

Abstract
Central nervous system (CNS) diseases, both traumatic and neurodegenerative, are characterized by impaired mitochondrial bioenergetics and often disturbed mitochondrial dynamics. The dysregulation observed in these pathologies leads to defective respiratory chain function and reduced ATP production, thereby promoting neuronal death. As such, attenuation of mitochondrial dysfunction through induction of mitochondrial biogenesis (MB) is a promising, though still underexplored, therapeutic strategy. MB is a multifaceted process involving the integration of highly regulated transcriptional events, lipid membrane and protein synthesis/assembly and replication of mtDNA. Several nuclear transcription factors promote the expression of genes involved in oxidative phosphorylation, mitochondrial import and export systems, antioxidant defense and mitochondrial gene transcription. Of these, the nuclear-encoded peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is the most commonly studied and is widely accepted as the ‘master regulator’ of MB. Several recent preclinical studies document that reestablishment of mitochondrial homeostasis through increased MB results in inhibited injury progression and increased functional recovery. This perspective will briefly review the role of mitochondrial dysfunction in the propagation of CNS diseases, while also describing current research strategies that mediate mitochondrial dysfunction and compounds that induce MB for the treatment of acute and chronic neuropathologies.
Keywords: Mitochondrial biogenesis, Mitochondrial dysfunction, Mitochondrial dynamics, Mitophagy, Neuroinflammation, Traumatic brain injury, Spinal cord injury, Stroke, Alzheimer’s disease, Parkinson’s disease
1. Introduction
Mitochondria are vital in maintaining cellular functions, such as amino acid synthesis, fatty acid metabolism, adenosine triphosphate (ATP) production, apoptosis, ion homeostasis, antioxidant defenses and reactive oxygen species (ROS) regulation (Golpich et al., 2017). As such, mitochondria play an extensive role in mediating metabolic pathways and tissue function. Mitochondria are highly dynamic organelles with the ability to routinely modify their size, shape and organization in response to internal and external stimuli (van der Bliek et al., 2013). Dynamic regulation of mitochondrial morphology and content, including fission/fusion, mitophagy and biogenesis, allow mitochondria the plasticity and protection necessary to meet the metabolic needs for cell repair and regeneration while in the presence of cellular stress (Friedman and Nunnari, 2014).
In the presence of toxins or other stressors, mitochondria can become damaged and dysfunctional, leading to a wide breadth of consequences, including hindered oxidative phosphorylation and ATP production, depolarization of the mitochondrial membrane, mitochondrial DNA (mtDNA) fragmentation, oxidative stress, impaired calcium homeostasis, altered mitochondrial dynamics and activation of apoptotic pathways (Funk and Schnellmann, 2012; Gibbs et al., 2016; Gibson et al., 2010; McGill et al., 2012; Morais and De Strooper, 2010; Pisano et al., 2016). Therefore, mitochondrial dysfunction is characteristic of a multitude of acute and chronic diseases, including those of the central nervous system (CNS).
Because disruption of mitochondrial quality control mechanisms has been implicated in various diseases within the CNS, therapeutic strategies aimed at maintaining and/or restoring mitochondrial homeostasis and related cellular processes are becoming increasingly popular. Specifically, pharmacological induction of mitochondrial biogenesis (MB), the generation of new, functional mitochondria, is a promising therapeutic target for a wide range of acute and chronic diseases characterized by mitochondrial dysfunction (Cameron et al., 2016; Scholpa and Schnellmann, 2017; Whitaker et al., 2016). Fortunately, there exist several pharmacological agents known to induce MB that are approved by the U.S. Food and Drug Administration (FDA) and repurposing these drugs for the treatment of various CNS pathologies could be a relatively expeditious process.
This perspective will review the role of mitochondrial dysfunction in the propagation of CNS diseases, as well as describe current research strategies that mediate mitochondrial dysfunction and compounds that induce MB for the treatment of acute and chronic CNS diseases (Scholpa and Schnellmann, 2017).
2. Mitochondrial function/dysfunction
Traumatic and neurodegenerative CNS pathologies are frequently characterized by impaired mitochondrial bioenergetics and disturbed mitochondrial dynamics (Bordone et al., 2019; Reddy and Beal, 2005; Scholpa and Schnellmann, 2017). As expected, the dysregulation observed in many these diseases leads to defective respiratory chain function and reduced ATP production, promoting neuronal death (Burte et al., 2015; Sebastian et al., 2017). A comprehensive review detailing mitochondrial function in the CNS can be found in Smith and Gallo (2018).
2.1. Mitophagy
Mitochondrial dysfunction leads to apoptotic cell death in the presence of increased ROS production, calcium accumulation, opening of mitochondrial permeability transition pore (mPTP) and release of cytochrome c (cyt c) (Rodriguez-Enriquez et al., 2009; Sims and Muyderman, 2010). Furthermore, loss of mitochondrial function can initiate mitochondrial autophagy, or mitophagy, a process of selective mitochondrial degeneration where mitochondrial derivatives are engulfed and transported to the lysosome or peroxisome for degradation (Pickrell and Youle, 2015). A well-studied pathway of mitophagy is the PINK1/Parkin-dependent mitophagy pathway. This pathway is activated in the presence of mitochondrial damage and destabilization of ubiquitin kinase-induce kinase 1 (PINK1). Once released from the outer membrane of the mitochondria, PINK1 will recruit, phosphorylate and activate E3 ubiquitin ligase Parkin. Both PINK and Parkin will then ubiqitinate mitochondrial proteins and promote the formation of autophagosomes. Finally, autophagosomes will fuse with a lysosome, where degradation will take place (Fig. 1) (Fivenson et al., 2017; Narendra et al., 2010; Pickrell and Youle, 2015).
Fig. 1. Mitochondrial dynamics and mitophagy.
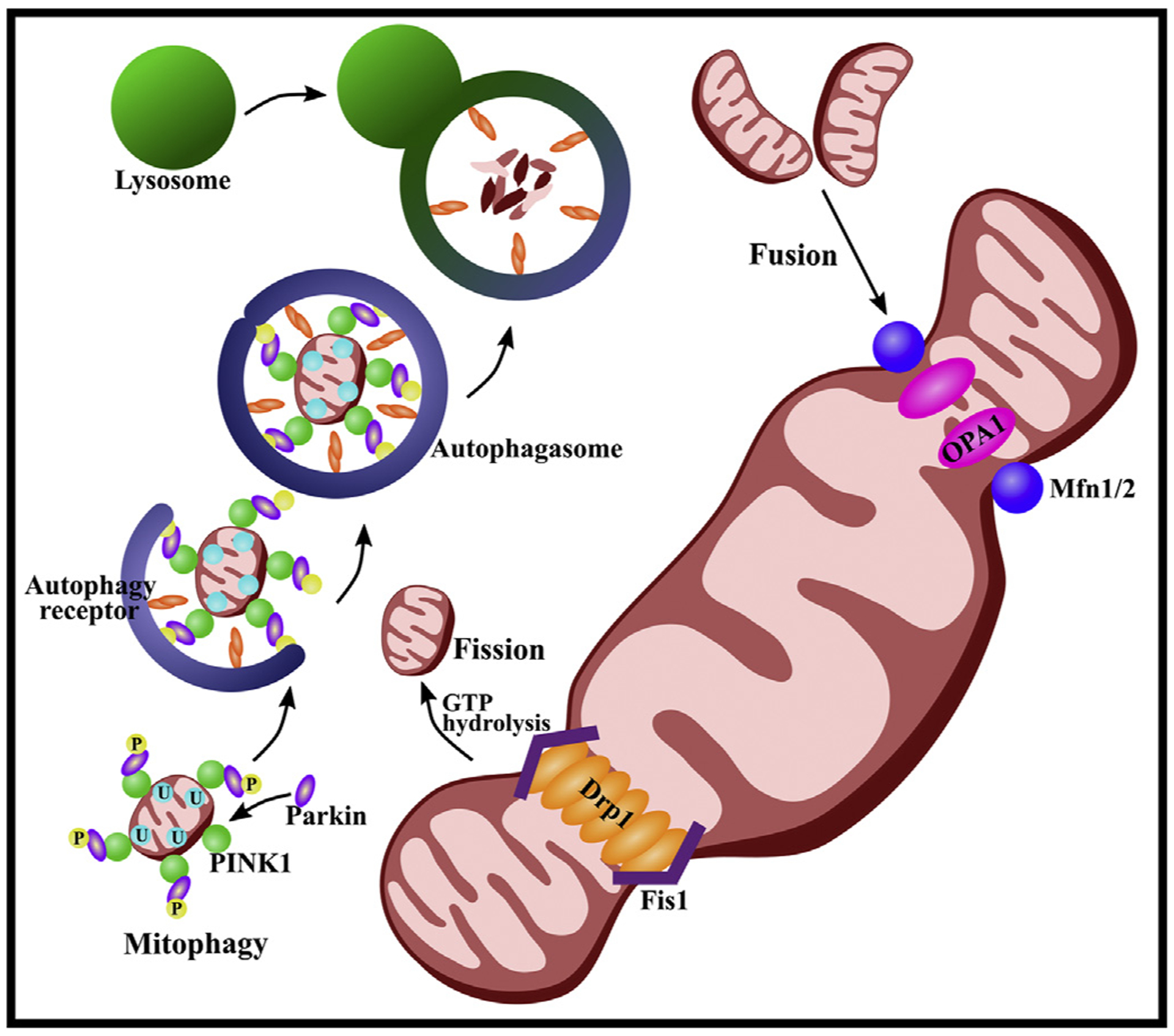
Three GTPases mediate the process of mitochondrial fusion: mitofusin (Mfn) 1 and 2, both existing on the outer membrane, and optic atrophy 1 (Opa1) located on the inner membrane. Mfn1 and Mfn2 initiate fusion of the outer membrane of two mitochondria by forming homo-oligomeric and hetero-oligomeric fusion complexes. Activation of the lipid-binding domain within Opa1 then create a pore in the membrane, completing mitochondrial fusion. The process of fission is initiated by recruitment of dynamin-related protein 1 (Drp1) from the cytosol to the mitochondrial membrane, where it can interact with Drp1 receptors on the outer mitochondrial membrane, including mitochondrial fission 1 protein (Fis1). Drp1 oligomerizes to form a ring-like structure around the mitochondria. The mitochondrial membrane can then be split via GTP hydrolysis. Excessive fission may promote mitophagy. The PINK1/Parkin-dependent mitophagy pathway is activated in the presence of mitochondrial damage and destabilization of ubiquitin kinase-induce kinase 1 (PINK1). Once released from the outer membrane of the mitochondria, PINK1 will recruit, phosphorylate (P) and activate E3 ubiquitin ligase Parkin. Both PINK and Parkin will then ubiqitinate (U) mitochondrial proteins and promote the formation of autophagosomes. Finally, autophagosomes will fuse with a lysosome where degradation will take place.
Mitophagy is essential in sustaining mitochondrial homeostasis, biogenesis and total number and quality of mitochondria (Golpich et al., 2017). Mitophagy has been shown to be neuroprotective and to reduce the production of ROS via clearance of dysfunctional mitochondria (Cao et al., 2017). Excessive ROS can exacerbate injuries of the brain, spinal cord and blood-CNS barriers by inducing apoptosis (Qu et al., 2016). Previous studies have shown defective mitophagy in neurodegenerative diseases, leading to the aggregation of autophagosomes, abnormal endosomes and abnormal lysosomes. This dysfunction hinders the mitophagy process, which likely contributes to the pathology of neural degeneration (Franco-Iborra et al., 2018; Ni et al., 2015). As such, insufficient or altered mitophagy can lead to cell death (Murphy, 2009) and may promote the development and propagation of many CNS-related diseases (Golpich et al., 2017).
2.2. Necrosis
Necrosis is an irreversible cell death process characterized by rapid loss of cellular membrane potential thereby leading to swelling of the cell, rupture and subsequent inflammation (Zong and Thompson, 2006). The mPTP is a key effector in the pathway to cell death (Baines et al., 2003; Karch et al., 2013; Karch and Molkentin, 2014; Xu et al., 2019) in that opening of the mPTP further exacerbates mitochondrial dysfunction (Baines et al., 2003; Karch et al., 2013; Karch and Molkentin, 2014). Recent studies suggest inhibition of mPTP opening may regulate programmed necrosis and limit neuronal loss in several CNS injuries (Xu et al., 2019; Ying and Padanilam, 2016). For additional information detailing the regulation of necrotic cell death, see Ying and Padanilam (2016).
2.3. Fission
Fusion and fission are two inverse processes encompassing mitochondrial dynamics that directly contribute to morphological changes of mitochondria (Fig. 1). Fusion mechanisms promote tethering and joining of two mitochondria, whereas fission initiates cleavage and division of mitochondria (Chan, 2006; Okamoto and Shaw, 2005; Westermann, 2008). Coordination of these processes maintain the growth, shape, distribution and structure of mitochondria, which is integral to homeostasis, cell stability and cell survival (Calo et al., 2013; Twig and Shirihai, 2011). Disruption to these processes may result in altered mitochondrial function and cellular bioenergetics, thereby contributing to the onset and propagation of several neuropathologies (Calkins et al., 2011; Filosto et al., 2011). Additionally, disruptions in mitochondrial dynamics may be a requirement for mitophagic mechanisms (Golpich et al., 2017).
Dysfunctional mitochondria contain impaired proteins, damaged membranes and mutated or fragmented mtDNA, all of which promote the division and fragmentation of mitochondria via activation of fission (Frank et al., 2012; Scott and Youle, 2010). Mitochondrial fission is initiated when cells require elimination of damaged mitochondria. The fission process is mediated by a family of dynamin-related proteins (Drps), particularly Drp1 (Boldogh and Pon, 2006; Hollenbeck and Saxton, 2005). This protein, once recruited from the cytosol to the mitochondrial membrane, can interact with Drp1 receptors on the outer mitochondrial membrane, including mitochondrial fission factor (Mff) and mitochondrial fission 1 protein (Fis1). Subsequently, Drp1 oligomerizes with Drp2 to form a ring-like structure around the mitochondria. The mitochondrial membrane is then able to be split via GTP hydrolysis (He et al., 2019; Lee et al., 2016; Loson et al., 2013; Otera et al., 2010; Palmer et al., 2011).
Defects in mitochondrial fission can induce mitochondrial dysfunction and related pathologies (Burte et al., 2015; Golpich et al., 2017). Accumulation of damaged or otherwise compromised mitochondria can have negative effects on electron transport chain (ETC) components and inhibit ATP production, resulting in cell death (Galloway et al., 2012; Twig et al., 2008; Twig and Shirihai, 2011). Furthermore, increased mitochondrial fragmentation has been reported in fibroblast cells of patients with neurodegenerative diseases, resulting in defective oxidative phosphorylation and ATP deficiency (Capaldi et al., 2004). Altered mitochondrial fission has also been linked with behavioral abnormalities and mood disorders. A recent study in mice revealed stress-induced triggers can promote severe mitochondrial fission in peripheral CD4+ T cells resulting in inflammation-induced anxiety-like behavior (Fan et al., 2019).
