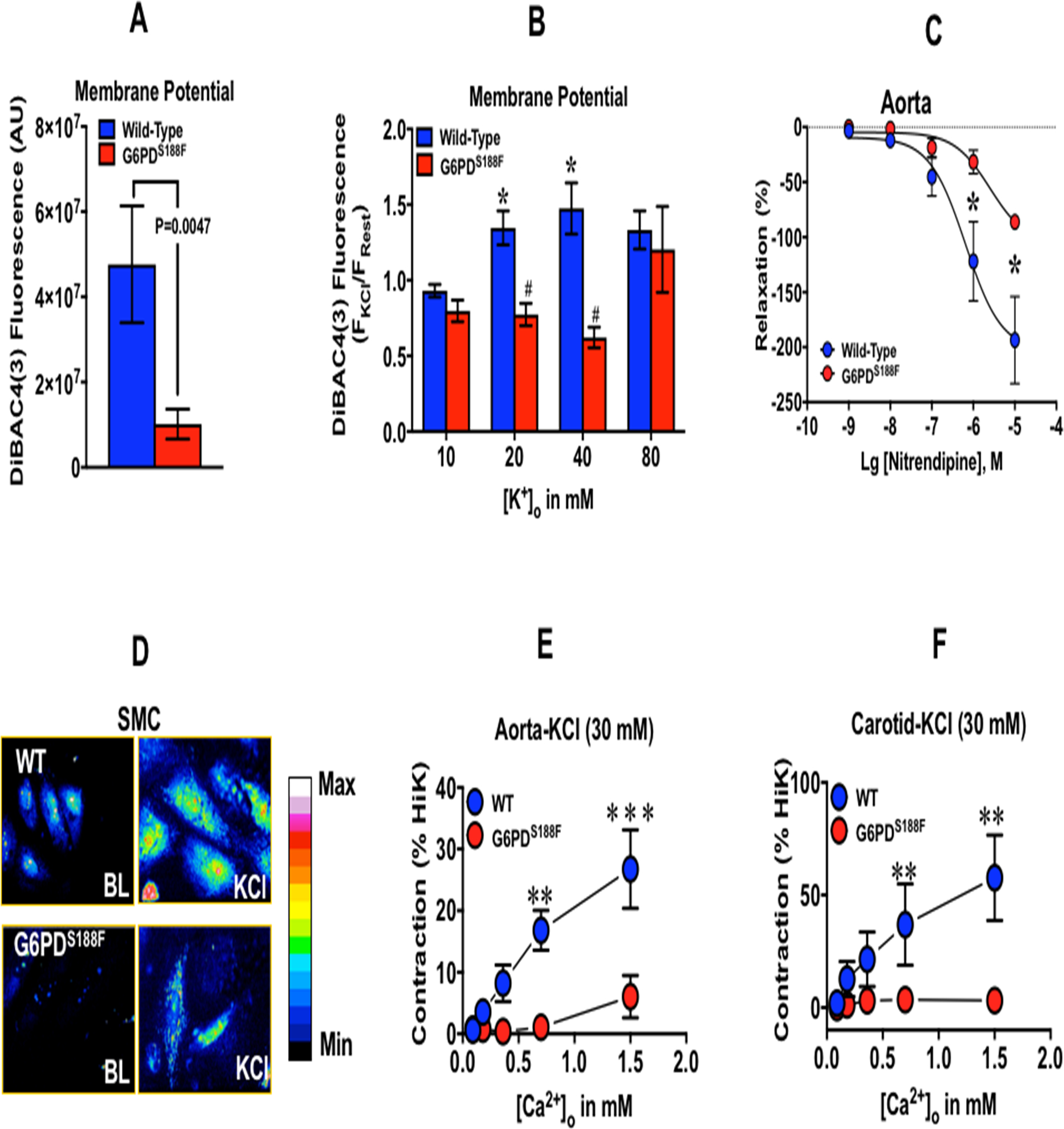
Membrane potential, nitrendipine-mediated relaxation, intracellular Ca2+, and Ca2+-mediated contraction of arteries isolated from WT and G6PDS188F rats. Membrane potential of (A) resting and (B) KCl-treated aorta was determined by the fluorescence (f480/520) of DiBAC4(3). N= 5 WT and G6PDS188F rats. (C) The effect of nitrendipine, a L-type Ca2+ channel blocker that inhibits channel activity at higher membrane potential, on aorta pre-contracted with KCl (30 mM). N= 5 WT and G6PDS188F rats. (D) A representative confocal image of four experiments demonstrating intracellular Ca2+ determined by Fluro-4 (f480/520) in aortic SMCs. Extracellular Ca2+-elicited contraction of aorta (E) and carotid artery (F) pre-contracted with KCl (30 mM) is shown. Aorta (N= 5 WT and 10 G6PDS188F rats) and carotid artery (N= 5 WT and 9 G6PDS188F rats).
