

Abstract
The tumor suppressor Merlin/NF2, a key activator of the Hippo pathway in growth control, is regulated by phosphorylation. However, it is uncertain whether additional post‐translational modifications regulate Merlin. Here, we show that ubiquitination is required to activate Merlin in the Hippo pathway. Ubiquitinated Merlin is mostly conjugated by one or two ubiquitin molecules. Such modification is promoted by serine 518 dephosphorylation in response to Ca2+ signaling or cell detachment. Merlin ubiquitination is mediated by the E3 ubiquitin ligase, NEDD4L, which requires a scaffold protein, AMOTL1, to approach Merlin. Several NF2‐patient‐derived Merlin mutations disrupt its binding to AMOTL1 and its regulation by the AMOTL1‐NEDD4L apparatus. Lysine (K) 396 is the major ubiquitin conjugation residue. Disruption of Merlin ubiquitination by the K396R mutation or NEDD4L depletion diminishes its binding to Lats1 and inhibits Lats1 activation. These effects are also accompanied by loss of Merlin's anti‐mitogenic and tumor suppressive properties. Thus, we propose that dephosphorylation and ubiquitination compose an intramolecular relay to activate Merlin functions in activating the Hippo pathway during growth control.
Keywords: AMOTL1, Hippo pathway, Merlin, NEDD4L, ubiquitination
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
The Hippo pathway tumor suppressor Merlin/NF2 is known to be regulated by phosphorylation. Here, the E3 ubiquitin ligase NEDD4L is shown to promote Hippo pathway activation via ubiquitination of Merlin.
Introduction
Merlin is a tumor suppressor encoded by the NF2 gene. Its inactivation causes the neurofibromatosis type 2 (NF2) familial cancer syndrome, an autosomal dominant genetic disorder characterized by the growth of multiple central and peripheral nervous system tumors. Merlin mutations have also been associated with sporadic tumors, such as schwannomas, meningiomas, malignant mesotheliomas, and thyroid carcinoma (Cooper & Giancotti, 2014; Petrilli & Fernandez‐Valle, 2016). In addition to its tumor suppressor functions, Merlin is important for maintaining neuron organization and integrity as well as regulating nerve regeneration after damage (Schulz et al, 2013, 2016; Mindos et al, 2017; Toledo et al, 2019). Recent studies have made significant progress toward understanding Merlin's molecular functions, but it remains unclear how Merlin is activated in response to growth suppressive cues or other signals.
Merlin's activation cycle can be regulated by phosphorylation of its serine 518 (Shaw et al, 2001; Kissil et al, 2002; Xiao et al, 2002; Surace et al, 2004; Jin et al, 2006). When phosphorylated, Merlin is inactive. The phosphorylation is regulated by p21‐activated kinase (PAK) (Kissil et al, 2002; Xiao et al, 2002), protein kinase A (PKA) (Alfthan et al, 2004), and MYPT1‐PP1δ phosphatase (Jin et al, 2006). Although it has been suggested that the phosphorylation status can affect its interaction with downstream effectors by regulating its conformation and subcellular localization (Cooper & Giancotti, 2014; Petrilli & Fernandez‐Valle, 2016), how dephosphorylation relays Merlin activation signals intramolecularly and regulates these properties remains elusive.
Previous reports of Merlin polyubiquitination (Tang et al, 2007; Huang & Chen, 2008; Verma et al, 2019) using recombinant Merlin found that polyubiquitination inhibits Merlin either through proteasome‐mediated degradation or by interrupting its interaction with a downstream effector, large tumor suppressor kinase 1 (Lats1). However, it remains uncertain whether endogenous Merlin is similarly regulated by polyubiquitination, since cycloheximide chase experiments indicated that endogenous Merlin is a relatively long‐lived protein (Li et al, 2010).
Merlin regulates growth control primarily through activation of the Hippo pathway. The pathway contains a core serine/threonine kinase cascade, which, in mammals, includes mammalian sterile 20‐like kinases 1 (MST1) and 2 (MST2) as well as their substrates, Lats1 and Lats2 (Lats1/2) (Yu et al, 2015). Once activated, Lats1/2 phosphorylate and inhibit two paralogous oncogenic transcriptional coactivators, Yes‐associated protein (YAP), and transcriptional co‐activator with PDZ‐binding motif (TAZ). In this study, we investigated ubiquitination of endogenous Merlin in the context of Hippo pathway regulation, and found Merlin ubiquitination to be common. Interestingly, modified Merlin is mostly conjugated by one or two ubiquitin molecules. Such ubiquitination is associated with Hippo pathway activation. It can be induced by Ca2+ signaling or cell detachment and promoted by dephosphorylation of serine 518. We identified NEDD4L as the E3 ubiquitin ligase responsible for Merlin ubiquitination and showed that this modification is important for Merlin to activate the Hippo pathway and control cell growth.
Results
Merlin ubiquitination is common and associated with Hippo pathway activation
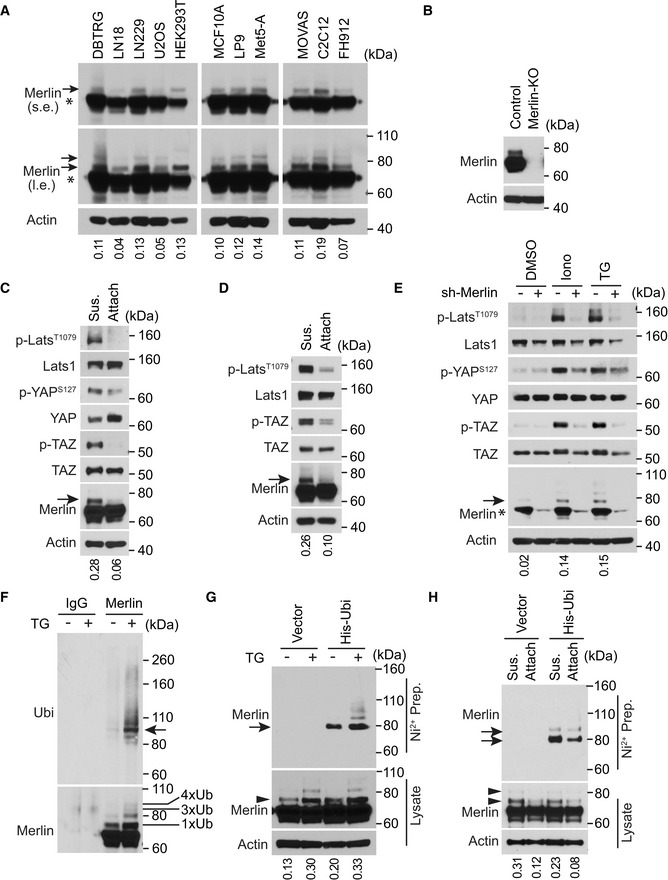
While studying Merlin regulation by Western blot, we noticed one or two protein species above the major Merlin band (Fig 1A, arrows and asterisk, respectively). This phenomenon did not appear to be cell type‐specific because it occurred in cell lines derived from various tissues, including DBTRG‐05MG, LN18 and LN229 human glioma cells, U2OS human osteosarcoma cells, HEK293T human embryonic kidney cells, MCF10A human mammary epithelial cells, LP9 and Met5‐A human mesothelial cells, MOVAS mouse vascular smooth muscle cells, C2C12 mouse myoblast cells, and FH‐912 mouse Schwann cells (Fig 1A). To examine whether these unidentified protein signals showing higher molecular weight were from Merlin, we used CRISPR to knock out Merlin in LN229 cells. Along with the major Merlin band, these higher molecular weight bands were also depleted (Fig 1B), suggesting that they are Merlin species. Cells examined in the above experiments were cultured in a steady state. In such conditions, the Hippo pathway effectors, such as phosphorylation of Lats1, YAP, and TAZ, did not appear to fully correlate with the higher molecular weight Merlin forms (Figs 1A and EV1A). To examine whether these Merlin species can be regulated, we employed two in vitro models involving Hippo pathway regulation. First, the Hippo pathway can be activated by cell detachment (Zhao et al, 2012). Consistent with this notion, we found that in both LN229 cells and Met5‐A cells, phosphorylation of Lats1, YAP, and TAZ was increased in detached cells compared to attached cells (Fig 1C and D). In these experiments, there was an increase in the amount of the higher molecular weight Merlin in detached cells (Fig 1C and D, arrows). In the second model, elevation of cytosolic Ca2+ through store‐operate calcium entry induced by thapsigargin (a pharmacologic inhibitor of the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase pump) (Takemura et al, 1989) or ionomycin (a Ca2+ ionophore) (Morgan & Jacob, 1994) markedly activated Lats1 (Fig 1E) (Liu et al, 2019). In this condition, the higher molecular weight Merlin was increased in response to ionomycin or thapsigargin treatment (Fig 1E, arrow). A similar effect was also observed in FH‐912 cells treated with thapsigargin (Fig EV1B), indicating that the effect was not limited to LN229 cells. To examine the role of Merlin in Ca2+‐induced Lats1 activation, we used an shRNA to silence Merlin expression in LN229 cells. Merlin depletion suppressed phosphorylation of Lats1 as well as its substrate YAP and TAZ under treatment with thapsigargin or ionomycin (Fig 1E). In addition, silencing Merlin expression in Met5‐A cells also inhibited phosphorylation of Lats1, YAP, and TAZ in detached cells (Fig EV1C). These results indicated that Merlin is important for Ca2+‐ or detachment‐induced Lats1 activation. They also demonstrated that higher molecular weight Merlin is associated with Hippo pathway activation in response to both cell detachment and increased Ca2+ signaling.
Figure 1. Merlin ubiquitination commonly exists and associates with activation of the Hippo pathway.
-
ATotal lysates from the indicated cell lines cultured in regular conditions at a steady state were subjected to Western blotting. l.e., long exposure; s.e., short exposure. The major Merlin band is indicated by an asterisk. The ratio of mono‐ubiquitinated to native Merlin in each lane of the short exposure blot was quantified by ImageJ and is shown under the blot. The same lysates were also used in Fig EV1A. Asterisks indicate native Merlin, arrows show ubiquitinated Merlin.
-
BTotal lysates from LN229 cells in which Merlin was or was not knocked out (KO) were subjected to Western blotting.
-
C, DLN229 (C) or Met5‐A (D) cells were detached (denoted by sus., referring to suspension) by trypsinization (see Materials and Methods) and reseeded (attach) for 2 (LN229) or 4 (Met5‐A) h. Total lysates from these cells were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot. Ubiquitinated Merlin is marked with arrows.
-
ELN229 cells transduced with a Merlin or scrambled shRNA were treated with DMSO, ionomycin (Iono) or thapsigargin (TG). Total lysates of these cells were subjected to Western blotting. The major Merlin band is indicated by an asterisk. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot. Ubiquitinated Merlin is marked with an arrow.
-
ELN229 cells treated with DMSO or thapsigargin (TG) were lysed and subjected to immunoprecipitation with a Merlin antibody or IgG. The immunoprecipitated products were subjected to Western blotting. The number of conjugated ubiquitin (Ub) moieties was marked. Tetra‐ubiquitinated Merlin is indicated by an arrow.
-
FLN229 cells stably transduced with 6×‐Histidine‐tagged ubiquitin (His‐Ubi) were treated with DMSO or thapsigargin (TG). These cells were lysed and subjected to nickel‐charged affinity purification followed by Western blotting for endogenous Merlin. Both arrowhead and arrow point to the mono‐ubiquitinated Merlin. The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
-
GLN229 cells stably transduced with 6×‐Histidine‐tagged ubiquitin (His‐Ubi) were detached or reseeded as in (C). These cells were lysed and subjected to nickel‐charged affinity purification followed by Western blotting for endogenous Merlin. Arrowheads and arrows point to the mono‐ or di‐ubiquitinated Merlin (see a longer exposed blotting results in Fig EV1D). The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
Source data are available online for this figure.
Figure EV1. Merlin ubiquitination associates with activation of the Hippo pathway.
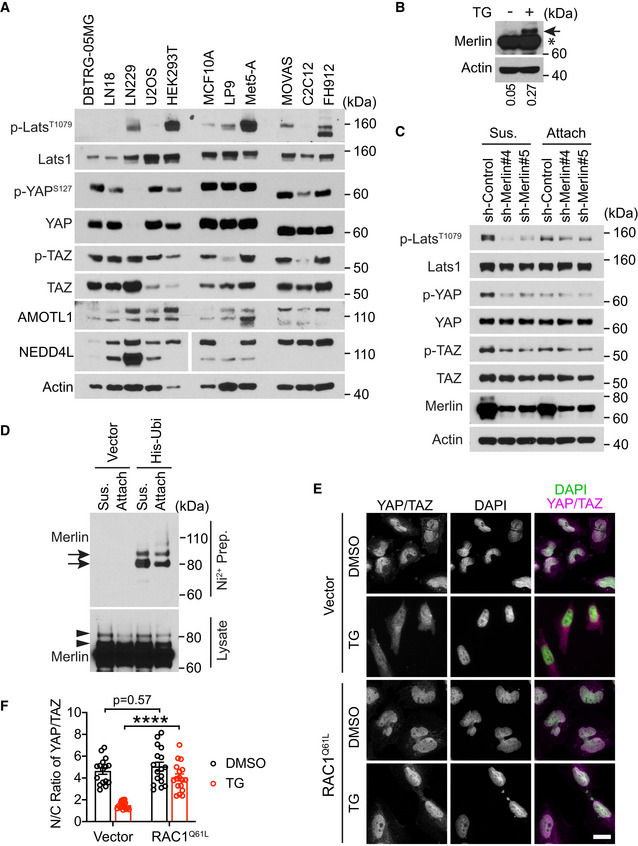
- Total lysates from indicated cell lines cultured in regular conditions at a steady state were subjected to Western blotting. The same lysates were also used in Fig 1A.
- FH‐912 cells treated with DMSO or thapsigargin and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot. Native Merlin (asterisk); ubiquitinated Merlin (arrow).
- Met5‐A cells stably transduced with indicated shRNAs against Merlin or a scrambled shRNA were detached (Sus., suspension) by trypsinization using the procedure described in Materials and Methods and reseeded (Attach, attached) for 4 h. Total lysates from these cells were subjected to Western blotting.
- Longer exposure of the blot in Fig 1H. Ubiquitinated Merlin (arrows and arrowheads).
- LN229 cells transduced with vector or EGFP‐RAC1(Q61L) were treated with DMSO or thapsigargin (TG) and subjected to immunofluorescent staining. Scale bar = 20 μm.
- The ratio of the relative amount of YAP/TAZ in the nucleus (N) compared to the cytoplasm (C) from panel (E) was quantified. Mean ± s.e.m, two‐way ANOVA. ****P < 0.0001. Each data point represents one cell. N Vector = 16 cells in each condition, N RAC1(Q61L) = 17 cells in each condition. All cells were from multiple random images collected from one experiment. Two independent experiments were performed and gave similar results.
Source data are available online for this figure.
