

Abstract
Peroxisome proliferator-activated receptors (PPARs) are fatty acid-activated transcription factors of nuclear hormone receptor superfamily that regulate energy metabolism. Currently, three PPAR subtypes have been identified: PPARα, PPARγ, and PPARβ/δ. PPARα and PPARδ are highly expressed in oxidative tissues and regulate genes involved in substrate delivery and oxidative phosphorylation (OXPHOS) and regulation of energy homeostasis. In contrast, PPARγ is more important in lipogenesis and lipid synthesis, with highest expression levels in white adipose tissue (WAT). In addition to tissues regulating whole body energy homeostasis, PPARs are expressed in immune cells and have an emerging critical role in immune cell differentiation and fate commitment. In this review, we discuss the actions of PPARs in the function of the innate and the adaptive immune system and their implications in immune-mediated inflammatory conditions.
Keywords: PPAR, metabolism, inflammation, T cells, myeloid cells
Introduction: Structure and Classification of PPARs
The transcription factors Peroxisome proliferator activated receptors (PPARs) were discovered 30 years ago in rodents [1] and belong to the subfamily 1 of the nuclear hormone receptor superfamily of transcription factors [2]. PPARs are the best-studied fatty acid-activated nuclear receptors comprising of the following three subtypes: PPARα, PPARγ, and PPARδ (also designated as PPARβ) [3–8]. Although all PPARs play a major regulatory role in energy homeostasis each of them has distinct functions and sites of expression [4–6,8,9].
All PPARs share the basic structural properties of the most nuclear receptors, consisting of four functional domains [10–12] named A/B, C, D and E/F (Figure 1A): The N-terminal A/B domain contains a ligand-independent activation function 1 (AF-1), which is responsible for PPAR phosphorylation. The conserved central DNA binding domain (DBD), also named C domain, is composed of two zinc fingers and is responsible for PPAR binding to the peroxisome proliferator response element (PPRE) in the promoter of PPAR target genes. The D domain is a docking site for various cofactors. The E domain is also named ligand-binding domain (LBD). This is a 12-helix that forms a large hydrophobic binding pocket in the shape of Y and gives the molecule that ability to bind endogenous or exogenous lipophilic ligands. By doing so, this domain provides ligand specificity. Ligand binding stabilizes LBD and promotes conformational changes that enable interaction with co-activator complexes. Recruitment of PPAR cofactors that participate in the transcription process is mediated by the ligand-dependent activation function-2 (AF-2), located in the E/F domain. The full transcriptional activity of PPARs requires binding of cognate lipid ligands, heterodimerization with another nuclear receptor (retinoid-X receptor, RXR), interaction with a number of transcriptional coactivators, including PPARγ coactivator-1 (PGC-1)α and PGC-1β, and binding of the complex to PPAR response elements (PPREs) in the promoter of target genes (Figure 1B) [7,9,13]. Each PPAR member has a different ligand preference that could be explained by differences in the size or the lipophilicity of their binding pockets [8]. Detailed studies using hydrogen-deuterium exchange mass spectrometry have identified how structurally and functionally distinct states of PPARs are induced by binding of agonists vs. antagonists or reverse agonists [14].
Figure 1. PPAR structure and mechanism of action.
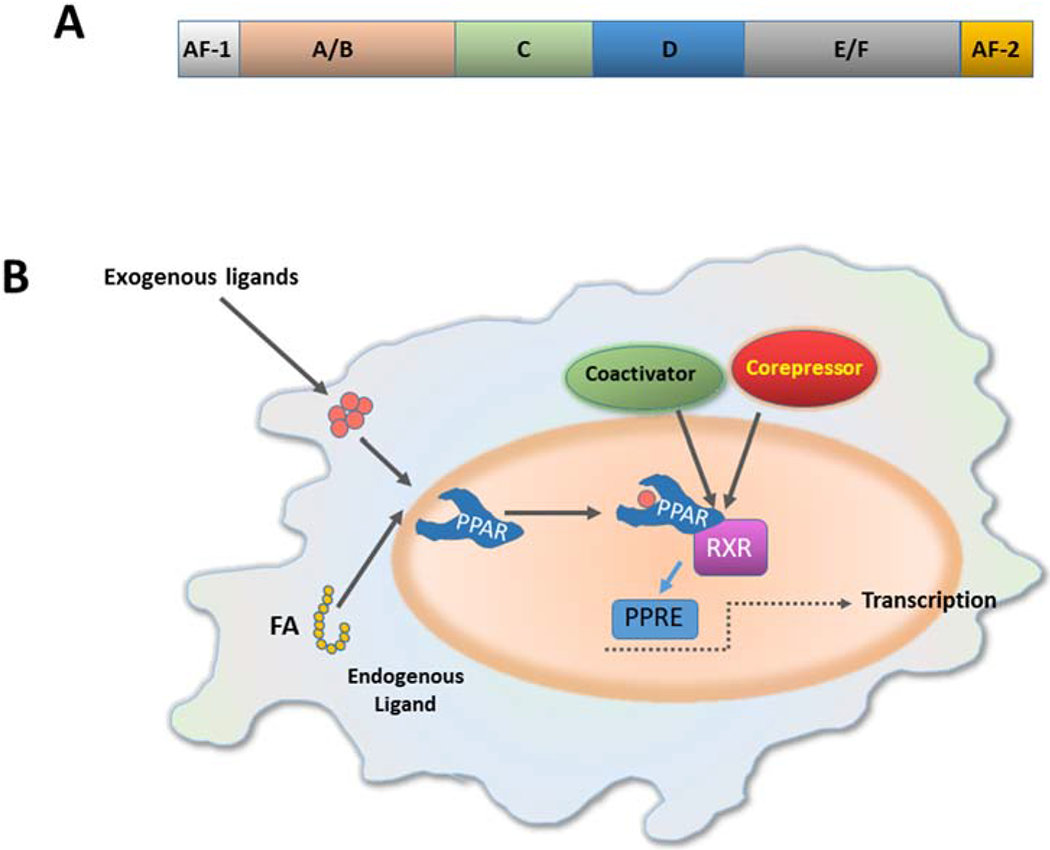
(A) All PPARs share the basic structural of the most nuclear receptors, consisting of four functional domains named A/B, C, D and E/F. The N-terminal A/B domain contains a ligand-independent activation function 1 (AF-1). The central DNA binding domain (DBD) or C domain is a conserved domain, composed of two zing fingers, and is responsible for the binding of PPAR to the peroxisome proliferator response element (PPRE) in the promoter of target genes. The D domain is a docking site for various cofactors. The E domain or ligand-binding domain (LBD) binds a variety of endogenous or exogenous lipophilic ligands and provides ligand specificity. Recruitment of PPAR cofactors that participate in the transcription process is mediated by the ligand-dependent activation function-2 (AF-2), located in the E/F domain. (B) Ligand binding promotes conformational changes that enable the interaction with co-activator complexes. The full transcriptional activity of PPARs requires binding of cognate lipid ligands, heterodimerization with another nuclear receptor (retinoid-X receptor, RXR), interaction with a number of transcriptional coactivators, and binding of the complex to PPAR response elements (PPREs) in the promotor of target genes. (FA, fatty acid)
PPARs are encoded by separate genes with distinct but overlapping interspecies sequences. The human PPARα gene is located on chromosome 22, PPARγ, for which three isoforms have been identified [15,16], is located on chromosome 3, and PPARβ/δ is located on chromosomes 6 [9,17,18]. PPAR natural ligands include lipid-derived metabolites such as fatty acids, acyl-CoAs, glycerol-phospholipids, and eicosanoids (Table 1). Dietary lipids activate PPAR, as evidenced by the increased expression of PPARα target genes in the liver and PPARβ/δ target genes in the skeletal muscle induced by high-fat diet (HFD) in mice [19,20].
Table 1:
PPAR subtypes, chromosome localization, natural ligands and biological effects.
| Subtype | Chromosome | Site of expression | Natural Ligands | Action |
|---|---|---|---|---|
| PPARγ | 3 | WAT, liver, skeletal muscles, intestine, immune cells | Linoleic acid Arachidonic acid Eicosatetraenoic acid PGJ2, Linoleic acid 9-HODE 13-HODE | • FA transport • Lipid synthesis • Adipogenesis • Energy storage • Thermogenesis (BAT) • Glucose homeostasis |
| PPARα | 22 | Liver, heart, skeletal muscles, BAT, intestine, kidneys | Palmitic acid Stearic acid Palmitoleic acid Oleic acid Linoleic acid Arachidonic acid Eicosatetraenoic acid Leukotriene B4 Pristanic acid | • FA transport • FA esterification • FAO • BAT browning • Energy dissipation |
| PPARβ/δ | 6 | Ubiquitous | FA Retinoic acid Carbaprostacyclin | • FAO • Glucose homeostasis |
WAT, white adipose tissue; BAT, brown adipose tissue; PGJ2, Prostaglandin J2; 9-HODE, 9 hydroxyoctadecadienoic acid; 13-HODE, 9-hydroxyoctadecadienoic acid; 15-HETE, 15-Hydroxyeicosatetraenoic acid; FA, Fatty acids; FAO, Fatty acid oxidation
Function of PPARs on lipid metabolism
PPARα and PPARδ are highly expressed in oxidative tissues and regulate genes involved in substrate delivery, substrate oxidation, and oxidative phosphorylation (OXPHOS) [3,9]. PPARα is expressed mainly in the liver, heart, skeletal muscles, brown adipose tissue (BAT), intestine and kidney and activates energy dissipation. PPARα mediates its functions by influencing fatty acid transport, esterification and oxidation. PPARβ/δ is expressed ubiquitously and participates in fatty acid oxidation, but also has a role in the regulation of blood glucose levels. In contrast, PPARγ contributes mainly to energy storage by promoting adipogenesis and lipid synthesis and displays highest expression levels in white adipose tissue (WAT). PPARγ is also expressed in the liver, skeletal muscles, intestine and immune cells [3,9] (Table 1).
Fatty acids involved in PPAR signaling originate from import of exogenous fatty acids or from endogenous de novo synthesis. Importantly, neither source of fatty acids can generate PPAR ligands directly. Instead, a cycle of fatty acid esterification and re-hydrolysis is required [21]. This process, termed lipolysis, involves the hydrolysis of triacylglycerols that generates free fatty acids and glycerol and involves three identified enzymes: adipose triglyceride lipase (ATGL) catalyzes the initial step of lipolysis, converting triacylglycerols to diacylglycerols; heat sensitive lipase (HSL) is hydrolyzes diacylglycerols to monoglycerols; and monoglycerol lipase (MGL), hydrolyzes monoglycerols to generate free fatty acids and glycerol. The indispensable role of lipolysis in PPAR signaling is evident by the fact that mice deficient for ATGL that controls the first step of lipolysis, exhibit a severe defect in PPARα signaling in oxidative tissues such as liver [22], macrophages [23], and BAT [24]. The most dramatic phenotype is observed in cardiac muscle. The reduced expression of PPARα target genes in ATGL knockout animals causes severe mitochondrial dysfu6nction, decreased substrate oxidation and OXPHOS, massive cardiac lipid accumulation, and lethal cardiomyopathy within few months after birth [21]. In addition to PPARα, this process is also required for PPARγ signaling [25].
PPARγ is essential for the development and function of the adipose tissue [26]. In vitro differentiation of adipose tissue from fibroblasts or embryonic stem cells relies on PPARγ [26,27]. Similarly, mice with PPARγ ablation failed to develop all types of fat [28,29]. The role of PPARγ on the adipose tissue is not restricted to its development but extends to lipid metabolism and energy homeostasis. In WAT, PPARγ controls uptake of fatty acids and lipogenesis. Several genes responsible for lipid uptake by adipocytes such as CD36 and fatty acid transport protein (FATP-1), or lipid metabolism such as adipocyte protein 2 (aP2), phosphoenolpyruvate carboxykinase (PEPCK), lipoprotein lipase (LPL) and acyl-CoA synthase, are directly regulated by PPARγ [9]. In BAT, PPARγ induces expression of target genes that regulate mitochondrial biogenesis, such as PGC-1α, and thermogenesis, such as uncoupling protein-1 (UCP-1, also known as thermogenin) [30]. PPARγ isoform 2 can also act as a transcription factor of the electron transport chain (ETC) genes on the mitochondrial DNA of brown adipose tissue cells, regulating cellular respiration [31].
