

Abstract
BACKGROUND:
Inactivating mutations in the gene for cartilage-associated protein (CRTAP) cause osteogenesis imperfecta type VII in humans, with a phenotype that can include craniofacial defects. Dental and craniofacial manifestations have not been a focus of case reports to date. We analyzed the craniofacial and dental phenotype of Crtap−/− mice by skull measurements, micro-computed tomography (micro-CT), histology, and immunohistochemistry.
RESULTS:
Crtap−/− mice exhibited a brachycephalic skull shape with fusion of the nasofrontal suture and facial bones, resulting in mid-face retrusion and a class III dental malocclusion. Loss of CRTAP also resulted in decreased dentin volume and decreased cellular cementum volume, though acellular cementum thickness was increased. Periodontal dysfunction was revealed by decreased alveolar bone volume and mineral density, increased periodontal ligament (PDL) space, ectopic calcification within the PDL, bone-tooth ankylosis, altered immunostaining of extracellular matrix proteins in bone and PDL, increased pSMAD5, and more numerous osteoclasts on alveolar bone surfaces.
CONCLUSIONS:
Crtap−/− mice serve as a useful model of the dental and craniofacial abnormalities seen in individuals with Osteogenesis Imperfecta type VII.
Keywords: Osteogenesis imperfecta, Phenotype, Dental, Craniofacial, Skull, Micro computed tomography, Bone
1 |. INTRODUCTION
Osteogenesis Imperfecta (OI) is a generalized connective tissue disorder that occurs in 0.3 to 0.8 per 10,000 births (Orioli et al., 1986; Stevenson et al., 2012; Lindahl et al., 2015). Individuals with OI can exhibit low bone mass with increased bone fragility, blue sclera, scoliosis, midface deficiency, class III malocclusion, facial dysmorphism, dentinogenesis imperfecta and decreased life span (O’Connell and Marini, 1999; Chang et al., 2007; Foster et al., 2014; Folkestad et al., 2016; Forlino and Marini, 2016; Marini et al., 2017). The majority of patients with OI have mutations in genes for type I collagen alpha chains, COL1A1 and COL1A2 (OMIM# 166200, 166210, 259420 and 166220). These mutations cause diminished collagen deposition and/or structural defects in collagen fibrils, as well as induce cell stress due to retention of the alpha chains in the endoplasmic reticulum (Marini et al., 2017). Other individuals with OI can have mutations in genes that regulate post-translational modification (Barnes et al., 2006; Morello et al., 2006; Cabral et al., 2007; van Dijk et al., 2009), secretion (Saga et al., 1987; Satoh et al., 1996; Christiansen et al., 2010) and processing (Canty and Kadler, 2005) of type I collagen. Mutations in genes that function in the collagen synthesis pathway are almost invariably recessive and can present with a phenotype that is equally or more severe than that of the dominant forms of OI. This can be explained in part by the fact that the synthesis of additional abnormal fibrillar collagens is affected by these mutations. Of relevance to this study, craniofacial dysmorphologies are more common in OI associated with structural as opposed to quantitative defects in collagen (Jensen and Lund, 1997; Jabbour et al., 2018).
Cartilage-associated protein (CRTAP) is a resident protein of the rough endoplasmic reticulum that forms a heterotrimeric complex with prolyl-3-hydroxylase-1 (P3H1) and cyclophilin B (CYPB). This complex facilitates the post-translational modification of procollagen chains and the folding of the triple helix (Morello et al., 2006; Baldridge et al., 2010; Barbirato et al., 2015). Biallelic mutations in CRTAP in humans cause osteogenesis imperfecta type VII (OMIM #610682), characterized by osteopenia, rib and long bone fractures, failure to thrive, hypermobility/joint laxity, rhizomelia (shortening of proximal limbs) and craniofacial defects (Ward et al., 2002; Valli et al., 2012). Mutations in genes for collagen processing proteins, including P4HB (Rauch et al., 2015), and CRTAP (Balasubramanian et al., 2015), can also cause Cole-Carpenter Syndrome. The craniofacial characteristics of this syndrome include frontal bossing, proptosis, maxillary hypoplasia, craniosynostosis and/or wormian bones (supernumerary bones that result from formation of ectopic ossification centers that develop in addition to those normally present, usually in conjunction with craniosynostosis) (Cole and Carpenter, 1987; Chang et al., 2007; Marini et al., 2010; Balasubramanian et al., 2015; Rauch et al., 2015).
Loss of CRTAP in mice causes decreases in collagen prolyl 3-hydroxylation, osteoid production, mineral apposition rate and bone formation rate and altered collagen fibrillogenesis (Morello et al., 2006). Crtap−/− mice develop progressive kyphoscoliosis, pre- and postnatal growth delay, rhizomelia, disorganization of proliferating chondrocytes and osteopenia. However, no studies have determined if dental and/or craniofacial skeletal defects were present in these mice. Treatment of Crtap−/− mice with a sclerostin neutralizing antibody improved vertebral and long bone cortical and trabecular parameters as well as improved mechanical properties (Grafe et al., 2016). The goal of this study was to analyze dental and craniofacial defects in the Crtap−/− mouse model of recessive osteogenesis imperfecta, to provide insights into functions of CRTAP in those tissues and point to potential dental and craniofacial manifestations in humans with OI type VII, allowing for future studies investigating potential treatments for these aspects of the phenotype.
2 |. RESULTS
Altered Craniofacial Shape and Bone Mineralization in Crtap−/− Mice
Initial qualitative analyses indicated that, compared to Crtap+/+ controls, Crtap−/− mice had shorter nasal bones and overall anterior/posterior skull lengths, and a wider skull with an open mandibular symphysis and abnormalities within nasofrontal, frontomaxillary and palatomaxillary sutures (Figure 1A–J). Fusion of the nasofrontal, frontomaxillary and palatomaxillary sutures was evident in all Crtap−/− mice but none of the Crtap+/+ mice (contingency P < 0.01). Crtap−/− mice also exhibited a class III malocclusion with maxillary molar teeth not anterior to the mandibular molar teeth and molar teeth not in a cusp to fossa relationship, when compared to Crtap+/+ littermates (Figure 1C, D). Linear skull measurements normalized to total skull lengths demonstrated significantly increased cranial width, inner canthal distance, parietal bone length, cranial vault lengths (two measurements) and cranial base bone lengths (presphenoid, basisphenoid and basioccipitus), with significantly diminished nasal bone length in Crtap−/− compared to WT mice (Figure 1K). Together these data indicate a brachycephalic (wide) skull shape with a longer (anterior-posterior) cranial vault and cranial base length but shorter midfacial length in the Crtap−/− mice.
Figure 1. Altered Craniofacial Skeletal Phenotype of Crtap−/− mice.

(A, B) Alizarin red staining of whole skulls reveals shorter nasal bones and overall skull length, as well as an open mandibular symphysis in Crtap−/− vs. Crtap+/+ mice. (C,D) Micro-CT lateral isosurface images including the mandible reveals a shorter nose length (nasale to bregma) and a class III malocclusion (pink vertical lines show maxillary molars to be anterior of mandibular molars in Crtap+/+ mice but not in Crtap−/− mice). (E-F) Micro-CT axial isosurface images from above reveal abnormalities within and surrounding the frontonasal and the frontomaxillary sutures (pink arrow in F). (G,H) Micro-CT axial isosurface images from below with mandible removed reveal abnormalities within and surrounding the palatomaxillary sutures (pink arrow in H). (I,J) Lateral slice micro-CT images again reveal abnormalities in the frontonasal suture (pink arrow in J). No evidence of craniosynostosis or wormian bones is evident. (K) Linear craniofacial measurements were taken using landmarks placed on micro-CT scans. Skulls of adult Crtap−/− mice are smaller than those of Crtap+/+ littermates, therefore measurements are presented as normalized to the total skull length. Normalized measurements show increased cranial width, increased infraorbital width, increased parietal bone length, increased cranial vault lengths and increased cranial base bone lengths (presphenoid, basisphenoid, basioccipitus), with decreased total skull length and nasal bone length when compared to Crtap+/+ littermates (n=5 per genotype at 2 months). Please note that two measurements of cranial vault length were taken (see landmarks and measurements shown in Figure 6). Results are shown as mean +/− standard deviation. *p < 0.05 between genotypes.
