

SUMMARY
Activating precursor B cell receptors of HIV-1 broadly neutralizing antibodies requires specifically-designed immunogens. Here, we compared the abilities of three such germline-targeting immunogens against the VRC01-class receptors to activate the targeted B cells in transgenic mice expressing the germline VH of the VRC01 antibody but diverse mouse light chains. Immunogen-specific VRC01-like B cells were isolated at different time points after immunization, their VH/VL genes were sequenced and the corresponding antibodies characterized. VRC01 B cell sub-populations with distinct cross-reactivity properties were activated by each immunogen, and these differences correlated with distinct biophysical and biochemical features of the germline-targeting immunogens. Our study indicates that the design of effective immunogens to activate B cell receptors leading to protective HIV-1 antibodies will require a better understanding of how the biophysical properties of the epitope and its surrounding surface on the germline-targeting immunogen influence its interaction with the available receptor variants in vivo.
Keywords: VRC01- antibodies, HIV, B cell receptors, eOD-GT8, 426c, epitope exposure, antibody affinity, germline antibody
eTOC blurb
Activating precursor B cell receptors of HIV-1 broadly neutralizing antibodies requires specifically-designed immunogens. Lin, Parks et al. compare three such germline-targeting immunogens against the VRC01-class receptors and find that each immunogen activates VRC01 B cell sub-populations with distinct cross-reactivity properties, and that these differences correlate with distinct biophysical and biochemical features of the germline-targeting immunogens.
Graphical Abstract
INTRODUCTION
The development of high-throughput assays to sequence VH/VL paired genes from individual B cells allowed the interrogation of large numbers of antigen-specific B cell receptors (BCRs), prior to and following vaccination or infection (Scheid et al., 2009). Such studies, combined with structural analysis of the antibody-epitope complexes, revealed that broadly neutralizing antibodies (bnAbs) recognize distinct Env regions, including the CD4-binding site (CD4-bs), the apex region, the N332 glycan patch and the interface between the gp120 and gp41 subunits (Burton and Hangartner, 2016; Kwong and Mascola, 2012; McCoy and Burton, 2017). Those that bind to the same viral Env epitope can be derived from the same, or very similar, VH/VL pair combinations and have common paratope structures (Chen et al., 2019; Zhou et al., 2013). For instance, the anti-CD4-bs VRC01-class bnAbs are derived from the pairing of VH1–2*02 heavy chains (HCs) and Vλ/Vκs (κ3–20, κ3–15, κ1–33, κ1–5, and λ2–14) light chains (LCs) expressing 5 amino acid-long CDRL3s (Huang et al., 2016; Kwong and Mascola, 2012; Sajadi et al., 2018; Scheid et al., 2011; Umotoy et al., 2019; Zhou et al., 2010; Zhou et al., 2013). They recognize their epitope on Env with similar angles of approach and their paratopes share similar structures, despite being up to 30% divergent in amino acid sequence. Several elements of their VH and VL domains contribute to the strength of their interaction with Env and their breadth of neutralization, but their gene-encoded CDRH2 domains contribute the majority of the antibody buried surface area with Env (Jardine et al., 2016; Zhou et al., 2010), similar to observations made with neutralizing antibodies against other viruses (Chen et al., 2019).
Natural virion-derived Env proteins capable of engaging the unmutated (commonly referred to as ‘germline’, gl) VRC01 BCRs expressed on naïve B cells are presently unknown, but specifically designed Env-derived recombinant proteins that bind glVRC01-class antibodies have been engineered (Jardine et al., 2015; McGuire et al., 2016; Medina-Ramirez et al., 2017). These proteins are employed as ‘gl-targeting’ immunogens to activate glVRC01-like BCRs in vivo (Dosenovic et al., 2015; Jardine et al., 2015; Parks et al., 2019). It is, however, expected that subsequent booster immunizations with heterologous, and more native-like, recombinant Env immunogens will be required to guide the maturation of glVRC01 BCRs towards their broadly neutralizing forms (Briney et al., 2016; Dosenovic et al., 2015; Tian et al., 2016).
Here, we examined whether the VRC01 epitope is equally effective in activating glVRC01-expressing B cells in vivo when expressed on three different gl-targeting immunogens, eOD-GT8 (Jardine et al., 2015), 426c Core (TM4ΔV1–3)(McGuire et al., 2016) and a newly designed 426c outer domain (426cOD). eOD-GT8 has entered phase I clinical evaluation (NCT03547245), while the 426c Core is scheduled for clinical evaluation in 2021. Despite the difference in binding affinities to glVRC01-class antibodies, all three immunogens were efficient in eliciting VRC01-like antibodies. However, whereas the epitope-specific antibodies elicited by the three immunogens utilized the same VH/VL pairings, they differed at key positions by only a few amino acids and as a result displayed distinct Env cross-reactive properties. Following that initial selection, the relative affinities of the selected BCRs dictated the fate of the corresponding activated B cells. Thus, the initial BCR selection by an immunogenic epitope is critically dependent on its biophysical properties and of its surrounding surface area. Our findings argue that gl-targeting immunogen-design efforts that are solely based on increasing the epitope-exposure on the immunogen or improving the affinity of an antigen are insufficient to accurately predict which target BCRs will be selected during immunization, and that the design of more effective immunogens that target the precursor BCRs of protective antibodies will need to take into account the biophysical properties of the epitope and its context.
RESULTS
Design and characterization of the 426c Env outer domain immunogen
The design of eOD-GT8 is based on the outer domain of the clade B HxB2 Env gp120 subunit (Jardine et al., 2015), while that of 426c Core is based on both the outer and inner domains of the clade C 426c gp120 subunit (McGuire et al., 2016). Here, we engineered a new gl-targeting protein that is derived from the 426c Core but lacks the inner domain (like eOD-GT8), termed 426c outer domain (426cOD). The design of 426cOD was based on that of eOD (which was the universal background of derived variants, such as eOD-GT8) (Jardine et al., 2015; Pejchal et al., 2011) (Fig 1a, Supplemental Table 1). An initial design (426c base) engineered to bind glVRC01-class mAbs was found to bind one of six gl antibodies (Fig 1b). Random library mutagenesis and yeast-display yielded 426cOD, which binds four of eight glVRC01-class mAbs tested (Fig 1c). We obtained a 3.4 Å X-ray structure of 426cOD bound to mature VRC01 (Fig 2a, Supplemental Table 2) which confirmed the proper folding of the designed 426cOD (Fig 1d). Comparison with previously published structures of eOD-GT8 and 426c Core (Fig 2b)(Scharf et al., 2016) indicated that VRC01 and glVRC01’s epitopes overlapped on each immunogen, and this comparison highlights differences in certain surface exposed residues between 426cOD and eOD-GT8 as follows: S278Rgp120, Q361Kgp120, I370Fgp120, L455Tgp120, D457Qgp120, D460Ygp120 (Fig 1a and Fig 2c). These differences translate into different electrostatic surface potentials for the 426c-sequence-based immunogens (negatively charged) compared to eOD-GT8 (positively charged), particularly in the gp120 V5 region which interacts with the CDRH2 and the N-terminus of glVRC01 LC (Fig 2c).
Figure 1. Characterization of 426cOD, related to supplemental figure 3 and supplemental table 1.

(a) Amino acid alignment of 426c Core, 426c base, 426cOD and eOD-GT8. HxB2 numbering is used. The sequences of 426c base, 426cOD and eOD-GT8 are reverted to align with that of the 426c Core, as their actual sequences from their N to C termini start with sequences highlighted in blue. Residues that differ between 426c base and 426cOD are shown in bold letters. The residues in eOD-GT8 that differ from those on 426c Core and 426cOD are underlined. (b-c) BLI was used to evaluate glVRC01-class antibodies binding to 426c base (b) and 426cOD (c). (d) 426c base design model (orange) superimposed onto the 426cOD crystal structure (green). The structural difference between the two is highlighted in red. All atom RMSD is 0.89 Å. (e-g). BLI was used to determine the affinity of germline VRC01 to the three germline-targeting immunogens. The binding curves use to calculate this value are shown. BLI data are representative of at least 2 independent experiments
Figure 2. Structures of three VRC01 germline-targeting immunogens, related to supplemental tables 2-4.

(a) Crystal structure of 426cOD (green) bound to human mature VRC01 Fab (light chain in gray, heavy chain in black) shown in cartoon representation. (b) Overlay of 426c Core (tan, PDB ID 5FA2) and 426c OD (green). (c) Structural comparison of the three germline-targeting immunogens. From left to right: Cartoon representation. Surface representation of immunogens with glVRC01 epitope highlighted in red. glVRC01 epitopes shown in stick representation: eOD-GT8 residues that differ from those on 426c Core and 426cOD are highlighted in magenta. Electrostatic potential of glVRC01 epitopes. The location of potential NLGS around the VRC01 epitope on the three antigens are indicated by green dots (electrostatic potentials do not consider the glycans as exact glycosylation at these positions is unknown).