Due to the PPARγ action on metabolism, PPARγ agonists have been used clinically for insulin sensitization (reviewed extensively elsewhere). Thiazolidinediones (TZD), the main medications for glucose homeostasis in patients with DM2, act as ligands for PPARγ [32], and upregulate the c-Cbl-associated protein (CAP) and the glucose transporter type 4 (GLUT4) [32,33]. The regulation of cytokines or hormones such as TNF-α, resistin and adiponectin also contributes to oxidation of fatty acids and to TZD-mediated insulin sensitization [34–36]. Notably, the insulin-sensitizing effect of PPARγ agonists is abolished in mice knocked out for Fibroblast Growth Factor-21 (FGF-21), suggesting that FGF-21 is a crucial mediator of TZD anti-diabetic activity [37]. Obesity-mediated insulin resistance and DM2 are modulated by posttranslational modification of PPARγ [38]. PPARγ phosphorylation reduces its activity in vitro whereas a genetic mutation that prevented PPARγ phosphorylation, enhanced PPARγ function and preserved insulin sensitivity in the context of diet-induced obesity [38]. These findings suggest that compounds that prevent PPARγ phosphorylation might restore insulin sensitivity by enhancing PPARγ function. Conversely, dominant negative PPARγ mutations result in DM2, hypertension and insulin resistance, demonstrating the relationship between PPARγ function and metabolic syndrome [39]. Besides ligand-dependent signaling, PPARγ expression level can be altered by cues of the microenvironment such as proinflammatory cytokines, thereby leading to altered PPAR-mediated events. For example, in patients with ulcerative colitis, PPARγ expression is significantly decreased in the intestinal mucosa and TZD-mediated PPARγ activation can mediate anti-inflammatory effects and improve the symptoms of patients with ulcerative colitis [40].
Studies have provided evidence that PPARα might exert anti-inflammatory action by mediating a direct effect on adipocytes. The proposed mechanism involves sirtuin1 (SIRT1), a NAD(+)-dependent deacetylase, that suppresses the inflammatory response by inhibiting TNF-α induced CD40 expression via the SIRT1-dependent signaling pathway [41,42]. PPARα also plays a role in the regulation of thermogenesis by promoting adipocyte browning and activating thermogenic beige adipocytes [43,44].
The mechanisms via which PPARβ/δ impacts lipid metabolism are less well investigated compared to other PPARs. Nevertheless, it is established that PPARβ/δ promotes lipid catabolism in various tissues and serves as mediator of fatty acid oxidation and fat burning [45]. PPARβ/δ activates heat producing uncoupling enzymes in brown adipose tissues and muscles and protects against obesity and fatty liver [46]. Alternatively, or in parallel, PPAR-β/δ might mediate its effects on fat metabolism by regulating the expression of adipokines [47] and by suppressing IL-6-induced STAT3 activation [48]. This effect might contribute to the prevention of IL-6-induced insulin resistance in adipocytes. Thus, therapeutic modulation of PPARβ/δ might be a promising approach for the treatment of obesity and metabolic syndrome. It should be noted that caution is required when pharmacologic modulators of PPARs are employed as several of these compounds may act on more than one PPAR subtypes albeit at different EC50 [49]
Function of PPARs on innate and adaptive immune cells
In the past few years, the role of PPARs in immune cells has been extensively studied. As key regulators of metabolism, PPARs guide the differentiation, expansion and fate commitment of various immune cell types. These effects have significant implications in organs that become targets of immunometabolic aberrations induced upon dysfunction of PPAR members.
PPARγ
This is, by far, the most extensively studied member of the PPAR family and the most common target for therapeutic intervention. PPARγ can control the homeostasis of the immune system by regulating the fate and function of various cell types. In macrophages, PPARγ regulates polarization, maturation, epigenetics and metabolism [50–54] (Figure 2). Macrophages adapt to distinct microenvironments by polarizing to either pro-inflammatory M1 macrophages or to anti-inflammatory M2 macrophages [55,56]. Under M2 conditions induced by IL-4, arginase activity, a classic marker of M2 polarization, is substantially diminished in bone marrow derived macrophages (BMDM) generated from mice with conditional deletion of PPARγ in macrophages, compared to WT counterparts [51]. Growth differentiation factor 3 (GDF3), a key mediator of muscle regeneration, is secreted by repair macrophages in response to cardiotoxin under the control of PPARγ [57]. On the other hand, under LPS culture conditions that drive M1 polarization, PPARγ KO macrophages have markedly elevated levels of proinflammatory cytokines such as IL-6, IL-12, IL-1β and TNF-α, that serve as markers of M1 polarization [58]. In contrast to macrophage polarization, PPARγ does not affect macrophage differentiation [59].
Figure 2. PPARs regulate macrophage polarization and function.
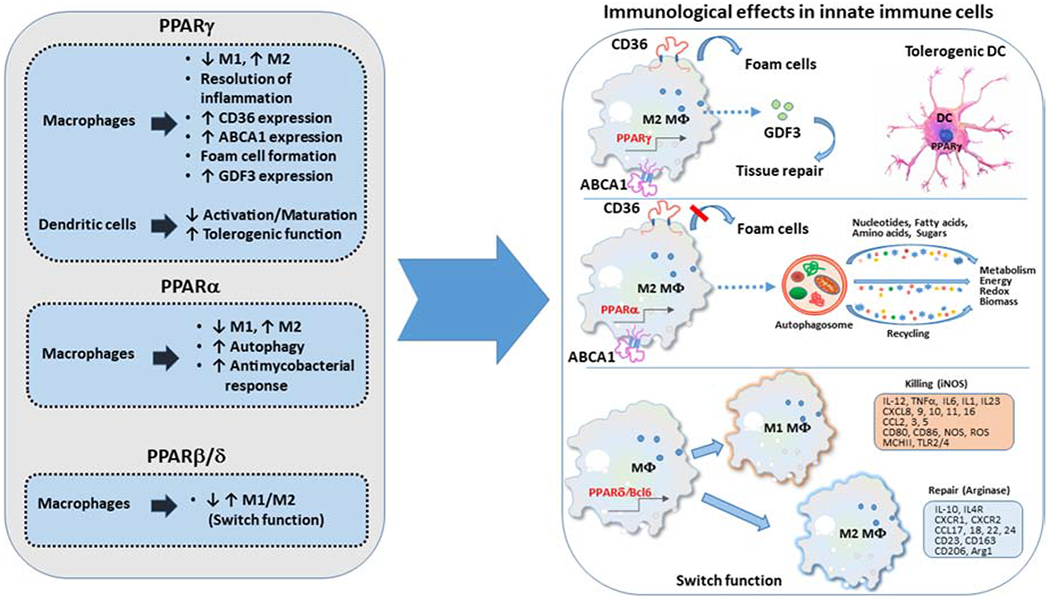
Signals mediated via PPARs regulate the differentiation and function of macrophages and dendritic cells.
In addition to regulating macrophage polarization, PPARγ has an important role in other macrophage functions, including formation of foam cells (Figure 2), i.e. macrophages that localize at the walls of blood vessels during lipid accumulation [60,61]. The expression of the macrophage scavenger receptor CD36, which is necessary for foam cell formation and subsequently atherogenesis [62], relies on PPARγ [60,61]. Cholesterol crystals can cause production of reactive oxygen species (ROS) through xanthine oxidase and NADPH, and ROS can activate BTK. The activated BTK phosphorylates p300 protein, leading to p300-STAT1-PPARγ interaction. Binding of the complex at −107 nucleotide on CD36 promoter results in enchased CD36 expression, absorption of oxLDL and formation of foam cells [61]. The significance of this mechanism is underlined by the diminished capacity of PPARγ deficient macrophages to uptake and degrade OxLDL [59]. Notably, not only cholesterol uptake but also efflux, mediated by the cholesterol efflux cassettes of the ABCA1 family, is under the transcriptional control of PPARγ [63]. Consistently, proline/serine-rich coiled-coil 1 (PSRC1) inhibits atherogenesis and foam cell formation by upregulating LXRα and PPARγ in vitro and in apoE−/− mice [64].
In dendritic cells (DC), which have a key role in regulating immunity vs. anergy (in vitro) and tolerance (in vivo), PPARγ is a central regulator of functional maturation, thereby guiding the ability to induce immunogenic T cell responses vs. immune tolerance (Figure 2). Specifically, sustained agonist-mediated activation of PPARγ in mouse DC reduced maturation-induced expression of costimulatory molecules and IL-12, and profoundly inhibited the ability of DC to prime naive CD4+ T cells in vitro [65]. These effects were abrogated when murine DCs were deficient for PPARγ gene, providing evidence that the effects of pharmacologic modulation of PPARγ-signaling were specific. Conversely, PPARγ ablation increased the immunogenicity of DC, indicating that PPARγ might function as a constitutive regulator of DC suppression [65]. In human monocyte-derived DC, activation of PPARγ with 15d-PGJ2 or troglitazone suppressed DC stimulation via the TLR ligands 2, 3, 4, and 7, as determined by down-regulation of costimulatory and adhesion molecules and reduced secretion of cytokines and chemokines [66]. These effects were mediated by inhibition of TLR-mediated activation of MAP kinase and NF-kB pathways and resulted in reduced capacity of DC to stimulate T-cell proliferation, emphasizing the inhibitory effect of PPARγ activation on DC maturation. PPARγ expression in DC also regulates immune tolerance in the airways and is indispensable for inhibition of Th17 hallmark cytokines and induction of Tregs [67].
PPARγ is also critical for the regulation of adaptive immune cells (Figure 3). In one study, PPARγ activation in mouse CD4+ T cells selectively suppressed Th17 differentiation [68]. Control of Th17 differentiation in T cells by PPARγ involved inhibition of TGF-β/IL-6-induced expression of RORγt. Pharmacologic activation of PPARγ also prevented RORγt transcription. Importantly this study confirmed the physiologic opposing role of PPARγ on Th17 differentiation by activation of endogenous ligand. In addition, human CD4+ T cells from healthy individuals and patients with multiple sclerosis were susceptible to PPARγ-mediated suppression of Th17 differentiation [68]. Further investigations also provided evidence for the role of endogenous PPARγ as a regulator of Th17 responses by showing that human CD4+ T cells with PPARγ siRNAs increased IL-17A production [69]. Interestingly, this effect was uniquely detected in female T cells. Consistently, the synthetic PPARγ ligand, rosiglitazone, reduced IL-17A production by CD4+ T cells of women but not men [69]. Notably, a different study observed that the signaling axis of TCR–mTORC1-PPARγ and TCR–mTORC1–SREBP1 is essential for the activation of the fatty acid metabolic program in activated CD4+ T cells [70]. In this context, PPARγ directly binds on DNA and regulates the induction of genes associated with fatty acid uptake and metabolism. Thus, although PPARγ opposes the generation of effector cells with Th17 properties, it may be involved in the generation of memory cells that depend on fatty acid metabolism.
Figure 3. PPARs regulate T cell differentiation and function.
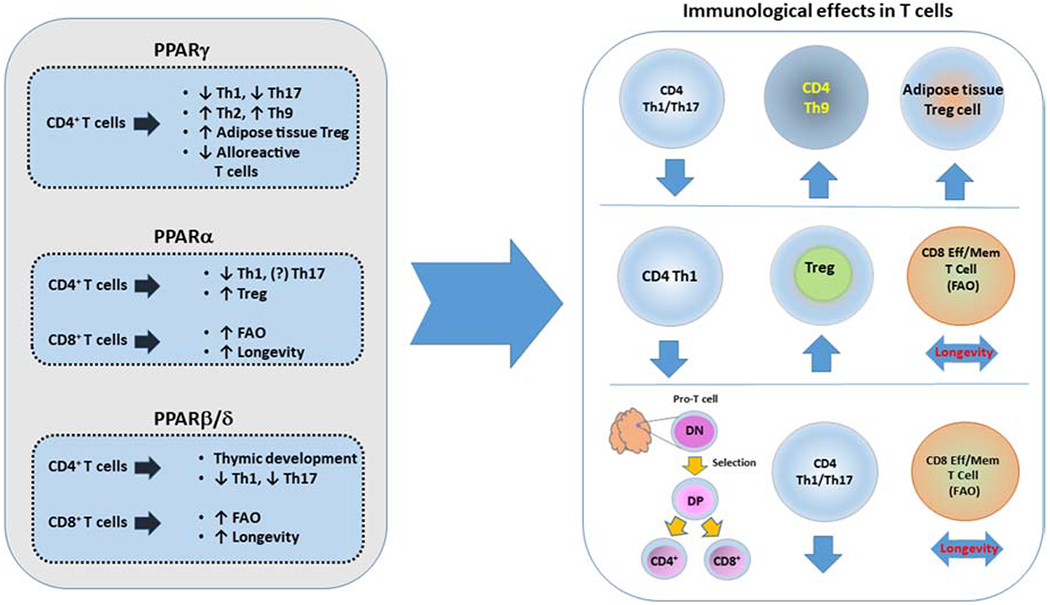
Signals mediated via PPARs regulate the development, differentiation and function of T cells.
PPARγ has an indispensable role in the differentiation of Treg cells in the visceral adipose tissue (VAT) [71] (Figure 3). Specifically, PPARγ is a major driver of a unique population of Treg that differentiate and accumulate in the VAT, and are implicated in the control of the inflammatory state of adipose tissue and, thereby, insulin sensitivity [71,72]. The transcriptional profile driving the differentiation of VAT Treg is under the control of PPARγ and depends on the exact same molecular regulation mediated by PPARγ Ser273, similarly to a PPARγ-mediated transcriptional differentiation of adipocytes [73]. Because Treg survival and function depends on lipid metabolism [74], the specific role of PPARs on Treg maintenance and function remains an open and intriguing question.