Histologic staining of skull sections revealed thickened and dysmorphic bone within anterior frontal and posterior nasal bones and a fused nasofrontal suture in all Crtap−/− mice but absent in Crtap+/+ mice (Figure 2; contingency P < 0.01). Micro-CT analyses demonstrated significantly diminished (−23 to −32% reduction; P < 0.05) bone volume, bone mineral content, and tissue mineral content in parietal and frontal bones in Crtap−/− as compared to Crtap+/+ mice (Table 1). Moreover, micro-CT analyses demonstrated significantly diminished (−19 to −38% reduction; P < 0.05) bone volume, bone volume fraction, bone mineral content, bone mineral density, and tissue mineral content in nasal bones (Table 1).
Figure 2. Crtap−/− Mice Exhibit Fusion of Frontal and Nasal Bones.
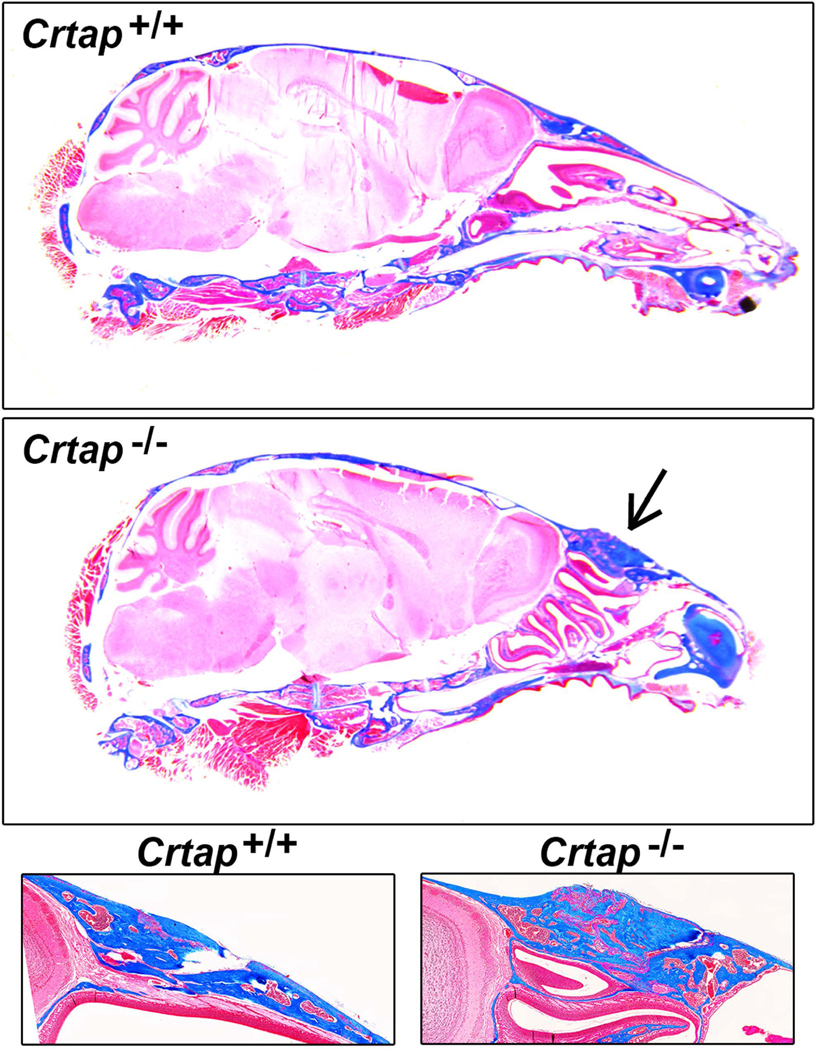
Histologic Masson’s trichrome stain on decalcified tissue sections reveals thickened and dysmorphic anterior frontal and posterior nasal bone in addition to fusion of the nasofrontal suture (black arrow). These defects were seen in all Crtap−/− mice (5/5) but no Crtap+/+ mice at 2 months.
Table 1.
Craniofacial bone analyses in Crtap−/− vs Crtap+/+ mice
| Frontal | Parietal | Nasal | ||||
|---|---|---|---|---|---|---|
| Crtap+/+ | Crtap−/− | Crtap+/+ | Crtap−/− | Crtap+/+ | Crtap−/− | |
| BV (mm3) | 0.033 ± −0.005 | 0.023 ± 0.002* (−30%) | 0.034 ± 0.002 | 0.024 ± 0.002* (−29%) | 0.039 ± 0.012 | 0.025 ± 0.011* (−36%) |
| BV/TV | 0.79 ± 0.10 | 0.86 ± 0.09 | 0.95 ± 0.02 | 0.91 ± 0.05 | 0.49 ± 0.01 | 0.34 ± 0.02* (−31%) |
| BMC (mg) | 0.022 ± 0.003 | 0.017 ± 0.002* (−23%) | 0.026 ± 0.002 | 0.018 ± 0.002* (−31%) | 0.41 ± 0.1 | 0.30 ± 0.01* (−27%) |
| BMD (mg/cc) | 680 ± 51 | 712 ± 60 | 769 ± 22 | 741 ± 30 | 518 ± 76 | 419 ± 98* (−19%) |
| TMC (mg) | 0.020 ± 0.002 | 0.015 ± 0.003* (−25%) | 0.025 ± 0.002 | 0.017 ± 0.002* (−32%) | 029 ± 0.010 | 0.18 ± 0.008* (−38%) |
| TMD (mg/cc) | 759 ± 30 | 753 ± 42 | 784 ± 18 | 771 ± 13 | 734 ± 40 | 707 ± 39 |
Note. MicroCT was used to measure total bone volume (BV). bone volume fraction (BV/TV). bone mi neral content (BMC), bone mineral density (BMD), tissue mineral content (TMC), and tissue mineral density (TMD) in cranial and nasal bones of Crtap+/+ and Crtap−/− mice.
indicate statistical significance between genotypes, P < .05. Percentage changes between genotypes are also shown.
Altered Dental and Periodontal Tissues in Crtap−/− Mice
Initial 3D and 2D micro-CT observations of mandibles revealed short and thin molar roots and reduced cellular cementum in Crtap−/− vs. control mice (Figure 3A–F). Heat maps of mineral density further supported reduced cellular cementum volume and suggested similar distribution of mineral density between genotypes, with perhaps more variability noted in Crtap−/− vs. Crtap+/+ dentin (Figure 3B, E). Crtap−/− mice exhibited reduced alveolar bone (# in Figure 3F) and despite reduced cellular cementum and bone volume, showed clear instances of ankylosis (indicated by arrow in Figure 3F). Quantitative micro-CT analysis confirmed, compared to controls, Crtap−/− mice had decreased volumes of dentin (−18%; P < 0.01), cellular cementum (−48%; P < 0.01) and alveolar bone (−28%; P < 0.01) (Figure 3G). Conversely, Crtap−/− mice exhibited increased volumes of PDL (+15%; P < 0.01) and dental pulp (+12%; P < 0.05). While dentin and cellular cementum densities were not different, alveolar bone density was decreased in Crtap−/− mice compared to controls (−2%; P < 0.05). Neither enamel volume nor density was affected in Crtap−/− mice. To determine whether dentin defects were localized to a specific region, dentin was subdivided into crown and root segments. Volume was significantly decreased in both components (P < 0.01–0.05) of Crtap−/− mouse molar dentin, while mineral density was not altered (Figure 4). Interestingly, both pulp and PDL densities were reduced in Crtap−/− vs. control mice (P < 0.01–0.001).
Figure 3. Altered Dental Development in Crtap−/− Mice.