Drp1 expression is upregulated during oxidative stress, disturbing the balance of mitochondrial dynamics, and leading to mitochondrial dysfunction and eventual cell death (Wu et al., 2011). Reports demonstrate a reduction in Drp1 expression and mitochondrial fragmentation with antioxidant treatment, such as vitamin E or the antioxidant CoQ10 (MitoQ) (de Arriba et al., 2013; Ferrari et al., 2011). Reports also show increased oxidative stress and ROS production during multiple injury types, which is reduced with knockdown of Drp1 (Ferrari et al., 2011; Kobashigawa et al., 2011; Peng et al., 2011). Dysregulation of fission can also propagate apoptosis, while its inhibition has been shown to hinder the release of cyt c, a potent mediator of cell death pathways, thereby delaying apoptosis (Herzig and Martinou, 2008). Additionally, mitochondrial dysfunction can activate Fis1, further enhancing mitochondrial fission (Loson et al., 2013). These published data, among others, indicate that maintenance of fission regulates mitochondrial quality and bioenergetic properties (Sebastian et al., 2017; Twig et al., 2008; Westermann, 2012), and that dysregulation can contribute to neurodegenerative diseases.
2.4. Fusion
Fusion is the process by which two mitochondria fuse together, merging their membranes and sharing intracellular contents, such as proteins, lipids, and growth factors. Sharing of these components is a crucial aspect to maintaining proper ETC function (Westermann, 2012). Three GTPases control the conservative process of mitochondrial fusion: mitofusin (Mfn) 1 and 2, which exist on the outer membrane, and optic atrophy 1 (Opa1) located on the inner membrane (Escobar-Henriques and Anton, 2013; Ishihara et al., 2006; Santel and Fuller, 2001). Mfn1 and Mfn2 initiate the fusion of the outer membrane of two mitochondria by forming homo-oligomeric and hetero-oligomeric fusion complexes (Chernomordik and Kozlov, 2005; Hoppins et al., 2007). Then, activation of the lipid-binding domain within Opa1 creates a pore in the membrane, completing mitochondrial fusion (Hoppins et al., 2007; Meglei and McQuibban, 2009) (Fig. 1). Fusion contributes to the balance of matrix metabolites, while also aiding in the equilibrium of mitochondrial membrane contents, particularly that of complex I of the ETC (Busch et al., 2006; Dimmer and Scorrano, 2006; Ono et al., 2001).
Like fission, regulation of this process is critical for cell survival during all stages of life, from embryonic and cortical development, to everyday homeostatic mitochondrial function. With impaired fusion, mitochondrial fragmentation increases, resulting in decreased expression and distribution of mitochondrial-encoded proteins of the ETC and inhibition of ATP synthesis, both of which lead to neuronal death (Westermann, 2012). Reports reveal that enhanced fusion aids in the maintenance of mitochondrial integrity in healthy mitochondria (Benard and Karbowski, 2009); however, hindered activity of fusion may contribute to the development of neurodegenerative disorders (Zuchner et al., 2006; Zuchner et al., 2004). Additional studies demonstrate enhanced mitochondrial fusion in response to stress stimuli (e.g. UV irradiation) or exposure to known stress-inducing drugs. In this hyperfusion state, mitochondria form highly interconnected and elongated networks of mitochondria and have increased ATP synthesis (Mouli et al., 2009; Westermann, 2012). Maintaining a balance between fission and fusion activity is crucial for ensuring mitochondrial homeostasis, while disturbance of mitochondrial dynamics promotes and exacerbates an array of CNS pathologies.
3. Mitochondrial biogenesis (MB)
Given the vital importance of mitochondria to a plethora of cellular functions, it is not surprising that dozens of diseases, including many within the CNS, are characterized by mitochondrial dysfunction (Bordone et al., 2019; Golpich et al., 2017; Sheng et al., 2012). While many mitochondrial genes are encoded by the nuclear genome, mitochondria also contain their own circular genome, which is composed of 13 essential genes required for successful mitochondrial function. As such, induction of MB is dependent on the coordinated activation of both the mitochondrial and nuclear genomes (Scarpulla, 2008).
MB is a multifaceted process involving the integration of highly regulated transcriptional events, lipid membrane and protein synthesis/assembly and replication of mtDNA (Fig 2) (Ventura-Clapier et al., 2008). Furthermore, impaired MB can contribute to mitochondrial and cellular dysfunction (Golpich et al., 2017). Several nuclear transcription factors promote the expression of genes involved in oxidative phosphorylation, mitochondrial import and export systems, antioxidant defense and mitochondrial gene transcription (Fig 2). Of these, the nuclear-encoded peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is the most commonly studied and is widely accepted as the ‘master regulator’ of MB (Kelly and Scarpulla, 2004; Ventura-Clapier et al., 2008).
Fig. 2. Mitochondrial dysfunction and biogenesis.
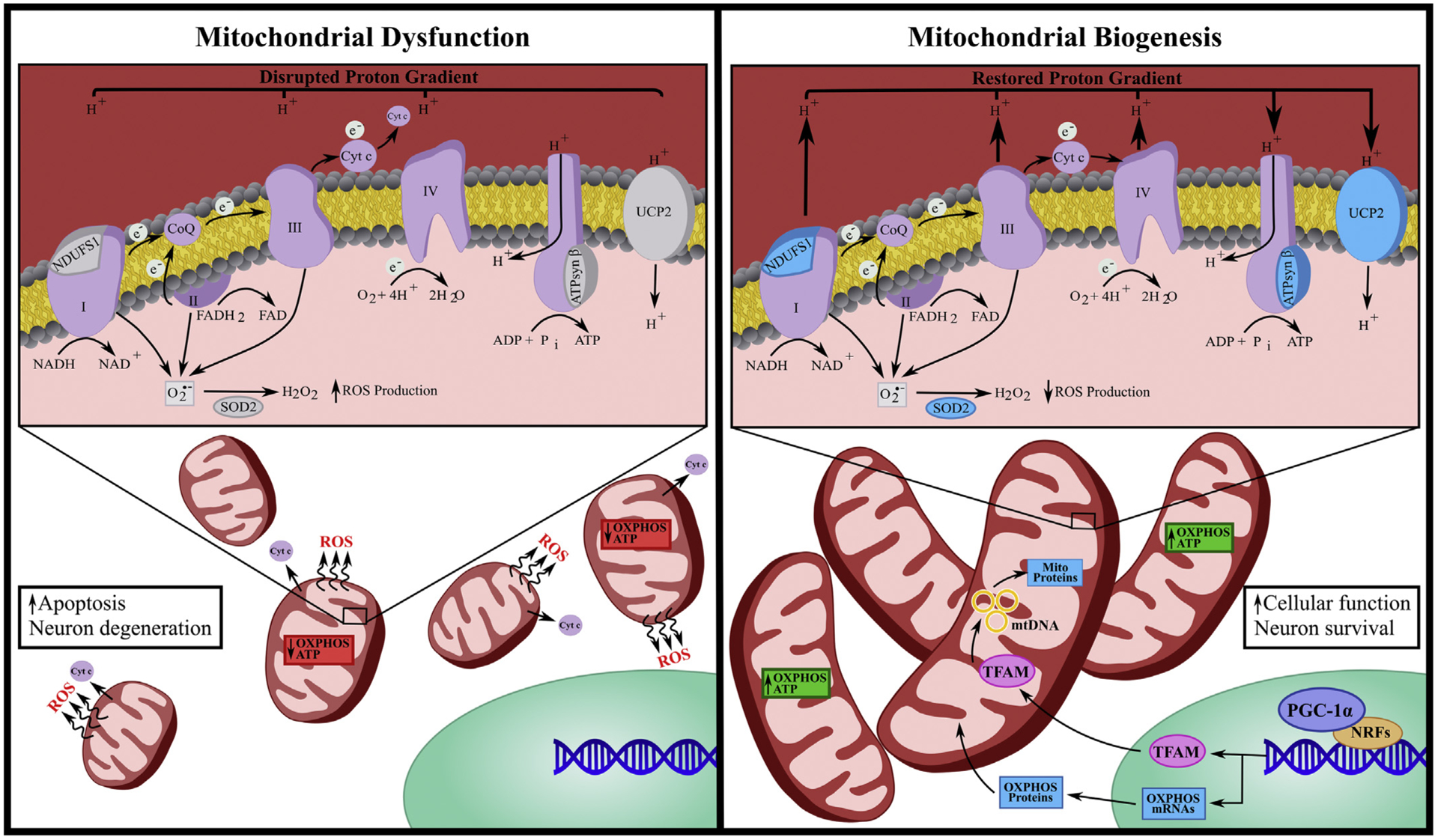
Mitochondrial dysfunction is characterized by decreased expression of oxidative phosphorylation (OXPHOS) proteins, impaired mitochondrial membrane potential, reduced ATP production as well as enhanced mitochondrial fission, reactive oxygen species (ROS) production and cytochrome c (cyt c) release. Mitochondrial dysfunction is often paired with increased apoptosis and neural degeneration. Conversely, mitochondrial biogenesis (MB) is characterized by mitochondrial fusion, reduced ROS, restoration of mitochondrial membrane potential and increased expression of OXPHOS proteins including ATP synthase β (ATPsyn β), NADH:Ubiquinone oxidoreductase core subunit 1 (NDUFS1) and uncoupling protein 2 (UCP2) as well as superoxide dismutase 2 (SOD2). MB induction often begins with activation of encoded peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) that co-activates nuclear respiratory factors (NRFs), which then promotes the transcription of several mitochondrial genes, including mitochondrial transcriptional factor a (TFAM). Once translated, TFAM translocates to the mitochondrial matrix and stimulates mtDNA replication and mitochondrial gene expression. MB is associated with enhanced cellular function and neuronal survival.
PGC-1α serves as a docking platform, recruiting additional transcription factors for the transcription of nuclear-encoded genes necessary for MB. Activation of PGC-1α initiates activation of nuclear respiratory factors (NRFs), which then promote the transcription of several mitochondrial genes, including various subunits of the ETC, such as ATP synthase, cyt c, cytochrome oxidase IV and mitochondrial transcriptional factor a (TFAM) (Cameron et al., 2016; Wu et al., 1999). Once translated, TFAM translocates to the mitochondrial matrix and stimulates mtDNA replication and mitochondrial gene expression (Ventura-Clapier et al., 2008). PGC-1α also has the capacity to interact with other transcription factors, such as peroxisome proliferator-activated receptors (PPARs), thyroid hormone, glucocorticoids, estrogen receptors, mitochondrial transporters, and antioxidant proteins (Ventura-Clapier et al., 2008). The transcription and translation of the aforementioned genes leads to MB and enhanced mitochondrial function. The expression of PGC-1α is highly inducible upon activation of physiological cues signaling for increased cellular metabolic needs, including oxidative stress and cell division (Handschin and Spiegelman, 2006). A variety of both central- and peripheral-related diseases are characterized by mitochondrial dysfunction, with several reports detailing a decrease in PGC-1α expression in both traumatic and neurodegenerative diseases (Scholpa et al., 2018a; Scholpa and Schnellmann, 2017). As such, current research aims to attenuate this dysfunction by targeting the activation of PGC-1α as a therapeutic strategy for inducing MB, as well as for evaluating mitochondrial homeostasis. Furthermore, ischemic injury, such as that which occurs with SCI, is also followed by reduced oxidative phosphorylation proteins as well as decreased PGC-1α and TFAM (Scholpa and Schnellmann, 2017; Whitaker et al., 2016). For more information detailing mechanisms and inducible compounds of MB induction, see Gibbs et al. (2018c) and Cameron et al. (2016). Reports discussed herein support the role of MB in restoring mitochondrial dynamics, increasing oxidative phosphorylation, attenuating expression of mitochondrial proteins and increasing cellular functions and neuronal survival, thereby promoting recovery in CNS diseases (Fig 2). See Table 1 for a partial list of CNS diseases characterized by mitochondrial dysfunction and evidence of pharmacological induction of MB as therapeutic interventions for each.
Table 1.
Partial list of CNS diseases characterized by mitochondrial dysfunction and evidence of pharmacological induction of MB as therapeutic interventions for each.
| Disease | Mitochondrial Dysfunction | Tested MB Treatments | References |
|---|---|---|---|
| Acute Diseases | |||
| Traumatic Brain Injury | Ischemia: ↓OXPHOS proteins, ↓ATP synthesis, ↓ATP-dependent cellular processes, ↓ion homeostasis Reperfusion: ↑ROS, ↑oxidative stress, ↑Ca2+, mPTP opening, ↑cell death |
Quercetin: ↑PGC-1α, ↓cell death DEX: ↑PGC-1α, ↓cell death, ↑behavior 7,8-DHF: ↑PGC-1α, ↑function SS-31: ↑PGC-1α, ↑function, ↑behavior |
(Li et al., 2018c) (Li et al., 2018a) (Krishna et al., 2017) (Zhu et al., 2018) |
| Spinal Cord Injury |
Formoterol: ↑PGC-1α, ↓cell death, ↑function LY344864: ↑PGC-1α, ↓cell death, ↑function Mitochondrial transplant: ↑pAKT, ↑function, ↓cell death |
(Scholpa et al., 2019a; Scholpa et al., 2019b) (Simmons et al., 2019) (Li et al., 2019) |
|
| Stroke |
Mitochondrial transplant: ↑pAKT, ↓cell death, ↑function Daidzein: ↓cell death, antioxidant Metformin: ↓cell death, ↑behavior Melatonin: ↑mitophagy, antioxidant |
(Chang et al., 2019) (Hayakawa et al., 2016) (Cao et al., 2017) (Andrzejewski et al., 2014; Owen et al., 2000) (Qi et al., 2017) |
|
| Neurodegenerative | |||
| Alzheimer’s Disease | ↓MB, ↑mitochondrial size and structural damage, ↓content, ↑fragmentation, ↑mtDNA mutations |
TZDs: ↑behavior Resveratrol: ↑PGC-1α, ↑behavior, ↑pAKT AICAR: ↑behavior, ↑pAKT Melatonin: ↑PGC-1α, ↑function, ↓Aβ |
(Cheng et al., 2016; Heneka et al., 2015; Risner et al., 2006; Watson et al., 2005) (Vingtdeux et al., 2010) (Wang et al., 2019) |
| Parkinson’s Disease | ↑mtDNA mutations (complex I), ↑oxidative damage, ↓PGC-1α, ↓OXPHOS, ↓homeostasis |
Bezafibrate: ↑PGC-1α, ↑behavior Resveratrol: ↑PGC-1α, ↑function, ↓cell death LY344864: ↑PGC-1α, ↑function, ↓cell death Triterpenoids: ↑Nrf2, antioxidant |
(Tufekci et al., 2011) (Corona and Duchen, 2016) (Peng et al., 2016) (Scholpa et al., 2018a) |
Adaption from (Gibbs et al., 2018c). Arrows indicated increase (↑) and decrease (↓). AICAR: 5-aminoimidazole-4-carboxamide ribonucleotide, ATP: adenosine triphosphate, Ca2+: calcium ion, DEX: dexmedetomidine, ETC: electron transport chain, MB: mitochondrial biogenesis, mPTP: membrane permeability transition pore, mtDNA: mitochondrial DNA, PGC-1α: peroxisome proliferator-activated receptor-γ coactivator 1-α, OXPHOS: oxidative phosphorylation, ROS: reactive oxygen species, TFAM: mitochondrial transcription factor A, TZDs: thiazolidinediones, 7,8-DHF: dihydroxyflavone.
