

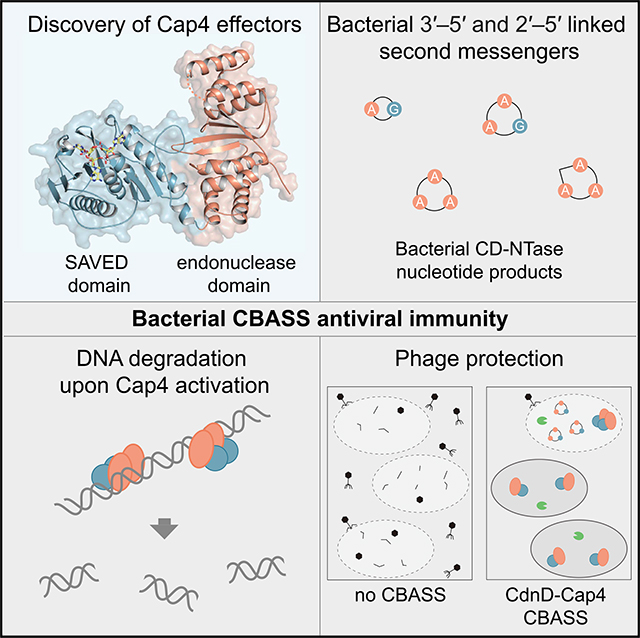
SUMMARY
cGAS/DncV-like nucleotidyltransferase (CD-NTase) enzymes are immune sensors that synthesize nucleotide second messengers and initiate antiviral responses in bacterial and animal cells. Here, we discover Enterobacter cloacae CD-NTase-associated protein 4 (Cap4) as a founding member of a diverse family of >2,000 bacterial receptors that respond to CD-NTase signals. Structures of Cap4 reveal a promiscuous DNA endonuclease domain activated through ligand-induced oligomerization. Oligonucleotide recognition occurs through an appended SAVED domain that is an unexpected fusion of two CRISPR-associated Rossman fold (CARF) subunits co-opted from type III CRISPR immunity. Like a lock and key, SAVED effectors exquisitely discriminate 2′–5′- and 3′–5′-linked bacterial cyclic oligonucleotide signals and enable specific recognition of at least 180 potential nucleotide second messenger species. Our results reveal SAVED CARF family proteins as major nucleotide second messenger receptors in CBASS and CRISPR immune defense and extend the importance of linkage specificity beyond mammalian cGAS-STING signaling.
Graphical Abstract
In Brief
A family of bacterial immune effectors responds to 3′–5′- and 2′–5′-linked nucleotide signals in CBASS antiviral immunity.
INTRODUCTION
cGAS/DncV-like nucleotidyltransferases (CD-NTases) are enzymes that synthesize specialized oligonucleotide signals to amplify pathway activation and control downstream effector responses. CD-NTases are conserved in animal and bacterial signaling systems and play a key role in innate immunity and phage defense (Ablasser and Chen, 2019; Bernheim and Sorek, 2020; Kranzusch, 2019). In human cells, the CD-NTase cyclic GMP-AMP synthase (cGAS) functions as a sensor for double-stranded DNA aberrantly localized in the cell cytosol during pathogen replication and cancer. When activated, cGAS produces the nucleotide second messenger 2′–5′, 3′–5′ cyclic GMP– AMP (2′3′-cGAMP) to induce antiviral immunity and interferon signaling (Ablasser et al., 2013; Diner et al., 2013; Gao et al., 2013a; Sun et al., 2013; Zhang et al., 2013). In an analogous system, the Vibrio cholerae CD-NTase dinucleotide cyclase in Vibrio (DncV) synthesizes the nucleotide second messenger 3′–5′, 3′–5′ cGAMP (3′3′-cGAMP) in response to an unknown stimulus during phage infection (Cohen et al., 2019; Davies et al., 2012; Kranzusch et al., 2014). Bacterial 3′3′-cGAMP activates a downstream effector response that results in cell death, limitation of phage replication through abortive infection, and protection of the remaining bacterial population (Cohen et al., 2019; Severin et al., 2018).
Bacterial CD-NTases include >5,600 unique enzymes (Whiteley et al., 2019) that control a diverse array of antiviral immune systems collectively called cyclic oligonucleotide-based antiphage signaling system (CBASS) immunity (Cohen et al., 2019; Ye et al., 2020). Synthesizing cyclic dinucleotide and cyclic trinucleotide products, bacterial CD-NTases are capable of using all four ribonucleotides as building blocks to dictate signal specificity and enable increased diversity of CBASS antiviral immune responses (Cohen et al., 2019; Whiteley et al., 2019). For example, in addition to the V. cholerae DncV product 3′3′-cGAMP, Escherichia coli CdnE synthesizes 3′3′ cyclic UMP-AMP (3′3′-cUA), and Enterobacter cloacae CdnD (CD-NTase in clade D) synthesizes the cyclic trinucleotide second messenger 3′3′3′ cyclic AMP-AMP-GMP (3′3′3′-cAAG) (Whiteley et al., 2019). In human cells, the non-canonical 2′–5′ linkage in the cGAS product 2′3′-cGAMP is critical for immune specificity and potent activation of the downstream receptor stimulator of interferon genes (STING) (Ablasser et al., 2013; Diner et al., 2013; Gao et al., 2013b; Zhang et al., 2013), but it is unknown whether phosphodiester linkage specificity is an additional determinant of receptor activation in bacterial CBASS antiviral immunity.
The best-characterized family of receptors that respond to bacterial CD-NTase nucleotide second messengers are patatin-like phospholipases, which are activated to degrade membrane phospholipids upon nucleotide signal binding. The first known example is the Vibrio cholerae protein cGAMP-activated phospholipase in Vibrio (CapV), which responds to 3′3′-cGAMP and causes membrane rupture and bacterial cell death (Severin et al., 2018). Similarly, the E. coli CD-NTase CdnE signals through CapE, a CapV homolog that specifically recognizes 3′3′-cUA, indicating that CBASS operons function with high specificity for a single-nucleotide second messenger (Whiteley et al., 2019). However, a majority of bacterial CD-NTase enzymes are encoded in CBASS operons that do not contain proteins with homology to CapV-like receptors (Burroughs et al., 2015; Cohen et al., 2019; Whiteley et al., 2019), suggesting that still-uncharacterized proteins must be responsible for downstream effector functions.
Here we reconstitute Enterobacter cloacae CdnD signaling in vitro and in vivo to discover CD-NTase-associated protein 4 (Cap4) as the founding member of a major family of downstream receptors that specifically respond to nucleotide second messenger signals in CBASS immunity. High-resolution crystal structures of Cap4 combined with negative-stain electron microscopy (EM) analysis reveals a DNA endonuclease effector domain activated through nucleotide second messenger-induced oligomerization. We demonstrate that a previously uncharacterized protein domain in Cap4 called SAVED is responsible for nucleotide second messenger recognition. Remarkably, SAVED is a fusion of two CRISPR-associated Rossman fold (CARF) domains derived from type III CRISPR immunity, revealing a common ancestry between these nucleotide second messenger-centric antiviral systems. Divergence in the SAVED nucleotide binding pocket enables recognition of an expanded range of CD-NTase products, including bacterial second messengers with alternative ring size, nucleobase, and 3′–5′ or 2′–5′ phosphodiester linkages. We further show that SAVED-containing effectors are essential for CBASS-mediated protection of bacteria from phage infection. Our results uncover a major family of nucleotide second messenger receptors and reveal a role of 2′–5′-linked nucleotide signals in bacterial resistance to phage infection.
RESULTS
Cap4 Proteins Are DNA Endonucleases that Respond to CD-NTase Nucleotide Second Messengers
The E. cloacae CD-NTase CdnD (EcCdnD) is constitutively active in vitro and synthesizes the nucleotide second messenger 3′3′3′-cAAG (Whiteley et al., 2019). EcCdnD is encoded in an operon containing three additional genes of unknown function, designated here CD-NTase-associated proteins 2, 3, and 4 (Cap2, Cap3, and Cap4) (Figure 1A). To define how bacterial CD-NTase enzymes control downstream signaling, we purified each protein from the E. cloacae CdnD02 operon and used a biochemical approach for receptor identification. Incubation of radiolabeled 3′3′3′-cAAG with purified EcCap4 (GenBank: WP_032676399.1) in an electrophoretic mobility shift assay resulted in complete shift and formation of a stable Cap4–3′3′3′-cAAG complex (Figures 1B and S1A). No interaction was observed between EcCap4 and the V. cholerae DncV product 3′3′-cGAMP, demonstrating that EcCap4 is a downstream receptor that specifically recognizes the cognate EcCdnD nucleotide second messenger.
Figure 1. Cap4 Proteins Are Endonucleases Activated by CD-NTase Nucleotide Second Messengers.

(A) Architectures of CBASS operons used for biochemical analysis. Enterobacter cloacae, Acinetobacter baumannii, and Moraxella osloensis systems lack a CapV phospholipase effector homolog and instead encode the protein CD-NTase-associated protein 4 (Cap4).
(B) Electrophoretic mobility shift assay measurement of Cap4–3′3′3′-cAAG complex formation. E. cloacae Cap2, Cap3, and Cap4 were incubated with 32P-labeled 3′3′-cGAMP or 3′3′3′-cAAG, and bound complexes were resolved by nondenaturing polyacrylamide gel electrophoresis.
(C) Cartoon schematic and crystal structure of apo MoCap4. MoCap4 contains an N-terminal DUF4297 domain (orange) with homology to restriction endonucleases and a C-terminal SAVED domain (blue) with homology to CARF domains found in type III CRISPR immunity. Red circles indicate locations of active-site residues.
(D) Structural comparison of the Cap4 DUF4297 domain and the restriction endonuclease HindIII, showing structural homology and shared catalytic residues. The core endonuclease domains in Cap4 and HindIII are highlighted in orange and green, respectively.
(E and F) Agarose gel analysis of plasmid DNA degradation by Cap4. (E) Cap4 proteins degrade target DNA only in the presence of activating nucleotide second messenger synthesized by the neighboring CD-NTase CdnD within the CBASS operon. (F) Activity is dependent on the conserved Cap4 catalytic active site, with no cleavage observed with the EcCap4 mutant K74A.
(G) Agarose gel analysis of Cap4 DNA cleavage promiscuity. In the presence of activating 3′3′3′-cAAG, EcCap4 is capable of degrading all sources of dsDNA, including plasmid DNA, E. coli genomic DNA, and synthetic 100-bp DNA. Biochemical data are representative of at least 3 independent experiments.
See also Figure S1.
To understand the function of Cap4, we screened homologs for suitability in a structural analysis and determined a 2.4 Å crystal structure of Cap4 from the bacterium Moraxella osloensis (MoCap4; GenBank: WP_060996052.1) and a 2.6 Å crystal structure of Cap4 from Acinetobacter baumannii (AbCap4; GenBank: WP_008942236.1) (Figures 1C and S1B; Table S1). The structure of Cap4 reveals a two-domain architecture with an N-terminal domain containing a mixed β sheet braced on either side with α-helical bundles and a globular C-terminal domain that contains internal two-fold pseudosymmetry (Figures 1C and S1B). Sequence alignment based on the MoCap4 structure demonstrates that all Cap4 homologs contain the same domain architecture with MoCap4 versus AbCap4 sharing ~80% identity and MoCap4 versus EcCap4 sharing ~20% identity at the amino acid level (Figure S1B). The Cap4 N-terminal domain is a member of an uncharacterized protein domain classification, domain of unknown function 4297 (DUF4297), that is widespread in bacteria. Comparative analysis using the MoCap4 structure reveals that the DUF4297 domain shares structural homology with type II restriction endonucleases, including the enzymes AgeI and HindIII (Figure 1D). Sequence alignment of Cap4 and type II restriction enzymes confirms conservation of all putative active-site residues required for metal coordination and suggests that Cap4 proteins are functional nuclease enzymes (Figure S1B; Burroughs et al., 2015; Watanabe et al., 2009). We therefore tested EcCap4 and AbCap4 proteins for the ability to cleave DNA substrates in vitro. In the presence of an activating nucleotide second messenger, Cap4 proteins catalyze complete degradation of plasmid DNA to small, <45 bp DNA fragments (Figure 1E). Cap4 alone exhibits no nuclease activity, indicating that recognition of a nucleotide second messenger is a strict requirement for nuclease activation. Cap4 activity is divalent cation metal dependent (Figure S1D), and mutation of a conserved lysine residue in the active site ablates nuclease activity (Figures 1F and S1E), further confirming that Cap4 proteins use a type II restriction enzyme-like reaction mechanism to degrade DNA.
Type II restriction endonucleases contain loops or helices that extend from the nuclease domain to form DNA major and minor groove contacts and control target sequence cleavage specificity (Tamulaitiene et al., 2017; Watanabe et al., 2009). The Cap4 N-terminal endonuclease domain is minimized, with nearly all extensions absent, suggesting relaxation of DNA targeting specificity (Figure 1D). Consistent with these structural observations, Ec-Cap4 degrades plasmid DNA, E. coli genomic DNA, and synthetic double-stranded DNA (dsDNA) in vitro with no apparent target sequence or DNA modification specificity (Figures 1G and S1F). In each case, Cap4 endonuclease activity remains strictly dependent on the presence of activating nucleotide second messenger. Together, these results demonstrate that Cap4 proteins are dsDNA endonucleases controlled through nucleotide second messenger-gated enzyme activation.