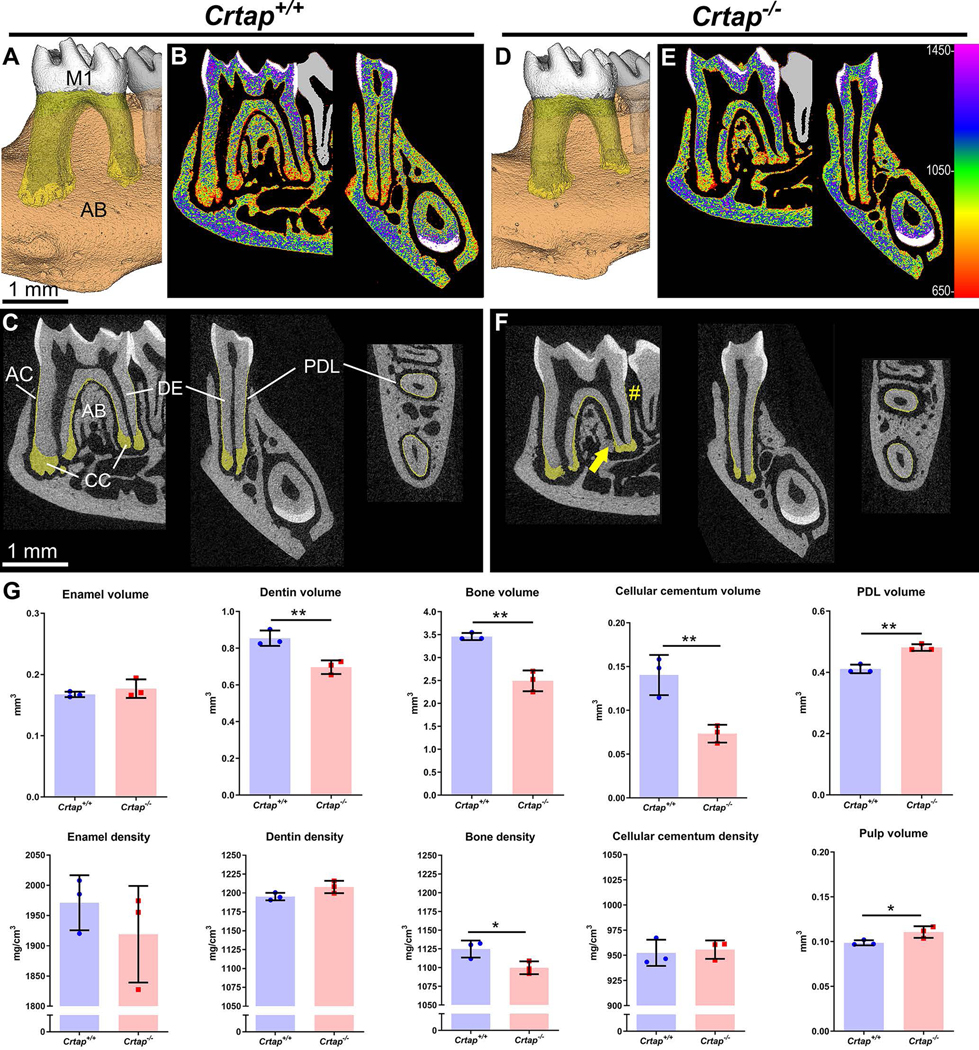
(A-F) 3D renders and 2D sagittal sections from micro-CT scans of Crtap+/+ and Crtap−/− mouse mandibles show first molar (M1), surrounding alveolar bone (AB; brown, transparent), acellular and cellular cementum (AC and CC; yellow, transparent), enamel (EN; white, opaque), and dentin (DE; gray, opaque). Heat map 2D images in panels B and E correspond to HA mineral densities (mg/cm3) defined by the color bar on righthand edge of E. (G) Quantification of micro-CT reveals that compared to Crtap+/+ controls (n=3 per genotype at 2 months). Crtap−/− mice feature significantly decreased volumes of DE, AB, and CC, and significantly increased PDL and dental pulp volumes. AB density is significantly decreased in Crtap−/− mice compared to Crtap+/+ controls. Results are shown as mean +/− standard deviation. *p < 0.05; ** p < 0.01 between genotypes.
Figure 4. Altered Dentin, Pulp, and PDL in Crtap−/− Mice.

Quantification of micro-CT reveals that compared to Crtap+/+ controls (n=3 per genotype at 2 months). Crtap−/− mice feature significantly decreased crown and root molar dentin volumes, while mineral density was different in neither. Both pulp and PDL densities were reduced in Crtap−/− vs. control mice. *p < 0.05; ** p < 0.01, ***p<.005 between genotypes.
Histology was performed to further explore differences revealed by micro-CT analysis. Histological organization of Crtap−/− mouse molars appeared grossly normal, though cellular cementum was dramatically reduced (Figure 5A–D; cellular cementum indicated by red dotted lines). Histomorphometry indicated no significant difference in predentin/dentin ratio, suggestive of delayed mineralization (Figure 5E). Acellular cementum thickness was increased in Crtap−/− vs. control mice (+100%; P < 0.01), while cellular cementum surface area was reduced on the lingual aspect (−40%; P < 0.05). PDL width was confirmed to be increased in Crtap−/− vs. control mice (+15%; P < 0.01), as indicated by micro-CT 3D volume analysis (Figure 5F–H).
Figure 5. Disrupted Periodontal Structure and Maintenance in Crtap−/− Mice.
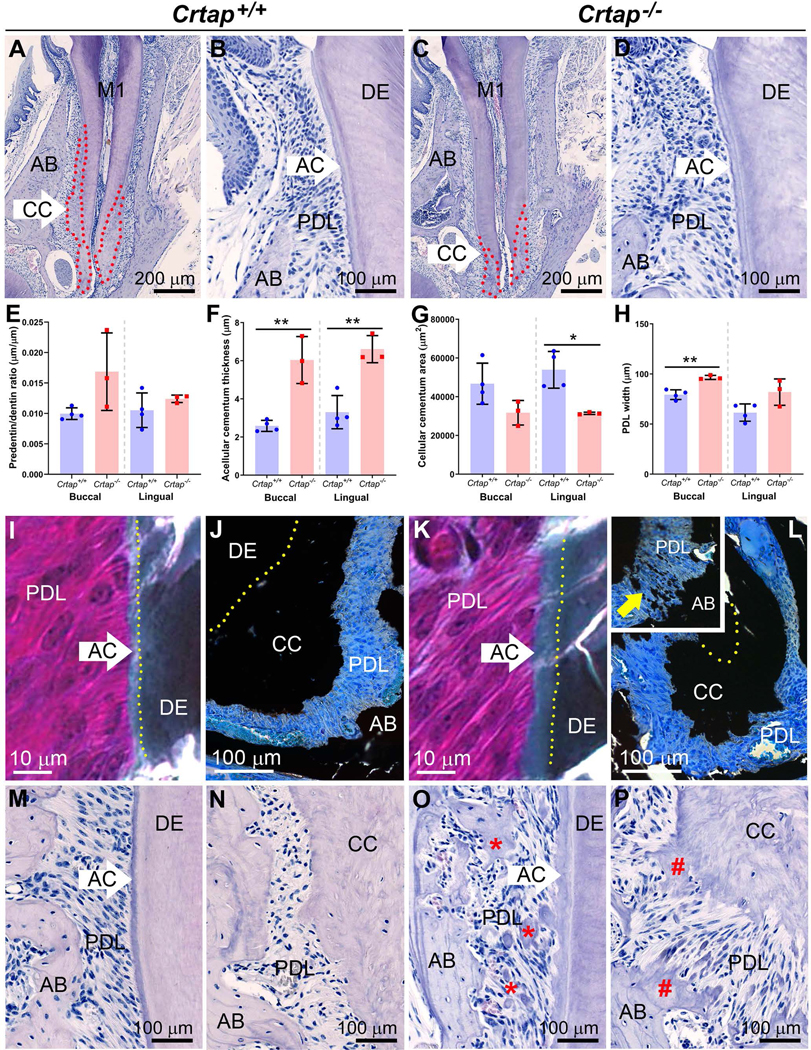
(A-D) H&E stained histological sections show Crtap+/+ and Crtap−/− mouse first mandibular molars (M1), acellular cementum (AC), cellular cementum (CC), periodontal ligament (PDL), and surrounding alveolar bone (AB). AC appears thicker while CC (outlined in red dotted lines in panels A and C) appears reduced in Crtap−/− mice. (E-H) Histomorphometric analyses reveal no significant difference in predentin/dentin ratio, but significant increases in AC and PDL thicknesses, and significantly reduced CC area in Crtap−/− vs. Crtap+/+ mice (n=3–4 per genotype at 2 months). Results are shown as mean +/− standard deviation. *p < 0.05; ** p < 0.01 between genotypes. (I, K) Goldner trichrome staining on undecalcified sections confirms presence of thicker AC (light green layer on darker green DE) in Crtap−/− mice. (J, L) Von Kossa staining on undecalcified sections reveals irregular borders on smaller CC, as well as evidence for ectopic calcification within the PDL (yellow arrow in inset). (M-P) Observation of dental tissues in a small number of older Crtap+/+ and Crtap−/− mice (n=2 per genotype at 3 and 10–11 months; 10–11 months shown here) reveals more severely disrupted periodontium in Crtap−/− mice featuring invasion of the cervical PDL space with extensive bone/cementum-like nodules (red * in O) and inappropriate ingrowth of both AB and CC into the apical PDL (red # in P). Panels I, J, M and N are from Crtap+/+ mice while panels K, L, O, and P are from Crtap−/− mice.
