

Abstract
Controlled release is an essential requirement for delivery of hydrogen sulfide (H2S) because of its reactive nature, short half-life in biological fluids, and toxicity at high concentrations. In this context, H2S delivery via hydrogels may be beneficial as they can deliver H2S locally at the site of interest. Herein, we employed hydrogels based on aromatic peptide amphiphiles (APAs) with tunable mechanical properties to modulate the rates of H2S release. The APAs contained an aromatic S-aroylthiooxime (SATO) H2S donor attached with a linker to a short IAVEEE hexapeptide. Linker units included carbonyl, substituted O-methylenes, alkenyl, and alkyl segments, with the goal of evaluating the role of linker structure on self-assembly, capacity for hydrogelation, and H2S release rate. We studied each peptide by transmission electron microscopy, circular dichroism spectroscopy, and rheology, and we measured H2S release rates from each gel, triggering SATO decomposition and release of H2S by addition of cysteine (Cys). Using an H2S-selective electrode probe as well as a turn-on fluorescent H2S probe in the presence of H9C2 cardiomyocytes, we found that the rate of H2S release from the hydrogels depended on the rate of Cys penetration into the nanofiber core, with stiffer gels showing longer overall release.
Keywords: Hydrogen sulfide, hydrogel, linker, aromatic peptide amphiphile, self-assembly, rheological behavior, controlled release
Graphical Abstract

1. INTRODUCTION
Supramolecular materials find extensive use in drug delivery, tissue engineering, 3-D cell culture, regenerative medicine, and several other fields of biomedical research.1,2 In particular, self-assembling amphiphilic peptides have garnered much attention due to their ease of synthesis, tunability, and biocompatibility in many contexts.3–5 Aromatic peptide amphiphiles (APAs) belong to a class of self-assembling peptides that bear an N-terminal aromatic unit and a short peptide chain attached with a linker. APAs self-assemble in aqueous solution as a result of hydrophobic and aromatic stacking interactions combined with H-bonding in the peptide chain, creating supramolecular nanoassemblies including spheres, cylinders, filaments, ribbons, coils, and others.6 Furthermore, the self-assembly and hydrogelation of APAs can be controlled by several factors, including pH, temperature, and concentration, with synergistic contributions from individual structural units (aromatic group, peptide chain, and linker).7–12 Therefore, APAs allow fine tuning of macroscopic properties through design modifications and can be potentially useful as efficient drug delivery systems.
APA hydrogels have been used as carriers of drugs, antigens, proteins, and other therapeutics.13,14 However, their ability to regulate payload delivery via small structural modifications in the linker segment has not been widely explored. The linker segment controls the spatial orientation of the aromatic unit relative to the peptide chain. As a result, subtle alterations in linker length and flexibility can influence both molecular packing and the ability of APAs to form gels.7,15–18 Herein, we investigate the effect of various linker segments on the rheological properties and drug release rates from a specific type of APA designed to release therapeutic doses of hydrogen sulfide (H2S).
H2S is a signalling gas (gasotransmitter) that regulates various (patho)physiological processes, including cardioprotection,19 vasodilation,20 neurotransmission,21 inflammation,22,23 and others.24 However, in order to study H2S biology and potentially use it therapeutically, delivery strategies are needed. Its acute toxicity at high concentrations and short in vivo half-life makes it crucial to precisely control and confine H2S release to the site of interest.25–29 While several polymeric and supramolecular H2S donors show controllable H2S release,30,31 most lack the capacity for localized release. Supramolecular hydrogels enable localized drug release,32 so it is beneficial to understand how small changes to APA chemical structures affect the macroscopic properties of H2S-releasing peptide hydrogels.
H2S-releasing APA hydrogels, first reported from our lab in 2015, consist of a small peptide chain (typically 2–7 amino acids) with an S-aroylthiooxime (SATO) covalently attached to the N-terminus via a linker segment.33 SATOs are thiol-triggered aromatic H2S donors,34 and in recent work, we have shown that substitution on the aromatic ring of H2S-releasing APAs can affect the rate of release of H2S from these hydrogels.26 Herein, we aimed to investigate how the chemical structure of the linker segment affects the self-assembly, rheological behavior, and H2S-releasing capacity of a series of H2S-releasing APA hydrogels. We reasoned that by controlling the stiffness of the hydrogel network and thereby the access of cysteine (Cys) into the nanofiber core, we could control the rate and duration of H2S release from these hydrogels. We hypothesized that stiffer hydrogels with restricted access of Cys would show more gradual H2S release compared to their weaker counterparts.
2. EXPERIMENTAL
Materials.
Rink Amide MBHA resin and 9-fluorenylmethoxy carbonyl (Fmoc) protected l-amino acids were purchased from P3Biosystems and used as received. HBTU, N-methylpiperidine, DBU and other reagents for peptide synthesis were purchased from commercial vendors and used as received unless otherwise noted. The solvents employed for peptide synthesis were reagent grade. The linkers were either synthesized (see supplementary information) or purchased commercially. DMEM, FBS, EDTA, trypsin, and other reagents for cell studies was purchased from VWR, Radnor, PA and streptomycin was purchased from MP Biomedicals.
Peptide synthesis and purification.
Peptides were synthesized either manually or using a Liberty 1 microwave-assisted peptide synthesizer (CEM) using solid-phase peptide synthesis (SPPS) via standard Fmoc protocol as described previously.35 Linkers were coupled to the N-terminus of the peptide on resin using HBTU and DIEA in DMF. After cleavage and isolation, peptides were dissolved in water containing 0.1% NH4OH and filtered through a 0.45 μm PTFE filter before purification. Purification by preparative-scale reverse phase-high performance liquid chromatography (RP-HPLC) was carried out on an Agilent Technologies 1260 Infinity HPLC system, eluting with a gradient of 2% ACN to 90% ACN in milliQ H2O over 33 min using an Agilent PLRP-S column (100Å particle size, 25 × 150 mm) and monitoring at 220 nm. To both mobile phases was added 0.1% NH4OH to aid in solubility. Fractions were analysed by mass spectrometry (Advion ExpressIon Compact Mass Spectrometer), and product-containing fractions were combined, rotovapped to remove ACN, and lyophilized (LabConco).
The lyophilized peptide was dissolved in dry DMSO and reacted with S-benzoylthiohydroxylamine (SBTHA) in the presence of catalytic TFA to afford the final SATO-containing APAs. Peptides were dissolved in a mixture of phosphate buffer (100 mM at pH 7.4) and acetonitrile (5:2 v/v) and filtered through a 0.45 μm PTFE filter before purification. Purification was carried out using RP-HPLC, eluting with a gradient of 2% ACN to 90% ACN in milliQ H2O without any additives. The protocol for analysing and recovering the peptides was the same as described above. The final peptides were dissolved in milliQ water and distributed into aliquots (100 μg each). Aliquots were frozen, lyophilized, and stored at −20 °C.
Critical aggregation concentration (CAC) measurements.
