

Abstract
With the advent of effective tools to study lipids, including mass spectrometry-based lipidomics, lipids are emerging as central players in cancer biology. Lipids function as essential building blocks for membranes, serve as fuel to drive energy-demanding processes and play a key role as signaling molecules and as regulators of numerous cellular functions. Not unexpectedly, cancer cells, as well as other cell types in the tumor microenvironment, exploit various ways to acquire lipids and extensively rewire their metabolism as part of a plastic and context-dependent metabolic reprogramming that is driven by both oncogenic and environmental cues. The resulting changes in the fate and composition of lipids help cancer cells to thrive in a changing microenvironment by supporting key oncogenic functions and cancer hallmarks, including cellular energetics, promoting feedforward oncogenic signaling, resisting oxidative and other stresses, regulating intercellular communication and immune responses. Supported by the close connection between altered lipid metabolism and the pathogenic process, specific lipid profiles are emerging as unique disease biomarkers, with diagnostic, prognostic and predictive potential. Multiple pre-clinical studies illustrate the translational promise of exploiting lipid metabolism in cancer, and critically, have shown context dependent actionable vulnerabilities that can be rationally targeted, particularly in combinatorial approaches. Moreover, lipids themselves can be used as membrane disrupting agents or as key components of nanocarriers of various therapeutics. With a number of pre-clinical compounds and strategies that are approaching clinical trials, we are at the doorstep of exploiting a hitherto underappreciated hallmark of cancer and promising target in the oncologist’s strategy to combat cancer.
Keywords: Fatty acids, Fatty acid synthesis, Lipid uptake, Lipid droplets, De novo lipogenesis, Membrane lipids, Reactive oxygen species, Lipidomics
1. Reprogramming of lipid metabolism as an emerging hallmark of cancer
With more than 17 million new cases per year worldwide and almost 10 million deaths, cancer remains one of the major health issues and societal burdens. According to current concepts, cancer is driven primarily by DNA mutations in genes that promote infinite growth, survival, and metastasis. This typically involves constitutive activation of growth factor receptors and downstream signaling events, but also a rewiring of metabolic processes that provide substrates and energy for cancer cells to thrive in a changing microenvironment [1]. One of the metabolic changes that was first reported almost 100 years ago is the altered usage of glucose. In fact, since the 1920’s it has been known that, in contrast to most normal tissues, cancer cells avidly take up glucose and convert it to lactate through the glycolytic pathway irrespective of whether oxygen is present. This phenomenon, known as aerobic glycolysis or the “Warburg effect” underpins modern-day imaging of cancer by FDG-PET. Aerobic glycolysis provides cancer cells with not only energy, but also carbon for the synthesis of cellular building blocks, including nucleotides and lipids [2, 3].
Lipids are a class of water-insoluble metabolites. Estimates of the number of molecular species range from 10,000s to millions [4, 5]. Despite this remarkable heterogeneity most lipids are composed of common building blocks such as fatty acids (FAs) and cholesterol. FAs are aliphatic hydrocarbons with a polar carboxylic headgroup. They differ in the number of carbons and hence acyl chain length and the number and position of double bonds or unsaturations. They are typically classified as saturated (SFA), mono-unsaturated (MUFA) and polyunsaturated FAs (PUFA). FAs are used as building blocks of more complex lipids including phospholipids (PL), which together with cholesterol and sphingolipids are the major constituents of membranes. Phospholipids typically consist of two fatty acyl chains and a polar phosphate head group with choline, ethanolamine, serine or inositol, linked by a glycerol molecule. Sphingolipids, such as sphingomyelins and ceramides, contain a sphingoid backbone instead of glycerol. Di- and triacylglycerides (DAG and TAG) consist of FAs linked to glycerol only. Triacylglycerides, together with cholesteryl esters form lipid stores in intracellular lipid droplets (LDs) and are used as a buffer to keep the cellular lipid composition in balance or as an energy source to drive cellular processes. Many lipids, including diglycerides, ceramides, sphingosines, and oxylipins, which are oxidized derivatives of PUFA such as prostaglandins, play important roles as intra- or extracellular signaling molecules [6].
Long before the discovery of DNA mutations as key drivers of the development and progression of cancer, lipids have been implicated in the etiology of this disease. Early studies revealed increased levels of cholesterol in tumor tissues as well as alterations in phospholipids. Studies with radiolabeled substrates as early as the 1960’s have brought to light that cancer cells display a dynamic lipid metabolism and actively synthesize and take up lipids [7, 8]. Other early indications for changes in lipid metabolism come from NMR studies of tumors revealing alterations in so called ‘mobile lipids’ [9]. One of the events that sparked more interest in the association between lipids and cancer is the discovery in the mid-1990s that OA-519, an oncogenic antigen that is highly expressed in human breast cancer (BC), encodes fatty acid synthase (FASN), a key enzyme involved in lipid metabolism [10]. In the meantime, numerous studies identifying differentially expressed proteins and genes, have almost invariably identified lipid metabolism as one of the main affected processes. Together with functional approaches in which specific pathways or enzymes are targeted, a complex picture of metabolic rewiring of lipid metabolism in cancer has emerged. This rewiring is not restricted to the cancer cells themselves, but also implies changes in other cell types in the tumor microenvironment, including stromal and endothelial cells. The full complexity of this rewiring is only just now being uncovered with the advent of more effective technologies to study lipids and lipid metabolic pathways in great detail (see Section 2 of this review).
A number of characteristic changes have emerged that may support cancer cells in a changing microenvironment and that contribute to key oncogenic processes and cancer hallmarks. One key characteristic is that cancer cells are notoriously dependent on a ready supply of FAs and cholesterol (see Section 3). This requirement has been linked to the increased need for membranes to support cell growth and division and provide energy to fuel cellular processes such as metastasis. Whereas most normal cells obtain the bulk of the required lipids from the circulation, cancer cells are known to synthesize a substantial fraction of their lipids de novo. Heightened de novo lipogenesis mediated through upregulation of the requisite enzymes, is considered a near-universal hallmark of human tumors and their precursor lesions. Nevertheless, cancer cells often do maintain their ability to take up lipids and in a context-dependent manner activate and exploit these mechanisms to acquire lipids from the circulation and adjacent adipose tissue. This provides an interesting link with diet and obesity, which has been supported by numerous epidemiological studies. Diets rich in saturated fat and cholesterol, as well as obesity have been associated with increased risk of many cancers and may lead to increased cancer-related mortality [11–13]. As lipids that are synthesized de novo are different in terms of saturation compared to FAs from the circulation, the balance between de novo lipogenesis and lipid uptake likely influences the ultimate lipid composition of a tumor.
A large body of evidence now shows that cancer cells also express a repertoire of enzymes that metabolize lipids modulating their lipid content (see Section 4). These processes include FA activation, desaturation and elongation. Depending on the tumor type, a substantial fraction of the lipids is oxidized through the beta-oxidation process, sequestered and stored in LDs or secreted or released as vesicles. This extensive rewiring of lipid metabolic pathways appears to be intricately linked to the oncogenic program that is driven by growth factor receptor signaling, as well as other oncogenic events including chromosomal rearrangements, mutations and epigenetic changes, and hormonal stimulation (see Section 5). These signaling pathways in part converge on a few transcriptional regulators that play a central role in lipid metabolism, including Sterol Regulatory Element Binding Proteins (SREBPs), Liver-X-Receptors (LXR) and peroxisome proliferator activated receptors (PPARs). Together with lipid sensors, these factors also play a central role in the adaptive regulation of lipid metabolism by providing the necessary plasticity to adapt to changing microenvironments. Such rewiring of lipid metabolism has long been connected to cell proliferation, cell fate, invasiveness and energy production (see Section 6). Through remodeling of membrane lipids, reprogramming of lipid metabolism also substantially changes the composition and biophysical properties of cellular membranes and affects membrane fluidity and microdomain formation. Evidence is emerging that this membrane lipid remodeling enhances oncogenic signal propagation by generating a feed-forward cycle of oncogenic growth factor signaling [14], in addition to the classical role of lipids as intracellular second messenger generation and membrane protein targeting by lipidation. As lipid metabolism pathways interlink with substrates such as acetyl-CoA and malonyl-CoA that are also used for protein modification, the impact of altered lipid metabolism may extend well beyond classical lipid-regulated pathways and affect numerous cellular processes. Another emerging role is the protection of cancer cells from oxidative and endoplasmic reticulum (ER) stress. In fact, de novo lipogenesis and the subsequent relative decrease in poly-unsaturation protects cancer cells from lipid peroxidation caused by reactive oxygen species (ROS) that are abundantly generated in tumors, and guards cancer cells from cell death inducing processes such as ferroptosis that is propagated by lipid peroxidation [15, 16]. FA oxidation may also play a role in this protective mechanism by producing NADH for redox balancing. In addition, LDs have also been implicated in this function and may function as a sink to sequester polyunsaturated FAs or other toxic lipids [17, 18]. Cancer cells are also known to release high amounts of extracellular vesicles, that are largely composed of lipids. This release may help cancer cells to balance their lipid content and moreover appear to function as critical lipid-based transport vesicles involved in intercellular communication [19]. Lipid signaling molecules are released from cells by specific phospholipases such as secreted phospholipase A enzymes (PLAs) and processed to oxidized lipid products or eicosanoids. These lipid-based products are involved in intercellular communication and play an important modulatory role in immune escape and tumor immunology [20].
Reprogramming of lipid metabolism and subsequent changes in lipid profiling can be leveraged for biomarker development. Technological advances enabling simultaneous, quantitative analysis of hundreds of lipids species have revealed the close interconnection between altered lipid metabolism and pathogenic processes. (see Section 7). Characteristic changes in lipid profiles and differential expression of lipid metabolic enzymes in tumor tissues versus non-malignant counterparts have been described as potential cancer biomarkers. Moreover, recent studies have revealed specific lipid signatures that correlate with disease state, prognosis, or therapy response (see Section 7). Alterations in lipid profiles form the basis for surgical decision making in intelligent surgical knife applications such as the iKnife [21]. Also in liquid biopsies, such as blood, saliva, sputum and urine samples, changes in lipid profiles have been linked with disease characteristics, including poor prognosis [22–26].
Multiple enzymes involved in lipid metabolism are potential targets for therapy (see Section 8). Several compounds are currently in preclinical and clinical trials. Whilst inhibition of individual enzymes may have limited therapeutic potential due to pathway plasticity, unique context-dependent vulnerabilities have been identified that can be targeted in smart combinatorial approaches. In this context, in view of the protective role of lipid saturation in therapy-induced stress and its involvement in therapy resistance, approaches aiming at increasing poly-unsaturation of lipids in cancer cells bear particular potential [16]. Interestingly, some of these approaches may also target other cell types in the tumor including endothelial cells. Evidence is also growing that lipids play a key modulatory role in immunotherapies, opening new avenues for lipid metabolism-mediated approaches and dietary interventions for immune therapy enhancement. Lipid analogues and lipid-based carriers of therapeutics may also find use in therapeutic approaches (see Section 8).
While we are at the doorstep of witnessing the clinical exploitation of this hallmark in a variety of tumor types, the changes in lipid metabolism known so far most likely present only the tip of the iceberg (see Section 9). Recent advances in analytical approaches, including sophisticated mass spectrometry-based lipidomics applications, mass spectrometry imaging and other spatial approaches have the potential to reveal the hitherto unknown complexity and heterogeneity of lipid metabolic rewiring in cancer and to further establish altered lipid metabolism as a central hallmark in cancer.
2. Profiling of alterations in lipid metabolism in cancer: the coming of age of technology
Although it has long been appreciated that lipids play key roles in cancer, the extent and complexity of the changes in profiles and their roles in cancer and physiology are only now coming to light. Historically, studying lipids has been challenging due to the difficulty in measuring them. In fact, many discoveries of changes in lipid metabolism in cancer have been made through the analysis of data sets other than lipid profiles and the use of other methods that indirectly infer changes in lipid metabolism. A typical example is the immunohistochemical detection of the overexpression of FASN as a surrogate of de novo lipogenesis. More recently, many insights into alterations in lipid metabolism have arisen from transcriptome analysis of cancer tissues. A recent Pan-cancer multi-omics analysis of The Cancer Genome Atlas Program (TCGA) datasets recapitulates the enormous complexity of alterations of lipid metabolism pathways in tumors [27]. Affected pathways, at least at the level of the transcriptome, include those involved in FA synthesis, uptake, activation, desaturation, elongation, oxidation and degradation. In addition, the expression of genes involved in the metabolism of more complex lipids including triacylglycerides (TAG), diacylglycerides (DAG), phospholipids (PL), sphingolipids, ceramides, and cholesterol is often altered. Some changes are observed in nearly all explored tumor types, whereas others are more cancer-type specific. Genes involved in de novo lipogenesis are upregulated in most tumor types. Conversely, genes regulating beta-oxidation appear to be downregulated. Changes in genes related to cholesterol metabolism display a high degree of specificity in different malignancies. Interestingly, the expression of genes involved in arachidonic acid metabolism (phospholipases, cycloxygenases, lipoxygenases) also shows substantial variation among cancer types. In situ expression analyses of lipid-related proteins also emphasize the inter- and often also intra-tumor heterogeneity of expression, recapitulating tissue heterogeneity that is characteristic of many tumors. Overexpression of FASN for instance is found in most tumor types, but the degree of expression may vary substantially from tumor to tumor and in many cases correlates with grade and stage of the disease.
Since levels of protein expression do not always correlate with activities, direct lipid analysis is of paramount importance. However, studies of the actual changes in the levels of lipids have long been hampered by the limitation of suitable tools that would allow the quantitative analysis of these molecules. Initial studies applied classical methods such as thin layer chromatography and high-performance liquid chromatography which are limited to the analysis of major lipid classes and phospholipid headgroup classes. Depending on the composition of the mobile phase, polar (phospholipid headgroup classes) or non-polar lipids (cholesterol, triacylglycerides, cholesterol esters) can be separated. Gas chromatography has been instrumental in the analysis of FA composition of lipids, but lacks the ability to analyze intact complex lipids. Further technological advances in lipid measurement and annotation have driven a recent explosion of lipidomic studies reported in experimental model systems of cancer and clinical specimens. As for other macromolecular “omics”, mass spectrometry (MS) plays a central analytical role in lipidomics, coupled predominantly with electrospray ionization (ESI). MS is either performed by direct infusion (known as shotgun lipidomics) or is combined with chromatographic separation techniques (most commonly ultra/high performance liquid chromatography UHPLC) aimed at reducing sample complexity and removing contaminants [28, 29]. Using these techniques, many hundreds of individual lipid species can now be successfully and accurately measured in biological samples, although this still falls short of the putative thousands of lipids present. The gold standard for precise lipid identification and quantification is tandem MS with low energy collision-induced fragmentation and the use of appropriate internal standards. Compared to UHPLC/MS, ultrahigh-performance supercritical fluid chromatography mass spectrometry (UHPSFC/MS) provides advantages in separation of both non-polar and polar lipid classes [30].
Recent developments in high-mass resolution instrumentation including Fournier-transformed MS and MRMS provide unprecedented mass resolution and accuracy. All of the above advances have been markedly assisted by the efforts of the LIPID MAPS consortium to standardize lipid nomenclature, pathway classification and data reporting, as well as generating tools for statistical analysis [31, 32]. Outstanding priorities for further developing lipidomic MS workflows include: improving the accuracy and precision of lipid quantitation through optimization of lipid standards, focus on detection of low-abundance but biologically important lipids, developing more rapid and high-throughput screening platforms, incorporating stable isotope analysis to assess lipid flux, increasing the structural information provided for the acyl chain component of parent lipids, and addressing inaccurate lipid identity assignments arising from ionization-inducted artefacts [33, 34]. Further, collaborative guidelines for lipidomic data curation and accurate identification of lipid species are being developed by the Lipidomic Standards Initiative to address common issues of lipid misidentification and data interpretation that have arisen in many published lipidomic studies. Going forward, this focus on standardization will continue to improve the reproducibility of lipidomics studies on a range of platforms, which is essential for precision medicine implementation [35].
Beyond advancements in mass spectrometry instruments, the recent growth in state-of-the-art analytical techniques in the lipidomics field has allowed the detection of very rare lipids and the identification of isometric lipids. A multitude of chemical derivatization protocols have been developed that enable sensitive detection of low abundant lipids. For example, boronic derivatization has been described for the detection of monoacylglycerol [36], the Girard reagent and d5-GP where successfully used to significantly increase the sensitivity for steroid hormones [37], while for the analysis of oxysterols, derivatization to oximes, Girard hydrazones and picolinyl or nicotinyl esters has been described (reviewed in [38]). Resolution of glucosylceramide and galactosylceramides isomers has been demonstrated with a HILIC based LC method and has revealed a remarkable isomeric preference of these lipids in different tissues [39]. Several methods have been described that allow the detection of C=C location isomers such as ozone-induced dissociation (OzID) [40] and high resolution ion mobility-mass spectrometry [41]. A recently published study demonstrated a large scale analysis of C=C location by combining the C=C specific Paternò–Büchi derivatization with LC-MS/MS and revealed that ratios of C=C isomers show much less interpersonal variability than their individual abundances [42]. By using a click-chemistry based alkyne labeling of lipids Thiele and colleagues were able to reach subfemtomole levels of sensitivity in detecting fatty acid incorporation into phospholipids and neutral lipids. In addition, they demonstrated that this technique can be applied at the single cell level [43].
Lipidomic analyses have been performed in a wide range of cancer and non-malignant cell lines and well as in clinical tissue specimens. These studies confirmed the extensive nature of lipid changes in many tumor types. In our own analysis, for instance, we discovered 91 differentially expressed phospholipid species in tumor versus non-malignant tissue homogenates from non-small cell lung cancer patients [44]. A major issue of all ‘omics’ approaches that utilize homogenized tissue samples is the loss of spatial information and the change in cellular tissue composition as a confounding factor. This is particularly critical in the context of heterogeneous and multifocal solid tumors containing multiple cell types including immune cell infiltrates. These appear to have a unique and potentially targetable lipid signature (reviewed in [45]). The advent of MS imaging (MSI) provided the opportunity to visualize lipid abundance in histological sections of tumors or needle biopsies, and relate the MS data to pathological features of the tissue [46–48]. MSI acquires mass spectra from material ablated from tissue sections using either a laser, particle beam or solvent spray. The x-, y- coordinates of each data point are recorded, and the spatial and mass spectral data can be used to build up a distribution map of a molecular ion of choice [49–51] containing spatial distributions and relative abundances of the sample ions. Importantly, the resultant lipid-ion image can be correlated with histological features of the tissue section [52]. Matrix-assisted laser desorption/ionization (MALDI) was introduced in the late 1980’s as a soft ionization technique for label-free MS analysis of large biomolecules. It has subsequently been developed into an imaging technology [53–55], applied to metabolomics and lipidomics in solid tissues [56–58]. More recently, desorption electrospray ionization (DESI) MSI, which uses a charged solvent rather than a laser for ionization, has allowed direct lipid analysis in tissues under ambient conditions with minimal pre-treatment. Notwithstanding the substantial progress that has been made in the field of MSI of lipids, a number of outstanding issues remain to be addressed [59]. These include the scope of evaluation of the lipidome produced and the overall quantitative capacity of lipid MSI-maps. One of the most important limitations of MALDI-MSI is the fact that it detects fewer lipid species than ESI-LC-MS, which may reflect ion suppression by highly abundant lipid species, uncontrolled in-source decay (ISD), specific matrix requirements for successful MSI and/or the ambiguity of some lipid species with respect to mass [52].
Taken together, the analysis of lipid metabolism pathways through various methods has revealed a complex rewiring in tumor tissue that is heterogeneous and that extends beyond the tumor cell compartment. Despite this heterogeneity, a number of characteristic and recurrent changes are emerging that we highlight in the next sections of this review.
3. Acquisition of lipids by cancer cells: the Yin and Yang of de novo lipogenesis versus exogenous lipid uptake
One of the earliest and best studied aspects of lipid metabolism in cancer is the notorious dependence of cancer cells on a supply of FAs and other lipids. This trait has been linked to the increased need of cancer cells to acquire lipids for membrane synthesis and energy production required for rapid cell proliferation. Generally, there are two main sources of lipids for mammalian cells: exogenously-derived (dietary) lipids and endogenously-synthesized lipids (Figure 1).
Figure 1: Currently recognized mechanisms of lipid acquisition by cancer cells.
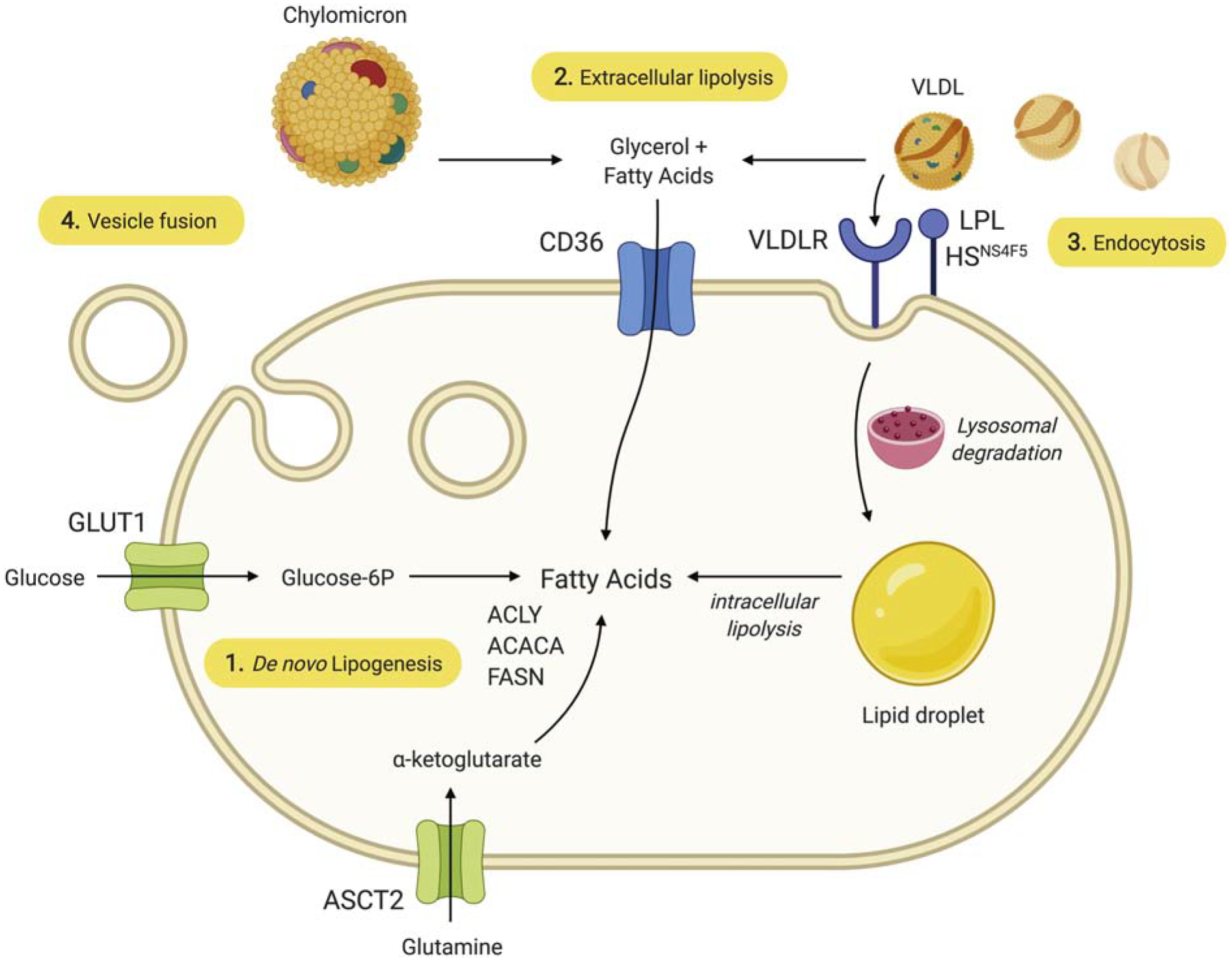
1) FA may be synthesized de novo using ATP citrate lyase (ACLY), acetyl-CoA carboxylase-α (ACACA), and fatty acid synthase (FASN). 2) FFA derived from the circulation or from local LPL-mediated extracellular lipolysis of TAG carried in lipoproteins such as VLDL enter the cell through CD36. 3) Alternatively, VLDL may dock on LPL that is bound to the cell surface heparan sulfate proteoglycan motif HSNS4F5, which optimizes positioning of the VLDL for endocytosis using the VLDL receptor (VLDLR). In this case, TAG are hydrolyzed intracellularly. Cholesterol-rich LDL particles may be similarly endocytosed using the LDL receptor without participation of LPL or HSNS4F5 (not shown). 4) Lipids and other molecules carried in exosomal vesicles may enter the cell through fusion with the plasma membrane.
In normal physiology, most lipids are derived from the diet. Dietary lipids are taken up by intestinal cells and packaged into chylomicrons (CMs), which are short-lived lipoprotein particles that enter the bloodstream and deliver FAs for oxidation in heart and skeletal muscle, and for storage in adipose tissue. The liver secretes a second type of TAG-rich lipoprotein particle, very low-density lipoproteins (VLDLs), which are much longer-lived in the bloodstream and serve to redistribute TAGs to peripheral tissues [60]. CMs and VLDLs are spherical particles that contain a core of neutral lipids, mainly TAGs. The surface of these particles contains polar lipids, including phospholipids, free cholesterol, and several exchangeable apolipoproteins [61]. Apolipoproteins can act as ligands for cell surface receptors enabling lipid uptake through receptor-mediated endocytosis mechanisms. They also function as cofactors for lipases, such as lipoprotein lipase (LPL), which is tethered to the luminal surface of capillary beds that perfuse LPL-secreting tissues and releases free fatty acids (FFA) from the complex lipids in lipoprotein particles [62]. FFA, but also more complex lipids, such as phospholipids, can be taken up by cells through both passive and active uptake mechanisms. One of the best studied mechanisms involves the FA translocase ‘Cluster of Differentiation 36’ or CD36. Other mechanisms involve FA transport proteins (FATPs)/SLC27A, and fatty acid binding proteins (FABPs). The remaining intermediate-density and low-density lipoproteins (IDL and LDL) are cholesterol-rich and are also taken up by specific receptors on the surface of cells, such as the LDL receptor (LDLR), providing cholesterol required for membrane formation or more specialized functions such as steroid or bile acid synthesis [63]. Recent evidence indicates that cells can also acquire lipids from circulating or locally produced extracellular vesicles which are taken up by endocytosis or membrane fusion (reviewed in [19]).
The second source of lipids is de novo lipogenesis, primarily from pyruvate, the end-product of glycolysis, and from glutamine [64]. The initial step in FA synthesis is the export of citrate from the mitochondrion to the cytosol. Three cytosolic enzymes then act sequentially to produce palmitic acid. ATP citrate lyase (ACLY) cleaves cytosolic citrate to yield acetyl-coenzyme A (acetyl-CoA), the basic building block for cholesterol through the mevalonate pathway and for FA and more complex lipids. Acetyl-CoA carboxylase-α (ACACA) catalyzes the carboxylation of the 2-carbon acetyl-CoA substrate to yield the 3-carbon product, malonyl-CoA, which forms the nidus for subsequent elongation by FASN, the core enzyme of FA synthesis. The direct product is palmitic acid, a fully saturated FA of 16 carbons, which can be further elongated by elongases (ELOVLs) and desaturated by stearoyl-CoA desaturases (SCDs), which introduce a double bond at the Δ9 position of palmitoyl-(16:0) or stearoyl-CoA (18:0) to generate the monounsaturated FAs [65–67]. The resulting FAs are used as substrates for synthesis of TAG, cholesterol esters, and plasma membrane phospholipids. In adult humans, de novo FA synthesis is restricted mainly to the liver, adipose tissue, and lactating mammary gland [68], and is considered to be of minor importance, owing to sufficient levels of dietary fat and the preferential use of these circulating lipids for structural synthesis [69]. This principle has been demonstrated in a variety of mouse models with tissue-specific knockout of FASN expression resulting in no detectable phenotype under non-stress conditions [70]. Studies have additionally shown that normal tissues (often compared to adjacent tumor tissue) rarely express significant amounts of FASN protein [71–75].
3.1. De novo lipogenesis in cancer cells: the lipogenic phenotype
In contrast to normal cells, most cancerous cells actively synthesize lipids such as FAs and cholesterol. Early studies using radioactivity-based methods to measure lipid synthesis showed that de novo FA synthesis accounts for 93% of the total cellular lipid content in certain cancer cell types [76]. FASN overexpression has been reported in a wide variety of human solid tumors [77]. Depending on the tumor type and on staining and scoring methods, overexpression has been observed in 15 to 95% of samples, often with inter- and intratumoral heterogeneity. Although this overexpression has been commonly linked to the increased need for membrane production required for rapid cell division, in several tumor types overexpression can be observed in precancerous lesions, as well as in invasive low-grade tumors like prostate cancer, where proliferation is limited. Indeed, synthesis of lipids is essential not only to form new cellular membrane during cell division, but also for the post-translational modification of signaling molecules, as well as for energy storage [78] (see also Section 6). FASN has been shown to facilitate oncogenesis when overexpressed in the mouse prostate [79] and has been proposed as a candidate oncogene [79–89]. Elevated levels of FASN were found in transgenic cancer models and its levels increased with the age, tumor progression and metastasis [90]. In prostate cancer, FASN is overexpressed especially in the metastatic, castration-resistant setting [89, 91–94] and is associated with poor prognosis [93, 94]. FASN inhibition with siRNA in LNCaP prostate cancer cells blocks cell proliferation and reduces pseudopodia and invadopodia formation, cell adhesion, migration and invasion [95–97].
The other lipogenic enzymes (ACLY and ACACA) are also overexpressed or activated as a near-universal metabolic feature of cancer [69, 98]. Blockade of these enzymes using RNA interference or chemical inhibitors reduces cell proliferation and evokes cell death in many cell line models and attenuates tumor growth in vivo [98–100]. Some of these studies, however, have to be interpreted with caution. In earlier studies promiscuous inhibitors such as cerulenin or TOFA were used, siRNAs were administered at high concentrations resulting in substantial off-target and nonspecific antiproliferative effects, and in many cases, cells were cultured with low levels of exogenous lipids, forcing them to depend on endogenous synthesis. Part of the growth inhibiting effects of lipogenesis inhibition may also be mediated by the accumulation of intermediates such as malonyl-CoA and subsequent protein modification as has been reported in endothelial cells [101]. More recently, it has been shown that suppression of de novo lipogenesis is the mechanism responsible for AMPK-mediated growth inhibition of prostate cancer growth, suggesting AMPK as a therapeutic target [102]. Finally, selective FASN inhibition with a potent, specific and irreversible inhibitor results in decreased growth of castration-resistant prostate cancer with downregulation of both full-length AR (AR-FL) and its ligand-independent splice variant [103].
Cancer cells also often show upregulation of enzymes involved in the synthesis of cholesterol, although this phenomenon appears to be more tumor-type specific. Blockage of cholesterol synthesis using inhibitors of HMG-CoA reductase (the rate-limiting enzyme of cholesterol synthesis) or of other downstream enzymes such as squalene synthase (farnesyl-diphosphate farnesyl transferase) reduces cell proliferation. Notably, the use of statins (inhibitors of HMG-CoA reductase) has been associated with a reduced risk of cancer development in large epidemiological studies, supporting a role for cholesterol synthesis in the development of cancer, although some controversy exists [104–107].
Cancer cells also show changes in the pathways that provide the building blocks for lipid synthesis. Besides the well-known Warburg-related increase in glucose uptake and glycolysis that is seen in many tumor types, cancer cells additionally rely on glutamine and acetate as carbon sources for lipid biosynthesis, particularly when access to glucose-derived acetyl-CoA is impaired [108–111] because pyruvate entry into the mitochondrion is curtailed as a manifestation of the Warburg Effect [112]. Under conditions of actual or pseudo-hypoxia or defective mitochondria, glutamine-derived α-ketoglutarate may be converted to citrate through reductive carboxylation and thereby contribute to de novo lipogenesis [113–117]. In cancer cells, acetyl-CoA can additionally be supplied via the ligation of acetate and CoA by acetyl-CoA synthetase (ACSS) in the cytoplasm [116, 118–122]. Interference with this enzyme can also block BC cell proliferation [120]. Recent evidence indicates that cancer cells can also use fructose as a source to produce FAs and more complex lipids [123], and the fructose transporter GLUT5 is induced by hypoxia [123, 124]. Overall, these findings support the importance of lipid synthesis for cancer cells and illustrate remarkable adaptability in the use of substrates for lipid production.
3.2. Lipid uptake by cancer cells
Despite the strong evidence for de novo lipogenesis as an important source of lipids for cancer cells, there is also solid body of evidence showing that exogenous lipid uptake remains an important route of lipid acquisition for many cancer cells. As early as the 1960’s pioneering work by Spector showed that FFA contained in the ascites fluid of Ehrlich ascites tumors could be esterified and catabolized by the tumor cells [125]. Almost a half century later, Louie et al. mapped palmitic acid incorporation into complex lipids, highlighting the ability of cancer cells to use exogenous FAs to generate lipids required for proliferation and oncogenic signaling [126]. Numerous studies over the past decade have supported the role of lipid uptake as an important route for lipid supply. One of the mechanisms that has been firmly established implies a critical role for LPL. LPL was found to be overexpressed in several tumor types including hepatocellular carcinoma, intrahepatic cholangiocarcinoma, and BC (see also Section 5). In chronic lymphocytic leukemia LPL was identified as one of the most differentially expressed genes [127] and as an independent predictor of reduced survival [128–133]. In hepatocellular carcinoma, high levels of LPL correlate with an aggressive tumor phenotype and shorter patient survival, supporting LPL expression as an independent prognostic factor [134]. Kuemmerle and colleagues showed that nearly all breast tumor tissues express LPL and that LPL-mediated uptake of TAG-rich lipoproteins accelerates cancer cell proliferation [135]. LPL is significantly upregulated in basal-like triple-negative breast cancer (TNBC) cell lines and tumors [135–137], most particularly in claudin-low TNBC [138, 139]. LPL and phospholipid transfer protein (PLTP) are upregulated in glioblastoma multiforme (GBM) compared to lower grade tumors, and are significantly associated with pathological grade as well as shortened survival of patients. Knockdown of LPL or related proteins [140] or culturing cancer cells in lipoprotein-depleted medium has been shown to result in significantly reduced cell proliferation and increased apoptosis in several cancer cell types [191]. Importantly, LPL may be produced locally or may be acquired from exogenous sources, such as human plasma or fetal bovine serum [141]. Besides the classical role of LPL in the release of FA from lipoprotein particles, recent work by Lupien and colleagues found that LPL-expressing BC cells display the enzyme on the cell surface, bound to a specific heparan sulfate proteoglycan (HSPG) motif. The failure to secrete LPL in this setting may arise from a lack of expression of heparanase, the enzyme required for secretion by non-cancer tissues. Cell surface LPL grossly enhanced binding of VLDL particles, which were then internalized by receptor-mediated endocytosis, using the VLDL receptor (VLDLR). Hydrolytic activity of LPL is not required for this process, and interestingly, BC cells that do not express the LPL gene do express the requisite HSPG motif and use it as “bait” to capture LPL secreted by other cells in the microenvironment. This was the first report of this nonenzymatic role for LPL in cancer cells, although elegant work by Menard and coworkers has shown brisk HSPG-dependent lipoprotein uptake by GBM cells that was upregulated by hypoxia [142]. This high capacity LPL-dependent mechanism for lipid acquisition appears to be of greater importance to certain BC cell lines in vitro than others, supporting previous descriptions of distinct metabolic features of different BC subtypes. In GBM there is also evidence [143] that LPL is secreted, captured by glycosylphosphatidylinositol high density lipoprotein binding protein 1 (GPIHBP1) rather than a HSPG, on the antiluminal aspect of the capillary endothelial cell, and shuttled to the inner capillary surface to create a “platform for lipolysis”. GPIHBP1 is present in glioblastoma tumor capillaries and, with LPL, facilitates the processing of TAG-rich lipoproteins [144]. Based on this and other work, it appears that cancer cells may employ LPL for both extracellular hydrolysis of TAG carried in lipoproteins as well as bulk lipoprotein endocytosis. Various other lipases, including endothelial lipase [145–149], and monoacylglycerol lipase [126, 144, 150–153], have been implicated in these mechanisms, as well as lipoprotein receptors.