Undecalcified histology was performed to investigate differences in acellular and cellular cementum. Goldner’s trichrome staining identified a thicker acellular cementum layer in Crtap−/− vs. control molar roots (Figure 5I, K). Von Kossa staining showed reduced cellular cementum layer in Crtap−/− compared to control mice, albeit with an unusual, splayed and disorganized border protruding into the PDL (Figure 5J, L). In some sections, ectopic mineral nodules were identified within Crtap−/− mouse PDL, which were never observed in the PDL region of Crtap+/+ mice (Figure 5L inset).
Based on morphological changes observed at 2 months, a small cohort of mice were examined to determine longer-term changes in periodontal tissues. Compared to controls, Crtap−/− mice at 3 or 10 to 11-month-old exhibited severely disrupted periodontal structures, with cervical alveolar bone breakdown and invasion of the PDL with numerous bone-like or cementum-like nodules, while apical bone and cellular cementum showed signs of invading the PDL space (Figure 5M–P). Acellular cementum of Crtap−/− mice showed increased width as well as a rough and irregular border protruding into the PDL, unlike the relatively smooth surfaces in controls.
To provide additional insights into morphological differences in Crtap−/− vs. control periodontia, immunostaining was performed for key mineralized tissue markers. Immunolocalizations for bone sialoprotein (BSP) and osteopontin (OPN) emphasized thicker acellular cementum and altered alveolar bone modeling/remodeling in in Crtap−/− vs. control molars (Figure 6A–D). Immunostaining for periostin (POSTN), a marker PDL fibers, indicated reduced numbers of POSTN-positive Sharpey’s fibers incorporated into Crtap−/− vs. control alveolar bone (Figure 6E, G). Increased TGFβ signaling has been reported in both autosomal dominant and recessive types of OI, including in skeletal elements of Crtap−/− mice (Grafe et al., 2014). IHC for the phosphorylated and active form of TGFβ receptor-regulated transcriptional mediator SMAD5 (pSMAD5), previously demonstrated in the PDL (Wang et al., 2014), suggested increased numbers of SMAD5-positive cells in the PDL of Crtap−/− vs. control mice (Figure 6F, H). Because of changes in bone volume and modeling/remodeling, we quantified tartrate-resistant acid phosphatase positive (TRAP+) osteoclast-like cells, finding more than 2-fold greater numbers of osteoclasts in Crtap−/− vs. control mice (p<0.01; Figure 6I–K).
Figure 6. Altered Periodontal Markers in in Crtap−/− Mice.
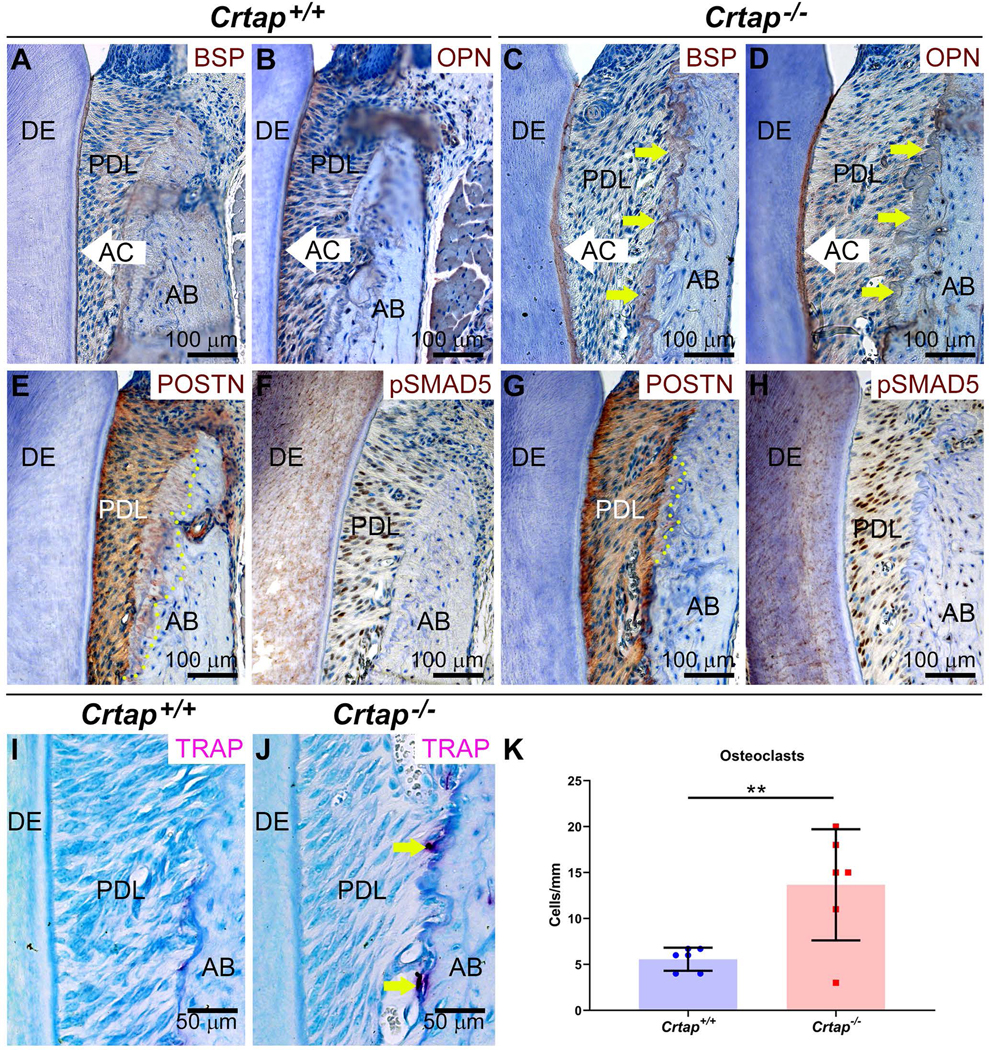
Immunohistochemistry reveals altered (A, C) bone sialoprotein (BSP) and (B, D) osteopontin (OPN) in Crap−/− vs. control alveolar bone (AB) adjacent to periodontal ligament (PDL) (indicated by yellow arrows), as well as a thicker acellular cementum (AC) layer. DE=dentin. (E, G) While periostin (POSTN) localizes to PDL and embedded Sharpey’s fibers (indicated by yellow dotted line) in AB of control mice, reduced numbers of POSTN-positive Sharpey’s fibers are included in Crtap−/− AB. (F, H) Increased numbers of cells positive for phosphorylated SMAD5 (pSMAD5) are present in the PDL of Crtap−/− vs. control mice. (I, J) Increased numbers of tartrate-resistant acid phosphatase positive (TRAP+) osteoclast-like cells are observed on Crtap−/− vs. control AB surfaces. (K) Quantification reveals more than 2-fold greater numbers of osteoclast-like cells in Crtap−/− vs. control mice (**p<0.01).