The beneficial role of PPARγ in suppressing T cell activation and generation of alloreactive T cells was reported in a model of cardiac transplantation. Macrophage and CD4+ T cell infiltration was significantly higher in cardiac allografts of recipient mice with T-cell-specific PPARγ deficiency, which displayed aggravated lesions of chronic allograft rejection. In contrast to WT recipients, infiltrating macrophages in mice with T-cell-specific PPARγ deficiency failed to polarize to M2. This was associated with the higher Thl/Th2 and Th17/Treg ratios among the infiltrating CD4+ T cells [75].
PPARα
Besides PPARγ, PPARα also appears to potentiate the polarization of macrophages towards an anti-inflammatory phenotype (Figure 2). Specifically, under LPS/IFN-γ culture conditions, peritoneal macrophages from PPARα deficient mice show a significant increase of various inflammatory parameters [76]. Similarly, treatment with the PPARα agonist WY14643 promotes the switch of peritoneal macrophages from Trypanosoma cruzi-infected mice from the M1 to M2 phenotype [77]. Notably, PPARα demonstrates an important role both in the inhibition of the excessive inflammatory response and the enhancement of innate host defense [78]. Bone marrow-derived macrophages infected with Mycobacterium Tuberculosis exhibit reduction of proinflammatory cytokines but also decreased intracellular mycobacterial levels when treated with PPARα agonists. Further in vitro studies revealed that PPARα, indeed, regulates macrophage antimycobacterial response through the transcriptional activation of autophagy, lysosomal biogenesis and enhancement of phagosome maturation mediated by the transcription factor EB (TFEB), a critical regulator of autophagy activation [78].
PPARα plays an important role in T cell responses and in the development of T cell-mediated autoimmune diseases, in a gender-specific manner. PPARα expression is higher in CD4+ T cells from male mice than female mice, which was found to be influenced by androgen levels. Additionally, genetic deletion of PPARα in male murine T cells resulted in augmented Th1 at the expense of Th2 responses through enhancement of NF-kB and c-Jun activity [79]. PPARα also affects the function of human T cells in a gender-specific manner [69]. Naive CD4+ T cells from peripheral blood of healthy women produce higher levels of IFNγ in response to anti-CD3 and anti-CD28 stimulation. PPARα, which is increased by androgens, represses IFNγ production in male T cells. Thus, PPARα, similarly to PPARγ may act as androgen-sensitive regulator driving the sex difference in Th cytokine production, and possibly the increased susceptibility of women to certain autoimmune diseases [69]. A proposed mechanism through which PPARα might inhibit IFNγ production involves the recruitment of nuclear receptor corepressor 1 to specific cis-regulatory elements in the Ifng locus and the subsequent reduction of histone acetylation at these sites [80]. In this context, a novel antagonist of PPARα, IS001, was found to increase IFNγ secretion by NK cells, CD4+, and CD8+ T cells and improve survival of male mice infected with Th1-associated pathogen Listeria monocytogenes. Although the role of PPARα on Th17 differentiation remains uncertain, its effect on Treg generation is better understood (Figure 3). Following antigen challenge, PPARα deficient mice produced lower levels of Tregs that also exhibited impaired suppressive capacity compared to WT mice [81]. PPARα agonists, bezafibrate, GW7647, and ETYA, increase Foxp3 expression in human iTreg through epigenetic modification of the Foxp3 promoter and induced functional Tregs with suppressive properties [82].
PPARβ/δ
The effects of PPARβ/δ on macrophages are less well established (Figure 2). Under M1 culture conditions, treatment of murine macrophages with PPARδ agonists could inhibit expression of proinflammatory mediators iNOS and COX2 [83]. However, other studies reported that peritoneal macrophages from PPARβ-deficient mice have reduced expression of proinflammatory genes [84]. PPARδ deletion renders adipose tissue resident murine macrophages incapable of transitioning to M2 phenotype [85]. In human macrophages, PPARδ activation did not influence polarization [86]. These apparently contradictory observations can be explained by the context-dependent effect of PPARβ/δ on macrophage polarization [87]. At least one proposed mechanism involves an inflammatory switch that is mediated through PPARβ/δ association and disassociation with transcriptional repressors such as Bc16. Unliganded PPARβ/δ has pro-inflammatory effects, while Bc16-ligated PPARβ/δ switches to an anti-inflammatory mediator by releasing Bc16 that suppresses transcription of proinflammatory genes [84]. The anti-inflammatory effects of PPARβ/δ are also exhibited in mast cells. Mast cells from PPARβ/δ KO mice express higher levels of inflammatory cytokines and lower levels of high-affinity IgE receptor compared to WT [88].
PPARβ/δ has implications on T cell development and function (Figure 3). By regulating the metabolic program of developing thymocytes, PPARβ/δ might enhance the proliferative burst of TCRβ-selected thymocytes and the growth of peripheral CD4+ T cells [89]. On the contrary, other studies support that activation or overexpression of PPARβ/δ decreases proliferation of double negative stage 4 (DN4) thymocytes through a switch of cellular fuel preference from glucose to fatty acid oxidation [90]. Regardless of the unresolved issues about the precise role of PPARδ in thymic development, the role of PPARδ in inducing tolerance and preventing autoimmunity is well established. In a murine EAE model, PPARδ deficiency augmented Th1 and Th17 polarization and inhibited Th2 and Treg differentiation, while the opposite results were induced with PPARδ activation [91–93]. Notably, mice lacking PPARδ expression only in the myeloid compartment also develop more severe EAE due to Th1/Th17 polarization of CD4+ T cells, highlighting the role of PPARδ in the crosstalk between innate and adaptive immune cells [94].
Role of PPARs in inflammatory diseases
PPARγ
The function of PPARγ has been associated with several inflammatory and autoimmune conditions. PPARγ null mice develop anti-phospholipid syndrome, an autoimmune disorder associated with glomerular injury and microthrombi [95]. The relationship between PPARγ and anti-phospholipid syndrome is not surprising since hemizygote T cell-specific [96] or myeloid cell-specific deletion [97] of PPARγ is associated with Lupus-like glomerular damage. Mice with PPARγ deletion in the T cell compartment develop splenomegaly and elevated plasma autoantibodies. Reduced expression of sphingosine-1-phosphate receptor 1, a molecule necessary for lymphocyte egress, is proposed to be the reason of splenic accumulation, while enhanced B-cell activation was attributed to increased Th17 polarization and IL-17 signaling [96].
By mediating effects in Kupffer cells, the resident macrophages of the liver, PPARγ can have implications on the development of several liver inflammatory conditions such as nonalcoholic fatty liver disease (NAFLD). High fat containing diet promotes liver steatosis and M1 polarization of the Kupffer cells. Conversely, polyunsaturated fatty acids (n3-PUFA) skew liver macrophages to M2 phenotype via upregulation of PPARγ. In a similar way, a PPARγ agonist converts M1 liver macrophages to M2 and reverses high fat-mediated steatosis [98]. n3-PUFA also promote Treg cell proliferation through upregulation of PPARγ and TGF-β, protecting mice from Con A-induced hepatotoxicity [99]. It is possible that there is an association between PUFA-mediated M2 polarization of Kupffer cells [98] and PUFA-mediated Treg polarization [99]. Tregs induced by PPARγ activation also alleviate Schistosoma mediated immunopathology in the spleen and liver [100]. Similarly to n3-PUFA, administration of 15d-PGJ2, a natural PPARγ ligand, partially prevents the development of hepatitis by attenuating NF-kB activation in macrophages and production of pro-inflammatory cytokines such as TNF-α, IL-2, IL-6 and IL-12 [101].
In the cardiac tissue, in addition to the direct effect of PPARγ deficiency in cardiomyocytes [25], the anti-inflammatory role of PPARγ relies also on its ability to influence macrophage polarization [102], consistent with observations in other tissues [98]. Upregulation of PPARγ, PPARα and Heme oxygenase 1 by epoxyeicosatrienoic acid (EETs) can skew macrophage polarization to M2, and inhibit LPS-induced M1 macrophage myocardial infiltration in vivo [102].
Th2 inflammation, an immune process related to allergic reactions and helminth infections, relies on PPARγ. Mice with conditional deletion of PPARγ in T cells fail to develop type 2 inflammation in response to HDM or nematode infection [103]. Recently, it was determined that Th9 cells, a subpopulation of human Th2 cells that produce IL-9 found in allergic skin conditions, are differentiated under the control of PPARγ [104]. Importantly, the role of PPARγ in the induction of type 2 inflammation is not mediated exclusively by a T cell-intrinsic mechanism. PPARγ controls the migration of CD11b+ DCs to the lymph nodes, promoting DC: T cell interaction [105].
PPARγ has a crucial role in regulating CNS immune responses and injury repair [106]. Polarization of microglial cells into anti-inflammatory M2 phenotype relies on PPARγ [107,108]. Adiponectin enhances microglia-mediated scavenging under amyloid β-induced toxicity by inducing M2 polarization through PPARγ, suggesting that PPARγ could have a potential role in the treatment of Alzheimer disease [107]. In an EAE model, PPARγ can protect CNS from autoimmunity by controlling the communication between self-antigen-presenting myeloid cells of the brain and autoreactive CD4+ T cells, which invade CNS [109]. In this model, CD4 T cell: myeloid cell interaction in the CNS resulted in CCL2 production by local astrocytes, which contributed to the recruitment of monocytes, that functioned as key mediators of EAE pathology. In mice with myeloid-specific PPARγ deficiency, monocyte invasion, neural demyelination and CCL2 production were higher than their wild type counterparts during EAE. In addition, there was a higher expression of CD40 and MHC-II in microglial cells and infiltrating macrophages, highlight ing the importance of PPARγ in attenuating the inflammatory response in the CNS. In a similar manner, PPARγ agonist-treated monocytes from multiple sclerosis patients demonstrate less prominent upregulation of activation markers after inflammatory stimulation [109].
PPARα
PPARα is involved in the regulation of inflammatory conditions. For example, PPARα attenuates acute liver inflammation. PPARα deficient mice exposed to high-fat diet have higher levels of acute liver inflammation and injury markers, upregulated inflammatory gene expression and increased liver infiltration of macrophages, compared to WT mice. Conversely, PPARα activation with synthetic agonist WY14643 under nonsteatotic conditions, directly downregulates inflammatory gene expression in a PPARα-dependent manner [110]. Similarly, treatment with fenofibrate, a PPARα agonist, in liver-specific PPARα-expressing mice inhibits the hepatic acute-phase response by lowering the levels of pro-inflammatory cytokines TNF, IL-1, and IL-6 [111]. PPARα also attenuates chronic liver inflammation. PPARα deletion in hepatocytes of high-fat fed mice promotes the development of liver inflammation and nonalcoholic fatty liver disease (NAFLD) [112]. Conversely, fenofibrate ameliorated the increased liver inflammatory gene expression and macrophage infiltration in mice with NAFLD [113]. Fenofibrate also improves liver inflammation in patients with NAFLD [114].
Similarly to its anti-inflammatory effects in liver biology, PPARα exhibits a protective role against renal inflammation. Transgenic mice with increased proximal tubule expression of PPARα develop less severe renal fibrosis and inflammation following unilateral urethral obstruction compared to WT mice. Further analyses reveal decreased production of TGF-β, IL-1b, IL-6 and TNF-α, reduced macrophage infiltration, and increased expression of anti-inflammatory cytokine IL-10 and arginase-1 [115]. In contrast, PPARα deficient mice develop increased inflammation, elevated IL-6 and worse sepsis-associated acute kidney injury [116]. An important mechanism by which PPARα protects against sepsis-induced injury and inflammation involves its regulation by the long non-coding RNA CRNDE and the microRNA miR-181-5p [117].
PPARα exhibits an indispensable role in cardiomyocyte metabolism and exhibits a protective role against cardiac inflammation [21]. In a mouse model, administration of the PPARα agonist, GW7647, attenuated cardiac ischemia/reperfusion injury and inflammation and decreased infarct size. This effect was accompanied by reduction in multiple proinflammatory cytokine levels, diminished neutrophil infiltration in the heart and reduced expression of myocardial matrix metalloproteinases-9 and −2. In this model, a tentative mechanism involves inhibition of NF-kB activation and enhanced levels of inhibitor-kBα [118]. In accordance with this, PPARα and PPARβ/δ activation inhibits LPS-induced expression of TNF-α in neonatal rat cardiomyocytes partly by antagonizing NF-kB [119]. Furthermore, fenofibrate treatment to type 1 diabetic mice significantly decreased cardiac dysfunction and inflammation via upregulating FGF21 ameliorating sirtuin1-mediated autophagy [120].