To compare the interactions of the 3 antigens (426c Core, eOD-GT8 and 426cOD) with glVRC01, we modeled their interactions by superimposing glVRC01-bound structure with WT 426c Core (PDB ID 6MFT) (Borst et al., 2018) and with eOD-GT6 (PDB ID 4JPK) (Jardine et al., 2013) onto our newly solved structure of 426cOD bound to VRC01. We also modeled glVRC01 binding to eOD-GT8 by superimposing glVRC01 onto VRC01c-HuGL2 bound to eOD-GT8 (PDB ID 5IES) (Jardine et al., 2016). 426cOD buries ~971 Å2 and ~1007 Å2 upon binding glVRC01 (modeled interactions onto 6MFT and 4JPK, respectively) while eOD-GT8 buries ~1142 Å2 (modeled interactions onto 5IES) and 426c Core buries ~947 Å2 when binding gl3BNC60 (PDB ID 5FEC) (Scharf et al., 2016). 6–15 estimated H-bonds are formed between modeled glVRC01 and 426cOD (modeled onto 6MFT and 4JPK, respectively). These differences reflect the different conformations adopted by glVRC01 when binding the WT 426 Core (with glycans present) versus the 426c Core (Borst et al., 2018). 21 estimated H-bonds are formed between glVRC01 modeled with eOD-GT8 while 20 estimated H-bonds are formed between 426c Core and gl3BNC60 (PDB ID 5FEC) (Scharf et al., 2016) (Supplemental Table 3).
The analysis also revealed differences in amino acid exposures between 426c Core and 426cOD when they interact with glVRC01: Val430, in the β20–21 region of gp120, present only in 426c Core showed some interactions with glVRC01 while Asp48 and Glu49, only present in 426cOD showed some interactions with VRC01.
The glVRC01 heavy chain buries a larger surface area with 426cOD than eOD-GT8 (Supplemental Table 4), while the light chain buries a larger surface area with eOD-GT8: this is likely because of the light chain interactions with bulkier residues present in eOD-GT8 compared to 426cOD: Arg278gp120 and Tyr460gp120 in eOD-GT8 in place of Ser278gp120 and Asp460gp120 in 426cOD. The affinity of glVRC01 mAb for 426cOD is in the same range as for 426c Core (289 nM and 217 nM, respectively), while it is <0.1 nM for eOD-GT8 (Fig 1e-1g).
We examined whether differences in affinity influence the efficiency by which the two immunogens select for naïve B cells that express VRC01 BCRs in vivo, and whether and how the presence of the gp120 inner domain of the 426c Core antigen influences this selection at the molecular BCR level.
Epitope affinity and the inner domain influences the efficiency by which epitope-specific naïve B cells can be isolated
As orthologs of the human VH1–2*02 gene are not expressed in animal species such as mice, rats, rabbits and non-human primates (West et al., 2012), the abilities of the three antigens to engage glVRC01 BCRs in vivo were compared in a well-defined, transgenic mouse model that is heterozygous for the human inferred gl heavy chain of the VRC01 mAb (VRC01glHC) (Jardine et al., 2015). Approximately 80% of B cells express the human transgene and all express mouse LCs (mLCs); only 0.01% of which contain 5 amino acid-long CDRL3s. As a result, approximately 0.08% of naïve B cells are expected to express VRC01-like BCRs, compared to 0.02% −0.012% of naïve human B cells (Abbott et al., 2018; Jardine et al., 2015).
Individual eOD-GT8+/eOD-GT8 CD4-bs KO-, 426c Core+/426c Core CD4-bs KO-, or 426cOD+/426cOD CD4-bs KO- B cells were isolated from the spleens of naïve mice. VH/VL genes were then amplified and sequenced. Approximately 62%, 89% and 61% of HCs isolated with 426c Core, eOD-GT8 and 426cOD, respectively, were VRC01glH (Fig 3a). The vast majority of VRC01glHC-paired mLCs that were selected by either antigen expressed 9 amino acid-long CDRL3s, but mLCs with 6–8 or 10 amino acid-long CDRL3s were also present (Fig 3b). Only the two ‘outer domain’ antigens selected for mLCs expressing five amino acid-long CDRL3s. We also sorted naïve B cells with the C4b-based form of 426c Core from three animals. Out of 282 VL genes that were sequenced only three LCs with 5 amino acid CDRL3s were identified (one from one animal (0.9%), two from another (1.9%) and none from the third animal (0%) (data not shown). The isolation of B cells expressing glVRC01-like BCRs by the two ‘outer domain-based’ antigens (eOD-GT8 and 426cOD), but not by 426c Core, despite the fact that 426cOD and 426c Core have similar affinities for glVRC01, suggests that the presence of the inner domain limits the accessibility of the VRC01 epitope by BCRs expressed on naïve B cells, but not by soluble antibodies. We next examined whether the differences observed in binding to naïve VRC01 B cells by the three immunogens had any effect on the ability of the immunogens to expand naïve B cells in vivo.
Figure 3. Vaccine elicited antibody and B cell responses, related to supplemental figure 2 and supplemental tables 5-7.

Antigen-specific B cells were sorted and sequenced from naïve and immunized animals. VH gene usage (VH sequences: 426c Core n = 21, eOD-GT8 n = 191, 426cOD n = 23) (a) and CDRL3 length (number of sequences: 426c Core n = 98, eOD-GT8 n = 225, 426cOD n = 125) (b) of naïve B cells sorted with 426c Core, eOD-GT8 or 426cOD . VH gene usage (number of sequences: 426c Core n = 89, eOD-GT8 n = 48, 426cOD n = 67) (c) and CDRL3 length (number of sequences: 426c Core n = 51, eOD-GT8 n = 59, 426cOD n = 110) (d) of antigen-specific B cells isolated 2 weeks post-immunization. (e) VL gene usage of 5 AA-long CDRL3 B cells isolated from immunized animals (number of sequences: 426c Core n = 12, eOD-GT8 n = 20, 426cOD n = 22) 2 weeks post-immunization. Percentages indicate the frequency of VH or VL genes with the indicated characteristics out of the total number of VH and VL genes sequenced. (f-h) ELISAs were performed with plasma from immunized mice. Plasma antibody endpoint titers 2 weeks post immunization with 426c Core (f), eOD-GT8 (g) or 426cOD (h) against 426c Core, eOD-GT8, and 426cOD. Data is shown as a heatmap, with the strongest response (greater endpoint titer) in red. No color indicates undetectable reactivity. X indicates not tested. 12–14 mice immunized with each immunogen were tested. The animal number is indicated to the left of the heat map. (i) Percent of plasma antibodies targeting the autologous CD4-BS, elicited by the indicated VRC01 germline-targeting immunogens. Each dot represents an animal. Lines represent mean and standard deviation.
Epitope affinity does not influence the efficiency by which epitope-specific B cells expand during immunization
C4b-based nanoparticle forms (heptameric) of the three antigens were used as immunogens (Hofmeyer et al., 2013). Negative stain electron microscopy analysis confirmed that all three preparations had similar oligomeric features (Supplemental Fig 1a-c). Two weeks post-immunization, CD4-bs-specific class-switched B cells were single cell-sorted from spleens and their VH/VL genes sequenced. Approximately 92%, 83% and 67% of the VHs sequenced from the 426c Core, eOD-GT8- and 426cOD-immunized animals, respectively, were VRC01glHC (Fig 3c). Approximately 24%, 34% and 20% of mLCs from the 426c Core-, eOD-GT8-, 426cOD-immunized animals, respectively, expressed 5 amino acid-long CDRL3s (Fig 3d). The majority of the mLCs with 5 amino acid-long CDRL3s were derived from κ8–30*1, but κ4–72*01, κV6–15*01, κ12-46*01, κ4-61∗01 and κV4–86*01 derived mLCs were also identified (Fig 3e). Thus, the three immunogens are effective in activating B cells expressing VRC01-like BCRs, despite differences in affinities, as others have reported in the case of model antigens (Kouskoff et al., 1998; Shih et al., 2002). Dosenovic et al., also reported that while tetrameric baits of eOD-GT8 do not identify glVRC01-class B cells in the gl3BNC60 mouse, immunization with the multimeric form of eOD-GT8 results in the activation of such cells (Dosenovic et al., 2015).
Different Env cross-reactivity properties of plasma antibodies
Robust autologous plasma antibody responses were elicited by all three immunogens in all animals (Fig 3f-h, Supplemental Fig 1d-f). A large proportion of plasma antibody responses targeted the autologous CD4-bs in all three cases (Fig 3i), although 426c Core was more consistent in eliciting such antibodies than the two outer domain immunogens. Thirteen of fourteen animals immunized with 426c Core elicited anti-eOD-GT8 plasma antibody responses (Fig 3f), which recognized the VRC01 epitope on eOD-GT8 (Supplemental Fig 1d), while only one of fourteen animals immunized with eOD-GT8 elicited (very weak) anti-426c Core plasma antibody responses (Fig 3g). All animals immunized with 426cOD elicited plasma antibodies against the VRC01 epitope expressed on eOD-GT8, and 4 of 12 animals elicited anti-426c Core plasma antibodies (Fig 3h and Supplemental Fig 1f). In contrast, only 4 of 14 animals immunized with eOD-GT8 generated anti-426cOD plasma antibodies and with only partial specificity to the CD4-bs of 426cOD (Fig 3g and Supplemental Fig 1e).