3.1. Limitations of pharmacological induction of MB
Though a multitude of studies support targeting MB for treatment of various CNS diseases, this therapeutic avenue is not without limitations. One potential limitation is that enhancing MB may increase unhealthy mitochondrial content. In diseases with disruption of mitochondrial homeostasis, such as the ones discussed in this review, it is possible that increasing MB will exacerbate the negative effects of mitochondrial dysfunction. However, defective or mutated mitochondria may not be able to undergo MB due to their dysfunctional state. In addition, MB and quality control are often upregulated in cancer, and mitochondria are reported to play a central and multifunctional role in malignant tumor progression (Zong et al., 2016). Targeting mitochondrial inhibition (i.e. inhibition of oxidative phosphorylation, promotion of ROS production, inhibition of mitophagy) may provide therapeutic opportunities in the treatment of various cancers (Zong et al., 2016). Therefore, while MB induction may prove an effective therapeutic avenue for the treatment of CNS-related diseases, systemic MB induction may also enhance tumor progression.
Fortunately, multiple pharmacological compounds that induce MB are already approved by the U.S. Food and Drug Administration for the treatment of various pathologies. Therefore, though limitations exist, attaining approval for the use of these drugs for the treatment of various CNS disease could be an expeditious process and provide meaningful treatment for populations that currently have limited therapeutic options.
4. Acute neurological diseases
While the canonical role for CNS mitochondria is the generation of energy, they are also involved in the homeostasis and degeneration of neurons (Dubinsky, 2005). In fact, minimal mitochondrial dysfunction can result in the development of neuropathologies (Dubinsky, 2005). Compromised perfusion is common among acute traumatic CNS injuries, including spinal cord injury (SCI), traumatic brain injury (TBI) and stroke, leading to localized ischemia and subsequent mitochondrial dysfunction. Neuronal cells are easily compromised following ischemic events due to their high reliance on ATP-driven processes, in combination with their inadequate energy reserves and limited capacity to buffer oxidative stress (Castro et al., 1997; Scholpa et al., 2018b; Tian et al., 2016). In addition, axons are more susceptible to the damage caused by ionic imbalance due to their high concentration of voltage gated sodium channels in the nodes of Ranvier (Oyinbo, 2011). Taken together, this manifests in the failure to generate and maintain adequate energy production, thereby exacerbating the pathology of acute CNS injuries, resulting in further cell dysfunction and death (Castro et al., 1997; Scholpa and Schnellmann, 2017; Scholpa et al., 2018b).
4.1. Neuroinflammation
During traumatic/ischemic injuries, several factors are released that initiate the immune response (DiSabato et al., 2016; Simon et al., 2017; Viviani et al., 2014). In the CNS, astrocytes and glial cells are activated in response to injury. These cells are involved in the maintenance and support of neurons and comprise a significant component of the blood-CNS barriers (Viviani et al., 2014). Both astrocytes and microglia, when activated, release cytokines and chemokines that modulate neuroinflammation and development. Astrocyte-derived cytokines including, in part, interleukins and tumor necrosis factor-α (TNF-α) promote neurotoxicity (Shabab et al., 2017). TNF-α can suppress expression of the excitatory amino acid transporter (EAAT) responsible for the routine uptake of glutamate at the synapse. This inhibited uptake exacerbates the already heightened levels of glutamate that were released by damaged and necrotic cells following injury. Glutamate excitotoxicity compromises the cellular membrane potential thereby facilitating calcium influx and overload to the cell. Calcium will then bind to and open the mPTP, promoting cyt c release and ROS production, ultimately inducing apoptosis (Viviani et al., 2014) (Fig 3). It should be noted that opening of mPTP can also promote necrosis via separate mechanisms (Baines et al., 2003; Karch et al., 2013; Karch and Molkentin, 2014). For a more comprehensive review of neuroinflammation see Shabab et al. (2017), Viviani et al. (2014) and DiSabato et al. (2016).
Fig. 3. Role of neuroinflammation on mitochondrial dysfunction.
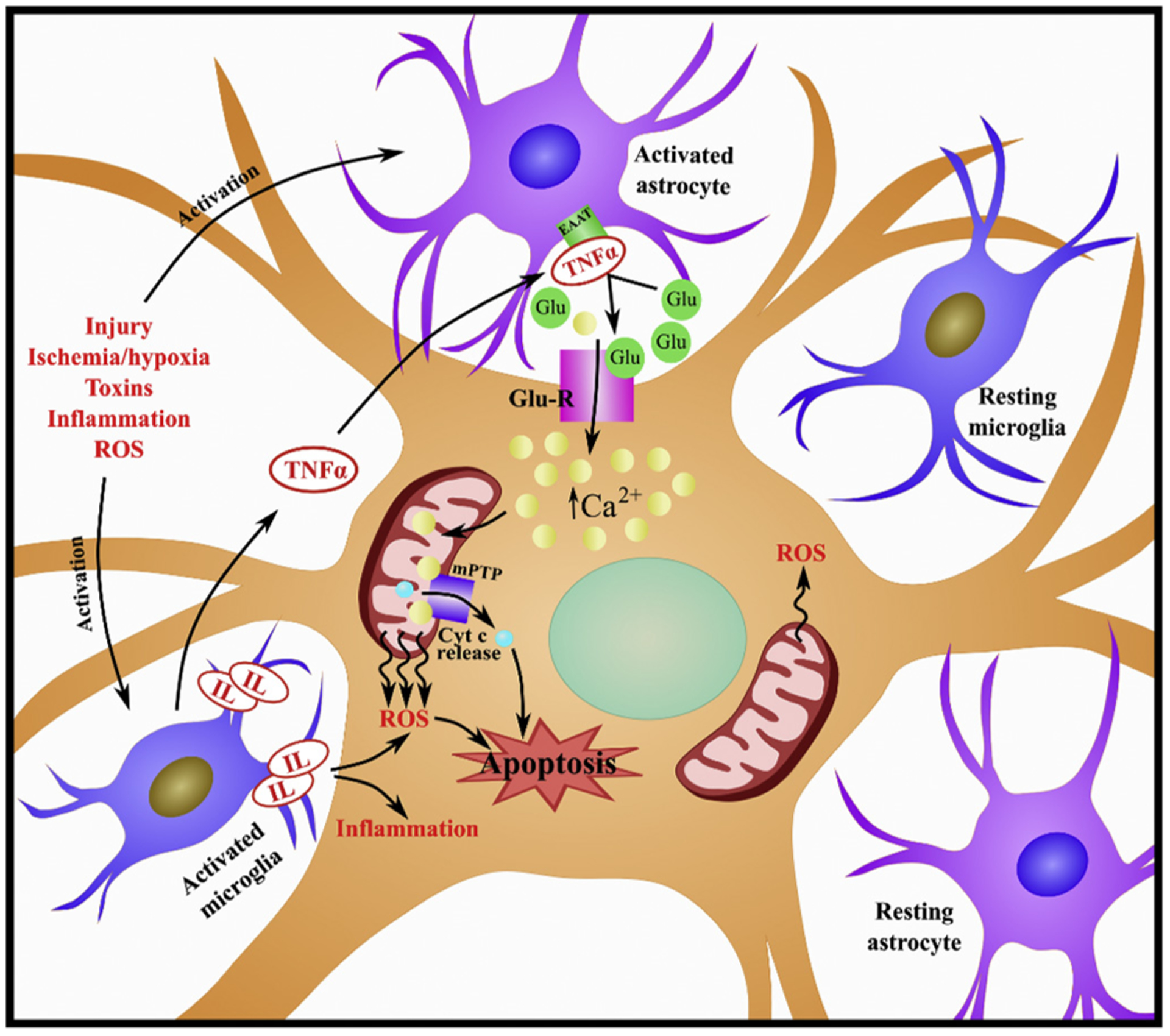
Injury, ischemia, hypoxia, toxins, inflammation and ROS can trigger activation of astrocytes and microglia. Both astrocytes and microglia, when activated, release cytokines and chemokines. Astrocyte-derived cytokines, including interleukins (IL) and tumor necrosis factor-α (TNF-α) can promote neurotoxicity. TNF-α can suppress expression of the excitatory amino acid transporter (EAAT) to exacerbate glutamate levels. Glutamate will bind to glutamate receptors (Glu-R), facilitating calcium influx and overload to the cell. Calcium will then bind to and open the mitochondrial permeability transition pore (mPTP), promoting cytochrome c (cyt c) release and ROS production, ultimately inducing apoptosis.
5. Traumatic brain injury
5.1. Background and pathology
TBI is a devastating injury, often resulting in long-term neuropsychological deficits, including, but not limited to, impaired attention, memory and cognitive functioning. TBI causes significant morbidity and mortality, largely due to minimally effective diagnostics and treatment options (Wang et al., 2017). TBI is initiated by mechanical damage immediately resulting in vascular injury and tissue destruction (Ladak et al., 2019). This primary phase of injury is followed by blood-brain barrier disruption and hypoxia, inducing glial cell activation, astrogliosis, inflammation and further cell death (Ladak et al., 2019; Wanner et al., 2013), all of which encompass a secondary wave of events characterized by a cascade of detrimental biochemical and pathophysiological stressors, referred to as secondary injury. These secondary stressors contribute to ongoing cell death and dysfunction, including glutamate excitotoxicity, mitochondrial dysfunction, free radical-mediated oxidative damage, inflammation and activation of necrotic and apoptotic cell death signaling pathways (Wang et al., 2017).
Numerous studies have documented considerable impairments in mitochondrial function in the injured brain following TBI, such as calcium overload, enhanced ROS production, decreased expression of PGC-1 α and damaged mtDNA (Clark et al., 2000; Gilmer et al., 2009; Raghupathi et al., 2000; Singh et al., 2006; Sullivan et al., 2002). Additionally, the magnitude of mitochondrial dysfunction post-TBI is a critical determinant of cell survival, tissue sparing and functional recovery (Lifshitz et al., 2004; Pandya et al., 2013; Sullivan et al., 1999). Moreover, Liftshitz et al., suggested the presence of a mixed mitochondrial response within the brain, such that structural changes in cortical regions post-TBI result in prominent loss of mitochondrial number, whereas hippocampal mitochondria are more prone to swelling and a transient loss of calcium buffering capacity (Lifshitz et al., 2003). Furthermore, reports document differences between neuronal and non-neuronal mitochondria (Bambrick et al., 2006). One study indicates isolated neuronal mitochondria have differing sensitivities to calcium-induced opening of the mPTP compared to isolated astrocytes, with the astrocytes displaying a fourfold higher calcium uptake capacity (Bambrick et al., 2006). Another study found that isolated neurons and astrocytes many have distinct mechanisms of neuroprotection in response to cyclosporine A treatment (Kahraman et al., 2011). This heterogeneity suggests that mitochondria have highly variable responses to injury across specific brain regions (Wang et al., 2017). With this in mind, combinatorial treatments may be required to address deficits seen in TBI.
Previous studies aimed at targeting mitochondrial dysfunction after TBI have proven successful in preclinical models. Mitochondrial uncoupler 2,4-dinitrophenol (DNP) was reported to have a neuroprotective role resulting in greater tissue and neuronal sparing, as well as improved behavioral outcomes following TBI (Korde et al., 2005; Maragos et al., 2003; Pandya et al., 2007; Pandya et al., 2009). Though preclinical data is promising, observed benefits have not yet been validated in the clinic. (Hubbard et al., 2018). Another mitochondrial uncoupler and prodrug of DNP, MP201, has shown therapeutic potential when delivered acutely after TBI in vivo. This study documented improved mitochondrial function, enhanced oxidative phosphorylation and restoration of ROS levels, paired with improved cognitive function and cortical sparing (Hubbard et al., 2018). Finally, upregulation of mitochondrial uncoupling protein 2 (UCP2) was shown to decrease ROS production and cell death, while reducing UCP2 levels elevated ROS (Sullivan et al., 2003; Sullivan et al., 2004). After TBI, overexpression of UCP2 also reduced ROS production and increased cortical sparing (Mattiasson et al., 2003). While the aforementioned preclinical studies have yielded promising data, currently no drugs have been FDA-approved for the treatment of TBI (Hubbard et al., 2018).
5.2. Current MB strategies
Given the detrimental ongoing effects of secondary injury post-TBI, mitigating this spread of injury via MB induction has been a focus for TBI research. Reports suggest that quercetin, a dietary flavonoid used as a food supplement, can induce MB via PGC-1α activation, thereby attenuating brain injury in a TBI mouse model (Li et al., 2018c). Quercetin reduced TBI-induced neuronal apoptosis in these studies and ameliorated mitochondrial lesions. Treatment with quercetin also restored the level of cyt c, and superoxide dismutase in mitochondria, suggesting that mitochondrial dysfunction was attenuated (Li et al., 2016; Li et al., 2018c).
Dexmedetomidine (DEX) is a selective α2-adrenergic receptor agonist associated with sedative and analgesic effects (Keating, 2015). Following a weight-drop model of TBI in rats, DEX treatment upregulated PGC-1α expression, while also attenuating encephala edema and apoptosis, resulting in increased behavioral function (Li et al., 2018a).
Tropomyosin receptor kinase B (TrkB) has recently emerged as a regulator of hippocampal long-term potentiation and learning (Seese et al., 2019). Treatment with the TrkB agonist, 7,8-dihydroxyflavone (7,8-DHF) following moderate fluid percussion injury in rats showed restored levels of PGC-1α and AMPK, while also augmenting synaptic plasticity and enhancing hippocampal functional connectivity (Krishna et al., 2017).
Finally, the antioxidant SS-31, a mitochondria-targeted peptide known to reduce ROS levels, provided neuroprotection in a variety of neurological diseases. In mouse models of TBI, SS-31 treatment beginning 30 min after injury restored expression of PGC-1α, increased activity of superoxide dismutase (SOD) and decreased cyt c release, all while attenuating neurological deficits, DNA damage and neural apoptosis (Zhu et al., 2019; Zhu et al., 2018). Taken together, these data demonstrate induction of PGC-1α and MB may be a viable treatment strategy to improve mitochondrial function, neuroprotection and behavioral recovery after TBI (Zhu et al., 2019; Zhu et al., 2018).
6. Spinal cord injury
6.1. Background and pathology
Spinal cord injury (SCI) is a traumatic event that can generate an array of impairments ranging from loss of function to complete paralysis below the injury site (Scholpa and Schnellmann, 2017). The clinical outcomes of SCI are diverse and may include loss of sensory and/or motor function, paraplegia or tetraplegia (Alizadeh et al., 2019). Similar to TBI, acute SCI is comprised of a primary injury with subsequent secondary injury resulting from a progressive local cascade of events promoting dysfunction and damage (Witiw and Fehlings, 2015). Primary injury results from a traumatic insult leading to mechanical damage of the spinal cord. As is seen following TBI, disruption of the vasculature causes hemorrhage and edema within seconds, impairing perfusion and ultimately facilitating a localized ischemic event (Witiw and Fehlings, 2015). Ischemia is a key mechanism of secondary injury post-SCI, with the degree of functional loss being proportional to the degree of ischemia (Tator and Fehlings, 1991). This sensitivity leads to compromised oxidative phosphorylation among other mitochondrial function in the presence of ischemia. Additional consequences of secondary injury include neuronal cell death (Anwar et al., 2016; Beattie et al., 2002), progressive axon demyelination (Totoiu and Keirstead, 2005), inflammation (Qiao et al., 2010; Qiao et al., 2015) and mitochondrial dysfunction (Scholpa and Schnellmann, 2017). Loss of mitochondrial homeostasis results in decreased ATP production and inactivation of ATP-dependent ion pumps that are essential for regulation of ion concentrations and reuptake of the excitatory neurotransmitter glutamate. This facilitates excitotoxicity, calcium overload, and the eventual initiation of cell death cascades, all of which are hallmarks of SCI that further exacerbate injury (Choi and Rothman, 1990; Oyinbo, 2011; Rowland et al., 2008).