Discussion
The results presented in this study provide the first experimental evidence that a mutation resulting in the replacement of a single amino acid (S188F) in G6PD, the rate-limiting enzyme in the PPP, protected the rat from developing large artery stiffness (compromised Windkessel function) and hypertension (HTN) that is associated with the metabolic syndrome and loss of nitric oxide. Furthermore, our results have illuminated new insight into the mechanisms through which the loss-of-function mutation in G6PD protects the rats from HTN – a major risk factor for coronary artery disease and stroke – and stiffness of large arteries – a critical risk for systolic HTN and heart failure in hypertensive patients.36, 37
G6PDS188F rat is an authentic experimental model of G6PD Mediterranean variant with genotype-phenotype similar to human, such as; X-linked c.563C>T (S188F) switch, severe loss of activity, increased GSSG-to-GSH ratio, decreased nucleotide levels, and little change in protein expression.4, 38 In contrast, although different inbred mouse strains exhibit variability in G6PD activity in RBCs, there are no differences in GSH levels reported in them.38, 39 Moreover, X-linked G6PD deficient mice, which was recovered from the offspring of male mice treated with 1-ethyl-1-nitrosourea, have normal protein synthesized in decreased quantity due to a mutation in the 5’ untranslated region.40 Thus, the latter mouse model is not a model of the human G6PD Mediterranean polymorphism. Because G6PD is coded by a gene on the X chromosome, X-linked G6PD deficiency is more common in males than in females. Generally, hemizygous males and females carrying two deficient variants (homozygous females) with defective G6PD alleles tend to have functionally faulty G6PD. Therefore, in this study hemizygous male and homozygous female rats were used as they are translationally relevant to humans.
HFD feeding, which produces metabolic syndrome-like symptoms in rats,41 increased pulse wave velocity and arterial elastance (markers of large artery stiffness), common carotid peak velocity (a marker of carotid artery stenosis), and systolic and diastolic BP, more in WT rats than G6PDS188F rats. High fat and HFD-induced hormonal changes, viz-a-viz increased insulin, augment the expression and activity of G6PD.29, 42, 43 Furthermore, G6PD is increased in various tissues of obese-diabetic individuals44–47 and the elevated G6PD has been associated with increased inflammation – a potential cause of HTN and large artery stiffness – in obese-diabetic mice.48 Our results support a mechanism(s) of preserved vasodilatory function and reduced arterial stiffness conferred by the G6PDS188F variant in rats fed a HFD.
Metabolic syndrome, a modifiable risk factor of cardiovascular diseases, is pandemic and a health concern in the US and worldwide.28 In metabolic syndrome patients, neurohumoral imbalance, endothelial dysfunction, loss of nitric oxide, and inflammation of blood vessels elicit constriction and contribute to the remodeling of small resistance arteries that increases BP.49 In rats, HFD feeding impairs endothelial function and contributes to increased vasoconstriction,50 thus raising BP. Decreased endothelial function and endothelial-derived nitric oxide bioavailability elicit vasoconstriction and remodeling of the arteries,51 and contributes to the pathogenesis of HTN.52, 53 Also, loss of nitric oxide causes and enhances arterial stiffness.54, 55 Consistently, L-NAME treatment reduced acetylcholine-elicited relaxation – an indicator of endothelial dysfunction – of carotid artery isolated from WT, but not G6PDS188F, rats. Furthermore, L-NAME increased BP as well as arterial elastance more in WT than G6PDS188F rats. These results suggest that arteries of WT rats are stiffer than G6PDS188F rats. This finding is indeed surprising and unexpected. Because, on the contrary, we expected that reduced levels of G6PD-derived NADPH, a critical co-factor for endothelial nitric oxide synthase, in arteries of G6PDS188F rats would augment, not reduce, L-NAME-induced vasoconstriction and BP. This paradox could have occurred if there was unmasking of a dominant compensatory hyperpolarization of the membrane potential, subsequent to inhibition of nitric oxide synthase, in arteries of G6PDS188F but not WT rats. Alternatively, since G6PD-derived NADPH fuels generation of superoxide anion instead of nitric oxide from inactivated nitric oxide synthase, which increases the Ca2+ sensitivity to the myofilament, we suggest that a loss-of-function mutation in G6PD might have decreased the Ca2+ sensitivity to the contractile apparatus and relaxed the vascular smooth muscle.