The absence of anti-426c Core antibodies in the plasmas of the eOD-GT8-immunized animals was unexpected, as VRC01-like B cells utilizing the same VH/VL pairings (VRC01glHC and κ8-30*1 mLCs) were isolated in both the 426c Core and eOD-GT8-immunized animals. Antibodies derived from this HC/LC pairing from the eOD-GT8- and 426c Core-immunized animals also displayed different cross-reactive properties. Those derived from the eOD-GT8-immunized animals did not bind 426c Core (but bound eOD-GT8 in a VRC01-epitope-dependent manner) (Supplemental Fig 1g), while those derived from the 426c Core-immunized animals recognized both 426c Core and eOD-GT8 (Supplemental Fig 1h). In sum, the cross-reactivities of VRC01-like antibodies elicited by 426c Core and by eOD-GT8 differ.
eOD-GT8 and 426c Core select for different variants of VRC01 BCRs
A key difference between the eOD-GT8- and 426c Core-selected κ8–30*01 mLCs with 5 amino acid-long CDRL3s is the selection of different amino acids at the C-terminus of the CDRL3 (at the junction of the V and J genes) (Fig 4a). The eOD-GT8-selected CDRL3s with aromatic residues at position 96LC followed by a threonine at position 97LC (W96T97or Y96T97). As a result, 30% of the 5 amino acid-long CDRL3s had the QQYWT motif and 60% had the QQYYT motif (Supplemental Table 5a). This preferential selection was reported by Jardine et al (Jardine et al., 2015). In contrast, the 426c Core-selected κ8–30*01 mLCs with 5 amino acid-long CDRL3s primarily expressed either a negatively charged amino acid at position 96LC (E96) or a positively charged amino acid at position 97LC (K97). As a result, 50% of the 5 amino acid-long CDRL3s had the QQYET motif and 50% had the QQYYK motif (Supplemental Table 5b). In addition to these differences in the mLCs, most VRC01glHC sequences from the 426c Core-immunized animals contained a H35N mutation in the CDRH1 (as it exists in mature VRC01), which was not present in the VRC01glHC sequences from the eOD-GT8-immunized animals. 426cOD displayed a B cell selection phenotype with some similarities to those of eOD-GT8 and 426c Core, as it selected for κ8–30*01 mLCs with 5 amino acid-long CDRL3s with (W96T97) (QQYWT; 20% of sequences), like eOD-GT8, but also with K97 (QQYYK; 20% of sequences), like 426c Core (Supplemental Table 5c). The QQYYS sequence was the most prevalent (60% of the sequences). We propose that the positive electrostatic surface around the VRC01 epitope on eOD-GT8 disfavors the selection of CDRL3s with a positively charged amino acid at position 97LC (K97) and that the presence of Y30 on eOD-GT8 (D460 on 426c Core) selects for CDRL3s with aromatic residues at position 96 to gain enthalpically favorable aromatic ring interaction (Sangesland et al., 2019). We note that the known human VRC01-class bnAbs do not express such bulky amino acids in their CDRL3s (Umotoy et al., 2019; Zhou et al., 2013).
Figure 4. VRC01 germline-targeting immunogens select for different amino acids within VRC01 BCRs, related to supplemental tables 5-7.

(a) Logo plots represent 5 AA CDRL3 sequences from antigen-specific B cells isolated 2 weeks post-immunization with the indicated VRC01 germline-targeting immunogens. The numbering of the CDRL3 region is based on the Kabat numbering system. Size of amino acid reflects frequency of use. (b-c) BLI was used to evaluate the binding abilities of mAbs, and of their indicated mutated forms, isolated from 426c core (b) and eOD-GT8 (b), to the indicated antigens. BLI data are representative of at least 2 independent experiments.
To examine whether the disparate cross-reactivities of the VRC01-like antibodies elicited by the germline-targeting immunogens are due to the selection of different amino acids in their CDRL3 and CDRH1 domains, we performed targeted mutagenesis on the VRC01glHC and κ8–30*01 genes of two antibodies, p2b4 and p2b9, derived from eOD-GT8-immunized animals and examined how the mutations affected the antibody-binding properties. To this end, we introduced H35N or W96E on p2b4 and T97K on p2b9. While neither p2b4 and p2b9 bound to 426c Core, the individual CDRH1 or CDRL3 substitutions resulted in weak but detectable binding (Fig 4c). Of note, both type of mutations decreased the binding off-rates to eOD-GT8 and p2b4 with the W96E mutation displayed negligible off-rate to eOD-GT8. When the CDRH1 and CDRL3 substitutions were simultaneously introduced, the binding of both antibodies to 426c Core became similar to that of VRC01-like antibodies isolated from 426 Core-immunized animals (Fig 4b). Conversely, when the reversed mutations K97T or E96W were introduced to two VRC01glHC and κ8–30*01 antibodies (p1d3 and p1e5) isolated from the 426c Core-immunized animals, their binding to 426c Core was drastically diminished, while their binding to eOD-GT8 was unaffected (Fig 4b).
We recently reported that VRC01-like antibodies elicited by 426c Core in this transgenic mouse model can bind to certain heterologous wild type (WT) gp120 core proteins, even in the presence of N276-associated glycans, which normally prevent the binding of germline VRC01-class mAbs to Env (Parks et al., 2019). These heterologous cores are derived from different clades (93TH057 is clade A/E, Q168a is clade A, and HxB2, 45_01 and QH0692 are clade B), they express a functional N276 glycan site and are not recognized by glVRC01 (Parks et al., 2019). In agreement, two of eight 426c Core-elicited VRC01-like Abs (p1a6 and p1a7) recognized two (45_01dH1 WT Core and HxB2 WT Core) of the five heterologous WT cores proteins tested (Fig 5a). In contrast, the eOD-GT8-elicited VRC01-like antibodies did not display such cross-reactive reactivities (Fig 5b). We tested the p2b4 and p2b9 mutants for binding to the heterologous WT core proteins and observed that the double substitution on the background of p2b4 (H35N, W96E) conferred cross-reactivity to the two heterologous core proteins recognized by the 426c Core-elicited VRC01-like antibodies (Fig 5c).
Figure 5. Vaccine elicited mAbs binding to multimeric (C4b-BP) heterologous Core Env, related to supplemental figure 1 and supplemental tables 5-7.

BLI was used to evaluate binding of mAbs elicited by 426c Core (a), eOD-GT8 (b) and 426cOD (d) to the indicated heterologous wild type (WT) Core proteins. BLI binding curves are shown. (c) BLI was used to evaluated binding of mAbs elicited by eOD-GT8 with CDRL3 and CDRH1 substitutions to multimeric heterologous Core Env. BLI binding curves (representative of at least two independent experiments) are shown.
Thus, even though eOD-GT8 and 426c Core effectively stimulate naïve B cells expressing BCRs composed of VRC01glHC paired with κ8–30*01 mLCs with 5 amino acid long CDRL3s, they select for different amino acids at positions 96 and 97 (Kabat numbering) in the CDRL3 (junction of V and J genes). Additionally, the VRC01glHC from the 426c Core-immunized animals almost universally acquired the H35N mutation. Those few amino acid differences are responsible for differences in the cross-Env recognition potentials of the VRC01-like antibodies elicited by eOD-GT8 and by 426c Core. Our results suggest therefore that VRC01-like antibodies expressing bulky amino acids at position 96 will not bind effectively the VRC01 epitope on heterologous proteins in the presence of the inner gp120 domain. In agreement with this, VRC01-like antibodies elicited by 426cOD, expressing tyrosine at position 96, have no binding to any heterologous WT gp120 core Env proteins (Fig 5d). As the first wave of VRC01-like antibodies elicited by these germline-targeting immunogens are minimally mutated, the antibodies did not display neutralizing activities, in agreement with previous reports (Briney et al., 2016; Parks et al., 2019)
Temporal evolution of epitope-specific VRC01-like B cell variants
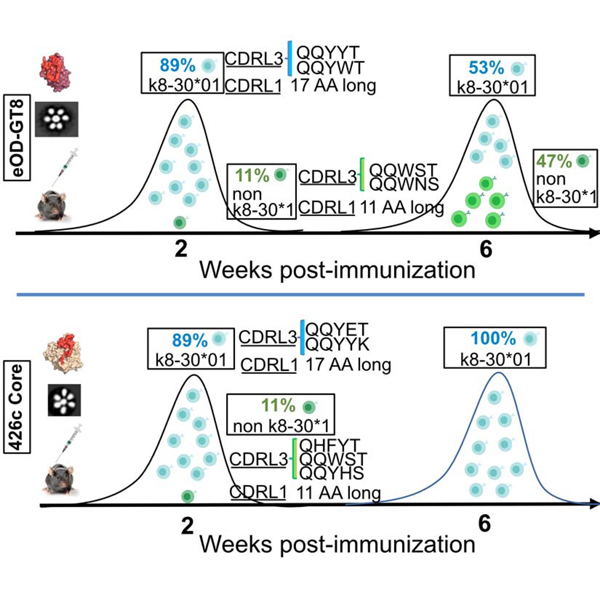
The germinal center reaction is a dynamic process during which B cell clones compete for antigen and T-cell help (Shih et al., 2002; Tas et al., 2016; Victora and Nussenzweig, 2012). To examine whether and how the VRC01-like B cell responses elicited by the three immunogens evolve over time, animals were immunized with the three immunogens, CD4-bs+/KO- B cells were isolated 6 weeks later, and their VH/VL genes were sequenced. The evolution of the plasma antibody responses to all three immunogens was also evaluated. The titers and epitope specificities of autologous and heterologous plasma antibodies elicited by all three immunogens remained stable over the 6-week observation period (Supplemental Fig 2a-2c). That is: (a) 426c Core generated autologous, anti-eOD-GT8 and anti-426cOD plasma antibodies (the heterologous antibody responses being exclusively directed to the CD4-bs); (b) eOD-GT8 generated autologous but not heterologous antibodies; and (c) 426cOD elicited strong autologous and anti-eOD-GT8 antibody responses, but weaker anti-426c Core antibody responses (a fraction of the heterologous antibody responses being exclusively directed to the CD4-bs).
