

Long-read analysis of 36 individual genomes revealed extensive structural diversity within human centromeric repeat arrays.
Abstract
Our understanding of centromere sequence variation across human populations is limited by its extremely long nested repeat structures called higher-order repeats that are challenging to sequence. Here, we analyzed chromosomes 11, 17, and X using long-read sequencing data for 36 individuals from diverse populations including a Han Chinese trio and 21 Japanese. We revealed substantial structural diversity with many previously unidentified variant higher-order repeats specific to individuals characterizing rapid, haplotype-specific evolution of human centromeric arrays, while frequent single-nucleotide variants are largely conserved. We found a characteristic pattern shared among prevalent variants in human and chimpanzee. Our findings pave the way for studying sequence evolution in human and primate centromeres.
INTRODUCTION
Centromeres have been one of the most mysterious parts of the human genome since they were characterized, in the 1970s, as large tracts of 171–base pair (bp) strings called alpha-satellite monomers (1, 2). With a growing body of evidence suggesting their relevance to human diseases as sources of genomic instability or as repositories of haplotypes containing causative mutations (3–8), it has become more important to investigate the underlying sequence variations in centromeric regions (9, 10).
Human centromeric regions have nested repeat structures. Namely, a series of distinctively divergent alpha-satellite monomers compose a larger unit called higher-order repeat (HOR) unit, and copies of an HOR unit are tandemly arranged thousands of times to form large, homogeneous HOR arrays. While HOR units are chromosome specific and consist of 2 to 34 alpha-satellite monomers, copies of an HOR unit are almost identical (95 to 100%) within a chromosome (Fig. 1A) (11–17).
Fig. 1. Comprehensive probing of variant HORs in centromeric arrays.
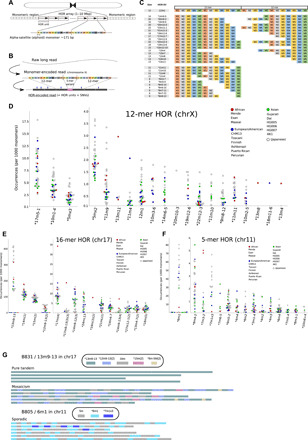
(A) Schematics of a typical DNA sequence structure of human centromeric regions. The entire region consists mostly of alphoid monomers of 171 bp long. The core centromeric regions (up to several million base pairs) with an HOR structure are sandwiched by the pericentromeric (monomeric) regions, where monomers are arranged tandemly without HOR. (B) Steps for HOR encoding of long reads. Monomer-encoded reads were obtained by aligning monomer sequences into raw long reads, and then frequent patterns of assigned monomers were considered HORs. The blue pins indicate the mismatches recorded in HOR-encoded reads, which contain both single-nucleotide variations (SNVs) and sequencing errors. (C) Structures of the canonical and some variant HORs detected in chromosome X. The rectangles represent the presence of corresponding alphoid monomers. No gap is allowed between two constituent alphoid monomers to be detected as HORs. All structures are shown in supplementary figures. (D to F) Relative frequencies (per 1000 monomers) of some detected variant HORs for 36 samples in (D) chromosome X, (E) chromosome 17, and (F) chromosome 11. (G) Example of the HOR-encoded long reads containing the variant HORs. Reads from a Japanese sample (B831) contain 13m9-13 (green rectangles), a variant found in chromosome 17. They typically showed mosaicism with other variant HORs (8-, 12-, 15-, and canonical 16-mers) or purely tandem structures. Detected HORs are represented as rectangles, placed proportionally to their actual positions within reads. Reads from a Japanese, B805, show the 6-mer variant 6m1 (light blue rectangles). While the variant seemed enriched in reads, their distribution was sporadic; at most three variants were found in tandem.
The total HOR array length of each chromosome differs markedly among individuals (7, 18) and human populations (19–21). Structural alterations such as unequal crossing over and/or gene conversion are thought to be among the major driving forces of this centromeric variation (22, 23). Other types of variation occur within HOR arrays, such as single-nucleotide variations (SNVs) between paralogous HOR units (21, 24, 25) and structurally variant HORs, which consist of different numbers and/or types of alpha-satellite monomers (21, 26–28). However, the importance of structurally variant HORs remains unknown because they are difficult to detect comprehensively via traditional approaches such as restriction enzymes sensitive to alpha-satellite monomers, Southern blotting, or the analysis of k-mers unique to centromeric regions in short reads obtained in the 1000 Genomes Project (29).
Recently, the advent of long-read sequencing technologies has paved the way for direct, comprehensive observation of sequence variations among various human populations (30–34). Long-read sequencing was capable of yielding contiguous reference sequences of centromeres for several species (35, 36), and reconstruction of whole centromeric sequences for a human haploid genome is now possible despite their idiosyncratic repeat structures (37–40). While reference-quality de novo assembly of such repetitive regions remains a demanding task involving substantial manual curation (38, 41, 42), the use of unassembled long reads has promise for investigating variations within centromeric regions of diploid genomes in a cost-effective manner (43).
Therefore, we exploited a strategy of HOR encoding of unassembled long reads for comprehensive detection and quantification of variant HORs. The use of unassembled reads enabled us to analyze diploid samples without the danger of collapsing them in assemblies. In addition, the uncorrected reads could address SNVs in the HORs in an unbiased way. Here, we revealed a hidden diversity of centromeric arrays in terms of variant HORs through analysis of long reads from 36 human samples of diverse origins. We identified many previously unidentified variant HORs including some specific to a few samples, and even when variants were shared, their observed frequencies were substantially different in general.
RESULTS
Direct detection and quantification of variant HORs through HOR encoding of long reads
To investigate interindividual variation within the centromeric array, we analyzed publicly available, single-molecule, real-time sequencing reads collected from 12 samples from geographically diverse origins, including three from Africa (Mende, Sierra Leone; Esan, Nigeria; and Maasai, Kenya), two from Europe (Toscani, Italy, and Finland), five from Asia (Gujarati, India; Dai, China; and three from Han, China), and two from Latin America (Puerto Rico and Peru). We also analyzed 21 newly sequenced Japanese datasets and three previously described samples: AK1 (Korea), HG002 (Ashkenazi), and CHM13 (Europe) (31, 32, 34). Thus, we analyzed a total of 36 samples (fig. S1).
First, the long reads were preprocessed in silico to filter out the noncentromeric fraction. The remaining reads were then interpreted as a series of alphoid monomers using a catalog of 58 monomers (i.e., they were represented as monomer-encoded reads) (Fig. 1B). Then, monomer-encoded reads were clustered on the basis of the composition of different monomer types. For each cluster of reads associated with one of the HOR arrays, a catalog of variant HORs was constructed by detection of frequent patterns in the monomer-encoded reads. Thus, HORs may or may not be arranged in tandems of the same type. Last, HOR-encoded reads were obtained by automatically replacing these patterns with symbols representing HORs (fig. S1).
In this analysis, we avoided chromosomes 5, 13, 14, 19, 21, and 22, in which the chromosome identity is obscured by shared HOR patterns. We mainly focused on the HOR arrays of chromosomes 11 (D11Z1), 17 (D17Z1), and X (DXZ1), which evolved from the archetypal 5-mer HOR, since the variations in these chromosomes are more divergent than those of other chromosomes associated with dimeric archetypes, whose variant HORs are more difficult to capture (16). We therefore excluded these other chromosomes to avoid drawing inaccurate conclusions.
Rapid evolution of variant HORs among 36 human samples
The detected variant HORs were diverse in terms of presence and abundance among the samples. In chromosome X, the canonical HOR consists of 12 monomers; this was the most frequent pattern found in reads across all of the datasets (96.2 to 98.4% of all HOR types). In addition to the canonical 12-mer HOR, 51 variant HORs were defined, ranging in size from 2- to 23-mer (Fig. 1, C and D, and fig. S4). While some variant HORs (e.g., 10m1-4 and 17m5-1) were shared by all 36 samples, others were specific to or missing from a few samples (Fig. 1D). For example, 18m1-6 was specific to CHM13. 13m11 was found only in five samples: Esan, Maasai, Toscani, and two Japanese (B480 and B700). The 11m9 variant was shared almost universally but was absent from HG005 and B402.
For chromosome 17, 91 distinct variants were detected, ranging in size from 5- to 39-mers (Fig. 1E and fig. S5). Notably, a 13-mer variant (13m9-13; the 10th, 11th, and 12th monomers had been deleted from the canonical 16-mer) was present at high frequency in approximately half of the samples, whereas it was generally missing from other samples. Samples with the characteristic 13-mer variant exhibited a so-called haplotype II, which has an estimated allele frequency of ∼35% for European populations (25, 44). Prevalent variant HORs were also observed, including a 15-mer [15m(2)] and a 14-mer [14m(1)], which suggested that the canonical 16-mer was less stable than canonical HORs in chromosomes X or 11. Consequently, unlike chromosome X, the relative frequencies of canonical 16-mer HORs were highly divergent among the samples, ranging from 21.6 to 76.0%. For the remaining variant HORs, the distribution of variant HORs across the individual samples was markedly nonuniform as well (data file S1).
In chromosome 11, where the 5-mer canonical HOR (16) was the most frequent (92.6 to 99.5% of all HOR types), 23 variant HORs were detected. As with the other chromosomes investigated, variant HORs were observed at substantially variable frequency across the 36 samples (Fig. 1F and fig. S2). The most prominent difference was observed for a 6-mer variant (6m1, a duplication of the first monomer), which existed at high frequency in Toscani, Puerto Rican, Peruvian, Korean, and 11 Japanese samples; however, it was generally missing from the remaining samples. Notably, a 7-mer variant (7m1x3, the first monomer is tripled) was found only in samples with the 6m1 variant, suggesting that 7m1x3 evolved from 6m1.
To evaluate the diversity of variant HORs within a population, we quantitatively measured variation among the 21 Japanese samples. The SD of variant HOR frequency was 45.05 events per megabase (Mb), which approximated the expected density of distinct variant HORs harbored by each individual genome. We then compared our results with a recent estimate of genome-wide structural variation (SV) detection from accurate circular-consensus long reads, which obtained a reliable set of ∼30,000 SVs for an individual genome, with respect to a reference genome (34). The average density of SVs for each of the 23 chromosomes (autosomes and X) was 21.16 SVs/Mb (SE = 4.45 SVs/Mb); a two-tailed one-sample t test confirmed that SVs were significantly more abundant in centromeric regions than in noncentromeric regions (P = 6.51 × 10−18). Therefore, the centromeric array appears to change rapidly in terms of variant HORs.
Together, although canonical HOR patterns were observed in all samples, noncanonical variant HORs were more dynamic overall, as they were likely to be specific to subsets of individuals across different populations or exhibited divergent frequencies even within a population, showing rapid evolution in the human centromeric arrays.
The modes of local expansion of variant HORs
We investigated the contexts in which variant HORs were found in long reads (Fig. 1G). For example, the characteristic 13-mer variant (13m9-13) of chromosome 17 was observed in tandem or interleaved with other HORs (Fig. 1G). In contrast, the 6-mer variant (6m1) of chromosome 11 was observed only sporadically. Therefore, unlike variant 13m9-13, 6m1 appeared incapable of independent tandem expansion; it may exhibit some preference (e.g., for length) with respect to the unit of expansion. Although modes of expansion were apparently distinct depending on the type of HOR variant, we found that the same type of HOR variant was significantly enriched locally (binomial test P < 10−100 for most samples with the focal variant). This finding suggests that the variant HORs had expanded locally through a series of duplication events, rather than occurring independently (data file S2).
Detection of ongoing evolution within an HOR array
Next, we used rare variant HORs to detect evolutionary events in human HOR arrays; these variant HORs exist at relatively low frequencies (e.g., <5 per 1000 monomers) but are shared among multiple samples. We typically observed similar HOR patterns around the same rare variant across multiple samples, which indicated that these rare variants were orthologous or paralogous (i.e., they shared the same original event that had given rise to the variant). Alternatively, these very similar patterns may have emerged independently in a recurrent manner, but this was much less plausible according to the maximum-parsimony criterion. Therefore, we compared patterns around the rare variants to understand local sequence evolution in centromeres.
