

Abstract
Circulating levels of endothelial miR-150 are reduced in pulmonary arterial hypertension (PAH) and act as an independent predictor of patient survival, but links between endothelial miR-150 and vascular dysfunction are not well understood. We studied the effects of endothelial miR-150 supplementation and inhibition in PAH mice and cells from patients with idiopathic PAH. The role of selected mediators of miR-150 identified by RNA sequencing was evaluated in vitro and in vivo. Endothelium-targeted miR-150 delivery prevented the disease in Sugen/hypoxia mice, while endothelial knockdown of miR-150 had adverse effects. miR-150 target genes revealed significant associations with PAH pathways, including proliferation, inflammation, and phospholipid signaling, with PTEN-like mitochondrial phosphatase (PTPMT1) most markedly altered. PTPMT1 reduced inflammation and apoptosis and improved mitochondrial function in human pulmonary endothelial cells and blood-derived endothelial colony-forming cells from idiopathic PAH. Beneficial effects of miR-150 in vitro and in vivo were linked with PTPMT1-dependent biosynthesis of mitochondrial phospholipid cardiolipin and reduced expression of pro-apoptotic, pro-inflammatory, and pro-fibrotic genes, including c-MYB, NOTCH3, transforming growth factor β (TGF-β), and Col1a1. In conclusion, we are the first to show that miR-150 supplementation attenuates pulmonary endothelial damage induced by vascular stresses and may be considered as a potential therapeutic strategy in PAH.
Keywords: endothelial, vascular, pulmonary hypertension, cardiolipin, mitochondria, microRNA, remodelling, PTPMT1, fibrosis, proliferation
Graphical Abstract
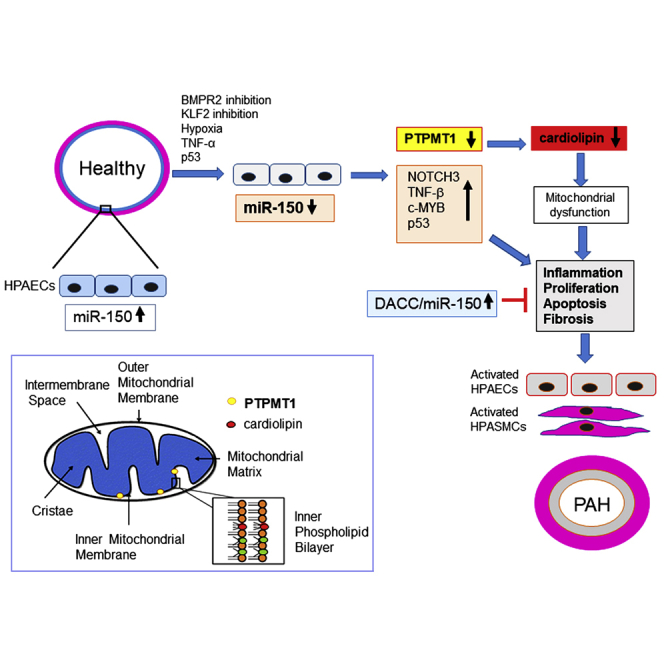
Pulmonary arterial hypertension is a severe disease with no cure. Loss of endothelial microRNA-150 is detrimental, while supplementation of microRNA-150 prevents the disease by reducing fibrosis, proliferation, and inflammation and improving mitochondrial energy production in lung and heart via increased production of the mitochondrial phospholipid cardiolipin.
Introduction
Pulmonary arterial hypertension (PAH) is a severe and currently incurable disease characterized by progressive thickening of small arteries in the lung, leading to increased pulmonary vascular resistance and right heart failure.1 Endothelial damage followed by proliferation of vascular smooth muscle cells underlie the disease pathology.2 The converging effects of hypoxia, inflammation, and oxidative and metabolic stress play a key contributory role.
At the cellular level, the arterial and right ventricular (RV) remodeling in PAH is associated with a shift from oxidative phosphorylation to glycolysis,3 which increases the availability of non-oxidized lipids, amino acids, and sugars essential for rapid cell proliferation.4 These changes are accompanied by inhibition of mitochondrial biogenesis, mitochondrial fragmentation, membrane hyperpolarization, and altered reactive oxygen species (ROS) production.4,5
MicroRNAs (miRNAs) have emerged as essential regulators of multiple cellular processes, simultaneously controlling mRNA processing, stability, and translation of multiple gene targets. Given the multifaceted nature of PAH pathology, there is interest in the role of miRNAs in the pathogenesis of this condition.6
We have previously shown that reduced miR-150-5p (referred to as miR-150) levels in plasma, circulating microvesicles, and the blood cell fraction from PAH patients are significant predictors of survival, independent of age, cardiac index, disease duration, and circulating lymphocyte count.7 While miR-150 is highly expressed in mature lymphocytes, circulating lymphocytes account for only about 6% of the variation in the miR-150 level, suggesting that endothelial cells are a likely source of this miRNA.8 The impact of variation in endothelial miR-150 expression on endothelial function or disease pathology has not yet been investigated.
Herein, we describe the effects of endothelial miR-150 supplementation and inhibition in experimental PAH, human pulmonary artery endothelial and smooth muscle cells, and blood-derived endothelial colony-forming cells (ECFCs) from PAH patients and identify the signaling mediators involved. We show that miR-150 has anti-apoptotic, anti-inflammatory, anti-proliferative, and anti-fibrotic effects and is required for mitochondrial adaptation to an increased energy demand in conditions of vascular stress.
Results
Endothelial supplementation of miR-150 improves pulmonary vascular hemodynamics and reduces vascular remodeling in Sugen/hypoxia mice
As a proof of concept, the potential therapeutic effects of miR-150 administration were evaluated in Sugen/hypoxia PAH mice (Figure 1A). miR-150 complexed with the lipid carrier DACC was delivered via tail vein injection at 4-day intervals throughout the 3-week period of study. The DACC liposomal formulation targets the vascular endothelium, with highest efficiency seen in the lung.9,10 Analysis of the distribution of fluorescently labeled RNA mimic duplex 1-Cy3 delivered in DACC liposomes 24 h post-injection confirmed that liposomal cargo accumulated in the lung, but also in other tissues, including heart and liver (Figure S1).