A role for CD36 in FFA uptake has been well established in several cancer types. CD36 is markedly upregulated in various tumor types including BC [135], GBM, gastric cancer [154], oral squamous cell carcinoma and ovarian cancer [155]. CD36 knockdown in BC cells abolished the capacity of FFA to stimulate proliferation [156]. Inhibition of CD36 in mouse models of BC grossly reduced metastasis, diminished the ability of a high fat diet to stimulate tumor growth [157] and impaired the growth of anti-Her2 therapy-resistant tumors in a mouse model of Her2+ BC. [158]. Prostate cancer is known to be highly lipogenic, and CD36 was found to play an important role in FA uptake and its deletion attenuated cancer progression [159]. CD36-mediated FFA uptake has been linked the epithelial to mesenchymal transition in hepatocellular carcinoma [160] and with metastasizing potential in several cancer types [157]. CD36 is also induced in therapy-resistant melanoma [161]. Taken together, this recent body of work establishes the uptake of FFA via CD36 as a significant mechanism for lipid acquisition by cancer cells. Besides CD36, the uptake of FAs is facilitated through the upregulation of fatty-acid-binding proteins 3 and 7 [162].
3.3. Lipid droplets as intracellular reservoirs of lipids: the lipid-accumulating phenotype
In several studies, lipid uptake and synthesis have been linked to the formation of LDs, which mainly consist of TAG and cholesterol esters and represent a third reservoir and source of lipids for cancer cells, particularly under stress conditions such as hypoxia. Increased abundance of LDs is a feature of many aggressive cancers [163–166]. This “lipid accumulating” phenotype, may enable cells to make use of lipid stores in conditions of stress or limiting access to lipids. The role of LDs, however, extends far beyond a reservoir function as is further discussed in Section 4.8 of this review.
3.4. Exploiting fat stores, associations with obesity and high fat diets
Interestingly, several tumor types exist in anatomic proximity to adipose cells, including primary BCs in the mammary fat pad, metastatic ovarian carcinomas that “home” to omental fat, prostate tumors adjacent to the peri-prostatic fat and a variety of metastases in fatty bone marrow [167]. Recent work has uncovered the ability of cancer cells to exploit the large quantity of fat stored in tumor-adjacent adipocytes. Traditionally considered an inert tissue whose sole function was energy storage, white adipose is now appreciated as an important endocrine and metabolic organ, as well as a key player in immunity and inflammation [168]. During early tumor invasion, BC cells enter the mammary fat pad [114]. Studies have shown that interaction with adipocytes augments the growth and survival of breast and other types of tumors. BC cells secrete a variety of factors, including cytokines and lipolytic enzymes that directly affect adipocytes. Reciprocally, tumor-associated adipocytes secrete adipokines, growth factors, proteases, and FAs that enhance tumor growth and survival [169–171]. The lipolysis of TAG stored in the lipid droplets of adipose tissue and subsequent release of FFA increases tumor growth and results in the reduction of adipose tissue mass observed in the BC microenvironment [172]. Adipocyte-derived FAs promote BC cell proliferation and triglyceride accumulation [169, 173]. Several studies have shown that BC cells increase exogenous FA uptake and utilization from adipocytes by upregulating genes involved in FA uptake, including FABP4 and CD36 [173–176].
3.5. Plasticity in lipid acquisition
Many tumor types show evidence for the activation of both lipid synthesis and uptake, either simultaneously or in a plastic, cell type- and context-dependent manner. Hepatocellular carcinoma for instance has been found to be both lipogenic and lipolytic. Similarly, BC cells can show both phenotypes, although the balance between the lipid acquisition modes appear to be subtype-dependent [137]. TNBC appears to be more dependent on the uptake and storage of exogenous lipids than estrogen receptor-positive BC, which differentially rely on FA synthesis, mobilization, and oxidation. In prostate cancer both lipid synthesis and uptake are increased, and are induced by androgen signaling [177]. Interestingly, many cancer cells seem to be able to adjust their reliance on de novo lipogenesis vs. lipid uptake both in response to exogenous nutrient availability and the functional status of cellular FA synthetic machinery. Several groups have now shown that prolonged incubation of cancer cells in lipoprotein-depleted media can elicit a significant upregulation of genes involved in de novo lipogenesis, FA or lipoprotein uptake, and cholesterol synthesis [15, 178, 179]. Studies have highlighted the upregulation of FA uptake channels [116, 157, 175, 176] and lipolytic enzymes, including monoacylglycerol lipase [144, 150, 151], endothelial lipase [145, 146], and LPL [135, 179] in a variety of tumor types. Moreover, several reports have shown that the cytotoxic effects of inhibiting de novo lipogenesis in vitro may be partially averted by supplying cancer cells with exogenous lipid [72, 98, 134, 135, 142, 180–182]. Other stress conditions may also affect the balance between liquid acquisition pathways. GBM cells for instance under hypoxic and acidic stress acquire a lipid droplet-loaded phenotype characterized by increased recruitment of all major lipoproteins (HDL, LDL and VLDL). Here, stress-mediated uptake of lipoproteins was shown to be mediated through heparan sulfate proteoglycan-dependent endocytosis (involving lipoprotein receptors VLDLR and SR-B1) [183], similar to the findings in BC [179]. Metabolic plasticity has also been observed upon exposure of cancer cells to adipose cells. BC cells for instance induced a lipolytic phenotype when co-cultured with adipocytes, and adjusted their metabolism to capitalize on the influx of FFA [169]. Metabolic plasticity in the context of the cancer cell-adipocyte interaction was further highlighted by the finding that prostate cancer cells cocultured with adipocytes enhance the expression of genes related to FA uptake and transport, including CD36 [167, 184]. Importantly, adipocytes “preloaded” with triglyceride using a high-fat diet accelerated the growth of bony prostate cancer xenografts in that study. Together, these observations support a model in which cancer cells adapt their metabolism in response to the availability of lipids in the microenvironment, including from the circulation and adjacent adipocytes, to support growth [171, 179, 185].
Interestingly, alterations in the balance between lipid uptake and synthesis have also been found in other cell types in the tumor microenvironment. In endothelial cells in tumor microvessels, CD36 expression is overall downregulated. Tumor-associated endothelial cells have been shown to rely on FA-derived carbon for DNA synthesis [186]. CD36 was also shown to be low in tumor-associated stroma and to be associated with tumor aggressiveness [187]. Significant upregulation of de novo lipogenesis has also been found to be essential for the differentiation of monocytes into macrophages. Upon stimulation with macrophage colony-stimulating factor, SREBP-1c driven upregulation of FASN, ELOVL6 and SCD leads to dramatic shift towards the synthesis of saturated and mono-unsaturated FAs. Moreover, knock-down of FASN was detrimental to macrophage’s ability to develop filopodia [188].
3.6. Consequences of lipid acquisition pathways for cellular lipid composition
Critically, the choice between synthesis vs. uptake pathways dramatically affects the lipid composition of membranes with important consequences for membrane and cancer cell biology. In fact, as mentioned before, de novo FA synthesis produces saturated and mono-unsaturated FAs as humans lack delta-12 and delta-15 desaturases required to produce PUFA. As a consequence, mammals cannot generate FAs that are unsaturated in the ω−3 or ω−6 position of the acyl chain. Hence, these essential FAs (α-linoleic acid and linoleic acid) must be obtained from the diet [67]. Lipidomics analysis in fact has demonstrated that a switch between FA synthesis and uptake dramatically affects the degree of lipid unsaturation in cancer cells and that this has major consequences for cancer cell biology (see Section 6).
4. Metabolism and fate of lipids in cancer cells
Concordant with the vast diversity of lipids and the many roles they play in cell biology, FAs and other lipid building blocks are extensively metabolized in diverse pathways leading to the synthesis of more complex lipids including PLs, TAGs or oxylipins, just to name a few, which play diverse roles ranging from membrane formation, lipid storage and cell signaling. Lipids are also an important source of fuel for energy production. Intriguingly, most of these pathways and enzymes are affected in cancer cells, illustrating the extent of the rewiring of lipid metabolism in cancer and the central role lipids play in cancer biology.
4.1. Activation of lipids
For FAs to be used in metabolic pathways (both anabolic and catabolic) they need to be activated by conversion to fatty acyl-CoAs. This is a process that is catalyzed by long chain Acyl-CoA Synthetases (ACSLs). In humans there are 5 ACSL isoforms, each of which has a different cellular and subcellular distribution, regulation, substrate specificity, and enzyme kinetics. Cumulative evidence from several studies indicates that nearly all members are dysregulated in cancer, depending on the tumor type. The strongest evidence for a role in cancer development and progression is available for ACSL1 and ACSL4. ACSL1 is found to be overexpressed in multiple types of cancer, including breast, myeloma, liver and colon [189–191]. In some cases, such as colon, overexpression of ACSL1 is correlated with a poor prognosis and is thought to play an oncogenic role. In lung squamous cell carcinoma, however, the expression is downregulated, suggesting a context-dependent tumor suppressing role [192]. Similarly, ACSL4 is upregulated in many cancers, including cancer of the liver, prostate, breast and colon, but is downregulated in gastric cancer. Interestingly, recent evidence indicates that ACSL4 is essential in the induction of ferroptosis, a form of regulated cell death propagated by toxic lipid peroxides [192]. Induction of ferroptotic cell death may represent a therapeutic strategy against various types of cancer with high levels of ACSL4 (see Section 8).
4.2. Intracellular transport of lipids
FAs serve several functions in the cell. They are extensively metabolized and used as an energy source or as building blocks to generate more complex derivatives. These processes may take place in different compartments of the cell, such as the endoplasmic reticulum, Golgi apparatus, peroxisomes or mitochondria. This requires an intensive transport of lipids that is mediated by a superfamily of lipid-binding proteins, including Fatty Acid Binding Proteins (FABPs). FABPs act as lipid chaperones that bind saturated and unsaturated FAs and other hydrophobic ligands such as eicosanoids, and monoacylglycerols. FABPs form a family of 12 members that exhibit unique patterns of tissue expression. Numerous reports mention changes in the expression of FABPs in various cancer types. FABP1 (also known as liver type FABP) is overexpressed in many tumor types while FABP4 (or adipocyte FABP) has been described as a tumor suppressor that correlates with tumor stage and is often downregulated in prostate and bladder cancer [193]. In the serum, on the other hand, FABP4 levels have been reported to be higher in patients with cancer, such as BC, than in healthy controls [194]. High extracellular FABP4 is correlated with tumor size and lymph node involvement. It is reported to promote metastasis of prostate cancer and is a risk factor for BC, linked with obesity [194]. FABP-4 interacts with hormone-sensitive lipase (HSL) and modulates several signaling pathways that regulate inflammatory responses mediated by JNK/inhibitor of kappa kinase (IKK) [195]. FABP5, or epidermal FABP, is also upregulated in many cancer types, including colon, pancreatic, endometrial, and gastric cancer, cancer of the bladder, skin, prostate, head and neck, hepatocellular carcinoma, and non-small cell lung cancer [193]. FABP5 has been shown to deliver ligands to PPAR-β/δ in the nucleus (see Section 5) and to increase angiogenesis through the PPAR-γ-VEGF signal transduction [193]. Knockdown of FABP5 inhibits cell proliferation, invasion and metastasis in several preclinical cancer models. Hepatocellular carcinoma patients with overexpressed FABP5 have a worse progression and higher relapse rates [196]. FABP6 or ileal bile acid binding protein (I-BABP), like FABP4, is mainly expressed in adipocytes and macrophages and is thought to be involved in the link between bile acids and colon cancer. FABP7, or Brain FABP (BFABP) expression is increased in renal cell carcinoma and in well- and moderately differentiated prostate cancer (Grade groups 1–3) and is down-regulated in poorly differentiated tumors (Grade groups 4–5) [197]. High expression was associated with proliferation and tumor size of melanoma biopsies and was shown to promote proliferation and invasion in melanoma cells [198]. Also FA binding protein 9 (FABP9), or Testis-FABP (T-FABP) is overexpressed in prostate cancer and is thought to play an important role in progression and development of prostate cancer [199].
4.3. Desaturation of lipids
FA desaturation is a process almost ubiquitously activated in tumors. Desaturation, or introduction of one or more double bonds, into FAs is catalyzed by a family of FA desaturases, which vary based on their substrate preferences. Stearoyl-CoA desaturases (SCD), for example, introduce a double bond at the cis-delta-9 position of saturated fatty acyl-CoAs, thereby converting stearoyl-CoA or palmitoyl-CoA to oleate or palmitoleate, respectively. Two human isoforms of SCD exist, SCD1 and SCD5, representing the final enzymes involved in the de novo FA synthetic pathway. FA desaturases, on the other hand (FADS1–3), primarily generate PUFAs from the dietary essential fatty acids, linoleic acid (LA, 18:2n-6) and α-linolenic acid (ALA, 18:3n-3). SCD1 is most widely expressed in human cells and is overexpressed in many tumors [200–202]. It has been reported that rapidly proliferating cancer cells have a greater demand for MUFAs, which are utilized mainly for the synthesis of membrane PLs and TAGs, and indeed most cancer cells are characterized by a higher relative proportion of MUFAs than corresponding normal tissues [203], a notable exception being colorectal cancer which is enriched in PUFA according to recent reports [204, 205]. Knockdown or chemical inhibition of SCD1 show promising efficacy and treatment sensitization in a range of cancers [206–209]. Although the underlying mechanism remains to be fully explored, interference with SCD1 in lipogenic cancer cells has been shown to disturb the balance between saturated and monounsaturated FAs, and leads to ER stress and changes in cardiolipins. As a result, cytochrome c release drives cells into apoptosis [210]. FA desaturation requires strong reducing equivalents and oxygen, which can be particularly challenging in the hypoxic conditions experienced particularly in solid tumors. However, tumors have developed approaches to overcome these limitations and maintain membrane desaturation. For example, in glioma models, the SREBP-dependent lipogenic program (see Section 5) and SCD are more highly expressed in hypoxia, and this is in part shown to compensate for the reduced oxygen availability [211]. In renal cell carcinoma models, TAGs provide a reservoir for MUFAs and are preferentially shunted to lipid droplets; the MUFAs can be subsequently hydrolyzed and assembled into phospholipids under hypoxic conditions [212].
Although far less-studied, the delta-6 desaturase FADS2 is also overexpressed/overactive in certain cancers [213–215] and can function as a compensatory pathway, which can generate the unusual FA sapienate rather than palmitoleate from palmitate, to bypass the cells’ reliance on SCDs for MUFA production [216]. Inhibition of one or both FADS enzymes has shown preclinical efficacy in intestinal cancer [217]. Given their respective roles in generation of MUFAs and PUFAs, it is likely that the balance between these two families of desaturases has a profound impact on membrane properties and therapy response/resistance of cancer cells. Membrane unsaturation mediated by SCD/FADS or the uptake of extracellular MUFAs/PUFAs markedly enhances the fluidity of cellular membranes, however PUFAs in particular are highly oxidizable and therefore make cells more susceptible to ferroptosis, an iron-dependent form of cell death induced by lipid peroxidation. Synthesis or uptake of MUFAs provides a robust protection from ferroptosis [218], however whether this is due solely to the relative depletion of membrane PUFA or includes multiple other mechanisms remains unclear (see also Section 6).
4.4. Elongation of lipids
Several studies show that membrane lipid elongation is a common feature in cancer when compared to matched normal tissue. Lipid elongation is catalyzed by a class of enzymes called elongases (ELOngation of Very Long fatty acids; ELOVLs), comprising 7 members (ELOVL 1–7). ELOVLs are key components of the elongation system that adds two carbon units to the carboxyl end of fatty acyl chains. While their precise specificities are not fully characterized, ELOVL1, 3 and 6 elongate saturated FAs and MUFAs, ELOVL2 and 4 elongate PUFAs, ELOVL5 elongates MUFAs and PUFAs, and ELOVL7 elongates saturated FAs and PUFAs [219, 220]. As targeting ELOVLs has revealed functional effects in cancer models [221–223], it is likely that membrane lipid elongation is more than just a consequence of enhanced de novo lipogenesis in cancer. In prostate cancer, knockdown of ELOVL7 has been shown to reduce saturated FAs in membrane phospholipids but also the levels of neutral lipids such as cholesterol, which in turn reduced synthesis of the androgen that drive prostate cancer growth [223]. A study in glioma models provides further mechanistic insights, where ELOVL2 alters membrane long-chain PUFAs in order to promote epidermal growth factor receptor (EGFR) signaling through membrane domains [224]. In addition to the role of ELOVLs in membrane lipid elongation, through the production of arachidonic acid, PUFA elongation via ELOVL2 and ELOVL5 is necessary for the generation of inflammatory and signaling lipids, many of which have potent signaling effects in cancer and on immune cells. Moreover, elongation generates NAD+ and may therefore contribute to sustaining glycolysis, a process analogous to the desaturation of FAs or lactate fermentation [225].
4.5. Hydroxylation of lipids
Hydroxylation of FAs is a process whereby a hydroxyl group is introduced in the fatty acyl chain and occurs naturally in microbial, plant and mammalian cells. Hydroxylation of FA in mammalian cells is catalyzed by several enzymes, including several members of the cytochrome P450 superfamily (CYPs) and FA 2-hydroxylase (FA2H). While some CYPs show high positional selectivity, others are highly relaxed in their regioselectivity and catalyze hydroxylation of FAs merely as a side reaction [226, 227]. A range of different CYP members catalyze the hydroxylation of PUFAs, a necessary step in the synthesis of signaling lipids such as HETEs and EETs (see Section 4.9). FA2H stereospecifically produces a hydroxyl (R)-enantiomer at the second carbon (ω−2) of long chain FAs [228]. Fa2h knockout in mice resulted in long-term demyelination and the myelin was found to be lacking in 2’-hydroxy galactosylceramides [229]. One recent study found that FA2H was one of the top 4 downregulated genes in a BC stem cell population when compared to non-stem cell populations, and reported under-expression of FA2H in TNBC [230]. Overexpression of FA2H in a BC cell line reduced the cancer cells stemness, reduced the growth and promoted apoptosis, suggesting a tumor suppressive role for FA2H in BC [230].
4.6. Phospholipid synthesis and membrane remodeling
Cancer cells also frequently show alterations in the expression of enzymes involved in the synthesis and remodeling of PLs. In line with these findings, a substantial fraction of the lipids acquired by cancer cells end up in PLs, which together with cholesterol and sphingolipids are the major constituents of membranes (see Section 6.1). This has been well documented in cancer cell lines with labeled substrates [231]. PLs can be synthesized de novo but are also dynamically remodeled. PLs synthesis involves many enzymes, some of these are redundant, that may have different substrate specificities and cell type distributions, leading to the well-known diversity of lipid composition in different tissues and/or cell types (reviewed in [232]). Lipid synthesis is also compartmentalized within cells, with different steps taking place in different organelles, mainly in the ER, Golgi and nuclear membrane compartment, resulting in subcellular differences in lipid compositions. For de novo PL synthesis, FAs are first incorporated in phosphatidic acid (PA) as the main precursor of PLs. The Kennedy pathway is the main route to synthesize Phosphatidylcholine (PC), the most abundant PL headgroup class in most mammalian cells. The second most abundant PLs are phosphatidylethanolamines (PE), which can be synthesized de novo, but can also be generated from phosphatidylserines (PS) by headgroup exchange. PS is synthesized in the ER by headgroup exchange from PC and PE. Phosphatidylinositol (PI) is synthesized de novo indirectly from PA. Cardiolipins (CL) are found mainly in the mitochondria where they are synthesized locally. These are important for energy production and the regulation of cell death mechanisms. Sphingosine and ceramides are formed in the ER and transferred to the Golgi where they are used to synthesize sphingolipids or glucosyl- and galactosylceramides. Another important class of lipids are the ether lipids such as plasmalogens, which are ether or vinyl-linked at the 1-position of the glycerophospholipid and of which plasmenylethanolamines are the most abundant. These lipids are synthesized in peroxisomes. Besides de novo synthesis and headgroup exchanges, acyl chains of phospholipids are also exchanged in a highly dynamic way. This FA remodeling involves a cycle of diacylation catalyzed by phospholipases which can release acyl chains at different positions depending on the subclass of enzymes (PLA, PLC, PLD), and reacylation or transacylation catalyzed by a class of acyltransferases such as lysophosphatidylcholine acyl transferases (LPCAT).
Intriguingly, many of the enzymes involved in PL synthesis and remodeling are overexpressed in cancer. Lipin-1, for instance, a phosphatidic acid phosphatase (PAP) controlling the rate-limiting step in PL synthesis and co-regulator of transcription factors such as PPARs and SREBPs (see Section 5), is up-regulated in a subset of diverse cancer types including high grade prostate cancer, colon cancer, lung cancer and TNBC [233–235]. High level Lipin-1 expression is associated with poor prognosis and inflammation and downregulation of the enzyme induces ER stress and apoptosis, and attenuates tumor growth in vivo in orthotopic xenograft mouse models [233–235].
Choline kinase alpha (ChoKα), the first committed enzyme in in the Kennedy pathway for PC and PE synthesis, is overexpressed in a variety of tumor types and activated by a wide range of oncogenic events. Activation and overexpression of ChoKα has been linked to the increased cellular need for PC, and is a potential biomarker. Knockdown or chemical inhibition of ChoKα causes cell death and attenuates tumor growth in vivo [236, 237].
Another class of PL metabolizing enzymes that is implicated in several aspects of tumor biology are the phospholipases. Members of all three subfamilies have been shown to be altered in many cancers. Some isoforms are overexpressed, others are decreased or mutated. Part of their role is related to lipid remodeling but also to the generation of lipids involved in signaling such as arachidonic acid (see Section 6) (reviewed in [238]). The other end product, lysophospholipids (LysoPLs), are elevated in many tumors and have been linked with tumor promotion [20]. LysoPLs are also the substrate for monoacylglycerol lipase (MAGL), which is additionally overexpressed in several tumor types and regenerates FAs (see also Section 3). A higher amount of secreted phospholipase A2 is associated with ovarian cancer [239], and phospholipase D mediated release of phosphatidic acid is shown to mediate cell invasiveness in BC models [240]. Intriguingly, a recent report revealed that PLA2G2A is associated with prostate cancer progression and confers ferroptosis resistance to prostate cancer cells by depleting membrane PUFA [241].
Another emerging class of enzymes that appear to be affected in many tumors are the lysophosphatylcholine acyl transferases (LPCATs) that play a central role in the reacylation of lysophospholipids. There are 4 members of this enzyme family, all of which have been implicated in cancer. LPCAT1 has been shown to be overexpressed and to function as a potential prognostic biomarker for many cancer types. LPCAT2 is found in aggressive prostate cancer, LPCAT4 is linked to intestinal stem cell proliferation and tumorigenesis and LPCAT4 is associated with high levels of PC(16:0/16:1) in colorectal cancer [237]. In hepatocellular carcinoma (HCC) cell line experiments, LPCAT1 overexpression enriched PCs and promoted cell proliferation, migration, and invasion, while LPCAT1 knockdown did the opposite (see also Section 5). Thus, LPCAT1 may be a potential target molecule to inhibit HCC progression because it modulates PL composition to create favorable conditions in HCC cells [242].
An intriguing finding is the loss of membrane lipid asymmetry in many cancers. In healthy cells, different headgroup classes of PLs show a differential distribution over the inner and outer membrane leaflet. PS for instance is mainly found in the inner membrane leaflet, where it plays a crucial role in signaling. Under certain conditions, such as induction of apoptosis, this membrane asymmetry is disturbed and PS is exposed on the cell surface where it attracts macrophages for clearance of dead cells. Intriguingly, in viable cancer cells a substantial fraction of PS is found in the outer plasma membrane leaflet and is thought to play a role in immune modulation. These changes are linked with the loss of expression of specific phospholipid scramblases (PLSCRs), enzymes that bidirectionally flip lipids across membranes. Elevated PLSCR1 expression has been found in liver and colorectal cancer for instance [243].
4.7. Lipid oxidation
Cancer cells frequently show changes in enzymes involved in fatty acid oxidation (FAO). The rate-limiting step in this process is the translocation of FA-CoAs across the outer mitochondrial membrane through conversion to FA-carnitine by carnitine palmitoyl transferase 1 (CPT-1). There are 3 paralogs of CPT-1 in mammals; CPT-1A (expressed mainly in liver, prostate), CPT-1B (skeletal muscle, breast) and CPT-1C (brain). In the FAO process, FAs are degraded to acetyl-CoAs that are used in the Krebs cycle for anabolic processes and the production of reducing equivalents to support redox homeostasis. FAO is transcriptionally regulated by the PPAR family of transcription factors (see Section 5), which activate expression of CPT1 and other FAO enzymes in response to glucose deficiency, and post-translationally via allosteric inhibition of CPT1 by malonyl-CoA. The latter is mediated by activation of the nutrient sensor AMPK, which in turn phosphorylates and inhibits ACACA, the enzyme that catalyzes production of malonyl-CoA. It is increasingly evident that, despite the widespread focus on so-called Warburg cancers, FAO is an important bioenergetic pathway in many cancers and promotes proliferation, metastasis, stemness and treatment resistance [244, 245] (see also Section 6). In hypoxic conditions or in response to treatment, tumor cells appear to favor FAO to rapidly generate ATP and NADPH and promote survival. Consistent with this concept, clinical BC tissues exhibit enhanced expression of the FAO enzyme CPT1B upon disease recurrence and in response to chemotherapy [149], while CPT1A is higher in chemoresistant pancreatic tumors [246] and associated with poorer outcomes in gastric cancer [247] and acute myeloid leukemia [248]. Moreover, FAO has been identified as a key upregulated pathway and therapeutic target in MYC-overexpressing TNBC [249], thereby linking FA metabolism to oncogenic signaling. It is important to note that oxidation of lipids also takes place in peroxisomes, involving both β-oxidation of very long chain FAs and α-oxidation of branched chain FAs. These processes, and their requisite enzymes, have not been thoroughly investigated in cancer cells and may offer novel opportunities for therapeutic intervention beyond CPT1 in certain cancers that rely on peroxisomal FAO pathways.
4.8. Lipid storage
Depending on the context, a substantial fraction of lipids can be found in lipid droplets (LDs), which are abundant in several cancer types. These organelles are made of deposits of TAGs and cholesterol esters, and are surrounded by a monolayer of PLs. In times of excess lipids, LDs are believed to originate from microdomains in the ER membrane that recruit TAGs. As the TAGs reach the maximum concentration the ER membrane can hold, they start to accumulate between the two membrane leaflets and ultimately separate from the ER through a budding process [250]. LDs are found in many tumor types and in part account for the ‘mobile lipid’ signals in NMR. Clear cell renal cell carcinoma (ccRCC) is one type of cancer that is characterized by a significant accumulation of TAGs in LDs which in addition to glycogen deposits results in its typical histological phenotype. This accumulation has remained poorly understood but one recent study described a role in resisting hypoxic stress. The authors discovered that under hypoxia ccRCC cells release oleate from their TAG stores in an effort to counter the toxic accumulation of saturated lipids which cannot be desaturated by SCD without sufficient oxygen [212]. Accumulation of lipid droplets is a characteristic phenotype of chemoresistant cancer cell lines [251–255], and co-treatment with triacsin C, a non-specific inhibitor of LD biogenesis, has been shown to chemo-sensitize cancer cells [255]. While LD accumulation may in part reflect treatment-induced autophagy, LDs may also serve as an extra source of lipids for FAO under nutrient stress conditions, or as a “sink” to sequester hydrophobic drugs. The terminal step in TAG biosynthesis is catalyzed by the DGAT enzymes, which transfer an acyl chain from fatty acyl CoA to DAG. DGAT1 and DGAT2 both catalyze this reaction but are unrelated genes that evolutionary converged. DGAT1 is overexpressed in prostate cancer compared to normal epithelium and a recent study demonstrated that inhibition of DGAT1 reduced tumor growth in an in vivo xenograft model [256]. Conversely, another study showed that DGAT1 overexpression in lung SV40-transformed fibroblasts reduced proliferation and anchorage-independent growth by reducing DAG and PL levels, suggesting that depending on the metabolic context DGAT could function as a negative regulator of tumor progression [257].
4.9. Signaling lipids
A diverse range of lipids and classes of lipids function as intra- and extracellular messengers to direct cell behavior under normal physiological conditions. When dysregulated in cancer, these signaling lipids can become potent mediators of malignant behavior. Sphingolipids are a class of lipids that contain a sphingoid backbone and important sphingolipid signal mediators are sphingosine, spingosine-1-phosphate (S1P), ceramide and ceramide-1-phosphate (C1P). S1P is produced from sphingosine by sphingosine kinases (SK1 and SK2) and several transcriptomic studies have linked high SK1 and SK2 expression to poor prognosis in BC [258, 259]. S1P can be secreted from cells as well as bind to intracellular targets such as HDAC1/2 and its functional roles in cancer have been described. These include promoting vascularization of tumors [260], progression promoting inflammation through STAT3 [261] and promoting the Warburg effect [262]. Ceramide is converted by ceramide kinase (CERK) into C1P. A BC study has shown that CERK is required for the development and survival of recurrent disease following Adriamycin treatment and that elevated CERK expression is linked with recurrent disease in patients [263]. Classically, ceramide is believed to induce senescence and growth inhibition in cancer, and while a recent study linked high ceramide levels to reduced aggressiveness of BC, other recent studies have suggested the effects of ceramide may be context dependent and rely on the presence of downstream effectors [264]. Both ceramide and C1P are activators of phospholipase A2 (PLA2), an enzyme that functions to release arachidonic acid (AA) for subsequent conversion to prostaglandins (vide infra).
Phosphoinositides are a class of lipid molecules that comprise phosphatidylinositol mono-, bis- and trisphosphate and are central mediators of the PI3K/Akt/mTORC1 signaling axis. Activation of PI3K results in the rapid conversion of PI(4,5)P2 into PI(3,4,5)P3 which leads to the activation of Akt. Conversely, the tumor suppressor PTEN dephosphorylates PI(3,4,5)P3 back to PI(4,5)P2 [265]. Recently there has been growing appreciation that PI(4,5)P2 does not only function as a substrate for the synthesis of the growth promoting PI(3,4,5)P3, but that PI(4,5)P2 itself has an important role as a lipid messenger in cancer [265]. Due to specific protein interactions, PI(4,5)P2 has a major role in recruiting cytosolic proteins, facilitating processes like fusion and budding of membranes and the formation of signaling platforms. Local reductions in PI(4,5)P2 are believed to be linked to the regulation of directional movement of cancer cells [266].
Eicosanoids are lipid signaling molecules that are derived from 20 carbon PUFAs, mainly AA and eicosapentaenoic acid (EPA). They function as both autocrine and paracrine signaling molecules to promote or inhibit inflammation or other immune responses. There exist many subfamilies of which prostaglandins, leukotrienes, lipoxins and resolvins are the most well studied. Prostaglandin E2 (PGE2) is the most abundant prostaglandin and is a strong mediator of inflammation through binding with the G-protein-coupled receptors EP1 to 4 [267]. Increased levels of PGE2 have been described in several cancers and are associated with a poor prognosis [268]. The prostaglandin PGD2 has been less extensively investigated in cancer, but most studies are reporting antitumor activity. A recent study in gastric cancer reported that PGD2 inhibited tumor growth and suppressed the ability to form metastases [269], while another study in prostate cancer concluded that PGD2 secreted by the stroma can suppress the growth of the tumor cells [270]. Leukotrienes are a type of eicosanoids produced mainly by leukocytes that function in a paracrine manner. Leukotriene LTB4 is one of the most well studied in cancers and is believed to induce a chronic tumor promoting inflammatory state. In medulloblastoma, blockage of leukotriene synthesis in 5-lipoxygenase–deficient mice dramatically reduced tumor growth in vivo [271]. Lipoxins are a type of pro-resolving, anti-inflammatory prostaglandins. Colorectal cancer was found to be associated with overall low levels of lipoxin A4 and in an in vivo xenograft model lipoxin A4 was able to reduce proliferation of cancer cells [272]. Furthermore in a hepatocarcinoma model lipoxin A4 reduced the production of vascular endothelial growth factor and reduced tumor growth in vivo by impairing tumor-related angiogenesis [273]. Resolvins are another type of eicosanoids with pro-resolving (restoration of tissue homeostasis) and anti-inflammatory action. The release of cytokines from tumor cell debris following therapy is known to stimulate tumor growth. A recent study however found that resolvins can counter the effect of cytokines by attracting debris clearing macrophages [274].
Glycerolipid derived mediators include DAG, LysoPA and LysoPC. DAG is formed during the phospholipase C catalyzed hydrolysis of PI(4,5)P2 and functions as a second messenger that triggers the activation of protein kinase C. In cancer cells, one study showed that resistance to FASN inhibition may be driven by maintaining PKC signaling through sustained DAG levels, and could be overcome by a combinatorial treatment with FASN and PKC inhibitors [275]. LysoPA is generated by the removal of the choline group from LysoPC by autotaxin (ATX), a secreted enzyme. LysoPA exhibits growth factor-like effects through a family of G-protein coupled LysoPA receptors (LPAR) that are highly expressed in several cancers, including lung [276], pancreatic [277] and ovarian cancer [278]. A recent study proposed ATX/LPA signaling as an interesting therapeutic target in liver cancer, as it is involved in both chronic liver inflammation diseases that can progress to cancer and the development of liver cancer itself [279].
4.10. Lipids as membrane anchors and modulators of protein functioning
Lipids also function as anchors to target proteins to membranes in specific locations in cells. Classical examples include palmitoylation, prenylation and farnesylation of proteins, and several key signaling oncoproteins such as Ras, Rho, Wnt and Hedgehog depend on these posttranslational modifications for their functioning [280–282]. In several reports, changes in lipid metabolism in cancer cells are linked to alterations in lipidation and anchoring of proteins. Interestingly, changes in the expression or activity of enzymes involved in protein lipidation have been found in many cancer types. This is evident in the case of farnesyltransferases, which are often overexpressed in tumor cells and are being explored as targets for therapy [283](see Section 8). Recently, also palmitoylation of proteins has become an intense area of research in cancer biology. Several hundreds of proteins that are linked to the oncogenic process are palmitoylated on a cysteine. Some proteins are autopalmitoylated and it has been suggested that overexpression of FASN may be a driver in this process [284–288]. Most palmitoylation processes are however catalyzed by zinc finger DHHC-type palmitoyl S-acyltransferases (PATs), comprising a family of 23 proteins. Acylprotein thioesterases (APT) remove palmitoyl groups from proteins. As nicely summarized by Ko and Dixon [282], some of these proteins are overexpressed in certain cancers and may function as oncogenes, whereas others are downregulated and are considered tumor suppressors. Other functions of lipids in the modulation of protein functioning include the modification of Hedgehog by cholesterol [289] and the role of PE as an anchor for LC3 to autophagosomal membranes.
5. Key drivers of alterations in lipid metabolism
In view of the complexity of lipid metabolism and its central role in many biological processes, it is not surprising that this pathway is under tight regulatory control. Aside from a small number of central transcription factors that coordinately regulate enzymes involved in lipid metabolism, this regulation is fine-tuned at several other levels and involves posttranslational and other mechanisms. In cancer, a dramatic rewiring of lipid metabolism takes place that is in part driven by oncogenes and tumor suppressors. Lipid metabolism is also highly adaptive and helps cancer cells to cope in a challenging and changing microenvironment (Figure 2).
Figure 2: Key drivers of alterations in lipid metabolism.
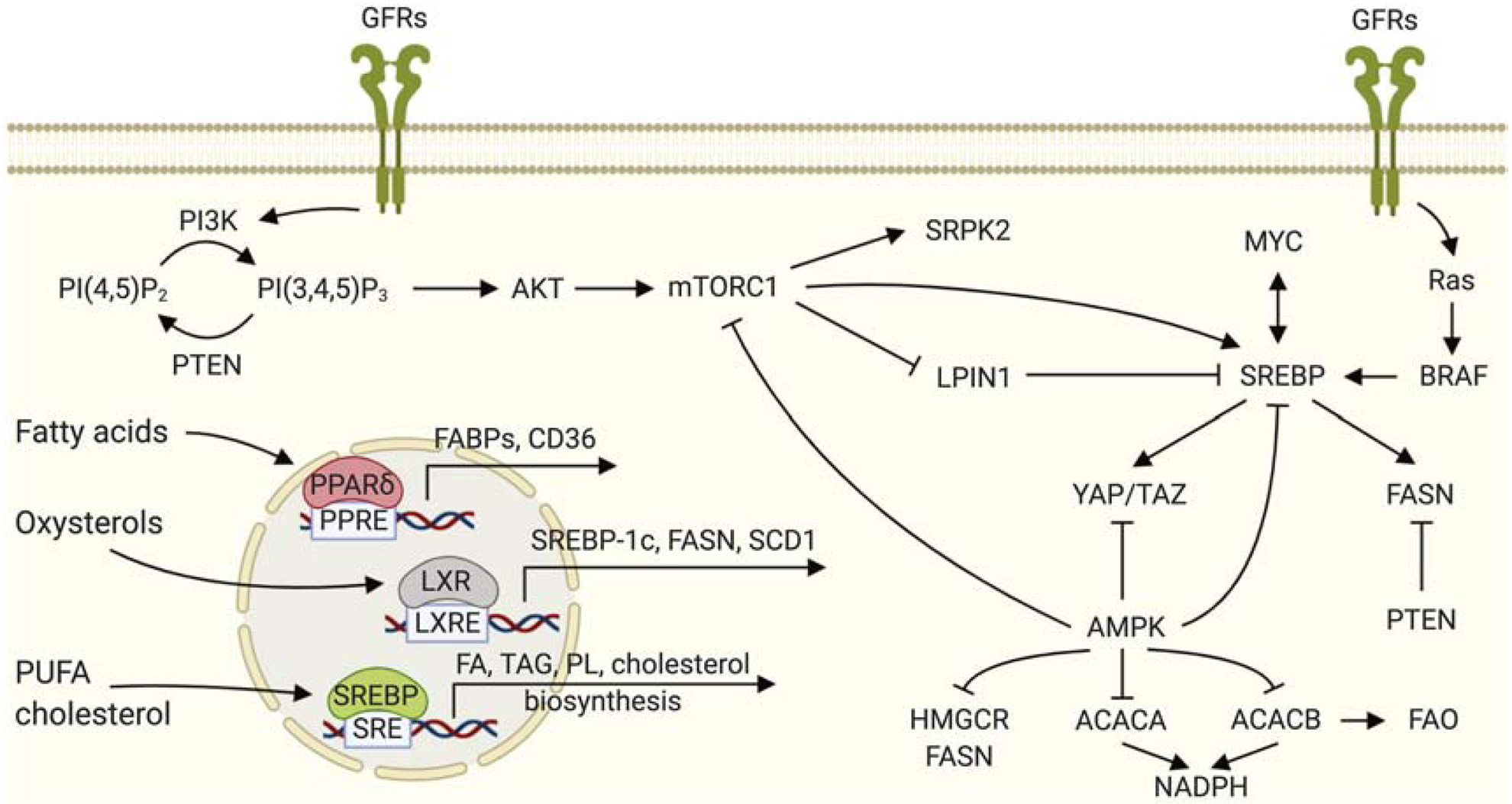
Cellular FA and cholesterol acquisition and metabolism are transcriptionally controlled and tightly regulated by Liver X Receptors (LXR) and Peroxisome Proliferator-Activated Receptors (PPARs) and by the basic-helix-loop-helix-leucine zipper (bHLH-LZ) transcription factors (TF) Sterol Regulatory Element Binding Proteins (SREBP). Multiple growth factor receptors (GFRs) regulate lipid metabolism through the PI3K/AKT/mTORC1 and MAPK signaling axes. SREBP and its downstream targets such as FASN and ACACA are regulated by a multitude of signaling molecules, including AMPK, MYC, PTEN and BRAF.
5.1. Critical transcription factors in lipid metabolism: SREBPs, LXR, PPARs
Cellular FA and cholesterol acquisition and metabolism are transcriptionally controlled and tightly regulated by two main members of the superfamily of nuclear receptors [290], Liver X Receptors (LXRs) and PPARs and by the basic-helix-loop-helix-leucine zipper (bHLH-LZ) transcription factors (TF) SREBPs [291].
LXRs are TFs of the nuclear receptor superfamily which upon activation form heterodimers with retinoid X receptor (RXR) and bind to LXR response element (LXRE) on the promoter region of target genes. The two isoforms, LXRα and LXRβ, are key transcriptional regulators of lipid and carbohydrate metabolism. LXRs act as sterol sensors protecting the cells from cholesterol overload. They ensure an adequate intracellular sterol content via activation or repression of their direct target genes (respectively ABCA1 and LDLR) [292]. The lipogenic action of LXRs is mediated by direct upregulation of SREBP-1c, FASN and SCD1 [293–296]. LXRs are activated by the oxysterols 22-hydroxycholesterol, 24-hydroxycholesterol, 25-hydroxycholesterol, 25,26-hydroxycholesterol, and 24,25-epoxycholesterol [292]. Apart from LXRs, other nuclear receptors have also been found to be regulated by specific oxysterols. For example, 27-hydroxycholesterol was shown to act as an endogenous selective estrogen receptor modulator (SERM) [297, 298]. LXR has been suggested to be involved in BC and prostate cancer progression [299, 300].
PPARs are part of the nuclear receptor family and play a major regulatory role in energy homeostasis and metabolism. Three nuclear receptor isoforms, PPARγ, PPARα, and PPARβ/δ are encoded by different genes and have different functions. Activation of PPAR-α reduces TAG levels and is involved in regulation of energy homeostasis. PPAR-γ causes insulin sensitization and enhances glucose utilization, whereas activation of PPAR-β/δ increases FA synthesis.
SREBPs are the master regulators of lipid biosynthesis [291]. These TFs regulate lipid homeostasis by controlling the expression of enzymes involved in endogenous cholesterol, FA, TAG and PL biosynthesis [291]. From yeasts to humans SREBPs are highly conserved, therefore the expression of lipogenic genes is regulated according to species-specific requirements [301]. As such, SREBP is regulated by palmitate in Drosophila [302], by hypoxia in fission yeast [303] and by sterols in mammals [304]. Different isoforms play different roles in the physiological modulation of lipid synthesis [291]. SREBP1a strongly activates global lipid synthesis and growth, whereas SREBP1c primarily controls energy storage through nutritional regulation of FA and TAG. SREBP2 mediates cholesterol metabolism-related gene expression [305, 306]. However, when overexpressed, the isoforms exhibit functional overlap.
Key events in the activation and regulation of SREBPs involve several steps of trafficking between cellular compartments such as cleavage, recycling and degradation. SREBPs are synthesized as inactive precursor proteins that normally reside in the ER in complex with SCAP (SREBP cleavage-activating protein) and INSIG (insulin-induced gene) [307–312]. In response to sterol depletion, SREBP-SCAP migrate to the Golgi and, through the sequential action of the Golgi-localized Site-1 and Site-2 Proteases, the N-terminal domain is proteolytically released [313]. The cleaved SREBP then translocates into the nucleus where it binds to the promoter of several genes involved in the synthesis, uptake and metabolism of cholesterol and FAs, thus restoring sterol homeostasis in a feedback regulation loop and regulating cellular lipid homeostasis [314]. SREBPs are also affected by FAs and are self-regulated by a transcriptional positive feedback [315–317]. In normal physiology, the SREBP pathways are mostly active in organs involved in the handling and control of lipids, such as the liver and are under tight control by hormones such as insulin. To date, a variety of TFs activated in response to extracellular stimuli has been reported to modulate SREBP transcriptional activity. For instance, LXR activated by oxysterols regulates SREBP activity by direct binding [294, 318, 319]. SREBPs further interact with various transcriptional co-activators such as CBP and p300, which acetylate and stabilize SREBPs by preventing ubiquitination [320, 321]. These modifications regulate the stability and/or transcriptional activity of the active TFs. Transcriptional coactivators and cooperating TFs provide yet another level of regulatory control of SREBP activity [301]. In human hepatocellular carcinoma cells, SREBP1 cooperates with its associated factors, nuclear factor Y (NFY) and simian-virus-40-protein-1 (SP1), to regulate the expression of a subset of target genes through direct interaction [315, 322].
More than 20 years ago SREBPs were shown to be activated in cancer and to contribute to lipid synthesis and uptake [323]. SREBPs are frequently activated through other mechanisms such as constitutive growth factor signaling that functions through the same signal transduction mechanism as insulin [324].
5.2. Growth factor signaling as key driver of lipid metabolism reprogramming
Uncontrolled proliferation is central to tumor development and is regulated by persistent growth factor (GF) signaling. After binding to their receptors generally residing on plasma membranes, GFs activate a signaling cascade triggering a variety of changes in cellular processes allowing growth, division and increase of biomass. Mutations or amplifications of GF genes lead to the constitutive activation of their pathways, further affected by the lipid composition of the membranes in which growth factor receptors (GFR) reside [325].
EGFR is one of the most commonly activated growth factor receptors in cancers. In prostate cancer cells, the epithelial growth factor activates de novo FA synthesis and increases the cellular pool of saturated FAs [80]. Importantly, the upregulation of FASN expression is mediated by EGF-induced activation of SREBP pathway [324]. In non-small cell lung cancer cells, mutated EGFR mediates tyrosine kinase inhibitor resistance through regulation of FASN [287]. Indeed, FASN-dependent palmitoylation of EGFR is needed for EGFR function and kinase activation [326]. EGFR signaling contributes to increased FASN expression in pancreatic ductal adenocarcinoma as well [327]. It has also been shown recently that genetic constitutive activation of EGFR activates LPCAT1, which regulates PL saturation and oncogenic growth factor signaling [14]. LPCAT1 is a key enzyme involved in membrane lipid remodeling that is frequently amplified in cancer and associated with poor patient survival. Using orthotopic glioma cell line xenograft models, as well as lung and renal cancer models, the authors show that knockdown of LPCAT1 suppresses tumor growth and prolongs the survival of tumor-bearing mice [14].
ERBB2 (Erb-B2 Receptor Tyrosine Kinase 2) is a member of the EGFR family of receptor tyrosine kinases. Commonly referred to as HER2, it enhances kinase-mediated activation of downstream signaling pathways, such as MAPK and PI3K–AKT. HER2 is amplified and/or overexpressed in 20–30% of invasive breast carcinomas characterizing a more aggressive disease. Sustained upregulation of de novo lipogenesis has been found to contribute to HER2-positive tumor aggressiveness [328]. Overexpression of HER2 in non-transformed epithelial cells induces a lipogenic phenotype similar to that of cancer cells and is dependent on FASN activation [328, 329]. Connections between FASN and HER2 overexpression have been described at a transcriptional level [330] with cellular localization of HER2 changing in response to FASN level and activity. Silencing FASN impinges on the appropriate localization and the membrane accumulation of HER2 altering also the cell morphology [330]. As a consequence, the correct dimerization of HER2 with EGFR is also impaired, blocking a mechanism driving targeted therapy resistance [329, 331]. Overexpression of HER2 has also been found in castration-resistant prostate cancer human samples where FASN is overexpressed. The study showed that progression of prostate cancer toward androgen independence is accompanied by an increase in Her2 expression [332].
Insulin-like growth factor 1 (IGF1) binds to its receptor IFGF-1R initiating a cascade of downstream signaling events leading to activation of the PI3K-AKT/PKB and the Ras-MAPK pathways with consequent increased proliferation and enhanced survival of both normal and cancer cells [333]. The mitogenic activity of the IGF-1R is also mediated by downregulation of cell cycle suppressors and PTEN [334, 335]. Reciprocal rescuing/activation occurs between IGF-1R, EGFR and HER2 thus conferring resistance to single-agent targeted therapy. In BC, the IGF-1R may contribute to tamoxifen resistance via either an IGF-mediated activation of AKT and subsequent estrogen-independent activation of ERα [336] or via a direct interaction between ERα and IGF-1R [337].
The phosphatidylinositol 3-OH-kinase/ protein kinase B (PI3K/AKT)-mTORC1 pathway is a well-known pro-survival axis constitutively activated in cancer with prominent roles in neoplastic transformation, growth, drug resistance and metastasis [338–340]. The activity of Akt via mammalian target of rapamycin complex 1 (mTORC1) is required for the nuclear accumulation of mature SREBP1, directly regulating its expression [341, 342]. SREBP1 function is also essential for Akt/mTORC1-dependent regulation of cell size [203, 341, 343]. In melanoma, the PI3K-AKT-mTORC1-SREBP axis can control cell growth independently of BRAF mutation [340, 344] while in prostate cancer the PI3K-PTEN-AKT pathway was linked to FASN overexpression [92]. The proto-oncogene B-RAF encodes a protein of the RAF family of serine/threonine protein kinases that plays a role in cell division and differentiation by regulating the MAP kinase/ERK signaling pathway. A recent study from our group showed that therapy resistance to vemurafenib in BRAF-mutant melanoma activates sustained SREBP1-driven de novo lipogenesis and that inhibition of SREBP-1 sensitizes melanoma to targeted therapy [16].
In breast epithelial cells, the oncogenic PI3K or K-Ras signaling converging on the activation of mTORC1 is sufficient to induce SREBP-driven de novo lipogenesis [345]. In addition, oncogenic stimulation of mTORC1 is associated with increased SREBP activity promoting aberrant growth and proliferation in primary human BC samples [345]. The mTORC1-S6K1 complex phosphorylates SRPK2 (SRSF Protein Kinase 2) to induce its nuclear translocation [346]. SRPK2, in turn, promotes splicing of lipogenesis-related transcripts. SRPK2 inhibition results in instability of mRNAs arising from lipogenesis-related genes, thus suppressing lipid metabolism and cancer cell growth. Thus, SRPK2 is a potential therapeutic target for mTORC1-driven tumors [346].
Overexpression of FASN and altered metabolism in prostate cancer cells is associated with the inactivation of the tumor suppressor PTEN [91, 347, 348]; accordingly, PTEN expression is inversely correlated with FASN expression in prostate cancer [349], while inhibition of PTEN leads to the overexpression of FASN in vitro [92]. PTEN is a lipid phosphatase and the second most commonly mutated tumor suppressor gene in human cancers. Deletions and mutations in PTEN, are among the most frequent alterations found in prostate cancer, particularly in the metastatic setting [339, 350, 351] suggesting a coordinated feedback between lipogenesis and oncogenic signals to promote tumor growth and progression [88, 350, 352–357].
A concomitant loss of Promyelocytic Leukemia (PML) in PTEN-null prostate cancer is found in 20% of metastatic androgen independent or castration-resistant prostate cancer (mCRPC). PML/PTEN-null promotes metastatic progression through reactivation of MAPK (Mitogen-Activated Protein Kinase) signaling and subsequent hyperactivation of an aberrant SREBP pro-metastatic lipogenic program [358]. Inhibition of SREBP using Fatostatin can block lipid synthesis and metastatic potential [358]. PTEN loss due to mutations or deletions results in PIP3 accumulation and activation of the PI3K/AKT pathway [359, 360]. The PI3K/Akt signaling axis increases the expression of enzymes required for FA synthesis including ACLY, the enzyme catalyzing the production of acetyl-CoA from cytoplasmic citrate, FASN and LDLR [361, 362]. This pathway is responsible for the increase in cell survival, metastasis and castration-resistant growth in prostate cancer. Studies on bone metastasis revealed elevated levels of LDLR that are responsible for LDL uptake and for maintenance of intracellular cholesterol homeostasis [81]. Prostate cancer cells esterify cholesterol in lipid droplets to avoid cellular toxicity due to high intracellular cholesterol levels and maintain cholesterol levels independently of the free cholesterol concentration. In this way, cancer cells can keep SREBP constantly active [363].
5.3. Other oncogenes and tumor suppressor genes as drivers of alterations in lipid metabolism in cancer
A range of other oncogenes and tumor suppressors is known to affect lipid metabolism in cancer. c-Myc is an important proto-oncogene TF regulating growth of both normal and cancer cells. c-Myc promotes tumor initiation, progression and survival. MYC is amplified in about 30% of prostate tumors, frequently in the late stages, but is also overexpressed in the absence of a genetic lesion [341, 364]. It has been reported that SREBP2 directly induces c-Myc activation to drive stemness and metastasis in prostate cancer [365] and that SREBP1 promotes reprogramming by interacting with c-Myc in a translocation-dependent manner [366]. SREBP1 interacts with c-Myc facilitating its binding to and promoting the expression of downstream pluripotent targets [366]. MYC regulates lipogenesis to promote tumorigenesis through SREBP1 [367]. Inhibition of FA synthesis blocked tumorigenesis and induced tumor regression in both xenograft and primary transgenic mouse models, revealing the vulnerability of MYC-induced tumors to the inhibition of lipogenesis. Extrinsic risk factors are also able to enrich for MYC signaling. Our group showed that the MYC-transcriptional program can be amplified by a high-fat diet through metabolic alterations contributing to cancer progression and lethality [367].
Upon MYC induction across different cancers, in vivo lipidomic changes have been described. We showed that MYC-driven prostate cancer cells are associated with deregulated lipid metabolism in vitro and in vivo, whereas AKT1 has been associated with enhanced aerobic glycolysis [368]. However, the human data in this study showed metabolic heterogeneity in addition to genetic and signaling pathway heterogeneity. Indeed, heterogeneity in human tumors makes this simplistic interpretation obtained from experimental models more challenging.
The Yes-associated protein (YAP) and Transcriptional coactivator with PDZ-binding motif (TAZ) proto-oncogenes are inhibited by the Hippo tumor-suppressor pathway. YAP/TAZ promote tissue proliferation, organ growth, cancer stem cell properties, metastatic potential and resistance to cancer therapy [369]. Emerging evidence indicates that deregulation of YAP and TAZ mediators of the Hippo pathway signaling may be a major mechanism of intrinsic and acquired resistance to various targeted and chemotherapies promoting tissue proliferation and organ growth [369, 370]. In response to various therapies, numerous upstream signals could impinge on components of the Hippo pathway to activate YAP/TAZ. It has been shown that the SREBP/mevalonate pathway promotes YAP/TAZ nuclear localization and transcriptional activity [371]. Mechanistically, geranylgeranyl pyrophosphate produced by the mevalonate cascade activates YAP/TAZ by inhibiting their phosphorylation and promoting their nuclear accumulation. Thus, these findings indicate that mevalonate–YAP/TAZ axis is required for proliferation and self-renewal of BC cells [371].
The tumor suppressor p53 is a TF that controls the expression of proteins involved in cell cycle arrest, DNA repair, apoptosis, and senescence. p53 also regulates cellular metabolism, which appears to play a key role in its tumor suppressive activities [372, 373]. p53 regulates lipid metabolism by transcriptional control or protein–protein interaction. Enzymes affecting lipogenesis whose activities are negatively regulated by p53 include glucose-6-phosphate dehydrogenase [374], which catalyzes the first step in the pentose phosphate pathway. Indeed, loss of p53 activates glucose-6-phosphate dehydrogenase and the pentose phosphate pathway, leading to lipid accumulation [374] while disruption of p53 in ob/ob mice restores the expression of lipogenic enzymes regulated by SREBP-1 [375]. p53 alters the membrane PL composition causing a shift towards a higher degree of saturation. This is mediated by decreased SCD expression through repression of SREBP1. As a consequence, p53-induced changes in PI lipid species attenuate AKT activation contributing to the p53-mediated control of cell survival [376]. More than 50% of human tumors are characterized by mutations of the TP53 gene [350, 377, 378]. Generally, wild type p53 inhibits FA synthesis and lipid accumulation. In contrast, mutant p53 enhances FA synthesis by inhibitory interaction with AMPKα [379]. Previous studies have also suggested that missense mutations confer tumor-promoting functions to p53 [379–381]. A possible mechanism has been proposed where the upregulation of the mevalonate pathway in breast tumors might be mediated by mutated p53 and SREBP and SCAP [382, 383]. Although a comprehensive understanding of the metabolic functions of p53 is yet to be achieved, perturbations of p53-mediated metabolic activities are pivotal during cancer progression as extensively reviewed elsewhere [384–388].
The tumor suppressor protein Retinoblastoma protein (Rb) activates SREBP, leading to activation of the DNA damage response and cellular senescence [389]. In 5% of primary and 37% of advanced prostate cancers, Rb is inactivated, enhancing N-Ras through induction of SREBP1 and 2 [341]. Rb suppresses the malignant progression of tumors in part by controlling the cellular lipid composition. Enzymes involved in elongation and desaturation of FAs, including ELOVL and SCD1, are upregulated by Rb possibly through SREBP. Depletion of ELOVL6 or SCD1 significantly suppresses tumor formation and growth in cell lines and xenografts of Rb-deficient tumor cells [390].
The 5’ adenosine monophosphate-activated protein kinase AMPK is a metabolic sensor and its activation leads to inhibition of metabolic pathways including lipogenesis and cholesterol synthesis. Decreased AMPK activation has been implicated in human metabolic disorders associated with increased cancer risk such as obesity and the metabolic syndrome [391]. AMPK is hypothesized to drive cancer progression by promoting metabolic plasticity, resistance to cellular stress and cell survival. Mechanisms by which the AMPK pathway supports cancer progression include promotion of FAO and increase of intracellular NADPH required to support lipogenesis. The intracellular NADPH level is determined by the difference between its production (generated from the PPP and mitochondrial metabolism) and its consumption (mainly during fatty acid synthesis). Under conditions of energy stress, when NADPH generation from the PPP is impaired, AMPK activation plays a critical role in cancer cell survival by maintaining NADPH levels through inhibition of ACACA and ACACB [392]. It has been shown that AMPK-mediated inhibition of ACACA decreases NADPH consumption in FAS whereas AMPK-mediated inhibition of ACACB increases NADPH generation by activating FAO. However, FAO could also increase the ATP level eventually inhibiting AMPK, therefore the hypothesis that NADPH maintenance rather than ATP maintenance is the predominant mechanism by which AMPK promotes cell survival during metabolic stress. In addition, a recently suggested spatiotemporal hypothesis could further explain the context-dependent and time-dependent effects of AMPK regulation in cancer (382). Early in tumorigenesis AMPK may act as a tumor suppressor, but in the advanced stages of the disease it may rather function as an oncogene contributing to therapy resistance and cancer recurrence [396].
In vitro and in pre-clinical models, drug-induced supra-physiological activation of AMPK reduces tumor growth through the suppression of key biosynthetic pathways, most notably lipogenesis [102, 393]. The tumor suppressor role of AMPK has been reported to act through several mechanisms: i) activated by either the STK11/LKB1 tumor suppressor pathway or p53, AMPK blocks de novo FA synthesis by phosphorylating acetyl-CoA carboxylase and inducing cell-cycle arrest (metabolic role), ii) induction of mitotic spindle assembly/chromosome segregation abnormalities (non-metabolic role), iii) suppression of the oncogenic MEK–ERK signaling and consequent impairment of cell proliferation and cell-cycle progression via phosphorylation of the oncogene BRAF, iv) counteraction of the epithelial-to-mesenchymal transition, v) loss of AMPK activity contributing to tumorigenesis through hyperactivation of YAP, vi) inactivation of AMPK via ubiquitination and degradation leading to inhibition of autophagy and activation of mTORC1 signaling [102, 393, 395, 397–399].
5.4. Genetic and epigenetic alterations leading to rewiring of lipid metabolism
Chromosome alterations have been proposed to drive cancer progression [400–402]. In particular, chromosome 8 is a hotspot for genomic aberrations comprising not only chromosomal rearrangements and deletions, but also amplifications in several cancer types. The short arm of chromosome 8 (8p) is one of the most frequently deleted genomic regions in a variety of human epithelial cancers [401]. While 8p loss is insufficient to transform cells, it results in the upregulation of the mevalonate and FA pathways. Loss of the 8p chromosome leads to the alteration of lipid metabolism and composition, increasing invasiveness and intravasation and protecting cancer cells from hypoxic stress due to increased autophagy [403]. The human LPL gene is located on 8p22 and plays an important role in lipid metabolism. Lowering or deficiency of LPL expression due to chromosome 8p loss, LPL gene polymorphism, and epigenetic changes in its promoter region are associated with hyperlipidemia and increased cancer risk, especially in the prostate [404–406]. In particular, biallelic inactivation of LPL by chromosomal deletion or promoter methylation has been suggested to contribute to prostate tumorigenesis [407]. Interestingly, an opposite role for LPL was described in cervical squamous cell carcinoma cells where an expressed fusion gene has been identified from novel t(8;12)(p21.3;p13.31) reciprocal translocation [408]. The rearrangement involved LPL and peroxisome biogenesis factor-5 (PEX5). The wild-type LPL overexpression was found to be common in both tissue samples and cell lines. Forced overexpression of wild-type LPL and PEX5–LPL fusion transcripts increased invasiveness in cervical squamous cell carcinoma cells [408].
Chromosome 8q is also the most commonly gained aneuploidy in cancer [401]. In both prostate and breast cancer, chromosome 8 amplification has been associated with increased proliferation rates, disease progression and reduced patient survival [409]. A study of 229 primary invasive BC cases identified substantial coamplification of the 8p11-p12 genomic region and the MYC oncogene (8q24.21), as well as aberrant methylation and transcriptional patterns for several genes spanning the 8q12.1- q24.22 genomic region of which one of the rate-limiting enzymes in sterol biosynthesis the squalene epoxidase (SQLE) [410]. MYC activity and DNA hypomethylation may therefore have a pivotal role in the aggressive tumor phenotype frequently observed in BC harboring 8p11-p12 amplification [410]. Another study, involving two independent patient cohorts of 160 patients each, showed that gains of chromosomes 7p and 8q are associated with poor prognosis among ERα positive early stage BC [411]. Whereas SQLE expression levels were not correlated with tumor size, grade, ER status and HER2 expression, there was a significant independent influence on prognosis of the stage I and II study population for SQLE [411]. The correlation between SQLE copy number and expression has been assessed in a large-scale study among more than 8000 cases from 22 cancer types. The authors found the highest prevalence and interaction of SQLE copy number amplification with its gene expression variation in breast, ovarian, and colorectal cancers with BC presenting the strongest association [412]. In particular, SQLE overexpression was more prevalent in aggressive BC suggesting SQLE as a bona fide metabolic oncogene by amplification and by being an independent prognostic factor of unfavorable outcome [412]. Overexpression of SQLE has also been found in hepatocellular carcinoma tissues. In hepatocellular carcinoma cells SQLE upregulation promoted cell proliferation and migration, while downregulation of SQLE inhibited tumorigenicity in vitro and in vivo [413].
An amplification and overexpression of the pyruvate dehydrogenase complex (PDC) has been recently reported in prostate cancer. PDC is responsible of converting pyruvate into acetyl-coA for entry into the TCA cycle in mitochondria [414]. The authors showed that the principal effect of targeting the PDC complex is tumor suppression by abrogating lipid biosynthesis [414].
Genetic alterations of members of the cytochrome P450 superfamily have also been described to play an important role in cancer. The aromatase enzyme CYP19A1 catalyzes the conversion of androgen to estrogen representing a rate-limiting step in estrogen biosynthesis. Aromatase Inhibitors (AI) are used in BC treatment as part of the gold standard endocrine therapy. Amplification of the CYP19A1 gene (CYP19A1amp) has been demonstrated to occur in 22% of AI-treated BC patients [415] causing increased aromatase activity, estrogen independent ERα binding to target genes and decreased sensitivity to AI therapy. These data indicate that endocrine therapy selects for acquired CYP19A1amp and promotes local autocrine estrogen signaling in AI-resistant metastatic patients [415]. De novo cholesterol synthesis is upregulated in LTED cells (long-term estradiol deprived ERα BC cell lines) via epigenetic reprogramming to promote autonomous ERα activation [416]. Indeed, LTED cells have a ten-fold increase in ERα protein expression [416] despite no evidence of ESR1 amplification or activating mutations [417]. This study suggests that hormone deprivation promotes acquired CYP19A1 amplification that, in turn, triggers ERα activity by converting testosterone possibly obtained via epigenetically driven de novo cholesterol biosynthesis. Similarly, CYP19A1 has also been reported to be upregulated in malignant versus benign prostate epithelium, and in metastatic prostate cancer compared to localized disease [418]. Another possible mechanism through which BC cells acquire resistance to AI is via a specific drug-induced epigenetic reprogramming that it has been revealed to be activated in AI resistant cells but not in cells developing resistance to Tamoxifen [416]. The AI-induced rewiring consisted in an epigenetic activation of the cholesterol biosynthesis pathway. This work supports the hypothesis that intratumoral de novo cholesterol biosynthesis is constitutively activated after chronic exposure to AI leading to the local accumulation of metabolic ligands for ERα such as 27-hydroxycholesterol [297–299, 419].
The cytochrome P450 enzymes sterol 27-hydroxylase (CYP27A1) and oxysterol 7α-hydroxylase (CYP7B1) are responsible for synthesis and degradation respectively of 27-hydroxycholesterol from cholesterol. Mounting evidence suggest that 27-hydroxycholesterol plays an important role in cancer progression. Nelson and colleagues revealed that 27-hydroxycholesterol promotes cell growth and metastasis in mouse models of BC. Dyslipidemia increases the risk of BC through 27-hydroxycholesterol stimulated cell proliferation in ESR-dependent and GDNF-RET (Glial cell line-derived neurotrophic factor-RET)-dependent manners, with 27-hydroxycholesterol accumulation in BC tissue [419]. Importantly, it has been demonstrated that 27-hydroxycholesterol is the first known endogenous SERM [297, 298]. The Nelson lab further showed that 27-hydroxycholesterol is a major biochemical mediator of BC metastasis by increasing the number of polymorphonuclear-neutrophils and γδ-T cells in patients with hypercholesterolemia [420]. CYP27A1 is detected in macrophages within human benign and malignant mammary tissue and in high-grade tumors, and both tumor cells and tumor-associated macrophages exhibited high expression of the enzyme [299]. The expression of CYP7B1 is down-regulated in human BC tissues and CYP7B1 is silenced by epigenetic mechanisms in BC cells [419, 421]. The hypermethylation of CYP7B1 and recruitment of monocytes likely contributes to the accumulation of 27-hydroxycholesterol in BC and the interaction of 27-HC with macrophages further promotes tumor development [419, 421].
Recent studies described the role of lipid biosynthesis-related epigenetic rewiring in cancer actively contributing to aging, therapy resistance, cellular structural changes and tumor invasive capacity. Methylation of ELOVL2 is highly correlated with the aging process. Impaired ELOVL2 function accelerates aging through disruption of lipid synthesis via increased endoplasmic reticulum stress and mitochondrial dysfunction [422]. In addition, epigenetically upregulated ELOVL2 in glioma stem cells contributes to poor prognosis by driving therapeutic resistance and maintenance of cellular heterogeneity. In contrast, ELOVL2 depletion altered cellular membrane PL composition disrupting membrane structural properties and inhibiting cell growth and tumor initiation [423]. In endocrine therapy-resistant BC cells, epigenetic reprogramming at the type II keratin locus leading to Keratin 80 upregulation drives active cytoskeleton re-organisation and F-Actin remodelling. Chromatin Immunoprecipitation coupled with Next Generation Sequencing (ChIP-seq) revealed that reprogramming is dependent on de novo SREBP1 binding to a single enhancer that is activated upon chronic AI treatment. With this study, Perone et al identified a novel and direct link between epigenetic and cytoskeletal reprogramming via SREBP1 promoting cancer cell invasive behavior [424].
5.5. Regulation by microRNAs
In recent years another layer of regulation of lipid metabolism in cancer involving microRNAs (miRs) has emerged. miRs are small endogenous RNA molecules measuring 18–24 nucleotides in length that occur in eukaryotes only. These regulate post-transcriptional and translational protein expression. Among the lipid-related microRNAs (miRs), miR-33a has been described to be located in the intron of SREBF2 and miR-33b in the human intron of SREBF [425–427]. The miR-33 system regulates lipid homeostasis by modulating HDL biogenesis and cholesterol efflux. miR-33 also targets SREBP1c and has been shown to inhibit BC metastasis [428, 429]. Furthermore, both miR33a [430] and miR-33b [431] were up-modulated after therapy with statins, thus down-regulating the oncogene c-Myc and cell proliferation. Statin treatment has been shown to alter more than 400 miRNAs using in-vitro assays (as reviewed in [432]). For instance, the well-known tumor suppressor miR-612 is upregulated after statin intervention, promoting cancer cell differentiation and response to chemotherapy [433]. Another statin-regulated miRNA, miR-182, inhibits the antiapoptotic Bcl-2 transcript consequently favoring cell apoptosis [434]. Two miRs, miR-185 and 342, control cholesterol and FA synthesis in prostate cancer cells by inhibiting SREBP1 and 2 expression and downregulating their target genes, including FASN and HMGCR. Both miRs inhibited tumorigenicity, cell growth, migration and invasion in prostate cancer [427]. Their expression was significantly decreased in tumor cells compared to non-cancerous epithelial cells. It has further been shown that restoring miR-185 and 342 led to caspase-dependent apoptotic death in prostate cancer cells [435]. In glioblastoma, SCAP/SREBP1 activates miR-29 by directly binding to its promoter. A negative feedback loop has been shown for miR-29 that is able to suppress SCAP/SREBP1 and inhibit tumor growth [436].
The miR-22 molecule is involved in the posttranscriptional regulation of ACLY thus supporting metastasis in breast, osteosarcoma, prostate, cervical and lung cancers [437]. A miRNA profile of pituitary oncocytoma reported that the tumor suppressors miR-127–3p and miR-744–5p influenced cell proliferation, carbohydrate and lipid metabolism. In particular, a central role has been proposed for miR-744–5p targeting Aconitase 2 in the regulation of TCA cycle in spindle cell oncocytomas [438].
MiR-497–5p is a known tumor suppressor. miR-497–5p overexpression in HCT116 cells modulated colorectal cancer malignancy via downregulation of IGF1/IGF1-R and inhibition of PI3K/Akt signaling pathway [439]. Another study found that overexpression of miR-497–5p modulates metabolism of the FAs via decreasing ACSL5 levels. The Acyl-CoA Synthetase Long Chain Family Member 5 plays a key role in lipid biosynthesis and FA degradation and is highly expressed in colon cancer cells. miR-497–5p prevents cancer colony formation and negatively regulates cell cycle progression whereas its upregulation increases apoptosis and modulates invasiveness and metastasis in colon cancer cells both in vitro and in vivo. In patients with colorectal cancer, miR-497–5p downregulation correlated with tumor differentiation, TNM staging, lymph node metastasis and poor survival [440]. Other miRNAs regulating FA biosynthesis identified in malignant pleural mesothelioma, miR-15b-5p and miR-185–5p, have been reported to regulate the target genes FASN, OXSM, ACACB [441].
In esophageal cancer, miR-142–5p suppresses tumorigenesis by targeting SREBP1. Treatment with Fatostatin in both 2D and 3D cell line models and in vivo, resulted in the reduced staining of SREBP1, increased miR-142–5p and suppressed tumor growth [442].
In gastric cancer, miR-671–5p directly interacted with another non-coding RNA, the circPIP5K1A. This is one of the circular RNAs (circRNAs) which have been shown to play a significant role in the initiation or development of human cancers. In vitro and in vivo experiments indicated that CircPIP5K1A plays an oncogenic role in gastric cancer enhancing cell proliferation, invasion and migration. Mechanistically, the interaction between circPIP5K1A and miR-671–5p modulates Keratin 80 expression forming an axis that contributes to cancer progression through PI3K/AKT pathway [443]. Interestingly, a direct link between SREBP1 activation and invasive behavior via upregulation of Keratin 80 has been previously shown in drug-resistant ER+ breast cancer (vide supra, [424]).
In a recent study, starting from metabolic and transcriptomic analysis of renal cell cancer patient tissues, the authors identified upregulated miR-146a-5p that altered the expression of key genes involved in the pentose phosphate pathway and the TCA cycle. They then extended the analysis to more than 6000 patients suggesting that miR-34a-5p, miR-106b-5p, miR-146a-5p, and miR-155–5p are pan cancer microRNAs involved in global regulation of cancer metabolism [444].
Finally, many inflammatory obesity-related miRNAs (inflammatory miRNAs involved in adipogenesis) have been demonstrated to play a role in several cancers (as reviewed in [445]).
5.6. Posttranslational regulation at the level of protein activity, stability and degradation
SREBPs and several other proteins involved in lipid metabolism are also potently regulated at the level of their stability and degradation. It has been demonstrated that the mature forms of SREBPs are modified by phosphorylation [446–450], acetylation [320, 451], sumoylation [320, 451], and ubiquitination [321, 452]. Not only mature, but also SREBP precursor forms are subject to proteasome-dependent degradation via ubiquitylation. Heat shock protein (HSP) 90 regulates SREBP by binding to and stabilizing the SCAP-SREBP complex; inhibition of HSP90 leads to proteasome-dependent degradation of SCAP-SREBP protein [453]. Furthermore, after dissociation from the complex SCAP/SREBP, Insig1 is ubiquitinated and degraded in proteasomes. Ubiquitination is not necessary for release of SCAP/SREBP from Insig1, but it establishes a requirement for synthesis of newly-synthetized Insig1 for feedback inhibition. When the new Insig1 and cholesterol converge on SCAP, SCAP/SREBP binds to Insig1, preventing ubiquitination [454]. As a result, treating cells with proteasome inhibitors increases nuclear levels of SREBPs and target gene expression.
Another mechanism of regulation is provided by ingestion of PUFA, which reduces hepatic SREBP1c activity, thereby decreasing lipogenesis and plasma TAG. PUFA-dependent inhibition occurs by accelerated mRNA decay and proteasomal degradation of nuclear SREBP1c [455–457].
Kinases play also an important role in posttranslational modulation of lipogenic homeostasis. Protein kinase A (PKA) is a family of enzymes whose activity is dependent on cellular levels of cyclic AMP. PKA inhibits lipogenesis by phosphorylating and disrupting the DNA-binding activity of SREBP1 [458, 459] and phosphorylating upstream LXR [460]. Phosphorylation of SREBP1c by AMPK is necessary for inhibition of its proteolytic processing and transcriptional activity [393, 461]. Moreover, AMPK is also able to block FA and cholesterol biosynthesis through direct phosphorylation of the enzymes HMGR and ACACA. ACACA phosphorylation levels have been found to be increased in invading cells and correlated with metastatic potential in breast and lung cancer patients [462]. In head and neck squamous cell carcinoma, phosphorylation and inhibition of ACACA is followed by a compensatory increase in total ACACA, which rewires cancer metabolism from glycolysis-dependent to lipogenesis-dependent allowing cells to survive cetuximab treatment [463].
Several other proteins involved in lipid metabolism are regulated at the posttranslational level by altering their activity and degradation. The key enzyme in sterol biosynthesis, HMGCR, is degraded by the ER-associated degradation (ERAD) pathway [464, 465]. HMGR degradation is a key aspect of feedback inhibition that is critical for sterol homeostasis in humans. In cancer, degradation of FASN is prevented at the pre-proteasomal level by the isopeptidase USP2a (ubiquitin-specific protease-2a). The deubiquitinating enzyme USP2a associates with and prolongs the half-life of FASN thus playing a critical role in prostate cancer cell survival through FASN stabilization. [466, 467]. The post translational regulation of FASN involves also other factors that promote proteasomal degradation, such as acetylation, thus inhibiting de novo lipogenesis and tumor cell growth. In human hepatocellular carcinoma samples, acetylation of FASN is downregulated and expression of the deacetylase HDAC3 and FASN protein levels are increased [468].
The metabolic enzyme ACLY, which plays a pivotal role in promoting cancer metabolism [469, 470], is activated by phosphorylation and acetylation and is degraded by ubiquitination. In cancer, fructose-6-phosphate, provided by glycolysis, promotes phosphorylation of ACLY, thereby enhancing its activity and ultimately contributing to the Warburg effect [471]. Increased phosphorylated ACLY was found in non-small cell lung cancer samples; the authors showed that ACLY phosphorylation, activation and subsequent stabilization is directly mediated by PI3K-Akt pathway [472]. ACLY can also be phosphorylated by other kinases, such as nucleoside diphosphate kinase and AMPK [469]. In lung cancer, acetylation at lysine residues blocks ACLY degradation by ubiquitination further stabilizing the enzymatic activity of ACLY promoting tumor growth and enhanced de novo lipid synthesis [473]. The ubiquitin ligase complex is responsible for degradation of ACLY and has often been reported to be down-regulated in lung cancer [474]. Moreover, ubiquitin-specific peptidase 13 (USP13) specifically inhibits degradation and thus upregulates ACLY in ovarian cancer [475].
5.7. Regulation by hormones
Hormones play a crucial role in regulating lipid synthesis in certain cancers. In particular, androgens have a striking effect on lipid metabolism in prostate cancer. It is well documented that the expression of more than 20 enzymes involved in lipid synthesis, binding, uptake, metabolism, and transport are regulated by androgens, thereby influencing the entire lipid profile of prostate cells [323, 341, 423, 476–482]. Prostate cancer cells exposed to androgens showed an accumulation of LDs, especially in aggressive metastatic deposits [483], and in circulating prostate tumor cells [484]. This lipogenesis is largely dependent upon increased synthesis of FA and cholesterol [479], is reversed by an AR antagonist and is not observed in AR-negative prostate cancer cells (also referred to as “the lipidic phenotype”).
Currently, the best-characterized mechanism by which androgens may stimulate de novo lipogenesis and lipid uptake is through indirect activation of SREBPs [323, 478], although there is evidence of AR binding sites in the vicinity of many lipid metabolic genes that suggest more direct transcriptional regulation [485]. In prostate cancer, SREBP1 plays a crucial role in the activation of the lipogenic phenotype through a described but still incompletely characterized interaction with androgens and AR [486]. Activation of AR by androgens increases expression of lipogenic enzymes in a SREBP1c-dependent manner [480]. A positive feedback loop promotes this signaling pathway since binding sites for SREBP1 are also found in the AR gene [478]. Androgens appear to activate the SREBP pathway with minor effects on SREBP precursor levels and a major increase in the expression of SCAP [477, 479, 487], which in turn plays a pivotal role in the lipogenic effects of androgens in tumor cells [488]. In this positive feedback loop, androgens stimulate the expression of SREBP1 through SCAP [480]. In turn, SREBP1 regulates the expression of the androgen receptor [478, 488].
Elevated levels of SREBP1 protein are found in prostate tumors compared with normal prostate tissue [489]. SREBP1 induces prostate cancer cell proliferation, migration and invasion in vitro and promotes prostate cancer tumor growth and castration-resistant progression in vivo [486]. In addition, many of the cholesterol synthesis enzymes downstream of SREBPs (HMGCS, SQLE, squalene monooxygenase, lanosterol synthase, farnesyl diphosphate synthase) are elevated during progression of the disease [489]. The clinical and animal data collectively indicate that SREBP1 expression and its nuclear translocation play a critical role in the regulation of prostate cancer development and progression to castration-resistance [486].
Progesterone is an alternative steroidal precursor of dihydrotestosterone [490]. It has been reported that CRPC tumors producing relatively high levels of progesterone are capable of de novo synthesis of androgenic steroids [491]. [489, 490]. Progesterone, like dihydrotestosterone, is known to induce cholesterol synthesis in prostate cancer cells [323]. Tumor progesterone levels are high after castration and enzymes necessary for progesterone synthesis from cholesterol (CYP11A1 (Cytochrome P450 family) and StAR (Steroidogenic Acute Regulatory Protein)) and steroids (CYP17A1 and SRD5A1 (Steroid 5 Alpha-Reductase 1)) are increased in CRPC tumors [492]. Compared with untreated primary prostate tumors, castration-resistant metastasis showed a significant increase in the expression of key enzymes required for metabolism of progestins to adrenal androgens and their subsequent conversion to testosterone [493].
Over the past decades, it has become clear that estrogens also affect lipoprotein metabolism. Estrogens usually decrease serum levels of total cholesterol and LDL-C, and increase HDL-C concentration, but induce an unfavorable increase in serum TG levels [494]. Estrogens can mediate their biologic effects in the liver through binding to the steroid nuclear hormone receptors, ERα or ERβ [495]. Interestingly, over 1000 human liver genes display a sex-driven difference in their expression of which the top biological pathways are in lipid metabolism [496]. [497]. E2 suppresses expression of high-fat diet dependent LXR target genes and TG accumulation through ERα-LXR interactions in mouse liver [498]. Upon E2 stimulation, ERα is recruited to the LXR responsive element in the SREBP-1c promoter in complex with LXRα/RXRα leading to TG reduction [498].
5.8. Adaptive processes and regulation by substrate fluxes
As mentioned in Section 3, lipid metabolism is highly adaptive. Conditions such as nutrient starvation and hypoxia dramatically rewire lipid metabolism. This also involves several of the mechanisms mentioned above. SREBP transcriptional activity, for instance, was shown to be induced by serum depletion both in normoxic and hypoxic cells and activation of SREBP was required to maintain the expression of FA and cholesterol metabolism genes under hypoxic conditions [211]. Additionally, hypoxia-induced expression of SCD, FABP3 and FABP7 was strongly dependent on SREBP function. Inhibition of SREBP blocked lipid biosynthesis and impaired cell survival in a three-dimensional spheroid hypoxic cancer cells model.
In liver and lung carcinomas desaturation of palmitate to the unusual FA sapienate, supports membrane biosynthesis during proliferation [216]. Sapienate biosynthesis enables cancer cells to bypass SCD-dependent FA desaturation. The authors reported that targeting both desaturation pathways was required to inhibit proliferation in vitro and in vivo. Consistent with these and other reports [15, 499, 500], Bi et al recently demonstrated that membrane lipid saturation is essential for oncogene-driven cancer development [14]. Finally, membrane phospholipid remodeling generates an actionable dependency across cancers.
Cancer cells grown in lipid-reduced conditions become more dependent on de novo lipid synthesis pathways and are more sensitive to inhibitors of lipogenic pathways [181]. Cancer cell lines like breast and prostate have more lipid rafts and are more sensitive to cell death induced by cholesterol depletion than their normal counterparts. Cholesterol-rich lipid rafts facilitate the accumulation of receptor tyrosine kinases, such as HER2 and IGF-1, to rapidly induce oncogenic signaling [501, 502].
At the intracellular level, cholesterol derivatives such as cholesteryl esters (CE) and oxysterols play important roles in cancer. The acetyl-CoA acetyltransferase 1 (ACAT1) is the key enzyme that converts cholesterol to CE, usually stored in lipid droplets [503]. ACAT1 appears to exert a pro-tumor function in many cancer cells, such as pancreatic [483] and breast cancer [504]. In xenograft models of pancreatic and prostate cancer, blocking ACAT1 markedly represses tumor growth [483, 505]. CE accumulation is a consequence of PTEN loss and subsequent activation of PI3K/AKT pathway in prostate cancer cells [483].
Other CE-metabolic enzymes are highly expressed and function as key players in controlling cholesterol esterification and storage in tumors, including sterol O-acyltransferase 1 (SOAT1) and lysosomal acid lipase. Targeting SOAT1 suppresses glioblastoma growth and prolongs survival in xenograft models via inhibition of SREBP-1-regulated lipid synthesis [506]. The knockdown of SOAT1 alters the distribution of cellular cholesterol, and effectively suppresses the proliferation and migration of hepatocellular carcinoma cells [507]. Lysosomal acid lipase is upregulated and promotes cell proliferation in clear cell renal cell carcinoma [508]. Interestingly, HIF has been reported to control FA metabolism contributing to renal cell carcinoma tumorigenesis [505]. HIF directly represses the rate-limiting component of mitochondrial FA transport, carnitine palmitoyltransferase 1A, therefore reducing FA transport into mitochondria and increasing lipid deposition in clear cell renal cell carcinoma [509]. Hypoxia-induced-lipid storage has also been demonstrated to serve as a protective barrier against oxidative stress-induced toxicity in breast and glioma cell lines due to a HIF1α-dependent increase of FA uptake via FA binding proteins FABP3 and FABP7 [510].
The PI3K-AKT-SREBP pathway controls de novo lipid biosynthesis through glucose and glutamine [203]. Rapidly proliferating tumor cells depend more on glucose and glutamine for extensive de novo lipogenesis because of the action of oncogenic growth signaling molecules. Some cancer cells preferentially use glutamine as the main precursor to synthesize FA by reprogramming glutamine metabolism (glutaminolysis). Previous findings showed oncogenic levels of MYC to be linked to increased glutaminolysis resulting in glutamine addiction of MYC-transformed cells [363, 511]. SREBP can directly induce glutamine-derived carbon flux into lipid precursors at the sacrifice of the normal tricarboxylic acid cycle. As a result, cancer cells do not simply shift to an anabolic phenotype, but they rather actively reprogram their metabolism via SREBP [344].
6. Lipids as central players in cancer biology
Lipids are involved in numerous cellular processes, many of which are linked to the oncogenic process. In addition to the classical roles of lipids as building blocks for membranes and energy sources, evidence is emerging that altered lipid metabolism plays a central role in supporting cancer cell growth and survival in a changing and hostile environment (Figure 3).
Figure 3: Lipids as central players in cancer biology.
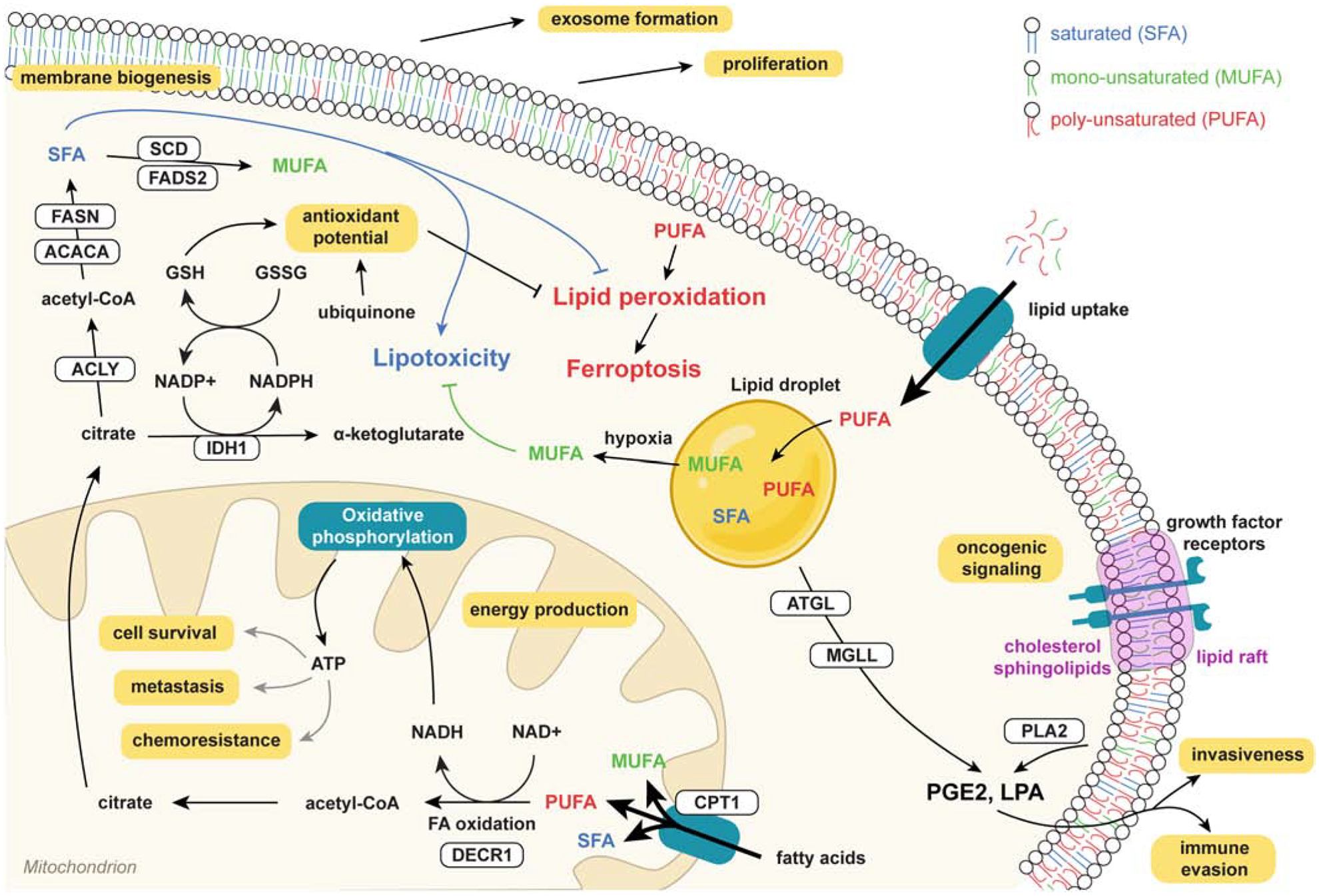
Main mechanisms by which lipids contribute to cancer biology. FA synthesis and uptake support membrane biogenesis to drive cell proliferation and exosome formation. However, since increased SFA levels induce lipotoxicity, desaturation to MUFA is required. As membrane-incorporated PUFA is prone to lipid peroxidation and underlies ferroptotic cell death, multiple mechanisms can be deployed to reduce relative PUFA abundance in the membrane. By increasing lipogenesis, relative SFA and MUFA levels are increased, decreasing relative PUFA levels. Furthermore, PUFAs can be stored in lipid droplets or catabolized through FAO. Besides generating ATP to drive cell survival, metastasis and chemoresistance, FAO can increase the antioxidant potential through GSH synthesis, further offering protection against lipid peroxidaton and ferroptotic cell death. Finally, lipids play crucial roles in cellular and intracellular signaling through regulation of membrane fluidity and lipid rafts, and the synthesis of lipid-derived mediators regulating important aspects in tumor biology such as immune evasion and cellular invasion.
6.1. Feeding membrane production and cell proliferation
Consistent with the intuitive concept that rapidly proliferating cancer cells require more lipids for membrane synthesis, lipids that are acquired by cancer cells from the microenvironment or by de novo synthesis largely end up in cellular membranes [15, 97, 478, 512–514]. That is the case for both phospholipids and cholesterol, which are the major building blocks of membranes. Early experiments with cell cultures indicated that inhibition of FA synthesis by knocking down or chemical inhibition of key lipogenic enzymes such FASN or ACACA dramatically decreases the size and membrane content of cancer cells to almost half of the original levels [15, 97, 514]. Concomitantly, cells stop proliferating and ultimately die [15, 97, 181, 513]. Supplementation with external lipids can, but not always [515], reverse these effects [515], indicating that while lipid acquisition either through de novo lipogenesis or uptake supports membrane formation and cell proliferation, de novo synthesis occurs in cancer cells even with an adequate extracellular lipid supply. Similar findings have been made in numerous other cell lines and models. Several studies have also found a link between lipogenic enzymes such as FASN and the proliferation marker Ki67 [89]. Nevertheless, activation of lipogenesis and other alterations in lipid metabolism are also observed in cancer precursor lesions where cell proliferation is low, pointing to other roles than mere bulk membrane synthesis for proliferation [89].
6.2. Balancing lipid content and protecting cells from lipotoxicity
Besides providing biomass for cell proliferation, lipid metabolism helps the cancer cell to adapt to a changing environment and to keep the lipid composition within a narrow range of variation. In fact, the lipid composition of membranes dramatically affects their biophysical properties and functioning. In this context, a plethora of reports highlight the frequency of membrane lipid desaturation in lipogenic cancers, which balances saturated and unsaturated fatty acid content to avoid lipotoxicity and ER stress [516]. In fact, saturated fatty acids, the direct end products of FASN, are toxic to cells at high levels and induce an ER stress response and cell death unless a substantial fraction is desaturated by desaturases such as SCD1, which accordingly are overexpressed along with FASN in lipogenic tumors [517, 518].
MUFA-PUFA balance is also suggested to be just as carefully controlled, since PUFAs render membranes more sensitive to lipid peroxidation and ferroptosis [519] (see Section 6.5). Recent evidence from cancer cell lines exposed to exogenous FAs confirms that membrane lipid composition can be modulated only to a certain extent and that excess lipids are stored in LDs. For instance, when cancer cells are challenged with exogenous DHA, a 2-fold increase in DHA levels is observed in membranes, whereas their levels in triacylglycerides are increased by more than 100-fold. Similar effects are observed in cells exposed to saturated FAs (JD, unpublished data). Furthermore, it was recently shown in prostate models that increased lipid uptake stimulates FA storage preferentially into DAGs and TAGs, and to a much lesser extent into phospholipids [159]. These findings are consistent with the emerging concept that LDs play a central role as a sink to keep the cellular balance of FAs in a narrow range and to sequester excess PUFA to protect cells from ROS, as detailed in 6.5.
This sequestration of lipid may also play an important role in the regulation of lipid composition under stress conditions such as hypoxia. For example, FA desaturation requires strong reducing equivalents and oxygen, which can be particularly limiting in the hypoxia characteristic of solid tumors. In glioma models, the SREBP dependent lipogenic program and SCD are more highly expressed in hypoxia, and this is in part shown to compensate for the reduced availability of oxygen [211]. Renal carcinoma cells store MUFAs such as oleate into TAGs in conditions of less extreme hypoxia. Under extreme hypoxia where desaturation becomes challenging, MUFA from TAGs can be hydrolyzed and assembled into PLs, providing a buffer to allow the cancer cell to maintain membrane desaturation [212].
A final example of how alterations in lipid metabolism function in the detoxification of certain lipids is the observed overexpression of the enzyme α-methylacyl-CoA racemase (AMACR) in prostate cancer [520, 521]. AMACR is involved in branched FA oxidation and is shown to be necessary for proliferation of prostate cancer cells in vitro [522] through detoxification of dietary methyl-branched lipids [523] and other mechanisms detailed below.
6.3. Empowering cellular processes
Since the 1920s it has been known that cancer cells largely rely on glucose for energy production [2]. However, it is now established that glucose is also preferentially used as a carbon source for anabolic processes [524–527]. Lipids can provide a large quantity of energy in addition to reductive and anabolic equivalents for a proliferating cell. Together with the observations that several cancers are shown to stimulate lipolysis and take up FAs from surrounding adipose tissue [528–532], this suggests that obtaining FAs for oxidation could be important for cancer cells.
There is substantial evidence that lipids are a relevant source for energy production. In physiology, FAO occurs in tissues with a high energy demand like heart and skeletal muscle [244]. Furthermore, in situations of increased ATP demand, such as loss of attachment to the extracellular matrix by cancer cells, FAO is required for ATP production and cell survival [533, 534]. Moreover, it was recently shown that upon acidosis stress, FAO is increased to generate ATP and drive metastasis through stimulation of early steps such as invasion and resistance to anoikis [535]. Interestingly, metastatic cells found in the lymph nodes were recently shown to have altered their metabolism towards FAO in order to adapt to the lymph node environment [369]. Furthermore, another study recently demonstrated the role of FAO-driven ATP production in glioblastoma tumorigenesis [536], and FAO-derived ATP synthesis has been shown to drive chemoresistance in BC and leukemic stem cells [537, 538]. FAO can also drive anabolic reactions through production of FA-derived carbon in the form of acetyl-CoA. Interestingly, it was shown that in endothelial cells, acetyl-CoA produced through FAO is essential for de novo nucleotide synthesis [539]. In this way, FAO drives pathological angiogenesis in vivo.
6.4. Membrane biophysics and oncogenic signaling and metastasis
Membrane lipid composition is known to dramatically alter membrane function [540] and, in particular, membrane fluidity. PLs containing saturated FAs have straight acyl chains that pack densely and thus decrease membrane fluidity. As double bonds result in a kink in the acyl chain, unsaturated FAs pack less densely and increase membrane fluidity. Also changes in cholesterol, which are often observed in tumors, dramatically affect membrane fluidity [541, 542]. Evidence from several teams, including ours, has shown that de novo FA synthesis and the subsequent changes in membrane lipid composition affect both lateral membrane fluidity (within a membrane leaflet) and transversal membrane fluidity (between leaflets). We previously showed that these changes in membrane fluidity also affect the uptake of certain chemotherapeutics such as doxorubicin that traverse the membranes through a flip-flop mechanism [15]. Moreover, increased membrane fluidity is shown to stimulate metastasis in lung cancer [543], and correlates with a poor prognosis [544]. These findings demonstrate that balancing saturated and unsaturated FAs in membrane lipids is not only important in preventing lipotoxicity and lipid peroxidation, but also affects biophysical properties of the membrane with far-reaching consequences.
Moreover, according to the current concepts, membrane lipids are not uniformly distributed but, depending on their biophysical properties, tend to cluster into specific microdomains. Although microdomains with several different compositions exist, they are overall enriched in sphingolipids and cholesterol [545]. By their specific lipid composition, these nano-scale subdomains in the plasma membrane create optimal biophysical conditions for certain signaling proteins to be recruited and to cluster [545]. Therefore, they often act as platforms for growth factor or cell death receptor signaling. Cellular signaling by receptor tyrosine kinases (RTKs) at the plasma membrane is facilitated by transient lipid microdomains termed lipid rafts [546, 547]. Furthermore, cholesterol-rich lipid rafts allow the accumulation of RTKs such as HER2 and IGF-1, to rapidly induce oncogenic signaling [501, 502]. Early evidence from one of our teams has shown that de novo synthesized FAs largely end up in detergent-resistant microdomains [548]. Together with the observation that acyl chains of phospholipids in lipid rafts are generally more saturated [549], this suggests a role for de novo lipogenesis in oncogenic signaling through lipid rafts. Furthermore, a recent study in glioma models shows that increased saturation of plasma membrane phosphatidylcholine species mediated by LPCAT1 enhances EGFR clustering and activation [14] (see also Section 5). Another recent study showed that ELOVL2-dependent accumulation of PUFA at the plasma membrane is required to promote EGFR signaling, also in glioma models [224]. Therefore, the contribution of membrane lipid changes to oncogenic signaling appears to be complex and multifactorial. As described in Section 4.10, lipids can also regulate signaling through post-translational modifications of proteins. It is well established that prenylation or palmitoylation of important oncogenes like EGFR and RAS is essential to their localization and function, and targeting these post-translational modifications holds promise in pre-clinical models, although only limited clinical efficacy was observed thus far [282, 550]. Overall a concept is emerging that alterations in lipid metabolism in cancer play a central role in feedforward oncogenic signaling. Moreover, altered sphingolipid metabolism, as occurs in many cancers, reduces the levels of the pro-apoptotic lipid ceramide and increases the levels of key proliferative signaling lipids such as sphingosine-1-phosphate (S1P), leading to extensive efforts to modify this pathway pharmacologically (reviewed in [551]). Recent observations suggest that lipid metabolism also contributes to cancer development by inducing epigenetic changes. In fact, FAO-derived acetyl-CoA is shown to be a carbon source for histone acetylation in octanoate-treated hepatocytes and BC cells [552]. However, this finding contradicts earlier claims that FAO does not result in nucleocytoplasmic acetyl-CoA and does not contribute to histone acetylation [553]. Therefore, there is a need for more research on the context-dependent role of FAO in epigenetic regulation.
6.5. Protection from oxidative stress
Cancer cells often contain high levels of reactive oxygen species (ROS), arising due to oncogenic transformation, altered metabolism, deregulated redox homeostasis and hypoxia. Increased ROS has been shown to contribute to genomic instability and tumorigenesis. However, a critical balance needs to be maintained as excess ROS can induce cell death [554–556]. It is well known that PUFAs are more susceptible to peroxidation than saturated or monounsaturated lipids [519]. In fact, peroxidation of PUFA is a key driver of ferroptosis, a newly-recognized form of cell programmed death [557, 558]. To protect cancer from the deleterious effects of ROS, a plethora of mechanisms employed by cancer cells have recently been described. One of these is the degradation of lipid hydroperoxides by GPX4, a lipid hydroperoxidase that can selectively degrade lipid hydroperoxides from the membrane. In multiple cancer models, GPX4 is a central driver of ferroptosis resistance [559, 560]. Although GPX4 is a key protective enzyme against ferroptosis, several reports have identified other players that are required for ferroptosis that are dominant over GPX4. A CRISPR screen of cells knocked out for GPX4 surprisingly found that cells lacking both GPX4 and ACSL4 were resistant to ferroptosis. Mechanistically, ACSL4 is required to enrich membranes with PUFA and thereby drives a vulnerability to membrane lipid peroxidation [561].
Another mechanism cancer cells use to decrease their levels of PUFA in membranes and to protect themselves from ROS is the activation of fatty acid synthesis. Since human cells lack the enzymes required to generate essential PUFAs, increased lipogenesis not only provides the cancer cell with membrane biomass but also increase its relative degree of saturation. We and others have shown that de novo lipogenesis effectively leads to membrane lipid saturation and depletes polyunsaturated FAs from the cell membrane, and thereby protects cancer cells from lipid peroxidation and ferroptosis [15, 16, 562]. Similarly, membrane mono-desaturation mediated by SCD in ovarian cancer models [206] or the uptake of MUFAs and incorporating them into membrane PLs has been shown to provide a robust protection from ferroptosis [218]. Along the same lines, it was recently shown that PUFA incorporation into TAGs can protect them from lipid peroxidation and ferroptosis [241, 563]. Furthermore, the rate-limiting enzyme for FAO of PUFAs, DECR1, promoted prostate cancer cell survival by protecting cells from lipid peroxidation and ferroptosis [564]. As mentioned above, FAO derived NADPH can be used to maintain antioxidant potential via the glutathione recycling system [392, 565]. For these reasons, in periods of nutrient deprivation or ROS stress, cancer cells may rely more heavily on FAO. A study in melanoma shows that under ROS stress and MAPK inhibition, FAO is required for melanoma cell survival [161, 566]. Moreover, FAO inhibition was shown to be toxic in an oxidative subset of diffuse large B-cell lymphoma cells where it interfered with glutathione generation [567]. Furthermore, sustained FAO drives metastatic colonization of BC via protection from oxidative stress [568]. It is therefore tempting to speculate that FAO plays a key role in ferroptosis resistance. Indeed, inhibition of FAO induced ferroptosis sensitivity in ccRCC, although the contribution of NADPH was not assessed [569]. Furthermore, in two back to back papers, screening for genes that can complement the loss of GPX4 further implicates the mevalonate pathway and NADPH generation in identifying FSP1 as a driver of ubiquinone recycling. Ubiquinone was identified as a potent antioxidant that was sufficient to compensate for GPX4 loss [570, 571]. Furthermore, anaplastic large cell lymphoma models and cell lines have been shown to generate high levels of squalene, which is identified as an endogenous antioxidant that protects the cells from ferroptosis. Interfering with squalene synthesis is therefore a promising strategy in this cancer [572].
The ability of lipid metabolism to regulate reductive equivalents is not restricted to the process of FAO. Interestingly, a recent finding shows that sustained membrane lipid desaturation is critical in physiology not merely due to its products, but due to the fact that the enzymatic reaction consumes NADH and generates NAD+ [225]. Much like lactate production, the increased availability of NAD+ is required to sustain glycolysis, although the contribution of this mechanism in cancer is unknown.
6.6. Signaling in the microenvironment
Lipids function as precursors for important intracellular signals such as diacylglycerol and phosphatidylinositol phosphates (PIPs), which are often deregulated in cancer and involved in cell motility and tumor progression [266] (See also Section 4 of this review). Furthermore, FAs are precursors of extracellular signaling lipids which include the diverse class of oxylipins, LPA, ceramide and sphingosine-1-phosphate. The intracellular pool of free FAs is very limited since the majority of FAs are rapidly incorporated into membranes and neutral fats. Therefore, the liberation of FAs from phospholipids or neutral fat is necessary in the generation of free FAs and lysophospholipids (LysoPLs). Compared to the metabolic contributions of lipids, the oncogenic roles of this source of FAs has only recently come to light [573].
FAs can also be released from neutral fat stores by the enzymes ATGL, HSL and MAGL [574]. ATGL in particular has been shown to have oncogenic roles in colorectal and lung cancer cells [575, 576], and may contribute to BC growth and invasiveness by releasing adipose derived FAs [577]. A pharmacological inhibitor of ATGL is available [578] and ATGL has been shown to have pro-tumorigenic roles in multiple cancer models; mice lacking ATGL spontaneously form tumors [576] and ATGL protects cells from lipid peroxidation and ferroptosis. MAGL, which hydrolyses monoacylglycerol, has been shown to contribute to cancer progression and aggressiveness, in driving an array of oncogenic signaling pathways including synthesis of prostaglandins, LysoPLs and ether lipids [579]. However, it can also play key immunosuppressive functions in tumor-associated macrophages (TAMs) [580]. Inhibition of MAGL by the small molecule JZL184 or knockdown suppresses tumorigenesis of melanoma and ovarian cancer cells [581]. However, not all studies support a pro-tumorigenic role of phospholipases in cancer. Indeed, their expression is often lowered in cancers [582], perhaps in a context-dependent manner. The lysis of adipose-derived FAs may also provide the cancer cells with free FAs and FA-derived signaling molecules that can drive cell invasiveness. In pancreatic cancer cells, the secretion of the extracellular autotaxin provides stromal-derived LPCs which can be used to generate LPA, thereby powering cancer cell invasiveness [583]
PUFAs such as arachidonic acid can be modified and oxygenated in order to generate a highly diverse and complex class of molecules termed oxylipins. These metabolites can have profound effects on multiple aspects of tumor biology, including mediating cell invasiveness and immune evasion as detailed below in Section 6.7.
Cancer cells have long been shown to generate lipid-enclosed microvesicles such as exosomes, microsomes or oncosomes. These microvesicles are taken up by nearby stroma and distant tissues and can exert potent effects at target sites [584]. In particular, an elegant study shows that the specific distribution of integrins found in exosomes dictates their binding to target organs and thereby results in inflammation, and prepares the site for the eventual establishment of metastases [585]. Although the biological role of exosomes in cancer biology remains underexplored, the unique RNA, protein and lipid cargo contained in these circulating vesicles can almost certainly have significant biological effects [586] (See also Section 8). The vesicles may also deliver enzymes involved in lipid metabolism [587].
6.7. Immune-modulation
One of the established hallmarks of cancer is the evasion from immune surveillance, a phenomenon that is successfully targeted using immune checkpoint inhibitors, which currently are revolutionizing cancer therapy. However, many patients fail to respond to this therapy through primary or acquired resistance mechanisms [485]. Findings showing the importance of lipid metabolism in functioning of the immune system therefore offer exciting new opportunities to address this issue. A recent report shows that interferon gamma induces cell death in cancer cells by inducing ferroptosis and points towards the importance of lipid metabolism in the context of clinical treatment with immune checkpoint inhibitors [588]. Cancer cells not only suppress immune cell function but can convert the immune system to sustain tumor growth. In ovarian cancer for instance, cancer cells are shown to promote the efflux of cholesterol from macrophages which in turn drives a pro-tumoral M2 phenotype [589]. PGE2 is the most well-described oxylipin in cancer, which has a dominant suppressive role on the immune environment and leads to the failure of immune-cell cancer clearance in addition to its pro-inflammatory and angiogenic roles. Although PGE2 can be produced by cancer cells, recent evidence shows that PGE2 is primarily produced by tumor-associated myeloid-derived suppressor cells in a FATP2 dependent manner [590]. The FATP2 FA transporter plays critical roles in tumor associated neutrophils to transport arachidonic acid for the synthesis of prostaglandin E2, as interference with this process abrogated tumor growth [590]. These and other findings suggest the importance of lipid metabolism in the clinical response to immunotherapy. Although the roles of other oxylipins such as leukotrienes and resolvins is well appreciated in the context of asthma and inflammation, their contribution to cancer remains less well understood. This is in part due to their low abundance and technical challenges in their measurement. With their potent effects on multiple aspects of biology including immune cell chemotaxis and function, this is likely to be an emerging and important field in the context of cancer biology and immunotherapy. Moreover, several recent studies have highlighted the critical role of lipid metabolism in immune cell functions and therefore caution against a systemic approach in targeting lipid metabolism. Both FA synthesis and oxidation are important regulators of immune responses. FA synthesis plays a role in antigen presentation and T cell activation, whereas FAO regulates hematopoietic stem cell maintenance. In a nutrient deficient tumor microenvironment, CD8+ T cells require FAO to efficiently clear melanoma cells [591]. This is compounded by further evidence showing that FAO may increase the prevalence of cancer neoantigen presentation and correlates with a good response to immune checkpoint inhibitors [592]. Also cholesterol metabolism may play key roles in the formation of an effective T-cell receptor complex and therapeutic interference may therefore be detrimental to T-cell function [593]. Moreover, there is evidence that the rapid expansion of T-cells requires SREBP mediated lipogenesis [594]. The externalization of phosphatidylserine, a hallmark of the apoptotic process, has also gained increasing interest recently due to its immunosuppressive action that promotes the tumor’s immune evasion [595]. Also acid sphingomyelinase (ASM) which is required for the conversion of the cell membrane component sphingomyelin into ceramide has been shown to elicit an effective antitumor immune response by the host [596]. As a significant portion of preclinical cancer models use immunodeficient mice, the potentially profound effects of lipogenic inhibitors on immune cell function is completely missed.
7. Lipids as biomarkers for cancer
A growing body of evidence indicates that the rewiring of lipid metabolism in cancer holds potential for the development and use of biomarkers. A large number of studies have shown that enzymes involved in lipid metabolism are differentially expressed in tumors and, depending on the enzyme and tumor type, correlate with stage or grade, or have diagnostic or prognostic potential. So far, few of these markers are currently being used in the clinic.
Driven by recent technological developments in the analysis of lipids and the recent explosion of lipidomic studies of experimental cancer models and clinical specimens, the potential of the lipidome to yield novel biomarkers in cancer has attracted considerable interest as a complement to other, predominantly genomic markers [33, 597, 598]. Initial studies focused on cancer biomarker discovery were largely qualitative and exploratory in nature with relatively small patient numbers, and sought to identify lipids associated with the presence of cancer, certain clinical features or patient outcomes (for a recent comprehensive overview of lipid biomarker studies undertaken in cancer the reader is referred to Bandu et al, 2018 [598]).
7.1. Lipid metabolism enzymes as potential biomarkers
As mentioned in the previous sections, there are numerous reports on the altered expression of enzymes involved in lipid metabolism in almost all tumor types. In some studies, claims have been made on the potential use of these proteins as biomarkers for diagnosis, stage or grade, or disease progression. Such claims have been made for instance for FASN, FABPs and many others. Caution has to be taken that few of these findings have been confirmed in large cohorts of patients and many of the claimed associations cannot be confirmed in TCGA data for several reasons. Several enzymes involved in lipid metabolism are also found in body fluids such as serum or plasma. That is for instance the case for FASN, FABPs and LSCR1, suggesting that these proteins might be used as a noninvasive serological diagnostic and prognostic biomarker for cancer [599]. However, none of these proteins are routinely used in the clinic as biomarkers.
7.2. Lipidomics in biofluids
The potential for lipids as biomarkers has come to the forefront thanks to great advances in technologies enabling the quantitative analysis of complex lipids, including mass spectrometry-based lipidomics as detailed in section 3, and fatty acids measured by GC-MS. So far, the majority of lipid profiling studies in patient samples have been undertaken in biofluids, which offer patient-friendly sources of biomarkers without the invasive nature of tissue biopsies or heterogeneity of tissue sampling. An ever-expanding number of studies have used MS-based lipidomics or metabolomics to identify individual or groups of lipid species in serum or plasma that can distinguish between cancer patients and cancer-free controls (reviewed in [597, 598]). While patient numbers are often low and factors such as patient fasting status or metabolic medications can be confounders, several recent larger-scale lipidomics studies have provided compelling evidence for the potential of the lipidome to provide diagnostic and clinically-actionable prognostic biomarkers in a range of cancers (Table 1 and Table 2). Identified signatures comprising relatively small numbers of circulating lipids or fatty acids had the capacity to distinguish breast [600, 601], ovarian [22], colorectal [602] liver [23], lung [24, 25] and prostate [26, 603] cancer patients from cancer-free controls. Of arguably greater clinical significance, lipid profiles have also been shown to have prognostic value for cancer development [604][603, 605, 606], aggressiveness [607], therapeutic response [608–610] and patient survival [611].
Table 1:
Lipidomic studies on biofluids reporting diagnostic lipid signatures
| Cancer type | Control samples | Cancer samples | MS Platform | Biofluid | Lipids altered | Reference |
|---|---|---|---|---|---|---|
| Ovarian | 212 | 211 | LC/ESI/MS/MS | Plasma | PPE(16:0,18:1) LPC15:0 LPA18:2 PPE(18:0,22:6) |
Shan et al, 2012 [1] |
| Liver | 90 | 82 | UPLC-MS | Serum | LPC16:0 LPC18:0 PS16:0 PC18:0 |
Wang et al, 2012 [2] |
| Liver | 37 | 37 | GC-MS | Plasma | 18:2n-6 18:1n-9 20:4n-6 16:0 |
Qiu et al, 2015 [3] |
| Lung | 495 | 58 | FTICR-MS | Serum | SM(16:0,16:1) LPC18:1 LPC20:4 LPC20:3 LPC22:6 |
Guo et al, 2012 [4] |
| Lung | 147 | 199 | ESI-MS | Plasma | LPE18:1 CE18:2 ePE40:4 SM22:0 |
Yu et al, 2017 [5] |
| Prostate | 76 | 57 | Q-TOF-MS | Plasma | ePC38:5 PC40:3 PC42:4 |
Patel et al, 2014 [6] |
| Breast | 74 (inc. 40 non-malignant lesions) | 40 | GC-MS | Serum | 16:0 18:0 18:2 18:3 20:5 22:5 |
Lv et al, 2012 [7] |
| Colorectal | 84 | 63 | GC-MS | Plasma | 10:0 | Crotti et al, 2016 [8] |
Table 2:
Lipidomic studies reporting prognostic lipid signatures in biofluids
| Cancer type | Samples | Outcome | MS | biofluid | Lipids altered | Reference |
|---|---|---|---|---|---|---|
| Measure | Platform | |||||
| Lung | 300 Pre-treatment | Risk of cancer development | LC//MS | Plasma | Shingosine 1-phosphate | Alberg et al, 2013 [9] |
| Prostate | 3,057 Matched case-controls (EPIC study) | Development of advanced or aggressive prostate cancer | MS) | Plasma | PCs, lysoPCs | Schmidt et al, 2020 [10] |
| Hepatocellular carcinoma | 43 | Responders vs non-responders to sorafenib | RPLC-MS | Plasma | PC34:2; PC34:3; PC35:2; PC36:4a; PC34:3e; acylcarnitine (18:0); cholesterol ester (20:2); DG34:2 | Saito et al, 2018 [11] |
| Ovarian | 101 | Recurrence vs non-recurrence following standard of care therapy | UPLC-MS | plasma | 31 lipids predicted recurrence; LPG20:5 increased predictive power of current clinical predictors; reduced triacylglycerides predicted early relapse | Li et al, 2017 [12] |
| Prostate | 159 | Overall survival | LC-MS/MS | plasma | Cer (d18:1/24:1); SM(d18:2/16:0); PC(16:0/16:0) | Lin et al, 2017 [13] |
While plasma lipidomics has not yet experienced widespread clinical implementation, the increasing use of accredited MS-based blood lipid profiling platforms for clinical diagnosis of inborn errors of metabolism and other metabolic disorders provides feasible opportunities for rapid clinical implementation of circulating lipid biomarkers in cancer. The current priority to develop guidelines for plasma lipid profiling will further assist in implementation and validation of such testing [612], as it is currently difficult to compare lipidomic data between studies due to variation in MS platforms, data normalization and processing. The next key conceptual step for plasma lipidomics is linking lipid-based risk profiles to an underlying biology in order to most appropriately design therapeutic or preventive strategies. Beyond plasma, there has been interest in lipidomic profiling of urine [613, 614] and extracellular vesicles [615] that may also prove informative as non-invasive sources of cancer biomarkers.
7.3. Tumor lipidomics
For clinical tissue specimens, instrument sensitivity initially constrained lipidomic analysis of the often limited quantities of cancer tissues available. This meant that early studies were mostly undertaken using cell line models. The numbers of different lines analyzed in these studies are often small, thus limiting their value for clinical biomarker discovery. Nonetheless, these studies have provided the first detailed information about the lipidomic features of cancer cells that impact on various aspects of cancer cell behavior, how these profiles change in response to treatment, and clues as to the initiating factors that drive certain cancer-related lipid profiles. For example, in 2010, Rysman et al. investigated phospholipid composition in prostate cancer cells using electrospray ionization (ESI) tandem mass spectrometry (ESI-MS/MS) and concluded that these cells commonly feature a lipogenic phenotype with a preponderance of saturated and mono-unsaturated acyl chains due to the promotion of de novo lipogenesis [15]. These features were associated with reduced plasma membrane permeability and resistance to chemotherapeutic agents. Sorvina et al showed using LC-ESI-MS/MS that lipid profiles could distinguish between different prostate cancer cell lines and a non-malignant line and, consistent with their MS data, staining for polar lipids showed enhanced signal in cancer versus non-malignant cells [616]. A study from 2015 by Burch et al. integrated lipidomic with metabolomics profiling (LC-ESI-MS/MS) using bioinformatics to identify and quantify differentially regulated molecules in five prostate cell lines. Their data revealed upregulation of multiple phospholipid classes and other metabolites in all malignant lines, but suggested that different lipogenic pathways are activated in metastatic cells as compared to non-metastatic and normal prostate cells [617]. Analysis of lipid and fatty acid content of breast [618] and melanoma [619] cell lines with differing metastatic potential revealed that higher levels of phospholipids containing SFA and MUFA chains (C16:0, C18:0, C18:1) were associated with greater metastatic potential. Importantly, the discovery by Roy et al (2019) of diacylglycerols being overexpressed in metastatic vs non-metastatic osteosarcoma lines allowed pharmacological targeting of diacylglycerol synthesis, which reduced cell viability and migration and provided proof of principle that certain lipidomic changes in cancer cells can support the cancer phenotype [620]. Moreover, analysis of treatment-related changes in lipid composition in cancer cells may give clues about sensitivity to novel agents, and potential adaptive metabolic changes that may underpin treatment resistance [621].
With successive gains in instrument sensitivity currently being achieved, cell line-based lipidomics has extended to pathologically annotated clinical specimens. Several studies have analyzed lipids in surgical tumor tissue or in needle biopsies, either on homogenates of the samples or by mass spectrometry imaging. For example, Marien et al. discovered 91 differently expressed phospholipid species in tumor versus non-malignant tissue homogenates from 162 non-small cell lung cancer patients [44], while Wang et al recently identified tumor-related changes in the abundance of several lysophospholipid classes compared to matched normal mucosa in colorectal cancer patients [622]. GC-MS analysis of fatty acid content in 25 matched normal and tumor samples from colorectal cancer patients revealed reduced TAG and oleate (C18:1) in tumor tissues while total phospholipids, sphingomyelin, SFAs, PUFAs and cholesterol were increased [623]. Nagai et al studied 38 cases of hepatocellular carcinoma and identified the triacylglyceride TAG(16:0/18:1/20:1) as being more abundant in tumor compared to non-tumor tissues, while TAG(16:0/18:1/18:2) was more abundant in non-tumor tissue, both alterations being validated using DESI-MSI [624]. Budhu et al studied a total of 386 hepatocellular carcinomas, including paired normal and tumor samples from 30 patients, and by integrating metabolomic and transcriptomic data identified a signature of lipid changes indicative of enhanced SCD1 activity that was associated with more aggressive cancer [625]. Increasingly, discovery studies have been performed using MSI, and tumor-specific lipid profiles have been identified using MSI in a range of cancers (key studies summarized in Table 3). In some cases, there is evidence of the lipid profile being linked to histological or pathological subtypes of cancer. It is evident from inspection of the identified lipid classifiers that certain similarities in lipid profile exist between different cancers, such as tumor specific abundance of lyso-phospholipids and PI species, although in many cases the precise identity of these lipids should be interpreted with caution unless verified using tandem MS.
Table 3:
Overview of MSI based lipidomics studies
| Cancer type | Samples | MS Platform | Main observations | Ref. |
|---|---|---|---|---|
| Bladder cancer | 20 tumors, 20 benign | DESI-MS | PS(18:0/18:1) and PI(18:0/20:4) upregulated in tumor tissue | [14] |
| Brain cancer | 36 | DESI-MSI | Lipid signatures varied between subtypes of glioma, PI(18:0/20:4) and PS(18:0/22:4) were the most significant discriminatory lipids | [15] |
| Breast cancer | 28 | MALDI-TOF/TOF | Cancerous regions enriched in mono-unsaturated PC species versus saturated | [16] |
| Breast cancer | 8 | MALDI-TOF/MS | A complex signature of +/− 20 PC and TAG species differentiates cancer from normal. | [17] |
| Breast cancer | 56 | MALDI-iMScope | Two different populations of cancer cells that predominantly expressed either PI(18:0/18:1) or PI(18:0/20:3). | [18] |
| Breast cancer | 67 Tumors, 55 containing matched benign | DESI-MSI | Highly saturated lipids could distinguish breast cancer from benign tissue. Distinct lipid profiles were evident across different molecular tumor subtypes, and PE38:6 and PS38:3 plus 2 fatty acids were able to distinguish ductal carcinoma in situ from invasive breast cancer | [19] |
| Breast cancer | 86 invasive tumors, 45 normal tissue | DESI-MSI | Complex signature of >40 lipid masses could distinguish cancer from normal, and was validated in two independent centers | [20] |
| Colorectal cancer | Training: 12 Validation: 40 | MALDI-TOF/TOF | Lipid signature correlates with prognosis | [21] |
| Colorectal cancer | 12 tumors, 12 matched benign tissue | MALDI-MSI | Cancerous tissues had higher levels of PC(16:0/18:1), lysoPC(16:0) and lysoPC(18:1) compared to matched normal tissue | [22] |
| Gastric cancer | 62 | DESI-MSI | Lipid signatures distinguished gastric cancer from normal tissue, and were validated on surgical margin samples | [23] |
| Lung adenocarcinoma | 25 | LC-MS + MALDI-iMScope | Enriched monounsaturated/saturated PC ratios in cancerous regions | [24] |
| Oral cancer | Training: 3 Validation: 1 | MALDI-TOF/TOF | Logistic regression classifier allowed for a high precision labeling of cancerous regions | [25] |
| Ovarian cancer | 107 | DESI-MSI | Lipid profiles were tissue type-dependent. PA, PS, PE, PG and ceramide species were discriminatory for cancer versus normal. | [26] |
| Medulloblastoma + Pineoblastoma | MB: 8 PB: 3 | MALDI-FTMS | Glycerophosphoglycerols and sphingolipids allow differentiation between MB and PB | [27] |
| Prostate cancer | 10 | MALDI-FTMS | 31 lipids correlated with Gleason score | [28] |
| Prostate cancer | 52 | MALDI-IMS | Cancerous tissues had higher levels of PI species (PI(18:0/18:1), PI(18:0/20:3), PI(18:0/20:2) | [29] |
| Prostate cancer | 31 | MALDI-IMS | Decreased levels of lyso PC (16:0/OH) and SM(d18:1/16:0) in tumor vs benign regions of tissues; lower tumoral lysoPC(16:0/OH) levels were associated with biochemical relapse | [30] |
| Prostate cancer | 18 | DESI-MSI | Benign tissue regions had higher levels of lysoPEs, PI and citrate; Malignant regions had higher levels of FAs, PE, PC, PI and glutamate | [31] |
| Prostate cancer | 68 | DESI-MSI | Cholesterol sulfate was higher in malignant and pre-malignant lesions compared to benign tissue | [32] |
| Non-small cell lung cancer | Discovery: 73 Validation: 89 | ESI-MS/MS + MALDI-FTMS | 40 and 42 carbon PC and PE species enriched in cancerous regions; decrease in SM and specific PI | [33] |
| Renal cell cancers | 20 normal 15 benign 46 tumors | DESI-MSI | Lipid profiles could distinguish normal kidney from benign neoplasms and different subtypes of cancer, increased cardiolipins were prominent features | [34] |
| Skin cancer (basal cell carcinoma) | 86 | DESI-MSI | PG(18:1/18:1), PS(18:0/20:4), PI(16:1/18:0) and PI(18:2/18:0) more abundant in carcinoma versus normal skin; cholesterol sulfate was higher in normal compared with tumor tissue | [35] |
Although not standardly used in the clinic so far, the distinctive lipid profile of cancer tissue may have interesting applications as diagnostic biomarkers. As modern mass spectrometry imaging can be performed in minutes, tumor-characteristic lipid profiles may yield important information for clinical decision making in the operating theater [626]. In another setting, a special surgical knife is used (referred to as iKnife) that evaporates the tissue, which is analyzed in line and in real time by mass spectrometry using the REIMS technology, providing instant instructions to the surgeon. In some cases lipid profiles have been identified related to therapeutic response [627, 628], which raises the possibility that lipids may have useful prognostic value. Similarly, the MasSpec Pen identifies cancerous tissue in real time based on profiles of lipids, proteins and metabolites.
8. Lipids and lipid metabolism as promising tools and targets for anti-cancer therapies.
In view of the central role of lipids and altered lipid metabolism in the pathophysiology of cancer, the exploitation of lipid metabolism as a potential target for antineoplastic therapy has become a very active domain of research. Since the discovery of the overexpression and dysregulation of enzymes involved in lipid metabolism in cancer, various natural and synthetic compounds targeting lipid metabolism have been reported to be effective against cancer cells with a significant degree of selectivity. Much of the effort has focused on compounds targeting liquid acquisition, particularly de novo lipogenesis, with some compounds making it to clinical trials. One of the challenges of this approach is the context dependence of lipid acquisition and the high degree of plasticity of cancer cells, leading to the exploration of lipid uptake as an alternative or complementary target. One particularly promising antineoplastic strategy in this context exploits this plasticity in lipid acquisition to increase cellular lipid polyunsaturation to induce a unique vulnerability to ROS and to a variety of therapeutics that evoke changes in the redox balance as part of their mechanism of action. This strategy has been successfully employed in preclinical models to (re)sensitize cancers to chemo- and targeted therapies. Other approaches aim at disbalancing lipid homeostasis by interfering with specific lipid metabolism enzymes, inducing lipid toxicity. Besides targeting lipid metabolism, lipids themselves have also shown promise as anti-cancer lipid drugs, and as critical components of nanomedicines and lipid-based vehicles of therapeutics. Here we provide a short overview of some of the most promising or emerging therapeutic strategies based on lipids and altered lipid metabolism in tumors.
8.1. Cutting off lipid supplies
Based on the high dependence of many cancers on lipids, several approaches have been explored to cut off lipid supplies to cancer cells. As detailed above, cancer cells can acquire lipids through various routes, including de novo lipogenesis and lipid uptake.
Since the discovery of the overexpression of FASN in many cancers, this enzyme has been a prime target for anti-lipogenic drug discovery (Table 4). Initial studies with the fungal antibiotic cerulenin showed promising anti-proliferative and death-inducing effects in many cell lines, but suffered from the poor selectivity of this compound. Other natural compounds, including flavonoids such as quercitin, luteolin and EGCG found in green tea, were shown to block lipogenesis in cancer cells, along with their many potential mechanisms of action. Orlistat, an approved anti-obesity drug that reduces fat uptake from the gut by inhibiting lipases, has also been shown to inhibit FASN and to attenuate tumor growth in preclinical models. The first synthetic anti-FASN compound C75 showed potent effects in several preclinical models in vivo, but also produced severe side effects, including a dramatic weight loss caused in part by accumulation of malonyl-CoA and by a proposed role for FASN in neuronal stem cell functioning [629, 630]. Next generation compounds targeting FASN such as C93, IPI-9119 and TVB-2640 appeared less toxic and showed significant potential in various preclinical models. One of the compounds that has progressed most is TVB-2640 which is being explored for colon and other cancers in a phase I study and has entered phase II clinical trials for HER2 -positive BC in combination with paclitaxel and trastuzumab [285, 631, 632]. Interestingly, inhibition of FASN has also been shown to impair angiogenesis through mTOR malonylation [101].
Table 4:
Overview of potential agents as modulators of lipid metabolism in cancer
| Target | Agent | Phase of Development | Comments | Reference |
|---|---|---|---|---|
| FASN | Orlistat | Approved | Antiobesity lipase and FASN inhibitor; not yet assessed clinically for cancer | [36, 37] |
| cerulenin | Preclinical | [37, 38] | ||
| C75 | Preclinical | [580, 581][37, 38] | ||
| C93 | Preclinical | [39] | ||
| IPI-9119 | Preclinical | [40] | ||
| TVB-2640 | Phase II | Trials in breast cancer +/− trastuzumab/docetaxel | [41–43] | |
| Fasnall | Preclinical | [44] | ||
| ACACA | TOFA | Preclinical | [45–47] | |
| Soraphen A | Preclinical | [48] | ||
| ND-646 | Preclinical | [49, 50] | ||
| HMGCR | Statins | Approved | Association studies between statin use and outcomes in many cancers, some randomized trials | [51–54] |
| CD36 | ABT-510 | Phase I | Trial in metastatic melanoma | [55] |
| SREBPs | Fatostatin | Preclinical | [56–59] [60, 61] [62] | |
| Betulin | Prelinical | [63–65] | ||
| Sibilin (milk thistle) | Clinical | Nutritional supplement in trials for improvement of toxicity/quality of life in acute lymphoblastic leukemia, head and neck cancer and BC | [66–68] | |
| AMPK | MT 63–78 | Preclinical | [69] | |
| JWH-133 | Preclinical | CB2R agonist | [70–75] | |
| GPR119 agonists (GSK1292263; MBX-2982) | Phase I/II for T2DM | Preclinical studies in cancer | [76, 77] | |
| Metformin | Approved for T2DM | Randomized clinical studies in lung and breast cancer | [78–81] | |
| AICAR | Approved for T2DM | Preclinical studies in cancer | [82, 83] | |
| LXR | LXR-623 | Phase I for atherosclerosis | Preclinical studies in cancer | [84] |
| ACSLs | Triacsin C | Preclinical | ||
| ATGL | atglistatin | Specific for murine ATGL | [85] | |
| MAGL | JZL184 | Preclinical | [86] | |
| SCD | A939572 | Preclinical | [87–89] | |
| T-3764518 | Preclinical | [90, 91] | ||
| SSI-4 | Preclinical | [92] | ||
| SW203668 | Preclinical | [93] | ||
| CPT1 | Etomoxir | Preclinical | [94–96] | |
| Perhexiline | Approved (Aust & NZ) | [97–99] | ||
| ST1326 | Preclinical | [100, 101] | ||
| PLD | FIPI | Preclinical | Targets immune infiltration into tumors | [102] |
| VU0155072–2 | Preclinical | Targets immune infiltration into tumors | [102] | |
| FABP | EI-05 | Preclinical | [103] | |
| SBFI-102, 103 | Preclinical | [104] | ||
| Choline kinase | HC-3, JCR and MN derivatives | Preclinical | [105] | |
| Triterpene quinone methides | Preclinical | Derived from natural products | [106] | |
| ICL-CCIC-0019 | Preclinical | [107] | ||
| EB-3D, EB-3P | Preclinical | [108–110] | ||
| Farnesyltransferase | tipifarnib | Phase I/II | Phase II studies in metastatic BC and squamous cell carcinoma; Phase I trial in glioblastoma | [111–113] |
| lonafarnib | Phase I/Ib | Largely combination studies in glioblastoma, BC | [114–116] | |
| BMS-214662 | Phase I | Single agent or in combination with paclitaxel in advanced solid tumors | [117, 118] | |
| Palmitoylation | 2-bromopalmitate | Preclinical | [119–121] | |
| Acid sphingomyelinase | Fluphenazine | Preclinical | [122] |
Other enzymes of the pathway that have been explored as potential targets are ACACA and ACLY. Early studies on ACACA inhibition were performed with TOFA, which upon conversion to TOFyl-CoA (5-tetradecyloxy-2-furoyl-CoA) exerts an allosteric inhibition on ACACA. These studies showed promising results with induction of apoptosis in many cancer cell lines, but were blurred by its poor efficacy and the concomitant depletion of cellular CoA stores. The natural compound soraphen A, a myxobacterial metabolite, appears to be very efficacious in cell lines in vitro, even at nanomolar concentrations. Its death-inducing potential seems to depend on the abundance of exogenous lipids. The applicability of this compound is also limited by low bioavailability in vivo. Promising candidate drugs from the ND-600 series that were developed in the context of other metabolic diseases including dyslipidemia, steatosis, and obesity, have brought the targeting of ACACs in the cancer field closer to the clinic [633]. ND-646, a small molecule allosteric inhibitor of both ACACA and ACACB that prevents enzyme dimerization, has shown efficacy in preclinical models of non-small-cell lung cancer and breast and liver cancer and is in clinical trials [634]. As a dual inhibitor of both ACAC enzymes, the compound both inhibits lipogenesis and enhances FAO (vide infra). In this sense, ACAC and FASN inhibition may not be equivalent. FASN inhibition results in an accumulation of Malonyl Co-A which is the final product of the upstream enzyme ACACA, but is also a potent inhibitor of beta oxidation, and therefore FASN inhibition also blocks beta oxidation [103]. Conversely, ACAC inhibition may have the opposite effect, leading to a depletion of malonyl Co-A and may further drive beta oxidation. Inhibition of ACLY also attenuates tumor growth by regulating levels of acetyl-CoA, which feeds both FA and cholesterol synthesis. It also affects acetylation of proteins and subsequently evokes changes in gene expression.
The cholesterol synthesis pathway is another potential target. Notably, the use of statins, which inhibit cholesterol synthesis by targeting the rate-limiting HMG-CoA reductase enzyme and which are widely used as cholesterol lowering drugs, has been associated with a reduced risk of cancer development in animal models and in some, but not all cancers in human epidemiological studies. In a treatment setting, statin use has been associated with reduced mortality or recurrence in a wide range of cancers [635], although a recent meta-analysis of randomized trials in cancer showed no significant effect of adding statins to therapy on progression-free or overall survival [636, 637]. Moreover, re-analyses of large scale association studies on statin use have revealed low levels of evidence for a protective effect of statins on cancer incidence [638] or overall survival [637, 639]; emphasizing the need for larger, randomized Phase III trials in cancers where the strongest epidemiological data exists- although the feasibility of such studies is compromised by the current widespread use of statins for hypercholesterolemia in Western countries. Any enhanced outcome due to statin use may be in part be mediated by the reduction of circulating cholesterol and by changes in protein isoprenylation, which is also affected. In experimental studies, statins reduce the viability of cancer cell lines. Further evidence for cholesterol synthesis as a potential target comes from studies targeting the first enzymes committed to cholesterol synthesis i.e. squalene synthase.
A possible limitation of targeting lipid synthesis is that cancer cells may be able to compensate by increasing lipid uptake. However, it is conceivable that the kinetics of lipid uptake in a poorly vascularized tumor may be insufficient to fully compensate. Nevertheless, targeting lipid uptake has provided beneficial effects in a number of pre-clinical models. A challenge in targeting lipid uptake is that there are multiple mechanisms that may compensate for each other, including other receptors, endocytosis, or tunneling nanotubes [640]. One of the mechanisms that is shown to play critical roles in lipid uptake in several models and that shows promise as a therapeutic target is CD36. Targeting CD36 is shown to be a promising avenue in several preclinical studies in various cancer types including glioblastoma, melanoma and prostate cancer [159]. Most of these targeting approaches are based on TSP-1 mimetics. Some of these, such as ABT-510 have reached phase I and II clinical trials. It should be noted that interference with CD36 does not exclusively affect lipid uptake [641]. Several FABP inhibitors have been developed and tested for the prevention and treatment of obesity, atherosclerosis, diabetes, and metabolic syndromes. In cancer, most studies have used knockdown of FABP5, but recently the FABP5 inhibitors SBFI-102 and 103 have been shown to suppress prostate cancer growth and synergize with taxane-based chemotherapeutics [642]. On the other hand, activation of epidermal FABP (E-FABP) by EI-05 suppresses mammary tumor growth by promoting the anti-tumor activity of macrophages [643].
Targeting transcription factors as regulators of lipid metabolism may be another interesting approach. As detailed above, many cancers show an activation of SREBP-1 but targeting transcription factors such as SREBPs remains challenging. Fatostatin is an inhibitor of SREBPs that was originally developed to block insulin-induced adipogenesis [644]. This compound directly binds SCAP at a site distinct from the sterol-binding domain and hinders ER-to-Golgi transport of the SREBP-SCAP complex. Fatostatin: i) blocks hepatic lipid accumulation and body weight gain in obese mice; ii) inhibits cell growth by impeding intracellular shuttling in a SCAP-independent manner [645]; iii) has antiproliferative effects that are mediated via inhibition of mitotic microtubule spindle assembly [646]; iv) has shown to be a promising anticancer agent in BC (induced a significant reduction of several key genes in the cholesterol biosynthesis pathway including HMGCR and SQLE and blocked cell invasion in ER+ BC but not TNBC and specifically in LTED cells [416, 647], prostate (both AR-positive and metastatic AR-negative prostate cancer cells) [381, 648] and pancreatic cancer [649]. Blocking SREBP translocation with Fatostatin in prostate cancer cell: i) suppressed cell proliferation and anchorage-independent colony formation in both androgen-responsive LNCaP and androgen-insensitive C4–2B prostate cancer cells, ii) reduced in vitro invasion and migration in both cell lines causing G2/M cell cycle arrest, iii) induced apoptosis by increasing caspase-3/7 activity and cleavage of caspase-3 and PARP (Poly (ADP-Ribose) Polymerase), iv) significantly inhibited subcutaneous C4–2B tumor growth and markedly decreased serum PSA level compared to the control group in vivo animal results, v) decreased the expression of AR and its target gene PSA in vitro and in vivo [358, 381, 648].
Betulin is a natural compound abundant in birch bark that inhibits the maturation of SREBPs by directly interacting with SCAP and improves hyperlipidemia, insulin resistance and atherosclerotic plaques [650]. Betulin decreases hepatocarcinoma development and progression through reduction of SREBP-driven lipogenesis and attenuated inflammatory responses by down-regulation of tumor-promoting cytokines, including interleukin 6 (IL6), tumor necrosis factor alpha and IL1b [651]. Nelfinavir and its analogues block S2P cleavage leading to suppression of proteolytic activity and accumulation of SREBP1 precursor and ATF6 [652]. Nelfinavir induces liposarcoma apoptosis and is able to inhibit castration resistant prostate cancer cells proliferation in vitro [652, 653].
Silibin is a natural compound isolated from the seeds of milk thistle plant (Silybum marianum) and widely consumed as a hepatoprotective agent. Through activation of AMPK, SREBP1 phosphorylation is increased in turn inhibiting SREBP1 nuclear translocation. In this way, Silibin decreases nuclear protein levels of SREBP1 and their target genes in prostate cancer cells leading to reduced lipid and cholesterol accumulation with consequent cell cycle arrest and inhibition of tumor cell proliferation. Silibin also blocked androgen-induced lipid accumulation and prevented the development of androgen-independent LNCaP cell clones via targeting SREBP1 [654].Other agents targeting SREBP through activation of cAMP-PKA or AMPK signaling pathways [391] include i) indirect: Metformin, Thiazolidinediones, Resveratrol, and agonists of GLP1R, the cannabinoid receptor CB2R, and GPR119; and ii) direct: AICAR (5-aminoimidazole-4-carboxamide ribo-nucleotide), PT-1, S396 (inhibits the transcriptional activity of SREBP) (reviewed in [396]) and MT 63–78 [102]. Metformin has received significant attention due to epidemiological associations between its use as an anti-diabetic and cancer incidence and/or outcomes (reviewed in [635]), although better designed studies have now weakened associations with cancer risk [655]). Randomized trials of metformin with TK inhibitors in lung cancer have yielded conflicting results [656, 657], while no effects were seen in BC for randomized trials of metformin added to chemotherapy [658] or endocrine therapy [659]. The results of a number of ongoing Phase II randomized trials are now awaited to reveal the potential of metformin to improve cancer patient outcomes. Agonists of the cannabinoid receptor CB2R have shown preclinical efficacy against growth and/or invasion of cancer lines in vitro [660–665] and suppress in vivo tumor growth and metastasis [660, 661, 664], while MT 63–78 is showing promise as a specific and potent direct AMPK activator able to inhibit prostate cancer cell growth both in androgen sensitive and CRPC models, inducing mitotic arrest, and apoptosis [102].
Beyond SREBP, many studies have reported the in vitro and in vivo antiproliferative effect of LXR activation in all types of cancers [666], and PPAR-γ activation induces cell cycle arrest in several malignant cell lineages [667]. However, animal studies and clinical trials have not been conclusive on the beneficial effect of PPAR agonism as antineoplastic therapy [667]. Further attention to these equally important regulators of lipid metabolic genes may yield novel agents and combinatorial strategies.
8.2. Blocking downstream lipid metabolism
Given the challenges outlined above in the targeting of transcription factors such as SREBPs and their upstream regulators, interest has also focused on directly targeting lipid metabolic enzymes themselves and, most notably, de novo lipogenesis via FASN. More recently, interest has broadened into inhibition of other key metabolic enzymes involved in synthesis, uptake and utilization of FAs. This has provided the opportunity not only to develop novel anticancer agents, but also to repurpose existing enzymatic inhibitors previously developed for metabolic disorders such as type II diabetes or hypercholesterolemia. Some promising new therapeutic targets are discussed below.
In the context of the cancer’s plasticity in lipid acquisition, ACSLs may be interesting targets to block the use of FAs irrespective of whether they are synthesized de novo or acquired exogenously. Although ACSL enzymes are required for the assembly and storage of FAs, they play complex biological roles in physiology and cancer means that the context dependent contribution of their roles should be carefully considered. Specifically, whereas ACSL3 may be a good target in the context of lipid uptake dependent tumors, ACSL4 inhibition may be detrimental in helping to saturate cell membranes and protect cells from ROS stress.
Besides membrane phospholipids as a source of FAs, FAs can be assembled from neutral fat stores by the enzymes ATGL, HSL and MAGL [574]. ATGL in particular has been shown to have oncogenic roles in colorectal and lung cancer cells [575, 576], and may contribute to BC growth and invasiveness by releasing adipose derived FAs [577]. A pharmacological inhibitor of ATGL is available [578]. Inhibition of MAGL by the small molecule JZL184 suppresses tumorigenesis of melanoma and ovarian cancer cells [581]. ATGL knockdown or chemical inhibitor such as atglistatin suppresses the growth of several types of cancer cells, although ATGL expression in human malignancies is lower than in adjacent normal tissues. Inhibitors (JZL184) or shRNA probes that target MAGL can impair prostate cancer cell aggressiveness.
Knockdown or chemical inhibition of SCD1 shows promising efficacy and treatment sensitization in a range of cancers [206–209], while inhibition of one or both FADS enzymes has shown preclinical efficacy in intestinal cancer [217]. Whereas the role of FA and membrane lipid desaturation in cancer is well-described, and novel agents are available that are currently being evaluated in preclinical cancer models (see Section 4.3), comparatively less progress has been made in targeting of membrane lipid elongation in cancer. However, as described in section 4.4, membrane lipid elongation is a common feature of many cancers. The main limitation of ELOVL targeting in cancer is a current lack of development of small molecule inhibitors, further complicated by the membrane-bound structure of the ELOVL enzymes. Nevertheless, inhibitors of ELOVL6 have been synthesized [668–672], some of which show cross-selectivity for ELOVL3, although these have not yet been studied for their anticancer properties. Hyperlipidemic agents bezafibril and gemfibrazil have been reported to inhibit ELOVL1 [673], but it would be difficult to mechanistically separate their effects on ELOVL1 from their effects on cholesterol and other lipids in any preclinical investigations. Overcoming the technical challenges of crystallizing and developing inhibitors of this intriguing enzyme family will allow selective inhibition of different elongation pathways in cancer cells, which will provide insight into the relative significance of each pathway and its various lipid products for tumorigenesis and metastasis.
Pharmacological inhibition of FAO using the CPT1 inhibitor etomoxir or perhexiline not only reveals single agent efficacy in cancer cell lines [674–678], but also sensitizes tumor cells to chemotherapy [149, 246, 247, 679], radiotherapy [680, 681] and endocrine therapies [682]. Notwithstanding the fact that at least some of the anticancer properties of etomoxir occur via non-CPT1A-related mechanisms [683], these reports highlight the importance of FAO not only for cancer cell survival, but also as a key mechanism of resistance to therapy.
FABP modulators include derivatives of niacin, quinoxaline, arylquinoline, and bicyclicpyridine. They modulate the interaction of FAs with FABPs and can have dual effects in a context dependent manner. For example, an activator of epidermal FA binding protein, EI-05, suppresses mammary tumor growth in mice [643], while inhibitors of FABP-5 are active alone or can synergize with taxanes to inhibit prostate tumor growth in mice [642].
Constitutive activation of choline kinase is a key metabolic feature of oncogene-driven cancers, resulting in increased cellular phosphocholine levels. A range of choline kinase inhibitors have been developed since the 1990s, and exhibit antiproliferative activity in cancer cells [684–688], however none have yet been investigated clinically.
Lipidation of oncoproteins presents a novel vulnerability for cancer therapy, as this post-translational modification can stabilize or activate a range of cancer cells [281]. Farnesylation in particular has experienced a strong focus for drug development in cardiovascular disease, and novel clinical agents (e.g. tipifarnib, lonafarnib, BMS-2154662) have recently been repurposed for cancer in a series of Phase I/II studies evaluating combinatorial efficacy, with promising results. Palmitoylation has been targeted using a preclinical agent, 2-bromopalmitate, which has demonstrated sensitization of osteosarcoma cells to the chemotherapeutic agent adriamycin [689] and revealed an intriguing role for palmitoylation of PD-L1 in enhancing its stability, with 2-bromopalmitate enhancing T-cell immune responses in colon and breast tumor models [690, 691]. Given the increasing interest in harnessing immunometabolism for cancer therapy, these agents afford an exciting new approach to immunotherapy beyond the current anti-PD-L1 antibody approaches.
8.3. Targeting lipid metabolism in combinatorial approaches as sensitizer to other therapies
A plethora of evidence points towards the contribution of lipid metabolism to multiple aspects of cancer. Although the contributions of blunt approaches such as blocking lipogenesis or lipid uptake have translational effects in preclinical models, they generally exert a cytostatic effect or reduce the metastatic disease burden, but they are not curative. A more rational and less complex approach is to exploit context and tissue dependent vulnerabilities acquired by cancer cells. In this way, the magnitude of the sum of multiple combined approaches that exploits acquired vulnerabilities is many times greater than the contribution of each separate approach. The concept of such approaches often termed ‘synthetic lethality’ is certainly not unique to metabolism, but may be particularly applicable to it, as in contrast to degenerate signaling pathways, lipid metabolic pathways often converge on a few key enzymes. Therefore, if a lipid metabolic pathway becomes less dispensable, it can be a potent antineoplastic target. For example, in a particularly lipid deficient environment such as in a solid tumor, lipogenesis will be required to generate membrane biomass, whereas in a lipid rich environment such as that of primary breast and prostate cancers, targeting lipid uptake may be more prudent. Combinatorial approaches in targeting lipid metabolism in cancer, often combined with standard of care therapies, is emerging as an immensely fruitful field in translational research.
The intimate link between growth factor and oncogenic signaling and lipogenesis is well-established, as cell proliferation requires the generation of biological membranes. Castration resistant metastatic prostate cancer re-activates endogenous androgen receptor signaling, and moreover rapidly develops resistance to antiandrogen compounds, often through amplification of the androgen receptor gene or the generation of novel splice variants such as the ARV7. Importantly, the androgen receptor promotes a program of SREBP-dependent lipogenesis, which contributes to therapy resistance to antiandrogens, whereby a combination of anti-androgens and FASN inhibition synergistically reduce disease burden [103]. Similarly, in liver cancer cells the combination of AMPK mediated ACACA inhibition synergizes with antiangiogenetic therapy, with a possible mechanism being that under a hypoxic and nutrient deprived setting, lipogenesis becomes increasingly critical in promoting cell proliferation [692]. In both these examples, the combinatorial approaches include an addition over the standard of care, allowing more rapid clinical adoption. In addition to the role of oncogenes and growth factor receptors in promoting lipogenesis to power membrane biogenesis, lipogenesis may in turn power oncogenic signaling. FASN mediated lipogenesis contributes to HER2 expression and clustering, and in turn the combined inhibition of HER2 and FASN induces apoptosis in BC cell lines in vitro [693]. Similarly, ELOVL2 mediated enrichment of PUFA containing membrane PL species promotes EGFR signaling in glioma, and the combination of ELOVL2 KO and anti-EGFR therapy exerts a dramatic antineoplastic effect [224]. This is further supported by a study in lung cancer showing that SREBP mediated changes to membrane fluidity promote EGFR phosphorylation and combined inhibition of SREBP and EGFR shows translational benefit [694]. Menendez and Lupu show that FASN regulates E2/ERα signaling in BC cells. Inhibition of FASN activity induced reduction of ERα protein, suppression of E2-stimulated BC cell proliferation and inactivation of AKT mediating E2-promoted anchorage-independent colony formation. Furthermore, treating BC cells with pure antiestrogen or inhibitor of MEK/ERK, blocked the interaction between FASN blockade and E2-stimulated ERα transactivation [695].
In tumors that are surrounded by a lipid rich environment, lipid uptake may play a dominant role over lipogenesis in establishing a similar phenotype. Although inhibiting lipid uptake remains challenging, combinatorial approaches for inhibiting CD36 mediated lipid uptake have been met with some success in preclinical models. In HER2 positive therapy resistant models, CD36 deletion can re-sensitize cancer cells to lapatinib [158]. Several lines of evidence point towards the context dependent requirement of FAO under conditions of increased lipid uptake. In non-solid leukemia cells which do not suffer from the drawback of a nutrient deficient environment, the oxidation of exogenous FAs drives energy generation. Leukemia cells are therefore sensitive to the combination of beta oxidation inhibitors and apoptosis inducers [696]. In melanoma cells, anti-MAP kinase therapy inhibits lipogenesis and kills the majority of melanoma cells, however a small subset of persister cells survive, and they in turn require CD36 mediated lipid uptake to drive beta oxidation. Combinatorial inhibition of beta oxidation and MAP Kinase inhibition significantly attenuated the ability of cells to develop therapy resistance [697].
Lipogenesis inhibition sensitizes lung cancer cells to carboplatin [634, 698]. The fact that several tumor types are sensitized to cytotoxic chemotherapy in combination with lipogenesis inhibitors hints at roles for lipids other than membrane biogenesis and energy production. A study showed that inhibition of lipogenesis promotes membrane lipid poly-unsaturation mediated by lipid uptake, and this in turn confers a sensitivity to ROS inducing agents such as chemotherapeutics [15]. Since this publication, further evidence supporting this claim has come to light. In BRAF mutant melanoma models, therapy resistance depends on sustained lipogenesis mediated by SREBP activity. Inhibition of SREBP by SCAP targeting compounds betulin or fatostatin drive membrane lipid poly-unsaturation and confer sensitivity to ROS elevation in melanoma. The combination of SREBP inhibition synergizes with BRAF inhibition to elevate ROS, and exerts a potent antineoplastic effect in therapy resistant melanoma [16, 699].
Besides chemotherapy, radiotherapy is an often-critical early therapeutic step in cancer treatment, and much like chemotherapy, its cytotoxic effects are in part mediated by ROS. Concordantly, the combination of radiotherapy and lipogenesis inhibition synergistically decreased tumor growth in mouse models of prostate cancer [700]. Recently, it is shown that under ionizing radiation, cancer cells increase the expression of ACSL4 which can act as a potent inducer of ferroptosis. Moreover, radiotherapy combined with ferroptosis inducers led to the radio-sensitization of cancer cells [701, 702]. Promisingly, radiotherapy can work in concert with immunotherapy to sensitize tumor cells to ferroptosis, and effect that can be further enhanced by ferroptosis inducers [703].
8.4. Dietary intervention of cancer
Since many cancers have the ability to take up lipids and since excessive caloric intake and obesity are associated with cancer aggressiveness, reoccurrence and resistance to therapy, diet adjustments could have significant benefits in some types of cancer. In a BRAF V600E mutant melanoma xenograft model in mice, a high fat diet resulted in enhanced tumor growth, while overall survival and response to dacarbazine in obese melanoma bearing mice could be improved by weight control intervention [704, 705]. Conversely, in so called ketogenic diets, which are high in fat but low in carbohydrates with an overall normal caloric intake, several studies have described anti-cancer effects such as reducing the growth of a glioblastoma PDX model [706] or sensitizing tumors to targeted therapies [707, 708]. These studies suggest that beyond the total lipid levels in the diet, the total caloric intake and the lipid composition of the diet play an important role.
Whereas saturated fat overall has been shown to increase the risk of several cancers, MUFA have been reported to be protective. Particularly olive oil appears to be effective in several studies [709, 710]. These effects may not be entirely attributed to its high content of MUFAs, but also its high content of lipid-soluble antioxidants such as alpha-tocopherol, which protects against free radical-induced lipid peroxidation [711]. High intake of omega-6 PUFAs has been linked with a poor outcome in cancer patients, whereas omega-3 lipids appear to ameliorate cancer. Multiple mechanisms have been reported, including a differential effect on the production of prostaglandins and other eicosanoids [712, 713]. Several studies have reported that supplementation of conjugated linoleic acid (CLA), can protect against cancer in animal models of chemical carcinogenesis [714]. CLA is thought to modulate prostaglandin metabolism, to affects growth factor signaling, activation of PPARs-alpha and several other mechanisms. Interestingly, a proof of principal biomarker study of CLA administration to newly-diagnosed BC patients showed encouraging results [715]. In view of the well-known role of eicosanoids and other oxylipins in the communication with the immune component, it will be interesting to see to what extent modulation of lipid composition by the diet affects the outcome of immunotherapy as a rapidly expanding therapy option for many cancers.
8.5. Transdifferentiation of cancer cells by modulating lipid metabolism
Another interesting therapeutic approach based on the modulation of lipid metabolism involves the transdifferentiation of cancer cells into fat cells. This concept has been proposed for cancer stem cell transdifferentiation by treating them with specific unsaturated FAs [716]. Recently, a similar strategy has been proposed to exploit the plasticity of cancer cells to undergo endothelial-mesenchymal transition to force them to transdifferentiate into post-mitotic adipocytes, thereby blocking primary tumor invasion and metastasis. In preclinical BC model systems this was achieved by a combination treatment with the antidiabetic drug rosiglitazone (a PPAR agonist) and inhibitors of MEK [717].
8.6. Anti-cancer lipid drugs
Since the 1970’s several synthetic lipids, mainly alkylphospholipids, have been shown to be effective towards several diseases, including cancer. These molecules resemble natural ether lipids and thus lack ester bonds, making them more resistant to the degradation by lipases. Alkylphospholipids incorporate into the cell membrane and exert their effects in part by targeting lipid microdomains, membrane disorganization and changes in signaling of specific proteins. Several alkylphospholipids are currently in clinical use, such as miltefosine, edelfosine and perifosine. In the context of cancer, edelfosine has been studied in a variety of tumor types. In cell line models, edelfosine inhibits survival pathways such as MAPK/ERK and Akt/PKB pathways and activates the Fas/CD95cell death receptor, and shows significant selectivity towards cancer cells. In animal models, edelfosine showed a higher accumulation in cancer tissues and was active against several cancer types. In phase I and II clinical trials, edelfosine was shown to be safe and possibly effective. Recently, alkylphospholipids such as edelfosine have been tested as conjugations with other drugs targeting lipid metabolism, including quercitin, and are being exploited in nanoassemblies with chemotherapeutics as nanomedicines (vide infra) [718]. Several other synthetic lipids have been tested in cancer models including minerval, a synthetic 2-hydroxyoleic acid (2OHOA) and propofol-DHA [719].
8.7. Lipid-directed drug targeting approaches in cancer
The differential lipid composition of cancer cells can also be exploited to target drugs to cancer cells. One of these strategies exploits the externalization of phosphatidylserine (PS) to the surface of cancer cells. Both in vitro and in vivo, it was shown that specific nanovesicles containing saposin C specifically target tumor cells by recognizing PS on the cell surface [720]. Tumor targeting is further enhanced by the fact that the affinity of saponin C for PS is the highest at acidic pH, this way exploiting the acidic microenvironment of tumors. Other approaches involve the targeting of PE by compounds such as duramycin, cinnamycin, cyclotides and ophiobolin A.
8.8. Lipid-based drug delivery systems for cancer therapeutics
Due to tumor-specific constraints including poor vascularization and high interstitial pressure, efficient drug delivery into tumors has remained a challenge. Lipid-based vesicles, including liposomes, microbubbles or nanoparticles have long been explored as carriers for therapeutics. Because of their ability to ‘shield’ toxic compounds, their small size favoring tissue penetration, high payload, long retention times and efficient uptake by cancer cells, lipid-based or lipoprotein-based vehicles are increasingly studied as drug delivery systems, with major advances in the last few years. Some of these carriers exploit the unique natural properties of lipoprotein particles, including their binding to lipoprotein receptors, which are frequently overexpressed in cancer cells to support lipid take up (vide supra). They are internalized through receptor-mediated mechanisms, upon which the therapeutic load is released, depending on the nature of the vehicle. Both natural and recombinant LDL-and HDL-derived particles and phospholipid-based nanovectors and nanodiscs, of which the lipid composition can be modulated, are being explored in combination with diverse groups of therapeutic agents such as chemotherapeutics (paclitaxel, hydroxycamptothecin), imaging agents, radioactive compounds, photodynamic agents, nucleic acids including siRNAs, proteins and carbohydrate complexes [721]. Currently some 50 nanoparticles are FDA approved including some for the treatment of cancer [722].
New players on the block are extracellular vesicles (EVs), which are derived from cells. As they are natural, they are thought to be less susceptible to the host immune system than artificial nanoparticles. Using various physical and chemical methods, EVs can be loaded with cancer drugs or other cancer targeting agents. Their surface can be decorated with specific homing peptides to enhance selective uptake by target cells through direct fusion with plasma membrane or through endocytosis pathways [723, 724]. The implementation of EVs as lipid-based drug delivery systems awaits however further preclinical developments, including maximization of drug loading, more selective targeting and optimization of large scale production and purification, and achieving safety requirements by FDA and EMA (reviewed in [725]).
9. Future perspectives
Although a link between lipids and cancer has been known for decades, recent years have witnessed an explosion of new findings portraying a complex and intricate network of alterations in lipid metabolism in cancer that involves nearly every lipid-related pathway and biological function. Recent advances in lipid analysis technologies predict that our current knowledge represents only the tip of the iceberg. Current lipidomics approaches cover only a small fraction of the more than 200,000 predicted lipid species. Many less abundant lipid species remain under the radar, yet may play important roles for instance in the intricate interplay between cancer and immune cells. In this context, recent developments in ionization methods including MALDI-2 and their application for instance in redox lipidomics, provide an exciting opportunity to study rare lipids [726, 727]. In view of the ongoing shift in cancer research towards single cell approaches revealing the importance of tissue heterogeneity in cancer progression and therapeutic outcome, we will witness a shift in the lipidomics field from lipidomics analyses on bulk tissue to single cell and spatial analysis by mass spectrometry imaging. New technological developments in this domain promise an unseen performance in terms of analytical aspects as well as spatial resolution, leading to novel insights in the role of lipids in the complex metabolic interplay between different cell types in the heterogenous tumor microenvironment. With novel technologies allowing imaging of lipids at the intracellular level including dynamic SIMS ion microcopy and Raman microscopy, a whole new area of lipid research is opening up, revealing changes in organellar lipidomes, trafficking pathways and membrane structures [728, 729]. The further development of stable isotope lipidomics will allow to follow changes in pathway fluxes instead of current steady state analysis and together with spatial multi-omics approaches will provide unprecedented insight in affected pathways and potential biomarkers. Along with these developments there is an urgent need for standardization of methods and technologies to allow future clinical implementations of the discovered biomarkers. In terms of therapeutic potential, current findings suggest that interference with lipid metabolism will have promising applications, particularly in combinatorial approaches[16]. With small molecules targeting enzymes in lipid metabolism entering clinical trials we are at the doorstep of witnessing the clinical exploitation of altered lipid metabolism as a hallmark of cancer. The link with the diet, including dietary lipids will also create unique opportunities for preventive strategies and therapy enhancement. Particularly in the field of tumor immunology, lipids hold great potential as modulators. In summary, although lagging behind compared to other omics approaches, the study of lipids in cancer is rapidly catching up and is establishing itself as a central hallmark of cancer with promising opportunities for clinical application.
Funding
This work was supported by an EU Interreg grant V-A EMR23 EURLIPIDS, KU Leuven grants C1 (C16/15/073) and C3 (C32/17/052), Research Foundation-Flanders (FWO), Stichting tegen Kanker, Kom op tegen Kanker, the Movember Foundation/Prostate Cancer Foundation of Australia (MRTA3), The Prostate Cancer Foundation of Australia (ID NDDA), the Cancer Council South Australia Beat Cancer Project, NIH grant RO1CA58961, a Norris Cotton Cancer Center grant, and the Dartmouth College Norris Cotton Cancer Center Support Grant P30CA023108. ML’s work is supported by NIH grants RO1CA131945, R01CA187918, DoD PC160357, DoD PC180582, P50CA211024, and the Prostate Cancer Foundation.
Abbreviations
- AA
arachidonic acid
- ACACA
acetyl-CoA carboxylase alpha
- ACACB
acetyl-CoA carboxylase beta
- ACLY
ATP-citrate lyase
- ACSL
Acyl-CoA Synthetase
- APT
acylprotein thioesterase
- AR
androgen receptor
- ASM
acid sphingomyelinase
- ATGL
adipose triglyceride lipase
- ATX
autotaxin
- BC
breast cancer
- ccRCC
clear cell renal cell carcinoma
- CE
cholesteryl ester
- ChoKα
Choline kinase alpha
- CM
chylomicron
- CoA
co-enzyme A
- CPT
carnitine palmitoyl transferase
- DAG
diacylglycerol
- EGFR
epidermal growth factor receptor
- ELOVLs
fatty acid elongases
- EPA
eicosapentaenoic acid
- ER
endoplasmic reticulum
- ER+/−
estrogen receptor positive/negative
- ESI
electrospray ionization
- FA
fatty acid
- FA2H
fatty acid 2-hydroxylase
- FABP
fatty acid binding protein
- FADS
fatty acid desturase
- FAO
fatty acid oxidation
- FASN
fatty acid synthase
- FDG
fluoro-deoxy glucose
- FFA
free fatty acid
- GBM
glioblastoma multiforme
- GPIHBP1
glycosylphosphatidylinositol high density lipoprotein binding protein 1
- HPLC
high performance liquid chromatography
- HSL
Hormone sensitive lipase
- HSPG
heparan sulfate proteoglycan
- INSIG
insulin-induced gene
- LDL
low-density lipoprotein
- LDLR
low-density lipoprotein receptor
- LPA
lysophosphatidic acid
- LPAR
LysoPA receptor
- LPCAT
lysophosphatidylcholine acyl transferase
- LPL
Lipoprotein Lipase
- LysoPA
lysophosphatidic acid
- LysoPC
lysophosphatidylcholine
- LysoPL
lysophospholipid
- LXR
Liver X receptor
- MAGL
monoacylglycerol lipase
- MALDI
matrix-assisted laser desorption/ionization
- MAPK
Mitogen-Activated Protein Kinase
- MUFA
monounsaturated fatty acid
- MS
mass spectrometry
- MSI
mass spectrometry imaging
- PA
phosphatidic acid
- PAP
phosphatidic acid phosphatase
- PAT
palmitoyl S-acyltransferase
- PC
phosphatidylcholine
- PDC
pyruvate dehydrogenase complex
- PE
phosphatidylethanolamine
- PET
positron emission tomography
- PGE2
prostaglandin E2
- PI
phosphatidylinositol
- PL
phospholipid
- PKC
protein kinase C
- PLA/C/D
phospholipase A, C or D
- PLSCR
phospholipid scramblase
- PPAR
peroxisome proliferator-activated receptor
- PS
phosphatidylserine
- PTEN
Phosphatase and Tensin homolog
- PUFA
polyunsaturated fatty acid
- ROS
reactive oxygen species
- RXR
retinoid X receptor
- S1P
sphingosine-1-phosphate
- SCAP
SREBP cleavage-activating protein
- SERM
selective estrogen receptor modulator
- SFA
saturated fatty acid
- SCD
stearoyl-CoA desaturase
- SREBP
sterol regulatory element-binding protein
- TAMs
tumor-associated macrophages
- TF
transcription factor
- TAG
triacylglyceride
- TNBC
triple-negative breast cancer
- VLDL
very low-density lipoprotein
- VLDLR
very low-density lipoprotein receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell, 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [2].Warburg O, Wind F, Negelein E, The Metabolism of Tumors in the Body, J Gen Physiol, 8 (1927) 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Swinnen J, Brusselmans K, Verhoeven G, Increased lipogenesis in cancer cells: new players, novel targets., Curr. Opin. Clin. Nutr. Metab. Care, 9 (2006) 358–365. [DOI] [PubMed] [Google Scholar]
- [4].Li L, Han J, Wang Z, Liu J, Wei J, Xiong S, Zhao Z, Mass spectrometry methodology in lipid analysis, Int J Mol Sci, 15 (2014) 10492–10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lydic TA, Goo YH, Lipidomics unveils the complexity of the lipidome in metabolic diseases, Clin Transl Med, 7 (2018) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].van Meer G, Voelker DR, Feigenson GW, Membrane lipids: where they are and how they behave, Nature reviews. Molecular cell biology, 9 (2008) 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Spector AA, Steinberg D, Relationship between fatty acid and glucose utilization in Ehrlich ascites tumor cells, J Lipid Res, 7 (1966) 657–663. [PubMed] [Google Scholar]
- [8].Tator CH, Evans JR, Olszewski J, Tracers for the detection of brain tumors. Evaluation of radioiodinated human serum albumin and radioiodinated fatty acid, Neurology, 16 (1966) 650–661. [DOI] [PubMed] [Google Scholar]
- [9].Mountford CE, Grossman G, Reid G, Fox RM, Characterization of transformed cells and tumors by proton nuclear magnetic resonance spectroscopy, Cancer Res, 42 (1982) 2270–2276. [PubMed] [Google Scholar]
- [10].Jensen V, Ladekarl M, Holm-Nielsen P, Melsen F, Soerensen F, The prognostic value of oncogenic antigen 519 (OA-519) expression and proliferative activity detected by antibody MIB-1 in node-negative breast cancer, J Pathol, 176 (1995) 343–352. [DOI] [PubMed] [Google Scholar]
- [11].Ferro M, Terracciano D, Buonerba C, Lucarelli G, Bottero D, Perdona S, Autorino R, Serino A, Cantiello F, Damiano R, Andras I, De Placido S, Di Lorenzo G, Battaglia M, Jereczek-Fossa BA, Mirone V, De Cobelli O, The emerging role of obesity, diet and lipid metabolism in prostate cancer, Future oncology (London, England), 13 (2017) 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hoy AJ, Balaban S, Saunders DN, Adipocyte-Tumor Cell Metabolic Crosstalk in Breast Cancer, Trends Mol Med, 23 (2017) 381–392. [DOI] [PubMed] [Google Scholar]
- [13].Tworoger SS, Huang T, Obesity and Ovarian Cancer, Recent results in cancer research. Fortschritte der Krebsforschung. Progres dans les recherches sur le cancer, 208 (2016) 155–176. [DOI] [PubMed] [Google Scholar]
- [14].Bi J, Ichu TA, Zanca C, Yang H, Zhang W, Gu Y, Chowdhry S, Reed A, Ikegami S, Turner KM, Zhang W, Villa GR, Wu S, Quehenberger O, Yong WH, Kornblum HI, Rich JN, Cloughesy TF, Cavenee WK, Furnari FB, Cravatt BF, Mischel PS, Oncogene Amplification in Growth Factor Signaling Pathways Renders Cancers Dependent on Membrane Lipid Remodeling, Cell Metab, 30 (2019) 525–538 e528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniels VW, Machiels J, Vanderhoydonc F, Smans K, Waelkens E, Verhoeven G, Swinnen JV, De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation, Cancer Res, 70 (2010) 8117–8126. [DOI] [PubMed] [Google Scholar]
- [16].Talebi A, Dehairs J, Rambow F, Rogiers A, Nittner D, Derua R, Vanderhoydonc F, Duarte JAG, Bosisio F, Van den Eynde K, Nys K, Perez MV, Agostinis P, Waelkens E, Van den Oord J, Fendt SM, Marine JC, Swinnen JV, Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy, Nat Commun, 9 (2018) 2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, Zhang Q, Wakelam MJO, Karpe F, Schulze A, Harris AL, Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation, Cell Rep, 9 (2014) 349–365. [DOI] [PubMed] [Google Scholar]
- [18].Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, Postle AD, Gould AP, Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila, Cell, 163 (2015) 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lazar I, Clement E, Attane C, Muller C, Nieto L, A new role for extracellular vesicles: how small vesicles can feed tumors’ big appetite, The Journal of Lipid Research, 59 (2018) 1793–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rolin J, Maghazachi AA, Effects of lysophospholipids on tumor microenvironment, Cancer Microenviron, 4 (2011) 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Balog J, Sasi-Szabo L, Kinross J, Lewis MR, Muirhead LJ, Veselkov K, Mirnezami R, Dezso B, Damjanovich L, Darzi A, Nicholson JK, Takats Z, Intraoperative tissue identification using rapid evaporative ionization mass spectrometry, Science translational medicine, 5 (2013) 194ra193. [DOI] [PubMed] [Google Scholar]
- [22].Shan L, Chen YA, Davis L, Han G, Zhu W, Molina AD, Arango H, LaPolla JP, Hoffman MS, Sellers T, Kirby T, Nicosia SV, Sutphen R, Measurement of phospholipids may improve diagnostic accuracy in ovarian cancer, PLoS One, 7 (2012) e46846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang X, Zeng C, Lin J, Chen T, Zhao T, Jia Z, Xie X, Qiu Y, Su M, Jiang T, Zhou M, Zhao A, Jia W, Metabonomics approach to assessing the modulatory effects of St John’s wort, ginsenosides, and clomipramine in experimental depression, J Proteome Res, 11 (2012) 6223–6230. [DOI] [PubMed] [Google Scholar]
- [24].Guo Y, Wang X, Qiu L, Qin X, Liu H, Wang Y, Li F, Wang X, Chen G, Song G, Li F, Guo S, Li Z, Probing gender-specific lipid metabolites and diagnostic biomarkers for lung cancer using Fourier transform ion cyclotron resonance mass spectrometry, Clin Chim Acta, 414 (2012) 135–141. [DOI] [PubMed] [Google Scholar]
- [25].Yu Z, Chen H, Ai J, Zhu Y, Li Y, Borgia JA, Yang JS, Zhang J, Jiang B, Gu W, Deng Y, Global lipidomics identified plasma lipids as novel biomarkers for early detection of lung cancer, Oncotarget, 8 (2017) 107899–107906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Patel N, Vogel R, Chandra-Kuntal K, Glasgow W, Kelavkar U, A novel three serum phospholipid panel differentiates normal individuals from those with prostate cancer, PLoS One, 9 (2014) e88841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hao Y, Li D, Xu Y, Ouyang J, Wang Y, Zhang Y, Li B, Xie L, Qin G, Investigation of lipid metabolism dysregulation and the effects on immune microenvironments in pan-cancer using multiple omics data, BMC bioinformatics, 20 (2019) 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Grant RP, High throughput automated LC-MS/MS analysis of endogenous small molecule biomarkers, Clin Lab Med, 31 (2011) 429–441. [DOI] [PubMed] [Google Scholar]
- [29].Dehairs J, Derua R, Rueda-Rincon N, Swinnen JV, Lipidomics in drug development, Drug Discov Today Technol, 13 (2015) 33–38. [DOI] [PubMed] [Google Scholar]
- [30].Wolrab D, Chocholouskova M, Jirasko R, Peterka O, Holcapek M, Validation of lipidomic analysis of human plasma and serum by supercritical fluid chromatography-mass spectrometry and hydrophilic interaction liquid chromatography-mass spectrometry, Analytical and bioanalytical chemistry, (2020). [DOI] [PubMed] [Google Scholar]
- [31].O’Donnell VB, Dennis EA, Wakelam MJO, Subramaniam S, LIPID MAPS: Serving the next generation of lipid researchers with tools, resources, data, and training, Sci Signal, 12 (2019). [DOI] [PubMed] [Google Scholar]
- [32].Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CR, Shimizu T, Spener F, van Meer G, Wakelam MJ, Dennis EA, Update of the LIPID MAPS comprehensive classification system for lipids, J Lipid Res, 50 Suppl (2009) S9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stephenson DJ, Hoeferlin LA, Chalfant CE, Lipidomics in translational research and the clinical significance of lipid-based biomarkers, Transl Res, 189 (2017) 13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hsu FF, Mass spectrometry-based shotgun lipidomics - a critical review from the technical point of view, Anal Bioanal Chem, 410 (2018) 6387–6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].O’Donnell VB, Ekroos K, Liebisch G, Wakelam M, Lipidomics: Current state of the art in a fast moving field, Wiley Interdiscip Rev Syst Biol Med, 12 (2020) e1466. [DOI] [PubMed] [Google Scholar]
- [36].Zhu M, Xu X, Hou Y, Han J, Wang J, Zheng Q, Hao H, Boronic Derivatization of Monoacylglycerol and Monitoring in Biofluids, Anal Chem, 91 (2019) 6724–6729. [DOI] [PubMed] [Google Scholar]
- [37].Guo N, Liu P, Ding J, Zheng SJ, Yuan BF, Feng YQ, Stable isotope labeling - Liquid chromatography/mass spectrometry for quantitative analysis of androgenic and progestagenic steroids, Anal Chim Acta, 905 (2016) 106–114. [DOI] [PubMed] [Google Scholar]
- [38].Griffiths WJ, Abdel-Khalik J, Crick PJ, Yutuc E, Wang Y, New methods for analysis of oxysterols and related compounds by LC-MS, The Journal of steroid biochemistry and molecular biology, 162 (2016) 4–26. [DOI] [PubMed] [Google Scholar]
- [39].von Gerichten J, Schlosser K, Lamprecht D, Morace I, Eckhardt M, Wachten D, Jennemann R, Grone HJ, Mack M, Sandhoff R, Diastereomer-specific quantification of bioactive hexosylceramides from bacteria and mammals, J Lipid Res, 58 (2017) 1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Thomas MC, Mitchell TW, Harman DG, Deeley JM, Nealon JR, Blanksby SJ, Ozone-induced dissociation: elucidation of double bond position within mass-selected lipid ions, Anal Chem, 80 (2008) 303–311. [DOI] [PubMed] [Google Scholar]
- [41].Groessl M, Graf S, Knochenmuss R, High resolution ion mobility-mass spectrometry for separation and identification of isomeric lipids, Analyst, 140 (2015) 6904–6911. [DOI] [PubMed] [Google Scholar]
- [42].Zhang W, Zhang D, Chen Q, Wu J, Ouyang Z, Xia Y, Online photochemical derivatization enables comprehensive mass spectrometric analysis of unsaturated phospholipid isomers, Nat Commun, 10 (2019) 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Thiele C, Wunderling K, Leyendecker P, Multiplexed and single cell tracing of lipid metabolism, Nat Methods, 16 (2019) 1123–1130. [DOI] [PubMed] [Google Scholar]
- [44].Marien E, Meister M, Muley T, Fieuws S, Bordel S, Derua R, Spraggins J, Van de Plas R, Dehairs J, Wouters J, Bagadi M, Dienemann H, Thomas M, Schnabel PA, Caprioli RM, Waelkens E, Swinnen JV, Non-small cell lung cancer is characterized by dramatic changes in phospholipid profiles, Int J Cancer, 137 (2015) 1539–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].O’Donnell VB, Rossjohn J, Wakelam MJ, Phospholipid signaling in innate immune cells, J Clin Invest, 128 (2018) 2670–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Goto-Inoue N, Hayasaka T, Zaima N, Setou M, Imaging mass spectrometry for lipidomics, Biochim Biophys Acta, 1811 (2011) 961–969. [DOI] [PubMed] [Google Scholar]
- [47].Fernandez JA, Ochoa B, Fresnedo O, Giralt MT, Rodriguez-Puertas R, Matrix-assisted laser desorption ionization imaging mass spectrometry in lipidomics, Anal Bioanal Chem, 401 (2011) 29–51. [DOI] [PubMed] [Google Scholar]
- [48].Amstalden van Hove ER, Smith DF, Heeren RM, A concise review of mass spectrometry imaging, J Chromatogr A, 1217 (2010) 3946–3954. [DOI] [PubMed] [Google Scholar]
- [49].Rohner TC, Staab D, Stoeckli M, MALDI mass spectrometric imaging of biological tissue sections, Mechanisms of Ageing and Development, 126 (2005) 177–185. [DOI] [PubMed] [Google Scholar]
- [50].Song X, He J, Pang X, Zhang J, Sun C, Huang L, Li C, Zang Q, Li X, Luo Z, Zhang R, Xie P, Liu X, Li Y, Chen X, Abliz Z, Virtual Calibration Quantitative Mass Spectrometry Imaging for Accurately Mapping Analytes across Heterogenous Biotissue, Anal Chem, 91 (2019) 2838–2846. [DOI] [PubMed] [Google Scholar]
- [51].Trim PJ, Snel MF, Small molecule MALDI MS imaging: Current technologies and future challenges, Methods, 104 (2016) 127–141. [DOI] [PubMed] [Google Scholar]
- [52].Sparvero LJ, Amoscato AA, Dixon CE, Long JB, Kochanek PM, Pitt BR, Bayir H, Kagan VE, Mapping of phospholipids by MALDI imaging (MALDI-MSI): realities and expectations, Chem Phys Lipids, 165 (2012) 545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Enomoto H, Sugiura Y, Setou M, Zaima N, Visualization of phosphatidylcholine, lysophosphatidylcholine and sphingomyelin in mouse tongue body by matrix-assisted laser desorption/ionization imaging mass spectrometry, Anal Bioanal Chem, 400 (2011) 1913–1921. [DOI] [PubMed] [Google Scholar]
- [54].Ishikawa S, Tateya I, Hayasaka T, Masaki N, Takizawa Y, Ohno S, Kojima T, Kitani Y, Kitamura M, Hirano S, Setou M, Ito J, Increased expression of phosphatidylcholine (16:0/18:1) and (16:0/18:2) in thyroid papillary cancer, PLoS One, 7 (2012) e48873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schwamborn K, Caprioli RM, Molecular imaging by mass spectrometry--looking beyond classical histology, Nat Rev Cancer, 10 (2010) 639–646. [DOI] [PubMed] [Google Scholar]
- [56].Pirman DA, Efuet E, Ding X-P, Pan Y, Tan L, Fischer SM, DuBois RN, Yang P, Changes in Cancer Cell Metabolism Revealed by Direct Sample Analysis with MALDI Mass Spectrometry, PLoS One, 8 (2013) e61379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Drake RR, Powers TW, Jones EE, Bruner E, Mehta AS, Angel PM, MALDI Mass Spectrometry Imaging of N-Linked Glycans in Cancer Tissues, Adv Cancer Res, 134 (2017) 85–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Pirman DA, Efuet E, Ding XP, Pan Y, Tan L, Fischer SM, DuBois RN, Yang P, Changes in cancer cell metabolism revealed by direct sample analysis with MALDI mass spectrometry, PLoS One, 8 (2013) e61379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lagarrigue M, Lavigne R, Guevel B, Com E, Chaurand P, Pineau C, Matrix-assisted laser desorption/ionization imaging mass spectrometry: a promising technique for reproductive research, Biol Reprod, 86 (2012) 74. [DOI] [PubMed] [Google Scholar]
- [60].Hegele RA, Multidimensional regulation of lipoprotein lipase: impact on biochemical and cardiovascular phenotypes, Journal of Lipid Research, 57 (2016) 1601–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Brunzell JD DS, Familial lipoprotein lipase deficiency, apo C-II deficiency, and hepatic lipase deficiency., McGraw-Hill, New York, 2001. [Google Scholar]
- [62].Mead JR, Irvine SA, Ramji DP, Lipoprotein lipase: structure, function, regulation, and role in disease, J Mol Med (Berl), 80 (2002) 753–769. [DOI] [PubMed] [Google Scholar]
- [63].Fielding PE, Fielding CJ, Chapter 15 Dynamics of lipoprotein transport in the circulatory system, in: Vance DE, Vance JE (Eds.) New Comprehensive Biochemistry, Elsevier; 1991, pp. 427–459. [Google Scholar]
- [64].DeBerardinis R, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thyompson C, Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis., PNAS, 104 (2007) 19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Enoch HG, Catala A, Strittmatter P, Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid, J Biol Chem, 251 (1976) 5095–5103. [PubMed] [Google Scholar]
- [66].Bai Y, McCoy JG, Levin EJ, Sobrado P, Rajashankar KR, Fox BG, Zhou M, X-ray structure of a mammalian stearoyl-CoA desaturase, Nature, 524 (2015) 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Baenke F, Peck B, Miess H, Schulze A, Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development, Disease models & mechanisms, 6 (2013) 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Aja S, Bi S, Knipp SB, McFadden JM, Ronnett GV, Kuhajda FP, Moran TH, Intracerebroventricular C75 decreases meal frequency and reduces AgRP gene expression in rats, American journal of physiology. Regulatory, integrative and comparative physiology, 291 (2006) R148–154. [DOI] [PubMed] [Google Scholar]
- [69].Menendez J, Lupu R, Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis, Nat. Rev. Cancer, 7 (2007) 763–777. [DOI] [PubMed] [Google Scholar]
- [70].Chirala S, Chang H, Matzuk M, Abu-Elheiga L, Mao J, Mahon K, Finegold M, Wakil S, Fatty acid synthesis is essential in embryonic development: fatty acid synthase null mutants and most heterozygotes die in utero., PNAS, 100 (2003) 6358–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bhatt AP, Jacobs SR, Freemerman AJ, Makowski L, Rathmell JC, Dittmer DP, Damania B, Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma, Proceedings of the National Academy of Sciences, 109 (2012) 11818–11823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ardecky R, Hedrick MP, Gosalia P, Yamamoto F, Milewski M, Barron N, Sun Q, Zhou J, Ganji S, Mehta A, Sugarman E, Nguyen K, Vasile S, Suyama E, Teriete P, Sheffler D, Lee PS, Mattmann M, De Ingeniis J, Stonich D, Mangravita-Novo A, Salaniwal S, Kung P, Diwan J, Smith LH, Sergienko E, Chung TDY, Pinkerton AB, Cosford NDP, Smith JW, Selective inhibitors of FAS-TE, Probe Reports from the NIH Molecular Libraries Program, National Center for Biotechnology Information (US), Bethesda (MD), 2010. [PubMed] [Google Scholar]
- [73].Cai Y, Wang J, Zhang L, Wu D, Yu D, Tian X, Liu J, Jiang X, Shen Y, Zhang L, Ren M, Huang P, Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer, Med Oncol, 32 (2015) 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Di Vizio D, Adam RM, Kim J, Kim R, Sotgia F, Williams T, Demichelis F, Solomon KR, Loda M, Rubin MA, Lisanti MP, Freeman MR, Caveolin-1 interacts with a lipid raft-associated population of fatty acid synthase, Cell cycle (Georgetown, Tex.), 7 (2008) 2257–2267. [DOI] [PubMed] [Google Scholar]
- [75].Gelebart P, Zak Z, Anand M, Belch A, Lai R, Blockade of fatty acid synthase triggers significant apoptosis in mantle cell lymphoma, PloS one, 7 (2012) e33738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ookhtens M, Kannan R, Lyon I, Baker N, Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor, The American journal of physiology, 247 (1984) R146–153. [DOI] [PubMed] [Google Scholar]
- [77].Turrado C, Puig T, Garcia-Carceles J, Artola M, Benhamu B, Ortega-Gutierrez S, Relat J, Oliveras G, Blancafort A, Haro D, Marrero PF, Colomer R, Lopez-Rodriguez ML, New synthetic inhibitors of fatty acid synthase with anticancer activity, Journal of medicinal chemistry, 55 (2012) 5013–5023. [DOI] [PubMed] [Google Scholar]
- [78].Zadra G, Loda M, Metabolic Vulnerabilities of Prostate Cancer: Diagnostic and Therapeutic Opportunities, Cold Spring Harb Perspect Med, 8 (2018) a030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, Inazuka F, Grisanzio C, Palescandolo E, Shin E, Fiore C, Xie W, Kung AL, Febbo PG, Subramanian A, Mucci L, Ma J, Signoretti S, Stampfer M, Hahn WC, Finn S, Loda M, Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer, Journal of the National Cancer Institute, 101 (2009) 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Swinnen JV, Vanderhoydonc F, Elgamal AA, Eelen M, Vercaeren I, Joniau S, Van Poppel H, Baert L, Goossens K, Heyns W, Verhoeven G, Selective activation of the fatty acid synthesis pathway in human prostate cancer, Int J Cancer, 88 (2000) 176–179. [DOI] [PubMed] [Google Scholar]
- [81].Baron A, Migita T, Tang D, Loda M, Fatty acid synthase: A metabolic oncogene in prostate cancer?, Journal of cellular biochemistry, 91 (2004) 47–53. [DOI] [PubMed] [Google Scholar]
- [82].Gansler T, Hardman W, Hunt D, Schaffel S, Hennigar R, Increased expression of fatty acid synthase (OA-519) in ovarian neoplasms predicts shorter survival., Human Pathol, 28 (1997) 686–692. [DOI] [PubMed] [Google Scholar]
- [83].Visca P, Sebastiani V, Botti C, Diodoro MG, Lasagni RP, Romagnoli F, Brenna A, Joannon BCD, Donnorso RP, Lombardi G, Alo PL, Fatty Acid Synthase (FAS) is a Marker of Increased Risk of Recurrence in Lung Carcinoma, Anticancer Research, 24 (2004) 4169–4174. [PubMed] [Google Scholar]
- [84].Sebastiani V, Fatty acid synthase is a marker of increased risk of recurrence in endometrial carcinoma, Gynecologic oncology, 92 (2004) 101–105. [DOI] [PubMed] [Google Scholar]
- [85].Ogino S, Kawasaki T, Ogawa A, Kirkner GJ, Loda M, Fuchs CS, Fatty acid synthase overexpression in colorectal cancer is associated with microsatellite instability, independent of CpG island methylator phenotype, Human Pathology, 38 (2007) 842–849. [DOI] [PubMed] [Google Scholar]
- [86].Ogino S, Nosho K, Meyerhardt JA, Kirkner GJ, Chan AT, Kawasaki T, Giovannucci EL, Loda M, Fuchs CS, Cohort Study of Fatty Acid Synthase Expression and Patient Survival in Colon Cancer, Journal of Clinical Oncology, 26 (2008) 5713–5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Agostini M, Silva SD, Zecchin KG, Coletta RD, Jorge J, Loda M, Graner E, Fatty acid synthase is required for the proliferation of human oral squamous carcinoma cells, Oral Oncology, 40 (2004) 728–735. [DOI] [PubMed] [Google Scholar]
- [88].Suburu J, Chen YQ, Lipids and prostate cancer, Prostaglandins & Other Lipid Mediators, 98 (2012) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Swinnen JV, Roskams T, Joniau S, Van Poppel H, Oyen R, Baert L, Heyns W, Verhoeven G, Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer, International Journal of Cancer. Journal International Du Cancer, 98 (2002) 19–22. [DOI] [PubMed] [Google Scholar]
- [90].Pflug B, Pecher S, Brink A, Nelson J, Foster B, Increased fatty acid synthase expression and activity during progression of prostate cancer in the TRAMP model, The Prostate, 57 (2003) 245–254. [DOI] [PubMed] [Google Scholar]
- [91].Epstein JI, Carmichael M, Partin AW, OA-519 (fatty acid synthase) as an independent predictor of pathologic stage in adenocarcinoma of the prostate, Urology, 45 (1995) 81–86. [DOI] [PubMed] [Google Scholar]
- [92].Van de Sande T, De Schrijver E, Heyns W, Verhoeven G, Swinnen JV, Role of the phosphatidylinositol 3’-kinase/PTEN/Akt kinase pathway in the overexpression of fatty acid synthase in LNCaP prostate cancer cells, Cancer Res, 62 (2002) 642–646. [PubMed] [Google Scholar]
- [93].Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, Bubley G, Balk S, Loda M, Fatty Acid Synthase Expression Defines Distinct Molecular Signatures in Prostate Cancer1 1 NCI (Director’s Challenge CA84995–04, SPORE in Prostate Cancer CA90381–01A1, and PO1 CA89021–02), Novartis Investigator, and CaPCURE awards, Molecular Cancer Research, 1 (2003) 707–715. [PubMed] [Google Scholar]
- [94].Shah U, Dhir R, Gollin S, Chandran U, Lewis D, Acquafondata M, Pflug B, Fatty acid synthase gene overexpression and copy number gain in prostate adenocarcinoma☆, Human Pathology, 37 (2006) 401–409. [DOI] [PubMed] [Google Scholar]
- [95].Scott KEN, Wheeler FB, Davis AL, Thomas MJ, Ntambi JM, Seals DF, Kridel SJ, Metabolic Regulation of Invadopodia and Invasion by Acetyl-CoA Carboxylase 1 and De novo Lipogenesis, PloS one, 7 (2012) e29761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yoshii Y, Furukawa T, Oyama N, Hasegawa Y, Kiyono Y, Nishii R, Waki A, Tsuji AB, Sogawa C, Wakizaka H, Fukumura T, Yoshii H, Fujibayashi Y, Lewis JS, Saga T, Fatty acid synthase is a key target in multiple essential tumor functions of prostate cancer: uptake of radiolabeled acetate as a predictor of the targeted therapy outcome, PloS one, 8 (2013) e64570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV, RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells, Cancer Res, 63 (2003) 3799–3804. [PubMed] [Google Scholar]
- [98].Zaidi N, Royaux I, Swinnen JV, Smans K, ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms, Mol Cancer Ther, 11 (2012) 1925–1935. [DOI] [PubMed] [Google Scholar]
- [99].Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen J, Chemical inhibition of acteyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells., Cancer Res, 67 (2007) 8180–8187. [DOI] [PubMed] [Google Scholar]
- [100].Brusselmans K, De Schrijver E, Verhoeven G, Swinnen J, RNA interference-mediated silencing of the acetyl-CoA-carboxylase-a gene induces growth inhibition and apoptosis of prostate cancer cells., Cancer Res, 65 (2005) 6719–6725. [DOI] [PubMed] [Google Scholar]
- [101].Bruning U, Morales-Rodriguez F, Kalucka J, Goveia J, Taverna F, Queiroz KCS, Dubois C, Cantelmo AR, Chen R, Loroch S, Timmerman E, Caixeta V, Bloch K, Conradi LC, Treps L, Staes A, Gevaert K, Tee A, Dewerchin M, Semenkovich CF, Impens F, Schilling B, Verdin E, Swinnen JV, Meier JL, Kulkarni RA, Sickmann A, Ghesquiere B, Schoonjans L, Li X, Mazzone M, Carmeliet P, Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation, Cell Metab, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zadra G, Photopoulos C, Tyekucheva S, Heidari P, Weng QP, Fedele G, Liu H, Scaglia N, Priolo C, Sicinska E, Mahmood U, Signoretti S, Birnberg N, Loda M, A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis, EMBO molecular medicine, 6 (2014) 519–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, Syamala S, Bango C, Photopoulos C, Huang Y, Tyekucheva S, Bastos DC, Tchaicha J, Lawney B, Uo T, D’Anello L, Csibi A, Kalekar R, Larimer B, Ellis L, Butler LM, Morrissey C, McGovern K, Palombella VJ, Kutok JL, Mahmood U, Bosari S, Adams J, Peluso S, Dehm SM, Plymate SR, Loda M, Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer, Proceedings of the National Academy of Sciences of the United States of America, 116 (2019) 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Dale K, Coleman C, Henyan N, Kluger J, White C, Statins and cancer risk: a meta-analysis, Jama, 295 (2006) 74–80. [DOI] [PubMed] [Google Scholar]
- [105].Ahern TP, Pedersen L, Tarp M, Cronin-Fenton DP, Garne JP, Silliman RA, Sorensen HT, Lash TL, Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study, Journal of the National Cancer Institute, 103 (2011) 1461–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Allott EH, Ebot EM, Stopsack KH, Gonzalez-Feliciano AG, Markt SC, Wilson KM, Ahearn TU, Gerke TA, Downer MK, Rider JR, Freedland SJ, Lotan TL, Kantoff PW, Platz EA, Loda M, Stampfer MJ, Giovannucci E, Sweeney CJ, Finn SP, Mucci LA, Statin Use Is Associated with Lower Risk of PTEN-Null and Lethal Prostate Cancer, Clinical cancer research : an official journal of the American Association for Cancer Research, 26 (2020) 1086–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Nielsen SF, Nordestgaard BG, Bojesen SE, Statin use and reduced cancer-related mortality, The New England journal of medicine, 367 (2012) 1792–1802. [DOI] [PubMed] [Google Scholar]
- [108].Deberardinis R, Chandel N, Fundamentals of cancer metabolism, Scientific Advances, (2016) 2016; 2012 : e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F, Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth, Molecular systems biology, 7 (2011) 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, The somatic genomic landscape of glioblastoma, Cell, 155 (2013) 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Beloribi-Djefaflia S, Vasseur S, Guillaumond F, Lipid metabolic reprogramming in cancer cells, Oncogenesis, 5 (2016) e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Amemori S, Ootani A, Aoki S, Fujise T, Shimoda R, Kakimoto T, Shiraishi R, Sakata Y, Tsunada S, Iwakiri R, Fujimoto K, Adipocytes and preadipocytes promote the proliferation of colon cancer cells in vitro, American journal of physiology. Gastrointestinal and liver physiology, 292 (2007) G923–929. [DOI] [PubMed] [Google Scholar]
- [113].Anastasiou D, Cantley LC, Breathless cancer cells get fat on glutamine, Cell research, 22 (2012) 443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Le Gonidec S, Garrido I, Escourrou G, Valet P, Muller C, Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion, Cancer Res, 71 (2011) 2455–2465. [DOI] [PubMed] [Google Scholar]
- [115].Cheng T, Sudderth J, Yang C, Mullen A, Jin E, Mates J, DeBerardinis R, Pyruvate carboxylase is required for glutamine-independent growth of tumor cells., PNAS, 108 (2011) 8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Cao Q, Ruan H, Wang K, Song Z, Bao L, Xu T, Xiao H, Wang C, Cheng G, Tong J, Meng X, Liu D, Yang H, Chen K, Zhang X, Overexpression of PLIN2 is a prognostic marker and attenuates tumor progression in clear cell renal cell carcinoma, International journal of oncology, 53 (2018) 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Altman BJ, Stine ZE, Dang CV, From Krebs to clinic: glutamine metabolism to cancer therapy, Nat Rev Cancer, 16 (2016) 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, Horton JD, Hammer RE, McKnight SL, Tu BP, Acetate dependence of tumors, Cell, 159 (2014) 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, Huang Z, Barnett S, Mickey BE, DeBerardinis RJ, Tu BP, Maher EA, Bachoo RM, Acetate is a bioenergetic substrate for human glioblastoma and brain metastases, Cell, 159 (2014) 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, McGarry L, James D, Shanks E, Kalna G, Saunders RE, Jiang M, Howell M, Lassailly F, Thin MZ, Spencer-Dene B, Stamp G, van den Broek NJ, Mackay G, Bulusu V, Kamphorst JJ, Tardito S, Strachan D, Harris AL, Aboagye EO, Critchlow SE, Wakelam MJ, Schulze A, Gottlieb E, Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress, Cancer cell, 27 (2015) 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Yoshii Y, Furukawa T, Yoshii H, Mori T, Kiyono Y, Waki A, Kobayashi M, Tsujikawa T, Kudo T, Okazawa H, Yonekura Y, Fujibayashi Y, Cytosolic acetyl-CoA synthetase affected tumor cell survival under hypoxia: the possible function in tumor acetyl-CoA/acetate metabolism, Cancer science, 100 (2009) 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Yoshii Y, Furukawa T, Saga T, Fujibayashi Y, Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: overview and application, Cancer letters, 356 (2015) 211–216. [DOI] [PubMed] [Google Scholar]
- [123].Chen W, Jin X, Wang M, Liu D, Luo Q, Xie X, Yu G, Li L, Dong C, Jia W, Wei W, Jia L, GLUT5-mediated fructose utilization drives lung cancer growth by stimulating fatty acid synthesis and AMPK/mTORC1 signaling, JCI Insight, 5 (2020) 10.1172/jci.insight.131596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Hamann I, Krys D, Glubrecht D, Bouvet V, Marshall A, Vos L, Mackey J, Wuest M, Wuest F, Expression and function of hexose transporters GLUT1, GLUT2, and GLUT5 in breast cancer—effects of hypoxia, FASEB Journal, 32 (2018) 10.1096/fj.201800360R. [DOI] [PubMed] [Google Scholar]
- [125].Spector AA, The importance of free fatty acid in tumor nutrition, Cancer Res, 27 (1967) 1580–1586. [PubMed] [Google Scholar]
- [126].Louie S, Roberts L, Mulvihill M, Luo K, Nomura D, Cancer cells incorporate and remodel exogenous palnmitate into structural and oncogenic signaling lipids., Biochimica Biophysica Acta, 1831 (2013) 566–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, Freedman A, Inghirami G, Cro L, Baldini L, Neri A, Califano A, Dalla-Favera R, Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells, J Exp Med, 194 (2001) 1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Rombout A, Stamatopoulos B, Lagneaux L, Lust S, Offner F, Naessens E, Vanderstraeten H, Verhasselt B, Philippe J, Lipoprotein lipase SNPs rs13702 and rs301 correlate with clinical outcome in chronic lymphocytic leukemia patients, PloS one, 10 (2015) e0121526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Heintel D, Kienle D, Shehata M, Krober A, Kroemer E, Schwarzinger I, Mitteregger D, Le T, Gleiss A, Mannhalter C, Chott A, Schwarzmeier J, Fonatsch C, Gaigner A, Dohner H, Stigenbauer S, Jager U, High expression of lipoprotein lipase in poor risk B-cell chronic lymphocytic leukemia., Leukemia and Lymphoma, 19 (2005) 1216–1223. [DOI] [PubMed] [Google Scholar]
- [130].Baba F, Swartz K, van Buren R, Eickhoff J, Zhang Y, Wolberg W, Friedl A, Syndecan-1 and syndecan-4 are overexpressed in an estrogen receptor-negative, highly proliferative breast cancer subtype, Breast Cancer Res. Treat, 98 (2006) 91–98. [DOI] [PubMed] [Google Scholar]
- [131].Van Bockstaele F, Pede V, Janssens A, Callewaert F, Offner F, Verhasselt B, Philippe J, Lipoprotein lipase mRNA expression in whole blood is a prognostic marker in B cell chronic lymphocytic leukemia, Clin Chem, 53 (2007) 204–212. [DOI] [PubMed] [Google Scholar]
- [132].Kristensen L, Kristensen T, Abildgaard N, Royo C, Frederiksen M, Mourits-Andersen T, Campo E, Moller MB, LPL gene expression is associated with poor prognosis in CLL and closely related to NOTCH1 mutations, Eur J Haematol, 97 (2016) 175–182. [DOI] [PubMed] [Google Scholar]
- [133].Matrai Z, Andrikovics H, Szilvasi A, Bors A, Kozma A, Adam E, Halm G, Karaszi E, Tordai A, Masszi T, Lipoprotein Lipase as a Prognostic Marker in Chronic Lymphocytic Leukemia, Pathology oncology research : POR, 23 (2017) 165–171. [DOI] [PubMed] [Google Scholar]
- [134].Cao D, Song X, Che L, Li X, Pilo MG, Vidili G, Porcu A, Solinas A, Cigliano A, Pes GM, Ribback S, Dombrowski F, Chen X, Li L, Calvisi DF, Both de novo synthetized and exogenous fatty acids support the growth of hepatocellular carcinoma cells, Liver international : official journal of the International Association for the Study of the Liver, 37 (2017) 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, Pettus JR, Froehlich HM, Memoli VA, Morganelli PM, Swinnen JV, Timmerman LA, Chaychi L, Fricano CJ, Eisenberg BL, Coleman WB, Kinlaw WB, Lipoprotein lipase links dietary fat to solid tumor cell proliferation, Mol Cancer Ther, 10 (2011) 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Camarda R, Zhou A, Kohnz R, Balakrishnan S, Mahieu C, Anderton B, Eyob H, Kajimura S, Tward A, Krings G, Nomura D, Goga A, Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer, Nat. Med, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Monaco M, Fatty acid metabolism in breast cancer subtypes., Oncotarget, 8 (2017) 29487–29500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Castellano-Castillo D, Moreno-Indias I, Fernandez-Garcia JC, Alcaide-Torres J, Moreno-Santos I, Ocana L, Gluckman E, Tinahones F, Queipo-Ortuno MI, Cardona F, Adipose Tissue LPL Methylation is Associated with Triglyceride Concentrations in the Metabolic Syndrome, Clin Chem, 64 (2018) 210–218. [DOI] [PubMed] [Google Scholar]
- [139].Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM, Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer, Breast Cancer Research : BCR, 12 (2010) R68–R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Dong L, Yuan Y, Opansky C, Cheng Y, Aguilera-Barrantes I, Wu S, Yuan R, Cheng Y, Sahoo D, Silverstein R, Ren B, Diet-induced obesity links to ER positive breast cancer progression via LPA/PKD-1-CD36 signaling-mediated microvascular remodeling, Oncotarget, 8 (2017) 22550–22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Prieto D, Seija N, Uriepero A, Souto-Padron T, Oliver C, Irigoin V, Guillermo C, Navarrete MA, Ines Landoni A, Dighiero G, Gabus R, Giordano M, Oppezzo P, LPL protein in Chronic Lymphocytic Leukaemia have different origins in Mutated and Unmutated patients. Advances for a new prognostic marker in CLL, British journal of haematology, 182 (2018) 521–525. [DOI] [PubMed] [Google Scholar]
- [142].Menard J, Christianson H, Kucharzewska P, Bourseau-Guilmain E, Svensson K, Lindqvist E, Indira-Chandran V, Johansson M, Beiting M, Metastasis stimulation by hypoxia and aciodosis-induced extracellular lipid uptake is mediated by proteoglycan-dependent endocytosis., Cancer Research, 76 (2016) 4828–4840. [DOI] [PubMed] [Google Scholar]
- [143].Davies B, Beigneux A, Barnes R, Tu Y, Gin P, Weinstein M, Nobumori C, Nyren R, Olivecrona G, Bensadoun A, Young S, Fong L, GPIHP1 is responsible for the entry of lipoprotein lipase into capillaries, Cell Metabolism, 12 (2010) 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Balaban S, Nassar ZD, Zhang AY, Hosseini-Beheshti E, Centenera MM, Schreuder M, Lin HM, Aishah A, Varney B, Liu-Fu F, Lee LS, Nagarajan SR, Shearer RF, Hardie RA, Raftopulos NL, Kakani MS, Saunders DN, Holst J, Horvath LG, Butler LM, Hoy AJ, Extracellular Fatty Acids are the Major Contributor to Lipid Synthesis in Prostate Cancer, Mol Cancer Res, (2019). [DOI] [PubMed] [Google Scholar]
- [145].Dong X, Wang G, Zhang G, Ni Z, Suo J, Cui J, Cui A, Yang Q, Xu Y, Li F, The endothelial lipase protein is promising urinary biomarker for diagnosis of gastric cancer, Diagnostic pathology, 8 (2013) 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Slebe F, Rojo F, Vinaixa M, Garcia-Rocha M, Testoni G, Guiu M, Planet E, Samino S, Arenas EJ, Beltran A, Rovira A, Lluch A, Salvatella X, Yanes O, Albanell J, Guinovart JJ, Gomis RR, FoxA and LIPG endothelial lipase control the uptake of extracellular lipids for breast cancer growth, Nat Commun, 7 (2016) 11199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Akutagawa T, Aoki S, Yamamoto-Rikitake M, Iwakiri R, Fujimoto K, Toda S, Cancer-adipose tissue interaction and fluid flow synergistically modulate cell kinetics, HER2 expression, and trastuzumab efficacy in gastric cancer, Gastric cancer : official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association, (2018). [DOI] [PubMed] [Google Scholar]
- [148].Cadenas C, Vosbeck S, Edlund K, Grgas K, Madjar K, Hellwig B, Adawy A, Glotzbach A, Stewart JD, Lesjak MS, Franckenstein D, Claus M, Hayen H, Schriewer A, Gianmoena K, Thaler S, Schmidt M, Micke P, Ponten F, Mardinoglu A, Zhang C, Kafferlein HU, Watzl C, Frank S, Rahnenfuhrer J, Marchan R, Hengstler JG, LIPG-promoted lipid storage mediates adaptation to oxidative stress in breast cancer, International journal of cancer, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, Zhang C, Lifshitz V, Song J, Yuan Y, Somlo G, Jandial R, Ann D, Hanash S, Jove R, Yu H, JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance, Cell Metab, 27 (2018) 136–150 e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Nomura D, Long J, Niessen S, Hoover H, Ng S, Cravatt B, Monoacylglycerol lipase regulates a fatty acid network the promotes cancer pathogenesis., Cell, 140 (2010) 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Pagano E, Borrelli F, Orlando P, Romano B, Monti M, Morbidelli L, Aviello G, Imperatore R, Capasso R, Piscitelli F, Buono L, Di Marzo V, Izzo AA, Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis, Pharmacological research, 119 (2017) 227–236. [DOI] [PubMed] [Google Scholar]
- [152].Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, Mai J, Shen H, Hu DZ, Adoro S, Hu B, Song M, Tan C, Landis MD, Ferrari M, Shin SJ, Brown M, Chang JC, Liu XS, Glimcher LH, XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway, Nature, 508 (2014) 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Zhu W, Zhao Y, Zhou J, Wang X, Pan Q, Zhang N, Wang L, Wang M, Zhan D, Liu Z, He X, Ma D, Liu S, Wang L, Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-kappaB-mediated epithelial-mesenchymal transition, Journal of hematology & oncology, 9 (2016) 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Pan J, Fan Z, Wang Z, Dai Q, Xiang Z, Yuan F, Yan M, Zhu Z, Liu B, Li C, CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3beta/beta-catenin pathway, J Exp Clin Cancer Res, 38 (2019) 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Ladanyi A, Mukherjee A, Kenny H, Johnson A, Mitra A.
- [156].Zhao J, Zhi Z, Wang C, Xing H, Song G, Yu X, Zhu Y, Wang X, Zhang X, Di Y, Exogenous lipids promote the growth of breast cancer cells via CD36, Oncology reports, 38 (2017) 2105–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, Bescos C, Di Croce L, Benitah SA, Targeting metastasis-initiating cells through the fatty acid receptor CD36, Nature, 541 (2017) 41–45. [DOI] [PubMed] [Google Scholar]
- [158].Feng W, Wilkens O, Bang S, Ung M, Li J, An J, del Genio C, Canfield K, D’iRenzo J, Wells W, Gaur A, Robey R, Guo J, Powles R, Sotiriuo C, Pusztal L, Febbrario M, Cheng C, Kinlaw W, Kurakowa M, CD36-mediated metabolic rewiring of breast cancer cells promotes resistance to HER2-targeted therapies, Cell reports, 29 (2019) 3405–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Watt MJ, Clark A, Selth L, Haynes V, Lister N, Rebello R, Porter L, Nitanjan B, Whitby S, Lo J, Huang C, Schittenhelm R, Anderson K, Furic L, Wijayaratne P, Maytzaris M, Montgomery M, Papargiris M, Norden S, Febbraio M, Risbridger G, Frydenberg M, Nomura D, Taylor RA, Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer, Sci. Transl. Med, 11 (2019) DOI: 10.1126/scitranslmed.aau5758. [DOI] [PubMed] [Google Scholar]
- [160].Nath A, Li I, Roberts LR, Chan C, Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma, Sci Rep, 5 (2015) 14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Aloia A, Mulhaupt D, Chabbert C, Eberhart T, Flueckiger S, Vukolic A, Eichoff O, Irmisch A, Alexander L, Scibona E, Frederick D, Miao B, Tian T, Cheng C, Kwong L, Wei Z, Sullivan R, Boland G, Herlyn M, Flaherty K, Zamboni N, D’ummer R, Zghang G, Levesque M, Krek W, Kocvacs W, A fatty acid oxidation-dependent metabolic shift regulates the adaptation of BRAF-mutated melanoma to MAPK inhibitors, Clin. Cancer Res, (2019) DOI: 10.1158/1078-0432.CCR-1119-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins J, Pinnick K, Wigfield S, Buffa F, Li J, Zhang Q, Wakelam M, Karpe F, Schulze A, Harris A, Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation, Cell Rep., 9 (2014) 349–365. [DOI] [PubMed] [Google Scholar]
- [163].Antalis C, Arnold T, Rasool T, Lee B, Buhman K, Siddiqui R, High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation., Breast Cancer Res. Treat, 122 (2010) 661–670. [DOI] [PubMed] [Google Scholar]
- [164].Chamras H, Ardashian A, Heber D, Glaspy JA, Fatty acid modulation of MCF-7 human breast cancer cell proliferation, apoptosis and differentiation, The Journal of nutritional biochemistry, 13 (2002) 711–716. [DOI] [PubMed] [Google Scholar]
- [165].Przybytkowski E, Joly E, Nolan CJ, Hardy S, Francoeur AM, Langelier Y, Prentki M, Upregulation of cellular triacylglycerol - free fatty acid cycling by oleate is associated with long-term serum-free survival of human breast cancer cells, Biochemistry and cell biology = Biochimie et biologie cellulaire, 85 (2007) 301–310. [DOI] [PubMed] [Google Scholar]
- [166].Shiu RP, Paterson JA, Alteration of cell shape, adhesion, and lipid accumulation in human breast cancer cells (T-47D) by human prolactin and growth hormone, Cancer Res, 44 (1984) 1178–1186. [PubMed] [Google Scholar]
- [167].Nassar ZD, Aref AT, Miladinovic D, Mah CY, Raj GV, Hoy AJ, Butler LM, Peri-prostatic adipose tissue: the metabolic microenvironment of prostate cancer, BJU international, 121 Suppl 3 (2018) 9–21. [DOI] [PubMed] [Google Scholar]
- [168].Fantuzzi G, Adipose tissue, adipokines, and inflammation, The Journal of allergy and clinical immunology, 115 (2005) 911–919; quiz 920. [DOI] [PubMed] [Google Scholar]
- [169].Balaban S, Shearer R, Lee L, van Geldermaisen M, Schreuder M, Shtein H, Cairns R, Thomas K, Fazakerley D, Grewal T, Holst J, Saunders D, Hoy A, Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration., Cancer and Metabolism, 5 (2017) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Go GW, Mani A, Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis, The Yale journal of biology and medicine, 85 (2012) 19–28. [PMC free article] [PubMed] [Google Scholar]
- [171].Blücher C, Stadler SC, Obesity and Breast Cancer: Current Insights on the Role of Fatty Acids and Lipid Metabolism in Promoting Breast Cancer Growth and Progression, Frontiers in endocrinology, 8 (2017) 293–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Nieman K, Romero I, Van Houten B, Lengyel E, Adipose tissue and adipocytes support tumorigenesis and metastasis, Biochimica et biophysica acta, 1831 (2013) 1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Nieman K, Kenny H, Penicka C, Ladanyi A, Buell-Gutbrod R, Zillhardt M, Romero I, Carey M, Mills G, Hotamisligil G, Yamada S, Peter M, Gwin K, Lengyel E, Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth, Nat Med, 17 (2011) 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Guaita-Esteruelas S, Bosquet A, Saavedra P, Guma J, Girona J, Lam EW, Amillano K, Borras J, Masana L, Exogenous FABP4 increases breast cancer cell proliferation and activates the expression of fatty acid transport proteins, Molecular carcinogenesis, 56 (2017) 208–217. [DOI] [PubMed] [Google Scholar]
- [175].Chiu AP, Wan A, Lal N, Zhang D, Wang F, Vlodavsky I, Hussein B, Rodrigues B, Cardiomyocyte VEGF Regulates Endothelial Cell GPIHBP1 to Relocate Lipoprotein Lipase to the Coronary Lumen During Diabetes Mellitus, Arterioscler Thromb Vasc Biol, 36 (2016) 145–155. [DOI] [PubMed] [Google Scholar]
- [176].Park JW, Zhao L, Willingham MC, Cheng SY, Inhibition of STAT3 signaling blocks obesity-induced mammary hyperplasia in a mouse model, American journal of cancer research, 7 (2017) 727–739. [PMC free article] [PubMed] [Google Scholar]
- [177].Heemers H, Maes B, Foufelle F, Heyns W, Verhoeven G, Swinnen J, Androgens stimulate lipogenic gene expression by activation of sterol regulatory element-binding protein cleavage activating protein/sterol regulatory element-binding pathway., Mol. Endocrinol, 15 (2001) 1817–1828. [DOI] [PubMed] [Google Scholar]
- [178].Andersen OM, Rudolph IM, Willnow TE, Risk factor SORL1: from genetic association to functional validation in Alzheimer’s disease, Acta neuropathologica, 132 (2016) 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Lupien L, Dehairs J, Bloch K, Feng W, Davis W, Zhao Y, Ung M, Cheng C, Swinnen J, Kinlaw W, Endocytosis of very low-density lipoproteins: an unexpected mechanism for lipid acquisition by breast cancer cells, Journal of Lipid Research, 61 (2019) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Olsen A, Eisenberg B, Flanagan A, Kuemmerle N, Morganelli P, Lombardo P, Kinlaw W, Fatty Acid Synthesis is a Therapeutic Target in Human Liposarcoma, Int. J. Oncol, 36 (2010) 1309–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Daniels V, Smans K, Royaux I, Chypre M, Swinnen J, Zaidi N, Cancer cells differentially activate and thrive on de novo lipid synthesis pathways in a low-lipid environment., PloS one, 9(9):e106913, 2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Zaidi N, Lupien L, Kuemmerle N, Kinlaw W, Swinnen J, Smans K, Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids, Progr. Lipid Res, 52 (2013) 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Menard J, Cerezo-Magana M, Belting M, Functional role of extracellular vesicles and lipoproteins in the tumour microenvironment., Phil. Trans. R. Soc., B 373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Herroon M, Rajagurubandara E, Hardaway A, Powell K, Turchick A, Feldmann D, Podgorski I, Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms, Oncotarget, 4 (2013) 2108–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Mentoor I, Engelbrecht AM, Nell T, Fatty acids: Adiposity and breast cancer chemotherapy, a bad synergy?, Prostaglandins, leukotrienes, and essential fatty acids, 140 (2019) 18–33. [DOI] [PubMed] [Google Scholar]
- [186].Schoors S, Brunning U, Missiaen R, Queiroz K, Borgers G, Elia I, Zecchin A, Cantelmo A, Christen S, Goveia J, Heggermont W, Godde L, Vinckier S, Van Veldhoven P, Eelen G, Schoonjans L, Gerhardt H, Dewerchin M, Baes M, De Bock K, Ghesquiere B, Lunt S, Fendt S, Carmeliet P, Fatty acid carbon is esential for dNTP synthesis in endothelial cells., Nature, 520 (2015) 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].DeFilippis R, Chang H, Dumont N, Rabban J, Chen Y, Fontenay G, Berman HA, Gauthier M, Zhao J, Hu DZ, Marx J, Tjoe J, Ziv E, Febbraio M, Kerlikowske K, Parvin B, Tlsty T, CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues, Cancer Discov, 2 (2012) 826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Ecker J, Liebisch G, Englmaier M, Grandl M, Robenek H, Schmitz G, Induction of fatty acid synthesis is a key requirement for phagocytic differentiation of human monocytes, Proceedings of the National Academy of Sciences of the United States of America, 107 (2010) 7817–7822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Yang G, Wang Y, Feng J, Liu Y, Wang T, Zhao M, Ye L, Zhang X, Aspirin suppresses the abnormal lipid metabolism in liver cancer cells via disrupting an NFkappaB-ACSL1 signaling, Biochem Biophys Res Commun, 486 (2017) 827–832. [DOI] [PubMed] [Google Scholar]
- [190].Vargas T, Moreno-Rubio J, Herranz J, Cejas P, Molina S, Mendiola M, Burgos E, Custodio AB, De Miguel M, Martin-Hernandez R, Reglero G, Feliu J, Ramirez de Molina A, 3’UTR Polymorphism in ACSL1 Gene Correlates with Expression Levels and Poor Clinical Outcome in Colon Cancer Patients, PloS one, 11 (2016) e0168423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Krammer J, Digel M, Ehehalt F, Stremmel W, Fullekrug J, Ehehalt R, Overexpression of CD36 and acyl-CoA synthetases FATP2, FATP4 and ACSL1 increases fatty acid uptake in human hepatoma cells, Int J Med Sci, 8 (2011) 599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Tang Y, Zhou J, Hooi SC, Jiang YM, Lu GD, Fatty acid activation in carcinogenesis and cancer development: Essential roles of long-chain acyl-CoA synthetases, Oncol Lett, 16 (2018) 1390–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Amiri M, Yousefnia S, Seyed Forootan F, Peymani M, Ghaedi K, Nasr Esfahani MH, Diverse roles of fatty acid binding proteins (FABPs) in development and pathogenesis of cancers, Gene, 676 (2018) 171–183. [DOI] [PubMed] [Google Scholar]
- [194].Hancke K, Grubeck D, Hauser N, Kreienberg R, Weiss JM, Adipocyte fatty acid-binding protein as a novel prognostic factor in obese breast cancer patients, Breast Cancer Res Treat, 119 (2010) 367–367. [DOI] [PubMed] [Google Scholar]
- [195].Lass A, Zimmermann R, Oberer M, Zechner R, Lipolysis - a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores, Prog Lipid Res, 50 (2011) 14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Ohata T, Yokoo H, Kamiyama T, Fukai M, Aiyama T, Hatanaka Y, Hatanaka K, Wakayama K, Orimo T, Kakisaka T, Kobayashi N, Matsuno Y, Taketomi A, Fatty acid-binding protein 5 function in hepatocellular carcinoma through induction of epithelial-mesenchymal transition, Cancer medicine, 6 (2017) 1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Das R, Hammamieh R, Neill R, Melhem M, Jett M, Expression pattern of fatty acid-binding proteins in human normal and cancer prostate cells and tissues, Clinical cancer research : an official journal of the American Association for Cancer Research, 7 (2001) 1706–1715. [PubMed] [Google Scholar]
- [198].de Wit NJ, Rijntjes J, Diepstra JH, van Kuppevelt TH, Weidle UH, Ruiter DJ, van Muijen GN, Analysis of differential gene expression in human melanocytic tumour lesions by custom made oligonucleotide arrays, British journal of cancer, 92 (2005) 2249–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Al Fayi MS, Gou X, Forootan SS, Al-Jameel W, Bao Z, Rudland PR, Cornford PA, Hussain SA, Ke Y, The increased expression of fatty acid-binding protein 9 in prostate cancer and its prognostic significance, Oncotarget, 7 (2016) 82783–82797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Wang M, Han J, Xing H, Zhang H, Li Z, Liang L, Li C, Dai S, Wu M, Shen F, Yang T, Dysregulated fatty acid metabolism in hepatocellular carcinoma, Hepatic oncology, 3 (2016) 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Holder A, Gonzalez-Angulo A, Chen H, Akcakanat A, Do K, Fraser Symmans W, Pusztai L, Hortobagyi G, Mills G, Meric-Bernstam F, High steroyl-CoA desaturase 1 expression is associated with shgorter survival in breast cancer patients., Breast Cancer Res. Treat, 137 (2013) 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Presler M, Wojtczyk-Miaskowska A, Schlichtholz B, Kaluzny A, Matuszewski M, Mika A, Sledzinski T, Swierczynski J, Increased expression of the gene encoding stearoyl-CoA desaturase 1 in human bladder cancer, Molecular and cellular biochemistry, 447 (2018) 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Guo D, Bell EH, Mischel P, Chakravarti A, Targeting SREBP-1-driven lipid metabolism to treat cancer, Current pharmaceutical design, 20 (2014) 2619–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Notarnicola M, Lorusso D, Tutino V, De Nunzio V, De Leonardis G, Marangelli G, Guerra V, Veronese N, Caruso MG, Giannelli G, Differential Tissue Fatty Acids Profiling between Colorectal Cancer Patients with and without Synchronous Metastasis, Int J Mol Sci, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Mika A, Kobiela J, Pakiet A, Czumaj A, Sokolowska E, Makarewicz W, Chmielewski M, Stepnowski P, Marino-Gammazza A, Sledzinski T, Preferential uptake of polyunsaturated fatty acids by colorectal cancer cells, Sci Rep, 10 (2020) 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, Furdui CM, Hegde P, Torti FM, Torti SV, Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death, Cancer Res, 79 (2019) 5355–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Piao C, Cui X, Zhan B, Li J, Li Z, Li Z, Liu X, Bi J, Zhang Z, Kong C, Inhibition of stearoyl CoA desaturase-1 activity suppresses tumour progression and improves prognosis in human bladder cancer, J Cell Mol Med, 23 (2019) 2064–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Pisanu ME, Maugeri-Sacca M, Fattore L, Bruschini S, De Vitis C, Tabbi E, Bellei B, Migliano E, Kovacs D, Camera E, Picardo M, Jakopin Z, Cippitelli C, Bartolazzi A, Raffa S, Torrisi MR, Fulciniti F, Ascierto PA, Ciliberto G, Mancini R, Inhibition of Stearoyl-CoA desaturase 1 reverts BRAF and MEK inhibition-induced selection of cancer stem cells in BRAF-mutated melanoma, Journal of experimental & clinical cancer research : CR, 37 (2018) 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Imamura K, Tomita N, Kawakita Y, Ito Y, Ono K, Nii N, Miyazaki T, Yonemori K, Tawada M, Sumi H, Satoh Y, Yamamoto Y, Miyahisa I, Sasaki M, Satomi Y, Hirayama M, Nishigaki R, Maezaki H, Discovery of Novel and Potent Stearoyl Coenzyme A Desaturase 1 (SCD1) Inhibitors as Anticancer Agents, Bioorg Med Chem, 25 (2017) 3768–3779. [DOI] [PubMed] [Google Scholar]
- [210].Peck B, Schug ZT, Zhang Q, Dankworth B, Jones DT, Smethurst E, Patel R, Mason S, Jiang M, Saunders R, Howell M, Mitter R, Spencer-Dene B, Stamp G, McGarry L, James D, Shanks E, Aboagye EO, Critchlow SE, Leung HY, Harris AL, Wakelam MJO, Gottlieb E, Schulze A, Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments, Cancer Metab, 4 (2016) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Lewis CA, Brault C, Peck B, Bensaad K, Griffiths B, Mitter R, Chakravarty P, East P, Dankworth B, Alibhai D, Harris AL, Schulze A, SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme, Oncogene, 34 (2015) 5128–5140. [DOI] [PubMed] [Google Scholar]
- [212].Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, Xie H, Simon MC, Kamphorst JJ, Triglycerides Promote Lipid Homeostasis during Hypoxic Stress by Balancing Fatty Acid Saturation, Cell reports, 24 (2018) 2596–2605.e2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Ge L, Gordon JS, Hsuan C, Stenn K, Prouty SM, Identification of the delta-6 desaturase of human sebaceous glands: expression and enzyme activity, J Invest Dermatol, 120 (2003) 707–714. [DOI] [PubMed] [Google Scholar]
- [214].Muir K, Hazim A, He Y, Peyressatre M, Kim DY, Song X, Beretta L, Proteomic and lipidomic signatures of lipid metabolism in NASH-associated hepatocellular carcinoma, Cancer Res, 73 (2013) 4722–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Pender-Cudlip MC, Krag KJ, Martini D, Yu J, Guidi A, Skinner SS, Zhang Y, Qu X, He C, Xu Y, Qian SY, Kang JX, Delta-6-desaturase activity and arachidonic acid synthesis are increased in human breast cancer tissue, Cancer science, 104 (2013) 760–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, Dehairs J, Escalona-Noguero C, Schmieder R, Cornfield T, Charlton C, Romero-Perez L, Rossi M, Rinaldi G, Orth MF, Boon R, Kerstens A, Kwan SY, Faubert B, Mendez-Lucas A, Kopitz CC, Chen T, Fernandez-Garcia J, Duarte JAG, Schmitz AA, Steigemann P, Najimi M, Hagebarth A, Van Ginderachter JA, Sokal E, Gotoh N, Wong KK, Verfaillie C, Derua R, Munck S, Yuneva M, Beretta L, DeBerardinis RJ, Swinnen JV, Hodson L, Cassiman D, Verslype C, Christian S, Grunewald S, Grunewald TGP, Fendt SM, Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity, Nature, 566 (2019) 403–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Hansen-Petrik MB, McEntee MF, Johnson BT, Obukowicz MG, Masferrer J, Zweifel B, Chiu CH, Whelan J, Selective inhibition of Delta-6 desaturase impedes intestinal tumorigenesis, Cancer letters, 175 (2002) 157–163. [DOI] [PubMed] [Google Scholar]
- [218].Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, Olzmann JA, Dixon SJ, Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State, Cell Chem Biol, 26 (2019) 420–432 e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Jump DB, Mammalian fatty acid elongases, Methods Mol Biol, 579 (2009) 375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Naganuma T, Sato Y, Sassa T, Ohno Y, Kihara A, Biochemical characterization of the very long-chain fatty acid elongase ELOVL7, FEBS letters, 585 (2011) 3337–3341. [DOI] [PubMed] [Google Scholar]
- [221].Su YC, Feng YH, Wu HT, Huang YS, Tung CL, Wu P, Chang CJ, Shiau AL, Wu CL, Elovl6 is a negative clinical predictor for liver cancer and knockdown of Elovl6 reduces murine liver cancer progression, Sci Rep, 8 (2018) 6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Marien E, Meister M, Muley T, Gomez Del Pulgar T, Derua R, Spraggins JM, Van de Plas R, Vanderhoydonc F, Machiels J, Binda MM, Dehairs J, Willette-Brown J, Hu Y, Dienemann H, Thomas M, Schnabel PA, Caprioli RM, Lacal JC, Waelkens E, Swinnen JV, Phospholipid profiling identifies acyl chain elongation as a ubiquitous trait and potential target for the treatment of lung squamous cell carcinoma, Oncotarget, 7 (2016) 12582–12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Tamura K, Makino A, Hullin-Matsuda F, Kobayashi T, Furihata M, Chung S, Ashida S, Miki T, Fujioka T, Shuin T, Nakamura Y, Nakagawa H, Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long-chain fatty acid metabolism, Cancer Research, 69 (2009) 8133–8140. [DOI] [PubMed] [Google Scholar]
- [224].Gimple RC, Kidwell RL, Kim LJY, Sun T, Gromovsky AD, Wu Q, Wolf M, Lv D, Bhargava S, Jiang L, Prager BC, Wang X, Ye Q, Zhu Z, Zhang G, Dong Z, Zhao L, Lee D, Bi J, Sloan AE, Mischel PS, Brown JM, Cang H, Huan T, Mack SC, Xie Q, Rich JN, Glioma Stem Cell-Specific Superenhancer Promotes Polyunsaturated Fatty-Acid Synthesis to Support EGFR Signaling, Cancer discovery, 9 (2019) 1248–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Kim W, Deik A, Gonzalez C, Gonzalez ME, Fu F, Ferrari M, Churchhouse CL, Florez JC, Jacobs SBR, Clish CB, Rhee EP, Polyunsaturated Fatty Acid Desaturation Is a Mechanism for Glycolytic NAD(+) Recycling, Cell Metab, 29 (2019) 856–870 e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Zimmerlin A, Salaun JP, Durst F, Mioskowski C, Cytochrome p-450-dependent hydroxylation of lauric Acid at the subterminal position and oxidation of unsaturated analogs in wheat microsomes, Plant Physiol, 100 (1992) 868–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Fang B, Xu H, Liu Y, Qi F, Zhang W, Chen H, Wang C, Wang Y, Yang W, Li S, Mutagenesis and redox partners analysis of the P450 fatty acid decarboxylase OleTJE, Sci Rep, 7 (2017) 44258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Guo L, Zhang X, Zhou D, Okunade AL, Su X, Stereospecificity of fatty acid 2-hydroxylase and differential functions of 2-hydroxy fatty acid enantiomers, J Lipid Res, 53 (2012) 1327–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Zoller I, Meixner M, Hartmann D, Bussow H, Meyer R, Gieselmann V, Eckhardt M, Absence of 2-hydroxylated sphingolipids is compatible with normal neural development but causes late-onset axon and myelin sheath degeneration, The Journal of neuroscience : the official journal of the Society for Neuroscience, 28 (2008) 9741–9754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Dai X, Zhang S, Cheng H, Cai D, Chen X, Huang Z, FA2H Exhibits Tumor Suppressive Roles on Breast Cancers via Cancer Stemness Control, Front Oncol, 9 (2019) 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Cheng M, Bhujwalla ZM, Glunde K, Targeting Phospholipid Metabolism in Cancer, Front Oncol, 6 (2016) 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Harayama T, Riezman H, Understanding the diversity of membrane lipid composition, Nat Rev Mol Cell Biol, 19 (2018) 281–296. [DOI] [PubMed] [Google Scholar]
- [233].Brohee L, Demine S, Willems J, Arnould T, Colige AC, Deroanne CF, Lipin-1 regulates cancer cell phenotype and is a potential target to potentiate rapamycin treatment, Oncotarget, 6 (2015) 11264–11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Meana C, Garcia-Rostan G, Pena L, Lorden G, Cubero A, Orduna A, Gyorffy B, Balsinde J, Balboa MA, The phosphatidic acid phosphatase lipin-1 facilitates inflammation-driven colon carcinogenesis, JCI Insight, 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].He J, Zhang F, Tay LWR, Boroda S, Nian W, Levental KR, Levental I, Harris TE, Chang JT, Du G, Lipin-1 regulation of phospholipid synthesis maintains endoplasmic reticulum homeostasis and is critical for triple-negative breast cancer cell survival, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 31 (2017) 2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Sanchez-Lopez E, Zimmerman T, Gomez del Pulgar T, Moyer MP, Lacal Sanjuan JC, Cebrian A, Choline kinase inhibition induces exacerbated endoplasmic reticulum stress and triggers apoptosis via CHOP in cancer cells, Cell death & disease, 4 (2013) e933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Arlauckas SP, Popov AV, Delikatny EJ, Choline kinase alpha-Putting the ChoK-hold on tumor metabolism, Prog Lipid Res, 63 (2016) 28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Park JB, Lee CS, Jang JH, Ghim J, Kim YJ, You S, Hwang D, Suh PG, Ryu SH, Phospholipase signalling networks in cancer, Nat Rev Cancer, 12 (2012) 782–792. [DOI] [PubMed] [Google Scholar]
- [239].Cai Q, Zhao Z, Antalis C, Yan L, Del Priore G, Hamed AH, Stehman FB, Schilder JM, Xu Y, Elevated and secreted phospholipase A(2) activities as new potential therapeutic targets in human epithelial ovarian cancer, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 26 (2012) 3306–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Henkels KM, Boivin GP, Dudley ES, Berberich SJ, Gomez-Cambronero J, Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model, Oncogene, 32 (2013) 5551–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Tousignant KD, Rockstroh A, Poad BL, Talebi A, Young RR, Fard AT, Gupta R, Zang T, Wang C, Lehman ML, Swinnen JV, Blanksby SJ, Nelson CC, Sadowski MC, Therapy-induced lipid uptake and remodeling underpin ferroptosis hypersensitivity in prostate cancer, bioRxiv, (2020) 2020.2001.2008.899609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Shi C, Qiao S, Wang S, Wu T, Ji G, Recent progress of lysophosphatidylcholine acyltransferases in metabolic disease and cancer, Int J Clin Exp Med, (2018) 8941–8953. [Google Scholar]
- [243].Cui W, Li SY, Du JF, Zhu ZM, An P, Silencing phospholipid scramblase 1 expression by RNA interference in colorectal cancer and metastatic liver cancer, Hepatobiliary Pancreat Dis Int, 11 (2012) 393–400. [DOI] [PubMed] [Google Scholar]
- [244].Carracedo A, Cantley LC, Pandolfi PP, Cancer metabolism: fatty acid oxidation in the limelight, Nat Rev Cancer, 13 (2013) 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, Fang X, Fatty acid oxidation: An emerging facet of metabolic transformation in cancer, Cancer Lett, 435 (2018) 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Luo J, Hong Y, Tao X, Wei X, Zhang L, Li Q, An indispensable role of CPT-1a to survive cancer cells during energy stress through rewiring cancer metabolism, Tumour Biol, (2016). [DOI] [PubMed] [Google Scholar]
- [247].He W, Liang B, Wang C, Li S, Zhao Y, Huang Q, Liu Z, Yao Z, Wu Q, Liao W, Zhang S, Liu Y, Xiang Y, Liu J, Shi M, MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer, Oncogene, 38 (2019) 4637–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Shi J, Fu H, Jia Z, He K, Fu L, Wang W, High Expression of CPT1A Predicts Adverse Outcomes: A Potential Therapeutic Target for Acute Myeloid Leukemia, EBioMedicine, 14 (2016) 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, Eyob H, Kajimura S, Tward A, Krings G, Nomura DK, Goga A, Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer, Nature medicine, 22 (2016) 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Cohen S, Lipid Droplets as Organelles, Int Rev Cell Mol Biol, 337 (2018) 83–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Sirois I, Aguilar-Mahecha A, Lafleur J, Fowler E, Vu V, Scriver M, Buchanan M, Chabot C, Ramanathan A, Balachandran B, Legare S, Przybytkowski E, Lan C, Krzemien U, Cavallone L, Aleynikova O, Ferrario C, Guilbert MC, Benlimame N, Saad A, Alaoui-Jamali M, Saragovi HU, Josephy S, O’Flanagan C, Hursting SD, Richard VR, Zahedi RP, Borchers CH, Bareke E, Nabavi S, Tonellato P, Roy JA, Robidoux A, Marcus EA, Mihalcioiu C, Majewski J, Basik M, A Unique Morphological Phenotype in Chemoresistant Triple-Negative Breast Cancer Reveals Metabolic Reprogramming and PLIN4 Expression as a Molecular Vulnerability, Mol Cancer Res, 17 (2019) 2492–2507. [DOI] [PubMed] [Google Scholar]
- [252].Schlaepfer IR, Hitz CA, Gijon MA, Bergman BC, Eckel RH, Jacobsen BM, Progestin modulates the lipid profile and sensitivity of breast cancer cells to docetaxel, Mol Cell Endocrinol, 363 (2012) 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Verbrugge SE, Al M, Assaraf YG, Kammerer S, Chandrupatla DM, Honeywell R, Musters RP, Giovannetti E, O’Toole T, Scheffer GL, Krige D, de Gruijl TD, Niessen HW, Lems WF, Kramer PA, Scheper RJ, Cloos J, Ossenkoppele GJ, Peters GJ, Jansen G, Multifactorial resistance to aminopeptidase inhibitor prodrug CHR2863 in myeloid leukemia cells: down-regulation of carboxylesterase 1, drug sequestration in lipid droplets and pro-survival activation ERK/Akt/mTOR, Oncotarget, 7 (2016) 5240–5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Hultsch S, Kankainen M, Paavolainen L, Kovanen RM, Ikonen E, Kangaspeska S, Pietiainen V, Kallioniemi O, Association of tamoxifen resistance and lipid reprogramming in breast cancer, BMC Cancer, 18 (2018) 850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Cotte AK, Aires V, Fredon M, Limagne E, Derangere V, Thibaudin M, Humblin E, Scagliarini A, de Barros JP, Hillon P, Ghiringhelli F, Delmas D, Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance, Nat Commun, 9 (2018) 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Nardi F, Franco OE, Fitchev P, Morales A, Vickman RE, Hayward SW, Crawford SE, DGAT1 Inhibitor Suppresses Prostate Tumor Growth and Migration by Regulating Intracellular Lipids and Non-Centrosomal MTOC Protein GM130, Sci Rep, 9 (2019) 3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Bagnato C, Igal RA, Overexpression of diacylglycerol acyltransferase-1 reduces phospholipid synthesis, proliferation, and invasiveness in simian virus 40-transformed human lung fibroblasts, J Biol Chem, 278 (2003) 52203–52211. [DOI] [PubMed] [Google Scholar]
- [258].Ohotski J, Edwards J, Elsberger B, Watson C, Orange C, Mallon E, Pyne S, Pyne NJ, Identification of novel functional and spatial associations between sphingosine kinase 1, sphingosine 1-phosphate receptors and other signaling proteins that affect prognostic outcome in estrogen receptor-positive breast cancer, International journal of cancer, 132 (2013) 605–616. [DOI] [PubMed] [Google Scholar]
- [259].Watson C, Long JS, Orange C, Tannahill CL, Mallon E, McGlynn LM, Pyne S, Pyne NJ, Edwards J, High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients, The American journal of pathology, 177 (2010) 2205–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].LaMontagne K, Littlewood-Evans A, Schnell C, O’Reilly T, Wyder L, Sanchez T, Probst B, Butler J, Wood A, Liau G, Billy E, Theuer A, Hla T, Wood J, Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization, Cancer Res, 66 (2006) 221–231. [DOI] [PubMed] [Google Scholar]
- [261].Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, Takabe K, Kordula T, Milstien S, Spiegel S, Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer, Cancer cell, 23 (2013) 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Watson DG, Tonelli F, Alossaimi M, Williamson L, Chan E, Gorshkova I, Berdyshev E, Bittman R, Pyne NJ, Pyne S, The roles of sphingosine kinases 1 and 2 in regulating the Warburg effect in prostate cancer cells, Cell Signal, 25 (2013) 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Payne AW, Pant DK, Pan TC, Chodosh LA, Ceramide kinase promotes tumor cell survival and mammary tumor recurrence, Cancer Res, 74 (2014) 6352–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Saddoughi SA, Ogretmen B, Diverse functions of ceramide in cancer cell death and proliferation, Adv Cancer Res, 117 (2013) 37–58. [DOI] [PubMed] [Google Scholar]
- [265].Thapa N, Tan X, Choi S, Lambert PF, Rapraeger AC, Anderson RA, The Hidden Conundrum of Phosphoinositide Signaling in Cancer, Trends in cancer, 2 (2016) 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Bunney TD, Katan M, Phosphoinositide signalling in cancer: beyond PI3K and PTEN, Nat Rev Cancer, 10 (2010) 342–352. [DOI] [PubMed] [Google Scholar]
- [267].Gomes RN, Felipe da Costa S, Colquhoun A, Eicosanoids and cancer, Clinics (Sao Paulo), 73 (2018) e530s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A, The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment, Carcinogenesis, 30 (2009) 377–386. [DOI] [PubMed] [Google Scholar]
- [269].Zhang B, Bie Q, Wu P, Zhang J, You B, Shi H, Qian H, Xu W, PGD2/PTGDR2 Signaling Restricts the Self-Renewal and Tumorigenesis of Gastric Cancer, Stem cells (Dayton, Ohio), 36 (2018) 990–1003. [DOI] [PubMed] [Google Scholar]
- [270].Kim J, Yang P, Suraokar M, Sabichi AL, Llansa ND, Mendoza G, Subbarayan V, Logothetis CJ, Newman RA, Lippman SM, Menter DG, Suppression of prostate tumor cell growth by stromal cell prostaglandin D synthase-derived products, Cancer Res, 65 (2005) 6189–6198. [DOI] [PubMed] [Google Scholar]
- [271].Du F, Yuelling L, Lee EH, Wang Y, Liao S, Cheng Y, Zhang L, Zheng C, Peri S, Cai KQ, Ng JMY, Curran T, Li P, Yang ZJ, Leukotriene Synthesis Is Critical for Medulloblastoma Progression, Clinical cancer research : an official journal of the American Association for Cancer Research, 25 (2019) 6475–6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Liu H, Zeng J, Huang W, Xu Q, Ye D, Sun R, Zhang D, Colorectal Cancer Is Associated with a Deficiency of Lipoxin A4, an Endogenous Anti-inflammatory Mediator, J Cancer, 10 (2019) 4719–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Chen Y, Hao H, He S, Cai L, Li Y, Hu S, Ye D, Hoidal J, Wu P, Chen X, Lipoxin A4 and its analogue suppress the tumor growth of transplanted H22 in mice: the role of antiangiogenesis, Mol Cancer Ther, 9 (2010) 2164–2174. [DOI] [PubMed] [Google Scholar]
- [274].Sulciner ML, Serhan CN, Gilligan MM, Mudge DK, Chang J, Gartung A, Lehner KA, Bielenberg DR, Schmidt B, Dalli J, Greene ER, Gus-Brautbar Y, Piwowarski J, Mammoto T, Zurakowski D, Perretti M, Sukhatme VP, Kaipainen A, Kieran MW, Huang S, Panigrahy D, Resolvins suppress tumor growth and enhance cancer therapy, J Exp Med, 215 (2018) 115–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Benjamin DI, Li DS, Lowe W, Heuer T, Kemble G, Nomura DK, Diacylglycerol Metabolism and Signaling Is a Driving Force Underlying FASN Inhibitor Sensitivity in Cancer Cells, ACS chemical biology, 10 (2015) 1616–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Magkrioti C, Oikonomou N, Kaffe E, Mouratis MA, Xylourgidis N, Barbayianni I, Megadoukas P, Harokopos V, Valavanis C, Chun J, Kosma A, Stathopoulos GT, Bouros E, Bouros D, Syrigos K, Aidinis V, The Autotaxin-Lysophosphatidic Acid Axis Promotes Lung Carcinogenesis, Cancer Res, 78 (2018) 3634–3644. [DOI] [PubMed] [Google Scholar]
- [277].Fukushima K, Takahashi K, Yamasaki E, Onishi Y, Fukushima N, Honoki K, Tsujiuchi T, Lysophosphatidic acid signaling via LPA1 and LPA3 regulates cellular functions during tumor progression in pancreatic cancer cells, Experimental cell research, 352 (2017) 139–145. [DOI] [PubMed] [Google Scholar]
- [278].Rogers LC, Davis RR, Said N, Hollis T, Daniel LW, Blocking LPA-dependent signaling increases ovarian cancer cell death in response to chemotherapy, Redox Biol, 15 (2018) 380–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Kaffe E, Magkrioti C, Aidinis V, Deregulated Lysophosphatidic Acid Metabolism and Signaling in Liver Cancer, Cancers (Basel), 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Wang M, Casey PJ, Protein prenylation: unique fats make their mark on biology, Nat Rev Mol Cell Biol, 17 (2016) 110–122. [DOI] [PubMed] [Google Scholar]
- [281].Chen B, Sun Y, Niu J, Jarugumilli GK, Wu X, Protein Lipidation in Cell Signaling and Diseases: Function, Regulation, and Therapeutic Opportunities, Cell Chem Biol, 25 (2018) 817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Ko PJ, Dixon SJ, Protein palmitoylation and cancer, EMBO Rep, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Waller DD, Park J, Tsantrizos YS, Inhibition of farnesyl pyrophosphate (FPP) and/or geranylgeranyl pyrophosphate (GGPP) biosynthesis and its implication in the treatment of cancers, Crit Rev Biochem Mol Biol, 54 (2019) 41–60. [DOI] [PubMed] [Google Scholar]
- [284].Fiorentino M, Zadra G, Palescandolo E, Fedele G, Bailey D, Fiore C, Nguyen PL, Migita T, Zamponi R, Di Vizio D, Priolo C, Sharma C, Xie W, Hemler ME, Mucci L, Giovannucci E, Finn S, Loda M, Overexpression of fatty acid synthase is associated with palmitoylation of Wnt1 and cytoplasmic stabilization of beta-catenin in prostate cancer, Lab Invest, 88 (2008) 1340–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Heuer TS, Ventura R, Mordec K, Lai J, Fridlib M, Buckley D, Kemble G, FASN Inhibition and Taxane Treatment Combine to Enhance Anti-tumor Efficacy in Diverse Xenograft Tumor Models through Disruption of Tubulin Palmitoylation and Microtubule Organization and FASN Inhibition-Mediated Effects on Oncogenic Signaling and Gene Expression, EBioMedicine, 16 (2017) 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Bollu LR, Ren J, Blessing AM, Katreddy RR, Gao G, Xu L, Wang J, Su F, Weihua Z, Involvement of de novo synthesized palmitate and mitochondrial EGFR in EGF induced mitochondrial fusion of cancer cells, Cell Cycle, 13 (2014) 2415–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Ali A, Levantini E, Teo JT, Goggi J, Clohessy JG, Wu CS, Chen L, Yang H, Krishnan I, Kocher O, Zhang J, Soo RA, Bhakoo K, Chin TM, Tenen DG, Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated non-small cell lung cancer, EMBO Mol Med, 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].De Piano M, Manuelli V, Zadra G, Otte J, Edqvist PD, Ponten F, Nowinski S, Niaouris A, Grigoriadis A, Loda M, Van Hemelrijck M, Wells CM, Lipogenic signalling modulates prostate cancer cell adhesion and migration via modification of Rho GTPases, Oncogene, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Riobo NA, Cholesterol and its derivatives in Sonic Hedgehog signaling and cancer, Curr Opin Pharmacol, 12 (2012) 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Huang P, Chandra V, Rastinejad F, Structural Overview of the Nuclear Receptor Superfamily: Insights into Physiology and Therapeutics, Annu. Rev. Physiol, 72 (2010) 247–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F, SREBP transcription factors: master regulators of lipid homeostasis, Biochimie, 86 (2004) 839–848. [DOI] [PubMed] [Google Scholar]
- [292].Baranowski M, Biological role of liver X receptors, J. Physiol. Pharmacol, 59 Suppl 7 (2008) 31–55. [PubMed] [Google Scholar]
- [293].Chu K, Miyazaki M, Man WC, Ntambi JM, Stearoyl-Coenzyme A Desaturase 1 Deficiency Protects against Hypertriglyceridemia and Increases Plasma High-Density Lipoprotein Cholesterol Induced by Liver X Receptor Activation, Molecular and Cellular Biology, 26 (2006) 6786–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Repa JJ, Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta, Genes & development, 14 (2000) 2819–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Bovenga F, Sabbà C, Moschetta A, Uncoupling Nuclear Receptor LXR and Cholesterol Metabolism in Cancer, Cell Metabolism, 21 (2015) 517–526. [DOI] [PubMed] [Google Scholar]
- [296].Tiano JP, Mauvais-Jarvis F, Molecular Mechanisms of Estrogen Receptors’ Suppression of Lipogenesis in Pancreatic β-Cells, Endocrinology, 153 (2012) 2997–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].DuSell CD, Umetani M, Shaul PW, Mangelsdorf DJ, McDonnell DP, 27-Hydroxycholesterol Is an Endogenous Selective Estrogen Receptor Modulator, Molecular Endocrinology, 22 (2008) 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, Korach KS, Shaul PW, Mangelsdorf DJ, 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen, Nature medicine, 13 (2007) 1185–1192. [DOI] [PubMed] [Google Scholar]
- [299].Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, Umetani M, Geradts J, McDonnell DP, 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology, Science, 342 (2013) 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].de Boussac H, Pommier AJ, Dufour J, Trousson A, Caira F, Volle DH, Baron S, Lobaccaro JM, LXR, prostate cancer and cholesterol: the Good, the Bad and the Ugly, American journal of cancer research, 3 (2013) 58–69. [PMC free article] [PubMed] [Google Scholar]
- [301].Osborne TF, Espenshade PJ, Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it’s been, Genes & development, 23 (2009) 2578–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Seegmiller AC, Dobrosotskaya I, Goldstein JL, Ho YK, Brown MS, Rawson RB, The SREBP Pathway in Drosophila, Developmental cell, 2 (2002) 229–238. [DOI] [PubMed] [Google Scholar]
- [303].Hughes AL, Todd BL, Espenshade PJ, SREBP Pathway Responds to Sterols and Functions as an Oxygen Sensor in Fission Yeast, Cell, 120 (2005) 831–842. [DOI] [PubMed] [Google Scholar]
- [304].Brown MS, Goldstein JL, The SREBP Pathway: Regulation of Cholesterol Metabolism by Proteolysis of a Membrane-Bound Transcription Factor, Cell, 89 (1997) 331–340. [DOI] [PubMed] [Google Scholar]
- [305].Horton J, Goldstein J, Brown M, SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver., J. Clin. Invest, 109 (2002) 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Horton JD, Sterol regulatory element-binding proteins: transcriptional activators of lipid synthesis, Biochemical Society transactions, 30 (2002) 1091–1095. [DOI] [PubMed] [Google Scholar]
- [307].Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, Goldstein JL, Cholesterol and 25-Hydroxycholesterol Inhibit Activation of SREBPs by Different Mechanisms, Both Involving SCAP and Insigs, Journal of Biological Chemistry, 279 (2004) 52772–52780. [DOI] [PubMed] [Google Scholar]
- [308].Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL, Cholesterol Addition to ER Membranes Alters Conformation of SCAP, the SREBP Escort Protein that Regulates Cholesterol Metabolism, Molecular cell, 10 (2002) 237–245. [DOI] [PubMed] [Google Scholar]
- [309].Sakai J, Nohturfft A, Goldstein JL, Brown MS, Cleavage of Sterol Regulatory Element-binding Proteins (SREBPs) at Site-1 Requires Interaction with SREBP Cleavage-activating Protein: EVIDENCE FROM IN VIVO COMPETITION STUDIES, Journal of Biological Chemistry, 273 (1998) 5785–5793. [DOI] [PubMed] [Google Scholar]
- [310].Yang T, Espensade P, Wright M, Yabe D, Aebersold R, Goldstein J, Brown M, Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a mambrane protein that facilitates retention of SREBPs in ER., Cell, 110 (2002) 489–500. [DOI] [PubMed] [Google Scholar]
- [311].Sakai J, Nohturfft A, Cheng D, Ho YK, Brown MS, Goldstein JL, Identification of Complexes between the COOH-terminal Domains of Sterol Regulatory Element-binding Proteins (SREBPs) and SREBP Cleavage-Activating Protein, Journal of Biological Chemistry, 272 (1997) 20213–20221. [DOI] [PubMed] [Google Scholar]
- [312].Nohturfft A, Brown MS, Goldstein JL, Topology of SREBP Cleavage-activating Protein, a Polytopic Membrane Protein with a Sterol-sensing Domain, Journal of Biological Chemistry, 273 (1998) 17243–17250. [DOI] [PubMed] [Google Scholar]
- [313].DeBose-Boyd RA, Brown MS, Li W-P, Nohturfft A, Goldstein JL, Espenshade PJ, Transport-Dependent Proteolysis of SREBP Relocation of Site-1 Protease from Golgi to ER Obviates the Need for SREBP Transport to Golgi, Cell, 99 (1999) 703–712. [DOI] [PubMed] [Google Scholar]
- [314].Osborne TF, LaMorte VJ, Molecular Aspects in Feedback Regulation of Gene Expression by Cholesterol in Mammalian Cells, Methods, 16 (1998) 42–48. [DOI] [PubMed] [Google Scholar]
- [315].Inoue J, Sato R, Maeda M, Multiple DNA Elements for Sterol Regulatory Element-Binding Protein and NF-Y Are Responsible for Sterol-Regulated Transcription of the Genes for Human 3-Hydroxy-3-Methylglutaryl Coenzyme A Synthase and Squalene Synthase, Journal of biochemistry, 123 (1998) 1191–1198. [DOI] [PubMed] [Google Scholar]
- [316].Wang X, Sato R, Brown M, Hua X, Goldstein J, SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis., Cell, 77 (1994) 53–62. [DOI] [PubMed] [Google Scholar]
- [317].Shao W, Espenshade PJ, Expanding Roles for SREBP in Metabolism, Cell Metabolism, 16 (2012) 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Chen G, Liang G, Ou J, Goldstein JL, Brown MS, Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver, Proceedings of the National Academy of Sciences, 101 (2004) 11245–11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [319].Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, Bashmakov Y, Goldstein JL, Brown MS, Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR, Proceedings of the National Academy of Sciences, 98 (2001) 6027–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Giandomenico V, Simonsson M, Gronroos E, Ericsson J, Coactivator-Dependent Acetylation Stabilizes Members of the SREBP Family of Transcription Factors, Molecular and Cellular Biology, 23 (2003) 2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Sundqvist A, Ericsson J, Transcription-dependent degradation controls the stability of the SREBP family of transcription factors, Proceedings of the National Academy of Sciences, 100 (2003) 13833–13838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M, Genome-Wide Occupancy of SREBP1 and Its Partners NFY and SP1 Reveals Novel Functional Roles and Combinatorial Regulation of Distinct Classes of Genes, PLoS Genet, 4 (2008) e1000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Swinnen J, Ulrix W, Heyns W, Verhoven G, Coordinate regulation of lipogenic gene expression by androgens: evidence for a cascade mechanism involving sterol regulatory element binding proteins., PNAS, 94 (1997) 12975–12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [324].Swinnen J, Heemers H, Deboel L, Foufelle F, Heyns W, Verhoeven G, Stimulation of tumor-associated fatty acid synthase expression by growth factor activation of the sterol regulatory element-binding protein pathway., Oncogene, 19 (2000) 5173–5181. [DOI] [PubMed] [Google Scholar]
- [325].Arkhipov A, Shan Y, Das R, Nicholas F, Eastwood Michael P., Wemmer David E., Kuriyan J, Shaw David E., Architecture and Membrane Interactions of the EGF Receptor, Cell, 152 (2013) 557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Bollu LR, Katreddy RR, Blessing AM, Pham N, Zheng B, Wu X, Weihua Z, Intracellular activation of EGFR by fatty acid synthase dependent palmitoylation, Oncotarget, 6 (2015) 34992–35003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Bian Y, Yu Y, Wang S, Li L, Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer, Biochem Biophys Res Commun, 463 (2015) 612–617. [DOI] [PubMed] [Google Scholar]
- [328].Menendez JA, Fine-tuning the lipogenic/lipolytic balance to optimize the metabolic requirements of cancer cell growth: Molecular mechanisms and therapeutic perspectives, Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, 1801 (2010) 381–391. [DOI] [PubMed] [Google Scholar]
- [329].Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R, Lupu R, Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells, Proceedings of the National Academy of Sciences, 101 (2004) 10715–10720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Menendez JA, Lupu R, Oncogenic properties of the endogenous fatty acid metabolism: molecular pathology of fatty acid synthase in cancer cells, Current opinion in clinical nutrition and metabolic care, 9 (2006) 346–357. [DOI] [PubMed] [Google Scholar]
- [331].Koundouros N, Poulogiannis G, Reprogramming of fatty acid metabolism in cancer, British Journal of Cancer, 122 (2020) 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [332].Signoretti S, Her-2-neu Expression and Progression Toward Androgen Independence in Human Prostate Cancer, Journal of the National Cancer Institute, 92 (2000) 1918–1925. [DOI] [PubMed] [Google Scholar]
- [333].Weroha SJ, Haluska P, The Insulin-Like Growth Factor System in Cancer, Endocrinology and Metabolism Clinics of North America, 41 (2012) 335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [334].Mairet-Coello G, Tury A, DiCicco-Bloom E, Insulin-Like Growth Factor-1 Promotes G1/S Cell Cycle Progression through Bidirectional Regulation of Cyclins and Cyclin-Dependent Kinase Inhibitors via the Phosphatidylinositol 3-Kinase/Akt Pathway in Developing Rat Cerebral Cortex, Journal of Neuroscience, 29 (2009) 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [335].Ma J, Sawai H, Matsuo Y, Ochi N, Yasuda A, Takahashi H, Wakasugi T, Funahashi H, Sato M, Takeyama H, IGF-1 Mediates PTEN Suppression and Enhances Cell Invasion and Proliferation via Activation of the IGF-1/PI3K/Akt Signaling Pathway in Pancreatic Cancer Cells, Journal of Surgical Research, 160 (2010) 90–101. [DOI] [PubMed] [Google Scholar]
- [336].Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H, Phosphatidylinositol 3-Kinase/AKT-mediated Activation of Estrogen Receptor α: A NEW MODEL FOR ANTI-ESTROGEN RESISTANCE, Journal of Biological Chemistry, 276 (2001) 9817–9824. [DOI] [PubMed] [Google Scholar]
- [337].Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R, Tamoxifen Resistance in Breast Tumors Is Driven by Growth Factor Receptor Signaling with Repression of Classic Estrogen Receptor Genomic Function, Cancer Research, 68 (2008) 826–833. [DOI] [PubMed] [Google Scholar]
- [338].Elfiky AA, Jiang Z, The PI3 Kinase Signaling Pathway in Prostate Cancer, CCDT, 13 (2013) 157–164. [DOI] [PubMed] [Google Scholar]
- [339].Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry Christopher D., Annala M, Aprikian A, Armenia J, Arora A, Auman JT, Balasundaram M, Balu S, Barbieri Christopher E., Bauer T, Benz Christopher C., Bergeron A, Beroukhim R, Berrios M, Bivol A, Bodenheimer T, Boice L, Bootwalla Moiz S., Borges dos Reis R, Boutros Paul C., Bowen J, Bowlby R, Boyd J, Bradley Robert K., Breggia A, Brimo F, Bristow Christopher A., Brooks D, Broom Bradley M., Bryce Alan H., Bubley G, Burks E, Butterfield Yaron S.N., Button M, Canes D, Carlotti Carlos G., Carlsen R, Carmel M, Carroll Peter R., Carter Scott L., Cartun R, Carver Brett S., Chan June M., Chang Matthew T., Chen Y, Cherniack Andrew D., Chevalier S, Chin L, Cho J, Chu A, Chuah E, Chudamani S, Cibulskis K, Ciriello G, Clarke A, Cooperberg Matthew R., Corcoran Niall M., Costello Anthony J., Cowan J, Crain D, Curley E, David K, Demchok John A., Demichelis F, Dhalla N, Dhir R, Doueik A, Drake B, Dvinge H, Dyakova N, Felau I, Ferguson Martin L., Frazer S, Freedland S, Fu Y, Gabriel Stacey B., Gao J, Gardner J, Gastier-Foster Julie M., Gehlenborg N, Gerken M, Gerstein Mark B., Getz G, Godwin Andrew K., Gopalan A, Graefen M, Graim K, Gribbin T, Guin R, Gupta M, Hadjipanayis A, Haider S, Hamel L, Hayes DN, Heiman David I., Hess J, Hoadley Katherine A., Holbrook Andrea H., Holt Robert A., Holway A, Hovens Christopher M., Hoyle Alan P., Huang M, Hutter Carolyn M., Ittmann M, Iype L, Jefferys Stuart R., Jones Corbin D., Jones Steven J.M., