

Abstract
Objective:
To elucidate molecular risk factors for posterior segment uveitis using a functional genomics approach.
Design:
Genetic Association Cohort Study.
Methods:
Setting:
Single-center study at an academic referral center.
Study Population:
164 patients with clinically diagnosed uveitis of the posterior segment.
Main Outcome Measures:
Exome sequencing was used to detect variants identified in 164 patients with posterior segment uveitis. A phenotype-driven analysis, protein structural modeling and in silico calculations were then used to rank and predict the functional consequences of key variants.
Results:
A total of 203 single nucleotide variants, in 23 genes across 164 patients, were included in this study. Both known and novel variants were identified in genes previously implicated in specific types of syndromic uveitis — such as NOD2 (Blau Syndrome) and CAPN5 NIV (Neovascular Inflammatory Vitreoretinopathy) — as well as variants in genes not previously linked to posterior segment uveitis. Based on a ranked list and protein-protein-interaction network, missense variants in NOD-like receptor family genes (NOD2, NLRC4, NLRP3, and NLRP1), CAPN5, and TYK2 were characterized via structural modeling and in silico calculations to predict how specific variants might alter protein structure and function. The majority of analyzed variants were notably different from wild type.
Conclusions:
This study implicates new pathways and immune signaling proteins that may be associated with posterior segment uveitis susceptibility. A larger cohort and functional studies will help validate the pathogenicity of the mutations identified. In specific cases, whole exome sequencing can help diagnose non-syndromic uveitis patients harboring known variants for syndromic inflammatory diseases.
Introduction
Uveitis is a diverse group of intraocular inflammatory disorders that causes significant visual morbidity and blindness1. Uveitis can be classified by anatomical location: anterior segment uveitis (iritis or iridocyclitis) or posterior segment uveitis: intermediate uveitis (pars planitis or vitritis), posterior uveitis (choroiditis or chorioretinitis), and panuveitis2. Its etiology can be traumatic, neoplastic, infectious, or non-infectious3. Non-infectious uveitis comprises a group of heterogenous autoinflammatory diseases that can be either confined to the eye or present with systemic symptoms4. A wide range of these autoinflammatory diseases are driven by genetic mutations. For example, hyperactivating CAPN5 mutations cause neovascular inflammatory vitreoretinopathy (NIV), a rare inflammatory condition that involves severe progressive retinal inflammation, neovascularization, degeneration, and fibrosis5,6. NOD2 hyperactivating mutations cause Blau Syndrome, a rare disease characterized by early-onset granulomatous inflammation, skin rash, and uveitis7. Heterozygous loss-of-function mutations of TNFAIP3, known as A20 Haploinsufficiency, can cause an inflammatory syndrome that may include uveitis8,9. Despite this knowledge, more than half of uveitis cases remain idiopathic and the genetic susceptibility factors for non-Mendelian causes of uveitis are poorly understood10,11.
Genomic analyses of uveitis patients and animal models have advanced our understanding of the genetic risk factors for intraocular inflammation. While the precise mechanisms are poorly understood, the amassing evidence suggests an interplay between complex genetic backgrounds, immune system dysregulation, and environmental triggering factors such as infection. Several large-scale genomic studies have been performed on patients with systemic diseases associated with uveitis including Behçet’s disease (BD)12–17, sarcoidosis18,19, Vogt-Koyanagi-Harada syndrome (VKH)20,21, as well as inflammatory diseases confined to the eye, like birdshot chorioretinopathy (BSCR)22. These studies have identified variants in specific immune system genes linked to uveitis. Human leukocyte antigen (HLA) genes are the most common example, with HLA-A29 variants being linked to BSCR23. Systemic causes of uveitis, including BD or VKH, have been associated with specific HLA alleles and genetic variants in immune signaling genes including NLRP3, IL6, IL10, PTPN2, and ICAM3,21,24–27. Similarly, CCR2 and CCL2 polymorphisms were associated with worse clinical features in posterior uveitis patients3,6,7. However, these studies have been limited to certain subsets of genes or patients with known uveitis etiologies.
Proteomic analysis of human vitreous has identified cytokine signatures common to various etiologies of posterior segment uveitis, including idiopathic posterior uveitis28. The presence of elevated interleukin 23 (IL-23), IL-1 receptor I (IL-1RI), IL-17R, tissue inhibitors of metalloproteinase 1 and 2 (TIMP-1 and −2), insulin-like growth factor-binding protein 2 (IGFBP-2), nerve growth factor (NGF), platelet-derived growth factor receptor β (PDGFRb), bone morphogenic protein 4 (BMP-4), and stem cell factor (SCF) in uveitic vitreous suggests that these proteins represent a common pathogenic link regardless of etiology28. The use of genomics analyses may similarly identify common genetic susceptibility factors for uveitis. To our knowledge, there have not been any genetic studies of uveitis using whole exome sequencing (WES). Here, we use a systematic informatic approach to uncover novel genetic susceptibility factors associated with posterior segment uveitis.
Methods
Patients volunteered for this exploratory genetic-association cohort study at a single academic referral center. The project received prospective IRB approval. The title of the primary IRB is “Molecular Genetics of Vitreoretinal Disease and Uveitis” and it approved the collection of peripheral blood samples from uveitis patients to be used for exome sequencing. A separate IRB titled “Retrospective Chart Review of Vitreoretinal Disease Patients” was used to collect clinical information about our uveitis patient cohort. The study included 164 patients with uveitis involving the posterior segment. Both syndromic and non-syndromic cases were included, and all diagnoses were made by experienced retina specialists. Specific exome sequencing results were not shared with patients. The IRB and consent specifically stated that genetic testing was for research purposes only and all patients received pre-test counseling around this topic.
Whole Exome Sequencing and Analysis—
Peripheral blood samples were obtained from 164 uveitis patients, following informed consent. Blood samples were processed using standard exome enrichment analysis before undergoing massively parallel (NextGen) exome sequencing on the Illumina Hiseq 2000 platform at the University of Iowa DNA Core Facility. Sequencing was performed using paired end 150bp reads. The mean depth of coverage was 100x. Reads were then aligned to the GRCh37/UCSC hg19 reference genome with BWA-MEM, a Burrows-Wheeler aligner, and results were stored in the SAM/BAM format29. Single nucleotide variants (SNVs) and small insertions/deletions were detected using five separate variant callers: HaplotypeCaller from Genome Analysis Toolkit (GATK-HC), mpileup from SAMtools, FreeBayes, VarScan2, and Pindel30–34. Variants were then filtered in VarSeq for quality control, a minor allele frequency of <1% in the ExAC and 1000 Genomes databases35, to predict loss-of-function or missense mutations (VarSeq™ (Version 2.0); Golden Helix, Inc., Bozeman, MT). Genes containing the remaining variants were then ranked using PhoRank, a tool available in VarSeq modeled on the Phevor algorithm, that combines phenotype, gene function, and disease information from multiple ontologies, to prioritize disease-causing alleles36. The PhoRank score assigned to each gene was weighted (multiplied) by the number of unique variants discovered within that gene. The final list of 203 missense variants used for downstream analysis had a weighted Phevor score of greater than 1.
Annotation of Variants—
Variant annotation was performed in VarSeq, using RefSeq 105 Interim v1. For each variant, functional prediction was determined using SIFT37, PolyPhen-238, MutationTaster39, MutationAssessor40, FATHMM41, and FATHMM-MKL42. Each variant was searched in ClinVar (Feb. 2018) and dbSNP149 to determine if it had been previously identified and whether it was known to be associated with a particular disease. Each gene was also queried in OMIM and the literature, to determine if it was implicated in any ocular or inflammatory diseases.
Structural modeling of proteins —
Each protein was queried in the Protein Data Bank (PDB; https://www.rcsb.org) for known high-resolution structures. All homology-based models were generated using MODELLER 9.14 unless stated otherwise43,44. NOD2: A homology-based model was generated using the crystal structure of inactive (ADP-bound) rabbit NOD2 lacking the CARD domain as a template (PDB: 5IRM; 86% sequence identity). A model of the CARD domain was generated using the NOD1 CARD domain as a template (PDB: 4JQW; 24% sequence identity). A full-length model of NOD2 was generated from the two models using ab initio domain assembly45. NLRC4: A homology-based model was generated using the crystal structure of murine NLRC4 in its closed form (PDB: 4KXF; 99% sequence identity). NLRP3: The structure of NLRP3 was generated using the cryo-EM structure of the NAIP5-NLRC4-flagellin inflammasome as a template (PDB: 6B5B, 23% sequence identity) along with the crystal structure of NALP3 PYD (PDB: 3QF2, 100% sequence identity for residues 5–110). NLRP1 The structure of NLRP1, was similarly generated. Structure predictions were templated on a flagellin derivative in a complex with the NLR protein NAIP5 (PDB: 5YUD, 100% sequence identity), and supported by NMR-study predictions of the NLRP1PYD (PDB: 1PN5, 100% sequence identity for residues 1–93) and X-ray diffraction studies of the NLRP1LRR (PDB: 4IM6, 100% sequence identity for residues 792–989). CAPN5: Structural predictions were modeled on the crystal structure of the CAPN5 protease core domain (CAPN5-PC; PDB: 6P3Q). The active (Ca2+-bound) CAPN5-PC structure was modeled using the structure of active CAPN9-PC as a template (PDB: 2P0R; 48% sequence identity). Homology models of CAPN5’s calpain β-sandwich domain (CBSW) were generated using CAPN2 as a template (PDB: 1KFU, 32% identity). Homology models of the CAPN5 C2 domain were generated using the structure of the Munc13–1 C2 domain (PDB: 2CJT, 30% identity). A full-length model of CAPN5 was generated from the models using ab initio domain assembly as previously described46. TYK2: Structural predictions were based on the crystal structure of nonphosphorylated human TYK2 kinase with CMP6 (PDB 3NZ0; 99.3% sequence identity). Predictions of mutational effects on protein stability (i.e. ΔΔGfold) were performed using SDM47. PyMOL generated all structural images. (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC).