Figure 1.

Effect of pulmonary endothelial miR-150 supplementation on development of PH in Sugen/hypoxia mice
(A) Experimental layout. (B–D) Right ventricular systolic pressure (RVSP) (B), right ventricular hypertrophy (RVH; right ventricle to left ventricle + septum ratio [RV/LV+S]) (C), and percentage (D) of muscularized vessels <50 μm in diameter/total number of vessels in the lungs of normoxia control mice and Sugen/hypoxia mice treated with scrambled control or miR-150 mimic delivered by intravenous (i.v.) administration of DACC lipoplex, as indicated. (E) Representative images of α-SMA staining in lung sections from Sugen/hypoxia mice treated with scrambled control or miR-150 mimic. (F and G) miR-150 expression in lung and heart, as indicated; fold change of normoxia control. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, comparisons with normoxia control; #p < 0.05, ###p < 0.001, ####p < 0.0001, comparisons with scrambled control (by one-way ANOVA with a Tukey’s post-test.). Bars are means ± SEM. n = 8 mice/group.
The control Sugen/hypoxia animals showed a significantly elevated right ventricular systolic pressure (RVSP), right ventricular hypertrophy (RVH), and increased vascular muscularization, marked by a prominent staining of α-smooth muscle actin (α-SMA) of pre-capillary arterioles (Figures 1B–1E). Lung and heart levels of miR-150 were significantly reduced in these mice when compared with healthy transfection controls (Figures 1F and 1G). Reduction in miR-150 expression was most prominent in the vascular endothelium while miR-150 expression levels in leukocytes and airway epithelium remained relatively unaffected (Figure S2).
Effective miR-150 delivery to lung and heart tissues was confirmed by qPCR (Figures 1F and 1G). The treatment reduced RVSP (p < 0.05), RVH (p < 0.05), and vascular muscularization (p < 0.0001; Figures 1B–1E). To evaluate potential liver toxicity of DACC/miR-150 delivery, aspartate aminotransferase (AST) assay was performed on liver tissues from different study groups (Figure S3). DACC-treated mice showed an ∼2-fold increase in AST activity, comparable to the levels seen in hyperglycemic mice.11 This mild hepatotoxic effect is more likely to be associated with liposomal liver clearance12 rather than miR-150, as no significant difference in AST activity was noted between DACC controls and DACC/miR-150-treated mice.
Heterozygous endothelial-specific deletion of miR-150 worsens the symptoms of pulmonary hypertension (PH)
Heterozygous miR-150iEC-knockout (KO) mice (miR-150fl/Cdh5(PAC)-iCreERT2) were used for experiments to mimic the reduction (but not complete depletion) of miR-150 content seen in human disease and pre-clinical models of PAH. Following tamoxifen administration, the efficiency of Cre-recombinase-mediated deletion of miR-150 was confirmed by qPCR (Figure 2A).
Figure 2.

Effect of endothelial miR-150 deletion on development of PH in Sugen/hypoxia mice
(A) Effect of tamoxifen administration on miR-150 levels in lungs of wild-type (WT) (150+/+) and miR-150iEC-HTKO mice (150+/−). Data are expressed as fold change of normoxia control. (B–D) RVSP (B), RV/LV+S (C), and percentage (D) of muscularized vessels <50 μm in diameter/total number of vessels in the lungs of normoxia wild-type control (+ tamoxifen), normoxia miR-150+/− control (without tamoxifen), and Sugen/hypoxia miR-150+/− mice with and without tamoxifen, as indicated. In (A)–(D), open bars mark miR-150-deficient animals. (E) Representative images of α-SMA staining. Scale bar, 25 μm. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗∗p < 0.0001, comparisons with normoxia control; #p < 0.05, ####p < 0.0001, comparisons with miR-150+/− control (by one-way ANOVA with a Tukey’s post-test). Bars are means ± SEM. n = 4–8 mice/group.
Sugen/hypoxia miR-150iEC-KO mice showed a substantial elevation of RVSP (∼2-fold increase, p < 0.0001), accompanied by a rise in RVH and pulmonary vascular muscularization (both p < 0.05), compared with Sugen/hypoxia wild-type littermates (Figures 2B–2E).
Identification of miR-150-regulated genes
In order to identify potential mediators of miR-150-induced effects, human pulmonary artery endothelial cells (HPAECs) transfected with miR-150 or non-targeting control miRNA were subjected to RNA profiling.
Out of the 13,767 genes identified, 180 genes were significantly upregulated (p < 0.01, fold change > 1.5) and 207 were downregulated (p < 0.01, fold change < -1.5) by miR-150 (Figure 3A). Heatmap and unsupervised hierarchical clustering of the top 26 differentially expressed genes (with adjusted p < 0.05) are shown in Figure 3B. A list of differentially expressed genes is provided in Table S3.
Figure 3.

RNA sequencing analysis in miR-150-overexpressing HPAECs shows PTPMT1 as the most upregulated gene
To identify miR-150 signaling mediators, HPAECs from three different donors were transfected with miR-150 or scrambled control (20 nM) and RNA was extracted for RNA sequencing in three independent experiments. (A) Volcano plot of differentially expressed genes (DEGs). Each point represents the difference in expression (log2 fold difference) between groups and the associated significance of this change (independent unpaired sample t test). PTPMT1 is highlighted as the most upregulated gene (fold change of 2.51, p = 3.98 × 10−11, adjusted p [p-adj] value = 5.48 × 10−7; n = 3). (B) Heatmap showing 26 most significant genes after multiple test correction using the Benjamini-Hochberg procedure (p-adj < 0.05). Green and red represent downregulation and upregulation, respectively. (C) miR-150 predicted binding sequence with the top minimum free energy (MFE) event (−31.3 kcal/mol) in the promoter (242 bp 5′ upstream) region of the PTPMT1 gene (chr11:47565430–47573461) using RNAhybrid. (D) PTPMT1 mRNA levels in cells transfected with control miRNA. Bars are mean fold changes of normoxia control ± SEM. n = 5. ∗∗∗∗p < 0.0001, comparisons with normoxia control (by unpaired Student’s t test). (E) Significantly enriched pathways (p < 0.05) regulated by miR-150; Ingenuity Pathway Analysis (IPA; version 01-12) of top 26 differentially expressed genes in cells transfected with miR-150.