3 |. DISCUSSION
Cartilage-associated protein (CRTAP) is a rough endoplasmic reticulum protein involved in posttranslational modifications of fibrillar collagens. Inactivating mutations in the CRTAP gene cause osteogenesis imperfecta (OI) type VII in humans, with a phenotype that can include craniofacial bone defects. In the few case reports on type VII OI to date, oral health status has not been reported. To provide further insights into the roles of CRTAP in the craniofacial region, we performed multimodal analysis of mice genetically ablated for Crtap. Crtap−/− mice exhibited craniofacial abnormalities in the form of a brachycephalic skull shape, thickening of anterior frontal and posterior nasal bones, fusion of the nasofrontal suture, and diminished bone volumes and densities in nasal and craniofacial base bones. Loss of CRTAP also resulted in decreased dentin volume and periodontal defects including decreased cellular cementum volume, decreased alveolar bone volume and mineral density, increased PDL space, ectopic calcification within the PDL, and bone-tooth ankylosis. These changes were associated with altered localization of periodontal markers, increase in TGFβ signaling mediator pSMAD5, and greater numbers of osteoclasts. This mouse phenotype represents a novel form of OI-associated dentinogenesis imperfecta (DI) that extends beyond dentin effects. Taking into account how other forms of OI impact dentoalveolar tissues in affected subjects, these findings suggest that the oral health of individuals with type VII OI should be evaluated carefully and followed closely.
3.1 |. Role of CRTAP in Craniofacial Morphogenesis
OI type VII classification was initially created to report the effect of a recessive, moderate to severe form of OI, which occurred in a large, consanguineous Quebec family (an isolated First Nations community) due to a hypomorphic mutation in CRTAP (Ward et al., 2002; Valli et al., 2012). While the reported craniofacial defects of this population were limited to evidence of wormian bones, case reports of more severely affected individuals with CRTAP null mutations (eg: those with Cole-Carpenter Syndrome) include craniofacial features such as craniosynostosis, midface hypoplasia, doming forehead/frontal bossing and wormian bones (Cole and Carpenter, 1987; Balasubramanian et al., 2015). We did not find evidence of cranial bone fusions or extracranial sutures (wormian bones) in Crtap−/− mice, but facial bone fusions, including fusion of the nasofrontal suture, were evident. The mice also exhibited an open mandibular symphysis suggestive of joint laxity, frontal bossing, midface hypoplasia and a class III malocclusion, in addition to abnormalities in bone mineral content, density and volume in cranial and nasal bones. The brachycephalic head shape with frontal bossing and class III malocclusion were likely caused by the facial bone fusions, leading to midface retrusion. In combination with previous case reports, these data indicate that OI type VII individuals should be referred to a craniofacial team for assessment, regardless of the presence of overt craniosynostosis, because surgical treatment for correction of facial bone fusions and malocclusion may be needed (Aizenbud et al., 2008; Rosen et al., 2011). It is also worth noting that we found a highly consistent craniofacial phenotype in Crtap−/− mice on a mixed C57BL6/J;129/SvEv genetic background. This signifies that phenotype severity is unlikely to be genetic background-dependent. It is therefore possible that CRTAP homozygous null mutations cause a less severe phenotype in mice than in humans, as was previously suggested by others (Marini et al., 2010). Overall, our craniofacial findings in Crtap−/− mice represent a more moderate craniofacial phenotype than that seen in individuals with Cole-Carpenter Syndrome, that appears to phenocopy humans with OI type VII.
3.2 |. Importance of CRTAP in Dental and Periodontal Development and Function
The more common forms of OI arising from mutations in COL1A1 and COL1A2 (types I-IV) can be accompanied by a well characterized dental phenotype sometimes called OI-related DI, distinct from DI caused by mutations in the dentin sialophosphoprotein (DSPP) gene. OI-affected teeth have been described in many case reports, with the phenotype including smaller tooth size, altered crown and/or root morphology, ultrastructural abnormalities in dentin, discoloration of dentin, and pulp calcifications leading to obliteration [summarized in (Foster et al., 2014)]. The description of periodontal phenotypes associated with OI is exceedingly rare, which is surprising considering that cementum, PDL, and alveolar bone extracellular matrices are approximately 90% type I collagen and would be expected to manifest direct effects of OI-associated mutations. A single report of type IV OI due to mutations in COL1A2 describes by scanning electron microscopy, “bush-like” ectopic calcifications on cementum surfaces of primary teeth of the affected child, the first such observation (Kantaputra et al., 2018).
Several mouse models of OI have been developed. Oim mice featuring a Col1a2 frameshift mutation, exhibit thin dentin, wide pulp chambers, and fragile teeth. Brtl mice, a model for moderately severe type IV OI due to a knock-in Col1a1 mutation, feature reduced dentin, wide pulp chambers, delayed dentin mineralization, and qualitative micro-CT findings revealed reduced and disorganized alveolar bone (Boskey et al., 2013). CypB (or Ppib) null mice, a model for OI type IX, were described with thin dentin, irregular predentin/dentin border, and smaller dentin collagen fibrils (Terajima et al., 2017). Detailed and quantitative analyses of periodontal tissues were lacking in those studies. The report here is the first to examine the dental phenotypes of Crtap null mice, a model for autosomal recessive OI type VII. Our dental analyses agree with and extend previous mouse OI findings on many points. Ablation of Crtap in mice reduced both dentin volume (crown and root) and alveolar bone volume. While dentin density was not grossly abnormal, increased variation in mineral density distribution was observed, and bone displayed significantly reduced mineralization. Pulp volume increased in Crtap−/− mice in association with dramatically thinner dentin. Importantly, no major alterations were observed in tooth enamel.
Two studies on mouse models of OI have examined periodontal tissues. Conditional deletion of both Bmp1 and Tll1 in mice was proposed to model a form of autosomal recessive OI (type XIII is caused by BMP1 mutations), and these mice showed short molars with thin root dentin (Muir et al., 2014). Periodontal involvement included reduced cellular cementum, derangement of PDL, and periodontal breakdown including alveolar bone resorption. Mice featuring an induced mutation in the Col1a1 gene (Col1a1Jrt/+) were proposed to replicate effects of OI and Ehlers Danlos syndrome (Eimar et al., 2016). Their incisors featured defective dentin mineralization and surrounding alveolar bone had smaller collagen fibrils.
Our analyses provide further insight into the effects of OI on periodontal tissues. Ablation of Crtap led to increased acellular cementum with an irregular surface topology at older ages, decreased cellular cementum, and dramatically decreased volume and mineral density of alveolar bone, and increased PDL space. The altered acellular cementum formation is consistent with electron microscopy findings in a human patient with type IV OI (Kantaputra et al., 2018), suggesting that multiple forms of OI may have parallel effects on periodontal tissues, a hypothesis that deserves further analysis. We observed numerous instances of ectopic calcification in the PDL and evidence of ankylosis in Crtap−/− mice, and alveolar bone showed signs of increased modeling/remodeling including altered localization of BSP, OPN, and POSTN and increased numbers of TRAP+ osteoclasts on alveolar bone surfaces. Increased numbers of pSMAD5-positive cells indicated increased TGFβ signaling, similar to findings in calvaria and long bones of Crtap−/− mice and other OI models (Grafe et al., 2014). Notably, increased pSMAD5 was found in periodontal tissues of mice dually ablated for proteoglycans fibromodulin and biglycan, in association with elevated numbers of osteoclasts and increased bone remodeling (Wang et al., 2014). Increased TGFβ signaling was also linked to compression-induced osteogenic differentiation of PDL cells in vitro (Manokawinchoke et al., 2019). The relationship between altered remodeling, ectopic nodules in the PDL, and tooth-bone ankylosis remains unclear, though all are suspected to be linked. The PDL is remarkably resistant to ectopic calcification and ankylosis, as demonstrated in several genetically engineered mouse models, however the carefully maintained homeostasis can be disrupted by substantial disruption of matrix production, mineralization, and/or resorption activities(Wesselink and Beertsen, 1994a; Wesselink and Beertsen, 1994b; Schatzle et al., 2005; Zweifler et al., 2015; Foster et al., 2018; Thumbigere-Math et al., 2018; Wolf et al., 2018).
