

Abstract
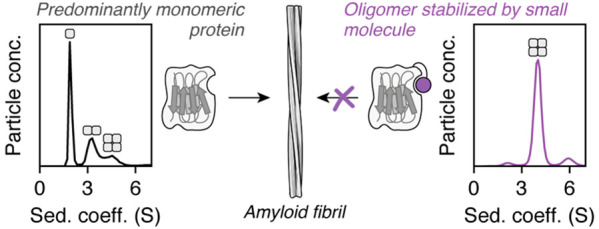
Protein–protein interactions (PPIs) are involved in many of life’s essential biological functions yet are also an underlying cause of several human diseases, including amyloidosis. The modulation of PPIs presents opportunities to gain mechanistic insights into amyloid assembly, particularly through the use of methods which can trap specific intermediates for detailed study. Such information can also provide a starting point for drug discovery. Here, we demonstrate that covalently tethered small molecule fragments can be used to stabilize specific oligomers during amyloid fibril formation, facilitating the structural characterization of these assembly intermediates. We exemplify the power of covalent tethering using the naturally occurring truncated variant (ΔN6) of the human protein β2-microglobulin (β2m), which assembles into amyloid fibrils associated with dialysis-related amyloidosis. Using this approach, we have trapped tetramers formed by ΔN6 under conditions which would normally lead to fibril formation and found that the degree of tetramer stabilization depends on the site of the covalent tether and the nature of the protein–fragment interaction. The covalent protein–ligand linkage enabled structural characterization of these trapped, off-pathway oligomers using X-ray crystallography and NMR, providing insight into why tetramer stabilization inhibits amyloid assembly. Our findings highlight the power of “post-translational chemical modification” as a tool to study biological molecular mechanisms.
Introduction
The regulated self-assembly of proteins into ordered complexes drives many biological processes, ranging from viral capsid formation and actin polymerization, to DNA maintenance and repair.1,2 However, self-assembly can also occur aberrantly as a result of changes in the concentration,3 environment,4−6 primary sequence,7,8 or post-translational processing9−11 of a protein. Aberrant assembly events are associated with a range of disorders, and they can involve polymerization of natively folded protein molecules,2 as in sickle cell anemia,12 or aggregation which is accompanied by significant conformational changes, as exemplified by amyloid diseases.13,14 Understanding the molecular basis and consequences of such protein–protein interaction (PPI) pathways and identifying methods for their modulation15−20 therefore have implications for the treatment of disease,21 as well as in the development of new biomaterials, where protein self-assembly can be exploited to yield structures with defined architectures or novel biomechanical properties.22,23 However, manipulating and defining the mechanisms of self-assembly is challenging, due to the transient nature and heterogeneity (in mass and structure) of oligomeric intermediates.24,25 The use of methods to trap specific oligomeric complexes can help overcome these challenges and offers the opportunity to structurally characterize otherwise transient intermediates, identify targets for drug discovery, and develop new scaffolds for protein-based nanostructures.
Here, we describe the use of disulfide tethering26 to rapidly explore chemical space and identify “post-translational chemical modifications”27,28 that stabilize specific PPIs associated with amyloid assembly. These site-specific, covalent modifications may act in one of two ways: altering the surface properties of the protein and/or covalently reinforcing a noncovalent protein–ligand interaction. Using a naturally occurring, amyloidogenic variant of β2-microglobulin (β2m)—the ΔN6 variant—as a model system, we show that covalently tethered fragments represent highly effective tools for tuning oligomer populations and stabilizing particular species in self-assembly pathways. By combining kinetic analysis with structural information obtained from NMR and X-ray crystallography, the covalent modifications identified here have led to structural and functional insights into the role of tetramers in ΔN6 amyloid formation.
Native monomeric β2m has a seven-stranded immunoglobulin fold and forms the noncovalently bound light chain of the major histocompatibility class I complex.29 Aberrant self-assembly of β2m molecules into amyloid fibrils30 is a hallmark of dialysis-related amyloidosis (DRA).31−33 The amyloidogenic variant of β2m which is the focus of this paper—ΔN6—is formed from the wild-type protein by proteolysis of its N-terminal six amino acids and makes up ∼20–30% of β2m molecules found in fibrils extracted from DRA patients.34,35 ΔN6 is capable of rapid assembly into amyloid fibrils in vitro at near-neutral pH, through the association of the dynamically structured monomers36 into dimers and hexamers that retain a nativelike fold37 (Figure 1A). Subsequent conformational rearrangement of these oligomers into cross-β structures leads to amyloid fibril formation and elongation.38
Figure 1.

(A) Native monomeric ΔN6 (PDB 2XKU(36)) contains seven antiparallel β-strands (labeled A–G), with a solvent-excluded disulfide bond between strands B and F (yellow). The monomeric protein is capable of associating into transient, nativelike dimers and hexamers en route to amyloid fibrils (structures not drawn to scale).37 The conversion of these nativelike oligomers into fibrils requires structural rearrangement of the existing β-strands38 and further self-assembly. ΔN6 tetramers have been observed, but their structure and role in fibril formation depend on the solution conditions.37,86 (B) The two most ligandable sites (pink) of ΔN6 identified by computational solvent mapping are located adjacent to the BC (target site 1) and DE (target site 2) loops. Three residues (S33, S52, and L65; purple) at these two sites were substituted with cysteine in order to target each pocket using the disulfide tethering strategy (see Figure 2A). The orientation of ΔN6 in the “Target site 1” panel is the same as for the monomer shown in (A).
Recent structural models of ΔN6 dimers and hexamers have shown that the DE and BC loops of the protein are involved in both oligomerization interfaces (Figure 1A).37 The same regions have also been identified at the interface of amyloid-competent ΔN6−β2m heterodimers,39 β2m−β2m homodimers,40 and inhibitory heterodimers formed between ΔN6 and a nonamyloidogenic β2m ortholog (murine β2m).39 These examples implicate the DE and BC loops, and thereby also the associated β-strands, as mediators of oligomerization, and thus targeting these regions (e.g., by small molecules) was anticipated to yield tools for controlling and studying ΔN6 self-assembly. The dynamic nature of ΔN6 monomers36 and oligomers,37 combined with the lack of an obvious ligand binding pocket, has thus far hindered the development of small molecule modulators of amyloid formation (although protein-based modulators have been identified41). In light of this challenge, we focused our efforts on the development of covalent ligands to manipulate ΔN6 self-assembly, based on the success of such compounds in targeting other challenging PPIs.42−44
Results and Discussion
Identifying Covalent Ligands for ΔN6 by Disulfide Tethering
Computational solvent mapping (using the FTMap server45) was performed against an NMR-derived conformational ensemble of ΔN636 in order to predict which regions of the protein are likely to be hot spots for small molecule binding. The resulting data (Figure S1) highlighted two pockets adjacent to the DE and BC loops as promising targets for ligand development (site 1, between the C and D strands; and site 2, beneath the DE loop; Figure 1B). To identify possible covalent ligands for these sites, a library of small molecules was screened using the “disulfide tethering” method26 (Figure 2A). First developed by Erlanson and co-workers as a site-directed screening strategy,46 this approach uses disulfide bonds to covalently trap and assess the interaction affinity of small molecules (typically fragments) which have bound noncovalently to the protein of interest. Libraries of disulfide-functionalized molecules are screened for their ability to form disulfide bonds with cysteine variants of the target protein, as small molecules which exhibit favorable noncovalent interactions near the free, solvent-exposed cysteine residue will undergo thiol–disulfide exchange more effectively due to their increased local concentration. The relative population of different covalent protein−ligand complexes at equilibrium can therefore be used as a proxy for the noncovalent affinity of a given fragment for a particular region of the target protein.
Figure 2.