PTEN-like mitochondrial phosphatase (PTPMT1), a mitochondrial protein tyrosine phosphatase essential for cardiolipin biosynthesis, was the most significantly altered gene, showing a 2.5-fold increase in expression (adjusted p value of 5.48 × 10−7), compared with controls. miRNAs can reduce gene expression by binding to the 3′ UTR of target mRNAs or increase target gene expression by binding to gene promoters.13 RNAhybrid analysis has identified 70 putative miR-150 predicted binding sites to PTPMT1, with free energy ranges between −20.2 and −38.1 kcal/mol. The top minimum free energy (MFE) event (−38.1 kcal/mole) occurred within the promoter region of PTPMT1 (Figure 3C; Figure S4). Predicted binding of miR-150 to PTPMT1 and transforming growth factor β1 (TGFB1) is illustrated in Figure S4. While previous studies showed an upregulation of mRNAs through miRNA binding to their promoter,14 this mechanism has been relatively less well investigated, compared to mRNA degradation and translational repression through binding to the 3′ UTR. Delineating specific mechanisms through which miR-150 regulates PTPMT1 and other gene targets will require further studies. The increase in the expression of PTPMT1 was validated by qPCR (Figure 3D) and western blotting (Figure S5), and reductions in the expression of other genes identified by RNA sequencing (RNA-seq), including SERPINE1, PERP, DUSP5, NOTCH3, and c-MYB, were validated by qPCR (Figure S5).
Pathway analysis of miR-150 gene targets showed significant associations with pathways regulating cell proliferation, inflammation, and oxidative metabolism, including NOTCH signaling (p = 0.029), cardiolipin biosynthesis (p = 0.014), and inositol phosphate signaling (p = 0.007) (Figure 3E).
Consistent with the findings in vitro, quantification of transcripts by qPCR and RNAscope fluorescent in situ hybridization, which allows specific identification of single transcripts,15 confirmed expression changes of miR-150 target genes (c-MYB and NOTCH3) in the lungs of miR-150-treated animals (Figures S6 and S7). Heart tissue from miR-150-treated Sugen/hypoxia mice showed significantly reduced expression of markers of cardiac hypertrophy and fibrosis, including TGFB1, alpha-1 type I collagen (Col1a1), and regulator of calcineurin 1 (Rcan1) (Figure S8). In contrast, heart tissues from Sugen/hypoxia miR-150iEC-KO mice showed significantly increased levels of Col1a1, compared with the corresponding wild-type disease controls (Figure S9).
PTPMT1 mediates homeostatic effects of miR-150
In order to study the regulatory role of miR-150 in endothelial cell responses, HPAECs were transfected with miR-150 mimic or miR-150 inhibitor. Changes in the intracellular miR-150 levels were confirmed by qPCR (Figure 4A). Transfection efficiency evaluated with the Cy5-labeled miR negative control was ∼85% (Figure S10). Supplementation of miR-150 markedly attenuated endothelial cell apoptosis, hypoxia-induced cell proliferation, and nuclear factor κB (NF-κB) activation (Figures 4B–4D). In contrast, inhibition of miR-150 markedly augmented endothelial damage and inflammatory activation (Figures 4B–4D).
Figure 4.

Endothelium-protective effects of miR-150 and PTPMT1
(A–D) Effect of miR-150 mimic and miR-150 inhibitor on (A) miR-150 expression levels in HPAECs, (B) starvation-induced apoptosis (caspase-3/7 activity assay), (C) hypoxia-induced proliferation (EdU incorporation assay), and (D) hypoxia (24 h)-induced NF-κB activity (luciferase reporter assay). (E–H) Effect of PTPMT1 overexpression (PTPMT1) or silencing (siPTPMT1) on (E) PTPMT1 mRNA expression, (F) apoptosis, (G) proliferation, and (H) hypoxia-induced NF-κB activity in HPAECs. In (A), n = 3; in (B)–(H), n = 6. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, comparisons with untreated transfection control; #p < 0.05, ##p < 0.001, ####p < 0.0001, comparisons with treated (starvation or hypoxia, as appropriate) transfection controls (one-way ANOVA with a Tukey’s post-test). Bars are mean fold changes of transfection control ± SEM.
To see whether manipulation of miR-150 levels in endothelial cells can affect smooth muscle cell proliferation, HPAECs and human pulmonary artery smooth muscle cells (HPASMCs) were seeded on the opposite sides of a porous membrane in Transwell dishes (Figure S11). Endothelial miR-150 overexpression significantly reduced hypoxia-induced proliferation of HPASMCs (Figure S11).
Hypoxic exposure significantly reduced PTPMT1 expression in HPAECs (1.5-fold decrease, p < 0.01) and HPASMCs (2.5-fold decrease, p < 0.001), compared with normoxic controls (Figure S12). Overexpression and silencing of PTPMT1 (Figure 4E) mimicked, to a large extent, changes induced by manipulation of miR-150 expression (Figures 4F–4H), suggesting that PTPMT1 acts as a key mediator of the anti-proliferative and anti-inflammatory effects of miR-150 in pulmonary endothelial cells.
miR-150 and PTPMT1 improve mitochondrial function in HPAECs
Energy metabolism constitutes an essential link between cell growth and apoptosis.16 In order to assess the effect of miR-150 and PTPMT1 on energy metabolism, HPAECs and HPAECs transfected with miR-150 or PTPMT1 were subjected to bioenergetic profiling. The extracellular acidification rate (ECAR), which reflects the level of glycolysis, was not significantly affected by either treatment, but mitochondrial oxygen consumption rate (OCR), reflective of the level of mitochondrial respiration, was significantly elevated in miR-150 and PTPMT1-overexpressing cells (Figures 5A–5C).
Figure 5.

Effect of miR-150 and PTPMT1 on metabolic potential in HPAECs
(A–F) Extracellular acidification rate (ECAR) (A), oxygen consumption rate (OCR) (B), energy map (C), proton leak (D), ECAR metabolic potential (% of basal control) (E), and OCR metabolic potential (% of basal control) (F) in control HPAECs (transfection control) and HPAECs transfected with miR-150 or PTPMT1, as indicated. n = 5. Bars are means ± SEM. ∗p < 0.05, ∗∗∗∗p < 0.0001, comparison with transfection control (basal); #p < 0.05, as indicated (by one-way ANOVA with a Tukey’s post-test).
The treatment of cells with miR-150 and PTPMT1 significantly reduced mitochondrial proton leak (Figure 5D). As proton leak depicts the protons that migrate into the matrix without producing ATP, a reduction in proton leak indicates an improvement in coupling of substrate oxygen and ATP generation.17
Measurement of metabolic potential helps to evaluate the capacity of cells to respond to stress conditions associated with increased energy demand via mitochondrial respiration and glycolysis. The results show that miR-150 and PTPMT1 significantly increased mitochondrial metabolic potential measured as fold increase in OCR over basal control levels, while glycolytic metabolic potential in cells remained relatively unaffected (Figures 5E and 5F).
miR-150 and PTPMT1 restore cardiolipin levels in Sugen/hypoxia lung and heart tissues and increase mitochondrial content in human PAH ECFCs
PTPMT1 is a mitochondrial tyrosine kinase, essential for the biosynthesis of cardiolipin, the main phospholipid component of mitochondrial membranes and a key regulator of mitochondrial structure and function.18,19 We evaluated the effect of miR-150 and PTPMT1 supplementation on cardiolipin levels in lungs and hearts from miR-150-treated Sugen/hypoxia mice, as well as HPAECs and ECFCs from idiopathic PAH (IPAH) patients.
PTPMT1 and cardiolipin levels were significantly reduced in Sugen/hypoxia mice, while miR-150 supplementation restored their expression to the level seen in healthy mice (Figures 6A–6C). Overexpression of PTPMT1 and miR-150 significantly elevated cardiolipin levels in cultured endothelial cells (p < 0.01 and p < 0.05, respectively) (Figure 6D).
Figure 6.

miR-150 and PTPMT1 modulate cardiolipin levels in tissues and cells
(A–C) Lung PTPMT1 mRNA (A), lung cardiolipin (B), and heart cardiolipin (C) levels in control and DACC/miR-150-treated Sugen/hypoxia mice, as indicated. (D) Cardiolipin levels in control HPAECs and HPAECs transfected with miR-150 and PTPMT1, as indicated. In (A), data are expressed as fold change of normoxia control. Bars are mean of ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, comparison with transfection control (by one-way ANOVA with a Tukey’s post-test). n = 4–8.
Blood-derived ECFCs are often used as surrogates for pulmonary endothelial cells in PAH.20 qPCR analysis showed that miR-150 and PTPMT1 expressions were markedly reduced in ECFCs from IPAH patients, compared with the cells from healthy individuals (p < 0.01, n = 12–14) (Figures 7A and 7B). IPAH cells also showed a marked (∼2-fold, p < 0.05) reduction in cardiolipin levels, which was restored upon treatment with miR-150 and PTPMT1 (Figure 7C).
Figure 7.

miR-150, PTPMT1, and cardiolipin levels and mitochondrial biogenesis in IPAH ECFCs
(A and B) miR-150 (A) and PTPMT1 (B) expression levels in ECFCs from healthy individuals and IPAH patients (fold change of control). (C–E) Cardiolipin levels (C), mitochondrial fragmentation (D), and mitochondrial content (mitochondrial coverage/cell) (E) in ECFCs treated, as indicated. (F) Representative images of mitochondrial fragmentation in healthy and IPAH ECFCs. Inset in the top right corner is an enlarged image of the boxed area. Mitochondria were immunolabeled with fluorescein isothiocyanate (FITC) (green) and F-actin with tetramethylrhodamine isothiocyanate (TRITC)-phalloidin (red). Scale bar, 10 μm. Data are expressed as means ± SEM. ∗p < 0.05, ∗∗p < 0.01, comparison with healthy control. In (A) and (B), n = 12–14; in (C)–(E), n = 4.
Reduction in mitochondrial oxidative phosphorylation in PAH is linked with an increase in mitochondrial fragmentation and a reduction in mitochondrial biomass.5 Accordingly, we observed increased mitochondrial fragmentation and reduced mitochondrial content in ECFCs from IPAH patients, compared with healthy controls (Figures 7D–7F). Overexpression of miR-150 and PTPMT1 restored healthy control phenotype in IPAH cells (Figures 7D–7F).
Discussion
This study highlights the key role of endothelial miR-150 in the regulation of pulmonary vascular homeostasis. We show that supplementation of miR-150 reduces expression of markers of inflammation, apoptosis, and fibrosis critical to the pathology of PAH, including c-MYB, NOTCH3, TGF-β and Col1a1, and it enhances mitochondrial metabolic potential via increased expression of PTPMT1, the key regulator of cardiolipin biosynthesis.
Downregulation of miR-150 in pulmonary endothelium and IPAH endothelial cells may result from transcriptional repression by apoptosis-inducer p53,21 which is highly expressed in endothelial cells in pulmonary hypertensive lung.22 Hypoxia and inhibition of Krüppel-like factor 2 (KLF-2) and bone morphogenetic protein receptor 2 (BMPR2) are also likely to play contributory roles.23, 24, 25, 26
RNA sequencing identified PTPMT1 as a key gene affected by miR-150 overexpression, and functional analysis showed that PTPMT1 has endothelium-protective effects. PTPMT1 is exclusively localized to the inner membrane of mitochondria, with close proximity to electron transport chain complexes and enzymes of the tricarboxylic acid cycle.19 Interestingly, PTPMT1-ablated cells show a marked decrease in aerobic metabolism, enhancement of glycolysis,19 and mitochondrial fragmentation,27 reminiscent of changes seen in PAH.28 Accordingly, we observed that miR-150 and PTPMT1 supplementation restored mitochondrial content and reduced mitochondrial fragmentation in PAH ECFCs to the level seen in healthy controls.