Administration of either PPARα or PPARγ agonists, but not PPARβ/δ, to LDL receptor deficient male mice exposed to high fat/high cholesterol diet results in significant reduction in atherosclerotic lesions in the aorta and inhibits the formation of macrophage foam cells in the peritoneal cavity [121]. PPARα and γ induce cholesterol efflux from foam cells through activation of the LXR-ABCA1 pathway [122], but can also inhibit foam cell formation through ABCA1-indepedent pathways [121]. PPARα and γ may also attenuate foam cell formation by inhibiting platelet-induced differentiation of human CD34+ progenitor cells into foam cells [123]. Thus, although the role of PPARγ in mediating foam cell formation is documented [60,61], under certain conditions, the cholesterol efflux mediated by the LXR-ABCA1 pathway might dominate leading to the opposite outcome.
PPARβ/δ
Consistent with its immunosuppressive effects in immune cells, PPARβ/δ attenuates acute liver inflammation. Administration of PPARβ/δ agonist protects mice against chemically induced hepatotoxicity and downregulates expression of proinflammatory mediators, including TNF-α and monocyte chemoattractant protein-1 (MCP-1), through inhibition of NF-kB [124]. Consistently, administration of the PPARβ/δ agonist, GW501516, to hepatoma HepG2 cells and rat primary hepatocytes reduced expression of various IL-6-mediated acute phase proteins by inhibiting the transcriptional activity of STAT [125]. PPARβ/δ also attenuates chronic liver inflammation. PPARβ/δ agonist treatment to high-fat diet-fed mice alleviates liver steatosis and inflammation and suppresses hepatic caspase-1 and IL-1β expression, while in vitro studies reveal that PPARβ/δ agonist administration to HepG2 cells inhibits palmitic acid and LPS-induced inflammasome activation [126]. Similarly, diabetic rats treated with GW0742, a PPARβ/δ agonist, have decreased fatty liver infiltration and significantly decreased hepatic expression of various inflammatory cytokines, TNF-α and MCP-1 compared to the diabetic control group [127].
PPARβ/δ exhibits a renoprotective role as reported in a number of studies. In a mouse model of protein-overload nephropathy, mice receiving the PPARδ agonist GW501516, developed less severe tubulointerstitial lesions, reduced macrophage infiltration, and decreased expression of MCP-1 and TNFα compared to experimental control mice. The anti-inflammatory effect of PPARδ in that model was mediated through direct inhibition of the TGF-β activated kinase 1 (TAK-1)-NF-kB pathway [128]. PPARβ/δ mediates a renoprotective effect on diabetic nephropathy via a similar mechanism [129]. In other studies, in response to IL-4 treatment, PPARδ drives the differentiation of M2 adipose tissue macrophages and liver Kupffer cells [130]. Conversely, PPARδ deficient macrophages treated with LPS have a highly inflammatory profile and adoptive transfer of PPARδ deficient bone marrow caused autoimmune hepatic dysfunction and systemic insulin resistance. In a different model, the importance of PPARδ on self-tolerance was confirmed by the observation that PPARδ-deficient female mice are much more likely to develop autoimmune kidney disease, a lupus-like autoimmunity, compared to their WT counterparts [131]. This was mediated by the effects of PPARβ/δ as a transcriptional sensor of apoptotic cells inducing recognition and phagocytosis by macrophages, thus limiting autoimmune responses. In accordance with these findings, treatment of lupus mice with a PPARβ/δ agonist resulted in improvement of renal inflammation, hypertrophy and injury with concomitant reduction in gene expression of the proinflammatory cytokines TNF-α, IL-1β, and IL-6 in the kidney [132].
PPARβ/δ mediated signals affect the vascular system. PPARβ/δ activation in endothelial cells upregulates antioxidant gene expression and inhibits TNF-α-induced expression of pro-inflammatory adhesion molecules, thus suppressing endothelial-leukocyte adhesion. The proposed mechanism involves the dissociation of Bc1-6 from PPARδ, which relocates to the promoter pro-inflammatory genes mediating transcriptional repression [133]. PPARβ/δ activation inhibits foam cell formation in THP-1 macrophages treated with VLDL by attenuating VLDL-stimulated lipid accumulation and downregulates expression of inflammatory cytokines and adhesion molecules by attenuating VLDL-stimulated ERK1/2 activation and reversing VLDL-mediated inhibition of AKT/FoxO1 phosphorylation [134]. On the contrary, other studies suggest that PPARβ/δ activation promotes foam cell formation and lipid accumulation via altering expression of genes involved in lipid uptake, storage, metabolism and efflux, including scavenger receptor class A and CD36 [135,136]. In accordance with the latter, genetic deletion of PPARβ/δ in hematopoietic cells of LDLR deficient mice results in significant reduction of aortic atherosclerotic lesions, pro-inflammatory gene expression and macrophage infiltration of atherosclerotic lesions [137].
Therapeutic exploitation of PPARs
As multifunctional molecules, PPARs are implicated in a variety of a human diseases such as cancer [138–140], metabolic [141] and autoimmune conditions [142]. Therapeutic targeting of PPARs has been attempted in several of these conditions. Several synthetic exogenous ligands of PPAR receptors have been developed and therapeutically exploited (Table 2). Fibrates consist of a large group of synthetic PPARα agonists used for the treatment of hypertriglyceridemia, while TZD is a group of PPARγ agonists used as insulin sensitization in patients with metabolic syndrome and Diabetes Mellitus type 2 (DM2) [32,143]. Furthermore, compounds that activate more than one PPAR members, such as dual agonists, or all PPAR members, such as pan-agonists (Table 3), could be beneficial in both lipid and glucose imbalances, minimizing the needs for dual pharmacological intervention, and potentially diminishing side effects that might arise from the administration of two individual pharmacologic compounds [144]. On the other hand, dual PPAR agonists or pan-agonists might be related with more adverse effects due to lower target selectivity [144,145]
Table 2:
Synthetic PPAR ligands and therapeutic exploitation.
| Subtype | Synthetic ligands (agonists) | Therapeutic exploitation |
|---|---|---|
| PPARγ | Thiazolidinediones (TZD) (pioglitazone, troglitazone, rosiglitazone) [32] Farglitazar [145] Ibuprofen [182] INT131 (CHS-131) [145] S26948 [183] GW7845 [184] Efatutazone (CS-7017) [145] GED 0507-34-Levo [145] OMS 405 [145] |
• DM2 • Metabolic syndrome • Complications of metabolic diseases, such as renal tubulointerstitial fibrosis and hypertension-induced renal fibrosis • Autoimmune diseases (SLE) • Cancer immunotherapy |
| PPARα | Fibrates (Fenofibrate, clofibrate, gemfibrozil) [143] Pirinixic acid (WY-14643) [185] GW7647 [82,186] |
• Dyslipidemia • Autoimmune diseases (EAE) • Cancer immunotherapy |
| PPARβ/δ |
GW501516 [128,145] GW0742 [127,145] Seladelpar (MBX-8025) [145] |
• Autoimmune diseases (SLE) • Cancer immunotherapy |
DM2, Diabetes mellitus type 2; SLE, Systemic lupus erythematosus; EAE, Experimental autoimmune encephalomyelitis.
Table 3:
Synthetic dual or pan-PPAR ligands and therapeutic exploitation.
| Subtype | Dual synthetic agonists | Therapeutic exploitation |
|---|---|---|
| PPARα/γ | Saroglitazar [144] Oxeglitazar [145] Lobeglitazone [145] |
• Dyslipidemia • DM2 |
| PPARα/β(δ) | Elafibranor (GFT505) [145] | •Hepatic steatosis • NASH • Dyslipidemia • Pre-diabetes • Insulin resistance • Obesity |
| PPARγ/δ | T3D 959 [187] | • Alzheimer’s Disease |
| Pan-PPAR | Chiglitazar [144] Lanifibranor (IVA337) [145] |
• Dyslipidemia in obese mice • DM2 • Improved skin fibrosis in mice • NASH |
DM2, Diabetes mellitus type 2; NASH, Nonalcoholic steatohepatitis
PPARα is involved in lipid and carbohydrate metabolism [146–148]. Synergistic and contrasting actions between PPARα and PPARγ have been reported [149,150]. Although most of the experimental studies have shown that PPARα agonists do not have such a beneficial effect on lipid metabolism and insulin resistance as PPARγ agonists, particularly TZD [151], novel approaches for obesity and metabolic syndrome have explored the effect of PPARα [41,152–155]. This is particularly significant because, despite their greater anti-diabetic potential, clinical application of PPARγ agonists is accompanied by detrimental side effects. Studies have exploited joint targeting of PPARγ and PPARα with a dual PPARα/γ agonists. These dual agonists improve lipid parameters and reduce cardiovascular complications through PPARα in addition to the insulin-sensitizing effects mediated via PPARγ [156]. Combined with a cutting-edge approach for cell-type specific uptake of PPARα/γ dual agonists by peptide-mediated internalization and controlled release into adipocytes, these compounds could potentially reduce the intolerable side effects of PPARγ agonists and provide major benefit for the treatment of DM2 [157].
Another promising approach in reducing the side effects of PPARγ agonists, was the development of selective PPARγ ligands that are structurally and pharmacologically distinct from glitazones. Efforts toward this goal have resulted in the development of non-thiazolidinedione compounds that belong to a distinct family of drugs named selective PPARγ modulators (SPPARMs). Such compounds mediate a distinct pattern of coregulator recruitment to PPARγ that allows improved specificity and diminished toxicity providing an opportunity for the treatment of DM2 and metabolic syndrome with limited side effects on heart and lung, weight gain, hemodilution, and plasma volume [158–161]. Using X-ray crystallography, it was determined that the SPPARM, INT131, forms hydrophobic contacts with the ligand-binding pocket of PPARγ without direct hydrogen-bonding interactions to residues in helix 12, as full agonists [160]. Because of the beneficial effects of classic PPARγ agonists on protection against neurotoxicity from Aβ amyloid oligomer in Alzheimer’s disease (AD), the role of INT131 in the central nervous system was studied in a mouse model of AD [162]. The study determined that INT131 increased dendritic branching, promoted neuronal survival against Aβ amyloid, increased expression of PGC1-1α and modulated neuronal mitochondrial dynamics. Thus SPPARMs, might have applications not only in DM2 and metabolic syndrome but also in other conditions in which PPARγ-mediated signaling might have beneficial effects.
Nuclear receptors have been associated with autoimmune diseases and PPAR agonists could be used for therapeutic purposes in autoimmunity [142]. PPARα agonists improve the clinical manifestation of EAE mice [163]. Similarly, administration of, the PPARβ/δ agonist GW0742 alleviates systemic lupus erythematosus’s (SLE) complications such as albuminuria, splenomegaly, hypertension, renal and cardiac hypertrophy [132]. These complications of SLE are also improved by rosiglitazone through adiponectin induction [164].
The role of PPAR on innate and adaptive immune cells in the context of cancer is extensively studied because factors that affect cancer development, evolution and therapeutic response might simultaneously affect the fate of immune cells via interfering with PPAR-mediated signaling. Global or conditional deficiencies of PPAR members affect tumor progression suggesting that PPARs are potential targets for cancer therapy. PPARα or PPARβ deletion in Tregs diminishes anergic properties in the tumor microenvironment [165,166]. Furthermore, combined activation of PPARα and PPARδ improves antitumor efficacy of adoptively transferred CD8+ T cells by reprogramming metabolism from aerobic glycolysis toward fatty acid oxidation, thereby increasing their in vivo longevity and enhancing inflammatory signals [167] (Figure 3). Similarly, PPARα-mediated fatty acid catabolism and uptake by CD8+ tumor-infiltrating T lymphocytes preserves their effector function and decelerates tumor growth [168]. PPAR activation can also enhance anti-tumor activity when combined with checkpoint inhibitors such as anti-PD-1 [169,170] and anti-CTLA-4 [171].
PPARγ also enhances the anti-tumor capacity of intratumoral iNKT cells. Cholesterol is one of the main precursors for IFNγ synthesis by the iNKT cells. The elevated lactic acid of the tumor microenvironment decreases PPARγ expression in iNKT cells resulting in decreased cholesterol synthesis, diminished IFNγ production, and weaker anti-tumor response. Under these conditions, a PPARγ agonist can restore cholesterol and IFNγ production, improving the antitumor activity of iNKT cells [172]. On the other hand, studies have shown that during obesity, a major risk factor for cancer development, NK cells undergo PPARα/δ-depended accumulation of lipids, resulting in metabolic defects that subsequently diminish their anti-tumor capacity [173]. These metabolic alternations can be restored after PPARα/δ inhibition, suggesting that PPARα/δ blockers could have a role in the treatment of cancer in obese patients, by enhancing the cytotoxic ability of NK cells.