(A) Schematic representation of the disulfide tethering method used to compare noncovalent affinities of different fragments for specific target sites on ΔN6. When using cocktails of fragments, the compound which forms the most favorable noncovalent interactions with the target protein while tethered by a disulfide bond will produce the most stable (i.e., highest populated) covalent protein–fragment adduct at equilibrium—this is reflected by a higher RZ score. (B) Data from a disulfide tethering screen against three ΔN6 cysteine variants (S33C, S52C, and L65C) normalized as RZ scores. For fragments which were present in more than one screening cocktail, data are shown as the mean ± one standard deviation. Each cysteine variant was present at 5 μM and was incubated for 24 h with 25 μM of each disulfide-linked fragment (in cocktails of five fragments) and 500 μM βME, in 25 mM sodium phosphate, pH 6.2, 2% v/v DMSO. Black circles are shown for fragments which were synthesized but not included in the screening library due to poor purity.
Three single cysteine variants of ΔN6 (S33C, S52C, and L65C; Figure 1B) were expressed and purified (Figure S2) in order to monitor small molecule binding at sites 1 and 2 by using the disulfide tethering approach. A library of 84 symmetrical disulfides (designed with the aid of molecular docking, as described in the Supporting Information) was prepared through the use of solid-phase synthesis, and 76 of these fragments were sufficiently pure for screening purposes (Figure S3) and were screened against each ΔN6 cysteine variant in cocktails of five, in the presence of excess reducing agent (β-mercaptoethanol, βME; Figure S4). The relative populations of the different protein–fragment adducts were assessed at 24 h by electrospray ionization mass spectrometry and normalized as robust Z (RZ) scores47 (Figure 2B; Figures S5–S7), where higher RZ scores were anticipated to reflect more favorable protein–fragment interactions (Figure 2A). Comparison of the distribution of protein–fragment adducts observed for the three ΔN6 cysteine variants to that of an unrelated, largely helical control protein, MCL-1, showed that there was poor correlation between the datasets for S52C– and L65C−ΔN6 with that for MCL-1 (median r = 0.37; Figure S6) and therefore that the identity of the protein affects which protein–fragment adducts dominate at equilibrium. The higher correlation between the datasets for S33C−ΔN6 and MCL-1 reflects the similar chemical nature of their binding pockets (Figure S6). These observations suggest that the preference of ΔN6 for particular fragments (as shown in Figure 2B) is a result of specific noncovalent interactions. RZ scores were therefore used to report on the relative noncovalent affinities of tethered fragments for a particular region on the surface of ΔN6.
Covalent Functionalization and Ligand Binding Drive ΔN6 Tetramerization
To interrogate how covalent modification of the different regions of ΔN6 affects aggregation, sedimentation velocity analytical ultracentrifugation (SV-AUC) was used to assess the oligomeric state of a series of individual protein–fragment adducts (Figure 3A; Figures S8–S10). Fragments with a range of RZ scores were selected for testing with each cysteine variant, so as to distinguish between changes in oligomeric state which are due to covalent modification of the protein (i.e., changes observed for all samples of a given cysteine variant) versus changes which arise from specific noncovalent protein–ligand interactions (i.e., those observed only for covalently tethered fragments with high RZ scores).
Figure 3.
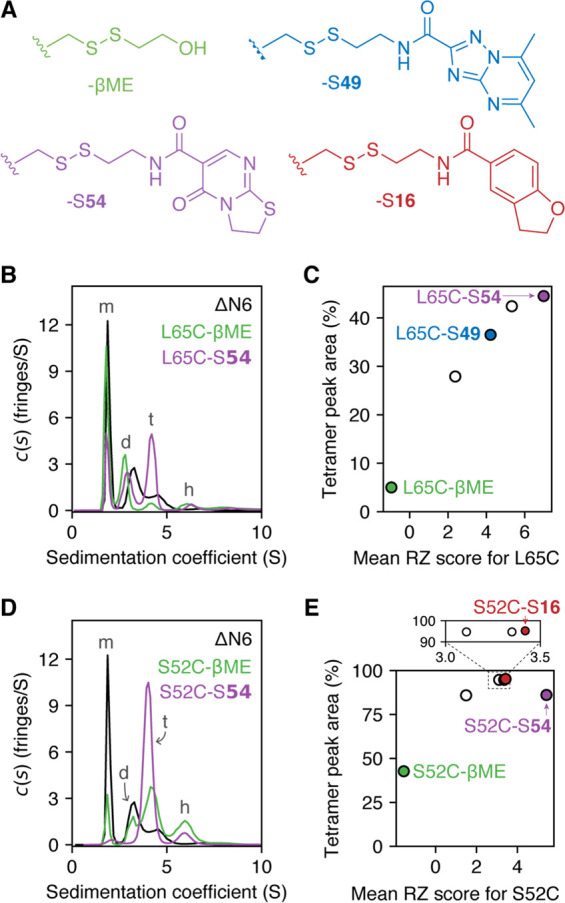
(A) Examples of fragments used to form the S52C– and L65C–fragment adducts studied by SV-AUC. (B–E) SV-AUC data collected for L65C–fragment adducts (B, C) or S52C–fragment adducts (D, E). Experiments were performed with 150 μM protein in 25 mM sodium phosphate at pH 6.2, 25 °C. Peaks in the c(s) distributions were assigned to monomer (m), dimer (d), tetramer (t), or hexamer (h), based on previous studies37 and predicted sedimentation coefficients for these oligomers (calculated using the Svedberg equation48). Tetramer peak areas for a range of L65C–fragment adducts were found to correlate with the RZ score of the attached fragment (C). The relationship between fragment RZ score and tetramer peak area for S52C–fragment adducts was less clear, and other properties of the fragments may play a role in modulating tetramer populations (E) (see Figure S14).
In the absence of covalent modification, ΔN6 is approximately 50% monomeric under the conditions employed (150 μM protein, pH 6.2, 25 °C), with dimers, tetramers, and hexamers representing the majority of faster sedimenting species (Figure S2), consistent with previous reports.37 Tethering of high RZ score fragments to all three ΔN6 cysteine variants was found to increase the population of tetramers (representative data in Figure 3B,D; see also Figures S11–S13). Tethering with low RZ score fragments, however, produced a variety of results, which depended both on the cysteine variant and the ligand employed.
For L65C–fragment adducts, the area of the tetramer peak in the continuous sedimentation coefficient (c(s)) distributions showed a positive correlation with the RZ score of the tethered fragment (Figure 3C; Figures S13, S14): a covalently attached fragment with a low RZ score (βME) produced an oligomer distribution which was similar (albeit not identical) to that of ΔN6 alone (tetramer peak areas of 5 and 18%, respectively), while tethered fragments with high RZ scores produced significantly larger tetramer peaks (e.g., a 45% tetramer peak area was observed for the adduct between L65C and disulfide 54, named L65C–S54; Figure 3B,C). The only predicted ligandable pocket that is near residue 65 of ΔN6 is target site 2 (Figure 1B), suggesting that the formation of tetramers by L65C–fragment adducts is driven by noncovalent binding to this pocket.
The nature of the relationship between tetramer population and fragment RZ score for the S52C– and S33C–fragment adducts was less clear. All S52C–fragment adducts produced tetramer peak areas of ≥43% (with most between 86 and 95%), regardless of RZ score (Figure 3D,E; Figures S12, S14), implying that a different property of the fragments was driving the changes in oligomeric state at this site. Although a limited range of fragment sizes were used in screening, the observed tetramer populations for the different S52C–fragment adducts are consistent with fragment size (and thus the surface topography of protein–fragment adducts) being a contributing factor (Figure S14). The S33C–fragment adducts analyzed were generally polydisperse, with only two samples (of seven analyzed) producing c(s) distributions which were readily interpretable (Figure S11): S33C−βME and S33C–S79, the latter of which showed an increased tetramer peak area (43%) over the former (16%). These data are insufficient to ascertain which characteristic (characteristics) of the covalent fragments influences (influence) the tetramer population of S33C–fragment adducts, but they show that, as for L65C and S52C, the addition of specific covalent fragments to S33C can be used to modulate oligomer distributions.