Based on temporal data conducted by Sullivan et al. (2007), restoring mitochondrial function acutely after injury may be an advantageous strategy for the treatment of SCI (McEwen et al., 2011; Rabchevsky et al., 2011; Sullivan et al., 2007). Many pharmaceuticals that have shown that beneficial outcomes for the treatment of SCI in vivo affect mitochondria or mitochondrial function. For example, treatment with the antibiotic minocycline after SCI stimulated mitochondrial stabilization, inhibited release of cyt c, increased antioxidant activity and impeded mitochondrial-dependent cell death (Aras et al., 2015b; Casha et al., 2012; Wells et al., 2003). Minocycline also displayed neuroprotective effects along with behavioral and cellular recovery when administered post-SCI in rats (Ahmad et al., 2016; Aras et al., 2015b; Casha et al., 2012; Sonmez et al., 2013; Teng et al., 2004; Wells et al., 2003).
Studies also support beneficial outcomes following SCI after restoring mitochondrial dynamics equilibrium. The mechanistic target of rapamycin (mTOR) pathway plays a role in modulating mitochondrial functions (Morita et al., 2015). Rapamycin, an inhibitor of mTOR, may reduce neuronal death by enhancing mitophagy, activating autophagy pathways and attenuating apoptosis in the injured spinal cord (Li et al., 2018b; Sekiguchi et al., 2012; Song et al., 2015). Mitochondrial division inhibitor-1 (Mdivi-1), a selective Drp1 inhibitor, has proven beneficial in models of various CNS pathologies (Luo et al., 2013; Wu et al., 2016). In rat studies, Mdivi-1 treatment prior to SCI increased ATP and mitochondrial membrane potential, decreased caspase-3 release and the number of apoptotic cells, all while increasing locomotor activity (Liu et al., 2015). Additional studies revealed Mdivi-1 treatment increased endogenous antioxidant activity, decreased ROS and decreased cyt c release in cultured spinal cord neurons with glutamate-induce injury (Liu et al., 2015; Scholpa and Schnellmann, 2017). Cyclosporine A (CsA) is an immunosuppressant that inhibits mPTP opening. Treatment with a derivative of CsA, NIM811, reduced oxidative damage and attenuated mitochondrial function and tissue sparring following SCI, while also significantly improving locomotion, tissue sparing and bladder control in rodents (Springer et al., 2018).
Acetyl-l-carnitine (ALC), a metabolite transported through the inner mitochondrial membrane capable of acetyl-CoA synthesis (Pettegrew and McClure, 2002), has shown beneficial effects in several pathologies, including Parkinson’s disease, Alzheimer’s disease, and multiple sclerosis (Pettegrew and McClure, 2002; Puca et al., 1990; Tomassini et al., 2004). Studies report ALC treatment after SCI in rats reduced neuronal degeneration and neuroinflammation (Karalija et al., 2012, 2014), maintained mitochondrial function, improved functional recovery, protected both white and gray matter (Cohen et al., 2009; Patel et al., 2012; Patel et al., 2010), and reduced the number of damaged mitochondria. Treatment also improved mitochondrial membrane potential, and decreased SCI-induced apoptosis (Zhang et al., 2015). These data, though preclinical, provide evidence for ALC as a potential therapeutic treatment of SCI.
6.2. Current MB strategies
Given the aforementioned studies as well as others that have targeted mitochondrial function post-SCI, pharmacological activation of MB represents a promising approach. In support of this, PGC-1α expression is decreased in the spinal cord after contusive SCI in rats (Hu et al., 2015; Hu et al., 2016) and mice (Scholpa et al., 2018b; Simmons et al., 2020). In addition, spinal lentiviral overexpression of PGC-1α immediately after injury attenuated neuronal cell death and promoted functional recovery (Hu et al., 2015; Hu et al., 2016). Evidence also indicates that pharmacological-induced restoration of mitochondrial homeostasis promptly following injury may improve neuronal survival and promote functional recovery (Rabchevsky et al., 2011; Scholpa and Schnellmann, 2017; Scholpa et al., 2019a; Sullivan et al., 2007), all of which is suggestive of the potential benefit of pharmacologically increasing PGC-1α and MB following injury.
Studies have also demonstrated a positive correlation between PGC-1α and angiogenesis (Arany et al., 2008; Saint-Geniez et al., 2013; Thom et al., 2014). As compromised vasculature is a delirious consequence of SCI, particularly blood-spinal cord barrier dysfunction, enhanced angiogenesis could serve as an effective treatment for SCI. Therefore, therapeutics targeting reestablishment of mitochondrial homeostasis through MB activation may unveil a therapeutic avenue for mediating several facets of secondary injury progression, improving functional and vascular recovery and neuronal survival following SCI (Scholpa and Schnellmann, 2017).
A recent report demonstrated transplantation of mitochondria from bone marrow mesenchymal stem cells to injured neurons may be neuroprotective in the injured spinal cord of rats. Transplantation resulted in improved bioenergetics, improved activation/phosphorylation of an intermediate signaling molecule of MB, protein kinase B (pAKT), and mitochondrial respiration, decreased apoptosis, enhanced motor neuron survival and improved locomotor recovery (Li et al., 2019). This study supports the theory that restored mitochondrial number and function promote recovery following SCI. However, pharmacological activation of MB may serve as a more efficient approach for promoting mitochondrial function and locomotor recovery.
Pharmacological activation of MB can be initiated via agonism of the G protein-coupled 5-hydroxytryptamine 1F receptor (5-HT1FR) and β2-adrenergic receptor (β2R). Formoterol is a well-studied agonist of the β2R and potent mediator of MB, (Gibbs et al., 2018a; Gibbs et al., 2018b; Wills et al., 2012) and is currently FDA-approved for the treatment of asthma and related disorders. Reports show that formoterol treatment post-SCI has prominent effects in the injured spinal cord, including enhanced PGC-1α, increased mitochondrial number, mediation of mitochondrial dysfunction and improved locomotor capabilities in mice (Scholpa et al., 2019a; Scholpa et al., 2019b). In these studies, formoterol treatment also increased body weight and skeletal muscle mass. Similar mitochondrial and locomotor recovery was seen with treatment of the selective 5-HT1FR agonist LY344864. In addition, treatment with this 5-HT1FR agonist improved integrity of the blood-spinal cord barrier after injury (Simmons et al., 2020). Though LY344864 failed phase II clinical trials, the more selective 5-HT1FR agonist lasmiditan was recently approved for the treatment of migraines (Brandes et al., 2019; Shapiro et al., 2019). With both of these drugs already FDA-approved for the treatment of other pathologies, repurposing these drugs for the treatment of SCI could accelerate approval for treatment of SCI.
7. Stroke
7.1. Background and pathology
Stroke is the leading cause of adult disability and mortality in most developing and developed countries (Guzik and Bushnell, 2017). Ischemic events occur when blood flow to brain tissue is decreased or blocked. In an ischemic stroke patient, a significant decline in the focal cerebral blood flow leads to deprivation of glucose and oxygen, which rapidly compromises biochemical functions, leading to cell death and ultimately brain damage. (Yang et al., 2018). As is common with acute/traumatic injuries, the downstream signaling pathways of stroke induce glutamate excitotoxicity and excessive calcium influx leading to mitochondrial dysfunction and ROS production (Galluzzi et al., 2012). Such events initiate various pathological processes such as mtDNA damage, mitochondrial fission and fusion changes, mitophagy and apoptosis (Dharmasaroja, 2016; Hofmeijer and van Putten, 2012; Lee et al., 2000).
The current therapy for treatment of ischemia is to quickly restore blood flow to the compromised tissue. As such, the standard of care for ischemic stroke is to remove the blockage from the cerebral artery via thrombolysis (Adeoye et al., 2011) or angiographic revascularization (Zaidat et al., 2013). Unfortunately, a major generator of ischemic injuries is the restoration of blood flow to the ischemic tissue, known as ischemia and reperfusion (IR) injury (Chouchani et al., 2016; Dawson and Dawson, 2017; Lesnefsky et al., 2017; Sanderson et al., 2013). Mitochondria and other systems have the potential to generate a considerable quantity of ROS during the reperfusion process, further compromising mitochondrial and cellular processes and functions. IR is a key driver of the pathology of stroke (Chouchani et al., 2016; Sanderson et al., 2013); therefore, therapeutic research now aims to minimize the effects of IR-induced injury following ischemia (He et al., 2019; Lesnefsky et al., 2017).
Inflammation is another pivotal mechanism in the pathology of ischemic stroke. Recent studies have recognized the roles of mitochondria in the regulation of the inflammatory response (DiSabato et al., 2016). MitoQ is capable of hindering production of cytokines in peripheral blood monocytes (Boldogh and Pon, 2006). Studies using MitoQ treatment for ischemic stroke, while limited, suggest a neuroprotective role for MitoQ in adult ischemic studies and restoration of mitochondrial function may play a role in promoting these observed effects (Calo et al., 2013; Loson et al., 2013; Thornton et al., 2018).
Since mitochondrial dysfunction is a known hallmark of ischemic stroke, restoring function is essential to allow for cell survival and neurological improvement after ischemia. Several studies report a neuroprotective effect of ALC, an acetylated derivative of L-carnitine, in ischemic stroke models. In an animal model of global cerebral ischemia and reperfusion, reports show treatment with ALC normalized brain energy metabolites and improved neurological outcomes (Rosenthal et al., 1992). ALC also protected against early changes in brain metabolites and mitochondrial function following a rat model of perinatal hypoxia ischemia of both male and female pups (Tang et al., 2016). In primary cultures of rat cortical neurons, ALC may inhibit both acute and delayed cell death following excitotoxic injury, potentially via inhibition of mPTP opening (Zanelli et al., 2005). Following calcium exposure in primary rat cortical neurons and astrocytes, PTP inhibitors such as CsA showed reduced calcium sensitivity of the mPTP and membrane depolarization, though the mechanism explaining this protective effect may not be through mPTP inhibition (Kahraman et al., 2011).
In addition, several reports demonstrated that treatment with Mdivi-1 improves outcomes after oxygen/glucose deprivation in the brain (Ma et al., 2016; Wang et al., 2014; Wappler et al., 2013; Zhao et al., 2014) and also attenuates brain injury in several models of stroke and TBI (Chuang et al., 2016; Thornton et al., 2018; Wu et al., 2017; Wu et al., 2016). Although there exists extensive promising data in preclinical models of stroke, no drug nor neuroprotective compound has been elucidated for the treatment of stroke (He et al., 2019).
7.2. Current MB strategies
Studies employing mitochondrial transplantation attenuated stroke-induced neuronal death (Chang et al., 2019). Furthermore, injection of astrocyte-derived mitochondria in an rat model of stroke increased levels of pAKT (Gibbs et al., 2018a), as well as B-cell lymphoma-extra large (BCL-XL), a transmembrane molecule in the mitochondria (Valentin et al., 2018). Enhanced expression of both pAKT and BCL-XL also promoted cell survival in mice (Hayakawa et al., 2016). Finally, injection of neuron-, astrocyte- and microglia-derived mitochondria facilitated enhanced mitochondrial function and locomotor recovery (Huang et al., 2016).
Recent studies revealed that oxidative stress in ischemic neurons may potentially involve PGC-1α and its pathways (Ham 3rd and Raju, 2017). Upon activation of the PGC-1α signaling pathway following global ischemia, expression of UCP2 and SOD2 increased and MB was induced (Chen et al., 2010; Su et al., 2017). Additionally, several studies show a reduction in ROS production with treatment with MB-inducible compounds. In a rat model of middle cerebral artery occlusion (MCAO), melatonin increased mitophagy, reduced ROS and inhibited inflammasome activation, thereby suppressing the inflammatory response (Cao et al., 2017). Daidzein is an MB agent and antioxidant (Rasbach and Schnellmann, 2008). Daidzein suppressed ROS production and mitochondrial swelling, while increasing antioxidant activities (Aras et al., 2015a; Liu et al., 2017; Stout et al., 2013; Thornton et al., 2018; Yuan et al., 2017). Metformin, a known inhibitor of interleukin 1β (IL1β), which is also a modulator of MB (Markowicz-Piasecka et al., 2017), reduced ROS production, hindering mitochondria-mediated apoptosis (Andrzejewski et al., 2014; Owen et al., 2000). Improved behavior assessments, promotion of oligodendrocyte survival and remyelination were also reported following metformin treatment in mice (Qi et al., 2017).
Restoration of impaired mitochondrial dynamics has also been investigated in in vivo stroke models. Decreased mitochondrial fission via downregulation of Drp1 resulted in a diminished degree of infarct size in ischemic stroke (Barsoum et al., 2006; Grohm et al., 2012; He et al., 2019; Zhao et al., 2014). Conversely, enhancing mitochondrial fusion with overexpression of Opa1 was shown to attenuate cerebral edema following cerebral ischemia in rats (Zhang et al., 2014). These data taken together support the concept that restoration of mitochondrial homeostasis and balance may generate neuroprotection against stroke (Thornton et al., 2018). Such effects may be possible with pharmacological activation of MB (He et al., 2019); however, cerebral ischemia is an intricate and complex pathology and effective and meaningful treatment may require a combinational approach.
8. Chronic neurological disease
A hallmark of several chronic neurological diseases is neurodegeneration characterized by an array of disruptions in neuronal systems (Golpich et al., 2017). Neurodegeneration is an umbrella term for the progressive death of nerve cells and loss of brain tissue (Golpich et al., 2017). Several lines of pathological and physiological evidence reveal that impaired mitochondrial function and dynamics play crucial roles in both aging and pathogenesis of neurodegenerative diseases. The transcriptional decline in genes that control MB frequently occurs with advancing age (Beckervordersandforth et al., 2017) and likely serves a key role in the pathogenesis of neurodegenerative disease. As mitochondria are the major intracellular organelles that regulate both cell survival and death, they are considered potential targets for pharmacological-based therapies in chronic neurological diseases (Golpich et al., 2017).
Mitochondrial dysfunction is implicated in multiple neurodegenerative diseases including Alzheimer’s disease (AD) and Parkinson’s disease (PD), Huntington’s disease (HD) and Multiple Sclerosis (MS), among others. This review, however, will focus on AD and PD. For comprehensive reviews detailing MB in neurodegenerative diseases, see Golpich et al. (2017) and Sebastian et al. (2017).
9. Alzheimer’s disease
9.1. Background and pathology
AD is the most common neurodegenerative disorder and is characterized by an accumulation of amyloid plaques (Aβ) and intraneurofibrillary tangles (NFT) made from hyperphosphorylated tau protein (pTau) in the brain (Golpich et al., 2017). Progressive loss of synapses and cholinergic fibers, as well as the proliferation of reactive astrocytes and microglia, also contribute to the pathology of AD (Lin and Beal, 2006). In addition, mitochondrial dysfunction is a hallmark of AD, even in its early stages (Golpich et al., 2017). Altered mitochondrial respiration in postmortem brains of AD patients has been reported in several studies (Chaturvedi and Flint Beal, 2013; Golpich et al., 2015), specifically diminished ATP production paired with increased oxidative stress. There also exist reports of damaged and mutated mtDNA, as well as compromised oxidative phosphorylation and loss of glucose metabolism (Lin and Beal, 2006; McFarland et al., 2010).
Aβ plaques and pTau aggregate in synaptic clefts, highly metabolic regions, thereby hindering synaptic mitochondria and neurotransmission. This hindrance leads to dysfunctional neurons and diminished cognition in AD patients (Lin and Beal, 2006). pTau selectively impairs complex I of the ETC, further hindering mitochondrial respiration (Golpich et al., 2017; Oliver and Reddy, 2019). Aβ aggregates also promote ROS production, further damaging mtDNA, while also propagating additional Aβ formation (Oliver and Reddy, 2019).