Phenylephrine, a selective α1-agonist that increases vasoconstriction through augmenting Rho kinase-mediated Ca2+ sensitivity to the myofilament,31 and KCl, which elicits vasoconstriction through depolarizing the membrane potential of SMCs, increased contraction of carotid arteries isolated from WT, but not G6PDS188F, rats treated with L-NAME. We interpret these results to signify reductions inCa2+ influx through the voltage-gated Ca2+ channels and Ca2+ sensitivity to the myofilament in arteries of G6PDS188F rats. However, BayK8644, but not phenylephrine, elicited less increase of BP in G6PDS188F than WT rats. These results suggest that Ca2+ channel activity and not the Ca2+ sensitivity to the contractile apparatus was reduced in arteries of G6PDS188F rats. Potentially, Bay K8644-induced increase of Ca2+ channel activity could be decreased if; 1) the α-subunit of L-type Ca2+ channel expression is decreased and 2) the membrane potential is more hyperpolarized. Because in both scenarios BayK8644, which binds to channels and increases the open channel probability in a membrane potential-dependent manner,32 cannot activate L-type Ca2+ channel. However, we rule out the first scenario because expression of Cacna1c, Cacnb3, and Cacna2d1 was higher (>5-fold) in arteries of G6PDS188F than WT rats. Therefore, the second possibility of more hyperpolarization of the membrane potential likely explains why actions of Bay K8644 were blunted in G6PDS188F rats. We suggest that: 1) augmented expression of Kcnmb1, which increases voltage- and Ca2+-sensitivity of BKCa channel, 2) decreased ancillary polyamine pathway (spermine and spermidine; Fig. 2B), which inhibits the activity of inward rectifier K+ channels,56, 57 and 3) elevated polyunsaturated fatty acids (EPA and DHA; Fig. 2C), which increases K+ channel activity;58 altogether significantly contributed to a more hyperpolarized membrane potential in arteries isolated from G6PDS188F versus WT rats. This notion is supported by our results indicating that the differences in the membrane potential observed between the arteries of WT and G6PDS188F rats was abolished by high extracellular KCl (80 mM), which blocks all outward K+ currents. Lower membrane potential decreases Ca2+ influx and contraction of the SMC. Consistently, in arteries of G6PDS188F rats as compared to WT rats, we found decreased: 1) Ca2+ influx-induced contraction of KCl- and Bay K8644-treated arteries; 2) intracellular Ca2+ in resting and KCl-treated SMCs; and 3) relaxation of pre-contracted arteries by nitrendipine, a Ca2+ channel blocker that inhibits channel activity by binding to a more depolarized SMC. These results suggest that lower resting membrane potential of SMCs moderated over excitement of the cell that elicits influx and overload of Ca2+, a second messenger that not only elicits contraction of SMCs but also mediates gene expression and stiffening of blood vessels.59, 60 This in turn would guard the G6PDS188F rats from abnormal vascular function and baneful vascular diseases often caused by HFD and loss of nitric oxide. Additionally, we propose that the lack of HFD-induced increase of Cacna1c, Cacnb1, and Cacnb3 expression in arteries of G6PDS188F mutants fortified these rats from developing large artery stiffness and HTN .