Tetramer Stabilization Inhibits Amyloid Assembly
The ability of covalently tethered fragments to generate defined oligomer populations offered the opportunity to explore the effect of tetramer stabilization on ΔN6 amyloid assembly. Protein–fragment adducts with a range of tetramer populations were added to preformed ΔN6 amyloid fibrils, and the ability of these samples to elongate the fibril seeds was analyzed using the fluorescent, fibril-binding dye, thioflavin T (ThT).49,50 By using this strategy, fibril elongation by each sample will be templated by seeding to a common fibril product, irrespective of the covalent ligand, enabling direct comparison of the initial rates of fibril growth for different protein–fragment adducts. It should be noted that, under the conditions employed, spontaneous (i.e., unseeded) fibril formation does not occur on the time scale of the experiment.
For all three ΔN6 cysteine variants, the observed initial elongation rates were lower for samples with higher tetramer populations (Figure 4; Figure S15A,B). The most dramatic change in fibril elongation was seen for S52C–S54 (86% tetramer peak area in the c(s) distribution), where the rate of elongation was reduced more than 30-fold relative to ΔN6 (Figure 4E,F). Global linear regression analysis across all samples showed that there is a negative correlation (r = −0.78) between the observed initial fibril elongation rate and tetramer population, and extrapolation of the linear regression line to 100% tetramer corresponds precisely to an elongation rate of zero (Figure S15B). Together, these observations indicate that the tetramers formed by the S33C–, S52C–, and L65C–fragment adducts lie off-pathway to amyloid fibril formation. Prediction of elongation rates using kinetic schemes in which tetramers lie on- or off-pathway to fibrils also supports this conclusion (Figure S16). The fibril elongation data thus highlight tetramer stabilization as a strategy to inhibit ΔN6 amyloid formation. In addition, they support a model in which covalent functionalization and covalent reinforcement of ligand binding around the DE and BC loops can be used to slow the progression of seeded amyloid formation by modulating the stability and population distribution of oligomers.
Figure 4.
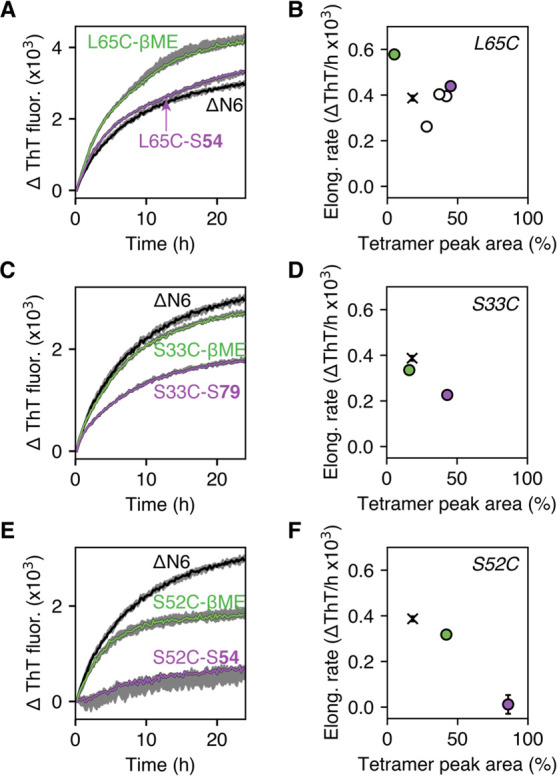
Change in ThT fluorescence over time for various protein–fragment adducts (150 μM) in the presence of ΔN6 fibril seeds (15 μM monomer equivalents) shows that the ability of samples to elongate fibrils depends on the tetramer population. ThT fluorescence curves in (A), (C), and (E) are shown as the median curve, with the highest and lowest values shaded in gray (n = 3). The relationship between the initial elongation rates calculated from these curves and the tetramer peak areas from the corresponding c(s) distributions are shown in (B), (D), and (F). Error bars (standard deviation) are shown for all data points (mean values) in (B), (D), and (F)—those error bars that are not visible are smaller than the displayed data point. Additional ThT fluorescence curves associated with (B) which were not shown in (A) (i.e., the white circles in (B)) are shown in Figure S15A. All experiments were performed under quiescent conditions in 25 mM sodium phosphate at pH 6.2, 25 °C. Elongation rates for unliganded ΔN6 are shown by black crosses in (B), (D), and (F).
Structural Characterization of Stabilized Tetramers
To understand how the tetramers generated by the covalent modification of ΔN6 are structurally related to the previously characterized ΔN6 dimers and hexamers,37 as well as how and why covalent modification around the DE and BC loops leads to tetramerization, X-ray crystallography and solution-state NMR structural studies were performed.
S52C–S54 was found to crystallize as a ring-shaped tetramer with a solvent-accessible central cavity, formed from two asymmetric units each containing two ΔN6 molecules in an antiparallel orientation (Figure 5A–C; Table S1). As seen for the on-pathway ΔN6 dimer and hexamer,37 the protein subunits in the crystallized tetramer are highly nativelike, but notably with perturbations to the DE and BC loops as well as a shift of the D strand from a β-bulge to a straight β-strand (Figure 5D; Figure S17A). These structural changes appear to be linked to each other and to tetramerization: straightening of the D strand allows for protein–protein interactions to occur via β-sheet augmentation, forming one of the two interaction interfaces in the tetramer (Figure 5B), and additionally results in a rearrangement of phenylalanine residues at the top of the D, E, and B strands (Phe56, Phe62, and Phe30, respectively) which is accommodated by rearrangement of the BC loop (Figure S17B). This movement of residues around the BC and DE loops allows several key protein–protein contacts to be made within the second interaction interface, which occurs through a face-to-face, antiparallel interaction of the ABED β-sheets (Figure 5C; Figure S17C).
Figure 5.

(A–C) Crystal structure of the S52C–S54 tetramer (diffracted to 2.4 Å; PDB 7AFV), formed from two asymmetric units: subunits 1a/1b (gray/pale blue) and subunits 2a/2b (pale green/dark green). Protein subunits interact via two interfaces: the D strand interface (B) and the ABED sheet interface (C). Four copies of the covalent −S54 fragment (shown as spheres in A and C) are present in this complex and lie within the ABED sheet interface, in a central solvent-accessible cavity (C). (D) Per-residue, pairwise RMSD values for the S52C–S54 tetramer crystal structure (subunit 1a/2a) compared with the monomeric ΔN6 NMR ensemble (30 structures; PDB 2XKU(36)), reported for all non-hydrogen atoms as the mean RMSD (±standard deviation). Residues which are ≤6 Å from another protein subunit or a −S54 fragment within the S52C–S54 tetramer structure are additionally highlighted by green or purple bars, respectively. The locations of the β-strands in the S52C–S54 tetramer crystal structure are shown above the plot. (E) Each −S54 fragment can bind in one of two binding sites around the site 2 pocket: by π stacking between two Tyr26 residues within the ABED sheet interface (top) or by π stacking against Tyr67 (bottom). The 2Fo–Fc electron density map (contoured at 1.1σ) is shown in cyan for the displayed amino acid side chains and organic molecules. (F) Combined 1H–15N chemical shift differences between S52C–S54 and L65C−βME HMQC NMR spectra—these samples had tetramer peak areas of 86 and 5%, respectively, in their c(s) distributions. Residues which were not visible (or could not be confidently assigned based on comparison with previous ΔN6 assignments36) for either sample are shown as black circles. Residues which were visible for L65C−βME but were broadened beyond detection for S52C–S54 are shown by purple bars. Resonances which were visible in both spectra are colored according to the magnitude of the chemical shift perturbation (CSP) relative to the standard deviation of the dataset (σ): CSP ≥ 2σ, red; σ ≤ CSP < 2σ, yellow; CSP < σ, gray. Four main regions (labeled 1–4) show either significant changes in the position of 1H–15N resonances or complete loss of these resonances in samples with higher tetramer populations (see also Figure S21).