None of the case reports on OI type VII that have been published to date have included dental observations. Based on data collected from Crtap−/− mice, we propose a novel form of OI-associated DI that deserves further study and suggests the oral health of individuals with type VII OI should be evaluated carefully and followed closely.
4 |. CONCLUSION
The findings presented here represent the first qualitative and quantitative analysis of the functional importance of CRTAP in craniofacial, dental and periodontal tissues, providing new insights into effects of OI on these tissues. Overall the results are consistent with craniofacial and dental phenotypes of Col1a1 (Col1a1Jrt/+) mutant mice, which are characterized by short skull length, malocclusion, widened periodontal space, and defects in the dentin matrix and mineralization(Eimar et al., 2016). Limitations of this study include a relatively small sample size; while dramatic differences were detected in a number of craniofacial and dentoalveolar measurements, more subtle differences may not reach significance. While n=5 mice per genotype at age 2 months were used for the quantitative analyses, additional mice at 1, 3, and 10–11 months manifested similar phenotypes. While our analysis was multimodal, additional studies may include electron microscopy or atomic force microscopy, Raman spectroscopy, or other approaches to reveal novel differences in craniofacial tissues in the absence of CRTAP. Challenge models such as bone defect healing and orthodontic tooth movement can also provide important insights in addition to developmental studies and should be considered as future steps.
5 |. EXPERIMENTAL PROCEDURES
5.1 |. Animals
Five 2-month-old male Crtap−/− mice and five 2-month-old male wild type littermates on a mixed C57BL6/J;129/SvEv background were analyzed in random order, with the dental phenotype additionally qualitatively observed in a small cohort of mice at 3 and 10–11 months (n=2 per genotype). Mice were housed in a pathogen-free facility, with unlimited access to water, standard rodent chow and a 12-hour light/dark cycle. Genotyping was performed by PCR using the primers: Forward 5’-TGACCGCTTCCTCGTGC-3’ and Reverse 5’-CCCGCCTATCACCAACC-3’ for detecting the mutant allele, and Forward 5’-GGCCAATGACCTCCCGAAG-3’ and Reverse 5’-AACTTCGGGGTAAAGCCAGAG-3’ for the wild type allele. Sample sizes were based upon previous long bone and connective tissue analyses showing high consistency of phenotype in Crtap−/− mice (Morello et al., 2006; Baldridge et al., 2010). Mice were euthanized to harvest relevant tissues according to the recommendations of the Guide for Care and Use of Laboratory Animals 8th Edition. All mice were healthy with no noted adverse effects in mutant mice. Mice were euthanized and tissues were fixed in 10% neutral buffered formalin. For initial phenotypic analysis, whole skulls of Crtap−/− mice and wild type littermates were dissected and stained with Alizarin Red. All animal work performed in this study was conducted under approval of the University of Arkansas animal use ethics committee and conforms to ARRIVE guidelines. Primary outcomes measures included skull morphology, craniofacial bone fusions, tooth and alveolar bone mineral volume and density, plus periodontal ligament structure. Secondary outcomes measures were craniofacial bone volume/density/mineral content.
5.2 |. Micro-Computed Tomography
Mouse skulls were scanned at an 18 μm isotropic voxel resolution using the eXplore Locus SP micro-computed tomography (micro-CT) imaging system (GE Healthcare Pre-Clinical Imaging). Regions of interest (ROI’s) for parietal and frontal bones were established as previously described (Liu et al., 2013; Liu et al., 2014). A region of interest (ROI) for the nasal bone included the entire nasal bone as outlined by custom spline. Density, volume and mineral content of cranial bones, nasal bones and cranial base bones from mice were measured using Microview version 2.2 software and established algorithms (Meganck et al., 2009; Umoh et al., 2009). Micro-CT bone data were analyzed and are reported in accordance with the recommendations of Bouxsein et al (Bouxsein et al., 2010).
Mandibles were scanned in a μCT 50 (Scanco Medical, Bassersdorf, Switzerland) at 70 kVp, 76 μA, 0.5 Al filter, 900 ms integration time, and 6 μm voxel dimension and analyzed using AnalyzePro (version 1.0; AnalyzeDirect, Overland Park, KS), as previously described (Thumbigere-Math et al., 2018). For isolation of cementum from dentin, images were first filtered using a median filter, 7 kernel size in all three axes, and a cementum “mask” was segmented from 450–1050 mg/cm3 HA. This “mask” was then loaded onto the original calibrated image and cementum was traced under the “mask” for any mineralized tissue above 650 mg/cm3 HA. Crown and root dentin were subdivided: “crown dentin” was designated as dentin coronal to the cementoenamel junction (CEJ), and “root dentin” was defined as dentin apical to the CEJ. Reconstructed images were analyzed using AnalyzePro (version 1.0; AnalyzeDirect, Overland Park, KS), as previously described (Thumbigere-Math et al., 2018).
5.3 |. Linear Skull Measurements
Craniofacial linear skeletal measurements were taken by placing landmarks on micro CT skull scans using Dolphing Imaging 11.0 software (Dolphin Imaging and Management Solutions, Catsworth, CA) using previously established landmarks (Figure 7) (Liu et al., 2013; Liu et al., 2014). This software displays scans in axial, sagittal and coronal slices for landmark verification in all views simultaneously. Linear measurements were normalized to total skull length (measured from nasale to paro) to account for size differences between Crtap+/+ and Crtap−/− mice. Measurements were performed twice and an average of the two measurements was utilized for statistical comparison by genotype.
Figure 7. Craniofacial Landmarks and Linear Distances.

3D landmarks placed on micro CT images of skulls to compare the craniofacial shape of Crtap−/− mice and wild type littermates. Micro-CT regions of interest for the parietal bone is outlined in black on axial image; Regions of interest for the frontal bone is outlined in blue on axial image; Regions of interest for the nasal bone is outlined in green on axial image. These regions of interest are custom splined to extend only enough to include the entire bone thickness, as described in experimental procedures.
5.4 |. Histological Analyses
Histologic sections from adult mice were taken to examine craniofacial bones, sutures and synchondroses of the cranial base. Dissected skulls were fixed, decalcified, incubated in xylene then embedded in paraffin. Paraffin blocks were cut in 4 uM sagittal sections using a Leica RM2255 microtome equipped with a tungsten carbide blade (Leica Microsystems Inc.). Sections were transferred to slides and dried at 42 °C in a slide press overnight, then stained with Masson’s trichrome stain.
Hemi-mandibles prepared for histology were decalcified in acetic acid/formalin/sodium chloride solution, and paraffin embedded for 6 um coronal sectioning and hematoxylin and eosin (H&E) staining (Thumbigere-Math et al., 2018). Histomorphometry was performed on H&E stained sections chosen from the center of the first molar mesial root. Measurements were performed on buccal and lingual aspects of the root at a distance 300 um apical to the CEJ: Predentin and dentin thickness, acellular cementum thickness, and periodontal ligament (PDL) thickness. Cellular cementum area was measured on buccal and lingual aspects of the apical root. Histomorphometric measurements were made using ImageJ software (version 1.49d; Bethesda, MD). Non-decalcified hemi-mandibles were embedded in methyl methacrylate for von Kossa and Goldner’s trichrome staining, as described previously (Foster et al., 2015).