Results
We sequenced the exomes of 164 patients with a spectrum of clinically diagnosed uveitis involving the posterior segment (Fig. 1; Fig. 2A; Table S1). The mean age of the cohort was 57 (SD 17.5, range 5–91) and 115 (70%) were female. One hundred forty-one patients (86%) were white, fourteen were Native American, two were Asian, one was Black, and six had unknown race. All single nucleotide variants were filtered for minor allele frequency (MAF) <1% and predicted loss-of-function (LOF) or missense mutations. Genes containing one or more of these filtered variants were ranked using a phenotype and ontology-driven analysis to prioritize disease-causing alleles, then weighted based on the number of variants in that gene (Fig. 1). A total of 203 missense variants in the 23 highest prioritized genes were included in the downstream analysis (Table 1). All variants were single nucleotide missense or nonsense mutations, with a mean MAF of 1.05 × 10−3 and median MAF of 1.30 × 10−4 (Table S2). Sixty-three variants were novel, and 140 were previously reported. One hundred and twenty-eight patients (78%) had one or more of these variants, and 55 patients (43%) had only one (Fig. 2B). No subtype of posterior segment uveitis was over-represented in the 36 patients (22%) without any significant variants. Idiopathic uveitis patients were as likely to harbor these variants as patients with other etiologies of uveitis. Three patients harbored variants in genes that correlated with their clinical phenotype (e.g. CAPN5 and NOD2). Otherwise, in general, there was no single variant that correlated with a specific type of posterior segment uveitis.
Fig. 1. Whole-exome sequencing and analysis pipeline:
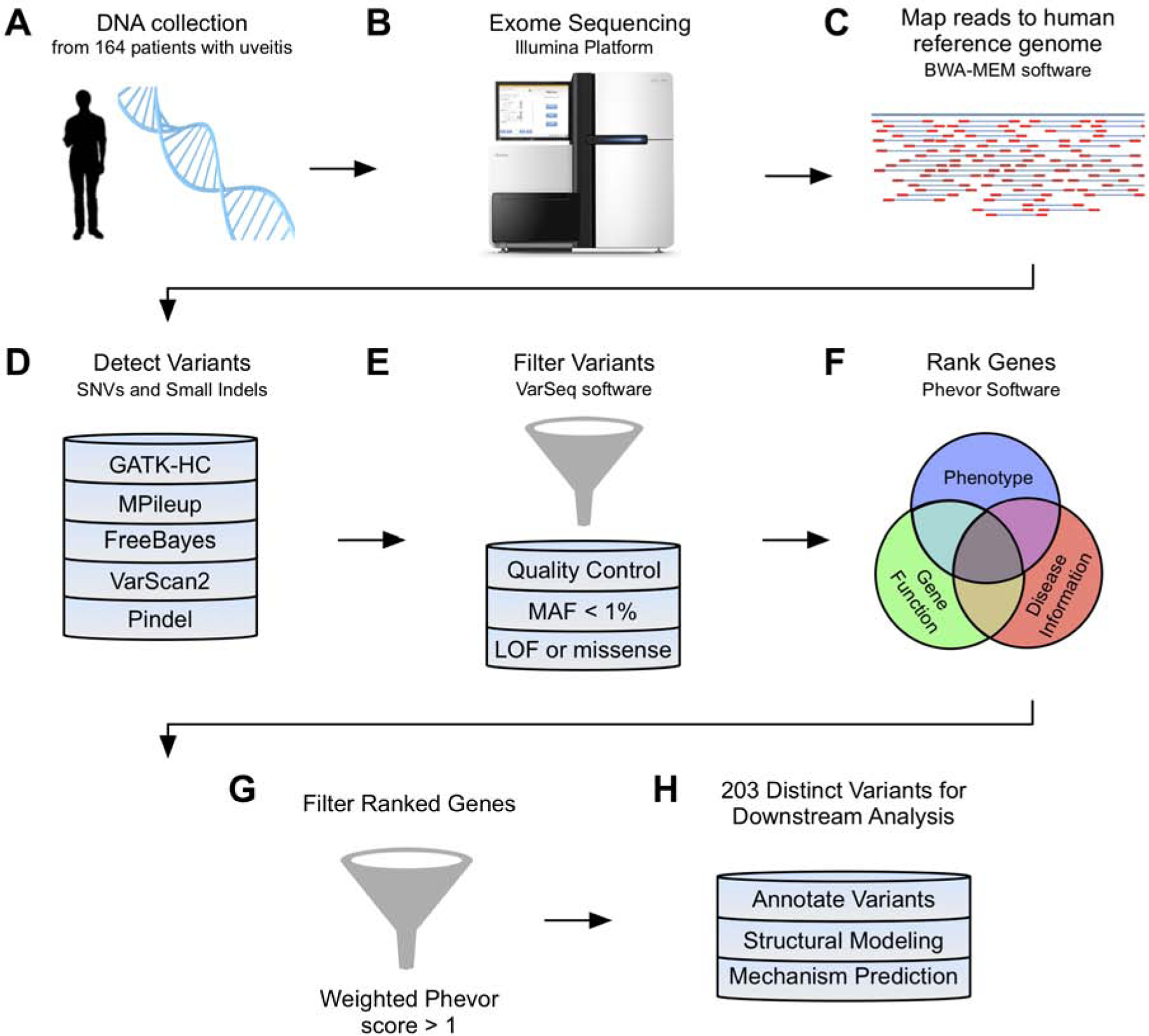
Overview of workflow used to sequence and analyze variants from 164 patients with uveitis. Five variant callers were used to detect SNVs and small insertions/deletions, and all variants were stringently filtered as described in the methods section. Genes containing the filtered variants were ranked to prioritize disease-causing alleles and weighted by the number of distinct variants in that gene. Variants were annotated using a combination of software programs (SIFT, PolyPhen-2, MutationTaster, MutationAssessor, FATHMM, and FATHMM-MKL), publicly available databases (ClinVar, dbSNP), and the literature.
Fig. 2. Network analysis highlights the inflammasome and other clusters of immune signaling genes:
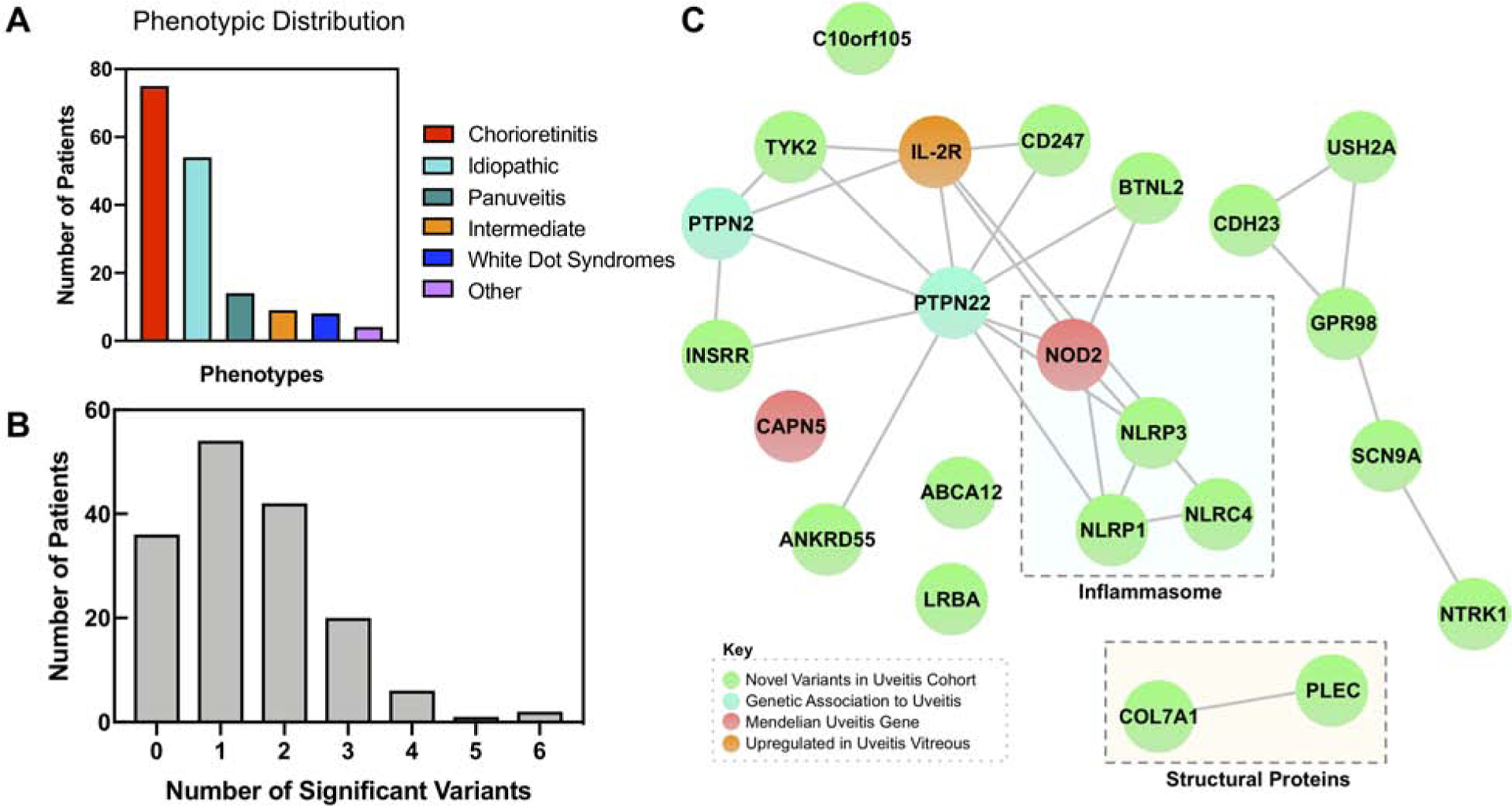
(A) Bar chart highlighting distribution of clinical phenotypes in the uveitis cohort. Uveitis patients were grouped into one of six categories based on their clinical phenotype, including the anatomic location and etiology. ‘Other’ includes cases such as uveitic glaucoma or uveitis after retinal tear. (B) Distribution of number of significant variants per patient. Histogram showing the number of significant variants per patient. Significant variants refer to the 203 variants in the 23 genes that were ranked to prioritize disease causing alleles and weighted based on the number of variants in that gene. The distribution has a mean of 1.49 and a standard deviation of 1.22. (C) A StringDB query was run on the 23 most prioritized genes from the exome analysis, resulting in this network. It demonstrated close relationships between members of the NLR inflammasome (NOD2, NLRP3, NLRP1, and NLRC4). In addition, there were connections between PTPN22 and other immune and signaling molecules, including TYK2 and BTNL2. There were also edges between CDH23, USH2A, GPR98, SCN9A, and NTRK1, and COL7A1 and PLEC. Proteins that did not cluster in the main network included CAPN5, ABCA12, LRBA, and C10orf105. There were nodes corresponding to novel associations to uveitis (green), previously identified genetic associations to uveitis (cyan), Mendelian forms of uveitis (red), and proteins upregulated in uveitis vitreous (orange).
