

Abstract
Hepcidin is central to regulation of iron metabolism. Its effect on a cellular level involves binding ferroportin, the main iron export protein, resulting in its internalization and degradation and leading to iron sequestration within ferroportin-expressing cells. Aberrantly increased hepcidin leads to systemic iron deficiency and/or iron restricted erythropoiesis. Furthermore, insufficiently elevated hepcidin occurs in multiple diseases associated with iron overload. Abnormal iron metabolism as a consequence of hepcidin dysregulation is an underlying factor resulting in pathophysiology of multiple diseases and several agents aimed at manipulating this pathway have been designed, with some already in clinical trials. In this chapter, we present an overview of and rationale for exploring the development of hepcidin agonists and antagonists in various clinical scenarios.
1. Introduction
Iron is an essential element for almost all living organisms, from mammals to unicellular organisms. It forms the core of molecules such as hemoglobin, myoglobin, cytochromes and nitric oxide synthase as well as in multiple enzymes required for the generation of ATP in all cells. Two to three million red blood cells (RBCs) are produced every second and require 30–40mg of iron delivered to the erythron to make 30pg of hemoglobin per cell, a total of 6g of hemoglobin daily. Thus, of the 3–4g of iron total in the human body, 2–2.5g are present with hemoglobin, 0.5–1g within macrophages and hepatocytes, and 0.5g total myoglobin, ferritin, and iron-containing enzymes in other cell types. Because the majority of iron is found in the hemoglobin compartment, erythropoiesis dominates iron metabolism and the two are inextricably intertwined, regulating iron absorption, recycling, and trafficking. Iron availability must be tightly regulated to prevent (1) shortfalls and iron deficiency, resulting in anemia, as well as (2) iron excess, resulting in the generation of free iron and consequent reactive oxygen species (ROS), causing tissue injury and organ failure.
As systemic iron loss is not modulated during iron deficiency or overload, the stable concentration of circulating iron is maintained by baseline dietary absorption, storage, and recycling of iron. Most of the iron found in circulation is recycled from erythrophagocytosis of RBCs within macrophages. Recycled iron within macrophages can be exported to carrier proteins in the circulation. Iron in circulation is bound to transferrin, the main iron transporter in circulation, and transferrin saturation of iron-binding sites, calculated as a ratio of serum iron to total transferrin iron-binding capacity, is approximately 20–40% under normal conditions. Of the 20–25mg of iron daily required for erythropoiesis, only 2–4mg is found in circulation at any one time. Thus, iron trafficking is a dynamic process, using transferrin to transport iron between sites of absorption, recycling, and storage to sites of iron utilization. Transferrin-iron is taken up by binding transferrin receptor 1 (TfR1) ubiquitously expressed on all cells. The highest concentration of TfR1 is found on erythroid precursors, hemoglobin producing cells with the highest iron requirements in the body.
Pathological blood loss and some forms of hemolysis are the main mechanisms of iron loss. As a consequence, phlebotomy is used as a therapeutic approach for non-anemic diseases of iron overload. In response to our enhanced understanding of the regulation of iron metabolism, additional strategies are being evaluated to expand therapeutic options for iron-loaded anemias. In addition, therapy targeting the iron regulatory pathway may be a useful alternative to the easy and efficacious but relatively ancient therapeutic phlebotomy approach for non-anemic diseases of iron overload. Because iron absorption is greatly increased in response to phlebotomy, a more mechanistic approach would circumvent the unintended physiological compensatory responses and enable more targeted effects. The goals of this chapter are to (1) describe our current understanding of iron metabolism and its mechanistic relationship with erythropoiesis, (2) elucidate abnormalities in regulation of iron metabolism in various diseases, and (3) discuss potential therapeutic modalities.
2. Regulation of iron metabolism
Complex living organisms have developed sophisticated mechanisms for obtaining, distributing, and sequestering iron. The peptide hormone hepcidin, secreted primarily by hepatocytes, is the principal regulator of iron homeostasis (Ganz, 2005; Krause, Neitz, Magert, et al., 2000; Park, Valore, Waring, & Ganz, 2001), including dietary iron absorption, iron recycling by macrophages, and the release of iron from hepatic stores (Fig. 1). Hepcidin down-regulates iron release into plasma by binding to and functionally down-regulating ferroportin 1, the sole iron exporter (Donovan, Lima, Pinkus, et al., 2005; Nemeth, Tuttle, Powelson, et al., 2004). Iron released from reticuloendothelial cells (i.e., splenic macrophages and Kupffer cells in the liver) is bound for intracellular storage in cytosolic ferritin or exported to bind circulating proteins (mainly transferrin). In addition, ferroportin 1 expression by macrophages is also transcriptionally regulated by heme (Marro et al., 2010) and under translational regulation (i.e., iron response elements on messenger RNA bound to iron response proteins) by iron (Zhang, Hughes, Ollivierre-Wilson, Ghosh, & Rouault, 2009) independent of hepcidin regulation.
Fig. 1.
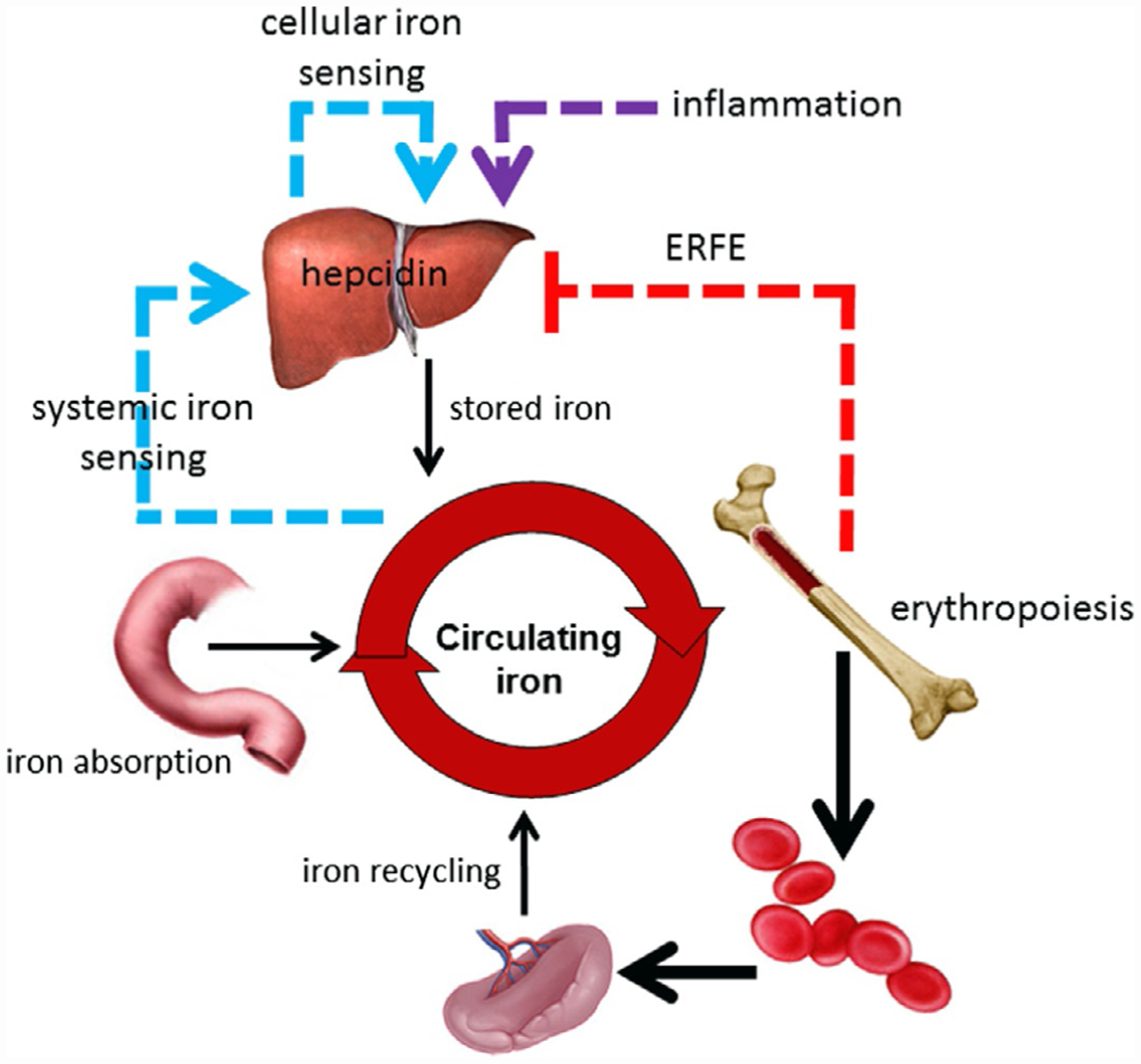
Central role of hepcidin in regulation of iron metabolism. Hepcidin regulates iron absorption, iron recycling and iron released from stores. Hepcidin expression is enhanced by iron regulatory pathway (blue arrow) and inflammatory cytokines (violet arrow) and suppressed by erythropoiesis (red arrow). ERFE, erythroferrone.
Hepcidin is a negative regulator of iron flows. Thus, high hepcidin concentration typically results in blockade of iron absorption and sequestration of iron in hepatocytes and macrophages. Decreased or insufficiently increased hepcidin concentration results in recovery from iron deficiency or iron overload, respectively, by increasing iron absorption to its maximal capacity as well as releasing iron from intracellular compartments in hepatocytes and macrophages. In some pathological conditions (see later), insufficiently increased hepcidin results in efflux of iron into the circulation, overwhelming transferrin’s iron binding capacity and resulting in the generation of non-transferrin bound iron (NTBI) (Esposito et al., 2003). NTBI, in particular its redox active form, labile plasma iron (LPI), is the cause of clinically significant iron overload (Cabantchik, Breuer, Zanninelli, & Cianciulli, 2005). NTBI/LPI is unavailable for erythropoiesis, is taken up by non-hematopoietic cells in a dysregulated manner, causes parenchymal iron deposition (Jenkitkasemwong et al., 2015), and can result in free radical damage leading to the morbidity and mortality of iron overload diseases.
3. Regulation of hepcidin expression
Hepcidin expression is predominantly regulated by iron in a feedback loop, a process that is modulated by inflammation and erythropoiesis (Fig. 1). Hepcidin regulation by iron involves multiple pathways through which hepatocytes sense circulating iron status. First, hepatocytes sense iron indirectly in response to iron-induced bone morphogenic protein (BMP) production by liver sinusoidal endothelial cells (Enns et al., 2013). The BMP pathway is critical for the regulation of hepcidin expression by iron (Babitt et al., 2007; Truksa, Peng, Lee, & Beutler, 2006). BMP6 and BMP2 stimulate hepcidin expression by binding BMP receptor and triggering phosphorylation and signaling via SMAD1/5/8 which, coupled with SMAD4, translocate to the nucleus to induce transcription of hepcidin (Fig. 2). Second, hepatocytes can also directly sense iron as a consequence of cell surface expression of TfR1, TfR2, and HFE. HFE associates with TfR1 under low iron conditions 0 and is displaced when TfR1 binds Tf-Fe(III) (Bennett, Lebrón, & Bjorkman, 2000; Giannetti & Björkman, 2004; Lebrón et al., 1998). As serum iron concentration increases, TfR2 expression exceeds that of TfR1 and Tf-Fe(III) binds both TfR1 and TfR2, increasing TfR2 stability (Johnson & Enns, 2004; Robb & Wessling-Resnick, 2004) on the membrane and induces HFE binding to TfR2. This HFE/TfR2 complex interacts with hemojuvelin (HJV), the iron-specific BMP co-receptor, and potentiates the BMP signaling pathway and hepcidin transcription 0 (Fig. 2). Thus, both TfR2 and HFE/TfR1 complex function as the main Tf-Fe(III) sensors (Goswami & Andrews, 2006; Robb & Wessling-Resnick, 2004) and communicate systemic iron status to the hepatocyte resulting in altered hepcidin secretion.