Nile red stock solution in acetone (1 mg/mL) was diluted in PB (1X) to a concentration of 0.01 mg/mL and was used to make all peptide solutions. A peptide stock solution was prepared at 4 mg/mL in the Nile red stock solution and was further diluted to the concentration of 3 mg/mL, 2 mg/mL, 1 mg/mL, 0.5 mg/mL, 0.25 mg/mL, 0.1 mg/mL, 0.01 mg/mL, 0.001 mg/mL, and 0.0001 mg/mL. All peptide dilutions were vortexed for a few seconds, then 300 μL of each was transferred to a 96-well plate, and the plate was allowed to sit in the dark for 15–20 min. Fluorescence spectra were recorded using a Varian Cary Eclipse fluorescence spectrophotometer (FL1105M003) with an excitation wavelength of 550 nm. Fluorescence intensity measured at 628 nm was plotted against log[concentration], and the final CAC values were estimated to be the point of intersection between the linear fits of high and low concentration regime.
Circular Dichroism (CD) Spectroscopy.
CD spectra were measured at room temperature using a Jasco J-815 CD spectrometer (Jasco Inc.) with a pre-set N2 flow at 120 mL/min. The range of wavelengths employed was 190 to 250 nm (50 nm/min) with a response time of 8 s. Samples for both APA solutions were freshly prepared at 0.25 and 10 mM (20 μL) in 1X PB (pH 7.4) and were analysed using a quartz cuvette with a path length of 1 mm with 3 iterations for each sample. Similarly, CD spectra for freshly prepared hydrogels (20 μL) were recorded using a dismountable cuvette with a path length of 0.2 mm. Raw spectra were converted to mean residual ellipticity for comparison.
Hydrogelation.
All APAs except Pep[–OC(CH3)2–] formed hydrogels in 1X PB at physiological pH upon addition of 10 μL of CaCl2 solution (200 mM in water) to 90 μL of peptide solution (10 mM in 1X PB). The final concentration of CaCl2 in the hydrogels was 20 mM. Hydrogels were allowed to sit for 10 min before any further analysis.
Morphological analyses.
Peptide solutions (10 mM in 0.05 M phosphate buffer at pH 7.4) were prepared and allowed to age overnight, then diluted with water to 500 μM. Next, 10 μL of peptide solution was deposited on a carbon-coated copper TEM grid (300 mesh, Electron Microscopy Sciences), allowed to sit for 5–6 min, and then gently blotted with filter paper. The grid was then washed by adding a drop of MilliQ water, allowed it to stand for 1 min, and then blotted with filter paper. Samples were stained with 10 μL of a 2% uranyl acetate aqueous solution for 5–6 min, blotted with filter paper, and allowed to dry in air before TEM observation. Images were taken on a Philips EM420 TEM with a slow scan CCD camera.
Rheology.
Rheological experiments were done on an AR-2000 (TA instruments) using a 25 mm parallel plate geometry. Buffered peptide solutions (225 μL, 10 mg/mL peptide, ~10 mM) were prepared for each peptide in 1X PB (pH 7.4) and quickly transferred to the rheometer’s bottom geometry. Gelation was initiated upon addition of 25 μL CaCl2 solution (200 mM in water) and the resulting solution was mixed thoroughly with a pipet tip to ensure homogeneity. After allowing the solution to gel for 10 min, the upper geometry was lowered to a pre-set gap of 500 μm, and a dynamic time sweep was performed at a frequency of 1 Hz and 0.5% strain to measure storage (G’) and loss (G”) moduli. Each time sweep was followed by a dynamic frequency sweep (0.010–100 Hz at 0.5% strain) and a strain sweep (1 Hz at 0.5–100% strain).
H2S release measurement.
H2S release from peptide gels and solutions was measured amperometrically using an electrode probe8. A solution of APA (90 μL of 10 mM solution in 1X PB) was placed in an inner well inside a specially designed glass vial equipped with a stir bar (Figure S11). 10 μL of CaCl2 was added to the inner well and mixed thoroughly with a pipette tip to form hydrogel. After 10 min, 4 μL of Cys solution (200 mM in water) was gently deposited on the top layer of the hydrogel. The final concentrations in the inner well were 10 mM in peptide and 20 mM (2 equiv) in Cys. The well was immediately covered with the gas-permeable membrane (Breathe easier, Diversified Bioteck), and PB buffer at pH 7.4 (4.95 mL) mixed with 50 μL of diethylenetriaminepentaacetic acid (DTPA) solution (10 mM in water) was added into the vial, covering the inner well. The H2S-selective microelectrode was then immersed in the PB solution, and the output signal was recorded. Similarly, H2S release from APA solutions was measured by transferring 90 μL of peptide solution (10 mM) to the inner well followed by addition of Cys solution (4 μL, 200 mM in water). Calibration was carried out as previously reported.36
In vitro studies using fluorescent probe.
Cell studies were conducted using an adherent H9C2 cell line of rat embryonic cardiomyocytes (ATCC, Manassas, VA, USA). Cells were cultured in 5% CO2-air atmosphere at 37 °C using Dulbecco’s Modified Eagle Medium (DMEM), fortified with 10 % fetal bovine serum (FBS), 50 IU/mL penicillin, and 50 g/mL streptomycin. Cell cultures were passaged after 70–85% confluence, washing in between with 1X PBS and releasing with trypsin and 0.25% EDTA solution.
H9C2 cells were plated at a density of 0.08 million per well in a 48-well plate and cultured for 48 h, followed by washing with 1X PBS three times and addition of 480 μL serum-free DMEM per well. Cells were incubated with peptide hydrogel (20 μL, 10 mM) and Cys solution (4 μL, 100 mM) for 2 h. Following treatment with either Gel[–OCH2–] or Gel[–CH=CH–], with or without additional Cys, media and gel were removed, and cells were washed with PBS three times. WSP-5 (10 μL, 2.5 mM), reported previously,37 and CTAB (10 μL, 5 mM) solutions were added to each well and total volume was brought up to 500 μL per well with serum-free DMEM. After 30 min incubation, the media was removed, and cells were washed with PBS three times. Fresh PBS (500 μL) was added to the wells, and the plate was visualized by bright-field and fluorescence microscopy (Nikon Eclipse Ti-U) with FITC filter set. The magnification was 20x. Corrected total cell fluorescence (CTCF) intensity was estimated using equation CTCF = integrated density – (area of selected cell) × (mean fluorescence of background). A rectangular drawing/selection tool in ImageJ was used to select a desired region with fluorescence, and its integrated density and area were measured. Mean fluorescence intensity of the background was calculated by averaging the intensities of the selected areas that had no fluorescence.
3. RESULTS AND DISCUSSION
Peptide design and synthesis.
Previously, we reported an APA with a short carbonyl linker (Pep[–C(O)–]) that formed a robust hydrogel.33 In order to investigate the effect of linker structure on self-assembly, hydrogelation, and H2S release profile, we synthesized five additional peptides with linkers O-methylene (Pep[–OCH2–]), O-methyl-methylene (Pep[–OCH(CH3)–]), ethyl (Pep[–(CH2)2–]), ethenyl (Pep[–CH=CH–]), and O-dimethyl-methylene (Pep [–OC(CH3)2–]) (Scheme 1). We speculated that the alkyl and O-methylene linkers would provide additional flexibility relative to the carbonyl linker, whereas the alkenyl linker would have restricted rotation compared with alkyl. We included O-methylene linkers with zero, one, or two pendant methyl groups to investigate the steric effects of methyl group substitution on the CH2 carbon.
Scheme 1.
Chemical structure of six APAs with different linker segments.
All six peptides were synthesized via solid-phase peptide synthesis as described in our previous work.35 S-Benzylthiohydroxylamine (SBTHA) was covalently attached to the peptide fragment IAVEEE via an aryl aldehyde group on each linker segment, generating the H2S-releasing SATO functional group and yielding the H2S-releasing APAs. The nomenclature of each peptide consists of two parts: the first part depicts the physical state of the peptide (Pep or Gel), and the second part depicts the type of linker (e.g., the soluble form of the peptide with the −OCH2− linker is referred to as Pep[−OCH2−], while its hydrogel is referred to as Gel[−OCH2−]).
Secondary structure of APAs.
The first step toward understanding the molecular packing of the APAs was the measurement of their critical aggregation concentration (CAC), a minimum concentration above which self-assembly occurs. CAC values for all APAs were in the range of 0.8–1.0 mg/mL (~1 mM) as measured by the Nile red assay (Figure S5). This range is consistent with CAC measurements for APAs with similar structures.26
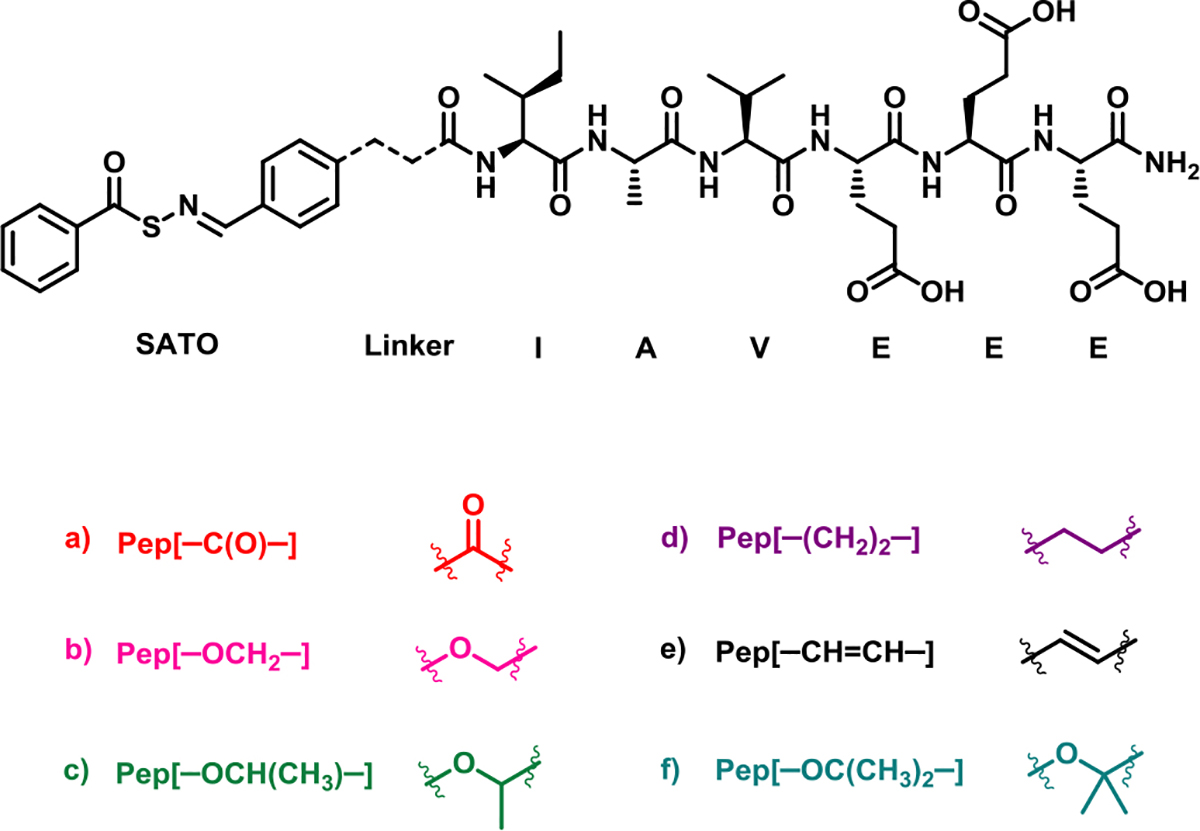
Next, we utilized circular dichroism (CD) spectroscopy to investigate the secondary structures of the APAs. Below the CAC (0.25 mg/mL), all peptides showed a characteristic random coil spectrum with a minimum around 200 nm (Figure 1A). However, above the CAC (10 mg/mL), differences in secondary structures of the APAs were observed (Figure 1B). Because the peptide backbone for all APAs is the same, any differences in their secondary structures should be a result of the linker segment and the role it plays in directing secondary structure. Linker [–C(O)–] showed a β-sheet signature with a minimum around 219 nm, but it lacked the typical peak near 197 nm. Similarly, Pep[–OCH2–] and Pep[–OCH(CH3)–], formed what appeared to be twisted β-sheets, characterized by red-shifted minima and maxima around 222 nm and 206 nm, respectively. Pep[–(CH2)2–] also showed a minimum at 214 nm along with an additional small negative peak around 195 nm suggesting that the observed structure may have some random coil character mixed with β-sheet. Pep[–OC(CH3)2–], on the other hand, showed an even stronger minimum around 199 nm suggesting a higher degree of random coil character. Interestingly, Pep[–CH=CH–] showed a small minimum at 208 nm along with a strong negative peak at 225 nm, indicating some α-helical character. Although definitive interpretations of the CD data are difficult, in part due to SATO absorptions in the 190–240 nm region, the differences observed in the secondary structures of APA hydrogels highlight the importance of the linker segment indicating the secondary structure of the APAs.
Figure 1.
Circular dichroism spectra of APA solutions at concentration of A) 0.25 mg/mL and B) 10 mg/mL in 1X phosphate buffer (PB).
Morphology of APAs.