Significantly more amino acid mutations accumulated in both the VH/VL genes 6 weeks post-immunization, irrespective of the immunogen (Supplemental Fig 2d and 2e and Supplemental Table 5). The H35N mutation was present in 90% of VRC01glHC sequences from 426c Core-immunized animals. Additional mutations were also selected in CDRH1, CDRH2 and FW3 of the VH gene (Supplemental Table 5b). The H35N mutation was also preferentially selected (71%) in the VRC01glHC sequences from 426cOD-immunized animals (Supplemental Table 5c). In contrast, no clear pattern of amino acid selection was evident in the VRC01glHC sequences from eOD-GT8 immunized animals (Supplemental Table 5a).
All VRC01glHC-associated mLCs from the 426c Core-immunized animals were derived from κ8–30*1 and all expressed the five amino acid CDRL3 QQYYK sequence (i.e., the E96 mutation was not evident) (Supplemental Table 5b). This sequence also dominated (64%) the mLCs sequences from the 426cOD-immunized animals (Supplemental Table 5c). The mLC mutations were primarily confined to the CDRL1s and resulted in the accumulation of negatively charged amino acids (Asp or Glu). A more complex pattern of mLCs with 5 AA CDRL3 paired with VRC01glHC was observed in the eOD-GT8-immunized animals (Supplemental Table 5a). Between weeks 2 and 6 post-immunization, the relative frequency of κ8–30*01 mLCs decreased by approximately half (from 89% to 47%) while the relative frequencies of non-κ8–30*01 mLCs increased by approximately five-fold (from 11% to 53%). While the κ8–30*01 mLCs isolated from the 426c Core-immunized animals were mutated from germline (Supplemental Table 5b), the majority of κ8–30*01 sequences isolated from the eOD-GT8-immunized animals remained unmutated (15/28 sequences had 0 amino acid mutations) (Supplemental Table 5a). In contrast, the non-κ8–30 mLCs isolated from eOD-GT8-immunized animals were extensively mutated at week 6 (up to 9 amino acid mutations).
Of note, all the non-κ8–30*01 mLCs have short CDRL1 domains, as is the case of all known VRC01-class bNAbs (Zhou et al., 2013). As observed at week 2 post-immunization, eOD-GT8 preferentially selected for QQYYT, QQYWT CDRL3s for all κ8–30*01 mLCs but in the case of κ4–61*01, the QQYET sequence was present in 5/6 mLCs sequenced and paired with VRC01glHC. The 426cOD selection pattern was intermediate to those of 426c Core as most of the κ8–30*01-associated 5 amino acid CDRL3s expressed the QQYYK sequence and the non-κ8–30*01 mLCs were unmutated from germline (Supplemental Table 5c).
VRC01-like mAbs were generated from paired VH/VL sequences isolated at week 6 from the eOD-GT8-immunized animals, and their binding to the three immunogens were evaluated (Fig 6). We included both κ8–30*01-derived and non-κ8–30*01-derived mAbs, some of which (κ4–61*01) expressed CDRL3s with the QQYET sequence that predominates in the 426c Core-elicited VRC01-like antibody responses (Supplemental Table 5). None bound the 426c Core and 426cOD proteins. Thus, the presence of shorter CDRL1s on VRC01-like antibodies with κ4–61*01 LCs (5 amino acid CDRL3) does not appear to improve their binding to the VRC01 epitope on heterologous Envs. We expect, however, that VRC01-like BCRs with short CDRL1s can more easily mature towards broadly neutralizing forms, as short CDRL1s are necessary to accommodate the N276 glycans and elements of the V5 gp120 domains (Zhou et al., 2013).
Figure 6. Binding properties of monoclonal antibodies isolated 6 weeks post immunization with eOD-GT8, related to supplemental tables 5-7.

Monoclonal antibodies isolated 6-weeks post-immunization with eOD-GT8 were evaluated for binding to 426c Core, eOD-GT8 or 426cOD using BLI. mAbs utilizing κ4–61*01 are noted with an asterisk. Data are representative of at least 2 independent experiments.
Interactions of human VRC01-like antibodies with ‘germline-targeting’ immunogens.
We generated VRC01-like antibodies from previously published VH/VL paired sequences from naïve human B cells sorted with eOD-GT8 (Havenar-Daughton et al., 2018) and examined their binding to the three ‘gl-targeting’ immunogens (Fig 7, Supplemental Table 5d). The antibodies only bound eOD-GT8. These results confirm preferential interaction of eOD-GT8-selected VRC01-like antibodies for eOD-GT8 and suggest that the three antigens evaluated here are not recognized by all available variations of human VRC01-like putative antibodies precursors.
Figure 7. Binding properties of human germline VRC01-like antibodies isolated with eOD-GT8, related to supplemental tables 5-8.
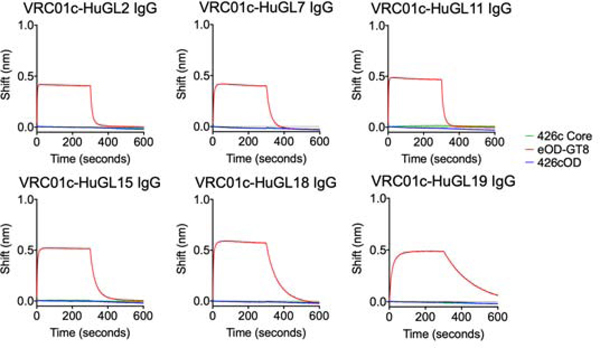
Six germline VRC01 antibodies derived from naïve human B cells that were sorted by eOD-GT8 were tested for binding against 426c Core, eOD-GT8 or 426cOD using BLI. BLI binding curves are shown. Data representative of 2 independent experiments
DISCUSSION
The development of VRC01-class bnAbs by vaccination will require the activation of their unmutated BCR precursors by immunizing with gl-targeting immunogens, and the accumulation of specific VH/VL amino acid mutations through sequential immunizations with heterologous Envs (Dosenovic et al., 2015). Although eOD-GT8 and 426c Core gl-targeting immunogens activate VRC01 precursor B cells in vivo, ‘boost’ immunogens that will guide the maturation of VRC01 antibodies towards their broad neutralizing forms have not been identified (Briney et al., 2016; Dosenovic et al., 2015; Parks et al., 2019; Tian et al., 2016). Our study revealed that depending on the background onto which the VRC01 epitope is expressed on the gl-targeting immunogen, B cells with different epitope-specific VH/VL pairs were selected to enter the germinal center reactions. That is, from the available pool of VRC01-like B cells available, each gl-targeting immunogen selected a different subpopulation (containing B cells with different affinities towards that immunogen). As a result, the VRC01 antibody responses elicited by different gl-targeting immunogens displayed distinct heterologous Env cross-recognition properties. We defined a set of mutations responsible for these cross-recognition properties confirming that our studies unveiled differences in the subpopulations of antibodies elicited by different immunogens. We expect therefore that following eOD-GT8 and 426c Core human immunizations, different booster immunogens will be required to further expand the initial waves of VRC01-like B cells stimulated by these two germline-targeting immunogens. Indeed, the VRC01-like antibodies elicited in this animal model by 426c Core are boosted by HxB2 WT Core (Parks et al., 2019) while those elicited by eOD-GT8 expand following a boost immunization with BG505 core-GT3 (Briney et al., 2016). Our data therefore reveal aspects of immunogen-BCR interactions that may have been underappreciated previously, but which we believe will contribute to the development of more effective prime-boost immunization protocols for the elicitation of bnAbs. Our results also highlight potential limitations of eOD-GT8 and 426c Core. eOD-GT8 may not stimulate VRC01 B cells that can bypass the N276 associated glycans, thus necessitating the use of booster immunogens also lacking the N276 glycan site and consequently further increasing the affinities of antibodies that cannot overcome this key obstacle, while 426c Core may activate VRC01 antibodies with long CDRL1 domains that will be less amenable to maturation towards their broadly neutralizing forms.
The observations reported here are applicable to gl-targeting immunogens against other classes of CD4-bs bnAbs (Bonsignori et al., 2016; Williams et al., 2017) and bnAbs against other Env regions such as the apex domain of the HIV-1 trimer (Andrabi et al., 2015), or the N322 glycan patch (Escolano et al., 2019; Steichen et al., 2019), but also to non-HIV bnAbs such as Influenza and HCV (Chen et al., 2019; Sangesland et al., 2019).
Subsequent to the initial glVRC01 BCR-selection by germline-targeting immunogens, the fate of the corresponding B cells was defined by the relative affinities of the BCR variants, as others have reported in studies conducted with model antigens (Dal Porto et al., 2002; Kouskoff et al., 1998; Silva et al., 2017). The relative numbers of the high affinity, epitope-specific glVRC01HC/κ8–30*01 B cells that were preferentially selected by eOD-GT8 and which predominated the early B cell response, decreased over the 6-week period of observation and their BCRs did not accumulate somatic hypermutations. During the same period, the frequencies of lower affinity VRC01-like B cells expressing different LCs (also paired with glVRC01HC) increased and their BCRs accumulated somatic hypermutations. In contrast, epitope-specific glVRC01HC/κ8–30*01 B cell variants selected by 426c Core had lower affinity for the VRC01 epitope and predominated both during the early and late B cell response and undergone somatic hypermutation.
