

Abstract
Melanoma is the most aggressive and dangerous form of skin cancer that develops from transformed melanocytes. It is crucial to identify melanoma at its early stages, in situ, as it is “curable” at this stage. However, after metastasis, it is difficult to treat and the five-year survival is only 25%. In recent years, a better understanding of the etiology of melanoma and its progression has made it possible for the development of targeted therapeutics, such as vemurafenib and immunotherapies, to treat advanced melanomas. In this review, we focus on the molecular mechanisms that mediate melanoma development and progression, with a special focus on the immune evasion strategies utilized by melanomas, to evade host immune surveillances. The proposed mechanism of action and the roles of immunotherapeutic agents, ipilimumab, nivolumab, pembrolizumab, and atezolizumab, adoptive T- cell therapy plus T-VEC in the treatment of advanced melanoma are discussed. In this review, we implore that a better understanding of the steps that mediate melanoma onset and progression, immune evasion strategies exploited by these tumor cells, and the identification of biomarkers to predict treatment response are critical in the design of improved strategies to improve clinical outcomes for patients with this deadly disease.
Keywords: melanoma, melanoma immune evasion, immunotherapy, immune checkpoint blockade therapy, anti-PD-1, anti-PD-L1, anti-CTLA-4, adoptive T-cell therapy, T-VEC
1. Introduction to Melanoma
1.1. Melanocyte Biology and its Role in Melanoma Etiology
Stochastic accumulation of somatic mutations or inherited genetic defects allow normal melanocytes to transform into malignant melanoma. Melanocytes are the pigment forming cells of the skin that are of neural crest origin and are distributed across the epidermis, uvea, hair follicles, inner ear, heart, and mucosal tissue [1,2]. Melanocytes through various biochemical steps produce melanin within melanosomes, which is responsible for pigmentation and protection from harmful UV radiation [3,4,5,6,7]. Skin pigmentation is associated with genetic polymorphisms, between and within races, dictated by the amount of melanin produced by melanocytes and the size of melanosomes in the skin, rather than the number of melanocytes [4,8,9,10,11]. Races with darker skin pigmentation show higher melanin concentrations in their epidermis, making them less susceptible to melanoma, while individuals with lighter skin pigmentation who have a lower melanin content correspond to an increased risk for melanoma [4,8,12,13,14,15,16].
1.2. Melanoma Statistics and Risk Factors
Melanoma only accounts for about 1% of all skin cancers, but it is the most aggressive and dangerous one and accounts for 90% of all skin cancer deaths [14]. In the United States, it was estimated that in 2020, approximately 100,000 new cases of invasive melanoma will be diagnosed, with approximately 7000 deaths from this disease [14]. Women in general show better prognosis and overall survival than men, possibly due to sex hormone interactions with melanoma cells. There are reports suggesting that women have a stronger immune system, but the precise mechanism of this sex bias is under investigation [17,18,19,20,21,22,23,24]. In addition to gender, other risk factors for melanoma include fair skin, number of moles, UV exposure, age, and family history of skin cancer.
Moles or nevi are clusters of benign melanocytes in the quiescence state. As the number of nevi increases, the risk of developing melanoma also increases. It is estimated that about 30%–50% of all melanomas arise from nevi and are associated with non-chronically sun damaged (non-CSD) melanomas [2,25,26]. It was established that the number of nevi increases one’s risk for malignant melanoma, however, it does not guarantee that one will develop melanoma [2,27].
Both intermittent and chronic sun (UV) exposure, increases somatic mutation rates and leads to de novo cutaneous melanomagenesis, and also promotes pre-existing nevi to transform into melanoma [2,28,29,30,31,32,33]. UV signatures were detected in cutaneous melanoma, and consist of C > T transitions at dipyrimidine sites and CC > TT or (C/T)C > (C/T)T mutations [2,34]. UV exposure shows a “one-two punch” effect, enabling a pre-cancerous melanocyte to transform into a tumor cell. UV increases the mutational burden in the cells, while also locally and systemically suppressing the immune system. The relationship of UV exposure and immune escape is further discussed in Section 2.
Melanoma is a disease of old age and the stochastic accumulation of mutations within melanocytes either inherited or acquired, result in melanocyte transformation into melanoma. The average age of melanoma diagnosis in the United States is 65 years-old and death is 71 years-old, however, worldwide, melanoma incidences peaks at 70–80 years of age [14,33].
Familial history of skin cancer increases the likelihood of developing melanomas and is even higher if you have many atypical moles, also known as familial atypical multiple mole melanoma syndrome (FAMM) [35,36]. Familial melanoma comprises of 5%–12% of all melanomas [35]. Inherited genetic defects in CDKN2A and CDK4 that deregulate cell cycle in melanocytes were linked with the development of familial melanoma. Another genetic disorder Xeroderma Pigmentosum (XP), is where patients have a reduced ability to repair DNA damage caused by UV [37,38,39,40,41,42,43,44,45].
1.3. Melanoma Diagnosis and Staging
In the clinical setting, a dermatologist diagnoses suspicious skin lesions using the ABCDE and the “ugly duckling” guidelines, also known as ABCDEF [46,47,48,49]. The ABCDEF criterion is as follows: Asymmetry, Border Irregularity, Color Variegation, Diameter Larger than 6 mm, Evolution of a lesion (changing in size, color, shape, or nevogenesis), and “Funny Looking”, where the “ugly duckling” nevi does not fit the common profiles of nevi found on a patient [46,47,48,49,50]. Dermoscope is used by dermatologists to identify possible malignant lesions [51,52,53]. Once a suspected lesion is identified, a biopsy is taken for pathohistological analysis to confirm or refute the initial diagnosis [53,54,55]. Accurate disease staging is essential for the correct diagnosis, prognosis, and treatment regimen provided to the patient. Both clinical and pathological data are assessed using the Tumor Thickness, Nodal Involvement, and Metastasis (TNM) system [56]. Tumor thickness in the TNM system considers both the thickness of the primary tumor(s) and the extent to which the tumor is ulcerated. Tumor thickness or Breslow measurement considers the depth of which the melanoma has invaded the skin, since a greater vertical depth is correlated with worse prognosis of melanoma and is associated with greater spread of the disease [56,57]. Furthermore, the extent of ulceration of melanomas gives insight to the spread of the disease, since it frees up the melanoma to grow horizontally or vertically [58,59,60]. Nodal involvement in the TNM criterion evaluates whether the melanoma has spread to the nearby lymph nodes [56]. The M in the TNM system addresses if the melanoma has spread to distant organs and lymph nodes [56]. The most common sites for melanoma dissemination are the lung, liver, brain, bones, and skin [61]. There are vast complexities in uniformly and accurately diagnosing human melanomas, therefore, the American Joint Committee on Cancer (AJCC) suggests categorizing the disease based on various permutations of the TNM system [56,62].
1.4. Melanoma Subtypes and Their Molecular Abnormalities
Based on primary tumor tissue location, melanoma can be broadly categorized into cutaneous and non-cutaneous melanoma. Cutaneous melanoma (sun exposed) makes up about 91.2% of melanoma cases, while non-cutaneous melanoma (sun-shielded) makes up less than 10% of all cases and have distinct genetic alterations between them [5,63,64]. Non-cutaneous melanoma has a worse prognosis than cutaneous melanoma, due to the delay in primary tumor diagnosis, the aggressive nature of these tumors, a high recurrence rate after treatment, and a poor overall survival [65,66]. Interestingly, for both cutaneous and non-cutaneous melanoma, post metastatic disease diagnosis show similar overall survival [65]. Epidemiology studies provided strong evidence that fair skinned individuals have a higher susceptibility to cutaneous melanoma, while darker skinned individuals have higher cases of non-cutaneous melanoma [22,67,68].
Cutaneous melanoma arises from transformed melanocytes on sun exposed skin and has the highest mutation burden (179 mutations per sun exposed tumor), compared to non-cutaneous melanoma (9 mutations per sun-shielded tumor) [34,69,70]. Non-cutaneous melanoma occurs in regions with low UV exposure, such as uvea, mucosal tissue, and acral tissue, and cutaneous melanoma occurs in regions more susceptible to damages by harmful UV radiation (Figure 1) [34,69,70]. Cutaneous melanoma can be further subdivided into chronically sun induced melanoma (CSID) and non-chronically sun induced melanoma (non-CSID) (Figure 1). CSID is associated with the head, neck, and the furthest extremities and individuals older than 55 years [2]. Non-CSID is associated with individuals 55 years or younger and is associated with the torso and proximal extremities [2]. The genetic abnormalities commonly associated with these two subtypes of cutaneous melanoma are neurofibromin 1 (NF1), NRAS, BRAF non-V600E mutations, or KIT in CSID, while non-CSID is associated with BRAF V600E mutations, suggesting that the non-CSID might originate from nevi (Figure 1) [2,71,72]. Four major genomic subtypes in cutaneous melanoma are BRAF, RAS, NF1, and triple wild-type (Figure 1) [34]. The BRAF subtype is characterized by the presence of BRAF hot-spot mutations (V600E, V600K, V600R, and K601E) and is mutually exclusive with NRAS hot spot mutations [34]. Additionally, non-hotspot mutations in BRAF occurred together with N/H/K-RAS hotspot mutations and NF1 mutations [34]. Hot-spot mutations in BRAF and N/H/K-RAS show increased MAPK and PI3K/AKT signaling cascade activation [34]. NF1 mutations are detected in 15% of melanoma and the majority of them are from older patients with a higher mutational burden [34]. More than half of the NF1 mutations are associated with a loss of function [34]. Mutations in NF1 also lead to the activation of the MAPK pathway [34]. The fourth subtype is triple wild-type with none of the mutations in the BRAF, NRAS, and NF1 genes. Interestingly, other mutated genes were also found in this subtype: GNAQ, GNA11, KIT, CTNNB1, and EZH2 [34]. This subtype lacks UV signatures, but the potential oncogenic drivers include structural genomic changes in copy number alterations and gene fusions [34]. Three out of the four genomic classifications engage in hyperactivating the MAPK pathway, which is involved in cell proliferation, supporting the notion that the MAPK pathway is a key player in melanoma development and progression (Figure 1) [34,73]. Other mutations that are commonly found in cutaneous melanoma are TERT promoter mutations, suggesting that enhanced telomerase activity leads to proliferative immortality [74,75].
Figure 1.

Melanoma can be segregated into distinct subtypes, based on anatomical location, sun exposure, and genetic profiles, which affect treatment responses to MAPK, PI3K/AKT, receptor tyrosine kinase inhibitors, and immunotherapies. CSID—Chronically Sun Induced melanoma; non-CSID—Non-Chronically Sun Induced melanoma; and triple WT—Triple Wild-Type. Created with BioRender.com.
Non-cutaneous melanoma arises from melanocytes located near the uvea, mucosal tissue, and acral regions. Mucosal melanoma is the rarest subtype of melanoma, comprising 1.3% of all melanoma cases, followed by acral melanoma at 2%–3%, and uveal melanoma at 5.2% (Figure 1) [5,63,76]. We recognize that acral melanoma could be considered as a subtype of cutaneous melanoma, however, the genomic profiles and risk factors/etiologies mirror other non-cutaneous melanoma subtypes, thus, we categorized it as a non-cutaneous melanoma in this review. Uveal melanoma originates from melanocytes located in the three segments of the eye—iris, choroid, and ciliary body, and is associated with older individuals, people who are fair skinned, welders, people who have light colored eyes, and is common amongst men [77,78,79,80]. Genomic abnormalities driving uveal melanoma are characterized by a low mutational burden (approximately 1.1 somatic mutations per Mb) and chromosomal gains/losses [70,81,82]. Common mutations found in uveal melanoma include CYSLTR2, PLCB4, and GNAQ/GNA11; all promote activation of the MAPK and PI3K/AKT signaling cascades (Figure 1) [83,84,85,86,87]. Activating mutations in GNAQ/GNA11 also lead to activation of several transcriptional factors associated with RNA splicing, DNA damage response, and cellular proliferation [88,89].
Acral melanoma usually occurs in older darker skinned individuals and frequently have a poorer prognosis than other subtypes of melanoma, possibly due to a delay in diagnosis [76,90,91]. Acral melanoma etiology, is associated with long-term trauma/physical stress/pressure in acral regions, as well as UV exposure, which might contribute to melanocyte transformation in these regions [69,76,92]. Acral melanoma is defined as a melanoma originating from non-hair bearing skin (glabrous tissue), such as the palms, soles, or under the finger and toe nails [90,93]. This subtype is characterized by gene amplification/losses and show a low mutational burden [64,69,82,90,94,95]. Mutated genes associated with this subtype are: KIT, PDGFRA, BRAF, NRAS, NF1, GNAQ, and the TERT promoter (Figure 1) [82,94,95,96,97]. Amplification or deletion of many genes is a common carcinogenic process involved in acral melanoma. Genetic alterations commonly found within acral melanoma are correlated with the signaling pathways associated with cell cycle progression and cell growth. Interestingly, like cutaneous melanoma, mutations in the TERT promoter and TERT amplification might upregulate telomerase activity in acral melanoma cells, allowing them to become replicative immortal (Figure 1) [69,75,95,96].
The rarest melanoma subtype is mucosal melanoma, which originates from melanocytes located in the mucous membranes of the gastrointestinal, genitourinary, and respiratory tracts [98]. Mucosal melanoma is most commonly found amongst older women and fair skinned individuals [80,98,99]. Similar to other non-cutaneous melanoma subtypes, mucosal melanoma is characterized by a low mutational burden (2.64 mutations per Mb compared to cutaneous melanoma with 49.17 mutations per Mb), high copy number variations, and increased chromosomal structural variations [64,69,70,100]. Several genes were identified that are commonly amplified in this subtype, including KIT, CCND1, and CDK4 (Figure 1) [64,69,97,101]. CDKN2A loss is often associated with mucosal melanoma [64,101]. Genes that are frequently mutated in mucosal melanoma are: BRAF (10%–17%), NRAS (5%–10%), SF3B1, NF1, KIT (activating mutations), GNAQ, GNA11, TPR, TTN, and PTEN (Figure 1) [69,101,102,103,104,105]. Additionally, Furney et al. identified mutations that were not previously identified in mucosal melanoma: MUC2, UBE4A, PTPRT, NRK, NALCN, MUC4, MAP4K4, LRRC7, LRP1B, FURIN, CNBD1, CDH13, CACNA1C, AHNAK, ABH1B, KIR2DL1, MGAM, and SELPLG [103]. Similar to acral melanoma, molecular abnormalities associated with mucosal melanoma converge on phenotypic profiles associated with hyperactivation of the MAPK and PI3K/AKT pathways, resulting in cell cycle progression and anti-apoptosis signals (Figure 1) [105]. Various subtypes of melanoma have different etiologies and genomic profiles but they converge into two major signaling pathways that were shown to play key roles in cell transformation and tumorigenesis—the MAPK and PI3K/AKT pathways [64,82].
Our group uncovered yet another partaker in melanomagenesis, the aberrant expression of a normal neuronal receptor, Metabotropic Glutamate Receptor 1 (mGluR1:protein, GRM1:gene) in melanocytes [106,107,108,109,110]. In the course of constructing transgenic mice with a fragment of genomic DNA (Clone B), which demonstrated adipocyte differentiation in vitro, resulted in concomitant deletion of 70 kb of host DNA and insertion of Clone B [106,107,108]. This disruption of host genome led to ectopic mGluR1 expression in melanocytes, in one out of five founder mice [106,107]. As this founder mouse aged, elevated pigmented lesions were detected throughout the body [106,107,111]. These pigmented lesions were histologically identified as melanocytes with high mitotic index. To confirm that the aberrant mGluR1 expression in melanocytes drives the tumor phenotype, a second transgenic line was made with GRM1 cDNA, under a melanocyte-specific promoter, dopachrome tautomerase (DCT) [108]. This second transgenic line displays similar tumor onset and progression as the first one, confirming that the aberrant expression of mGluR1 in melanocytes was sufficient to promote melanocyte hyperplasia and transformation into malignant melanoma, similar to human melanoma development [108,111,112]. Our findings that mGluR1 plays a role in melanomagenesis in mice, prompted us to examine human melanoma cell lines and biopsies for mGluR1 expression. We found 23 of 25 cell lines and approximately 60% of melanoma biopsies expressed mGluR1 at both mRNA and protein levels, independent of the BRAF/NRAS genotypes [113]. mGluR1 is a G-protein coupled receptor (GPCR) activated by L-glutamate, this receptor is normally expressed in the central nervous system and is involved in memory and learning [73]. Activation of the receptor led to stimulation of downstream effectors and hyperactivation of the MAPK and PI3K pathways [73,114,115]. Taken together, results from these studies point to the importance of delineating the stepwise molecular evolution process in the transformation of normal melanocytes into metastatic melanoma.
2. Conventional Melanoma Therapies
2.1. Surgery
Regardless of the stage at which the melanoma is diagnosed, the primary tumor is excised by local wide excision surgery, if possible, to control local disease and prevent further spread of the cancer [55,116,117,118,119]. For melanoma in situ, surgery is considered to be curative. Before or during primary tumor excision, sentinel lymph node biopsy is performed to see if the cancer cells migrated to the lymph nodes or beyond [55,116,120,121]. If cancer cells are found in the local lymph nodes, the lymph nodes surrounding the tumor area is removed (lymphadenectomy) [55,116]. Some doctors recommend the use of imiquimod cream for early stage melanoma patients who are unable to undergo surgery, in order to control the local melanoma [122,123]. The imiquimod cream promotes local anti-tumor innate and adaptive immune responses, in addition to the induction of tumor apoptosis [124,125]. If surgery can be performed, radiation therapy is sometimes recommended to prevent recurrence, by killing the remaining undetectable melanoma cells (Figure 2) [116].
Figure 2.

Various melanoma therapies used in the clinic. These therapies include the standard treatments along with the precursors for modern-day immunotherapies, followed by “targeted” immunotherapy. Created with BioRender.com.
2.2. Radiation Therapy
Melanoma is historically considered a radiation-resistant tumor type, but under certain circumstances, radiation can be used to treat melanoma [126]. These circumstances include—if a patient cannot undergo surgery, palliative therapy for late stage melanoma, and radiation given at the site of lymphadenectomy [126,127]. Melanoma is radioresistant due to the robust intrinsic DNA damage repair mechanisms [127]. Radiation therapy was shown to not only kill the targeted melanomas but also induce a systemic anti-tumor immune response against metastatic lesions, a phenomenon known as the abscopal effect [128,129]. The abscopal effect was characterized in many cancers; it is the ability of localized radiation to trigger systemic anti-tumor effect (Figure 2) [130].
2.3. Chemotherapy
Up until 2011, there were only two FDA-approved therapies for metastatic melanoma—dacarbazine, a chemotherapeutic agent, and high dose IL-2, an immunotherapeutic agent (Figure 2) [131]. The goal of every cytotoxic chemotherapeutic agent is to inhibit actively dividing cells, by targeting cell division or inducing DNA damage. Chemotherapy is used as a palliative/salvage therapy for late stage melanoma patients with refractory, progressive, or recurrent melanoma [132,133]. To date, no combinatorial chemotherapy regimen successfully improved the response rates, or survival, compared to monotherapy [132]. Chemotherapy induces mitotic catastrophe in cancer cells and if not repaired rapidly, it can induce apoptosis or necrosis, depending on the extent of the damage [134,135,136,137].
2.4. Targeted Therapy
As the cost of DNA sequencing decreases, the feasibility of genomics in cancer therapeutics increases. In the last two decades, the rapid sequencing of patient tumors unveiled a myriad set of molecular targets that were used to successfully treat malignant melanoma and other cancers. Targeted therapy is the concept of developing agents specifically attacking the drivers of carcinogenesis, to improve outcomes and reduce toxicity (Figure 2). Targeted therapy can be given as a first line therapy or as an adjuvant therapy to melanoma patients, based on their melanoma genotype and stage. The components of the MAPK pathway, specifically RAF and MEK, are druggable, and melanoma patients derived therapeutic benefits with low toxicity (Figure 2) [131,138]. In 2011, the FDA approved the first in-class targeted therapy for melanoma, vemurafenib. [131,139]. Vemurafenib was specifically approved for melanomas with BRAF V600E-activating mutation, this agent inhibits the kinase activity that is responsible for hyperactivating the MAPK pathway [139,140,141]. In clinical trials, vemurafenib was shown to improve survival with response rates at 48%, with manageable toxicities, when compared to the standard of care, dacarbazine [139,142]. The results of the vemurafenib trial mirrored the second-in-class BRAF inhibitor, dabrafenib [143,144,145]. Dabrafenib is approved for melanoma tumors with BRAF V600E/K mutations [144]. Furthermore, in clinical trials, trametinib, an MEK inhibitor, showed genotypic differences in response rates. The mutated BRAF melanomas had a 33% response while the wild-type BRAF tumors only had a 10% response [146]. In 2012, the FDA approved trametinib for mutated BRAF melanoma, based on the improved patient survival in the trametinib arm, compared to the standard of care [147]. Evidence suggests that in patients with mutated BRAF, the combination of BRAF and MEK inhibitors yields greater benefit and prolongs the development of resistance [148,149,150,151]. Recently, additional new combinations of BRAF and MEK inhibitors were approved for their usage in malignant melanoma, such as the BRAF inhibitor, encorafenib, and MEK inhibitors, cobimetinib, and binimetinib [152,153,154,155]. Like all therapeutics, patients receiving BRAF and MEK inhibitors eventually develop resistance and their disease progresses, due to acquired and tumor-intrinsic resistance mechanisms [131,138].
Another targeted therapy utilized for the treatment of melanoma are KIT inhibitors (Figure 2). A small subset of melanoma patients have DNA alterations in KIT that manifest as point mutations and amplifications in less than 7% of cutaneous melanoma patients, and in approximately 40% of mucosal and acral melanoma patients [97,138,156]. KIT, a tyrosine kinase, when stimulated by binding of stem cell factor (SCF) or when mutated, activates the MAPK and PI3K/AKT pathway [138]. Melanoma patients benefit from the off-label use of KIT inhibitors, imatinib and nilotinib, only after stratification based on their KIT mutation status [138]. KIT amplifications do not confer sensitivity to KIT inhibitors [138,157,158,159]. Growing evidence suggest that depending on the melanoma subtype, KIT can act as an oncogene or a tumor suppressor [160,161]. It is, therefore, important to elucidate the functions of cancer associated genes under various contexts, including tumor microenvironment, cancer types, anatomical locations, and splicing variants, since the context of a cancer associated gene affects the therapeutic viability of a targeted therapy agent [160,162].
In the era of precision medicine, it is important to characterize melanoma tumors through molecular subtypes and to identify targeted therapies that are best suited for these subtypes—BRAF, RAS, NF1, and triple wild-type (Figure 1) [34]. As we learn more about the genomic profiles of melanomas, these molecular subtypes must be redefined and tested against current and upcoming agents targeting oncogenes and tumor suppressors. In Section 4, we discuss immunotherapeutic agents and propose that a better understanding of each melanoma subtype biology would improve the response and survival rates for patients, given immune checkpoint blockade therapy or adoptive T-cell therapy.
3. Mechanisms of Immune Evasion in Melanoma
Cancer cells are constantly adapting to its host defenses, by manipulating the intrinsic and extrinsic biological pathways. Hanahan and Weinberg classified these manipulations into eight biological components—sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, reprogramming of energy metabolism, and evading immune destruction [163]. Within the last few years, immune evasion by cancer cells has become a popular and valuable therapeutic target to study. In this section, we explore the various strategies that melanoma cells utilize to escape detection by the immune system (Figure 3).
Figure 3.

A summary of the immune dysfunctions that contribute to melanoma development and progression. Created with BioRender.com.
