

Abstract
As tetrazines are important tools to the field of bioorthogonal chemistry, there is a need for new approaches to synthesize unsymmetrical and 3-monosubstituted tetrazines. Described here is a general, one-pot method for converting (3-methyloxetan-3-yl)methyl carboxylic esters into 3-thiomethyltetrazines. These versatile intermediates were applied as a platform for the synthesis of unsymmetrical tetrazines via Pd-catalyzed cross-coupling and in the first example of catalytic thioether reduction to access monosubstituted tetrazines. The method enables the development of new tetrazines possessing a favorable combination of kinetics, small size and hydrophilicity. The chemistry was applied to a broad range of aliphatic and aromatic ester precursors and to the synthesis of heterocycles including BODIPY fluorophores and biotin. In addition, a series of tetrazine probes for monoacylglycerol lipase (MAGL) were synthesized and the most reactive one was applied in labeling of endogenous MAGL in live cells.
Keywords: tetrazine, bioorthogonal chemistry, minimal probe, divergent synthesis, orthoester
Graphical Abstract
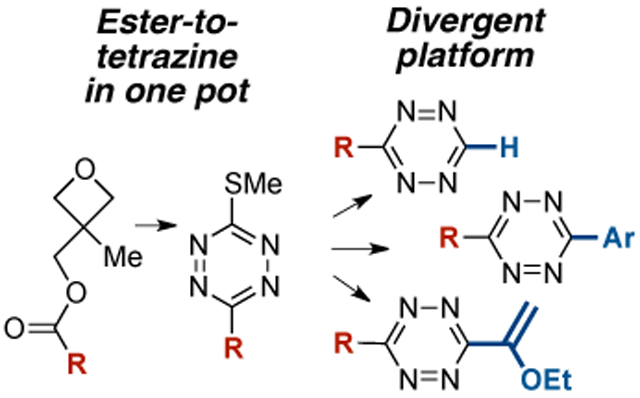
Divergent Tetrazine Synthesis: Described is a new strategy for the one-pot synthesis of 3-thiomethyltetrazines from carboxylic ester precursors, providing a platform for the synthesis of unsymmetrical tetrazines via Pd-catalyzed cross-coupling and the first catalytic thioether reduction to access monosubstituted tetrazines.
Introduction
Bioorthogonal reactions have become increasingly important to chemistry and biology since the term was initially coined early in this century.[1] The bioorthogonal reactions of tetrazines have become widely used in various fields,[2] including natural product synthesis,[3] cargo delivery,[4] genetic code expansion,[5] fluorogenic labeling,[6] radiochemistry,[7] coordination chemistry,[8] and material science.[9] Coupled to the growing utility of tetrazinebased bioorthogonal chemistry is the need for mild, safe and general methods of introducing tetrazine groups to complex molecules. Carboxylic esters are ubiquitous functional groups that have been used as handles for the introduction of preformed tetrazine groups via amide bond forming reactions.[10] For this approach, linkers are required which can add bulk and hydrophilicity to the conjugate.[11] The direct preparation of tetrazines from carboxylic precursors could greatly expand access to functionalized derivatives. Described here is a new strategy for the one-pot synthesis of 3-thiomethyltetrazines from carboxylic esters of commercially available 3-methyl-3-oxetanemethanol. The compounds serve as versatile intermediates for the divergent synthesis of a broad range of functionalized tetrazines via Pd-catalyzed cross-coupling and in the first example of catalytic thioether reduction to form monosubstituted tetrazines (Figure 1). This ‘carboxylate-totetrazine’ strategy complements known approaches of tetrazine construction,[12] tolerates a range of heterocycles and functional groups, and generates classes of tetrazines that are most useful to bioorthogonal chemistry.
Figure 1.

Direct approach for the introduction of tetrazine functionality from carboxylic ester precursors.
The most widely used method of preparing tetrazines involves condensation of nitriles and hydrazine followed by oxidation (Figure 2A).[13] Devaraj’s method for Zn(OTf)2 or Ni(OTf)2 catalyzed condensation of nitriles in neat anhydrous hydrazine has greatly improved access to unsymmetrical tetrazines.[13d] More recently, thiols have been used as organocatalysts that function with hydrazine hydrate which is safer and more broadly available than anhydrous hydrazine.[13e] Mono-functional tetrazines have been prepared from nitriles, hydrazine and formamidine acetate under Lewis acid catalyzed conditions[13d, 13f, 13g] or with dichloromethane under sulfur-catalyzed conditions (Figure 2B).[14] Safety considerations for these procedures include the formation of volatile tetrazine byproducts with high nitrogen content and the direct addition of an oxidant to reaction mixtures containing hydrazine. Tetrazines have been used in metal catalyzed Heck-type reactions,[6c] CH activations[15] and crosscouplings.[6d, 6e, 16] 3,6-Dichlorotetrazine,[2, 17] 3,6-bis(3,5-dimethylpyrazol-l-yl) tetrazine[18], and 3-bromotetrazine[19] have been used to create thiol, alcohol and amine functionalized tetrazines by nucleophilic aromatic substitution. While these methods for tetrazine synthesis have been enabling, there is a continuing need to broaden the types of precursors that can serve for the direct installation of tetrazines with improved functional group tolerance.
Figure 2.

Selected methods of tetrazine synthesis
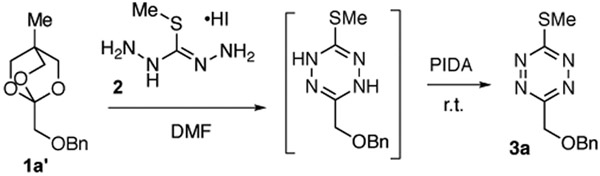
Carboxylic esters have played only a limited role as precursors to tetrazines.[12] Unsymmetrical 3-thiomethyltetrazines were synthesized in low yield through the condensation of Smethylisothiocarbonohydrazidium iodide (2) with either trimethylorthoacetate or trimethylorthoformate.[20] Recently, our group described a procedure for the conversion of trimethylorthoacetate to a 3-methyl-6-thioalkyltetrazine that can in turn be used for the installation of minimal tetrazines through the silvermediated Liebeskind-Srogl coupling with arylboronic acids.[21] This method was limited to trimethylorthoacetate and triethylorthobenzoate, and we hoped to develop a general approach for directly converting ester precursors into tetrazines. However, trimethyl- and triethylorthoesters cannot be prepared directly from esters but instead require nitrile precursors and harshly acidic conditions.[22] Thus, our previous method did not enable the use of carboxylic acids or esters as precursors to tetrazines. Here, we describe a one-pot procedure for the conversion of (3-methyloxetan-3-yl)methyl carboxylic esters (1) to unsymmetrical 3-thiomethyltetrazines (3) via condensation of oxabicyclo[2.2.2] octyl (OBO) orthoester[23] intermediates with 2 and subsequent oxidation (Figure 2C). The activated esters 3 can be prepared simply and in high yield from the corresponding carboxylic acids and inexpensive 3-methyl-3-oxetanemethanol. These compounds provide a divergent platform for 3,6-disubstituted tetrazine synthesis and a new approach to monosubstituted tetrazines (Figure 2C).
Results and Discussion
Compound 2 is available commercially or can be prepared in one step from iodomethane and thiocarbohydrazide.[24] The differential scanning calorimetry (DSC) profile of 2 has an onset temperature of 135 °C and a transition enthalpy of 875 J/g (Figure S3). The OBO orthoester 1a’ was prepared by treating its (3-methyloxetan-3-yl) methyl ester with BF3·OEt2. Table 1 shows conditions for optimizing the condensation of 1a’ with 2, with subsequent oxidation by phenyliodosodiacetate (PIDA)[25] to provide thiomethyltetrazine 3a. DMF was the only effective solvent likely due to the limited solubility of salt 2 in other solvents. Simple combination of 1a’ and 2 at r.t. gave 3a in only 18% yield (Table 1, entry 1). Given the acid-lability of orthoesters, a number of basic additives were explored (entries 2-6). Pyridine was found to be uniquely effective, and further optimization showed that the temperature could be raised to 80 °C and reaction time shortened without sacrificing yield (entry 7). Using 3 equiv. of pyridine and increasing the concentration to 1 M had a significant impact and raised the yield to 54% (entry 8). Using 1.4 equiv. of 1a’ further raised the yield to 87% (entry 10).