Overall, our study indicates that the development of effective gl-targeting immunogens to target specific BCRs, such as those known to give rise to bnAbs, will require a better understanding of how the biophysical properties of the epitope and its surrounding surface on the immunogen influences its interaction with the available BCR variants in vivo, as structural information or affinity of the immunogen alone are insufficient to accurately predict which BCRs will initially be selected.
LIMITATIONS OF THE STUDY
Orthologs of the human VH1–2*02 gene are not expressed in animal species such as mice, rats, rabbits and non-human primates. Thus, the study was conducted in transgenic mice expressing the pre-arranged V, D, J segment of the inferred germline VRC01 antibody. We expect that the differential selection of VRC01-like antibody subpopulations by the germline-targeting immunogens tested here will be even more pronounced in humans or transgenic animals expressing broader VRC01-class B cell receptors.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents should be directed to and will be fulfilled by Leonidas Stamatatos (lstamata@fredhutch.org).
Materials Availability
All reagents generated in this study can be made available upon request through Materials Transfer Agreements. Genbank numbers for the mAbs: MT585207-MT585272.
Data and Code Availability
The sequences of monoclonal antibodies reported will be deposited on GenBank and in the GitHub repository: https://github.com/krparks/epitope_presentation. The sequence of 426cOD is reported here and is deposited on GenBank with the accession number: MT671449. Coordinates and structure factors for 426cOD in complex with mature VRC01 Fab have been deposited with the Protein Data Bank under accession code 6VLW. Data collection and refinement statistics can be found in Supplemental Table 2.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Immunizations were performed with the C4b-based nanoparticles (heptameric forms) (Hofmeyer et al., 2013). Immunizations were performed in the glH-VRC01 mouse (Jardine et al., 2015). It is heterozygous for the transgene (glVRC01 HC) and expresses mouse light chains. Details on the number of animals used are presented in Supplemental Table 6. 6–12 week old glH-VRC01 knock-in mice were immunized intramuscularly with 60 μg or 50 μg of recombinant Env along with 60μg of polyinosinic:polycytidylic acid (poly (I:C)) or 50 ul GLA-LSQ (IDRI) split between the rear hind legs. Male and female mice were included in this study. Blood was collected via cardiac puncture two- or six-weeks following immunization, at which time spleen and lymph nodes were also harvested. Spleen and lymph nodes were processed into a single cell suspension separately and frozen in FBS + 10% DMSO and stored in liquid nitrogen until further use. Blood, spleens and lymph nodes were also isolated from non-immunized animals. The Fred Hutch Institutional Animal Care and Use Committee approved all mouse studies.
METHODS DETAILS
Computational design of the clade C 426c outer domain
RosettaRemodel (RosettaRemodel: A Generalized Framework for Flexible Backbone Protein Design; https://doi.org/10.1371/journal.pone.0024109) was used to engineer a gp120 outer domain of 426c based on 426c Core (426c TM4Δ1–3) (McGuire et al., 2016). To isolate the outer domain from inner domain, the C terminus of the α5 helix and N terminus of C2 region was closed with RosettaRemodel and a new N and C terminus of the outer domain was introduced before V4. To improve antibody interaction, asparagine(N)-linked glycan groups shielding the CD4-bs, at residues 289gp120, 332gp120, and 391gp120, were mutated to glutamines. A glycan site at position 448gp120 was re-introduced to the 426c sequence, as it was previously identified to be kinetically important for proper folding of the engineered outer domain. RosettaRemodel was used to introduce mutations in the residues adjacent the CD4-bs.
Comparison of the 426c outer domain with eOD identified differences in the position and rigidity of a helix important in CD4 and antibody binding (Supplemental Fig 3). Therefore, this helix was lengthened by two residues on the C-terminal end to extend it into the binding region and RosettaRemodel’s -build_disulf was used to introduce disulfide bonds and lock the helix into the conformation compatible with glVRC01-binsing. The space behind the helix in 426cOD had increased polarity and reduced packing compared to that of eOD (Supplemental Fig 3b and 3c), which includes many nonpolar and bulky residues in the pocket. Therefore, we replaced the incompatible residues in this region using RosettaRemodel’s manual design features. S2, which was among the lowest-energy and most-populated clusters of the resulting models, was selected for further testing (Supplemental Fig 3d). The sequence of S2 is reported in Supplemental Table 1, with the mutations from 426c highlighted. S2 was able to bind a subset of mature bnAbs: VRC-PG20, VRC01, and 3BNC60; but not germline ones (Supplemental Fig 3e and 3f, respectively).
In S2, the helix and the V5 loop were subsequently, brought closer together by extending the helix by one turn, allowing the two structures to better interact (Supplemental Fig 3g and 3h). To increase the packing and stability of the helix and V5 loop, we created an extended hydrogen bond network in the space (Supplemental Fig 3i). As a result, S2 gained the ability to bind a germline VRC01 antibody, glVRC-PG20 (Supplemental Fig 3k). This binding is dependent upon the residue at position 51 (Supplemental Fig 3I). In S2 this residue is an aspartic acid which hydrogen bonds with residues in V5, but when it is mutated to an isoleucine such interactions are not there and the protein is not recognized by glVRC-PG20 (Supplemental Fig 3j).
Construction of the clade C 426c outer domain
We ordered 426cOD-base gene segments with flanking segments homologous to pTT3 vector from Integrated DNA Technologies (IDT). We then used In-Fusion HD Cloning Plus system (Takara Bio USA, Inc.) to clone the gene construct into pTT3 vector with C-terminal 6x His-Avi-Tag.
Construction of yeast library
PCR and sequencing primers were obtained from IDT (Supplemental Table 7). The 426cOD-base gene segment was amplified using extension primers to add pETcon(−) yeast homologous recombination segments to each end. The GeneMorph II Random Mutagenesis Kit (Agilent) was used to generate the library which was then run on a 1% agarose gel and the gene was gel extracted and purified (Qiagen QIAquick Gel Extraction Kit). The purified library pool was then amplified with the extension primers to generate 12μg of DNA. Yeast EBY100 cells were transformed with library DNA and linearized pETcon(−) plasmid (McGuire et al., 2016). After transformation (minimum of 1E7 transformants), cells were grown for two days in SDCAA medium at 30 °C. Cells can then be galactose induced with SGCAA media for yeast display experiment or stored in SDCAA with 20% glycerol (w/v) at 1E7 cells per aliquot at −80 °C.
Yeast display
Cell aliquots were thawed on ice and centrifuged at 13,000 RPM for 30s, resuspended at 1E7 cells per ml of C-Trp-Ura medium, and grown at 30°C overnight. Cells were then centrifuged at 13,000 RPM for 1 min and resuspended (1E7 per ml) in SGCAA medium and induced at 30°C for 16–20 hrs. glVRC01 IgG was biotinylated with EZ-Link NHS-PEG4-Biotin and labeling kit (Thermo Fisher Scientific) at a one to one ratio of protein to biotin and tetramerized with streptavidin-phycoerythrin. Yeast cells expressing Env on their surface (based on glVRC01-SAPE and anti-c-Myc fluorescein isothiocyanate (FITC, Immunology Consultant Lab) staining) were sorted using a Sony SH800. Sorted cells were grown for two days in C-Trp-Ura media at 30°C. After 4 rounds of sorting the cells were plated on a C-Trp-Ura agarose plate. DNA from individual colonies was isolated and sequenced (GENEWIZ) to identify potential amino acid mutations that lead to glVRC01-recognition. Mutations were then introduced into the mammalian pTT3 426c base plasmid by site-directed mutagenesis using Quick Change II system (Agilent Technologies, Inc.) and conventional primers ordered from IDT (Supplemental Table 7).
Protein expression and purification
All the recombinant envelopes were expressed and purified as previously described(McGuire et al., 2016; Parks et al., 2019) Briefly, Envs were produced in 293E or 293F. Cell supernatants were purified by lectin affinity chromatography (Galanthus nivalis, Vector Labs), then subjected to Superdex 200 size exclusion chromatography (GE Healthcare).
Monomeric proteins were produced by transfecting 293E cells with Env encoding plasmid. Cells are cultured for 6 days at 37°C, 5% CO2, 80% humidity, and shaking at 125 RPM. Cells were centrifuged for 3000 RPM for 30 minutes and the supernatant sterile filtered through at .22 uM filter. Protein was purified by passing the cellular supernatant over a 5ml HisTrap Fast Flow column. The eluted protein was purified on SEC as described above.
426c Core and 426cOD CD4-bs KO Env constructs contain the D368R, E370A, and D279A mutations, while the eOD-GT8 KO contains the D368R mutation and the amino acids at positions 276–279 have been mutated to “NFTA”.
Crystallization
The VRC01 Fab and 426cOD complex was formed by mixing equal molar parts of VRC01 Fab and 426cOD for 1 hour at room temperature. The complex was concentrated to ~10 mgs/mL for crystallization trials.
Crystallization, data collection and refinement
Crystallization conditions were screened and monitored with an NT8 drop setter and Rock Imager (Formulatrix). Screening was done with Rigaku Wizard Precipitant Synergy block no. 2, Molecular Dimensions Proplex screen HT-96, and Hampton Research Crystal Screen HT using the sitting drop vapor diffusion method. Final crystals were grown in a solution of 18.43% PEG 3350, 11- .01%, Lithium Sulfate, 0.11 M Imidazole pH 6.5. Crystals were cryoprotected in a solution of 30% excess of the crystallization condition and 20% glycerol. Crystals were sent to ALS 5.0.1 and diffraction data was collected to 3.42 Å. Data was processed using HKL-2000 (Otwinowski and Minor, 1997). The structure was solved using molecular replacement using PDB ID: 5IFA as a search model in Phaser in Phenix (Adams et al., 2010). The structure was further refined with COOT (Emsley and Cowtan, 2004) and Phenix (Adams et al., 2010). The refinement statistics are summarized in Supplemental Table 2.