An imbalance in mitochondrial dynamics also aids in the pathogenesis of AD, namely upregulation of mitochondrial fission, downregulation of mitochondrial fusion, as well as a reduction in mitophagy (Kerr et al., 2017). This altered balance of mitochondrial morphology leads to considerable neuronal loss, brain volume shrinkage and cognitive decline (Oliver and Reddy, 2019). Evidence suggests that interaction between Aβ and Drp1 is a critical component of mitochondrial dysfunction and impaired dynamics in AD (Reddy et al., 2018). It has been suggested that pTau can interact with and increase expression of Drp1, effectively enhancing mitochondrial fission, compromising mtDNA and damaging synaptic activity in AD neurons(Calkins et al., 2011; Manczak and Reddy, 2012), all leading to cognitive impairments (Reddy et al., 2018; Reiss et al., 2018). The gradual loss of mtDNA may also contribute to increased fission and decreased mitophagic activity seen in AD (Oliver and Reddy, 2019). These reports implicate mitochondrial dysfunction in the pathogenesis of AD, while also revealing a potential target for therapeutic strategies via MB induction.
9.2. Current MB strategies
In mouse models of AD, overexpression of PGC-1α mediated mitochondrial dysfunction (Sheng et al., 2012) and improved cognition (Dumont et al., 2012). Treatment with thiazolidinediones (TZDs), a PPAR activator, was shown to induce MB and improve cognitive capabilities in early mild to moderate cases of AD (Cheng et al., 2016; Heneka et al., 2015; Risner et al., 2006; Watson et al., 2005). Similar protective effects were observed with resveratrol and AICAR (5-aminoimidazole-4-carboxamide ribonucleotide), both activators of the MB signaling molecule, AMP-activated protein kinase (AMPK) (Vingtdeux et al., 2010). Treatment with resveratrol also activates PGC-1α and has been investigated in animal models of PD, HD and ALS, leading to marked decreases in neuronal degeneration (Blanchet et al., 2008; Ho et al., 2010; Kim et al., 2007; Long et al., 2009; Whitaker et al., 2016). Additionally, melatonin increased PGC-1α, Nrf2 and mtDNA, among other mitochondrial genes, while also restoring mitochondrial structure and function in in vitro models of AD (Wang et al., 2019). The generation of Aβ was also mediated with melatonin treatment, all of which suggest a neuroprotective effect via MB induction (Wang et al., 2019). These observations further propose MB induction as a therapeutic avenue for the treatment of mild to moderate cases of AD.
10. Parkinson’s disease
10.1. Background and pathology
Precise etiology has yet to be elucidated for PD, but it is suggested that genetic and environmental factors contribute (Golpich et al., 2017). Cellular mechanisms resulting in nigrostriatal cell death are also unclear (Golpich et al., 2017); however, mitochondrial dysfunction and oxidative stress are known hallmarks of PD (Yan et al., 2013). Like with many diseases characterized by mitochondrial dysfunction, brains from PD patients show increased oxidative stress, chronic inflammation, aberrant protein folding and abnormal protein aggregation (Golpich et al., 2017; Harischandra et al., 2019; Pickrell and Youle, 2015; Truban et al., 2017). Postmortem studies observed decreased activity in complex I, antioxidant coenzyme Q10 and complex IV in the substantia nigra (Hyman et al., 2012; Kilbride et al., 2011). This decreased activity, particularly in complex I, may encourage the accumulation of protein inclusions or lewy bodies (LB) containing alpha-synuclein (α-Syn), both of which are known to be key drivers for the progression of PD (Bengoa-Vergniory et al., 2017; Golpich et al., 2017).
α-Syn was implicated in the maintenance of mitochondrial dynamics in neurons, with aggregation present in mitochondria from the substantia nigra (Devi and Anandatheerthavarada, 2010; Devi et al., 2008; Rani and Mondal, 2019). Vacuolar protein sorting associated protein 35 (VPS35) is another modulator of mitochondrial dynamic activity (Wang et al., 2016a) that encodes α-Syn. In PD, mutated VPS35 depicts increased interaction with Drp1, thereby enhancing mitochondrial fission. This imbalance in mitochondrial dynamics then facilitates mitochondrial fragmentation and neuronal cell death (Wang et al., 2016b). Additionally, PD-associated α-Syn mutations have also been shown to compromise mitophagy. (Rani and Mondal, 2019).
As stated in other sections, mutated and fragmented mtDNA lead to reduced oxidative phosphorylation. Such impairments are known to occur in early stages of PD (Coskun et al., 2012). Compared to other neuronal cells, dopaminergic neurons of the substantia nigra are particularly vulnerable to mtDNA mutations and oxidative damage (Bender et al., 2006; Juarez Olguin et al., 2016). These neurons are charged with creating dopamine (DA). If not properly stored, DA will be metabolized into 3,4-Dihydroxyphenylacetaldehyde (DOPAL), which is highly toxic to dopaminergic neurons (Gandhi et al., 2012). DOPAL can easily modify proteins in such a way as to promote aggregation, resulting in enhanced ROS production and opening of the mPTP, all of which leads to mitochondrial dysfunction and neurodegeneration in PD (Chiu et al., 2015; Coelho-Cerqueira et al., 2019; Kristal et al., 2004; Rani and Mondal, 2019; Zhou and Lim, 2009).
A variety of drugs targeting mitochondrial quality control have been studied in both in vivo and in vitro models of PD. Oral administration of CoQ10 in mice attenuated the loss of DA neurons in PD. However, CoQ10 has yielded conflicting evidence in clinical trials for PD (Raizner, 2019; Rani and Mondal, 2019). Another mitochondrial-targeted antioxidant, MitoQ, has shown positive outcomes in numerous cell and animal models of mitochondrial dysfunction (Ramis et al., 2015), though it did not slow progression of PD in clinical trials (Snow et al., 2010). Finally, P110 is a peptide derived from the interaction site of Drp1 and Fis1. P110 was able to effectively block the localization of Drp1 to mitochondria (Qi et al., 2013), thereby hindering fusion and providing neuroprotection in several rodent models of neurodegeneration, including PD (Disatnik et al., 2013; Disatnik et al., 2016; Guo et al., 2013; Qi et al., 2013; Thornton et al., 2018). Treatment with neuroprotective peptides has also been explored. Small molecule antioxidant peptides SS31 and SS20 showed neuroprotection in PD models by targeting various mitochondrial aberrations and oxidative stress (Yang et al., 2009). Finally, isolated mitochondria were delivered systemically in a mouse model of Parkinson’s disease and found to increase behavioral outcomes (Shi et al., 2017), thereby supporting the theory that restored mitochondrial number and function promote recovery following SCI.
The most potent treatment available for PD consists of a DA precursor levodopa (Tambasco et al., 2018). Though currently serving as the standard treatment for locomotor deficits in PD, several adverse side effects exist, including motor fluctuations and dyskinesia (Virmani et al., 2016). To date, there exist no efficient drugs for repressing the loss of dopaminergic neurons in PD (Rani and Mondal, 2019).
10.2. Current MB strategies
Several studies employing pharmacological-induction of MB reveal beneficial outcomes in animal models of PD. Triterpenoids have been shown to activate Nrf2 pathways in mouse models of PD, thereby promoting expression of genes involved in MB and antioxidative defense (Tufekci et al., 2011). Treatment with benzafibrate, a PPAR agonist and known activator of PGC-1α and inducer MB, has yielded beneficial outcomes in rodents (Corona and Duchen, 2016). Reports in both in vitro and in vivo models of PD showed that PGC-1α overexpression rescued the loss of the DA-ergic neurons and mitochondrial homeostasis, while also enhancing expression of antioxidants (Di Giacomo et al., 2017). Rotenone, a commonly used pesticide, is considered an environmental risk factor for the pathogenesis of non-familial PD. Ongoing exposure to low doses of rotenone results in compromised ETC function and oxidative stress. Following rotenone exposure, treatment with resveratrol improved mitochondrial homeostasis in both cell and animal models, correlating to increased TFAM and PGC-1α expression, indicating improved MB (Peng et al., 2016). Treatment with the mitochondrially biogenic agent LY344864 enhanced expression of mtDNA and PGC-1α, and induced MB in various brain regions in a PD mouse model. LY344864 also attenuated tyrosine hydroxylase immune-reactivity, while improving locomotor activity (Scholpa et al., 2018a). These data indicate that restoring PGC-1α and/or inducing MB may be a promising approach for the development of effective drugs for the treatment of PD (Golpich et al., 2017; Rani and Mondal, 2019).
11. Conclusion
Mitochondrial dysfunction serves as a hallmark in many diseases and injuries of the CNS. Such dysfunction is often characterized by decreased PGC-1α and suppressed MB (Chen et al., 2011; Kim et al., 2010; Li et al., 2018c; Scholpa et al., 2019b). Studies discussed in this review demonstrated that activation of MB restores mitochondrial function and homeostasis and promotes meaningful recovery across multiples CNS related pathologies. As such, inducing MB and attenuating mitochondrial dysfunction may be an effective therapeutic strategy for a multitude of CNS diseases.
Funding
This study was supported by the National Institutes of HealthNational Institute of General Medical Sciences: GM084147 (R.G.S), and the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs: BX: 000851 (R.G.S.).
Footnotes
Declaration of Competing Interest
No disclosures or competing interests.
References
- Adeoye O, Hornung R, Khatri P, Kleindorfer D, 2011. Recombinant tissue-type plasminogen activator use for ischemic stroke in the United States: a doubling of treatment rates over the course of 5 years. Stroke 42, 1952–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad M, Zakaria A, Almutairi KM, 2016. Effectiveness of minocycline and FK506 alone and in combination on enhanced behavioral and biochemical recovery from spinal cord injury in rats. Pharmacol. Biochem. Behav 145, 45–54. [DOI] [PubMed] [Google Scholar]
- Alizadeh A, Dyck SM, Karimi-Abdolrezaee S, 2019. Traumatic spinal cord injury: an overview of pathophysiology, models and acute injury mechanisms. Front. Neurol 10, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrzejewski S, Gravel SP, Pollak M, St-Pierre J, 2014. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar MA, Al Shehabi TS, Eid AH, 2016. Inflammogenesis of secondary spinal cord injury. Front. Cell. Neurosci 10, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM, 2008. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 451, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Aras AB, Guven M, Akman T, Ozkan A, Sen HM, Duz U, Kalkan Y, Silan C, Cosar M, 2015a. Neuroprotective effects of daidzein on focal cerebral ischemia injury in rats. Neural Regen. Res 10, 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aras M, Altas M, Motor S, Dokuyucu R, Yilmaz A, Ozgiray E, Seraslan Y, Yilmaz N, 2015b. Protective effects of minocycline on experimental spinal cord injury in rats. Injury 46, 1471–1474. [DOI] [PubMed] [Google Scholar]
- Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P, 2003. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res 92, 873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambrick LL, Chandrasekaran K, Mehrabian Z, Wright C, Krueger BK, Fiskum G, 2006. Cyclosporin A increases mitochondrial calcium uptake capacity in cortical astrocytes but not cerebellar granule neurons. J. Bioenerg. Biomembr 38, 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E, 2006. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25, 3900–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie MS, Hermann GE, Rogers RC, Bresnahan JC, 2002. Cell death in models of spinal cord injury. Prog. Brain Res 137, 37–47. [DOI] [PubMed] [Google Scholar]
- Beckervordersandforth R, Ebert B, Schaffner I, Moss J, Fiebig C, Shin J, Moore DL, Ghosh L, Trinchero MF, Stockburger C, Friedland K, Steib K, von Wittgenstein J, Keiner S, Redecker C, Holter SM, Xiang W, Wurst W, Jagasia R, Schinder AF, Ming GL, Toni N, Jessberger S, Song H, Lie DC, 2017. Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron 93 560–573.e566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard G, Karbowski M, 2009. Mitochondrial fusion and division: Regulation and role in cell viability. Semin. Cell Dev. Biol 20, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM, 2006. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet 38, 515–517. [DOI] [PubMed] [Google Scholar]
- Bengoa-Vergniory N, Roberts RF, Wade-Martins R, Alegre-Abarrategui J, 2017. Alpha-synuclein oligomers: a new hope. Acta Neuropathol. 134, 819–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet J, Longpre F, Bureau G, Morissette M, DiPaolo T, Bronchti G, Martinoli MG, 2008. Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 32, 1243–1250. [DOI] [PubMed] [Google Scholar]
- Boldogh IR, Pon LA, 2006. Interactions of mitochondria with the actin cytoskeleton. Biochim. Biophys. Acta 1763, 450–462. [DOI] [PubMed] [Google Scholar]
- Bordone MP, Salman MM, Titus HE, Amini E, Andersen JV, Chakraborti B, Diuba AV, Dubouskaya TG, Ehrke E, Espindola de Freitas A, Braga de Freitas G, Goncalves RA, Gupta D, Gupta R, Ha SR, Hemming IA, Jaggar M, Jakobsen E, Kumari P, Lakkappa N, Marsh APL, Mitlohner J, Ogawa Y, Kumar PR, Ribeiro FC, Salamian A, Saleem S, Sharma S, Silva JM, Singh S, Sulakhiya K, Tefera TW, Vafadari B, Yadav A, Yamazaki R, Seidenbecher CI, 2019. The energetic brain - A review from students to students. J. Neurochem 151, 139–165. [DOI] [PubMed] [Google Scholar]
- Brandes JL, Klise S, Krege JH, Case M, Khanna R, Vasudeva R, Raskin J, Pearlman EM, Kudrow D, 2019. Interim results of a prospective, randomized, open-label, Phase 3 study of the long-term safety and efficacy of lasmiditan for acute treatment of migraine (the GLADIATOR study). Cephalalgia 39, 1343–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P, 2015. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol 11, 11–24. [DOI] [PubMed] [Google Scholar]
- Busch KB, Bereiter-Hahn J, Wittig I, Schagger H, Jendrach M, 2006. Mitochondrial dynamics generate equal distribution but patchwork localization of respiratory Complex I. Mol. Membr. Biol 23, 509–520. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH, 2011. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet 20, 4515–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo L, Dong Y, Kumar R, Przyklenk K, Sanderson TH, 2013. Mitochondrial dynamics: an emerging paradigm in ischemia-reperfusion injury. Curr. Pharm. Des 19, 6848–6857. [DOI] [PubMed] [Google Scholar]
- Cameron RB, Beeson CC, Schnellmann RG, 2016. Development of therapeutics that induce mitochondrial biogenesis for the treatment of acute and chronic degenerative diseases. J. Med. Chem 59, 10411–10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Shrestha S, Li J, Yu X, Chen J, Yan F, Ying G, Gu C, Wang L, Chen G, 2017. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci. Rep 7, 2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaldi RA, Murray J, Byrne L, Janes MS, Marusich MF, 2004. Immunological approaches to the characterization and diagnosis of mitochondrial disease. Mitochondrion 4, 417–426. [DOI] [PubMed] [Google Scholar]
- Casha S, Zygun D, McGowan MD, Bains I, Yong VW, Hurlbert RJ, 2012. Results of a phase II placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain 135, 1224–1236. [DOI] [PubMed] [Google Scholar]
- Castro ME, Pascual J, Romon T, del Arco C, del Olmo E, Pazos A, 1997. Differential distribution of [3H]sumatriptan binding sites (5-HT1B, 5-HT1D and 5-HT1F receptors) in human brain: focus on brainstem and spinal cord. Neuropharmacology 36, 535–542. [DOI] [PubMed] [Google Scholar]