3.1. T-Cell Dysfunction
Cancer cells use the programmed cell death protein 1 (PD-1)/programmed death ligand 1 (PD-L1) immune-checkpoint axis to their advantage. PD-1/PD-L1 axis acts as a negative regulator of immune response and protects the host against autoimmunity [164,165,166,167]. When a pathogen infects the host, T-lymphocytes are recruited to the site of infection and exert their anti-pathogenic response. Cells in the surrounding healthy tissues protect themselves from T-lymphocytes, by expressing PD-L1, which interacts with PD-1 receptor on T-lymphocytes and prevent further T-cell activation [168,169,170,171,172,173]. In cancer, the interactions between PD-1 on cytotoxic T-lymphocytes and PD-L1 on tumor cells or tumor macrophages, NK cells, dendritic cells, and various other immune cells, result in an exhausted T-cell phenotype, rendering the immune system unable to detect and eliminate tumors via epigenetic changes within T-cells (Figure 3) [168,171,174,175,176,177,178,179,180,181,182]. In addition to the PD-1/PD-L1 interactions in the tumor microenvironment, the importance of these interactions were observed in the tumor draining lymph nodes, between the PD-1 expressing T-cells and PD-L1 expressing dendritic cells, which contribute to the anergic/exhausted T-cell phenotype [183]. Program cell death protein 2 (PD-L2) is the second ligand for PD-1 and is expressed on antigen presenting cells and melanoma cells. PD-L2 has overlapping functions with PD-L1, as a negative regulatory of cytotoxic T-cell activity [173,184,185,186]. Melanoma was one of the first solid tumors where high PD-L1 expression was detected with different expression levels, depending on the melanoma subtypes (62% in cutaneous melanoma, 44% in mucosal melanoma, 31% in acral melanoma, and 10% in uveal melanoma) [187,188]. Accumulating evidences proposes that in human melanomas, PD-L2 is more abundant than PD-L1, with a greater affinity towards PD-1, suggesting differential contribution by PD-L1/PD-L2, in regulating immune response [189,190]. Furthermore, glycosylation in PD-L1 was shown to improve the half-life of PD-L1, and strengthen its engagement with PD-1, thereby improving its ability to exhaust T-cells [191,192,193,194]. It was proposed that in the periphery tissues, cytotoxic T-cells undergoing continuous exposure to tumor antigens become activated and produce interferon-γ (IFN-γ) [187,195]. IFN-γ then interacts with the IFN- γ receptor on melanoma cells, which activates the downstream signal cascade of the JAK/STAT/IRF1 axis, stimulates the transcription factors, IRF1 and MYC, to bind to the PD-L1 promoter, and PD-L2 requires the participation of transcription factors STAT3 and IRF1 [196,197,198]. In melanomas, numerous transcription factors—HIF-1, AP-1, and NF-қβ—were implicated in regulating PD-L1 expression, albeit by varying mechanisms, as a result of differing mutational landscape [194,199,200]. In T-cells, when the T-cell receptor (TCR) is activated as a result of TCR engagement with the antigen/MHC complex, it activates the MAPK and PI3K/AKT pathway and result in T-cell activation phenotypes—transcriptional activation, cytokine production, T-cell survival, and proliferation [201,202,203,204,205]. However, in the context of cancer, when PD-L1/PD-L2 interact with PD-1 on cytotoxic T-cells, it leads to SHP1/2 recruitment to the TCR and modulates numerous phosphorylation activities, resulting in defective cytolytic T-cell function and metabolism [169,171,195,204,206,207,208,209,210,211,212,213,214,215]. In addition to PD-1, there are other immune suppressive checkpoint molecules that dampen cytotoxic T- cell activity against cancer cells—NRP-1, cytotoxic T-lymphocyte associated protein -4 (CTLA-4), TIM-3, LAG 3, and VISTA [216,217,218,219,220,221,222,223,224]. Disruption between PD-1 and one or more of these molecules result in defective development of cytotoxic memory T-cells and exhaustion of CD8+ T cells. Results from several studies propose that for early stage melanoma, anti-PD1/anti-PD-L1 therapy should not be given, because it would impede the development of long-term immunity. The importance of sequential treatment of anti-PD-1/anti-PD-L1 therapy with cancer vaccines was also suggested as another approach to release the brakes of the exhausted immune cells.
CTLA-4 is the second most well-known immune suppressive checkpoint regulator (Figure 2 and Figure 3). Its role as a negative regulator of the immune system is supported by the development of severe autoimmune diseases in CTLA-4 knockout mice, stemmed from unrestrained T-cell activity [217,218]. CTLA-4 expression on T-cells, exerts its immune suppressive activities by inhibiting T-cell activation by outcompeting CD28 for the ligands, CD80/CD86—a process defined as T cell anergy [225,226,227,228,229,230].
Interestingly, the role of PD-1, PD-L1, and CTLA-4 might extend beyond their canonical functions as a negative regulator of the immune system. In melanomas, it was shown that PD-1, PD-L1, and CTLA-4 signaling might be responsible for tumor intrinsic cell proliferation, survival, growth, and metastatic signals, in addition to establishing an immunosuppressive shield around the tumor cell [231,232,233,234,235,236]. In conclusion, the notion of blocking the interactions between the immune checkpoint receptor (PD-1 and CTLA-4) and its ligand (PD-L1/PD-L2 and CD80/CD86, respectively) to reinvigorate the immune system to attack cancer cells, led to the development of anit-CTLA-4, anti-PD-1, and anti-PD-L1 checkpoint inhibitors, which is discussed further in Section 4 (Figure 2).
3.2. Melanoma Microenvironment Contains Many Immune Suppressive Immune Cells: Regulatory T-cells, Myeloid Derived Suppressor Cells, and Tumor Associated Macrophages
3.2.1. Role of Regulatory T-cells in Melanoma Immune Evasion
CD4+ regulatory T-cells (Tregs) actively participate in reigning in an overactive immune response from damaging the host [237]. These same protective features are used by cancer cells for immune evasion. In melanoma, Tregs increase in the peripheral blood, lymph nodes, and tumor microenvironment, which corresponds with reduced cytolytic function of anti-tumor immune cells (Figure 3) [238,239,240,241,242,243,244,245,246,247,248]. Melanoma’s recruit/induce Tregs by secreting H-ferratin and chemoattractant cytokines/chemokines, which modulates the Treg function within the tumor microenvironment [246,249,250,251,252,253,254]. Tregs have four distinct mechanisms through which they suppress the immune system and was extensively reviewed by Vuganali et al. and Shevach [237,255]. In brief, Tregs induce immune suppression by: (1) releasing immune suppressive cytokines, IL-10, IL-35, and TGF-β, which inhibit the cytotoxic activities of immune cells, (2) induce cytolysis of immune cells, (3) target dendritic cells (antigen presenting cells), and (4) metabolically disrupt the immune cell function [237,255]. To note, in a murine melanoma model, Tregs did not affect the dendritic cell function, therefore, additional studies are needed to further delineate the mechanism(s) of Treg suppression in melanoma [256].
3.2.2. Role of Myeloid Derived Suppressor Cells in Melanoma Immune Evasion
Myeloid cells are a major component of the innate immune system. These cells are responsible for protecting the host against foreign invaders, by phagocytosing pathogens and eliciting inflammatory responses to recruit other immune cells [257]. Cancer cells transform the myeloid cells found in the bone marrow, into myeloid derived suppressor cells (MDSC) [258]. MDSCs are critical in cancer progression, as they support tumor cell dissemination, and inhibit T-cell function [258,259,260,261]. In melanoma, an increase in MDSCs in the peripheral blood and tumor microenvironment is associated with disease progression, reduced T-cell function, and prognostic value (Figure 3) [260,262,263,264,265,266]. MDSCs reduce cytotoxic T-cell function in the tumor microenvironment by disrupting key metabolic pathways required for proper T cell function, which eventually result in T-cell apoptosis [267,268,269,270,271,272]. Depletion of MDSCs might improve anti-melanoma immunity, since MDSCs were shown to negative correlate with survival [263].
3.2.3. Tumor Associated Macrophages in Melanoma Immune Evasion
MDSCs can differentiate into tumor associated macrophages (TAMs) and oscillate between an M1- or M2-like macrophage phenotype [273,274,275,276,277] in the tumor microenvironment [275,278]. Hypoxic regions within the tumor, push TAMs towards an M2-like phenotype, while under normoxic conditions they are pushed towards an M1-like phenotype [275,276,279]. Increased infiltration of TAMs is found within the melanoma microenvironment, as the disease progresses, specifically in M2-like TAMs, the ratio of M1/M2 TAMs are a valuable prognostic marker (Figure 3) [273,280,281,282,283,284,285]. M1 TAMs are associated with anti-tumor effects, while M2 TAMs support tumor progression. In melanomas, the enrichment of M1 gene signatures showed better prognosis than patients with enriched M2 gene signatures [286,287].
Blockade of M-CSF receptors on MDSCs preferentially direct TAMs into an M1 phenotype and GM-CSF signaling is responsible for reinforcing this phenotype [288]. Similar to the blockade of the M-CSF receptor on MDSCs, Georgoudaki et al. demonstrated that preferentially blocking the MARCO receptor by an antibody, could promote TAMs to differentiate into an M1 phenotype [289]. The MARCO receptor, a pattern recognition scavenger receptor, was associated with a gene expression profile resembling an M2-like TAM phenotype [289]. By developing an antibody against MARCO, this group was able to drive M2 TAMs into an M1 phenotype in the experimental models of melanoma and breast carcinoma [289]. Results from several studies suggest that M-CSF and MARCO receptors could regulate the PI3K/AKT/mTOR axis and polarization of M1 or M2 TAMs, however, further studies are necessary to elucidate the precise mechanisms [288,289,290,291,292,293,294].
Helper T cells (Th) regulate adaptive immune response by activating cytotoxic T-cells and phagocytic/digestive properties of the macrophages [295]. Two subclasses of the helper T cells, Th1 and Th2 are responsible for M1 and M2 polarization, respectively [286,296]. In healthy individuals and patients with surgically resected melanomas, there is a Th1 bias [297]. In melanoma patients, the Th2 subclass is predominant and leads to systemic chronic inflammation and support melanoma progression, attributed to Th2′s ability to polarize TAMs into an M2 phenotype [297,298,299]. M1 TAMs exert their anti-tumor properties by releasing proinflammatory cytokines, ROS, NO, and act as efficient antigen presenting cells, to support adaptive anti-tumor immune responses [300,301]. M2 macrophages support melanoma growth by strengthening tumor angiogenesis, inducing the Treg function to reduce cytolytic T-cell activities, and express soluble factors to dampen anti-tumor immune response [273,302,303]. As melanoma progresses into advanced stages, M1 TAMs shift to M2 TAM phenotype, to support tumor growth and tumor immune evasion (Figure 3). Developing therapeutics that can mediate the switch of M2 TAMs back to M1 TAMs might be a valuable tool to add to the arsenal of immunotherapeutic agents to improve melanoma treatment outcomes [286,289].
3.3. Defective Immune Recognition of Melanomas by the Immune System
Melanomas establish a process of immune editing by selecting subclones based on their capability of evading immune detection—elimination, equilibrium, and escape phases [304,305]. During melanoma progression, there is a progressive loss of antigen presentation capacity to cytotoxic T-cells by the dendritic cells, thus, reducing their immunogenicity (Figure 3) [304]. During the first phase, surveying professional antigen presenting cells, dendritic cells, detect immunogenic melanoma clones and capture these melanoma antigens. These cells then process the melanoma antigens and place them onto their Major Histocompatibility Complex II (MHC II), so they can present these peptides to naïve T-cells in the lymph nodes, resulting in the activation/expansion of melanoma-specific cytotoxic CD8+ T cells [304]. The equilibrium phase is when the immune system eliminates highly immunogenic melanoma clones, however, there are clones that escape anti-tumor immune responses [304]. Establishment of low immunogenic melanoma clones enables melanomas to rapidly proliferate and disseminate, a phase known as the escape phase [304].
During melanoma progression, there are various soluble factors released by tumor and immunosuppressive immune cells, which disrupt the proper function of dendritic cells in priming naïve T-cells into effector CD8+ T-cells in the lymph nodes (Figure 3) [256,304,306,307]. The immunosuppressive cytokine, IL-10, released by the regulatory T-cells and tumor cells, can lead to a defective antigen presentation capacity of dendritic cells (or macrophages), corresponding to reduced T-cell activation [308,309,310]. Defective antigen presentation by dendritic cells occurs as a result of the downregulation of cell surface expression of MHC II and the co-stimulatory molecules, CD80/CD86 on dendritic cells, which are essential molecules required for T cell activation [308,309]. Evidence suggest that expression of immune checkpoint molecules—CTLA-4, PD-1, PD-L1, and PD-L2 on dendritic cells, disrupts the innate immune functions and affects T-cell activation [305,311,312,313,314,315,316]. During genetic/epigenetic changes or immune editing, melanoma subclones can successfully downregulate key components of their MHC I antigen presentation pathways, and effectively escape immune surveillance (Figure 3) [317,318,319,320,321,322,323]. In concordance with this, it was observed that non-mutated melanoma associated antigens, MART-1/Melan-A, gp100, and tyrosinase, are heterogeneously expressed across melanoma cells [324,325,326,327]. During the melanoma immune editing phase, once a tumor is recognized by the immune system, it is eliminated and the subclones that have successfully downregulated MHC I or expression of another melanoma antigen, can lead to an immune refractory tumor (Figure 3). Interestingly, it was shown that melanoma cells express MHC II on the cell surface, which allows them to attract tumor-specific CD4+ T-cells [328]. These CD4+ T cells are shown to suppress the anti-tumor cytotoxic T-cell activity, by counteracting IFN-γ mediated immune response [328].
3.4. Spontaneous Melanoma-Prone Mouse Model Mimics Immune Dysfunction in Humans
Our lab developed various spontaneous melanoma-prone mouse models, driven by aberrant mGluR1 expression in melanocytes, which mimic melanoma development and progression in humans [107,108,329,330,331]. Two independent groups showed that these melanoma-prone transgenic mouse models accurately depict the immunological profiles of human melanoma patients [248,262,332]. Stoitzeners et al. demonstrated that an increase in immunosuppressive MDSCs within the tumor microenvironment is associated with the presence of anergic gp100-melanoma specific cytotoxic CD8+ T cells, consistent with melanoma patient data [262]. Further studies by this group showed an inverse relationship between the levels of DCs and the tumor burden in these mice [332]. They went on to show that rescuing DC populations within these mice by Flt3L, an endogenous small molecule that functions as a cytokine and growth factor, was able to restore cytotoxic cytokine production by T-cells [332]. Another study performed by Schrama’s group showed that as melanoma progressed in these melanoma-prone mice, there was an increase in Tregs, a decrease in CD8+ T cells within tumor tissues, and an increase in immunosuppressive cytokines, IL-10 and TGF-β [248]. Furthermore, as tumor burden increases in these mice, there is a decrease in CD8+ T-cell activation markers and lymphocyte proliferative capacity [248]. Taken together, the immune profiling data of these melanoma-prone mice, point to the notion that these mice accurately mimic the dysfunctional immune system in melanoma patients and is a good model to predict the treatment response to immunotherapy.
3.5. Ultraviolet (UV) Radiation-Induced Immune Suppression in Melanomagenesis
UV radiation is considered to be one of the biggest risk factors in the development of cutaneous melanoma. UV-induced accumulation of stochastic mutations in melanocytes leads to cell transformation and tumor formation. UV radiation has the capacity to induce local and systemic antigen-specific immune responses, thereby, enabling transformed melanocytes to escape immune surveillance (Figure 3) [333]. Kripke and Fisher demonstrated immune tolerance of highly antigenic UV-induced murine tumors, when allografted into syngeneic mice plus UV treatment. In contrast, in the absence of UV, the inoculated tumors were rejected [334,335,336,337]. Similar observations were made in immune-suppressed mice, suggesting that UV exposure mediates immune suppression [334,335,336,337]. Several mechanisms mediating UV-induced immune suppression were proposed, including defective antigen presentation, release of immunosuppressive cytokines, and apoptosis of immune cells [338]. UV exposure of the skin is associated with a reduction of the Langerhan cells, a type of dermal dendritic cell, at the site of exposure [333,336]. UV-exposed Langerhan cells migrate to the lymph nodes, where they are unable to activate Th1 cells, which is important in mounting an effective immune response. Instead, the Th2 cells are stimulated, which instill immune suppression by activating the regulatory T cells [338,339,340]. In addition, UV-exposed Langerhan cells are defective in antigen presentation in the lymph nodes, and undergo apoptosis when exposed to higher doses of UV, suggesting that UV exposure has the capacity to reduce the tumor antigen presentation to the immune system [333]. In addition to the modulation of immune cells into an immunosuppressive phenotype, it also showed an increase in immunosuppressive cytokines, IL-10, IL-4, and TNF-α, which were present both locally and systemically [338,340,341,342]. Furthermore, UV exposure reduced the cytokine IL-12, leading to an imbalance of Th1 and Th2 cells, with an increase in the latter [343,344]. These results propose that UV-induced activation of Th2 cells might contribute to the increased M2-like TAMs in melanoma patients. Taken together, these results suggest that when a patient receives immunotherapeutic agents, an agent that rejuvenates the immune system, the amount of sun exposure should be limited, since high UV exposure during the treatment regimen might render immunotherapeutic agent less effective.
3.6. Exosomes
Exosomes are nano-sized vesicles ranging from 30–120 nm, with cargo representing the cell membrane and cytoplasm (DNA, RNA, proteins, and lipids) components of the cell they originate from [5]. These are released by both normal and cancer cells, but cancer cells release greater amounts of exosomes, compared to their normal counterparts [5]. The cargo found within tumor exosomes are responsible for priming the pre-metastatic niche and suppressing anti-tumor immune response, thereby, enabling cancer cell metastasis [5]. In this subsection, we focus on the several mechanisms through which melanoma exosomes suppress anti-tumor immune responses, both locally and systemically (Figure 3).
Melanoma-derived exosomes were shown to migrate to the lymph nodes and induce tumor tolerance to prepare the lymph nodes for arrival of the melanoma cells [345,346]. Within the lymph nodes, exosomes can modulate antigen presentation, inhibit antigen-specific immune response, and upregulate immunosuppressive cytokines [345]. Melanoma exosomes were shown to transfer melanoma-derived MHC I into antigen presenting cells, downregulate the co-stimulatory molecules CD80/CD86, and upregulate immunosuppressive cytokines IL-6 and TGF-β, resulting in the defective function of antigen presenting cells, correlating with reduced T-cell proliferation [347]. Furthermore, tumor-derived exosomes, through cell surface interactions with T-cells, inhibit cell activation and induce apoptosis [348,349,350]. In addition to modulating the function of T-cells, tumor-derived exosomes reduce the cytolytic function of natural killer cells (NK) [350,351]. Tumor-derived exosomes carrying TGF-β and prostaglandin E2 induce MDSC formation, resulting in MDSC accumulation within the tumor microenvironment, and a suppressed immune response [352]. MiR-125b-5p detected in the melanoma exosome cargo can induce TAM formation, to support melanoma growth [353]. Furthermore, melanoma exosomes can induce a mixed population of M1- and M2-like TAMs, and the tumor microenvironment is responsible for reinforcing the M2-like TAM phenotype [274,276,279,354]. Melanoma and prostate cancer derived exosomes were shown to express the immune checkpoint molecule, PD-L1 on their surface, resulting in suppressed immune response, both locally and systemically [355,356].
Our lab demonstrated that inhibition of mGluR1 expression or function by genetic or pharmacological inhibitors in melanoma cells, did not modulate the number of exosomes released, but rather reduced the functions of the exosomes on the recipient cells in cell migration, invasion, and anchorage-independent growth, perhaps through cargo sorting into exosomes [357].
4. Understanding Melanoma Subtype Etiology and Biology to Better Treat Patients with Immunotherapies: Identification of Patient Biomarkers, Characteristics, and Combination Therapies to Improve Response Rates and Survival
Immunotherapy, a therapy that reinvigorates a patient’s own immune system to exert an anti-tumor immune response, has been around for over 130 years [358]. Early immunotherapies used biological molecules such as Coley’s toxin, IFN, and a high dose of IL-2, as well as the cancer vaccine Bacillus–Calmette–Guerin (BCG), to treat melanoma patients (Figure 2) [358,359,360]. In general, these agents were marked by low response rates but those who responded, showed a durable response [131,358,359,360,361]. Newer immunotherapies such as immune checkpoint blockade therapy and adoptive cell therapy showed remarkable anti-tumor response, corresponding to long-term durable survival, but not all patients responded to these therapies [131,172,358,362,363,364]. The inability for all patients to benefit from immunotherapy agents, suggests that better patient stratification based on patient characteristics, molecular biomarkers, and melanoma subtype, is required, to improve response rates. In light of these new immunotherapy modalities, immune checkpoint blockade therapy and adoptive cell therapy, the Response Evaluation Criteria in Solid Tumors (RECIST) guidelines that were initially developed to unbiasedly determine tumor response to cytotoxic agents, were modified to reflect the delay in adaptive anti-tumor immune response [365,366,367]. These new guidelines are known as immune-related RECIST (iRECIST) that help clinicians across multiple centers to consistently design, and manage data related to immunotherapy modalities, to ensure accurate data interpretation and analysis of the efficacy across studies [366,367]. In this section, we describe the various immunotherapeutic agents along with their known mechanism of action, molecular markers, and patient demographics, which is required to better identify patients who respond to adoptive cell therapy and immune checkpoint blockade therapy (Figure 4). Furthermore, we discuss that stratification based on melanoma subtype might improve response rates to immune checkpoint blockade therapy (anti-PD-1/anti-PD-L1/anti-CTLA-4) and adoptive cell therapy.
Figure 4.

Biomarkers for immune checkpoint blockade therapy (A) and adoptive T-cell therapy (B) that can improve stratification of melanoma patients into responders and non-responders. Created with BioRender.com.
4.1. Immune Checkpoint Blockade Therapy
Antibodies that block CTLA-4, PD-1, and PD-L1 are widely used for the treatment of various cancers, including melanoma (Figure 2). These molecules are known as immune checkpoints that normally reign in an overactive immune response during infections, and reduce the likelihood of developing an autoimmune disorder [166,167,218,368]. Cancer being a chronic condition enables these immune checkpoints to be upregulated on immune cells, hence, protecting cancer cells from immune detection. Ipilimumab (yervoy), the monoclonal anti-CTLA-4 antibody, was first approved for unresectable and metastatic melanoma in 2011, soon followed by its approval as an adjuvant therapy [369]. Treatment with ipilimumab helps overcome T-cell anergy within lymph nodes and allows proper anti-tumor T-cell cytotoxicity [172]. In brief, within the lymph nodes, when CD8+ T-cells interact with dendritic cells, two signals are required for proper T-cell activation—TCR interaction with the MHC/peptide complex found on dendritic cells and the secondary signal of CD28 on T-cells, binding with CD80/CD86 on dendritic cells [172]. However, CTLA-4 outcompetes CD28 for CD80/CD86 binding, thereby, inhibiting the downstream TCR signaling and hampering anti-tumor CD8+ T cell function [226,228,370,371]. Ipilimumab binds to CTLA-4 on T-cells, which inhibits its ability to bind to CD80/CD86, allowing for the expansion of a repertoire of antigen-specific anti-tumor cytotoxic CD8+ T-cells and CD4+ T-cells, which corresponds to an improved anti-tumor immune response [372,373,374,375,376,377,378]. Interestingly, it was shown that the Fc portion of the ipilimumab antibody could deplete Tregs in the tumor microenvironment, by activating the Fcγ expressing macrophages, supporting an anti-tumor immune response [379,380,381,382]. Unfortunately, it was documented that ipilimumab treatment leads to higher immune-related adverse effects on organs, due to the promiscuous expansion of both normal and tumor cytotoxic T-cells [383,384,385]. The response rates for metastatic melanoma patients receiving ipilimumab is approximately 11%, albeit low frequency, but patients who respond display durable survival [386,387].
Monoclonal antibodies against PD-1 and PD-L1, pembrolizumab (keytruda)/nivolumab (opdivo) and atezolizumab (tecentriq), respectively, were approved for the treatment of unresectable/metastatic melanoma. Additionally, the anti-PD-1 antibodies were also approved for usage in the adjuvant setting [369,388]. The triple combination of atezolizumab, vemurafenib (BRAF V600E inhibitor), and cobimetnib (MEK inhibitor) was approved by the FDA as a first line therapy for BRAF V600 unresectable or metastatic melanoma [388]. It was demonstrated that the PD-1 and PD-L1 checkpoint axis acts as a negative regulator of immune response [389]. Cancer cells use this axis to their advantage to escape immune system surveillance [389,390]. Cancer cells including melanoma cells, upregulate PD-L1 expression on the cell surface. T-lymphocytes with surface expression of PD-1, interacts with PD-L1 on the tumor, leading to T-cell exhaustion, thereby, causing dysfunction of the immune system in detecting and eliminating the tumor cells [390,391]. Monoclonal antibody binding of the PD-1 or PD-L1 protein, disrupts the ligand–receptor interactions, subsequently releasing the “brakes” of quiescent cytotoxic T-cells and NK cells, to exert an anti-tumor immune response [177,392,393,394,395]. PD-1 antibodies show response rates in the range of 30%-40% which are higher than ipilimumab and showed improved overall survival [172,358,396]. Interestingly, additional studies revealed that PD-L1 blocking antibodies have a higher potency in blocking the PD-1/PD-L1 signaling than PD-1 antibodies, and pembrolizumab is more potent than nivolumab [397].
Immunotherapies, such as anti-CTLA-4 or anti-PD-1/anti-PD-L1 antibodies, have better response rates and improvement in patient survival with advanced melanoma, but clinical trial data suggest that not all melanoma patients are responsive to single agent monoclonal antibodies for CTLA-4/PD-1/PD-L1 [386,398]. Utilizing various permutations of drugs, including other immunotherapies in combination with immune checkpoint blockade therapy, might yield improvement in patient responsiveness. Furthermore, detailed characterization of melanoma patients who are sensitive to immune checkpoint therapy would improve response rates and survival outcomes for future patients, given anti-CTLA-4/anti-PD-1/anti-PD-L1 antibodies.
Patient Characteristics that can Improve Response Rates to Immune Checkpoint Blockade Therapy
In this subsection, we discuss patient characteristics and molecular markers that render a patient’s melanoma susceptible to immune checkpoint blockade therapy (Figure 4A). We first discuss the value of individual biomarkers in predicting treatment response, followed by the validity of testing various permutations of these markers to improve the predictive power for treatment response. The major goal of biomarker studies is to identify patient characteristics at baseline, to predict treatment response and survival outcomes [399]. Accurately identifying cancer patients who respond to therapy would improve their quality of life, reduce financial burden on non-responding patients, and most importantly, precisely identify therapies that would benefit a cancer patient who does not have the luxury of time. Meta-analysis of immune checkpoint blockade trials showed that male melanoma patients derive greater benefit than female patients from immune checkpoint blockade therapies [400]. These differences are associated with sex differences in molecular markers, such as tumor mutational burden, neoantigen load, PD-L1 expression, and density of both anti- and pro- tumor immune cells (Figure 4A) [400]. In line with this, older cancer patients and males have a better antigen presentation on their tumor cells than female and younger patients, resulting in better immune system detection and response to immune checkpoint blockade therapies [401].
Biomarkers that are associated with treatment response to ipilimumab are—densities of immune cells, molecular markers, and serum cytokine levels (Figure 4A) [399,402]. Ipilimumab treatment was shown to increase ICOS+ CD4+ T cells and enhance the ratio of effector T-cell/regulatory T-cells, correlating with clinical benefit in patients [402,403,404]. Low serum concentrations of IL-15 and TIM-3 expression on circulating T and NK cells, along with increased circulating mature NK cells, correlated with improved survival in melanoma patients treated with ipilimumab [402,405]. High baseline expression of indoleamine 2,3-dioxygenase (IDO) and FOXP3 expression in melanoma biopsies was correlated with treatment response and increase in tumor infiltrating lymphocytes [402,406]. Interestingly, CTLA-4 is not only expressed on the cell surface of T-cells but also found in the serum of metastatic melanoma patients [407]. High concentrations of soluble CTLA-4 at baseline was associated with improved overall survival but also an increased risk of immune-related adverse events [407]. Baseline levels of both anti-tumor and pro-tumor immune cell infiltrates were responsible for predicting treatment response to ipilimumab [402]. Immune-sensitive melanomas that respond to ipilimumab were marked by high baseline expression of immune associated genes and linked with pathways involved in “inflammatory response, cytotoxic T-cell mediated apoptosis of target cells, immune cell activation and migration and antigen presentation pathways” [408].
Molecular markers that are associated with monoclonal anti-PD-1 and anti-PD-L1 antibody treatment response are—PD-L1 expression, tumor mutational burden, tumor infiltrating lymphocytes, and soluble molecular factors (Figure 4A) [399,402]. Stratifying melanoma patients based on PD-L1 IHC expression on tumor cells and immune cells, is crucial in identifying responders to anti-PD-1/anti-PD-L1 therapies. Anti-PD-1-treated patients who have PD-L1 positive melanomas, have response rates of 50%–60%, and show better survival than PD-L1 negative melanomas, which have response rates of 10%–20% [179,194,409,410,411]. A similar trend was observed for atezolizumab, anti-PD-L1-antibody-treated patients [179]. However, in some patients with PD-L1-positive tumors, they do not respond to anti-PD-1 therapy, while some patients with PD-L1-negative tumors respond to therapy, suggesting that other factors also mediate the treatment responses [194,412]. PD-L1 is considered a dynamic biomarker and might not be a valuable tool to use by itself, since its expression changes can be modulated, depending on treatment, inflammation, intratumorally/intertumoral heterogeneity, temporal heterogeneity, and expression differences between the primary and metastatic melanomas [194,413]. Genomic instability is associated with treatment responses to anti-PD-1/anti-PD-L1 antibodies in melanoma (and other cancers), specifically by examining the tumor mutational burden. This concept revolves around the fact that these tumors have a higher mutation rate that increases their likelihood of presenting neoepitopes for surveying immune cells, to recognize and mount an anti-tumor immune response [414,415]. In melanoma, patients with a high tumor mutational burden responded to anti-PD-1/anti-PD-L1 therapies with improved survival, however, some patients with a high tumor mutational burden did not respond [416,417]. Panda et al. proposed that if there are specific mutations that make a tumor more responsive to immune checkpoint therapy, “immune checkpoint activating mutation threshold (iCAM)” [418]. iCAM positive tumors are defined as tumors with gene expression profiles associated with increased immune cell infiltrations, high CD8+ T-cells, and upregulation of immune checkpoint pathway genes. Immune cell infiltration was confirmed by histological analysis, and the iCAM-positive tumors were correlated with improved responses to immune checkpoint therapy (anti-PD-1/anti-CTLA-4) [418]. As eluded to, increased T-cell infiltration defined as a “hot tumor”, renders a melanoma susceptible to anti-PD-1/anti-PD-L1 therapy, as compared to a “cold tumor”, which has a low T-cell infiltration [419,420]. Furthermore, high eosinophil and lymphocyte counts with low lactate dehydrogenase (LDH) are associated with improved response rates and survival outcomes [402,421]. Circulating soluble PD-L1 and exosomal PD-L1 were found to be higher in melanoma patients, associated with an immune suppressed tumor microenvironment, compared to healthy donors [355,422]. Preclinical and clinical evidence propose that an imbalance of the gut microbiome with respect to presence or absence of specific microbes can modulate treatment response to immune checkpoint inhibitors [423,424,425].
Melanoma is a genetically heterogenous tumor that thrives in an environment composed of numerous cell types with various metabolic profiles, including immune cells supporting its development, growth, and progression. All these factors that participate in melanomagenesis are heterogenous, within and between patients. This might confound the use of individual biomarkers to predict treatment response. The study of biomarkers might unravel the mechanism of action or biological pathway(s) targeted by the drug(s). This helps identify why certain therapies benefit certain patients but not others. Cristescu et al. showed that a combined stratification of high tumor mutational burden plus high T-cell inflamed gene expression profiles in melanomas and head and neck squamous cell carcinoma patients, improved the identification of responders and non-responders to anti-PD-1 therapy [426,427]. Furthermore, patients within this classification also display improved progression-free survival, compared to those patients who deviated from this classification [426,427]. Similar results were observed when the patients were stratified based on the PD-L1 positivity of their tumors, high tumor mutational burden or high T-cell inflamed gene expression profiles showed improved responses and survival [426,427]. The ratio of reinvigorated exhausted T-cells in the blood to tumor burden, was proposed as an important predictor for treatment response and survival [428]. In addition to the various cellular and molecular markers used to identify responders and non-responders to immune checkpoint blockade therapy, a crucial characteristic that is not commonly addressed is melanoma subtype.
The four melanoma subtypes show distinct tumor mutational burdens, mutational profiles, PD-L1 expression, and microenvironments that impact the differential responses to immune checkpoint blockade therapies [81,188,429,430]. These subtypes can be divided into cutaneous melanoma; CSID and non-CSID with a higher mutational burden, as compared to non-cutaneous melanoma that include acral, mucosal, and uveal melanoma. In general, cutaneous melanoma shows better response rates to immune checkpoint blockade therapies than non-cutaneous melanoma (Figure 4 and Figure 5) [81,188,429,430]. Multiple factors contribute to the variance in responses to immune checkpoint blockade therapies, profiling neoepitopes and anergic/exhausted T-cells across various melanoma subtypes provide additional clues to this very complicated puzzle (Figure 5). The distinct melanoma subtype profiles suggest that cutaneous melanoma might benefit from immune checkpoint blockade therapy, while non-cutaneous melanoma benefit from adoptive cell therapy (Figure 2, Figure 4, Figure 5 and Figure 6).
Figure 5.
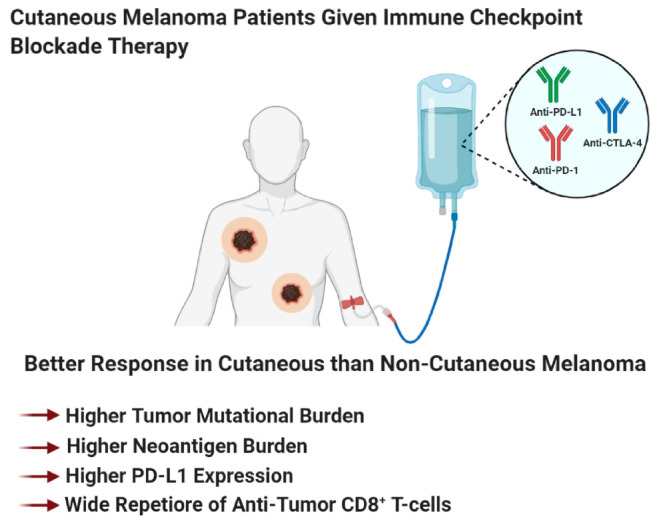
Cutaneous melanoma patients benefit from immune checkpoint blockade therapy better than non-cutaneous melanoma patients. Created with BioRender.com.
Figure 6.
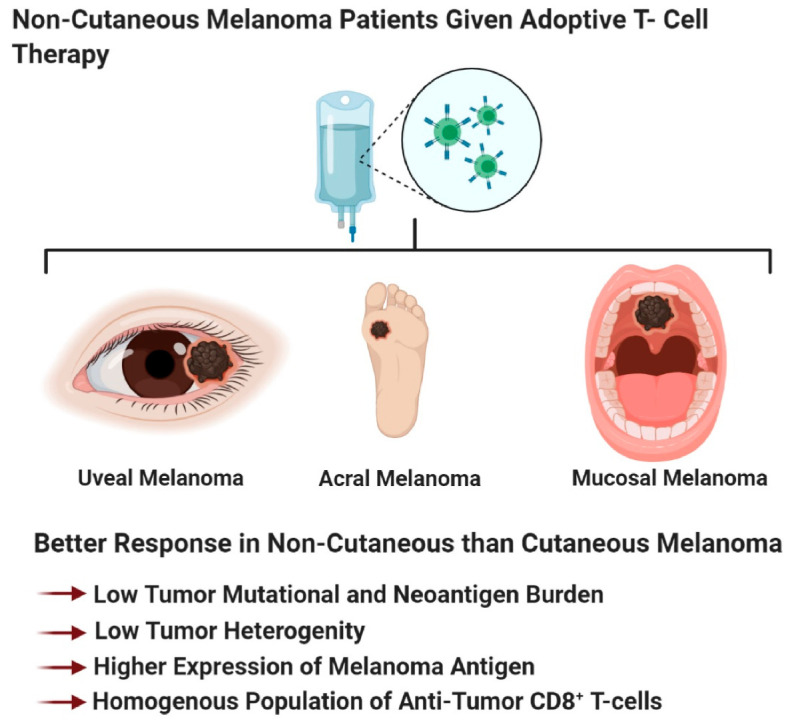
Non-cutaneous melanoma patients benefit from adoptive T-cell therapy better than cutaneous melanoma patients. Created with BioRender.com.
4.2. Adoptive T Cell Therapy
The three major types of adoptive T-cell therapies are—tumor infiltrating lymphocyte therapy, engineered TCR therapy, and chimeric antigen receptor (CAR) therapy (Figure 2) [431]. These therapies subtly differ from each other, but their main goal is to enhance the cytotoxicity of cytotoxic T-cells, and other immune cells ex vivo, followed by infusion back into patients, to induce tumor regression [431,432]. In this sub-section, we discuss the different types of adoptive cell therapies for the treatment of melanoma, followed by identification of the characteristics of the patient and markers that renders a melanoma sensitive to this therapy.
4.2.1. Tumor Infiltrating Lymphocyte Therapy
Tumor infiltrating lymphocyte therapy is the foundation for engineered TCR therapy and CAR therapy (Figure 2) [364]. This therapy requires the isolation of tumor infiltrating lymphocytes (TIL) from excised tumors [358,431,433]. This assumes that these lymphocytes can induce an anti-tumor immune response [358,431,433]. Isolated TILs are expanded by IL-2 treatment and then reinfused back into lympho-depleted patients, with additional treatment of IL-2 [431,434,435]. IL-2 treatment, in conjunction with TIL therapy, supports persistence of reintroduced TILs in patients, in addition to supporting cytotoxic abilities of anti-tumor CD8 + T-cells and NK cells [436]. TIL therapy in metastatic melanoma patients showed remarkable response rates ≥50%, with 22% among these patients showing complete remission [434,435]. The downsides of TIL therapy is that the tumors must be resectable, the resected tumor needs to have TILs that can be isolated and expanded, plus the overall health of the patient [358].
4.2.2. Engineered TCR Therapy
To overcome the barriers of TIL isolation from tumors as well as availability of tumor-specific T-cells in patients, the engineered TCR therapy was developed (Figure 2) [358]. This therapy utilizes T-cells isolated from a patient, and genetically engineered these T-cells to express tumor-antigen-specific TCR [431]. Once modified, these cells can be expanded and infused back into the patients [432]. A major pitfall for this therapy is the reliance on the surface expression of tumor antigens on MHC I [431]. Frequently, cell surface presentation of antigens in melanoma is down-regulated and reduces its ability to induce the cytotoxic T-cell response [317,318,323]. In line with this, the response rates for engineered TCR therapy is only 13% (2/15 patients) [437]. To overcome this, CAR therapy was developed to bypass barriers imposed by tumor-surface expression of MHC I and enable T-cells to directly bind to tumor surface antigens (Figure 2) [431].
4.2.3. Chimeric Antigen Receptor Therapy
The chimeric antigen receptor introduced into T-cells is engineered by the fusion of an antigen-specific recognition of monoclonal antibody domain, linked through a transmembrane domain to the components of the intracellular TCR, and the co-stimulatory molecules required for T-cell activation (Figure 2) [431,438]. This fusion receptor enables T-cells to recognize tumor antigens on the tumor surface, independent of MHC binding, enabling CAR T cells to eliminate cancer cells, regardless of MHC status [431,438]. CAR T cell therapy was shown to be remarkably successful in treating patients with B-cell malignancies, however, for solid tumors like melanoma, it was met with low response rates (19% for CARs targeting gp100 and 30% for CARs targeting DMF5), and the toxicities associated with the destruction of normal melanocytes [358,431,439]. Disadvantages in CAR T cell therapy is the time it takes to develop CAR T cells from a patient’s T-cell, cost, and toxicities [431,440].
4.2.4. Patient Characteristics that can Improve Response Rates to Adoptive T cell Therapy
As we improve our understanding of adoptive T-cell therapy, the identification of prognostic biomarkers that can predict responders, non-responders, and patients who would develop resistance to therapy, has improved (Figure 4B). Patients with metastatic melanoma and have failed prior therapies, demonstrated improved survival to adoptive T cell therapy if they have a high tumor mutational burden and neoantigen load (Figure 4B) [441]. We propose that non-cutaneous melanoma, with a low tumor mutational burden, low neo-antigen load, or those that express higher levels of melanoma-associated antigens (gp100, MART1, tyrosinase, and TRP-1) would be intratumoral homogenous, rendering these tumors susceptible to elimination of antigen-specific CAR T cells (recognizes one antigen) (Figure 4B and Figure 6). This contrasts with cutaneous melanoma which has a higher neoantigen load, and is intratumoral heterogeneous, making this type of melanoma non-responsive to a single-type of antigen-specific CAR T cells (Figure 4B and Figure 6).
4.3. Oncolytic Viruses for the Treatment of Malignant Melanoma
Talimogene Laherparepevec (T-VEC) is the only FDA-approved oncolytic virus for the treatment of unresectable and metastatic (stage III-IV) melanoma (Figure 2). T-VEC is administered by intralesional injection directly to melanomas on the skin, or in the lymph nodes [442]. T-VEC preferentially targets, infects, and replicates within melanoma cells, without infecting healthy tissues [442,443]. T-VEC is a genetically engineered hepatitis simplex virus 1 that expresses GM-CSF [442,443]. GM-CSF facilitates the recruitment and activation of antigen-presenting cells, following T-VEC mediated lysis of melanoma cells [442,443]. Oncolytic viruses upon infection, replicate within the tumor and produce viral-associated GM-CSF, subsequently leading to tumor lysis [443]. Viral-induced tumor lysis releases tumor immunogens and GM-CSF, inducing an innate and adaptive anti-tumor immune response [443]. Preclinical studies suggest that oncolytic viruses can induce an abscopal-like effect; with tumor regression occurring at the site of injection, and induction of a systemic anti-tumor immune response that affects distant tumors [443,444]. T-VEC in the clinic showed a strong durable and objective response, with improved survival by 4.4 months, compared to the GM-CSF only treatment. Interestingly, some treated patients showed oncolytic-induced abscopal-like effects for the not-treated lesions [442,445,446,447,448]. The goal of oncovirus therapy is to convert an immunologically “cold” tumor into a “hot” tumor, or make the tumor microenvironment more immunologically active to induce a local and systemic anti-tumor immune response [443,449]. It was proposed that this therapy should be combined with different immunotherapeutic agents, cytotoxic agents, or radiation therapy, to improve clinical outcomes of metastatic melanoma patients [443,449].
5. Mechanistic Driven Design of Combination Therapies with Immunotherapy
In recent years, various combinatorial approaches with immunotherapeutic agents or molecular inhibitors targeting single or multiple pathway(s) entered the clinical trials for human cancers, including melanoma. Combinatorial strategies using inhibitors of non-redundant, independent pathways, without toxicity to overcome the suppressive tumor microenvironment, would be an ideal approach to improve disease treatment outcome and reduce resistance. A mechanistic-driven design of combination modalities is crucial in therapeutic treatment design, as it allows various drugs to work in concert with each other, to improve clinical response, survival, and overcome resistance. FDA approved the combination therapy of anti-CTLA-4 (ipilimumab) with anti-PD-1 (nivolumab), for the treatment of unresectable or metastatic melanoma, and resulted in improved response rates by 50%–60%, with enhanced durable survival compared to either agent alone, however, greater toxicities are detected with this combination [450,451,452,453,454,455]. The rationale for combining these two immune checkpoint blockade therapies is to expand anti-tumor cytotoxic T-cells within the lymph nodes through anti-CTLA-4 treatment, and anti-PD-1 releases the “breaks” of these effector T-cells at the tumor site to overcome the immune suppressive environment created by tumor cells (Figure 7) [450]. This combination also leads to distinct genetic and functional immune changes, as compared to anti-PD-1 or anti-CTLA-4 monotherapies [456,457].
Figure 7.
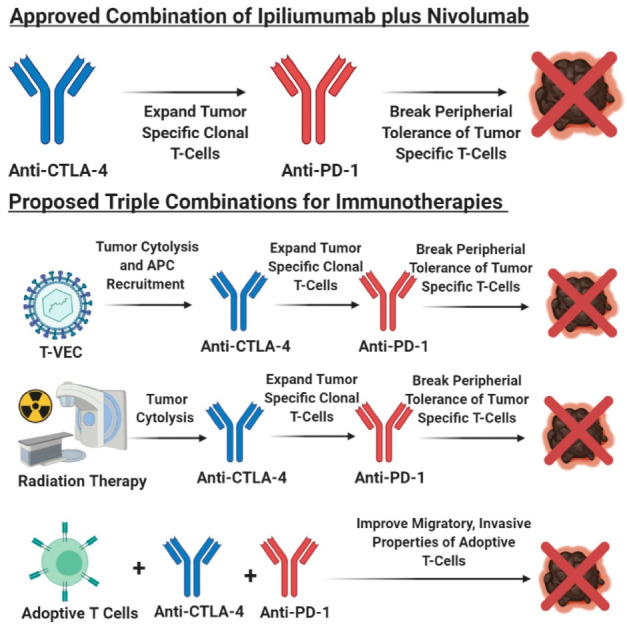
Mechanistic-driven design of combination immunotherapies. Ipilimumab plus nivolumab are approved for the treatment of metastatic melanoma, while the other three are currently under investigation or are proposed in this review. Created with BioRender.com.
T-VEC in combination with immune checkpoint blockade therapy has shown promising results, with improved response rates in combination therapies, compared to monotherapies in phase I and II trials [443,450]. A triple combination utilizes T-VEC plus anti-PD-1 and anti-CTLA-4, but should be staggered to reduce toxicities. A proposed treatment regimen could be, T-VEC is given initially to induce cytolysis of tumor cells releasing tumor immunogens and GM-CSF, where GM-CSF would recruit antigen presenting cells. The next treatment would be anti-CTLA-4 to induce expansion of clonal anti-tumor CD8+ T- cells, and finally anti-PD-1 would be administered to reduce peripheral tolerance and induce a robust anti-tumor immune response against the various clonal melanoma cell populations (Figure 7). The importance of determining the appropriate dosing schedule is essential to reduce toxicities, while also providing enough lead time to develop an immune response for the respective treatments to synergistically support each treatment modality.
Radiation therapy in combination with either anti-PD-1 or anti-CTLA-4 antibodies yielded mixed outcomes with better response in some cases, while others did not show improvement [450]. Similarly, the rational of sequential administration of T-VEC, anti-CTLA-4, and anti-PD-1, radiation therapy could be given first to promote tumor necrosis, and to induce an anti-tumor adaptive immune response. Subsequently, this could be followed by anti-CTLA-4 to induce expansion of tumor-specific cytotoxic T-cells and finally anti-PD-1 to disrupt peripheral tolerance (Figure 7). In this proposed triple combination, addition of T-VEC or GM-CSF after radiation therapy, could enhance recruitment and activation of the antigen presenting cells, therefore, strengthening the tumor-specific clonal expansion of cytotoxic T-cells [443].
Preclinical evidence suggest that adoptive T-cell transfer along with the dual treatment of anti-CTLA-4 and anti-PD-1, can improve tumor-antigen-specific cytotoxic T-cell infiltration and function within the tumor site, corresponding to the improved survival in experimental animal models (Figure 7) [458]. Adoptive T-cell transfer as a monotherapy show low response rates based on the poor infiltration of cytotoxic T-cells and function within the tumor microenvironment [458]. Albeit not directly tested, adoptively transferred T-cells along with anti-CTLA-4 treatment, mediates the expansion and improves T-cell function at the tumor site, followed by anti-PD-1 interrupts peripheral tolerance (Figure 7) [458]. Clinical evaluation of this triple combination is yet to be tested, however, in ovarian cancer, patients treated with adoptive T-cell therapy plus ipilimumab generated promising results, 1/6 patients showed partial response and 5/6 patients showed stable disease for up to 1 year [459]. Interestingly, vemurafenib, the inhibitor for mutated BRAF, paradoxically activates the MAPK pathway in adoptively transferred T-cells in a mutant BRAF-driven mouse melanoma model [460]. In this model, the inhibitor, vemurafenib, acts within its canonical function to inhibit mutant BRAF in melanoma cells but also paradoxically activates the MAPK pathway in T-cells to enhance the anti-tumor cytotoxic function of the tumor recognizing T-cells [460]. Future combination therapies can focus on how to preferentially deplete immune suppressive cells in the tumor microenvironment, to enhance the efficacy/response of immune checkpoint blockade therapy, adoptive T-cell therapy, or T-VEC therapy.
6. Conclusions and Future Directions
Characterizing the intrinsic and extrinsic mechanism that underlie melanoma pathology and progression, is crucial in improving the clinical outcome of patients with this deadly disease. A better understanding of melanoma biology would improve the design of novel combination therapies to improve response rate, promote tumor remission, and increase survival with a reduction in resistance development. In-depth characterization of patients who show complete, partial, or no response would unravel patient characteristics or molecular markers, which makes a melanoma (or other cancers) susceptible or unresponsive to treatments. Improved identification of responders or non-responders would improve a patient’s quality of life and ease the financial burden of cancer treatment.
Immunotherapy revolutionized the field of cancer therapeutics by its ability to induce long-term clinical response in patients who responded to treatment. Unfortunately, response rates to immunotherapy are low. However, with the ongoing biomarker studies uncovering key molecular markers such as tumor mutational burden, molecular marker expression on tumor or immune cells, circulating soluble markers, and pro- or anti-tumor immune cell populations, at baseline or on treatment, would advance the identification of responders vs. non-responders. Not surprisingly, the most common adverse event associated with immunotherapy are autoimmune disorders. Therefore, studies are being conducted to identify treatment biomarkers for immune-related adverse events, to detect and eliminate therapy-associated toxicities. In addition to molecular markers to predict treatment response, we believe that melanoma subtype should be used to stratify patients into either immune checkpoint blockade therapy or adoptive T-cell therapy (Figure 4, Figure 5 and Figure 6). Cutaneous melanoma patients including both CSID and non-CSID melanomas would benefit from immune checkpoint blockade therapy, due to it being a heterogenous tumor, with a wide repertoire of tumor-specific cytotoxic-T-cells that are restrained by immune checkpoint molecule expression (Figure 4 and Figure 5). Non-cutaneous melanoma would benefit from adoptive T-cell therapy, because of its low tumor mutation burden, and would have a higher expression of melanoma-associated antigen (Figure 4 and Figure 6). Therefore, there would be a higher response to adoptive T-cell transfer because this melanoma subtype contains a more homogenous T-cell population (Figure 6). Multiple biomarkers should be used to predict treatment response or identify immune-related adverse events. Biomarker identification would help tease out the mechanism of action of these immunotherapeutic agents. Better understanding of the mechanism of action of immune checkpoint blockade therapy, adoptive T-cell therapy, and T-VEC therapy would improve the design of combination therapies with other immunotherapy agents, targeted therapies, radiation therapy, or chemotherapy.
Abbreviations
| ABCDEF | Asymmetry, Border irregularity, Color variegation, Diameter larger than 6 mm, Evolution of a lesion, Funny looking |
| TNM | Tumor thickness, Nodal Involvement, Metastasis |
| CSID | Chronically Sun Induced Melanoma |
| Non-CSID | Non- Chronically Sun Induced Melanoma |
| mGluR | Metabotropic Glutamate Receptor |
| PD-1 | Program Cell Death Protein 1 |
| PD-L1 | Programmed Death Ligand 1 |
| PD-L2 | Programmed Death Ligand 2 |
| CTLA-4 | Cytotoxic T-Lymphocyte Associated Protein- 4 |
| Tregs | Regulatory T-cells |
| MDSC | Myeloid Derived Suppressor Cells |
| TAMs | Tumor Associated Macrophages |
| Th | Helper T-cells |
| MHC I | Major Histocompatibility I |
| MHC II | Major Histocompatibility II |
| IFN | Interferon |
| IL-2 | Interleukin-2 |
| BCG | Bacillus-Calmette-Guerin |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| iRECIST | Immune related Response Evaluation Criteria in Solid Tumors |
| iCR | Immune Complete Response |
| iPR | Immune Partial Response |
| iSD | Immune Stable Disease |
| iUPD | Immune Unconfirmed Progressive Disease |
| iCPD | Immune Confirmed Progressive Disease |
| NLR | Neutrophil to Lymphocyte Ratio |
| iCAM | Immune Checkpoint Activating Mutation |
| CAR | Chimeric Antigen Receptor |
| TIL | Tumor Infiltrating Lymphocytes |
| T-VEC | Talimogene Laherparepevec |
| TCR | T-Cell Receptor |
Author Contributions
Conceptualization, K.E. and S.C.; Writing—Original Draft Preparation, K.E.; Writing—Review and Editing, K.E. and S.C.; Supervision, S.C.; Funding Acquisition, K.E. and S.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by New Jersey Commission on Cancer Research (NJCCR) Pre-Doctoral Fellowship (DCHS19PPC027) to K.E. National Cancer Institute Small Business Innovation Research (NCI SBIR) (R44CA156781-04) to S.C.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mort R.L., Jackson I.J., Patton E.E. The melanocyte lineage in development and disease. Development. 2015;142:620–632. doi: 10.1242/dev.106567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shain A.H., Bastian B.C. From melanocytes to melanomas. Nat. Rev. Cancer. 2016;16:345–358. doi: 10.1038/nrc.2016.37. [DOI] [PubMed] [Google Scholar]
- 3.Seiji M., Fitzpatrick T.B. The Reciprocal Relationship between Melanization and Tyrosinase Activity in Melanosomes (Melanin Granules) J. Biochem. 1961;49:700–706. doi: 10.1093/oxfordjournals.jbchem.a127360. [DOI] [PubMed] [Google Scholar]
- 4.Brenner M., Hearing V.J. The Protective Role of Melanin Against UV Damage in Human Skin†. Photochem. Photobiol. 2008;84:539–549. doi: 10.1111/j.1751-1097.2007.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isola A.L., Eddy K., Chen S. Biology, Therapy and Implications of Tumor Exosomes in the Progression of Melanoma. Cancers. 2016;8:110. doi: 10.3390/cancers8120110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seiji M., Fitzpatrick T., Birbeck M. The Melanosome: A Distinctive Subcellular Particle of Mammalian Melanocytes and the Site of Melanogenesis1. J. Investig. Dermatol. 1961;36:243–252. doi: 10.1038/jid.1961.42. [DOI] [PubMed] [Google Scholar]
- 7.Fitzpatrick T.B., Breathnach A.S. The Epidermal Melanin Unit System. Dermatol. Wochenschr. 1963;147:481–489. [PubMed] [Google Scholar]
- 8.Alaluf S., Atkins D., Barrett K., Blount M., Carter N., Heath A. Ethnic Variation in Melanin Content and Composition in Photoexposed and Photoprotected Human Skin. Pigment. Cell Res. 2002;15:112–118. doi: 10.1034/j.1600-0749.2002.1o071.x. [DOI] [PubMed] [Google Scholar]
- 9.Crawford N., Kelly D.E., Hansen M.E.B., Beltrame M.H., Fan S., Bowman S.L., Jewett E., Ranciaro A., Thompson S., Lo Y., et al. Loci associated with skin pigmentation identified in African populations. Science. 2017;358:eaan8433. doi: 10.1126/science.aan8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu F., Visser M., Duffy D.L., Hysi P.G., Jacobs L.C., Lao O., Zhong K., Walsh S., Chaitanya L., Wollstein A., et al. Genetics of skin color variation in Europeans: Genome-wide association studies with functional follow-up. Hum. Genet. 2015;134:823–835. doi: 10.1007/s00439-015-1559-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sturm R.A. Molecular genetics of human pigmentation diversity. Hum. Mol. Genet. 2009;18:R9–R17. doi: 10.1093/hmg/ddp003. [DOI] [PubMed] [Google Scholar]
- 12.Thody A.J., Higgins E.M., Wakamatsu K., Ito S., Burchill S.A., Marks J.M. Pheomelanin as well as Eumelanin Is Present in Human Epidermis. J. Investig. Dermatol. 1991;97:340–344. doi: 10.1111/1523-1747.ep12480680. [DOI] [PubMed] [Google Scholar]
- 13.Hennessy A., Oh C., Diffey B., Wakamatsu K., Ito S., Rees J. Eumelanin and pheomelanin concentrations in human epidermis before and after UVB irradiation. Pigment Cell Res. 2005;18:220–223. doi: 10.1111/j.1600-0749.2005.00233.x. [DOI] [PubMed] [Google Scholar]
- 14.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 15.Yamaguchi Y., Takahashi K., Zmudzka B.Z., Kornhauser A., Miller S.A., Tadokoro T., Berens W., Beer J.Z., Hearing V.J. Human skin responses to UV radiation: Pigment in the upper epidermis protects against DNA damage in the lower epidermis and facilitates apoptosis. FASEB J. 2006;20:1486–1488. doi: 10.1096/fj.06-5725fje. [DOI] [PubMed] [Google Scholar]
- 16.Tadokoro T., Kobayashi N., Zmudzka B.Z., Ito S., Wakamatsu K., Yamaguchi Y., Korossy K.S., Miller S.A., Beer J.Z., Hearing V.J. UV-induced DNA damage and melanin content in human skin differing in racial/ethnic origin. FASEB J. 2003;17:1177–1179. doi: 10.1096/fj.02-0865fje. [DOI] [PubMed] [Google Scholar]
- 17.Scoggins C.R., Ross M.I., Reintgen D.S., Noyes R.D., Goydos J.S., Beitsch P.D., Urist M.M., Ariyan S., Sussman J.J., Edwards M.J., et al. Gender-Related Differences in Outcome for Melanoma Patients. Ann. Surg. 2006;243:693–700. doi: 10.1097/01.sla.0000216771.81362.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw H.M., McGovern V.J., Milton G.W., Farago G.A., McCarthy W.H. Histologic features of tumors and the female superiority in survival from malignant melanoma. Cancer. 1980;45:1604–1608. doi: 10.1002/1097-0142(19800401)45:7<1604::AID-CNCR2820450715>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 19.Natale C.A., Duperret E.K., Zhang J., Sadeghi R., Dahal A., O’Brien K.T., Cookson R., Winkler J.D., Ridky T.W. Sex steroids regulate skin pigmentation through nonclassical membrane-bound receptors. eLife. 2016;5 doi: 10.7554/eLife.15104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White L.P. Studies on Melanoma. N. Engl. J. Med. 1959;260:789–797. doi: 10.1056/NEJM195904162601601. [DOI] [PubMed] [Google Scholar]
- 21.Natale C.A., Li J., Zhang J., Dahal A., Dentchev T., Stanger B.Z., Ridky T.W. Activation of G protein-coupled estrogen receptor signaling inhibits melanoma and improves response to immune checkpoint blockade. eLife. 2018;7:e31770. doi: 10.7554/eLife.31770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu X.-C., Eide M.J., King J., Saraiya M., Huang Y., Wiggins C., Barnholtz-Sloan J.S., Martin N., Cokkinides V., Miller J., et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999-2006. J. Am. Acad. Dermatol. 2011;65:S26.e1–S26.e13. doi: 10.1016/j.jaad.2011.05.034. [DOI] [PubMed] [Google Scholar]
- 23.Kemeny M., Busch E., Stewart A.K., Menck H.R. Superior Survival of Young Women with Malignant Melanoma. Am. J. Surg. 1998;175:437–445. doi: 10.1016/S0002-9610(98)00070-1. [DOI] [PubMed] [Google Scholar]
- 24.Roh M.R., Eliades P., Gupta S., Grant-Kels J.M., Tsao H. Cutaneous melanoma in women. Int. J. Women’s Dermatol. 2017;3:S11–S15. doi: 10.1016/j.ijwd.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shitara D., Nascimento M.M., Puig S., Yamada S., Enokihara M.M., Michalany N., Bagatin E. Nevus-associated melanomas: Clinicopathologic features. Am. J. Clin. Pathol. 2014;142:485–491. doi: 10.1309/AJCP4L5CJGKTJVDD. [DOI] [PubMed] [Google Scholar]
- 26.Elder D.E., Bastian B.C., Cree I.A., Massi D., Scolyer R.A. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch. Pathol. Lab. Med. 2020;144:500–522. doi: 10.5858/arpa.2019-0561-RA. [DOI] [PubMed] [Google Scholar]
- 27.Holly E.A., Kelly J.W., Shpall S.N., Chiu S.-H. Number of melanocytic nevi as a major risk factor for malignant melanoma. J. Am. Acad. Dermatol. 1987;17:459–468. doi: 10.1016/S0190-9622(87)70230-8. [DOI] [PubMed] [Google Scholar]
- 28.Harrison S.L., Speare R., Wronski I., MacLennan R. Sun exposure and melanocytic naevi in young Australian children. Lancet. 1994;344:1529–1532. doi: 10.1016/S0140-6736(94)90348-4. [DOI] [PubMed] [Google Scholar]
- 29.Aalborg J., Morelli J.G., Mokrohisky S.T., Asdigian N.L., Byers T.E., Dellavalle R.P., Box N.F., Crane L.A. Tanning and Increased Nevus Development in Very-Light-Skinned Children Without Red Hair. Arch. Dermatol. 2009;145:989–996. doi: 10.1001/archdermatol.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A.J.R., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.-L., et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shain A.H., Yeh I., Kovalyshyn I., Sriharan A., Talevich E., Gagnon A., Dummer R., North J., Pincus L., Ruben B., et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015;373:1926–1936. doi: 10.1056/NEJMoa1502583. [DOI] [PubMed] [Google Scholar]
- 32.Elwood J.M. Melanoma and sun exposure: Contrasts between intermittent and chronic exposure. World J. Surg. 1992;16:157–165. doi: 10.1007/BF02071515. [DOI] [PubMed] [Google Scholar]
- 33.Matthews N.H., Li W.Q., Qureshi A.A., Weinstock M.A., Cho E. Epidemiology of Melanoma. In: Ward W.H., Farma J.M., editors. Cutaneous Melanoma: Etiology and Therapy. Codon Publications; Brisbane, Australia: 2017. [DOI] [PubMed] [Google Scholar]
- 34.Akbani R., Akdemir K.C., Aksoy B.A., Albert M., Ally A., Amin S.B., Arachchi H.M., Arora A., Auman J.T., Ayala B., et al. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eckerle Mize D., Bishop M., Resse E., Sluzevich J. Familial Atypical Multiple Mole Melanoma Syndrome. In: Riegert-Johnson D.L., Boardman L.A., Hefferon T., Roberts M., editors. Cancer Syndromes. National Center for Biotechnology Information (US); Bethesda, MD, USA: 2009. [PubMed] [Google Scholar]
- 36.Ford D., Bliss J.M., Swerdlow A.J., Armstrong B.K., Franceschi S., Green A., Holly E.A., Mack T., MacKie R.M., Osterlind A., et al. Risk of cutaneous melanoma associated with a family history of the disease. The International Melanoma Analysis Group (IMAGE) Int. J. Cancer. 1995;62:377–381. doi: 10.1002/ijc.2910620403. [DOI] [PubMed] [Google Scholar]
- 37.Hussussian C.J., Struewing J.P., Goldstein A.M., Higgins P.A., Ally D.S., Sheahan M.D., Clark W.H., Jr., Tucker M.A., Dracopoli N.C. Germline p16 mutations in familial melanoma. Nat. Genet. 1994;8:15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 38.Kamb A., Shattuck-Eidens D., Eeles R., Liu Q., Gruis N.A., Ding W., Hussey C., Tran T., Miki Y., Weaver-Feldhaus J., et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat. Genet. 1994;8:22–26. doi: 10.1038/ng0994-22. [DOI] [PubMed] [Google Scholar]
- 39.Zuo L., Weger J., Yang Q., Goldstein A.M., Tucker M.A., Walker G.J., Hayward N., Dracopoli N.C. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat. Genet. 1996;12:97–99. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]
- 40.Soufir N., Avril M.-F., Chompret A., Demenais F., Bombled J., Spatz A., Stoppa-Lyonnet D., Bénard J., Paillerets B.B.-D. Prevalence of p16 and CDK4 germline mutations in 48 melanoma-prone families in France. Hum. Mol. Genet. 1998;7:209–216. doi: 10.1093/hmg/7.2.209. [DOI] [PubMed] [Google Scholar]
- 41.Molven A., Grimstvedt M.B., Steine S.J., Harland M., Avril M.-F., Hayward N.K., Akslen L.A. A large Norwegian family with inherited malignant melanoma, multiple atypical nevi, and CDK4 mutation. Genes Chromosom. Cancer. 2005;44:10–18. doi: 10.1002/gcc.20202. [DOI] [PubMed] [Google Scholar]
- 42.Goldstein A.M., Chan M., Harland M., Gillanders E.M., Hayward N.K., Avril M.-F., Azizi E., Bianchi-Scarra G., Newton-Bishop J., Paillerets B.B.-D., et al. High-risk Melanoma Susceptibility Genes and Pancreatic Cancer, Neural System Tumors, and Uveal Melanoma across GenoMEL. Cancer Res. 2006;66:9818–9828. doi: 10.1158/0008-5472.CAN-06-0494. [DOI] [PubMed] [Google Scholar]
- 43.Black J.O. Xeroderma Pigmentosum. Head Neck Pathol. 2016;10:139–144. doi: 10.1007/s12105-016-0707-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tagliabue E., Gandini S., Bellocco R., Maisonneuve P., Newton-Bishop J., Polsky D., Lazovich D., Kanetsky P.A., Ghiorzo P., Gruis N.A., et al. MC1R variants as melanoma risk factors independent of at-risk phenotypic characteristics: A pooled analysis from the M-SKIP project. Cancer Manag. Res. 2018;10:1143–1154. doi: 10.2147/CMAR.S155283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puntervoll H.E., Yang X.R., Vetti H.H., Bachmann I.M., Avril M.F., Benfodda M., Catricalà C., Dalle S., Duval-Modeste A.B., Ghiorzo P., et al. Melanoma prone families with CDK4 germline mutation: Phenotypic profile and associations with MC1R variants. J. Med. Genet. 2013;50:264–270. doi: 10.1136/jmedgenet-2012-101455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daniel Jensen J., Elewski B.E. The ABCDEF Rule: Combining the “ABCDE Rule” and the “Ugly Duckling Sign” in an Effort to Improve Patient Self-Screening Examinations. J. Clin. Aesthet. Dermatol. 2015;8:15. [PMC free article] [PubMed] [Google Scholar]
- 47.Friedman R.J., Rigel D.S., Kopf A.W. Early detection of malignant melanoma: The role of physician examination and self-examination of the skin. CA Cancer J. Clin. 1985;35:130–151. doi: 10.3322/canjclin.35.3.130. [DOI] [PubMed] [Google Scholar]
- 48.Abbasi N.R., Shaw H.M., Rigel D.S., Friedman R.J., McCarthy W.H., Osman I., Kopf A.W., Polsky D. Early diagnosis of cutaneous melanoma: Revisiting the ABCD criteria. JAMA. 2004;292:2771–2776. doi: 10.1001/jama.292.22.2771. [DOI] [PubMed] [Google Scholar]
- 49.Grob J.J., Bonerandi J.J. The ‘ugly duckling’ sign: Identification of the common characteristics of nevi in an individual as a basis for melanoma screening. Arch. Dermatol. 1998;134:103–104. doi: 10.1001/archderm.134.1.103-a. [DOI] [PubMed] [Google Scholar]
- 50.Ward W.H., Lambreton F., Goel N., Yu J.Q., Farma J.M. Clinical Presentation and Staging of Melanoma. In: Ward W.H., Farma J.M., editors. Cutaneous Melanoma: Etiology and Therapy. Codon Publications; Brisbane, Australia: 2017. [DOI] [PubMed] [Google Scholar]
- 51.Dinnes J., Deeks J.J., Chuchu N., Ferrante di Ruffano L., Matin R.N., Thomson D.R., Wong K.Y., Aldridge R.B., Abbott R., Fawzy M., et al. Dermoscopy, with and without visual inspection, for diagnosing melanoma in adults. Cochrane Database Syst. Rev. 2018;12:CD011902. doi: 10.1002/14651858.CD011902.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herschorn A. Dermoscopy for melanoma detection in family practice. Can. Fam. Physician. 2012;58:740–745. [PMC free article] [PubMed] [Google Scholar]
- 53.Holmes G.A., Vassantachart J.M., Limone B.A., Zumwalt M., Hirokane J., Jacob S.E. Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination. Fed. Pract. 2018;35:S39–S45. [PMC free article] [PubMed] [Google Scholar]
- 54.Sonthalia S., Kaliyadan F. StatPearls. StatPearls Publishing LLC.; Treasure Island, FL, USA: 2020. Dermoscopy Overview and Extradiagnostic Applications. [PubMed] [Google Scholar]
- 55.Davis L.E., Shalin S.C., Tackett A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019;20:1366–1379. doi: 10.1080/15384047.2019.1640032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gershenwald J.E., Scolyer R.A., Hess K.R., Sondak V.K., Long G.V., Ross M.I., Lazar A.J., Faries M.B., Kirkwood J.M., McArthur G.A., et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017;67:472–492. doi: 10.3322/caac.21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Breslow A. Thickness, cross-sectional areas and depth of invasion in the prognosis of cutaneous melanoma. Ann. Surg. 1970;172:902–908. doi: 10.1097/00000658-197011000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bønnelykke-Behrndtz M.L., Steiniche T. Ulcerated Melanoma: Aspects and Prognostic Impact. In: Ward W.H., Farma J.M., editors. Cutaneous Melanoma: Etiology and Therapy. Codon Publications; Brisbane, Australia: 2017. [DOI] [PubMed] [Google Scholar]
- 59.Urso C. Are growth phases exclusive to cutaneous melanoma? J. Clin. Pathol. 2004;57:560. doi: 10.1136/jcp.2003.014852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clark W.H., Jr., Elder D.E., Guerry D.T., Epstein M.N., Greene M.H., Van Horn M. A study of tumor progression: The precursor lesions of superficial spreading and nodular melanoma. Hum. Pathol. 1984;15:1147–1165. doi: 10.1016/S0046-8177(84)80310-X. [DOI] [PubMed] [Google Scholar]
- 61.Damsky W.E., Rosenbaum L.E., Bosenberg M. Decoding melanoma metastasis. Cancers. 2010;3:126–163. doi: 10.3390/cancers3010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thompson J.F., Soong S.J., Balch C.M., Gershenwald J.E., Ding S., Coit D.G., Flaherty K.T., Gimotty P.A., Johnson T., Johnson M.M., et al. Prognostic significance of mitotic rate in localized primary cutaneous melanoma: An analysis of patients in the multi-institutional American Joint Committee on Cancer melanoma staging database. J. Clin. Oncol. 2011;29:2199–2205. doi: 10.1200/JCO.2010.31.5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilkins D.K., Nathan P.D. Therapeutic opportunities in noncutaneous melanoma. Ther. Adv. Med. Oncol. 2009;1:29–36. doi: 10.1177/1758834009337664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Curtin J.A., Fridlyand J., Kageshita T., Patel H.N., Busam K.J., Kutzner H., Cho K.-H., Aiba S., Bröcker E.-B., LeBoit P.E., et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 65.Kuk D., Shoushtari A.N., Barker C.A., Panageas K.S., Munhoz R.R., Momtaz P., Ariyan C.E., Brady M.S., Coit D.G., Bogatch K., et al. Prognosis of Mucosal, Uveal, Acral, Nonacral Cutaneous, and Unknown Primary Melanoma from the Time of First Metastasis. Oncologist. 2016;21:848–854. doi: 10.1634/theoncologist.2015-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bennett D.R., Wasson D., MacArthur J.D., McMillen M.A. The effect of misdiagnosis and delay in diagnosis on clinical outcome in melanomas of the foot. J. Am. Coll. Surg. 1994;179:279–284. [PubMed] [Google Scholar]
- 67.Cress R.D., Holly E.A. Incidence of cutaneous melanoma among non-Hispanic whites, Hispanics, Asians, and blacks: An analysis of california cancer registry data, 1988–1993. Cancer Causes Control. 1997;8:246–252. doi: 10.1023/A:1018432632528. [DOI] [PubMed] [Google Scholar]
- 68.Cormier J.N., Xing Y., Ding M., Lee J.E., Mansfield P.F., Gershenwald J.E., Ross M.I., Du X.L. Ethnic Differences Among Patients with Cutaneous Melanoma. Arch. Intern. Med. 2006;166:1907–1914. doi: 10.1001/archinte.166.17.1907. [DOI] [PubMed] [Google Scholar]
- 69.Hayward N.K., Wilmott J.S., Waddell N., Johansson P.A., Field M.A., Nones K., Patch A.-M., Kakavand H., Alexandrov L.B., Burke H., et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545:175–180. doi: 10.1038/nature22071. [DOI] [PubMed] [Google Scholar]
- 70.Krauthammer M., Kong Y., Ha B.H., Evans P., Bacchiocchi A., McCusker J.P., Cheng E., Davis M.J., Goh G., Choi M., et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pollock P.M., Harper U.L., Hansen K.S., Yudt L.M., Stark M., Robbins C.M., Moses T.Y., Hostetter G., Wagner U., Kakareka J., et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 72.Menzies A.M., Haydu L.E., Visintin L., Carlino M.S., Howle J.R., Thompson J.F., Kefford R.F., Scolyer R.A., Long G.V. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2012;18:3242–3249. doi: 10.1158/1078-0432.CCR-12-0052. [DOI] [PubMed] [Google Scholar]
- 73.Teh J.L., Chen S. Glutamatergic signaling in cellular transformation. Pigment Cell Melanoma Res. 2012;25:331–342. doi: 10.1111/j.1755-148X.2012.00983.x. [DOI] [PubMed] [Google Scholar]
- 74.Horn S., Figl A., Rachakonda P.S., Fischer C., Sucker A., Gast A., Kadel S., Moll I., Nagore E., Hemminki K., et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 75.Chiba K., Lorbeer F.K., Shain A.H., McSwiggen D.T., Schruf E., Oh A., Ryu J., Darzacq X., Bastian B.C., Hockemeyer D. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science. 2017;357:1416–1420. doi: 10.1126/science.aao0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bradford P.T., Goldstein A.M., McMaster M.L., Tucker M.A. Acral lentiginous melanoma: Incidence and survival patterns in the United States, 1986–2005. Arch. Dermatol. 2009;145:427–434. doi: 10.1001/archdermatol.2008.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weis E., Shah C.P., Lajous M., Shields J.A., Shields C.L. The Association Between Host Susceptibility Factors and Uveal Melanoma: A Meta-analysis. Arch. Ophthalmol. 2006;124:54–60. doi: 10.1001/archopht.124.1.54. [DOI] [PubMed] [Google Scholar]
- 78.Shah C.P., Weis E., Lajous M., Shields J.A., Shields C.L. Intermittent and Chronic Ultraviolet Light Exposure and Uveal Melanoma: A Meta-analysis. Ophthalmology. 2005;112:1599–1607. doi: 10.1016/j.ophtha.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 79.Kaliki S., Shields C.L. Uveal melanoma: Relatively rare but deadly cancer. Eye. 2017;31:241–257. doi: 10.1038/eye.2016.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McLaughlin C.C., Wu X.C., Jemal A., Martin H.J., Roche L.M., Chen V.W. Incidence of noncutaneous melanomas in the U.S. Cancer. 2005;103:1000–1007. doi: 10.1002/cncr.20866. [DOI] [PubMed] [Google Scholar]
- 81.Rossi E., Schinzari G., Zizzari I.G., Maiorano B.A., Pagliara M.M., Sammarco M.G., Fiorentino V., Petrone G., Cassano A., Rindi G., et al. Immunological Backbone of Uveal Melanoma: Is There a Rationale for Immunotherapy? Cancers. 2019;11:1055. doi: 10.3390/cancers11081055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rabbie R., Ferguson P., Molina-Aguilar C., Adams D.J., Robles-Espinoza C.D. Melanoma subtypes: Genomic profiles, prognostic molecular markers and therapeutic possibilities. J. Pathol. 2019;247:539–551. doi: 10.1002/path.5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Johansson P., Aoude L.G., Wadt K., Glasson W.J., Warrier S.K., Hewitt A.W., Kiilgaard J.F., Heegaard S., Isaacs T., Franchina M., et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2016;7:4624–4631. doi: 10.18632/oncotarget.6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Van Raamsdonk C.D., Griewank K.G., Crosby M.B., Garrido M.C., Vemula S., Wiesner T., Obenauf A.C., Wackernagel W., Green G., Bouvier N., et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010;363:2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moore A.R., Ceraudo E., Sher J.J., Guan Y., Shoushtari A.N., Chang M.T., Zhang J.Q., Walczak E.G., Kazmi M.A., Taylor B.S., et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016;48:675–680. doi: 10.1038/ng.3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Raamsdonk C.D., Bezrookove V., Green G., Bauer J., Gaugler L., O’Brien J.M., Simpson E.M., Barsh G.S., Bastian B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Khalili J.S., Yu X., Wang J., Hayes B.C., Davies M.A., Lizee G., Esmaeli B., Woodman S.E. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin. Cancer Res. 2012;18:4345–4355. doi: 10.1158/1078-0432.CCR-11-3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu F.X., Luo J., Mo J.S., Liu G., Kim Y.C., Meng Z., Zhao L., Peyman G., Ouyang H., Jiang W., et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell. 2014;25:822–830. doi: 10.1016/j.ccr.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Feng X., Degese M.S., Iglesias-Bartolome R., Vaque J.P., Molinolo A.A., Rodrigues M., Zaidi M.R., Ksander B.R., Merlino G., Sodhi A., et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25:831–845. doi: 10.1016/j.ccr.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Toshiaki S.H.K., Yoriko Y., Masaru T. In: Acral Melanoma In Color Atlas of Melanocytic Lesions of the Skin. Soyer H.P., Argenziano G., Hofmann-Wellenhof R., Johr R.H., editors. Volume 1. Springer; Berlin, Germany: New York, NY, USA: 2007. pp. 196–203. [Google Scholar]
- 91.Huang K., Fan J., Misra S. Acral Lentiginous Melanoma: Incidence and Survival in the United States, 2006–2015, an Analysis of the SEER Registry. J. Surg. Res. 2020;251:329–339. doi: 10.1016/j.jss.2020.02.010. [DOI] [PubMed] [Google Scholar]
- 92.Jung H.J., Kweon S.-S., Lee J.-B., Lee S.-C., Yun S.J. A Clinicopathologic Analysis of 177 Acral Melanomas in Koreans: Relevance of Spreading Pattern and Physical Stress. JAMA Dermatol. 2013;149:1281–1288. doi: 10.1001/jamadermatol.2013.5853. [DOI] [PubMed] [Google Scholar]
- 93.Feibleman C.E., Stoll H., Maize J.C. Melanomas of the palm, sole, and nailbed: A clinicopathologic study. Cancer. 1980;46:2492–2504. doi: 10.1002/1097-0142(19801201)46:11<2492::AID-CNCR2820461130>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 94.Moon K.R., Choi Y.D., Kim J.M., Jin S., Shin M.H., Shim H.J., Lee J.B., Yun S.J. Genetic Alterations in Primary Acral Melanoma and Acral Melanocytic Nevus in Korea: Common Mutated Genes Show Distinct Cytomorphological Features. J. Investig. Dermatol. 2018;138:933–945. doi: 10.1016/j.jid.2017.11.017. [DOI] [PubMed] [Google Scholar]
- 95.Liang W.S., Hendricks W., Kiefer J., Schmidt J., Sekar S., Carpten J., Craig D.W., Adkins J., Cuyugan L., Manojlovic Z., et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 2017;27:524–532. doi: 10.1101/gr.213348.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De Lima Vazquez V., Vicente A.L., Carloni A., Berardinelli G., Soares P., Scapulatempo C., Martinho O., Reis R.M. Molecular profiling, including TERT promoter mutations, of acral lentiginous melanomas. Melanoma Res. 2016;26:93–99. doi: 10.1097/CMR.0000000000000222. [DOI] [PubMed] [Google Scholar]
- 97.Curtin J.A., Busam K., Pinkel D., Bastian B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006;24:4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 98.Mihajlovic M., Vlajkovic S., Jovanovic P., Stefanovic V. Primary mucosal melanomas: A comprehensive review. Int. J. Clin. Exp. Pathol. 2012;5:739–753. [PMC free article] [PubMed] [Google Scholar]
- 99.Altieri L., Eguchi M., Peng D.H., Cockburn M. Predictors of mucosal melanoma survival in a population-based setting. J. Am. Acad. Dermatol. 2019;81:136–142.e132. doi: 10.1016/j.jaad.2018.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lerner B.A., Stewart L.A., Horowitz D.P., Carvajal R.D. Mucosal Melanoma: New Insights and Therapeutic Options for a Unique and Aggressive Disease. Oncology. 2017;31:e23–e32. [PubMed] [Google Scholar]
- 101.Kabbarah O., Chin L. Revealing the genomic heterogeneity of melanoma. Cancer Cell. 2005;8:439–441. doi: 10.1016/j.ccr.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 102.Hintzsche J.D., Gorden N.T., Amato C.M., Kim J., Wuensch K.E., Robinson S.E., Applegate A.J., Couts K.L., Medina T.M., Wells K.R., et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017;27:189–199. doi: 10.1097/CMR.0000000000000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Furney S.J., Turajlic S., Stamp G., Nohadani M., Carlisle A., Thomas J.M., Hayes A., Strauss D., Gore M., van den Oord J., et al. Genome sequencing of mucosal melanomas reveals that they are driven by distinct mechanisms from cutaneous melanoma. J. Pathol. 2013;230:261–269. doi: 10.1002/path.4204. [DOI] [PubMed] [Google Scholar]
- 104.Sheng X., Kong Y., Li Y., Zhang Q., Si L., Cui C., Chi Z., Tang B., Mao L., Lian B., et al. GNAQ and GNA11 mutations occur in 9.5% of mucosal melanoma and are associated with poor prognosis. Eur. J. Cancer. 2016;65:156–163. doi: 10.1016/j.ejca.2016.06.019. [DOI] [PubMed] [Google Scholar]
- 105.Si L., Wang X., Guo J. Genotyping of mucosal melanoma. Chin. Clin. Oncol. 2014;3:34. doi: 10.3978/j.issn.2304-3865.2014.07.03. [DOI] [PubMed] [Google Scholar]
- 106.Chen S., Zhu H., Wetzel W.J., Philbert M.A. Spontaneous melanocytosis in transgenic mice. J. Investig. Dermatol. 1996;106:1145–1151. doi: 10.1111/1523-1747.ep12340194. [DOI] [PubMed] [Google Scholar]
- 107.Zhu H., Reuhl K., Zhang X., Botha R., Ryan K., Wei J., Chen S. Development of heritable melanoma in transgenic mice. J. Investig. Dermatol. 1998;110:247–252. doi: 10.1046/j.1523-1747.1998.00133.x. [DOI] [PubMed] [Google Scholar]
- 108.Pollock P.M., Cohen-Solal K., Sood R., Namkoong J., Martino J.J., Koganti A., Zhu H., Robbins C., Makalowska I., Shin S.S., et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat. Genet. 2003;34:108–112. doi: 10.1038/ng1148. [DOI] [PubMed] [Google Scholar]
- 109.Ohtani Y., Harada T., Funasaka Y., Nakao K., Takahara C., Abdel-Daim M., Sakai N., Saito N., Nishigori C., Aiba A. Metabotropic glutamate receptor subtype-1 is essential for in vivo growth of melanoma. Oncogene. 2008;27:7162–7170. doi: 10.1038/onc.2008.329. [DOI] [PubMed] [Google Scholar]
- 110.Wangari-Talbot J., Wall B.A., Goydos J.S., Chen S. Functional effects of GRM1 suppression in human melanoma cells. Mol. Cancer Res. 2012;10:1440–1450. doi: 10.1158/1541-7786.MCR-12-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhu H., Reuhl K., Botha R., Ryan K., Wei J., Chen S. Development of early melanocytic lesions in transgenic mice predisposed to melanoma. Pigment Cell Res. 2000;13:158–164. doi: 10.1034/j.1600-0749.2000.130307.x. [DOI] [PubMed] [Google Scholar]
- 112.Shin S.S., Namkoong J., Wall B.A., Gleason R., Lee H.J., Chen S. Oncogenic activities of metabotropic glutamate receptor 1 (Grm1) in melanocyte transformation. Pigment. Cell Melanoma Res. 2008;21:368–378. doi: 10.1111/j.1755-148X.2008.00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Namkoong J., Shin S.S., Lee H.J., Marin Y.E., Wall B.A., Goydos J.S., Chen S. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007;67:2298–2305. doi: 10.1158/0008-5472.CAN-06-3665. [DOI] [PubMed] [Google Scholar]
- 114.Shin S.S., Wall B.A., Goydos J.S., Chen S. AKT2 is a downstream target of metabotropic glutamate receptor 1 (Grm1) Pigment. Cell Melanoma Res. 2010;23:103–111. doi: 10.1111/j.1755-148X.2009.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Marín Y.E., Namkoong J., Cohen-Solal K., Shin S.S., Martino J.J., Oka M., Chen S. Stimulation of oncogenic metabotropic glutamate receptor 1 in melanoma cells activates ERK1/2 via PKCepsilon. Cell. Signal. 2006;18:1279–1286. doi: 10.1016/j.cellsig.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 116.Rutkowski P., Zdzienicki M., Nowecki Z.I., Van Akkooi A.C. Surgery of primary melanomas. Cancers. 2010;2:824–841. doi: 10.3390/cancers2020824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tyrell R., Antia C., Stanley S., Deutsch G.B. Surgical resection of metastatic melanoma in the era of immunotherapy and targeted therapy. Melanoma Manag. 2017;4:61–68. doi: 10.2217/mmt-2016-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Leung A.M., Hari D.M., Morton D.L. Surgery for distant melanoma metastasis. Cancer J. 2012;18:176–184. doi: 10.1097/PPO.0b013e31824bc981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang D., Dong Y., Sun X., Yuan S., Yu J. Surgery of primary tumor improves the survival of newly diagnosed metastatic melanoma: A population-based, propensity-matched study. Cancer Manag. Res. 2019;11:339–346. doi: 10.2147/CMAR.S187208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gannon C.J., Rousseau D.L., Jr., Ross M.I., Johnson M.M., Lee J.E., Mansfield P.F., Cormier J.N., Prieto V.G., Gershenwald J.E. Accuracy of lymphatic mapping and sentinel lymph node biopsy after previous wide local excision in patients with primary melanoma. Cancer. 2006;107:2647–2652. doi: 10.1002/cncr.22320. [DOI] [PubMed] [Google Scholar]
- 121.Swetter S.M., Tsao H., Bichakjian C.K., Curiel-Lewandrowski C., Elder D.E., Gershenwald J.E., Guild V., Grant-Kels J.M., Halpern A.C., Johnson T.M., et al. Guidelines of care for the management of primary cutaneous melanoma. J. Am. Acad. Dermatol. 2019;80:208–250. doi: 10.1016/j.jaad.2018.08.055. [DOI] [PubMed] [Google Scholar]
- 122.Smyth E.C., Flavin M., Pulitzer M.P., Gardner G.J., Costantino P.D., Chi D.S., Bogatch K., Chapman P.B., Wolchok J.D., Schwartz G.K., et al. Treatment of locally recurrent mucosal melanoma with topical imiquimod. J. Clin. Oncol. 2011;29:e809–e811. doi: 10.1200/JCO.2011.36.8829. [DOI] [PubMed] [Google Scholar]
- 123.Turza K., Dengel L.T., Harris R.C., Patterson J.W., White K., Grosh W.W., Slingluff C.L., Jr. Effectiveness of imiquimod limited to dermal melanoma metastases, with simultaneous resistance of subcutaneous metastasis. J. Cutan. Pathol. 2010;37:94–98. doi: 10.1111/j.1600-0560.2009.01290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nanda J., Bermudez R. StatPearls. StatPearls Publishing LLC.; Treasure Island, FL, USA: 2020. Imiquimod. [Google Scholar]
- 125.Schön M., Bong A.B., Drewniok C., Herz J., Geilen C.C., Reifenberger J., Benninghoff B., Slade H.B., Gollnick H., Schön M.P. Tumor-selective induction of apoptosis and the small-molecule immune response modifier imiquimod. J. Natl. Cancer Inst. 2003;95:1138–1149. doi: 10.1093/jnci/djg016. [DOI] [PubMed] [Google Scholar]
- 126.Shi W. Radiation Therapy for Melanoma. In: Ward W.H., Farma J.M., editors. Cutaneous Melanoma: Etiology and Therapy. Codon Publication; Brisbane, Australia: 2017. [DOI] [PubMed] [Google Scholar]
- 127.Strojan P. Role of radiotherapy in melanoma management. Radiol. Oncol. 2010;44:1–12. doi: 10.2478/v10019-010-0008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Postow M.A., Callahan M.K., Barker C.A., Yamada Y., Yuan J., Kitano S., Mu Z., Rasalan T., Adamow M., Ritter E., et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012;366:925–931. doi: 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.D’Andrea M.A., Reddy G.K. Systemic Antitumor Effects and Abscopal Responses in Melanoma Patients Receiving Radiation Therapy. Oncology. 2020;98:202–215. doi: 10.1159/000505487. [DOI] [PubMed] [Google Scholar]
- 130.Mole R.H. Whole body irradiation; radiobiology or medicine? Br. J. Radiol. 1953;26:234–241. doi: 10.1259/0007-1285-26-305-234. [DOI] [PubMed] [Google Scholar]
- 131.Lo J.A., Fisher D.E. The melanoma revolution: From UV carcinogenesis to a new era in therapeutics. Science. 2014;346:945–949. doi: 10.1126/science.1253735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wilson M.A., Schuchter L.M. Chemotherapy for Melanoma. Cancer Treat. Res. 2016;167:209–229. doi: 10.1007/978-3-319-22539-5_8. [DOI] [PubMed] [Google Scholar]
- 133.Chang W., Lee S.J., Park S., Choi M.K., Hong J.Y., Kim Y.S., Maeng C.H., Jung H.A., Kim S., Lee J. Effect of paclitaxel/carboplatin salvage chemotherapy in noncutaneous versus cutaneous metastatic melanoma. Melanoma Res. 2013;23:147–151. doi: 10.1097/CMR.0b013e32835efd8d. [DOI] [PubMed] [Google Scholar]
- 134.Vakifahmetoglu H., Olsson M., Zhivotovsky B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008;15:1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 135.Morse D.L., Gray H., Payne C.M., Gillies R.J. Docetaxel induces cell death through mitotic catastrophe in human breast cancer cells. Mol. Cancer Ther. 2005;4:1495–1504. doi: 10.1158/1535-7163.MCT-05-0130. [DOI] [PubMed] [Google Scholar]
- 136.Guerriero J.L., Ditsworth D., Fan Y., Zhao F., Crawford H.C., Zong W.X. Chemotherapy induces tumor clearance independent of apoptosis. Cancer Res. 2008;68:9595–9600. doi: 10.1158/0008-5472.CAN-08-2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ricci M.S., Zong W.X. Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 2006;11:342–357. doi: 10.1634/theoncologist.11-4-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shtivelman E., Davies M.Q., Hwu P., Yang J., Lotem M., Oren M., Flaherty K.T., Fisher D.E. Pathways and therapeutic targets in melanoma. Oncotarget. 2014;5:1701–1752. doi: 10.18632/oncotarget.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chapman P.B., Hauschild A., Robert C., Haanen J.B., Ascierto P., Larkin J., Dummer R., Garbe C., Testori A., Maio M., et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tsai J., Lee J.T., Wang W., Zhang J., Cho H., Mamo S., Bremer R., Gillette S., Kong J., Haass N.K., et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Søndergaard J.N., Nazarian R., Wang Q., Guo D., Hsueh T., Mok S., Sazegar H., MacConaill L.E., Barretina J.G., Kehoe S.M., et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J. Transl. Med. 2010;8:39. doi: 10.1186/1479-5876-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Flaherty K.T., Puzanov I., Kim K.B., Ribas A., McArthur G.A., Sosman J.A., O’Dwyer P.J., Lee R.J., Grippo J.F., Nolop K., et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Falchook G.S., Long G.V., Kurzrock R., Kim K.B., Arkenau T.H., Brown M.P., Hamid O., Infante J.R., Millward M., Pavlick A.C., et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet. 2012;379:1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ascierto P.A., Minor D., Ribas A., Lebbe C., O’Hagan A., Arya N., Guckert M., Schadendorf D., Kefford R.F., Grob J.J., et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J. Clin. Oncol. 2013;31:3205–3211. doi: 10.1200/JCO.2013.49.8691. [DOI] [PubMed] [Google Scholar]
- 145.Hauschild A., Grob J.J., Demidov L.V., Jouary T., Gutzmer R., Millward M., Rutkowski P., Blank C.U., Miller W.H., Jr., Kaempgen E., et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 146.Falchook G.S., Lewis K.D., Infante J.R., Gordon M.S., Vogelzang N.J., DeMarini D.J., Sun P., Moy C., Szabo S.A., Roadcap L.T., et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–789. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Flaherty K.T., Robert C., Hersey P., Nathan P., Garbe C., Milhem M., Demidov L.V., Hassel J.C., Rutkowski P., Mohr P., et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012;367:107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 148.Eroglu Z., Ribas A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016;8:48–56. doi: 10.1177/1758834015616934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Flaherty K.T., Infante J.R., Daud A., Gonzalez R., Kefford R.F., Sosman J., Hamid O., Schuchter L., Cebon J., Ibrahim N., et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Long G.V., Stroyakovskiy D., Gogas H., Levchenko E., de Braud F., Larkin J., Garbe C., Jouary T., Hauschild A., Grob J.J., et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014;371:1877–1888. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 151.Robert C., Karaszewska B., Schachter J., Rutkowski P., Mackiewicz A., Stroiakovski D., Lichinitser M., Dummer R., Grange F., Mortier L., et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 152.Larkin J., Ascierto P.A., Dréno B., Atkinson V., Liszkay G., Maio M., Mandalà M., Demidov L., Stroyakovskiy D., Thomas L., et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014;371:1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 153.Ascierto P.A., McArthur G.A., Dréno B., Atkinson V., Liszkay G., Di Giacomo A.M., Mandalà M., Demidov L., Stroyakovskiy D., Thomas L., et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248–1260. doi: 10.1016/S1470-2045(16)30122-X. [DOI] [PubMed] [Google Scholar]
- 154.Dummer R., Ascierto P.A., Gogas H.J., Arance A., Mandala M., Liszkay G., Garbe C., Schadendorf D., Krajsova I., Gutzmer R., et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603–615. doi: 10.1016/S1470-2045(18)30142-6. [DOI] [PubMed] [Google Scholar]
- 155.Dummer R., Ascierto P.A., Gogas H.J., Arance A., Mandala M., Liszkay G., Garbe C., Schadendorf D., Krajsova I., Gutzmer R., et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315–1327. doi: 10.1016/S1470-2045(18)30497-2. [DOI] [PubMed] [Google Scholar]
- 156.Beadling C., Jacobson-Dunlop E., Hodi F.S., Le C., Warrick A., Patterson J., Town A., Harlow A., Cruz F., III, Azar S., et al. KIT gene mutations and copy number in melanoma subtypes. Clin. Cancer Res. 2008;14:6821–6828. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- 157.Lebbe C., Chevret S., Jouary T., Dalac S., Dalle S., Guillot B., Arnault J.-P., Avril M.-F., Bedane C., Bens G., et al. Phase II multicentric uncontrolled national trial assessing the efficacy of nilotinib in the treatment of advanced melanomas with c-KIT mutation or amplification. J. Clin. Oncol. 2014;32:9032. doi: 10.1200/jco.2014.32.15_suppl.9032. [DOI] [Google Scholar]
- 158.Hodi F.S., Corless C.L., Giobbie-Hurder A., Fletcher J.A., Zhu M., Marino-Enriquez A., Friedlander P., Gonzalez R., Weber J.S., Gajewski T.F., et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013;31:3182–3190. doi: 10.1200/JCO.2012.47.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Guo J., Carvajal R.D., Dummer R., Hauschild A., Daud A., Bastian B.C., Markovic S.N., Queirolo P., Arance A., Berking C., et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: Final results from the global, single-arm, phase II TEAM trial. Ann. Oncol. 2017;28:1380–1387. doi: 10.1093/annonc/mdx079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Slipicevic A., Herlyn M. KIT in melanoma: Many shades of gray. J. Investig. Dermatol. 2015;135:337–338. doi: 10.1038/jid.2014.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dahl C., Abildgaard C., Riber-Hansen R., Steiniche T., Lade-Keller J., Guldberg P. KIT is a frequent target for epigenetic silencing in cutaneous melanoma. J. Investig. Dermatol. 2015;135:516–524. doi: 10.1038/jid.2014.372. [DOI] [PubMed] [Google Scholar]
- 162.Shen L., Shi Q., Wang W. Double agents: Genes with both oncogenic and tumor-suppressor functions. Oncogenesis. 2018;7:25. doi: 10.1038/s41389-018-0034-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hanahan D., Weinberg R.A. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 164.Nishimura H., Nose M., Hiai H., Minato N., Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/S1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 165.Freeman G.J., Long A.J., Iwai Y., Bourque K., Chernova T., Nishimura H., Fitz L.J., Malenkovich N., Okazaki T., Byrne M.C., et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nishimura H., Honjo T. PD-1: An inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 2001;22:265–268. doi: 10.1016/S1471-4906(01)01888-9. [DOI] [PubMed] [Google Scholar]
- 167.Nishimura H., Okazaki T., Tanaka Y., Nakatani K., Hara M., Matsumori A., Sasayama S., Mizoguchi A., Hiai H., Minato N., et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 168.Barber D.L., Wherry E.J., Masopust D., Zhu B., Allison J.P., Sharpe A.H., Freeman G.J., Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 169.Chemnitz J.M., Parry R.V., Nichols K.E., June C.H., Riley J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004;173:945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 170.Blackburn S.D., Shin H., Haining W.N., Zou T., Workman C.J., Polley A., Betts M.R., Freeman G.J., Vignali D.A., Wherry E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Baumeister S.H., Freeman G.J., Dranoff G., Sharpe A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016;34:539–573. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 172.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Latchman Y., Wood C.R., Chernova T., Chaudhary D., Borde M., Chernova I., Iwai Y., Long A.J., Brown J.A., Nunes R., et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 174.Ghoneim H.E., Fan Y., Moustaki A., Abdelsamed H.A., Dash P., Dogra P., Carter R., Awad W., Neale G., Thomas P.G., et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell. 2017;170:142–157. doi: 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pauken K.E., Sammons M.A., Odorizzi P.M., Manne S., Godec J., Khan O., Drake A.M., Chen Z., Sen D.R., Kurachi M., et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pereira R.M., Hogan P.G., Rao A., Martinez G.J. Transcriptional and epigenetic regulation of T cell hyporesponsiveness. J. Leukoc. Biol. 2017;102:601–615. doi: 10.1189/jlb.2RI0317-097R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Dong W., Wu X., Ma S., Wang Y., Nalin A.P., Zhu Z., Zhang J., Benson D.M., He K., Caligiuri M.A., et al. The Mechanism of Anti-PD-L1 Antibody Efficacy against PD-L1-Negative Tumors Identifies NK Cells Expressing PD-L1 as a Cytolytic Effector. Cancer Discov. 2019;9:1422–1437. doi: 10.1158/2159-8290.CD-18-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hartley G., Regan D., Guth A., Dow S. Regulation of PD-L1 expression on murine tumor-associated monocytes and macrophages by locally produced TNF-α. Cancer Immunol. Immunother. 2017;66:523–535. doi: 10.1007/s00262-017-1955-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., Sosman J.A., McDermott D.F., Powderly J.D., Gettinger S.N., et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hartley G.P., Chow L., Ammons D.T., Wheat W.H., Dow S.W. Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol. Res. 2018;6:1260–1273. doi: 10.1158/2326-6066.CIR-17-0537. [DOI] [PubMed] [Google Scholar]