The incidence of vascular diseases is less in the Mediterranean region as compared to Western countries. Some epidemiological studies suggest that individuals with the G6PD Mediterranean variant modeled here are even less susceptible to cardiovascular diseases.12–14 Others suggest that the same loss-of-function SNP in G6PD is detrimental and contribute to the worsening of cardiovascular pathology/disease.61, 62 Our findings support the epidemiological studies that suggest G6PD Mediterranean variant protects individuals from cardiovascular diseases,12–14 and provides insight into the molecular mechanisms that shield the organism from vascular diseases. Many polymorphic variants of G6PD exist worldwide. The polymorphic G6PD African variant has less severe G6PD deficiency as compared to G6PD Mediterranean variant.4 Therefore, it will be interesting to develop an experimental rat model of G6PD African variant and determine whether they are or are not protected from cardiovascular diseases. Nonetheless, based on our current findings, we propose that a modest inhibition of G6PD expression and/or activity may be beneficial, and not harmful, in reducing severity of vascular diseases associated with metabolic syndrome as compared to persons without the G6PD Mediterranean mutation.
Perspectives:
G6PD, an enzyme in a major metabolic pathway, unexpectedly contribute to the expression of genes, which encode ion channels that are critically involved in the regulation of membrane potential and Ca2+ homeostasis in SMCs, and to vascular function. A loss-of-function Mediterranean mutation in G6PD protected from arterial diseases like, large artery stiffness and HTN, elicited by HFD and loss of nitric oxide. Therefore, these findings give us a new opportunity to exploit this enzyme as a novel pharmacotherapeutic target, in the future, to reduce the load of all forms of vascular diseases.
Supplementary Material
Novelty and Significance:
What is new:
Epidemiological studies suggest that individuals in the Mediterranean region with a loss-of-function, non-synonymous single nucleotide polymorphism (SNP; S188F), in glucose-6-phosphate dehydrogenase (G6pd) are less susceptible to vascular diseases. Therefore, we developed the first rat model with the Mediterranean SNP in G6PD using CRISPR-Cas9 genome editing.
What is relevant:
In this rat model, we determined whether Mediterranean G6PD variant is or is not protective to cardiovascular system.
Summary:
The results presented in this study provide the intriguing experimental evidence that a mutation resulting in the replacement of a single amino acid (S188F) in G6PD, the rate-limiting enzyme in the PPP, ascribed properties to the vascular smooth muscle that shields the organism from risk factors associated with vascular diseases.
Sources of Funding:
This study was supported NIHLBI R0HL132574 (to SAG and JMM), NHLBI R01HL146442 and R01HL148151 (to ADA).
Footnotes
Conflicts of interest/Disclosures: None
References:
- 1.Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, Olin-Sandoval V, Gruning NM, Kruger A, Tauqeer Alam M, Keller MA, Breitenbach M, Brindle KM, Rabinowitz JD and Ralser M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. 2015;90:927–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puleston DJ, Villa M and Pearce EL. Ancillary Activity: Beyond Core Metabolism in Immune Cells. Cell Metab. 2017;26:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nkhoma ET, Poole C, Vannappagari V, Hall SA and Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. 2009;42:267–78. [DOI] [PubMed] [Google Scholar]
- 4.Vulliamy TJ, D’Urso M, Battistuzzi G, Estrada M, Foulkes NS, Martini G, Calabro V, Poggi V, Giordano R, Town M and et al. Diverse point mutations in the human glucose-6-phosphate dehydrogenase gene cause enzyme deficiency and mild or severe hemolytic anemia. Proc Natl Acad Sci U S A. 1988;85:5171–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tzounakas VL, Kriebardis AG, Georgatzakou HT, Foudoulaki-Paparizos LE, Dzieciatkowska M, Wither MJ, Nemkov T, Hansen KC, Papassideri IS, D’Alessandro A and Antonelou MH. Glucose 6-phosphate dehydrogenase deficient subjects may be better “storers” than donors of red blood cells. Free Radic Biol Med. 2016;96:152–65. [DOI] [PubMed] [Google Scholar]
- 6.Tiwari M. Glucose 6 phosphatase dehydrogenase (G6PD) and neurodegenerative disorders: Mapping diagnostic and therapeutic opportunities. Genes Dis. 2017;4:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murry CE, Gipaya CT, Bartosek T, Benditt EP and Schwartz SM. Monoclonality of smooth muscle cells in human atherosclerosis. Am J Pathol. 1997;151:697–705. [PMC free article] [PubMed] [Google Scholar]
- 8.Long WK, Wilson SW and Frenkel EP. Associations between red cell glucose-6-phosphate dehydrogenase variants and vascular diseases. Am J Hum Genet. 1967;19:35–53. [PMC free article] [PubMed] [Google Scholar]
- 9.Yang HC, Wu YH, Yen WC, Liu HY, Hwang TL, Stern A and Chiu DT. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dore MP, Davoli A, Longo N, Marras G and Pes GM. Glucose-6-phosphate dehydrogenase deficiency and risk of colorectal cancer in Northern Sardinia: A retrospective observational study. Medicine (Baltimore). 2016;95:e5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nobrega-Pereira S, Fernandez-Marcos PJ, Brioche T, Gomez-Cabrera MC, Salvador-Pascual A, Flores JM, Vina J and Serrano M. G6PD protects from oxidative damage and improves healthspan in mice. Nat Commun. 2016;7:10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cocco P, Todde P, Fornera S, Manca MB, Manca P and Sias AR. Mortality in a cohort of men expressing the glucose-6-phosphate dehydrogenase deficiency. Blood. 1998;91:706–9. [PubMed] [Google Scholar]
- 13.Pinna A, Contini EL, Carru C and Solinas G. Glucose-6-phosphate dehydrogenase deficiency and diabetes mellitus with severe retinal complications in a Sardinian population, Italy. Int J Med Sci. 2013;10:1907–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meloni L, Manca MR, Loddo I, Cioglia G, Cocco P, Schwartz A, Muntoni S and Muntoni S. Glucose-6-phosphate dehydrogenase deficiency protects against coronary heart disease. J Inherit Metab Dis. 2008;31:412–7. [DOI] [PubMed] [Google Scholar]
- 15.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Council on E, Prevention Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 16.Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life. 2012;64:362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miano JM, Zhu QM and Lowenstein CJ. A CRISPR Path to Engineering New Genetic Mouse Models for Cardiovascular Research. Arterioscler Thromb Vasc Biol. 2016;36:1058–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, Joly JS and Concordet JP. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao S, Ho D, Vatner DE and Vatner SF. Echocardiography in Mice. Curr Protoc Mouse Biol. 2011;1:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams DS and Levin M. Measuring resting membrane potential using the fluorescent voltage reporters DiBAC4(3) and CC2-DMPE. Cold Spring Harb Protoc. 2012;2012:459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI and Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105:9627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D’Alessandro A, Nemkov T, Yoshida T, Bordbar A, Palsson BO and Hansen KC. Citrate metabolism in red blood cells stored in additive solution-3. Transfusion. 2017;57:325–336. [DOI] [PubMed] [Google Scholar]
- 23.Nemkov T, Hansen KC and D’Alessandro A. A three-minute method for high-throughput quantitative metabolomics and quantitative tracing experiments of central carbon and nitrogen pathways. Rapid Commun Mass Spectrom. 2017;31:663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joshi SR, Dhagia V, Gairhe S, Edwards JG, McMurtry IF and Gupte SA. MicroRNA-140 is elevated and mitofusin-1 is downregulated in the right ventricle of the Sugen5416/hypoxia/normoxia model of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2016;311:H689–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chettimada S, Ata H, Rawat DK, Gulati S, Kahn AG, Edwards JG and Gupte SA. Contractile protein expression is upregulated by reactive oxygen species in aorta of Goto-Kakizaki rat. Am J Physiol Heart Circ Physiol. 