- Chan DC, 2006. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol 22, 79–99. [DOI] [PubMed] [Google Scholar]
- Chang CY, Liang MZ, Chen L, 2019. Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl Neurodegener 8, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Flint Beal M, 2013. Mitochondrial diseases of the brain. Free Radic. Biol. Med 63, 1–29. [DOI] [PubMed] [Google Scholar]
- Chen SD, Lin TK, Lin JW, Yang DI, Lee SY, Shaw FZ, Liou CW, Chuang YC, 2010. Activation of calcium/calmodulin-dependent protein kinase IV and peroxisome proliferator-activated receptor gamma coactivator-1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res 88, 3144–3154. [DOI] [PubMed] [Google Scholar]
- Chen SD, Yang DI, Lin TK, Shaw FZ, Liou CW, Chuang YC, 2011. Roles of oxidative stress, apoptosis, PGC-1alpha and mitochondrial biogenesis in cerebral ischemia. Int. J. Mol. Sci 12, 7199–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Shang Y, Jiang L, Shi T-L, Wang L, 2016. The peroxisome proliferators activated receptor-gamma agonists as therapeutics for the treatment of Alzheimer’s disease and mild-to-moderate Alzheimer’s disease: a meta-analysis. Int. J. Neurosci 126, 299–307. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Kozlov MM, 2005. Membrane hemifusion: crossing a chasm in two leaps. Cell 123, 375–382. [DOI] [PubMed] [Google Scholar]
- Chiu CC, Yeh TH, Lai SC, Wu-Chou YH, Chen CH, Mochly-Rosen D, Huang YC, Chen YJ, Chen CL, Chang YM, Wang HL, Lu CS, 2015. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Exp. Neurol 263, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Rothman SM, 1990. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci 13, 171–182. [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP, 2016. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23, 254–263. [DOI] [PubMed] [Google Scholar]
- Chuang YC, Lin TK, Yang DI, Yang JL, Liou CW, Chen SD, 2016. Peroxisome proliferator-activated receptor-gamma dependent pathway reduces the phosphorylation of dynamin-related protein 1 and ameliorates hippocampal injury induced by global ischemia in rats. J. Biomed. Sci 23, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, Melick J, Loeffert JE, Nathaniel PD, Jin KL, Graham SH, 2000. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J. Neurochem 74, 740–753. [DOI] [PubMed] [Google Scholar]
- Coelho-Cerqueira E, de Araujo Correia Campos C, Follmer C, 2019. Formation of large oligomers of DOPAL-modified alpha-synuclein is modulated by the oxidation of methionine residues located at C-terminal domain. Biochem. Biophys. Res. Commun 509, 367–372. [DOI] [PubMed] [Google Scholar]
- Cohen DM, Patel CB, Ahobila-Vajjula P, Sundberg LM, Chacko T, Liu SJ, Narayana PA, 2009. Blood-spinal cord barrier permeability in experimental spinal cord injury: dynamic contrast-enhanced MRI. NMR Biomed. 22, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona JC, Duchen MR, 2016. PPARgamma as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic. Biol. Med 100, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, Laferla F, Wallace DC, 2012. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta 1820, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL, 2017. Mitochondrial mechanisms of neuronal cell death: potential therapeutics. Annu. Rev. Pharmacol. Toxicol 57, 437–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Arriba G, Calvino M, Benito S, Parra T, 2013. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicol. Lett 218, 30–38. [DOI] [PubMed] [Google Scholar]
- Devi L, Anandatheerthavarada HK, 2010. Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. Biochim. Biophys. Acta 1802, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK, 2008. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem 283, 9089–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmasaroja PA, 2016. Fluid intake related to brain edema in acute middle cerebral artery infarction. Transl. Stroke Res 7, 49–53. [DOI] [PubMed] [Google Scholar]
- Di Giacomo E, Benedetti E, Cristiano L, Antonosante A, d’Angelo M, Fidoamore A, Barone D, Moreno S, Ippoliti R, Ceru MP, Giordano A, Cimini A, 2017. Roles of PPAR transcription factors in the energetic metabolic switch occurring during adult neurogenesis. Cell Cycle 16, 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Scorrano L, 2006. (De)constructing mitochondria: what for? Physiology (Bethesda) 21, 233–241. [DOI] [PubMed] [Google Scholar]
- DiSabato DJ, Quan N, Godbout JP, 2016. Neuroinflammation: the devil is in the details. J. Neurochem 139 (Suppl. 2), 136–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X, Mochly-Rosen D, 2013. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc 2, e000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disatnik MH, Joshi AU, Saw NL, Shamloo M, Leavitt BR, Qi X, Mochly-Rosen D, 2016. Potential biomarkers to follow the progression and treatment response of Huntington’s disease. J. Exp. Med 213, 2655–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky JM, 2005. CNS mitochondria in neurodegenerative disorders. Antioxid. Redox Signal 7, 1089–1091. [DOI] [PubMed] [Google Scholar]
- Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova N, Calingasan NY, Yang L, Tampellini D, Starkov AA, Chan RB, Di Paolo G, Pujol A, Beal MF, 2012. Bezafibrate administration improves behavioral deficits and tau pathology in P301S mice. Hum. Mol. Genet 21, 5091–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Henriques M, Anton F, 2013. Mechanistic perspective of mitochondrial fusion: tubulation vs. fragmentation. Biochim. Biophys. Acta 1833, 162–175. [DOI] [PubMed] [Google Scholar]
- Fan KQ, Li YY, Wang HL, Mao XT, Guo JX, Wang F, Huang LJ, Li YN, Ma XY, Gao ZJ, Chen W, Qian DD, Xue WJ, Cao Q, Zhang L, Shen L, Tong C, Zhong JY, Lu W, Lu L, Ren KM, Zhong G, Wang Y, Tang M, Feng XH, Chai RJ, Jin J, 2019. Stress-induced metabolic disorder in peripheral CD4(+) T cells leads to anxiety-like behavior. Cell 179 864–879.e819. [DOI] [PubMed] [Google Scholar]
- Ferrari LF, Chum A, Bogen O, Reichling DB, Levine JD, 2011. Role of Drp1, a key mitochondrial fission protein, in neuropathic pain. J. Neurosci 31, 11404–11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosto M, Scarpelli M, Cotelli MS, Vielmi V, Todeschini A, Gregorelli V, Tonin P, Tomelleri G, Padovani A, 2011. The role of mitochondria in neurodegenerative diseases. J. Neurol 258, 1763–1774. [DOI] [PubMed] [Google Scholar]
- Fivenson EM, Lautrup S, Sun N, Scheibye-Knudsen M, Stevnsner T, Nilsen H, Bohr VA, Fang EF, 2017. Mitophagy in neurodegeneration and aging. Neurochem. Int 109, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Iborra S, Vila M, Perier C, 2018. Mitochondrial quality control in neurodegenerative diseases: focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci 12, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS, 2012. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 1823, 2297–2310. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Nunnari J, 2014. Mitochondrial form and function. Nature 505, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk JA, Schnellmann RG, 2012. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Ren. Physiol 302, F853–F864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway CA, Lee H, Yoon Y, 2012. Mitochondrial morphology-emerging role in bioenergetics. Free Radic. Biol. Med 53, 2218–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Kroemer G, 2012. Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol 13, 780–788. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Vaarmann A, Yao Z, Duchen MR, Wood NW, Abramov AY, 2012. Dopamine induced neurodegeneration in a PINK1 model of Parkinson’s disease. PLoS One 7, e37564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs WS, Weber RA, Schnellmann RG, Adkins DL, 2016. Disrupted mitochondrial genes and inflammation following stroke. Life Sci. 166, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs WS, Collier JB, Morris M, Beeson CC, Megyesi J, Schnellmann RG, 2018a. 5-HT1F receptor regulates mitochondrial homeostasis and its loss potentiates acute kidney injury and impairs renal recovery. Am. J. Physiol. Ren. Physiol 315, F1119–f1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs WS, Garrett SM, Beeson CC, Schnellmann RG, 2018b. Identification of dual mechanisms mediating 5-hydroxytryptamine receptor 1F-induced mitochondrial biogenesis. Am. J. Physiol. Ren. Physiol 314, F260–f268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs WS, Scholpa NE, Beeson CC, Schnellmann RG, 2018c. Pharmacological activation of mitochondrial biogenesis for the treatment of various pathologies In: Will Y, Dykens JA (Eds.), Mitochondrial Dysfunction Caused by Drugs and Environmental Toxicants. Wiley Online Books. [Google Scholar]
- Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF, 2010. Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim. Biophys. Acta 1802, 122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmer LK, Roberts KN, Joy K, Sullivan PG, Scheff SW, 2009. Early mitochondrial dysfunction after cortical contusion injury. J. Neurotrauma 26, 1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golpich M, Amini E, Hemmati F, Ibrahim NM, Rahmani B, Mohamed Z, Raymond AA, Dargahi L, Ghasemi R, Ahmadiani A, 2015. Glycogen synthase kinase-3 beta (GSK-3beta) signaling: Implications for Parkinson’s disease. Pharmacol. Res 97, 16–26. [DOI] [PubMed] [Google Scholar]
- Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, Ahmadiani A, 2017. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neurosci Ther 23, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J, Plesnila N, Culmsee C, 2012. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 19, 1446–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X, 2013. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Invest 123, 5371–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik A, Bushnell C, 2017. Stroke epidemiology and risk factor management. Continuum (Minneap Minn) 23, 15–39. [DOI] [PubMed] [Google Scholar]
- Ham PB 3rd, Raju R, 2017. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol 157, 92–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Spiegelman BM, 2006. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev 27, 728–735. [DOI] [PubMed] [Google Scholar]
- Harischandra DS, Ghaisas S, Zenitsky G, Jin H, Kanthasamy A, Anantharam V, Kanthasamy AG, 2019. Manganese-induced neurotoxicity: new insights into the triad of protein misfolding, mitochondrial impairment, and neuroinflammation. Front. Neurosci 13, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH, 2016. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Ning N, Zhou Q, Khoshnam SE, Farzaneh M, 2019. Mitochondria as a therapeutic target for ischemic stroke. Free Radic. Biol. Med 146, 45–58. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Fink A, Doblhammer G, 2015. Effect of pioglitazone medication on the incidence of dementia. Ann. Neurol 78, 284–294. [DOI] [PubMed] [Google Scholar]
- Herzig S, Martinou JC, 2008. Mitochondrial dynamics: to be in good shape to survive. Curr. Mol. Med 8, 131–137. [DOI] [PubMed] [Google Scholar]
- Ho DJ, Calingasan NY, Wille E, Dumont M, Beal MF, 2010. Resveratrol protects against peripheral deficits in a mouse model of Huntington’s disease. Exp. Neurol 225, 74–84. [DOI] [PubMed] [Google Scholar]
- Hofmeijer J, van Putten MJ, 2012. Ischemic cerebral damage: an appraisal of synaptic failure. Stroke 43, 607–615. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM, 2005. The axonal transport of mitochondria. J. Cell Sci 118, 5411–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J, 2007. The machines that divide and fuse mitochondria. Annu. Rev. Biochem 76, 751–780. [DOI] [PubMed] [Google Scholar]
- Hu J, Lang Y, Cao Y, Zhang T, Lu H, 2015. The neuroprotective effect of tetramethylpyrazine against contusive spinal cord injury by activating PGC-1α in rats. Neurochem. Res 40, 1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Lang Y, Zhang T, Ni S, Lu H, 2016. Lentivirus-mediated PGC-1α overexpression protects against traumatic spinal cord injury in rats. Neuroscience 328, 40–49. [DOI] [PubMed] [Google Scholar]
- Huang PJ, Kuo CC, Lee HC, Shen CI, Cheng FC, Wu SF, Chang JC, Pan HC, Lin SZ, Liu CS, Su HL, 2016. Transferring xenogenic mitochondria provides neural protection against ischemic stress in ischemic rat brains. Cell Transplant. 25, 913–927. [DOI] [PubMed] [Google Scholar]
- Hubbard WB, Harwood CL, Geisler JG, Vekaria HJ, Sullivan PG, 2018. Mitochondrial uncoupling prodrug improves tissue sparing, cognitive outcome, and mitochondrial bioenergetics after traumatic brain injury in male mice. J. Neurosci. Res 96, 1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ, 2012. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K, 2006. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25, 2966–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez Olguin H, Calderon Guzman D, Hernandez Garcia E, Barragan Mejia G, 2016. The role of dopamine and its dysfunction as a consequence of oxidative stress. Oxidative Med. Cell. Longev 2016, 9730467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahraman S, Bambrick LL, Fiskum G, 2011. Effects of FK506 and cyclosporin a on calcium ionophore-induced mitochondrial depolarization and cytosolic calcium in astrocytes and neurons. J. Neurosci. Res 89, 1973–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karalija A, Novikova LN, Kingham PJ, Wiberg M, Novikov LN, 2012. Neuroprotective effects of N-acetyl-cysteine and acetyl-L-carnitine after spinal cord injury in adult rats. PLoS One 7, e41086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karalija A, Novikova LN, Kingham PJ, Wiberg M, Novikov LN, 2014. The effects of N-acetyl-cysteine and acetyl-L-carnitine on neural survival, neuroinflammation and regeneration following spinal cord injury. Neuroscience 269, 143–151. [DOI] [PubMed] [Google Scholar]
- Karch J, Molkentin JD, 2014. Identifying the components of the elusive mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. U. S. A 111, 10396–10397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EH, Robbins J, Kinnally KW, Molkentin JD, 2013. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife 2, e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating GM, 2015. Dexmedetomidine: a review of its use for sedation in the intensive care setting. Drugs 75, 1119–1130. [DOI] [PubMed] [Google Scholar]
- Kelly DP, Scarpulla RC, 2004. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 18, 357–368. [DOI] [PubMed] [Google Scholar]
- Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF, 2017. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 40, 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilbride SM, Gluchowska SA, Telford JE, O’Sullivan C, Davey GP, 2011. High-level inhibition of mitochondrial complexes III and IV is required to increase glutamate release from the nerve terminal. Mol. Neurodegener 6, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH, 2007. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Moody JP, Edgerly CK, Bordiuk OL, Cormier K, Smith K, Beal MF, Ferrante RJ, 2010. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet 19, 3919–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobashigawa S, Suzuki K, Yamashita S, 2011. Ionizing radiation accelerates Drp1-dependent mitochondrial fission, which involves delayed mitochondrial reactive oxygen species production in normal human fibroblast-like cells. Biochem. Biophys. Res. Commun 414, 795–800. [DOI] [PubMed] [Google Scholar]
- Korde AS, Sullivan PG, Maragos WF, 2005. The uncoupling agent 2,4-dinitrophenol improves mitochondrial homeostasis following striatal quinolinic acid injections. J. Neurotrauma 22, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Krishna G, Agrawal R, Zhuang Y, Ying Z, Paydar A, Harris NG, Royes LFF, Gomez-Pinilla F, 2017. 7,8-Dihydroxyflavone facilitates the action exercise to restore plasticity and functionality: Implications for early brain trauma recovery. Biochim. Biophys. Acta Mol. basis Dis 1863, 1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristal BS, Stavrovskaya IG, Narayanan MV, Krasnikov BF, Brown AM, Beal MF, Friedlander RM, 2004. The mitochondrial permeability transition as a target for neuroprotection. J. Bioenerg. Biomembr 36, 309–312. [DOI] [PubMed] [Google Scholar]