Table 1. Summary of identified uveitis variants.
A total of 23 genes that met the threshold for significance and were included in the final analysis. Most variants were single nucleotide variants that caused missense or nonsense mutations, and several included small insertions or deletions in the coding region of the gene. On average, each patient had variants in more than one of these genes.
| Gene | Number of Variants Found | Number of Patients with Mutations |
|---|---|---|
| NOD2/NLRC2 | 10 | 10 |
| CAPN5 | 6 | 7 |
| TYK2 | 5 | 12 |
| NLRC4 | 3 | 3 |
| PTPN22 | 2 | 2 |
| NLRP3 | 3 | 3 |
| NLRP1 | 3 | 5 |
| IL-2RB | 3 | 6 |
| ANKRD55 | 3 | 3 |
| PLEC | 32 | 42 |
| CD247 | 1 | 1 |
| COL7A1 | 15 | 22 |
| NTRK1 | 5 | 11 |
| BTNL2 | 1 | 1 |
| PTPN2 | 1 | 1 |
| LRBA | 13 | 14 |
| ADGRV1/GPR98 | 27 | 32 |
| USH2A | 23 | 29 |
| SCN9A | 10 | 11 |
| CDH23 | 17 | 24 |
| ABCA12 | 9 | 10 |
| INSRR | 8 | 8 |
| C10orf105 | 3 | 4 |
To hone our analysis, we queried known and predicted protein-protein interactions48. The proteins encoded by the top 23 affected genes were assembled into a functional protein-association network (Fig. 2C). The high degree of relatedness among proteins in the association network indicated that the variants identified in our uveitis cohort affected related genes and signaling pathways. The most striking cluster in the network contained four members of the Nod-like receptor (NLR) family: NOD2, NLRP3, NLRP1, and NLRC4. Other notable interactions in the network were between two protein tyrosine phosphatases, non-receptor type 22 (PTPN22) and tyrosine kinase 2 (TYK2). PTPN22 variants have been previously associated with anterior uveitis while TYK2 has not been implicated before in uveitis49. To determine if the protein products of these 23 genes were expressed in uveitis patients, we interrogated our published vitreous, retina, and RPE-choroid proteomics datasets, and found that 7 of the gene products (TYK2, PLEC, CDH23, COL7A1, LRBA, CAPN5, and IL-2R) were expressed28,50–52. IL-2 and IL-2R were significantly elevated in patients with posterior segment uveitis compared to controls (p = 0.006 and 0.0002, respectively; Fig. S1). Elevated serum IL-2R levels have been previously detected in posterior segment uveitis patients; anti-IL-2 therapies (e.g. daclizumab) have been used in the management of refractory uveitis53. Activated inflammasomes cleave caspase-1, mediating an IL-1β-driven inflammatory response54,55. We similarly detected elevated IL-1β levels in uveitis vitreous (p = 0.02; Fig. S1). Taken together, these results suggest that variants in related immune signaling pathways may be risk factors for posterior segment uveitis.
CAPN5 –
Six known CAPN5 coding variants cause a Mendelian posterior segment uveitis. Our WES pipeline confirmed known familial variants in two cases and identified variants in five other patients. One patient without Neovascular Inflammatory Vitreoretinopathy harbored a p.R243L mutation, but their clinical diagnosis was multifocal choroiditis (MFC). A novel variant (p.R289W) was identified in the first reported pediatric patient (Table S1; Fig. 3A–B)56,57. Four of the six CAPN5 variants (p.D72N, p.Q77R, p.R408Q, and p.L632I) were novel. We mapped these novel CAPN5 mutations on structure of the wild-type CAPN5 protease core domain (CAPN5-PC; PDB 6P3Q; Fig. 3C). The p.D72N mutation was identified in a chorioretinitis patient and predicted to be damaging by SIFT and PolyPhen. To date, most NIV-causing mutations mapped to the PC2 subdomain. However, this mutation mapped to the PC1 subdomain, near a putative Ca2+-coordination site. Since our CAPN5-PC structure lacked bound-Ca2+, the active (Ca2+-bound) CAPN5-PC was modelled using the structure of active CAPN9 as a template. Inspection of PC1 revealed that Asp72 contributes two side-chain oxygens to the Ca2+-coordination site. Substitution of this residue would remove a negative charge, disrupting the PC1 binding site (Fig. 3D). It is unclear if this substitution would hyperactivate CAPN5 since cooperative Ca2+ binding to PC1 and PC2 is required for formation of the active site58,59. Thus, further experimentation is required to determine if this variant is a disease-causing mutation. The p.Q77R variant was identified in a patient with recurrent, bilateral panuveitis beginning in adolescence and was predicted to be damaging by PolyPhen. The variant was located on a loop facing the substrate-binding pocket (Fig. 3E), where substitution of Gln77 with Arg would introduce a positive charge potentially altering CAPN5 substrate specificity. Two patients in our cohort harbored variants located in CAPN5’s non-catalytic domains: p.R408Q and p.L632I (located in the CBSW and C2 domains, respectively). Both of these mutations were predicted to be tolerated (SIFT and PolyPhen), but models of these two domains suggested potential functional effects. The p.R408Q variant was identified in a patient with bilateral panuveitis and was located in the CBSW domain, a putative autoinhibitory domain (Fig. 3F)60. The p.L632I variant was identified in a patient with choroiditis, and this region of the protein is thought to associate with the membrane in the inactive state5.
Fig. 3. Sequence and structure-based pathogenicity predictions of CAPN5 variants:
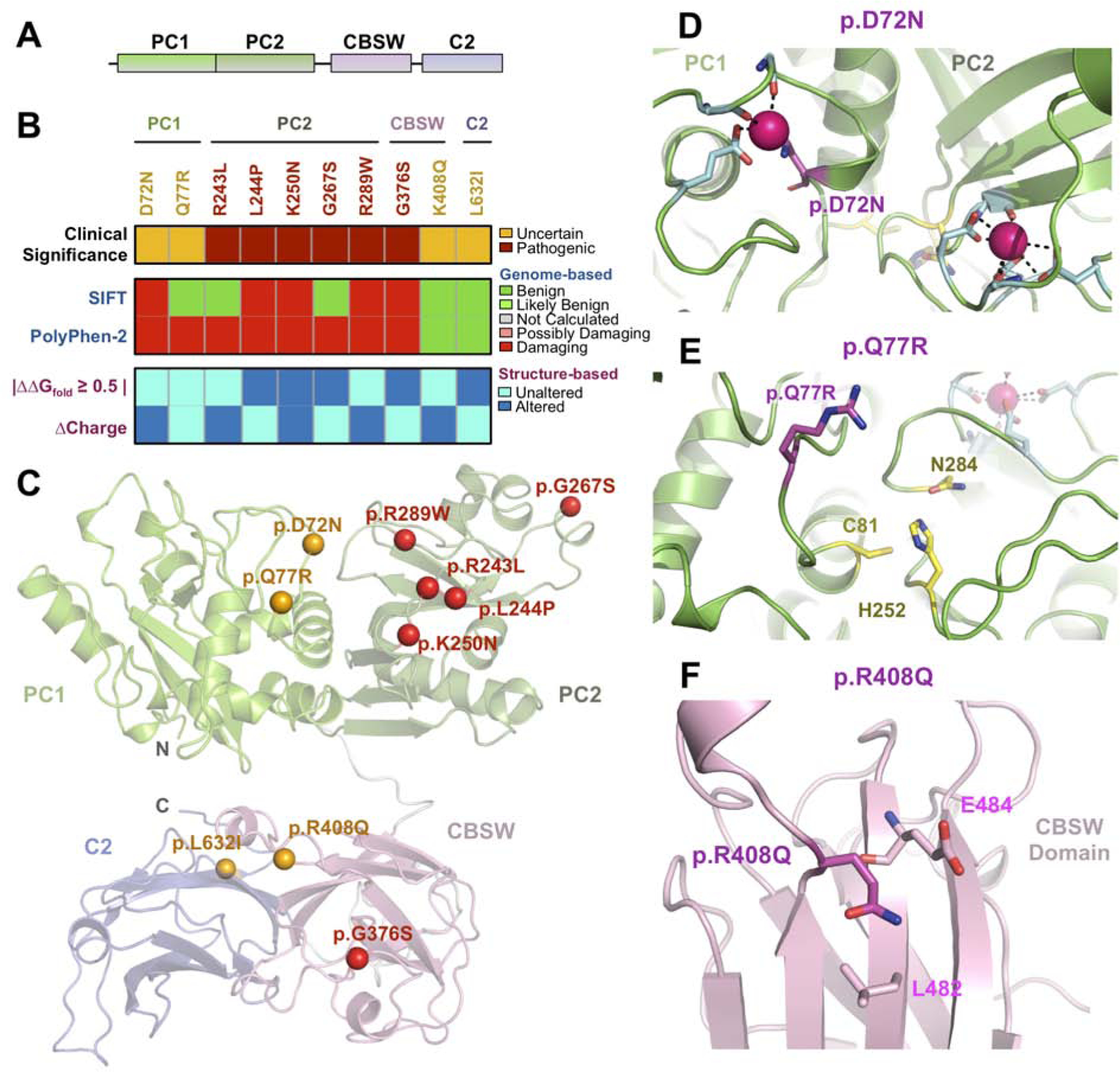
(A) Schematic representation of calpain-5 (CAPN5) domains. (B) Sequence-and structure-based pathogenicity predictions for CAPN5 variants identified in uveitis patients. Variants are annotated by their clinical significance (uncertain or pathogenic). Sequence-based predictors (SIFT, PolyPhen-2) provide predictions of degree of damage for individual variants, while structure-based predictors provide information on specific mechanistic alterations. (C) Ribbon trace diagram of the full-length CAPN5 structural model highlighting the different functional domains. Variants identified in the WES screen are represented as spheres and are colored according to their clinical significance – orange, unknown; red – confirmed pathogenic. (D) Close-up view of PC1 and −2 Ca2+-binding sites. The p.D72N variant (magenta) likely disrupts a calcium coordination site. (E) Close-up view of the CAPN5-PC active site. The Q77 residue side chain points towards the substrate binding pocket. The p.Q77R variant likely affects CAPN5-PC substrate specificity by introducing a basic residue into the binding pocket. (F) Close-up view of the p.R408Q variant in the CBSW domain.
NOD2 –
NOD2 belongs to the nucleotide-binding and oligomerization domain (NOD)-like receptor (NLR) family, which recognizes pathogen- and damage-associated molecular pattern molecules such as bacterial lipopolysaccharide61. Ten patients in our uveitis cohort harbored 10 different NOD2 variants: four variants were novel and five were previously identified in patients with Crohn’s Disease, ulcerative colitis, spondyloarthropathy, and orofacial granulomatosis62–65 (Table S2; Fig. 4A–B). One patient harbored the canonical Blau syndrome mutation (p.R334W)66,67. Although this is a dominant disease, the parents of this 28-year-old patient were not affected. Instead, she had been diagnosed with juvenile idiopathic arthritis. Although the other five patients did not have syndromic uveitis, they appeared to harbor known variants for syndromic diseases.
Fig. 4. Sequence and structure-based pathogenicity predictions of NOD2 variants:
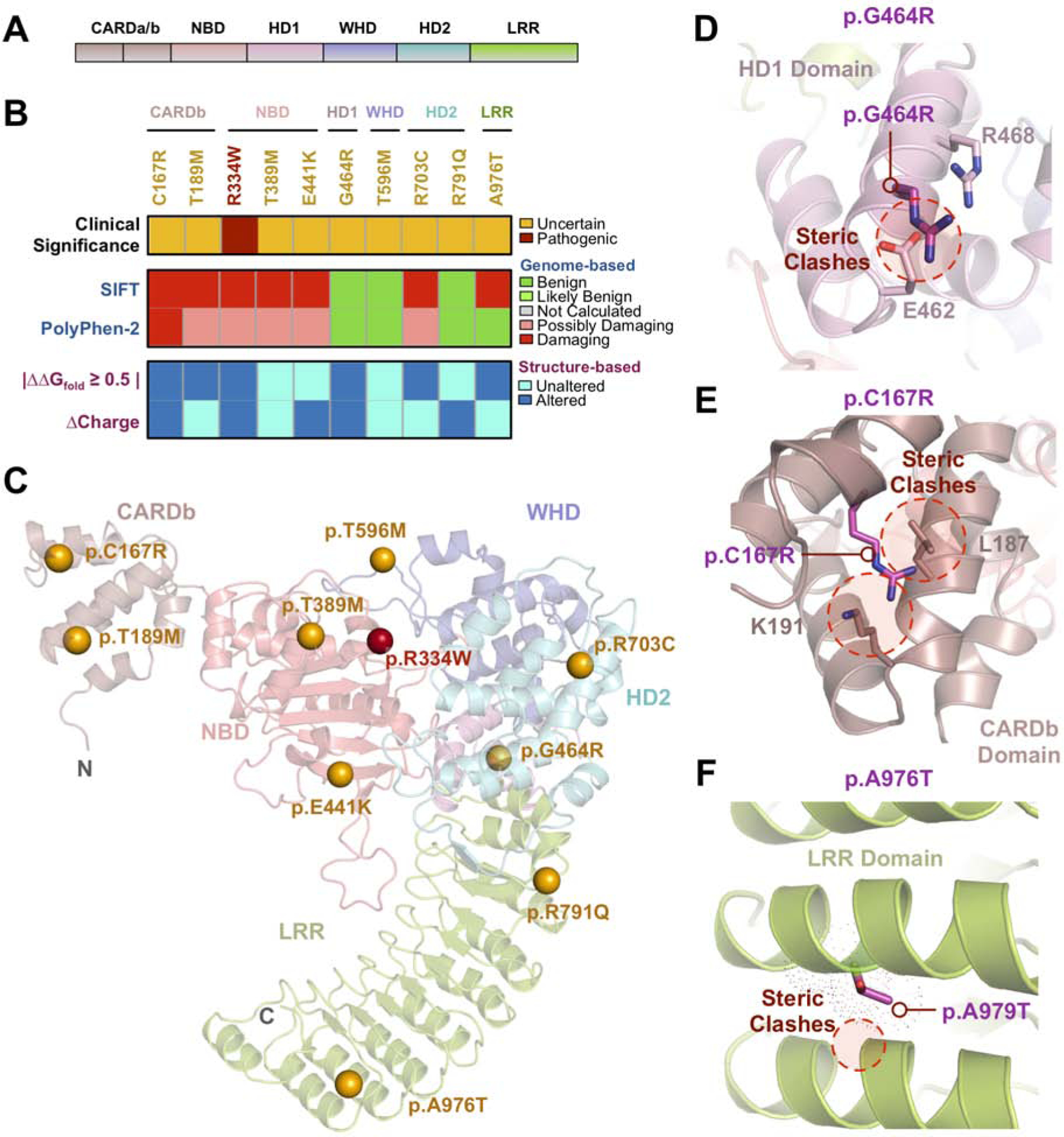
(A) Schematic representation of NOD2 domains. (B) Sequence- and structure-based pathogenicity predictions for NOD2 variants identified in uveitis patients. Variants are annotated by their clinical significance (uncertain or pathogenic). Sequence-based predictors (SIFT, PolyPhen-2) provide predictions of degree of damage for individual variants, while structure-based predictors provide information on specific mechanistic alterations. (C) Ribbon trace diagram of the NOD2 structural model highlighting the different functional domains. Variants identified in the WES screen are represented as spheres and are colored according to their clinical significance – orange, unknown; red – confirmed pathogenic. (D) Close-up view of the p.G464R variant in the HD1 region. This substitution introduces steric clashes with nearby residues, E462 and R488. (E) Close-up view of the p.C167R variant in the CARDb domain. This variant introduces steric clashes with nearby residues, K191 and L187. (F) Close-up view of the p.A976T variant in the LRR domain. This substitution likely introduces steric clashes in the neighboring α-helix.
Presumed ocular histoplasmosis syndrome (POHS) is a chorioretinitis characterized by chorioretinal lesions, peri-papillary atrophy, and the absence of vitreous inflammation2,68. Despite its name, there is considerable controversy over the precise cause of POHS and its link to H. capsulatum infection68. A vast majority of POHS patients have no history of systemic histoplasmosis infection and do not present with acute infection68. We found an association between NOD2 and POHS (p=0.016, q=0.131), comprising 50% of the identified NOD2 variants (5 patients). One POHS patient carried the p.G464R variant. Although this variant was predicted to be benign (SIFT and PolyPhen), mutations in this residue have been previously associated with Crohn’s Disease (CD) and are predicted to alter proper folding of the NOD2 HD1 region61. We therefore used structural modeling to gain insight into this variant’s pathogenicity (Fig. 4C). Our NOD2 model revealed that p.G464R introduced structural inflexibility and altered the charge at a specific α-helical turn (Fig. 4D), likely disrupting HD1 folding. Another POHS patient harbored a p.C167R variant. Structural modeling revealed that the variant was located on the regulatory CARDb domain and resulted in destabilization of this region through steric clashes and introduction of a positive charge (ΔΔGfold of −1.8 kcal/mol; Fig. 4E; Table S3)69.