As an example of the rare variants, we selected 27m12-1(2) in chromosome 17 (Fig. 1E). This variant existed in a number of contexts, although Han Chinese trio samples (HG005, HG006, and HG007) shared a homologous pattern with other variants: 14m(1), 14m10(2), and 15m(2) (Fig. 2A). The patterns, which appeared downstream from the 27-mer variant, differed slightly between HG006 (father) and HG007 (mother) by one unit of the 15-mer variant; this suggested an indel event. Of note, both patterns were observed in HG005 (son), consistent with the Mendelian inheritance of the locus.
Fig. 2. Tracing sequence evolution within an HOR array via analysis of variant HORs found in long reads.
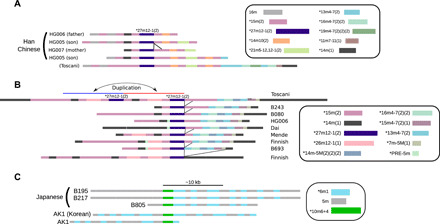
Each variant HOR is differently colored. (A) The pattern with four SVs, 14m(1), 14m10(2), 15m(2), and 27m12-1(2), was found only in the Chinese trio (HG005 to HG007), and both maternal and paternal patterns were observed in the son. The lines between the haplotype structures indicate the position of insertion/deletion events. (B) Other distinct patterns around a rare variant, 27m12–1(2). A total of nine patterns are shown. Blue and red lines represent a duplication event found within the pattern observed in Toscani samples. (C) A variant HOR, 10m6+4 (light green), is found only in four Asian samples (three Japanese and a Korean). The patterns downstream of the focal SV retained homology among five loci found in the four samples.
For the same variant, 27m12-1(2), another homologous pattern was observed in eight samples (Fig. 2B). There was considerable variation downstream from the variant, which could have occurred through a series of indel events. The variation upstream appeared more complex; however, a local duplication of ∼20 kb was suggested within the pattern found in Toscani samples.
Furthermore, 10m6+4 in chromosome 11 was another rare variant, found only in four Asian samples (Fig. 2C). The variant shared a subsequence with the characteristic variant 6m1; it always appeared along with 6m1, suggesting that 10m6+4 had recently evolved from 6m1. We identified five loci with the variant among the four samples; the patterns downstream indicated a single indel event between loci. Two loci found in a Korean (AK1) sample seemed to be divergent from the other three Japanese loci, according to the upstream patterns.
The above examples demonstrated that we could detect evolutionary events through analysis of variant HORs and that SV was abundant within centromeric arrays. Together, we observed ongoing evolution in the human centromeric arrays, generating rare, specific, HOR patterns.
SNV landscape on canonical HORs
Next, we analyzed the SNV landscape among orthologous/paralogous copies of canonical HORs: 5-mers in chromosome 11, 12-mers in chromosome X, and 16-mers in chromosome 17. Here, we did not consider indels because they cannot be called confidently using long reads. Although most of the alternative bases were observed at a low frequency ∼3% owing to substitution errors in the long reads, we could identify prevalent SNV sites as prominent peaks in the plots (Fig. 3, A to C; figs. S6 to S9; and data file S3). Notably, those SNVs were often shared among the samples, and their frequencies were strongly correlated (Fig. 3, D to F, and figs. S10 to S13). Although SNV frequencies typically showed stronger correlations within the trio samples or within Japanese samples (fig. S14), they did not appear to reflect a geographical pattern otherwise. This finding suggests that these prevalent SNVs were present in the ancestral human population and were relatively conserved, or that a process such as gene conversion may have substantially reduced SNV diversity, in contrast to the greater structural diversity in terms of variant HORs.
Fig. 3. Comparison of SNV frequencies on the canonical 12-mer HOR (chromosome X) among three samples.
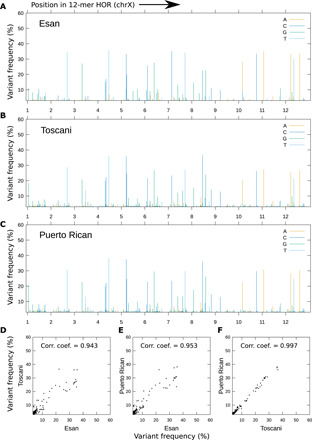
(A to C) SNV landscape over the 12-mer canonical HOR in chromosome X. SNVs with a frequency of >3% are shown. The x axis is labeled with monomer index, but the actual coordinate represents position and base; for example, the alternative base G at the 20th base of the 2nd monomer is plotted at x = 3 + (20 × 4) + (2 × 800) = 1683. The y axis is the observed frequency in percentage. Four colors are used to distinguish the alternative (nonreference) bases. (D to F) Correlation of SNV frequencies. Each dot represents a single SNV (designated by a position and an alternative base). SNVs with frequencies >3% in both samples in x and y axes are shown.
Within the set of observed paralogous SNVs on canonical HORs across our dataset (36 individuals, four types of canonical HORs in chromosomes 1, 11, 17, and X), we did not observe enrichment of transitions (A/G or C/T) over transversions ([A or G]/[C or T]) or a preference of variants for CpG sites (data file S4). These rather unexpected patterns may be partly explained by the fact that these paralogous SNVs were generated not only via original spontaneous mutations but also via a series of expansion events including crossing over and gene conversion. Notably, we confirmed that the representative HOR unit sequences were already AT-rich (GC rate = 40.24 to 41.05%) and contained fewer CpG sites (fig. S15). For example, CpG was the least frequent 2-mer in all cases, at about half of the frequency of GpC. The transition of methylated CpG to TpG may have contributed to this observed pattern.
Haplotype-specific evolution of the centromeric array
For chromosome 17, the correlation of SNV frequencies was considerably diverse, depending on the pair of samples (Fig. 4A). Samples with highly correlated SNV frequencies often shared a similar set of variant HORs (Fig. 4B). For example, 10 samples (Maasai, Esan, and 8 Japanese) were strongly correlated in terms of SNV frequencies; they also shared a characteristic pattern of variant HORs, such as the presence of the 13m9-13 variant or the absence of the 14m6-9 variant. Another 13 samples (Mende, Toscani, CHM13, Ashkenazi, Finnish, Dai Chinese, Han Chinese trio, Peruvian, and 3 Japanese) with shared SNVs exhibited the reverse pattern in terms of variant HORs. The 13m9-13 variant is a marker for a well-known alternative allele (haplotype II) for the chromosome 17 centromere in contrast to the wild-type allele (haplotype I) (25, 44). Below, we refer to haplotypes I and II as haplotypes A and B, respectively, just for a better readability. Our analysis indicated that many other variant HORs exhibited positive or negative correlations with the marker variant 13m9-13. The haplotype combination in each sample (AA, BB, or AB) was also evident in the pairwise correlation of SNV frequencies (Fig. 4, A and B). Similarly, for chromosome 11, the presence of the 6-mer variant 6m1 defined two distinct clusters of samples, which were confirmed by SV and SNV analysis (fig. S16). This clear difference between alternative haplotypes suggested that minimal or no recombination occurred between the distinct haplotypes. Thus, they act as a single genetic locus while their internal sequences undergo rapid haplotype-specific evolution.
