

Abstract
ANKS6 is a ciliary protein that localizes to the proximal compartment of the primary cilium, where it regulates signaling. Mutations in the ANKS6 gene cause multiorgan ciliopathies in humans, which include laterality defects of the visceral organs, renal cysts as part of nephronophthisis and congenital hepatic fibrosis (CHF) in the liver. Although CHF together with liver ductal plate malformations are common features of several human ciliopathy syndromes, including nephronophthisis-related ciliopathies, the mechanism by which mutations in ciliary genes lead to bile duct developmental abnormalities is not understood. Here, we generated a knockout mouse model of Anks6 and show that ANKS6 function is required for bile duct morphogenesis and cholangiocyte differentiation. The loss of Anks6 causes ciliary abnormalities, ductal plate remodeling defects and periportal fibrosis in the liver. Our expression studies and biochemical analyses show that biliary abnormalities in Anks6-deficient livers result from the dysregulation of YAP transcriptional activity in the bile duct-lining epithelial cells. Mechanistically, our studies suggest, that ANKS6 antagonizes Hippo signaling in the liver during bile duct development by binding to Hippo pathway effector proteins YAP1, TAZ and TEAD4 and promoting their transcriptional activity. Together, this study reveals a novel function for ANKS6 in regulating Hippo signaling during organogenesis and provides mechanistic insights into the regulatory network controlling bile duct differentiation and morphogenesis during liver development.
Introduction
Congenital hepatic fibrosis (CHF) is a developmental disorder of the liver that affects the portobiliary system and is associated with a spectrum of biliary abnormalities related to ductal plate remodeling defects in affected humans (1). The majority of CHF cases occur as part of various ciliopathy syndromes, such as Meckel-Gruber syndrome (OMIM #249000), Joubert syndrome (OMIM #213300), Bardet-Biedl syndrome (OMIM #209900), nephronophthisis (NPHP) (OMIM #256100) or oral-facial-digital syndrome (OMIM #311200), suggesting that ciliary dysfunction underpins the fibrogenic pathology and ductal plate remodeling abnormalities of the portal tracts (2–7). These monogenetic disorders have also commonly been referred to as hepatorenal fibrocystic syndromes, on the basis of the observation that mutations in the underlying ciliary genes cause fibrocystic changes both in the kidney and liver in the affected individual (8–11). In the liver, the disease is characterized by portal fibrosis in association with biliary cysts and a variable degree of intrahepatic biliary tract dilatation (1). Although primary cilia decorate virtually every cell in the human body, in the liver they are found only on differentiated cholangiocytes that form the epithelial lining of the intrahepatic bile ducts (IHBD) (12,13). The biliary primary cilia are solitary, microtubule-based, hair-like cell surface organelles that are involved in extracellular signal transduction including the mechanosensation of bile flow in the IHBD. These functions are critical for normal bile duct development and cholangiocyte differentiation, as well as for regulating cholangiocyte secretory and absorptive actions (3,12,14,15). Although there is overwhelming evidence, that cilia play an important role in IHBD development, the underlying mechanism(s) by which mutations in ciliary genes lead to ductal plate malformation (DPM) and congenital liver fibrosis is not understood.
IHBD assembly takes place in several distinct developmental steps, starting with the commitment of bipotential hepatoblasts to the biliary fate, followed by the formation of a single-layered ductal plate, acquisition of a second ductal layer, bile duct lumen formation and finally incorporation of the differentiated bile ducts into portal mesenchyme (16). These processes are regulated by the integrated actions of multiple signaling activities, including the evolutionarily conserved Notch and Hippo signaling pathways (17,18). In the developing IHBD, Notch signaling is activated by its ligand JAGGED1 (JAG1) that is expressed in the portal mesenchyme. This leads to the transactivation of Notch target genes, including Jagged1 and Hes1 in the ductal plate (19,20). Loss-of-function studies using transgenic mouse models revealed an essential function of Notch signaling in all stages of IHBD development, including biliary fate commitment, cholangiocyte differentiation and bile duct morphogenesis (21,22). Moreover, mutations in the Notch pathway genes Jagged1 or Notch2 cause Alagille syndrome, a congenital disorder that is primarily characterized by a paucity of bile ducts in affected individuals (23–26). Although our understanding of the regulation of Notch signaling during IHBD development is still incomplete, recent evidence suggests that Notch activity is regulated in part by the Hippo signaling effector YAP1. The Hippo pathway is a highly conserved kinase cascade that regulates cell growth and cell fate determination (27). The activation of Hippo signaling leads to inhibition of its downstream effectors YAP1 and TAZ by promoting their phosphorylation and cytoplasmic sequestration (28). In contrast, when the Hippo pathway is repressed, YAP1 and TAZ translocate to the nucleus, where they bind to various DNA-binding proteins, such as TEAD-family proteins, KLF4 or RBPJ transcription factors to activate gene expression (29–32). Liver-specific inactivation of YAP1 in mice has been demonstrated to cause postnatal cholestasis and progressive portal bridging fibrosis due to impaired IHBD development (33). Although mutations in ciliary genes do not completely recapitulate the clinical phenotypes caused by deficiencies in Notch or YAP1 signaling activity, several of the liver-specific developmental anomalies, including DPM, CHF and neonatal cholestasis are shared by individuals with hepatorenal fibrocystic disease (34–37). Accumulating evidence also suggests that ciliary proteins, such as INVS/NPHP2, NPHP3, NPHP4 and NEK8/NPHP9 directly interact with the Hippo pathway effectors YAP1 and TAZ (38–40).
We and others have recently reported mutations in the NPHP gene ankyrin repeat and sterile alpha motif domain containing 6 (ANKS6) as causing pleiotropic congenital abnormalities, including renal cysts, cardiovascular anomalies and laterality defects in humans (37,41). One of the most frequently affected organs in ANKS6-deficient patients besides the kidney and heart is liver, which features congenital periportal liver fibrosis and neonatal cholestasis (37). In ciliated cells, ANKS6 localizes to the inversin compartment within the proximal portion of the primary cilium that consists of INVS/NPHP2, NPHP3 and NEK8/NPHP9 proteins (37). Using biochemical analysis, it has been previously demonstrated that ANKS6 interacts with and regulates the NEK8 kinase activity in the INVS-compartment (42), suggesting ANKS6 has a regulatory role in ciliary signaling. However, not all ANKS6 pathogenic alleles regulate NEK8 activity (42), suggesting that ANKS6 may have additional previously unrecognized functions in the cell.
To study the role of ANKS6 in murine liver development, we generated an Anks6 transgenic mouse model. Here, we report that abrogation of Anks6 in mice leads to phenotypic changes that recapitulate the range of defects observed in ANKS6-deficient humans including severe bile duct morphogenesis and cholangiocyte differentiation defects, and fibrogenic processes in the periportal mesenchyme in the liver. Our molecular and biochemical analyses demonstrate that ANKS6 binds to the transcriptional co-activator YAP1 and regulates its transcriptional activity in the developing biliary tract. In the absence of ANKS6, YAP1 transcriptional activity is reduced.
Results
Anks6 mutant mice display perinatal lethality and laterality defects
Rodent models with missense mutations in the Anks6 gene have been used to study the role of ANKS6 in organ development and pathogenesis (42–45). Depending on the specific allele, these mutations affect Anks6 expression levels and subcellular localization (46), or interfere with its interaction with other proteins (42,44). As a result, there is a considerable variability in the reported organ involvement and phenotypic severity between these different genetic models. Importantly, none of them fully recapitulates the human ANKS6-deficiency phenotype that we have reported on recently (37). In order to study the full spectrum of ANKS6 function in development and pathogenesis, we generated an Anks6-deficient mouse line, using targeted Anks6tm1a embryonic stem cells from the European Conditional Mouse Mutagenesis Program (47). Allele-specific primers were used to genotype the mice (Supplementary Material, Fig. S1ac–). To ensure complete the inactivation of the Anks6 gene expression, we crossed Anks6tm1a mice first with the FLPo-deleter mice (48) and the resulting Anks6tm1b offspring with the EIIa-cre mice, thus removing the entire targeting cassette and Anks6 exon2 from the transgenic animals. The resulting Anks6tm1d/tm1d mice are referred to as Anks6−/− or mutant mice, hereafter. The complete the loss of ANKS6 expression from Anks6tm1d/tm1d mouse embryonic fibroblasts (MEFs) was confirmed by western blot analysis (Supplementary Material, Fig. S1d). We confirmed by quantitative reverse transcription PCR (qRT-PCR) that Cre-mediated recombination in the Anks6 locus did not inadvertently affect the expression of its neighboring genes—Gabbr2 and Galnt12 (Supplementary Material, Fig. S1e and f).
To characterize the role of ANKS6 in embryonic development, we collected littermate Anks6 mutant embryos at E (embryonic day) 18.5 or P (postnatal day) 0 and examined them for gross morphological and histological abnormalities (Fig. 1). None of the mutant pups survived beyond P0, and careful examination of the newborn mice showed that the mutants were either stillborn or died within hours after birth. Genotypic analysis of prenatal (E18.5), neonatal (P0) and postnatal (>P0) animals demonstrated that Anks6 mutant mice were present at Mendelian ratios before birth (n = 153, Anks6−/− n = 34), at P0 (n = 30, Anks6−/− n = 6), but none of the mutant mice survived beyond P0 (n = 72, Anks6−/− n = 0) (Fig. 1B).
Figure 1.
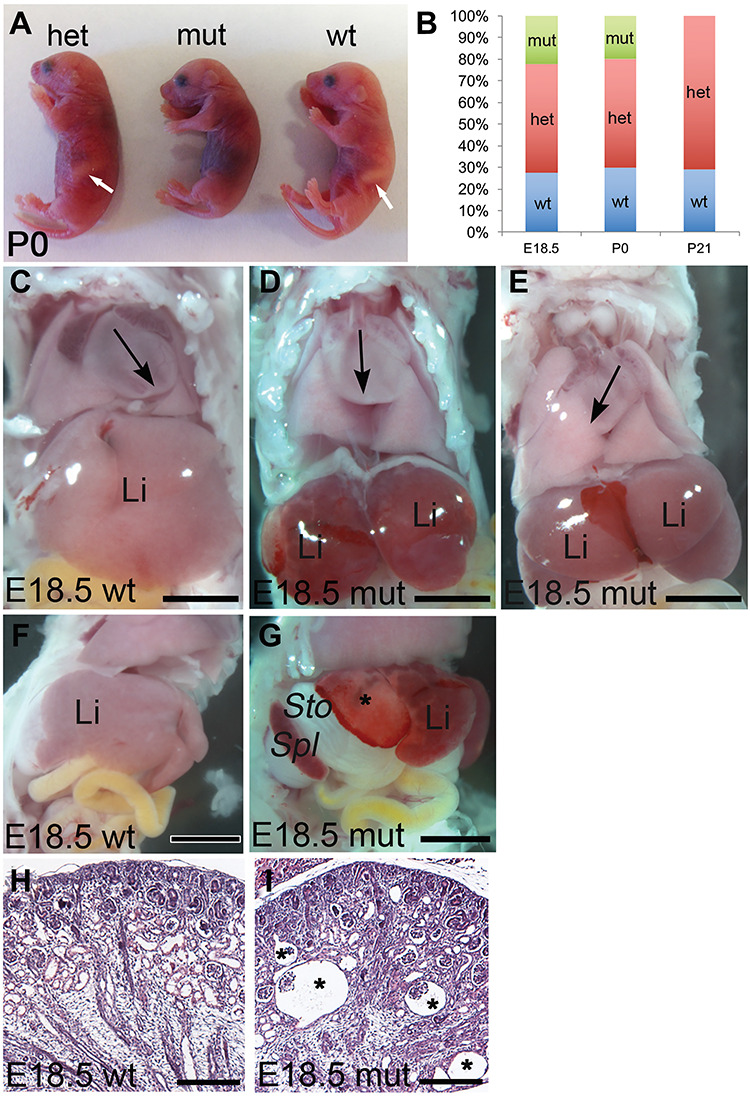
Anks6 mutant mice exhibit perinatal lethality and situs defects. (A) Lateral view of neonatal P0 littermates. Anks6 mutant newborns died shortly after birth. Note the absence of a stomach on the left side of the body in mutant pups, indicating a defect in visceral organ arrangement. White arrow indicates a stomach in wild-type and heterozygous newborn pups. (B) Distribution of mouse genotypes from Anks6 heterozygous crosses at prenatal, P0 and postnatal stages. Blue, red and green designate Anks6 wild type, Anks6 heterozygous and Anks6 mutant embryos, respectively (E18.5 n = 153, Anks6 mutant n = 34; P0 n = 30, Anks6 mutant n = 6; P21 n = 72, Anks6 mutant n = 0). χ2 test was used to test whether the distribution of genotypes at E18.5 and P0 corresponds to expected Mendelian ratio. (C, D and E) Frontal views of wild type (C) and mutant (D and E) E18.5 embryos. In wild-type embryos, the apex of the heart is pointing to the left (C). Anks6 mutant embryos develop either heterotaxia (D), or situs inversus with a rotation of the heart to the right (E). Note the reduced size of the liver in mutant animals (D and E). Black arrow points toward the apex of the left ventricle. Li, liver. Scale bar, 4.5 mm. (F and G) Lateral views of a wild type (F) and Anks6 mutant (G) E18.5 embryos illustrate the abdominal situs defect in mutant embryos. In contrast to wild-type embryos, stomach and spleen are positioned on the right body side in the mutants. Notice the necrotic white areas on the mutant liver (asterisk), indicating a lack of proper vascularization in the underling parenchymal tissue. St, stomach; Spl, spleen. Scale bar, 1.5 mm. (H and I) Comparison of kidney sagittal sections from wild type (J) and mutant (K) newborn mice reveals the formation of corticomedullary glomerular cysts (asterisk) in the mutant kidneys. Scale bar, 100 μm.
To investigate the cause of Anks6−/− lethality we performed a necropsy on E18.5 embryos. In contrast to the normal situs of wild-type mice (Fig. 1C and F), most of the Anks6−/− littermates (90%) displayed heterotaxia and situs inversus phenotypes with the apex of the heart (Fig. 1D and E) and the stomach (Fig. 1G) rotated to the right side of the body (Table 1). Previous report analyzing the Anks6Streaker mice had revealed a spectrum of congenital cardiac abnormalities at birth, which underlay the perinatal lethality in heterotaxic Anks6Streaker mice (42). To define the cause of perinatal lethality in Anks6−/− mice, we analyzed the mutant heart morphology using high-throughput magnetic resonance imaging (MRI) and hematoxylin eosin staining. Regardless of the situs phenotype, all Anks6−/− mice displayed a range of complex congenital heart defects, including atrioventricular septal defects, ventricular hypertrophy and overriding aorta (Supplementary Material, Fig. S2a,b and c), indicating that heart abnormalities are the primary cause of lethality in Anks6−/− mice around birth. In the abdominal cavity, the liver appeared reduced in size in E18.5 mutant embryos (Fig. 1C, D and E), and necrotic areas were frequently observed in the periphery (Fig. 1G). In addition, histological analysis of the mutant P0 pups revealed the formation of glomerular cysts in the kidneys (Fig. 1H and I).
Table 1.
Summary of situs phenotypes in ANKS6 mutant embryos (n = 29)
| Body situs | Heart situs | Lung situsa | Abdominal situs | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SS | SI | SA | Left | Right | Normal | SI | Right isomersim | Left isomersim | Normal | SI |
| 3 (10%) | 11 (38%) | 15 (52%) | 4 (14%) | 25 (86%) | 2 (7%) | 15 (52%) | 8 (28%) | 3 (10%) | 10 (34%) | 19 (66%) |
SS, situs solitus; SI, situs inversus; SA, situs am biguus or heterotaxia
aone case of polycystic lung.
Taken together, our morphological and histological analysis of the mutant embryos revealed a broad range of morphogenetic abnormalities in the visceral organs in mutant embryos, which recapitulate the spectrum of human ANKS6-deficiency phenotypes (41,42,49). MRI analysis of the mutant mice revealed complex congenital heart defects in all Anks6−/− mice regardless of the situs, explaining the perinatal lethality in these mice.
Loss of Anks6 causes liver developmental abnormalities
Congenital periportal liver fibrosis and cholestasis have been reported in individuals with ANKS6 mutations (37), but the role of ANKS6 in murine liver development has not been studied. To address this question, we analyzed the histology of mutant livers from E15.5, E18.5 and P0 animals by hematoxylin and eosin staining. We did not observe gross histological abnormalities in E15.5 mutant livers, a timepoint when mouse biliary development is initiated (Supplementary Material, Fig. S3). At E18.5 and at birth, the portal tracts in the central and peripheral regions of wild-type livers appeared well-differentiated—the bile ducts were lined by a characteristic columnar epithelium and enveloped in periportal mesenchyme (Fig. 2A and B and Supplementary Material, Fig. S4a). The biliary epithelium stained positive for Dolichos biflorus agglutinin lectin (DBA), a marker of differentiated cholangiocytes (Fig. 2C). In contrast, the biliary epithelium appeared low cuboidal and flattened in the mutant livers in the central and peripheral regions (Fig. 2D and E) and showed scattered DBA labeling (Fig. 2F), suggesting a defect in cholangiocyte differentiation. In addition, the peribiliary mesenchyme was absent and portal mesenchyme reduced in mutant livers (Fig. 2D and E and Supplementary Material, Fig. S4a). At birth (P0), the wild-type bile ducts were mostly embedded in the periportal mesenchyme both in the hilar (Fig. 2G) and peripheral (Fig. 2H) portal areas and showed strong staining for DBA (Fig. 2I), whereas the portal tracts in the mutant livers remained undifferentiated and incompletely remodeled—consisting of multiple undifferentiated and atrophic bile ducts per portal vein (Fig. 2J–L). The portal areas in the mutant livers displayed a spectrum of degenerative phenotypes, including collapsed bile ducts, surrounding an obliterated portal vein (Supplementary Material, Fig. S4b–e), or enlarged heavily fibrotic portal areas (Supplementary Material, Fig. S4e) that were not observed in wild-type livers. Together, our data demonstrate that the loss of Anks6 causes ductal plate remodeling defects in the liver characterized by bile duct morphogenesis defects and maintenance of multiple undifferentiated bile ducts per portal vein in mutant animals.
Figure 2.
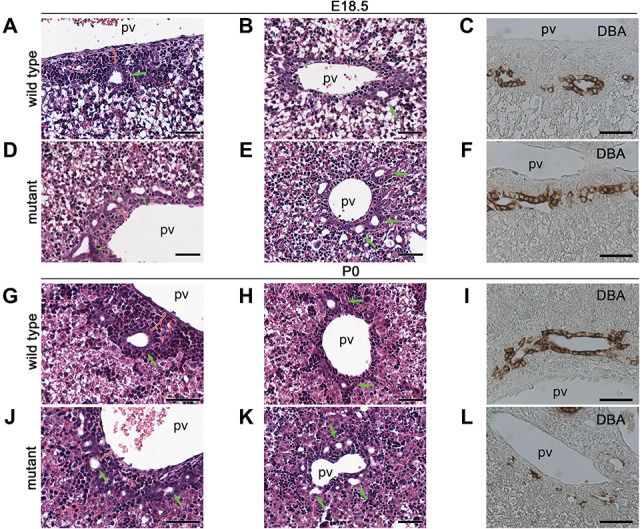
Anks6 deficiency causes bile duct abnormalities. (A, B, D and E) Hematoxylin and eosin-stained liver sections of E18.5 wild type (A and B) and Anks6 mutant (D and E) embryos. In wild-type liver, bile ducts are lined by cuboidal epithelium and surrounded by portal mesenchyme in the hilar region (A). Less differentiated bile ducts are observed in the peripheral area (B). In the Anks6−/− liver, note the presence of multiple, low-cuboidal bile ducts in the hilar (D), as well as in the peripheral (E) area, indicative of a failure in ductal plate remodeling. Green arrows indicate bile ducts; pv, portal vein. Scale bar, 50 μm. (G, H, J and K) Hematoxylin and eosin-stained liver sections of neonatal wild type (G and H) and mutant (J and K) mice. Both hilar and peripheral bile ducts are embedded in the periportal mesenchyme in wild-type liver at this stage (G and H). In contrast, the bile ducts remain in a ductal plate conformation in the mutant livers (J and K). Green arrows indicate bile ducts. Scale bar, 50 μm. (C, F, I and L) DBA expression in E18.5 (C and F) and neonatal (I and L) liver. DBA is expressed in the entire biliary epithelium in wild-type livers at E18.5 (C) and P0 (I). In the mutant liver, DBA expression is limited to the portal side of the bile duct at E18.5 (F) and has scattered biliary expression at P0 (L), indicative of a failure in biliary epithelium differentiation in the mutant liver. Scale bar, 50 μm.
Loss of Anks6 causes periportal fibrosis
To investigate whether Anks6 mutant livers develop periportal fibrosis, we stained E15.5 and E18.5 livers with antibodies against alpha smooth muscle actin (α-SMA), a marker of activated myofibroblasts. No significant changes in α-SMA expression were observed in E15.5 Anks6−/− livers compared with wild-type controls (Supplementary Material, Fig. S5a and b). At E18.5 α-SMA expression was restricted to the pericyte cell layer underneath the portal endothelium in wild-type livers (Fig. 3A). In contrast, E18.5 mutant livers expressed α-SMA throughout the portal region and α-SMA+ myofibroblasts extended into the liver parenchyma between albumin-positive hepatocytes, indicating a widespread fibrogenic process (Fig. 3B). Co-staining of the livers with a bile duct marker pan-CK further revealed that in contrast to the situation in wild type (Fig. 3C), myofibroblasts surrounded the developing bile ducts in Anks6 mutant livers (Fig. 3D). Taken together, our data demonstrate that Anks6 mutant mice develop congenital periportal liver fibrosis, replicating the human ANKS6-deficient liver phenotype.
Figure 3.
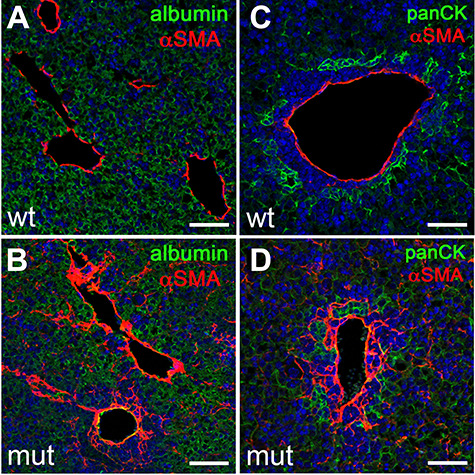
Loss of Anks6 causes portal fibrosis. (A–D) Immunofluorescent analysis of αSMA expression in E18.5 wild type (A and C) and Anks6 mutant (B and D) livers. αSMA is expressed in a layer of perivascular cells underneath the portal endothelium in wild-type livers (A and C). In Anks6 mutant livers, αSMA expression extends throughout the periportal mesenchyme and αSMA-positive cells can be seen in the albumin-positive (green) liver parenchyma (B), and they envelope the pan-cytokeratin-positive bile ducts (panCK, green) (D), indicating a conversion of the periportal mesenchyme to myofibroblasts. Scale bar, 75 μm.