Based on their higher molecular weights, we speculated that these Merlin species might be mono‐ or di‐ubiquitinated. To examine this notion, we used two approaches to detect Merlin ubiquitination in LN229 cells. First, endogenous Merlin was immunoprecipitated with an anti‐Merlin antibody. The immunoprecipitated products were then probed with an anti‐ubiquitin antibody. We found that there were more ubiquitinated species in the immunoprecipitated products from cells treated with thapsigargin (Fig 1F). Notably, the band showing the strongest ubiquitination signal appeared to correspond to Merlin conjugated by four, but not one, ubiquitin moieties (Fig 1F, arrow), which may reflect increased binding of the anti‐ubiquitin antibody when more ubiquitin moieties are conjugated to Merlin. To faithfully reflect the stoichiometry of Merlin ubiquitination, a second approach was employed. In LN229 cells, we stably expressed 6×‐histidine‐tagged ubiquitin and conducted nickel‐charged affinity purification under guanidine/urea‐denaturing conditions. In the purified ubiquitinated proteins, endogenous Merlin detected by the anti‐Merlin antibody was more abundant in cells treated with thapsigargin than with in DMSO control (Fig 1G, upper panel). The difference is clearer for di‐ or tri‐ubiquitinated Merlin than for mono‐ubiquitinated Merlin (Fig 1G, arrow). This approach revealed that Merlin is mostly mono‐ubiquitinated when undergoing ubiquitination. This observation is consistent with that from probing total lysates (Fig 1G, arrowhead). Using this histidine‐tagged ubiquitin‐mediated co‐purification approach, we examined endogenous Merlin in the detachment condition. As seen in probing of total lysates, enhanced mono‐ and di‐ubiquitination was observed by this ubiquitination‐detecting assay when cells were detached (Figs 1H and EV1D, arrowheads and arrows, respectively). Overall, the above analyses demonstrated that Merlin ubiquitination is associated with its active state in regulating the Hippo pathway by Ca2+ signaling and cell detachment.
Dephosphorylation of serine 518 promotes Merlin ubiquitination
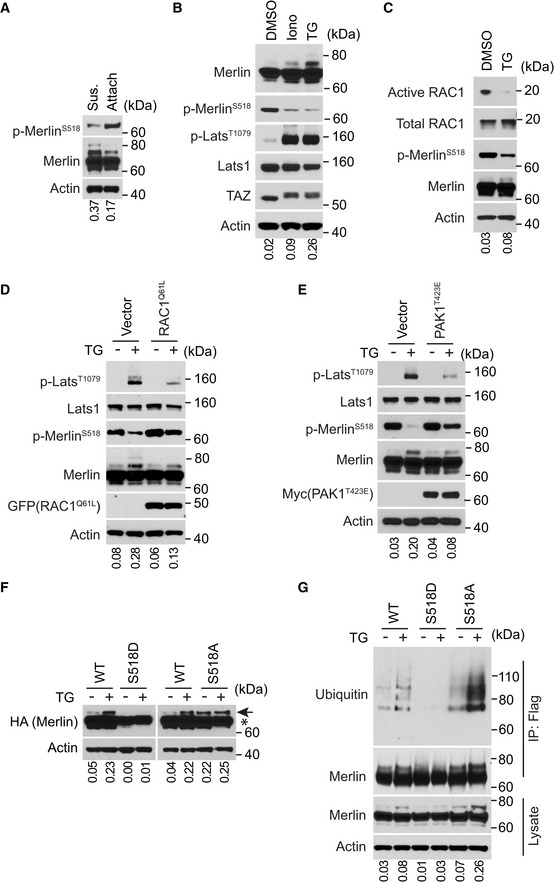
Given the importance of serine 518 phosphorylation in regulating Merlin activities, we examined whether there is a correlation between serine 518 phosphorylation and Merlin ubiquitination. In contrast to the increase of ubiquitination, detachment of LN229 cells reduced serine 518 phosphorylation (Fig 2A). Treatment by thapsigargin or ionomycin also reduced serine 518 phosphorylation in LN229 cells (Fig 2B). These regulations were accompanied with increased Merlin ubiquitination and Lats1 phosphorylation. Because signaling from the small GTPase RAC1 is able to regulate Merlin phosphorylation (Shaw et al, 2001) and because cell detachment can inactivate RAC1 (del Pozo et al, 2000), we examined the activation status of RAC1 under thapsigargin treatment. During this treatment, it appeared that the amount of active RAC1 decreased (Fig 2C). To examine whether inactivation of RAC1 causes Merlin dephosphorylation, we utilized a constitutively active RAC1 mutant (RAC1Q61L) (Kraynov et al, 2000). Stably expressing RAC1Q61L in LN229 cells can partially suppress Merlin dephosphorylation in response to thapsigargin (Fig 2D). Under this condition, thapsigargin‐induced Merlin ubiquitination and Lats1 phosphorylation were also partially suppressed (Fig 2D). In addition, thapsigargin‐induced YAP/TAZ cytoplasmic translocation was also inhibited (Fig EV2E and F). Promoting Merlin phosphorylation by stably expressing an active PAK mutant T423E in LN229 cells showed a similar inhibitory effect on thapsigargin‐induced Merlin ubiquitination and Lats1 phosphorylation (Fig 2E). These results suggested that Merlin dephosphorylation promotes its ubiquitination and activation. To further examine this, we utilized Merlin S518D phosphomimetic and S518A phospho‐blocking mutants. As indicated by the higher molecular weight Merlin, thapsigargin‐induced Merlin ubiquitination was inhibited by S518D mutation, whereas S518A mutation enhanced ubiquitination even in the absence of thapsigargin (Fig 2F). This observation was further confirmed by the aforementioned ubiquitination‐detection assay (Fig 2G). Therefore, the above results supported that dephosphorylation of serine 518 promotes Merlin ubiquitination.
Figure 2. Dephosphorylation of serine 518 promotes Merlin ubiquitination.
-
ALN229 cells were detached (denoted by sus., referring to suspension) by trypsinization and reseeded (denoted by attach) for 2 h. Total lysates from these cells were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
BLN229 cells treated with DMSO, ionomycin (Iono) or thapsigargin (TG) were lysed and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
CLN229 cells treated with DMSO or thapsigargin (TG) were subjected to active RAC1 pull‐down assay followed by Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
D, ELN229 cells transduced with vector or EGFP‐RAC1(Q61L) (D) or Myc‐PAK1(T423E) (E) were treated with DMSO or thapsigargin (TG) and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
FLN229 cells stably transduced with wild‐type Merlin or the indicated mutants were treated with DMSO or thapsigargin (TG) and analyzed by Western blot. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot. Native Merlin (asterisk); ubiquitinated Merlin (arrow).
-
FMerlin‐depleted LN229 cells stably transduced with Flag‐tagged wild‐type Merlin or the indicated mutants were treated with DMSO or thapsigargin (TG). The cells were lysed and subjected to immunoprecipitation with a Flag antibody. The lysate and immunoprecipitated products were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
Source data are available online for this figure.
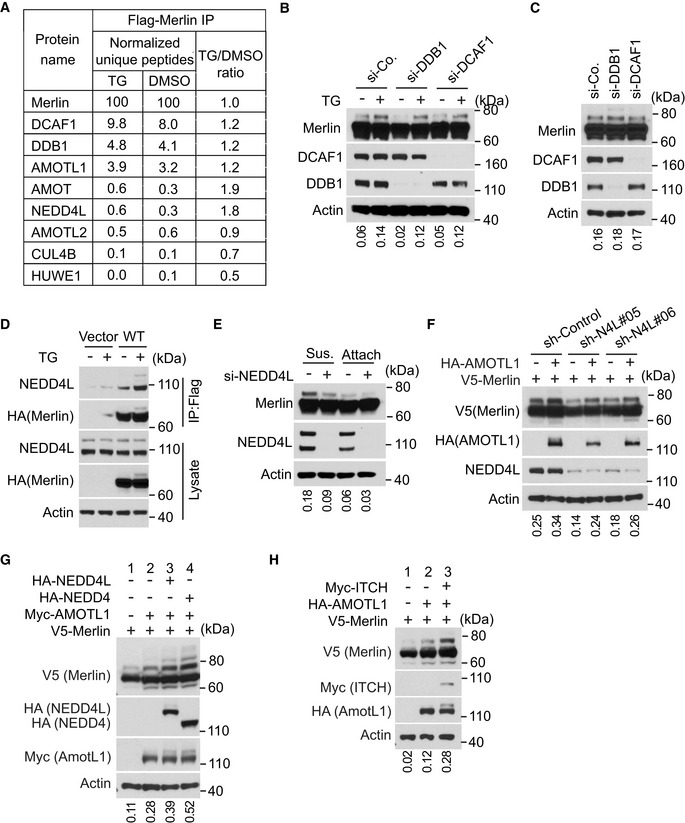
Figure EV2. NEDD4L is required for Merlin ubiquitination.
-
ANumbers of unique peptides of each identified protein shown in Fig 3A were normalized to those of Merlin from the indicated treatment condition. The ratio of these normalized peptides from TG‐treated cells to those from DMSO‐treated cells is shown.
-
BLN229 cells transfected with indicated siRNAs were treated with DMSO or thapsigargin (TG) and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
CLN229 cells transfected with indicated siRNAs were detached using the procedure described in Materials and Methods and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
DMerlin‐depleted LN229 cells stably transduced with Flag‐tagged wild‐type Merlin were treated with DMSO or thapsigargin (TG). The cells were lysed and subjected to immunoprecipitation with a Flag antibody. The lysate and immunoprecipitated products were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
-
EMet5‐A cells transfected with a pool of four siRNAs targeting NEDD4L (+) or a scrambled siRNA (−) were detached (denoted by sus., referring to suspension) by using the procedure described in Materials and Methods and reseeded (attached) for 4 h. Total lysates from these cells were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
FHEK293T cells stably transduced with indicated shRNAs targeting NEDD4L or a scrambled shRNA were transfected with indicated genes. The cells were then lysed and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
G, HHEK293T cells were transfected with indicated genes and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
Source data are available online for this figure.
NEDD4L is the E3 ubiquitin ligase of Merlin
To identify the E3 ubiquitin ligase responsible for Merlin ubiquitination, we analyzed proteins co‐immunopurified with Merlin using mass spectrometry. DDB1‐ and Cul4‐associated factor 1 (DCAF1, also named VprBP) and DNA damage‐binding protein 1 (DDB1) were two of the most abundant proteins (Figs 3A and EV2A). DCAF1 and DDB1 are two components of the Cullin‐RING E3 ubiquitin ligase complex CRL4DCAF1, which uses Cullin 4 as the scaffold protein (Lee & Zhou, 2007). In the co‐immunoprecipitated products, we also found Cullin4B (Cul4B; Fig 3A). Previous studies showed that Merlin interacts with CRL4DCAF1 through binding to DCAF1 (Huang & Chen, 2008; Li et al, 2010). This interaction leads to inhibition of the E3 ligase (Li et al, 2010, 2014). On the other hand, CRL4DCAF1 may promote Merlin degradation by mediating its polyubiquitination following serum stimulation (Huang & Chen, 2008). We found that depletion of DCAF1 or DDB1 did not inhibit thapsigargin‐ or detachment‐induced Merlin ubiquitination (Fig EV2B and C). Therefore, CRL4DCAF1 is unlikely to be responsible for the ubiquitination. In addition to the CRL4DCAF1 complex, we found two other E3 ubiquitin ligases in the co‐immunopurified products. These are neural precursor cell‐expressed developmentally downregulated protein 4 like (NEDD4L) and HECT, UBA, and WWE domain containing E3 ubiquitin protein ligase 1 (HUWE1; Fig 3A). There was more NEDD4L in the co‐immunopurified products from cells treated with thapsigargin than in the products from DMSO‐treated cells (Figs 3A and EV2A). In addition, because NEDD4L was much more abundant than HUWE1 in the co‐immunopurified products, we focused on testing NEDD4L. Western blotting confirmed the mass spectrometry results, suggesting that the interaction between Merlin and NEDD4L is enhanced by thapsigargin treatment (Fig EV2D). In LN229 cells, knockdown of NEDD4L with a pool of four different siRNAs or two different shRNAs reduced thapsigargin‐induced Merlin ubiquitination (Fig 3B and C). Merlin ubiquitination under the detachment condition was also reduced when NEDD4L expression was silenced by two different shRNAs (Fig 3D). In Met5‐A cells, depletion of NEDD4L by a pool of siRNAs also reduced Merlin ubiquitination in the detachment condition (Fig EV2E). In these experiments, we used the higher molecular weight Merlin as surrogates for ubiquitinated Merlin. To confirm these observations, we conducted histidine‐tagged ubiquitin‐mediated co‐purification as above under denaturing conditions. This assay also showed that thapsigargin‐induced Merlin ubiquitination was reduced when NEDD4L was knocked down by the shRNAs (Fig 3E). The above results suggested that NEDD4L is responsible for Merlin ubiquitination. To further test this, we used recombinant NEDD4L, Merlin, and angiomotin like 1 (AMOTL1) to construct an enzyme‐substrate regulatory system in HEK293T cells. The reason for including AMOTL1 is explained in the following section. In this system, we found that overexpression of NEDD4L markedly increased Merlin ubiquitination (Fig 3F, comparing lanes 3 and 2), which was indicated by the higher molecular weight Merlin. To confirm this observation, we purified the recombinant Merlin by immunoprecipitation and probed the products with an ubiquitin antibody. This approach confirmed that Merlin ubiquitination can be induced by NEDD4L (Fig 3G, comparing lanes 3 and 2). To examine whether the enzymatic activity of NEDD4L is required, we employed an E3 ligase dead NEDD4L mutation (C942A) (Chung et al, 2008). Compared to wild‐type NEDD4L, the C942A mutant was unable to ubiquitinate Merlin (Fig 3F and G, comparing lanes 3 and 4 in each panel). Notably, AMOTL1 expression on its own slightly increased Merlin's ubiquitination, which is presumably because it recruits endogenous NEDD4L (Fig 3F and G, comparing lanes 2 and 1 in each panel). We tested this by knocking down NEDD4L. Merlin ubiquitination was reduced upon NEDD4L knockdown in cells with and without recombinant AMOTL1. AMOTL1 expression can still slightly increase the ubiquitination in NEDD4L‐silenced cells (Fig EV2F). This effect may be due to incomplete NEDD4L depletion or other E3 ligases recruited by overexpressed AMOTL1. Previous studies found that NEDD4 and Itchy E3 ubiquitin protein ligase (ITCH) can also bind to AMOTL1 (Skouloudaki & Walz, 2012). We observed that upon co‐expressing with AMOTL1 and Merlin, both of these ligases induce Merlin ubiquitination (Fig EV2G and H). The observation suggested that NEDD4 and ITCH may have similar functions to NEDD4L in this condition. To further establish a physiological link between Merlin and NEDD4L, we examined whether endogenous NEDD4L interacts with Merlin. Co‐immunoprecipitation in LN229 cells showed that each protein can be mutually co‐precipitated by the other, indicating that NEDD4L and Merlin interact with each other under physiological conditions (Fig 3H). Overall, these results supported that NEDD4L is the E3 ubiquitin ligase of Merlin in Ca2+‐ or cell detachment‐induced ubiquitination.
Figure 3. NEDD4L is the E3 ubiquitin ligase of Merlin.
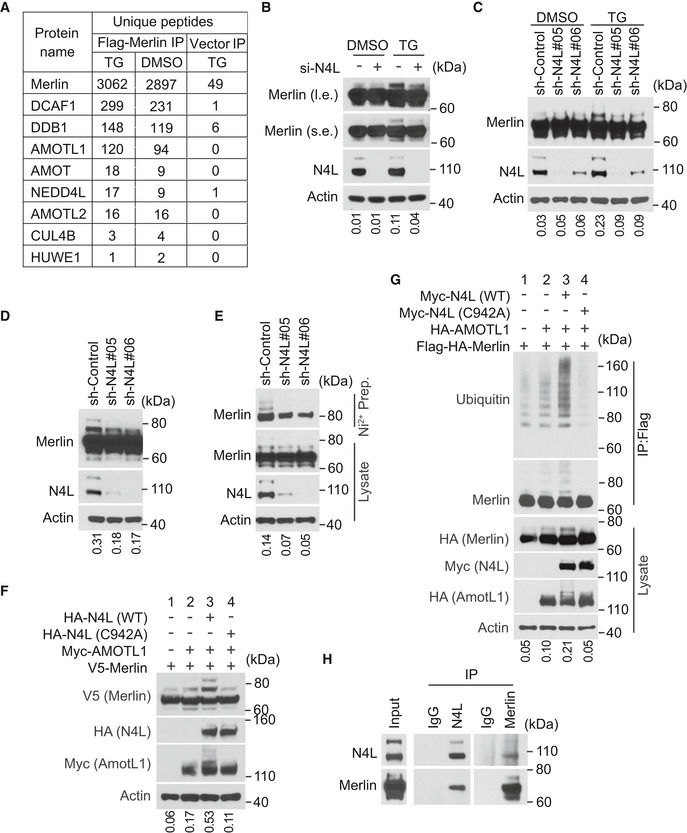
- LN229 cells stably transduced with Flag‐tagged Merlin or empty vector were treated with DMSO or thapsigargin (TG) and subjected to Flag immunoprecipitation followed by mass spectrometry. Numbers of unique peptides of each protein in the immunoprecipitated products are shown.
- LN229 cells transduced with a pool of four siRNAs against NEDD4L (+) or a scrambled siRNA (−) were treated with DMSO or thapsigargin (TG) and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane of the short exposure blot was quantified by ImageJ and is shown under the blot.
- LN229 cells stably transduced with indicated shRNAs targeting NEDD4L or a scrambled shRNA were treated with DMSO or thapsigargin (TG) and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
- LN229 cells stably transduced with indicated shRNAs targeting NEDD4L or a scrambled shRNA were detached by trypsinization (see Materials and Methods) and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
- LN229 cells stably transduced with 6×‐Histidine‐tagged Ubiquitin (His‐Ubi) were then stably transduced with indicated shRNAs targeting NEDD4L or a scrambled shRNA. These cells were detached as in (D), lysed and subjected to nickel‐charged affinity purification followed by Western blotting for endogenous Merlin. The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
- HEK293T cells were transfected with indicated genes and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
- HEK293T cells were transfected with indicated genes. The cells were lysed and subjected to immunoprecipitation with a Flag antibody. The immunoprecipitated products were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
- LN229 cells were lysed and subjected to immunoprecipitation with a Merlin antibody, a NEDD4L antibody, or IgG. The immunoprecipitated products were subjected to Western blotting.
Source data are available online for this figure.
AMOTL1 promotes Merlin ubiquitination by mediating the interaction between NEDD4L and Merlin
In the products co‐immunopurified with Merlin, we also found proteins of the angiomotin (AMOT) family, among which angiomotin like 1 (AMOTL1) was most abundant (Fig 3A). Interactions between Merlin and AMOT family proteins have been reported previously (Yi et al, 2011; Couderc et al, 2016). Such interactions promote release of a small GTPase, Rich1, from the inhibition by angiomotin (Yi et al, 2011) or induce AMOTL1 degradation (Couderc et al, 2016). We examined whether AMOTL1 is involved in regulating Merlin ubiquitination. In LN229 cells, knockdown of AMOTL1 with a pool of four different siRNAs markedly reduced thapsigargin‐induced Merlin ubiquitination (Fig 4A). In addition, depletion of AMOTL1 with two different shRNAs also reduced cell detachment‐induced Merlin ubiquitination (Fig 4B). Changes in ubiquitination were confirmed by the aforementioned histidine‐tagged ubiquitin‐mediated co‐purification assay conducted under denaturing conditions (Fig 4B). The above results indicated that AMOTL1 is important for Merlin ubiquitination. It was previously reported that NEDD4L is able to bind AMOTL1 (Skouloudaki & Walz, 2012). Considering the relative amounts of AMOTL1 and NEDD4L co‐immunopurified with Merlin (Fig 3A), we hypothesized that the interaction between NEDD4L and Merlin might be mediated by AMOTL1 as illustrated in Fig 4C. Consistent with this hypothesis, co‐immunoprecipitation experiments demonstrated that the NEDD4L‐Merlin interaction was reduced when AMOTL1 expression was silenced by two different shRNAs (Figs 4D and EV3A). To further examine whether AMOTL1 mediates the interaction between NEDD4L and Merlin, we reconstituted these proteins in HEK293T cells. Without AMOTL1, a modest amount of Merlin was co‐precipitated with NEDD4L (Fig EV3B, lane 3). In contrast, AMOTL1 expression markedly increased the amount of co‐precipitated Merlin (Fig EV3B, lane 6). This result indicated that AMOTL1 promotes the interaction between NEDD4L and Merlin. AMOTL1 contains two PPXY motifs and one LPXY motif, while NEDD4L contains four WW domains (Skouloudaki & Walz, 2012). It was suggested that the two PPXY motifs in AMOTL1 are important for binding to the WW domains in NEDD4L (Skouloudaki & Walz, 2012). We constructed a mutation (2PY*) disrupting these two PPXY motifs. Unexpectedly, AMOTL1 containing this mutation did not show a reduced ability to interact with NEDD4L (Fig 4E, comparing lanes 2 and 4). We then constructed a mutation (2PY* + LY*) disrupting the LPXY motif in addition to the two PPXY motifs. AMOTL1 with this 2PY* + LY* mutation failed to bind NEDD4L (Fig 4E, lane 3). This loss of interaction is unlikely to be caused by a non‐specific disruption of AMOTL1 structure by this mutation, because the mutant can still interact with Merlin similarly to wild‐type AMOTL1 (Fig 4E, comparing lanes 2 and 3). In this experiment, we found that AMOTL1 markedly enhances Merlin ubiquitination, as indicated by increased amounts of the higher molecular weight Merlin in total lysates (Fig 4E, comparing lanes 1 and 2). In contrast, the 2PY* + LY* AMOTL1 mutant has no such ability (Fig 4E, comparing lanes 2 and 3), suggesting that interaction between AMOTL1 and NEDD4L is important for promoting Merlin ubiquitination. To examine whether binding between Merlin and AMOTL1 is essential for such regulation, we performed mutagenesis analysis. Since Merlin and AMOTL1 binding has not been characterized molecularly, we looked at the studies of Merlin's binding to AMOT. It was shown that an NF2 patient‐derived Merlin mutation, Δ513–521, disrupts the binding between Merlin and the AMOT coiled‐coil domain (Li et al, 2015). In addition, studies suggested that several other patient‐derived Merlin mutations, such as R346S, E463K, M514V, L517P, and L535P, may disrupt Merlin's binding to AMOT (Yi et al, 2011; Li et al, 2014). We tested these Merlin mutants by co‐immunoprecipitation. Δ513–521, L517P, and L535P markedly disrupted Merlin's binding to AMOTL1. While M514V partially reduced their interaction, R346S and E463K appeared to have no impact (Fig EV3C). We then examined ubiquitination of Merlin Δ513–521, L517P, and L535P mutants. L535P and Δ513–521 markedly inhibited thapsigargin‐induced Merlin ubiquitination, while L517P partially reduced such ubiquitination (Fig 4F). We used L535P to further examine Merlin ubiquitination induced by the AMOTL1‐NEDD4L apparatus in the above reconstituted system. Although ubiquitination of wild‐type Merlin can be robustly induced by AMOTL1 and NEDD4L, this effect was suppressed by the Merlin L535P mutation (Fig 4G). Because AMOT and AMOTL2 were also seen in the precipitate co‐purified with Merlin (Fig 3A), we tested whether they have a similar ability to mediate Merlin ubiquitination. Overexpression of each protein with NEDD4L was able to induce Merlin ubiquitination similarly to AMOTL1 (Fig EV3D), suggesting that the AMOT family proteins may have similar properties in this setting. Of note, we saw that overexpression of NEDD4L also induced ubiquitination and likely degradation of the AMOT family proteins, especially AMOTL1 and AMOTL2 (Fig EV3E). This observation is consistent with previous reports (Skouloudaki & Walz, 2012; Wang et al, 2012). Overall, these results supported that AMOTL1 is responsible for recruiting Merlin to NEDD4L for ubiquitination, and other AMOT family members may also play a similar role (Fig 4C).
Figure 4. AMOTL1 promotes Merlin ubiquitination by mediating the interaction between NEDD4L and Merlin.
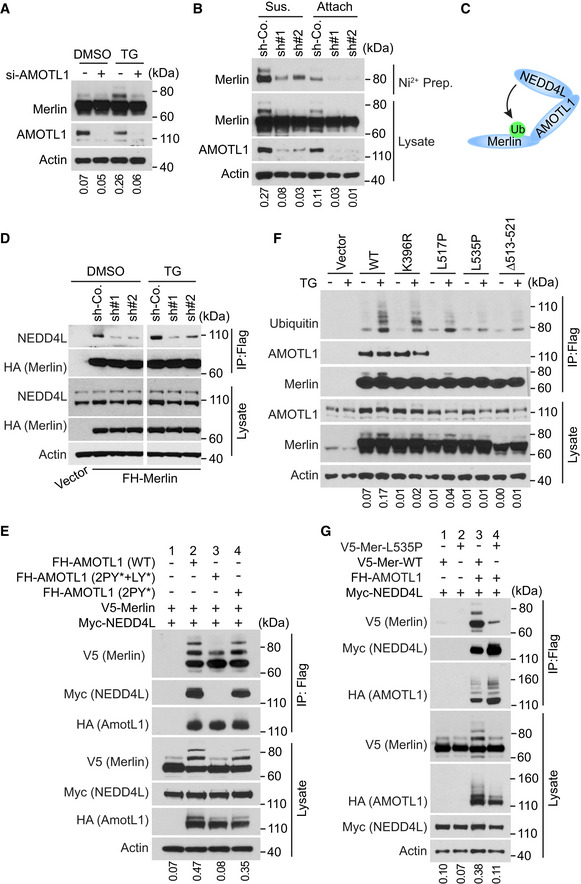
- LN229 cells transduced with a pool of four siRNAs against AMOTL1 (+) or a scrambled siRNA (−) were treated with DMSO or thapsigargin (TG) and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
- LN229 cells stably transduced with 6×‐Histidine‐tagged Ubiquitin (His‐Ubi) were then stably transduced with indicated shRNAs against AMOTL1 or a scrambled shRNA control (sh‐Co.). These cells were detached, lysed, and subjected to nickel‐charged affinity purification followed by Western blotting for endogenous Merlin. The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
- A diagram illustrating that AMOTL1 mediates the NEDD4L‐Merlin interaction.
- Merlin‐depleted LN229 cells stably expressing Flag‐HA-tagged Merlin were transduced with scrambled shRNA (sh‐Co.) or two distinct shRNAs targeting AMOTL1, treated with DMSO or thapsigargin (TG) and subjected to Flag immunoprecipitation followed by Western blotting.
- HEK293T cells were transfected with the indicated constructs. The cells were lysed and subjected to immunoprecipitation with a Flag antibody. The lysate and immunoprecipitated products were subjected to Western blotting. LY, LPTY motif; PY, PPEY motif. The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
- Merlin‐depleted LN229 cells stably transduced with Flag‐tagged wild‐type Merlin or the indicated mutants were treated with DMSO or thapsigargin (TG). The cells were lysed and subjected to immunoprecipitation with a Flag antibody. The lysate and immunoprecipitated products were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
- HEK293T cells were transfected with the indicated constructs. The cells were lysed and subjected to immunoprecipitation with a Flag antibody. The lysate and immunoprecipitated products were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
Source data are available online for this figure.
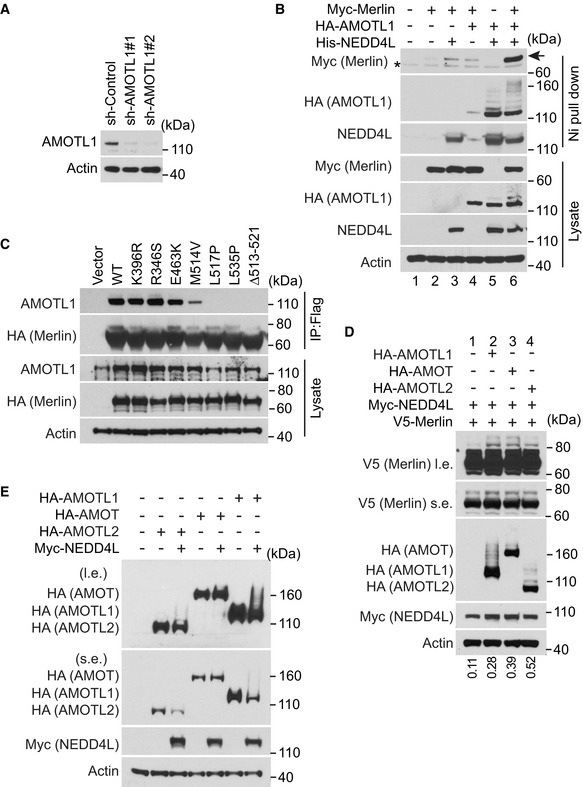
Figure EV3. AMOTL1 promotes Merlin ubiquitination.
-
AMerlin‐depleted LN229 cells stably expressing Flag‐HA-tagged Merlin used in Fig 4D were stably transduced with indicated shRNAs targeting AMOTL1 or a scrambled shRNA and subjected to Western blotting.
-
BHEK293T cells were transfect with indicated genes and subjected to nickel pull‐down in a native condition followed by Western blotting. Myc‐Merlin (arrow); non‐specific bands (asterisk).
-
CMerlin‐depleted LN229 cells stably transduced with Flag‐tagged wild‐type Merlin or the indicated mutants were subjected to immunoprecipitation with a Flag antibody. The lysate and immunoprecipitated products were subjected to Western blotting.
-
D, EHEK293T cells were transfected with indicated genes and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot in (D).
Source data are available online for this figure.