The effects of PTPMT1 can be linked to its role in the biosynthesis of cardiolipin, a mitochondrial-specific phospholipid regulating mitochondrial membrane integrity and function. Interaction with cardiolipin is required for optimal activity of several inner mitochondrial membrane proteins, including the enzyme complexes of the electron transport chain and ATP production.18
miR-150 and PTPMT1 may reduce maladaptive right ventricular remodeling by augmentation of glucose oxidation and prevention of capillary rarefaction.29 In addition to the increased PTPMT1 levels, miR-150-treated Sugen/hypoxia mice showed reduced expression of cardiac hypertrophy and the fibrosis markers Col1a1, TGFB1, and Rcan1.30 Reduction in Col1a1 expression is likely to be mediated by direct targets of miR-150, c-MYB, Sp1, or β3 integrin.31,32 miR-150 interaction with TGFB1 can potentially occur through multiple locations, with the top predicted site within the first intron of TGFB1. Interestingly, TGF-β signaling can also block miR-150 expression,31,33 suggesting the existence of a feedback regulatory mechanism. Further studies are required to establish the precise nature of miR-150 interactions with its target genes.
Endothelial apoptosis, increased ROS generation and reduction in mitochondrial cardiolipin contribute to right ventricular failure in PAH.34,35 Electron leak is the major causative factor for production of mitochondrial superoxide, and hence the reduction in mitochondrial proton leak by PTPMT1 may account for the beneficial effects of miR-150 treatment.
Anti-remodeling effects of miR-150 are likely to result from the cumulative changes in expression of multiple genes. Besides PTPMT1, other signaling mediators, including c-MYB, NOTCH3, activin receptors 1 and 2, and matrix metalloproteinases, are likely to play a role. c-MYB stimulates cell migration, increases recruitment of endothelial progenitor cells,36 and promotes cardiac hypertrophy and fibrosis.37 NOTCH3 is a marker and predictor of PAH, and its blockade is sufficient to reverse experimental PAH.38,39 Consistently, we observed contemporaneous, opposing changes in the expression of miR-150 and its targets, c-MYB and NOTCH3, in human cells and lung tissues from PAH mice. While the overall effect of DACC-mediated miR-150 supplementation was beneficial, it showed a mild hepatotoxic effect, possibly as a result of liposomal clearance by the liver. While miRNAs can pleiotropically target multiple disease pathways and miRNA drugs start to enter clinical medicine, it is important to consider their potential off-target effects.40 Therefore, organ-specific drug delivery and new platforms evaluating toxicity and therapeutic efficacy of miRNA candidates ought to be developed. The use of other pre-clinical models of PAH in addition to Sugen/hypoxia mice would be needed for the full evaluation of miR-150 and PTPMT1-based therapies.
To summarize, we show that reduction in endothelial miR-150 levels has adverse effects on pulmonary hemodynamics in PAH mice, while endothelium-targeted delivery of miR-150 is protective. In addition to the anti-proliferative and anti-fibrotic actions of miR-150, activation of PTPMT1-cardiolipin signaling by this miRNA may facilitate adaptation of lung and heart to high energy demand in stress conditions induced by mechanical workload, hypoxia, or inflammation.
Materials and methods
Animal experiments
All studies were conducted in accordance with UK Home Office Animals (Scientific Procedures) Act 1986. To induce PAH, 8- to 12-week-old C57BL/6 male mice (20 g; Charles River Laboratories, UK) were injected subcutaneously with Sugen (SU5416; 20 mg/kg) and housed in hypoxia (10% O2) for 3 weeks (n = 8/group).41 mirVana hsa-miR-150-5p (ID MC10070) mimic or scrambled miRNA control (Ambion) in complex with DACC lipoplex preparation (Silence Therapeutics, London, UK)9 was administered intravenously once every fourth day at 1.5 mg/kg/day for 3 weeks, on five occasions to PAH mice and normoxic healthy controls. The first injection was given 1 day before Sugen/hypoxia administration. At 3 weeks, the mice were anesthetized by intraperitoneal injection of ketamine/Domitor (75 mg/kg + 1 mg/kg). The development of PAH was verified as previously described.41
Mice with inducible, conditional, endothelium-specific deletion of miR-150 were obtained by crossing floxed miR-150 mice (stock Mir150tm1Mtm/Mmjax mice from Jackson Laboratory) on a C57BL/6 background with C57BL/6 mice carrying tamoxifen-inducible Cre recombinase under the control of the Cdh5 promoter (Cdh5(PAC)-iCreERT2).42 Deletion of miR-150 in miR-150fl/fl/Cdh5(PAC)-iCreERT2 mice (referred to as miR-150iEC-KO) induced by tamoxifen was confirmed by PCR.
A detailed description of experimental procedures is available in the Supplemental materials and methods.
AST activity assay
An AST assay - (MAK055, Sigma-Aldrich, Darmstadt, Germany) was performed on frozen mouse liver tissues, according to the manufacturer’s guidelines.
RNAscope
For formalin-fixed, paraffin-embedded lung sections, an RNAscope multiplex fluorescent reagent kit v2 (Advanced Cell Diagnostics, Newark, CA, USA) and a TSA cyanine 3 & 5, tetramethylrhodamine (TMR), fluorescein evaluation kit system (PerkinElmer, Waltham, MA, USA) were used according to the manufacturers’ protocols.
A detailed description of experimental procedures is available in the Supplemental materials and methods.
Cell culture
HPAECs and HPASMCs were cultured as previously described.41 Cells were exposed to hypoxia (5% CO2, 2% O2) for 24–72 h.
A detailed description of non-contact cell culture and cell treatments is available in the Supplemental materials and methods.
Blood-derived human endothelial cells and human lung samples
All investigations were conducted in accordance with the Declaration of Helsinki. Venous blood samples were obtained with the approval of the Brompton Harefield & NHLI and Hammersmith Hospitals Research Ethics Committees, and informed written consent was received from healthy volunteers (n = 14) and patients with IPAH (n = 12). Human ECFCs were derived from peripheral blood samples as previously described.41 Clinical information and experimental procedures are provided in the Supplemental materials and methods.
Cell transfection
Detailed descriptions of transfection procedures and cell treatments are available in the Supplemental materials and methods.