Fig. 4. Haplotype-specific evolution of chromosome 17 centromeric arrays among 36 samples.
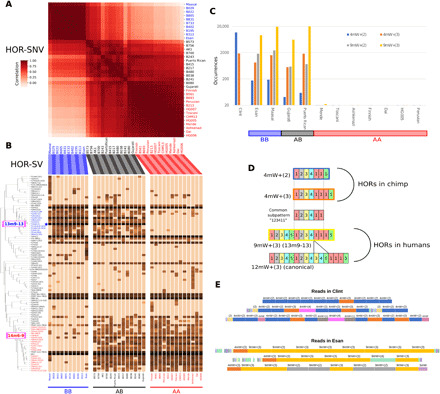
(A) Correlation of SNV frequencies among samples on the canonical 16-mer HOR units for chromosome 17. Sample labels are colored blue (BB), black (AB), or red (AA) according to the haplotype combination inferred by SV analysis. (B) Occurrence of variant HORs in each sample serves as a fingerprint of the haplotype. SVs were clustered by co-occurrence over the samples. A-specific and B-specific variant HORs are labeled with red and blue, respectively. Blue star: The marker variant HOR for the haplotype B, 13m9-13. Darker cells indicate that they are observed with higher frequency. Sample labels are colored according to the haplotype combination (blue, BB; black, AB; red, AA). (C) Frequencies of B-specific variant HORs (in terms of generic monomers) detected in chimpanzee and humans. (D) Schematic representations of the HORs with the B-specific pattern. The numbered blocks represent the alphoid monomers (of suprachromosomal family 3), which constitute HOR patterns in humans and chimpanzees. (E) Visualization of HOR-encoded reads with the B-specific breakpoints, 9mW+(n) and 4mW+(n), n = 1,2,3,⋯. HORs and monomers are shown according to the actual coordinates found within reads.
These haplotypes, once established, seem to follow an expected pattern. The 21 Japanese samples included 3 homozygous AA, 10 heterozygous AB, and 8 homozygous BB observed genotypes for the chromosome 17 centromere; the allele frequencies of the A and B haplotypes were 38.1 and 61.9%, respectively. According to the Hardy-Weinberg equilibrium, the expected genotype combinations for the 21 individuals are 3.05 AA, 9.90 AB, and 8.05 BB; our observed combinations exhibited almost perfect adherence to the Hardy-Weinberg equilibrium, although the sample size (n = 21) may be too small to represent a rigorous test. The allele frequency of haplotype B in the Japanese population, 26 of 42 (61.9%), was significantly higher (P = 0.000341, binomial test) than the estimated frequency for the European population (∼35%) (25); this might be explained by a founder effect in the Japanese population.
Distribution of haplotype B–specific patterns in a chimpanzee centromeric array
To determine which haplotype, A or B, was ancestral in terms of centromere sequence evolution, we performed corresponding HOR analysis using a chimpanzee (Clint) as the outgroup (45). Although chimpanzee centromeric arrays share some HOR structures with humans, we did not rely on existing information regarding HOR patterns (16). We used a set of 10 generic monomers including five monomers (W1 to W5) of suprachromosomal family 3 so that we could equally capture HOR patterns present both in chimpanzee and in humans.
Using the generic monomers, we identified HOR patterns that were shared by the human samples with haplotype B (homozygous or heterozygous) but were absent from those homozygous for haplotype A (Fig. 4C and figs. S17 and S18). These characteristic patterns shared an HOR subpattern (123411), which served as a haplotype B–specific marker. Notably, this pattern was frequently observed in the chimpanzee (Fig. 4C and fig. S18), although the contexts in which the breakpoints occurred differed slightly in humans and the chimpanzee (Fig. 4, D and E). These findings implied that the pattern found in haplotype B was originally shared by both species, but they might have evolved into distinct HOR arrays in each species. Subsequently, haplotype A (in which the pattern was lost) had spread within the human population.
DISCUSSION
Through an analysis of centromeric arrays, we found great diversity in minor variations and widespread characteristics that are presumably of ancient origin. Collectively, these observations demonstrated the rapid, ongoing evolution of human centromeres.
The studies of variations in the centromeric arrays at the sequence level remain preliminary in a sense. For example, although we conveniently referred 5-, 16-, and 12-mer arrays as chr11, chr17, and chrX arrays, respectively, these traditional assignments may not always be true for all individual genomes. Therefore, chromosome-level reconstruction of individual genomes is crucial as well as the analysis of local variants. Because of the limited availability of sequencing data, much of our analyses relied on cell culture, where we do not know yet how stable the centromeric arrays would be. Thus, it is possible that we have overestimated the rate of change there. Ideally for understanding the biology of the centromeric arrays, it is important to use nonculture samples and to determine the presence of somatic variations precisely.
In analyzing long-read data, it is crucial to control for data errors and biases. The detection of variant HORs was less affected by sequencing errors in this study because they were characterized by a difference of at least one alphoid monomer (171 bp). In contrast, SNV quantification may have been affected by indel errors around the sites and suffered from a low signal-to-noise ratio, especially in regions with fewer variants. The recent improvement in accuracy provided by PacBio circular-consensus sequencing technology promises more faithful observation of SNVs that occur less frequently (34).
We detected variant HORs in the diploid human centromeric arrays of chromosomes 11, 17, and X using long-read data without explicit sequence assembly. We substantially increased the knowledge of variant HORs (21, 26, 27), thereby revealing unexpected diversity in human centromeric arrays through analysis of 36 individuals. Conserved homologous regions around rare variant HORs enabled us to detect ongoing structural changes among sequences in multiple samples. Similar structural changes may occur within the sea of tandem replicates of canonical HORs. Therefore, even greater hidden diversity may be present there, compared to the conservative estimates we have described. With such diversity in centromeric arrays, we hypothesize that the tandem nature of those arrays makes them extremely variable; moreover, there is sufficient information to identify individuals, similar to the use of microsatellites. Our analysis of Han Chinese trio samples and 21 Japanese samples indicated that the HOR array structure is diverse within a single population, supporting this hypothesis.
Although the centromeric arrays showed great diversity with minor SV, there were relatively conserved characteristics among samples from geographically distant populations. For example, the frequent SNVs in the most abundant HOR units were conserved across all samples; moreover, the segregation of haplotypes A and B in chromosome 17 was recapitulated in both the African samples and the Japanese population. These universal features might have spread before the relatively recent expansion of the human population out of Africa (46), unless they were acquired independently. Investigating the evolution of the segregating haplotypes more robustly would require much denser samples of human genomes including those from sub-Saharan Africa; in the present study, we focused on analyzing an available chimpanzee long-read dataset as an outgroup for the human population. Although the majority of the HOR patterns showed divergence between humans and chimpanzees, we found some common repetitive patterns. Thus, the comparison of variant HORs, not limited to canonical HORs, is useful for analysis of human and primate centromere evolution when more human and primate samples will be available.
What does it mean to have such large structural diversity in centromeric arrays? Because centromeres have a fundamental importance to proper chromosome segregation during cell division, it was once considered unusual to observe great diversity in centromeric sequences across different eukaryotic taxa (“centromere paradox”) (47). Centromere drive theory explained the rapid evolvability of centromeres via genetic conflict during female meiosis I, rendering the centromeres as a crux of the molecular identity of species (48). Nevertheless, growing evidence suggests that centromeres can be highly variable within a single species (5, 10, 21, 24), and our findings of diverse variant HORs add another layer of diversification. With a more comprehensive catalog of variations, we have better chances to extract new information from existing or upcoming sequencing data. If specific types of variants turn out to have functional implication, then these variants can be useful as biomarkers. Also, we expect that such markers would be helpful for tracing evolutionary events within the centromeric satellite arrays, leading to better understanding of their formation.
This great diversity suggests that centromere function may be highly robust with respect to the underlying sequence, although some variant HORs have been associated with centromere functional abnormality (25, 49). Transcription from the centromeric arrays is another intriguing phenomenon (50); we wonder whether structurally different HORs may affect transcription processes and/or functions. At the very least, we believe that a comprehensive understanding of sequence variants would improve the mapping of genomic/transcriptomic short-read data, which would ultimately benefit future studies of centromere function.
Several mechanisms can contribute to such structural diversity within centromeric sequences: unequal crossover between sister chromatids, meiotic unequal crossover, gene conversion, and homologous recombination resulting in noncrossover products, to name a few. Among them, meiotic crossovers might arguably be excluded as a major driving force because they are suppressed near centromeric regions (7, 51), and consequently, centromeric regions are reported to form large conserved linkage-disequilibrium blocks (10). On the one hand, the structural diversity within centromeric arrays can be best explained by frequent unequal crossovers between sister chromatids and gene conversions. On the other hand, centromere integrity in a human population might have been maintained through occasional gene conversions and infrequent meiotic crossovers, both of which can counteract the diversification processes by effectively homogenizing sequences among different alleles. Notably, all these mechanisms are consistent with the local, progressive expansion suggested in this study as well as in previous evolutionary analyses (52). We speculate that all these mechanisms might have contributed to the current landscape of human centromeric arrays.
Recently, a number of whole centromeric arrays reconstructed with ultralong nanopore reads and/or accurate PacBio HiFi read have been reported for a haploid genome, showing that, at last, the time is ripe to investigate centromeres in terms of sequencing technology (37–40). While de novo assemblies of centromeric arrays provide unique information, it remains a nontrivial task to validate them especially for diploids. Meanwhile, the SV analysis can be a faithful representation of local features and complements the process of de novo assembly, which must be able to recover the same types and frequencies of HORs found in reads. Notably, it requires only a single SMRT Cell per sample to obtain the amount of data (10× to 40× of 3Gb human genome) used in this study. Cost-effectiveness is an important characteristic of SV analysis, making it easier to consider the scale-up.
With an increasing number of individual genomes from the same or closely related populations sequenced by long reads, one would be able to precisely observe the processes of diversification and homogenization that occur within human centromeric arrays. Therefore, such a study should provide a basis to delineate the complex mechanisms involved and to understand the true nature of centromere evolution.
MATERIALS AND METHODS
Preparation of long-read sequencing data
In this study, we used B cells derived from Japanese people, which was distributed by the National Institute of Biomedical Innovation, Health and Nutrition, and the study was approved by The Research Ethics Committee of the Faculty of Medicine of the University of Tokyo (Human Genome/Gene Analysis Research Ethics Review; review number 19-323). For SMRTbell library preparation, B cell DNA (Japanese samples in the main text) was sheared using a Diagenode’s Megaruptor 2 with software setting 75 kb and purified using a 0.6× volume ratio of AMPure beads (Pacific Biosciences, Menlo Park, CA, USA). SMRTbell libraries for sequencing were prepared using the “Procedure & Checklist-Preparing >30 kb Libraries Using SMRTbell Express Template Preparation Kit” protocol. Briefly, the steps included (i) DNA repair, (ii) blunt ligation with hairpin adapters with the SMRTbell Express Template Preparation Kit (Pacific Biosciences), (iii) 15-kb cutoff size selection using the BluePippin DNA Size Selection System by Sage Science, and (iv) binding to polymerase using Sequel Binding Kit 2.1, later Sequel Binding Kit 3.0 (Pacific Biosciences). SMRTbell libraries were sequenced on Sequel SMRT Cells (Pacific Biosciences) using diffusion loading, 30-kb insert size, and 600-min movies. All the other long-read data including AK1 (31), CHM13 (32), and HG002 (Ashkenazi) (34) were obtained via a public repository (Sequence Read Archive; table S1).
Filtering out noncentromeric reads
To enrich the centromeric reads in silico, we calculated the reference 6-mer frequency vector with the 14 typical alphoid monomers: A, B, D1, D2, J1, J2, W1 to W5, R1, R2, and M1 (table S3). We also calculated the “query” 6-mer frequency vector (normalized by length in base pair) and its dot product with the reference for each long read. The dot products exhibited a bimodal distribution, which represents the mixture of centromeric and noncentromeric reads. Thus, only reads with the dot product greater than a specified threshold were included in later analysis. We modified squeakr (53) to perform these steps.
Determination of chromosome-specific monomer sequences
To enhance the sensitivity in detection of HOR in noisy long reads, we defined chromosome-specific monomer sequences (table S2 and fig. S19). First, 10 generic monomers (the typical alphoid monomers aforementioned excluding A, B, R1, and R2) were mapped to long reads with the same parameter as described in the next subsection. Then, the reads were segregated according to chromosomes. For example, the reads from chromosome X were identified as those that contained tandems of the pattern: W1, W2, W3, W4, W5, W1, W2, W3, W4, W3, W4, and W5. Last, corresponding subsequences were extracted from the long reads, and then we took the consensus of them to obtain chromosome-specific monomer sequences. For chromosome 17, the three characteristic arrays (D17Z1, D17Z1B, and D17Z1C) were collectively analyzed because they were not distinguished from each other at our resolution. Also, noisy long reads could not clearly segregate arrays evolved from dimeric patterns by means of the generic dimeric monomers (J1 and J2 and D1 and D2). We suspect that this is because the possible combinations of those monomers were limited compared to the pentameric case (W1 to W5).
Monomer encoding of long reads
The distinct 58 monomers (table S2) were mapped by blastn (version 2.4.0+) to long reads with the following parameters:
-max_target_seqs 1000000 -word_size 7 -qcov_hsp_perc 60
Optimal assignment was calculated via dynamic programming procedure, maximizing the following quantity Σi(si − 50) − Σi, j, bi < bj max (0,2(ei − bj)), where i indexes monomers assigned to the read, si is the BLAST (Basic Local Alignment Search Tool) score of the hit, and (bi, ei) is the region covered by the monomer. Intuitively, it tries to assign as many monomers with acceptable scores as possible, because of the first term. The second term penalized the overlaps (cf. gaps were not penalized) so that each segment of the read be assigned at most one monomer.
As related tools for analyzing centromeric repeats, there are Alpha-CENTAURI (43) and StringDecomposer (39, 54), but they serve rather different purposes; Alpha-CENTAURI detects regular and irregular HOR patterns in individual long reads, but it does not aggregate data across the reads; StringDecomposer gives us an essentially gapless decomposition of long read into a series of monomers, but it does not summarize the data as variant HORs.
HOR encoding
The reads from chromosomes 1, 11, 17, and X were identified as those that contained >5 chromosome-specific alphoid monomers. For the analysis including the chimpanzee, the set of 10 generic monomers, D1, D2, J1, J2, W1 to W5, and M1 (16), were used instead of the chromosome-specific alphoid monomers, as the chromosome-specific monomers (derived from human samples) were not able to capture HOR structure in chimpanzee.
Then, recurrent combinations of monomers were identified as HORs. No gap of >100 bp was allowed between neighboring monomers within the detected HORs. With the list of identified HORs, reads were processed again to be encoded as series of assigned HORs plus the mismatches (SNVs) against the reference monomers. Then, these HOR-encoded reads were analyzed as described in the main text. To confirm that noisy long reads can robustly capture the characteristics of the samples, we used the HiFi data available for the CHM13 sample. The numbers of (each type of) detected variant HORs in CHM13 HiFi have higher correlations with those in CHM13 CLR (Continuous Long Read) (0.818, 0.934, and 0.860 for 12-, 16-, and 5-mer arrays, Spearman), but lower correlations with the other 35 samples that ranged from 0.306, 0.084, and 0.075 to 0.707, 0.797, and 0.701 for 12-, 16-, and 5-mer arrays, respectively. We also confirmed that the noisy long reads can detect frequent SNVs by comparing HiFi and CLR data for the CHM13 (fig. S20).
A measurement of diversity in variant HORs
For each chromosome, we have Mi, the total number of detected monomers in individual i, and , the frequency of variant HOR v in individual i. Then, is the normalized frequency v of i per 1 Mbp (million base pairs). Then, we calculated σv to be the SD of over the set of individuals, which served as a measure of typical variation of variant v. Last, we approximated the total variation (per 1 Mbp) for the chromosome by V = Σvσv.
Statistical analysis of local expansion
We calculated the frequency of patterns where (i) the variant is followed by the same type of variant or (ii) the variant is followed by the canonical HOR. Then, we performed binomial test against the null hypothesis where they occur randomly according to the observed frequency of HORs.