Negative-stain EM sample preparation
All nanoparticle constructs used in this study (3 μL) were negatively stained at final concentrations between 10 μg/mL and 20 μg/mL using Gilder Grids overlaid with a thin layer of amorphous carbon and 2% uranyl formate, as previously described (Veesler et al., 2014).
Negative-Stain EM data collection and processing
Data were collected on an FEI Technai 12 Spirit 120kV electron microscope equipped with a Gatan Ultrascan 4000 CCD camera. A total of 150–200 images were collected per sample by using a random defocus range of 1.5–2.5 μm with a total exposure of 45 e−/A2. Data were automatically acquired using Leginon (Suloway et al., 2005), and data processing was carried out using Appion (Lander et al., 2009). The parameters of the contrast transfer function (CTF) were estimated using CTFFIND4 (Mindell and Grigorieff, 2003), and particles were picked in a reference-free manner using DoG picker (Voss et al., 2009). Particles were extracted with a binning factor of 2 after correcting for the effect of the CTF by flipping the phases of each micrograph with EMAN 1.9 (Ludtke et al., 1999). Particle stacks were pre-processed in RELION/3.0 (Zivanov et al., 2018; Kimanius et al., 2016; Scheres, 2012b; a) with an additional binning factor of 2 applied, resulting in a final pixel size of 6.4 Å. Resulting particles were sorted by reference-free 2D classification over 25 iterations.
Biolayer interferometry (BLI)
Antibody binding to recombinant Env proteins was determined using biolayer interferometry on the Octet Red 96 (ForteBio, Inc, Menlo Park, CA). Briefly, anti-human Fc capture biosensors (ForteBio, Cat#: 18–5063) were activated by immersion into 1x Kinetics Buffer (1x PBS, 0.1% BSA, 0.02% Tween-20, 0.005% NaN3) for 10 minutes. Antibodies were loaded onto an anti-human Fc capture probe at 20 ug/ml. The probes were then dipped into solutions containing 2 uM recombinant Env. Parameters for all BLI assays were: 30 seconds of baseline measurement, 240 seconds to load the antibody onto the anti-human Fc capture probe, 60 seconds of baseline measurement, 300 seconds of association, 300 seconds of dissociation. All measurements of Env-Ab binding were corrected by subtracting the background signal obtained from Env traces generated with an irrelevant negative control IgG.
Kinetic analyses were performed by BLI using recombinant Fabs loaded onto FAB2G biosensors (ForteBio, Cat#: 18–5126) (@ 40 ug/ in 1XPBS) and 2-fold dilutions of Env monomers. The assay parameters are the same as for measuring the binding of IgG, but with an extended dissociation phase of 600 seconds. Curve fitting was used to determine relative apparent antibody affinities for envelope was performed using a 1:1 binding model and the data analysis software (ForteBio). Mean kon, koff, and KD values were determined by averaging all binding curves within a dilution series having R2 values of greater than 95% confidence level.
Plasma and antibody ELISA
Plasma samples were heated inactivated at 56° C for 1 hour, centrifuged at 13,000 RPM for 10 min at stored at 4° C or −20° C. ELISAs were done in a 384 well plate format. 30 ul of protein at 0.1 uM in coating buffer (0.1M sodium bicarbonate, pH: 9.4–9.6) was added to each well and incubated a room temperature overnight. Plates were washed 4x with ELISA wash buffer (1XPBS + .2% Tween-20). 80 ul of blocking buffer (1X PBS + 10% non-fat milk + 0.03% Tween20) was added to the plates and they were incubated at 37°C for 1–2 hours. Plates were then washed 4x with ELISA wash buffer. Plasma was diluted 1:10 in blocking buffer and diluted 1:3 across or down the plate. His tag control started at 1 mg/ml. The plates were incubated for 1 hour at 37°C. The plates were washed 4x with ELISA was buffer. 30 ul of secondary antibody was added to each well. The plates were incubated for an hour at 37° C. Plates were washed 4x with ELISA wash buffer. Following washing 30 ul of SureBlue Reserve TMB Microwell Peroxidase Substrate: KPL (Cat #: 53–00-02) was added to each well. The plates were incubated for 5 minutes at room temperature. 30 ul of 1N H2SO4 was added to each well. Plates were read immediately on the SpectraMax M2 microplate reader (Molecular Devices) at 450 nm. Blank wells were used to subtract the background signal in the analysis.
Single cell sorting
Spleen samples from −80°C were thawed in 37°C water bath until a small ice pellet remained in the sample tube. Then warm (37°C) RPMI was added dropwise to the samples. Cells were then spun down at 300xg for 5 mins and washed with 20ml FACs buffer (1x PBS containing 2% FBS and 1mM EDTA) once and then re-suspended in the remaining drop of FACs buffer after decantation.
Cells were then incubated with 1 pmol PE-DL650 and 3 pmols APC-DL755(decoys) for 5 mins at room temperature and then 1 pmol PE-antigen tetramer and 1 pmol APC-antigen_knock out (KO) tetramer for 25 mins on ice in dark. Tetramers were made by combining biotinylated Env with Streptavidin conjugated to either a PE or APC fluorophore (Prozyme, PJFS25 and PJ27S). Cells were then washed with FACs buffer once and then re-suspended in remaining drop of FACs buffer after decantation. 50 ul anti-PE MicroBeads and 50 ul anti-APC MicroBeads (Miltenyi Biotec) were then added to the cells and they were incubated for 15–30 mins on ice. Cells bound with PE and/or APC were then enriched based on MACS cell separation manual.
Antigen-enriched samples were then stained with the following list of antibody-fluorophore conjugates: IgG1 FITC (BD Biosciences Cat#: 553443, 1:100 dilution), IgG2b FITC (BD Biosciences Cat#: 553395, 1:50 dilution), IgG2c FITC (Bio-Rad Cat#: STAR135F, 1:100 dilution), IgG3 FITC (BD Biosciences Cat#: 553403, 1:400 dilution), IgD PerCP-Cy5.5 (Biolegend Cat#: 405710, 1:180 dilution), Fixable viability stain BV510 (Affymetrix Cat#: 65-0866-14, 1:200 dilution), CD3 BV510 (BD Biosceinces Ct#: 564024, 1:50 dilution), CD4 BV510 (BD Biosciences Cat#: 563106, 1:100 dilution), Gr-1 BV510 (BD Biosciences Cat#: 563040, 1:200 dilution), F4/80 BV510 (BD Biosciences Cat#: 123135, 1:50 dilution), IgM BV605 (Biolegend Cat#: 406523, 1:50 dilution), CD19 BV650 (BD Bioscience Cat#: 563235, 1:100 dilution), and B220 BV786 (BD Bioscience Cat#: 563894, 1:100 dilution). Cells were stained for 30 min on ice in the dark. Cells were then washed 3 times with 2 ml of FACs buffer wash. Samples were then re-suspended in 1–2 ml of FACs buffer.
Naïve, antigen-specific B cells were sorted as CD3-, CD4-, Gr-1-, F4/80-, B220+, CD19+, antigen+/antigen KO-. Class-switched IgG B cells from immunized animals were sorted based on the following markers: CD3-, CD4-, Gr-1-, F4/80-, B220+, CD19+, IgM-, IgD-, IgG1+, IgG2b+, IgG2c+, IgG3+, antigen+/antigen KO- (Supplemental Fig 4). Individual B cells were sorted using the FACS ARIA II into a 96 well plate containing lysis buffer, as previously reported (Parks et al., 2019). The number of cells sorted from each immunization group is now indicated in Supplemental Table 6.
VH/VL amplification and sequencing
The amplification and sequencing of VH and VL genes were performed as previously described (Parks et al., 2019). For light chain PCR reactions, 15 ul of cDNA and 15 ul of first round PCR product were used. 7 ul of cDNA or PCR product was used for the heavy chain PCR reactions. Samples were sequenced using Sanger sequencing (Genewiz, Seattle, WA) and sequence analysis was performed as previously described (Parks et al., 2019). Amino acid and nucleotide mutations were identified by aligning the VH/VL gene sequences to the corresponding germline genes (IMGT Repertoire) using the Geneious Software (Version 8.1.9). For VL, mutations were counted beginning at the CDR1 of the V-gene to the 3’ end of FW3. For VH, mutations were counted beginning at the CDR1 of the V-gene to the end of the CDR3. The number of sequences obtained from each immunization group is summarized in Supplemental Table 6.