Lysine 396 is the major ubiquitin conjugation residue on Merlin
To further investigate Merlin ubiquitination, we used mass spectrometry to map amino acid residues conjugated by ubiquitin. Since the S518D mutation reduces Merlin ubiquitination (Fig 2F and G), we used it as a negative control. Wild‐type Merlin or the S518D mutant were stably expressed in LN229 cells. After treatment with thapsigargin, these Merlin forms were immunoprecipitated and resolved by SDS–PAGE (Fig 5A). Mass spectrometry identified fewer peptides from ubiquitin in the S518D mutant‐immunoprecipitated products compared to wild‐type Merlin (Fig 5B). This result is consistent with our previous observation in Western blotting (Fig 2F and G). The analysis further revealed that five lysine (K) residues on wild‐type Merlin, but not on the S518D mutant, could be conjugated by ubiquitin (Fig 5C–E). We compared the relative modification frequency among these lysine residues by calculating the ubiquitinated peptides and found that K396 (0.46%) and K159 (0.1%) were more frequently conjugated by ubiquitin than the others (Fig 5C). This suggested that K396 and K159 might be the major ubiquitin conjugation sites. To test this, we constructed individual and combinational lysine to arginine (K–R) mutations on these two residues and examined the effect on thapsigargin‐induced Merlin ubiquitination. Although the K159R mutation partially reduced Merlin ubiquitination, the K396R mutation more strongly abolished Merlin ubiquitination (Fig 5F, arrows). The 159/396 KR double mutation did not further reduce Merlin ubiquitination. These results suggested that K396 is the major ubiquitination residue on Merlin in response to thapsigargin treatment. We further examined whether K396 is also responsible for ubiquitination in the detachment condition. Consistently, the K396R Merlin mutant showed markedly reduced ubiquitination when cells are detached. This observation was confirmed by probing purified Merlin with a ubiquitin antibody (Fig 5G). Since S518 phosphorylation reduces Merlin ubiquitination, we examined whether reduced ubiquitination by the K396R mutation is due to effects on S518 phosphorylation. The results showed that the K396R mutation did not change S518 phosphorylation in response to either thapsigargin or detachment (Fig 5H and I). Therefore, the reduced ubiquitination on the K396R mutant is unlikely due to a non‐specific disruption of Merlin regulation, such as S518 phosphorylation.
Figure 5. Lysine 396 is the Ubiquitin conjugation residue on Merlin.
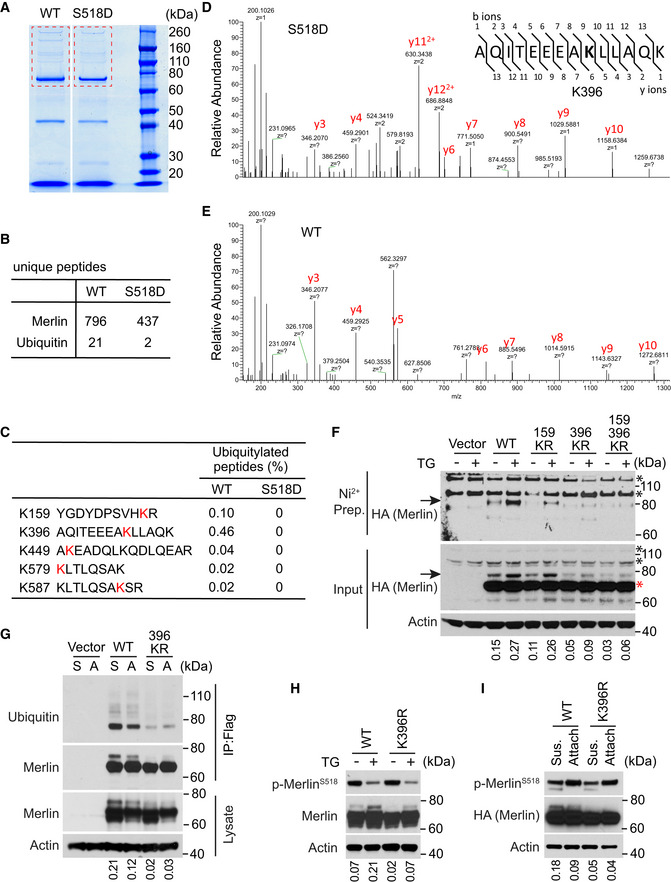
-
ALN229 cells stably transduced with Flag‐tagged wild‐type Merlin or the S518D mutant were treated with thapsigargin and subjected to immunoprecipitation with a Flag antibody and followed by SDS–PAGE. Outlined gel pieces were analyzed by mass spectrometry.
-
BNumbers of unique peptides of Merlin or Ubiquitin from each outlined gel pieces in (A) are shown.
-
CThe percentage of ubiquitinated forms for each peptide from each immunoprecipitated product is shown.
-
D, EMass spectrometry spectrums of the indicated peptides from each immunoprecipitated product are shown.
-
FMerlin‐depleted LN229 cells expressing His‐Ubiquitin were stably transduced with the indicated HA‐tagged Merlin forms or an empty vector. These cells treated with DMSO or thapsigargin were subjected to nickel affinity purification in a denaturing step followed by Western blotting for HA. Arrows indicate mono‐ubiquitinated Merlin. The red asterisk indicates native Merlin and black asterisks indicate non‐specific signals. The ratio of mono‐ubiquitinated to native Merlin in each lane of the input blot was quantified by ImageJ and is shown under the blot.
-
FMerlin‐depleted LN229 cells were stably transduced with an empty vector, Flag‐tagged wild‐type (WT) Merlin or its K396R mutant. These cells were detached (S, suspension) by trypsinization using the procedure described in the Materials and Methods section and reseeded (A, attached) for 2 h. Cell lysates were subjected to immunoprecipitation with a Flag antibody followed by Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane of the lysate blot was quantified by ImageJ and is shown under the blot.
-
GMerlin‐depleted LN229 cells stably transduced with wild‐type (WT) Merlin or its K396R mutant were treated with DMSO or thapsigargin (TG) and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
HMerlin‐depleted LN229 cells stably transduced with HA‐tagged wild‐type (WT) Merlin or its K396R mutant were treated as described in (G) and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
Source data are available online for this figure.
Merlin ubiquitination is required for activating Lats1
The above studies indicated a correlation between Merlin ubiquitination and Lats1 activation in response to Ca2+ signaling and cell detachment (Fig 1C–E). To further examine the link between the two signaling events, we conducted time‐course studies. Both Merlin ubiquitination and Lats1 phosphorylation can be detected after 5 min of treatment with thapsigargin (Fig 6A). Their temporal kinetics appeared similar, suggesting a link between the two processes. Similar temporal kinetics between these two events were also seen under the cell reattachment condition (Fig EV4A). Since Merlin is important for Lats1 phosphorylation in the presence of ionomycin or thapsigargin (Fig 1E) or when cells are detached (Fig EV1C), we hypothesized that ubiquitination might be important for Merlin to activate Lats1. To test this, we used CRISPR to knock out Merlin in LN229 cells (Fig 1B) and reconstituted with wild‐type Merlin or its K396R or S518D mutants (Fig 6B). Compared to wild‐type Merlin, both K396R and S518D mutants were less capable of inducing Lats1 phosphorylation in the presence of thapsigargin (Fig 6B). We further tested this using the cells reconstituted by Merlin Δ513–521, L517P, and L535P mutants. Thapsigargin‐induced Lats1 and TAZ phosphorylation was also compromised in these cells when compared to wild‐type Merlin‐reconstituted cells (Fig 6C). This result suggested that dephosphorylation and ubiquitination are important for Merlin to have full capability of inducing Lats1 phosphorylation. We identified NEDD4L as the E3 ubiquitination ligase for Merlin, so to further examine the impact of ubiquitination, we silenced the expression of NEDD4L in LN229 cells using a pool of four siRNAs. Depletion of NEDD4L markedly reduced thapsigargin‐induced Lats1 and TAZ phosphorylation (Fig 6D), indicating that NEDD4L is important for activating Lats1. We further examined whether Merlin ubiquitination is also important for activating Lats1 by cell detachment. When Merlin was depleted from LN229 cells, both Lats1 and TAZ phosphorylation were markedly reduced when cells were detached. Merlin reconstitution in these cells induced Lats1 and TAZ phosphorylation (Fig 6E). However, the K396R mutant lacked this ability. This result further supported that Merlin ubiquitination is important for Hippo pathway activation. To further examine this, we used a pool of four siRNAs to silence the expression of NEDD4L. Depletion of NEDD4L reduced detachment‐induced Lats1 and TAZ phosphorylation both in LN229 cells (Fig 6F) and Met5‐A cells (Fig EV4B). To examine whether the NEDD4L‐Merlin signaling axis regulates transcriptional targets of the Hippo pathway, we analyzed the expressions of CTGF and CYR61, two well characterized genes regulated by the Hippo pathway. Silencing the expression of NEDD4L in LN229 cells with a pool of four different of siRNAs increased the expressions of CTGF and CYR61 (Fig 6G). Similar results were also observed when Merlin was depleted using the same assay (Fig 6G). Overall, the above results indicated that NEDD4L‐mediated Merlin ubiquitination is important for Lats1 activation.
Figure 6. Merlin ubiquitination is required for activating Lats1.
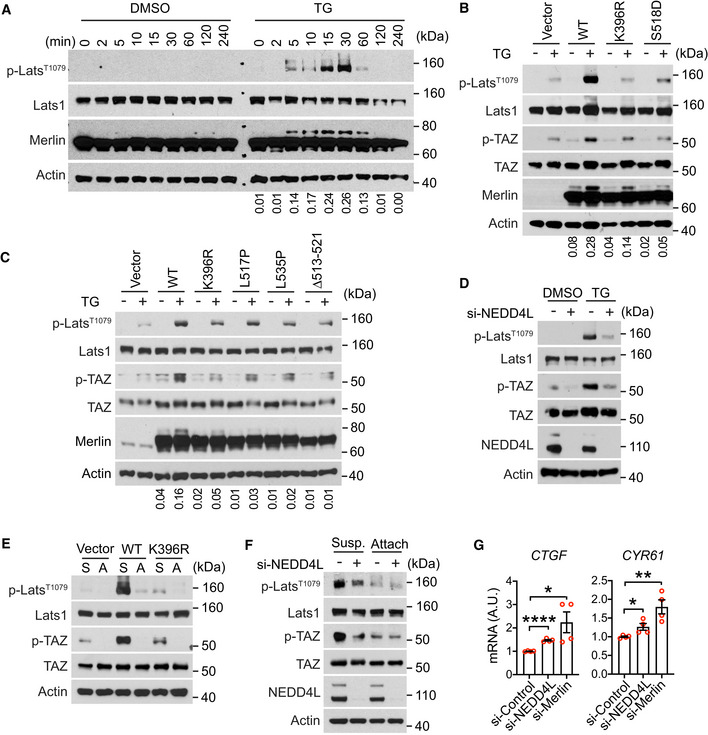
-
ALN229 cells were treated with DMSO or thapsigargin (TG) for the indicated time and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
B, CMerlin‐depleted LN229 cells were reconstituted with wild‐type (WT) Merlin or its mutants, treated with DMSO or thapsigargin (TG), and subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
DLN229 cells transfected with a pool of four siRNAs targeting NEDD4L (+) or a scrambled siRNA (−) were treated with DMSO or thapsigargin (TG) and subjected to Western blotting.
-
DMerlin‐depleted LN229 cells were stably transduced with empty vector, Flag‐tagged wild‐type (WT) Merlin, or its K396R mutant. These cells were detached (S, suspension) by trypsinization using the procedure described in Materials and Methods and reseeded (A, attached) for 2 h. Cell lysates were subjected to Western blotting.
-
ELN229 cells transfected with a pool of four siRNAs targeting NEDD4L (+) or a scrambled siRNA (−) were detached (Susp., suspension) by trypsinization using the procedure described in Materials and Methods and reseeded (Attach, attached) for 2 h. Total lysates from these cells were subjected to Western blotting.
-
FLN229 cells transfected with indicated siRNAs were seeded for 3 h. CTGF or CYR61 mRNA in these cells was quantified by qRT–PCR. *P < 0.05, **P < 0.01, ****P < 0.0001. Mean ± s.e.m, N = 4 biological repeats, unpaired t‐test.
Source data are available online for this figure.
Figure EV4. Merlin ubiquitination associates with Lats1 activation.
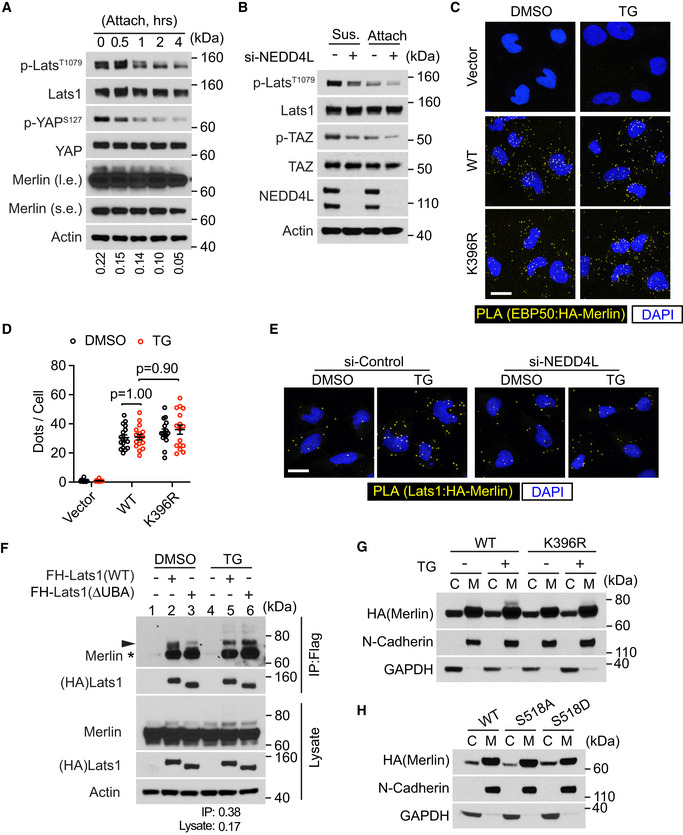
-
AMet5‐A cells were detached by using the procedure described in Materials and Methods and reseeded (attach) for 4 h. Total lysates from these cells were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
-
BMet5‐A cells transfected with a pool of four siRNAs against NEDD4L (+) or a scrambled siRNA (−) were detached (Sus., suspension) by using the procedure described in Materials and Methods and reseeded (Attach, attached) for 4 h. Total lysates from these cells were subjected to Western blotting.
-
CMerlin‐depleted LN229 cells were stably transduced with HA‐tagged wild‐type (WT) Merlin or its K396R mutant. These cells were treated with thapsigargin (TG) and subjected to PLA using HA and EBP50 antibodies. Scale bar = 20 μm.
-
DPLA signals (dots) in each cell from the results in (C) were quantified. Mean ± s.e.m, two‐way ANOVA. Each data point represents an image field containing averagely 10 cells. N Vector/DMSO = 12, N Vector/TG = 12, N WT/DMSO = 20, N WT/TG = 16, N K396R/DMSO = 20, N K396R/TG = 20 images in each condition. All images were collected from one experiment. Two independent experiments were performed and showed similar results.