To further understand the self-assembling behavior and to probe supramolecular structures formed by these APAs, we used transmission electron microscopy (TEM) (Figure 2). APA solutions well above their CAC (10 mg/mL) were aged overnight, diluted to 0.5 mg/mL, and quickly cast onto grids before staining with uranyl acetate. Pep[–C(O)–] formed long continuous fibrils, which were a few micrometres in length with average widths around 5 nm (Figure 2A). In the case of the O-methylene linkers, both Pep[–OCH2–] and Pep[–OCH(CH3)–] formed long and wide twisted nanoribbons with widths around 9 nm (Figure 2B and C), similar to literature reports on APAs containing similar linkers.15–17 Pep[–(CH2)2–], on the other hand, formed short twisted nanoribbons with an average width of around 7 nm (Figure 2D). Several short fragments were also observed suggesting some disrupted packing, which is in agreement with the presence of a substantial random coil peak in the CD spectrum for this peptide. Lastly, Pep[–CH=CH–] showed narrow, twisted nanoribbons with widths around 6 nm (Figure 2E). Although both [–CH=CH–] and [–C(O)–] are considered rigid, the observed differences in their morphologies may result from the hydrogen-bond accepting capability of the carbonyl in Pep[–C(O)–]. Furthermore, APAs with flexible linkers like Pep[–OCH2–], Pep[–(CH2)2–], and Pep[–OCH(CH3)–] formed twisted nanoribbons with fairly similar widths. However, there were notable differences in the pitch lengths of these three APAs, with values of 350 ± 50 nm, 220 ± 40 nm, and 100 ± 10 nm, respectively (Figure S6). The differences in the pitch lengths for Pep[–OCH2–] and Pep[–(CH2)2–] may be due to the additional O atom in Pep[–OCH2–], which may facilitate long range packing through additional H-bonding, as has been observed in related APAs.16 On the other hand, the nanoribbons formed by Pep[–OCH(CH3)–] showed smaller pitch lengths relative to Pep[–OCH2–] and Pep[–(CH2)2–], possibly due to steric repulsions caused by the additional methyl group, which may drive the nanoribbons to twist at shorter intervals. This was further validated by examining the TEM image of Pep[–OC(CH3)2–] (Figure 2F); in this APA with two methyl groups on the linker carbon, no well-defined morphology was observed.
Figure 2.

TEM micrographs of self-assembling APAs. A) Pep [–C(O)–], B) Pep [–OCH2–], C) Pep [–OCH(CH3)–], D) Pep [–(CH2)2–), E) Pep [–(CH=CH)–], and F) Pep [–OC(CH3)2–]. The APAs were aged for 14 h in 1X PB (10 mM), diluted to 0.5 mM before casting onto grids, and stained with uranyl acetate. Twisted points in ribbons are indicated by red arrows.
The differences in twisting pitch among the self-assembled structures of the APAs appears to be a result of how linker flexibility drives packing. Aggeli et al. determined in 2001 that in the case of long pitch length, β-sheets form closely packed assemblies and stack to form ribbons; in contrast, if the natural pitch length is small, then more loosely packed assemblies like fibrils are preferred.38 More recently, Cui et al. observed similar effects in drug-conjugated peptides.38,39 This is consistent with our observations that the flexible linkers, ([–OCH2–], [–OCH(CH3)–], and [–(CH2)2–]) formed less twisted and hence comparatively broader nanoribbons compared to rigid linkers ([–C(O)–] and [–CH=CH–]), which formed narrow, fibril-like structures. The observed differences in the morphology of the APAs further confirms the role played by subtle alterations in the linker segment in controlling the self-assembly of APAs.
Gelation and rheological properties.
As observed from the TEM micrographs, all APAs except Pep[–OC(CH3)2–] self-assembled into one dimensional nanostructures. We previously found that 10 mg/mL solutions of APAs at physiological pH formed hydrogels upon addition of 20 mM CaCl2 via coordinate bridging of glutamic acid units in the peptide backbone.26,33 Similarly, in this work, we mixed APA solutions with CaCl2 to initiate gelation. All APAs except Pep[–OC(CH3)2–] formed self-supporting hydrogels.
To evaluate the rheological properties of the APA hydrogels, all gels were prepared on a rheometer and allowed to sit for 10 min before applying oscillatory shear. Dynamic time sweep experiments under constant strain (0.5%) and frequency (1 Hz) showed that APA hydrogels were fairly stable to the applied force with storage moduli (G’) higher than loss moduli (G”) for all hydrogels in the studied range (Figure S7A). This behaviour is typical of amphiphilic peptide hydrogels.11,40 Furthermore, after each time sweep, hydrogels were subjected to an oscillatory frequency sweep (Figure S7B). Nearly flat traces in the frequency sweep experiments in the low frequency regime (< 10 Hz) indicated frequency-independent rheological properties for all APA hydrogels.
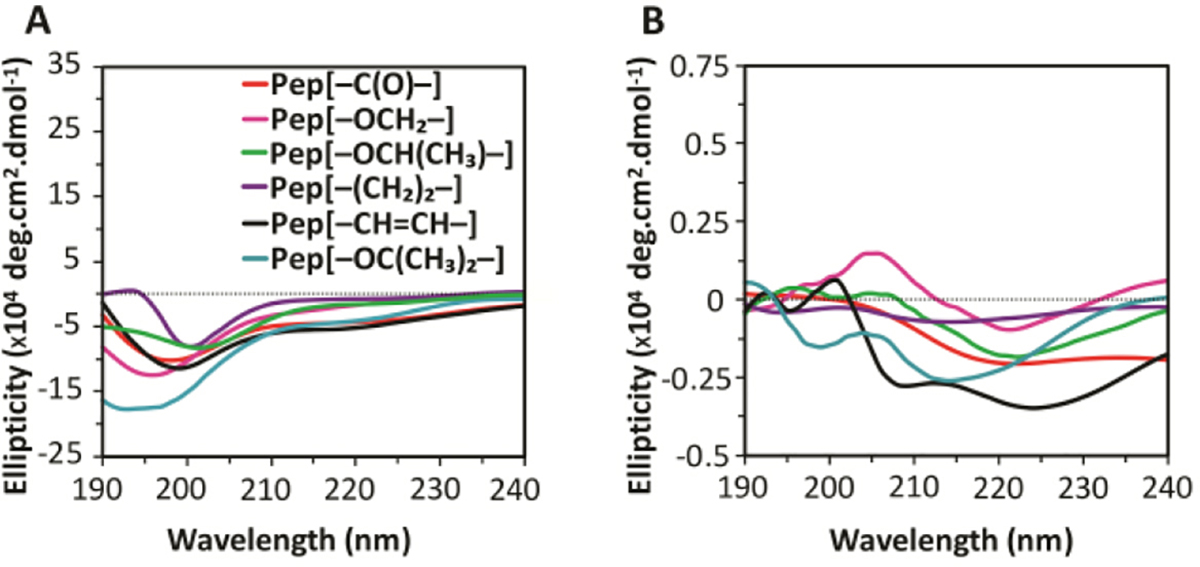
Although all five APA hydrogels showed relatively constant rheological behavior, their storage moduli were affected by the linker unit (Figure 3). For example, Gel[–C(O)–] had a storage modulus of 250 ± 60 Pa, while Gel[–OCH2–] formed a significantly stronger hydrogel with a storage modulus of 1400 ± 200 Pa. Furthermore, Gel[–OCH(CH3)–] was also weaker than Gel[–OCH2–] at 560 ± 50 Pa likely due to steric hindrance caused by the additional methyl group, which may hinder close packing of fibers essential for strong coordination with Ca2+. The influence of steric effects on hydrogelation was further confirmed when Pep[–OC(CH3)2–] failed to form a hydrogel with Ca2+, which we attribute to additional steric congestion caused by two methyl groups. Gel[–(CH2)2–] exhibited similar rheological behaviour to Gel[–OCH(CH3)–] with G’ at 570 ± 70 Pa. Gel[–CH=CH–] showed the lowest equilibrium storage modulus of all APA hydrogels tested at 40 ± 10 Pa.