Monoclonal antibody production
Gene-specific PCR was used to amplify the DNA product from the first round PCR using primers designed to anneal to the gene of interest as well as add ligation sites to facilitate insertion of the DNA fragment into the human IgG1 vector [ptt3 k for kappa (Snijder et al., 2018), and PMN 4–341 for gamma (Mouquet et al., 2010)]. Each gene specific PCR reaction contained 0.5 ul each of 10 uM 5’ and 3’ primer, 22.5 ul Accuprime Pfx Supermix (Cat#: 12344040), and 1.5 ul of 1st or 2nd round PCR product. The gene-specific PCR product was infused into the cut IgG1 vector in a reaction containing 12.5 ng of cut vector, 50 ng of insert, 0.5 ul of 5X Infusion enzyme (InFusion HD Cloning Kit, Cat#: 639649), and nuclease-free water to bring volume to 2.5 ul. The entire reaction was used to transform competent E. coli cells and plated on agar plates containing ampicillin. DNA was prepared using QIAprep Spin Miniprep Kit (Qiagen, Cat#: 27106). Equal amounts of heavy and light chain DNA and 293F transfection reagent (Millipore Sigma, Cat#: 72181) were used to transfect 293E cells. 5–7 days post transfection cell supernatants were collected, and the antibodies were purified using Pierce Protein A agarose beads (ThermoFisher, Cat#: 20334). The antibodies were eluted using 0.1 M Citric Acid into a tube containing 1 M Tris. The antibodies were buffer exchanged into 1XPBS using an amicon centrifugal filter.
To make Fabs, the IgG was cleaved overnight at 37°C to generate antigen binding fragment (Fab) with Endoproteinase Lys-C (NEB). To remove undigested IgG and IgG Fc fragments, the mixture was incubated with Protein A Agarose Resin for 1 hour at room temperature. Beads were washed with 1X PBS to remove excess Fab. Fab was further purified on SEC using a HiLoad 16/600 Superdex 200 pg (GE) column.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mean and standard deviations were calculated using R (Version 3.4.1). Statistical analyses were calculated using R (Version 3.4.1) (R Core Team, 2018) and GraphPad Prism. Descriptions of the statistical methods used for each data set are described in the figure legends. The tidyverse packages (Wickham, 2017) were used in R to manipulate data and create graphs in addition to GraphPad Prism. WebLogo was used to generate the logo plots (Crooks et al., 2004).
Supplementary Material
Supplemental Table 5. VH/VL sequences isolated following eOD-GT8 immunization, related to figure 4 and supplemental figures 2 and 4.
Amino acid sequences are aligned to the V gene from which they are derived. For sequences where the PCR product did not contain the 5’ end, the sequence is shown beginning at CDR1. Sequences from eOD-GT8 (a), 426c Core (b) and 426c OD (c) immunized animals. Sequences of human antibodies used (d).
Supplemental Table 3. Per-residue buried surface area (BSA) and interactions of glVRC01 bound to different antigens calculated by PDBePISA, related to figure 2.
Comparison of glVRC01 interactions with different HIV-1 antigens: a) bound to 426cOD (modeled onto PDB ID 6MFT) and WT 426c Core (PDB ID 6MFT), b) bound to 426cOD (modeled onto PDB ID 4JPK) and WT 426c Core (PDB ID 6MFT), c) bound to 426cOD (modeled) and gl3BNC60 bound to 426c core (PDB ID 5FEC) and d) bound to 426cOD and to eOD-GT8 (modeled usingPDB ID 4JPK). Residues with non-zero BSA are shown. Structurally aligned residues in the various antigens are shown in the same row. H: Hydrogen-bond, S: Salt-bridge, D: Disulfide bond, BSA: Buried Surface Area (Ų). aResidues that are mutated from WT 426c Core. bResidues are part of the gp120 inner domain. cResidues that differ in sequence in eOD-GT8 relative to 426cOD.
Highlights.
An epitope present on different immunogens selects for different targeted B cells
Each selected subpopulation comprises B cells with diverse affinities
The relative affinity dictates the fate of B cells following the initial selection
Developed 426cOD, a novel VRC01-class germline targeting immunogen
ACKNOWLEDGEMENTS
This study was supported by NIH/NIAID grants R01 AI104384 and P01 AI138212. We thank the James B. Pendleton Charitable Trust for the support of the Formulatrix robotic instrumentation. X-ray diffraction data were collected at the Berkeley Center for Structural Biology beamline 5.0.1, which is supported in part by the National Institute of General Medical Sciences. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under contract DE-AC02-05CH11231. P.-S.H. is supported by the U.S. Department of Energy, Office of Science, Office of Advanced Scientific Computing Research, Scientific Discovery through Advanced Computing (SciDAC) program and Stanford’s Bio-X Interdisciplinary Initiatives Seed Grants Program. We would also like to thank Cassie Brian from the Baker lab at the University of Washington for her help with the yeast display experiments.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests. P.-S.H. is an inventor on the patent for eOD (US Patent 10,421,789). L.S. is an inventor on US Patent US 2018/0117140.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abbott RK, Lee JH, Menis S, Skog P, Rossi M, Ota T, Kulp DW, Bhullar D, Kalyuzhniy O, Havenar-Daughton C, et al. (2018). Precursor Frequency and Affinity Determine B Cell Competitive Fitness in Germinal Centers, Tested with Germline-Targeting HIV Vaccine Immunogens. Immunity 48, 133–146 e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi R, Voss JE, Liang CH, Briney B, McCoy LE, Wu CY, Wong CH, Poignard P, and Burton DR (2015). Identification of Common Features in Prototype Broadly Neutralizing Antibodies to HIV Envelope V2 Apex to Facilitate Vaccine Design. Immunity 43, 959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsignori M, Zhou T, Sheng Z, Chen L, Gao F, Joyce MG, Ozorowski G, Chuang GY, Schramm CA, Wiehe K, et al. (2016). Maturation Pathway from Germline to Broad HIV-1 Neutralizer of a CD4-Mimic Antibody. Cell 165, 449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst AJ, Weidle CE, Gray MD, Frenz B, Snijder J, Joyce MG, Georgiev IS, Stewart-Jones GB, Kwong PD, McGuire AT, et al. (2018). Germline VRC01 antibody recognition of a modified clade C HIV-1 envelope trimer and a glycosylated HIV-1 gp120 core. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briney B, Sok D, Jardine JG, Kulp DW, Skog P, Menis S, Jacak R, Kalyuzhniy O, de Val N, Sesterhenn F, et al. (2016). Tailored Immunogens Direct Affinity Maturation toward HIV Neutralizing Antibodies. Cell 166, 1459–1470 e1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochet X, Lefranc MP, and Giudicelli V. (2008). IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 36, W503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DR, and Hangartner L. (2016). Broadly Neutralizing Antibodies to HIV and Their Role in Vaccine Design. Annu Rev Immunol 34, 635–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charif D, and Lobry JR (2007). SeqinR 1.0–2: a contributed package to the R Project for statistical computing devoted to biological sequences retrieval and analysis In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations, Bastolla U, Porto M, Roman E, and Vendruscolo M, eds. (Springer; ), pp. 207–232. [Google Scholar]
- Chen F, Tzarum N, Wilson IA, and Law M. (2019). VH1–69 antiviral broadly neutralizing antibodies: genetics, structures, and relevance to rational vaccine design. Curr Opin Virol 34, 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Porto JM, Haberman AM, Kelsoe G, and Shlomchik MJ (2002). Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J Exp Med 195, 1215–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosenovic P, von Boehmer L, Escolano A, Jardine J, Freund NT, Gitlin AD, McGuire AT, Kulp DW, Oliveira T, Scharf L, et al. (2015). Immunization for HIV-1 Broadly Neutralizing Antibodies in Human Ig Knockin Mice. Cell 161, 1505–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Escolano A, Gristick HB, Abernathy ME, Merkenschlager J, Gautam R, Oliveira TY, Pai J, West AP Jr., Barnes CO, Cohen AA, et al. (2019). Immunization expands B cells specific to HIV-1 V3 glycan in mice and macaques. Nature 570, 468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicelli V, Brochet X, and Lefranc M-P (2011). IMGT/V-QUEST: IMGT Standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb. Protoc 2011, 695–715. [DOI] [PubMed] [Google Scholar]
- Havenar-Daughton C, Sarkar A, Kulp DW, Toy L, Hu X, Deresa I, Kalyuzhniy O, Kaushik K, Upadhyay AA, Menis S, et al. (2018). The human naive B cell repertoire contains distinct subclasses for a germline-targeting HIV-1 vaccine immunogen. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmeyer T, Schmelz S, Degiacomi MT, Dal Peraro M, Daneschdar M, Scrima A, van den Heuvel J, Heinz DW, and Kolmar H. (2013). Arranged sevenfold: structural insights into the C-terminal oligomerization domain of human C4b-binding protein. J Mol Biol 425, 1302–1317. [DOI] [PubMed] [Google Scholar]
- Huang J, Kang BH, Ishida E, Zhou T, Griesman T, Sheng Z, Wu F, Doria-Rose NA, Zhang B, McKee K, et al. (2016). Identification of a CD4-Binding-Site Antibody to HIV that Evolved Near-Pan Neutralization Breadth. Immunity 45, 1108–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PS, Ban YE, Richter F, Andre I, Vernon R, Schief WR, and Baker D. (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine J, Julien JP, Menis S, Ota T, Kalyuzhniy O, McGuire A, Sok D, Huang PS, MacPherson S, Jones M, et al. (2013). Rational HIV immunogen design to target specific germline B cell receptors. Science 340, 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine JG, Kulp DW, Havenar-Daughton C, Sarkar A, Briney B, Sok D, Sesterhenn F, Ereno-Orbea J, Kalyuzhniy O, Deresa I, et al. (2016). HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science 351, 1458–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine JG, Ota T, Sok D, Pauthner M, Kulp DW, Kalyuzhniy O, Skog PD, Thinnes TC, Bhullar D, Briney B, et al. (2015). HIV-1 VACCINES. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 349, 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D, Forsberg BO, Scheres SH, and Lindahl E. (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouskoff V, Famiglietti S, Lacaud G, Lang P, Rider JE, Kay BK, Cambier JC, and Nemazee D. (1998). Antigens varying in affinity for the B cell receptor induce differential B lymphocyte responses. J Exp Med 188, 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong PD, and Mascola JR (2012). Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity 37, 412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, et al. (2009). Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol 166, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, and Chiu W. (1999). EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128, 82–97. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy LE, and Burton DR (2017). Identification and specificity of broadly neutralizing antibodies against HIV. Immunol Rev 275, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire AT, Gray MD, Dosenovic P, Gitlin AD, Freund NT, Petersen J, Correnti C, Johnsen W, Kegel R, Stuart AB, et al. (2016). Specifically modified Env immunogens activate B-cell precursors of broadly neutralizing HIV-1 antibodies in transgenic mice. Nat Commun 7, 10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Ramirez M, Garces F, Escolano A, Skog P, de Taeye SW, Del Moral-Sanchez I, McGuire AT, Yasmeen A, Behrens AJ, Ozorowski G, et al. (2017). Design and crystal structure of a native-like HIV-1 envelope trimer that engages multiple broadly neutralizing antibody precursors in vivo. J Exp Med 214, 2573–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, and Grigorieff N. (2003). Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142, 334–347. [DOI] [PubMed] [Google Scholar]
- Mouquet H, Scheid JF, Zoller MJ, Krogsgaard M, Ott RG, Shukair S, Artyomov MN, Pietzsch J, Connors M, Pereyra F, et al. (2010). Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature 467, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W. (1997). Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pages G, and Grudinin S. (2018). Analytical symmetry detection in protein assemblies. II. Dihedral and cubic symmetries. J Struct Biol 203, 185–194. [DOI] [PubMed] [Google Scholar]
- Parks KR, MacCamy AJ, Trichka J, Gray M, Weidle C, Borst AJ, Khechaduri A, Takushi B, Agrawal P, Guenaga J, et al. (2019). Overcoming Steric Restrictions of VRC01 HIV-1 Neutralizing Antibodies through Immunization. Cell Rep 29, 3060–3072 e3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejchal R, Doores KJ, Walker LM, Khayat R, Huang PS, Wang SK, Stanfield RL, Julien JP, Ramos A, Crispin M, et al. (2011). A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 334, 1097–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018). R: The R Project for Statistical Computing. https://www.R-project.org/. RosettaRemodel: a generalized framework for flexible backbone protein design. PLoS One 6, e24109.
- Sajadi MM, Dashti A, Rikhtegaran Tehrani Z, Tolbert WD, Seaman MS, Ouyang X, Gohain N, Pazgier M, Kim D, Cavet G, et al. (2018). Identification of Near-Pan-neutralizing Antibodies against HIV-1 by Deconvolution of Plasma Humoral Responses. Cell 173, 1783–1795 e1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangesland M, Ronsard L, Kazer SW, Bals J, Boyoglu-Barnum S, Yousif AS, Barnes R, Feldman J, Quirindongo-Crespo M, McTamney PM, et al. (2019). Germline-Encoded Affinity for Cognate Antigen Enables Vaccine Amplification of a Human Broadly Neutralizing Response against Influenza Virus. Immunity 51, 735–749 e738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf L, West AP, Sievers SA, Chen C, Jiang S, Gao H, Gray MD, McGuire AT, Scheid JF, Nussenzweig MC, et al. (2016). Structural basis for germline antibody recognition of HIV-1 immunogens. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Feldhahn N, Walker BD, Pereyra F, Cutrell E, Seaman MS, Mascola JR, Wyatt RT, Wardemann H, et al. (2009). A method for identification of HIV gp140 binding memory B cells in human blood. J Immunol Methods 343, 65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TY, Pietzsch J, Fenyo D, Abadir A, Velinzon K, et al. (2011). Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333, 1633–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). A Bayesian view on cryo-EM structure determination. J Mol Biol 415, 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, and Chen S. (2012). Prevention of overfitting in cryo-EM structure determination. Nat Methods 9, 853–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih TA, Meffre E, Roederer M, and Nussenzweig MC (2002). Role of BCR affinity in T cell dependent antibody responses in vivo. Nat Immunol 3, 570–575. [DOI] [PubMed] [Google Scholar]
- Silva M, Nguyen TH, Philbrook P, Chu M, Sears O, Hatfield S, Abbott RK, Kelsoe G, and Sitkovsky MV (2017). Targeted Elimination of Immunodominant B Cells Drives the Germinal Center Reaction toward Subdominant Epitopes. Cell Rep 21, 3672–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder J, Ortego MS, Weidle C, Stuart AB, Gray MD, McElrath MJ, Pancera M, Veesler D, and McGuire AT (2018). An Antibody Targeting the Fusion Machinery Neutralizes Dual-Tropic Infection and Defines a Site of Vulnerability on Epstein-Barr Virus. Immunity 48, 799–811 e799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steichen JM, Lin YC, Havenar-Daughton C, Pecetta S, Ozorowski G, Willis JR, Toy L, Sok D, Liguori A, Kratochvil S, et al. (2019). A generalized HIV vaccine design strategy for priming of broadly neutralizing antibody responses. Science 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B. (2005). Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Tas JM, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM, Chen CS, Weill JC, Reynaud CA, Browne EP, et al. (2016). Visualizing antibody affinity maturation in germinal centers. Science 351, 1048–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Cheng C, Chen X, Duan H, Cheng HL, Dao M, Sheng Z, Kimble M, Wang L, Lin S, et al. (2016). Induction of HIV Neutralizing Antibody Lineages in Mice with Diverse Precursor Repertoires. Cell 166, 1471–1484 e1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiller T, Busse CE, and Wardemann H. (2009). Cloning and expression of murine Ig genes from single B cells. J Immunol Methods 350, 183–193. [DOI] [PubMed] [Google Scholar]
- Umotoy J, Bagaya BS, Joyce C, Schiffner T, Menis S, Saye-Francisco KL, Biddle T, Mohan S, Vollbrecht T, Kalyuzhniy O, et al. (2019). Rapid and Focused Maturation of a VRC01-Class HIV Broadly Neutralizing Antibody Lineage Involves Both Binding and Accommodation of the N276- Glycan. Immunity 51, 141–154 e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora GD, and Nussenzweig MC (2012). Germinal centers. Annu Rev Immunol 30, 429–457. [DOI] [PubMed] [Google Scholar]
- West AP Jr., Diskin R, Nussenzweig MC, and Bjorkman PJ (2012). Structural basis for germ-line gene usage of a potent class of antibodies targeting the CD4-binding site of HIV-1 gp120. Proc Natl Acad Sci U S A 109, E2083–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2017). Tidyverse: easily install and load the “tidyverse.”. https://cran.r-project.org/web/packages/tidyverse/index.html. [Google Scholar]
- Williams WB, Zhang J, Jiang C, Nicely NI, Fera D, Luo K, Moody MA, Liao HX, Alam SM, Kepler TB, et al. (2017). Initiation of HIV neutralizing B cell lineages with sequential envelope immunizations. Nat Commun 8, 1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, Kwon YD, Scheid JF, Shi W, Xu L, et al. (2010). Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329, 811–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Zhu J, Wu X, Moquin S, Zhang B, Acharya P, Georgiev IS, Altae-Tran HR, Chuang GY, Joyce MG, et al. (2013). Multidonor Analysis Reveals Structural Elements, Genetic Determinants, and Maturation Pathway for HIV-1 Neutralization by VRC01-Class Antibodies. Immunity 39, 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 5. VH/VL sequences isolated following eOD-GT8 immunization, related to figure 4 and supplemental figures 2 and 4.
Amino acid sequences are aligned to the V gene from which they are derived. For sequences where the PCR product did not contain the 5’ end, the sequence is shown beginning at CDR1. Sequences from eOD-GT8 (a), 426c Core (b) and 426c OD (c) immunized animals. Sequences of human antibodies used (d).
Supplemental Table 3. Per-residue buried surface area (BSA) and interactions of glVRC01 bound to different antigens calculated by PDBePISA, related to figure 2.
Comparison of glVRC01 interactions with different HIV-1 antigens: a) bound to 426cOD (modeled onto PDB ID 6MFT) and WT 426c Core (PDB ID 6MFT), b) bound to 426cOD (modeled onto PDB ID 4JPK) and WT 426c Core (PDB ID 6MFT), c) bound to 426cOD (modeled) and gl3BNC60 bound to 426c core (PDB ID 5FEC) and d) bound to 426cOD and to eOD-GT8 (modeled usingPDB ID 4JPK). Residues with non-zero BSA are shown. Structurally aligned residues in the various antigens are shown in the same row. H: Hydrogen-bond, S: Salt-bridge, D: Disulfide bond, BSA: Buried Surface Area (Ų). aResidues that are mutated from WT 426c Core. bResidues are part of the gp120 inner domain. cResidues that differ in sequence in eOD-GT8 relative to 426cOD.
Data Availability Statement
The sequences of monoclonal antibodies reported will be deposited on GenBank and in the GitHub repository: https://github.com/krparks/epitope_presentation. The sequence of 426cOD is reported here and is deposited on GenBank with the accession number: MT671449. Coordinates and structure factors for 426cOD in complex with mature VRC01 Fab have been deposited with the Protein Data Bank under accession code 6VLW. Data collection and refinement statistics can be found in Supplemental Table 2.