Fig. 2.
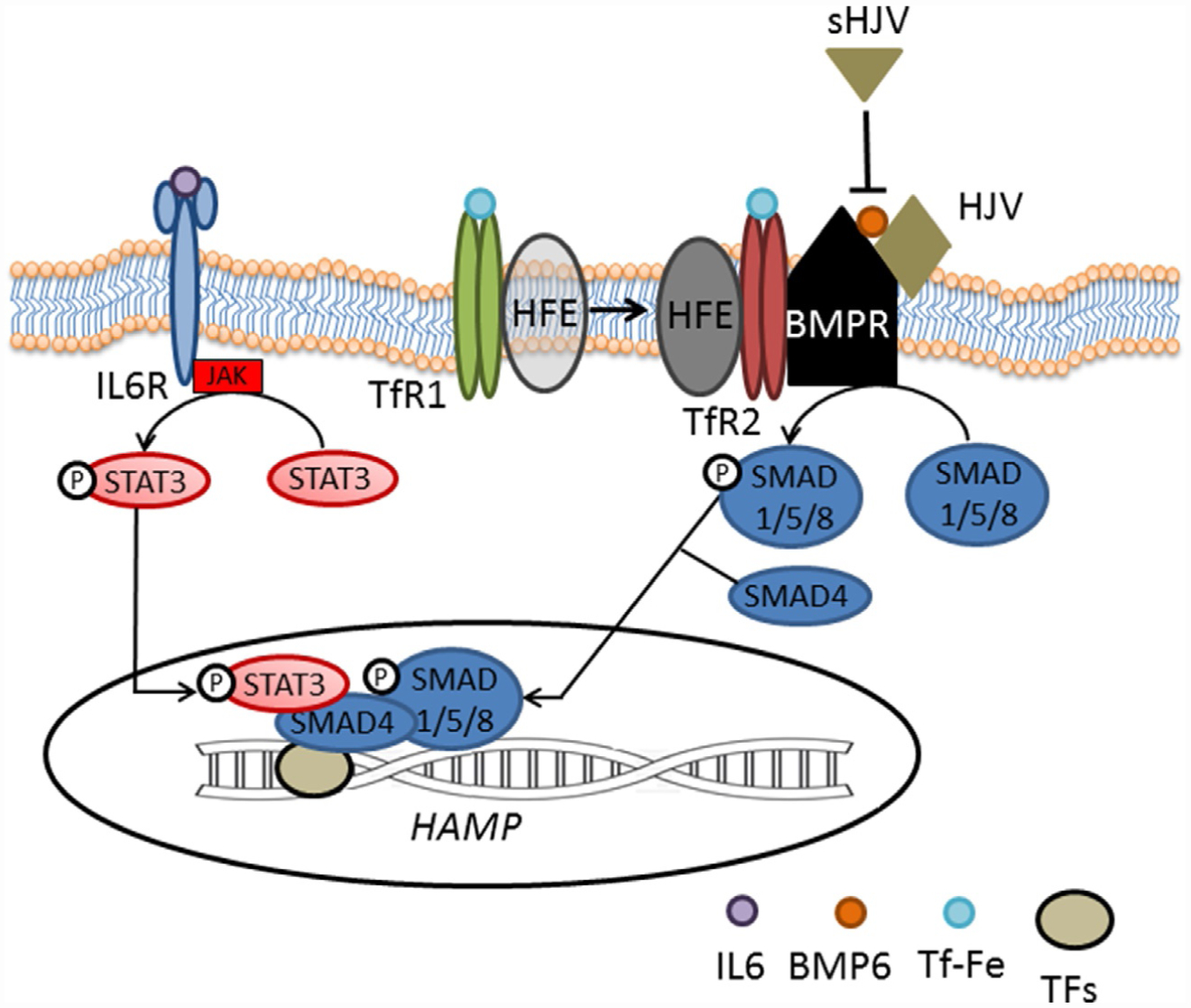
Regulation of hepcidin expression. Hepcidin expression involves iron sensing by hepatocytes. BMP6 and possibly BMP2 bind at BMP receptor, initiating a signaling cascade of SMAD1/5/8 phosphorylation which, couples with SMAD4 and translocates into the nucleus to initiate hepcidin transcription. As part of the inflammatory cascade, IL6 binds IL6 receptor, initiating a JAK/STAT signaling cascade of STAT3 phosphorylation which translocates into the nucleus and, in a SMAD-dependent manner, initiate hepcidin expression. BMP, bone morphogenic protein; BMPR, BMP receptor; Fe, iron; HFE, high iron Fe; HJV, hemojuvulin; IL6, interleukin 6; IL6R, IL6 receptor; JAK, janus kinase; sHJV, soluble HJV; SMAD, single mother against decapentaplegic; STAT, signaling transducer and activator of transcription; Tf, transferrin; TFs, transcription factors.
4. Hepcidin regulation by inflammation
Hepcidin induction during inflammation is typically mediated by IL-6 (but also IL-1β and IL-22 (Lee et al., 2006), signaling via STAT3 pathway in hepatocytes (Fig. 2), and results in iron sequestration within iron-recycling macrophages, hypoferremia, and restricts iron availability for erythropoiesis (Smith et al., 2013; Wrighting & Andrews, 2006). More recent data suggests that IL-6 may have a secondary suppressive effect on erythroid precursors (McCranor, Kim, Cruz, et al., 2014). Some crosstalk between IL-6 induced STAT3 signaling and BMP/SMAD signaling to hepcidin has been documented. For example, the SMAD binding site on the hepcidin promoter remains essential for IL-6 mediated hepcidin expression (Fleming, 2007; Huang, Constante, Layoun, & Santos, 2009; Verga Falzacappa, Casanovas, Hentze, & Muckenthaler, 2008), and liver-specific Smad4 knockout mice demonstrate diminished hepcidin responsiveness to IL-6 (Wang, Li, Xu, et al., 2005). In vitro experiments reveal that methods blocking BMP receptor signaling inhibit IL-6-mediated hepcidin expression (Babitt, Huang, Wrighting, et al., 2006; Babitt et al., 2007), providing a rationale for modulating the BMP pathway to alter hepcidin expression in the context of ACI.
5. Hepcidin regulation by erythropoiesis
Because of the high iron requirements for hemoglobin synthesis, erythropoiesis dominates regulation of iron metabolism requiring significant crosstalk. For instance, iron absorption increases, often dramatically, during stress erythropoiesis to accommodate the higher iron demand. Recent data provides mechanistic evidence of an iron restriction response, demonstrating regulation of erythroid precursor proliferation and differentiation during iron restriction (Bullock et al., 2010; Khalil et al., 2018), in addition to an expected decrease in per cell and total hemoglobin synthesis. Conversely, disease states of excess iron are often associated with expanded RBC size and higher cellular hemoglobin concentrations as a way of sequestering iron into a non-toxic compartment (McLaren et al., 2007). Furthermore, diseases in which anemia and excess iron coexist exhibit complicated regulation schema that remain incompletely understood. Such diseases of concurrent iron overload and expanded or ineffective erythropoiesis (e.g., β-thalassemia, myelodysplastic syndromes, and dyserythropoietic anemias) exhibit lower than expected hepcidin expression, despite increased iron stores. In fact, sub-optimal hepcidin expression is implicated in iron overload in these diseases and predicted the existence of an “erythroid factor” regulating iron metabolism (Gardenghi et al., 2007; Ginzburg et al., 2009).
To explore mechanisms by which erythropoiesis regulates hepcidin and thus iron metabolism required separating whether Epo, hypoxia, anemia, reticulocytosis, or erythropoiesis itself are involved. Prior experiments demonstrate that phlebotomy, Epo administration, and hemolysis all resulted in decreased hepcidin expression (Nicolas, Chauvet, et al., 2002; Nicolas, Viatte, et al., 2002; Vokurka, Krijt, Sulc, & Necas, 2006). To separate the effect of erythropoiesis from that of anemia and iron stores, ablation of erythropoiesis, accomplished by chemotherapeutic agents, radiation, and Epo-blocking antibodies, was compared with compensated hemolysis (without ablation of erythropoiesis). Results revealed that bone marrow ablation prevents hepcidin suppression in response to hemolysis, bleeding, or Epo injection (Pak, Lopez, Gabayan, Ganz, & Rivera, 2006; Vokurka et al., 2006). Although ablation of erythropoiesis results in increased hepcidin expression, compensated hemolysis (without ablation of erythropoiesis) does not affect hepcidin expression despite an equivalent degree of anemia and hepatic iron deposition. These results strongly support the expectation that erythroid regulation of hepcidin is a consequence of expanded number of erythroid precursors during stress or ineffective erythropoiesis.
Furthermore, multiple pieces of data predict that an erythroid factor is secreted by erythroid precursors, functioning as a hormone to suppress hepcidin expression in the liver. Several candidates have been proposed. Circulating growth differentiation factor 15 (GDF15) is increased in patients with several congenital and acquired anemias and correlates with concurrent low hepcidin (Tanno et al., 2007). However, studies in phlebotomized mice (Casanovas et al., 2013) and in MDS patients (Santini et al., 2011) have shown poor correlation between GDF15 and hepcidin levels. Thus, mechanisms of hepcidin suppression may be disease specific. Recently, a potential erythroid regulator of hepcidin, erythroferrone (ERFE), has been identified (Kautz et al., 2014). However, ERFE appears to play a minimal role in baseline erythropoiesis and iron regulation with ERFE knockout mice exhibiting only mild anemia during the postnatal period (Kautz et al., 2014); its main function is likely to facilitate iron mobilization during recovery from anemia. ERFE receptor and signaling pathways involved in ERFE regulation of hepcidin are active areas of exploration.
6. Hypoxia mediated hepcidin-dependent and independent regulation of iron absorption
Hypoxia regulates hepcidin in both an Epo-dependent and Epo-independent manner. Physiological responses to hypoxia are regulated by transcription factors known as hypoxia-inducible factors (HIFs). HIFs bind hypoxia-response elements to regulate genes central to erythropoiesis (e.g., Epo), angiogenesis, and metabolism. HIFs are degraded in the proteasome as a consequence of HIF hydroxylation by propyl hydroxylases in normoxic conditions. Propyl hydroxylase function requires iron and inactivating mutations in propyl hydroxylases result in increased HIF concentration and induction of erythrocytosis, overriding the upregulation of hepcidin associated with inflammation (Peyssonnaux et al., 2007), resulting in hepcidin suppression (Piperno et al., 2011). Several in vitro studies suggest direct hepcidin regulation by HIFs (Braliou et al., 2008; Peyssonnaux et al., 2007). However, subsequent in vivo studies identified Epo-dependent pathways (e.g., via ERFE) as critical for HIF-induced hepcidin suppression (Liu, Davidoff, Niss, & Haase, 2012; Mastrogiannaki et al., 2012). In addition, hypoxia induces iron absorption directly, in part independently of regulation by hepcidin and ferroportin. The typically hypoxic environment and increased HIF2α in the small intestine which induced iron uptake at the apical side of enterocytes and enhances iron absorption at the basolateral side (via ferroportin) independently of hepcidin (Shah, Matsubara, Ito, Yim, & Gonzalez, 2009) (Fig. 3). Lastly, recent in vivo data indicates that iron restriction enhances hypoxia responsiveness (Frise et al., 2016) by regulating HIF2α (Asshoff et al., 2017).