PPAR can also affect the generation and function of myeloid-derived suppressor cells (MDSCs), a population of immature myeloid cells derived from myeloid progenitors during cancer-mediated emergency myelopoiesis, which have immunosuppressive properties and promote tumor progression [174]. Tumor-infiltrating MDSCs preferentially depend on fatty acid uptake and fatty acid oxidation over glycolysis for their survival and function [175]. Detailed studies have shown that upregulation of fatty-acid synthase (FASN) by M-CSF is mandatory for the differentiation of tumor infiltrating myeloid cells into MDSC, expression of MDSC hallmark genes such as IL-10, Arg1 and VEGF, and tumor progression. These events depend on fatty acid-mediated signaling via PPARβ/δ [176]. PPARγ also plays an important role in MDSC differentiation by regulating neutral lipid metabolism that depends on the generation of endogenous fatty acids via lipolysis mediated by lysosomal acid lipase (LAL). In this context, signaling via PPARγ impairs MDSC development and function and suppresses tumor growth [177]. The opposite outcomes were observed when PPARγ signaling in myeloid cells was abrogated by expressing a dominant negative PPARγ construct in a myeloid-specific bitransgenic mouse model [178]. Under these conditions, there was a massive expansion of PPARγ deficient MDSC that mediated potent suppression expansion and function of CD4+ and CD8+ T cells and resulted in the development of multiple forms of cancers in various organs. Notably, PPARγ expression is elevated in myeloid cells and tumor associated macrophages (TAMs) in PD-1 KO mice or mice with myeloid-specific PD-1 deletion [179]. These results indicate that PPARγ-mediated signals during PD-1 ablation might specifically regulate the effector function of myeloid cells and prevent the generation of MDSC in the context of cancer.
Together these studies indicate that increase of fatty acid availability might promote the generation and function of immunosuppressive and tumor promoting myeloid cells by altering PPAR-mediating signaling. This mechanism might provide a mechanistic explanation for the fact that obesity is a risk factor for carcinogenesis [180,181]. However, as outlined above, it becomes increasingly apparent that PPAR subtypes have distinct and opposing roles in the generation of MDSC [176–178]. Thus, it is unpredictable how metabolic adaptation of immune cells in the context of altered host metabolism, as in diabetes, metabolic syndrome and obesity, might affect distinct PPAR subtypes under patient-specific conditions. Therefore, development of markers related to PPAR signaling in innate and adaptive immune cells might provide critical patient-specific information that might be helpful for guiding selection of treatment strategies exploiting the potential of PPAR signaling in cancer therapy.
Concluding remarks
Members of the PPAR family of nuclear hormone receptors are well-established regulators of lipid metabolism, mitochondrial biogenesis and energy homeostasis. Their activation has central implications in the function of oxidative tissues and organs such as cardiomyocytes, liver and muscle. For many years, PPARs have been attractive therapeutic targets for the treatment of metabolic disorders. The role of various members of this nuclear receptor family is currently emerging in the differentiation and function of immune cells as they guide metabolism-mediated immune cell fate commitment. Well-established as mediators of macrophage polarization, PPARs are also key determinants of T cell activation, expansion and differentiation as well as regulators of MDSC generation. The cell-specific temporal expression and activation of PPARs provide new opportunities for therapeutic intervention to alter the function of innate and adaptive immune cells with the goal to modulate immune cell activation for the treatment of autoimmune diseases, organ transplantation, neurodegenerative disorders, and cancer. Discoveries on the posttranslational regulation of PPARs, will assist the development of new compounds to target for cell-specific activation for therapeutic intervention. New compounds that function as agonists for more than one PPAR subtypes will allow the simultaneous therapeutic exploitation of these nuclear receptors to maximize therapeutic benefit while minimizing toxicity. Cutting edge approaches, which allow the identification of how structurally and functionally distinct states of PPARs are induced by binding of agonists vs. antagonists or reverse agonists, will provide guidance for the design of compounds to promote the desired activation state. Moreover, novel technologies for targeted delivery of PPAR-modulating compounds will provide unprecedented opportunities for targeted therapeutic interventions in the context of immune-mediated diseases.
Highlights.
PPAR in the function on innate immune cells
PPAR function on adaptive immune cells
PPAR role in immune-mediated inflammatory conditions
PPAR therapeutic options in immunology
Acknowledgments:
This work was supported by the National Institutes of Health awards: RO1CA212605, RO1CA238263 and RO1CA229784 (VAB)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest/financial disclosure: The authors do not have relevant conflicts of interest to disclose.
References
- 1.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature, 1990;347:645–50. 10.1038/347645a0 [DOI] [PubMed] [Google Scholar]
- 2.Nuclear Receptors Nomenclature C A unified nomenclature system for the nuclear receptor superfamily. Cell, 1999;97:161–3. 10.1016/s0092-8674(00)80726-6 [DOI] [PubMed] [Google Scholar]
- 3.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science, 2001;294:1866–70. 10.1126/science.294.5548.1866 [DOI] [PubMed] [Google Scholar]
- 4.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem, 2000;43:527–50. 10.1021/jm990554g [DOI] [PubMed] [Google Scholar]
- 5.Grygiel-Gorniak B Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications--a review. Nutr J, 2014; 13:17 10.1186/1475-2891-13-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desvergne B, Michalik L, Wahli W. Transcriptional regulation of metabolism. Physiol Rev, 2006;86:465–514. 10.1152/physrev.00025.2005 [DOI] [PubMed] [Google Scholar]
- 7.Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell, 1992;68:879–87. 10.1016/0092-8674(92)90031-7 [DOI] [PubMed] [Google Scholar]
- 8.Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev, 2006;58:726–41. 10.1124/pr.58.4.5 [DOI] [PubMed] [Google Scholar]
- 9.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med, 2002;53:409–35. 10.1146/annurev.med.53.082901.104018 [DOI] [PubMed] [Google Scholar]
- 10.Kota BP, Huang TH, Roufogalis BD. An overview on biological mechanisms of PPARs. Pharmacol Res, 2005;51:85–94. 10.1016/j.phrs.2004.07.012 [DOI] [PubMed] [Google Scholar]
- 11.Werman A, Hollenberg A, Solanes G, Bjorbaek C, Vidal-Puig AJ, Flier JS. Ligand-independent activation domain in the N terminus of peroxisome proliferator-activated receptor gamma (PPARgamma). Differential activity of PPARgamma1 and −2 isoforms and influence of insulin. J Biol Chem, 1997;272:20230–5. 10.1074/jbc.272.32.20230 [DOI] [PubMed] [Google Scholar]
- 12.Nielsen R, Grontved L, Stunnenberg HG, Mandrup S. Peroxisome proliferator-activated receptor subtype- and cell-type-specific activation of genomic target genes upon adenoviral transgene delivery. Mol Cell Biol, 2006;26:5698–714. 10.1128/MCB.02266-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci, 2004;29:317–24. 10.1016/j.tibs.2004.04.006 [DOI] [PubMed] [Google Scholar]
- 14.Heidari Z, Chrisman IM, Nemetchek MD, Novick SJ, Blayo AL, Patton T, et al. Definition of functionally and structurally distinct repressive states in the nuclear receptor PPARgamma. Nat Commun, 2019;10:5825 10.1038/s41467-019-13768-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fajas L, Auboeuf D, Raspe E, Schoonjans K, Lefebvre AM, Saladin R, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem, 1997;272:18779–89. 10.1074/jbc.272.30.18779 [DOI] [PubMed] [Google Scholar]
- 16.Fajas L, Fruchart JC, Auwerx J. PPARgamma3 mRNA: a distinct PPARgamma mRNA subtype transcribed from an independent promoter. FEBS Lett, 1998;438:55–60. 10.1016/s0014-5793(98)01273-3 [DOI] [PubMed] [Google Scholar]
- 17.Greene ME, Blumberg B, McBride OW, Yi HF, Kronquist K, Kwan K, et al. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: expression in hematopoietic cells and chromosomal mapping. Gene Expr, 1995;4:281–99. [PMC free article] [PubMed] [Google Scholar]
- 18.Moreno M, Lombardi A, Silvestri E, Senese R, Cioffi F, Goglia F, et al. PPARs: Nuclear Receptors Controlled by, and Controlling, Nutrient Handling through Nuclear and Cytosolic Signaling. PPAR Res, 2010;201010 1155/2010/435689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patsouris D, Reddy JK, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology, 2006;147:1508–16. 10.1210/en.2005-1132 [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Roves P, Huss JM, Han DH, Hancock CR, Iglesias-Gutierrez E, Chen M, et al. Raising plasma fatty acid concentration induces increased biogenesis of mitochondria in skeletal muscle. Proc Natl Acad Sci USA, 2007;104:10709–13. 10.1073/pnas.0704024104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med, 2011;17:1076–85. 10.1038/nm.2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology, 2011;53:116–26. 10.1002/hep.24006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandak PG, Radovic B, Aflaki E, Kolb D, Buchebner M, Frohlich E, et al. Efficient phagocytosis requires triacylglycerol hydrolysis by adipose triglyceride lipase. J Biol Chem, 2010;285:20192–201. 10.1074/jbc.M110.107854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab, 2011;13:739–48. 10.1016/j.cmet.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Festuccia WT, Laplante M, Berthiaume M, Gelinas Y, Deshaies Y. PPARgamma agonism increases rat adipose tissue lipolysis, expression of glyceride lipases, and the response of lipolysis to hormonal control. Diabetologia, 2006;49:2427–36. 10.1007/s00125-006-0336-y [DOI] [PubMed] [Google Scholar]
- 26.Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell, 1999;4:611–7. 10.1016/s1097-2765(00)80211-7 [DOI] [PubMed] [Google Scholar]
- 27.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell, 1994;79:1147–56. 10.1016/0092-8674(94)90006-x [DOI] [PubMed] [Google Scholar]
- 28.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell, 1999;4:585–95. 10.1016/s1097-2765(00)80209-9 [DOI] [PubMed] [Google Scholar]
- 29.Rosen ED, Hsu CH, Wang X, Sakai S, Freeman MW, Gonzalez FJ, et al. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev, 2002;16:22–6. 10.1101/gad.948702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seale P Transcriptional Regulatory Circuits Controlling Brown Fat Development and Activation. Diabetes, 2015;64:2369–75. 10.2337/db15-0203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang JS, Ha K. A truncated PPAR gamma 2 localizes to mitochondria and regulates mitochondrial respiration in brown adipocytes. PLoS One, 2018;13:e0195007 10.1371/journal.pone.0195007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yki-Jarvinen H Thiazolidinediones. N Engl J Med, 2004;351:1106–18. 10.1056/NEJMra041001 [DOI] [PubMed] [Google Scholar]
- 33.Ribon V, Johnson JH, Camp HS, Saltiel AR. Thiazolidinediones and insulin resistance: peroxisome proliferatoractivated receptor gamma activation stimulates expression of the CAP gene. Proc Natl Acad Sci U S A, 1998;95:14751–6. 10.1073/pnas.95.25.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature, 2001;409:307–12. 10.1038/35053000 [DOI] [PubMed] [Google Scholar]
- 35.Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med, 2001;7:947–53. 10.1038/90992 [DOI] [PubMed] [Google Scholar]
- 36.Peraldi P, Xu M, Spiegelman BM. Thiazolidinediones block tumor necrosis factor-alpha-induced inhibition of insulin signaling. J Clin Invest, 1997;100:1863–9. 10.1172/JCI119715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, et al. Fibroblast growth factor-21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell, 2012;148:556–67. 10.1016/j.cell.2011.11.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rangwala SM, Rhoades B, Shapiro JS, Rich AS, Kim JK, Shulman GI, et al. Genetic modulation of PPARgamma phosphorylation regulates insulin sensitivity. Dev Cell, 2003;5:657–63. 10.1016/s1534-5807(03)00274-0 [DOI] [PubMed] [Google Scholar]