Table 1.
Optimization of condensation between 1a’ and 2.
 | ||||||
|---|---|---|---|---|---|---|
| entry | additive | equiv of 1a' |
temp. | time | conc. | yield of 3a |
| 1 | none | 1.0 | r.t. | 6 h | 0.2 M | 18% |
| 2 | NEt3 (1 equiv) | 1.0 | r.t. | 6 h | 0.2 M | 0 |
| 3 | DMAP (1 equiv) | 1.0 | r.t. | 6 h | 0.2 M | 0 |
| 4 | Cs2CO3 (1 equiv) | 1.0 | r.t. | 7 h | 0.2 M | 0 |
| 5 | KOtBu (1 equiv) | 1.0 | r.t. | 6 h | 0.2 M | 0 |
| 6 | pyridine (1 equiv) | 1.0 | r.t. | 7 h | 0.2 M | 24% |
| 7 | pyridine (3 equiv) | 1.0 | 80 °C | 1 h | 0.2 M | 23% |
| 8 | pyridine (3 equiv) | 1.0 | 80 °C | 20 min | 1.0 M | 54% |
| 9 | pyridine (3 equiv) | 1.25 | 80 °C | 20 min | 1.0 M | 75% |
| 10 | pyridine (3 equiv) | 1.4 | 80 °C | 20 min | 1.0 M | 87% |
We next developed a one-pot method for the synthesis of 3-thiomethyltetrazines 3 from esters 1, which can be easily prepared in high yield (>90%) and inexpensive 3-methyl-3-oxetanemethanol from the corresponding acids by Steglich esterification. After initial treatment of 1 in CH2Cl2 with BF3·OEt2 to form the OBO orthoester, pyridine and 2 were added. The solvent was exchanged with DMF, and after heating at 80 °C, the dihydrotetrazine solution was cooled and directly treated with PIDA to provide tetrazines 3 in 50–79% yield (Scheme 1). Aromatic and alkyl substituents with a variety of functional groups were tolerated including Boc-amino (3b-c, 3n), nitro (3d) and methoxy (3e) groups. The method also tolerates nitrile (3h) and ester (3g, 3k-m) groups which are generally not compatible with tetrazine synthesis. Protected amino acids (3o-p), biotin (3q), BODIPY-FL (3w) and a number of heterocycles (3i-j, 3s-v) were also successful and highlighted the ability to directly conjugate biologically relevant compounds. While the ester of 2-fluoro benzoic acid provides 3y in 50% yield, esters with bulkier ortho-substituents (2-nitro or N-Boc-2-amino) were unsuccessful.
Scheme 1.

One-pot synthesis of thiomethyltetrazines from oxetane esters.
Monosubstituted tetrazines are valued in bioorthogonal chemistry for their rapid kinetics and minimal size.[26] We sought to prepare monosubstituted tetrazines through thiomethyltetrazine reduction (Scheme 2). After optimization (see Table S1), it was found that efficient reduction could be realized with catalytic PdCl2 (10 mol %) and triethylsilane [27] (3 equiv) in THF at 45 °C followed by treatment with PIDA to oxidize the initially formed dihydrotetrazine. The method provides monosubstituted tetrazines derived from aliphatic and aromatic esters with a variety of functional groups, including Boc-amino (4a–b, 4g), methoxy (4c), ester (4d, 4f) nitrile (4e), protected amino acids (4h-i), and heterocycles (4j-k). Reduction of 3w gave BODIPY-FL derivative (4m), which shows a 15-fold turn-on of fluorescence upon reaction with eq-5-hydroxy-trans-cyclooctene (5-hydroxyTCO).[6a-e] Pyridine derivatives (3i-j, 3t, 3v) gave low conversions and attempted reduction of 3q was unsuccessful, possibly due to overreduction of the biotin core.