Additionally, we identified two variants in LRR region of NOD2, an important domain that directly activates NOD2 and facilitates inflammasome oligomerization (Table S2)70. The first novel LRR variant, p.R791Q, was identified in a patient with idiopathic posterior uveitis. Previous studies demonstrated that disrupting this residue destabilizes autoinhibitory LRR and HD2 domain interactions70. The p.A976T variant, which has been previously reported in patients with Crohn’s Disease and orofacial granulomatosis, was identified in patient with panuveitis. Modeling revealed that this variant introduced steric clashes in the LRR domain, resulting in reduction in the fold energy of the protein (ΔΔGfold = −2.5 kcal/mol; Fig. 4F; Table S3)71. Structural modeling of the remaining NOD2 variants (p.T189M, p.T389M, p.E441K, p.T596M, and p.R703C) did not display clear functional consequences, although all but one (p.E441K) were predicted to be pathogenic (Table S2).
NLR inflammasome –
NLR-subset inflammasome is implicated in uveitis and mutations in NLR genes are implicated in systemic inflammatory conditions72–80. Across the three NLR-subset inflammasome genes (NLRP1, NLRP3, and NLRC4), ten distinct variants were identified in 12 patients (Table S2). There was an association between NLRP1 and intermediate uveitis (p = 0.025, q = 0.099). Two of the five patients with NLRP1 variants had vitritis or pars planitis. Although intermediate uveitis has been linked to IL2RA, IL6, and TNFA polymorphisms, it has never been associated with the inflammasome81–83. The p.Q533H, a previously reported NLRP1 variant, was identified in three unrelated patients (with retinal vasculitis, pars planitis, and idiopathic posterior uveitis respectively) and was predicted to be pathogenic (SIFT and PolyPhen). Structural modeling of NLRP1 revealed that this mutation was located in the NACHT domain (Fig. 5A). The other two variants, p.H92Q and p.G102R, were identified in patients with vitritis and panuveitis, respectively. Although these variants were predicted to be tolerated by sequence-based methods, structural modeling suggested that they might be gain-of-function variants. The p.H92Q variant, for example, was located in the C-terminal end of the PYD autoinhibitory domain and had a destabilizing effect on this region (ΔΔGfold = −0.9 kcal/mol; Table S3). The p.G102R variant was located in the conserved linker region between the PYD and NBD domains, which is cleaved by bacterial toxins during activation of the NLRP1 inflammasome (Fig. 5A). Polymorphisms in this region have been previously demonstrated to upregulate IL-1β, without exogenous stimulation84,85.
Fig. 5. Sequence and structure-based pathogenicity predictions of NLR inflammasome variants.
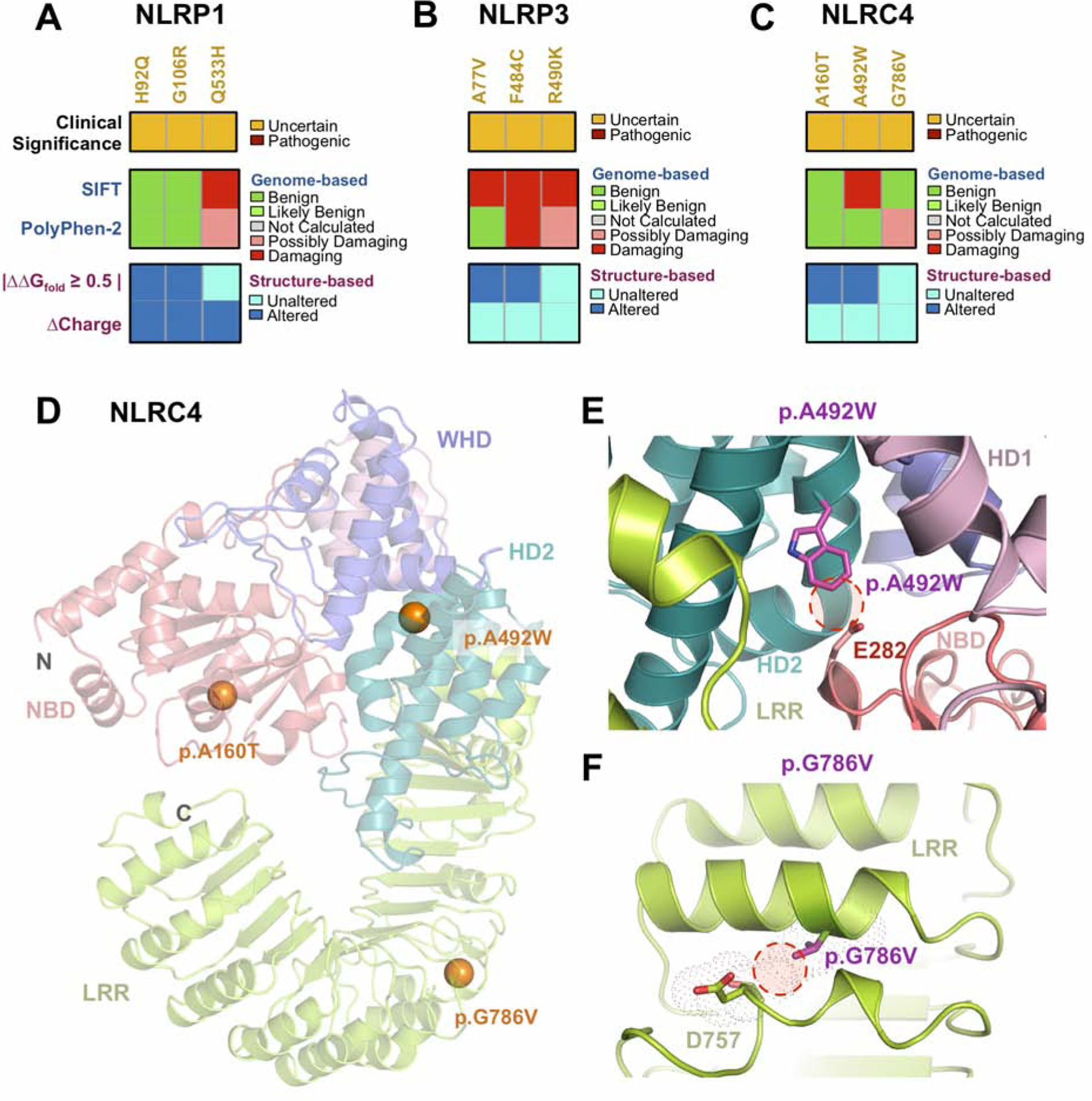
Sequence- and structure-based pathogenicity predictions for (A) NLRP1, (B) NLRP3, and (C) NLRC4 variants identified in uveitis patients. Variants are annotated by their clinical significance (uncertain or pathogenic). Sequence-based predictors (SIFT, PolyPhen-2) provide predictions of degree of damage for individual variants, while structure-based predictors provide information on specific mechanistic alterations. (D) Ribbon trace diagram of the murine NLRC4 structure (PDB 4KXF) highlighting the different functional domains. Variants identified in the WES screen are represented as spheres and are colored according to their clinical significance – orange, unknown; red – confirmed pathogenic. (E) Close up view of the p.A492W variant in the HD2 domain. Introduction of a Trp residue results in steric clashes between residues of the HD2 and NBD domain. (F) Close up view of the p.G786V variant in the LRR domain. This substitution likely introduces steric clashes in the neighboring α-helix.
Three NLRP3 variants were identified in three patients with chorioretinitis: acute posterior multifocal placoid pigment epitheliopathy (APMPPE; p.R490K), multi-focal choroiditis (p.F484C), and POHS (p.A77V). The p.R90K variant was predicted to be damaging (SIFT and PolyPhen) but did not display obvious structural changes in our NLRP3 model (ΔΔGfold = −0.3 kcal/mol; Table S3). The p.F484C and p.A77V variants were both predicted by SIFT to be damaging and had destabilizing effects on the NLRP3 model (ΔΔGfold = −0.8 and −0.9 kcal/mol, respectively; Fig. 5B; Table S3). We identified three patients in our uveitis cohort with NLRC4 variants: one with pars planitis (p.A160T), one with uveitis following a retinal tear (p.R492W), and another with POHS (p.G786V). Although the p.A160T variant was predicted to be tolerated by SIFT and PolyPhen-2, structural modeling suggested that it could destabilize the NLRC4 structure (ΔΔGfold = −1.6 kcal/mol; Table S3). Structural modeling also suggested that the other two NLRC4 variants could be gain-of-function mutations (Fig. 5C–D). The p.R492W variant, located in the NLRC4 HD2 domain, was predicted to be damaging by SIFT. The HD2 domain negatively regulates the function of the NBD domain by sterically masking the conserved α-helices required for NLRC4 oligomerization86. The substitution of Arg with Trp in this region led to steric clashes that may disrupt the inhibitory interactions between the HD2 and NBD domains (Fig. 5E). The previously reported p.G786V variant fell in the LRR domain and led to steric clashes that could destabilize this domain and disrupt its autoinhibitory function (Fig. 5F)87. These results suggest that rare variants in NLRP3 may be a risk factor for chorioretinitis.