Fig. 3.
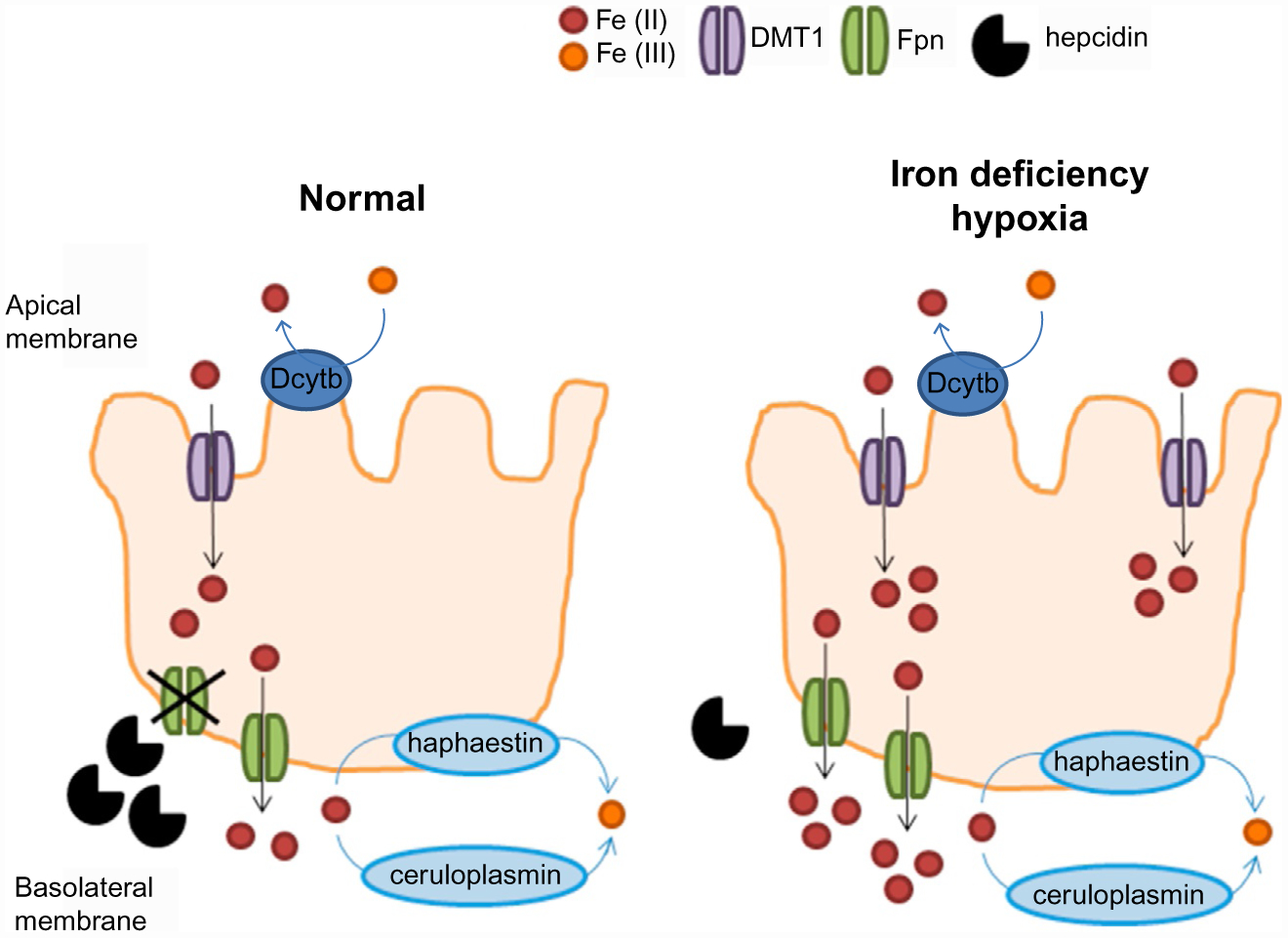
Regulating iron absorption by hepcidin-dependent and -independent mechanisms. At the duodenal enterocyte, where iron is absorbed, hepcidin functions as a negative regulator at the basolateral surface, where ferroportin exports iron from duodenal enterocytes into the circulation. Hypoxia functions as a positive regulator at the apical surface where DMTI imports iron into duodenal enterocytes. Together, the presence of hypoxia in the gastrointestinal tract with suppression of hepcidin would provide the most potent stimulus for iron absorption. DMTI, divalent metal transporter 1; Fpn, ferroportin.
7. Hepcidin-ferroportin axis regulates iron flows
Iron absorption is predominantly regulated at the basolateral surface of the duodenal enterocyte by control of iron export through ferroportin into plasma. Iron is take up into the duodenal enterocyte on the apical membrane via DMT1 and is stored or exported during its life span of a few days (Fig. 3). If iron retained within the enterocyte is no exported, it will be lost as duodenal enterocytes are shed in the gastrointestinal tract. Ferroportin is also expressed on macrophages. As a consequence of their involvement in erythrophagocytosis of senescent RBCs, macrophages break down hemoglobin and recycle iron. Kupffer cells, residing in the liver sinusoids, and red pulp macrophages in the spleen are the major populations of macrophages responsible for steady-state erythrophagocytosis. As in duodenal enterocytes, iron liberated from hemoglobin is either exported via ferroportin or sequestered within cytosolic ferritin. Ferroportin is the receptor for hepcidin the binding of which results in the endocytosis and degradation of ferroportin and consequently reduced iron export (Fig. 4). As ferroportin mediates all cellular iron export, hepcidin regulates both the acquisition of dietary iron and the release of iron from macrophages and in conditions associated with high hepcidin (e.g., ACI), iron absorption and recycling are decreased, resulting in a decrease in circulating serum iron concentration and transferrin saturation and an increase in serum ferritin concentration (Fig. 4), the later proportional to the increased intracellular ferritin, classically driven by increased intracellular iron through the IRE:IRP post-transcriptional regulation.
Fig. 4.
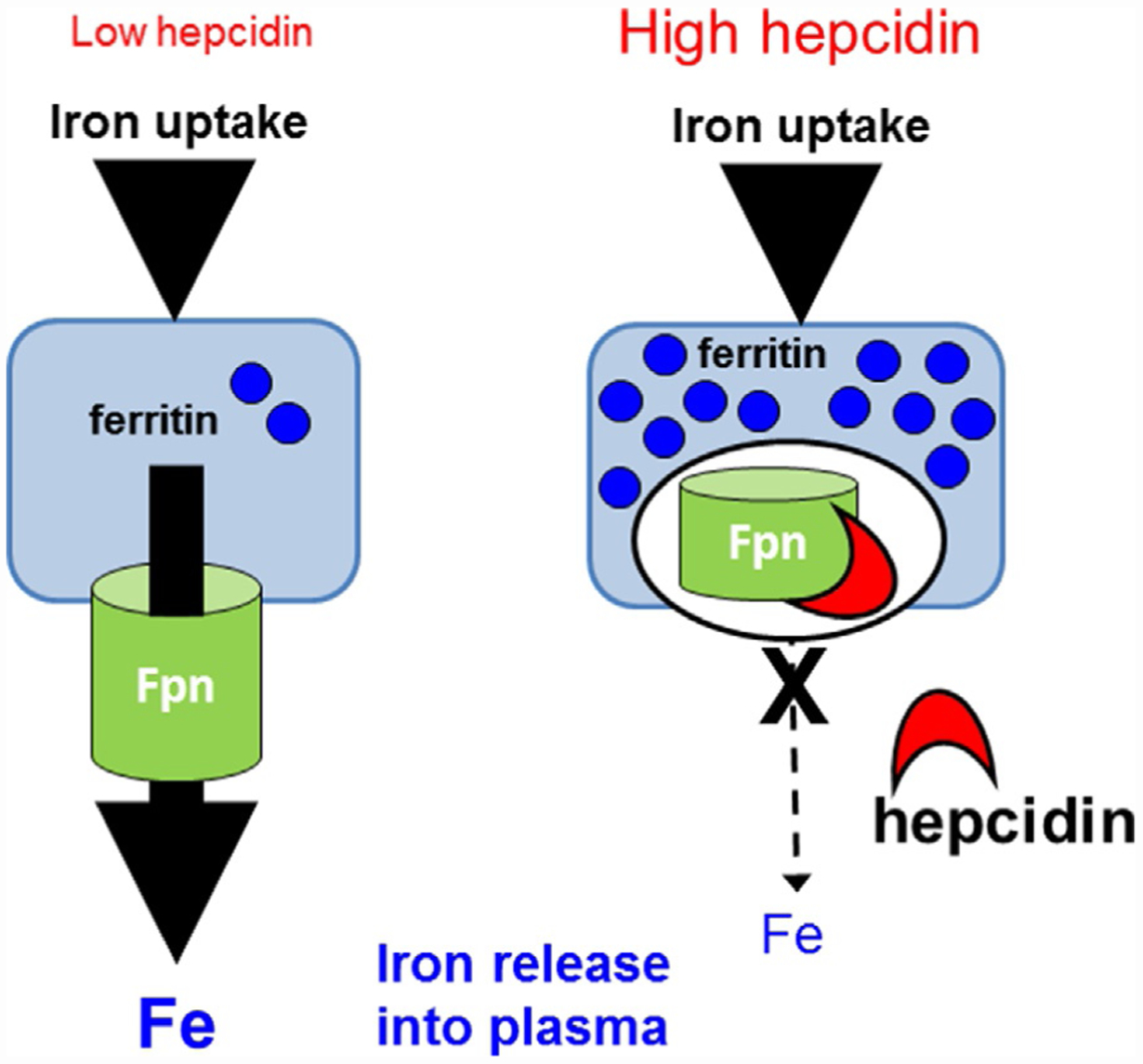
Hepcidin functions via effects on ferroportin mediated iron efflux. Hepcidin binds to and results in the internalization and degradation of ferroportin, leading to a block in iron efflux from ferroportin expressing cells (duodenal enterocytes, hepatocytes, macrophages, and placenta). In low hepcidin states, iron taken up by these cells is exported via ferroportin. Conversely, in high hepcidin states, the loss of membrane ferroportin results in iron sequestration, typically within cytosolic ferritin. Fe, iron; Fpn, ferroportin.
The discovery of hepcidin as a central regulator of iron metabolism and erythroid regulation of hepcidin by ERFE has enabled a more comprehensive exploration of aberrant iron metabolism and molecular mechanism underlying this effect in many diseases.
8. Hereditary hemochromatosis
Hereditary hemochromatosis (HH) is a genetically inherited disorder of iron metabolism. Although gene frequency is as high as 5–7%, low penetrance results in only 1:300 to 1:400 affected individuals. Four types of disorders exist, all of which result in increased intestinal iron absorption as a consequence of inadequate hepcidin, or hepcidin insensitivity, relative to the degree of systemic iron (Bridle et al., 2003; Muckenthaler et al., 2003; Nicolas et al., 2003). The most common type of HH, Type I HH, results from a mutation in the HFE gene. Homozygous C282Y mutation accounts for more than 80% HH patients. Disease is occasionally present as a compound heterozygote mutation with a second synergistic H63D mutation. Both mutations lead to the reduction of cell surface HFE on hepatocytes. HFE is one of several membrane proteins involved in communicating systemic iron status to the hepatocyte and affects HJV/BMP signaling pathway to positively influence hepcidin production. Suppressed HFE levels in HH prevent appropriate sensing and result in a dampened hepcidin response to iron load, consequent increased iron absorption and transferrin saturation.