- 39.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature, 1999;402:880–3. 10.1038/47254 [DOI] [PubMed] [Google Scholar]
- 40.Pedersen G, Brynskov J. Topical rosiglitazone treatment improves ulcerative colitis by restoring peroxisome proliferator-activated receptor-gamma activity. Am J Gastroenterol, 2010;105:1595–603. 10.1038/ajg.2009.749 [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Teng R, Di L, Rogers H, Wu H, Kopp JB, et al. PPARalpha and Sirt1 mediate erythropoietin action in increasing metabolic activity and browning of white adipocytes to protect against obesity and metabolic disorders. Diabetes, 2013;62:4122–31. 10.2337/db13-0518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang W, Lin Q, Lin R, Zhang J, Ren F, Zhang J, et al. PPARαlpha agonist fenofibrate attenuates TNF-alpha-induced CD40 expression in 3T3-L1 adipocytes via the SIRT1-dependent signaling pathway. Exp Cell Res, 2013;319:1523–33. 10.1016/j.yexcr.2013.04.007 [DOI] [PubMed] [Google Scholar]
- 43.Walden TB, Petrovic N, Nedergaard J. PPARαlpha does not suppress muscle-associated gene expression in brown adipocytes but does influence expression of factors that fingerprint the brown adipocyte. Biochem Biophys Res Commun, 2010;397:146–51. 10.1016/j.bbrc.2010.05.053 [DOI] [PubMed] [Google Scholar]
- 44.Lee YK, Sohn JH, Han JS, Park YJ, Jeon YG, Ji Y, et al. Perilipin 3 Deficiency Stimulates Thermogenic Beige Adipocytes Through PPARαlpha Activation. Diabetes, 2018;67:791–804. 10.2337/db17-0983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coll T, Rodriguez-Calvo R, Barroso E, Serrano L, Eyre E, Palomer X, et al. Peroxisome proliferator-activated receptor (PPAR) beta/delta: a new potential therapeutic target for the treatment of metabolic syndrome. Curr Mol Pharmacol, 2009;2:46–55. 10.2174/1874467210902010046 [DOI] [PubMed] [Google Scholar]
- 46.Wolf G The function of the nuclear receptor peroxisome proliferator-activated receptor delta in energy homeostasis. Nutr Rev, 2003;61:387–90. 10.1301/nr.2003.nov.387-390 [DOI] [PubMed] [Google Scholar]
- 47.Choi KC, Lee SY, Yoo HJ, Ryu OH, Lee KW, Kim SM, et al. Effect of PPAR-delta agonist on the expression of visfatin, adiponectin, and resistin in rat adipose tissue and 3T3-L1 adipocytes. Biochem Biophys Res Commun, 2007;357:62–7. 10.1016/j.bbrc.2007.03.114 [DOI] [PubMed] [Google Scholar]
- 48.Serrano-Marco L, Rodriguez-Calvo R, El Kochairi I, Palomer X, Michalik L, Wahli W, et al. Activation of peroxisome proliferator-activated receptor-beta/-delta (PPAR-beta/-delta) ameliorates insulin signaling and reduces SOCS3 levels by inhibiting STAT3 in interleukin-6-stimulated adipocytes. Diabetes, 2011;60:1990–9. 10.2337/db10-0704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nandhikonda P, Yasgar A, Baranowski AM, Sidhu PS, McCallum MM, Pawlak AJ, et al. Peroxisome proliferation-activated receptor delta agonist GW0742 interacts weakly with multiple nuclear receptors, including the vitamin D receptor. Biochemistry, 2013;52:4193–203. 10.1021/bi400321p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity, 2010;33:699–712. 10.1016/j.immuni.2010.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature, 2007;447:1116–20. 10.1038/nature05894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daniel B, Nagy G, Czimmerer Z, Horvath A, Hammers DW, Cuaranta-Monroy I, et al. The Nuclear Receptor PPARgamma Controls Progressive Macrophage Polarization as a Ligand-Insensitive Epigenomic Ratchet of Transcriptional Memory. Immunity, 2018;49:615–26 e6 10.1016/j.immuni.2018.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol, 2014;15:1026–37. 10.1038/ni.3005 [DOI] [PubMed] [Google Scholar]
- 54.Stafeev YS, Michurina SS, Zubkova ES, Beloglazova IB, Ratner EI, Parfenova EV, et al. Modulation of the Inflammatory Status of Macrophages and Their Paracrine Effect on the Sensitivity of Adipocytes to Insulin with Sirtuin and PPARgamma Receptor Activators. Bull Exp Biol Med, 2018;165:429–33. 10.1007/s10517-018-4186-7 [DOI] [PubMed] [Google Scholar]
- 55.Murray PJ. Macrophage Polarization. Annu Rev Physiol, 2017;79:541–66. 10.1146/annurev-physiol-022516-034339 [DOI] [PubMed] [Google Scholar]
- 56.Bashir S, Sharma Y, Elahi A, Khan F. Macrophage polarization: the link between inflammation and related diseases. Inflamm Res, 2016;65:1–11. 10.1007/s00011-015-0874-1 [DOI] [PubMed] [Google Scholar]
- 57.Varga T, Mounier R, Patsalos A, Gogolak P, Peloquin M, Horvath A, et al. Macrophage PPARgamma, a Lipid Activated Transcription Factor Controls the Growth Factor GDF3 and Skeletal Muscle Regeneration. Immunity, 2016;45:1038–51. 10.1016/j.immuni.2016.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heming M, Gran S, Jauch SL, Fischer-Riepe L, Russo A, Klotz L, et al. Peroxisome Proliferator-Activated Receptor-gamma Modulates the Response of Macrophages to Lipopolysaccharide and Glucocorticoids. Front Immunol, 2018;9:893 10.3389/fimmu.2018.00893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore KJ, Rosen ED, Fitzgerald ML, Randow F, Andersson LP, Altshuler D, et al. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat Med, 2001;7:41–7. 10.1038/83328 [DOI] [PubMed] [Google Scholar]
- 60.Kotla S, Rao GN. Reactive Oxygen Species (ROS) Mediate p300-dependent STAT1 Protein Interaction with Peroxisome Proliferator-activated Receptor (PPAR)-gamma in CD36 Protein Expression and Foam Cell Formation. J Biol Chem, 2015;290:30306–20. 10.1074/jbc.M115.686865 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Kotla S, Singh NK, Rao GN. ROS via BTK-p300-STAT1-PPARgamma signaling activation mediates cholesterol crystals-induced CD36 expression and foam cell formation. Redox Biol, 2017;11:350–64. 10.1016/j.redox.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab, 2006;4:211–21. 10.1016/j.cmet.2006.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell, 2001;7:161–71. 10.1016/s1097-2765(01)00164-2 [DOI] [PubMed] [Google Scholar]
- 64.Guo K, Hu L, Xi D, Zhao J, Liu J, Luo T, et al. PSRC1 overexpression attenuates atherosclerosis progression in apoE(−/−) mice by modulating cholesterol transportation and inflammation. J Mol Cell Cardiol, 2018;116:69–80. 10.1016/j.yjmcc.2018.01.013 [DOI] [PubMed] [Google Scholar]
- 65.Klotz L, Dani I, Edenhofer F, Nolden L, Evert B, Paul B, et al. Peroxisome proliferator-activated receptor gamma control of dendritic cell function contributes to development of CD4+ T cell anergy. J Immunol, 2007;178:2122–31. 10.4049/jimmunol.178.4.2122 [DOI] [PubMed] [Google Scholar]
- 66.Appel S, Mirakaj V, Bringmann A, Weck MM, Grunebach F, Brossart P. PPAR-gamma agonists inhibit toll-like receptor-mediated activation of dendritic cells via the MAP kinase and NF-kappaB pathways. Blood, 2005;106:3888–94. 10.1182/blood-2004-12-4709 [DOI] [PubMed] [Google Scholar]
- 67.Khare A, Chakraborty K, Raundhal M, Ray P, Ray A. Cutting Edge: Dual Function of PPARgamma in CD11c+ Cells Ensures Immune Tolerance in the Airways. J Immunol, 2015;195:431–5. 10.4049/jimmunol.1500474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med, 2009;206:2079–89. 10.1084/jem.20082771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang MA, Rego D, Moshkova M, Kebir H, Chruscinski A, Nguyen H, et al. Peroxisome proliferator-activated receptor (PPAR)alpha and -gamma regulate IFNgamma and IL-17A production by human T cells in a sex-specific way. Proc Natl Acad Sci US A, 2012;109:9505–10. 10.1073/pnas.1118458109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, et al. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun, 2016;7:13683 10.1038/ncomms13683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature, 2012;486:549–53. 10.1038/nature11132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med, 2009;15:930–9. 10.1038/nm.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cipolletta D, Cohen P, Spiegelman BM, Benoist C, Mathis D. Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: age, diet, and PPARgamma effects. Proc Natl Acad Sci U S A, 2015;112:482–7. 10.1073/pnas.1423486112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol, 2011;186:3299–303. 10.4049/jimmunol.1003613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang X, Ren L, Ye P, Cheng C, Wu J, Wang S, et al. Peroxisome proliferator-activated receptor gamma deficiency in T cells accelerates chronic rejection by influencing the differentiation of CD4+ T cells and alternatively activated macrophages. PLoS One, 2014;9:e112953 10.1371/journal.pone.0112953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crisafulli C, Cuzzocrea S. The role of endogenous and exogenous ligands for the peroxisome proliferator-activated receptor alpha (PPAR-alpha) in the regulation of inflammation in macrophages. Shock, 2009;32:62–73. 10.1097/shk.0b013e31818bbad6 [DOI] [PubMed] [Google Scholar]
- 77.Penas F, Mirkin GA, Vera M, Cevey A, Gonzalez CD, Gomez MI, et al. Treatment in vitro with PPARalpha and PPARgamma ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim Biophys Acta, 2015;1852:893–904. 10.1016/j.bbadis.2014.12.019 [DOI] [PubMed] [Google Scholar]
- 78.Kim YS, Lee HM, Kim JK, Yang CS, Kim TS, Jung M, et al. PPAR-alpha Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J Immunol, 2017;198:3283–95. 10.4049/jimmunol.1601920 [DOI] [PubMed] [Google Scholar]
- 79.Dunn SE, Ousman SS, Sobel RA, Zuniga L, Baranzini SE, Youssef S, et al. Peroxisome proliferator-activated receptor (PPAR)alpha expression in T cells mediates gender differences in development of T cell-mediated autoimmunity. J Exp Med, 2007;204:321–30. 10.1084/jem.20061839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang MA, Ahn JJ, Zhao FL, Selvanantham T, Mallevaey T, Stock N, et al. Antagonizing Peroxisome Proliferator-Activated Receptor alpha Activity Selectively Enhances Th1 Immunity in Male Mice. J Immunol, 2015;195:5189–202. 10.4049/jimmunol.1500449 [DOI] [PubMed] [Google Scholar]
- 81.Dubrac S, Elentner A, Schoonjans K, Auwerx J, Schmuth M. Lack of IL-2 in PPAR-alpha-deficient mice triggers allergic contact dermatitis by affecting regulatory T cells. Eur J Immunol, 2011;41:1980–91. 10.1002/eji.201041357 [DOI] [PubMed] [Google Scholar]
- 82.Lei J, Hasegawa H, Matsumoto T, Yasukawa M. Peroxisome proliferator-activated receptor alpha and gamma agonists together with TGF-beta convert human CD4+CD25-T cells into functional Foxp3+ regulatory T cells. J Immunol, 2010;185:7186–98. 10.4049/jimmunol.1001437 [DOI] [PubMed] [Google Scholar]
- 83.Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proc Natl Acad Sci U S A, 2003;100:6712–7. 10.1073/pnas.1031789100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM, et al. Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science, 2003;302:453–7. 10.1126/science.1087344 [DOI] [PubMed] [Google Scholar]
- 85.Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocytederived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab, 2008;7:485–95. 10.1016/j.cmet.2008.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bouhlel MA, Brozek J, Derudas B, Zawadzki C, Jude B, Staels B, et al. Unlike PPARgamma, PPARalpha or PPARbeta/delta activation does not promote human monocyte differentiation toward alternative macrophages. Biochem Biophys Res Commun, 2009;386:459–62. 10.1016/j.bbrc.2009.06.047 [DOI] [PubMed] [Google Scholar]
- 87.Adhikary T, Wortmann A, Schumann T, Finkernagel F, Lieber S, Roth K, et al. The transcriptional PPARbeta/delta network in human macrophages defines a unique agonist-induced activation state. Nucleic Acids Res, 2015;43:5033–51. 10.1093/nar/gkv331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yao PL, Morales JL, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-beta/delta modulates mast cell phenotype. Immunology, 2017;150:456–67. 10.1111/imm.12699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhao FL, Ahn JJ, Chen ELY, Yi TJ, Stickle NH, Spaner D, et al. Peroxisome Proliferator-Activated Receptor-delta Supports the Metabolic Requirements of Cell Growth in TCRbeta-Selected Thymocytes and Peripheral CD4(+) T Cells. J Immunol, 2018;201:2664–82. 10.4049/jimmunol.1800374 [DOI] [PubMed] [Google Scholar]