TYK2 –
There were 12 unrelated patients harboring one of five different TYK2 variants (Table S2). TYK2 has not previously been implicated in uveitis, but TYK2 mutations are associated with an increased risk of other autoimmune disorders, as well as increased susceptibility to infections (e.g. mycobacteria, HSV, and Candida infections)88,89. The TYK2 gene encodes an intracellular receptor-associated tyrosine kinase involved in the JAK-STAT pathway (Fig. 6A)90–92. The p.V15A variant was identified in a patient with APMPPE (Table S2; Fig. 6B–C). The p.A53T mutation was identified in five patients with chorioretinitis that had been diagnosed sympathetic uveitis, panuveitis, birdshot chorioretinitis, multi-focal choroiditis, and autoimmune retinopathy, respectively. Substitution of Ala with Thr in our TYK2 model resulted in steric clashes that led to a significant destabilizing effect in the FERM domain (ΔΔGfold = −2.8 kcal/mol; Fig. 6D; Table S3). The p.G496S mutation, which was identified in a patient with birdshot chorioretinitis led to significant stability changes in the structural model (ΔΔGfold = −3.6 kcal/mol; Fig. 6E; Table S3). The p.D810V variant was identified in unrelated two patients with idiopathic uveitis and panuveitis, respectively. Modeling indicated that this variant could stabilize the protein’s pseudo-kinase domain (ΔΔGfold = 0.9 kcal/mol; Fig. 6F; Table S3)93. Finally, the p.A928V mutation was identified in three patients in our uveitis cohort: one with bilateral panuveitis, one with presumed sarcoidosis, and another with chorioretinitis and posterior scleritis. These results suggest that TYK2 variants may be associated with susceptibility to chorioretinal uveitis.
Fig. 6. Sequence and structure-based pathogenicity predictions of TYK2 inflammasome variants.
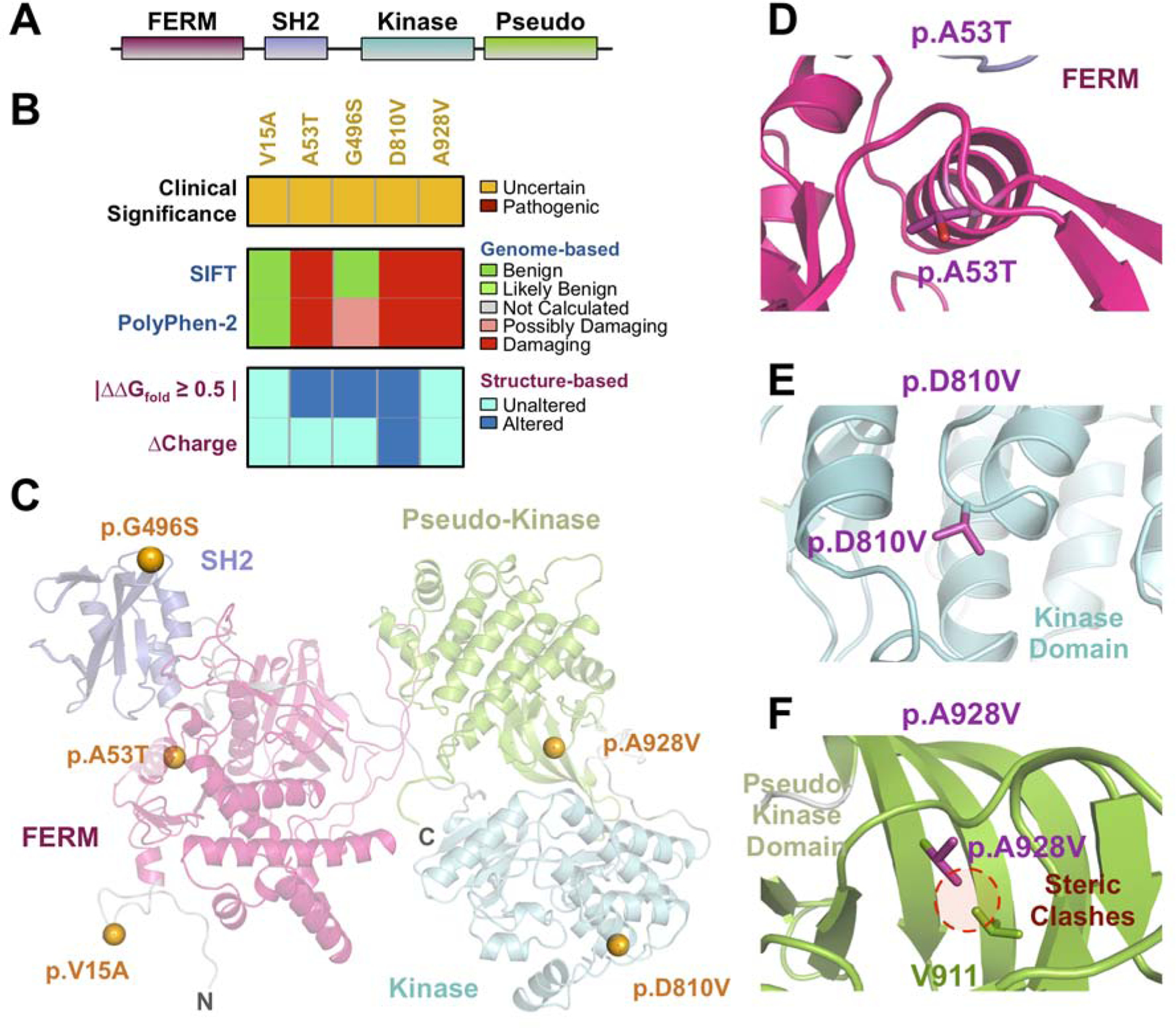
(A) Schematic representation of TYK2 domains. (B) Sequence- and structure-based pathogenicity predictions for TYK2 variants identified in uveitis patients. Variants are annotated by their clinical significance (uncertain or pathogenic). Sequence-based predictors (SIFT, PolyPhen-2) provide predictions of damaging for individual variants, while structure-based predictors provide information on specific mechanistic alterations. (C) Ribbon trace diagram of the TYK2 structural model highlighting the different functional domains. Variants identified in the WES screen are represented as spheres and are colored according to their clinical significance – orange, unknown; red – confirmed pathogenic. (D) Close up view of the p.A53T variant in the FERM domain. (E) Close up view of the p.D810V variant in the TYK2 kinase domain. (F) Close up view of the p.A928V variant in the pseudo-kinase domain.
Discussion
Whole-exome sequencing is a powerful approach that allows for a comprehensive analysis of rare coding variants and suggests potential susceptibility factors likely to drive uveitis. Despite the small sample size, statistical analyses suggest that coding variants in inflammatory genes may confer susceptibility to autoinflammatory diseases of the eye. In addition, our analysis identified pathogenic mutations known to cause non-syndromic uveitis and in patients with an atypical presentation. This suggests that in addition to identifying novel variants, WES can serve as a valuable diagnostic tool in specific cases of posterior segment uveitis, although limitations remain including cost, timeliness, and identification of unrelated and false-positive variants. Furthermore, we have found that three-dimensional modeling of coding variants provides valuable insights into pathogenicity compared to conventional prediction algorithms43,94,95. For example, NOD2 or CAPN5 variants occurred in highly conserved residues that appeared to affect protein structure and stability. These predictions offer insight into the mechanisms of NOD2 and CAPN5 hyperactivity.
The identification of NOD2, NLRP1, NLRP3, and NLRC4 variants in our cohort implicates the inflammasome in posterior segment uveitis pathogenesis. There is some precedent for this as NOD2 has been implicated in uveitis. NOD2 mutations cause Blau syndrome and can increase susceptibility to Crohn’s Disease, psoriatic arthritis, and sarcoidosis96. Conversely, NOD2 deficiency is implicated in the development of peptidoglycan-triggered uveitis in mice97,98. NLRP1 has previously been associated with acute glaucoma and inhibition of caspase-8, the upstream regulator of NLRP1, has shown to reduce the severity of glaucoma in mice99. More broadly, NLRP1 mutations are associated with Crohn’s Disease, rheumatoid arthritis, and vitiligo-associated multiple autoimmune disease susceptibility100,101. NLRP3 inflammasome activation has been associated with a variety of ophthalmic diseases including acute glaucoma, diabetic retinopathy, age-related macular degeneration, and keratoendotheliitis fugax hereditaria102–105. Furthermore, NLRP3 overexpression and the subsequent IL-1β hyperactivation is associated with anterior uveitis and Behçet’s disease27,106,107. Thus, therapies targeting the inflammasome can be considered in patients with posterior segment uveitis.
Posterior segment uveitis is rare, and validation of these findings will require a larger cohort of patients in different populations. For example, 86% of our patients were of European descent, and 18% of our cohort (30/164 patients) had POHS. Thus, it would be helpful to validate the presence of these variants in a larger, more diverse population. Future directions would also include functionally validating these variants in laboratory experiments.