Clear electron density indicated the presence of four covalently bound −S54 fragments within the central cavity of the tetramer (Figure 5A; bottom structure) which make intrasubunit and/or intersubunit contacts with the site 2 pocket of surrounding protein subunits (Figure 5E; Figure S17C). The protein–ligand interactions with the strongest electron density, and which are therefore the interactions which presumably contribute most to the stabilization of the tetramer, involve π stacking between two Tyr26 residues within the ABED sheet interface and the bicyclic ring system of a fragment 54 molecule (gray ligand in Figure 5E)—this arrangement is seen for two of the four fragments in a tetramer. These protein–ligand interactions may additionally stabilize the straight conformation of the D strand: residue 52 lies too far from Tyr26 in the monomeric ΔN6 structure for fragment 54 to interact with Tyr26, and it thus appears that this protein–ligand interaction can only form when the D strand is straight (Figure S17D). Therefore, while no direct protein–ligand contacts occur with either the DE or BC loops, contacts with the associated β-strands appear to have driven conformational changes in these regions, and together the observed structural changes have altered the ΔN6 self-assembly landscape in favor of the tetramer. The electron density associated with the remaining two fragments in the tetramer is not as well-defined as for those intercalated between Tyr26 residues, but it suggests a binding mode whereby the fragments can form intrasubunit π–π interactions with Tyr67 residues (Figure 5E, pale blue ligand).
1H–15N heteronuclear multiple quantum coherence (HMQC) NMR spectra and 15N-relaxation measurements acquired for various S52C– and L65C–fragment adducts were consistent with the tetramer observed within the S52C–S54 crystal lattice, implying that this crystal structure reflects the nature of these off-pathway ΔN6 tetramers in solution (Figures S18–S21). Comparison of the chemical shifts and intensities of 1H–15N backbone amide resonances between samples with higher tetramer populations (e.g., S52C–S54 and L65C–S54) and those with lower tetramer populations (e.g., ΔN6, L65C−βME) showed differences across four regions of primary sequence (labeled 1–4 in Figure 5F and Figure S21). Regions 1–3 contain residues that either make up the tetramer interaction interfaces and/or have shifted significantly (≥4 Å root-mean-square deviation, RMSD) in the crystal structure relative to monomeric ΔN6 (Figure 5D). While region 4 does not undergo any structural changes itself in the crystal structure, it is adjacent to the BC loop, and hence residues in region 4 will experience a change in chemical environment upon tetramerization (Figure 5D).
In addition to the NMR data supporting the relevance of the S52C–S54 crystal structure to oligomerization in solution, the NMR data also indicate that the tetramers formed by the S52C– and L65C–fragment adducts are structurally similar. The ability of S52C– and L65C–fragment adducts to adopt similar tetrameric species can be rationalized in the context of the crystal structure. D strand straightening results in residues 52 and 65 now lying adjacent to one another (Figure S17A, inset), so we anticipate that fragments tethered to the L65C variant of ΔN6 can access the same binding pockets which −S54 is observed to occupy in the S52C–S54 crystal structure.
Structural Similarities of Off-Pathway ΔN6 Tetramers with Full-Length β2m Oligomers
While similar regions of ΔN6 are involved in the formation of the off-pathway tetramer and the previously identified on-pathway dimers/hexamers (which were characterized under conditions similar to those employed here),37 the PPIs themselves are significantly different and mutually exclusive (Figure S22A). This observation rationalizes the off-pathway behavior of the tetramer, as it would need to completely dissociate to form the on-pathway hexamer. However, although the tetramer is different from previously identified ΔN6 oligomers,37,41,51 its interfaces resemble those observed in oligomers of the full-length β2m protein.52−55 The most striking similarities are those with a crystallographic tetramer formed by the P32A variant (PDB 2F8O;52Figure S22B,C). P32A β2m is largely monomeric in solution52 (in the absence of divalent copper56), but at the high concentrations within the crystal lattice the protein molecules can interact via the same D strand and ABED interfaces shown in Figure 5 (Figure S22B). Although the solution relevance of the crystallographic P32A tetramer was not shown, the NMR data obtained for S52C–S54 confirm that this arrangement of protomers is possible in solution and stable in the presence of suitable ligands.
While the crystallographic P32A β2m tetramer possesses the same straight D strand as the S52C–S54 ΔN6 tetramer, other β2m oligomers for which high-resolution structural information is available lack straight D strands and instead involve DE and BC loop-mediated PPIs.53−55 Since these oligomers which lack straight D strands have been shown (or proposed) to be on-pathway species, we hypothesize that formation of an eight-stranded ABED–DEBA β-sheet is an off-pathway PPI which can be driven by ligand binding across the ABED sheet (as seen for ΔN6 S52C–S54) or modulation of the BC loop (as for β2m P32A).
Conclusions
Covalent small molecules are becoming increasingly attractive tools to modulate specific protein–protein interactions,43,44,57−59 to interrogate the role of different proteins in biological processes,44,57,60−62 and to facilitate structural studies of challenging targets.42,63 The results presented here expand this covalent chemical tool approach, highlighting its power to facilitate analysis of the structure and the role of specific oligomers in self-assembly pathways. We have demonstrated that covalently reinforced protein–ligand interactions64,65 can be used to manipulate heterogeneous and dynamic PPIs, exemplified by those formed in the initial stages of amyloid formation, which are notoriously difficult to target selectively when purely noncovalent approaches are used.66 Specifically, covalent ligands identified by disulfide tethering allowed a transient off-pathway tetramer (as shown by kinetic data and models) formed by the amyloidogenic ΔN6 variant of β2m to be trapped and characterized in atomic detail for the first time. These results highlight the power of the chemical modification of proteins as a general strategy to manipulate complex self-assembling systems and to drive the formation of defined oligomers for detailed study.
Experimental Section
Safety
No unexpected, new, and/or significant hazards or risks were associated with this research.
Disulfide Tethering Screening
Screening cocktails were prepared by combining five disulfide-linked fragments (1.25 mM each; all present as symmetrical disulfides) in the presence of βME (25 mM) in DMSO. These initial cocktail mixtures were incubated at ambient temperature (∼18 °C) for 12 h to allow the formation of mixed disulfides with βME (which generally had improved aqueous solubility over the symmetrical disulfides).
Screening cocktails were diluted further into each ΔN6 cysteine variant (5 μM protein, with 25 μM each disulfide, 500 μM βME, 2% v/v DMSO) in 25 mM sodium phosphate at either pH 6.2 (main text data) or pH 8.0 (Supporting Information), and the resulting screening mixtures were incubated at ambient temperature. The distribution of covalent protein–fragment adducts over time was analyzed by a Bruker maXis Impact QTOF mass spectrometer, with an electrospray ionization source. Samples (1 μL injections) were desalted prior to mass spectrometry by an in-line Dionex UltiMate 3000 liquid chromatography system (Thermo Scientific), equipped with an Aeris Widepore C4 column (Phenomenex), running a gradient between water and acetonitrile, both supplemented with 0.1% formic acid.
MCL-1 (residues 172–327; purified as described previously67) was screened using the same procedure, but in 25 mM sodium phosphate, pH 7.4.
Mass spectra were deconvoluted with the use of a maximum entropy algorithm (DataAnalysis, Bruker). For each protein–fragment adduct in each cocktail, the intensity of the deconvoluted peak at 24 h was measured relative to the protein−βME adduct peak. These relative intensities were then converted to RZ scores.47 The RZ score for a given fragment can be described by eq 1:
| 1 |
where xi is the relative intensity of the protein–fragment adduct peak for fragment i and xall is a list of relative peak intensities for all protein–fragment adducts in that dataset (i.e., for a given cysteine variant at a given pH). In all cases, peak intensities were measured relative to the βME adduct. The medium absolute deviation (MAD)68 is defined as
| 2 |
RZ scores for the disulfide analogue of βME could not be determined by this method, due to the presence of βME as a reducing agent in all screening mixtures. Instead, its RZ score was estimated through competition experiments, as shown in Figure S7.