Figure 3.
Storage moduli of 10 mg/mL APA hydrogels at 1 Hz frequency and 0.5% strain with 20 mM CaCl2 at physiological pH at 25 °C.
Taken together, the TEM micrographs along with rheological data suggest that closely packed structures (twisted ribbons) formed stronger hydrogels with Ca2+ relative to loosely packed structures (fibrils). This observation is consistent with related materials in the literature.11,14,40 Additionally, the observed differences in the widths of the ribbons can be correlated to the hydrogel storage moduli, with wider nanoribbons forming stiffer hydrogels relative to their counterparts. This observation is likely due to strong matrices formed by inter-fibre calcium coordination as observed previously.11, 36 Collectively, these results support our hypothesis that the linker segment affects both self-assembly and gelation behaviour of APAs by controlling the orientation of the aromatic component relative to the peptide chain.
H2S release from APA hydrogels.
Next, we explored whether the observed differences in molecular packing and viscoelastic properties of these APA hydrogels influenced their H2S release behavior. H2S release from APA hydrogels was triggered by Cys, which led to decomposition of the SATO group with H2S release (Scheme S3). H2S release from 10 mg/mL hydrogels at pH 7.4 was measured by an H2S-sensitive electrochemical probe and a specially designed vial (Figure S11).12,26 The vial contained an inner well, which was loaded with APA hydrogel, onto which was added a solution of Cys. H2S, formed by the reaction of Cys with SATO groups, percolated through a gas-permeable membrane into a PBS trapping solution in the outer well of the vial, where it was measured as an output current by the electrochemical probe. We compared the peaking times of APA hydrogels, which is the time required to reach a maximum H2S concentration from the time of addition of Cys.
We speculated that stiffer gels would limit the access of Cys to SATOs in the nanofiber core and release H2S more slowly than weaker gels because the molecular packing in stiff peptide-based hydrogels, such as Gel[–OCH2–], is typically more compact compared to weak peptide-based hydrogels, such as Gel[–CH=CH–]. As Cys passes through the hydrogel network, it must penetrate the nanofiber core and react with SATO groups to release H2S. In addition to triggering H2S release, this process also results in hydrogel degradation (APAs without SATOs do not form gels). In order to ascertain whether the rate of Cys diffusion through hydrogel network would affect the rate of H2S release, we studied cumulative release of Rhodamine B encapsulated within the hydrogel network of the weakest and stiffest gels (Figure S8). No significant differences in the release profiles were observed, suggesting that diffusion of Cys though the hydrogel network does not affect the H2S release rate, indicating that any observed differences in release rate would more likely be due to different rates of Cys penetration into the nanofiber core. Therefore, we expected that in situations where Cys diffuses quickly into the nanofiber core, as is the case for weak hydrogels, it would react quickly with SATOs, resulting in fast H2S release and fast degradation of the hydrogel. Alternatively, for stiff hydrogels, Cys would diffuse slowly into the tightly packed nanofibers in the hydrogel network, resulting in slow H2S release and slow gel degradation.
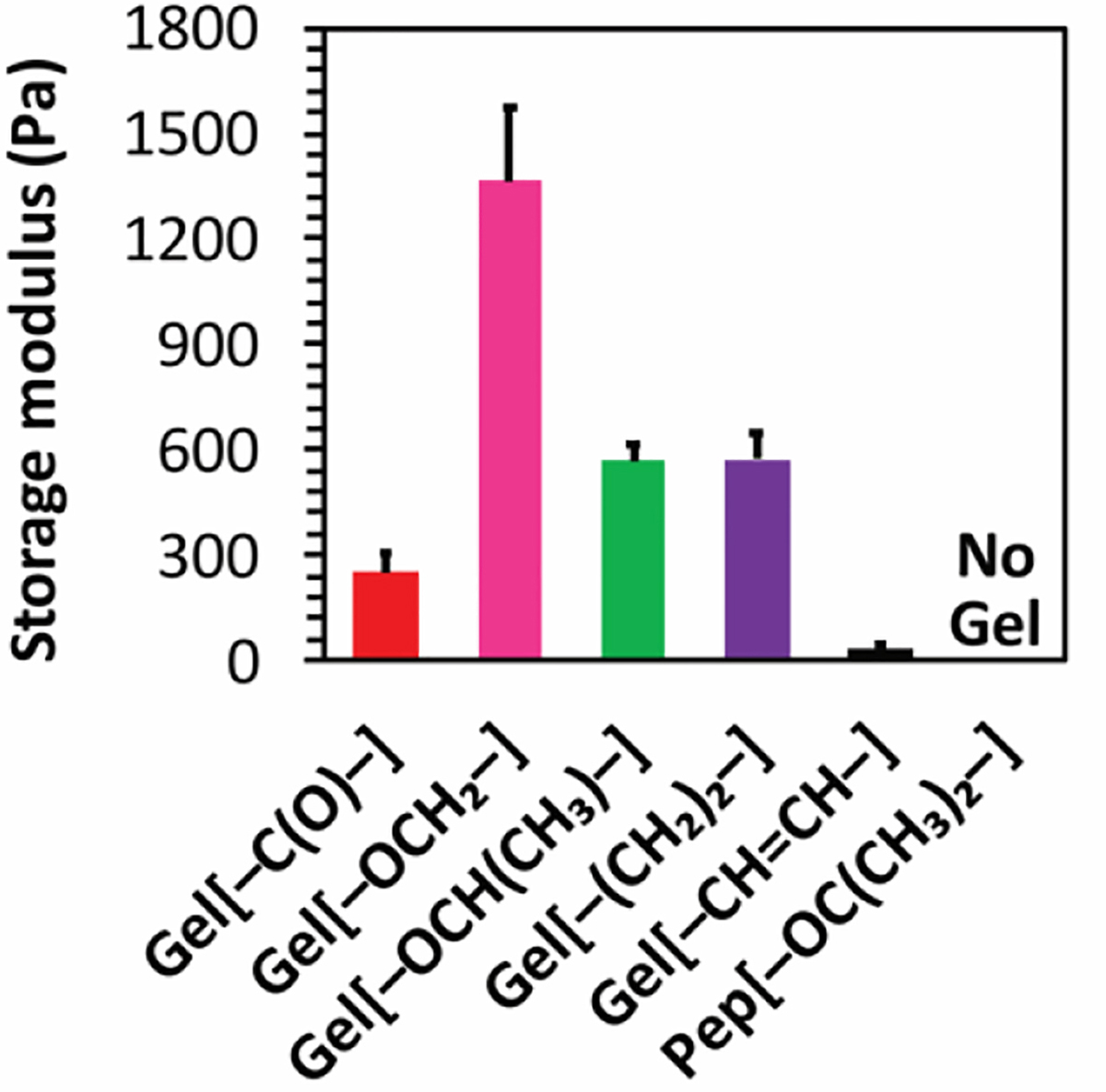
Interestingly, H2S release peaking times, as measured by the H2S probe, showed the opposite trend (Figure 4A and S9A). The observed H2S release peaking times were shorter for stiff Gel[–OCH2–] (22 ± 4 min) and longer for weaker Gel[–CH=CH–] (80 ± 6 min), with gels of intermediate stiffness falling in between. Moreover, Pep[–OC(CH3)2–], which formed a viscous solution upon addition of CaCl2, showed longest H2S release profile with a peaking time above 200 min (Figure S9A). The inverse correlation between stiffness and H2S release rate (stiff gels release H2S faster than weak gels) may be a result of our heavily engineered H2S release setup involving a specially designed vial (described above). The output signal depends on several kinetic factors, including diffusion of Cys through the gel network and to the nanofiber core, diffusion of H2S through the gel, permeability of H2S through the membrane, and oxidation/volatilization of H2S from the PBS trapping solution in the outer well. We reason that when H2S release is slow, the loss of H2S due to volatilization and oxidation is comparable to the rate at which H2S enters the trapping solution. As a result, the output signal peaks quickly (early peaking time). In contrast, for fast releasing gels, the rate of H2S entering the trapping solution is greater than the rate of H2S than being lost due to volatilization and oxidation. Therefore, the H2S concentration in the trapping solution increases gradually, and the output signal slowly reaches a peak (late peaking time). In other words, our experimental setup, necessitated by the limited analytical tools available for studying H2S release rates, showed a correlation that appeared at first to be the opposite of the expected result. However, based on a careful consideration of all the rates involved, we believe that the data support our initial hypothesis, that stiff gels release H2S slower than weak gels.
Figure 4.
H2S release from A) 10 mg/mL APA hydrogels and B) APA solutions at 10 mg/mL concentration, both in 1X PB with 2 equiv Cys added to trigger H2S release. * indicates p<0.05, ** indicates p<0.01, and *** indicates p<0.001 for comparisons among the groups indicated, as determined by a one-way analysis of variance (ANOVA) with a Student-Newman-Keuls comparison posthoc test (n=3).
From our previous work on related SATO peptides, we have observed that H2S release rates depend primarily on nanofiber geometry and are not affected by the sequence or structure of the peptide in its monomeric form (i.e., its unassembled form, below the CAC).12,34,35 Therefore, We measured the H2S release from 10 mg/mL APA solutions under similar conditions but without Ca2+ (Figure 4B and S9B). In the absence of Ca2+, the inter-chain coordinate bridges between the nanofibers cannot be formed, and some repulsion exists in the nanofibers due to negative charges present on their surface. Therefore, we expected that Cys may be able to diffuse into the nanofiber cores more easily in solution than in the gel state, where nanofibers are bundled together through inter-chain coordinate bridges.
We observed that H2S release peaking times observed for all APA solutions (near 200 min) were much longer compared to APA hydrogels. Only Pep[–(CH2)2–] showed a significantly shorter peaking time than the others (150 min). Interestingly, peaking times observed for Pep[–OC(CH3)2–] both in the presence and absence of CaCl2 were very close, suggesting that Ca2+ fails to form strong coordinate bonds to create a network, consistent with the inability of this APA to gel. Overall, the solution H2S release results for the five APAs that do form gels suggest that the observed H2S release trends for APA hydrogels may be attributed to a differential rate of Cys penetration into the hydrophobic core of the nanofibers in the gel phase.
H2S release experiments described above provide insight into the short-term H2S release behavior of these APA hydrogels and solutions. However, due to baseline fluctuations in the electrode probe, reliably measuring H2S release beyond a few hours is difficult. Therefore, we evaluated gel degradation in response to Cys addition visually over several hours (Figure S12). As mentioned earlier, H2S release from hydrogels in the presence of Cys results in hydrogel dissolution because the peptide aldehyde cannot form a hydrogel. As expected, we observed that the strongest hydrogel, Gel[–OCH2–], took several minutes for any degradation to become visible, and even after 7 h, translucent structures could still be seen. In contrast, a weak hydrogel, Gel[–CH=CH–], began to degrade within the first 30 min. These results suggest that stiff APA hydrogels may release H2S for longer periods of time by shielding SATOs inside their stiffer network. Thus, our observations support that stiff Gel[–OCH2–] may be a suitable candidate for localized H2S release over hours to days.
In vitro H2S release from APA hydrogels
To test the H2S release behavior of hydrogels under physiologically relevant conditions in the presence of cells, we compared the in vitro H2S release from stiff Gel[–OCH2–] and weak Gel[–CH=CH–]. H9C2 cardiomyocytes were incubated with these two selected hydrogels and Cys for 2 h followed by incubation with WSP-5 fluorescent probe37 to visually assess and quantify H2S release via fluorescence microscopy. As expected, cells incubated with Cys only showed negligible fluorescence intensity (Figure 5A, first row), consistent with the fact that WSP-5 responds to H2S much more strongly than Cys. Cells incubated with hydrogels alone (without Cys) showed a small fluorescence signal (Figure 5A, rows 2 and 3), likely because the low thiol concentration in the media triggers a small amount of H2S release over the timescale of the experiment. Treatment of cells in the presence of additional Cys with stiff Gel[–OCH2–] (Figure 5A, row 4) showed moderate fluorescence, while those incubated with weak Gel[–CH=CH–] (Figure 5A, row 5) showed a dramatic increase in fluorescence intensity compared with the other treatment groups. We quantified these observations by measuring corrected total cell fluorescence intensity in each group (Figure 5B), with results showing that the intensity of weak Gel[–CH=CH–] was about three times greater than that of stiff Gel[–OCH2–]. These results indicate that H2S release from the stiffer hydrogel is indeed slower than release from the weaker hydrogel under conditions where Cys can trigger release throughout the entire gel and where cumulative H2S release can be measured.
Figure 5.