Cap4 Proteins Respond to Specific 3′–5′- and 2′–5′-Linked Nucleotide Second Messengers
To determine the specificity of CD-NTase-Cap4 signaling, we next compared cross-activation between E. cloacae and A. baumannii CdnD operons. Although each CdnD activates the robust endonuclease activity of its cognate Cap4 protein in vitro, heterologous reactions with one CdnD or Cap4 component derived from each operon fail to reconstitute signaling and do not result in DNA degradation (Figures 2A and S2A). Selective Cap4 activation reveals that AbCdnD must synthesize a nucleotide second messenger distinct from the EcCdnD product 3′3′3′-cAAG. We tested nucleotide combinations and observed that ATP is necessary and sufficient to allow AbCdnD to synthesize the activating nucleotide second messenger signal (Figure S2B). However, all known canonically linked cyclic di-, tri-, and tetra-AMP RNA products failed to reconstitute activation of AbCap4 DNA degradation activity (Figure 2B). We therefore hypothesized that, like human cGAS-STING, AbCdnD-Cap4 signaling may require a noncanonical 2′–5′-linked second messenger. To assess whether the AbCdnD nucleotide product contains a non-canonical linkage, we analyzed the sensitivity of CD-NTase reaction products to digestion by nuclease P1, which specifically hydrolyzes 3′–5′-linked phosphodiester bonds and is unable to cleave non-canonical 2′–5′ phosphodiester bonds (Ablasser et al., 2013; Diner et al., 2013; Whiteley et al., 2019). Unlike the V. cholerae DncV and EcCdnD products 3′3′-cGAMP and 3′3′3′-cAAG, the mammalian cGAS 2′3′-cGAMP and the AbCdnD reaction products contain phosphates resistant to nuclease P1 digestion, confirming the presence of a 2′–5′-linked bond (Figure 2C).
Figure 2. Cap4 Proteins Respond to Specific 3′–5′ - and 2′–5′-Linked Nucleotide Second Messengers.

(A) Analysis of Cap4 activation specificity using nucleotide second messengers synthesized with CD-NTase enzymes (DncV reaction, 3′3′-cGAMP; EcCap4 reaction, 3′3′3′-cAAG). EcCap4 and AbCap4 activation requires the specific nucleotide second messenger synthesized by the cognate CD-NTase, with no cross-activation occurring between distantly related operons.
(B) Activation of EcCap4 and AbCap4 DNA degradation activity with synthetic nucleotide second messengers. Synthetic 3′3′3′-cAAA is able to activate the EcCap4 enzyme normally responsive to 3′3′3′-cAAG, but no tested canonically 3′–5′ -linked cyclic oligonucleotide species is able to activate AbCap4.
(C) P1 nuclease digestion and thin-layer chromatography analysis of CD-NTase products. Nuclease P1 specifically degrades 3′–5′ bonds, leaving non-canonical 2′–5′ bonds intact and resistant to phosphatase treatment. The human enzyme cGAS product 2′3′-cGAMP and the bacterial AbCdnD product contain nuclease P1-resistant 2′–5′ linkages.
(D and E) Large-scale synthesis and purification of the AbCdnD nucleotide second messenger products. AbCdnD synthesizes two closely related cyclic oligoadenylate products that co-migrate as (D) a single peak on anion-exchange (IEX) chromatography but can be separated into (E) major and minor product species with C18 chromatography.
(F) Cartoon schematic and crystal structure of AbCap4 bound to the AbCdnD major product 2′3′3′-cAAA. The cyclic trinucleotide 2′3′3′-cAAA binds within the Cap4 SAVED domain (blue), indicating that this domain is responsible for CD-NTase signal recognition.
(G) Simulated-annealing Fo-Fc omit map (contoured at 3 σ) of the ligand density, demonstrating unambiguous assignment of the major AbCdnD nucleotide second messenger as 2′3′3′-cAAA. Biochemical data are representative of at least 3 independent experiments.
See also Figure S2.
We next isolated the AbCdnD nucleotide second messenger directly from enzymatic reactions for further characterization. AbCdnD synthesizes two closely related products that co-elute with nearly all ion-exchange purification steps (Figure 2D) but can be separated on a C18 reverse-phase column (Figure 2E). Biochemical analysis demonstrates that the major (~65%) and minor (~35%) AbCdnD products are cyclic oligoadenylate species containing P1-resistant 2′ –5′ phosphodiester linkages (Figure S2C). Mass spectrometry analysis demonstrates that the major product is a cyclic trinucleotide and that the minor product peak is a mixture of cAAA and cA4 species (Figure S2G). Interestingly, the minor product most potently activates AbCap4, with full enzyme activation and DNA degradation requiring low nanomolar concentrations of nucleotide second messenger (Figure S2D). Although the major product does not activate AbCap4 DNA degradation activity, this AbCdnD product is still capable of binding and stabilizing the AbCap4 enzyme, and we were able to determine a 2.2 Å co-crystal structure of the AbCap4-nucleotide second messenger complex (Figures 2F and S2E). Clear ligand density was observed for a cyclic trinucleotide bound within the AbCap4 globular C-terminal domain, allowing unambiguous assignment of this AbCdnD product as the cyclic trinucleotide 2′–5′, 3′ –5′, 3′–5′ c-AMP-AMP-AMP (2′3′3′-cAAA) and direct structural confirmation of the ability of a bacterial CD-NTase to synthesize noncanonical 2′–5′-linked RNAs (Figures 2G and S2F). To further confirm these findings, we verified, using mass spectrometry fragmentation analysis, that the AbCdnD major product matches a chemically synthesized 2′3′3′-cAAA standard (Figure S2G). 2′–5′ phosphodiester linkages are rare in biology, and their role in nucleotide signaling has been suggested previously to be a unique adaptation evolved within eukaryotic innate immune signaling (Danilchanka and Mekalanos, 2013; Kranzusch et al., 2015; Margolis et al., 2017). These results demonstrate that 2′–5′-linked products are also involved in bacterial antiviral signaling and that Cap4 nucleases function as selective sensors that can use linkage specificity to adapt to distinct CD-NTase nucleotide second messenger signals.
Cap4 Contains a C-Terminal CARF Family Domain that Controls Ligand Specificity
The structure of the AbCap4–2′3′3′-cAAA complex reveals that the Cap4 C-terminal domain is responsible for nucleotide second messenger recognition. Previously, the Cap4 C-terminal SAVED domain was identified bioinformatically as enriched in CD-NTase-containing operons (Burroughs et al., 2015). Surprisingly, analysis of the AbCap4 SAVED domain structure reveals clear structural homology with CARF proteins, including Csm6 (DALI Z score, 4.2), with each half of the pseudo-symmetric SAVED domain containing homology to a single CARF domain (Figures 3A and 3B). The CARF domain of Csm6 from different CRISPR systems binds cA4 or cA6 cyclic oligoadenylate signals made by Cas10 following target recognition in type III CRISPR systems (Kazlauskiene et al., 2017; Niewoehner et al., 2017). Canonical CARF domain proteins like Csm6 homodimerize through CARF-CARF interactions to form a two-fold symmetric binding surface for nucleotide second messenger recognition (Jia et al., 2019; Niewoehner and Jinek, 2016). In contrast, the Cap4 SAVED domain is comprised of two individual CARF-like subunits joined with an ~25-amino acid internal linker between β strands 4 and 5. Using a thermal shift assay to measure EcCap4 and AbCap4 nucleotide second messenger complex formation, we confirmed that the SAVED domain alone is sufficient to specifically recognize the activating signal (Figure S3A).
Figure 3. Cap4 Ligand Specificity Is Controlled by the SAVED Domain, a Divergent CARF Family Domain.

(A) Structure of the AbCap4 SAVED domain, colored in blue and gray to highlight internal two-fold pseudosymmetry. Each half of the SAVED domain monomer has homology to a CARF subunit. Within the SAVED domain, these two CARF subunits are fused into a single chain with an ~25-amino acid linker.
(B) Topology diagram of AbCap4 and comparison with the T. onnurineus Csm6 CARF domain from type III CRISPR immunity. Each half of the AbCap4 SAVED domain shares the common core CARF domain topology, including a β strand that leads into a bracing outer helix (β1-to-α2 and β7-to-α6) and a second β strand and central helix (β2-to-α3 and β8-to-α7) at the normal CARF dimerization interface. All CARF family proteins contain these conserved structural features, including Csm6 (PDB: 606V), Csx3 (PDB: 3WZI), and Csx1 (PDB: 2I71).
(C) Structural comparison of the AbCap4 SAVED-2′3′3′-cAAA and ToCsm6 CARF-cA4 complexes. SAVED and CARF domains recognize nucleotide ligands with a similar binding surface. The single-domain architecture of the SAVED domain breaks the restriction of two-fold symmetry (dashed line) and enables recognition of diverse CD-NTase nucleotide second messengers. In contrast, the homo-dimeric architecture of canonical CARF proteins necessitates ligands with two-fold symmetry (solid line).
(D and E) Structure of AbCap4 adenosine 1 (A1) pocket interactions and conservation of key residues shared in the SAVED and CARF domains. (E) Strict conservation of the A1 pocket with an aromatic residue and a tyrosine residue further supports emergence of SAVED domains through duplication and fusion of an ancient CARF family protein.
See also Figures S3 and S4.
Comparison of the AbCap4 SAVED-2′3′3′-cAAA structure with previous CARF domain structures bound to RNA ligands reveals a mixture of shared and divergent features required for nucleotide second messenger recognition. The Csm6 homodimer has two-fold symmetry, with each monomer recognizing two nucleobases of the cA4 signal (Jia et al., 2019; Molina et al., 2019). Unlike the larger cA4 or cA6 signals in type III CRISPR immunity, the majority of CD-NTase enzymes synthesize asymmetric nucleotide signals that lack internal two-fold symmetry (Whiteley et al., 2019). Pseudo-symmetry of the AbCap4 SAVED domain permits the existence of three unique pockets and specific recognition of each base of 2′3′3′-cAAA (Figures 3C, 3D, and S3B). The “A1 pocket” in AbCap4 is formed through a highly conserved aromatic W449 position that stacks against the nucleobase and Y454, which hydrogen-bonds with the adenine N7 nitrogen (Figures 3D and 3E). Nucleobase A2 is recognized through base-stacking interactions within a pocket formed by K299 and R301 (Figure S3B).
The A1 pocket is conserved in Cap4 and Csm6 homologs, demonstrating that nucleobase interactions at this site have been maintained throughout divergence of SAVED proteins from a CARF protein ancestor (Figure 3E). Mutagenesis confirms the importance of the A1 pocket in AbCap4 and EcCap4, with Cap4 proteins containing mutations within this pocket requiring >10-fold more ligand to induce similar levels of activation (Figure S3C). In contrast, the contacts to nucleobases A2 and A3 occur in pockets that do not exist in homodimeric CARF proteins. We determined an additional 2.4 Å structure of the AbCap4–3′3′3′-cAAA complex to further guide analysis of Cap4-ligand interactions and phosphodiester linkage specificity (Figure S4A). Comparison of the AbCap4–2′3′3′-cAAA and −3′3′3′-cAAA complexes demonstrates that a flat, parallel orientation of the nucleotide ribose at the A2 position along the SAVED domain surface allows the neighboring gap between the A1 and A2 pockets to accommodate a 3′–5′ or 2′–5′ linkage. In contrast, the perpendicular orientation of the A3 ribose constrains the nucleotide backbone to permit only a 3′–5′ linkage and explains how the architecture within the SAVED domain binding pocket can dictate signal specificity. Diversification of the ligand binding interface through genetic fusion of two CARF-like subunits into a single-chain SAVED domain was likely a key evolutionary intermediate enabling specific recognition of diverse CD-NTase nucleotide second messengers that lack two-fold symmetry. Together, these data demonstrate that SAVED domains are divergent members of the CARF protein family and that the single-chain architecture of the Cap4 SAVED domain allows recognition of diverse asymmetric nucleotide recognition signals.
Cap4 Proteins Are Activated through Ligand-Dependent Oligomerization
To define the mechanism of Cap4 ligand-dependent activation, we next analyzed EcCap4 and AbCap4 proteins using negative-stain EM. In the presence of activating nucleotide ligand, Cap4 proteins oligomerize and form higher-order complexes (Figures 4A and S5A). The activated Cap4 complexes primarily adopt a dimeric state with two Cap4 proteins stacked against each other in an SS-shaped configuration. We also observed Cap4 particles that correspond to higher-order oligomeric complexes (Figure S5B), and we confirmed, with size-exclusion chromatography-multiangle light scattering (SEC-MALS) analysis, that activated Cap4 in solution can oligomerize into multiple higher-order oligomeric states (Figures 4B and S5C). We classified the oligomerization status of ~20,000 particles from each condition to quantify the frequency of ligand-induced oligomerization. In agreement with Cap4-ligand interactions functioning as a requirement for high-order complex formation, <1% of the Cap4 particles occupied the dimeric or oligomeric state in the absence of an activating nucleotide signal (Figure 4A). The same higher-order oligomerization is observed in the presence of 45-bp target DNA, suggesting that nucleotide second messenger binding alone allows assembly of Cap4 into a fully active oligomeric state.
Figure 4. Cap4 Proteins Are Activated through Ligand-Dependent Oligomerization.

(A) Negative-stain EM class averages of EcCap4 (K74A) with or without activating 3′3′3′-cAAG and 45 bp DNA. Particle classification and quantification for each condition demonstrates that Cap4 oligomerization only occurs in the presence of activating nucleotide second messenger.
(B) Size-exclusion chromatography-multiangle light scattering (SEC-MALS) analysis of EcCap4 in the presence or absence of activating ligand 3′3′3′-cAAG. Absolute molecular masses of apo and ligand-bound complexes confirms EcCap4 oligomerization in the presence of activating nucleotide second messenger (EcCap4 expected molecular weight [MW], 56.1 kDa).
(C) Electrophoretic mobility shift assay assessing DNA binding of EcCap4 (K74A). Cap4 was incubated with 45-bp DNA and activating ligand 3′3′3′-cAAG as indicated. Stable EcCap4-DNA complex formation only occurs in the presence of activating nucleotide second messenger.
(D) Deep sequencing of EcCap4 DNA fragments and analysis of EcCap4 target specificity and distribution of DNA fragment sizes following E. coli genomic DNA or plasmid DNA degradation by EcCap4 (left). Cut site mapping demonstrates a minimal 5′ CNG cut site preference (right).
(E) Mapping of EcCap4 cleaved DNA fragments across the E. coli genomic DNA confirms the relaxed targeting specificity of EcCap4 endonuclease activity. y axis positive and negative values indicate strand-specific cutting. Data are representative of at least 3 independent experiments.
See also Figures S5 and S6.
3D reconstruction at ~15 Å of the activated dimeric MoCap4 complex allowed docking of the high-resolution AbCap4–2′3′3′-cAAA crystal structure and further analysis of the mechanism of activation (Figure S5D). In the docked assembly, two SAVED domains stack against each other and result in alignment of the Cap4 endonuclease active sites. Docking of Cap4 within the 3D reconstructions suggests that no major domain rearrangement is required for activation (Figure S5B). In agreement, all four crystal structures of AbCap4 and MoCap4 share a fixed, rigid orientation of the Cap4 N-terminal endonuclease and C-terminal SAVED domains despite distinct packing and crystal forms (Figures S4B and S4C). These results support a model where recognition of the activating nucleotide second messenger likely induces a local conformational change in the SAVED domain that drives oligomerization and endonuclease activation. Oligomerization and positioning of two adjacent Cap4 endonuclease domains creates a singular surface to engage target DNA (Figure S5D). Using an electrophoretic mobility shift assay, we observed that Cap4 alone is unable to bind DNA and that ligand recognition is a pre-requisite for target DNA interaction (Figures 4C and S5E). An ~10- to 20-amino acid extension is conserved at the N terminus of each Cap4 effector but is not ordered in any of our AbCap4 or MoCap4 crystal structures (Figures S1B and S1C). Biochemical analysis of Cap4 proteins with N-terminal deletions shows that the N-terminal extension is dispensable for nucleotide signal recognition and oligomerization but required for DNA binding and endonuclease domain activation (Figure S6). Together, these results suggest a two-step model of Cap4 activation where nucleotide second messenger recognition in the SAVED domain induces Cap4 oligomerization and subsequent DNA binding and target degradation.
To further define the result of ligand-induced Cap4 activation, we next developed a deep sequencing approach to map the cleavage specificity and fragment distribution of Cap4 nuclease activity. Sequencing of DNA fragments remaining following degradation of genomic or plasmid DNA with EcCap4 reveals a consistent fragment length of ~17 bp and further suggests that DNA degradation occurs through defined oligomerization of multiple nuclease active sites (Figure 4D). In agreement with the promiscuous nuclease activity observed in biochemical assays (Figure 1), analysis of EcCap4 cut sites from mapped DNA fragments demonstrates a highly degenerate recognition sequence distinct from the strict sequence preference characteristic of type II restriction enzymes. EcCap4 exhibits preference for a minimal recognition sequence, 5′ CNG, whereas Ab-Cap4 is less specific, targeting a 5′ C or G and a final average fragment size of only ~6 bp (Figures 4D and S5F). Mapping of the observed Cap4 cut sites in E. coli genomic DNA demonstrates complete degradation across nearly all regions of the genome (Figures 4E and S5G). Together, these data support a model of Cap4 regulation where ligand-induced oligomerization activates the endonuclease domain and results in promiscuous cleavage of DNA to minimal fragments.
SAVED Domain-Containing Proteins Are a Major Form of Viral Defense in Diverse Bacteria
SAVED domain-containing proteins occur in 29.8% of sequenced CD-NTase-containing operons and comprise one of the most prevalent effector modules in CBASS phage immunity (Figure 5A; Burroughs et al., 2015; Cohen et al., 2019; Whiteley et al., 2019). In addition to the endonuclease-SAVED architecture in Cap4 proteins, CBASS operons encode SAVED domains fused to additional protein partners, including putative HNH nucleases, caspase-like proteases, calcineurin-like phosphatases, Toll/interleukin-1 receptor (TIR) NAD+ hydrolase enzymes, and transmembrane-containing segments (Figure 5B). Nucleotide second messenger-induced oligomerization of SAVED domains is likely a general strategy to activate diverse effector functions and coordinate abortive infection systems to limit phage replication.
Figure 5. SAVED Domain-Containing Proteins Are a Major Form of Viral Defense in Diverse Bacteria.

(A) Phylogenetic tree showing distribution of SAVED-containing effectors in bacterial CD-NTase operons. Bacterial CD-NTase clades are displayed as an unrooted tree (Whiteley et al., 2019), with clades containing SAVED effectors (~30% of all bacterial CD-NTase operons) outlined in bold (clades B, C, D, G, and H). Dots indicate locations of CD-NTases of interest.
(B) Domain organization of diverse SAVED-containing effectors (Burroughs et al., 2015). The prevalence of each domain architecture in sequenced bacterial genomes is listed as a percentage of all SAVED-containing effectors (REase, restriction endonuclease).
(C) A genetic screen to identify SAVED effectors responsive to 3′3′-cGAMP. SAVED effectors were co-expressed with the 3′3′-cGAMP-producing CD-NTase DncV, and a spot-dilution assay with quantification of recovered colony-forming units (CFUs) was used to assess SAVED activation and cytotoxicity. SAVED domain effectors from CD-NTase clades B and G respond to 3′3′-cGAMP, demonstrating that SAVED domains are capable of responding to cyclic dinucleotide and cyclic trinucleotide second messengers. Data represent the mean ± SD of 3 independent experiments. See STAR Methods for CBASS effector species designations.
(D) In vitro reconstitution of nucleotide second messenger specificity for divergent Cap4 proteins and the HNH endonuclease-SAVED fusion B. pseudomallei CD-NTase-associated protein 5 (Cap5). DNA cleavage by BpCap5 demonstrates that SAVED domain-dependent nucleotide second messenger sensing is capable of activating structurally distinct enzymatic domains. Data are representative of at least 3 independent experiments.
Analysis of the bacterial CD-NTase phylogenetic tree reveals that SAVED domain-containing proteins are encoded near vastly divergent CD-NTase enzymes from across clades B, C, D, G, and H (Figure 5A; Whiteley et al., 2019). The broad distribution of SAVED domain-containing effectors suggests that this domain can recognize diverse nucleotide second messengers. To test this hypothesis, we conducted a genetic screen to identify SAVED-containing proteins that specifically respond to the cyclic dinucleotide 3′3′-cGAMP. Co-expression analysis in E. coli with the promiscuously active CD-NTase V. cholerae DncV identified multiple SAVED domain-containing proteins in clades B and G that respond to 3′3′-cGAMP and induce cell death (Figure 5C). These effectors contain an HNH-SAVED architecture structurally distinct from the Cap4 DUF4297 endonuclease-SAVED architecture, and we therefore designated genes with this architecture CD-NTase-associated protein 5 (Cap5). Purified Burkholderia pseudomallei Cap5 (GenBank: WP_004556385.1) is active in the presence of 3′3′-cGAMP and catalyzes robust dsDNA degradation in vitro (Figure 5D). Activation of each tested SAVED domain-containing protein requires recognition of a specific nucleotide second messenger, further demonstrating the specificity of CD-NTase signaling pathways (Figure 5D). Notably, the 3′3′-cGAMP-responsive SAVED domain effector BpCap5 failed to respond to the common bacterial second messengers 3′3′ cyclic di-GMP and 3′3′ cyclic di-AMP. Exquisite nucleotide ligand specificity therefore insulates CBASS immunity from other host signals and limits inappropriate activation.
CD-NTase operons protect bacterial populations during phage infection (Cohen et al., 2019; Ye et al., 2020). Interestingly, SAVED domain-containing proteins are also present within type III CRISPR operons, suggesting that the shared mechanism of SAVED- and CARF-dependent activation allows exchange of CBASS and CRISPR components and further diversification of antiviral immune defenses (Figure 6A). To verify that CD-NTase operons with SAVED domain-containing effectors also restrict phage replication, we used the E. cloacae operon containing CdnD, Cap2, Cap3, and Cap4 and an E. coli operon containing CdnG (GenBank: WP_000064266.1), Cap2, Cap3, and Cap5 to analyze phage resistance (Figure 6B). Introduction of plasmids encoding one of these operons in E. coli mediated a >3-log decrease in efficiency of plaque formation for phage T2. Expression of the CdnG operon resulted in an ~1-log decrease in phage T5 plaque formation, whereas the CdnD operon had no effect. No effect was observed on replication of phage T7 (Figure 6B; Cohen et al., 2019; Ye et al., 2020). Restriction of phage replication is strictly dependent on the catalytic activity of the CD-NTase and the nuclease active site of Cap4 (Figure 6B). Together, these data demonstrate a broad strategy in bacteria that couples CD-NTase signaling to activation of downstream SAVED-containing proteins to protect against viral infection.
Figure 6. SAVED Effectors Are Essential for CBASS Antiviral Defense.

(A) Example CBASS and CRISPR immunity operons encoding SAVED domain-containing effector proteins: Vibrio cholerae (WP_001901330), Enterobacter cloacae (WP_032676400), Citrobacter freundii (WP_032942206), Methylibium petroleiphilum (WP_011829962), Escherichia coli (WP_000058223), Myxococcus xanthus (YP_635404), and Fervidobacterium nodosum (WP_011994539).
(B) CBASS operons encoding SAVED domain effectors protect E. coli from phage replication. E. coli containing either an empty vector or a vector encoding E. cloacae CdnD or E. coli CdnG CBASS operons, as indicated, were infected with phage, and efficiency of plating was quantified by plaque-forming units. Data represent the mean ± SEM of 3 independent experiments.
DISCUSSION
Here we identify Cap4 as the founding member of a widespread family of antiviral effector proteins in bacteria. We show that Cap4 proteins are nucleotide second messenger sensors that function as downstream DNA endonuclease effectors in CBASS immunity. Following phage infection, CBASS immunity begins with activation of a CD-NTase enzyme to catalyze nucleotide second messenger synthesis (Cohen et al., 2019; Whiteley et al., 2019). Although the mechanism of phage recognition remains unclear, many bacterial CD-NTase enzymes are constitutively active in vitro and may therefore function similar to guards in plant immunity and sense phage replication indirectly by detecting changes in metabolite concentrations or loss of homeostasis (Dodds and Rathjen, 2010; Kranzusch, 2019; Whiteley et al., 2019). After CD-NTase activation, Cap4 recognizes the resulting nucleotide signal and couples ligand binding with induction of a promiscuous DNA cleavage response that restricts phage replication. SAVED domain-containing proteins like Cap4 are found in ~30% of CBASS operons and provide a clear mechanism for how these systems mediate phage resistance.
Structures of Cap4 explain a molecular mechanism for ligand recognition and effector function activation. Following nucleotide second messenger recognition in the SAVED domain, Cap4 oligomerizes to closely stack two nuclease domains (Figures 3, 4, and S4D). The Cap4 nuclease effector domain is therefore maintained in an inactive monomeric state until detection of the correct CD-NTase signal enables higher-order complex assembly and target DNA degradation. The Cap4 SAVED domain is sufficient to recognize the activating nucleotide signal (Figure S3A), indicating that the effector domain does not participate in ligand specificity. Additionally, we characterize an HNH endonuclease-SAVED effector Cap5 from B. pseudomallei and demonstrate that SAVED domains can activate structurally divergent effector domains (Figure 5D). These results support a sensor-effector model where fusion of a SAVED domain allows nucleotide second messenger-gated control of diverse effectors. SAVED domains are fused to a wide variety of effector domains, including proteases, phosphodiesterases, and potentially pore-forming transmembrane proteins (Burroughs et al., 2015; Cohen et al., 2019), supporting that the generalizability of SAVED-dependent activation allows diverse domains to be co-opted as modules for viral defense.
We show that the SAVED domain formed through fusion of two CARF-like subunits into a single-chain nucleotide second messenger sensor (Figure 3). These results explain how SAVED domains recognize diverse asymmetric ligands and reveal an unexpected shared evolutionary history of CBASS and CRISPR antiviral systems. Nearly all characterized type III CRISPR systems function through homodimeric CARF proteins that recognize a nucleotide second messenger with two-fold symmetry (Kazlauskiene et al., 2017; Niewoehner et al., 2017). Fusion of two CARF-like subunits into a SAVED domain breaks the requirement for two-fold symmetrical ligands and explains a key evolutionary event that enabled CD-NTase systems to signal with diverse asymmetric ligands. A similar evolutionary event was recently observed with fusion of two CARF domains in a Thermus thermophilus type III CRISPR effector (McMahon et al., 2020; Figure S4D), further highlighting how genetic fusion of multiple CARF domains can provide an evolutionary advantage in antiviral immunity. Interestingly, a structurally unrelated 3′3′3′-cAAA-activated homotrimeric DNA endonuclease, NucC, was identified in an E. coli CdnC CD-NTase system (Lau et al., 2020). NucC uses a three-fold symmetric ligand-binding pocket, suggesting that SAVED and NucC effectors are alternative evolutionary paths that enabled nucleotide second messenger diversification within CBASS immunity. CD-NTase enzymes synthesize nucleotide second messengers using all four nucleobases as building blocks (Whiteley et al., 2019), and we now show that CD-NTase product second messengers are further specified with incorporation of 2′–5′ and 3′–5′ phosphodiester linkages. These findings dramatically expand the known nucleotide second messenger signaling space and indicate that CD-NTase enzymes are theoretically capable of synthesizing at least 180 distinct oligonucleotide variants. Combined with cA4 and cA6 species in type III CRISPR immunity and linear 2′–5′-linked chains produced by mammalian oligoadenylate synthase enzymes (Hornung et al., 2014), these results reveal a vast diversity of natural nucleotide second messenger signals in viral defense.
The enormous diversity of CD-NTase nucleotide second messenger signals likely enables CBASS antiviral systems to specialize and adapt to phage resistance. Mammalian poxviruses encode a 2′3′-cGAMP-specific nuclease that degrades the cGAS product and prevents STING-dependent immune responses (Eaglesham et al., 2019). Likewise, a phage protein was recently identified that degrades cA4 to evade type III CRISPR immunity (Athukoralage et al., 2020). Viral nucleases that specifically degrade second messengers likely create evolutionary pressure to diversify antiviral signaling systems. The ability of CBASS immunity to morph between cyclic dinucleotide and cyclic trinucleotide signals may represent larger “leaps” that allow escape and temporary relief from an evolutionary arms race (Daugherty and Malik, 2012). Our discovery of a role for 2′–5′ linkages in A. baumannii CBASS immunity demonstrates that incorporation of noncanonical linkages is another mechanism that evolved in bacteria to subvert viral resistance. Along with the previous identification of diverse cGAS-like enzymes in bacteria and effector proteins with sequence homology to mammalian STING (Cohen et al., 2019; Whiteley et al., 2019), the discovery of bacterial signals with 2′–5′ linkages demonstrates that all fundamental components of mammalian cGAS-STING signaling are functionally shared within ancestrally related bacterial CBASS immunity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philip J. Kranzusch (philip_kranzusch@dfci.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Coordinates of the Cap4 structures have been deposited in the RCSB Protein Data Bank under the following accession numbers: MoCap4 (6VM5), AbCap4 (6WAM), AbCap4–2′3′3′-cAAA (6VM6), AbCap4–3′3′3′-cAAA (6WAN).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Escherichia coli strains
Recombinant CD-NTase and Cap4 proteins were expressed in E. coli BL21-RIL DE3 (Agilent) bacteria harboring a pRARE2 tRNA plasmid. Single transformants were inoculated into a 20 mL MDG media starter culture (0.5% glucose, 25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 0.25% aspartic acid, 100 mg ml−1 ampicillin, 34 mg ml−1 chloramphenicol, and trace metals), grown overnight at 37°C, and used to seed 1 L M9ZB media cultures (0.5% glycerol, 1% Cas-Amino Acids, 47.8 mM Na2HPO4, 22 mM KH2PO4, 18.7 mM NH4Cl, 85.6 mM NaCl, 2 mM MgSO4, 100 mg ml−1 ampicillin, 34 mg ml−1 chloramphenicol, and trace metals) grown at 37°C, 230 RPM until an OD600 of ~2.5. Cultures were induced by chilling flasks on ice for 20 min, supplementing cultures with to 0.5 mM IPTG, and then incubating at 16°C, 230 RPM for ~15 h before harvest.
Phage challenge experiments were performed with electrocompetent E. coli MG1655 recovered and plated on selected LB media (carbenicillin 100 μg ml−1). Single colonies were inoculated into selective MMCG medium (1 × M9 Minimal Salts, 0.4% glucose, 0.02% MgSO4, 0.001% CaCl2, plus 100 μg ml−1 carbenicillin) and cultivated at 37°C shaking for ~20 h. Cultures were diluted 1:100 into selective MMCG medium and cultivated at 37°C shaking for 4 h to harvest mid-log cultures.
Bacteriophage strains
High-titer T2 phage (Coli Genetic Stock Center CGSC12141), T5 (CGSC12144), or T7 (CGSC12146) lysate stocks were purified from E. coli MG1655 cultures grown in LB.
METHOD DETAILS
Protein expression and purification
Recombinant CD-NTase and effector proteins were purified from E. coli as previously described (Whiteley et al., 2019; Zhou et al., 2018). Briefly, effector and CD-NTase enzymes were cloned from synthetic DNA (Integrated DNA Technologies) into a custom pET vector encoding a 6× His-SUMO2-tagged fusion protein and expressed in BL21-RIL E. coli (Agilent) containing the rare tRNA plasmid pRARE2. E. coli from large-scale M9ZB cultures (typically 2 L for each construct) were pelleted, washed with 1× PBS, and lysed by sonication in 1× Lysis Buffer (20 mM HEPES-KOH pH 7.5, 400 mM NaCl, 30 mM imidazole, 10% glycerol and 1 mM DTT). Recombinant protein was purified by gravity chromatography and binding to Ni-NTA resin (QIAGEN). Resin was washed with 1× Lysis Buffer supplemented to 1 M NaCl and then eluted with 1× Lysis Buffer supplemented to 300 mM imidazole. Purified protein was supplemented with recombinant human SENP2 protease (D364–L589, M497A) to remove the SUMO2 tag, and dialyzed overnight against 20 mM HEPES-KOH pH 7.5, 250 mM KCl, 1 mM DTT. Proteins were concentrated using a 30K-cutoff concentrator (Millipore) and further purified by size-exclusion chromatography on a 16/600 Superdex 200 column in 20 mM HEPES-KOH pH 7.5, 250 mM KCl, 1 mM TCEP. Final purified fractions were analyzed by SDS-PAGE and Coomassie staining, concentrated to > 30 mg ml−1, flash-frozen in liquid nitrogen, and stored as 35 μL aliquots at −80°C.
Electrophoretic mobility shift assay
Radiolabeled nucleotide products were enzymatically synthesized with purified V. cholerae DncV or EcCdnD in reactions containing 25 mM ATP, 25 μM GTP, trace α32P-GTP in 13 Reaction Buffer (5 mM MgCl2, 50 mM KCl, 1 mM TCEP and either 50 mM Tris-HCl pH 7.5 (DncV) or 50 mM CAPSO pH 9.4 (EcCdnD)) in a final reaction volume of 20 μl. Synthesis reactions were incubated ~15 h at 37°C and then inactivated by treating with 1 μL of 5 units μl−1 Calf Intestinal Phosphatase (New England Biolabs) and incubating at 37°C for 1 h. Radiolabeled products were then diluted 1:20 into electrophoretic mobility shift reactions containing either no protein, ~1 μM 6× His-SUMO2-EcCap2, 6× His-SUMO2-EcCap3, or 6× His-SUMO2-EcCap4 and 50 mM KCl, 50 mM Tris-HCl pH 7.5, 5 mM MgCl2, and 1 mM TCEP. Reactions were incubated for 20 min at 25°C, then separated on an 8 cm 10% nondenaturing polyacrylamide gel run at 100 V for 45 min. The gel was dried and then exposed to a phosphor-screen and imaged with a Typhoon Trio Variable Mode Imager (GE Healthcare).
Crystallization and structure determination
Cap4 proteins were crystallized at 18°C using the hanging drop method. Concentrated protein stocks were thawed on ice and diluted in buffer (25 mM HEPES-KOH pH 7.5, 1 mM TCEP). Drops were set by mixing protein stock and reservoir solution in 2 μl drops over a 350 μl reservoir in Easy-Xtal 15-well trays (QIAGEN). Each protein was crystallized as follows: 1) Apo MoCap4: Native or selenomethionine MoCap4 protein was diluted to 10 mg ml−1, supplemented with 10.5 mM MgCl2, mixed at a 1:1 ratio of protein:reservoir solution (110 mM Tris-HCl pH 8.5, 21% PEG-400), crystals were allowed to grow for 5 days and then were cryoprotected using reservoir solution supplemented with 10% ethylene glycol. 2) AbCap4–2′3′3′-cAAA: Native AbCap4 protein was diluted to 10 mg ml−1 and pre-incubated with 10.5 mM MgCl2 and ~200 μM purified AbCdnD product, mixed at a 1:1 ratio of protein:reservoir solution (10% PEG-3350, 200 mM lithium sulfate, 100 mM imidazole pH 8.0), crystals were allowed to grow for 25 days and then were cryoprotected using reservoir solution supplemented with 25% ethylene glycol; Selenomethionine AbCap4 was diluted to 10 mg ml−1 and pre-incubated with 10.5 mM MgCl2 and ~200 μM purified AbCdnD product, mixed at a 1:1 ratio of protein:reservoir solution (0.2 M lithium sulfate, 0.1 M Tris-HCl pH 8.5, 25% PEG-400), crystals were allowed to grow for 13 days and then were cryoprotected using reservoir solution supplemented with 10% PEG-400. 3) Apo AbCap4: Native AbCap4 protein was diluted to 10 mg ml−1 and mixed at a 1:1 ratio of protein:reservoir solution (0.1 M Tris pH 8.5, 0.2 M LiSO4, 16% PEG-3350), crystals were allowed to grow for 6 days and then were cryoprotected using reservoir solution supplemented with 30% ethylene glycol. 4) AbCap4–3′3′3′-cAAA: Native AbCap4 protein was diluted to 10 mg ml−1 and pre-incubated with 500 μM 3′3′3′-cAAA (Biolog), mixed at a 1:1 ratio of protein:reservoir solution (0.1 M Tris pH 8.5, 0.2 M LiSO4, 16% PEG-3350), crystals were allowed to grow for 6 days and then were cryoprotected using reservoir solution supplemented with 25% ethylene glycol.
X-ray data were collected at the Lawrence Berkeley National Laboratory Advanced Light Source beamline 8.2.2 and at the Northeastern Collaborative Access Team beamlines 24-ID-C and 24-ID-E (P30 GM124165), and used a Pilatus detector (S10RR029205), an Eiger detector (S10OD021527) and the Argonne National Laboratory Advanced Photon Source (DE-AC02–06CH11357). X-ray crystallography data were processed with XDS and AIMLESS (Kabsch, 2010) using the SSRL autoxds script (A. Gonzalez, Stanford SSRL). Experimental phase information for MoCap4 and AbCap4 was determined using data collected from selenomethionine-substituted crystals. In total, 5 and 36 sites respectively were identified with HySS in PHENIX (Adams et al., 2010) and an initial map was produced using SOLVE/RESOLVE (Terwilliger, 1999). Model building was performed using Coot (Emsley and Cowtan, 2004), before refinement in PHENIX. Statistics were analyzed as described in Table S1 (Chen et al., 2010; Karplus and Diederichs, 2012; Weiss, 2001).
DNA degradation assays
DNA degradation assays were performed by incubating 50 nM Cap4 protein with 10 ng μl−1 pGEM9z plasmid (Promega), E. coli genomic DNA, or 100 bp synthetic DNA as specified in a 25 μl reaction for 20 min at 37°C with 10 mM Tris-HCl pH 7.5, 25 mM NaCl, 5 mM MgCl2, and 1 mM TCEP. Degradation reactions were supplemented with activating nucleotide second messenger product at 50 nM synthetic or purified product (AbCdnD product), or unpurified cognate CD-NTase reaction (see below). Reactions were stopped by addition of 6× loading buffer (60 mM EDTA, 20 mM Tris-HCl pH 7.5, 30% glycerol, 0.1% SDS), and then 10 μl was separated on a 1.5% or 2% TAE agarose gel. Gels were run at 100 V for 20 min, then stained by rocking at room-temperature in buffer with 10 μg ml−1 ethidium bromide for 30 min. Gels were de-stained in water for ~10 min and then imaged with a ChemiDoc MP Imaging System.
AbCdnD product purification
Large scale AbCdnD product synthesis was carried out in 250 mL reactions with 500 nM purified AbCdnD and 250 μM ATP in 13 reaction buffer (50 mM KCl, 20 mM MgCl2, 1 mM MnCl2, 1 mM TCEP, 40 mM Tris pH 9.0), incubated overnight at 37°C with gentle shaking. The reaction was then treated with 20 μl of 5 units μl−1 Calf Intestinal Phosphatase (New England Biolabs) at 37°C for 3 h, then 0.2 mm filtered. The reaction was diluted to 12.5 mM salt with water, then loaded on to a HiTrap Q column (GE Healthcare) at 5 mL min−1 with a peristaltic pump and eluted on an AKTA FPLC (GE Healthcare) with a linear gradient of ammonium acetate from 0 to 2 M over 50 min. The AbCdnD product elutes as a single major peak at approximately 1.5 M ammonium acetate. Fractions containing the major product were identified by absorbance at 254 nm, pooled and dried by speedvac, and then re-suspended in approximately 1 mL of water. Initial ion exchange-purified product was desalted by gel filtration on a Superdex 30 Increase 10/300 GL column (GE Healthcare) run at 0.5 mL min−1 with water as running buffer.
Second-step purification was carried out using a C18 column (Agilent Zorbax Bonus-RP 4.6 3 150 mm, 3.5-micron). The column was heated to 50°C and run at 1 mL min−1 with a mobile phase of 50 mM NaH2PO4 (pH 6.8 with NaOH) supplemented with 3% acetonitrile. The sample was injected with ~10 μg per run to ensure separation of major and minor products. Fractions from several runs were pooled and dried by speedvac overnight, then desalted using C18 cartridges (Thermo Scientific Hypersep C18 100 mg columns). Speedvac-dried fractions were re-suspended in nuclease-free water with 2% acetic acid. The C18 cartridge was pre-washed with 500 μl MeOH, then 2× with 500 μl water with 2% acetic acid before loading the sample over the cartridge 3×. The cartridge was washed 2× with 500 μl water with 2% acetic acid, then eluted with 500 μl MeOH. Sample was dried by speedvac for approximately 2 h, then re-suspended in nuclease-free water. Concentrations were estimated using an extinction coefficient of ε = 40,500 l mole−1 cm−1.
CD-NTase product thin layer chromatography
CD-NTase reactions were performed with 5 μM recombinant enzyme in 20 μl reactions with 25 μM NTPs and trace α32P-ATP (mcGAS: ATP, GTP; DncV: ATP, GTP; EcCdnD: ATP, GTP; AbCdnD: ATP) overnight at 37°C with 50 mM KCl, 10 mM MgCl2, 1 mM TCEP, and 50 mM CAPSO pH 9.4 (CdnD) or 50 mM Tris-HCl pH 7.5 (cGAS, DncV). cGAS reactions were additionally supplemented with 2 μM 45-bp DNA, and AbCdnD reactions were additionally supplemented with 1 mM MnCl2. Reactions were terminated with addition of 1 μ of 5 units μl−1 Calf Intestinal Phosphatase (New England Biolabs) and incubating at 37°C for 1 h. For P1 degradation assays, 10 μl of each reaction was treated with 1 μl P1 nuclease (Sigma Product N8630) for 1 h at 37°C. Reactions were analyzed by thin-layer chromatography by diluting each reaction 1:10 in 100 mM sodium acetate pH 5.2 and then spotting 1 μl of each reaction on a PEI-cellulose plate (Millipore) developed in ~1 cm of 1.5 M KH2PO4 pH 3.8 until buffer was 1 cm from the top of plate. Plates were dried then expose to a phosphor-screen and imaged with a Typhoon Trio Variable Mode Imager (GE Healthcare).
AbCdnD product degradation analysis
Synthetic controls (3′3′3′-cAAA and 2′3′3′-cAAA) or purified AbCdnD product were diluted to 25 μM in 100 μl reactions and supplemented with 10× P1 buffer (final 1× concentration: 30 mM NaOAc pH 5.3, 5 mM ZnSO4, 50 mM NaCl) or snake venom phosphodiesterase buffer (final 1× concentration: 50 mM Tris pH 9, 10 mM MgCl2, 50 mM NaCl). Snake venom phosphodiesterase (0.8 mU) (Sigma, Phosphodiesterase I from Crotalus adamanteus venom, Product P3243) or Nuclease P1 (80 mU) (Sigma Product N8630) was added and reactions were incubated for 1 h or 5 h at 37°C, respectively. Reactions were diluted 1:2 in nuclease-free water, then filtered through a 10-kDa filter (Millipore) by centrifugation. The degradation products were analyzed using a C18 column (Agilent Zorbax Bonus-RP 4.6 × 150 mm, 3.5-micron) on HPLC held at 50°C and run at 1 mL min−1 with 50 mM NaH2PO4 (pH 6.8 with NaOH) supplemented with 3% acetonitrile.
Chemical synthesis of 2′3′3′-cAAA
All reagents and solvents for chemical operations were of analytical grade or the best grade available from commercial suppliers. Solvents for chromatographic operations were specified as analytical grade, HPLC grade, or gradient HPLC grade. YMC*Gel SIL (6 nm, S-75 μm) was used for preparative flash chromatography and TLC was performed with Merck 60 F254 silica gel plates. All chromatographic operations were performed at ambient temperature. Evaporation of solvents was accomplished by rotary evaporation in-vacuo either with membrane pump vacuum or oil pump high vacuum with water bath temperatures not exceeding 30–33°C.
UV-spectra were recorded on a JASCO V-650 spectrometer in phosphate buffered aqueous solution (pH 7). Mass spectra were generated with a Bruker Esquire LC 6000 spectrometer in the electrospray ionization mass spectrometry (ESI-MS) mode with 50% water / 49.5% methanol / 0.5% NH3 (pH 9–10) as matrix. Nuclear magnetic resonance (NMR) spectra were recorded with a 400 MHz Bruker Avance III HD and chemical shifts are expressed in parts per million (ppm). Chemical shifts were referenced to the DMSO solvent signal, 2.50 ppm for 1H. 85% phosphoric acid was used as external standard for 31P NMR spectra with 0 ppm. All 31P NMR spectra were recorded with proton decoupling. VWR / Hitachi: LaChromElite L-2130 Pump; VWR / Hitachi: LaChromElite L-2420 UV/Vis detector; VWR / Hitachi: LaChromElite organizer, Agilent Technologies: OpenLAB Control Panel A.02.01.
3 mmol of cyanoethyl phosphoramidite 5′-DMTr-2′-TBDMS-3′-CEP-N6-Bz-adenosine (ChemGenes, Wilmington, MA, USA, Cat. No. ANP-5671) were used as starting material for the synthesis of the protected dimeric linear precursor 5′-OH-2′-TBDMS-N6-Bz-adenosine-(3′ /5′)-cyanoethyl-phosphate-2′-TBDMS-3′-H-phosphonate-N6-Bz-adenosine with a standard oligonucleotide coupling protocol, originally developed for cyclic dinucleotides (Gaffney et al., 2010). After preparative flash chromatography purification on silica gel with chloroform / methanol (1:1), the linear dimer was evaporated to dryness. 7.5 mmol (2.5 eq.) 5-ethylthio-tetrazole as coupling reagent were added and the resulting mixture was evaporated 4 times from 40 mL absolute acetonitrile. The last evaporation was stopped at ~15 mL total volume, 0.5 g molecular sieves 3 Å were added and the mixture was stirred at room temperature for 30 min. 6 mmol (2 eq.) cyanoethyl phosphoramidite 5′-DMTr-2′-CEP-3′-TBDMS-N6-Bz-adenosine (ChemGenes, Wilmington, MA, USA, Cat. No. ANP-5681) were added and the protected trimeric linear precursor 5′-OH-3′-TBDMS-N6-Bz-adenosine-(2′/5′)-cyanoethyl-phosphate-2′-TBDMS-N6-Bz-adenosine-(3′/5′)-cyanoethyl-phosphate-2′-TBDMS-3′-H-phosphonate-N6-Bz-adenosine was prepared as previously described (Gaffney et al., 2010). After preparative flash chromatography purification on silica gel with chloroform / methanol (1:1), the linear trimer was evaporated to dryness. The final cyclization step and the release of protection groups was performed according to the standard protocol in Gaffney et al. (2010), leading to the raw product 2′3′3′-cAAA after evaporation of solvents.
100 mL water was added and the resulting suspension was placed in an ultrasonic bath at room temperature for 15 min, followed by 3 extraction cycles with 50 mL chloroform each. The combined organic phases were extracted with 50 mL water and the combined product-containing aqueous phase was filtered with a 0.45 μm regenerated cellulose (RC) filter and partially concentrated under reduced pressure to remove traces of chloroform. The complex product solution was diluted with water to 1000 mL and applied to a Q Sepharose Fast Flow anion exchange column (40–165 μm; 380 × 50 mm) Cl−-form (Sigma), previously regenerated with 2 M sodium chloride and washed with water. The column was washed with water (1000 ml), followed by a gradient of 0–600 mM triethylammonium bicarbonate buffer (TEAB, pH 7, 7500 ml) in water, 500 mL 600 mM TEAB and 1500 mL 1 M TEAB (detection wavelength 254 nm). The title compound eluted with ~400 mM TEAB. Product-containing fractions were carefully concentrated to a final volume of approximately 20 mL with a rotary evaporator equipped with a drop catcher in-vacuo. Subsequent purification of 2′3′3′-cAAA were accomplished by repeated preparative reversed phase medium pressure liquid chromatography (MPLC). The product solution was applied to a LiChroprep RP-18 column (15–25 μm; 450 × 50 mm) (Merck) previously equilibrated with 100 mM triethylammonium formate (TEAF, pH 6.8) in water. Elution was performed with 100 mM TEAF, 20 mM TEAF, followed by a step-gradient of 1%, 2%, 3%, 4% and 10% 2-propanol, 20 mM TEAF (pH 6.8) in water. Final purification of product containing fractions was accomplished by a LiChroprep RP-18 column (15–25 mm; 430 × 25 mm) (Merck) with a similar step-gradient of 2-propanol, 20 mM TEAF as organic modifier. For desalting, 2′3′3′-cAAA fractions of sufficient purity were applied to an in-line set-up of two LiChroprep RP-18 columns (15–25 μm; 450 × 50 mm each) (Merck), previously equilibrated with water. The columns were washed with water to remove excess TEAF buffer. Afterward, 2% 2-propanol in water was used to elute the desalted 2′3′3′-cAAA. To generate the sodium salt form of 2′3′3′-cAAA, pooled product-containing fractions were partially concentrated under reduced pressure and subsequently applied to a Toyopearl SP-650M cation exchange column (65 μm; 90 × 35 mm) Na+-form (Sigma), previously regenerated with 2 M sodium chloride and washed with water. For elution the column was washed with water until no UV-absorbance was detectable at 254 nm anymore. After filtration and careful evaporation under reduced pressure, 845.6 μmol 2′3′3′-cAAA, sodium salt was isolated with a purity of 99.02% by HPLC (theoretical yield: 28.2%).
Formula (free acid): C30H36N15O18P3 (MW 987,63 g/mol)
UV-Vis (water pH 7.0): λmax 259 nm; ε 40500.
ESI-MS pos. mode: m/z 988 (M+H)+, m/z 1010 (M+Na)+, m/z 1032 (M-H+2Na)+.
ESI-MS neg. mode: m/z 986 (M-H)−, m/z 1008 (M-2H+Na)−, m/z 1030 (M-3H+2Na)−.
1H NMR (400 MHz, Deuterium Oxide) δ 8.51 (s, 1H), 8.47 (s, 1H), 8.24 (s, 1H), 8.22 (s, 1H), 8.21 (s, 1H), 8.18 (s, 1H), 6.34 (d, J = 7.8 Hz, 1H), 6.20 (d, J = 7.0 Hz, 1H), 6.17 (d, J = 4.2 Hz, 1H), 5.25 – 5.18 (m, 1H), 4.89 – 4.71 (m, 7H), 4.58 – 4.54 (m, 2H), 4.37 – 4.20 (m, 6H) ppm.
31P NMR (162 MHz, Deuterium Oxide) δ 0.25 (s, 1P), 0.02 (s, 1P), −0.29(s, 1P) ppm.
Analytical HPLC: (Kromasil 100–10, RP-8 (10 μm; 250 × 4 mm)) 3% acetonitrile, 50 mM sodium dihydrogen phosphate buffer, pH 6.8; 1.5 mL/min; UV 259 nm; tRET 5.84 min.
Liquid chromatography-mass spectrometry
To characterize the enzymatic products of AbCdnD, LC-MS/MS analysis was performed as previously described (Lau et al., 2020; Ye et al., 2020). LC-MS/MS was performed on a Thermo Vanquish UHPLC coupled to a Thermo QExactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific). Chromatography utilized a Sequant ZIC-pHILIC polymeric column (100 mm × 2.1 mm, 5 μm) (EMD Millipore) maintained at 45°C and a flow rate of 0.4 mL min−1. AbCdnD products and cyclic oligonucleotide standards were separated by injecting 2 μl of sample and eluting on the following linear gradient: (A) 20 mM ammonium bicarbonate in water, pH 9.6, and (B) acetonitrile; 90% B for 0.25 min, a linear gradient to 55% B at 4 min, and sustained until 6 min. Column was re-equilibrated for 2.5 min at 90% B. Detection of cyclic oligonucleotides was performed in positive ionization mode using an heated electrospray ionization (HESI) source with the following parameters: spray voltage of 3.5 kV; sheath gas, auxiliary gas, and sweep gas flow rates of 40, 20, and 2 AU, respectively; capillary and auxiliary gas heater temperature of 275 and 350°C, respectively. Profile MS1 spectra were acquired under the following parameters: mass resolution of 35,000, AGC volume of 1 × 106 maximum IT of 75 ms and a scan range from 450 to 1400 m/z to account for z = 1 and z = 2 ions of cyclic tri- and tetraadenosine nucleotides. Data-dependent tandem mass spectra were acquired using CID of the following settings: mass resolution of 17,500, AGC volume of 1 × 105, maximum IT of 50 ms; a loop count of 5, isolation window of 1.5 m/z; normalized collision energy of 25 eV; dynamic exclusion was not used. Data reported are for the most common ion for each indicated cyclic oligonucleotide.
Thermal shift assay
Proteins were mixed with 3× Sypro dye and 100 μM purified nucleotide ligand (3′3′3′-cAAG or IEX-purified AbCnD product) in 20 mM HEPES-KOH pH 7.5 and 75 mM KCl, with a final protein concentration of 10 μM. Using a BioRad CFX96 thermocycler, samples were brought from 25 to 95°C, reading fluorescence in HEX channel every 0.5°C. The derivative of each curve over time was calculated using BioRad CFX Manager, then normalized as a percent maximum change in fluorescence for each sample.
Negative stain electron microscopy
0.05–0.1 mM purified Cap4 proteins were mixed with equimolar amounts of cyclic trinucleotide and DNA as indicated. For negative staining, all samples were adjusted to 0.0025–0.005 mg ml−1 in 20 mM HEPES-KOH pH 7.5, 150 mM KCl, 1 μM TCEP without or with 1 mM activating ligand. Negative stain grids were prepared by applying the sample (3 μl) to a glow-discharged (30 s, 30 mA) 400-mesh Cu grid (Electron Microscopy Sciences) coated with an ~15 nm layer of continuous carbon (Safematic CCU-010). After 30 s, the grid was blotted from the side, immediately stained with 1.5% uranyl formate, and blotted again. The staining procedure was repeated two times, with a 30 s incubation with uranyl formate before the final blotting step. The grid was allowed to dry for at least 15 min before imaging. Samples were imaged with a Tecnai T12 (FEI) transmission electron microscope operated at 120 keV and equipped with a Gatan 4K × 4K CCD camera. Images were collected at a nominal magnification of 67,000× and pixel size of 1.68 Å with defocus values of ~1.0–2.0 μm. Between 80 and 333 micrograph images were collected for each of the 9 datasets. Image processing was done in RELION-3.0 (Zivanov et al., 2018). After CTF estimation with GCTF (Zhang, 2016), particle picking was carried out with gautomatch (Kai Zhang; https://www.mrc-lmb.cam.ac.uk/kzhang/) or LoG-based autopicking (Zivanov et al., 2018) and manually inspected. Particles were extracted with a box size of 156 pixels and subjected to reference-free 2D classification. Particles in the best-resolved classes were selected and subjected to an additional one to two rounds of 2D classification. In addition, for each dataset, random subsets of 10,000 particles were subjected to 2D classification to compare the distributions of particles in different image groups. For datasets with more than 10,000 particles, the standard error of mean of the distributions between four independent subsets of 10,000 particles ranged from 0.1 to 1.1%. Particles retained after 2D classification were used to generate a de novo initial model using the stochastic gradient descent algorithm in RELION, followed by 3D refinement and classification. Overall resolutions of EM maps of dimer complexes were ~15 Å. Rigid body docking was performed using Chimera (Pettersen et al., 2004).
SEC-MALS
Cap4 and Cap4–nucleotide second messenger complex samples were prepared by diluting in SEC-MALS running buffer (150 mM KCl, 20 mM HEPES-KOH pH 7.5 and 1 mM TCEP) to 2 mg ml−1 and incubating on ice for 5 min with 100 μM nucleotide second messenger and then separated on an SRT SEC-300 column (Sepax). Protein concentration was calculated using refractive index on a Wyatt Optilab T-rex Refractive Index Detector assuming dn dc−1 of 0.185 and a molar mass was calculated using a Wyatt Dawn Heleos II Multi-Angle Light Scattering detector and ASTRA software.
DNA EMSA assay
Catalytically inactive Cap4 proteins (AbCap4 K69A, EcCap4 K74A) were mixed on ice at a concentration of 10 μM with 1 μM 45 bp DNA and 50 μM 3′3′3′-cAAG (EcCap4) or IEX-purified AbCdnD product (AbCap4) in a final reaction volume of 20 μL containing 20 mM HEPES-KOH pH 7.5, 50 mM KCl, 5 mM MgCl2 and 1 mM TCEP. Reactions were incubated on ice for 5 min and then supplemented with to a final concentration of 5% glycerol (v/v). Samples were separated on a 2% native agarose TB gel containing 100 mM Tris and 45 mM boric acid by running at 250 V for 45 min at 4°C. The gel was stained by soaking in buffer with 10 μg ml−1 ethidium bromide solution for 20 min and then imaged with ChemiDoc MP Imaging System.
DNA fragment sequencing
Cap4 proteins were incubated with 400 ng plasmid or genomic DNA in 80 μl reactions for 2 h (500 nM protein, 100 nM activating ligand, 10 mM Tris-HCl pH 7.5, 25 mM NaCl, 1 mM TCEP, 5 mM MgCl2), then DNA fragments were purified by phenol-chloroform extraction. Sequencing libraries were made from single stranded DNA fragments using an Accel-NGS 1S Plus DNA Library kit (Swift Biosciences) as previously described (Lau et al., 2020) and sequenced on a NextSeq500. For bioinformatics analyses, given that Cap4 digestion results in small fragments, we took advantage of that fact that reads containing the 3′ adaptor sequence have the full fragment sequence. Reads containing the 3′ adaptor were selected and adaptor trimmed using Cutadapt (Martin, 2011). The 8 nucleotide low complexity sequence was trimmed by Cutadapt, and reads were mapped to the pGEM9z(−) plasmid (Promega) or the E. coli K12 genomic sequence (GenBank U00096.3) using Bowtie2 (Langmead and Salzberg, 2012). The location of the 5′ end of each read was used as the cut site and extracted from the output sam files. The 10 nucleotide sequence upstream and downstream of the cut sites were compiled and used to identify the consensus cut site using WebLogo3 (Crooks et al., 2004). The lengths of the mapped reads were extracted using custom python scripts and histogram plots were made using ggplot2 (Wickham, 2016).
DncV co-expression with CBASS effectors
A plasmid expressing dncV (pAW1371-pBAD33-dncV) and a second plasmid expressing a predicted CBASS effector were electroporated into competent Escherichia coli BL21-DE3 (Invitrogen). Bacteria were recovered for 1 h shaking at 37°C in Super Optimal Broth with Catabolite repression (SOC), then plated onto selective LB Agar with glucose to repress dncV expression (LB: 10 g L−1 tryptone, 5 10 g L−1 yeast extract, 5 10 g L−1 NaCl, 20 μg ml−1 chloramphenicol, 100 μg ml−1 carbenicillin, 0.2% w/v glucose). Single colonies were inoculated into selective LB Agar plus glucose medium and cultured at 37°C shaking for ~16 h. 5 μl spots of 10-fold serial dilutions in LB were pipetted onto selective high-salt LB (20 μg ml−1 chloramphenicol, 100 μg ml−1 carbenicillin, NaCl concentration adjusted to 2% w/v) under inducing conditions (0.2% arabinose, 5 μM IPTG) or repressive conditions (0.2% glucose). Data are measured as CFU per ml and the ratio of inducing to repressive conditions and are the mean of three independent experiments. The predicted effector plasmids are pETSUMO2 expressing either: green fluorescent protein (gfp, negative control); Vibrio dncV native effector capV (WP_001133548.1, positive control); Desulfotomaculum alkaliphilium CD-NTase005 effector Saf2TM-SAVED (WP_031517735.1); Escherichia coli CD-NTase010 effector Saf2TM-SAVED (WP_001057904.1); Acinetobacter baumannii CD-NTase011 effector AbCap5 (WP_031984940.1); Geobacillus sp. CD-NTase012 effector GsCap5 (WP_013400843.1); Myxococcus xanthus CD-NTase022 effector caspase-SAVED (WP_020479061.1); Acinetobacter baumannii CD-NTase037 effector a.k.a. AbCdnD effector AbCap4 (WP_008942236.1); Enterobacter cloacae CD-NTase038 effector a.k.a. EcCdnD effector EcCap4 (WP_032676399.1); Bradyrhizobium japonicum CD-NTase039 effector BjCap5 (WP_011082906.1); Burkholderia pseudomallei CD-NTase041 a.k.a. bpCdnG effector bpCap5 (WP_004556385.1); Citrobacter freundii CD-NTase042 effector highly similar to EcCdnG effector CfCap5 (ETX65525.1); Acinetobacter baumannii CD-NTase043 effector AbCap5 (WP_000539314.1); Pseudomonas aeruginosa CD-NTase044 effector PaCap5 (WP_023082129.1); Bacillus coagulans CD-NTase046 effector BcCap5 (WP_013858316.1); Vibrio cholerae CD-NTase053 effector VcCap5 (WP_000259919.1).
Phage resistance assays
Electrocompetent E. coli MG1655 was electroporated with individual medium-copy plasmids encoding entire CBASS operons under their native promoters and analyzed similarly to previously described methods (Cohen et al., 2019; Doron et al., 2018). E. coli were recovered and plated on selective LB (carbenicillin 100 μg ml−1). Single colonies were inoculated into selective MMCG medium (1× M9 Minimal Salts, 0.4% glucose, 0.02% MgSO4, 0.001% CaCl2, plus 100 μg ml−1 carbenicillin) and cultivated at 37°C shaking for ~20 h. Cultures were diluted 1:100 into selective MMCG medium and cultivated at 37°C shaking for 4 h to harvest mid-log cultures. Phage resistance was measured by a modified double-agar overlay technique. Bacteria were immobilized in soft-agar overlays by thoroughly mixing 400 μl of mid-log culture with 3.5 mL molten MMCG top agar (MMCG medium plus 0.35% agar, 5 mM MgCl2, 0.1 mM CaCl2, 1 mM MnCl2). Bacteria combined with top agar was immediately poured onto a 100 × 15 mm Petri dish containing 20 mL solidified MMCG Agar (1.6%) and allowed to cool for 10 min at room temperature. A high-titer T2 phage lysate (Coli Genetic Stock Center CGSC12141), T5 lysate (CGSC12144), or T7 lysate (CGSC12146) prepared from MG1655 E. coli was 10-fold serially diluted into SM Buffer (100 mM NaCl, 8 mM MgSO4, 50 mM Tris-HCl pH 7.5). 3 μl spots of dilutions were pipetted onto the solidified double-agar overlays. Spots were allowed to dry for 20 min at room temperature. Plates were incubated at 37°C for ~16 h and plaques were quantified to compare efficiency of plating. Data are the mean of at least three independent experiments. Plasmids expressing CBASS operons were pLOCO2-dncV operon (Genome AE003852.1, 178097–185003), pLOCO2-Ec-cdnD02 operon (Locus JCKK01000002, 2261880–2268858), and pLOCO2-Ec-cdnG operon a.k.a. CD-NTase042 operon (Locus JSNY01000125, 10563–17743).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details for each experiment can be found in the figure legends and outlined in the corresponding methods details section. Data are plotted with error bars representing the standard deviation (SD) or standard error of the mean (SEM) as indicated.
Supplementary Material
Figure S1. Biochemical Reconstitution of Cap4 Endonuclease Activity, Related to Figure 1
(A) SDS-PAGE and Coomassie stain analysis of purified recombinant Cap4 and CdnD proteins.
(B) Cartoon schematics and crystal structures of apo MoCap4 and AbCap4 enzymes.
(C) Alignment of divergent Cap4 homologs. Endonuclease active site residues are indicated with a red circle, and the known secondary structure of the Cap4 endonuclease domain (orange) and SAVED sensor domain (blue) determined from the AbCap4 crystal structure is displayed below.
(D) EcCap4 and AbCap4 endonuclease activity is inhibited by the divalent metal chelator EDTA. A requirement of divalent cation metals for catalysis agrees with structural analysis of the Cap4 active site and homology with Type II restriction endonucleases.
(E) Like EcCap4, AbCap4 mutation of the putative catalytic residue K69A ablates endonuclease activity.
(F) Agarose gel analysis of AbCap4 endonuclease cleavage activity. Like EcCap4, AbCap4 is a promiscuous DNA endonuclease that degrades plasmid DNA, E. coli genomic DNA, and synthetic DNA but requires recognition of the activating nucleotide second messenger synthesized by the cognate CdnD CD-NTase. Biochemical data are representative of at least 3 independent experiments.
Figure S2. Purification and Characterization of the AbCdnD Nucleotide Second Messenger Product, Related to Figure 2
(A) DNA cleavage analysis of MoCap4 endonuclease activity. The AbCdnD nucleotide second messenger product is able to activate both AbCap4 and the more closely related MoCap4 (~80% identity) but not the distantly related EcCap4 enzyme (~20% identity).
(B) Analysis of AbCdnD nucleotide specificity. AbCap4 endonuclease activity was used to detect the presence of activating nucleotide second messenger signal and determine which nucleotides are required for AbCdnD product formation. ATP is alone necessary and sufficient for AbCdnD-dependent activation demonstrating that the nucleotide second messenger is an oligoadenylate species.
(C) Enzymatic degradation and HPLC analysis of synthetic control cyclic oligoadenylate species and AbCdnD products. Synthetic and AbCdnD product nucleotide second messengers were analyzed following cleavage with Nuclease P1 (P1, cleaves 3′ –5′ phosphodiester bonds) and snake venom phosphodiesterase (SVPD, cleaves both 3′ –5′ and 2′ –5′ phosphodiester bonds) by HPLC and C18 chromatography. Degradation of the AbCdnD major product matches that of synthetic 2′3′3′-cAAA further confirming the structural assignment of this nucleotide second messenger species. The AbCdnD minor product that potently activates AbCap4 activity is partially P1-resistant confirming the presence of a 2′ –5′ phosphodiester bond in this closely related species.
(D) Assessment of DNA degradation after activation of AbCap4 with either the major or minor AbCdnD products from Figure 2E.
(E) Thermal denaturation assay to measure nucleotide second messenger-dependent stabilization of AbCap4. AbCap4 is stabilized in the presence of the AbCdnD major product 2′3′3′-cAAA but not in presence of the unrelated DncV product 3′3′-cGAMP demonstrating specific recognition of 2′3′3′-cAAA.
(F) AbCap4 DNA degradation assay comparing candidate activating ligands. The AbCdnD product mixture, but not synthetic 2′3′3′-cAAA, is capable of activating AbCap4 further confirming that the minor 2′ –5′-linked product is primarily responsible for AbCap4 activation.