In addition to HFE, iron overload diseases are also associated with mutation in genes coding for other proteins involved in iron sensing and hepcidin regulation. Type II HH results from HJV or hepcidin mutations. As those with HFE mutations, patients with mutated HJV/HAMP also exhibit enhanced iron absorption and rapid iron accumulation at a young age. HJV mutations, like mutations in hepcidin itself, result in nearly absent hepcidin expression and clinically result in the most severe form of HH termed Juvenile Hemochromatosis. Type III HH results from a mutation in the gene encoding TfR2. Lastly, type IV HH is an autosomal dominant mutation in ferroportin. Because mutations in the ferroportin gene may affect its membrane concentration as well as hepcidin binding potential, there are two clinical features of this genetic disorder. Patients with mutation leading to reduced ferroportin membrane expression (loss-of-function mutation) develop low transferrin saturation and Kupffer cell iron loading due to limited iron export and become anemic when treated with phlebotomy. These patients are unable to mobilize their iron stores. Although ferroportin on duodenal enterocytes is likely also affected, the transport of 2–4mg of iron during iron absorption may be easier to accomplish by compensatory mechanisms than the 20mg of iron recycled daily by macrophages. The other type of ferroportin mutation is characterized by hepcidin insensitivity (gain-of-function mutation). This mutation is associated with high transferrin saturation and hepatocyte iron loading. Supporting evidence from a ferroportin mutated cell line demonstrates normal iron efflux activity but no respond to hepcidin. Thus, “hepcidin-resistant” HH is phenotypically similar to hepcidin deficiency in other types of HH.
Overall, HH mutations are associated with hepcidin suppression or hepcidin insensitivity. Hepcidin injections inhibit the increased iron absorption in the duodena of Hfe−/− mice and forced expression of hepcidin corrects the hemochromatosis phenotype. Furthermore, sera from patients with HH do not result in hepcidin suppression in HepG2 cells and induce an increased hepcidin gene expression in normal hepatocytes. These findings demonstrate mutant HFE on hepatocytes results in insufficient hepcidin stimulation in HH patients leading to increased iron absorption and iron overload in this disease. As a consequence, hepcidin-mimetic agents may be applicable as novel therapies in HH patients either in conjunction or in place of therapeutic phlebotomy.
9. β-thalassemia syndromes
β-thalassemias are caused by mutations in the β-globin gene resulting in reduced or absent β-chain synthesis. A relative excess of α-globin chain synthesis leads to increased erythroid precursor apoptosis, causing ineffective erythropoiesis which in turn results in extramedullary expansion and splenomegaly. Together with shortened RBC survival, these abnormalities result in anemia. The clinical phenotype is heterogeneous. Patients with β-thalassemia major, the most severe form of β-thalassemia, require life-long RBC transfusions to ameliorate anemia and suppress extramedullary erythropoiesis. Without transfusions, expanded erythropoiesis results in progressive hepatosplenomegaly and bone deformities, due to expansion of extramedullary and intra-osseous erythropoiesis, respectively. In contrast, patients with β-thalassemia intermedia show a milder clinical picture requiring only intermittent transfusions. Both β-thalassemia major and especially intermedia have increased intestinal iron absorption which, in addition to transfusion, contributes to iron overload. If left untreated, iron overload results in progressive iron deposition, leading to multiple organ dysfunction and accounts for the majority of deaths in this disease.
Insufficient hepcidin expression, relative to the degree of iron overload, is implicated as the cause of iron overload observed in β-thalassemia. Increased hepcidin levels are expected in diseases of iron overload to prevent continued iron absorption and exacerbation of iron loading. Thus, hepcidin suppression in diseases of iron overload with ineffective erythropoiesis exacerbates the degree of iron overload by increasing iron absorption. This excess iron deposits in the parenchyma of non-hematopoietic tissue. Because further iron absorption ultimately exceeds transferrin iron-carrying capacity, suppressed hepcidin results in the formation of NTBI which is not available for erythropoiesis.
Serum hepcidin concentration in β-thalassemic patients is increased in correlation with hemoglobin and increases in response to RBC transfusion, suggesting that enhanced erythropoietic activity induces hepcidin suppression. Furthermore, the exposure of HepG2 cells to sera from patients with β-thalassemia major after transfusion resulted in higher hepcidin levels relative to the cells exposed to sera of the same patients prior to the next transfusion (Kemna et al., 2008; Weizer-Stern et al., 2006). These findings suggest that hepcidin suppression in β-thalassemic patients results from the secretion of a soluble factor the concentration of which is proportional to the degree of erythroid activity. Recent evidence supports the proposal that ERFE is this erythroid regulator of hepcidin which in β-thalassemia is stronger than the regulation of hepcidin by iron and leads to the exacerbation of iron overload, the very complication associated with clinical deterioration and mortality in this disease. Supporting evidence reveals loss of hepcidin suppression after phlebotomy in ERFE knockout mice, increased ERFE in mouse models of β-thalassemia, and relatively increased hepcidin expression and decreased iron overload in β-thalassemic/ERFE knockout relative to β-thalassemic mice (Kautz et al., 2014).
Manipulating the ERFE:hepcidin:ferroportin axis to increase hepcidin or block ERFE or ferroportin could help limit intestinal iron absorption and sequester iron in macrophages and hepatocytes to reverse or prevent iron overload. Furthermore, multiple lines of evidence support the beneficial effects of iron deficiency on ineffective erythropoiesis which could reverse splenomegaly and decrease RBC transfusion requirements by reducing RBC destruction in the spleen or improve erythroid differentiation and enucleation in β-thalassemia (Gardenghi et al., 2010; Gelderman et al., 2015; Li et al., 2017, 2010). Thus, modulation of the ERFE: hepcidin:ferroportin axis may provide significant iron restriction within the erythroid compartment and enable more effective erythropoiesis in iron loading anemias.
10. Anemia of chronic inflammation (iron restricted anemia due to infection, inflammatory disease, and/or cancer)
Anemia of chronic inflammation (ACI) is characterized as a mild, normocytic normochromic anemia with hemoglobin between 8 and 10g/dL, elevated inflammatory markers (e.g., CRP and ESR), and decreased serum iron concentration and transferrin saturation despite ample iron stores (e.g., serum ferritin >100ng/mL). The pathophysiology of ACI is multifactorial, involving inflammatory cytokines which induce erythrophagocytosis by splenic macrophages, suppresses erythroid precursor differentiation and Epo responsiveness (Grigorakaki, Morceau, Chateauvieux, Dicato, & Diederich, 2011; Means, Dessypris, & Krantz, 1992; Wang, Udupa, & Lipschitz, 1995), and decrease Epo production and renal excretion of hepcidin (Jelkmann, 1998; Krajewski, Batmunkh, Jelkmann, & Hellwig-Burgel, 2007; Vannucchi et al., 1994). Recent consensus regarding the pathophysiology underlying ACI supports altered iron metabolism at its center. Within hours of infection or inflammatory stimuli, plasma iron concentration decreases in humans, other mammals, and lower vertebrates. As a consequence of immune mediators, such as IL-6 and possibly other cytokines involved in host defense, induced as part of the underlying disease, hepcidin-induced hypoferremia results in iron sequestration within the reticuloendothelial system, decreasing iron availability for erythropoiesis and causing anemia (Fig. 5). In support of this, the loss of IL-6 or hepcidin in mice results in absence or attenuation of hypoferremia, a milder anemia, and more rapid recovery of hemoglobin in a well-established mouse model of ACI (Gardenghi, Renaud, Meloni, et al., 2014). Furthermore, IL-6 knockout animals exhibited faster bone marrow recovery relative to hepcidin knockout animals. Taken together, although treatment of the underlying disease remains at the center of therapy for ACI, recent advances in molecular understanding of the hepcidin-ferroportin axis are stimulating the development of hepcidin antagonists that would be potentially beneficial in select ACI cases.
Fig. 5.
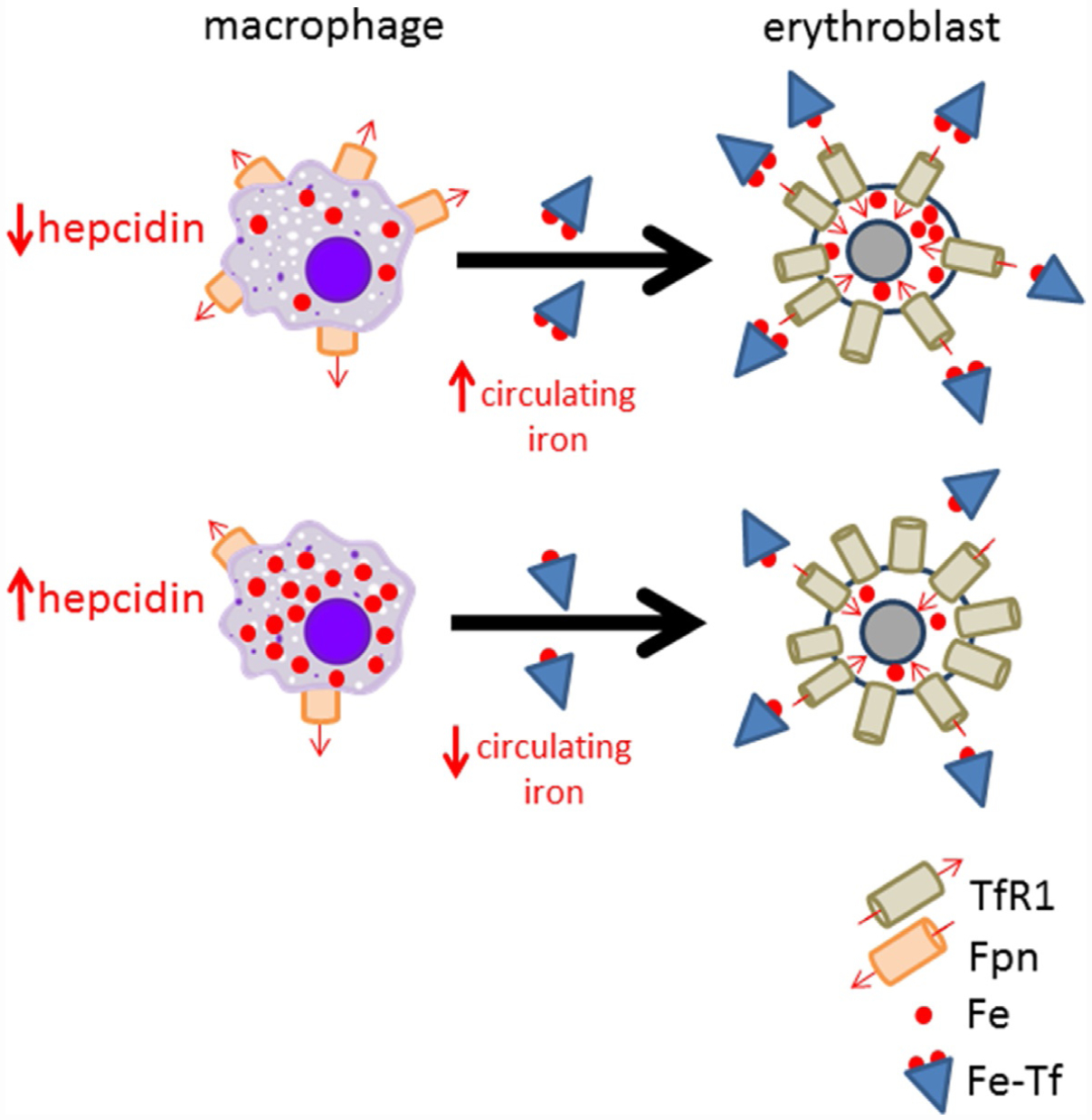
Model of hepcidin’s effect on iron availability for erythropoiesis. Decreased hepcidin, as in systemic iron deficiency, leads to more ferroportin and consequently more iron efflux into circulation where it is available for uptake by erythroblasts via Fe-Tf binding to TfRl. Alternatively, hepcidin elevation, as in anemia of chronic disease, leads to hepcidin:Fpn binding, preventing iron egress, resulting in iron sequestration within macrophages (e.g., splenic macrophages, involved in iron recycling from senescent red blood cells), and leads to iron restricted erythropoiesis. Fe, iron; Fe-Tf, Fe loaded transferrin; Fpn, ferroportin; TfRl, transferrin receptor 1.