- 181.Ahmadzadeh M., Johnson L.A., Heemskerk B., Wunderlich J.R., Dudley M.E., White D.E., Rosenberg S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Curiel T.J., Wei S., Dong H., Alvarez X., Cheng P., Mottram P., Krzysiek R., Knutson K.L., Daniel B., Zimmermann M.C., et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 183.Dammeijer F., van Gulijk M., Mulder E.E., Lukkes M., Klaase L., van den Bosch T., van Nimwegen M., Lau S.P., Latupeirissa K., Schetters S., et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell. 2020;38:685–700.e8. doi: 10.1016/j.ccell.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 184.Yearley J.H., Gibson C., Yu N., Moon C., Murphy E., Juco J., Lunceford J., Cheng J., Chow L.Q.M., Seiwert T.Y., et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017;23:3158–3167. doi: 10.1158/1078-0432.CCR-16-1761. [DOI] [PubMed] [Google Scholar]
- 185.Obeid J.M., Erdag G., Smolkin M.E., Deacon D.H., Patterson J.W., Chen L., Bullock T.N., Slingluff C.L. PD-L1, PD-L2 and PD-1 expression in metastatic melanoma: Correlation with tumor-infiltrating immune cells and clinical outcome. Oncoimmunology. 2016;5:e1235107. doi: 10.1080/2162402X.2016.1235107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rodig N., Ryan T., Allen J.A., Pang H., Grabie N., Chernova T., Greenfield E.A., Liang S.C., Sharpe A.H., Lichtman A.H., et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur. J. Immunol. 2003;33:3117–3126. doi: 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]
- 187.Taube J.M., Anders R.A., Young G.D., Xu H., Sharma R., McMiller T.L., Chen S., Klein A.P., Pardoll D.M., Topalian S.L., et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012;4:127ra137. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kaunitz G.J., Cottrell T.R., Lilo M., Muthappan V., Esandrio J., Berry S., Xu H., Ogurtsova A., Anders R.A., Fischer A.H., et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Investig. 2017;97:1063–1071. doi: 10.1038/labinvest.2017.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Morales-Betanzos C.A., Lee H., Gonzalez Ericsson P.I., Balko J.M., Johnson D.B., Zimmerman L.J., Liebler D.C. Quantitative Mass Spectrometry Analysis of PD-L1 Protein Expression, N-glycosylation and Expression Stoichiometry with PD-1 and PD-L2 in Human Melanoma. Mol. Cell. Proteomics. 2017;16:1705–1717. doi: 10.1074/mcp.RA117.000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Youngnak P., Kozono Y., Kozono H., Iwai H., Otsuki N., Jin H., Omura K., Yagita H., Pardoll D.M., Chen L., et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem. Biophys. Res. Commun. 2003;307:672–677. doi: 10.1016/S0006-291X(03)01257-9. [DOI] [PubMed] [Google Scholar]
- 191.Li C.W., Lim S.O., Xia W., Lee H.H., Chan L.C., Kuo C.W., Khoo K.H., Chang S.S., Cha J.H., Kim T., et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Maher C.M., Thomas J.D., Haas D.A., Longen C.G., Oyer H.M., Tong J.Y., Kim F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2018;16:243–255. doi: 10.1158/1541-7786.MCR-17-0166. [DOI] [PubMed] [Google Scholar]
- 193.Hsu J.M., Li C.W., Lai Y.J., Hung M.C. Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy. Cancer Res. 2018;78:6349–6353. doi: 10.1158/0008-5472.CAN-18-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zerdes I., Matikas A., Bergh J., Rassidakis G.Z., Foukakis T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: Biology and clinical correlations. Oncogene. 2018;37:4639–4661. doi: 10.1038/s41388-018-0303-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dong H., Strome S.E., Salomao D.R., Tamura H., Hirano F., Flies D.B., Roche P.C., Lu J., Zhu G., Tamada K., et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 196.Garcia-Diaz A., Shin D.S., Moreno B.H., Saco J., Escuin-Ordinas H., Rodriguez G.A., Zaretsky J.M., Sun L., Hugo W., Wang X., et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017;19:1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Thiem A., Hesbacher S., Kneitz H., Di Primio T., Heppt M.V., Hermanns H.M., Goebeler M., Meierjohann S., Houben R., Schrama D. IFN-gamma-induced PD-L1 expression in melanoma depends on p53 expression. J. Exp. Clin. Cancer Res. 2019;38:397. doi: 10.1186/s13046-019-1403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Casey S.C., Tong L., Li Y., Do R., Walz S., Fitzgerald K.N., Gouw A.M., Baylot V., Gütgemann I., Eilers M., et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–231. doi: 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Atefi M., Avramis E., Lassen A., Wong D.J., Robert L., Foulad D., Cerniglia M., Titz B., Chodon T., Graeber T.G., et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin. Cancer Res. 2014;20:3446–3457. doi: 10.1158/1078-0432.CCR-13-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Jiang X., Zhou J., Giobbie-Hurder A., Wargo J., Hodi F.S. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin. Cancer Res. 2013;19:598–609. doi: 10.1158/1078-0432.CCR-12-2731. [DOI] [PubMed] [Google Scholar]
- 201.Alcázar I., Marqués M., Kumar A., Hirsch E., Wymann M., Carrera A.C., Barber D.F. Phosphoinositide 3-kinase gamma participates in T cell receptor-induced T cell activation. J. Exp. Med. 2007;204:2977–2987. doi: 10.1084/jem.20070366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.D’Souza W.N., Chang C.F., Fischer A.M., Li M., Hedrick S.M. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J. Immunol. 2008;181:7617–7629. doi: 10.4049/jimmunol.181.11.7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Adachi K., Davis M.M. T-cell receptor ligation induces distinct signaling pathways in naive vs. antigen-experienced T cells. Proc. Natl. Acad. Sci. USA. 2011;108:1549–1554. doi: 10.1073/pnas.1017340108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Boussiotis V.A., Chatterjee P., Li L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014;20:265–271. doi: 10.1097/PPO.0000000000000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Arasanz H., Gato-Cañas M., Zuazo M., Ibañez-Vea M., Breckpot K., Kochan G., Escors D. PD1 signal transduction pathways in T cells. Oncotarget. 2017;8:51936–51945. doi: 10.18632/oncotarget.17232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Parry R.V., Chemnitz J.M., Frauwirth K.A., Lanfranco A.R., Braunstein I., Kobayashi S.V., Linsley P.S., Thompson C.B., Riley J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Patsoukis N., Duke-Cohan J.S., Chaudhri A., Aksoylar H.I., Wang Q., Council A., Berg A., Freeman G.J., Boussiotis V.A. Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun. Biol. 2020;3:128. doi: 10.1038/s42003-020-0845-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Quigley M., Pereyra F., Nilsson B., Porichis F., Fonseca C., Eichbaum Q., Julg B., Jesneck J.L., Brosnahan K., Imam S., et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010;16:1147–1151. doi: 10.1038/nm.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gibbons R.M., Liu X., Pulko V., Harrington S.M., Krco C.J., Kwon E.D., Dong H. B7-H1 limits the entry of effector CD8(+) T cells to the memory pool by upregulating Bim. Oncoimmunology. 2012;1:1061–1073. doi: 10.4161/onci.20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Fife B.T., Pauken K.E., Eagar T.N., Obu T., Wu J., Tang Q., Azuma M., Krummel M.F., Bluestone J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009;10:1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Patsoukis N., Bardhan K., Chatterjee P., Sari D., Liu B., Bell L.N., Karoly E.D., Freeman G.J., Petkova V., Seth P., et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yokosuka T., Takamatsu M., Kobayashi-Imanishi W., Hashimoto-Tane A., Azuma M., Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012;209:1201–1217. doi: 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Okazaki T., Maeda A., Nishimura H., Kurosaki T., Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA. 2001;98:13866–13871. doi: 10.1073/pnas.231486598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Patsoukis N., Li L., Sari D., Petkova V., Boussiotis V.A. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol. Cell. Biol. 2013;33:3091–3098. doi: 10.1128/MCB.00319-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Qin W., Hu L., Zhang X., Jiang S., Li J., Zhang Z., Wang X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019;10:2298. doi: 10.3389/fimmu.2019.02298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Leclerc M., Voilin E., Gros G., Corgnac S., de Montpréville V., Validire P., Bismuth G., Mami-Chouaib F. Regulation of antitumour CD8 T-cell immunity and checkpoint blockade immunotherapy by Neuropilin-1. Nat. Commun. 2019;10:3345. doi: 10.1038/s41467-019-11280-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tivol E.A., Borriello F., Schweitzer A.N., Lynch W.P., Bluestone J.A., Sharpe A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 218.Waterhouse P., Penninger J.M., Timms E., Wakeham A., Shahinian A., Lee K.P., Thompson C.B., Griesser H., Mak T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 219.Leach D.R., Krummel M.F., Allison J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 220.Sakuishi K., Apetoh L., Sullivan J.M., Blazar B.R., Kuchroo V.K., Anderson A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Fourcade J., Sun Z., Benallaoua M., Guillaume P., Luescher I.F., Sander C., Kirkwood J.M., Kuchroo V., Zarour H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010;207:2175–2186. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Grosso J.F., Kelleher C.C., Harris T.J., Maris C.H., Hipkiss E.L., De Marzo A., Anders R., Netto G., Getnet D., Bruno T.C., et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Investig. 2007;117:3383–3392. doi: 10.1172/JCI31184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Huang C.T., Workman C.J., Flies D., Pan X., Marson A.L., Zhou G., Hipkiss E.L., Ravi S., Kowalski J., Levitsky H.I., et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 224.Lines J.L., Pantazi E., Mak J., Sempere L.F., Wang L., O’Connell S., Ceeraz S., Suriawinata A.A., Yan S., Ernstoff M.S., et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014;74:1924–1932. doi: 10.1158/0008-5472.CAN-13-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sansom D.M. CD28, CTLA-4 and their ligands: Who does what and to whom? Immunology. 2000;101:169–177. doi: 10.1046/j.1365-2567.2000.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Azuma M., Ito D., Yagita H., Okumura K., Phillips J.H., Lanier L.L., Somoza C. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. 1993;366:76–79. doi: 10.1038/366076a0. [DOI] [PubMed] [Google Scholar]
- 227.Greene J.L., Leytze G.M., Emswiler J., Peach R., Bajorath J., Cosand W., Linsley P.S. Covalent dimerization of CD28/CTLA-4 and oligomerization of CD80/CD86 regulate T cell costimulatory interactions. J. Biol. Chem. 1996;271:26762–26771. doi: 10.1074/jbc.271.43.26762. [DOI] [PubMed] [Google Scholar]
- 228.Linsley P.S., Greene J.L., Brady W., Bajorath J., Ledbetter J.A., Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1:793–801. doi: 10.1016/S1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- 229.van der Merwe P.A., Bodian D.L., Daenke S., Linsley P., Davis S.J. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med. 1997;185:393–403. doi: 10.1084/jem.185.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Linsley P.S., Brady W., Urnes M., Grosmaire L.S., Damle N.K., Ledbetter J.A. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 1991;174:561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kleffel S., Posch C., Barthel S.R., Mueller H., Schlapbach C., Guenova E., Elco C.P., Lee N., Juneja V.R., Zhan Q., et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell. 2015;162:1242–1256. doi: 10.1016/j.cell.2015.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Clark C.A., Gupta H.B., Sareddy G., Pandeswara S., Lao S., Yuan B., Drerup J.M., Padron A., Conejo-Garcia J., Murthy K., et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016;76:6964–6974. doi: 10.1158/0008-5472.CAN-16-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Mo X., Zhang H., Preston S., Martin K., Zhou B., Vadalia N., Gamero A.M., Soboloff J., Tempera I., Zaidi M.R. Interferon-γ Signaling in Melanocytes and Melanoma Cells Regulates Expression of CTLA-4. Cancer Res. 2018;78:436–450. doi: 10.1158/0008-5472.CAN-17-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Contardi E., Palmisano G.L., Tazzari P.L., Martelli A.M., Falà F., Fabbi M., Kato T., Lucarelli E., Donati D., Polito L., et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer. 2005;117:538–550. doi: 10.1002/ijc.21155. [DOI] [PubMed] [Google Scholar]
- 235.Seliger B., Maio M., Cuaia O., Calabro L. Expression and function of CTLA4 in melanoma. J. Clin. Oncol. 2013;31:e20040. doi: 10.1200/jco.2013.31.15_suppl.e20040. [DOI] [Google Scholar]
- 236.Pistillo M.P., Carosio R., Grillo F., Fontana V., Mastracci L., Morabito A., Banelli B., Tanda E., Cecchi F., Dozin B., et al. Phenotypic characterization of tumor CTLA-4 expression in melanoma tissues and its possible role in clinical response to Ipilimumab. Clin. Immunol. 2020;215:108428. doi: 10.1016/j.clim.2020.108428. [DOI] [PubMed] [Google Scholar]
- 237.Vignali D.A., Collison L.W., Workman C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Viguier M., Lemaître F., Verola O., Cho M.S., Gorochov G., Dubertret L., Bachelez H., Kourilsky P., Ferradini L. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J. Immunol. 2004;173:1444–1453. doi: 10.4049/jimmunol.173.2.1444. [DOI] [PubMed] [Google Scholar]
- 239.Gray C.P., Arosio P., Hersey P. Association of increased levels of heavy-chain ferritin with increased CD4+ CD25+ regulatory T-cell levels in patients with melanoma. Clin. Cancer Res. 2003;9:2551–2559. [PubMed] [Google Scholar]
- 240.Fujii H., Josse J., Tanioka M., Miyachi Y., Husson F., Ono M. Regulatory T Cells in Melanoma Revisited by a Computational Clustering of FOXP3+ T Cell Subpopulations. J. Immunol. 2016;196:2885–2892. doi: 10.4049/jimmunol.1402695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Magnuson A.M., Kiner E., Ergun A., Park J.S., Asinovski N., Ortiz-Lopez A., Kilcoyne A., Paoluzzi-Tomada E., Weissleder R., Mathis D., et al. Identification and validation of a tumor-infiltrating Treg transcriptional signature conserved across species and tumor types. Proc. Natl. Acad. Sci. USA. 2018;115:E10672–E10681. doi: 10.1073/pnas.1810580115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Ladányi A., Mohos A., Somlai B., Liszkay G., Gilde K., Fejos Z., Gaudi I., Tímár J. FOXP3+ cell density in primary tumor has no prognostic impact in patients with cutaneous malignant melanoma. Pathol. Oncol. Res. 2010;16:303–309. doi: 10.1007/s12253-010-9254-x. [DOI] [PubMed] [Google Scholar]
- 243.Leslie C., Bowyer S.E., White A., Grieu-Iacopetta F., Trevenen M., Iacopetta B., Amanuel B., Millward M. FOXP3+ T regulatory lymphocytes in primary melanoma are associated with BRAF mutation but not with response to BRAF inhibitor. Pathology. 2015;47:557–563. doi: 10.1097/PAT.0000000000000314. [DOI] [PubMed] [Google Scholar]
- 244.Klages K., Mayer C.T., Lahl K., Loddenkemper C., Teng M.W., Ngiow S.F., Smyth M.J., Hamann A., Huehn J., Sparwasser T. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res. 2010;70:7788–7799. doi: 10.1158/0008-5472.CAN-10-1736. [DOI] [PubMed] [Google Scholar]
- 245.Rasku M.A., Clem A.L., Telang S., Taft B., Gettings K., Gragg H., Cramer D., Lear S.C., McMasters K.M., Miller D.M., et al. Transient T cell depletion causes regression of melanoma metastases. J. Transl. Med. 2008;6:12. doi: 10.1186/1479-5876-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Klarquist J., Tobin K., Farhangi Oskuei P., Henning S.W., Fernandez M.F., Dellacecca E.R., Navarro F.C., Eby J.M., Chatterjee S., Mehrotra S., et al. Ccl22 Diverts T Regulatory Cells and Controls the Growth of Melanoma. Cancer Res. 2016;76:6230–6240. doi: 10.1158/0008-5472.CAN-16-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Ahmadzadeh M., Felipe-Silva A., Heemskerk B., Powell D.J., Jr., Wunderlich J.R., Merino M.J., Rosenberg S.A. FOXP3 expression accurately defines the population of intratumoral regulatory T cells that selectively accumulate in metastatic melanoma lesions. Blood. 2008;112:4953–4960. doi: 10.1182/blood-2008-06-163048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Alb M., Sie C., Adam C., Chen S., Becker J.C., Schrama D. Cellular and cytokine-dependent immunosuppressive mechanisms of grm1-transgenic murine melanoma. Cancer Immunol. Immunother. 2012;61:2239–2249. doi: 10.1007/s00262-012-1290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Gray C.P., Franco A.V., Arosio P., Hersey P. Immunosuppressive effects of melanoma-derived heavy-chain ferritin are dependent on stimulation of IL-10 production. Int. J. Cancer. 2001;92:843–850. doi: 10.1002/ijc.1269. [DOI] [PubMed] [Google Scholar]
- 250.Gray C.P., Arosio P., Hersey P. Heavy chain ferritin activates regulatory T cells by induction of changes in dendritic cells. Blood. 2002;99:3326–3334. doi: 10.1182/blood.V99.9.3326. [DOI] [PubMed] [Google Scholar]
- 251.Deng G. Tumor-infiltrating regulatory T cells: Origins and features. Am. J. Clin. Exp. Immunol. 2018;7:81–87. [PMC free article] [PubMed] [Google Scholar]
- 252.Fan X., Allison J.P. Chemokines and recruitment of regulatory T cells to the tumor. J. Immunol. 2009;182:40.14. [Google Scholar]
- 253.Chaudhary B., Elkord E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines. 2016;4:28. doi: 10.3390/vaccines4030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Baumgartner J., Wilson C., Palmer B., Richter D., Banerjee A., McCarter M. Melanoma induces immunosuppression by up-regulating FOXP3(+) regulatory T cells. J. Surg. Res. 2007;141:72–77. doi: 10.1016/j.jss.2007.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Shevach E.M. Foxp3(+) T Regulatory Cells: Still Many Unanswered Questions-A Perspective After 20 Years of Study. Front. Immunol. 2018;9:1048. doi: 10.3389/fimmu.2018.01048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Ataera H., Hyde E., Price K.M., Stoitzner P., Ronchese F. Murine melanoma-infiltrating dendritic cells are defective in antigen presenting function regardless of the presence of CD4CD25 regulatory T cells. PLoS ONE. 2011;6:e17515. doi: 10.1371/journal.pone.0017515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kawamoto H., Minato N. Myeloid cells. Int. J. Biochem. Cell Biol. 2004;36:1374–1379. doi: 10.1016/j.biocel.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 258.Gabrilovich D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017;5:3–8. doi: 10.1158/2326-6066.CIR-16-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Condamine T., Ramachandran I., Youn J.I., Gabrilovich D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015;66:97–110. doi: 10.1146/annurev-med-051013-052304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.mansky V., Sevko A., Gebhardt C., Utikal J. Myeloid-derived suppressor cells in malignant melanoma. J. Dtsch. Dermatol. Ges. 2014;12:1021–1027. doi: 10.1111/ddg.12411. [DOI] [PubMed] [Google Scholar]
- 261.Schlecker E., Stojanovic A., Eisen C., Quack C., Falk C.S., Umansky V., Cerwenka A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J. Immunol. 2012;189:5602–5611. doi: 10.4049/jimmunol.1201018. [DOI] [PubMed] [Google Scholar]
- 262.Mairhofer D.G., Ortner D., Tripp C.H., Schaffenrath S., Fleming V., Heger L., Komenda K., Reider D., Dudziak D., Chen S., et al. Impaired gp100-Specific CD8(+) T-Cell Responses in the Presence of Myeloid-Derived Suppressor Cells in a Spontaneous Mouse Melanoma Model. J. Investig. Dermatol. 2015;135:2785–2793. doi: 10.1038/jid.2015.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Weide B., Martens A., Zelba H., Stutz C., Derhovanessian E., Di Giacomo A.M., Maio M., Sucker A., Schilling B., Schadendorf D., et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: Comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin. Cancer Res. 2014;20:1601–1609. doi: 10.1158/1078-0432.CCR-13-2508. [DOI] [PubMed] [Google Scholar]
- 264.Jordan K.R., Amaria R.N., Ramirez O., Callihan E.B., Gao D., Borakove M., Manthey E., Borges V.F., McCarter M.D. Myeloid-derived suppressor cells are associated with disease progression and decreased overall survival in advanced-stage melanoma patients. Cancer Immunol. Immunother. 2013;62:1711–1722. doi: 10.1007/s00262-013-1475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Meyer C., Sevko A., Ramacher M., Bazhin A.V., Falk C.S., Osen W., Borrello I., Kato M., Schadendorf D., Baniyash M., et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc. Natl. Acad. Sci. USA. 2011;108:17111–17116. doi: 10.1073/pnas.1108121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Poschke I., Mougiakakos D., Hansson J., Masucci G.V., Kiessling R. Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010;70:4335–4345. doi: 10.1158/0008-5472.CAN-09-3767. [DOI] [PubMed] [Google Scholar]
- 267.Raber P.L., Thevenot P., Sierra R., Wyczechowska D., Halle D., Ramirez M.E., Ochoa A.C., Fletcher M., Velasco C., Wilk A., et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int. J. Cancer. 2014;134:2853–2864. doi: 10.1002/ijc.28622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Kusmartsev S., Gabrilovich D.I. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J. Immunol. 2005;174:4880–4891. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- 269.Mazzoni A., Bronte V., Visintin A., Spitzer J.H., Apolloni E., Serafini P., Zanovello P., Segal D.M. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J. Immunol. 2002;168:689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 270.Apolloni E., Bronte V., Mazzoni A., Serafini P., Cabrelle A., Segal D.M., Young H.A., Zanovello P. Immortalized myeloid suppressor cells trigger apoptosis in antigen-activated T lymphocytes. J. Immunol. 2000;165:6723–6730. doi: 10.4049/jimmunol.165.12.6723. [DOI] [PubMed] [Google Scholar]
- 271.Gabrilovich D.I., Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kusmartsev S., Nefedova Y., Yoder D., Gabrilovich D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 273.Jayasingam S.D., Citartan M., Thang T.H., Mat Zin A.A., Ang K.C., Ch’ng E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019;9:1512. doi: 10.3389/fonc.2019.01512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Fujimura T., Kambayashi Y., Fujisawa Y., Hidaka T., Aiba S. Tumor-Associated Macrophages: Therapeutic Targets for Skin Cancer. Front. Oncol. 2018;8:3. doi: 10.3389/fonc.2018.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Tcyganov E., Mastio J., Chen E., Gabrilovich D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018;51:76–82. doi: 10.1016/j.coi.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Movahedi K., Laoui D., Gysemans C., Baeten M., Stangé G., Van den Bossche J., Mack M., Pipeleers D., In’t Veld P., De Baetselier P., et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 277.Laoui D., Van Overmeire E., Di Conza G., Aldeni C., Keirsse J., Morias Y., Movahedi K., Houbracken I., Schouppe E., Elkrim Y., et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74:24–30. doi: 10.1158/0008-5472.CAN-13-1196. [DOI] [PubMed] [Google Scholar]