Immunohistochemistry (IHC) was performed on deparaffinized tissue sections using an avidin-biotinylated peroxidase-based kit with a 3-amino-9-ethylcarbazole substrate (Vector Labs, Burlingame, CA) to produce a red-brown product (n = 3–4 mice/genotype). Primary antibodies included: rabbit polyclonal anti-bone sialoprotein (BSP; courtesy of Dr. Renny Franceschi, University of Michigan, Ann Arbor, MA) (Foster et al., 2013); rabbit polyclonal anti-mouse osteopontin (LF-175; OPN, courtesy of Dr. Larry Fisher, NIDCR, Bethesda, MD) (Foster et al., 2018); rabbit polyclonal anti-periostin (POSTN; Abcam, Cambridge, MA); and rabbit monoclonal anti-phospho-SMAD5 (phospho-S463 and S465; Abcam) (Wang et al., 2014). Tartrate-resistant acid phosphatase (TRAP) staining was performed to identify by a red-purple stain multinucleated osteoclast-like cells on alveolar bone surfaces (Foster et al., 2018). Enumeration of TRAP+ osteoclasts and measurement of bone surfaces, was performed using ImageJ software.
5.5 |. Statistical Analyses
Comparisons of craniofacial parameters between genotypes were performed using Fisher’s exact test and two-way Student’s t-test. Micro-CT data analyses of teeth between genotypes were performed using ANOVA and post-hoc Tukey test for multiple comparisons. Statistical tests were performed using GraphPad Prism version 8.0. Groups were considered to be significantly different at P < 0.05, though actual P values as low as P < 0.001 are reported. For consistency throughout the manuscript, statistical abbreviations are: a indicates statistical significance between genotypes at P < 0.001, b indicates statistical significance between genotypes at P < 0.005, c indicates statistical significance between genotypes at P < 0.01, d indicates statistical significance between genotypes at P < 0.05.
6 |. ACKNOWLEDGEMENTS
The authors thank Dr. Evelyn Ralston and Dr. Kristina Zaal (Light Imaging Section, NIAMS/NIH) for assistance in slide scanning and Daniel Leigh and Alyssa Coulter for micro-CT and histological assistance. The authors thank Hwa Kyung Nam (University of Michigan) for her assistance with tissue sectioning and staining.
This research was supported by: NIDCR/NIIH DE025827 (to NEH), NIAMS/NIH AR066110 and NIDCR/NIH DE027639 (to BLF), NIAMS/NIH AR060823 (to RM), NIAMS/NIH Intramural Research Program (to MJS), NIAMS/NIH AR069620 and NIGMS/NIH GM125503.
Footnotes
Disclosure Statement: The authors report no conflicts of interest.
References
- Aizenbud D, Peled M, Figueroa AA. 2008. A combined orthodontic and surgical approach in osteogenesis imperfecta and severe Class III malocclusion: case report. J Oral Maxillofac Surg 66:1045–1053. [DOI] [PubMed] [Google Scholar]
- Balasubramanian M, Pollitt RC, Chandler KE, Mughal MZ, Parker MJ, Dalton A, Arundel P, Offiah AC, Bishop NJ. 2015. CRTAP mutation in a patient with Cole-Carpenter syndrome. Am J Med Genet A 167A:587–591. [DOI] [PubMed] [Google Scholar]
- Baldridge D, Lennington J, Weis M, Homan EP, Jiang MM, Munivez E, Keene DR, Hogue WR, Pyott S, Byers PH, Krakow D, Cohn DH, Eyre DR, Lee B, Morello R. 2010. Generalized connective tissue disease in Crtap−/− mouse. PLoS One 5:e10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbirato C, Trancozo M, Almeida MG, Almeida LS, Santos TO, Duarte JC, Reboucas MR, Sipolatti V, Nunes VR, Paula F. 2015. Mutational characterization of the P3H1/CRTAP/CypB complex in recessive osteogenesis imperfecta. Genet Mol Res 14:15848–15858. [DOI] [PubMed] [Google Scholar]
- Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR, Leikin S, Makareeva E, Kuznetsova N, Uveges TE, Ashok A, Flor AW, Mulvihill JJ, Wilson PL, Sundaram UT, Lee B, Marini JC. 2006. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med 355:2757–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boskey AL, Verdelis K, Spevak L, Lukashova L, Beniash E, Yang X, Cabral WA, Marini JC. 2013. Mineral and matrix changes in Brtl/+ teeth provide insights into mineralization mechanisms. Biomed Res Int 2013:295812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. 2010. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 25:1468–1486. [DOI] [PubMed] [Google Scholar]
- Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, Makareeva E, Kuznetsova NV, Rosenbaum KN, Tifft CJ, Bulas DI, Kozma C, Smith PA, Eyre DR, Marini JC. 2007. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 39:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canty EG, Kadler KE. 2005. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci 118:1341–1353. [DOI] [PubMed] [Google Scholar]
- Chang PC, Lin SY, Hsu KH. 2007. The craniofacial characteristics of osteogenesis imperfecta patients. Eur J Orthod 29:232–237. [DOI] [PubMed] [Google Scholar]
- Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, Pepin MG, Weis MA, Eyre DR, Byers PH. 2010. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet 86:389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DE, Carpenter TO. 1987. Bone fragility, craniosynostosis, ocular proptosis, hydrocephalus, and distinctive facial features: a newly recognized type of osteogenesis imperfecta. J Pediatr 110:76–80. [DOI] [PubMed] [Google Scholar]
- Eimar H, Tamimi F, Retrouvey JM, Rauch F, Aubin JE, McKee MD. 2016. Craniofacial and Dental Defects in the Col1a1Jrt/+ Mouse Model of Osteogenesis Imperfecta. J Dent Res 95:761–768. [DOI] [PubMed] [Google Scholar]
- Folkestad L, Hald JD, Canudas-Romo V, Gram J, Hermann AP, Langdahl B, Abrahamsen B, Brixen K. 2016. Mortality and Causes of Death in Patients With Osteogenesis Imperfecta: A Register-Based Nationwide Cohort Study. J Bone Miner Res 31:2159–2166. [DOI] [PubMed] [Google Scholar]
- Forlino A, Marini JC. 2016. Osteogenesis imperfecta. Lancet 387:1657–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Ao M, Salmon CR, Chavez MB, Kolli TN, Tran AB, Chu EY, Kantovitz KR, Yadav M, Narisawa S, Millan JL, Nociti FH Jr., Somerman MJ. 2018. Osteopontin regulates dentin and alveolar bone development and mineralization. Bone 107:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Ao M, Willoughby C, Soenjaya Y, Holm E, Lukashova L, Tran AB, Wimer HF, Zerfas PM, Nociti FH Jr., Kantovitz KR, Quan BD, Sone ED, Goldberg HA, Somerman MJ. 2015. Mineralization defects in cementum and craniofacial bone from loss of bone sialoprotein. Bone 78:150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Ramnitz MS, Gafni RI, Burke AB, Boyce AM, Lee JS, Wright JT, Akintoye SO, Somerman MJ, Collins MT. 2014. Rare bone diseases and their dental, oral, and craniofacial manifestations. J Dent Res 93:7S–19S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Soenjaya Y, Nociti FH Jr., Holm E, Zerfas PM, Wimer HF, Holdsworth DW, Aubin JE, Hunter GK, Goldberg HA, Somerman MJ. 2013. Deficiency in acellular cementum and periodontal attachment in bsp null mice. J Dent Res 92:166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafe I, Alexander S, Yang T, Lietman C, Homan EP, Munivez E, Chen Y, Jiang MM, Bertin T, Dawson B, Asuncion F, Ke HZ, Ominsky MS, Lee B. 2016. Sclerostin Antibody Treatment Improves the Bone Phenotype of Crtap(−/−) Mice, a Model of Recessive Osteogenesis Imperfecta. J Bone Miner Res 31:1030–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafe I, Yang T, Alexander S, Homan EP, Lietman C, Jiang MM, Bertin T, Munivez E, Chen Y, Dawson B, Ishikawa Y, Weis MA, Sampath TK, Ambrose C, Eyre D, Bachinger HP, Lee B. 2014. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat Med 20:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbour Z, Al-Khateeb A, Eimar H, Retrouvey JM, Rizkallah J, Glorieux FH, Rauch F, Tamimi F. 2018. Genotype and malocclusion in patients with osteogenesis imperfecta. Orthod Craniofac Res 21:71–77. [DOI] [PubMed] [Google Scholar]