-
EMerlin‐depleted LN229 cells were stably transduced with HA‐tagged Merlin. These cells were then transduced with a pool of four siRNAs against NEDD4L or a scrambled siRNA and treated with DMSO or thapsigargin (TG). PLA was performed in these cells using HA and Lats1 antibodies. Scale bar = 20 μm.
-
FLN229 cells stably transduced with Flag‐HA (FH)‐tagged wild‐type Lats1 or its UBA domain deletion (ΔUBA) mutant were treated with DMSO or thapsigargin (TG). The cells were lysed and subjected to immunoprecipitation with a Flag antibody. The lysate and immunoprecipitated products were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in lane 5 of IP and lysate blots was quantified by ImageJ and is shown under the blot, respectively. Native Merlin (asterisk); ubiquitinated Merlin (arrowhead).
-
G, HMerlin‐depleted LN229 cells stably transduced with Flag‐tagged wild‐type Merlin or its mutants were treated with DMSO or thapsigargin (TG). The cells were subjected to cytosolic (C)/membrane (M) fractionation followed by Western blotting.
Source data are available online for this figure.
Merlin ubiquitination is important for its interaction with Lats1
It was reported that Merlin plays a central role in Lats1/2 activation by recruiting Lats1/2 to the kinases MST1/2 (Yin et al, 2013). In this process, binding of Merlin to Lats1/2 is essential. Using the proximity ligation assay (PLA) (Soderberg et al, 2006), we found that the interaction between Merlin and Lats1 was enhanced by thapsigargin (Fig 7A and B). Time‐course analysis indicated that the kinetics of Merlin‐Lats1 interaction were similar to those of Merlin ubiquitination and Lats1 phosphorylation (comparing Figs 6A and 7B). To examine whether Merlin ubiquitination affects its interaction with Lats1, we compared wild‐type Merlin and its ubiquitination‐deficient K396R mutant in their interactions with Lats1 using PLA. Comparing to wild‐type Merlin, the K396R mutant showed markedly reduced PLA signals (Fig 7C and D), suggesting diminished interaction with Lats1. Interestingly, the K159R mutant still bound to Lats1 similarly to wild‐type Merlin, suggesting that the reduced Lats1 binding is specific for the K396R mutant. Our above results (Fig 5H and I) suggested that the K396R mutation specifically disrupts Merlin ubiquitination. To further examine whether this mutation non‐specifically affects Merlin interaction with other proteins, we assessed the mutant's interaction with NEDD4L and AMOTL1 by co‐immunoprecipitation. It appeared that the K396R mutant interacted with these proteins similarly to wild‐type Merlin (Fig 7E and F). The Merlin FERM domain is important for its binding to Lats1 (Yin et al, 2013). To examine whether Merlin ubiquitination could regulate other FERM domain binding partners, we assessed its interaction with EBP50 (a.k.a. NHE‐RF) (Murthy et al, 1998). PLA confirmed their interaction (Fig EV4C and D). Unlike Lats1, such interaction was not enhanced by thapsigargin or inhibited by the K396R mutation (Fig EV4C and D). Therefore, the impact of ubiquitination may not apply to all Merlin FERM domain binding proteins. To further examine the impact of Merlin ubiquitination on its interaction with Lats1, NEDD4L in LN229 cells was depleted by the siRNA pool. PLA showed that although the interaction between Merlin and Lats1 was enhanced by thapsigargin in control cells, such enhancement was diminished upon NEDD4L depletion (Figs 7G and EV4E).
Figure 7. Merlin ubiquitination is important for its interaction with Lats1.
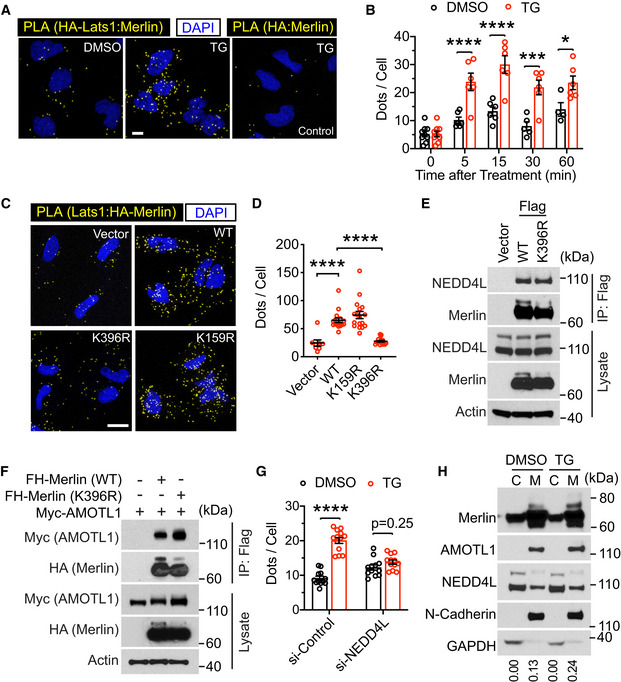
- LN229 cells stably transduced with HA‐Lats1 (two leftmost panels) or vector only (right panel) were treated with DMSO or thapsigargin (TG) and subjected to PLA using HA and Merlin antibodies. Scale bar = 10 μm.
- PLA signals (dots) in each cell from the results in (A) were quantified. Mean ± s.e.m, two‐way ANOVA. *P < 0.05, ***P < 0.001, ****P < 0.0001. Each data point represents an image field containing an average of 10 cells. N = 4–9 images for each condition as indicated. All images were collected from one experiment. Two independent experiments were performed and gave similar results.
- Merlin‐depleted LN229 cells were stably transduced with HA‐tagged wild‐type (WT) Merlin or its mutants. These cells were treated with thapsigargin (TG) for 15 min and subjected to PLA using HA and Lats1 antibodies. Scale bar = 20 μm.
- PLA signals (dots) in each cell from the results in (C) were quantified. Mean ± s.e.m, Ordinary one‐way ANOVA. ****P < 0.0001. Each data point represents an image field containing an average of 10 cells. N Vector = 8, N WT = 16, N K396R = 18, N K159R = 17 images. All images were collected from one experiment. Two independent experiments were performed and gave similar results.
- Merlin‐depleted LN229 cells stably transduced with empty vector, Flag‐tagged wild‐type (WT) Merlin, or its K396R mutant were subjected to immunoprecipitation with a Flag antibody followed by Western blotting.
- HEK293T cells were transfected with indicated proteins and subjected to immunoprecipitation with a Flag antibody. The lysates and immunoprecipitated products were subjected to Western blotting.
- Merlin‐depleted LN229 cells were stably transduced by HA‐tagged Merlin. These cells were then transduced with a pool of four siRNAs against NEDD4L or a scrambled control siRNA and treated with DMSO or thapsigargin (TG). PLA was performed in these cells using HA and Lats1 antibodies, and the PLA signals (dots, showed in Fig EV5E) in each cell were quantified. Mean ± s.e.m, two‐way ANOVA. ****P < 0.0001. Each data point represents an image field containing averagely 10 cells. N = 12 images in all conditions, except for N si‐Control:TG = 13 images. All images were collected from one experiment. Two independent experiments were performed and showed similar results.
- LN229 cells treated with DMSO or thapsigargin (TG) were subjected to cytosolic (C)/membrane (M) fractionation followed by Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
Source data are available online for this figure.
There is a ubiquitin‐associated (UBA) domain close to the Lats1 amino terminus. To examine whether it is involved in binding to the conjugated ubiquitin on Merlin, we constructed a UBA domain deletion (ΔUBA) Lats1 mutation and examined its interaction with Merlin through co‐immunoprecipitation. In DMSO‐treated cells, the Lats1 ΔUBA mutant was weaker than wild‐type Lats1 in binding to ubiquitinated Merlin (Fig EV4F, arrowhead, comparing lane 3 to 2). However, in thapsigargin‐treated cells, the ΔUBA mutant bound to ubiquitinated Merlin similarly to wild‐type Lats1 (Fig EV4F, arrowhead, comparing lane 6 to 5). This result suggested that the Lats1 UBA domain is unnecessary for Merlin's binding to Lats1, at least when Merlin ubiquitination is induced by thapsigargin. In this experiment, although both native (asterisk) and ubiquitinated (arrowhead) Merlin co‐precipitated with Lats1, densitometry quantification indicated that the ratio of the upper band (arrowhead) to the lower band (asterisk) in the co‐immunoprecipitated product was greater than that ratio for the lysate (Fig EV4F, lane 5). Such enrichment of the ubiquitinated Merlin in the co‐precipitated product suggested that Lats1 binds stronger to ubiquitinated Merlin than non‐ubiquitinated forms. It was reported that Merlin recruits Lats to the plasma membrane (Yin et al, 2013). To examine whether Merlin ubiquitination affects its membrane association, we conducted subcellular fractionation analysis. Interestingly, ubiquitinated Merlin was largely in the membrane fraction (Fig 7H). While NEDD4L localized in both cytosolic and membrane fractions, AMOTL1 was mostly in the membrane fraction. Merlin's cytosolic and membrane distribution was not affected by its K396R and S518A mutations, although the S518D mutant was slightly more cytosolic (Fig EV4G and H). These results suggested that Merlin may be ubiquitinated at the plasma membrane, where it binds to Lats1. Overall, the above results supported that Merlin ubiquitination promotes its binding to Lats1 and is important for Lats1 activation.
Merlin ubiquitination contributes to growth control and tumor suppression
The above studies found that Merlin ubiquitination is associated with its activation. Given that Merlin is important for growth control and tumor suppression, we examined whether ubiquitination is involved in these functions. First, we assessed the anti‐mitogenic function by a BrdU incorporation assay. Our studies have used LN299 and Met5‐A cells to study Merlin regulation. Given that Met5‐A is a non‐transformed cell line, it is more appropriate to use these cells to study the potentially uncontrolled proliferation due to Merlin dysfunction. In Met5‐A cells, silencing Merlin expression by two different shRNAs increased BrdU incorporation (Figs 8A and B, and EV5A). This result indicated that Merlin is involved in suppressing the G1‐S transition in these cells. Since NEDD4L is responsible for Merlin ubiquitination, we conducted a similar analysis by silencing the expression of NEDD4L with two different shRNAs in these cells. Consistently, depletion of NEDD4L promoted BrdU incorporation (Figs 8C and D, and EV5B). These results supported that NEDD4L‐mediated Merlin ubiquitination is important for controlling the G1‐S transition. To further examine this, we conducted a gain of function study in Merlin‐deficient cells. The Meso‐33 human mesothelioma cell line has lost Merlin expression and was shown to respond to recombinant Merlin in proliferation control (Li et al, 2010, 2014). As reported previously, ectopic expression of Merlin in Meso‐33 cells inhibited the G1‐S transition, whereas the S518D mutant showed a dampened inhibitory effect (Fig 8E and F) (Li et al, 2010, 2014). In these cells, we found that the K396R mutant failed to inhibit the G1‐S transition (Fig 8E and F). Since the K396R mutation disrupts Merlin ubiquitination and its ability to interact with and activate Lats1, this result suggested that these properties are important for Merlin's anti‐mitogenic function. Consistently, Merlin expression in Meso‐33 cells induced phosphorylation of Lats1, YAP, and TAZ. However, its S518D and K396R mutants were less capable of this induction (Fig EV5C). To further examine the requirement of Merlin ubiquitination, NEDD4L expression was silenced in Meso‐33 cells using two shRNAs. In contrast to Met5‐A cells, NEDD4L‐depletion in Meso‐33 cells did not increase BrdU incorporation (Fig 8G) or decrease phosphorylation of Lats1, YAP, or TAZ (Fig EV5D). These results suggested that the presence of Merlin is required for manifesting NEDD4L's loss of function effect. To test this, Merlin was ectopically expressed in Meso‐33 cells transduced by either of the two NEDD4L shRNAs or a scrambled control. NEDD4L‐depleted Meso‐33 cells showed expression of recombinant Merlin similar to control cells transduced by the scrambled shRNA (Fig EV5E), indicating that depletion of NEDD4L does not affect the expression of Merlin. In NEDD4L‐depleted cells, Merlin introduction showed less inhibition of BrdU incorporation compared to Merlin expression in control cells transduced by the scrambled shRNA (Fig 8G). This result confirmed our prediction and supported that NEDD4L is important for Merlin to function as a mitotic inhibitor. Meso‐33 cells are not tumorigenic (Li et al, 2010), so to further examine the tumor suppressive function of Merlin, we employed the FC‐1801 mouse schwannoma cell line, which does not express Merlin, and was previously used to study Merlin's tumor suppressive functions (Li et al, 2010, 2014). First, we examined the anti‐oncogenic function of Merlin by performing a tumor sphere assay. When different forms of Merlin (wild‐type, K396R, or S518D) were stably expressed in these cells (Fig EV5F), both K396R and S518D mutants showed less inhibition of tumor sphere formation compared to wild‐type Merlin (Fig 8H and I). FC‐1801 cells can grow into tumors subcutaneously (Li et al, 2010, 2014), so we next examined the tumor suppressive function by subcutaneously inoculating these cells transduced with different forms of Merlin into mice. Expression of wild‐type Merlin markedly inhibited tumor growth (Fig 8J and K). However, tumors arising from the cells expressing either K396R or S518D mutant grew faster than those expressing wild‐type Merlin (Fig 8J and K). Collectively, these results supported that Merlin ubiquitination is important for growth control and contributes to its function as a tumor suppressor.
Figure 8. Merlin ubiquitination contributes to growth control and tumor suppression.
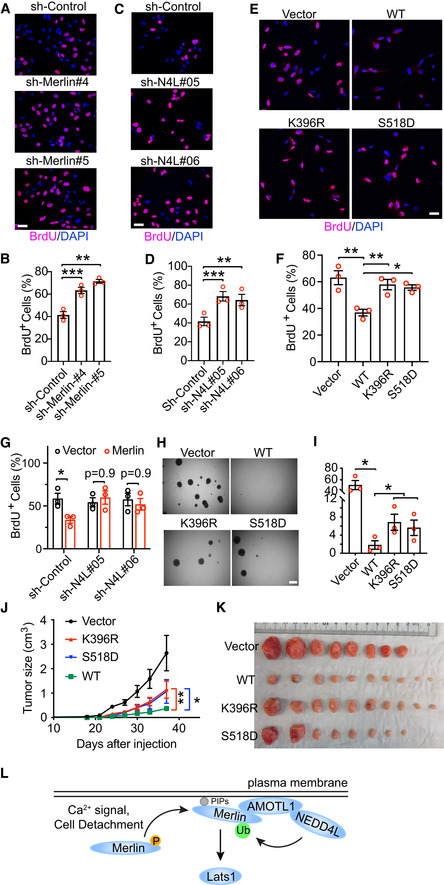
- Met5‐A cells stably transduced with the indicated shRNAs targeting Merlin or a scrambled shRNA were subjected to the BrdU incorporation assay. Scale bar = 40 μm.
- The percentage of BrdU‐positive cells from (A) was quantified. Mean ± s.e.m, N = 3 biological repeats, paired t‐test. **P < 0.01, ***P < 0.001.