- 90.Mothe-Satney I, Murdaca J, Sibille B, Rousseau AS, Squillace R, Le Menn G, et al. A role for Peroxisome Proliferator-Activated Receptor Beta in T cell development. Sci Rep, 2016;6:34317 10.1038/srep34317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kanakasabai S, Chearwae W, Walline CC, Iams W, Adams SM, Bright JJ. Peroxisome proliferator-activated receptor delta agonists inhibit T helper type 1 (Th1) and Th17 responses in experimental allergic encephalomyelitis. Immunology, 2010;130:572–88. 10.1111/j.1365-2567.2010.03261.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kanakasabai S, Walline CC, Chakraborty S, Bright JJ. PPARdelta deficient mice develop elevated Th1/Th17 responses and prolonged experimental autoimmune encephalomyelitis. Brain Res, 2011;1376:101–12. 10.1016/j.brainres.2010.12.059 [DOI] [PubMed] [Google Scholar]
- 93.Dunn SE, Bhat R, Straus DS, Sobel RA, Axtell R, Johnson A, et al. Peroxisome proliferator-activated receptor delta limits the expansion of pathogenic Th cells during central nervous system autoimmunity. J Exp Med, 2010;207:1599–608. 10.1084/jem.20091663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Drohomyrecky PC, Doroshenko ER, Akkermann R, Moshkova M, Yi TJ, Zhao FL, et al. Peroxisome Proliferator-Activated Receptor-delta Acts within Peripheral Myeloid Cells to Limit Th Cell Priming during Experimental Autoimmune Encephalomyelitis. J Immunol, 2019;203:2588–601. 10.4049/jimmunol.1801200 [DOI] [PubMed] [Google Scholar]
- 95.Toffoli B, Gilardi F, Winkler C, Soderberg M, Kowalczuk L, Arsenijevic Y, et al. Nephropathy in Pparg-null mice highlights PPARgamma systemic activities in metabolism and in the immune system. PLoS One, 2017;12:e0171474 10.1371/journal.pone.0171474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu YH, Tsai YS, Lin SC, Liao NS, Jan MS, Liang CT, et al. Quantitative PPARgamma expression affects the balance between tolerance and immunity. Sci Rep, 2016;6:26646 10.1038/srep26646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roszer T, Menendez-Gutierrez MP, Lefterova MI, Alameda D, Nunez V, Lazar MA, et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol, 2011;186:621–31. 10.4049/jimmunol.1002230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Luo W, Xu Q, Wang Q, Wu H, Hua J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep, 2017;7:44612 10.1038/srep44612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lian M, Luo W, Sui Y, Li Z, Hua J. Dietary n-3 PUFA Protects Mice from Con A Induced Liver Injury by Modulating Regulatory T Cells and PPAR-gamma Expression. PLoS One, 2015;10:e0132741 10.1371/journal.pone.0132741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhu Y, Ni Y, Liu R, Hou M, Yang B, Song J, et al. PPAR-gamma Agonist Alleviates Liver and Spleen Pathology via Inducing Treg Cells during Schistosoma japonicum Infection. J Immunol Res, 2018;2018:6398078 10.1155/2018/6398078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen K, Li J, Wang J, Xia Y, Dai W, Wang F, et al. 15-Deoxy- gamma 12,14-prostaglandin J2 Reduces Liver Impairment in a Model of ConA-Induced Acute Hepatic Inflammation by Activation of PPAR gamma and Reduction in NF- kappa B Activity. PPAR Res, 2014;2014:215631 10.1155/2014/215631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dai M, Wu L, He Z, Zhang S, Chen C, Xu X, et al. Epoxyeicosatrienoic acids regulate macrophage polarization and prevent LPS-induced cardiac dysfunction. J Cell Physiol, 2015;230:2108–19. 10.1002/jcp.24939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen T, Tibbitt CA, Feng X, Stark JM, Rohrbeck L, Rausch L, et al. PPAR-gamma promotes type 2 immune responses in allergy and nematode infection. Sci Immunol, 2017;210.1126/sciimmunol.aa15196 [DOI] [PubMed] [Google Scholar]
- 104.Micosse C, von Meyenn L, Steck O, Kipfer E, Adam C, Simillion C, et al. Human “TH9” cells are a subpopulation of PPAR-gamma(+) TH2 cells. Sci Immunol, 2019;410.1126/sciimmunol.aat5943 [DOI] [PubMed] [Google Scholar]
- 105.Nobs SP, Natali S, Pohlmeier L, Okreglicka K, Schneider C, Kurrer M, et al. PPARgamma in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J Exp Med, 2017;214:3015–35. 10.1084/jem.20162069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cai W, Yang T, Liu H, Han L, Zhang K, Hu X, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma): A master gatekeeper in CNS injury and repair. Prog Neurobiol, 2018;163–164:27–58. 10.1016/j.pneurobio.2017.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Song J, Choi SM, Kim BC. Adiponectin Regulates the Polarization and Function of Microglia via PPAR-gamma Signaling Under Amyloid beta Toxicity. Front Cell Neurosci, 2017;11:64 10.3389/fncel.2017.00064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Askari VR, Shafiee-Nick R. The protective effects of beta-caryophyllene on LPS-induced primary microglia M1/M2 imbalance: A mechanistic evaluation. Life Sci, 2019;219:40–73. 10.1016/j.lfs.2018.12.059 [DOI] [PubMed] [Google Scholar]
- 109.Hucke S, Flossdorf J, Grutzke B, Dunay IR, Frenzel K, Jungverdorben J, et al. Licensing of myeloid cells promotes central nervous system autoimmunity and is controlled by peroxisome proliferator-activated receptor gamma. Brain, 2012;135:1586–605. 10.1093/brain/aws058 [DOI] [PubMed] [Google Scholar]
- 110.Stienstra R, Mandard S, Patsouris D, Maass C, Kersten S, Muller M. Peroxisome proliferator-activated receptor alpha protects against obesity-induced hepatic inflammation. Endocrinology, 2007;148:2753–63. 10.1210/en.2007-0014 [DOI] [PubMed] [Google Scholar]
- 111.Mansouri RM, Bauge E, Staels B, Gervois P. Systemic and distal repercussions of liver-specific peroxisome proliferator-activated receptor-alpha control of the acute-phase response. Endocrinology, 2008;149:3215–23. 10.1210/en.2007-1339 [DOI] [PubMed] [Google Scholar]
- 112.Regnier M, Polizzi A, Smati S, Lukowicz C, Fougerat A, Lippi Y, et al. Hepatocyte-specific deletion of PPARalpha promotes NAFLD in the context of obesity. Sci Rep, 2020;10:6489 10.1038/s41598-020-63579-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shiri-Sverdlov R, Wouters K, van Gorp PJ, Gijbels MJ, Noel B, Buffat L, et al. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J Hepatol, 2006;44:732–41. 10.1016/j.jhep.2005.10.033 [DOI] [PubMed] [Google Scholar]
- 114.El-Haggar SM, Mostafa TM. Comparative clinical study between the effect of fenofibrate alone and its combination with pentoxifylline on biochemical parameters and liver stiffness in patients with non-alcoholic fatty liver disease. Hepatol Int, 2015;9:471–9. 10.1007/s12072-015-9633-1 [DOI] [PubMed] [Google Scholar]
- 115.Li S, Mariappan N, Megyesi J, Shank B, Kannan K, Theus S, et al. Proximal tubule PPARalpha attenuates renal fibrosis and inflammation caused by unilateral ureteral obstruction. Am J Physiol Renal Physiol, 2013;305:F618–27. 10.1152/ajprenal.00309.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Iwaki T, Bennion BG, Stenson EK, Lynn JC, Otinga C, Djukovic D, et al. PPARalpha contributes to protection against metabolic and inflammatory derangements associated with acute kidney injury in experimental sepsis. Physiol Rep, 2019;7:e14078 10.14814/phy2.14078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang J, Song J, Li Y, Shao J, Xie Z, Sun K. Down-regulation of LncRNA CRNDE aggravates kidney injury via increasing MiR-181a-5p in sepsis. Int Immunopharmacol, 2020;79:105933 10.1016/j.intimp.2019.105933 [DOI] [PubMed] [Google Scholar]
- 118.Yue TL, Bao W, Jucker BM, Gu JL, Romanic AM, Brown PJ, et al. Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation, 2003;108:2393–9. 10.1161/01.CIR.0000093187.42015.6C [DOI] [PubMed] [Google Scholar]
- 119.Takano H, Nagai T, Asakawa M, Toyozaki T, Oka T, Komuro I, et al. Peroxisome proliferator-activated receptor activators inhibit lipopolysaccharide-induced tumor necrosis factor-alpha expression in neonatal rat cardiac myocytes. Circ Res, 2000;87:596–602. 10.1161/01.res.87.7.596 [DOI] [PubMed] [Google Scholar]
- 120.Zhang J, Cheng Y, Gu J, Wang S, Zhou S, Wang Y, et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin Sci (Lond), 2016;130:625–41. 10.1042/CS20150623 [DOI] [PubMed] [Google Scholar]
- 121.Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest, 2004;114:1564–76. 10.1172/JCI18730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med, 2001;7:53–8. 10.1038/83348 [DOI] [PubMed] [Google Scholar]
- 123.Daub K, Langer H, Seizer P, Stellos K, May AE, Goyal P, et al. Platelets induce differentiation of human CD34+ progenitor cells into foam cells and endothelial cells. FASEB J, 2006;20:2559–61. 10.1096/fj.06-6265fje [DOI] [PubMed] [Google Scholar]
- 124.Shan W, Palkar PS, Murray IA, McDevitt EI, Kennett MJ, Kang BH, et al. Ligand activation of peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) attenuates carbon tetrachloride hepatotoxicity by downregulating proinflammatory gene expression. Toxicol Sci, 2008;105:418–28. 10.1093/toxsci/kfn142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kino T, Rice KC, Chrousos GP. The PPARdelta agonist GW501516 suppresses interleukin-6-mediated hepatocyte acute phase reaction via STAT3 inhibition. Eur J Clin Invest, 2007;37:425–33. 10.1111/j.1365-2362.2007.01796.x [DOI] [PubMed] [Google Scholar]
- 126.Lee HJ, Yeon JE, Ko EJ, Yoon EL, Suh SJ, Kang K, et al. Peroxisome proliferator-activated receptor-delta agonist ameliorated inflammasome activation in nonalcoholic fatty liver disease. World J Gastroenterol, 2015;21:12787–99. 10.3748/wjg.v21.i45.12787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lee MY, Choi R, Kim HM, Cho EJ, Kim BH, Choi YS, et al. Peroxisome proliferator-activated receptor delta agonist attenuates hepatic steatosis by anti-inflammatory mechanism. Exp Mol Med, 2012;44:578–85. 10.3858/emm.2012.44.10.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang X, Kume S, Tanaka Y, Isshiki K, Araki S, Chin-Kanasaki M, et al. GW501516, a PPARδelta agonist, ameliorates tubulointerstitial inflammation in proteinuric kidney disease via inhibition of TAK1-NFkappaB pathway in mice. PLoS One, 2011;6:e25271 10.1371/journal.pone.0025271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liang YJ, Chen SA, Jian JH. Peroxisome proliferator-activated receptor delta downregulates the expression of the receptor for advanced glycation end products and pro-inflammatory cytokines in the kidney of streptozotocin-induced diabetic mice. Eur J Pharm Sci, 2011;43:65–70. 10.1016/j.ejps.2011.03.011 [DOI] [PubMed] [Google Scholar]
- 130.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARδelta ameliorates obesity-induced insulin resistance. Cell Metab, 2008;7:496–507. 10.1016/j.cmet.2008.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med, 2009;15:1266–72. 10.1038/nm.2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Romero M, Toral M, Robles-Vera I, Sanchez M, Jimenez R, O’Valle F, et al. Activation of Peroxisome Proliferator Activator Receptor beta/delta Improves Endothelial Dysfunction and Protects Kidney in Murine Lupus. Hypertension, 2017;69:641–50. 10.1161/HYPERTENSIONAHA.116.08655 [DOI] [PubMed] [Google Scholar]
- 133.Fan Y, Wang Y, Tang Z, Zhang H, Qin X, Zhu Y, et al. Suppression of pro-inflammatory adhesion molecules by PPAR-delta in human vascular endothelial cells. Arterioscler Thromb Vasc Biol, 2008;28:315–21. 10.1161/ATVBAHA.107.149815 [DOI] [PubMed] [Google Scholar]
- 134.Bojic LA, Sawyez CG, Telford DE, Edwards JY, Hegele RA, Huff MW. Activation of peroxisome proliferator-activated receptor delta inhibits human macrophage foam cell formation and the inflammatory response induced by very low-density lipoprotein. Arterioscler Thromb Vasc Biol, 2012;32:2919–28. 10.1161/ATVBAHA.112.255208 [DOI] [PubMed] [Google Scholar]
- 135.Vosper H, Patel L, Graham TL, Khoudoli GA, Hill A, Macphee CH, et al. The peroxisome proliferator-activated receptor delta promotes lipid accumulation in human macrophages. J Biol Chem, 2001;276:44258–65. 10.1074/jbc.M108482200 [DOI] [PubMed] [Google Scholar]