Supplementary Material
Acknowledgments
We thank W. Qu, J. Yoshimura, C. Owa, T. Sugai, and Y. Saito for sequencing B cells using PacBio Sequel. Funding: This study was supported, in part, by the Advanced Genome Research and Bioinformatics Study to Facilitate Medical Innovation from Japan Agency for Medical Research and Development (AMED) to S.M. Author contributions: Y.S. conceived and conducted the study. Y.S., E.W.M., and S.M. analyzed the results and wrote the manuscript. All authors reviewed the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. All sequencing data for 21 Japanese samples are available in the DDBJ Japanese Genotype-phenotype Archive for genetic and phenotypic human data under accession number JGAS00000000173. The snapshot of all custom codes used for the study is available at https://github.com/hacone/hc_temporary/releases/tag/submission-v.1.0
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabd9230/DC1
REFERENCES AND NOTES
- 1.Manuelidis L., Repeating restriction fragments of human DNA. Nucleic Acids Res. 3, 3063–3076 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manuelidis L., Wu J. C., Homology between human and simian repeated DNA. Nature 276, 92–94 (1978). [DOI] [PubMed] [Google Scholar]
- 3.Black E. M., Giunta S., Repetitive fragile sites: Centromere satellite DNA as a source of genome instability in human diseases. Genes 9, 615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saha A. K., Mourad M., Kaplan M. H., Chefetz I., Malek S. N., Buckanovich R., Markovitz D. M., Contreras-Galindo R., The genomic landscape of centromeres in cancers. Sci. Rep. 9, 11259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barra V., Fachinetti D., The dark side of centromeres: Types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 9, 4340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amberger J., Bocchini C. A., Scott A. F., Hamosh A., McKusick’s Online Mendelian Inheritance in Man (OMIM®). Nucleic Acids Res. 37, D793–D796 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wevrick R., Willard H. F., Long-range organization of tandem arrays of alpha satellite DNA at the centromeres of human chromosomes: High-frequency array-length polymorphism and meiotic stability. Proc. Natl. Acad. Sci. U.S.A. 86, 9394–9398 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langley S. A., Miga K. H., Karpen G. H., Langley C. H., Haplotypes spanning centromeric regions reveal persistence of large blocks of archaic DNA. eLife 8, e42989 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aldrup-MacDonald M. E., Sullivan B. A., The past, present, and future of human centromere genomics. Genes 5, 33–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miga K. H., Centromeric satellite DNAs: Hidden sequence variation in the human population. Genes 10, 352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waye J. S., Willard H. F., Chromosome-specific alpha satellite DNA: Nucleotide sequence analysis of the 2.0 kilobasepair repeat from the human X chromosome. Nucleic Acids Res. 13, 2731–2743 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willard H. F., Chromosome-specific organization of human alpha satellite DNA. Am. J. Hum. Genet. 37, 524–532 (1985). [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell A. R., Gosden J. R., Miller D. A., A cloned sequence, p82H, of the alphoid repeated DNA family found at the centromeres of all human chromosomes. Chromosoma 92, 369–377 (1985). [DOI] [PubMed] [Google Scholar]
- 14.Willard H. F., Waye J. S., Hierarchical order in chromosome-specific human alpha satellite DNA. Trends Genet. 3, 192–198 (1987). [Google Scholar]
- 15.Jørgensen A. L., Bostock C. J., Bak A. L., Homologous subfamilies of human alphoid repetitive DNA on different nucleolus organizing chromosomes. Proc. Natl. Acad. Sci. U.S.A. 84, 1075–1079 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexandrov I., Kazakov A., Tumeneva I., Shepelev V., Yurov Y., Alpha-satellite DNA of primates: Old and new families. Chromosoma 110, 253–266 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Hayden K. E., Human centromere genomics: Now it’s personal. Chromosome Res. 20, 621–633 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Mahtani M. M., Willard H. F., Pulsed-field gel analysis of α-satellite DNA at the human X chromosome centromere: High-frequency polymorphisms and array size estimate. Genomics 7, 607–613 (1990). [DOI] [PubMed] [Google Scholar]
- 19.Oakey R., Tyler-Smith C., Y chromosome DNA haplotyping suggests that most European and Asian men are descended from one of two males. Genomics 7, 325–330 (1990). [DOI] [PubMed] [Google Scholar]
- 20.Mitchell R. J., Fricke B., Y-chromosome specific alleles and haplotypes in European and Asian populations: Linkage disequilibrium and geographic diversity. Am. J. Phys. Anthropol. 104, 167–176 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Miga K. H., Newton Y., Jain M., Altemose N., Willard H. F., Kent W. J., Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 24, 697–707 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith G. P., Evolution of repeated DNA sequences by unequal crossover. Science 191, 528–535 (1976). [DOI] [PubMed] [Google Scholar]
- 23.Dover G., Molecular drive: A cohesive mode of species evolution. Nature 299, 111–117 (1982). [DOI] [PubMed] [Google Scholar]
- 24.Roizès G., Human centromeric alphoid domains are periodically homogenized so that they vary substantially between homologues. Mechanism and implications for centromere functioning. Nucleic Acids Res. 34, 1912–1924 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aldrup-MacDonald M. E., Kuo M. E., Sullivan L. L., Chew K., Sullivan B. A., Genomic variation within alpha satellite DNA influences centromere location on human chromosomes with metastable epialleles. Genome Res. 26, 1301–1311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durfy S. J., Willard H. F., Patterns of intra- and interarray sequence variation in alpha satellite from the human X chromosome: Evidence for short-range homogenization of tandemly repeated DNA sequences. Genomics 5, 810–821 (1989). [DOI] [PubMed] [Google Scholar]
- 27.Warburton P. E., Waye J. S., Willard H. F., Nonrandom localization of recombination events in human alpha satellite repeat unit variants: Implications for higher-order structural characteristics within centromeric heterochromatin. Mol. Cell. Biol. 13, 6520–6529 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santos F. R., Pandya A., Kayser M., Mitchell R. J., Liu A., Singh L., Destro-Bisol G., Novelletto A., Qamar R., Mehdi S. Q., Adhikari R., de Knijff P., Tyler-Smith C., A polymorphic L1 retroposon insertion in the centromere of the human Y chromosome. Hum. Mol. Genet. 9, 421–430 (2000). [DOI] [PubMed] [Google Scholar]