- 278.Corzo C.A., Condamine T., Lu L., Cotter M.J., Youn J.I., Cheng P., Cho H.I., Celis E., Quiceno D.G., Padhya T., et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Palazón A., Aragonés J., Morales-Kastresana A., de Landázuri M.O., Melero I. Molecular pathways: Hypoxia response in immune cells fighting or promoting cancer. Clin. Cancer Res. 2012;18:1207–1213. doi: 10.1158/1078-0432.CCR-11-1591. [DOI] [PubMed] [Google Scholar]
- 280.Salmi S., Siiskonen H., Sironen R., Tyynelä-Korhonen K., Hirschovits-Gerz B., Valkonen M., Auvinen P., Pasonen-Seppänen S. The number and localization of CD68+ and CD163+ macrophages in different stages of cutaneous melanoma. Melanoma Res. 2019;29:237–247. doi: 10.1097/CMR.0000000000000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Falleni M., Savi F., Tosi D., Agape E., Cerri A., Moneghini L., Bulfamante G.P. M1 and M2 macrophages’ clinicopathological significance in cutaneous melanoma. Melanoma Res. 2017;27:200–210. doi: 10.1097/CMR.0000000000000352. [DOI] [PubMed] [Google Scholar]
- 282.Herwig M.C., Bergstrom C., Wells J.R., Höller T., Grossniklaus H.E. M2/M1 ratio of tumor associated macrophages and PPAR-gamma expression in uveal melanomas with class 1 and class 2 molecular profiles. Exp. Eye Res. 2013;107:52–58. doi: 10.1016/j.exer.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Bronkhorst I.H., Ly L.V., Jordanova E.S., Vrolijk J., Versluis M., Luyten G.P., Jager M.J. Detection of M2-macrophages in uveal melanoma and relation with survival. Invest. Ophthalmol. Vis. Sci. 2011;52:643–650. doi: 10.1167/iovs.10-5979. [DOI] [PubMed] [Google Scholar]
- 284.Mäkitie T., Summanen P., Tarkkanen A., Kivelä T. Tumor-infiltrating macrophages (CD68(+) cells) and prognosis in malignant uveal melanoma. Invest. Ophthalmol. Vis. Sci. 2001;42:1414–1421. [PubMed] [Google Scholar]
- 285.Jensen T.O., Schmidt H., Møller H.J., Høyer M., Maniecki M.B., Sjoegren P., Christensen I.J., Steiniche T. Macrophage markers in serum and tumor have prognostic impact in American Joint Committee on Cancer stage I/II melanoma. J. Clin. Oncol. 2009;27:3330–3337. doi: 10.1200/JCO.2008.19.9919. [DOI] [PubMed] [Google Scholar]
- 286.Ley K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017;199:2191–2193. doi: 10.4049/jimmunol.1701135. [DOI] [PubMed] [Google Scholar]
- 287.Buscher K., Ehinger E., Gupta P., Pramod A.B., Wolf D., Tweet G., Pan C., Mills C.D., Lusis A.J., Ley K. Natural variation of macrophage activation as disease-relevant phenotype predictive of inflammation and cancer survival. Nat. Commun. 2017;8:16041. doi: 10.1038/ncomms16041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Van Overmeire E., Stijlemans B., Heymann F., Keirsse J., Morias Y., Elkrim Y., Brys L., Abels C., Lahmar Q., Ergen C., et al. M-CSF and GM-CSF Receptor Signaling Differentially Regulate Monocyte Maturation and Macrophage Polarization in the Tumor Microenvironment. Cancer Res. 2016;76:35–42. doi: 10.1158/0008-5472.CAN-15-0869. [DOI] [PubMed] [Google Scholar]
- 289.Georgoudaki A.M., Prokopec K.E., Boura V.F., Hellqvist E., Sohn S., Östling J., Dahan R., Harris R.A., Rantalainen M., Klevebring D., et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016;15:2000–2011. doi: 10.1016/j.celrep.2016.04.084. [DOI] [PubMed] [Google Scholar]
- 290.Kaneda M.M., Messer K.S., Ralainirina N., Li H., Leem C.J., Gorjestani S., Woo G., Nguyen A.V., Figueiredo C.C., Foubert P., et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539:437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Vergadi E., Ieronymaki E., Lyroni K., Vaporidi K., Tsatsanis C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017;198:1006–1014. doi: 10.4049/jimmunol.1601515. [DOI] [PubMed] [Google Scholar]
- 292.Mouchemore K.A., Sampaio N.G., Murrey M.W., Stanley E.R., Lannutti B.J., Pixley F.J. Specific inhibition of PI3K p110δ inhibits CSF-1-induced macrophage spreading and invasive capacity. FEBS J. 2013;280:5228–5236. doi: 10.1111/febs.12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Huang S.C., Smith A.M., Everts B., Colonna M., Pearce E.L., Schilling J.D., Pearce E.J. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity. 2016;45:817–830. doi: 10.1016/j.immuni.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Arredouani M.S. Is the scavenger receptor MARCO a new immune checkpoint? Oncoimmunology. 2014;3:e955709. doi: 10.4161/21624011.2014.955709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. 4th ed. Garland Science; New York, NY, USA: 2002. Helper T Cells and Lymphocyte Activation. [Google Scholar]
- 296.Mills C.D., Kincaid K., Alt J.M., Heilman M.J., Hill A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000;164:6166–6173. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 297.Nevala W.K., Vachon C.M., Leontovich A.A., Scott C.G., Thompson M.A., Markovic S.N. Evidence of systemic Th2-driven chronic inflammation in patients with metastatic melanoma. Clin. Cancer Res. 2009;15:1931–1939. doi: 10.1158/1078-0432.CCR-08-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Lauerova L., Dusek L., Simickova M., Kocák I., Vagundová M., Zaloudík J., Kovarík J. Malignant melanoma associates with Th1/Th2 imbalance that coincides with disease progression and immunotherapy response. Neoplasma. 2002;49:159–166. [PubMed] [Google Scholar]
- 299.Enninga E.A., Nevala W.K., Holtan S.G., Leontovich A.A., Markovic S.N. Galectin-9 modulates immunity by promoting Th2/M2 differentiation and impacts survival in patients with metastatic melanoma. Melanoma Res. 2016;26:429–441. doi: 10.1097/CMR.0000000000000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Biswas S.K., Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 301.Massi D., Marconi C., Franchi A., Bianchini F., Paglierani M., Ketabchi S., Miracco C., Santucci M., Calorini L. Arginine metabolism in tumor-associated macrophages in cutaneous malignant melanoma: Evidence from human and experimental tumors. Hum. Pathol. 2007;38:1516–1525. doi: 10.1016/j.humpath.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 302.Wang H., Yang L., Wang D., Zhang Q., Zhang L. Pro-tumor activities of macrophages in the progression of melanoma. Hum. Vaccines Immunother. 2017;13:1556–1562. doi: 10.1080/21645515.2017.1312043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Chen P., Huang Y., Bong R., Ding Y., Song N., Wang X., Song X., Luo Y. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin. Cancer Res. 2011;17:7230–7239. doi: 10.1158/1078-0432.CCR-11-1354. [DOI] [PubMed] [Google Scholar]
- 304.Tucci M., Passarelli A., Mannavola F., Felici C., Stucci L.S., Cives M., Silvestris F. Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front. Oncol. 2019;9:1148. doi: 10.3389/fonc.2019.01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Escors D. Tumour immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J. Sci. 2014;2014 doi: 10.1155/2014/734515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Stoitzner P., Green L.K., Jung J.Y., Price K.M., Atarea H., Kivell B., Ronchese F. Inefficient presentation of tumor-derived antigen by tumor-infiltrating dendritic cells. Cancer Immunol. Immunother. 2008;57:1665–1673. doi: 10.1007/s00262-008-0487-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Itakura E., Huang R.R., Wen D.R., Paul E., Wünsch P.H., Cochran A.J. IL-10 expression by primary tumor cells correlates with melanoma progression from radial to vertical growth phase and development of metastatic competence. Mod. Pathol. 2011;24:801–809. doi: 10.1038/modpathol.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Chattopadhyay G., Shevach E.M. Antigen-specific induced T regulatory cells impair dendritic cell function via an IL-10/MARCH1-dependent mechanism. J. Immunol. 2013;191:5875–5884. doi: 10.4049/jimmunol.1301693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Mittal S.K., Roche P.A. Suppression of antigen presentation by IL-10. Curr. Opin. Immunol. 2015;34:22–27. doi: 10.1016/j.coi.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Groux H., Bigler M., de Vries J.E., Roncarolo M.G. Inhibitory and stimulatory effects of IL-10 on human CD8+ T cells. J. Immunol. 1998;160:3188–3193. [PubMed] [Google Scholar]
- 311.Laurent S., Carrega P., Saverino D., Piccioli P., Camoriano M., Morabito A., Dozin B., Fontana V., Simone R., Mortara L., et al. CTLA-4 is expressed by human monocyte-derived dendritic cells and regulates their functions. Hum. Immunol. 2010;71:934–941. doi: 10.1016/j.humimm.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 312.Yao S., Wang S., Zhu Y., Luo L., Zhu G., Flies S., Xu H., Ruff W., Broadwater M., Choi I.H., et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. 2009;113:5811–5818. doi: 10.1182/blood-2009-02-203141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Hobo W., Maas F., Adisty N., De Witte T., Schaap N., Van der Voort R., Dolstra H. siRNA silencing of PD-L1 and PD-L2 on dendritic cells augments expansion and function of minor histocompatibility antigen-specific CD8+ T cells. Blood. 2010;116:4501–4511. doi: 10.1182/blood-2010-04-278739. [DOI] [PubMed] [Google Scholar]
- 314.Oh S.A., Wu D.-C., Cheung J., Navarro A., Xiong H., Cubas R., Totpal K., Chiu H., Wu Y., Comps-Agrar L., et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer. 2020;1:681–691. doi: 10.1038/s43018-020-0075-x. [DOI] [PubMed] [Google Scholar]
- 315.Wang X.B., Fan Z.Z., Anton D., Vollenhoven A.V., Ni Z.H., Chen X.F., Lefvert A.K. CTLA4 is expressed on mature dendritic cells derived from human monocytes and influences their maturation and antigen presentation. BMC Immunol. 2011;12:21. doi: 10.1186/1471-2172-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Zhang W., Song Z., Xiao J., Liu X., Luo Y., Yang Z., Luo R., Li A. Blocking the PD-1/PD-L1 axis in dendritic cell-stimulated Cytokine-Induced Killer Cells with pembrolizumab enhances their therapeutic effects against hepatocellular carcinoma. J. Cancer. 2019;10:2578–2587. doi: 10.7150/jca.26961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Alexander M.A., Bennicelli J., Guerry D.T. Defective antigen presentation by human melanoma cell lines cultured from advanced, but not biologically early, disease. J. Immunol. 1989;142:4070–4078. [PubMed] [Google Scholar]
- 318.Kageshita T., Hirai S., Ono T., Hicklin D.J., Ferrone S. Down-regulation of HLA class I antigen-processing molecules in malignant melanoma: Association with disease progression. Am. J. Pathol. 1999;154:745–754. doi: 10.1016/S0002-9440(10)65321-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Chang C.C., Pirozzi G., Wen S.H., Chung I.H., Chiu B.L., Errico S., Luongo M., Lombardi M.L., Ferrone S. Multiple structural and epigenetic defects in the human leukocyte antigen class I antigen presentation pathway in a recurrent metastatic melanoma following immunotherapy. J. Biol. Chem. 2015;290:26562–26575. doi: 10.1074/jbc.M115.676130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Khan A.N., Gregorie C.J., Tomasi T.B. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol. Immunother. 2008;57:647–654. doi: 10.1007/s00262-007-0402-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Serrano A., Tanzarella S., Lionello I., Mendez R., Traversari C., Ruiz-Cabello F., Garrido F. Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2’-deoxycytidine treatment. Int. J. Cancer. 2001;94:243–251. doi: 10.1002/ijc.1452. [DOI] [PubMed] [Google Scholar]
- 322.Del Campo A.B., Kyte J.A., Carretero J., Zinchencko S., Méndez R., González-Aseguinolaza G., Ruiz-Cabello F., Aamdal S., Gaudernack G., Garrido F., et al. Immune escape of cancer cells with beta2-microglobulin loss over the course of metastatic melanoma. Int. J. Cancer. 2014;134:102–113. doi: 10.1002/ijc.28338. [DOI] [PubMed] [Google Scholar]
- 323.Hicklin D.J., Wang Z., Arienti F., Rivoltini L., Parmiani G., Ferrone S. beta2-Microglobulin mutations, HLA class I antigen loss, and tumor progression in melanoma. J. Clin. Investig. 1998;101:2720–2729. doi: 10.1172/JCI498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Cormier J.N., Hijazi Y.M., Abati A., Fetsch P., Bettinotti M., Steinberg S.M., Rosenberg S.A., Marincola F.M. Heterogeneous expression of melanoma-associated antigens and HLA-A2 in metastatic melanoma in vivo. Int. J. Cancer. 1998;75:517–524. doi: 10.1002/(SICI)1097-0215(19980209)75:4<517::AID-IJC5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 325.Marincola F.M., Hijazi Y.M., Fetsch P., Salgaller M.L., Rivoltini L., Cormier J., Simonis T.B., Duray P.H., Herlyn M., Kawakami Y., et al. Analysis of expression of the melanoma-associated antigens MART-1 and gp100 in metastatic melanoma cell lines and in in situ lesions. J. Immunother. Emphasis Tumor Immunol. 1996;19:192–205. doi: 10.1097/00002371-199605000-00004. [DOI] [PubMed] [Google Scholar]
- 326.Jäger E., Ringhoffer M., Karbach J., Arand M., Oesch F., Knuth A. Inverse relationship of melanocyte differentiation antigen expression in melanoma tissues and CD8+ cytotoxic-T-cell responses: Evidence for immunoselection of antigen-loss variants in vivo. Int. J. Cancer. 1996;66:470–476. doi: 10.1002/(SICI)1097-0215(19960516)66:4<470::AID-IJC10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 327.Cormier J.N., Abati A., Fetsch P., Hijazi Y.M., Rosenberg S.A., Marincola F.M., Topalian S.L. Comparative analysis of the in vivo expression of tyrosinase, MART-1/Melan-A, and gp100 in metastatic melanoma lesions: Implications for immunotherapy. J. Immunother. 1998;21:27–31. doi: 10.1097/00002371-199801000-00003. [DOI] [PubMed] [Google Scholar]
- 328.Donia M., Andersen R., Kjeldsen J.W., Fagone P., Munir S., Nicoletti F., Andersen M.H., Thor Straten P., Svane I.M. Aberrant Expression of MHC Class II in Melanoma Attracts Inflammatory Tumor-Specific CD4+ T- Cells, Which Dampen CD8+ T-cell Antitumor Reactivity. Cancer Res. 2015;75:3747–3759. doi: 10.1158/0008-5472.CAN-14-2956. [DOI] [PubMed] [Google Scholar]
- 329.Cohen-Solal K.A., Reuhl K.R., Ryan K.B., Roberts K.G., Chen S. Development of cutaneous amelanotic melanoma in the absence of a functional tyrosinase. Pigment Cell Res. 2001;14:466–474. doi: 10.1034/j.1600-0749.2001.140607.x. [DOI] [PubMed] [Google Scholar]
- 330.Schiffner S., Chen S., Becker J.C., Bosserhoff A.K. Highly pigmented Tg(Grm1) mouse melanoma develops non-pigmented melanoma cells in distant metastases. Exp. Dermatol. 2012;21:786–788. doi: 10.1111/j.1600-0625.2012.01560.x. [DOI] [PubMed] [Google Scholar]
- 331.Shah R., Singh S.J., Eddy K., Filipp F.V., Chen S. Concurrent Targeting of Glutaminolysis and Metabotropic Glutamate Receptor 1 (GRM1) Reduces Glutamate Bioavailability in GRM1+ Melanoma. Cancer Res. 2019;79:1799–1809. doi: 10.1158/0008-5472.CAN-18-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Prokopi A., Tripp C.H., Tummers B., Komenda K., Hutter K., Cappellano G., Bellmann L., Efremova M., Trajanoski Z., Chen S., et al. Abstract A102: Rescue of lost skin dendritic cells in melanoma is key for the resuscitation of antitumor T-cell responses. Cancer Immunol. Res. 2019;7:A102. doi: 10.1158/2326-6074.CRICIMTEATIAACR18-A102. [DOI] [Google Scholar]
- 333.Schwarz T. Mechanisms of UV-induced immunosuppression. Keio J. Med. 2005;54:165–171. doi: 10.2302/kjm.54.165. [DOI] [PubMed] [Google Scholar]
- 334.Kripke M.L., Fisher M.S. Immunologic parameters of ultraviolet carcinogenesis. J. Natl. Cancer Inst. 1976;57:211–215. doi: 10.1093/jnci/57.1.211. [DOI] [PubMed] [Google Scholar]
- 335.Kripke M.L., Fisher M.S. Immunologic aspects of tumor induction by ultraviolet radiation. Natl. Cancer Inst. Monogr. 1978;50:179–183. [PubMed] [Google Scholar]
- 336.Hart P.H., Norval M. Ultraviolet radiation-induced immunosuppression and its relevance for skin carcinogenesis. Photochem. Photobiol. Sci. 2018;17:1872–1884. doi: 10.1039/C7PP00312A. [DOI] [PubMed] [Google Scholar]
- 337.Fortner G.W., Kripke M.L. In vitro reactivity of splenic lymphocytes from normal and UV-irradiated mice against syngeneic UV-induced tumors. J. Immunol. 1977;118:1483–1487. [PubMed] [Google Scholar]
- 338.Sun X., Zhang N., Yin C., Zhu B., Li X. Ultraviolet Radiation and Melanomagenesis: From Mechanism to Immunotherapy. Front. Oncol. 2020;10:951. doi: 10.3389/fonc.2020.00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Schwarz A., Noordegraaf M., Maeda A., Torii K., Clausen B.E., Schwarz T. Langerhans cells are required for UVR-induced immunosuppression. J. Investig. Dermatol. 2010;130:1419–1427. doi: 10.1038/jid.2009.429. [DOI] [PubMed] [Google Scholar]
- 340.Shreedhar V.K., Pride M.W., Sun Y., Kripke M.L., Strickland F.M. Origin and characteristics of ultraviolet-B radiation-induced suppressor T lymphocytes. J. Immunol. 1998;161:1327–1335. [PubMed] [Google Scholar]
- 341.Rivas J.M., Ullrich S.E. The role of IL-4, IL-10, and TNF-alpha in the immune suppression induced by ultraviolet radiation. J. Leukoc. Biol. 1994;56:769–775. doi: 10.1002/jlb.56.6.769. [DOI] [PubMed] [Google Scholar]
- 342.Granstein R.D., Matsui M.S. UV radiation-induced immunosuppression and skin cancer. Cutis. 2004;74:4–9. [PubMed] [Google Scholar]
- 343.Ullrich S.E. Does exposure to UV radiation induce a shift to a Th-2-like immune reaction? Photochem. Photobiol. 1996;64:254–258. doi: 10.1111/j.1751-1097.1996.tb02454.x. [DOI] [PubMed] [Google Scholar]
- 344.Schmitt D.A., Walterscheid J.P., Ullrich S.E. Reversal of ultraviolet radiation-induced immune suppression by recombinant interleukin-12: Suppression of cytokine production. Immunology. 2000;101:90–96. doi: 10.1046/j.1365-2567.2000.00084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Hood J.L., San R.S., Wickline S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011;71:3792–3801. doi: 10.1158/0008-5472.CAN-10-4455. [DOI] [PubMed] [Google Scholar]
- 346.Hu L., Wickline S.A., Hood J.L. Magnetic resonance imaging of melanoma exosomes in lymph nodes. Magn. Reson. Med. 2015;74:266–271. doi: 10.1002/mrm.25376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Düchler M., Czernek L., Peczek L., Cypryk W., Sztiller-Sikorska M., Czyz M. Melanoma-Derived Extracellular Vesicles Bear the Potential for the Induction of Antigen-Specific Tolerance. Cells. 2019;8:665. doi: 10.3390/cells8070665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Taylor D.D., Gerçel-Taylor C. Tumour-derived exosomes and their role in cancer-associated T-cell signalling defects. Br. J. Cancer. 2005;92:305–311. doi: 10.1038/sj.bjc.6602316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Muller L., Mitsuhashi M., Simms P., Gooding W.E., Whiteside T.L. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci. Rep. 2016;6:20254. doi: 10.1038/srep20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Sharma P., Diergaarde B., Ferrone S., Kirkwood J.M., Whiteside T.L. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci. Rep. 2020;10:92. doi: 10.1038/s41598-019-56542-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Liu C., Yu S., Zinn K., Wang J., Zhang L., Jia Y., Kappes J.C., Barnes S., Kimberly R.P., Grizzle W.E., et al. Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function. J. Immunol. 2006;176:1375–1385. doi: 10.4049/jimmunol.176.3.1375. [DOI] [PubMed] [Google Scholar]
- 352.Xiang X., Poliakov A., Liu C., Liu Y., Deng Z.B., Wang J., Cheng Z., Shah S.V., Wang G.J., Zhang L., et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer. 2009;124:2621–2633. doi: 10.1002/ijc.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Gerloff D., Lützkendorf J., Moritz R.K.C., Wersig T., Mäder K., Müller L.P., Sunderkötter C. Melanoma-Derived Exosomal miR-125b-5p Educates Tumor Associated Macrophages (TAMs) by Targeting Lysosomal Acid Lipase A (LIPA) Cancers. 2020;12:464. doi: 10.3390/cancers12020464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Bardi G.T., Smith M.A., Hood J.L. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine. 2018;105:63–72. doi: 10.1016/j.cyto.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Chen G., Huang A.C., Zhang W., Zhang G., Wu M., Xu W., Yu Z., Yang J., Wang B., Sun H., et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–386. doi: 10.1038/s41586-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Poggio M., Hu T., Pai C.C., Chu B., Belair C.D., Chang A., Montabana E., Lang U.E., Fu Q., Fong L., et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell. 2019;177:414–427.e413. doi: 10.1016/j.cell.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Isola A.L., Eddy K., Zembrzuski K., Goydos J.S., Chen S. Exosomes released by metabotropic glutamate receptor 1 (GRM1) expressing melanoma cells increase cell migration and invasiveness. Oncotarget. 2017;9:1187–1199. doi: 10.18632/oncotarget.23455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Green J., Ariyan C. Update on immunotherapy in melanoma. Surg. Oncol. Clin. N. Am. 2015;24:337–346. doi: 10.1016/j.soc.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 359.Di Trolio R., Simeone E., Di Lorenzo G., Buonerba C., Ascierto P.A. The use of interferon in melanoma patients: A systematic review. Cytokine Growth Factor Rev. 2015;26:203–212. doi: 10.1016/j.cytogfr.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 360.Morton D.L., Eilber F.R., Holmes E.C., Hunt J.S., Ketcham A.S., Silverstein M.J., Sparks F.C. BCG immunotherapy of malignant melanoma: Summary of a seven-year experience. Ann. Surg. 1974;180:635–643. doi: 10.1097/00000658-197410000-00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Choudhry H., Helmi N., Abdulaal W.H., Zeyadi M., Zamzami M.A., Wu W., Mahmoud M.M., Warsi M.K., Rasool M., Jamal M.S. Prospects of IL-2 in Cancer Immunotherapy. BioMed Res. Int. 2018;2018:9056173. doi: 10.1155/2018/9056173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Lee S., Margolin K. Tumor-infiltrating lymphocytes in melanoma. Curr. Oncol. Rep. 2012;14:468–474. doi: 10.1007/s11912-012-0257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Hou B., Tang Y., Li W., Zeng Q., Chang D. Efficiency of CAR-T Therapy for Treatment of Solid Tumor in Clinical Trials: A Meta-Analysis. Dis. Markers. 2019;2019:3425291. doi: 10.1155/2019/3425291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Morgan R.A., Dudley M.E., Rosenberg S.A. Adoptive cell therapy: Genetic modification to redirect effector cell specificity. Cancer J. 2010;16:336–341. doi: 10.1097/PPO.0b013e3181eb3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Eisenhauer E.A., Therasse P., Bogaerts J., Schwartz L.H., Sargent D., Ford R., Dancey J., Arbuck S., Gwyther S., Mooney M., et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur. J. Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 366.Wolchok J.D., Hoos A., O’Day S., Weber J.S., Hamid O., Lebbé C., Maio M., Binder M., Bohnsack O., Nichol G., et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 367.Seymour L., Bogaerts J., Perrone A., Ford R., Schwartz L.H., Mandrekar S., Lin N.U., Litière S., Dancey J., Chen A., et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18:e143–e152. doi: 10.1016/S1470-2045(17)30074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Klocke K., Sakaguchi S., Holmdahl R., Wing K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc. Natl. Acad. Sci. USA. 2016;113:E2383–E2392. doi: 10.1073/pnas.1603892113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Zhang J., Medeiros L.J., Young K.H. Cancer Immunotherapy in Diffuse Large B-Cell Lymphoma. Front. Oncol. 2018;8:351. doi: 10.3389/fonc.2018.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Guntermann C., Alexander D.R. CTLA-4 suppresses proximal TCR signaling in resting human CD4(+) T cells by inhibiting ZAP-70 Tyr(319) phosphorylation: A potential role for tyrosine phosphatases. J. Immunol. 2002;168:4420–4429. doi: 10.4049/jimmunol.168.9.4420. [DOI] [PubMed] [Google Scholar]