- 136.Shi H, Mao X, Zhong Y, Liu Y, Zhao X, Yu K, et al. Lanatoside C Promotes Foam Cell Formation and Atherosclerosis. Sci Rep, 2016;6:20154 10.1038/srep20154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li G, Chen C, Laing SD, Ballard C, Biju KC, Reddick RL, et al. Hematopoietic knockdown of PPARδelta reduces atherosclerosis in LDLR−/− mice. Gene Ther, 2016;23:78–85. 10.1038/gt.2015.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gou Q, Gong X, Jin J, Shi J, Hou Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget, 2017;8:60704–9. 10.18632/oncotarget.19610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tachibana K, Yamasaki D, Ishimoto K, Doi T. The Role of PPARs in Cancer. PPAR Res, 2008;2008:102737 10.1155/2008/102737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fanale D, Amodeo V, Caruso S. The Interplay between Metabolism, PPAR Signaling Pathway, and Cancer. PPAR Res, 2017;2017:1830626 10.1155/2017/1830626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Botta M, Audano M, Sahebkar A, Sirtori CR, Mitro N, Ruscica M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int J Mol Sci, 2018;1910.3390/ijms19041197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Choi JM, Bothwell AL. The nuclear receptor PPARs as important regulators of T-cell functions and autoimmune diseases. Mol Cells, 2012;33:217–22. 10.1007/s10059-012-2297-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Staels B, Maes M, Zambon A. Fibrates and future PPARalpha agonists in the treatment of cardiovascular disease. Nat Clin Pract Cardiovasc Med, 2008;5:542–53. 10.1038/ncpcardio1278 [DOI] [PubMed] [Google Scholar]
- 144.Dubois V, Eeckhoute J, Lefebvre P, Staels B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J Clin Invest, 2017;127:1202–14. 10.1172/JCI88894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cheng HS, Tan WR, Low ZS, Marvalim C, Lee JYH, Tan NS. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int J Mol Sci, 2019;2010.3390/ijms20205055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yu S, Rao S, Reddy JK. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr Mol Med, 2003;3:561–72. 10.2174/1566524033479537 [DOI] [PubMed] [Google Scholar]
- 147.Motojima K, Passilly P, Peters JM, Gonzalez FJ, Latruffe N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor alpha and gamma activators in a tissue- and inducer-specific manner. J Biol Chem, 1998;273:16710–4. 10.1074/jbc.273.27.16710 [DOI] [PubMed] [Google Scholar]
- 148.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci U S A, 1999;96:7473–8. 10.1073/pnas.96.13.7473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Teruel T, Smith SA, Peterson J, Clapham JC. Synergistic activation of UCP-3 expression in cultured fetal rat brown adipocytes by PPARalpha and PPARgamma ligands. Biochem Biophys Res Commun, 2000;273:560–4. 10.1006/bbrc.2000.2982 [DOI] [PubMed] [Google Scholar]
- 150.Teruel T, Clapham JC, Smith SA. PPARalpha activation by Wy 14643 induces transactivation of the rat UCP-1 promoter without increasing UCP-1 mRNA levels and attenuates PPARgamma-mediated increases in UCP-1 mRNA levels induced by rosiglitazone in fetal rat brown adipocytes. Biochem Biophys Res Commun, 1999;264:311–5. 10.1006/bbrc.1999.1526 [DOI] [PubMed] [Google Scholar]
- 151.Smith U, Gogg S, Johansson A, Olausson T, Rotter V, Svalstedt B. Thiazolidinediones (PPARgamma agonists) but not PPARalpha agonists increase IRS-2 gene expression in 3T3-L1 and human adipocytes. FASEB J, 2001;15:215–20. 10.1096/fj.00-0020com [DOI] [PubMed] [Google Scholar]
- 152.Zhang Z, Li Q, Liu F, Sun Y, Zhang J. Prevention of diet-induced obesity by safflower oil: insights at the levels of PPARalpha, orexin, and ghrelin gene expression of adipocytes in mice. Acta Biochim Biophys Sin (Shanghai), 2010;42:202–8. 10.1093/abbs/gmq010 [DOI] [PubMed] [Google Scholar]
- 153.Ming GF, Li X, Yin JY, Ai YH, Xu DM, Ma XH, et al. JAZF1 regulates visfatin expression in adipocytes via PPARalpha and PPARbeta/delta signaling. Metabolism, 2014;63:1012–21. 10.1016/j.metabol.2014.05.006 [DOI] [PubMed] [Google Scholar]
- 154.Zhang YM, Li MX, Tang Z, Wang CH. Wogonin suppresses osteopontin expression in adipocytes by activating PPARalpha. Acta Pharmacol Sin, 2015;36:987–97. 10.1038/aps.2015.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhou L, Yu M, Arshad M, Wang W, Lu Y, Gong J, et al. Coordination Among Lipid Droplets, Peroxisomes, and Mitochondria Regulates Energy Expenditure Through the CIDE-ATGL-PPARalpha Pathway in Adipocytes. Diabetes, 2018;67:1935–48. 10.2337/db17-1452 [DOI] [PubMed] [Google Scholar]
- 156.Balakumar P, Rose M, Ganti SS, Krishan P, Singh M. PPAR dual agonists: are they opening Pandora’s Box? Pharmacol Res, 2007;56:91–8. 10.1016/j.phrs.2007.03.002 [DOI] [PubMed] [Google Scholar]
- 157.Wittrisch S, Kloting N, Morl K, Chakaroun R, Bluher M, Beck-Sickinger AG. NPY1R-targeted peptide-mediated delivery of a dual PPARalpha/gamma agonist to adipocytes enhances adipogenesis and prevents diabetes progression. Mol Metab, 2020;31:163–80. 10.1016/j.molmet.2019.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Balint BL, Nagy L. Selective modulators of PPAR activity as new therapeutic tools in metabolic diseases. Endocr Metab Immune Disord Drug Targets, 2006;6:33–43. 10.2174/187153006776056620 [DOI] [PubMed] [Google Scholar]
- 159.Li Y, Wang Z, Furukawa N, Escaron P, Weiszmann J, Lee G, et al. T2384, a novel antidiabetic agent with unique peroxisome proliferator-activated receptor gamma binding properties. J Biol Chem, 2008;283:9168–76. 10.1074/jbc.M800104200 [DOI] [PubMed] [Google Scholar]
- 160.Motani A, Wang Z, Weiszmann J, McGee LR, Lee G, Liu Q, et al. INT131: a selective modulator of PPAR gamma. J Mol Biol, 2009;386:1301–11. 10.1016/j.jmb.2009.01.025 [DOI] [PubMed] [Google Scholar]
- 161.Gilardi F, Giudici M, Mitro N, Maschi O, Guerrini U, Rando G, et al. LT175 is a novel PPARalpha/gamma ligand with potent insulin-sensitizing effects and reduced adipogenic properties. J Biol Chem, 2014;289:6908–20. 10.1074/jbc.M113.506394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Godoy JA, Zolezzi JM, Inestrosa NC. INT131 increases dendritic arborization and protects against Abeta toxicity by inducing mitochondrial changes in hippocampal neurons. Biochem Biophys Res Commun, 2017;490:955–62. 10.1016/j.bbrc.2017.06.146 [DOI] [PubMed] [Google Scholar]
- 163.Yang Y, Gocke AR, Lovett-Racke A, Drew PD, Racke MK. PPAR Alpha Regulation of the Immune Response and Autoimmune Encephalomyelitis. PPAR Res, 2008;2008:546753 10.1155/2008/546753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Aprahamian T, Bonegio RG, Richez C, Yasuda K, Chiang LK, Sato K, et al. The peroxisome proliferator-activated receptor gamma agonist rosiglitazone ameliorates murine lupus by induction of adiponectin. J Immunol, 2009;182:340–6. 10.4049/jimmunol.182.1.340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hichami A, Yessoufou A, Ghiringhelli F, Salvadori F, Moutairou K, Zwetyenga N, et al. Peroxisome proliferator-activated receptor alpha deficiency impairs regulatory T cell functions: Possible application in the inhibition of melanoma tumor growth in mice. Biochimie, 2016;131:1–10. 10.1016/j.biochi.2016.09.001 [DOI] [PubMed] [Google Scholar]
- 166.Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol, 2020;21:298–308. 10.1038/s41590-019-0589-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Saibil SD, St Paul M, Laister RC, Garcia-Batres CR, Israni-Winger K, Elford AR, et al. Activation of Peroxisome Proliferator-Activated Receptors alpha and delta Synergizes with Inflammatory Signals to Enhance Adoptive Cell Therapy. Cancer Res, 2019;79:445–51. 10.1158/0008-5472.CAN-17-3053 [DOI] [PubMed] [Google Scholar]
- 168.Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell, 2017;32:377–91 e9 10.1016/j.ccell.2017.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci U S A, 2017;114:E761–E70. 10.1073/pnas.1620433114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8(+) T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol Res, 2018;6:1375–87. 10.1158/2326-6066.CIR-18-0095 [DOI] [PubMed] [Google Scholar]
- 171.Goyal G, Wong K, Nirschl CJ, Souders N, Neuberg D, Anandasabapathy N, et al. PPARgamma Contributes to Immunity Induced by Cancer Cell Vaccines That Secrete GM-CSF. Cancer Immunol Res, 2018;6:723–32. 10.1158/2326-6066.CIR-17-0612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fu S, He K, Tian C, Sun H, Zhu C, Bai S, et al. Impaired lipid biosynthesis hinders anti-tumor efficacy of intratumoral iNKT cells. Nat Commun, 2020;11:438 10.1038/s41467-020-14332-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol, 2018;19:1330–40. 10.1038/s41590-018-0251-7 [DOI] [PubMed] [Google Scholar]
- 174.Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res, 2017;5:3–8. 10.1158/2326-6066.CIR-16-0297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res, 2015;3:1236–47. 10.1158/2326-6066.CIR-15-0036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Park J, Lee SE, Hur J, Hong EB, Choi JI, Yang JM, et al. M-CSF from Cancer Cells Induces Fatty Acid Synthase and PPARbeta/delta Activation in Tumor Myeloid Cells, Leading to Tumor Progression. Cell Rep, 2015;10:1614–25. 10.1016/j.celrep.2015.02.024 [DOI] [PubMed] [Google Scholar]
- 177.Zhao T, Du H, Blum JS, Yan C. Critical role of PPARgamma in myeloid-derived suppressor cell-stimulated cancer cell proliferation and metastasis. Oncotarget, 2016;7:1529–43. 10.18632/oncotarget.6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wu L, Yan C, Czader M, Foreman O, Blum JS, Kapur R, et al. Inhibition of PPARgamma in myeloid-lineage cells induces systemic inflammation, immunosuppression, and tumorigenesis. Blood, 2012;119:115–26. 10.1182/blood-2011-06-363093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol, 2020;510.1126/sciimmunol.aay1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, Inflammation, and Cancer. Annu Rev Pathol, 2016;11:421–49. 10.1146/annurev-pathol-012615-044359 [DOI] [PubMed] [Google Scholar]
- 181.Porta C, Marino A, Consonni FM, Bleve A, Mola S, Storto M, et al. Metabolic influence on the differentiation of suppressive myeloid cells in cancer. Carcinogenesis, 2018;39:1095–104. 10.1093/carcin/bgy088 [DOI] [PubMed] [Google Scholar]
- 182.Khan MA, Alam Q, Haque A, Ashafaq M, Khan MJ, Ashraf GM, et al. Current Progress on Peroxisome Proliferator-activated Receptor Gamma Agonist as an Emerging Therapeutic Approach for the Treatment of Alzheimer’s Disease: An Update. Curr Neuropharmacol, 2019;17:232–46. 10.2174/1570159X16666180828100002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sohn KA, Cruciani-Guglielmacci C, Kassis N, Clement L, Ouali F, Cauzac M, et al. S26948, a new specific peroxisome proliferator activated receptor gamma modulator improved in vivo hepatic insulin sensitivity in 48 h lipid infused rats. Eur J Pharmacol, 2009;608:104–11. 10.1016/j.ejphar.2009.02.033 [DOI] [PubMed] [Google Scholar]
- 184.Wang D, Ning W, Xie D, Guo L, DuBois RN. Peroxisome proliferator-activated receptor delta confers resistance to peroxisome proliferator-activated receptor gamma-induced apoptosis in colorectal cancer cells. Oncogene, 2012;31:1013–23. 10.1038/onc.2011.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Michaelis M, Rothweiler F, Wurglics M, Aniceto N, Dittrich M, Zettl H, et al. Substrate-specific effects of pirinixic acid derivatives on ABCB1-mediated drug transport. Oncotarget, 2016;7:11664–76. 10.18632/oncotarget.7345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Patil R, Mohanty B, Liu B, Chandrashekaran IR, Headey SJ, Williams ML, et al. A ligand-induced structural change in fatty acid-binding protein 1 is associated with potentiation of peroxisome proliferator-activated receptor alpha agonists. J Biol Chem, 2019;294:3720–34. 10.1074/jbc.RA118.006848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tong M, Deochand C, Didsbury J, de la Monte SM. T3D-959: A Multi-Faceted Disease Remedial Drug Candidate for the Treatment of Alzheimer’s Disease. J Alzheimers Dis, 2016;51:123–38. 10.3233/JAD-151013 [DOI] [PMC free article] [PubMed] [Google Scholar]