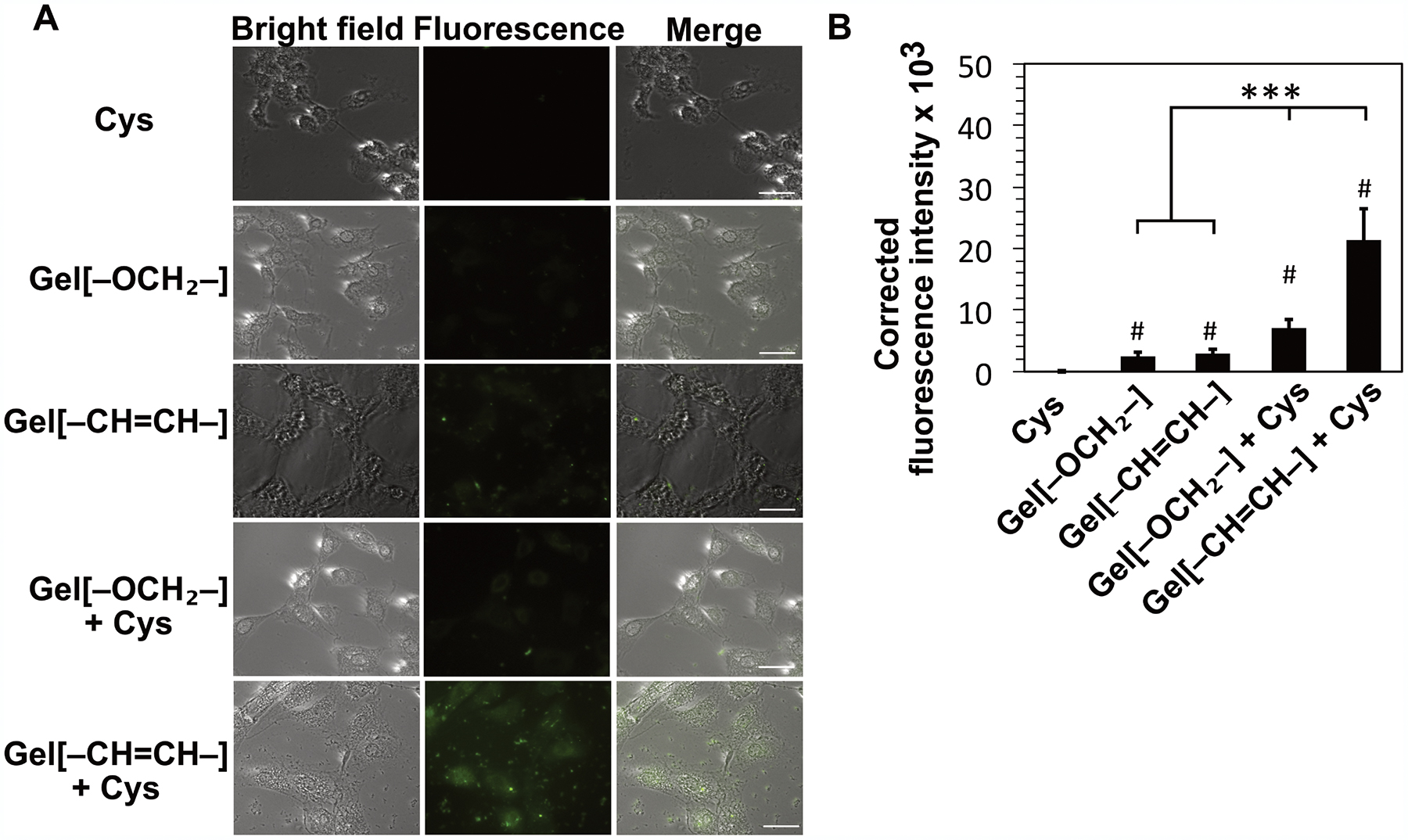
A) Bright field, fluorescence, and merged images of fluorescence intensity in H9C2 cells. Cells were incubated with WSP-5 fluorescent probe for 30 min after pre-treatment with Cys (800 μM), Gel[–OCH2–] (400 μM), and Gel[–CH=CH–] (400 μM) for 2 h. Scale bar is 100 μm. B) Quantified fluorescence intensity of the five respective treatment groups. Fluorescence intensity values were averaged over 30 measurements from images from two separate wells for each treatment group. # indicates p<0.001 with respect to Cys only treatment group and *** indicates p<0.001 for comparisons among the groups indicated, as determined by a one-way analysis of variance (ANOVA) with a Student-Newman-Keuls comparison posthoc test.
4. CONCLUSION
Self-assembling peptide hydrogels may be versatile and powerful drug delivery vehicles, especially for therapeutics such as H2S where localized delivery is crucial. APA-based hydrogels allow tunability of H2S release through minimal structural modifications. We synthesized six APAs, five of which formed hydrogels with Ca2+ ions at 10 mg/mL. The rheological behavior of the APA hydrogels varied depending on the flexibility of the linker. While peptides with flexible linkers formed stiffer hydrogels, peptides containing a relatively rigid linker formed weaker hydrogels. Furthermore, the APA hydrogels showed varying release of H2S regulated by modulating the access of Cys into the hydrophobic nanofiber core. The stiffer hydrogels showed overall longer release times compared to weaker hydrogels by retarding the Cys penetration into nanofiber core. These hydrogels may be applied in tissue engineering and regenerative medicine, where slow, sustained delivery of H2S could be beneficial to a number of disease states.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Science Foundation (DMR-1454754) and the National Institutes of Health (R01GM123508). We are grateful to Prof. Tijana Grove and Dr. Ann Norris for help with instrumentation. We also thank Ryan Carrazzone for reading of the manuscript and Yun Qian for several helpful discussions.
Footnotes
Supporting Information
Synthetic procedures, NMR spectra, and additional characterization, including CAC measurements, circular dichroism, rheology, fluorescence, and TEM analysis.
The authors declare no competing financial interest.
REFERENCES
- (1).Aida T; Meijer EW; Stupp SI Functional Supramolecular Polymers. Science 2012, 335, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Webber MJ; Appel EA; Meijer EW; Langer R Supramolecular biomaterials. Nat. Mater 2016, 15, 13–26. [DOI] [PubMed] [Google Scholar]
- (3).Acar H; Srivastava S; Chung EJ; Schnorenberg MR; Barrett JC; LaBelle JL; Tirrell M Self-assembling peptide-based building blocks in medical applications. Adv. Drug. Deliv. Rev 2017, 110–111, 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cavalli S; Albericio F; Kros A Amphiphilic peptides and their cross-disciplinary role as building blocks for nanoscience. Chem. Soc. Rev 2010, 39, 241–263. [DOI] [PubMed] [Google Scholar]
- (5).Sato K; Hendricks MP; Palmer LC; Stupp SI Peptide supramolecular materials for therapeutics. Chem. Soc. Rev 2018, 47, 7539–7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fleming S; Ulijn RV Design of nanostructures based on aromatic peptide amphiphiles. Chem. Soc. Rev 2014, 43, 8150–8177. [DOI] [PubMed] [Google Scholar]
- (7).Bowerman CJ; Liyanage W; Federation AJ; Nilsson BL Tuning beta-sheet peptide self-assembly and hydrogelation behavior by modification of sequence hydrophobicity and aromaticity. Biomacromolecules 2011, 12, 2735–2745. [DOI] [PubMed] [Google Scholar]
- (8).Castelletto V; Moulton CM; Cheng G; Hamley IW; Hicks MR; Rodger A; López-Pérez DE; Revilla-López G; Alemán C Self-assembly of Fmoc-tetrapeptides based on the RGDS cell adhesion motif. Soft Matter 2011, 7, 11405–11415. [Google Scholar]
- (9).Sahoo JK; Nazareth C; VandenBerg MA; Webber MJ Self-assembly of amphiphilic tripeptides with sequence-dependent nanostructure. Biomater. Sci 2017, 5, 1526–1530. [DOI] [PubMed] [Google Scholar]
- (10).Sahoo JK; Nazareth C; VandenBerg MA; Webber MJ Aromatic identity, electronic substitution, and sequence in amphiphilic tripeptide self-assembly. Soft Matter 2018, 14, 9168–9174. [DOI] [PubMed] [Google Scholar]
- (11).Shi J; Gao Y; Zhang Y; Pan Y; Xu B Calcium ions to cross-link supramolecular nanofibers to tune the elasticity of hydrogels over orders of magnitude. Langmuir 2011, 27, 14425–14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang Y; Kaur K; Scannelli SJ; Bitton R; Matson JB Self-Assembled Nanostructures Regulate H2S Release from Constitutionally Isomeric Peptides. J. Am. Chem. Soc 2018, 140, 14945–14951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Singh N; Kumar M; Miravet JF; Ulijn RV; Escuder B Peptide-Based Molecular Hydrogels as Supramolecular Protein Mimics. Chemistry 2017, 23, 981–993. [DOI] [PubMed] [Google Scholar]