- 371.Chambers C.A., Kuhns M.S., Egen J.G., Allison J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001;19:565–594. doi: 10.1146/annurev.immunol.19.1.565. [DOI] [PubMed] [Google Scholar]
- 372.He M., Chai Y., Qi J., Zhang C.W.H., Tong Z., Shi Y., Yan J., Tan S., Gao G.F. Remarkably similar CTLA-4 binding properties of therapeutic ipilimumab and tremelimumab antibodies. Oncotarget. 2017;8:67129–67139. doi: 10.18632/oncotarget.18004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Kvistborg P., Philips D., Kelderman S., Hageman L., Ottensmeier C., Joseph-Pietras D., Welters M.J., Van der Burg S., Kapiteijn E., Michielin O., et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci. Transl. Med. 2014;6:254ra128. doi: 10.1126/scitranslmed.3008918. [DOI] [PubMed] [Google Scholar]
- 374.Cha E., Klinger M., Hou Y., Cummings C., Ribas A., Faham M., Fong L. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci. Transl. Med. 2014;6:238ra270. doi: 10.1126/scitranslmed.3008211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Shin J.H., Park H.B., Oh Y.M., Lim D.P., Lee J.E., Seo H.H., Lee S.J., Eom H.S., Kim I.H., Lee S.H., et al. Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood. 2012;119:5678–5687. doi: 10.1182/blood-2011-09-380519. [DOI] [PubMed] [Google Scholar]
- 376.Friese C., Harbst K., Borch T.H., Westergaard M.C.W., Pedersen M., Kverneland A., Jönsson G., Donia M., Svane I.M., Met Ö. CTLA-4 blockade boosts the expansion of tumor-reactive CD8(+) tumor-infiltrating lymphocytes in ovarian cancer. Sci. Rep. 2020;10:3914. doi: 10.1038/s41598-020-60738-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Kearney E.R., Walunas T.L., Karr R.W., Morton P.A., Loh D.Y., Bluestone J.A., Jenkins M.K. Antigen-dependent clonal expansion of a trace population of antigen-specific CD4+ T cells in vivo is dependent on CD28 costimulation and inhibited by CTLA-4. J. Immunol. 1995;155:1032–1036. [PubMed] [Google Scholar]
- 378.Ng Tang D., Shen Y., Sun J., Wen S., Wolchok J.D., Yuan J., Allison J.P., Sharma P. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol. Res. 2013;1:229–234. doi: 10.1158/2326-6066.CIR-13-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Simpson T.R., Li F., Montalvo-Ortiz W., Sepulveda M.A., Bergerhoff K., Arce F., Roddie C., Henry J.Y., Yagita H., Wolchok J.D., et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Romano E., Kusio-Kobialka M., Foukas P.G., Baumgaertner P., Meyer C., Ballabeni P., Michielin O., Weide B., Romero P., Speiser D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA. 2015;112:6140–6145. doi: 10.1073/pnas.1417320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Ingram J.R., Blomberg O.S., Rashidian M., Ali L., Garforth S., Fedorov E., Fedorov A.A., Bonanno J.B., Le Gall C., Crowley S., et al. Anti-CTLA-4 therapy requires an Fc domain for efficacy. Proc. Natl. Acad. Sci. USA. 2018;115:3912–3917. doi: 10.1073/pnas.1801524115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Arce Vargas F., Furness A.J.S., Litchfield K., Joshi K., Rosenthal R., Ghorani E., Solomon I., Lesko M.H., Ruef N., Roddie C., et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell. 2018;33:649–663.e644. doi: 10.1016/j.ccell.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Subudhi S.K., Aparicio A., Gao J., Zurita A.J., Araujo J.C., Logothetis C.J., Tahir S.A., Korivi B.R., Slack R.S., Vence L., et al. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc. Natl. Acad. Sci. USA. 2016;113:11919–11924. doi: 10.1073/pnas.1611421113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Nishida N., Kudo M. Liver damage related to immune checkpoint inhibitors. Hepatol. Int. 2019;13:248–252. doi: 10.1007/s12072-018-9921-7. [DOI] [PubMed] [Google Scholar]
- 385.Ramos-Casals M., Brahmer J.R., Callahan M.K., Flores-Chávez A., Keegan N., Khamashta M.A., Lambotte O., Mariette X., Prat A., Suárez-Almazor M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Prim. 2020;6:38. doi: 10.1038/s41572-020-0160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Hodi F.S., O’Day S.J., McDermott D.F., Weber R.W., Sosman J.A., Haanen J.B., Gonzalez R., Robert C., Schadendorf D., Hassel J.C., et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Eroglu Z., Kim D.W., Wang X., Camacho L.H., Chmielowski B., Seja E., Villanueva A., Ruchalski K., Glaspy J.A., Kim K.B., et al. Long term survival with cytotoxic T lymphocyte-associated antigen 4 blockade using tremelimumab. Eur. J. Cancer. 2015;51:2689–2697. doi: 10.1016/j.ejca.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Gutzmer R., Stroyakovskiy D., Gogas H., Robert C., Lewis K., Protsenko S., Pereira R.P., Eigentler T., Rutkowski P., Demidov L., et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;395:1835–1844. doi: 10.1016/S0140-6736(20)30934-X. [DOI] [PubMed] [Google Scholar]
- 389.Okazaki T., Honjo T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 390.Alsaab H.O., Sau S., Alzhrani R., Tatiparti K., Bhise K., Kashaw S.K., Iyer A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017;8:561. doi: 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Haanen J.B. Immunotherapy of melanoma. Eur. J. Cancer Suppl. 2013;11:97–105. doi: 10.1016/j.ejcsup.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Lee H.T., Lee S.H., Heo Y.S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules. 2019;24:1190. doi: 10.3390/molecules24061190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Iwai Y., Ishida M., Tanaka Y., Okazaki T., Honjo T., Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Iwai Y., Terawaki S., Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int. Immunol. 2005;17:133–144. doi: 10.1093/intimm/dxh194. [DOI] [PubMed] [Google Scholar]
- 395.Hirano F., Kaneko K., Tamura H., Dong H., Wang S., Ichikawa M., Rietz C., Flies D.B., Lau J.S., Zhu G., et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–1096. [PubMed] [Google Scholar]
- 396.Li J., Gu J. Efficacy and safety of ipilimumab for treating advanced melanoma: A systematic review and meta-analysis. J. Clin. Pharm. Ther. 2019;44:420–429. doi: 10.1111/jcpt.12802. [DOI] [PubMed] [Google Scholar]
- 397.De Sousa Linhares A., Battin C., Jutz S., Leitner J., Hafner C., Tobias J., Wiedermann U., Kundi M., Zlabinger G.J., Grabmeier-Pfistershammer K., et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci. Rep. 2019;9:11472. doi: 10.1038/s41598-019-47910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Carretero-González A., Lora D., Ghanem I., Zugazagoitia J., Castellano D., Sepúlveda J.M., López-Martin J.A., Paz-Ares L., de Velasco G. Analysis of response rate with ANTI PD1/PD-L1 monoclonal antibodies in advanced solid tumors: A meta-analysis of randomized clinical trials. Oncotarget. 2018;9:8706–8715. doi: 10.18632/oncotarget.24283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Kitano S., Nakayama T., Yamashita M. Biomarkers for Immune Checkpoint Inhibitors in Melanoma. Front. Oncol. 2018;8:270. doi: 10.3389/fonc.2018.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Ye Y., Jing Y., Li L., Mills G.B., Diao L., Liu H., Han L. Sex-associated molecular differences for cancer immunotherapy. Nat. Commun. 2020;11:1779. doi: 10.1038/s41467-020-15679-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Castro A., Pyke R.M., Zhang X., Thompson W.K., Day C.P., Alexandrov L.B., Zanetti M., Carter H. Strength of immune selection in tumors varies with sex and age. Nat. Commun. 2020;11:4128. doi: 10.1038/s41467-020-17981-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Nakamura Y. Biomarkers for Immune Checkpoint Inhibitor-Mediated Tumor Response and Adverse Events. Front. Med. 2019;6:119. doi: 10.3389/fmed.2019.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Liakou C.I., Kamat A., Tang D.N., Chen H., Sun J., Troncoso P., Logothetis C., Sharma P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl. Acad. Sci. USA. 2008;105:14987–14992. doi: 10.1073/pnas.0806075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Carthon B.C., Wolchok J.D., Yuan J., Kamat A., Ng Tang D.S., Sun J., Ku G., Troncoso P., Logothetis C.J., Allison J.P., et al. Preoperative CTLA-4 blockade: Tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin. Cancer Res. 2010;16:2861–2871. doi: 10.1158/1078-0432.CCR-10-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Tallerico R., Cristiani C.M., Staaf E., Garofalo C., Sottile R., Capone M., Pico de Coaña Y., Madonna G., Palella E., Wolodarski M., et al. IL-15, TIM-3 and NK cells subsets predict responsiveness to anti-CTLA-4 treatment in melanoma patients. Oncoimmunology. 2017;6:e1261242. doi: 10.1080/2162402X.2016.1261242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Hamid O., Schmidt H., Nissan A., Ridolfi L., Aamdal S., Hansson J., Guida M., Hyams D.M., Gómez H., Bastholt L., et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011;9:204. doi: 10.1186/1479-5876-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Pistillo M.P., Fontana V., Morabito A., Dozin B., Laurent S., Carosio R., Banelli B., Ferrero F., Spano L., Tanda E., et al. Soluble CTLA-4 as a favorable predictive biomarker in metastatic melanoma patients treated with ipilimumab: An Italian melanoma intergroup study. Cancer Immunol. Immunother. 2019;68:97–107. doi: 10.1007/s00262-018-2258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Ji R.R., Chasalow S.D., Wang L., Hamid O., Schmidt H., Cogswell J., Alaparthy S., Berman D., Jure-Kunkel M., Siemers N.O., et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012;61:1019–1031. doi: 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Sunshine J.C., Nguyen P.L., Kaunitz G.J., Cottrell T.R., Berry S., Esandrio J., Xu H., Ogurtsova A., Bleich K.B., Cornish T.C., et al. PD-L1 Expression in Melanoma: A Quantitative Immunohistochemical Antibody Comparison. Clin. Cancer Res. 2017;23:4938–4944. doi: 10.1158/1078-0432.CCR-16-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Daud A.I., Wolchok J.D., Robert C., Hwu W.J., Weber J.S., Ribas A., Hodi F.S., Joshua A.M., Kefford R., Hersey P., et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J. Clin. Oncol. 2016;34:4102–4109. doi: 10.1200/JCO.2016.67.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Taube J.M., Klein A., Brahmer J.R., Xu H., Pan X., Kim J.H., Chen L., Pardoll D.M., Topalian S.L., Anders R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014;20:5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Cottrell T.R., Taube J.M. PD-L1 and Emerging Biomarkers in Immune Checkpoint Blockade Therapy. Cancer J. 2018;24:41–46. doi: 10.1097/PPO.0000000000000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Madore J., Vilain R.E., Menzies A.M., Kakavand H., Wilmott J.S., Hyman J., Yearley J.H., Kefford R.F., Thompson J.F., Long G.V., et al. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: Implications for anti-PD-1/PD-L1 clinical trials. Pigment Cell Melanoma Res. 2015;28:245–253. doi: 10.1111/pcmr.12340. [DOI] [PubMed] [Google Scholar]
- 414.Wolf Y., Bartok O., Patkar S., Eli G.B., Cohen S., Litchfield K., Levy R., Jiménez-Sánchez A., Trabish S., Lee J.S., et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell. 2019;179:219–235.e221. doi: 10.1016/j.cell.2019.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Gros A., Parkhurst M.R., Tran E., Pasetto A., Robbins P.F., Ilyas S., Prickett T.D., Gartner J.J., Crystal J.S., Roberts I.M., et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016;22:433–438. doi: 10.1038/nm.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Goodman A.M., Kato S., Bazhenova L., Patel S.P., Frampton G.M., Miller V., Stephens P.J., Daniels G.A., Kurzrock R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017;16:2598–2608. doi: 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Chalmers Z.R., Connelly C.F., Fabrizio D., Gay L., Ali S.M., Ennis R., Schrock A., Campbell B., Shlien A., Chmielecki J., et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Panda A., Betigeri A., Subramanian K., Ross J.S., Pavlick D.C., Ali S., Markowski P., Silk A., Kaufman H.L., Lattime E., et al. Identifying a Clinically Applicable Mutational Burden Threshold as a Potential Biomarker of Response to Immune Checkpoint Therapy in Solid Tumors. JCO Precis. Oncol. 2017;1:1–13. doi: 10.1200/PO.17.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Plesca I., Tunger A., Müller L., Wehner R., Lai X., Grimm M.O., Rutella S., Bachmann M., Schmitz M. Characteristics of Tumor-Infiltrating Lymphocytes Prior to and During Immune Checkpoint Inhibitor Therapy. Front. Immunol. 2020;11:364. doi: 10.3389/fimmu.2020.00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V., et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Weide B., Martens A., Hassel J.C., Berking C., Postow M.A., Bisschop K., Simeone E., Mangana J., Schilling B., Di Giacomo A.M., et al. Baseline Biomarkers for Outcome of Melanoma Patients Treated with Pembrolizumab. Clin. Cancer Res. 2016;22:5487–5496. doi: 10.1158/1078-0432.CCR-16-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Zhou J., Mahoney K.M., Giobbie-Hurder A., Zhao F., Lee S., Liao X., Rodig S., Li J., Wu X., Butterfield L.H., et al. Soluble PD-L1 as a Biomarker in Malignant Melanoma Treated with Checkpoint Blockade. Cancer Immunol. Res. 2017;5:480–492. doi: 10.1158/2326-6066.CIR-16-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Gopalakrishnan V., Spencer C.N., Nezi L., Reuben A., Andrews M.C., Karpinets T.V., Prieto P.A., Vicente D., Hoffman K., Wei S.C., et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Sivan A., Corrales L., Hubert N., Williams J.B., Aquino-Michaels K., Earley Z.M., Benyamin F.W., Lei Y.M., Jabri B., Alegre M.L., et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Routy B., Le Chatelier E., Derosa L., Duong C.P.M., Alou M.T., Daillère R., Fluckiger A., Messaoudene M., Rauber C., Roberti M.P., et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 426.Cristescu R., Mogg R., Ayers M., Albright A., Murphy E., Yearley J., Sher X., Liu X.Q., Lu H., Nebozhyn M., et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362 doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer. 2018;6:157. doi: 10.1186/s40425-018-0479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Huang A.C., Postow M.A., Orlowski R.J., Mick R., Bengsch B., Manne S., Xu W., Harmon S., Giles J.R., Wenz B., et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Rossi E., Schinzari G., Maiorano B.A., Indellicati G., Di Stefani A., Pagliara M.M., Fragomeni S.M., De Luca E.V., Sammarco M.G., Garganese G., et al. Efficacy of immune checkpoint inhibitors in different types of melanoma. Hum. Vaccin. Immunother. 2020:1–10. doi: 10.1080/21645515.2020.1771986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Liu D., Schilling B., Liu D., Sucker A., Livingstone E., Jerby-Arnon L., Zimmer L., Gutzmer R., Satzger I., Loquai C., et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019;25:1916–1927. doi: 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Waldman A.D., Fritz J.M., Lenardo M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020;20:651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Perica K., Varela J.C., Oelke M., Schneck J.P. Adoptive T Cell Immunotherapy for Cancer. Rambam Maimonides Med. J. 2015;6:e0004. doi: 10.5041/RMMJ.10179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Rosenberg S.A., Packard B.S., Aebersold P.M., Solomon D., Topalian S.L., Toy S.T., Simon P., Lotze M.T., Yang J.C., Seipp C.A., et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988;319:1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 434.Rosenberg S.A., Yang J.C., Sherry R.M., Kammula U.S., Hughes M.S., Phan G.Q., Citrin D.E., Restifo N.P., Robbins P.F., Wunderlich J.R., et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Besser M.J., Shapira-Frommer R., Treves A.J., Zippel D., Itzhaki O., Hershkovitz L., Levy D., Kubi A., Hovav E., Chermoshniuk N., et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2010;16:2646–2655. doi: 10.1158/1078-0432.CCR-10-0041. [DOI] [PubMed] [Google Scholar]
- 436.Rohaan M.W., Van den Berg J.H., Kvistborg P., Haanen J. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option. J. Immunother. Cancer. 2018;6:102. doi: 10.1186/s40425-018-0391-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Morgan R.A., Dudley M.E., Wunderlich J.R., Hughes M.S., Yang J.C., Sherry R.M., Royal R.E., Topalian S.L., Kammula U.S., Restifo N.P., et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Ott P.A., Dotti G., Yee C., Goff S.L. An Update on Adoptive T-Cell Therapy and Neoantigen Vaccines. Am. Soc. Clin. Oncol. Educ. Book. 2019;39:e70–e78. doi: 10.1200/EDBK_238001. [DOI] [PubMed] [Google Scholar]
- 439.Johnson L.A., Morgan R.A., Dudley M.E., Cassard L., Yang J.C., Hughes M.S., Kammula U.S., Royal R.E., Sherry R.M., Wunderlich J.R., et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Pfefferle A., Huntington N.D. You Have Got a Fast CAR: Chimeric Antigen Receptor NK Cells in Cancer Therapy. Cancers. 2020;12:706. doi: 10.3390/cancers12030706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Lauss M., Donia M., Harbst K., Andersen R., Mitra S., Rosengren F., Salim M., Vallon-Christersson J., Törngren T., Kvist A., et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2017;8:1738. doi: 10.1038/s41467-017-01460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Pol J., Kroemer G., Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology. 2016;5:e1115641. doi: 10.1080/2162402X.2015.1115641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Bommareddy P.K., Shettigar M., Kaufman H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018;18:498–513. doi: 10.1038/s41577-018-0014-6. [DOI] [PubMed] [Google Scholar]
- 444.Zamarin D., Holmgaard R.B., Subudhi S.K., Park J.S., Mansour M., Palese P., Merghoub T., Wolchok J.D., Allison J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014;6:226ra232. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Orloff M. Spotlight on talimogene laherparepvec for the treatment of melanoma lesions in the skin and lymph nodes. Oncolytic Virother. 2016;5:91–98. doi: 10.2147/OV.S99532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S., et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 447.Masoud S.J., Hu J.B., Beasley G.M., Stewart J.H.t., Mosca P.J. Efficacy of Talimogene Laherparepvec (T-VEC) Therapy in Patients with In-Transit Melanoma Metastasis Decreases with Increasing Lesion Size. Ann. Surg. Oncol. 2019;26:4633–4641. doi: 10.1245/s10434-019-07691-3. [DOI] [PubMed] [Google Scholar]
- 448.Andtbacka R.H., Ross M., Puzanov I., Milhem M., Collichio F., Delman K.A., Amatruda T., Zager J.S., Cranmer L., Hsueh E., et al. Patterns of Clinical Response with Talimogene Laherparepvec (T-VEC) in Patients with Melanoma Treated in the OPTiM Phase III Clinical Trial. Ann. Surg. Oncol. 2016;23:4169–4177. doi: 10.1245/s10434-016-5286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Achard C., Surendran A., Wedge M.E., Ungerechts G., Bell J., Ilkow C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. EBioMedicine. 2018;31:17–24. doi: 10.1016/j.ebiom.2018.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Eggermont A.M.M., Crittenden M., Wargo J. Combination Immunotherapy Development in Melanoma. Am. Soc. Clin. Oncol. Educ. Book. 2018;38:197–207. doi: 10.1200/EDBK_201131. [DOI] [PubMed] [Google Scholar]
- 451.Wolchok J.D., Kluger H., Callahan M.K., Postow M.A., Rizvi N.A., Lesokhin A.M., Segal N.H., Ariyan C.E., Gordon R.A., Reed K., et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Wolchok J.D., Chiarion-Sileni V., Gonzalez R., Rutkowski P., Grob J.J., Cowey C.L., Lao C.D., Wagstaff J., Schadendorf D., Ferrucci P.F., et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017;377:1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J.J., Rutkowski P., Lao C.D., Cowey C.L., Schadendorf D., Wagstaff J., Dummer R., et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019;381:1535–1546. doi: 10.1056/NEJMoa1910836. [DOI] [PubMed] [Google Scholar]
- 454.Hodi F.S., Chiarion-Sileni V., Gonzalez R., Grob J.J., Rutkowski P., Cowey C.L., Lao C.D., Schadendorf D., Wagstaff J., Dummer R., et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480–1492. doi: 10.1016/S1470-2045(18)30700-9. [DOI] [PubMed] [Google Scholar]
- 455.Carlino M.S., Menzies A.M., Atkinson V., Cebon J.S., Jameson M.B., Fitzharris B.M., McNeil C.M., Hill A.G., Ribas A., Atkins M.B., et al. Long-term Follow-up of Standard-Dose Pembrolizumab Plus Reduced-Dose Ipilimumab in Patients with Advanced Melanoma: KEYNOTE-029 Part 1B. Clin. Cancer Res. 2020;26:5086–5091. doi: 10.1158/1078-0432.CCR-20-0177. [DOI] [PubMed] [Google Scholar]
- 456.Das R., Verma R., Sznol M., Boddupalli C.S., Gettinger S.N., Kluger H., Callahan M., Wolchok J.D., Halaban R., Dhodapkar M.V., et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J. Immunol. 2015;194:950–959. doi: 10.4049/jimmunol.1401686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Wei S.C., Anang N.A.S., Sharma R., Andrews M.C., Reuben A., Levine J.H., Cogdill A.P., Mancuso J.J., Wargo J.A., Pe’er D., et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA. 2019;116:22699–22709. doi: 10.1073/pnas.1821218116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Shi L.Z., Goswami S., Fu T., Guan B., Chen J., Xiong L., Zhang J., Ng Tang D., Zhang X., Vence L., et al. Blockade of CTLA-4 and PD-1 Enhances Adoptive T-cell Therapy Efficacy in an ICOS-Mediated Manner. Cancer Immunol. Res. 2019;7:1803–1812. doi: 10.1158/2326-6066.CIR-18-0873. [DOI] [PubMed] [Google Scholar]
- 459.Kverneland A.H., Pedersen M., Westergaard M.C.W., Nielsen M., Borch T.H., Olsen L.R., Aasbjerg G., Santegoets S.J., van der Burg S.H., Milne K., et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget. 2020;11:2092–2105. doi: 10.18632/oncotarget.27604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Koya R.C., Mok S., Otte N., Blacketor K.J., Comin-Anduix B., Tumeh P.C., Minasyan A., Graham N.A., Graeber T.G., Chodon T., et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012;72:3928–3937. doi: 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]