Lastly, evidence of susceptibility to demise with siderophilic bacteria (e.g., Vibrio vulnificus and Yersinia enterocolitica but also the significantly more common Klebsiella pneumonia and Escherichia coli) as a consequence of circulating NTBI in diseases of iron overload highlights the potential clinical application of hepcidin agonists as protection from such susceptibility (Ganz, 2018; Stefanova et al., 2018). However, because some intracellular pathogens (e.g., Salmonella) target macrophages, it would be reasonable to consider possible increase in susceptibility to injection in response to hepcidin agonists. In vivo and in vitro studies have not yet provided definitive resolution of this question (Chlosta et al., 2006; Stefanova et al., 2017; Willemetz et al., 2017) but it remains clear that hepcidin agonists may play a significant clinical role in preventing demise from some severe acute systemic infections.
11. Polycythemia vera
Polycythemia vera (PV), essential thrombocytosis, and primary myelofibrosis are the classical BCR-ABL negative myeloproliferative neoplasms (MPNs). These blood cancers represent a heterogeneous group of clonal hematopoietic stem cell disorders with constitutively activated physiologic signal-transduction pathways (Spivak, 2017). PV is characterized by erythrocytosis, bone marrow hyperplasia, fatigue, microvascular symptoms and symptomatic splenomegaly. Complications of PV include a significantly increased risk of thrombosis (41%) and the potential for evolution to myelofibrosis (10–20%) and MPN-blast phase (3–10%), significantly reducing survival (Gruppo Italiano Studio Policitemia, 1995; Passamonti et al., 2004; Stein et al., 2015). The most common JAK2 driver mutation is JAK2 V617F which results in constitutive Epo-independent JAK-STAT signaling and upregulation of genes downstream of the JAK-STAT pathway (Akada et al., 2010; Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Lu, Huang, & Lodish, 2008; Rampal et al., 2014). The primary goals of treating PV patients are to ameliorate symptoms, reduce the risk of thrombosis, and prevent transformation to MF and/or MPN-blast phase. Although several therapeutic approaches have led to significant improvement in clinical symptoms and reduce the risk of thrombosis, no currently available therapy alters the natural history of disease progression.
Most patients with PV present with iron deficiency at diagnosis (Gianelli et al., 2008; Thiele et al., 2001), even prior to the onset of therapeutic phlebotomy, the mainstay of treatment, and iron deficiency is often exacerbated by repeated phlebotomies. Cytoreductive therapy may be associated with correction of the iron deficiency in PV patients. Whether this effect is achieved by decreasing phlebotomy requirement or by directly influencing regulators of iron metabolism is incompletely understood. For example, a recent phase III clinical trial in PV patients demonstrated significant reductions in HCT levels, splenomegaly, and PV-related symptoms in the ruxolitinib-treated group relative to those receiving best available therapy (Verstovsek et al., 2017). Notably, ruxolitinib treatment resulted in normalization of standard iron-related parameters in PV patients with baseline iron deficiency and hepcidin increased to a greater extent in ruxolitinib-treated PV patients relative to those treated with best available therapy. However, analysis of HepG2 cells treated with ruxolitinib in vitro led to decreased hepcidin expression at high concentration mainly by decreasing signaling via STAT3 (Asshoff et al., 2017). These findings suggest that enhanced hepcidin expression in ruxolitinib-treated PV patients may be due to suppression of erythropoiesis and thus a presumed reduction in ERFE levels. It is conceivable that reversal of PV-related symptoms in ruxolitinib-treated patients is a consequence solely of suppressing clonal and/or normal erythropoiesis. However, it is also possible that, because iron deficiency- and PV-related symptoms often overlap, ruxolitinib-treated patients improve symptomatically due to the consequent decreasing in phlebotomy requirements, reversing iron deficiency in general, or specifically increasing hepcidin levels, resulting in iron sequestration without exacerbating systemic iron deficiency. For example, administration of exogenous hepcidin (Casu et al., 2016) and ferroportin inhibitor (Kubovcakova et al., 2018) has been shown to reverse erythrocytosis and splenomegaly in JAK2 V617F mice; the mechanism of action is likely sequestration of iron in splenic macrophages, inducing iron restricted erythropoiesis (Fig. 5).
The mechanism by which PV patients present with iron deficiency prior to the onset of therapeutic phlebotomy is incompletely understood. Several explanations are possible. Previous reports suggest that hepcidin expression is higher than expected in PV patients (Tarkun et al., 2013), preventing recovery from iron deficiency. We previously demonstrate that despite increased ERFE and relative suppression of hepcidin, JAK2 mutated patients with erythrocytosis exhibit relatively more iron restricted erythropoiesis (Ginzburg et al., 2018). This is surprising because hepcidin suppression is expected to result in iron influx into the circulation to enable recovery from iron deficiency. If iron restriction normally serves as a brake on erythropoiesis when iron availability is limited, aberrant inflammation-insensitive erythropoiesis in PV may hijack iron for hemoglobin synthesis at the expense of iron requirements for all other cell functions, depleting iron stores. Thus, hepcidin suppression without recovery from iron deficiency suggests that aberrant regulation of the iron restriction response occurs in PV patients (Fig. 6). Iron restriction is thought to involve decreased erythroid precursor sensitivity to inflammation in an Epo-independent manner, and selectively enable cell survival without inducing differentiation (Bullock et al., 2010; Khalil et al., 2018).
Fig. 6.
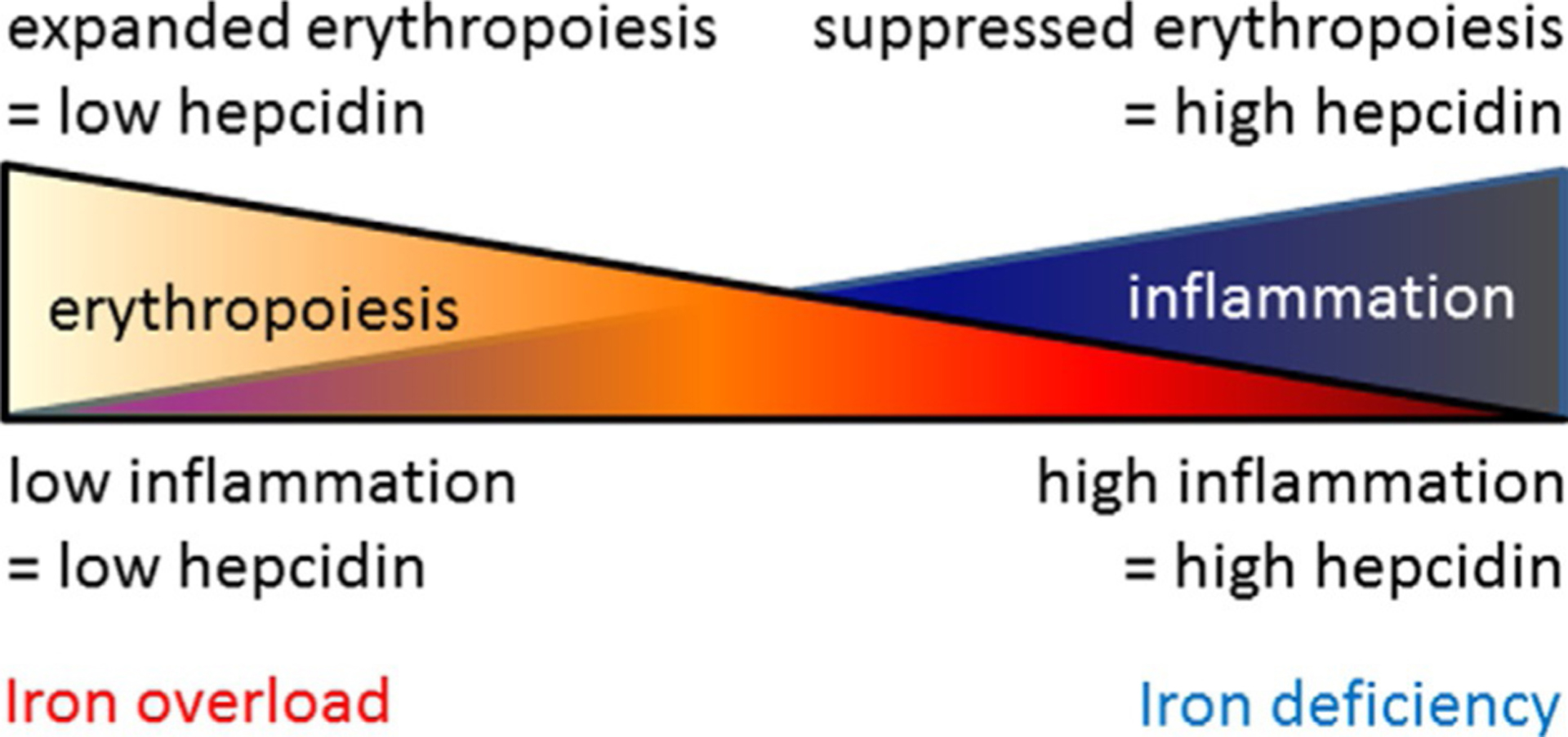
Competitive hepcidin regulation. Hepcidin expression is enhanced by inflammation and suppressed by erythropoiesis such that the combination of both conditions would theoretically lead to equal opposite directional effects on hepcidin regulation in PV, a disease of concurrent expanded erythropoiesis and inflammation.
As a consequence of systemic iron restriction, the use of hepcidin mimetics may appear counter intuitive. However, because PV is associated with increased red cell mass and a larger than typical proportion of body iron is contained within hemoglobin, using hepcidin mimetics would enable a redistribution of iron by sequestering it within hepatocytes and macrophages, preventing depletion of iron stores, restricting iron availability for erythropoiesis, and reversing symptoms of systemic iron deficiency. Studies in JAK2 V617F mice demonstrate the potential of exogenous hepcidin to reverse erythrocytosis, decrease splenomegaly, and sequester iron in splenic macrophages (Casu et al., 2016) and suggest that the use of such “hepcidin mimetic agents” may be beneficial in low risk PV patients.
12. Targeting the hepcidin:ferroportin axis for therapeutic purposes
Increasing hepcidin would be useful in diseases of primary iron overload or iron-loading anemias (e.g., β-thalassemia and myelodysplastic syndrome). Studies in mouse models of HH and β-thalassemia syndromes demonstrate that hepcidin overexpression prevents iron overload and improves erythropoiesis, respectively (Gardenghi et al., 2010; Nicolas et al., 2003). In addition, silencing Tmprss6, important for physiological hepcidin suppression, enhances hepcidin expression, preventing iron overload and improving erythropoiesis in β-thalassemic mice (Finberg, Whittlesey, & Andrews, 2011; Nai et al., 2012). These preclinical experiments stimulated the pursuit of generating agents targeting the hepcidin: ferroportin axis ultimately intended for clinical use. To this end, several strategies are being used to induce endogenous hepcidin production or to exogenously mimic its activity and clinical trials have already been initiated using hepcidin mimetics, stimulators of hepcidin production, and ferroportin inhibitors (Casu, Nemeth, & Rivella, 2018).