- Met5‐A cells stably transduced with the indicated shRNAs targeting NEDD4L or a scrambled shRNA were subjected to the BrdU incorporation assay. Scale bar = 40 μm.
- The percentage of BrdU‐positive cells from (C) was quantified. Mean ± s.e.m, N = 3 biological repeats, paired t‐test. **P < 0.01, ***P < 0.001.
- Meso‐33 cells transduced with vector or the indicated Merlin forms were subjected to the BrdU incorporation assay. Scale bar = 40 μm.
- The percentage of BrdU‐positive cells from (E) was quantified. Mean ± s.e.m, N = 3 biological repeats, ordinary one‐way ANOVA. *P < 0.05, **P < 0.01.
- Meso‐33 cells stably transduced with the indicated shRNAs targeting NEDD4L or a scrambled shRNA were then transduced with vector or Merlin prior to the BrdU incorporation assay. The percentage of BrdU‐positive cells was quantified. Mean ± s.e.m, N = 3 biological repeats, two‐way ANOVA.
- FC‐1801 cells stably transduced with vector or the indicated Merlin forms were subjected to the sphere formation assay. Scale bar = 500 μm.
- The number of spheres in each well from (H) was quantified. Mean ± s.e.m, N = 3 biological repeats, paired t‐test. **P < 0.05.
- FC‐1801 cells stably transduced with vector or the indicated Merlin forms were subcutaneously injected into nude mice. Tumor sizes in each group were quantified and plotted. Mean ± s.e.m, N Vector = 8 mice, N WT = 10 mice, N K396R = 10 mice, N S518D = 8 mice, two‐way ANOVA. P values of the last measurements (day 37 after injection) are shown. *P < 0.05, **P < 0.01.
- Tumors were collected when the vector group in (J) reached the end point (day 37 after injection).
- A proposed model showing how Merlin is activated to induce Lats1 phosphorylation on the plasma membrane.
Figure EV5. Depletion and overexpression of Merlin in various cells.
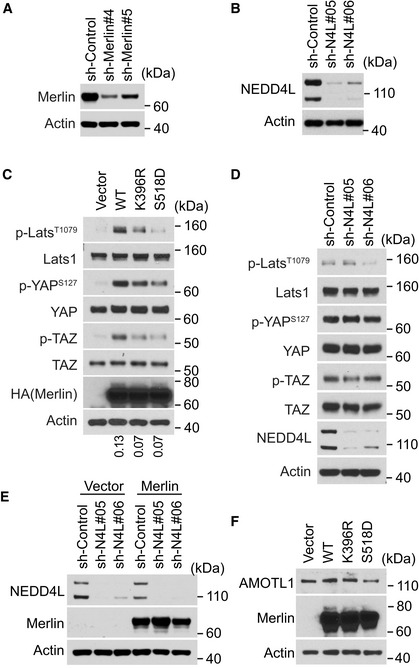
- Met5‐A cells stably transduced with the indicated shRNAs against Merlin or a scrambled shRNA were subjected to Western blotting.
- Met5‐A cells stably transduced with the indicated shRNAs against NEDD4L or a scrambled shRNA were subjected to Western blotting.
- Meso‐33 cells transduced with vector or the indicated Merlin forms were subjected to Western blotting. The ratio of mono‐ubiquitinated to native Merlin in each lane was quantified by ImageJ and is shown under the blot.
- Meso‐33 cells stably transduced with the indicated shRNAs targeting NEDD4L or a scrambled shRNA were detached by using the procedure described in the Materials and Methods and subjected to Western blotting.
- Meso‐33 cells stably transduced with the indicated shRNAs against NEDD4L or a scrambled shRNA were then transduced with vector or Merlin prior to Western blotting.
- FC‐1801 cells stably transduced with vector or the indicated Merlin forms were subjected to Western blotting.
Source data are available online for this figure.
Discussion
In this study, we found that Merlin ubiquitination is common under physiological conditions, with mono‐ and di‐ubiquitinated being the major ubiquitinated forms. In response to Ca2+ signaling and cell detachment, such limited ubiquitination and serine 518 dephosphorylation constitute an activation cascade, with dephosphorylation promoting ubiquitination. We identified NEDD4L as the E3 ubiquitin ligase for Merlin. The interaction between NEDD4L and Merlin is mediated by AMOTL1. Several NF2‐patient‐derived Merlin mutations disrupt Merlin's binding to AMOTL1 and regulation by the AMOTL1‐NEDD4L apparatus. Furthermore, we identified lysine 396 as the major ubiquitin‐conjugating residue on Merlin. Mutagenic analyses suggested that ubiquitination on lysine 396 is important for Merlin to interact with and activate Lats1. This ubiquitination is also involved in inhibiting cell proliferation and suppressing tumor growth.
Previous crystal structure studies found that Merlin's carboxyl terminus blocks its Lats‐binding site on the FERM domain. By binding to AMOT, such auto‐inhibition is released, and Merlin's binding to Lats is enhanced (Li et al, 2015). Whether AMOT binding induces any post‐translational modification on Merlin was unknown. Our results indicated that AMOTL1, and likely other AMOT family proteins, promotes Merlin ubiquitination, which is required for Merlin activation. This finding provides a further mechanism for AMOT family proteins in activating Merlin. The cycle of phosphorylation and dephosphorylation of serine 518 is an important regulatory modification of Merlin. It was reported that the S518A mutant more strongly binds to Lats1 and induces Lats1 phosphorylation (Yin et al, 2013), suggesting that the phosphorylation weakens Merlin‐Lats1 interaction. Our results are consistent with this and further provide a mechanistic insight by showing that dephosphorylation promotes Merlin ubiquitination, which is required for promoting Merlin‐Lats1 interaction and inducing Lats1 phosphorylation (Fig 8L). Beyond this, we showed that the phosphomimetic S518D mutation and the ubiquitination‐blocking K396R mutation similarly diminish anti‐mitogenic and tumor suppressive abilities of Merlin. Therefore, dephosphorylation followed by ubiquitination appears to be an intramolecular activation cascade for Merlin to activate the Hippo pathway and function as a tumor suppressor. How this regulation cascade is operated remains unknown. Phosphorylation of serine 518 was found to block Merlin from binding to AMOT (Li et al, 2015). This may explain why the S518D mutant showed less ubiquitination in our observation. Our results suggested that ubiquitinated Merlin largely localizes to the plasma membrane (Fig 7H). Since AMOTL1 is also enriched in this location, it is possible that translocation to the membrane is required for Merlin to be ubiquitinated. Crystal structure studies found that lipid binding promotes Merlin to adopt the open conformation and activation (Chinthalapudi et al, 2018). In addition, it was suggested that serine 518 phosphorylation status can affect Merlin conformation (Sher et al, 2012; Ali Khajeh et al, 2014). Because both lipid binding and serine 518 phosphorylation modulate Merlin ubiquitination, whether the conformation change associated with either of these processes is a cause or consequence of Merlin ubiquitination remains to be determined.
Our studies identified Merlin as a substrate of NEDD4L. By inducing Merlin ubiquitination, NEDD4L promotes Merlin's binding to Lats1, thereby activating the Hippo pathway. In addition to NEDD4L, we found that NEDD4 and ITCH can also induce Merlin ubiquitination when overexpressed with AMOTL1 and Merlin. Although this result suggested that these two ligases may have similar functions to NEDD4L in this case, it needs to be cautiously interpreted because our initial mass spectrometry result did not find these proteins in the precipitates co‐purified with Merlin (Fig 3A). In addition, ITCH and NEDD4 have been found to inactivate Lats kinases by promoting their degradation (Ho et al, 2011; Salah et al, 2011, 2013; Bae et al, 2015). The contrast effects on Lats by NEDD4L versus ITCH and NEDD4 suggested that different members in this E3 ligase family may regulate the Hippo pathway in opposite manners. It was shown that NEDD4, NEDD4L, and ITCH can promote ubiquitination and degradation of the AMOT family proteins (Skouloudaki & Walz, 2012; Wang et al, 2012), and Merlin can promote this phenomenon (Couderc et al, 2016). In our experiments, overexpression of NEDD4L with the AMOT family proteins in 293T cells showed that ubiquitination of AMOTL1 and AMOTL2 can be markedly induced. Such ubiquitination was accompanied by a decrease of their expression (Fig EV3E), which is consistent with previous reports. We did not observe a decrease of AMOTL1 expression when Merlin was expressed in FC1801 cells (Fig EV5F), suggesting that Merlin does not regulate AMOTL1 stability in this cell line. It is possible that although the AMOT family proteins are able to recruit NEDD4L to activate Merlin, such recruitment may trigger its own ubiquitination and degradation. This may constitute a negative feedback loop to avoid uncontrolled Merlin activation. Of note, AMOTL2 mono‐ubiquitination was found to promote Lats activation (Kim et al, 2016). Although the E3 ligase for this specific AMOTL2 modification has not been identified, it is conceivable that AMOTL1 and Merlin may both be ubiquitinated by NEDD4L. AMOTL1 ubiquitination induced by NEDD4L may also be involved in Lats1 activation. In this case, whether AMOTL1 ubiquitination activates Lats on its own or through activating Merlin warrants further study.
Although the genetic role of NEDD4L in the Hippo pathway has not been previously reported, recent studies of a human lung disease provided certain clues. In lungs of idiopathic pulmonary fibrosis (IPF) patients, NEDD4L expression is reduced. Conditional knockout of Nedd4 l in lung epithelial cells leads to pulmonary fibrosis in mice (Duerr et al, 2020). Notably, YAP is also activated in lung epithelial cells from IPF patients (Gokey et al, 2018). With our current results showing the NEDD4L function in the Hippo pathway, it is likely that YAP activation may be involved in Nedd4 l knockout‐induced pulmonary fibrosis.
Overall, our study has uncovered a novel Merlin post‐translational regulation operated by the NEDD4L‐AMOTL1 apparatus. Such regulation is important for Merlin to activate the Hippo pathway in response to Ca2+ signaling and cell detachment. In the future, it would be interesting to study how the interaction between Merlin and NEDD4L is regulated in response to upstream signals and to determine whether similar regulation is involved in other Hippo pathway‐controlled situations.
Materials and Methods
Mice
For tumorigenesis experiments, 6‐ to 8‐week‐old female nude mice (Nu(NCr)‐Foxn1nu, from Charles River, Strain Code: 490) were used. FC1801 cells were transduced in vitro with a retrovirus vector only or expressing indicated cDNAs. 2 × 105 cells were injected subcutaneously. Tumor size was measured by digital caliper twice per week. All experimental protocols were approved by the Penn State University Institutional Animal Care and Use Committee. All methods were performed in accordance with the relevant guidelines and regulations.
Cells
LN229 (CRL‐2611), LN18 (CRL‐2610), DBTRG‐05MG (CRL‐2020), MOVAS (CRL‐2797) and C2C12 (CRL‐1772) cells were from ATCC. U2OS cells were generously provided by Hong‐Gang Wang (Penn State College of Medicine). The above cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Corning, 10‐013‐CV) supplemented with 10% Fetal Bovine Serum (Gibco, 10437028) and 1% Antibiotic‐Antimycotic Solution (Corning, 30‐004‐CI) at 37°C with 5% CO2. HEK293T, MCF10A, LP9, Met5‐A, Meso‐33 cells were generously provided by Filippo Giancotti (MD Anderson Cancer Center). FC‐912 and FC‐1801 cells were generously provided by Marco Giovannini (UCLA Health). These cells were cultured in the conditions as reported previously (Li et al, 2010). None of these cell lines were listed in the database of misidentified cell lines maintained by ICLAC and NCBI Biosample. These cell lines were not authenticated in this study. All cell lines were examined to be mycoplasma negative before experiments.
Unless otherwise indicated, experiments were performed with cells grown to 50% confluency. For compound treatment experiments, cells were seeded in complete medium (10% FBS) for overnight and treated with the DMEM containing 2% FBS and indicated compounds for indicated times before harvest. For cell suspension and attachment experiments, cells were rinsed with Dulbecco's Phosphate‐Buffered Salt Solution (DPBS; MT21031CV, Corning) and detached with 0.05% trypsin/EDTA (25‐052‐CI, Corning). After collecting the cells and washing off trypsin/EDTA by DPBS, cells were incubated in DPBS for 15 min at 37°C in a conical tube. To prevent aggregation, cells were occasionally and gently agitated. These cells were then either lysed to be used as suspended cells or reseeded to the culture dish with regular growth medium. To be used as attached cells, the reseeded cells were allowed to attach to the dish for 2 (LN229) or 4 (Met5‐A) h before being lysed.
Antibodies and compounds
Anti‐Phospho‐Lats1 (Thr1079; 8654), anti‐Phospho‐YAP (Ser127; 13008), anti‐YAP (12395), anti‐TAZ (4883), anti‐Lats1 (3477), anti‐Merlin (12888), anti‐Phospho‐Merlin (Ser518; 13281), anti‐Phospho‐TAZ (Ser89; 59971), anti‐DCAF1 (14966), anti‐DDB1 (6998), anti‐NEDD4L (4013), anti‐mouse Ubiquitin(3936), anti‐rabbit Ubiquitin(3933), anti‐T7 (13246), anti‐Myc (2272), anti‐EBP50 (8601), and anti‐GFP (2956) antibodies were from Cell Signaling Technology. Anti‐HA (MMS‐101P) antibody was from BioLegend. Anti‐β‐Actin (A5316) and anti‐AMOTL1 (HPA001196) antibodies were from Sigma‐Aldrich. anti‐mouse IgG‐HRP (7076), anti‐rabbit IgG‐HRP (7074) antibodies were from Cell Signaling Technology. Ionomycin (I9657), and Thapsigargin (586005) were from Sigma‐Aldrich.
Vectors
pCAGGS‐HA‐Amot,Amotl1,Amotl2 were described previously (Sugihara‐Mizuno et al, 2007) and generously provided by Makoto Adachi (Kyoto University). pcDNA3‐EGFP‐RAC1(Q61L) was described preciously (Kraynov et al, 2000) and a gift from Klaus Hahn via Addgene (#13720). pCMV6M‐PAK1(T423E) was described preciously (Sells et al, 1997) and a gift from Jonathan Chernoff via Addgene (#12208). pSpCas9(BB)‐2A-Puro was described preciously (Ran et al, 2013) and a gift from Feng Zhang via Addgene (#48139). pCI‐HA‐NEDD4L was described preciously (Gao et al, 2009) and a gift from Joan Massague via Addgene (#27000). pcDNA4 HisMax‐hNEDD4‐2 was described preciously (Itani et al, 2005) and a gift from Christie Thomas via Addgene (#83433). pcDNA‐HA‐Nedd4 and pCINeo‐myc‐Itch were described preciously (Magnifico et al, 2003) and a gift from Allan Weissman via Addgene (#11426 and #11427). pRK5‐Flag‐HA-Merlin, pRK5‐Myc‐Merlin, pHis‐Myc‐Ubiquitin, and pQCXIN‐Flag‐HA-LATS1 were generously provided by Filippo Giancotti (MD Anderson Cancer Center). To generate pRK5‐myc‐NEDD4L, pRK5‐myc‐Amotl1, pRK5‐V5‐Merlin, and pRK5‐Flag‐HA-Amotl1, the target genes from above described plasmids were subcloned into the pRK5 vector individually. To generate pBabe‐Flag‐HA-Merlin, Flag‐HA‐tagged Merlin from pRK5‐Flag‐HA-Merlin was subcloned into the pBabe‐puro vector. To generate pBabe‐Flag‐HA-Merlin mutations, pRK5‐myc‐NEDD4L‐C942A, pRK5‐Flag‐HA-LATS1‐ΔUBA and pRK5‐Flag‐HA-Amotl1‐PY*, PY* + LY* mutations, pBabe‐Flag‐HA-Merlin, pRK5‐myc‐NEDD4L, pRK5‐Flag‐HA-LATS1 and pRK5‐Flag‐HA-Amotl1 plasmids were subjected to site‐directed mutagenesis using the Quickchange mutagenesis Kit (Stratagene).