- Ladak AA, Enam SA, Ibrahim MT, 2019. A review of the molecular mechanisms of Traumatic Brain Injury. World Neurosurg. 131, 126–132. [DOI] [PubMed] [Google Scholar]
- Lee JM, Grabb MC, Zipfel GJ, Choi DW, 2000. Brain tissue responses to ischemia. J. Clin. Invest 106, 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Westrate LM, Wu H, Page C, Voeltz GK, 2016. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540, 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL, 2017. Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annu. Rev. Pharmacol. Toxicol 57, 535–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang H, Gao Y, Li L, Tang C, Wen G, Yang Y, Zhuang Z, Zhou M, Mao L, Fan Y, 2016. Quercetin induces mitochondrial biogenesis in experimental traumatic brain injury via the PGC-1alpha signaling pathway. Am. J. Transl. Res 8, 3558–3566. [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang X, Deng Z, Zhang X, Gao P, Liu H, 2018a. Dexmedetomidine reduces oxidative stress and provides neuroprotection in a model of traumatic brain injury via the PGC-1alpha signaling pathway. Neuropeptides 72, 58–64. [DOI] [PubMed] [Google Scholar]
- Li Q, Gao S, Kang Z, Zhang M, Zhao X, Zhai Y, Huang J, Yang GY, Sun W, Wang J, 2018b. Rapamycin enhances mitophagy and attenuates apoptosis after spinal ischemia-reperfusion injury. Front. Neurosci 12, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang H, Wen G, Li L, Gao Y, Zhuang Z, Zhou M, Mao L, Fan Y, 2018c. Neuroprotection by quercetin via mitochondrial function adaptation in traumatic brain injury: PGC-1alpha pathway as a potential mechanism. J. Cell. Mol. Med 22, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang C, He T, Zhao T, Chen YY, Shen YL, Zhang X, Wang LL, 2019. Mitochondrial transfer from bone marrow mesenchymal stem cells to motor neurons in spinal cord injury rats via gap junction. Theranostics 9, 2017–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz J, Friberg H, Neumar RW, Raghupathi R, Welsh FA, Janmey P, Saatman KE, Wieloch T, Grady MS, McIntosh TK, 2003. Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: evidence for differentially sensitive populations in the cortex and hippocampus. J. Cereb. Blood Flow Metab 23, 219–231. [DOI] [PubMed] [Google Scholar]
- Lifshitz J, Sullivan PG, Hovda DA, Wieloch T, McIntosh TK, 2004. Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion 4, 705–713. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF, 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. [DOI] [PubMed] [Google Scholar]
- Liu JM, Yi Z, Liu SZ, Chang JH, Dang XB, Li QY, Zhang YL, 2015. The mitochondrial division inhibitor mdivi-1 attenuates spinal cord ischemia-reperfusion injury both in vitro and in vivo: Involvement of BK channels. Brain Res. 1619, 155–165. [DOI] [PubMed] [Google Scholar]
- Liu R, Zhong X, Zeng J, Huang Z, Li X, Xiao H, Chen Q, Li D, 2017. 3’-Daidzein sulfonate sodium inhibits neuronal apoptosis induced by cerebral ischemia-reperfusion. Int. J. Mol. Med 39, 1021–1028. [DOI] [PubMed] [Google Scholar]
- Long J, Gao H, Sun L, Liu J, Zhao-Wilson X, 2009. Grape extract protects mitochondria from oxidative damage and improves locomotor dysfunction and extends lifespan in a Drosophila Parkinson’s disease model. Rejuvenation Res. 12, 321–331. [DOI] [PubMed] [Google Scholar]
- Loson OC, Song Z, Chen H, Chan DC, 2013. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G, Yi J, Ma C, Xiao Y, Yi F, Yu T, Zhou J, 2013. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS One 8, e82112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Xie Y, Chen Y, Han B, Li J, Qi S, 2016. Post-ischemia mdivi-1 treatment protects against ischemia/reperfusion-induced brain injury in a rat model. Neurosci. Lett 632, 23–32. [DOI] [PubMed] [Google Scholar]
- Manczak M, Reddy PH, 2012. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet 21, 2538–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragos WF, Rockich KT, Dean JJ, Young KL, 2003. Pre- or post-treatment with the mitochondrial uncoupler 2,4-dinitrophenol attenuates striatal quinolinate lesions. Brain Res. 966, 312–316. [DOI] [PubMed] [Google Scholar]
- Markowicz-Piasecka M, Huttunen KM, Mateusiak L, Mikiciuk-Olasik E, Sikora J, 2017. Is metformin a perfect drug? updates in pharmacokinetics and pharmacodynamics. Curr. Pharm. Des 23, 2532–2550. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T, 2003. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med 9, 1062–1068. [DOI] [PubMed] [Google Scholar]
- McEwen ML, Sullivan PG, Rabchevsky AG, Springer JE, 2011. Targeting mitochondrial function for the treatment of acute spinal cord injury. Neurotherapeutics 8, 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland R, Taylor RW, Turnbull DM, 2010. A neurological perspective on mitochondrial disease. Lancet Neurol. 9, 829–840. [DOI] [PubMed] [Google Scholar]
- McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H, 2012. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Invest 122, 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meglei G, McQuibban GA, 2009. The dynamin-related protein Mgm1p assembles into oligomers and hydrolyzes GTP to function in mitochondrial membrane fusion. Biochemistry 48, 1774–1784. [DOI] [PubMed] [Google Scholar]
- Morais VA, De Strooper B, 2010. Mitochondria dysfunction and neurodegenerative disorders: cause or consequence. J. Alzheimers Dis 20 (Suppl. 2), S255–S263. [DOI] [PubMed] [Google Scholar]
- Morita M, Gravel SP, Hulea L, Larsson O, Pollak M, St-Pierre J, Topisirovic I, 2015. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 14, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouli PK, Twig G, Shirihai OS, 2009. Frequency and selectivity of mitochondrial fusion are key to its quality maintenance function. Biophys. J 96, 3509–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, 2009. How mitochondria produce reactive oxygen species. Biochem. J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ, 2010. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Williams JA, Ding WX, 2015. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Shaw JM, 2005. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet 39, 503–536. [DOI] [PubMed] [Google Scholar]
- Oliver DMA, Reddy PH, 2019. Molecular Basis of Alzheimer’s Disease: focus on mitochondria. J. Alzheimers Dis s1, S95–S116. [DOI] [PubMed] [Google Scholar]
- Ono T, Isobe K, Nakada K, Hayashi JI, 2001. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet 28, 272–275. [DOI] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K, 2010. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol 191, 1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MR, Doran E, Halestrap AP, 2000. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J 348 (Pt 3), 607–614. [PMC free article] [PubMed] [Google Scholar]
- Oyinbo CA, 2011. Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol. Exp 71, 281–299. [DOI] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT, 2011. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12, 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Nukala VN, Sebastian AH, Day KM, Korde AS, Maragos WF, Hall ED, Sullivan PG, 2007. Post-Injury administration of mitochondrial uncouplers increases tissue sparing and improves behavioral outcome following traumatic brain injury in rodents. J. Neurotrauma 24, 798–811. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Sullivan PG, 2009. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Exp. Neurol 218, 381–389. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Nukala VN, Sullivan PG, 2013. Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Front. Neuroenerg 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Lyttle TS, Rabchevsky AG, 2010. Acetyl-L-carnitine ameliorates mitochondrial dysfunction following contusion spinal cord injury. J. Neurochem 114, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Lyttle TS, Magnuson DS, Rabchevsky AG, 2012. Acetyl-L-carnitine treatment following spinal cord injury improves mitochondrial function correlated with remarkable tissue sparing and functional recovery. Neuroscience 210, 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L, Men X, Zhang W, Wang H, Xu S, Xu M, Xu Y, Yang W, Lou J, 2011. Dynamin-related protein 1 is implicated in endoplasmic reticulum stress-induced pancreatic beta-cell apoptosis. Int. J. Mol. Med 28, 161–169. [DOI] [PubMed] [Google Scholar]
- Peng K, Tao Y, Zhang J, Wang J, Ye F, Dan G, Zhao Y, Cai Y, Zhao J, Wu Q, Zou Z, Cao J, Sai Y, 2016. Resveratrol regulates mitochondrial biogenesis and fission/fusion to attenuate rotenone-induced neurotoxicity. Oxidative Med. Cell. Longev 2016, 6705621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettegrew JW, McClure RJ, 2002. Acetyl-l-carnitine as a possible therapy for Alzheimer’s disease. Expert. Rev. Neurother 2, 647–654. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ, 2015. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisano A, Cerbelli B, Perli E, Pelullo M, Bargelli V, Preziuso C, Mancini M, He L, Bates MG, Lucena JR, Della Monica PL, Familiari G, Petrozza V, Nediani C, Taylor RW, d’Amati G, Giordano C, 2016. Impaired mitochondrial biogenesis is a common feature to myocardial hypertrophy and end-stage ischemic heart failure. Cardiovasc. Pathol 25, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puca FM, Genco S, Specchio LM, Brancasi B, D’Ursi R, Prudenzano A, Miccoli A, Scarcia R, Martino R, Savarese M, 1990. Clinical pharmacodynamics of acetyl-L-carnitine in patients with Parkinson’s disease. Int. J. Clin. Pharmacol. Res 10, 139–143. [PubMed] [Google Scholar]
- Qi X, Qvit N, Su YC, Mochly-Rosen D, 2013. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci 126, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi B, Hu L, Zhu L, Shang L, Sheng L, Wang X, Liu N, Wen N, Yu X, Wang Q, Yang Y, 2017. Metformin attenuates cognitive impairments in hypoxia-ischemia neonatal rats via improving remyelination. Cell. Mol. Neurobiol 37, 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Qiao F, Atkinson C, Kindy MS, Shunmugavel A, Morgan BP, Song H, Tomlinson S, 2010. The alternative and terminal pathways of complement mediate post-traumatic spinal cord inflammation and injury. Am. J. Pathol 177, 3061–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao H, Zhang Q, Yuan H, Li Y, Wang D, Wang R, He X, 2015. Elevated neuronal alpha-synuclein promotes microglia activation after spinal cord ischemic/reperfused injury. Neuroreport 26, 656–661. [DOI] [PubMed] [Google Scholar]
- Qu J, Chen W, Hu R, Feng H, 2016. The injury and therapy of reactive oxygen species in intracerebral hemorrhage looking at mitochondria. Oxidative Med. Cell. Longev 2016, 2592935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, Patel SP, Springer JE, 2011. Pharmacological interventions for spinal cord injury: where do we stand? How might we step forward? Pharmacol. Ther 132, 15–29. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, Graham DI, McIntosh TK, 2000. Apoptosis after traumatic brain injury. J. Neurotrauma 17, 927–938. [DOI] [PubMed] [Google Scholar]
- Raizner AE, 2019. Coenzyme Q10. Methodist Debakey Cardiovasc J 15, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramis MR, Esteban S, Miralles A, Tan DX, Reiter RJ, 2015. Protective effects of melatonin and mitochondria-targeted antioxidants against oxidative stress: a review. Curr. Med. Chem 22, 2690–2711. [DOI] [PubMed] [Google Scholar]
- Rani L, Mondal AC, 2019. Emerging concepts of mitochondrial dysfunction in Parkinson’s disease progression: Pathogenic and therapeutic implications. Mitochondrion 50, 25–34. [DOI] [PubMed] [Google Scholar]
- Rasbach KA, Schnellmann RG, 2008. Isoflavones promote mitochondrial biogenesis. J. Pharmacol. Exp. Ther 325, 536–543. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Beal MF, 2005. Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res. Brain Res. Rev 49, 618–632. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Manczak M, Yin X, Grady MC, Mitchell A, Tonk S, Kuruva CS, Bhatti JS, Kandimalla R, Vijayan M, Kumar S, Wang R, Pradeepkiran JA, Ogunmokun G, Thamarai K, Quesada K, Boles A, Reddy AP, 2018. Protective effects of indian spice curcumin against amyloid-beta in Alzheimer’s Disease. J. Alzheimers Dis 61, 843–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss AB, Arain HA, Stecker MM, Siegart NM, Kasselman LJ, 2018. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci 29, 613–627. [DOI] [PubMed] [Google Scholar]
- Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD, 2006. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. The Pharmacogen J. 6, 246–254. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Kai Y, Maldonado E, Currin RT, Lemasters JJ, 2009. Roles of mitophagy and the mitochondrial permeability transition in remodeling of cultured rat hepatocytes. Autophagy 5, 1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal RE, Williams R, Bogaert YE, Getson PR, Fiskum G, 1992. Prevention of postischemic canine neurological injury through potentiation of brain energy metabolism by acetyl-L-carnitine. Stroke 23, 1312–1317 discussion 1317–1318. [DOI] [PubMed] [Google Scholar]
- Rowland JW, Hawryluk GW, Kwon B, Fehlings MG, 2008. Current status of acute spinal cord injury pathophysiology and emerging therapies: promise on the horizon. Neurosurg. Focus 25, E2. [DOI] [PubMed] [Google Scholar]
- Saint-Geniez M, Jiang A, Abend S, Liu L, Sweigard H, Connor KM, Arany Z, 2013. PGC-1alpha regulates normal and pathological angiogenesis in the retina. Am. J. Pathol 182, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Huttemann M, 2013. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol 47, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A, Fuller MT, 2001. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci 114, 867–874. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC, 2008. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev 88, 611–638. [DOI] [PubMed] [Google Scholar]
- Scholpa NE, Schnellmann RG, 2017. Mitochondrial-based therapeutics for the treatment of spinal cord injury: mitochondrial biogenesis as a potential pharmacological target. J. Pharmacol. Exp. Ther 363, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholpa NE, Lynn MK, Corum D, Boger HA, Schnellmann RG, 2018a. 5-HT1F receptor-mediated mitochondrial biogenesis for the treatment of Parkinson’s disease. Br. J. Pharmacol 175, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholpa NE, Williams H, Wang W, Corum D, Narang A, Tomlinson S, Sulllivan PG, Rabchevsky AGPD, Schnellmann RG, 2018b. Pharmacological stimulation of mitochondrial biogenesis using the FDA-approved beta < sub > 2 < /sub > - adrenoreceptor agonist formoterol for the treatment of spinal cord injury. J. Neurotrauma 6, 962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholpa NE, Simmons EC, Tilley DG, Schnellmann RG, 2019a. beta2-adrenergic receptor-mediated mitochondrial biogenesis improves skeletal muscle recovery following spinal cord injury. Exp. Neurol 322, 113064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholpa NE, Williams H, Wang W, Corum D, Narang A, Tomlinson S, Sullivan PG, Rabchevsky AG, Schnellmann RG, 2019b. Pharmacological stimulation of mitochondrial biogenesis using the food and drug administration-approved beta2-adrenoreceptor agonist formoterol for the treatment of spinal cord injury. J. Neurotrauma 36, 962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott I, Youle RJ, 2010. Mitochondrial fission and fusion. Essays Biochem. 47, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian D, Palacin M, Zorzano A, 2017. Mitochondrial dynamics: coupling mitochondrial fitness with healthy aging. Trends Mol. Med 23, 201–215. [DOI] [PubMed] [Google Scholar]