Quantitative real-time PCR
RNA was extracted from cultured cells or tissue using a Monarch total RNA miniprep kit (New England Biolabs, Ipswich, MA, USA). Input RNA was reverse transcribed using a LunaScript RT supermix kit (New England Biolabs) or a TaqMan miRNA reverse transcription kit (Thermo Fisher Scientific, Waltham, MA, USA) and a custom multiplex RT primer pool in a SimpliAmp thermal cycler (Applied Biosystems, Foster City, CA, USA) according to the manufacturers’ instructions. A list of TaqMan miRNA and gene expression assays and additional methodological information are available in the Supplemental materials and methods.
Western blotting
Protein levels of PTPMT1 and β-actin in HPAECs transfected with scrambled miRNA and miR-150 mimic obtained 24 h post-transfection were determined by western blotting. Blots were probed with mouse monoclonal anti-β-actin (Sigma-Aldrich, A1978; 1:3,000), mouse monoclonal anti-PTPMT1 (Santa Cruz Biotechnology, sc-390901; 1:500) and secondary antibodies, and horseradish peroxidase (HRP)-linked sheep anti-mouse immunoglobulin G (IgG) (GE Healthcare, NAG31V; 1:1,000). The relative intensity of the immunoreactive bands was determined by densitometry using ImageJ software (National Institutes of Health, https://imagej.nih.gov/ij/), and PTPMT1 expression was normalized to β-actin.
RNA sequencing and identification of signaling mediators of miR-150
Next-generation RNA sequencing was carried out as previously described.24 Genes were considered differentially expressed when the adjusted p value was greater than 0.05 and there was at least a 1.5-fold change in expression. miRNA target prediction was carried out with TargetScan Human, miRecords, and Ingenuity Expert Findings. Gene enrichment was carried out using Ingenuity Pathway Analysis (IPA, version 01-12, QIAGEN, Hilden, Germany).
The RNA sequencing data generated and analyzed during this study are available in the BioProject repository at the following link: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA645887 (BioProject ID PRJNA645887; BioSamples SAMN15518378, SAMN15518379, SAMN5518380, SAMN15518381, SAMN15518382, and SAMN15518383; SRA accession nos. SRR12210268, SRR12210267, SRR12210266, SRR12210265, and SRR12210264).
RNAhybrid43 was used to identify the MFE hybridization of the mature miR-150 (MIMAT0000451) sequence against the DNA sequence within a 2-kbp window inclusive of the gene body for PTPMT1 (chr11:47563600–47575461) and TGFB1 (chr19:41328324–41355922). A maximum of 100 hits per target and a maximum MFE threshold of −20 with approximate p values estimated from the 3utr_human for the target sequence were applied.
Measurement of cell apoptosis, proliferation, and inflammatory activation
A 5-ethynyl-2′-deoxyuridine (EdU) proliferation assay, NF-κB activity, and caspase-3/7 apoptosis assays were carried out as previously described.41 Additional details are provided in the Supplemental materials and methods.
Seahorse bioenergetics assay
OCRs and ECARs were measured in a Seahorse extracellular flux analyzer using XF24 (Seahorse Bioscience, North Billerica, MA, USA) and with a Seahorse XF mito stress test kit (Agilent Technologies, Santa Clara, CA, USA, 103015-100).
Immunostaining
A detailed description of experimental procedures is available in the Supplemental materials and methods.
Cardiolipin measurement
Quantification of cardiolipin was carried out with cardiolipin assay kit (BioVision, Milpitas, CA, USA, K944-100).
Mitochondrial fragmentation count and mitochondrial content
Mitochondrial fragmentation (area taken by mitochondrial particles <2 μm in length)44 and total mitochondrial coverage (area taken by all mitochondria) were determined in confocal images using NIP2 image software.45 A detailed description of experimental procedures is available in the Supplemental materials and methods.
Statistical analysis
All experiments were performed at least in triplicate, and measurements were taken from distinct samples. Data are presented as mean ± SEM. Normality of data distribution was assessed with a Shapiro-Wilk test in GraphPad Prism 7.03. Comparisons between two groups were made with a Student’s t test or Mann-Whitney’s U test, whereas three or more groups were compared by use of ANOVA with a Tukey’s post hoc test or Kruskal-Wallis test with Dunn’s post hoc test, as appropriate. Statistical significance was accepted at p < 0.05.
Acknowledgments
We thank the staff of the Imperial NIHR/Imperial Clinical Research Facility, Hammersmith Hospital (London, UK) and Dr. John Wharton (NHLI) for help in acquiring cells from IPAH patients. We also thank Prof. Anna Randi and Dr. Graeme Birdsay (NHLI, Imperial College London) for providing Cdh5(PAC)-iCreERT2 mice. This research was supported by PhD studentships from the Government of Saudi Arabia (to M.M.A.) and by British Heart Foundation project grant PG/16/4/31849.
Author contributions
G.R., K.B.J., V.B.A.-S., M.M.A., C.C.M., A.H.O., J.E., U.S., and B.W.-S. performed in vitro experiments and analyzed data; K.B.J., G.R., and V.B.A.-S. performed in vitro and in vivo experiments, immunohistochemistry, and analyzed data; M.M.A. provided analysis of mitochondrial fragmentation; M.R.W. critically analyzed the manuscript; C.M. analyzed RNA sequencing data; M.R.W. provided IPAH patient data; and B.W.-S. secured funding, performed experiments, and wrote the manuscript.
Declaration of interests
This work involved the collaboration of B.W.-S. with Silence Therapeutics (London, UK), who provided DACC/miRNA preparations. The remaining authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.10.042.