Cell fractionation
To collect the cytosolic soluble and membrane fractions, a protocol reported previously (Hergovich et al, 2005) was used. Briefly, after washing by PBS buffer, cells were collected in ice‐cold S100/P100 buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 2.5 mM EDTA, 1 mM EGTA and 1× Proteinase inhibitors) and incubated on ice for 20 min. Cells were then homogenized by passing through a 26‐gauge needle for 20 times. The homogenate was centrifuged at 1,000 g at 4°C for 4 min. After discarding the pellet, supernatant was further centrifuged at 21,000 g for 60 min at 4°C. The new supernatant was collected as cytosolic soluble fraction. The new pellet (membrane fraction) was washed once with S100/P100 buffer and resuspend by the SDS lysis buffer (10 mM Tris pH 7.5, 1% SDS, 50 mM NaF, 1 mM NaVO4).
Immunoblotting
Immunoblotting procedure was described previously (Li et al, 2014). Briefly, cells were lysed in SDS lysis buffer (10 mM Tris pH 7.5, 1% SDS, 50 mM NaF, 1 mM NaVO4) and subjected to SDS–PAGE on 4–12% Bis‐Tris SDS–PAGE gels (Invitrogen) and transferred to Immobilon‐P membranes (Millipore). Membranes were incubated in blocking buffer: 5% skim milk/TBST (0.1% Tween, 10 mM Tris at pH 7.6, 100 mM NaCl) for 1 h at room temperature and then with primary antibodies diluted in 5% BSA/TBST overnight at 4°C. After three washes, the membranes were incubated with anti‐rabbit HRP‐conjugated antibody (1:5,000) or anti‐mouse HRP‐conjugated antibody (1:10,000) at room temperature for 2 h and subjected to chemiluminescence using ECL (Pierce #1856136).
Immunofluorescence staining
Immunofluorescence staining was described previously (Li et al, 2014). Briefly, cells were fixed with 4% paraformaldehyde in PBS for 20 min and incubated in permeabilization buffer (PDT: 0.3% sodium deoxycholate, 0.3% Triton X‐100 in PBS) for 30 min on ice. They were then blocked with 5% BSA/PBS at 4°C for 1 hr and followed by incubating overnight at 4°C with primary antibodies diluted in 2.5% BSA/0.05% Triton X‐100/PBS. After washing with 0.1% Triton X‐100/PBS, cells were incubated with secondary antibodies diluted in 2.5% BSA/0.05% Triton X‐100/PBS for 2 h at 4°C. Cells were washed with 0.1% Triton X‐100/PBS, rinsed with PBS, and mounted in ProLong Gold Mountant (Invitrogen #P10144). When indicated, nuclei were stained with DAPI. The PLA was performed using the Duolink II Red Starter Kit (Sigma). Briefly, cells were prepared as above for immunofluorescence staining until being incubated overnight at 4°C with primary antibodies against Lats1 (1:50) and HA (1:100). Cells were then incubated for 1 h at 37°C with a mixture of the MINUS and PLUS PLA probes. Hybridized probes were ligated using the Ligation‐Ligase solution for 30 min at 37°C and then amplified utilizing the Amplification‐Polymerase solution for 100 min at 37°C. Cells were finally mounted using Duolink II Mounting Medium containing DAPI.
Gene expression and silencing
Transient transfections were performed using Fugene 6 (Promega), Lipofectamine 2000 (Invitrogen) or DharmaFECT 1 (Dharmacon) by following the manufacturer recommended protocols. Retroviral generation and infection were described previously (Li et al, 2014). Lentiviral vectors encoding shRNAs targeting AMOTL1 (#1: TRCN0000130879; #2: TRCN0000150112; #3: TRCN0000147555), targeting NEDD4L (#05: TRCN0000000905; #06:TRCN0000000906), were from Sigma‐Aldrich, targeting Merlin (#4, #5, RHS3979‐201798933) was from Dharmacon. ON‐TARGETplus SMARTpool siRNAs against AMOTL1, NEDD4L, DCAF1, DDB1 or ON‐TARGETplus siCONTROL were from Dharmacon. To knock out Merlin using CRISPR, LN229 cells were transiently transfected with pSpCas9(BB)‐2A‐Puro with a guide RNA targeting the NF2 locus. The guide sequence is GTCCATGGTGACGATCCTCA and was designed by using the Optimized CRISPR Design tool at http://crispr.mit.edu. After transfection, LN229 cells were selected with puromycin for 2 days. Merlin knockout was confirmed by Western blotting.
Quantitative RT–PCR
qRT–PCR was carried out according to standard protocols. Briefly, total RNA was extracted using TRIzol reagent (Invitrogen). cDNAs were synthesized using iScript cDNA Synthesis kit (Bio‐Rad, 1708891), and qPCR was carried out on a CFX96 Touch Real‐Time PCR Detection System with SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad, 1725271). GAPDH was used as an internal reference to normalize the input cDNA. Primer sequences used: CTGF forward, GCAGGCTAGAGAAGCAGAGC, reverse, ATGTCTTCATGCTGGTGCAG; CYR61 forward, ACTTCATGGTCCCAGTGCTC, reverse, TGGTCTTGCTGCATTTCTTG; AMOTL1 forward, CGGGGAACTTGTGAGCCTG, reverse, CTGGGGAAAAGTAGGTGGAGT; GAPDH forward, GGAGCGAGATCCCTCCAAAAT, reverse, GGCTGTTGTCATACTTCTCATGG. The primers were designed by using PrimerBank.
Immunoprecipitation of Flag‐tagged proteins
3 × 106 LN229 cells stably expressing Flag‐HA‐Merlin or Flag‐HA‐Lats1 were seeded in a 10 cm plate for overnight and treated with DMSO or thapsigargin for 15 min. Cells were then lysed in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 4 mM EDTA, 1 mM EGTA, 1% Triton X‐100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 10% glycerol, 1× phosphatase inhibitor [Roche] and 1× proteinase inhibitor [Roche]) or RIPA buffer without SDS as indicated. To isolate Flag‐HA‐Merlin or Flag‐HA‐Lats1, cell extracts were incubated with anti‐FLAG M2 Affinity Gel (Sigma) at 4°C for 2 h. The precipitates were washed three times with IP2 buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 4 mM EDTA, 1 mM EGTA, 0.5% NP‐40, 10% glycerol, 1× phosphatase inhibitor [Roche] and 1× proteinase inhibitor [Roche]), and eluted with 3xFLAG Peptide (Sigma # F329020) dissolved in TBS (10 mM Tris–HCl, 150 mM NaCl, pH 7.4).
Immunoprecipitation of endogenous protein
3 × 106 LN229 cells were seeded in a 10 cm plate for overnight and treated with DMSO or thapsigargin for 15 min. Cells were then lysed in RIPA buffer as described above. Cell extracts were cleared by centrifugation, incubated with anti‐Merlin (CST, #12888) antibody, anti‐NEDD4L (CST, #4013) antibody, or rabbit IgG (Sigma‐Aldrich, #I5006) at 4°C overnight, and further incubated with protein A/G agarose beads (Thermo Fisher, #20423) for 2 h. The precipitates were washed with RIPA buffer for three times. Bound proteins were dissociated in 20 μl of SDS sample buffer (25 mM Tris, pH 6.8, 4% SDS, 5% Glycerol, and bromophenol blue). The proteins were separated on 4–12% Bis‐Tris SDS–PAGE gels (Invitrogen) and subjected to standard immunoblotting.
Mass spectrometry
To identify proteins interacting with Merlin, Flag‐tagged Merlin was stably transduced into LN229 cells. These cells and empty vector‐transduced LN229 cells were treated with DMSO or thapsigargin for 15 min and subjected to immunoprecipitation with a Flag antibody. Immunoprecipitated products were resolved using SDS polyacrylamide gel electrophoresis, followed by staining with GelCode Blue Stain Reagent (Thermo Scientific). For each sample/gel lane, all protein bands were separated into two groups along the 80 kDa protein weight marker, and separately excised from the gel. In each gel slice, protein bands were subjected to reduction with 10 mM Dithiothreitol for 30 min at 60°C, alkylation with 20 mM iodoacetamide for 45 min at room temperature in the dark and digestion with trypsin (sequencing grade, Thermo Scientific, Cat# 90058) overnight at 37°C. Peptides were extracted twice with 5% formic acid, 60% acetonitrile and dried under vacuum. Samples were analyzed by nano‐liquid chromatography coupled to tandem mass spectrometry (nano LC‐MS/MS) using a Q‐Exactive HF mass spectrometer interfaced with an Ultimate 3000 RSLCnano chromatography system (Thermo Scientific). Samples were loaded on to a fused silica trap column (Acclaim PepMap 100, 75 μm × 2 cm, Thermo Scientific). After washing for 5 min at 5 μl/min with 0.1% TFA, the trap column was brought in‐line with an analytical column (Nanoease MZ peptide BEH C18, 130A, 1.7 μm, 75 μm × 250 mm, Waters) for LC‐MS/MS. Peptides were fractionated at 300 nl/min using a segmented linear gradient 4–15% B in 30 min (A: 0.2% formic acid; B: 0.16% formic acid, 80% acetonitrile), 15–25% B in 40 min, 25–50% B in 44 min, and 50–90% B in 11 min. Mass spectrometry data were acquired using a data‐dependent acquisition procedure with an MS1 scan (resolution 120,000) followed by MS/MS (resolution 30,000; HCD relative collision energy 27%) on the 20 most intense ions with a dynamic exclusion duration of 20 s. To identify the ubiquitination sites on Merlin, Flag‐tagged wild‐type Merlin or the S518D mutant were stably transduced into LN229 cells. These cells were treated with thapsigargin for 15 min and subjected to immunoprecipitation with a Flag antibody. Immunoprecipitated products were resolved using SDS polyacrylamide gel electrophoresis, followed by staining with GelCode Blue Stain Reagent. Protein bands as indicated in Fig 5A were excised from the gel and subjected to mass spectrometry as described above. To perform database search, peak lists (mgf files) were generated using Thermo Proteome Discoverer (v. 2.1) and searched against the Uniprot Database and a database composed of common contaminants (cRAP) using version local implementation of X! Tandem (GPM Furry). Search parameters were as follows: protein and peptide log10 expectation scores < −4 and −2, respectively; fragment mass error: 20 ppm, parent mass error: ± 7 ppm; fixed modification: Cys carboxymethylation; potential modifications during initial search: GlyGly tag on internal lysine for ubiquitination and monooxidation on methionine; potential modifications during refinement: round 1, Met and Trp monooxidation, Asn and Gln deamidation; round 2, Met and Trp dioxidation. Peptide false‐positive rates were 0.4%. The spectra that were identified as ubiquitinated were further manually inspected.
Native nickel‐His pull‐down
1 × 106 HEK293T cells were transfected with 1 μg of pcDNA4 HisMax‐hNEDD4‐2, 0.5 μg of pRK5‐myc‐Merlin, and 0.5 μg of pCAGGS‐HA‐AMOTL1. Cells were lysed 24 h after transfection in RIPA buffer without SDS. To isolate His‐NEDD4L, cell extracts were incubated with Ni‐NTA agarose beads (Qiagen) at 4°C for 2 h. The precipitates were washed three times with IP2 buffer and dissociated in 20 μl of SDS sample/elution buffer (25 mM Tris, pH 6.8, 4% SDS, 5% Glycerol, 250 mM Imidazole and bromophenol blue). Eluted proteins were separated on 4–12% Bis‐Tris SDS–PAGE gels (Invitrogen) and subjected to standard immunoblotting.
In vivo ubiquitination assay
In vivo Ubiquitination Assay was performed as described previously (Li et al, 2014). Briefly, LN229 cells stably expressing His‐Myc‐Ubiquitin were seeded in a 10 cm plate for overnight and treated with DMSO or thapsigargin for 15 min or were detached (suspension) by trypsinization and reseeded (attached) in a 10 cm plate for 2 h. When testing Merlin mutations, wild‐type Merlin and its mutants were stably expressed in LN229 cells. Cells were then lysed in 1 ml of Guanidinium chloride lysis buffer (6 M guanidinium‐HCl, 0.1 M NaHPO4, 0.01 M Tris/HCl, pH 8.0, 20 mM Imidazole, 10 mM beta‐mercaptoethanol) or IP2 buffer without EDTA/EGTA as indicated. 500 μg of protein lysates were incubated with 50 μl of Ni‐NTA agarose beads (Qiagen) for 3 h at room temperature. After incubation, Ni‐NTA agarose beads were washed once with each of guanidinium washing buffer (6 M guanidinium‐HCl, 0.1 M NaHPO4, 0.01 M Tris/HCl, pH 8.0, 10 mM beta‐mercaptoethanol, 0.2% Triton X‐100), Urea washing buffer I (8 M Urea, 0.1 M NaHPO4, 0.01M Tris/HCl, pH 8.0, 10 mM beta‐mercaptoethanol, 0.2% Triton X‐100), Urea washing buffer II (8 M Urea, 0.1 M NaHPO4, 0.01 M Tris/HCl, pH 6.3, 10 mM beta‐mercaptoethanol, 0.2% Triton X‐100) and Urea washing buffer III (8 M Urea, 0.1 M NaHPO4, 0.01 M Tris/HCl, pH 6.3, 10 mM beta‐mercaptoethanol, 0.1% Triton X‐100). Bound proteins were dissociated in 20 μl of SDS sample/elution buffer (25 mM Tris, pH 6.8, 4% SDS, 5% Glycerol, 250 mM Imidazole and bromophenol blue). Eluted proteins were separated on 4–12% Bis‐Tris SDS–PAGE gels (Invitrogen) and subjected to standard immunoblotting.
RAC1 activation assay
RAC1 activation assay was performed by using the RAC1 Activation Assay Biochem Kit (Cytoskeleton, Inc. # BK035) according to the protocol recommended by the manufacturer. Briefly, LN229 cells treated with DMSO or thapsigargin were lysed in ice‐cold cell lysis buffer. Lysates containing 500 μg total cell protein were incubated with 10 μl of PAK‐PBD beads at 4°C on a rotator for 1 h. The PAK‐PBD beads were washed once with 500 μl of wash buffer for 1 min. After washing, precipitated proteins were eluted by adding 15 μl of SDS sample buffer and boiling the beads for 5 min. Eluted proteins were separated on 4–12% Bis‐Tris SDS–PAGE gels (Invitrogen) and subjected to standard immunoblotting.