(G) LC-MS/MS analysis of AbCdnD major (top) and minor (bottom) enzymatic products. Parent mass extracted ion trace (left) and tandem mass spectra comparison (right) of the major AbCdnD product confirms 2′3′3′-cAAA. Distinct patterning in tandem mass spectra (black arrows) in the minor product relative to 3′3′3′3′-cA4 indicates a cA4 molecule of distinct phosphodiester linkages. Ions detected: m/z 988.1648 [M+H] + (cAAA); m/z 659.1123 [M+2H] 2+ (cA4).
Figure S3. Biochemical and Mutagenesis Analysis of Cap4-Ligand Interactions, Related to Figure 3
(A) Thermal denaturation assay measuring nucleotide second messenger-dependent stabilization of EcCap4 and AbCap4. Activating nucleotide second messengers bind and stabilize both full-length Cap4 and isolated SAVED-domain only constructs, confirming the SAVED domain is the ligand-binding domain. EcCap4 SAVED domain (258–end), AbCap4 SAVED domain (235–end).
(B) Zoom-in cutaways showing A2 and A3 nucleotide pocket interactions in the AbCap4–2′3′3′-cAAA complex structure, and alignments of key pocket residues in various Cap4 homologs.
(C) DNA degradation assays with AbCap4 and EcCap4 proteins with point mutations within the SAVED domain verifies the importance of individual contacts for nucleotide second messenger recognition. Cap4 mutants were activated with increasing concentrations of activating ligand (1 nM, 10 nM, 100 nM, 1 μM). Biochemical data are representative of at least 3 independent experiments.
Figure S4. Analysis of Cap4 Ligand-Bound and Apo Structures, Related to Figure 3
(A) Comparison of apo AbCap4, AbCap4–3′3′3′-cAAA, and AbCap4–2′3′3′-cAAA crystal structures. Each AbCap4 structure exhibits the same overall conformation supporting that the AbCap4-ligand bound structures represent an inactive-state.
(B) Schematic of packing observed of each of the six chains in the AbCap4 asymmetric unit.
(C) Packing of MoCap4 monomers with symmetry-related chains.
(D) Structural comparison of Cap4 and Can1 from the Thermus thermophilus Type III CRISPR immune system (PDB 6SCE). Cap4 and Can1 each contain two CARF domains in a single polypeptide, but the proteins have distinct domain organizations and represent independent evolutionary events.
Figure S5. Oligomerization of Cap4 Proteins upon Nucleotide Second Messenger Recognition, Related to Figure 4
(A) Negative stain EM class averages of AbCap4 (K69A) and MoCap4 (K69A) apo proteins, with activating ligand (purified using ion exchange chromatography from AbCdnD reactions), or with ligand and 45 bp DNA. Cap4 proteins were incubated with components as indicated, then analyzed by negative stain EM (left). Fractions of averaged classes of each oligomeric state were calculated (right).
(B) Micrograph image of a larger filament (highlighted with arrow) formed by EcCap4 in the presence of activating ligand 3′3′3′-cAAG. Higher-order oligomerization supports larger complexes observed in SEC-MALS experiments (Figure 4B). Scale bar represents 200 Å.
(C) SEC-MALS analysis of AbCap4 in the presence or absence of either purified AbCdnD product or synthetic 2′3′3′-cAAA. Absolute molecular masses of apo and ligand-bound complexes were quantified by multiangle light scattering (AbCap4 expected MW: 52.1 kDa).
(D) 3D reconstruction of the activated MoCap4 dimeric complex from negative stain EM data with high-resolution AbCap4–2′3′3′-cAAA structures docked within the density. The location of the Cap4 N-terminal extension and truncation point used for mutagenesis (G19) is denoted with an arrow and supports a possible role for this extension in further controlling endonuclease domain DNA binding and activation. See Figure S6.
(E) Electrophoretic mobility shift assay assessing DNA binding of AbCap4 (K69A). Cap4 was incubated with 45 bp DNA and activating ligand as indicated. Like EcCap4, stable AbCap4–DNA complex formation only occurs in the presence of activating nucleotide second messenger.
(F,G) Analysis of DNA fragments remaining after complete digestion with AbCap4. Distribution of fragments sizes (left), cut site consensus sequences (center), and location of cuts within E. coli genomic DNA showing relaxation of cut site sequence preference. Biochemical data are representative of at least 3 independent experiments.
Figure S6. Characterization of the Cap4 N Terminus in DNA Binding, Ligand Binding, and Oligomerization, Related to Figure 4
(A) Agarose gel analysis of Cap4 activity and plasmid DNA degradation. Full-length Cap4 proteins degrade target DNA in the presence of activating nucleotide second messenger, but N-terminal truncations of AbCap4 (G19–end) and EcCap4 (G11–end) do not.
(B) Electrophoretic mobility shift assay assessing DNA binding of N-terminal truncations of AbCap4 (G19–end) and EcCap4 (G11–end). The conserved N-terminal extension not visible in the AbCap4 or MoCap4 crystal structures is required for DNA recognition.
(C) Thermal denaturation assay measuring nucleotide second messenger-dependent stabilization of N-terminal truncations of EcCap4 and AbCap4. Activating nucleotide second messengers bind and stabilize truncated Cap4 proteins, indicating the N terminus is not required for ligand-binding.
(D) SEC-MALS analysis of N-terminal truncations of EcCap4 and AbCap4 in the presence or absence of activating ligand. Quantification of absolute molecular masses of EcCap4– and AbCap4–ligand complexes confirms the endonuclease domain N-terminal extension does not impact ligand-binding (DN EcCap4 expected MW: 55.1 kDa; ΔN AbCap4 expected MW: 50.4 kDa). Biochemical data are representative of at least 3 independent experiments.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| E. coli BL21-DE3 RIL | Agilent | 230245 |
| E. coli MG1655 | Coli Genetic Stock Center | CGSC6300 |
| E. coli BL21-DE3 | Invitrogen | C600003 |
| T2 phage | Coli Genetic Stock Center | CGSC12141 |
| T5 phage | Coli Genetic Stock Center | CGSC12144 |
| T7 phage | Coli Genetic Stock Center | CGSC12146 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ni-NTA Agarose | QIAGEN | 30250 |
| HiTrap Q HP Column | GE Healthcare | 17115401 |
| HiLoad 16/600 Superdex 75 PG | GE Healthcare | 28989333 |
| Zorbax Bonus-RP | Agilent | 863668–901 |
| Superdex 30 Increase 10/300 GL | GE Healthcare | 29219757 |
| SRT SEC-300 | Sepax | 215300–7830 |
| SeQuant ZIC-pHILIC | EMD-Millipore | 1504620001 |
| [α−32P] ATP | Perkin Elmer | BLU003H250UC |
| [α−32P] GTP | Perkin Elmer | BLU006H250UC |
| PEI-Cellulose F TLC plate | EMD Biosciences | EM1.05579.0001 |
| Nuclease P1 from Penicillium citrinum | Sigma-Aldrich | N8630 |
| Snake Venom Phosphodiesterase | Sigma-Aldrich | P3243 |
| Alkaline Phosphatase, Calf Intestinal (CIP) | New England Biolabs | M0290S |
| DNA Extraction Kit | QIAGEN | 69504 |
| ATP, GTP | New England Biolabs | N0450S |
| SYPRO Orange | ThermoFisher | S6650 |
| HEPES | VWR | 97061–824 |
| Tris base | VWR | 97062–416 |
| PEG-400 | Sigma-Aldrich | 202398 |
| PEG-3350 | Sigma-Aldrich | 202444 |
| Lithium sulfate | Sigma-Aldrich | 62609 |
| Imidazole | VWR | 97065–016 |
| Tris[−2carboxyethyl] phosphine hydrochloride (TCEP) | GoldBio | TCEP50 |
| Ethylene glycol | VWR | 97061–964 |
| 3′3′3′-cAAA | Biolog Life Science Institute GmbH & Co. KG | C 362 |
| 3′3′3′-cAAG | Biolog Life Science Institute GmbH & Co. KG | C 361 |
| Deposited Data | ||
| MoCap4 | This paper | PDB: 6VM5 |
| AbCap4–2′3′3′-cAAA | This paper | PDB: 6VM6 |
| AbCap4–3′3′3′-cAAA | This paper | PDB: 6WAN |
| AbCap4 | This paper | PDB: 6WAM |
| Oligonucleotides | ||
| 5′ -TACAGATCTACTAGTGATCTATGACTG ATCTGTACATGAT CTACA-3′ | Integrated DNA Technologies | DNA45 sense |
| 5′ -TGTAGAT CATGTACAGAT CAGTCATAG ATCACTAGTAGATCTGTA-3′ | Integrated DNA Technologies | DNA45 antisense |
| Software and Algorithms | ||
| Phenix 1.13–2998 | Adams et al., 2010 | https://www.phenix-online.org/ |
| Coot 0.8.9 | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Pymol vl.7.4.4 | Schrödinger | https://pymol.org/2/ |
| Prism 7.0d | GraphPad software | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
Cap4 proteins are a major family of nucleotide second messenger receptors in bacteria
Cap4 receptors degrade DNA and mediate phage resistance by CBASS operons
The Cap4 ligand-binding SAVED domain evolved from CRISPR CARF proteins
As in cGAS-STING signaling, bacteria use 2′–5′-linked signals for antiviral immunity
ACKNOWLEDGMENTS
The authors are grateful to Kevin Corbett (University of California, San Diego) and members of the Kranzusch lab for helpful comments and discussions, Kelly Arnett and Harvard University’s Center for Macromolecular Interactions, and the Molecular Electron Microscopy Suite at Harvard Medical School. The work was funded by the Richard and Susan Smith Family Foundation (to P.J.K. and S.S.); a Cancer Research Institute CLIP grant (to P.J.K.); the Parker Institute for Cancer Immunotherapy (to P.J.K.); the Pew Biomedical Scholars Program (to P.J.K. and A.S.Y.L.); the Searle Scholars Program (to A.S.Y.L.); a Sloan research fellowship (to A.S.Y.L.); the Vallee Foundation (to S.S.); NIH S10OD020025, R01ES027595, and P42ES010337 (to M.J.); and NIH R01AI018045 and R01AI026289 (to J.J.M.). B.L. is supported as a Herchel Smith Graduate Research Fellow. A.T.W. was supported as a fellow of The Jane Coffin Childs Memorial Fund for Medical Research. B.R.M. was supported as a Ruth L. Kirschstein NRSA postdoctoral fellow (NIH F32GM133063). I.T.M. was supported by NIH F31CA236405.
Footnotes
DECLARATION OF INTERESTS
D.K., P.N., and F.S. are employed at Biolog Life Science Institute GmbH & Co. KG, which sells 3′3′3′-cAAG and 3′3′3′-cAAA and may sell 2′3′3′-cAAA as research tools.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.05.019.
REFERENCES
- Ablasser A, and Chen ZJ (2019). cGAS in action: Expanding roles in immunity and inflammation. Science 363, eaat8657. [DOI] [PubMed] [Google Scholar]
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner KP, Ludwig J, and Hornung V (2013). cGAS produces a 2′−5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athukoralage JS, McMahon SA, Zhang C, Grüschow S, Graham S, Krupovic M, Whitaker RJ, Gloster TM, and White MF (2020). An anti-CRISPR viral ring nuclease subverts type III CRISPR immunity. Nature 577, 572–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheim A, and Sorek R (2020). The pan-immune system of bacteria: antiviral defence as a community resource. Nat. Rev. Microbiol 18, 113–119. [DOI] [PubMed] [Google Scholar]
- Burroughs AM, Zhang D, Schäffer DE, Iyer LM, and Aravind L (2015). Comparative genomic analyses reveal a vast, novel network of nucleotidecentric systems in biological conflicts, immunity and signaling. Nucleic Acids Res 43, 10633–10654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D, Melamed S, Millman A, Shulman G, Oppenheimer-Shaanan Y, Kacen A, Doron S, Amitai G, and Sorek R (2019). Cyclic GMP-AMP signalling protects bacteria against viral infection. Nature 574, 691–695. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilchanka O, and Mekalanos JJ (2013). Cyclic dinucleotides and the innate immune response. Cell 154, 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, and Malik HS (2012). Rules of engagement: molecular insights from host-virus arms races. Annu. Rev. Genet 46, 677–700. [DOI] [PubMed] [Google Scholar]
- Davies BW, Bogard RW, Young TS, and Mekalanos JJ (2012). Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149, 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, and Vance RE (2013). The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 3, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds PN, and Rathjen JP (2010). Plant immunity: towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet 11, 539–548. [DOI] [PubMed] [Google Scholar]
- Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, and Sorek R (2018). Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, eaar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaglesham JB, Pan Y, Kupper TS, and Kranzusch PJ (2019). Viral and metazoan poxins are cGAMP-specific nucleases that restrict cGAS-STING signalling. Nature 566, 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Gaffney BL, Veliath E, Zhao J, and Jones RA (2010). One-flask syntheses of c-di-GMP and the [Rp,Rp] and [Rp,Sp] thiophosphate analogues. Org. Lett 12, 3269–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. (2013a). Cyclic [G(2′, 5′ )pA(3′, 5′ ) p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al. (2013b). Structure-function analysis of STING activation by c[G(2′, 5′ )pA(3′, 5′ )p] and targeting by antiviral DMXAA. Cell 154, 748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Hartmann R, Ablasser A, and Hopfner KP (2014). OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol 14, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia N, Jones R, Yang G, Ouerfelli O, and Patel DJ (2019). CRISPR-Cas III-A Csm6 CARF Domain Is a Ring Nuclease Triggering Stepwise cA4 Cleavage with ApA > p Formation Terminating RNase Activity. Mol. Cell 75, 944–956.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). Xds. Acta Crystallogr. D Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus PA, and Diederichs K (2012). Linking crystallographic model and data quality. Science 336, 1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskiene M, Kostiuk G, Venclovas Č, Tamulaitis G, and Siksnys V (2017). A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science 357, 605–609. [DOI] [PubMed] [Google Scholar]
- Kranzusch PJ (2019). cGAS and CD-NTase enzymes: structure, mechanism, and evolution. Curr. Opin. Struct. Biol 59, 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee ASY, Wilson SC, Solovykh MS, Vance RE, Berger JM, and Doudna JA (2014). Structure-guided reprogramming of human cGAS dinucleotide linkage specificity. Cell 158, 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Wilson SC, Lee AS, Berger JM, Doudna JA, and Vance RE (2015). Ancient Origin of cGAS-STING Reveals Mechanism of Universal 2′, 3′ cGAMP Signaling. Mol. Cell 59, 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau RK, Ye Q, Birkholz EA, Berg KR, Patel L, Mathews IT, Watrous JD, Ego K, Whiteley AT, Lowey B, et al. (2020). Structure and Mechanism of a Cyclic Trinucleotide-Activated Bacterial Endonuclease Mediating Bacteriophage Immunity. Mol. Cell 77, 723–733.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis SR, Wilson SC, and Vance RE (2017). Evolutionary Origins of cGAS-STING Signaling. Trends Immunol 38, 733–743. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet.journal 17, 10–12. [Google Scholar]
- McMahon SA, Zhu W, Graham S, Rambo R, White MF, and Gloster TM (2020). Structure and mechanism of a Type III CRISPR defence DNA nuclease activated by cyclic oligoadenylate. Nat. Commun 11, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina R, Stella S, Feng M, Sofos N, Jauniskis V, Pozdnyakova I, López-Méndez B, She Q, and Montoya G (2019). Structure of Csx1-cOA4 complex reveals the basis of RNA decay in Type III-B CRISPR-Cas. Nat. Commun 10, 4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewoehner O, and Jinek M (2016). Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein Csm6. RNA 22, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewoehner O, Garcia-Doval C, Rostøl JT, Berk C, Schwede F, Bigler L, Hall J, Marraffini LA, and Jinek M (2017). Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature 548, 543–548. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Severin GB, Ramliden MS, Hawver LA, Wang K, Pell ME, Kieninger AK, Khataokar A, O’Hara BJ, Behrmann LV, Neiditch MB, et al. (2018). Direct activation of a phospholipase by cyclic GMP-AMP in El Tor Vibrio cholerae. Proc. Natl. Acad. Sci. USA 115, E6048–E6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamulaitiene G, Jovaisaite V, Tamulaitis G, Songailiene I, Manakova E, Zaremba M, Grazulis S, Xu SY, and Siksnys V (2017). Restriction endonuclease AgeI is a monomer which dimerizes to cleave DNA. Nucleic Acids Res 45, 3547–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC (1999). Reciprocal-space solvent flattening. Acta Crystallogr. D Biol. Crystallogr 55, 1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Takasaki Y, Sato C, Ando S, and Tanaka I (2009). Structures of restriction endonuclease HindIII in complex with its cognate DNA and divalent cations. Acta Crystallogr. D Biol. Crystallogr 65, 1326–1333. [DOI] [PubMed] [Google Scholar]
- Weiss MS (2001). Global indicators of X-ray data quality. J. Appl. Cryst. 34, 130–135. [Google Scholar]
- Whiteley AT, Eaglesham JB, de Oliveira Mann CC, Morehouse BR, Lowey B, Nieminen EA, Danilchanka O, King DS, Lee ASY, Mekalanos JJ, and Kranzusch PJ (2019). Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature 567, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer; ). [Google Scholar]
- Ye Q, Lau RK, Mathews IT, Birkholz EA, Watrous JD, Azimi CS, Pogliano J, Jain M, and Corbett KD (2020). HORMA Domain Proteins and a Trip13-like ATPase Regulate Bacterial cGAS-like Enzymes to Mediate Bacteriophage Immunity. Mol. Cell 77, 709–722.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, Gray NS, Mekalanos JJ, and Kranzusch PJ (2018). Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 174, 300–311.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Biochemical Reconstitution of Cap4 Endonuclease Activity, Related to Figure 1
(A) SDS-PAGE and Coomassie stain analysis of purified recombinant Cap4 and CdnD proteins.
(B) Cartoon schematics and crystal structures of apo MoCap4 and AbCap4 enzymes.
(C) Alignment of divergent Cap4 homologs. Endonuclease active site residues are indicated with a red circle, and the known secondary structure of the Cap4 endonuclease domain (orange) and SAVED sensor domain (blue) determined from the AbCap4 crystal structure is displayed below.
(D) EcCap4 and AbCap4 endonuclease activity is inhibited by the divalent metal chelator EDTA. A requirement of divalent cation metals for catalysis agrees with structural analysis of the Cap4 active site and homology with Type II restriction endonucleases.
(E) Like EcCap4, AbCap4 mutation of the putative catalytic residue K69A ablates endonuclease activity.
(F) Agarose gel analysis of AbCap4 endonuclease cleavage activity. Like EcCap4, AbCap4 is a promiscuous DNA endonuclease that degrades plasmid DNA, E. coli genomic DNA, and synthetic DNA but requires recognition of the activating nucleotide second messenger synthesized by the cognate CdnD CD-NTase. Biochemical data are representative of at least 3 independent experiments.
Figure S2. Purification and Characterization of the AbCdnD Nucleotide Second Messenger Product, Related to Figure 2
(A) DNA cleavage analysis of MoCap4 endonuclease activity. The AbCdnD nucleotide second messenger product is able to activate both AbCap4 and the more closely related MoCap4 (~80% identity) but not the distantly related EcCap4 enzyme (~20% identity).
(B) Analysis of AbCdnD nucleotide specificity. AbCap4 endonuclease activity was used to detect the presence of activating nucleotide second messenger signal and determine which nucleotides are required for AbCdnD product formation. ATP is alone necessary and sufficient for AbCdnD-dependent activation demonstrating that the nucleotide second messenger is an oligoadenylate species.
(C) Enzymatic degradation and HPLC analysis of synthetic control cyclic oligoadenylate species and AbCdnD products. Synthetic and AbCdnD product nucleotide second messengers were analyzed following cleavage with Nuclease P1 (P1, cleaves 3′ –5′ phosphodiester bonds) and snake venom phosphodiesterase (SVPD, cleaves both 3′ –5′ and 2′ –5′ phosphodiester bonds) by HPLC and C18 chromatography. Degradation of the AbCdnD major product matches that of synthetic 2′3′3′-cAAA further confirming the structural assignment of this nucleotide second messenger species. The AbCdnD minor product that potently activates AbCap4 activity is partially P1-resistant confirming the presence of a 2′ –5′ phosphodiester bond in this closely related species.
(D) Assessment of DNA degradation after activation of AbCap4 with either the major or minor AbCdnD products from Figure 2E.
(E) Thermal denaturation assay to measure nucleotide second messenger-dependent stabilization of AbCap4. AbCap4 is stabilized in the presence of the AbCdnD major product 2′3′3′-cAAA but not in presence of the unrelated DncV product 3′3′-cGAMP demonstrating specific recognition of 2′3′3′-cAAA.
(F) AbCap4 DNA degradation assay comparing candidate activating ligands. The AbCdnD product mixture, but not synthetic 2′3′3′-cAAA, is capable of activating AbCap4 further confirming that the minor 2′ –5′-linked product is primarily responsible for AbCap4 activation.
(G) LC-MS/MS analysis of AbCdnD major (top) and minor (bottom) enzymatic products. Parent mass extracted ion trace (left) and tandem mass spectra comparison (right) of the major AbCdnD product confirms 2′3′3′-cAAA. Distinct patterning in tandem mass spectra (black arrows) in the minor product relative to 3′3′3′3′-cA4 indicates a cA4 molecule of distinct phosphodiester linkages. Ions detected: m/z 988.1648 [M+H] + (cAAA); m/z 659.1123 [M+2H] 2+ (cA4).
Figure S3. Biochemical and Mutagenesis Analysis of Cap4-Ligand Interactions, Related to Figure 3
(A) Thermal denaturation assay measuring nucleotide second messenger-dependent stabilization of EcCap4 and AbCap4. Activating nucleotide second messengers bind and stabilize both full-length Cap4 and isolated SAVED-domain only constructs, confirming the SAVED domain is the ligand-binding domain. EcCap4 SAVED domain (258–end), AbCap4 SAVED domain (235–end).
(B) Zoom-in cutaways showing A2 and A3 nucleotide pocket interactions in the AbCap4–2′3′3′-cAAA complex structure, and alignments of key pocket residues in various Cap4 homologs.
(C) DNA degradation assays with AbCap4 and EcCap4 proteins with point mutations within the SAVED domain verifies the importance of individual contacts for nucleotide second messenger recognition. Cap4 mutants were activated with increasing concentrations of activating ligand (1 nM, 10 nM, 100 nM, 1 μM). Biochemical data are representative of at least 3 independent experiments.
Figure S4. Analysis of Cap4 Ligand-Bound and Apo Structures, Related to Figure 3
(A) Comparison of apo AbCap4, AbCap4–3′3′3′-cAAA, and AbCap4–2′3′3′-cAAA crystal structures. Each AbCap4 structure exhibits the same overall conformation supporting that the AbCap4-ligand bound structures represent an inactive-state.
(B) Schematic of packing observed of each of the six chains in the AbCap4 asymmetric unit.
(C) Packing of MoCap4 monomers with symmetry-related chains.
(D) Structural comparison of Cap4 and Can1 from the Thermus thermophilus Type III CRISPR immune system (PDB 6SCE). Cap4 and Can1 each contain two CARF domains in a single polypeptide, but the proteins have distinct domain organizations and represent independent evolutionary events.
Figure S5. Oligomerization of Cap4 Proteins upon Nucleotide Second Messenger Recognition, Related to Figure 4
(A) Negative stain EM class averages of AbCap4 (K69A) and MoCap4 (K69A) apo proteins, with activating ligand (purified using ion exchange chromatography from AbCdnD reactions), or with ligand and 45 bp DNA. Cap4 proteins were incubated with components as indicated, then analyzed by negative stain EM (left). Fractions of averaged classes of each oligomeric state were calculated (right).
(B) Micrograph image of a larger filament (highlighted with arrow) formed by EcCap4 in the presence of activating ligand 3′3′3′-cAAG. Higher-order oligomerization supports larger complexes observed in SEC-MALS experiments (Figure 4B). Scale bar represents 200 Å.
(C) SEC-MALS analysis of AbCap4 in the presence or absence of either purified AbCdnD product or synthetic 2′3′3′-cAAA. Absolute molecular masses of apo and ligand-bound complexes were quantified by multiangle light scattering (AbCap4 expected MW: 52.1 kDa).
(D) 3D reconstruction of the activated MoCap4 dimeric complex from negative stain EM data with high-resolution AbCap4–2′3′3′-cAAA structures docked within the density. The location of the Cap4 N-terminal extension and truncation point used for mutagenesis (G19) is denoted with an arrow and supports a possible role for this extension in further controlling endonuclease domain DNA binding and activation. See Figure S6.
(E) Electrophoretic mobility shift assay assessing DNA binding of AbCap4 (K69A). Cap4 was incubated with 45 bp DNA and activating ligand as indicated. Like EcCap4, stable AbCap4–DNA complex formation only occurs in the presence of activating nucleotide second messenger.
(F,G) Analysis of DNA fragments remaining after complete digestion with AbCap4. Distribution of fragments sizes (left), cut site consensus sequences (center), and location of cuts within E. coli genomic DNA showing relaxation of cut site sequence preference. Biochemical data are representative of at least 3 independent experiments.
Figure S6. Characterization of the Cap4 N Terminus in DNA Binding, Ligand Binding, and Oligomerization, Related to Figure 4
(A) Agarose gel analysis of Cap4 activity and plasmid DNA degradation. Full-length Cap4 proteins degrade target DNA in the presence of activating nucleotide second messenger, but N-terminal truncations of AbCap4 (G19–end) and EcCap4 (G11–end) do not.
(B) Electrophoretic mobility shift assay assessing DNA binding of N-terminal truncations of AbCap4 (G19–end) and EcCap4 (G11–end). The conserved N-terminal extension not visible in the AbCap4 or MoCap4 crystal structures is required for DNA recognition.
(C) Thermal denaturation assay measuring nucleotide second messenger-dependent stabilization of N-terminal truncations of EcCap4 and AbCap4. Activating nucleotide second messengers bind and stabilize truncated Cap4 proteins, indicating the N terminus is not required for ligand-binding.
(D) SEC-MALS analysis of N-terminal truncations of EcCap4 and AbCap4 in the presence or absence of activating ligand. Quantification of absolute molecular masses of EcCap4– and AbCap4–ligand complexes confirms the endonuclease domain N-terminal extension does not impact ligand-binding (DN EcCap4 expected MW: 55.1 kDa; ΔN AbCap4 expected MW: 50.4 kDa). Biochemical data are representative of at least 3 independent experiments.
Data Availability Statement
Coordinates of the Cap4 structures have been deposited in the RCSB Protein Data Bank under the following accession numbers: MoCap4 (6VM5), AbCap4 (6WAM), AbCap4–2′3′3′-cAAA (6VM6), AbCap4–3′3′3′-cAAA (6WAN).