- Seese RR, Le AA, Wang K, Cox CD, Lynch G, Gall CM, 2019. A TrkB agonist and ampakine rescue synaptic plasticity and multiple forms of memory in a mouse model of intellectual disability. Neurobiol. Dis 104604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi A, Kanno H, Ozawa H, Yamaya S, Itoi E, 2012. Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J. Neurotrauma 29, 946–956. [DOI] [PubMed] [Google Scholar]
- Shabab T, Khanabdali R, Moghadamtousi SZ, Kadir HA, Mohan G, 2017. Neuroinflammation pathways: a general review. Int J Neurosci 127, 624–633. [DOI] [PubMed] [Google Scholar]
- Shapiro RE, Hochstetler HM, Dennehy EB, Khanna R, Doty EG, Berg PH, Starling AJ, 2019. Lasmiditan for acute treatment of migraine in patients with cardiovascular risk factors: post-hoc analysis of pooled results from 2 randomized, double-blind, placebo-controlled, phase 3 trials. J Headache Pain 20, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng B, Wang X, Su B, Lee H.g., Casadesus G, Perry G, Zhu X, 2012. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem 120, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Zhao M, Fu C, Fu A, 2017. Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 34, 91–100. [DOI] [PubMed] [Google Scholar]
- Simmons EC, Scholpa NE, Cleveland KH, Schnellmann RG, 2020. 5-HT1F receptor agonist induces mitochondrial biogenesis and promotes recovery from spinal cord injury. J. Pharmacol. Exp. Ther 2, 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM, 2017. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol 13, 171–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR, Muyderman H, 2010. Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta 1802, 80–91. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED, 2006. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J. Cereb. Blood Flow Metab 26, 1407–1418. [DOI] [PubMed] [Google Scholar]
- Smith GM, Gallo G, 2018. The role of mitochondria in axon development and regeneration. Developmental neurobiology 78, 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM, 2010. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord 25, 1670–1674. [DOI] [PubMed] [Google Scholar]
- Song Y, Xue H, Liu TT, Liu JM, Chen D, 2015. Rapamycin plays a neuroprotective effect after spinal cord injury via anti-inflammatory effects. J. Biochem. Mol. Toxicol 29, 29–34. [DOI] [PubMed] [Google Scholar]
- Sonmez E, Kabatas S, Ozen O, Karabay G, Turkoglu S, Ogus E, Yilmaz C, Caner H, Altinors N, 2013. Minocycline treatment inhibits lipid peroxidation, preserves spinal cord ultrastructure, and improves functional outcome after traumatic spinal cord injury in the rat. Spine 38, 1253–1259. [DOI] [PubMed] [Google Scholar]
- Springer JE, Prajapati P, Sullivan PG, 2018. Targeting the mitochondrial permeability transition pore in traumatic central nervous system injury. Neural Regen. Res 13, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout JM, Knapp AN, Banz WJ, Wallace DG, Cheatwood JL, 2013. Subcutaneous daidzein administration enhances recovery of skilled ladder rung walking performance following stroke in rats. Behav. Brain Res 256, 428–431. [DOI] [PubMed] [Google Scholar]
- Su J, Liu J, Yan XY, Zhang Y, Zhang JJ, Zhang LC, Sun LK, 2017. Cytoprotective effect of the UCP2-SIRT3 signaling pathway by decreasing mitochondrial oxidative stress on cerebral ischemia-reperfusion injury. Int. J. Mol. Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW, 1999. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp. Neurol 160, 226–234. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Keller JN, Bussen WL, Scheff SW, 2002. Cytochrome c release and caspase activation after traumatic brain injury. Brain Res. 949, 88–96. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Dube C, Dorenbos K, Steward O, Baram TZ, 2003. Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann. Neurol 53, 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Springer JE, Hall ED, Scheff SW, 2004. Mitochondrial uncoupling as a therapeutic target following neuronal injury. J. Bioenerg. Biomembr 36, 353–356. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Krishnamurthy S, Patel SP, Pandya JD, Rabchevsky AG, 2007. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. J. Neurotrauma 24, 991–999. [DOI] [PubMed] [Google Scholar]
- Tambasco N, Murgia N, Nigro P, Paoletti FP, Romoli M, Brahimi E, Filidei M, Simoni S, Muzi G, Calabresi P, 2018. Levodopa-responsive breathing discomfort in Parkinson’s disease patients. J. Neural Transm. (Vienna) 125, 1033–1036. [DOI] [PubMed] [Google Scholar]
- Tang S, Xu S, Lu X, Gullapalli RP, McKenna MC, Waddell J, 2016. Neuroprotective effects of acetyl-L-carnitine on neonatal hypoxia ischemia-induced brain injury in rats. Dev. Neurosci 38, 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tator CH, Fehlings MG, 1991. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J. Neurosurg 75, 15–26. [DOI] [PubMed] [Google Scholar]
- Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM, 2004. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc. Natl. Acad. Sci. U. S. A 101, 3071–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom R, Rowe GC, Jang C, Safdar A, Arany Z, 2014. Hypoxic induction of vascular endothelial growth factor (VEGF) and angiogenesis in muscle by truncated peroxisome proliferator-activated receptor gamma coactivator (PGC)-1alpha. J. Biol. Chem 289, 8810–8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton C, Jones A, Nair S, Aabdien A, Mallard C, Hagberg H, 2018. Mitochondrial dynamics, mitophagy and biogenesis in neonatal hypoxic-ischaemic brain injury. FEBS Lett. 592, 812–830. [DOI] [PubMed] [Google Scholar]
- Tian B, Wang XL, Huang Y, Chen LH, Cheng RX, Zhou FM, Guo R, Li JC, Liu T, 2016. Peripheral and spinal 5-HT receptors participate in cholestatic itch and antinociception induced by bile duct ligation in rats. Sci. Rep 6, 36286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassini V, Pozzilli C, Onesti E, Pasqualetti P, Marinelli F, Pisani A, Fieschi C, 2004. Comparison of the effects of acetyl L-carnitine and amantadine for the treatment of fatigue in multiple sclerosis: results of a pilot, randomised, double-blind, crossover trial. J. Neurol. Sci 218, 103–108. [DOI] [PubMed] [Google Scholar]
- Totoiu MO, Keirstead HS, 2005. Spinal cord injury is accompanied by chronic progressive demyelination. J. Comp. Neurol 486, 373–383. [DOI] [PubMed] [Google Scholar]
- Truban D, Hou X, Caulfield TR, Fiesel FC, Springer W, 2017. PINK1, Parkin, and mitochondrial quality control: what can we learn about parkinson’s disease pathobiology? J. Parkinsons Dis 7, 13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufekci KU, Civi Bayin E, Genc S, Genc K, 2011. The Nrf2/ARE pathway: a promising target to counteract mitochondrial dysfunction in Parkinson’s Disease. Parkinsons Dis 2011, 314082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Shirihai OS, 2011. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal 14, 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS, 2008. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin R, Grabow S, Davids MS, 2018. The rise of apoptosis: targeting apoptosis in hematologic malignancies. Blood 132, 1248–1264. [DOI] [PubMed] [Google Scholar]
- van der Bliek AM, Shen Q, Kawajiri S, 2013. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura-Clapier R, Garnier A, Veksler V, 2008. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc. Res 79, 208–217. [DOI] [PubMed] [Google Scholar]
- Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, Marambaud P, 2010. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem 285, 9100–9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virmani T, Tazan S, Mazzoni P, Ford B, Greene PE, 2016. Motor fluctuations due to interaction between dietary protein and levodopa in Parkinson’s disease. J Clin Mov Disord 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Boraso M, Marchetti N, Marinovich M, 2014. Perspectives on neuroinflammation and excitotoxicity: a neurotoxic conspiracy? Neurotoxicology 43, 10–20. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang P, Li S, Wang S, Li Y, Liang N, Wang M, 2014. Mdivi-1 prevents apoptosis induced by ischemia-reperfusion injury in primary hippocampal cells via inhibition of reactive oxygen species-activated mitochondrial pathway. J. Stroke Cerebrovasc. Dis 23, 1491–1499. [DOI] [PubMed] [Google Scholar]
- Wang C, Niu M, Zhou Z, Zheng X, Zhang L, Tian Y, Yu X, Bu G, Xu H, Ma Q, Zhang YW, 2016a. VPS35 regulates cell surface recycling and signaling of dopamine receptor D1. Neurobiol. Aging 46, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, Cullen PJ, Liu J, Zhu X, 2016b. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med 22, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Sullivan PG, Springer JE, 2017. Mitochondria and microRNA crosstalk in traumatic brain injury. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 73, 104–108. [DOI] [PubMed] [Google Scholar]
- Wang CF, Song CY, Wang X, Huang LY, Ding M, Yang H, Wang P, Xu LL, Xie ZH, Bi JZ, 2019. Protective effects of melatonin on mitochondrial biogenesis and mitochondrial structure and function in the HEK293-APPswe cell model of Alzheimer’s disease. Eur. Rev. Med. Pharmacol. Sci 23, 3542–3550. [DOI] [PubMed] [Google Scholar]
- Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV, 2013. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci 33, 12870–12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wappler EA, Institoris A, Dutta S, Katakam PV, Busija DW, 2013. Mitochondrial dynamics associated with oxygen-glucose deprivation in rat primary neuronal cultures. PLoS One 8, e63206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, Fishel MA, Kulstad JJ, Green PS, Cook DG, Kahn SE, Keeling ML, Craft S, 2005. Preserved cognition in patients with early alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am. J. Geriatr. Psychiatry 13, 950–958. [DOI] [PubMed] [Google Scholar]
- Wells JE, Hurlbert RJ, Fehlings MG, Yong VW, 2003. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain 126, 1628–1637. [DOI] [PubMed] [Google Scholar]
- Westermann B, 2008. Molecular machinery of mitochondrial fusion and fission. J. Biol. Chem 283, 13501–13505. [DOI] [PubMed] [Google Scholar]
- Westermann B, 2012. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 1817, 1833–1838. [DOI] [PubMed] [Google Scholar]
- Whitaker RM, Corum D, Beeson CC, Schnellmann RG, 2016. Mitochondrial biogenesis as a pharmacological target: a new approach to acute and chronic diseases. Annu. Rev. Pharmacol. Toxicol 56, 229–249. [DOI] [PubMed] [Google Scholar]
- Wills LP, Trager RE, Beeson GC, Lindsey CC, Peterson YK, Beeson CC, Schnellmann RG, 2012. The beta2-adrenoceptor agonist formoterol stimulates mitochondrial biogenesis. J. Pharmacol. Exp. Ther 342, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witiw CD, Fehlings MG, 2015. Acute Spinal Cord Injury. J. Spinal Disord. Tech 28, 202–210. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM, 1999. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124. [DOI] [PubMed] [Google Scholar]
- Wu S, Zhou F, Zhang Z, Xing D, 2011. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J. 278, 941–954. [DOI] [PubMed] [Google Scholar]
- Wu Q, Xia SX, Li QQ, Gao Y, Shen X, Ma L, Zhang MY, Wang T, Li YS, Wang ZF, Luo CL, Tao LY, 2016. Mitochondrial division inhibitor 1 (Mdivi-1) offers neuroprotection through diminishing cell death and improving functional outcome in a mouse model of traumatic brain injury. Brain Res. 1630, 134–143. [DOI] [PubMed] [Google Scholar]
- Wu P, Li Y, Zhu S, Wang C, Dai J, Zhang G, Zheng B, Xu S, Wang L, Zhang T, Zhou P, Zhang JH, Shi H, 2017. Mdivi-1 alleviates early brain injury after experimental subarachnoid hemorrhage in rats, possibly via inhibition of Drp1-activated mitochondrial fission and oxidative stress. Neurochem. Res 42, 1449–1458. [DOI] [PubMed] [Google Scholar]
- Xu T, Ding W, Ao X, Chu X, Wan Q, Wang Y, Xiao D, Yu W, Li M, Yu F, Wang J, 2019. ARC regulates programmed necrosis and myocardial ischemia/reperfusion injury through the inhibition of mPTP opening. Redox Biol. 20, 414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan MH, Wang X, Zhu X, 2013. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med 62, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF, 2009. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid. Redox Signal 11, 2095–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JL, Mukda S, Chen SD, 2018. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 16, 263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Y, Padanilam BJ, 2016. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cell. Mol. Life Sci 73, 2309–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Chen Q, Zeng J, Xiao H, Huang ZH, Li X, Lei Q, 2017. 3’-Daidzein sulfonate sodium improves mitochondrial functions after cerebral ischemia/reperfusion injury. Neural Regen. Res 12, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidat OO, Yoo AJ, Khatri P, Tomsick TA, von Kummer R, Saver JL, Marks MP, Prabhakaran S, Kallmes DF, Fitzsimmons BF, Mocco J, Wardlaw JM, Barnwell SL, Jovin TG, Linfante I, Siddiqui AH, Alexander MJ, Hirsch JA, Wintermark M, Albers G, Woo HH, Heck DV, Lev M, Aviv R, Hacke W, Warach S, Broderick J, Derdeyn CP, Furlan A, Nogueira RG, Yavagal DR, Goyal M, Demchuk AM, Bendszus M, Liebeskind DS, 2013. Recommendations on angiographic revascularization grading standards for acute ischemic stroke: a consensus statement. Stroke 44, 2650–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanelli SA, Solenski NJ, Rosenthal RE, Fiskum G, 2005. Mechanisms of ischemic neuroprotection by acetyl-L-carnitine. Ann. N. Y. Acad. Sci 1053, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, He Z, Zhang Q, Wu Y, Yang X, Niu W, Hu Y, Jia J, 2014. Exercise pretreatment promotes mitochondrial dynamic protein OPA1 expression after cerebral ischemia in rats. Int. J. Mol. Sci 15, 4453–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZY, Fan ZK, Cao Y, Jia ZQ, Li G, Zhi XD, Yu DS, Lv G, 2015. Acetyl-L-carnitineameliorates mitochondrial damage and apoptosis following spinal cord injury in rats. Neurosci. Lett 604, 18–23. [DOI] [PubMed] [Google Scholar]
- Zhao YX, Cui M, Chen SF, Dong Q, Liu XY, 2014. Amelioration of ischemic mitochondrial injury and Bax-dependent outer membrane permeabilization by Mdivi-1. CNS Neurosci Ther 20, 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZD, Lim TM, 2009. Roles of glutathione (GSH) in dopamine (DA) oxidation studied by improved tandem HPLC plus ESI-MS. Neurochem. Res 34, 316–326. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Wang H, Fang J, Dai W, Zhou J, Wang X, Zhou M, 2018. SS-31 provides neuroprotection by reversing mitochondrial dysfunction after traumatic brain injury. Oxidative Med. Cell. Longev 2018, 4783602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Yang LK, Chen WL, Lin W, Wang YH, Chen T, 2019. Activation of SK/KCa channel attenuates spinal cord ischemia-reperfusion injury via anti-oxidative activity and inhibition of mitochondrial dysfunction in rabbits. Front. Pharmacol 10, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Thompson CB, 2006. Necrotic death as a cell fate. Genes Dev. 20, 1–15. [DOI] [PubMed] [Google Scholar]
- Zong WX, Rabinowitz JD, White E, 2016. Mitochondria and cancer. Mol. Cell 61, 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schroder JM, Vance JM, 2004. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet 36, 449–451. [DOI] [PubMed] [Google Scholar]
- Zuchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, Hamilton SR, Van Stavern G, Krajewski KM, Stajich J, Tournev I, Verhoeven K, Langerhorst CT, de Visser M, Baas F, Bird T, Timmerman V, Shy M, Vance JM, 2006. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol 59, 276–281. [DOI] [PubMed] [Google Scholar]