2014;306:H214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chettimada S, Joshi SR, Dhagia V, Aiezza A, 2nd, Lincoln TM, Gupte R, Miano JM and Gupte SA. Vascular smooth muscle cell contractile protein expression is increased through protein kinase G-dependent and -independent pathways by glucose-6-phosphate dehydrogenase inhibition and deficiency. Am J Physiol Heart Circ Physiol. 2016;311:H904–H912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 28.Saklayen MG. The Global Epidemic of the Metabolic Syndrome. Curr Hypertens Rep. 2018;20:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kletzien RF, Harris PK and Foellmi LA. Glucose-6-phosphate dehydrogenase: a “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J. 1994;8:174–81. [DOI] [PubMed] [Google Scholar]
- 30.Thomas G, Chung M and Cohen CJ. A dihydropyridine (Bay k 8644) that enhances calcium currents in guinea pig and calf myocardial cells. A new type of positive inotropic agent. Circ Res. 1985;56:87–96. [DOI] [PubMed] [Google Scholar]
- 31.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M and Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–4. [DOI] [PubMed] [Google Scholar]
- 32.Hockerman GH, Peterson BZ, Sharp E, Tanada TN, Scheuer T and Catterall WA. Construction of a high-affinity receptor site for dihydropyridine agonists and antagonists by single amino acid substitutions in a non-L-type Ca2+ channel. Proc Natl Acad Sci U S A. 1997;94:14906–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bechem M and Hoffmann H. The molecular mode of action of the Ca agonist (−) BAY K 8644 on the cardiac Ca channel. Pflugers Arch. 1993;424:343–53. [DOI] [PubMed] [Google Scholar]
- 34.Fleischmann BK, Murray RK and Kotlikoff MI. Voltage window for sustained elevation of cytosolic calcium in smooth muscle cells. Proc Natl Acad Sci U S A. 1994;91:11914–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanguinetti MC and Kass RS. Voltage-dependent block of calcium channel current in the calf cardiac Purkinje fiber by dihydropyridine calcium channel antagonists. Circ Res. 1984;55:336–48. [DOI] [PubMed] [Google Scholar]
- 36.Wu J, Montaniel KR, Saleh MA, Xiao L, Chen W, Owens GK, Humphrey JD, Majesky MW, Paik DT, Hatzopoulos AK, Madhur MS and Harrison DG. Origin of Matrix-Producing Cells That Contribute to Aortic Fibrosis in Hypertension. Hypertension. 2016;67:461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McMaster WG, Kirabo A, Madhur MS and Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. 2015;116:1022–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Francis RO, Jhang JS, Pham HP, Hod EA, Zimring JC and Spitalnik SL. Glucose-6-phosphate dehydrogenase deficiency in transfusion medicine: the unknown risks. Vox Sang. 2013;105:271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hutton JJ. Genetic regulation of glucose 6-phosphate dehydrogenase activity in the inbred mouse. Biochem Genet. 1971;5:315–31. [DOI] [PubMed] [Google Scholar]
- 40.Pretsch W, Charles DJ and Merkle S. X-linked glucose-6-phosphate dehydrogenase deficiency in Mus musculus. Biochem Genet. 1988;26:89–103. [DOI] [PubMed] [Google Scholar]
- 41.Panchal SK, Poudyal H, Iyer A, Nazer R, Alam MA, Diwan V, Kauter K, Sernia C, Campbell F, Ward L, Gobe G, Fenning A and Brown L. High-carbohydrate, high-fat diet-induced metabolic syndrome and cardiovascular remodeling in rats. J Cardiovasc Pharmacol. 2011;57:611–24. [DOI] [PubMed] [Google Scholar]
- 42.Talukdar I, Szeszel-Fedorowicz W and Salati LM. Arachidonic acid inhibits the insulin induction of glucose-6-phosphate dehydrogenase via p38 MAP kinase. J Biol Chem. 2005;280:40660–7. [DOI] [PubMed] [Google Scholar]
- 43.Wagle A, Jivraj S, Garlock GL and Stapleton SR. Insulin regulation of glucose-6-phosphate dehydrogenase gene expression is rapamycin-sensitive and requires phosphatidylinositol 3-kinase. J Biol Chem. 1998;273:14968–74. [DOI] [PubMed] [Google Scholar]
- 44.Steer KA, Sochor M and McLean P. Renal hypertrophy in experimental diabetes. Changes in pentose phosphate pathway activity. Diabetes. 1985;34:485–90. [DOI] [PubMed] [Google Scholar]
- 45.Serpillon S, Floyd BC, Gupte RS, George S, Kozicky M, Neito V, Recchia F, Stanley W, Wolin MS and Gupte SA. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am J Physiol Heart Circ Physiol. 2009;297:H153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hicks S, Labinskyy N, Piteo B, Laurent D, Mathew JE, Gupte SA and Edwards JG. Type II diabetes increases mitochondrial DNA mutations in the left ventricle of the Goto-Kakizaki diabetic rat. Am J Physiol Heart Circ Physiol. 2013;304:H903–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carley AN and Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochimica et biophysica acta. 2005;1734:112–26. [DOI] [PubMed] [Google Scholar]
- 48.Ham M, Choe SS, Shin KC, Choi G, Kim JW, Noh JR, Kim YH, Ryu JW, Yoon KH, Lee CH and Kim JB. Glucose-6-Phosphate Dehydrogenase Deficiency Improves Insulin Resistance With Reduced Adipose Tissue Inflammation in Obesity. Diabetes. 2016;65:2624–38. [DOI] [PubMed] [Google Scholar]
- 49.Stapleton PA, James ME, Goodwill AG and Frisbee JC. Obesity and vascular dysfunction. Pathophysiology. 2008;15:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Erdei N, Toth A, Pasztor ET, Papp Z, Edes I, Koller A and Bagi Z. High-fat diet-induced reduction in nitric oxide-dependent arteriolar dilation in rats: role of xanthine oxidase-derived superoxide anion. Am J Physiol Heart Circ Physiol. 2006;291:H2107–15. [DOI] [PubMed] [Google Scholar]
- 51.Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS and Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest. 1998;101:731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ritchie RH, Drummond GR, Sobey CG, De Silva TM and Kemp-Harper BK. The opposing roles of NO and oxidative stress in cardiovascular disease. Pharmacol Res. 2017;116:57–69. [DOI] [PubMed] [Google Scholar]
- 53.Gamboa A, Shibao C, Diedrich A, Choi L, Pohar B, Jordan J, Paranjape S, Farley G and Biaggioni I. Contribution of endothelial nitric oxide to blood pressure in humans. Hypertension. 2007;49:170–7. [DOI] [PubMed] [Google Scholar]
- 54.Wilkinson IB, Franklin SS and Cockcroft JR. Nitric oxide and the regulation of large artery stiffness: from physiology to pharmacology. Hypertension. 2004;44:112–6. [DOI] [PubMed] [Google Scholar]
- 55.Fitch RM, Vergona R, Sullivan ME and Wang YX. Nitric oxide synthase inhibition increases aortic stiffness measured by pulse wave velocity in rats. Cardiovasc Res. 2001;51:351–8. [DOI] [PubMed] [Google Scholar]
- 56.Ishihara K, Hiraoka M and Ochi R. The tetravalent organic cation spermine causes the gating of the IRK1 channel expressed in murine fibroblast cells. J Physiol. 1996;491 ( Pt 2):367–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ficker E, Taglialatela M, Wible BA, Henley CM and Brown AM. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science. 1994;266:1068–72. [DOI] [PubMed] [Google Scholar]
- 58.Elinder F and Liin SI. Actions and Mechanisms of Polyunsaturated Fatty Acids on Voltage-Gated Ion Channels. Front Physiol. 2017;8:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lyle AN and Raaz U. Killing Me Unsoftly: Causes and Mechanisms of Arterial Stiffness. Arterioscler Thromb Vasc Biol. 2017;37:e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wamhoff BR, Bowles DK and Owens GK. Excitation-transcription coupling in arterial smooth muscle. Circ Res. 2006;98:868–78. [DOI] [PubMed] [Google Scholar]
- 61.Pes GM, Parodi G and Dore MP. Glucose-6-phosphate dehydrogenase deficiency and risk of cardiovascular disease: A propensity score-matched study. Atherosclerosis. 2019;282:148–153. [DOI] [PubMed] [Google Scholar]
- 62.Thomas JE, Kang S, Wyatt CJ, Kim FS, Mangelsdorff AD and Weigel FK. Glucose-6-Phosphate Dehydrogenase Deficiency is Associated with Cardiovascular Disease in U.S. Military Centers. Tex Heart Inst J. 2018;45:144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.