Supplementary Material
Acknowledgements
A. Funding/Support: VBM and AGB are supported by NIH grants [R01EY026682, R01EY024665, R01EY025225, R01EY024698, R21AG050437, and P30EY026877], Research to Prevent Blindness (RPB), New York, NY. GV is supported by NIH grants [F30EYE027986 and T32GM007337]. The Barbara & Donald Jonas Laboratory of Regenerative Medicine and Bernard & Shirlee Brown Glaucoma Laboratory are supported by the National Institute of Health [5P30EY019007, R01EY018213, R01EY024698, R21AG050437], National Cancer Institute Core [5P30CA013696], the Research to Prevent Blindness (RPB) Physician-Scientist Award, unrestricted funds from RPB, New York, NY, USA. SHT is a member of the RD-CURE Consortium and is supported by the Tistou and Charlotte Kerstan Foundation, the Schneeweiss Stem Cell Fund, New York State [C029572], the Foundation Fighting Blindness New York Regional Research Center Grant [C-NY05-0705-0312], the Joel Hoffman Fund, the Professor Gertrude Rothschild Stem Cell Foundation, and the Gebroe Family Foundation. PFJ is supported by R01AR059703 and by the Marjorie K. Lamb Professorship.
Role of the Sponsor: The funding organizations had no role in design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures:
The following authors have no financial disclosures: Angela S. Li, Gabriel Velez, Benjamin Darbro, Marcus A. Toral, Jing Yang, Stephen H. Tsang, James C. Folk, Alexander G. Bassuk, Vinit B. Mahajan. Polly J. Ferguson is a consultant to Novartis for work that is unrelated to this study.
References
- 1.Suttorp-Schulten MS, Rothova A. The possible impact of uveitis in blindness: a literature survey. Br J Ophthalmol. 1996;80(9):844–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jabs DA, Nussenblatt RB, Rosenbaum JT, Standardization of Uveitis Nomenclature Working G. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140(3):509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang MM, Lai TY, Luk FO, Pang CP. The roles of genetic factors in uveitis and their clinical significance. Retina. 2014;34(1):1–11. [DOI] [PubMed] [Google Scholar]
- 4.Lee RW, Nicholson LB, Sen HN, et al. Autoimmune and autoinflammatory mechanisms in uveitis. Semin Immunopathol. 2014;36(5):581–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahajan VB, Skeie JM, Bassuk AG, et al. Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet. 2012;8(10):e1003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wert KJ, Bassuk AG, Wu WH, et al. CAPN5 mutation in hereditary uveitis: the R243L mutation increases calpain catalytic activity and triggers intraocular inflammation in a mouse model. Hum Mol Genet. 2015;24(16):4584–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahad MA, Missotten T, Abdallah A, Lympany PA, Lightman S. Polymorphisms of chemokine and chemokine receptor genes in idiopathic immune-mediated posterior segment uveitis. Mol Vis. 2007;13:388–396. [PMC free article] [PubMed] [Google Scholar]
- 8.Kone-Paut I, Georgin-Laviallec S, Galeotti C, et al. New data in causes of autoinflammatory diseases. Joint Bone Spine. 2019;86(5):554–561. [DOI] [PubMed] [Google Scholar]
- 9.Jamilloux Y, Belot A, Magnotti F, et al. Geoepidemiology and Immunologic Features of Autoinflammatory Diseases: a Comprehensive Review. Clin Rev Allergy Immunol. 2018;54(3):454–479. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez A, Calonge M, Pedroza-Seres M, et al. Referral patterns of uveitis in a tertiary eye care center. Arch Ophthalmol. 1996;114(5):593–599. [DOI] [PubMed] [Google Scholar]
- 11.Gritz DC, Wong IG. Incidence and prevalence of uveitis in Northern California; the Northern California Epidemiology of Uveitis Study. Ophthalmology. 2004;111(3):491–500; discussion 500. [DOI] [PubMed] [Google Scholar]
- 12.Remmers EF, Cosan F, Kirino Y, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet’s disease. Nat Genet. 2010;42(8):698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizuki N, Meguro A, Ota M, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behcet’s disease susceptibility loci. Nat Genet. 2010;42(8):703–706. [DOI] [PubMed] [Google Scholar]
- 14.Kirino Y, Bertsias G, Ishigatsubo Y, et al. Genome-wide association analysis identifies new susceptibility loci for Behcet’s disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. 2013;45(2):202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kappen JH, Medina-Gomez C, van Hagen PM, et al. Genome-wide association study in an admixed case series reveals IL12A as a new candidate in Behcet disease. PLoS One. 2015;10(3):e0119085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou S, Yang Z, Du L, et al. Identification of a susceptibility locus in STAT4 for Behcet’s disease in Han Chinese in a genome-wide association study. Arthritis Rheum. 2012;64(12):4104–4113. [DOI] [PubMed] [Google Scholar]
- 17.Fei Y, Webb R, Cobb BL, Direskeneli H, Saruhan-Direskeneli G, Sawalha AH. Identification of novel genetic susceptibility loci for Behcet’s disease using a genome-wide association study. Arthritis Res Ther. 2009;11(3):R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofmann S, Franke A, Fischer A, et al. Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat Genet. 2008;40(9):1103–1106. [DOI] [PubMed] [Google Scholar]
- 19.Fischer A, Ellinghaus D, Nutsua M, et al. Identification of Immune-Relevant Factors Conferring Sarcoidosis Genetic Risk. Am J Respir Crit Care Med. 2015;192(6):727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou S, Du L, Lei B, et al. Genome-wide association analysis of Vogt-Koyanagi-Harada syndrome identifies two new susceptibility loci at 1p31.2 and 10q21.3. Nat Genet. 2014;46(9):1007–1011. [DOI] [PubMed] [Google Scholar]
- 21.Alaez C, del Pilar Mora M, Arellanes L, et al. Strong association of HLA class II sequences in Mexicans with Vogt-Koyanagi-Harada’s disease. Hum Immunol. 1999;60(9):875–882. [DOI] [PubMed] [Google Scholar]
- 22.Kuiper JJ, Van Setten J, Ripke S, et al. A genome-wide association study identifies a functional ERAP2 haplotype associated with birdshot chorioretinopathy. Hum Mol Genet. 2014;23(22):6081–6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah KH, Levinson RD, Yu F, et al. Birdshot chorioretinopathy. Surv Ophthalmol. 2005;50(6):519–541. [DOI] [PubMed] [Google Scholar]
- 24.Pichi F, Carrai P, Srivastava SK, Lowder CY, Nucci P, Neri P. Genetic of uveitis. Int Ophthalmol. 2016;36(3):419–433. [DOI] [PubMed] [Google Scholar]
- 25.Takeuchi M, Kastner DL, Remmers EF. The immunogenetics of Behcet’s disease: A comprehensive review. J Autoimmun. 2015;64:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng J, Tan H, Hu J, et al. Genetic aspects of idiopathic paediatric uveitis and juvenile idiopathic arthritis associated uveitis in Chinese Han. Br J Ophthalmol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuksel S, Eren E, Hatemi G, et al. Novel NLRP3/cryopyrin mutations and pro-inflammatory cytokine profiles in Behcet’s syndrome patients. Int Immunol. 2014;26(2):71–81. [DOI] [PubMed] [Google Scholar]
- 28.Velez G, Roybal CN, Colgan D, Tsang SH, Bassuk AG, Mahajan VB. Precision Medicine: Personalized Proteomics for the Diagnosis and Treatment of Idiopathic Inflammatory Disease. JAMA Ophthalmol. 2016;134(4):444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25(21):2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sudmant PH, Rausch T, Gardner EJ, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 10 11–11 10 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singleton MV, Guthery SL, Voelkerding KV, et al. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am J Hum Genet. 2014;94(4):599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–576. [DOI] [PubMed] [Google Scholar]
- 40.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shihab HA, Gough J, Cooper DN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34(1):57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shihab HA, Rogers MF, Gough J, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015;31(10):1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bassuk AG, Yeh S, Wu S, et al. Structural modeling of a novel CAPN5 mutation that causes uveitis and neovascular retinal detachment. PLoS One. 2015;10(4):e0122352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gakhar L, Bassuk AG, Velez G, et al. Small-angle X-ray scattering of calpain-5 reveals a highly open conformation among calpains. J Struct Biol. 2016;196(3):309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu D, Jaroszewski L, Li Z, Godzik A. AIDA: ab initio domain assembly server. Nucleic Acids Res. 2014;42(Web Server issue):W308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wert KJ, Koch SF, Velez G, et al. CAPN5 genetic inactivation phenotype supports therapeutic inhibition trials. Hum Mutat. 2019;40(12):2377–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pandurangan AP, Ochoa-Montano B, Ascher DB, Blundell TL. SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017;45(W1):W229–W235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–452. s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin TM, Bye L, Modi N, et al. Genotype analysis of polymorphisms in autoimmune susceptibility genes, CTLA-4 and PTPN22, in an acute anterior uveitis cohort. Mol Vis. 2009;15:208–212. [PMC free article] [PubMed] [Google Scholar]
- 50.Velez G, Machlab DA, Tang PH, et al. Proteomic analysis of the human retina reveals region-specific susceptibilities to metabolic- and oxidative stress-related diseases. PLoS One. 2018;13(2):e0193250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Skeie JM, Roybal CN, Mahajan VB. Proteomic insight into the molecular function of the vitreous. PLoS One. 2015;10(5):e0127567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skeie JM, Mahajan VB. Proteomic landscape of the human choroid-retinal pigment epithelial complex. JAMA Ophthalmol. 2014;132(11):1271–1281. [DOI] [PubMed] [Google Scholar]