Anks6 is expressed in the developing liver and mature bile duct epithelium
To trace the primary site of ANKS6 function in the liver, we examined the spatial distribution of Anks6 expression in embryonic (E15.5, E18.5) and postnatal (P0, P21, P50) livers by RNA in situ hybridization analysis. In E15.5 livers Anks6 expression was found uniformly throughout the liver (Fig. 4A), which around birth became restricted to the biliary epithelium, periportal mesenchyme and hepatocytes (Fig. 4B and C). In fully developed liver, at postnatal days P21 and P50, Anks6 expression was maintained only in the portal region—in the biliary epithelium and surrounding mesenchyme and was absent from the hepatocytes (Fig. 4D–F). To validate the in situ hybridization data in the postnatal liver, we isolated hepatocyte and biliary fractions from adult livers using Percoll gradient centrifugation and fluorescence activated cell sorting (FACS) for EpCAM+ cells (Fig. 4G) (50). Using a qRT-PCR analysis against hepatocyte-specific genes (albumin, Fabp2) or cholangiocyte-specific genes (Sox9, Ck19), we found Anks6 expressed in the EpCAM+ cholangiocytes, but not in hepatocytes in the adult liver (Fig. 4H–L). Together, our data demonstrate that Anks6 expression in the liver is developmentally regulated; its embryonic liver-wide expression becomes gradually restricted to the biliary cholangiocytes and peribiliary mesenchyme cells as the liver matures.
Figure 4.
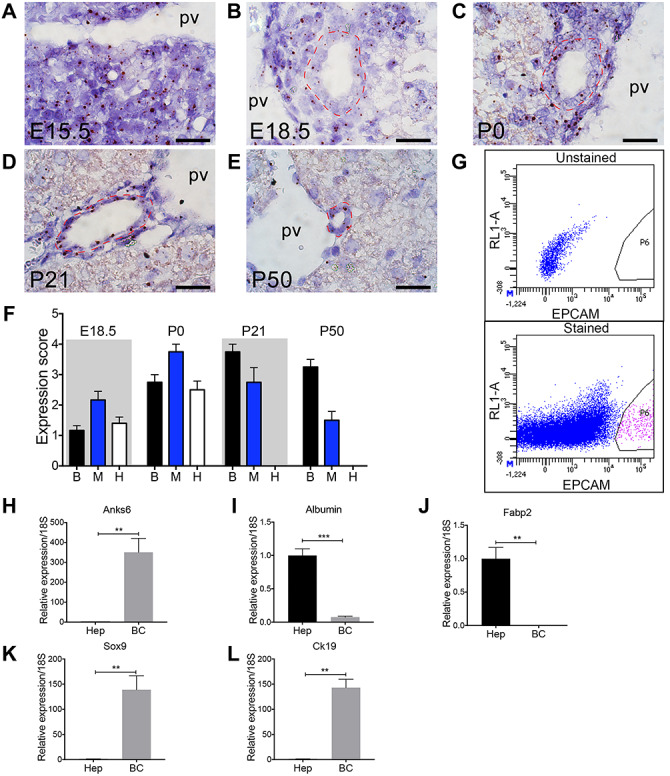
Anks6 is expressed in the biliary epithelium and portal mesenchyme. (A–E) Anks6 expression analysis using in situ hybridization in the liver at E15.5 (A), E18.5 (B), P0 (C), P21 (D), and P50 (E). Representative images of Anks6 staining reveal its expression in the hepatoblast progenitor cells at E15.5 (A), portal mesenchyme, bile duct epithelium and hepatocytes around birth (B and C), and in the biliary epithelium and surrounding mesenchyme at adult stage (D and E). Red dashed line surrounds bile ducts. pv, portal vein. Scale bar, 22.5 μm. (F) Anks6 expression scoring in wild-type liver during development and postnatal stages (0–4 arbitrary units). See representative images in (A). B, biliary cells; H, hepatocytes; M, portal mesenchyme. (G) Gating strategy for isolation of EpCAM+ biliary cells from the livers of 6-week old wild-type mice. Total single-cell suspensions of hepatocyte-depleted biliary and non-parenchymal cell fraction were sorted from the P6 population of EpCAM-stained samples. (H–L) Anks6 is expressed in biliary cells of adult liver. Hepatocytes (Hep) and biliary cells (bc) were isolated and relative mRNA expression of noted genes was quantitated via qRT-PCR. Hepatocytes are normalized to one and bc expression is quantitated as a ratio to its expression. Data represent the average of 3 biological samples ± s.e.m. Two-tailed unpaired t-test was used for statistical analysis, *P < 0.05, **P < 0.01, ***P < 0.001.
Bile duct morphogenesis is defective in Anks6 mutant livers
To characterize bile duct differentiation and morphogenesis defects in Anks6 mutant livers in greater detail, we examined the expression of known regulators and markers of biliary development. One of the earliest definitive cholangiocyte differentiation markers is the transcription factor Sry-box 9 (SOX9). SOX9 expression in the liver is initially activated around E15.5 in the single-layered ductal plate that surrounds the portal vein (51). Subsequently, a second layer of SOX9-negative hepatoblasts condenses on top of the first ductal layer, resulting in a two-layered organization. Following remodeling, this intermediate ductal structure gives rise to bile ducts, whose entire luminal epithelium will acquire SOX9 expression as the duct lining epithelium undergoes differentiation to cholangiocytes (51). As expected, E15.5 wild-type livers contained well-organized Sox9+ ductal plate structures in the hilar portal areas representing various developmental stages of bile duct (stained with anti-pan-cytokeratin (panCK) antibody) morphogenesis, including single-layered ductal plate and double-layered epithelial formations (Fig. 5A). Occasionally, bile duct lumen formation was observed, with SOX9+ cells surrounding the nascent tubule (Fig. 5B). In Anks6 mutant livers, the SOX9+ cells surrounding the portal vein appeared more scattered and discontinuous, and gave rise to tubule-like aggregates that were less organized at this stage (Fig. 5C and D). By E18.5, the bile duct morphogenesis was completed in the hilar region of wild-type livers—the biliary epithelium had a uniform SOX9 expression, and a distinct apico-basal polarity, defined by the expression of the basolateral marker β-catenin (Fig. 5E) and the apical marker osteopontin (Fig. 5H). In contrast, the mutant bile ducts appeared dilated and were lined by a poorly differentiated epithelium—SOX9+ cholangiocytes were present on the portal but not on the parenchymal side of the ducts (Fig. 5F and G). Defective cholangiocyte differentiation was accompanied by aberrant duct morphogenesis (Fig. 5F and G), and the lack of establishment of apical polarity in the parenchymal side of the biliary epithelium (Fig. 5I) in the mutant livers, whereas the localization of the basolateral polarity marker, β-catenin did not seem to be affected in mutant bile ducts at E18.5 (Fig. 5F and G). HNF1 Homeobox B (HNF1β) is a key regulator of IHBD morphogenesis that controls cholangiocyte differentiation from hepatoblast precursor cells (52). The inactivation of HNF1β causes a failure in bile duct organogenesis (52). Since HNF1β expression is not regulated by SOX9 and HNF1β is thought to control a different transcriptional circuitry than SOX9 during bile duct development (51), we examined weather the loss of Anks6 affects also HNF1β expression in the developing bile ducts. Immunohistochemical staining of E18.5 livers showed a uniform HNF1β expression in wild-type bile ducts (Fig. 5J). In contrast, HNF1β expression was detected only in the cells lining the portal side of the Anks6 mutant bile ducts and was absent from the parenchymal side (Fig. 5K), reminiscent of SOX9 expression, indicating that the loss of Anks6 affects both SOX9 and HNF1β controlled cholangiocyte differentiation programs. Although molecular expression studies have not been performed on ANKS6 patient liver biopsies, it was recently demonstrated that canonical Wnt signaling is aberrantly activated in ANKS6 patient kidneys (49). To examine whether Wnt signaling is also dysregulated in the Anks6−/− liver, we studied the expression of canonical (Axin2, cMyc and Ccnd1) and non-canonical (Wnt5a, Dkk1) Wnt signaling targets in E18.5 mutant livers using qRT-PCR. In contrast to the situation in human kidneys, none of the Wnt signaling target genes were significantly altered in the mutant livers at E18.5 (Supplementary Material, Fig. S6a and b), suggesting that neither canonical nor non-canonical Wnt signaling plays an essential role in the pathogenesis of Anks6−/− livers.
Figure 5.
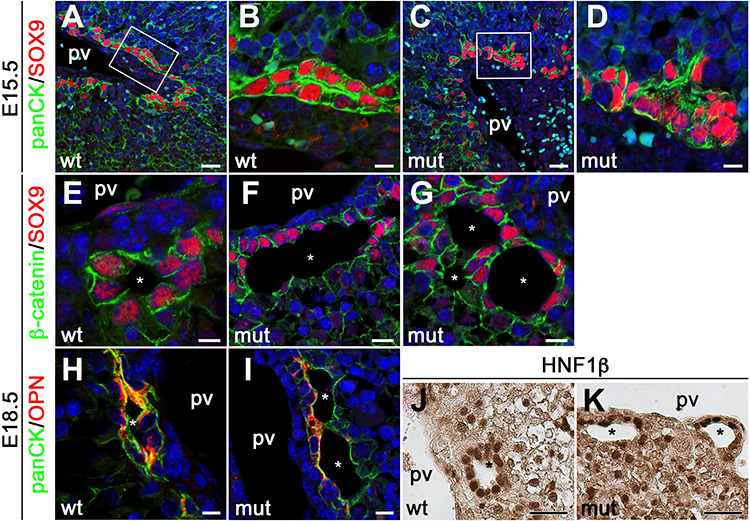
Anks6 is required for bile duct morphogenesis and cholangiocyte differentiation. (A–D) Immunofluorescent analysis of developing bile ducts in wild type (A and B), and mutant (C and D), livers with antibodies against panCK and Sox9 at E15.5. Scale bar, (A and C) 25 μm; (B and D) 7.5 μm. (E–G) Immunofluorescent analysis of E18.5 bile ducts with antibodies against β-catenin and Sox9. Sox9 is expressed in the entire epithelium of differentiated bile ducts in wild-type livers (E). Only the portal side of the ducts (first ductal layer) is positive for Sox9 in mutant livers (F and G), indicating that cholangiocyte differentiation is impaired in Anks6 mutant livers. In addition to biliary differentiation defects the mutant bile ducts demonstrate abnormal lumen formation (G). pv, portal vein, white asterisk indicates bile duct lumen. Scale bar, (E) 7.5 μm; (F and G) 10 μm. (H and I) Antibody staining against the apical marker osteopontin, demonstrates normal apical polarization of E18.5 wild-type biliary epithelial cells (H). In mutant bile ducts, osteopontin is present in the portal side, but missing from the parenchymal side of the ducts (I). pv, portal vein, white asterisk indicates bile duct lumen. Scale bar, (H) 7.5 μm; (I) 10 μm. (J and K) Immunohistochemistry against the cholangiocyte differentiation marker Hnf1β. Hnf1β is present in both layers of the bile duct in wild-type livers (J), but restricted to the portal layer in mutant livers (K). pv, portal vein, black asterisk indicates bile duct lumen. Scale bar, 30 μm.
Together, these data demonstrate that ANKS6 function is essential at several stages of bile duct development, including the formation of the ductal plates, differentiation of hepatoblasts to biliary epithelial cells and tubulogenesis of the bile duct, but does not involve Wnt signaling pathway.