Sedimentation Velocity Analytical Ultracentrifugation
Experiments were carried out at 48 000 rpm, 25 °C, with an An50-Ti rotor in a Beckman Coulter Optima XL-I ultracentrifuge. Samples and reference buffer (400 μL) were loaded into 12 mm aluminum centerpieces, with sapphire windows. Samples were thermally equilibrated for 2–3 h at 0 rpm and were subsequently analyzed using the interference detection system at a protein concentration of 150 μM in 25 mM sodium phosphate, pH 6.2. All samples had been exhaustively dialyzed in this buffer prior to loading, and the dialysate was used as the reference buffer.
Data were analyzed in SEDFIT (version 12.44)69 with a continuous c(s) distribution model and with a maximum entropy regularization confidence interval of 0.95. Values for the partial specific volume (ν̅) of ΔN6, buffer viscosity (η), and buffer density (ρ) were all calculated with SEDNTERP70 (ν̅ = 0.72832 mL/g; η = 0.00898 P and ρ = 0.99954 g/mL for 25 mM sodium phosphate, pH 6.2). The final fitting was performed with the Levenberg–Marquardt algorithm, although initial fitting was performed with both the Simplex and Levenberg–Marquardt algorithms. For c(s) distributions with significant peak broadening or overlap, further processing was subsequently performed to permit baseline resolution of these peaks: multiple Gaussian functions were fit to the initial c(s) distribution (using the curve_fit function from SciPy.optimize), and then the positions of these best fit Gaussians were used in SEDFIT as Bayesian prior expectations71 (modeled again as Gaussians, located at the best fit positions identified by curve_fit, with widths set to 0.2 and amplitudes set to 0.1). Refitting the data with the Levenberg–Marquardt algorithm led to improved or equally good root-mean-square deviations (compared to the original c(s) distribution) for all datasets to which this processing method had been applied, confirming the validity of this approach.
Peaks in the c(s) distributions were assigned to specific oligomers based on comparison to published ΔN6 SV-AUC data37 and predicted sedimentation coefficients for these oligomers. Predicted sedimentation coefficients were calculated with the Svedberg equation48 at a range of possible frictional ratios, so as to estimate the range of possible sedimentation coefficients which could describe a particle of a given molecular weight.
Thioflavin T Aggregation Assays
Protein–fragment adducts (150 μM) were incubated in 25 mM sodium phosphate, pH 6.2, with 10 μM ThT, in the presence or absence of 15 μM (monomer equivalent) fibril seeds prepared from ΔN6, in 96-well untreated half-area plates (Corning; 100 μL sample per well). In the same plate, 15 μM (monomer equivalent) fibril seeds were incubated alone in 25 mM sodium phosphate, pH 6.2. Details concerning the preparation of fibril seeds can be found in the Supporting Information.
Fibril elongation was allowed to occur quiescently, with brief agitation (10 s at 600 rpm) only occurring prior to each reading (every 12 min). Experiments were performed for 24 h at 25 °C in a FLUOstar OPTIMA plate reader (BMG Labtech), exciting ThT at 444 nm and measuring its fluorescence emission intensity at 480 nm.
Data were processed by subtracting two datasets from each seeded dataset to remove changes in ThT fluorescence which had occurred independently of fibril elongation: (a) data obtained from the same protein–fragment adduct in the absence of fibril seeds; (b) data obtained for fibril seeds alone in buffer. The absence of fibrillar aggregates in the unseeded samples after 24 h was confirmed by negative-stain electron microscopy (see Supporting Information). Initial elongation rates from the processed seeded datasets were measured by fitting a straight line to the first 3 h of data and determining the gradient.
Protein Crystallography and Data Processing
A stock solution of S52C–S54 was prepared by incubation of S52C (1.5 mM) and the symmetrical disulfide of fragment 54 (Di-S54; 1.5 mM) in 25 mM sodium phosphate, pH 8.0, 725 μM βME, 20% v/v DMSO, for 40 h at room temperature, before exhaustive dialysis against 25 mM sodium phosphate, pH 6.2, at 4 °C, using dialysis tubes with a 3.5 kDa molecular weight cutoff (Generon). The concentration of the dialyzed sample was determined through a bicinchoninic acid (BCA) assay.72 Crystals were grown by mixing 0.2 μL of the protein–fragment complex (1.1 mM) and 0.1 μL of the crystallization solution in hanging drop plates at 293 K. The crystallization solution (39 mM bicine, 61 mM Tris, pH 8.5, 7.8% w/v PEG 3350, 7.8% w/v PEG 1000, 7.8% v/v 2-methyl-2,4-pentanediol, and 6.7 mM each of 1,6-hexanediol, 1-butanol, 1,2-propanediol, 2-propanol, 1,4-butanediol, and 1,3-propanediol) was prepared from Morpheus Buffer System 3 at pH 8.5, Morpheus Precipitant Mix 4, and Morpheus Alcohols Mix (all from Molecular Dimensions). After 2 weeks, crystals were fished and flash-frozen in liquid nitrogen. The diffraction data were collected at 0.9795 Å, with 0.025 s exposition and 0.15° oscillation, for a total of 2400 images on beamline I24 at Diamond Light Source (U.K.). The data were processed using the xia273 bundle, with DIALS74 for integration and using Pointless/Aimless75,76 for scaling and merging. The data were processed using CC1/2 and completeness as cutoff criteria.77 The structure of S52C–S54 was solved by molecular replacement, using full-length β2m (PDB 5CS7(78)) with the first six amino acids removed as the search model in PHASER.79 COOT80 and REFMAC581 were used for refinement. Parameterization of Cys−S54 (as a non-natural amino acid) was carried out in XPLOR-NIH,82 followed by refinement of its structure using the fixupCovalentGeometry function while satisfying the electron density. The quality of the final structure was assessed with MolProbity.83 Data collection and refinement statistics are shown in Table S1. Figures were prepared using PyMOL (version 2.4, Schrödinger).
Protein NMR
All spectra were acquired by using a 750 MHz Bruker Avance III HD spectrometer equipped with a TCI cryoprobe, at a protein concentration of 150 μM in 25 mM sodium phosphate, pH 6.2, at 25 °C. 1H–15N SOFAST HMQC spectra were processed with the use of NMRPipe,84 and calculation of peak intensities was performed in CcpNmr Analysis (version 2.4).851H–15N peaks were assigned to specific backbone resonances for each protein–fragment adduct (L65C−βME, L65C–S54, S52C−βME, and S52C–S54) by comparison to existing ΔN6 assignments,36 only considering peaks with intensities at least 3-fold greater than the spectral noise. Backbone chemical shift perturbations (CSPs) between samples were calculated with eq 3:
| 3 |
where ΔδHN and ΔδN represent the difference in peak position in the direct and indirect dimensions, respectively.
L65C−βME and L65C–S54T2 experiments were performed by acquiring a series of sensitivity-enhanced 1H–15N heteronuclear single quantum coherence (HSQC) spectra in an interleaved fashion at a range of delay times (0.017–0.136 ms for L65C−βME, 0.017–0.119 ms for L65C–S54). Peak intensities (I) were extracted from each spectrum with the series.tab NMRPipe module, and per-residue 15N relaxation rates (R2) were calculated by fitting the peak intensity at different delay times (t) to a two-parameter exponential function (eq 4).
| 4 |
For S52C−βME and S52C–S54, sensitivity limitations did not permit the determination of 15N R2 rates on a per-residue basis. Instead, the first increment of the standard T2 experiment at each delay time (0.017–0.102 ms for S52C−βME, 0.017–0.085 ms S52C–S54) was used to determine the R2 rate of the whole amide region (7.6–9.2 ppm). Mean 15N R2 rates for the whole amide region were calculated in the same way for ΔN6 (0.017–0.085 ms) and the two L65C adducts (L65C−βME and L65C–S54, using the same datasets acquired for determining per-residue 15N R2 rates).