Scheme 2.
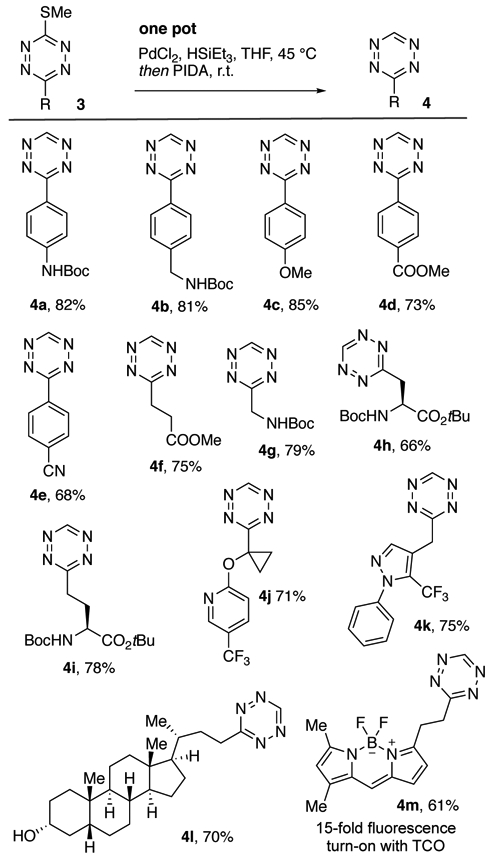
Synthesis of 3-monosubstituted tetrazines. Conditions: 3 (1 equiv), PdCl2 (10 mol %), HSiEt3 (3 equiv.), THF, 45 °C, 24 h; PIDA (1.2 equiv.), r.t., 1h.
Thiomethyltetrazines 3 also served as electrophiles for Ag-mediated Liebeskind-Srogl coupling reactions [21] with thioethers bearing either aliphatic or aromatic groups (Scheme 3). Tolerated functional groups include ether, chloride, ester, nitrile, trifluoromethyl, alcohol, Boc-amino, and protected amino acids as well as pyridyl and triazole groups. Additionally, the 3-furyl group was readily introduced to a range of thiomethyltetrazines via Ag-mediated Liebeskind-Srogl coupling. In line with our previous observations, electron rich arylboronic acids were the most efficient nucleophiles.
Scheme 3.

Synthesis of unsymmetrical tetrazines via Ag-mediated Pd-catalyzed Liebeskind-Srogl coupling. [a] ArB(OH)2 (1.9 equiv.), 60 °C, 20 h. [b] ArB(OH)2 (3 equiv.), 60 °C, 20 h. [c] ArB(OH)2 (3 equiv.), microwave 100 °C, 3 h. [d] Pd(dppf)Cl2 (30 mol%), Ag2O (5 equiv.), ArB(OH)2 (6 equiv.), microwave 100 °C, 3h.
3,6-Disubstituted aromatic tetrazines have been used broadly for bioorthogonal chemistry due to their combination of stability and rapid kinetics but can be limited by their hydrophobicity. We anticipated that furyl or vinylether groups introduced through cross-coupling could serve as small and solubilizing alternatives to phenyl groups. Shown in Scheme 4A are the experimental logP values for three tetrazine analogs and their rate constants in reactions with 5-hydroxyTCO (Figure S2A-C). Furyl analog 5I (logP 1.2) is considerably more hydrophilic albeit 2.9 times less reactive than phenyl derivative 5b (logP 2.2). Vinylether 6c is the most reactive and hydrophilic member of the series with logP 0.89 and reactivity that is 4.2 times faster than 5b. Vinylether-substituted tetrazines represent a new class of dienophile with a favorable combination of kinetics, small size and hydrophilicity. Tetrazines 5b, 5I and 6c all display >96% stability after 24 h incubation in PBS at 25 °C (Figure S1A). To introduce the 2-ethoxyvinyl group, we employed commercially available stannane 7. Here, coupling reactions were most efficient under conventional CuTc-promoted conditions with a brief reaction time (see Table S2). As shown in Scheme 4B, these cross-coupling procedures can tolerate a broad range of functional groups and heterocycles of biological interest.