Loss of ANKS6 causes ciliary defects
ANKS6’s known function in the cell is related to the primary cilium where it localizes to the inversin compartment (IC) and interacts with other nephronophthisis proteins—NPHP3, INVS/NPHP2, NEK8/NPHP9 and ANKS3 (37,42,53,54). In the liver, the main cell type to grow a primary cilium is the cholangiocyte (55). To characterize primary cilia in Anks6 mutant bile ducts, we stained E18.5 livers with antibodies against acetylated α-tubulin which labels the ciliary axoneme. As expected, primary cilia decorated the apical surface of every biliary epithelial cell in a wild-type liver (Fig. 6A). In contrast, only cells lining the portal side of the bile duct expressed cilia in Anks6 mutant livers (Fig. 6B), corroborating our finding that cholangiocyte differentiation is impaired in the mutant mice, as cholangiocyte differentiation precedes their ciliogenesis (3). Using antibodies against ANKS6, we found that ANKS6, indeed, localizes to the cholangiocyte cilia in wild-type liver (Fig. 6c, c′ and c″), but is absent from the mutant cilia (Fig. 6D, D′ and d″). Since ANKS6 is a component of the IC within the primary cilium, we next asked whether its loss affects cholangiocyte cilia ultrastructure in the mutant liver. Using a scanning electron microscopy (SEM), we observed fewer ciliated cholangiocytes in the mutant biliary epithelium compared with wild-type bile ducts, confirming the light microscopy findings. In addition, SEM revealed a variety of structural abnormalities in the mutant cilia. In contrast to wild-type cilia that were solitary and long (Fig. 6E), the mutant cilia appeared shorter (Fig. 6F), displayed bulbous formations (Fig. 6G), or appeared duplicated and had abnormal kinks (Fig. 6H) at the proximal portion of the cilium. Intriguingly, the distortions in cilia morphology seemed to localize to the first third of the cilium, a region where IC is located. These findings suggest that the loss of ANKS6 disrupts the IC composition, which in turn affects the ciliary architecture. To assess if this is the case, we stained wild type and Anks6 mutant ciliated MEFs with antibodies against acetylated α-tubulin and ANKS6 (Fig. 7A and B). After demonstrating the absence of ANKS6 expression in mutant cilia, we stained the ciliated MEFs with antibodies against acetylated α-tubulin (axonemal marker) or γ-tubulin (basal body marker) and INVS/NPHP2, which revealed a significantly reduced NPHP2 expression domain proximal to the basal body in the mutant cilia compared with wild-type cilia (Fig. 7C–G), confirming, that ANKS6 is critical for the formation or stability of the IC. On the basis of our data that Anks6 is expressed in the hepatoblasts at E15.5 (Fig. 3), we next asked whether hepatoblasts are ciliated, and if so, whether ANKS6 localizes to the hepatoblast cilia. To address this question, we knocked down Anks6 using shRNA in the hepatoblast-like BMEL (bipotential mouse embryonic liver) cells (Fig. 7H and I), which have been widely used to model hepatoblast differentiation in vitro (56). Both control (shNS) and Anks6 knockdown (shAnks6) BMEL cells formed a cilium after serum starvation, which stained positive for ANKS6 in shNS cells (Fig. 7H) but not in shAnks6 cells (Fig. 7I), demonstrating that hepatoblasts are ciliated and express ANKS6.
Figure 6.
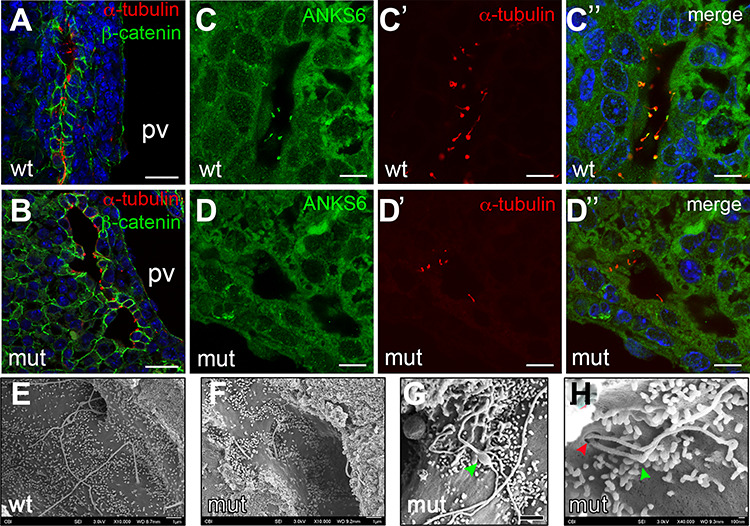
Loss of Anks6 causes structural abnormalities in bile duct cilia. (A and B) Immunofluorescent staining of wild type (A) and Anks6 mutant (B) bile ducts with acetylated α-tubulin (red, cilia) and β-catenin (green, bile ducts) demonstrates the lack of cilia from the parenchymal side of the bile duct in the mutant liver at E18.5. Scale bar, 25 μm. (C–D″) Anks6 localizes to cholangiocyte cilia in E18.5 livers. Antibody staining against Anks6 (green) and acetylated α-tubulin (red, cilia) shows Anks6 ciliary localization in wild-type cholangiocyte cilia (C, C′ and C″), and its absence from the mutant cilia (D, D′ and D″). Scale bar, 10 μm. (E–H) Scanning electron microscopy images reveal ciliary abnormalities in Anks6 mutant livers. Wild-type cilia are long and solitary (E), whereas mutant cilia are short (F), with bulbous structures (green arrowhead) (G), or are duplicated at the basal body (red arrowhead), and appear kinked (green arrowhead) (H). Scale bar, (e, F and G) 1 μm, (h) 100 nm.
Figure 7.
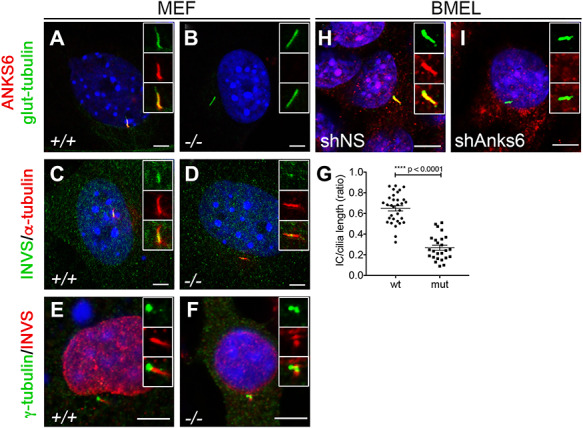
Anks6 localizes to the primary cilium and is required for the formation inversin compartment. (A and B) Anks6 mutant MEFs lack Anks6 ciliary expression. Immunofluorescent staining of wild type (A) and mutant (B) serum starved MEFs with acetylated α-tubulin (green, cilia) and Anks6 (red) shows complete absence of Anks6 protein from the mutant cilia. Scale bar, 5 mm. (C and D) IC is reduced in mutant cilia. Immunofluorescent staining of wild type (C) and mutant (C) MEFs with acetylated a-tubulin (red, cilia) and INVS (green) shows that mutant cilia have dramatically reduced INVS expression in the cilium. Scale bar, 5 mm. (E and F) IC localizes proximal to basal body. Immunofluorescent staining of wild type (E) and mutant (F) MEFs with INVS (red) and γ-tubulin (green, basal body) shows INVS localization next to basal body in the cilium. Scale bar, 5 mm. (G) Quantification of the INVS expression domain or IC in MEF cilia. A ratio of the IC length to ciliary length was measured on the basis of INVS immunostaining in MEFs (C and D). Differences in the IC length between wild type (n = 32) and mutant (n = 25) cilia were highly significant (0.65 ± 0.03 mm vs. 0.27 ± 0.02 mm, ****P < 0.0001). Two-tailed unpaired t-test was used for statistical analysis. Data are mean and SEM. (H and I) Serum starvation induces ciliation in BMEL cells. Antibody staining against acetylated a-tubulin and Anks6 demonstrates that BMEL cells grow a cilium (H), and Anks6 knockdown with shRNA effectively abolishes Anks6 expression from BMEL cilia (I). Scale bar, 5 mm.
Together, our data demonstrate that ANKS6 localizes to the cholangiocyte cilia in vivo, is required for proper cilia architecture and for the structural maintenance of the INVS compartment within the primary cilium. Nevertheless, the downstream effect of ANKS6’s absence from the cilium, as it relates to ANKS6’s role in biliary differentiation and morphogenesis remained uncovered.
ANKS6 binds to YAP1 and regulates its expression in the biliary epithelium
Hippo signaling pathway through its transcriptional effectors YAP1 and TAZ has recently emerged as a critical regulator of liver development and bile duct morphogenesis (33,57). Furthermore, it has been demonstrated that Hippo pathway components interact with ciliopathy proteins, providing an explanation for the phenotypic overlap of kidney and liver abnormalities between Hippo pathway deficient and ciliopathy disease models (38–40). We thus hypothesized that Anks6−/− liver phenotype may be caused by a dysregulation in YAP1 expression or activity in the mutant bile duct. To address this possibility, we first examined the expression of YAP1 in E18.5 mutant livers by immunofluorescence staining. We observed that YAP1 is expressed strongly in the nuclei of the wild-type bile duct epithelium marked by panCK staining (Fig. 8A and a′), whereas its expression was absent or very low in the mutant panCK+ biliary cells (Fig. 8B and B′), suggesting that ANKS6 regulates YAP1 expression in the developing biliary epithelium during cholangiocyte differentiation. Quantification of YAP1 immunofluorescent staining signal intensities confirmed a reduction in its expression levels in the Anks6−/− bile duct nuclei (Fig. 8C). To investigate the functional effect of reduced YAP1 expression in the mutant biliary epithelium, we stained livers with antibodies against the YAP1 transcriptional target CYR61 and quantified the signal intensities. Indeed, CYR61 expression levels were significantly reduced in the mutant bile duct cells compared with control livers (Fig. 8D–F), demonstrating that the loss of Anks6 leads to reduced YAP1 transcriptional activity in the biliary epithelial cells. To further address the relationship between ANKS6 and YAP1 in cholangiocyte differentiation, we knocked down Anks6 in BMEL and small mouse cholangiocyte cells (Smcc), which were originally isolated from the small bile ducts of BALB/c mice (58), using shRNA (BMEL) or CRISPR/Cas9 (Smcc) technology. Both methods effectively reduced the expression of Anks6 by 80% (shAnks6 BMEL) (Fig. 8G) or 100% (Smcc clones A6 1–18 and A6 2–15) (Fig. 8I) at mRNA level; Anks6 protein was undetectable by western blot analysis in both cases, whereas the control cells (BMEL, shNS or parental Smcc) demonstrated normal Anks6 expression levels (Fig. 8H and J). The inactivation of Anks6 significantly reduced the total YAP1 protein levels in BMEL (Fig. 8K) and Smcc cell lines (Fig. 8L and M), supporting our in vivo findings that ANKS6 regulates YAP1 expression in cholangiocytes during bile duct development. Densitometric analysis of YAP1 and serine-127 phosphorylated YAP1 western blot band intensities of the parental and Anks6 CRISPR knockout clones demonstrated that the loss of Anks6 causes increased YAP1 phosphorylation (Fig. 8N). Since YAP1 serine-127 phosphorylation leads to YAP1 nuclear exclusion and degradation via ubiquitination pathway, we expected to observe reduced expression of YAP1 target genes in Anks6 knockout cells. Indeed, the mRNA levels of two cholangiocyte-specific YAP1 targets, Ctgf and Cyr61, were significantly reduced in Anks6 knockdown cells (Fig. 8O and P).
Figure 8.
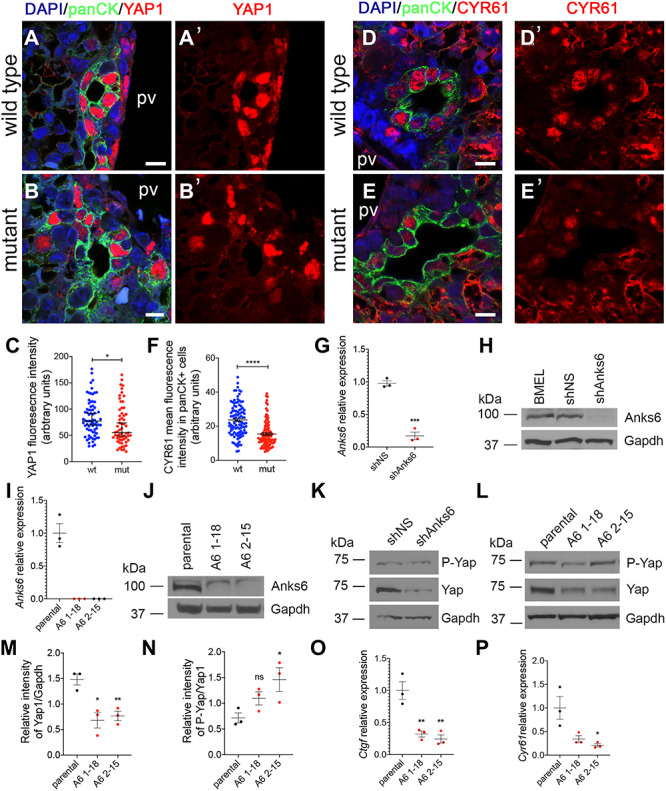
YAP1 expression is defective in ANKS6-deficient biliary epithelium. (A–B′) YAP1 expression is present in almost every cell in the wild-type bile duct lining epithelium (A and A′), whereas ANKS6 mutant bile ducts are mostly deficient in YAP1 expression (A and B′). panCK is used to mark the bile duct epithelium. Scale bar, 10 μm. (C) Quantification of YAP1 expression intensity in panCK-positive cells from wild type (n = 73 nuclei) and ANKS6 mutant (n = 60 nuclei) livers, as shown in (A) and (B). Data are median with 95% CI; mean of wild type—83.13 vs. mean of mutant—69.67, *P < 0.05. (D–E′) CYR61 expression is present in almost every cell in the wild-type bile duct lining epithelium (D and D′), whereas ANKS6 mutant bile ducts are mostly deficient in YAP1 expression (E and E′). panCK is used to mark the bile duct epithelium. Scale bar, 10 μm. (F) Quantification of CYR61 expression intensity in panCK-positive cells from wild type (n = 103 nuclei) and ANKS6 mutant (n = 102 nuclei) livers, as shown in (D) and (E). Data are median with 95% CI; mean of wild type—23.64 vs. mean of mutant—15.38, ****P < 0.0001. (G) qRT-PCR analysis of ANKS6 expression in shNS (control) and shANKS6 (ANKS6 knockdown) BMEL cells. ANKS6 expression is reduced by ~ 83% in shANKS6 BMEL cells. All experiments are n = 3. Two-tailed unpaired t-test was used for statistical analysis. Data are mean and SEM. ***P < 0.001. Mean of shNS—0.98 vs. mean of shANKS6–0.17. (H) Western blot analysis of ANKS6 protein levels in BMEL, shNS (control) and shANKS6 (ANKS6 knockdown) BMEL cells. ANKS6 protein levels are virtually undetectable in shAnks6 BMEL cells. (I) qRT-PCR analysis of ANKS6 expression in Smcc control (parental) and two different ANKS6 CRISPR knockout clones (A6 1–18 and A6 2–15). ANKS6 expression is completely absent in ANKS6 knockout cells. All experiments are n = 3. Two-tailed unpaired t-test was used for statistical analysis. Data are mean and SEM. *** P < 0.001. Mean of parental—1.00 vs. mean of A6 1–18—0.00 and mean of A6 2–15—0.00. (J) Western blot analysis of ANKS6 protein levels in Smcc (parental), ANKS6 knockout clone 1–18 and 2–15 cells. ANKS6 protein levels are undetectable in ANKS6 knockout cells. (k) YAP1 protein levels are reduced in shAnks6 BMEL cells compared with shNS control cells. GAPDH is a loading control. (L) YAP1 protein levels are reduced in 2 different Anks6 knockout clones 1–18 and 2–15 compared with parental Smcc cells. GAPDH is a loading control. (m) Densitometry of the data in (H). Intensity of YAP1 was normalized to GAPDH (n = 3). Error bars represent SEM. (N) Densitometry of the data in (H). Intensity of S127P-YAP1 was normalized to YAP1 (n = 3). Error bars represent SEM. (O) qRT-PCR analysis of Ctgf expression in parental Smcc and ANKS6 knockout clones 1–18 and 2–15. Ctgf expression is reduced in ANKS6-deficient cells. All experiments are n = 3. Two-tailed unpaired t-test was used for statistical analysis. Data are mean and SEM. **P < 0.01. (P) qRT-PCR analysis of Cyr61 expression in parental Smcc and ANKS6 knockout clones 1–18 and 2–15. Cyr61 expression is reduced in ANKS6-deficient cells. All experiments are n = 3. Two-tailed unpaired t-test was used for statistical analysis. Data are mean and SEM. **P < 0.01.
To measure YAP1 activity in Anks6 knockout cells, we transfected the cells with a TEAD-responsive element luciferase reporter (8xGTIIC-luc). In agreement with our western blot and qRT-PCR studies, we observed a significant downregulation of the reporter activity in Anks6 knockout clones compared with the parental cell line (Fig. 9A). It has been shown that two NPHP-proteins which interact with ANKS6—NPHP3 and NEK8/NPHP9, bind to the YAP1 paralogue—TAZ and modulate its transcriptional activity in a cell culture model (38). On the basis of these data, we wondered whether ANKS6 might regulate YAP1 stability or transcriptional activity through binding to YAP1 or the other Hippo effectors—TAZ and TEAD4. Indeed, we found that all three proteins co-immunoprecipitate with ANKS6 when overexpressed in HEK293 cells (Fig. 9B–D). Furthermore, the immunoprecipitation of endogenous ANKS6 from BMEL cell lysates co-precipitated endogenous YAP1 (Fig. 9E), demonstrating that ANKS6 and YAP1 proteins bind to each other under physiological conditions. In contrast, normal IgG failed to immunoprecipitate YAP1 (Fig. 9E), validating the specificity of ANKS6 and YAP1 interaction. Since TEAD4 is a nuclear protein, we next asked if ANKS6 localizes to the nucleus by performing a subcellular fractionation in BMEL cells. Indeed, we found that a fraction of ANKS6 protein localized to the nucleus (Fig. 9F), suggesting that ANKS6 may regulate YAP1/TAZ transcriptional activity by interacting with the YAP1/TAZ/TEAD transcriptional complex in the nucleus.
Figure 9.
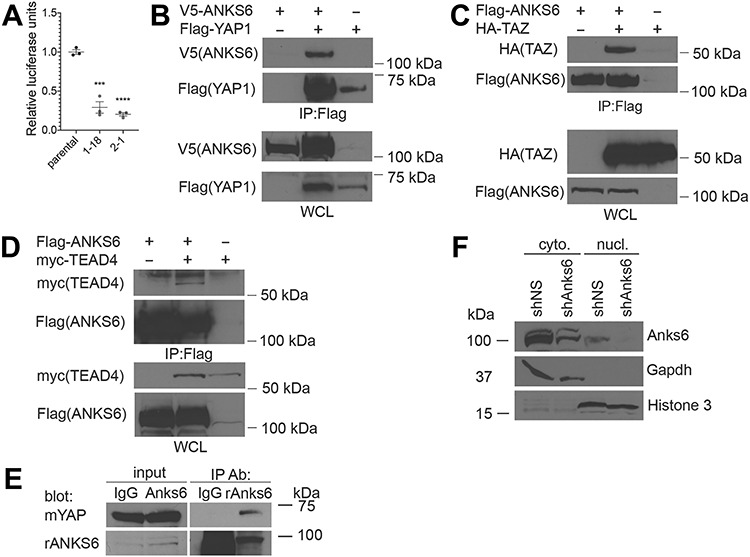
ANKS6 binds to YAP and TAZ in vitro. (A) ANKS6 promotes YAP1 transcriptional activity. Control and ANKS6-deficient Smcc clones 1–18, and 2–15 were cotransfected with 8xGTIIC-Luc reporter and Renilla luciferase control plasmid for luciferase activity. Reporter luciferase activity was normalized to Renilla luciferase activity. (B) ANKS6 binds to YAP1. V5-ANKS6 and Flag-YAP1 were overexpressed in HEK293 cells, and V5-ANKS6 was co-immunoprecipitated using Flag-beads. IP, antibody used for immunoprecipitation; WCL, whole cell lysate. (C) ANKS6 binds to TAZ. Flag-ANKS6 and HA-TAZ were overexpressed in HEK293 cells, and HA-TAZ was co-immunoprecipitated using Flag-beads. IP, antibody used for immunoprecipitation; WCL, whole cell lysate. (D) ANKS6 binds to TEAD4. Flag-ANKS6 and myc-TEAD4 were overexpressed in HEK293 cells, and myc-TEAD4 was co-immunoprecipitated using Flag-beads. IP, antibody used for immunoprecipitation; WCL, whole cell lysate. (E) Coimmunoprecipitation of endogenous ANKS6 and YAP1. Proteins were extracted from cultured bipotential cells (BMEL) and immunoprecipitated (IP) with anti-ANKS6 or IgG control antibodies, and analyzed by western blot with mouse anti-YAP or rabbit anti-ANKS6 antibodies. (F) ANKS6 localizes to the nucleus. Cell fractionation analysis of shNS and shANKS6 BMEL cells demonstrates that ANKS6 protein localizes to the nucleus. GAPDH is expressed in the cytoplasmic fraction. Cyto, cytoplasmic fraction; nucl, nuclear fraction.
Together, our in vivo and in vitro data reveal that Anks6 is required for YAP1 transcriptional activity in the biliary epithelium and hepatoblast precursor cells, probably by increasing YAP1 protein stability. We further demonstrate that ANKS6 promotes the expression of YAP1 target genes through its association with YAP1 and TEAD, thus providing a mechanism of pathogenicity for the loss of ANKS6 in the liver.
Discussion
Anks6 deficiency was first shown to lead to hepato-renal tubular pathology when a missense mutation in the conserved ANKS6 sterile alpha domain was identified as causing renal cyst formation and infrequent hepatic abnormalities in aged (21 months) Han:SPRD (cy/+) rat model of polycystic kidney disease (44,59). Since then, two other mouse models generated by using ENU mutagenesis have been described, one harboring I747N mutation in the Anks6 sterile alpha domain (43) and the other M187K mutation in an ANK-domain of the Anks6Streaker mouse (42). The analysis of the pathogenicity of these alleles has underscored the critical requirement of ANKS6 in the kidney, which is invariably affected in all Anks6-murine models (42–44) and humans (37,41,49). Less was known about the role of ANKS6 in other organs that are affected in patients with ANKS6-mutations (37). Here we showed that Anks6 mutant mice closely recapitulate the spectrum of phenotypes observed in ANKS6-deficient individuals, including congenital liver abnormalities that have not been examined in other Anks6-mouse models, thus providing us new insights into the molecular pathways in which ANKS6 functions during development. In addition, our analysis revealed that whole body deletion of Anks6 in a mouse causes a wide spectrum of severe congenital heart deficiencies (CHD), which together led to perinatal lethality regardless of the body situs. This agrees well with and extends the findings of Czarnecki et al. (42), who diagnosed a spectrum of complex CHD in heterotaxic Anks6Streaker mice, who succumbed to perinatal lethality; whereas mice with situs solitus or situs inversus displayed normal cardiovascular anatomy, and survived postnatally (42). Moreover, the comparison of the phenotypic severity between different Anks6 mouse models (42,43) and ANKS6-patients with different mutations (37,41,49) suggests a genotype–phenotype correlation, in which the presence of 2 loss-of-function alleles lead to a wider spectrum of organ involvement with more severe phenotypes than 2 hypomorphic alleles with missense mutations.