Novel therapeutic approaches are needed for patients with β-thalassemia for whom state-of-the-art therapy includes chronic iron chelation therapy and splenectomy in addition to lifelong RBC transfusion. Iron chelation therapy has been shown to reduce mortality from secondary iron overload due to recurrent RBC transfusion. Although RBC transfusions suppress ineffective erythropoiesis, thus decreasing ERFE expression and increasing serum hepcidin concentration relative to pre-transfusion levels, the effects are transient (Pasricha, Frazer, Bowden, & Anderson, 2013). Thus, hepcidin suppression, even in transfusion-dependent β-thalassemia major patients, may contribute somewhat to iron overload, both by enabling increased iron absorption between RBC transfusions as well as preventing sequestration of iron within macrophages and hepatocytes, resulting in increased circulating iron, generation of NTBI, and parenchymal iron overload. Furthermore, multiple lines of investigation suggest that increasing hepcidin and the consequent reduction of iron availability for erythropoiesis improves ineffective erythropoiesis, reversing splenomegaly and increasing hemoglobin, in mouse models of β-thalassemia intermedia (Casu et al., 2016; Guo et al., 2013; Li et al., 2010). Additional studies are needed to ascertain whether increasing hepcidin would also ameliorate ineffective erythropoiesis in transfusion-dependent β-thalassemia major as in transfusion-independent β-thalassemia intermedia.
Although phlebotomy therapy in HH is relatively easy, it is not free. The cost can be circumvented by enabling these patients to donate blood at donor centers but may be limited at some donor centers only to patients who would otherwise be eligible to donate blood (i.e., meeting all blood donor criteria for sexual preference, vital signs, concurrent illnesses (e.g., HCV), travel history, and medications). In addition, a small fraction of patients are intolerant to phlebotomy. Lastly, there is clear evidence that relative hepcidin deficiency leading to iron overload increases susceptibility of HH patients to siderophilic bacteria such as Vibrio vulnificus and Yersinia enterocolitica (Khan, Fisher, & Khakoo, 2007; Vadillo, Corbella, Pac, Fernandez-Viladrich, & Pujol, 1994). Mouse models of HH and iron loading in wild type mice revealed increased susceptibility to these infections as well as Klebsiella pneumoniae (Arezes et al., 2015; Horseman & Surani, 2011; Michels et al., 2017; Stefanova et al., 2017). We and others anticipate that increasing hepcidin to sequester iron within macrophages would decrease pathogen access to circulating iron and increase survival in infected iron overloaded patients (Brissot, Ropert, Le Lan, & Loréal, 2012). It is yet unclear how targeting the hepcidin:ferroportin axis would affect pathogenesis of intracellular organisms during infection.
In addition, iron sequestration as a consequence of increased hepcidin may be clinically beneficial for PV patients with high phlebotomy requirements. Although the pathophysiological cause for iron deficiency in newly diagnosed PV remains to be clarified, increasing hepcidin would be expected to induce a kind of “chemical phlebotomy” by preventing iron accessibility for erythropoiesis. Because increased hepcidin would also block further systemic iron absorption, there is a theoretical concern that increased hepcidin would induce further exacerbation of systemic iron deficiency. To support the potential clinical utility of increasing hepcidin in PV, we look to another disease of systemic iron deficiency caused by excess hepcidin, iron-refractory iron deficiency anemia (IRIDA) (Heeney & Finberg, 2014). IRIDA presents in childhood as an extremely microcytic anemia with very low serum ferritin and transferrin saturation. Despite this, IRIDA patients have significantly fewer signs and symptoms relative to true iron deficiency, and long-term follow-up reveals normal growth and cognitive development (unlike the concerns in systemic iron deficiency, leading to universal infant screening programs) (Baker & Greer, 2010; Melis et al., 2008; Pearson & Lukens, 1999). These differences suggest that iron sequestered in macrophages is available for cellular function and homeostasis, even during periods of rapid growth, but the quantities of available iron are insufficient to enable robust erythropoiesis. We hypothesize that redistribution of iron that is expected to result from increased hepcidin in PV would be expected to sequester iron within macrophages to halt constitutive and aberrant erythropoiesis, decrease phlebotomy requirements, and together reduce systemic symptoms of iron deficiency.
Hepcidin antagonists have been postulated as potentially effective novel therapy for ACI and anemia of cancer, diseases in which ESAs and IV iron are either scarcely effective or contraindicated. This category of potential therapeutics includes hepcidin sequestering antibodies, anticalins, and aptamers, BMP/SMAD and IL6/STAT3 inhibitors, hepcidin mRNA degrading and protein sequestering agents, or ferroportin stabilizers (Fung & Nemeth, 2013; Leung, Luan, Manetta, Tang, & Witcher, 2012). Specifically, first-in-human phase 1 study of fully humanized neutralizing anti-hepcidin monoclonal antibody, LY2787106, in anemia of cancer was recently published (Vadhan-Raj et al., 2017). Results demonstrate that LY2787106 is well tolerated and leads to increased serum iron and transferrin saturation within 24h, returning to baseline after 8 days. In addition, pre-clinical studies in cynomolgus monkeys using the small protein-based anticalin PRS-080 demonstrate high affinity and specificity for hepcidin, 43h half-life, and dose-dependent hepcidin suppression and iron mobilization (Hohlbaum et al., 2018). Furthermore, the anti-hepcidin Spiegelmer, lexaptepid, a synthetic compound designed to inhibit hepcidin without nuclease and immune system clearance susceptibility, resulted in a dose-dependent increase in serum iron and transferrin saturation and was well tolerated in healthy subjects (Boyce et al., 2016). This agent is being developed for ACI.
Other approaches, such as anti-BMP 6 antibody, were ineffective as a consequence presumably of compensation by other BMPs (Corradini et al., 2010) while inhibitors of the HJV blocked SMAD phosphorylation and decreased hepcidin expression in normal rodents (Andriopoulos et al., 2009; Babitt et al., 2007) as well as an ACD rat model, inducing recovery from anemia (Theurl et al., 2011). Anti-IL6 (siltuximab) and anti-IL6 receptor (tocilizumab) antibodies demonstrate decreased hepcidin in patients with Castleman’s disease, improving anemia (van Rhee et al., 2010; Song et al., 2010). In addition, several agents that prevent STAT3 phosphorylation decreased IL6-mediated hepcidin expression in mice (Fatih et al., 2010; Zhang, Wang, Wang, & Liu, 2011). An excellent review was recently published on this topic (Poli, Asperti, Ruzzenenti, Regoni, & Arosio, 2014). In addition, silencing of HJV and TfR2 by siRNA as well as induction of purported erythroid regulators of hepcidin (e.g., GDF15 or ERFE) may be additional future targets to explore.
13. Conclusion
The discovery of hepcidin as a central regulator of iron metabolism and erythroid regulation of hepcidin by ERFE enabled a deeper exploration of aberrant iron metabolism and molecular mechanism underlying this effect in many hematopoietic and non-hematopoietic diseases. This enhanced understanding has within a relatively short timeframe lead to the development of novel compounds manipulating this pathway to support both exogenous agonist and antagonist function with multiple agents already undergoing clinical trials for several indications.
References
- Akada H, Yan D, Zou H, Fiering S, Hutchison RE, & Mohi MG (2010). Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood, 115(17), 3589–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriopoulos BJ, Corradini E, Xia Y, Faasse S, Chen S, Grgurevic L, et al. (2009). BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nature Genetics, 41, 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arezes J, Jung G, Gabayan V, Valore E, Ruchala P, Gulig PA, et al. (2015). Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host & Microbe, 17(1), 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asshoff M, Petzer V, Warr MR, Haschka D, Tymoszuk P, Demetz E, et al. (2017). Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood, 129(13), 1823–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babitt JL, Huang FW, Wrighting DM, et al. (2006). Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nature Genetics, 38, 531–539. [DOI] [PubMed] [Google Scholar]
- Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, & Lin HY (2007). Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. The Journal of Clinical Investigation, 117(7), 1933–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RD, & Greer FR (2010). Committee on Nutrition American Academy of Pediatrics. Diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0–3 years of age). Pediatrics, 126(5), 1040–1050. [DOI] [PubMed] [Google Scholar]
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. (2005). Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet, 365(9464), 1054–1061. [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Lebrón JA, & Bjorkman PJ (2000). Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor. Nature, 403(6765), 46–53. [DOI] [PubMed] [Google Scholar]
- Boyce M, Warrington S, Cortezi B, Zöllner S, Vauléon S, Swinkels DW, et al. (2016). Safety, pharmacokinetics and pharmacodynamics of the anti-hepcidin Spiegelmer lexaptepid pegol in healthy subjects. British Journal of Pharmacology, 173(10), 1580–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braliou GG, Verga Falzacappa MV, Chachami G, Casanovas G, Muckenthaler MU, & Simos G (2008). 2-Oxoglutarate-dependent oxygenases control hepcidin gene expression. Journal of Hepatology, 48(5), 801–810. [DOI] [PubMed] [Google Scholar]
- Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, et al. (2003). Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet, 361(9358), 669–673. [DOI] [PubMed] [Google Scholar]
- Brissot P, Ropert M, Le Lan C, & Loréal O (2012). Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochimica et Biophysica Acta, 1820(3), 403–410. [DOI] [PubMed] [Google Scholar]
- Bullock GC, Delehanty LL, Talbot AL, Gonias SL, Tong WH, Rouault TA, et al. (2010). Iron control of erythroid development by a novel aconitase-associated regulatory pathway. Blood, 116(1), 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantchik ZI, Breuer W, Zanninelli G, & Cianciulli P (2005). LPI-labile plasma iron in iron overload. Best Practice & Research. Clinical Haematology, 18(2), 277–287. [DOI] [PubMed] [Google Scholar]
- Casanovas G, Vujić Spasic M, Casu C, Rivella S, Strelau J, Unsicker K, et al. (2013). The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematologica, 98(3), 444–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casu C, Nemeth E, & Rivella S (2018). Hepcidin agonists as therapeutic tools. Blood, 131(16), 1790–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casu C, Oikonomidou PR, Chen H, Nandi V, Ginzburg Y, Prasad P, et al. (2016). Minihepcidin peptides as disease modifiers in mice affected by β-thalassemia and polycythemia vera. Blood, 128(2), 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlosta S, Fishman DS, Harrington L, Johnson EE, Knutson MD, Wessling-Resnick M, et al. (2006). The iron efflux protein ferroportin regulates the intracellular growth of Salmonella enterica. Infection and Immunity, 74(5), 3065–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradini E, Schmidt PJ, Meynard D, Garuti C, Montosi G, Chen S, et al. (2010). BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology, 139, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan A, Lima CA, Pinkus JL, et al. (2005). The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metabolism, 1, 191–200. [DOI] [PubMed] [Google Scholar]