Acknowledgments
The authors wish to thank Rodolfo Ghirlando for advice on SV-AUC procedures, data analysis, and interpretation, Pallavi Ramsahye for purification of the MCL-1 protein, Honghao Zhang and Zsófia Hegedüs for their work performing the MCL-1 tethering experiments, Arnout Kalvedra for assistance with NMR experiments, and Rodrigo Gallardo and Sabine Ulamec for their help obtaining EM micrographs. We thank all our group members, especially those in the “amyloid team”, for many helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10629.
Supplementary materials and methods (including protein expression and purification, computational solvent mapping, design and synthesis of the disulfide fragment library, preparation of individual protein–fragment adducts, preparation of ΔN6 fibril seeds for thioflavin T aggregation assays, and negative-stain transmission electron microscopy), and characterization of synthesized compounds (PDF)
Accession Codes
Coordinates of the ΔN6 S52C–S54 tetramer crystal structure have been deposited in the Protein Data Bank with the PDB code 7AFV.
Author Contributions
E.E.C., S.E.R, and A.J.W. conceived and designed the research program; E.E.C. designed studies and performed the research with support from N.G. (crystallography), J.S.E. (AUC), and T.K.K. (NMR). The manuscript was written by E.E.C., S.E.R., and A.J.W. with contributions from all authors. E.E.C. prepared all figures.
This work was supported by The Wellcome Trust (109154/Z/15/A, 204963, 109984, 097827/Z/11/A, WT094232MA, 108466/Z/15/Z, and 094232/Z/10/Z), EPSRC (EP/N035267/1, EP/KO39292/1, EP/N013573/1), BBSRC (BB/M011151/1), and ERC (322408). A.J.W. holds a Royal Society Leverhulme Trust Senior Fellowship (SRF/R1/191087).
The authors declare no competing financial interest.
Supplementary Material
References
- Pieters B. J. G. E.; van Eldijk M. B.; Nolte R. J. M.; Mecinović J. Natural supramolecular protein assemblies. Chem. Soc. Rev. 2016, 45, 24–39. 10.1039/C5CS00157A. [DOI] [PubMed] [Google Scholar]
- Garcia-Seisdedos H.; Villegas J. A.; Levy E. D. Infinite assembly of folded proteins in evolution, disease, and engineering. Angew. Chem., Int. Ed. 2019, 58, 5514–5531. 10.1002/anie.201806092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novo M.; Freire S.; Al-Soufi W. Critical aggregation concentration for the formation of early amyloid-β (1–42) oligomers. Sci. Rep. 2018, 8, 1783. 10.1038/s41598-018-19961-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen M. C.; Gnutt D.; Gao M.; Wärmländer S. K. T. S.; Jarvet J.; Gräslund A.; Winter R.; Ebbinghaus S.; Strodel B. Effects of in vivo conditions on amyloid aggregation. Chem. Soc. Rev. 2019, 48, 3946–3996. 10.1039/C8CS00034D. [DOI] [PubMed] [Google Scholar]
- Stephens A. D.; Zacharopoulou M.; Kaminski Schierle G. S. The cellular environment affects monomeric α-synuclein structure. Trends Biochem. Sci. 2019, 44, 453–466. 10.1016/j.tibs.2018.11.005. [DOI] [PubMed] [Google Scholar]
- Rodriguez Camargo D. C.; Tripsianes K.; Buday K.; Franko A.; Göbl C.; Hartlmüller C.; Sarkar R.; Aichler M.; Mettenleiter G.; Schulz M.; Böddrich A.; Erck C.; Martens H.; Walch A. K.; Madl T.; Wanker E. E.; Conrad M.; Hrabě de Angelis M.; Reif B. The redox environment triggers conformational changes and aggregation of hIAPP in type II diabetes. Sci. Rep. 2017, 7, 44041. 10.1038/srep44041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Seisdedos H.; Empereur-Mot C.; Elad N.; Levy E. D. Proteins evolve on the edge of supramolecular self-assembly. Nature 2017, 548, 244–247. 10.1038/nature23320. [DOI] [PubMed] [Google Scholar]
- De Baets G.; Van Doorn L.; Rousseau F.; Schymkowitz J. Increased aggregation is more frequently associated to human disease-associated mutations than to neutral polymorphisms. PLoS Comput. Biol. 2015, 11, e1004374. 10.1371/journal.pcbi.1004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Lee J. H.; Jeon J. H.; Lee M. J. Degradation or aggregation: the ramifications of post-translational modifications on tau. BMB Rep. 2018, 51, 265–273. 10.5483/BMBRep.2018.51.6.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barykin E. P.; Mitkevich V. A.; Kozin S. A.; Makarov A. A. Amyloid β modification: a key to the sporadic Alzheimer’s disease?. Front. Genet. 2017, 8, 58. 10.3389/fgene.2017.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambataro F.; Pennuto M. Post-translational modifications and protein quality control in motor neuron and polyglutamine diseases. Front. Mol. Neurosci. 2017, 10, 82. 10.3389/fnmol.2017.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton W. A.; Hofrichter J. Sickle cell hemoglobin polymerization. Adv. Protein Chem. 1990, 40, 63–279. 10.1016/S0065-3233(08)60287-9. [DOI] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- Iadanza M. G.; Jackson M. P.; Hewitt E. W.; Ranson N. A.; Radford S. E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. 10.1038/s41580-018-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Henning-Knechtel A.; Magzoub M.; Hamilton A. D. Peptidomimetic-based multidomain targeting offers critical evaluation of Aβ structure and toxic function. J. Am. Chem. Soc. 2018, 140, 6562–6574. 10.1021/jacs.7b13401. [DOI] [PubMed] [Google Scholar]
- Hoffmann W.; Folmert K.; Moschner J.; Huang X.; von Berlepsch H.; Koksch B.; Bowers M. T.; von Helden G.; Pagel K. NFGAIL amyloid oligomers: the onset of beta-sheet formation and the mechanism for fibril formation. J. Am. Chem. Soc. 2018, 140, 244–249. 10.1021/jacs.7b09510. [DOI] [PubMed] [Google Scholar]
- Economou N. J.; Giammona M. J.; Do T. D.; Zheng X.; Teplow D. B.; Buratto S. K.; Bowers M. T. Amyloid β-protein assembly and Alzheimer’s disease: dodecamers of Aβ42, but not of Aβ40, seed fibril formation. J. Am. Chem. Soc. 2016, 138, 1772–1775. 10.1021/jacs.5b11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh M.-C.; Liang C.; Mehta A. K.; Lynn D. G.; Grover M. A. Multistep conformation selection in amyloid assembly. J. Am. Chem. Soc. 2017, 139, 17007–17010. 10.1021/jacs.7b09362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salveson P. J.; Haerianardakani S.; Thuy-Boun A.; Kreutzer A. G.; Nowick J. S. Controlling the oligomerization state of Aβ-derived peptides with light. J. Am. Chem. Soc. 2018, 140, 5842–5852. 10.1021/jacs.8b02658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.; Jacoby G.; Sawaya M. R.; Arnon Z. A.; Adler-Abramovich L.; Rehak P.; Vuković L.; Shimon L. J. W.; Král P.; Beck R.; Gazit E. Transition of metastable cross-α crystals into cross-β fibrils by β-turn flipping. J. Am. Chem. Soc. 2019, 141, 363–369. 10.1021/jacs.8b10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele Y. S.; Monteiro C.; Fearns C.; Encalada S. E.; Wiseman R. L.; Powers E. T.; Kelly J. W. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discovery 2015, 14, 759–780. 10.1038/nrd4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seroski D. T.; Hudalla G. A.. Self-assembled peptide and protein nanofibers for biomedical applications. In Biomedical Applications of Functionalized Nanomaterials: Concepts, Development and Clinical Translation; Elsevier Inc.: Amsterdam, 2018; pp 569–598. [Google Scholar]