Our observations in Anks6 mutant mice and in cell culture studies demonstrate that ANKS6 is a critical regulator of cholangiocyte differentiation and bile duct morphogenesis in the liver. Our data support earlier reports that ANKS6 localizes to the IC in the cilium (37,60) and further reveal that ANKS6 is essential for the integrity of the IC signaling module. Its absence abolishes Inversin localization to this subciliary structure and causes ultrastructural deformities in the ciliary axoneme. Although, not essential for cilia growth, ANKS6 appears to regulate cilia formation, since its absence leads to shorter or duplex cilia in cholangiocytes. The mechanisms of these abnormalities were not studied in this manuscript. ANKS6 expression and primary cilium coincide in postnatal liver in the biliary epithelium, demonstrating that ANKS6 cellular function is relevant for the morphogenesis of this tissue. Consistent with this idea, the primary abnormalities in the mutant mice were confined to the portal region, i.e. failure to induce cholangiocyte differentiation and defective remodeling of the ductal plates into functional bile ducts. In addition, our histologic analysis revealed a dramatic reduction in the thickness of the periportal mesenchyme, and lack of peribiliary fibroblasts around the bile ducts in Anks6 mutant livers, suggesting that impaired cross talk between the portal mesenchyme and biliary epithelium may underlie the portal morphogenesis defects, as observed in mouse models deficient in Notch signaling (61–63).
Cholangiocyte lineage marker analysis in Anks6 mutant livers suggests that although ANKS6 function is not essential for the induction of cholangiocyte-fate in nascent hepatoblasts, it is critical for maintaining the activities of the signaling pathways that regulate hepatoblast differentiation into cholangiocytes. In the absence of Anks6 expression, hepatoblasts do activate SOX9- and HNF1b-controlled transcriptional pathways but fail to maintain YAP1 transcriptional activity, resulting in global ductal plate abnormalities and failure to acquire definitive epithelial characteristics, including the establishment of apical polarity and formation of primary cilia in the cells lining the bile duct. Importantly, the liver developmental defects observed in Anks6 mutant livers are similar to the defects described in Yap1 mutant livers including bile duct differentiation defects (33) and progressive liver fibrosis (33). ANKS6 role in positively modulating YAP1 transcriptional activity was also demonstrated by functional assays and gene expression studies in BMEL and Smcc cells, in which Anks6 knockout invariably led to reduced YAP1 protein levels and target gene expression. Moreover, our biochemical studies demonstrated that ANKS6 binds to the Hippo downstream effectors YAP1, TAZ and TEAD4 in an overexpression system and to YAP1 under physiological conditions, providing a mechanism by which ANKS6 modulates YAP1 activity in the hepatoblasts during ductal plate formation and later in biliary morphogenesis and cholangiocyte differentiation.
It has been reported that the expression of β-catenin, a transcriptional effector of canonical Wnt signaling, was upregulated in the tubules of ANKS6-deficient kidneys, suggesting a pathological role for Wnt signaling in ANKS6-patients (49). However, our analyses of the expression levels of canonical (Axin2, c-Myc and Ccnd1), and non-canonical (Wnt5a, Dkk1) Wnt signaling targets (64) did not reveal significant changes in Anks6−/− livers at E18.5, a timepoint when morphological and molecular changes were discernible. This suggests that Wnt signaling does not play an important role in the pathogenesis of Anks6-deficient livers. Moreover, the biliary abnormalities caused by dysregulated canonical (β-catenin inactivation or overexpression) (65), or non-canonical Wnt signaling (Wnt5a inactivation) (66) are dissimilar from the DPMs observed in Anks6−/− livers.
Overall, our work demonstrates that the loss of Anks6 causes CHF with associated DPM via dysregulation of the Hippo signaling in the portal tract. Our findings provide further mechanistic understandings to the regulation of normal bile duct development and reveal a novel role for the ciliary protein Anks6 in integrating Hippo signaling in this process.
Materials and Methods
Mouse breeding and maintenance
Targeted Anks6 embryonic stem cell clone EPD0109_5_E04 was obtained from the Knockout Mouse Project Repository. Chimeric mice were prepared by blastocyst microinjection and bred with C57BL/6J mice to obtain germline transmission. Genotyping primers are shown on Supplementary Material, Figure S1. Anks6 wild-type littermates were used as controls for mutant mice. For timed mating, noon on the day a plug was found was designated as E0.5. The person who genotyped the embryos or new-born animals was not involved in collecting the material. FLPo-deleter mouse strain (B6.Cg-Tg(Pgk1-flpo)10Sykr/J, Stock #011065) and EIIa-cre mouse strain (B6.FVB-Tg(EIIa-cre)C5379Lmgd/J, Stock #003724) were obtained from the Jackson Laboratory. The experimental protocol was reviewed and approved by the Animal Care Committees of Boston Children’s Hospital and the University of Pittsburgh.
Immunohistochemistry and antibodies
E10.5 embryos were fixed in 4% (w/v) paraformaldehyde (PFA) in phosphate-buffered saline (PBS) at 4°C. Embryos were then immersed in 15% and 30% sucrose and embedded in Tissue Freezing Medium (Triangle Biomedical Sciences, Inc.). Sections were taken at 8 μm. For immunostaining sections were blocked in 10% donkey serum/1% BSA and permeabilized in 0.1% Tween-20. Primary antibodies are listed in Supplementary Material, Table S1. Secondary antibodies were donkey anti-mouse Alexa Fluor 488, donkey anti-mouse Alexa Fluor 594, donkey anti-rabbit Alexa Fluor 488, donkey anti-rabbit Alexa Fluor 594 (Molecular Probes), and goat anti-chicken Alexa Fluor 488 (Molecular Probes). Samples were mounted in ProlongGold (molecular probes) and images captured on a Leica TSC 5SP X confocal microscope (Leica Microsystems).
Cell culture and transfections
HEK293 (ATC; CRL-1573) cells were cultured in DMEM (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS, L-glutamine (Gibco, Thermo Fisher Scientific) and 100 U/ml penicillin–streptomycin (Gibco, Thermo Fisher Scientific). Plasmid transfections were done using Lipofectamine 3000 (Life Technologies, Thermo Fisher Scientific) according to the manufacturers’ instructions or with PEI (HEK293 cells). BMEL cells (56) were cultured on collagen I (Fischer) coated dishes using RPMI 1640 (invitrogen) supplemented with 10% fetal bovine serum (Atlas Biologicals), 50 ng/ml Epidermal Growth Factor (PeproTech), 30 ng/ml Insulin-like Growth Factor II (PeproTech), 10 μg/ml human insulin (Sigma-Aldrich) and 100 U/ml penicillin–streptomycin (Gibco, Thermo Fisher Scientific). BMEL cells were passaged every 3–4 days and never let sit at confluence. MEF were established from wild-type and Anks6−/− E13.5 embryos and cultured in DMEM with 10% FBS L-glutamine (Gibco, Thermo Fisher Scientific), and 100 U/ml penicillin–streptomycin (Gibco, Thermo Fisher Scientific). Cell lines were tested for mycoplasma contamination on a quarterly basis.
shRNA and CRISPR/Cas9-driven genome editing
Anks6 mRNA level was stably knocked down in BMEL cells using a pLKO (MISSION® shRNA) lentivirus (Cat. # TRCN0000267514, Sigma-Aldrich). MISSION® pLKO.1-puro (SHC016-1EA, Sigma) was used as a non-targeting shRNA control plasmid. BMELs were transduced with shRNA-expressing lentiviruses and selected for 1 wk with 2 μg/ml puromycin. sgRNAs targeting mouse Anks6 were designed using the CRISPRtool (http://crispr.mit.edu). The annealed oligos of sgRNA guide sequence were cloned into the plasmid pSpCas9(BB)-2A-GFP (px458), a gift from Dr Feng Zhang (Addgene plasmid #48138) (67). The sequence of the constructs was verified by Sanger sequencing. CRISPR single cell clones were obtained by transient transfection using Lipofectamine 3000 (Thermo Fisher Scientific). Transfected single cells, positive for green fluorescent protein, were sorted 48 h after transfection using a FACS ARIA II cell sorter (BD Biosciences). Isolated clones were screened for CRISPR-mediated genome editing at the target region by sequencing of genomic DNA. The loss of the corresponding mRNA transcripts or protein level was used to validate knockout. Sequences of shRNAs and sgRNAs are listed in Supplementary Material, Table S2.
Histological analyses
Tissues were fixed in 4% (w/v) PFA in PBS at 4°C. All tissues were then dehydrated through a methanol series and embedded in paraffin. Sections were taken at 5 μm. Hematoxylin and eosin staining followed standard protocols. Phenotyping in mouse embryos was performed blinded. More than 5 regions were selected at random and analyzed using Adobe Photoshop.
Scanning electron microscopy
For SEM, livers were perfused with a mixture of 4% PFA and 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 5–10 min, cut into small pieces (2–4 mm3), and then immersed in 2% phosphate-buffered glutaraldehyde for overnight. Then, liver was rinsed with phosphate buffer three times, and postfixed in 1% osmium tetroxide for 1 h on ice. After fixation, the samples were rinsed in distilled water, dehydrated in serial ethanol, dried in a critical point dryer, sputter coated with gold–palladium and examined using a Hitachi 4700 SEM.
High-throughput MRI
E18.5 fixed embryos were soaked in 0.5% gadolinium (Multihance, gadobenate dimeglumine injection solution, 529 mg/ml, Bracco Diagnostics, Inc. Monroe Twp, NJ) in PBS at 4C° for 2 days before MRI. Multiple embryos were secured on a wooden popsicle stick with Webglue Surgical Adhesive (n-butyl cyanoacrylate, Patterson Veterinary, Devens, MA), then were immersed in Fomblin-Y perfluoropolyether oil (Aldrich) to reduce susceptibility artifact at the air-tissue interface. High-resolution MRI was performed on a Bruker BioSpec 70/30 USR spectrometer (Bruker BioSpin MRI, Billerica, MA, USA) operating at 7-Tesla field strength, equipped with an actively shielded gradient system and a quadrature radio-frequency volume coil with an inner-diameter of 35 mm. 3D high-resolution MRI was acquired with Rapid Imaging with Refocused Echoes (RARE) pulse sequence with the following parameters: field of view = 4.2 cm × 2.39 cm × 1.19 cm, matrix = 1024 × 500 × 256, voxel size = 41 μm × 46 μm × 46 μm, RARE factor = 8, echo time (TE) = 12.35 msec, effective TE = 24.69 msec, repetition time (TR) = 900 msec, flip angle (FA) = 180°.
In situ hybridization
ISH for Anks6 transcripts was performed on formalin-fixed liver using RNAscope (ACDBio, Hayward, CA) according to the manufacturer’s instructions and developed with the Diaminobenzidine substrate. After development, slides were counterstained with hematoxylin. Scoring was performed using the following criteria at ×40 magnification: 0, no staining, or <1 dot/10 cells; 1, 1–3 dots/cell; 2, 4–10 dots/cell, no or very few dot clusters; 3, > 10 dots/cell, <10% positive cells have dot clusters; 4, > 10 dots/cell, >10% positive cells have dot clusters.