- Enns CA, Ahmed R, Wang J, Ueno A, Worthen C, Tsukamoto H, et al. (2013). Increased iron loading induces Bmp6 expression in the non-parenchymal cells of the liver independent of the BMP-signaling pathway. PLoS One, 8(4), e60534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito BP, Breuer W, Sirankapracha P, Pootrakul P, Hershko C, & Cabantchik ZI (2003). Labile plasma iron in iron overload: Redox activity and susceptibility to chelation. Blood, 102(7), 2670–2677. [DOI] [PubMed] [Google Scholar]
- Fatih N, Camberlein E, Island M, Corlu A, Abgueguen E, Détivaud L, et al. (2010). Natural and synthetic STAT3 inhibitors reduce hepcidin expression in differentiated mouse hepatocytes expressing the active phosphorylated STAT3 form. Journal of Molecular Medicine, 88, 477–486. [DOI] [PubMed] [Google Scholar]
- Finberg KE, Whittlesey RL, & Andrews NC (2011). Tmprss6 is a genetic modifier of the Hfe-hemochromatosis phenotype in mice. Blood, 117(17), 4590–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming RE (2007). Hepcidin activation during inflammation: Make it STAT. Gastroenterology, 132, 447–449. [DOI] [PubMed] [Google Scholar]
- Frise MC, Cheng HY, Nickol AH, Curtis MK, Pollard KA, Roberts DJ, et al. (2016). Clinical iron deficiency disturbs normal human responses to hypoxia. The Journal of Clinical Investigation, 126(6), 2139–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung E, & Nemeth E (2013). Manipulation of the hepcidin pathway for therapeutic purposes. Haematologica, 98, 1667–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T (2005). Hepcidin–a regulator of intestinal iron absorption and iron recycling by macrophages. Best Practice & Research. Clinical Haematology, 18, 171–182. [DOI] [PubMed] [Google Scholar]
- Ganz T (2018). Iron and infection. International Journal of Hematology, 107(1), 7–15. [DOI] [PubMed] [Google Scholar]
- Gardenghi S, Marongiu MF, Ramos P, Guy E, Breda L, Chadburn A, et al. (2007). Ineffective erythropoiesis in β-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood, 109(11), 5027–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardenghi S, Ramos P, Marongiu MF, Melchiori L, Breda L, Guy E, et al. (2010). Hepcidin as a therapeutic tool to limit iron overload and improve anemia in β-thalassemic mice. The Journal of Clinical Investigation, 120(12), 4466–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardenghi S, Renaud TM, Meloni A, et al. (2014). Distinct roles for hepcidin and interleukin-6 in the recovery from anemia in mice injected with heat-killed Brucella abortus. Blood, 123, 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderman MP, Baek JH, Yalamanoglu A, Puglia M, Vallelian F, Burla B, et al. (2015). Reversal of hemochromatosis by apotransferrin in non-transfused and transfused Hbbth3/+ (heterozygous B1/B2 globin gene deletion) mice. Haematologica, 100(5), 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianelli U, Iurlo A, Vener C, Moro A, Fermo E, Bianchi P, et al. (2008). The significance of bone marrow biopsy and JAK2V617F mutation in the differential diagnosis between the “early” prepolycythemic phase of polycythemia vera and essential thrombocythemia. American Journal of Clinical Pathology, 130(3), 336–342. [DOI] [PubMed] [Google Scholar]
- Giannetti AM, & Björkman PJ (2004). HFE and transferrin directly compete for transferrin receptor in solution and at the cell surface. The Journal of Biological Chemistry, 279(24), 25866–25875. [DOI] [PubMed] [Google Scholar]
- Ginzburg YZ, Feola M, Zimran E, Varkonyi J, Ganz T, & Hoffman R (2018). Dysregulated iron metabolism in polycythemia vera: Etiology and consequences. Leukemia, 32, 2105–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginzburg YZ, Rybicki AC, Suzuka SM, Hall CB, Breuer W, Cabantchik ZI, et al. (2009). Exogenous iron increases hemoglobin in beta-thalassemic mice. Experimental Hematology, 37(2), 172–183. [DOI] [PubMed] [Google Scholar]
- Goswami T, & Andrews NC (2006). Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. The Journal of Biological Chemistry, 281(39), 28494–28498. [DOI] [PubMed] [Google Scholar]
- Grigorakaki C, Morceau F, Chateauvieux S, Dicato M, & Diederich M (2011). Tumor necrosis factor alpha-mediated inhibition of erythropoiesis involves GATA-1/GATA-2 balance impairment and PU.1 over-expression. Biochemical Pharmacology, 82, 156–166. [DOI] [PubMed] [Google Scholar]
- Gruppo Italiano Studio Policitemia. (1995). Polycythemia vera: The natural history of 1213 patients followed for 20 years. Annals of Internal Medicine, 123(9), 656–664. [DOI] [PubMed] [Google Scholar]
- Guo S, Casu C, Gardenghi S, Booten S, Aghajan M, Peralta R, et al. (2013). Reducing TMPRSS6 ameliorates hemochromatosis and β-thalassemia in mice. The Journal of Clinical Investigation, 123(4), 1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeney MM, & Finberg KE (2014). Iron-refractory iron deficiency anemia (IRIDA). Hematology/Oncology Clinics of North America, 28(4), 637–652. [DOI] [PubMed] [Google Scholar]
- Hohlbaum AM, Gille H, Trentmann S, Kolodziejczyk M, Rattenstetter B, Laarakkers CM, et al. (2018). Sustained plasma hepcidin suppression and iron elevation by Anticalin-derived hepcidin antagonist in cynomolgus monkey. British Journal of Pharmacology, 175(7), 1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horseman MA, & Surani S (2011). A comprehensive review of Vibrio vulnificus: An important cause of severe sepsis and skin and soft-tissue infection. International Journal of Infectious Diseases, 15(3), e157–e166. [DOI] [PubMed] [Google Scholar]
- Huang H, Constante M, Layoun A, & Santos MM (2009). Contribution of STAT3 and SMAD4 pathways to the regulation of hepcidin by opposing stimuli. Blood, 113, 3593–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, et al. (2005). A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature, 434(7037), 1144–1148. [DOI] [PubMed] [Google Scholar]
- Jelkmann W (1998). Proinflammatory cytokines lowering erythropoietin production. Journal of Interferon & Cytokine Research, 18, 555–559. [DOI] [PubMed] [Google Scholar]
- Jenkitkasemwong S, Wang CY, Coffey R, Zhang W, Chan A, Biel T, et al. (2015). SLC39A14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis. Cell Metabolism, 22(1), 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, & Enns CA (2004). Diferric transferrin regulates transferrin receptor 2 protein stability. Blood, 104(13), 4287–4293. [DOI] [PubMed] [Google Scholar]
- Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, & Ganz T (2014). Identification of erythroferrone as an erythroid regulator of iron metabolism. Nature Genetics, 46(7), 678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemna EH, Kartikasari AE, van Tits LJ, Pickkers P, Tjalsma H, & Swinkels DW (2008). Regulation of hepcidin: Insights from biochemical analyses on human serum samples. Blood Cells, Molecules & Diseases, 40(3), 339–346. [DOI] [PubMed] [Google Scholar]
- Khalil S, Delehanty L, Grado S, Holy M, White Z 3rd, Freeman K, et al. (2018). Iron modulation of erythropoiesis is associated with Scribble-mediated control of the erythropoietin receptor. The Journal of Experimental Medicine, 215(2), 661–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan FA, Fisher MA, & Khakoo RA (2007). Association of hemochromatosis with infectious diseases: Expanding spectrum. International Journal of Infectious Diseases, 11(6), 482–487. [DOI] [PubMed] [Google Scholar]
- Krajewski J, Batmunkh C, Jelkmann W, & Hellwig-Burgel T (2007). Interleukin-1beta inhibits the hypoxic inducibility of the erythropoietin enhancer by suppressing hepatocyte nuclear factor-4alpha. Cellular and Molecular Life Sciences, 64, 989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. (2005). A gain-of-function mutation of JAK2 in myeloproliferative disorders. The New England Journal of Medicine, 352(17), 1779–1790. [DOI] [PubMed] [Google Scholar]
- Krause A, Neitz S, Magert HJ, et al. (2000). LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Letters, 480, 147–150. [DOI] [PubMed] [Google Scholar]
- Kubovcakova L, Manolova V, Nyffenegger N, Doucerain C, Flace A, Dürrenberger F, et al. (2018). Efficacy of oral Ferroportin Inhibitor in a mouse model of polycythemia vera. In Abstract, European Iron Club Meeting, Zurich, Switzerland, February 08–11. [Google Scholar]
- Lebrón JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, et al. (1998). Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell, 93(1), 111–123. [DOI] [PubMed] [Google Scholar]
- Lee C, Lim HK, Sakong J, Lee YS, Kim JR, & Baek SH (2006). Janus kinase-signal transducer and activator of transcription mediates phosphatidic acid-induced interleukin (IL)-1beta and IL-6 production. Molecular Pharmacology, 69(3), 1041–1047. [DOI] [PubMed] [Google Scholar]
- Leung DDM, Luan P, Manetta JV, Tang Y, & Witcher DR (2012). Anti-ferroportin 1 monoclonal antibodies and uses thereof. US 8183346. Washington, DC: U.S. Patent and Trademark Office. [Google Scholar]
- Li H, Choesang T, Bao W, Chen H, Feola M, Garcia-Santos D, et al. (2017). Decreasing TfR1 expression reverses anemia and hepcidin suppression in β-thalassemic mice. Blood, 129(11), 1514–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Rybicki AC, Suzuka SM, von Bonsdorff L, Breuer W, Hall CB, et al. (2010). Transferrin therapy ameliorates disease in beta-thalassemic mice. Nature Medicine, 16(2), 177–182. [DOI] [PubMed] [Google Scholar]
- Liu Q, Davidoff O, Niss K, & Haase VH (2012). Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. The Journal of Clinical Investigation, 122(12), 4635–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Huang LJ, & Lodish HF (2008). Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. The Journal of Biological Chemistry, 283(9), 5258–5266. [DOI] [PubMed] [Google Scholar]
- Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, et al. (2010). Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position −7007 of the FPN1 promoter. Haematologica, 95(8), 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrogiannaki M, Matak P, Mathieu JR, Delga S, Mayeux P, Vaulont S, et al. (2012). Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica, 97(6), 827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCranor BJ, Kim MJ, Cruz NM, et al. (2014). Interleukin-6 directly impairs the erythroid development of human TF-1 erythroleukemic cells. Blood Cells, Molecules & Diseases, 52, 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren CE, Barton JC, Gordeuk VR, Wu L, Adams PC, Reboussin DM, et al. (2007). Determinants and characteristics of mean corpuscular volume and hemoglobin concentration in white HFE C282Y homozygotes in the hemochromatosis and iron overload screening study. American Journal of Hematology, 82(10), 898–905. [DOI] [PubMed] [Google Scholar]
- Means RT Jr., Dessypris EN, & Krantz SB (1992). Inhibition of human erythroid colony-forming units by interleukin-1 is mediated by gamma interferon. Journal of Cellular Physiology, 150, 59–64. [DOI] [PubMed] [Google Scholar]
- Melis MA, Cau M, Congiu R, Sole G, Barella S, Cao A, et al. (2008). A mutation in the TMPRSS6 gene, encoding a transmembrane serine protease that suppresses hepcidin production, in familial iron deficiency anemia refractory to oral iron. Haematologica, 93(10), 1473–1479. [DOI] [PubMed] [Google Scholar]
- Michels KR, Zhang Z, Bettina AM, Cagnina RE, Stefanova D, Burdick MD, et al. (2017). Hepcidin-mediated iron sequestration protects against bacterial dissemination during pneumonia. JCI Insight, 2(6), e92002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muckenthaler M, Roy CN, Custodio AO, Miñana B, deGraaf J, Montross LK, et al. (2003). Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nature Genetics, 34(1), 102–107. [DOI] [PubMed] [Google Scholar]