Scheme 4.

Furyl- and vinylether-subsituted tetrazines with favorable kinetics, small size and hydrophilicity. (A) Relative reactivity and logP value. (B) Synthesis via CuTc-mediated Liebeskind-Srogl coupling.
This divergent approach to synthesizing tetrazine-coupled probes was applied to the study of MAGL, a serine hydrolase involved in endocannabinoid signaling.[28] Our previous approach to studying this drug target was limited by the need for a scaffold with a relatively uncommon boronic acid group, thus restricting the types of drug candidates that could be studied.[21] We hypothesized that a divergent approach could be used to tune reaction kinetics and/or inhibitory affinity of a new class of MAGLtetrazine probes. As shown in Scheme 5, probes 10a-c were designed based on a 4-(arylsulfonamidomethyl)piperidine scaffold bearing an electrophilic hexafluoroisopropyl (HFIP) carbamate warhead.[29] Thus, reaction of 2 with the (3-methyloxetane-3-yl) methyl ester 8 gave 72% yield of thiomethyltetrazine 9, which served as common intermediate for the synthesis of furyl-substituted 10a, vinylether-substituted 10b, and monosubstituted tetrazine 10c. Stopped flow kinetics with 5-hydroxyTCO showed 10c was 6.5-times more reactive than 10b, which in turn was 8.6-fold more reactive than 10a. All of these probes were 3–162 times more reactive than a previously described MAGL probe based on a different drug scaffold (12[21] Figure S4).
Scheme 5.

Divergent synthesis of tetrazine-coupled MAGL-probes 10a-c from common ester starting material and thiomethyltetrazine intermediate.
In an in vitro assay,[30] probes 10a-c inhibited MAGL activity with IC50’s of 12 nM (10a), 46 nM (10b) and 16 nM (10c). The most reactive probe 10c was further investigated for labeling of endogenous MAGL in live cells. Human brain vascular pericytes were treated with probe 10c for 1 h, followed by labeling with 2 μM TCO-TAMRA for 30 min. After cell lysis, gel-based activitybased protein profiling[31] (ABPP, Figure 3A) showed strong labeling of MAGL. The labeling was dose responsive (Figure 3B), and treating cells with 10c (1 μM, 1 h) followed by incubation with 200 nM TCO-TAMRA gave complete labeling with t1/2 of 6.1 min (Figure 3C). Similar kinetics were observed when the concentration of TCO-TAMRA was dropped to 50 nM (Figure S7), and with a higher concentration of TCO-TAMRA (2 μM), labeling was complete when the first data point was collected after 2 min. By contrast, our previous 6-methyltetrazin-3-yl probe for MAGL displayed slower labeling kinetics (t1/2 of 13 min) with 2 μM TCO-TAMRA (Figure S8).[21] Together, these experiments illustrate how modifying the structure of tetrazine-coupled probes can be used to tune the IC50 and labeling kinetics.
Figure 3.

(A) Live cells were treated with probe 10c for 1 h, followed by TCO-TAMRA treatment for 30 min, cell lysis, and analysis by gel-based ABPP. (B) In-gel fluorescence and dose response fitting of probe 10c. (C) Cellular labeling kinetics of 10c
Conclusion
In conclusion, a one-pot method is described for the conversion of (3-methyloxetan-3-yl)methyl carboxylic esters into 3 thiomethyltetrazines that can subsequently serve as a platform for divergent tetrazine synthesis via Pd-catalyzed cross-coupling and the first example of monosubstituted tetrazine synthesis via catalytic thioether reduction. The utility of the method was demonstrated through the synthesis of aliphatic, aromatic and heterocycle substituted tetrazines and to the development and biological evaluation of a new series of tetrazine-coupled probes for MAGL. We anticipate that the greater access to tetrazine-small molecule conjugates available by this method will serve as an enabling tool in chemical biology.
Supplementary Material
Acknowledgements
We would like to thank Jeffery Sperry for helpful discussions, Remzi Duzguner for DSC analysis, Edelweiss Evrard for outsourcing support and Lucy Stevens for the in vitro potency data on probes 10a-c. This work was supported by NIH GM132460 and Pfizer. Instrumentation was supported by NIH awards P20GM104316,P30GM110758,S10RR026962, and S10OD016267 and NSF awards CHE-0840401, CHE-1229234, and CHE-1048367.
References
- [1].a) Hang HC, Yu C, Kato DL, Bertozzi CR, Proc. Natl. Acad. Sci. USA 2003, 100, 14846–14851; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Patterson DM, Nazarova LA, Prescher JA, ACS Chem. Biol 2014, 9, 592–605. [DOI] [PubMed] [Google Scholar]
- [2].a) Clavier G, Audebert P, Chem. Rev 2010, 110, 3299–3314. [DOI] [PubMed] [Google Scholar]; b) Saracoglu N, N. Tetrahedron 2007, 63, 4199–4236. [Google Scholar]
- [3].Zhang J, Shukla V, Boger DL, J. Org. Chem 2019, 84, 9397–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Rossin R, Robillard MS, Curr. Opin. Chem. Biol 2014, 21, 161–169; [DOI] [PubMed] [Google Scholar]; b) Li J, Jia S, Chen PR, Nat. Chem. Biol 2014, 10, 1003–1005; [DOI] [PubMed] [Google Scholar]; c) Carlson JCT, Mikula H, Weissleder R, J. Am. Chem. Soc 2018, 140, 3603–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tu JL, Xu MH, Franzini RM, ChemBioChem 2019, 20, 1615–1627 [DOI] [PubMed] [Google Scholar]
- [5].a) Wang K, Sachdeva A, Cox DJ, Wilf NM, Lang K, Wallace S, Mehl RA, Chin JW, Nat. Chem 2014, 6, 393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lang K, Chin JW, Chem. Rev 2014, 114, 4764–4806; [DOI] [PubMed] [Google Scholar]; c) Nikić I, Lemke EA, Curr. Opin. Chem. Biol 2015, 28, 164–173. [DOI] [PubMed] [Google Scholar]
- [6].a) Devaraj NK, Hilderbrand S, Upadhyay R, Mazitschek R, Weissleder R, Angew. Chem. Int. Ed 2010, 49, 2869–2872; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Carlson JCT, Meimetis LG, Hilderbrand SA, Weissleder R, Angew. Chem. Int. Ed 2013, 52, 6917–6920; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wu H, Yang J, Šečkutė J, Devaraj NK, Angew. Chem. Int. Ed 2014, 53, 5805–5809; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wieczorek A, Werther P, Euchner J, Wombacher R, Chem. Sci 2017, 8, 1506–1510; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wieczorek A, Buckup T, Wombacher R, Org. Biomol. Chem 2014, 12, 4177–4185 [DOI] [PubMed] [Google Scholar]; f) Meimetis LG, Carlson JCT, Giedt RJ, Kohler RH, Weissleder R, Angew. Chem. Int. Ed 2014, 53, 7531–7534; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Lee Y, Cho W, Sung J, Kim E, Park SB, J. Am. Chem. Soc 2018, 140, 974–983. [DOI] [PubMed] [Google Scholar]
- [7].a) van de Watering FCJ, Rijpkema M, Robillard M, Oyen WJG, Boerman OC, Front. Med 2014, 1, 1–11; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meyer J-P, Adumeau P, Lewis JS, Zeglis BM, Bioconjugate Chem. 2016, 27, 2791–2807; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang M, Vannam R, Lambert WD, Xie Y, Wang H, Giglio B, Ma X, Wu Z, Fox J, Li Z, Chem. Commun 2019, 55, 2485–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Glockle M, Kaim W, Angew. Chem. Int. Ed 1999, 38, 3072–3074; [PubMed] [Google Scholar]; b) Kaim W, Coord. Chem. Rev 2002, 230, 127–139. [Google Scholar]
- [9].a) Zhang H, Dicker KT, Xu X, Jia X, Fox JM, ACS Macro Lett. 2014, 3, 727–731 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu S, Zhang H, Remy RA, Deng F, Mackay ME, Fox JM, Jia X, Adv. Mater 2015, 27, 2783–2790; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Alge DL, Azagarsamy MA, Donohue DF, Anseth KS, Biomacromolecules 2013, 14, 949–953 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhou H, Johnson JA, Angew. Chem. Int. Ed 2013, 52, 2235–2238. [DOI] [PubMed] [Google Scholar]
- [10].Wu H, Devaraj NK, Acc. Chem. Res 2018, 51, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Cañeque T, Müller S, Rodriguez R, Nat. Rev. Chem 2018, 2, 202–215; [Google Scholar]; b) Baalmann M, Ziegler MJ, Werther P, Wilhelm J, Wombacher R, Bioconjugate Chem. 2019, 30, 1405–1414. [DOI] [PubMed] [Google Scholar]
- [12].Stollé R, J. Prakt. Chem 1906, 73, 277–287. [Google Scholar]
- [13].a) Pinner A, Ber. Dtsch. Chem. Ges 1893, 26, 2126–2135; [Google Scholar]; b) Hofmann KA, Ehrhart O, Ber. Dtsch. Chem. Ges 1912, 45, 2731–2740; [Google Scholar]; c) Curtius T, Hess A, J. Prakt. Chem 1930, 125, 40–53 [Google Scholar]; d) Yang J, Karver MR, Li W, Sahu S, Devaraj NK, Angew. Chem. Int. Ed 2012, 124, 5312–5315 [Google Scholar]; e) Mao W, Shi W, Li J, Su D, Wang X, Zhang L, Pan L, Wu X, Wu H, Angew. Chem. Int. Ed 2019, 58, 1106–1109; [DOI] [PubMed] [Google Scholar]; f) S. A. L. Jr, Johnson BD, Cohen E, J. Heterocycl. Chem 1975, 12, 1143–1153 [Google Scholar]; g) Karver MR, Weissleder R, Hilderbrand SA, Bioconjugate Chem. 2011, 22, 2263–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Qu Y, Sauvage FX, Clavier G, Miomandre F, Audebert P, Angew. Chem. Int. Ed 2018, 57, 12057–12061. [DOI] [PubMed] [Google Scholar]
- [15].a) Mboyi CD, Testa C, Reeb S, Genc S, Cattey H, Fleurat-Lessard P, Roger J, Hierso J-C, ACS Catal. 2017, 7, 8493–8501 [Google Scholar]; b) Xiong H, Gu Y, Zhang S, Lu F, Ji Q, Liu L, Ma P, Yang G, Hou W, Xu H, Chem. Commun 2020; [Google Scholar]; c) Testa C, Gigot É, Genc S, Decréau R, Roger J, Hierso J-C, Angew. Chem. Int. Ed 2016, 55, 5555–5559; [DOI] [PubMed] [Google Scholar]; d) Mboyi CD, Vivier D, Daher A, Fleurat-Lessard P, Cattey H, Devillers CH, Bernhard C, Denat F, Roger J, Hierso J-C, Angew. Chem. Int. Ed 2020, 59, 1149–1154. [DOI] [PubMed] [Google Scholar]
- [16].a) Novák Z, Kotschy A, Org. Lett 2003, 5, 3495–3497; [DOI] [PubMed] [Google Scholar]; b) Nicolas Leconte AK-W, Suzenet Franck, Guillaumet Gérald, Synlett 2007, 2, 204–210 [Google Scholar]; c) Li Z, Ding J, Song N, Lu J, Tao Y, J. Am. Chem. Soc 2010, 132, 13160–13161; [DOI] [PubMed] [Google Scholar]; d) Pellegatti L, Vedrenne E, Leger J-M, Jarry C, Routier S, Tetrahedron 2010, 66, 4383–4389; [DOI] [PubMed] [Google Scholar]; e) Quinton C, Alain-Rizzo V, Dumas-Verdes C, Clavier G, Vignau L, Audebert P, New J. Chem 2015, 39, 9700–9713; [DOI] [PubMed] [Google Scholar]; f) Bender AM, Chopko TC, Bridges TM, Lindsley CW, Org. Lett 2017, 19, 5693–5696. [DOI] [PubMed] [Google Scholar]