Supplemental information
References
- 1.Schermuly R.T., Ghofrani H.A., Wilkins M.R., Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat. Rev. Cardiol. 2011;8:443–455. doi: 10.1038/nrcardio.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ranchoux B., Harvey L.D., Ayon R.J., Babicheva A., Bonnet S., Chan S.Y., Yuan J.X., Perez V.J. Endothelial dysfunction in pulmonary arterial hypertension: an evolving landscape (2017 Grover Conference Series) Pulm. Circ. 2018;8 doi: 10.1177/2045893217752912. 2045893217752912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freund-Michel V., Khoyrattee N., Savineau J.P., Muller B., Guibert C. Mitochondria: roles in pulmonary hypertension. Int. J. Biochem. Cell Biol. 2014;55:93–97. doi: 10.1016/j.biocel.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Paulin R., Michelakis E.D. The metabolic theory of pulmonary arterial hypertension. Circ. Res. 2014;115:148–164. doi: 10.1161/CIRCRESAHA.115.301130. [DOI] [PubMed] [Google Scholar]
- 5.Culley M.K., Chan S.Y. Mitochondrial metabolism in pulmonary hypertension: beyond mountains there are mountains. J. Clin. Invest. 2018;128:3704–3715. doi: 10.1172/JCI120847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Negi V., Chan S.Y. Discerning functional hierarchies of microRNAs in pulmonary hypertension. JCI Insight. 2017;2:e91327. doi: 10.1172/jci.insight.91327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhodes C.J., Wharton J., Boon R.A., Roexe T., Tsang H., Wojciak-Stothard B., Chakrabarti A., Howard L.S., Gibbs J.S., Lawrie A. Reduced microRNA-150 is associated with poor survival in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2013;187:294–302. doi: 10.1164/rccm.201205-0839OC. [DOI] [PubMed] [Google Scholar]
- 8.Gubrij I.B., Pangle A.K., Pang L., Johnson L.G. Reversal of microRNA dysregulation in an animal model of pulmonary hypertension. PLoS ONE. 2016;11:e0147827. doi: 10.1371/journal.pone.0147827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fehring V., Schaeper U., Ahrens K., Santel A., Keil O., Eisermann M., Giese K., Kaufmann J. Delivery of therapeutic siRNA to the lung endothelium via novel Lipoplex formulation DACC. Mol. Ther. 2014;22:811–820. doi: 10.1038/mt.2013.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdul-Salam V.B., Russomanno G., Chien-Nien C., Mahomed A.S., Yates L.A., Wilkins M.R., Zhao L., Gierula M., Dubois O., Schaeper U. CLIC4/Arf6 pathway. Circ. Res. 2019;124:52–65. doi: 10.1161/CIRCRESAHA.118.313705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka K., Nanbara S., Tanaka T., Koide H., Hayashi T. Aminotransferase activity in the liver of diabetic mice. Diabetes Res. Clin. Pract. 1988;5:71–75. doi: 10.1016/s0168-8227(88)80081-0. [DOI] [PubMed] [Google Scholar]
- 12.Sercombe L., Veerati T., Moheimani F., Wu S.Y., Sood A.K., Hua S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015;6:286. doi: 10.3389/fphar.2015.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaschetto L.M. miRNA activation is an endogenous gene expression pathway. RNA Biol. 2018;15:826–828. doi: 10.1080/15476286.2018.1451722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y., Fan M., Zhang X., Huang F., Wu K., Zhang J., Liu J., Huang Z., Luo H., Tao L., Zhang H. Cellular microRNAs up-regulate transcription via interaction with promoter TATA-box motifs. RNA. 2014;20:1878–1889. doi: 10.1261/rna.045633.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang F., Flanagan J., Su N., Wang L.C., Bui S., Nielson A., Wu X., Vo H.T., Ma X.J., Luo Y. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012;14:22–29. doi: 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mason E.F., Rathmell J.C. Cell metabolism: an essential link between cell growth and apoptosis. Biochim. Biophys. Acta. 2011;1813:645–654. doi: 10.1016/j.bbamcr.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng J., Nanayakkara G., Shao Y., Cueto R., Wang L., Yang W.Y., Tian Y., Wang H., Yang X. Mitochondrial proton leak plays a critical role in pathogenesis of cardiovascular diseases. Adv. Exp. Med. Biol. 2017;982:359–370. doi: 10.1007/978-3-319-55330-6_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudek J. Role of cardiolipin in mitochondrial signaling pathways. Front. Cell Dev. Biol. 2017;5:90. doi: 10.3389/fcell.2017.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen J., Liu X., Yu W.M., Liu J., Nibbelink M.G., Guo C., Finkel T., Qu C.K. A critical role of mitochondrial phosphatase Ptpmt1 in embryogenesis reveals a mitochondrial metabolic stress-induced differentiation checkpoint in embryonic stem cells. Mol. Cell. Biol. 2011;31:4902–4916. doi: 10.1128/MCB.05629-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duong H.T., Comhair S.A., Aldred M.A., Mavrakis L., Savasky B.M., Erzurum S.C., Asosingh K. Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulm. Circ. 2011;1:475–486. doi: 10.4103/2045-8932.93547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghose J., Bhattacharyya N.P. Transcriptional regulation of microRNA-100, -146a, and -150 genes by p53 and NFκB p65/RelA in mouse striatal STHdhQ7/ HdhQ7 cells and human cervical carcinoma HeLa cells. RNA Biol. 2015;12:457–477. doi: 10.1080/15476286.2015.1014288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z., Yang K., Zheng Q., Zhang C., Tang H., Babicheva A., Jiang Q., Li M., Chen Y., Carr S.G. Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019;316:L216–L228. doi: 10.1152/ajplung.00538.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hergenreider E., Heydt S., Tréguer K., Boettger T., Horrevoets A.J., Zeiher A.M., Scheffer M.P., Frangakis A.S., Yin X., Mayr M. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012;14:249–256. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 24.Sindi H.A., Russomanno G., Satta S., Abdul-Salam V.B., Jo K.B., Qazi-Chaudhry B., Ainscough A.J., Szulcek R., Jan Bogaard H., Morgan C.C. Therapeutic potential of KLF2-induced exosomal microRNAs in pulmonary hypertension. Nat. Commun. 2020;11:1185. doi: 10.1038/s41467-020-14966-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen M., Shen C., Zhang Y., Shu H. MicroRNA-150 attenuates hypoxia-induced excessive proliferation and migration of pulmonary arterial smooth muscle cells through reducing HIF-1α expression. Biomed. Pharmacother. 2017;93:861–868. doi: 10.1016/j.biopha.2017.07.028. [DOI] [PubMed] [Google Scholar]