- Adler-Abramovich L.; Gazit E. The physical properties of supramolecular peptide assemblies: from building block association to technological applications. Chem. Soc. Rev. 2014, 43, 6881–6893. 10.1039/C4CS00164H. [DOI] [PubMed] [Google Scholar]
- Breydo L.; Uversky V. N. Structural, morphological, and functional diversity of amyloid oligomers. FEBS Lett. 2015, 589, 2640–2648. 10.1016/j.febslet.2015.07.013. [DOI] [PubMed] [Google Scholar]
- Kjaergaard M.; Dear A. J.; Kundel F.; Qamar S.; Meisl G.; Knowles T. P. J.; Klenerman D. Oligomer diversity during the aggregation of the repeat region of tau. ACS Chem. Neurosci. 2018, 9, 3060–3071. 10.1021/acschemneuro.8b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. G.; Arkin M. R. Probing structural adaptivity at PPI interfaces with small molecules. Drug Discovery Today: Technol. 2013, 10, e501–e508. 10.1016/j.ddtec.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Krall N.; da Cruz F. P.; Boutureira O.; Bernardes G. J. L. Site-selective protein-modification chemistry for basic biology and drug development. Nat. Chem. 2016, 8, 103–113. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- Wright T. H.; Bower B. J.; Chalker J. M.; Bernardes G. J. L.; Wiewiora R.; Ng W. L.; Raj R.; Faulkner S.; Vallée M. R. J.; Phanumartwiwath A.; Coleman O. D.; Thézénas M.-L.; Khan M.; Galan S. R. G.; Lercher L.; Schombs M. W.; Gerstberger S.; Palm-Espling M. E.; Baldwin A. J.; Kessler B. M.; Claridge T. D. W.; Mohammed S.; Davis B. G. Posttranslational mutagenesis: a chemical strategy for exploring protein side-chain diversity. Science 2016, 354, aag1465. 10.1126/science.aag1465. [DOI] [PubMed] [Google Scholar]
- van Hateren A.; Bailey A.; Elliott T. Recent advances in Major Histocompatibility Complex (MHC) class I antigen presentation: plastic MHC molecules and TAPBPR-mediated quality control. F1000Research 2017, 6, 158. 10.12688/f1000research.10474.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadanza M. G.; Silvers R.; Boardman J.; Smith H. I.; Karamanos T. K.; Debelouchina G. T.; Su Y.; Griffin R. G.; Ranson N. A.; Radford S. E. The structure of a β2-microglobulin fibril suggests a molecular basis for its amyloid polymorphism. Nat. Commun. 2018, 9, 4517. 10.1038/s41467-018-06761-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner T.; Radford S. E. Understanding the complex mechanisms of β2-microglobulin amyloid assembly. FEBS J. 2011, 278, 3868–3883. 10.1111/j.1742-4658.2011.08186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpioni R.; Ricardi M.; Albertazzi V.; De Amicis S.; Rastelli F.; Zerbini L. Dialysis-related amyloidosis: challenges and solutions. Int. J. Nephrol. Renovasc. Dis. 2016, 9, 319–328. 10.2147/IJNRD.S84784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gejyo F.; Yamada T.; Odani S.; Nakagawa Y.; Arakawa M.; Kunitomo T.; Kataoka H.; Suzuki M.; Hirasawa Y.; Shirahama T.; Cohen A. S.; Schmid K. A new form of amyloid protein associated with chronic hemodialysis was identified as β2-microglobulin. Biochem. Biophys. Res. Commun. 1985, 129, 701–706. 10.1016/0006-291X(85)91948-5. [DOI] [PubMed] [Google Scholar]
- Bellotti V.; Stoppini M.; Mangione P.; Sunde M.; Robinson C.; Asti L.; Brancaccio D.; Ferri G. B2-microglobulin can be refolded into a native state from ex vivo amyloid fibrils. Eur. J. Biochem. 1998, 258, 61–67. 10.1046/j.1432-1327.1998.2580061.x. [DOI] [PubMed] [Google Scholar]
- Stoppini M.; Arcidiaco P.; Mangione P.; Giorgetti S.; Brancaccio D.; Bellotti V. Detection of fragments of β2-microglobulin in amyloid fibrils. Kidney Int. 2000, 57, 349–350. 10.1046/j.1523-1755.2000.00851.x. [DOI] [PubMed] [Google Scholar]
- Eichner T.; Kalverda A. P.; Thompson G. S.; Homans S. W.; Radford S. E. Conformational conversion during amyloid formation at atomic resolution. Mol. Cell 2011, 41, 161–172. 10.1016/j.molcel.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamanos T. K.; Jackson M. P.; Calabrese A. N.; Goodchild S. C.; Cawood E. E.; Thompson G. S.; Kalverda A. P.; Hewitt E. W.; Radford S. E. Structural mapping of oligomeric intermediates in an amyloid assembly pathway. eLife 2019, 8, e46574. 10.7554/eLife.46574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.; Sarell C. J.; Eddy M. T.; Debelouchina G. T.; Andreas L. B.; Pashley C. L.; Radford S. E.; Griffin R. G. Secondary structure in the core of amyloid fibrils formed from human β2m and its truncated variant ΔN6. J. Am. Chem. Soc. 2014, 136, 6313–6325. 10.1021/ja4126092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall Z.; Schmidt C.; Politis A. Uncovering the early assembly mechanism for amyloidogenic β2-microglobulin using cross-linking and native mass spectrometry. J. Biol. Chem. 2016, 291, 4626–4637. 10.1074/jbc.M115.691063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamanos T. K.; Kalverda A. P.; Thompson G. S.; Radford S. E. Visualization of transient protein-protein interactions that promote or inhibit amyloid assembly. Mol. Cell 2014, 55, 214–226. 10.1016/j.molcel.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennella E.; Cutuil T.; Schanda P.; Ayala I.; Gabel F.; Forge V.; Corazza A.; Esposito G.; Brutscher B. Oligomeric states along the folding pathways of β2-microglobulin: kinetics, thermodynamics, and structure. J. Mol. Biol. 2013, 425, 2722–2736. 10.1016/j.jmb.2013.04.028. [DOI] [PubMed] [Google Scholar]
- Domanska K.; Vanderhaegen S.; Srinivasan V.; Pardon E.; Dupeux F.; Marquez J. A.; Giorgetti S.; Stoppini M.; Wyns L.; Bellotti V.; Steyaert J. Atomic structure of a nanobody-trapped domain-swapped dimer of an amyloidogenic β2-microglobulin variant. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1314–1319. 10.1073/pnas.1008560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Majmudar C. Y.; Pomerantz W. C.; Gagnon J. K.; Sadowsky J. D.; Meagher J. L.; Johnson T. K.; Stuckey J. A.; Brooks C. L. III; Wells J. A.; Mapp A. K. Ordering a dynamic protein via a small molecule stabilizer. J. Am. Chem. Soc. 2013, 135, 3363–3366. 10.1021/ja3122334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijbesma E.; Hallenbeck K. K.; Leysen S.; de Vink P. J.; Skóra L.; Jahnke W.; Brunsveld L.; Arkin M. R.; Ottmann C. Site-directed fragment-based screening for the discovery of protein-protein interaction stabilizers. J. Am. Chem. Soc. 2019, 141, 3524–3531. 10.1021/jacs.8b11658. [DOI] [PubMed] [Google Scholar]
- Lee S.; Wales T. E.; Escudero S.; Cohen D. T.; Luccarelli J.; Gallagher C. G.; Cohen N. A.; Huhn A. J.; Bird G. H.; Engen J. R.; Walensky L. D. Allosteric inhibition of antiapoptotic MCL-1. Nat. Struct. Mol. Biol. 2016, 23, 600–607. 10.1038/nsmb.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D.; Grove L. E.; Hall D. R.; Bohnuud T.; Mottarella S. E.; Luo L.; Xia B.; Beglov D.; Vajda S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. 10.1038/nprot.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson D. A.; Braisted A. C.; Raphael D. R.; Randal M.; Stroud R. M.; Gordon E. M.; Wells J. A. Site-directed ligand discovery. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 9367–9372. 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goktug A. N.; Chai S. C.; Chen T.. Data analysis approaches in high throughput screening. In Drug Discovery; IntechOpen: 2013; pp 201–226. [Google Scholar]