BrdU incorporation assay
Meso‐33 cells or Met5‐A cells seeded at 2 × 104 cells per cm2 on coverslips were deprived of growth factors for 24 h. They were then incubated with complete medium supplemented with BrdU for 24 h (Meso‐33) or 16 h (Met5‐A). After fixation with 100% cold methanol, cells were stained with BrdU Labeling and Detection Kit I (Roche).
Tumor sphere assay
FC‐1801 cells were seeded at a density of 500 cells/well in the FC‐1801 growth medium (Dulbecco's Modified Eagle's Medium [DMEM]/F12 (15‐090‐CV, Corning) supplemented with 2 mM forskolin (F6886, Sigma), 10 ng/ml Neuregulin (396‐HB‐050, R&D), 1 × N‐2 supplement (17502048, Thermo Fisher), and 1% Antibiotic‐Antimycotic Solution [30‐004‐CI, Corning]) in 96‐well plates without adding agarose. The cells were incubated at 37°C with 5% CO2 for 2–3 weeks. Tumor spheres were counted manually under microscope.
Statistical methods
For statistical analyses (including animal studies), samples sizes were chosen based on if the differences between groups are biologically meaningful and are statistically significant. No data were excluded from the analyses. For cell experiments, all cells in one experiment were from the same pooled parental cells. All mice were from the same cohort. The mice were randomly picked to implant different types of cells. For data collection relying on objective instruments, such as plate reader, qPCR, Microscopy software, animal IVIS system, and Western blotting, the investigators were not blinded to group allocation during data collection. For animal studies, the investigators were not blinded to group allocation during data collection. The variance is similar between the groups that are being statistically compared. All center values shown were mean, and all error bars shown were standard error of the mean (s.e.m). All statistical calculations and plotting were performed using GraphPad Prism 8.
Author contributions
YW and WL conceived the project. YW performed most of the in vitro experiments with assistance from ZL, PPY, LZ, HG, BA, MG, and WL. YW, PPY, BA, and WL performed the animal experiments. HZ performed mass spectrometry analysis. YW and WL wrote an original manuscript. All authors provided intellectual inputs and edited the manuscript. WL supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We would like to thank Hong‐Gang Wang, Filippo Giancotti, Marco Giovannini, Makoto Adachi, Klaus Hahn, Jonathan Chernoff, Feng Zhang, Joan Massague, Christie Thomas and Allan Weissman for reagents; members of the Li laboratory for discussions; Thomas Abraham and Wade Edris from the Microscopy Imaging Core (Leica SP8 Confocal: 1S10OD010756‐01A1 CB); and Katherine Aird from the shRNA library core. This work was supported by DOD CDMRP Grant (W81XWH1810209 to W.L.) and the Four Diamonds Fund for Pediatric Cancer Research (to PSU).
EMBO Reports (2020) 21: e50642
Data availability
The mass spectrometry data from this publication have been deposited to the ProteomeXchange Consortium via the PRIDE (https://www.ebi.ac.uk/pride/) (Perez‐Riverol et al, 2019) partner repository with the dataset identifier PXD021057 (http://www.ebi.ac.uk/pride/archive/projects/PXD021057) and PXD021058 (http://www.ebi.ac.uk/pride/archive/projects/PXD021058).
References
- Alfthan K, Heiska L, Gronholm M, Renkema GH, Carpen O (2004) Cyclic AMP‐dependent protein kinase phosphorylates merlin at serine 518 independently of p21‐activated kinase and promotes merlin‐ezrin heterodimerization. J Biol Chem 279: 18559–18566 [DOI] [PubMed] [Google Scholar]
- Ali Khajeh J, Ju JH, Atchiba M, Allaire M, Stanley C, Heller WT, Callaway DJ, Bu Z (2014) Molecular conformation of the full‐length tumor suppressor NF2/Merlin–a small‐angle neutron scattering study. J Mol Biol 426: 2755–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae SJ, Kim M, Kim SH, Kwon YE, Lee JH, Kim J, Chung CH, Lee WJ, Seol JH (2015) NEDD4 controls intestinal stem cell homeostasis by regulating the Hippo signalling pathway. Nat Commun 6: 6314 [DOI] [PubMed] [Google Scholar]
- Chinthalapudi K, Mandati V, Zheng J, Sharff AJ, Bricogne G, Griffin PR, Kissil J, Izard T (2018) Lipid binding promotes the open conformation and tumor‐suppressive activity of neurofibromin 2. Nat Commun 9: 1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HY, Morita E, von Schwedler U, Muller B, Krausslich HG, Sundquist WI (2008) NEDD4L overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking PTAP and YPXL late domains. J Virol 82: 4884–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper J, Giancotti FG (2014) Molecular insights into NF2/Merlin tumor suppressor function. FEBS Lett 588: 2743–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couderc C, Boin A, Fuhrmann L, Vincent‐Salomon A, Mandati V, Kieffer Y, Mechta‐Grigoriou F, Del Maestro L, Chavrier P, Vallerand D et al (2016) AMOTL1 promotes breast cancer progression and is antagonized by Merlin. Neoplasia 18: 10–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr J, Leitz DHW, Szczygiel M, Dvornikov D, Fraumann SG, Kreutz C, Zadora PK, Seyhan Agircan A, Konietzke P, Engelmann TA et al (2020) Conditional deletion of Nedd4‐2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nat Commun 11: 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Alarcon C, Sapkota G, Rahman S, Chen PY, Goerner N, Macias MJ, Erdjument‐Bromage H, Tempst P, Massague J (2009) Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF‐beta signaling. Mol Cell 36: 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokey JJ, Sridharan A, Xu Y, Green J, Carraro G, Stripp BR, Perl AT, Whitsett JA (2018) Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight 3: e98738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergovich A, Bichsel SJ, Hemmings BA (2005) Human NDR kinases are rapidly activated by MOB proteins through recruitment to the plasma membrane and phosphorylation. Mol Cell Biol 25: 8259–8272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho KC, Zhou Z, She Y‐M, Chun A, Cyr TD, Yang X (2011) Itch E3 ubiquitin ligase regulates large tumor suppressor 1 tumor‐suppressor stability. Proc Natl Acad Sci USA 108: 4870–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Chen J (2008) VprBP targets Merlin to the Roc1‐Cul4A‐DDB1 E3 ligase complex for degradation. Oncogene 27: 4056–4064 [DOI] [PubMed] [Google Scholar]
- Itani OA, Stokes JB, Thomas CP (2005) Nedd4‐2 isoforms differentially associate with ENaC and regulate its activity. Am J Physiol Renal Physiol 289: F334–F346 [DOI] [PubMed] [Google Scholar]
- Jin H, Sperka T, Herrlich P, Morrison H (2006) Tumorigenic transformation by CPI‐17 through inhibition of a merlin phosphatase. Nature 442: 576–579 [DOI] [PubMed] [Google Scholar]
- Kim M, Kim M, Park SJ, Lee C, Lim DS (2016) Role of Angiomotin‐like 2 mono‐ubiquitination on YAP inhibition. EMBO Rep 17: 64–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissil JL, Johnson KC, Eckman MS, Jacks T (2002) Merlin phosphorylation by p21‐activated kinase 2 and effects of phosphorylation on merlin localization. J Biol Chem 277: 10394–10399 [DOI] [PubMed] [Google Scholar]
- Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM (2000) Localized Rac activation dynamics visualized in living cells. Science 290: 333–337 [DOI] [PubMed] [Google Scholar]
- Lee J, Zhou P (2007) DCAFs, the missing link of the CUL4‐DDB1 ubiquitin ligase. Mol Cell 26: 775–780 [DOI] [PubMed] [Google Scholar]
- Li W, You L, Cooper J, Schiavon G, Pepe‐Caprio A, Zhou L, Ishii R, Giovannini M, Hanemann CO, Long SB et al (2010) Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell 140: 477–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cooper J, Zhou L, Yang C, Erdjument‐Bromage H, Zagzag D, Snuderl M, Ladanyi M, Hanemann CO, Zhou P et al (2014) Merlin/NF2 loss‐driven tumorigenesis linked to CRL4(DCAF1)‐mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 26: 48–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhou H, Li F, Chan SW, Lin Z, Wei Z, Yang Z, Guo F, Lim CJ, Xing W et al (2015) Angiomotin binding‐induced activation of Merlin/NF2 in the Hippo pathway. Cell Res 25: 801–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wei Y, Zhang L, Yee PP, Johnson M, Zhang X, Gulley M, Atkinson JM, Trebak M, Wang HG et al (2019) Induction of store‐operated calcium entry (SOCE) suppresses glioblastoma growth by inhibiting the Hippo pathway transcriptional coactivators YAP/TAZ. Oncogene 38: 120–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnifico A, Ettenberg S, Yang C, Mariano J, Tiwari S, Fang S, Lipkowitz S, Weissman AM (2003) WW domain HECT E3s target Cbl RING finger E3s for proteasomal degradation. J Biol Chem 278: 43169–43177 [DOI] [PubMed] [Google Scholar]
- Mindos T, Dun XP, North K, Doddrell RD, Schulz A, Edwards P, Russell J, Gray B, Roberts SL, Shivane A et al (2017) Merlin controls the repair capacity of Schwann cells after injury by regulating Hippo/YAP activity. J Cell Biol 216: 495–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AJ, Jacob R (1994) Ionomycin enhances Ca2+ influx by stimulating store‐regulated cation entry and not by a direct action at the plasma membrane. Biochem J 300(Pt 3): 665–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy A, Gonzalez‐Agosti C, Cordero E, Pinney D, Candia C, Solomon F, Gusella J, Ramesh V (1998) NHE‐RF, a regulatory cofactor for Na(+)‐H+ exchange, is a common interactor for merlin and ERM (MERM) proteins. J Biol Chem 273: 1273–1276 [DOI] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrilli AM, Fernandez‐Valle C (2016) Role of Merlin/NF2 inactivation in tumor biology. Oncogene 35: 537–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo MA, Price LS, Alderson NB, Ren XD, Schwartz MA (2000) Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J 19: 2008–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salah Z, Melino G, Aqeilan RI (2011) Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Res 71: 2010–2020 [DOI] [PubMed] [Google Scholar]
- Salah Z, Cohen S, Itzhaki E, Aqeilan RI (2013) NEDD4 E3 ligase inhibits the activity of the Hippo pathway by targeting LATS1 for degradation. Cell Cycle 12: 3817–3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz A, Baader SL, Niwa‐Kawakita M, Jung MJ, Bauer R, Garcia C, Zoch A, Schacke S, Hagel C, Mautner VF et al (2013) Merlin isoform 2 in neurofibromatosis type 2‐associated polyneuropathy. Nat Neurosci 16: 426–433 [DOI] [PubMed] [Google Scholar]
- Schulz A, Buttner R, Toledo A, Baader SL, von Maltzahn J, Irintchev A, Bauer R, Morrison H (2016) Neuron‐specific deletion of the Nf2 tumor suppressor impairs functional nerve regeneration. PLoS ONE 11: e0159718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J (1997) Human p21‐activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol 7: 202–210 [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Paez JG, Curto M, Yaktine A, Pruitt WM, Saotome I, O'Bryan JP, Gupta V, Ratner N, Der CJ et al (2001) The Nf2 tumor suppressor, merlin, functions in Rac‐dependent signaling. Dev Cell 1: 63–72 [DOI] [PubMed] [Google Scholar]
- Sher I, Hanemann CO, Karplus PA, Bretscher A (2012) The tumor suppressor merlin controls growth in its open state, and phosphorylation converts it to a less‐active more‐closed state. Dev Cell 22: 703–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouloudaki K, Walz G (2012) YAP1 recruits c‐Abl to protect angiomotin‐like 1 from Nedd4‐mediated degradation. PLoS ONE 7: e35735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG et al (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3: 995–1000 [DOI] [PubMed] [Google Scholar]
- Sugihara‐Mizuno Y, Adachi M, Kobayashi Y, Hamazaki Y, Nishimura M, Imai T, Furuse M, Tsukita S (2007) Molecular characterization of angiomotin/JEAP family proteins: interaction with MUPP1/Patj and their endogenous properties. Genes Cells 12: 473–486 [DOI] [PubMed] [Google Scholar]
- Surace EI, Haipek CA, Gutmann DH (2004) Effect of merlin phosphorylation on neurofibromatosis 2 (NF2) gene function. Oncogene 23: 580–587 [DOI] [PubMed] [Google Scholar]
- Takemura H, Hughes AR, Thastrup O, Putney JW (1989) Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. Evidence that an intracellular calcium pool, and not an inositol phosphate, regulates calcium fluxes at the plasma membrane. J Biol Chem 264: 12266–12271 [PubMed] [Google Scholar]
- Tang X, Jang SW, Wang X, Liu Z, Bahr SM, Sun SY, Brat D, Gutmann DH, Ye K (2007) Akt phosphorylation regulates the tumour‐suppressor merlin through ubiquitination and degradation. Nat Cell Biol 9: 1199–1207 [DOI] [PubMed] [Google Scholar]
- Toledo A, Lang F, Doengi M, Morrison H, Stein V, Baader SL (2019) Merlin modulates process outgrowth and synaptogenesis in the cerebellum. Brain Struct Funct 224: 2121–2142 [DOI] [PubMed] [Google Scholar]
- Verma S, Yeddula N, Soda Y, Zhu Q, Pao G, Moresco J, Diedrich JK, Hong A, Plouffe S, Moroishi T et al (2019) BRCA1/BARD1‐dependent ubiquitination of NF2 regulates Hippo‐YAP1 signaling. Proc Natl Acad Sci USA 116: 7363–7370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, An J, Zhang P, Xu C, Gao K, Wu D, Wang D, Yu H, Liu JO, Yu L (2012) The Nedd4‐like ubiquitin E3 ligases target angiomotin/p130 to ubiquitin‐dependent degradation. Biochem J 444: 279–289 [DOI] [PubMed] [Google Scholar]
- Xiao GH, Beeser A, Chernoff J, Testa JR (2002) p21‐activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem 277: 883–886 [DOI] [PubMed] [Google Scholar]
- Yi C, Troutman S, Fera D, Stemmer‐Rachamimov A, Avila JL, Christian N, Persson NL, Shimono A, Speicher DW, Marmorstein R et al (2011) A tight junction‐associated Merlin‐angiomotin complex mediates Merlin's regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell 19: 527–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D (2013) Spatial organization of hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 154: 1342–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL (2015) Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163: 811–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL (2012) Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev 26: 54–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The mass spectrometry data from this publication have been deposited to the ProteomeXchange Consortium via the PRIDE (https://www.ebi.ac.uk/pride/) (Perez‐Riverol et al, 2019) partner repository with the dataset identifier PXD021057 (http://www.ebi.ac.uk/pride/archive/projects/PXD021057) and PXD021058 (http://www.ebi.ac.uk/pride/archive/projects/PXD021058).