- (14).Chakroun RW; Wang F; Lin R; Wang Y; Su H; Pompa D; Cui H Fine-Tuning the Linear Release Rate of Paclitaxel-Bearing Supramolecular Filament Hydrogels through Molecular Engineering. ACS Nano 2019, 13, 7780–7790. [DOI] [PubMed] [Google Scholar]
- (15).Fleming S; Debnath S; Frederix PW; Tuttle T; Ulijn RV Aromatic peptide amphiphiles: significance of the Fmoc moiety. Chem. Commun 2013, 49, 10587–10589. [DOI] [PubMed] [Google Scholar]
- (16).Yang Z; Liang G; Ma M; Gao Y; Xu B Conjugates of naphthalene and dipeptides produce molecular hydrogelators with high efficiency of hydrogelation and superhelical nanofibers. J. Mater. Chem 2007, 17, 850–854. [Google Scholar]
- (17).Yang Z; Liang G; Xu B Supramolecular hydrogels based on beta-amino acid derivatives. Chem. Commun 2006, 7, 738–740. [DOI] [PubMed] [Google Scholar]
- (18).Chen L; Revel S; Morris K; L, C. S.; Adams, D. J. Effect of molecular structure on the properties of naphthalene-dipeptide hydrogelators. Langmuir 2010, 26, 13466–13471. [DOI] [PubMed] [Google Scholar]
- (19).Barr LA; Calvert JW Discoveries of hydrogen sulfide as a novel cardiovascular therapeutic. Circ. J 2014, 78, 2111–2118. [DOI] [PubMed] [Google Scholar]
- (20).Zhao WZ, J Lu Y Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang X; Bian JS Hydrogen sulfide: a neuromodulator and neuroprotectant in the central nervous system. ACS Chem. Neurosci 2014, 5, 876–883. [DOI] [PubMed] [Google Scholar]
- (22).Bhatia M H2S and Inflammation: An Overview. Handb. Exp. Pharmacol 2015, 230, 165–180. [DOI] [PubMed] [Google Scholar]
- (23).Whiteman M; Winyard PG Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expert. Rev. Clin. Pharmacol 2011, 4, 13–32. [DOI] [PubMed] [Google Scholar]
- (24).Wang R Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev 2012, 92, 791–896. [DOI] [PubMed] [Google Scholar]
- (25).Longchamp A; Kaur K; Macabrey D; Dubuis C; Corpataux JM; Deglise S; Matson JB; Allagnat F Hydrogen sulfide-releasing peptide hydrogel limits the development of intimal hyperplasia in human vein segments. Acta. Biomater 2019, 97, 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Qian Y; Kaur K; Foster JC; Matson JB Supramolecular Tuning of H2S Release from Aromatic Peptide Amphiphile Gels: Effect of Core Unit Substituents. Biomacromolecules 2019, 20, 1077–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Qian Y; Matson JB Gasotransmitter delivery via self-assembling peptides: Treating diseases with natural signaling gases. Adv. Drug Deliv. Rev 2017, 110–111, 137–156. [DOI] [PubMed] [Google Scholar]
- (28).Connal LA The benefits of macromolecular hydrogen sulfide prodrugs. J. Mater. Chem. B 2018, 6, 7122–7128. [DOI] [PubMed] [Google Scholar]
- (29).Urquhart MC; Ercole F; Whittaker MR; Boyd BJ; Davis TP; Quinn JF Recent advances in the delivery of hydrogen sulfide via a macromolecular approach. Polym. Chem 2018, 9, 4431–4439. [Google Scholar]
- (30).Foster JC; Carrazzone RJ; Spear NJ; Radzinski SC; Arrington KJ; Matson JB Tuning H2S Release by Controlling Mobility in a Micelle Core. Macromolecules 2019, 52, 1104–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hasegawa U; van der Vlies AJ Polymeric micelles for hydrogen sulfide delivery. MedChemCommun 2015, 6, 273–276. [Google Scholar]
- (32).Fan DY; Tian Y; Liu ZJ Injectable Hydrogels for Localized Cancer Therapy. Front. Chem 2019, 7, 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Carter JM; Qian Y; Foster JC; Matson JB Peptide-based hydrogen sulphide-releasing gels. Chem. Commun 2015, 51, 13131–13134. [DOI] [PubMed] [Google Scholar]
- (34).Foster JC; Powell CR; Radzinski SC; Matson JB S-aroylthiooximes: a facile route to hydrogen sulfide releasing compounds with structure-dependent release kinetics. Org. Lett 2014, 16, 1558–1561. [DOI] [PubMed] [Google Scholar]
- (35).Kaur K; Qian Y; Gandour RD; Matson JB Hydrolytic Decomposition of S-Aroylthiooximes: Effect of pH and N-Arylidene Substitution on Reaction Rate. J. Org. Chem 2018, 83, 13363–13369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Powell CR; Kaur K; Dillon KM; Zhou M; Alaboalirat M; Matson JB Functional N-Substituted N-Thiocarboxyanhydrides as Modular Tools for Constructing H2S Donor Conjugates. ACS Chem. Biol 2019, 14, 1129–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Peng B; Chen W; Liu C; Rosser EW; Pacheco A; Zhao Y; Aguilar HC; Xian M Fluorescent probes based on nucleophilic substitution-cyclization for hydrogen sulfide detection and bioimaging. Chem. Eur. J 2014, 20, 1010–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Aggeli A; Nyrkova IA; Bell M; Harding R; Carrick L; McLeish TC; Semenov AN; Boden N Hierarchical self-assembly of chiral rod-like molecules as a model for peptide beta-sheet tapes, ribbons, fibrils, and fibers. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hu Y; Lin R; Zhang P; Fern J; Cheetham AG; Patel K; Schulman R; Kan C; Cui H Electrostatic-Driven Lamination and Untwisting of beta-Sheet Assemblies. ACS Nano 2016, 10, 880–888. [DOI] [PubMed] [Google Scholar]
- (40).Greenfield MA; Hoffman JR; de la Cruz MO; Stupp SI Tunable mechanics of peptide nanofiber gels. Langmuir 2010, 26, 3641–3647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.