- 29.1000 Genomes Project Consortium , An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaisson M. J. P., Huddleston J., Dennis M. Y., Sudmant P. H., Malig M., Hormozdiari F., Antonacci F., Surti U., Sandstrom R., Boitano M., Landolin J. M., Stamatoyannopoulos J. A., Hunkapiller M. W., Korlach J., Eichler E. E., Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seo J.-S., Rhie A., Kim J., Lee S., Sohn M.-H., Kim C.-U., Hastie A., Cao H., Yun J.-Y., Kim J., Kuk J., Park G. H., Kim J., Ryu H., Kim J., Roh M., Baek J., Hunkapiller M. W., Korlach J., Shin J.-Y., Kim C., De novo assembly and phasing of a Korean human genome. Nature 538, 243–247 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Huddleston J., Chaisson M. J. P., Steinberg K. M., Warren W., Hoekzema K., Gordon D., Graves-Lindsay T. A., Munson K. M., Kronenberg Z. N., Vives L., Peluso P., Boitano M., Chin C.-S., Korlach J., Wilson R. K., Eichler E. E., Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 27, 677–685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain M., Koren S., Miga K. H., Quick J., Rand A. C., Sasani T. A., Tyson J. R., Beggs A. D., Dilthey A. T., Fiddes I. T., Malla S., Marriott H., Nieto T., O’Grady J., Olsen H. E., Pedersen B. S., Rhie A., Richardson H., Quinlan A. R., Snutch T. P., Tee L., Paten B., Phillippy A. M., Simpson J. T., Loman N. J., Loose M., Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wenger A. M., Peluso P., Rowell W. J., Chang P.-C., Hall R. J., Concepcion G. T., Ebler J., Fungtammasan A., Kolesnikov A., Olson N. D., Töpfer A., Alonge M., Mahmoud M., Qian Y., Chin C.-S., Phillippy A. M., Schatz M. C., Myers G., De Pristo M. A., Ruan J., Marschall T., Sedlazeck F. J., Zook J. M., Li H., Koren S., Carroll A., Rank D. R., Hunkapiller M. W., Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.VanBuren R., Bryant D., Edger P. P., Tang H., Burgess D., Challabathula D., Spittle K., Hall R., Gu J., Lyons E., Freeling M., Bartels D., Ten Hallers B., Hastie A., Michael T. P., Mockler T. C., Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 527, 508–511 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Ichikawa K., Tomioka S., Suzuki Y., Nakamura R., Doi K., Yoshimura J., Kumagai M., Inoue Y., Uchida Y., Irie N., Takeda H., Morishita S., Centromere evolution and CpG methylation during vertebrate speciation. Nat. Commun. 8, 1833 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jain M., Olsen H. E., Turner D. J., Stoddart D., Bulazel K. V., Paten B., Haussler D., Willard H. F., Akeson M., Miga K. H., Linear assembly of a human centromere on the Y chromosome. Nat. Biotechnol. 36, 321–323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miga K. H., Koren S., Rhie A., Vollger M. R., Gershman A., Bzikadze A., Brooks S., Howe E., Porubsky D., Logsdon G. A., Schneider V. A., Potapova T., Wood J., Chow W., Armstrong J., Fredrickson J., Pak E., Tigyi K., Kremitzki M., Markovic C., Maduro V., Dutra A., Bouffard G. G., Chang A. M., Hansen N. F., Wilfert A. B., Thibaud-Nissen F., Schmitt A. D., Belton J.-M., Selvaraj S., Dennis M. Y., Soto D. C., Sahasrabudhe R., Kaya G., Quick J., Loman N. J., Holmes N., Loose M., Surti U., Risques R. A., Graves Lindsay T. A., Fulton R., Hall I., Paten B., Howe K., Timp W., Young A., Mullikin J. C., Pevzner P. A., Gerton J. L., Sullivan B. A., Eichler E. E., Phillippy A. M., Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bzikadze A. V., Pevzner P. A., Automated assembly of centromeres from ultra-long error-prone reads. Nat. Biotechnol. 38, 1309–1316 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nurk S., Walenz B. P., Rhie A., Vollger M. R., Logsdon G. A., Grothe R., Miga K. H., Eichler E. E., Phillippy A. M., Koren S., HiCanu: Accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koren S., Phillippy A. M., Simpson J. T., Loman N. J., Loose M., Reply to ‘errors in long-read assemblies can critically affect protein prediction’. Nat. Biotechnol. 37, 127–128 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Yoshimura J., Ichikawa K., Shoura M. J., Artiles K. L., Gabdank I., Wahba L., Smith C. L., Edgley M. L., Rougvie A. E., Fire A. Z., Morishita S., Schwarz E. M., Recompleting the Caenorhabditis elegans genome. Genome Res. 29, 1009–1022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sevim V., Bashir A., Chin C.-S., Miga K. H., Alpha-CENTAURI: Assessing novel centromeric repeat sequence variation with long read sequencing. Bioinformatics 32, 1921–1924 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warburton P. E., Willard H. F., Interhomologue sequence variation of alpha satellite DNA from human chromosome 17: Evidence for concerted evolution along haplotypic lineages. J. Mol. Evol. 41, 1006–1015 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Kronenberg Z. N., Fiddes I. T., Gordon D., Murali S., Cantsilieris S., Meyerson O. S., Underwood J. G., Nelson B. J., Chaisson M. J. P., Dougherty M. L., Munson K. M., Hastie A. R., Diekhans M., Hormozdiari F., Lorusso N., Hoekzema K., Qiu R., Clark K., Raja A., Welch A. E., Sorensen M., Baker C., Fulton R. S., Armstrong J., Graves-Lindsay T. A., Denli A. M., Hoppe E. R., Hsieh P., Hill C. M., Pang A. W. C., Lee J., Lam E. T., Dutcher S. K., Gage F. H., Warren W. C., Shendure J., Haussler D., Schneider V. A., Cao H., Ventura M., Wilson R. K., Paten B., Pollen A., Eichler E. E., High-resolution comparative analysis of great ape genomes. Science 360, eaar6343 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horai S., Hayasaka K., Kondo R., Tsugane K., Takahata N., Recent African origin of modern humans revealed by complete sequences of hominoid mitochondrial DNAs. Proc. Natl. Acad. Sci. U.S.A. 92, 532–536 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henikoff S., Ahmad K., Malik H. S., The Centromere Paradox: Stable inheritance with rapidly evolving DNA. Science 293, 1098–1102 (2001). [DOI] [PubMed] [Google Scholar]
- 48.H. S. Malik, in Centromere (Springer, 2009), pp. 33–52. [Google Scholar]
- 49.Sullivan L. L., Chew K., Sullivan B. A., α satellite DNA variation and function of the human centromere. Nucleus 8, 331–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Z. Duda, S. Trusiak, R. O’Neill, in Centromeres and Kinetochores (Springer, 2017), pp. 257–281. [Google Scholar]
- 51.Talbert P. B., Henikoff S., Centromeres convert but don’t cross. PLOS Biol. 8, e1000326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schueler M. G., Dunn J. M., Bird C. P., Ross M. T., Viggiano L.; NISC Comparative Sequencing Program, Rocchi M., Willard H. F., Green E. D., Progressive proximal expansion of the primate X chromosome centromere. Proc. Natl. Acad. Sci. U.S.A. 102, 10563–10568 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pandey P., Bender M. A., Johnson R., Patro R., Squeakr: An exact and approximate k-mer counting system. Bioinformatics 34, 568–575 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Dvorkina T., Bzikadze A. V., Pevzner P. A., The string decomposition problem and its applications to centromere analysis and assembly. Bioinformatics 36, i93–i101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabd9230/DC1