- 26.Eichstaedt C.A., Song J., Viales R.R., Pan Z., Benjamin N., Fischer C., Hoeper M.M., Ulrich S., Hinderhofer K., Grünig E. First identification of Krüppel-like factor 2 mutation in heritable pulmonary arterial hypertension. Clin. Sci. (Lond.) 2017;131:689–698. doi: 10.1042/CS20160930. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J., Guan Z., Murphy A.N., Wiley S.E., Perkins G.A., Worby C.A., Engel J.L., Heacock P., Nguyen O.K., Wang J.H. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 2011;13:690–700. doi: 10.1016/j.cmet.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dasgupta A., Wu D., Tian L., Xiong P.Y., Dunham-Snary K.J., Chen K.H., Alizadeh E., Motamed M., Potus F., Hindmarch C.C.T., Archer S.L. Mitochondria in the pulmonary vasculature in health and disease: oxygen-sensing, metabolism, and dynamics. Compr. Physiol. 2020;10:713–765. doi: 10.1002/cphy.c190027. [DOI] [PubMed] [Google Scholar]
- 29.Ryan J.J., Archer S.L. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 2014;115:176–188. doi: 10.1161/CIRCRESAHA.113.301129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voelkel N.F., Quaife R.A., Leinwand L.A., Barst R.J., McGoon M.D., Meldrum D.R., Dupuis J., Long C.S., Rubin L.J., Smart F.W., National Heart, Lung, and Blood Institute Working Group on Cellular and Molecular Mechanisms of Right Heart Failure Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114:1883–1891. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 31.Zheng J., Lin Z., Dong P., Lu Z., Gao S., Chen X., Wu C., Yu F. Activation of hepatic stellate cells is suppressed by microRNA-150. Int. J. Mol. Med. 2013;32:17–24. doi: 10.3892/ijmm.2013.1356. [DOI] [PubMed] [Google Scholar]
- 32.Honda N., Jinnin M., Kira-Etoh T., Makino K., Kajihara I., Makino T., Fukushima S., Inoue Y., Okamoto Y., Hasegawa M. miR-150 down-regulation contributes to the constitutive type I collagen overexpression in scleroderma dermal fibroblasts via the induction of integrin β3. Am. J. Pathol. 2013;182:206–216. doi: 10.1016/j.ajpath.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 33.Davoodian P., Ravanshad M., Hosseini S.Y., Khanizadeh S., Almasian M., Nejati Zadeh A., Esmaiili Lashgarian H. Effect of TGF-β/smad signaling pathway blocking on expression profiles of miR-335, miR-150, miR-194, miR-27a, and miR-199a of hepatic stellate cells (HSCs) Gastroenterol. Hepatol. Bed Bench. 2017;10:112–117. [PMC free article] [PubMed] [Google Scholar]
- 34.Rawat D.K., Alzoubi A., Gupte R., Chettimada S., Watanabe M., Kahn A.G., Okada T., McMurtry I.F., Gupte S.A. Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension. 2014;64:1266–1274. doi: 10.1161/HYPERTENSIONAHA.114.03261. [DOI] [PubMed] [Google Scholar]
- 35.Saini-Chohan H.K., Dakshinamurti S., Taylor W.A., Shen G.X., Murphy R., Sparagna G.C., Hatch G.M. Persistent pulmonary hypertension results in reduced tetralinoleoyl-cardiolipin and mitochondrial complex II + III during the development of right ventricular hypertrophy in the neonatal pig heart. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H1415–H1424. doi: 10.1152/ajpheart.00247.2011. [DOI] [PubMed] [Google Scholar]
- 36.Wang W., Li C., Li W., Kong L., Qian A., Hu N., Meng Q., Li X. miR-150 enhances the motility of EPCs in vitro and promotes EPCs homing and thrombus resolving in vivo. Thromb. Res. 2014;133:590–598. doi: 10.1016/j.thromres.2013.12.038. [DOI] [PubMed] [Google Scholar]
- 37.Deng P., Chen L., Liu Z., Ye P., Wang S., Wu J., Yao Y., Sun Y., Huang X., Ren L. MicroRNA-150 inhibits the activation of cardiac fibroblasts by regulating c-Myb. Cell. Physiol. Biochem. 2016;38:2103–2122. doi: 10.1159/000445568. [DOI] [PubMed] [Google Scholar]
- 38.Havrda M.C., Johnson M.J., O’Neill C.F., Liaw L. A novel mechanism of transcriptional repression of p27kip1 through Notch/HRT2 signaling in vascular smooth muscle cells. Thromb. Haemost. 2006;96:361–370. doi: 10.1160/TH06-04-0224. [DOI] [PubMed] [Google Scholar]
- 39.Li X., Zhang X., Leathers R., Makino A., Huang C., Parsa P., Macias J., Yuan J.X., Jamieson S.W., Thistlethwaite P.A. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 2009;15:1289–1297. doi: 10.1038/nm.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanna J., Hossain G.S., Kocerha J. The potential for microRNA therapeutics and clinical research. Front. Genet. 2019;10:478. doi: 10.3389/fgene.2019.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wojciak-Stothard B., Abdul-Salam V.B., Lao K.H., Tsang H., Irwin D.C., Lisk C., Loomis Z., Stenmark K.R., Edwards J.C., Yuspa S.H. Aberrant chloride intracellular channel 4 expression contributes to endothelial dysfunction in pulmonary arterial hypertension. Circulation. 2014;129:1770–1780. doi: 10.1161/CIRCULATIONAHA.113.006797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y., Nakayama M., Pitulescu M.E., Schmidt T.S., Bochenek M.L., Sakakibara A., Adams S., Davy A., Deutsch U., Lüthi U. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 43.Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farrand L., Kim J.Y., Im-Aram A., Suh J.Y., Lee H.J., Tsang B.K. An improved quantitative approach for the assessment of mitochondrial fragmentation in chemoresistant ovarian cancer cells. PLoS ONE. 2013;8:e74008. doi: 10.1371/journal.pone.0074008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez K., Cupitt J. 2005. VIPS—a highly tuned image processing software architecture. In Proceedings of the IEEE International Conference on Image Processing 2005 (IEEE) p. II-574. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.