- Nai A, Pagani A, Mandelli G, Lidonnici MR, Silvestri L, Ferrari G, et al. (2012). Deletion of TMPRSS6 attenuates the phenotype in a mouse model of β-thalassemia. Blood, 119(21), 5021–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E, Tuttle MS, Powelson J, et al. (2004). Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science, 306, 2090–2093. [DOI] [PubMed] [Google Scholar]
- Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, et al. (2002). The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. The Journal of Clinical Investigation, 110(7), 1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas G, Viatte L, Bennoun M, Beaumont C, Kahn A, & Vaulont S (2002). Hepcidin, a new iron regulatory peptide. Blood Cells, Molecules & Diseases, 29(3), 327–335. [DOI] [PubMed] [Google Scholar]
- Nicolas G, Viatte L, Lou DQ, Bennoun M, Beaumont C, Kahn A, et al. (2003). Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nature Genetics, 34(1), 97–101. [DOI] [PubMed] [Google Scholar]
- Pak M, Lopez MA, Gabayan V, Ganz T, & Rivera S (2006). Suppression of hepcidin during anemia requires erythropoietic activity. Blood, 108(12), 3730–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CH, Valore EV, Waring AJ, & Ganz T (2001). Hepcidin, a urinary antimicrobial peptide synthesized in the liver. The Journal of Biological Chemistry, 276, 7806–7810. [DOI] [PubMed] [Google Scholar]
- Pasricha SR, Frazer DM, Bowden DK, & Anderson GJ (2013). Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with β-thalassemia major: A longitudinal study. Blood, 122(1), 124–133. [DOI] [PubMed] [Google Scholar]
- Passamonti F, Rumi E, Pungolino E, Malabarba L, Bertazzoni P, Valentini M, et al. (2004). Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. The American Journal of Medicine, 117(10), 755–761. [DOI] [PubMed] [Google Scholar]
- Pearson HA, & Lukens JN (1999). Ferrokinetics in the syndrome of familial hypoferremic microcytic anemia with iron malabsorption. Journal of Pediatric Hematology/Oncology, 21(5), 412–417. [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, et al. (2007). Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). The Journal of Clinical Investigation, 117(7), 1926–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piperno A, Galimberti S, Mariani R, Pelucchi S, Ravasi G, Lombardi C, et al. (2011). Modulation of hepcidin production during hypoxia-induced erythropoiesis in humans in vivo: Data from the HIGHCARE project. Blood, 117(10), 2953–2959. [DOI] [PubMed] [Google Scholar]
- Poli M, Asperti M, Ruzzenenti P, Regoni M, & Arosio P (2014). Hepcidin antagonists for potential treatments of disorders with hepcidin excess. Frontiers in Pharmacology, 5, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, et al. (2014). Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood, 123(22), e123–e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb A, & Wessling-Resnick M (2004). Regulation of transferrin receptor 2 protein levels by transferrin. Blood, 104(13), 4294–4299. [DOI] [PubMed] [Google Scholar]
- Santini V, Girelli D, Sanna A, Martinelli N, Duca L, Campostrini N, et al. (2011). Hepcidin levels and their determinants in different types of myelodysplastic syndromes. PLoS One, 6(8), e23109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah YM, Matsubara T, Ito S, Yim SH, & Gonzalez FJ (2009). Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metabolism, 9(2), 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Arvedson TL, Cooke KS, Dickmann LJ, Forte C, Li H, et al. (2013). IL-22 regulates iron availability in vivo through the induction of hepcidin. Journal of Immunology, 191(4), 1845–1855. [DOI] [PubMed] [Google Scholar]
- Song SN, Tomosugi N, Kawabata H, Ishikawa T, Nishikawa T, & Yoshizaki K (2010). Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multi-centric Castleman disease. Blood, 116, 3627–3634. [DOI] [PubMed] [Google Scholar]
- Spivak JL (2017). Myeloproliferative neoplasms. The New England Journal of Medicine, 377(9), 895–896. [DOI] [PubMed] [Google Scholar]
- Stefanova D, Raychev A, Arezes J, Ruchala P, Gabayan V, Skurnik M, et al. (2017). Endogenous hepcidin and its agonist mediate resistance to selected infections by clearing non-transferrin-bound iron. Blood, 130(3), 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanova D, Raychev A, Deville J, Humphries R, Campeau S, Ruchala P, et al. (2018). Hepcidin protects against lethal E. coli sepsis in mice inoculated with isolates from septic patients. Infection and Immunity, 86(7), e00253–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein BL, Oh ST, Berenzon D, Hobbs GS, Kremyanskaya M, Rampal RK, et al. (2015). Polycythemia vera: An appraisal of the biology and management 10 years after the discovery of JAK2 V617F. Journal of Clinical Oncology, 33(33), 3953–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al. (2007). High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nature Medicine, 13(9), 1096–1101. [DOI] [PubMed] [Google Scholar]
- Tarkun P, Mehtap O, AtesogȈlu EB, Geduk A, Musul MM, & Hacihanefioglu A (2013). Serum hepcidin and growth differentiation factor-15 (GDF-15) levels in polycythemia vera and essential thrombocythemia. European Journal of Haematology, 91(3), 228–235. [DOI] [PubMed] [Google Scholar]
- Theurl I, Schroll A, Sonnweber T, Nairz M, Theurl M, Willenbacher W, et al. (2011). Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood, 118, 4977–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele J, Kvasnicka HM, Muehlhausen K, Walter S, Zankovich R, & Diehl V (2001). Polycythemia rubra vera versus secondary polycythemias. A clinicopathological evaluation of distinctive features in 199 patients. Pathology, Research and Practice, 197(2), 77–84. [DOI] [PubMed] [Google Scholar]
- Truksa J, Peng H, Lee P, & Beutler E (2006). Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proceedings of the National Academy of Sciences of the United States of America, 103(27), 10289–10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadhan-Raj S, Abonour R, Goldman JW, Smith DA, Slapak CA, Ilaria RL Jr., et al. (2017). A first-in-human phase 1 study of a hepcidin monoclonal antibody, LY2787106, in cancer-associated anemia. Journal of Hematology & Oncology, 10(1), 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadillo M, Corbella X, Pac V, Fernandez-Viladrich P, & Pujol R (1994). Multiple liver abscesses due to Yersinia enterocolitica discloses primary hemochromatosis: Three cases reports and review. Clinical Infectious Diseases, 18(6), 938–941. [DOI] [PubMed] [Google Scholar]
- van Rhee F, Fayad L, Voorhees P, Furman R, Lonial S, Borghaei H, et al. (2010). Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. Journal of Clinical Oncology, 28, 3701–3708. [DOI] [PubMed] [Google Scholar]
- Vannucchi AM, Grossi A, Rafanelli D, Statello M, Cinotti S, & Rossi-Ferrini P (1994). Inhibition of erythropoietin production in vitro by human interferon gamma. British Journal of Haematology, 87, 18–23. [DOI] [PubMed] [Google Scholar]
- Verga Falzacappa MV, Casanovas G, Hentze MW, & Muckenthaler MU (2008). A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. Journal of Molecular Medicine (Berlin, Germany), 86, 531–540. [DOI] [PubMed] [Google Scholar]
- Verstovsek S, Harrison CN, Kiladjian JJ, Miller C, Naim AB, Paranagama DC, et al. (2017). Markers of iron deficiency in patients with polycythemia vera receiving ruxolitinib or best available therapy. Leukemia Research, 56, 52–59. [DOI] [PubMed] [Google Scholar]
- Vokurka M, Krijt J, Sulc K, & Necas E (2006). Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiological Research, 55(6), 667–674. [DOI] [PubMed] [Google Scholar]
- Wang RH, Li C, Xu X, et al. (2005). A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metabolism, 2, 399–409. [DOI] [PubMed] [Google Scholar]
- Wang CQ, Udupa KB, & Lipschitz DA (1995). Interferon-gamma exerts its negative regulatory effect primarily on the earliest stages of murine erythroid progenitor cell development. Journal of Cellular Physiology, 162, 134–138. [DOI] [PubMed] [Google Scholar]
- Weizer-Stern O, Adamsky K, Amariglio N, Levin C, Koren A, Breuer W, et al. (2006). Downregulation of hepcidin and haemojuvelin expression in the hepatocyte cell-line HepG2 induced by thalassaemic sera. British Journal of Haematology, 135(1), 129–138. [DOI] [PubMed] [Google Scholar]
- Willemetz A, Beatty S, Richer E, Rubio A, Auriac A, Milkereit RJ, et al. (2017). Iron- and hepcidin-independent downregulation of the iron exporter ferroportin in macrophages during salmonella infection. Frontiers in Immunology, 8, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrighting DM, & Andrews NC (2006). Interleukin-6 induces hepcidin expression through STAT3. Blood, 108, 3204–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, & Rouault TA (2009). A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metabolism, 9(5), 461–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SP, Wang Z, Wang LX, & Liu SJ (2011). AG490: An inhibitor of hepcidin expression in vivo. World Journal of Gastroenterology, 17, 5032–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further reading
- Camaschella C, & Poggiali E (2009). Rare types of genetic hemochromatosis. Acta Haematologica, 122(2–3), 140–145. [DOI] [PubMed] [Google Scholar]
- De Domenico I, Ward DM, Musci G, & Kaplan J (2006). Iron overload due to mutations in ferroportin. Haematologica, 91(1), 92–95. [PMC free article] [PubMed] [Google Scholar]
- De Domenico I, Ward DM, Nemeth E, Vaughn MB, Musci G, Ganz T, et al. (2005). The molecular basis of ferroportin-linked hemochromatosis. PNAS, 102(25), 8955–8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. (1996). A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nature Genetics, 13(4), 399–408. [DOI] [PubMed] [Google Scholar]
- Feder JN, Penny DM, Irrinki A, Lee VK, Lebrón JA, Watson N, et al. (1998). The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. PNAS, 95(4), 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder JN, Tsuchihashi Z, Irrinki A, Lee VK, Mapa FA, Morikang E, et al. (1997). The hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression. The Journal of Biological Chemistry, 272(22), 14025–14028. [DOI] [PubMed] [Google Scholar]
- Fernandes A, Preza GC, Phung Y, De Domenico I, Kaplan J, Ganz T, et al. (2009). The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood, 114(2), 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattamis A, Papassotiriou I, Palaiologou D, Apostolakou F, Galani A, Ladis V, et al. (2006). The effects of erythropoetic activity and iron burden on hepcidin expression in patients with thalassemia major. Haematologica, 91(6), 809–812. [PubMed] [Google Scholar]
- Kearney SL, Nemeth E, Neufeld EJ, Thapa D, Ganz T, Weinstein DA, et al. (2007). Urinary hepcidin in congenital chronic anemias. Pediatric Blood & Cancer, 48(1), 57–63. [DOI] [PubMed] [Google Scholar]
- Laftah AH, Ramesh B, Simpson RJ, Solanky N, Bahram S, Schümann K, et al. (2004). Effect of hepcidin on intestinal iron absorption in mice. Blood, 103(10), 3940–3944. [DOI] [PubMed] [Google Scholar]
- Piperno A, Girelli D, Nemeth E, Trombini P, Bozzini C, Poggiali E, et al. (2007). Blunted hepcidin response to oral iron challenge in HFE-related hemochromatosis. Blood, 110(12), 4096–4100. [DOI] [PubMed] [Google Scholar]
- Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, & Andrews NC (2008). The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metabolism, 7(3), 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk BA, Laarakkers CM, Klaver SM, Jacobs EM, van Tits LJ, Janssen MC, et al. (2008). Serum hepcidin levels are innately low in HFE-related haemochromatosis but differ between C282Y-homozygotes with elevated and normal ferritin levels. British Journal of Haematology, 142(6), 979–985. [DOI] [PubMed] [Google Scholar]