- Svedberg T.; Pedersen K. O.. The Ultracentrifuge; Oxford University Press: Oxford, U.K., 1940. [Google Scholar]
- Xue C.; Lin T. Y.; Chang D.; Guo Z. Thioflavin T as an amyloid dye: fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017, 4, 160696. 10.1098/rsos.160696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancalana M.; Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 1405–1412. 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D.; Zhao R.; So M.; Adachi M.; Rivas G.; Carver J. A.; Goto Y. Recognizing and analyzing variability in amyloid formation kinetics: Simulation and statistical methods. Anal. Biochem. 2016, 510, 56–71. 10.1016/j.ab.2016.07.013. [DOI] [PubMed] [Google Scholar]
- Eakin C. M.; Berman A. J.; Miranker A. D. A native to amyloidogenic transition regulated by a backbone trigger. Nat. Struct. Mol. Biol. 2006, 13, 202–208. 10.1038/nsmb1068. [DOI] [PubMed] [Google Scholar]
- Calabrese M. F.; Eakin C. M.; Wang J. M.; Miranker A. D. A regulatable switch mediates self-association in an immunoglobulin fold. Nat. Struct. Mol. Biol. 2008, 15, 965–971. 10.1038/nsmb.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M.; De Rosa M.; Bellotti V.; Ricagno S.; Bolognesi M. A recurrent D-strand association interface is observed in β-2 microglobulin oligomers. FEBS J. 2012, 279, 1131–1143. 10.1111/j.1742-4658.2012.08510.x. [DOI] [PubMed] [Google Scholar]
- Halabelian L.; Relini A.; Barbiroli A.; Penco A.; Bolognesi M.; Ricagno S. A covalent homodimer probing early oligomers along amyloid aggregation. Sci. Rep. 2015, 5, 14651. 10.1038/srep14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaho D. V.; Miranker A. D. Delineating the conformational elements responsible for Cu 2+-induced oligomerization of β-2 microglobulin. Biochemistry 2009, 48, 6610–6617. 10.1021/bi900540j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Lodge J. M.; Fierke C. A.; Mapp A. K. Dissecting allosteric effects of activator-coactivator complexes using a covalent small molecule ligand. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 12061–12066. 10.1073/pnas.1406033111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz J.; Boehringer R.; Grogg M.; Raya J.; Schirer A.; Crucifix C.; Hellwig P.; Schultz P.; Torbeev V. Covalent tethering and residues with bulky hydrophobic side chains enable self-assembly of distinct amyloid structures. ChemBioChem 2016, 17, 2274–2285. 10.1002/cbic.201600440. [DOI] [PubMed] [Google Scholar]
- Cardoso R.; Love R.; Nilsson C. L.; Bergqvist S.; Nowlin D.; Yan J.; Liu K. K.-C.; Zhu J.; Chen P.; Deng Y.-L.; Dyson H. J.; Greig M. J.; Brooun A. Identification of Cys255 in HIF-1α as a novel site for development of covalent inhibitors of HIF-1α/ARNT PasB domain protein-protein interaction. Protein Sci. 2012, 21, 1885–1896. 10.1002/pro.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus K. M.; Correia B. E.; Lum K. M.; Forli S.; Horning B. D.; González-Páez G. E.; Chatterjee S.; Lanning B. R.; Teijaro J. R.; Olson A. J.; Wolan D. W.; Cravatt B. F. Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowsky J. D.; Burlingame M. A.; Wolan D. W.; McClendon C. L.; Jacobson M. P.; Wells J. A. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6056–6061. 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichert D.; Kruse A. C.; Manglik A.; Hiller C.; Zhang C.; Hübner H.; Kobilka B. K.; Gmeiner P. Covalent agonists for studying G protein-coupled receptor activation. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 10744–10748. 10.1073/pnas.1410415111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy V. M.; Semetey V.; Bracher P. J.; Shen N.; Whitesides G. M. Dependence of effective molarity on linker length for an intramolecular protein-ligand system. J. Am. Chem. Soc. 2007, 129, 1312–1320. 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.-X. Quantitative relation between intermolecular and intramolecular binding of Pro-rich peptides to SH3 domains. Biophys. J. 2006, 91, 3170–3181. 10.1529/biophysj.106.090258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L. M.; Ashcroft A. E.; Radford S. E. Small molecule probes of protein aggregation. Curr. Opin. Chem. Biol. 2017, 39, 90–99. 10.1016/j.cbpa.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles J. A.; Yeo D. J.; Rowell P.; Rodriguez-Marin S.; Pask C. M.; Warriner S. L.; Edwards T. A.; Wilson A. J. Hydrocarbon constrained peptides – understanding preorganisation and binding affinity. Chem. Sci. 2016, 7, 3694–3702. 10.1039/C5SC04048E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung N.; Zhang X. D.; Kreamer A.; Locco L.; Kuan P. F.; Bartz S.; Linsley P. S.; Ferrer M.; Strulovici B. Median absolute deviation to improve hit selection for genome-scale RNAi screens. J. Biomol. Screening 2008, 13, 149–158. 10.1177/1087057107312035. [DOI] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000, 78, 1606–1619. 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurton T.; Wright A.; Deubler G.; Bashir B.. SEDNTERP. 2012. http://bitc.unh.edu.
- Brown P. H.; Balbo A.; Schuck P. Using prior knowledge in the determination of macromolecular size-distributions by analytical ultracentrifugation. Biomacromolecules 2007, 8, 2011–2024. 10.1021/bm070193j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P. K.; Krohn R. I.; Hermanson G. T.; Mallia A. K.; Gartner F. H.; Provenzano M. D.; Fujimoto E. K.; Goeke N. M.; Olson B. J.; Klenk D. C. Measurement of protein using bicinchroninic acid. Anal. Biochem. 1985, 150, 76–85. 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Winter G. Xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010, 43, 186–190. 10.1107/S0021889809045701. [DOI] [Google Scholar]
- Winter G.; Waterman D. G.; Parkhurst J. M.; Brewster A. S.; Gildea R. J.; Gerstel M.; Fuentes-Montero L.; Vollmar M.; Michels-Clark T.; Young I. D.; Sauter N. K.; Evans G. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 85–97. 10.1107/S2059798317017235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus P. A.; Diederichs K. Linking Crystallographic Model and Data Quality. Science 2012, 336, 1030–1033. 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Marchand T.; de Rosa M.; Salvi N.; Sala B. M.; Andreas L. B.; Barbet-Massin E.; Sormanni P.; Barbiroli A.; Porcari R.; Mota C. S.; de Sanctis D.; Bolognesi M.; Emsley L.; Bellotti V.; Blackledge M.; Camilloni C.; Pintacuda G.; Ricagno S. Conformational dynamics in crystals reveal the molecular bases for D76N beta-2 microglobulin aggregation propensity. Nat. Commun. 2018, 9, 1658. 10.1038/s41467-018-04078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters C. D.; Bermejo G. A.; Clore G. M. Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Sci. 2018, 27, 26–40. 10.1002/pro.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Vranken W. F.; Boucher W.; Stevens T. J.; Fogh R. H.; Pajon A.; Llinas M.; Ulrich E. L.; Markley J. L.; Ionides J.; Laue E. D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins: Struct., Funct., Genet. 2005, 59, 687–696. 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.