- Jensen BL, Lund AM. 1997. Osteogenesis imperfecta: clinical, cephalometric, and biochemical investigations of OI types I, III, and IV. J Craniofac Genet Dev Biol 17:121–132. [PubMed] [Google Scholar]
- Kantaputra PN, Sirirungruangsarn Y, Intachai W, Ngamphiw C, Tongsima S, Dejkhamron P. 2018. Osteogenesis imperfecta with ectopic mineralizations in dentin and cementum and a COL1A2 mutation. J Hum Genet 63:811–820. [DOI] [PubMed] [Google Scholar]
- Lindahl K, Astrom E, Rubin CJ, Grigelioniene G, Malmgren B, Ljunggren O, Kindmark A. 2015. Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur J Hum Genet 23:1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nam HK, Campbell C, Gasque KC, Millan JL, Hatch NE. 2014. Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl(−/−) mouse model of infantile hypophosphatasia. Bone 67:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nam HK, Wang E, Hatch NE. 2013. Further analysis of the Crouzon mouse: effects of the FGFR2(C342Y) mutation are cranial bone-dependent. Calcif Tissue Int 92:451–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manokawinchoke J, Pavasant P, Sawangmake C, Limjeerajarus N, Limjeerajarus CN, Egusa H, Osathanon T. 2019. Intermittent compressive force promotes osteogenic differentiation in human periodontal ligament cells by regulating the transforming growth factor-beta pathway. Cell Death Dis 10:761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini JC, Cabral WA, Barnes AM. 2010. Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res 339:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini JC, Forlino A, Bachinger HP, Bishop NJ, Byers PH, Paepe A, Fassier F, Fratzl-Zelman N, Kozloff KM, Krakow D, Montpetit K, Semler O. 2017. Osteogenesis imperfecta. Nat Rev Dis Primers 3:17052. [DOI] [PubMed] [Google Scholar]
- Meganck JA, Kozloff KM, Thornton MM, Broski SM, Goldstein SA. 2009. Beam hardening artifacts in micro-computed tomography scanning can be reduced by X-ray beam filtration and the resulting images can be used to accurately measure BMD. Bone 45:1104–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, Castagnola P, Rauch F, Glorieux FH, Vranka J, Bachinger HP, Pace JM, Schwarze U, Byers PH, Weis M, Fernandes RJ, Eyre DR, Yao Z, Boyce BF, Lee B. 2006. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 127:291–304. [DOI] [PubMed] [Google Scholar]
- Muir AM, Ren Y, Butz DH, Davis NA, Blank RD, Birk DE, Lee SJ, Rowe D, Feng JQ, Greenspan DS. 2014. Induced ablation of Bmp1 and Tll1 produces osteogenesis imperfecta in mice. Hum Mol Genet 23:3085–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell AC, Marini JC. 1999. Evaluation of oral problems in an osteogenesis imperfecta population. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 87:189–196. [DOI] [PubMed] [Google Scholar]
- Orioli IM, Castilla EE, Barbosa-Neto JG. 1986. The birth prevalence rates for the skeletal dysplasias. J Med Genet 23:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch F, Fahiminiya S, Majewski J, Carrot-Zhang J, Boudko S, Glorieux F, Mort JS, Bachinger HP, Moffatt P. 2015. Cole-Carpenter syndrome is caused by a heterozygous missense mutation in P4HB. Am J Hum Genet 96:425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen A, Modig M, Larson O. 2011. Orthognathic bimaxillary surgery in two patients with osteogenesis imperfecta and a review of the literature. Int J Oral Maxillofac Surg 40:866–873. [DOI] [PubMed] [Google Scholar]
- Saga S, Nagata K, Chen WT, Yamada KM. 1987. pH-dependent function, purification, and intracellular location of a major collagen-binding glycoprotein. J Cell Biol 105:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Hirayoshi K, Yokota S, Hosokawa N, Nagata K. 1996. Intracellular interaction of collagen-specific stress protein HSP47 with newly synthesized procollagen. J Cell Biol 133:469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatzle M, Tanner SD, Bosshardt DD. 2005. Progressive, generalized, apical idiopathic root resorption and hypercementosis. J Periodontol 76:2002–2011. [DOI] [PubMed] [Google Scholar]
- Stevenson DA, Carey JC, Byrne JL, Srisukhumbowornchai S, Feldkamp ML. 2012. Analysis of skeletal dysplasias in the Utah population. Am J Med Genet A 158A:1046–1054. [DOI] [PubMed] [Google Scholar]
- Terajima M, Taga Y, Cabral WA, Nagasawa M, Sumida N, Hattori S, Marini JC, Yamauchi M. 2017. Cyclophilin B Deficiency Causes Abnormal Dentin Collagen Matrix. J Proteome Res 16:2914–2923. [DOI] [PubMed] [Google Scholar]
- Thumbigere-Math V, Alqadi A, Chalmers NI, Chavez MB, Chu EY, Collins MT, Ferreira CR, FitzGerald K, Gafni RI, Gahl WA, Hsu KS, Ramnitz MS, Somerman MJ, Ziegler SG, Foster BL. 2018. Hypercementosis Associated with ENPP1 Mutations and GACI. J Dent Res 97:432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umoh JU, Sampaio AV, Welch I, Pitelka V, Goldberg HA, Underhill TM, Holdsworth DW. 2009. In vivo micro-CT analysis of bone remodeling in a rat calvarial defect model. Phys Med Biol 54:2147–2161. [DOI] [PubMed] [Google Scholar]
- Valli M, Barnes AM, Gallanti A, Cabral WA, Viglio S, Weis MA, Makareeva E, Eyre D, Leikin S, Antoniazzi F, Marini JC, Mottes M. 2012. Deficiency of CRTAP in non-lethal recessive osteogenesis imperfecta reduces collagen deposition into matrix. Clin Genet 82:453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PG, Piersma SR, Fratantoni SA, Jimenez CR, Huizer M, Morsman AC, Cobben JM, van Roij MH, Elting MW, Verbeke JI, Wijnaendts LC, Shaw NJ, Hogler W, McKeown C, Sistermans EA, Dalton A, Meijers-Heijboer H, Pals G. 2009. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet 85:521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Foster BL, Kram V, Nociti FH Jr., Zerfas PM, Tran AB, Young MF, Somerman MJ. 2014. Fibromodulin and Biglycan Modulate Periodontium through TGFbeta/BMP Signaling. J Dent Res 93:780–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LM, Rauch F, Travers R, Chabot G, Azouz EM, Lalic L, Roughley PJ, Glorieux FH. 2002. Osteogenesis imperfecta type VII: an autosomal recessive form of brittle bone disease. Bone 31:12–18. [DOI] [PubMed] [Google Scholar]
- Wesselink PR, Beertsen W. 1994a. Ankylosis of the mouse molar after systemic administration of 1-hydroxyethylidene-1,1-bisphosphonate (HEBP). J Clin Periodontol 21:465–471. [DOI] [PubMed] [Google Scholar]
- Wesselink PR, Beertsen W. 1994b. Repair processes in the periodontium following dentoalveolar ankylosis: the effect of masticatory function. J Clin Periodontol 21:472–478. [DOI] [PubMed] [Google Scholar]
- Wolf M, Ao M, Chavez MB, Kolli TN, Thumbigere-Math V, Becker K, Chu EY, Jager A, Somerman MJ, Foster BL. 2018. Reduced Orthodontic Tooth Movement in Enpp1 Mutant Mice with Hypercementosis. J Dent Res 97:937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifler LE, Patel MK, Nociti FH Jr., Wimer HF, Millan JL, Somerman MJ, Foster BL. 2015. Counter-regulatory phosphatases TNAP and NPP1 temporally regulate tooth root cementogenesis. Int J Oral Sci 7:27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]