Liver cell isolation and FACS
Liver cell isolation was performed via collagenase perfusion as previously described (50). Briefly, the livers of terminally anesthetized mice given ketamine/xylazine were perfused with Hepatocyte Perfusion Buffer followed by 0.5 mg/ml collagenase (Sigma-Aldrich) in Hepatocyte Digestion Buffer. To maximally liberate biliary cells and other non-parenchymal cells, collagenase-digested livers were subsequently subjected to serial digestions with Accutase (EMD Millipore, Billerica, MA) and Trypsin/0.1% EDTA for 20 min and 10 min, respectively. Cells were collected at each step. Cells were filtered through a 40 mm sieve, washed and stored in PBS with 2% FCS and penicillin/streptomycin before sorting. Biliary cells were enriched by staining and sorting for EpCAM (CD326, Cat#118205, 1:100. BioLegend) for 30 min at 4°C.
Luciferase assay
To measure the activity of YAP1 signaling, cells were seeded in 12-well plates. YAP1-luciferase reporter activity was measured from cells co-transfected with the following plasmids, pGL3b.8xGTIIC-Luc (Addgene plasmid #34615 (68)) and pTK-Renilla (Promega). Luciferase activity was measured with a dual-luciferase reporter assay kit (Promega) according to the manufacturer’s instructions. Renilla activity was used to normalize luciferase reporter activity.
RNA extraction and qRT–PCR
RNA was isolated from BMEL and Smcc cells using Quick-RNA Miniprep Plus (Zymo Research). Reverse transcription was performed using iScript cDNA Synthesis Kit (Bio-Rad). Quantitative real-time PCR was carried out using iTaq Universal SYBR Green Supermix (Bio-Rad) and run on a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad). Data were normalized to Gapdh or 18S as indicated. Real-time PCR primers are listed in Supplementary Material, Table SS2.
Co-immunoprecipitation Assay
A total of 36 h after transfection, HEK293 or BMEL cells were washed in ice-cold PBS and lysed in the IP Lysis Buffer (25 mM Tris–HCl, 400 mM NaCl, 1% NP-40). Cleared cell lysates were produced by centrifugation of the resulting samples at 16 000 × g for 30 min at 4°C and subjected to immunoprecipitation using anti-Flag M2 beads (Sigma) or GFP-nAb Magnetic beads (Allele Biotechnology). Typically, 1 mg of protein sample was incubated with 20 μl bed volume of anti-FLAG M2 or GFP-nAb Magnetic beads in a volume of 1 ml for 16 h at 4°C. Gel electrophoresis of immunoprecipitation eluents was performed using the NuPAGE system (invitrogen). Samples were resolved on 4–12% Bis-Tris gels in MOPS buffer, transferred to a nitrocellulose membrane which was then probed for the protein of interest using antibodies diluted in TBS containing 5% milk and 0.1% Tween-20 (Sigma).
Statistics
χ2 test was used to test whether the distribution of mouse genotypes corresponds to Mendelian ratio. t-test was used to compare data between two groups. Significance was determined at P < 0.05 and represented by * to denote P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. All experiments were carried out three times or more (n ≥ 3). Data were analyzed using Prism 6 software (GraphPad Software, San Diego, CA) and are given as the mean ± SEM.
Study approval
The procedures for all animal experiments were reviewed and approved by the IACUC of the University of Pittsburgh.
Supplementary Material
Acknowledgements
We thank Dr M.C. Weiss for BMEL cells and Dr Gianfranco Alpini for Smcc cells.
Conflict of Interest statement. F.H. is a cofounder of Goldfinch-Bio. No other authors have competing financial interests.
Contributor Information
Merlin Airik, Division of Nephrology, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, USA.
Markus Schüler, Division of Nephrology and Internal Intensive Care Medicine, Charite University, Berlin, Germany; Division of Nephrology, Boston Children’s Hospital, Boston, MA, USA.
Blake McCourt, Division of Nephrology, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, USA.
Anna-Carina Weiss, Department for Molecular Biology, Hannover, Germany.
Nathan Herdman, Division of Nephrology, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, USA.
Timo H Lüdtke, Department for Molecular Biology, Hannover, Germany.
Eugen Widmeier, Division of Nephrology, Boston Children’s Hospital, Boston, MA, USA; Department of Medicine, Renal Division, Medical Center - University of Freiburg, Freiburg, Germany.
Donna B Stolz, Department of Cell Biology, University of Pittsburgh, Pittsburgh, PA, USA.
Kari N Nejak-Bowen, Division of Experimental Pathology, Department of Pathology, University of Pittsburgh, Pittsburgh, PA, USA.
Dean Yimlamai, Division of Gastroenterology and Nutrition, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, USA.
Yijen L Wu, Department of Developmental Biology, University of Pittsburgh, Pittsburgh, PA, USA.
Andreas Kispert, Department for Molecular Biology, Hannover, Germany.
Rannar Airik, Division of Nephrology, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, USA; Department of Developmental Biology, University of Pittsburgh, Pittsburgh, PA, USA.
Friedhelm Hildebrandt, Division of Nephrology, Boston Children’s Hospital, Boston, MA, USA.
Data Availability
The authors declare that all other data supporting the findings of this study are available within the article and its Supplementary Information files, or from the corresponding author on request.
Funding
National Institutes of Health (DK099434, P30DK079307, DK115403 to R.A., DK068306 to F.H.). German National Academy of Sciences Leopoldina (LPDS-2015-07 to E.W.). German Research Foundation for the Cluster of Excellence REBIRTH (From Regenerative Biology to Reconstructive Therapy) at Hannover Medical School (A.K.).
Author Contributions
R.A., M.A., D.Y., M.S. and F.H. designed the experiments. R.A., M.S., M.A., A.C.W., N.H., E.W., T.H.L., D.B.S., B.M., K.N.N.B., and Y.L.W. performed genetic crosses, tissue sampling, and experiments. R.A., D.Y., M.S., M.A., A.C.W., A.K., Y.L.W., F.H. and E.W. interpreted the data. R.A., M.A., M.S., D.Y., A.K., and F.H. wrote and reviewed the manuscript. R.A. and F.H. supervised the project.
References
- 1. Gunay-Aygun M. (2009) Liver and kidney disease in ciliopathies. Am. J. Med. Genet. C Semin. Med. Genet., 151C, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Desmet V.J. (1998) Ludwig symposium on biliary disorders--part I. Pathogenesis of ductal plate abnormalities. Mayo Clin. Proc., 73, 80–89. [DOI] [PubMed] [Google Scholar]
- 3. Clotman F., Libbrecht L., Killingsworth M.C., Loo C.C., Roskams T. and Lemaigre F.P. (2008) Lack of cilia and differentiation defects in the liver of human foetuses with the Meckel syndrome. Liver Int., 28, 377–384. [DOI] [PubMed] [Google Scholar]
- 4. Parisi M.A., Doherty D., Chance P.F. and Glass I.A. (2007) Joubert syndrome (and related disorders) (OMIM 213300). Eur. J. Hum. Genet., 15, 511–521. [DOI] [PubMed] [Google Scholar]
- 5. Forsythe E. and Beales P.L. (2013) Bardet-Biedl syndrome. Eur. J. Hum. Genet., 21, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hildebrandt F., Benzing T. and Katsanis N. (2011) Ciliopathies. N. Engl. J. Med., 364, 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chetty-John S., Piwnica-Worms K., Bryant J., Bernardini I., Fischer R.E., Heller T., Gahl W.A. and Gunay-Aygun M. (2010) Fibrocystic disease of liver and pancreas; under-recognized features of the X-linked ciliopathy oral-facial-digital syndrome type 1 (OFD I). Am. J. Med. Genet. A, 152A, 2640–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bernstein J., Chandra M., Creswell J., Kahn E., Malouf N.N., McVicar M., Weinberg A.G. and Wybel R.E. (1987) Renal-hepatic-pancreatic dysplasia: a syndrome reconsidered. Am. J. Med. Genet., 26, 391–403. [DOI] [PubMed] [Google Scholar]
- 9. Brueton L.A., Dillon M.J. and Winter R.M. (1990) Ellis-van creveld syndrome, Jeune syndrome, and renal-hepatic-pancreatic dysplasia: separate entities or disease spectrum? J. Med. Genet., 27, 252–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guay-Woodford L.M., Galliani C.A., Musulman-Mroczek E., Spear G.S., Guillot A.P. and Bernstein J. (1998) Diffuse renal cystic disease in children: morphologic and genetic correlations. Pediatr. Nephrol., 12, 173–182. [DOI] [PubMed] [Google Scholar]
- 11. Sergi C., Adam S., Kahl P. and Otto H.F. (2000) Study of the malformation of ductal plate of the liver in Meckel syndrome and review of other syndromes presenting with this anomaly. Pediatr. Dev. Pathol., 3, 568–583. [DOI] [PubMed] [Google Scholar]
- 12. Masyuk A.I., Masyuk T.V., Splinter P.L., Huang B.Q., Stroope A.J. and LaRusso N.F. (2006) Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology, 131, 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Masyuk T.V., Huang B.Q., Ward C.J., Masyuk A.I., Yuan D., Splinter P.L., Punyashthiti R., Ritman E.L., Torres V.E., Harris P.C. et al. (2003) Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology, 125, 1303–1310. [DOI] [PubMed] [Google Scholar]
- 14. Masyuk A.I., Gradilone S.A., Banales J.M., Huang B.Q., Masyuk T.V., Lee S.O., Splinter P.L., Stroope A.J. and Larusso N.F. (2008) Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors. Am. J. Physiol. Gastrointest. Liver Physiol., 295, G725–G734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roelandt P., Antoniou A., Libbrecht L., Van Steenbergen W., Laleman W., Verslype C., Van der Merwe S., Nevens F., De Vos R., Fischer E. et al. (2012) HNF1B deficiency causes ciliary defects in human cholangiocytes. Hepatology, 56, 1178–1181. [DOI] [PubMed] [Google Scholar]
- 16. Lemaigre F.P. (2003) Development of the biliary tract. Mech. Dev., 120, 81–87. [DOI] [PubMed] [Google Scholar]
- 17. Boulter L., Lu W.Y. and Forbes S.J. (2013) Differentiation of progenitors in the liver: a matter of local choice. J. Clin. Invest., 123, 1867–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yimlamai D., Fowl B.H. and Camargo F.D. (2015) Emerging evidence on the role of the hippo/YAP pathway in liver physiology and cancer. J. Hepatol., 63, 1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flynn D.M., Nijjar S., Hubscher S.G., Goyet Jde V., Kelly D.A., Strain A.J. and Crosby H.A. (2004) The role of notch receptor expression in bile duct development and disease. J. Pathol., 204, 55–64. [DOI] [PubMed] [Google Scholar]
- 20. Geisler F., Nagl F., Mazur P.K., Lee M., Zimber-Strobl U., Strobl L.J., Radtke F., Schmid R.M. and Siveke J.T. (2008) Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology, 48, 607–616. [DOI] [PubMed] [Google Scholar]
- 21. Zong Y., Panikkar A., Xu J., Antoniou A., Raynaud P., Lemaigre F. and Stanger B.Z. (2009) Notch signaling controls liver development by regulating biliary differentiation. Development, 136, 1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tchorz J.S., Kinter J., Muller M., Tornillo L., Heim M.H. and Bettler B. (2009) Notch2 signaling promotes biliary epithelial cell fate specification and tubulogenesis during bile duct development in mice. Hepatology, 50, 871–879. [DOI] [PubMed] [Google Scholar]
- 23. Emerick K.M., Rand E.B., Goldmuntz E., Krantz I.D., Spinner N.B. and Piccoli D.A. (1999) Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology, 29, 822–829. [DOI] [PubMed] [Google Scholar]
- 24. Li L., Krantz I.D., Deng Y., Genin A., Banta A.B., Collins C.C., Qi M., Trask B.J., Kuo W.L., Cochran J. et al. (1997) Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet., 16, 243–251. [DOI] [PubMed] [Google Scholar]
- 25. McDaniell R., Warthen D.M., Sanchez-Lara P.A., Pai A., Krantz I.D., Piccoli D.A. and Spinner N.B. (2006) NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the NOTCH signaling pathway. Am. J. Hum. Genet., 79, 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oda T., Elkahloun A.G., Pike B.L., Okajima K., Krantz I.D., Genin A., Piccoli D.A., Meltzer P.S., Spinner N.B., Collins F.S. et al. (1997) Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat. Genet., 16, 235–242. [DOI] [PubMed] [Google Scholar]
- 27. Yu F.X. and Guan K.L. (2013) The hippo pathway: regulators and regulations. Genes Dev., 27, 355–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu F.X., Zhao B. and Guan K.L. (2015) Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell, 163, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vassilev A., Kaneko K.J., Shu H., Zhao Y. and DePamphilis M.L.T.E.A.D. (2001) TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev., 15, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ota M. and Sasaki H. (2008) Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development, 135, 4059–4069. [DOI] [PubMed] [Google Scholar]
- 31. Imajo M., Ebisuya M. and Nishida E. (2015) Dual role of YAP and TAZ in renewal of the intestinal epithelium. Nat. Cell Biol., 17, 7–19. [DOI] [PubMed] [Google Scholar]
- 32. Rayon T., Menchero S., Nieto A., Xenopoulos P., Crespo M., Cockburn K., Cañon S., Sasaki H., Hadjantonakis A.K., Pompa J.L. et al. (2014) Notch and hippo converge on Cdx2 to specify the trophectoderm lineage in the mouse blastocyst. Dev. Cell, 30, 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang N., Bai H., David K.K., Dong J., Zheng Y., Cai J., Giovannini M., Liu P., Anders R.A. and Pan D. (2010) The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev. Cell, 19, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Otto E.A., Schermer B., Obara T., O'Toole J.F., Hiller K.S., Mueller A.M., Ruf R.G., Hoefele J., Beekmann F., Landau D. et al. (2003) Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet., 34, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Olbrich H., Fliegauf M., Hoefele J., Kispert A., Otto E., Volz A., Wolf M.T., Sasmaz G., Trauer U., Reinhardt R. et al. (2003) Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet., 34, 455–459. [DOI] [PubMed] [Google Scholar]
- 36. Calinescu-Tuleasca A.M., Bottani A., Rougemont A.L., Birraux J., Gubler M.C., Le Coultre C., Majno P., Mentha G., Girardin E., Belli D. et al. (2013) Caroli disease, bilateral diffuse cystic renal dysplasia, situs inversus, postaxial polydactyly, and preauricular fistulas: a ciliopathy caused by a homozygous NPHP3 mutation. Eur. J. Pediatr., 172, 877–881. [DOI] [PubMed] [Google Scholar]
- 37. Hoff S., Halbritter J., Epting D., Frank V., Nguyen T.M., Reeuwijk J., Boehlke C., Schell C., Yasunaga T., Helmstädter M. et al. (2013) ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet., 45, 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Habbig S., Bartram M.P., Sägmüller J.G., Griessmann A., Franke M., Müller R.U., Schwarz R., Hoehne M., Bergmann C., Tessmer C. et al. (2012) The ciliopathy disease protein NPHP9 promotes nuclear delivery and activation of the oncogenic transcriptional regulator TAZ. Hum. Mol. Genet., 21, 5528–5538. [DOI] [PubMed] [Google Scholar]
- 39. Frank V., Habbig S., Bartram M.P., Eisenberger T., Veenstra-Knol H.E., Decker C., Boorsma R.A., Göbel H., Nürnberg G., Griessmann A. et al. (2013) Mutations in NEK8 link multiple organ dysplasia with altered hippo signalling and increased c-MYC expression. Hum. Mol. Genet., 22, 2177–2185. [DOI] [PubMed] [Google Scholar]
- 40. Habbig S., Bartram M.P., Müller R.U., Schwarz R., Andriopoulos N., Chen S., Sägmüller J.G., Hoehne M., Burst V., Liebau M.C. et al. (2011) NPHP4, a cilia-associated protein, negatively regulates the hippo pathway. J. Cell Biol., 193, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kulkarni S., Abro B., Duque Lasio M.L., Stoll J., Grange D.K. and Clinical H.M. (2020) Pathological features of a newborn with compound heterozygous ANKS6 variants. Pediatr. Dev. Pathol., 23, 235–239. [DOI] [PubMed] [Google Scholar]
- 42. Czarnecki P.G., Gabriel G.C., Manning D.K., Sergeev M., Lemke K., Klena N.T., Liu X., Chen Y., Li Y., San Agustin J.T. et al. (2015) ANKS6 is the critical activator of NEK8 kinase in embryonic situs determination and organ patterning. Nat. Commun., 6, 6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bakey Z., Bihoreau M.T., Piedagnel R., Delestré L., Arnould C., Ad D.V., Devuyst O., Hoffmann S., Ronco P., Gauguier D. et al. (2015) The SAM domain of ANKS6 has different interacting partners and mutations can induce different cystic phenotypes. Kidney Int., 88, 299–310. [DOI] [PubMed] [Google Scholar]
- 44. Brown J.H., Bihoreau M.T., Hoffmann S., Kränzlin B., Tychinskaya I., Obermüller N., Podlich D., Boehn S.N., Kaisaki P.J., Megel N. et al. (2005) Missense mutation in sterile alpha motif of novel protein SamCystin is associated with polycystic kidney disease in (cy/+) rat. J. Am. Soc. Nephrol., 16, 3517–3526. [DOI] [PubMed] [Google Scholar]
- 45. Neudecker S., Walz R., Menon K., Maier E., Bihoreau M.T., Obermüller N., Kränzlin B., Gretz N. and Hoffmann S.C. (2010) Transgenic overexpression of Anks6(p.R823W) causes polycystic kidney disease in rats. Am. J. Pathol., 177, 3000–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nagao S., Morita M., Kugita M., Yoshihara D., Yamaguchi T., Kurahashi H., Calvet J.P. and Wallace D.P. (2010) Polycystic kidney disease in Han:SPRD Cy rats is associated with elevated expression and mislocalization of SamCystin. Am. J. Physiol. Renal Physiol., 299, F1078–F1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Skarnes W.C., Rosen B., West A.P., Koutsourakis M., Bushell W., Iyer V., Mujica A.O., Thomas M., Harrow J., Cox T. et al. (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature, 474, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu Y., Wang C., Sun H., LeRoith D. and Yakar S. (2009) High-efficient FLPo deleter mice in C57BL/6J background. PLoS One, 4, e8054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Taskiran E.Z., Korkmaz E., Gucer S., Kosukcu C., Kaymaz F., Koyunlar C., Bryda E.C., Chaki M., Lu D., Vadnagara K. et al. (2014) Mutations in ANKS6 cause a nephronophthisis-like phenotype with ESRD. J. Am. Soc. Nephrol., 25, 1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yimlamai D., Christodoulou C., Galli G.G., Yanger K., Pepe-Mooney B., Gurung B., Shrestha K., Cahan P., Stanger B.Z. and Camargo F.D. (2014) Hippo pathway activity influences liver cell fate. Cell, 157, 1324–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Antoniou A., Raynaud P., Cordi S., Zong Y., Tronche F., Stanger B.Z., Jacquemin P., Pierreux C.E., Clotman F. and Lemaigre F.P. (2009) Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology, 136, 2325–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coffinier C., Gresh L., Fiette L., Tronche F., Schütz G., Babinet C., Pontoglio M., Yaniv M. and Barra J. (2002) Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development, 129, 1829–1838. [DOI] [PubMed] [Google Scholar]
- 53. Delestre L., Bakey Z., Prado C., Hoffmann S., Bihoreau M.T., Lelongt B. and Gauguier D. (2015) ANKS3 co-localises with ANKS6 in mouse renal cilia and is associated with vasopressin signaling and apoptosis in vivo in mice. PLoS One, 10, e0136781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yakulov T.A., Yasunaga T., Ramachandran H., Engel C., Müller B., Hoff S., Dengjel J., Lienkamp S.S. and Walz G. (2015) Anks3 interacts with nephronophthisis proteins and is required for normal renal development. Kidney Int., 87, 1191–1200. [DOI] [PubMed] [Google Scholar]
- 55. Huang B.Q., Masyuk T.V., Muff M.A., Tietz P.S., Masyuk A.I. and Larusso N.F. (2006) Isolation and characterization of cholangiocyte primary cilia. Am. J. Physiol. Gastrointest. Liver Physiol., 291, G500–G509. [DOI] [PubMed] [Google Scholar]
- 56. Strick-Marchand H. and Weiss M.C. (2002) Inducible differentiation and morphogenesis of bipotential liver cell lines from wild-type mouse embryos. Hepatology, 36, 794–804. [DOI] [PubMed] [Google Scholar]
- 57. Lee D.H., Park J.O., Kim T.S., Kim S.K., Kim T.H., Kim M.C., Park G.S., Kim J.H., Kuninaka S., Olson E.N. et al. (2016) LATS-YAP/TAZ controls lineage specification by regulating TGFbeta signaling and Hnf4alpha expression during liver development. Nat. Commun., 7, 11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ueno Y., Alpini G., Yahagi K., Kanno N., Moritoki Y., Fukushima K., Glaser S., LeSage G. and Shimosegawa T. (2003) Evaluation of differential gene expression by microarray analysis in small and large cholangiocytes isolated from normal mice. Liver Int., 23, 449–459. [DOI] [PubMed] [Google Scholar]
- 59. Kranzlin B., Schieren G. and Gretz N. (1997) Azotemia and extrarenal manifestations in old female Han:SPRD (cy/+) rats. Kidney Int., 51, 1160–1169. [DOI] [PubMed] [Google Scholar]
- 60. Nakajima Y., Kiyonari H., Mukumoto Y. and Yokoyama T. (2018) The Inv compartment of renal cilia is an intraciliary signal-activating center to phosphorylate ANKS6. Kidney Int., 93, 1108–1117. [DOI] [PubMed] [Google Scholar]
- 61. Kodama Y., Hijikata M., Kageyama R., Shimotohno K. and Chiba T. (2004) The role of notch signaling in the development of intrahepatic bile ducts. Gastroenterology, 127, 1775–1786. [DOI] [PubMed] [Google Scholar]
- 62. Tanimizu N. and Miyajima A. (2004) Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors. J. Cell Sci., 117, 3165–3174. [DOI] [PubMed] [Google Scholar]
- 63. Zhao R. and Duncan S.A. (2005) Embryonic development of the liver. Hepatology, 41, 956–967. [DOI] [PubMed] [Google Scholar]
- 64. Park H.W., Kim Y.C., Yu B., Moroishi T., Mo J.S., Plouffe S.W., Meng Z., Lin K.C., Yu F.X., Alexander C.M., Wang C.Y. et al. (2015) Alternative Wnt signaling activates YAP/TAZ. Cell, 162, 780–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cordi S., Godard C., Saandi T., Jacquemin P., Monga S.P., Sabine Colnot S. and Lemaigre F.P. (2016) Role of beta-catenin in development of bile ducts. Differentiation, 91, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kiyohashi K., Kakinuma S., Kamiya A., Sakamoto N., Nitta S., Yamanaka H., Yoshino K., Fujiki J., Murakawa M., Kusano-Kitazume A. et al. (2013) Wnt5a signaling mediates biliary differentiation of fetal hepatic stem/progenitor cells in mice. Hepatology, 57, 2502–2513. [DOI] [PubMed] [Google Scholar]
- 67. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A. and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc., 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., Elvassore N. et al. (2011) Role of YAP/TAZ in mechanotransduction. Nature, 474, 179–183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all other data supporting the findings of this study are available within the article and its Supplementary Information files, or from the corresponding author on request.