- [17].a) Stetsiuk O, El-Ghayoury A, Lloret F, Julve M, Avarvari N, Eur. J. Inorg. Chem 2018, 2018, 449–457; [Google Scholar]; b) Chavez DE, Parrish DA, Mitchell L, Angew. Chem. Int. Ed 2016, 55, 8666–8669; [DOI] [PubMed] [Google Scholar]; c) Audebert P, Miomandre F, Clavier G, Vernieres M-C, Badre S, Meallet-Renault R, Chem. Eur. J 2005, 11, 5667–5673. [DOI] [PubMed] [Google Scholar]
- [18].a) Chavez DE, Hiskey MA, Gilardi RD, Angew. Chem. Int. Ed 2000, 39, 1791–1793; [DOI] [PubMed] [Google Scholar]; b) Huynh MHV, Hiskey MA, Chavez DE, Naud DL, Gilardi RD, J. Am. Chem. Soc 2005, 127, 12537–12543. [DOI] [PubMed] [Google Scholar]
- [19].a) Schnell SD, Hoff LV, Panchagnula A, Wurzenberger MHH, Klapotke TM, Sieber S, Linden A, Gademann K, Chem. Sci 2020, 11, 3042–3047; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ros E, Bellido M, Verdaguer X, Ribas de Pouplana L, Riera A, Bioconjugate Chem. 2020, 31, 933–938. [DOI] [PubMed] [Google Scholar]
- [20].Fields SC, Parker MH, Erickson WR, J. Org. Chem 1994, 59, 8284–8287. [Google Scholar]
- [21].Lambert WD, Fang Y, Mahapatra S, Huang Z, am Ende CW, Fox JM, J. Am. Chem. Soc 2019, 141, 17068–17074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang Z. Pinner Reaction In Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2010, pp. 2237–2240. [Google Scholar]
- [23].Corey EJ, Raju N, Tetrahedron Lett. 1983, 24, 5571–5574. [Google Scholar]
- [24].Scott ES, Audrieth LF, J. Org. Chem 1954, 19, 1231–1237. [Google Scholar]
- [25].Selvaraj R, Fox JM, Tetrahedron Lett. 2014, 55, 4795–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Sauer J, Chem. Heterocycl. Compd 1995, 31, 1140–1154; [Google Scholar]; b) Devaraj NK, Weissleder R, Acc. Chem. Res 2011, 44, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Graham TH, Liu W, Shen D-M, Org. Lett 2011, 13, 6232–6235; [DOI] [PubMed] [Google Scholar]; b) Modak A, Maiti D, Org. Biomol. Chem 2016, 14, 21–35; [DOI] [PubMed] [Google Scholar]; c) Matsumura T, Niwa T, Nakada M, Tetrahedron Lett. 2012, 53, 4313–4316. [Google Scholar]
- [28].Blankman JL, Cravatt BF, Pharmacol. Rev 2013, 65, 849–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chang Jae W., Niphakis Micah J., Lum Kenneth M., Cognetta Armand B., Wang C, Matthews Megan L., Niessen S, Buczynski Matthew W., Parsons Loren H., Cravatt Benjamin F., Chem. Biol 2012, 19, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Butler CR, Beck EM, Harris A, Huang Z, McAllister LA, am Ende CW, Fennell K, Foley TL, Fonseca K, Hawrylik SJ, Johnson DS, Knafels JD, Mente S, Noell GS, Pandit J, Phillips TB, Piro JR, Rogers BN, Samad TA, Wang J, Wan S, Brodney MA, J. Med. Chem 2017, 60, 9860–9873. [DOI] [PubMed] [Google Scholar]
- [31].Chang JW, Cognetta AB, Niphakis MJ, Cravatt BF, ACS Chem. Biol 2013, 8, 1590–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.