- 53.Nussenblatt RB, Thompson DJ, Li Z, et al. Humanized anti-interleukin-2 (IL-2) receptor alpha therapy: long-term results in uveitis patients and preliminary safety and activity data for establishing parameters for subcutaneous administration. J Autoimmun. 2003;21(3):283–293. [DOI] [PubMed] [Google Scholar]
- 54.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Platnich JM, Muruve DA. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys. 2019;670:4–14. [DOI] [PubMed] [Google Scholar]
- 56.Velez G, Sun YJ, Khan S, et al. Structural Insights into the Unique Activation Mechanisms of a Non-classical Calpain and Its Disease-Causing Variants. Cell Rep. 2020;30(3):881–892 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Velez G, Bassuk AG, Schaefer KA, et al. A novel de novo CAPN5 mutation in a patient with inflammatory vitreoretinopathy, hearing loss, and developmental delay. Cold Spring Harb Mol Case Stud. 2018;4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moldoveanu T, Jia Z, Davies PL. Calpain activation by cooperative Ca2+ binding at two non-EF-hand sites. J Biol Chem. 2004;279(7):6106–6114. [DOI] [PubMed] [Google Scholar]
- 59.Moldoveanu T, Hosfield CM, Lim D, Elce JS, Jia Z, Davies PL. A Ca(2+) switch aligns the active site of calpain. Cell. 2002;108(5):649–660. [DOI] [PubMed] [Google Scholar]
- 60.Campbell RL, Davies PL. Structure-function relationships in calpains. Biochem J. 2012;447(3):335–351. [DOI] [PubMed] [Google Scholar]
- 61.Parkhouse R, Boyle JP, Monie TP. Blau syndrome polymorphisms in NOD2 identify nucleotide hydrolysis and helical domain 1 as signalling regulators. FEBS Lett. 2014;588(18):3382–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lesage S, Zouali H, Cezard JP, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70(4):845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lappalainen M, Paavola-Sakki P, Halme L, et al. Novel CARD15/NOD2 mutations in Finnish patients with Crohn’s disease and their relation to phenotypic variation in vitro and in vivo. Inflamm Bowel Dis. 2008;14(2):176–185. [DOI] [PubMed] [Google Scholar]
- 64.Mentzer A, Nayee S, Omar Y, et al. Genetic Association Analysis Reveals Differences in the Contribution of NOD2 Variants to the Clinical Phenotypes of Orofacial Granulomatosis. Inflamm Bowel Dis. 2016;22(7):1552–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miceli-Richard C, Zouali H, Lesage S, et al. CARD15/NOD2 analyses in spondylarthropathy. Arthritis Rheum. 2002;46(5):1405–1406. [DOI] [PubMed] [Google Scholar]
- 66.Okafuji I, Nishikomori R, Kanazawa N, et al. Role of the NOD2 genotype in the clinical phenotype of Blau syndrome and early-onset sarcoidosis. Arthritis Rheum. 2009;60(1):242–250. [DOI] [PubMed] [Google Scholar]
- 67.Rose CD, Pans S, Casteels I, et al. Blau syndrome: cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatology (Oxford). 2015;54(6):1008–1016. [DOI] [PubMed] [Google Scholar]
- 68.Thuruthumaly C, Yee DC, Rao PK. Presumed ocular histoplasmosis. Curr Opin Ophthalmol. 2014;25(6):508–512. [DOI] [PubMed] [Google Scholar]
- 69.Coussens NP, Mowers JC, McDonald C, Nunez G, Ramaswamy S. Crystal structure of the Nod1 caspase activation and recruitment domain. Biochem Biophys Res Commun. 2007;353(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maekawa S, Ohto U, Shibata T, Miyake K, Shimizu T. Crystal structure of NOD2 and its implications in human disease. Nat Commun. 2016;7:11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chamaillard M, Philpott D, Girardin SE, et al. Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc Natl Acad Sci U S A. 2003;100(6):3455–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Romberg N, Al Moussawi K, Nelson-Williams C, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet. 2014;46(10):1135–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Canna SW, de Jesus AA, Gouni S, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46(10):1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med. 2014;211(12):2385–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29(3):301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dode C, Le Du N, Cuisset L, et al. New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet. 2002;70(6):1498–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Feldmann J, Prieur AM, Quartier P, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71(1):198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oberg TJ, Vitale AT, Hoffman RO, Bohnsack JF, Warner JE. Cryopyrin-associated periodic syndromes and the eye. Ocul Immunol Inflamm. 2013;21(4):306–309. [DOI] [PubMed] [Google Scholar]
- 79.Finetti M, Omenetti A, Federici S, Caorsi R, Gattorno M. Chronic Infantile Neurological Cutaneous and Articular (CINCA) syndrome: a review. Orphanet J Rare Dis. 2016;11(1):167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46(12):3340–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ewald L, Beate LW, Stephanie S, Wilfried R, Yosuf el S. Analysis of a Functional IL-6 Gene Polymorphism in HLAB27 Associated and Intermediate Uveitis Gives New Insight in Disease Pathogenesis and Commonality with Other Autoimmune Diseases. J Immunol Res. 2015;2015:174062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lindner E, Weger M, Ardjomand N, Renner W, El-Shabrawi Y. Associations of Independent IL2RA Gene Variants with Intermediate Uveitis. PLoS One. 2015;10(7):e0130737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Atan D, Heissigerova J, Kuffova L, et al. Tumor necrosis factor polymorphisms associated with tumor necrosis factor production influence the risk of idiopathic intermediate uveitis. Mol Vis. 2013;19:184–195. [PMC free article] [PubMed] [Google Scholar]
- 84.Levandowski CB, Mailloux CM, Ferrara TM, et al. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1beta processing via the NLRP1 inflammasome. Proc Natl Acad Sci U S A. 2013;110(8):2952–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alkhateeb A, Jarun Y, Tashtoush R. Polymorphisms in NLRP1 gene and susceptibility to autoimmune thyroid disease. Autoimmunity. 2013;46(3):215–221. [DOI] [PubMed] [Google Scholar]
- 86.Lechtenberg BC, Mace PD, Riedl SJ. Structural mechanisms in NLR inflammasome signaling. Curr Opin Struct Biol. 2014;29:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kilic SS, Hacimustafaoglu M, Boisson-Dupuis S, et al. A patient with tyrosine kinase 2 deficiency without hyper-IgE syndrome. J Pediatr. 2012;160(6):1055–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kreins AY, Ciancanelli MJ, Okada S, et al. Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med. 2015;212(10):1641–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Strobl B, Stoiber D, Sexl V, Mueller M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front Biosci (Landmark Ed). 2011;16:3214–3232. [DOI] [PubMed] [Google Scholar]
- 91.Tao JH, Zou YF, Feng XL, et al. Meta-analysis of TYK2 gene polymorphisms association with susceptibility to autoimmune and inflammatory diseases. Mol Biol Rep. 2011;38(7):4663–4672. [DOI] [PubMed] [Google Scholar]
- 92.Wallweber HJ, Tam C, Franke Y, Starovasnik MA, Lupardus PJ. Structural basis of recognition of interferon-alpha receptor by tyrosine kinase 2. Nat Struct Mol Biol. 2014;21(5):443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lupardus PJ, Ultsch M, Wallweber H, Bir Kohli P, Johnson AR, Eigenbrot C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc Natl Acad Sci U S A. 2014;111(22):8025–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Oh JK, Lima de Carvalho JR Jr., Sun YJ, et al. Novel mutations in the 3-box motif of the BACK domain of KLHL7 associated with nonsyndromic autosomal dominant retinitis pigmentosa. Orphanet J Rare Dis. 2019;14(1):295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu KY, Sengillo JD, Velez G, et al. Missense mutation in SLIT2 associated with congenital myopia, anisometropia, connective tissue abnormalities, and obesity. Orphanet J Rare Dis. 2018;13(1):138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29(1):19–20. [DOI] [PubMed] [Google Scholar]
- 97.Blau EB. Familial granulomatous arthritis, iritis, and rash. J Pediatr. 1985;107(5):689–693. [DOI] [PubMed] [Google Scholar]
- 98.Rosenzweig HL, Galster K, Vance EE, et al. NOD2 deficiency results in increased susceptibility to peptidoglycan-induced uveitis in mice. Invest Ophthalmol Vis Sci. 2011;52(7):4106–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chi W, Li F, Chen H, et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc Natl Acad Sci U S A. 2014;111(30):11181–11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grandemange S, Sanchez E, Louis-Plence P, et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann Rheum Dis. 2017;76(7):1191–1198. [DOI] [PubMed] [Google Scholar]
- 101.Jin Y, Mailloux CM, Gowan K, et al. NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med. 2007;356(12):1216–1225. [DOI] [PubMed] [Google Scholar]
- 102.Yang X, Luo C, Cai J, et al. Neurodegenerative and inflammatory pathway components linked to TNF-alpha/TNFR1 signaling in the glaucomatous human retina. Invest Ophthalmol Vis Sci. 2011;52(11):8442–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Devi TS, Lee I, Huttemann M, Kumar A, Nantwi KD, Singh LP. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: implications for diabetic retinopathy. Exp Diabetes Res. 2012;2012:438238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tseng WA, Thein T, Kinnunen K, et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2013;54(1):110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells--implications for age-related macular degeneration (AMD). Immunol Lett. 2012;147(1–2):29–33. [DOI] [PubMed] [Google Scholar]
- 106.Liang L, Tan X, Zhou Q, et al. IL-1beta triggered by peptidoglycan and lipopolysaccharide through TLR2/4 and ROS-NLRP3 inflammasome-dependent pathways is involved in ocular Behcet’s disease. Invest Ophthalmol Vis Sci. 2013;54(1):402–414. [DOI] [PubMed] [Google Scholar]
- 107.Kim EH, Park MJ, Park S, Lee ES. Increased expression of the NLRP3 inflammasome components in patients with Behcet’s disease. J Inflamm (Lond). 2015;12:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.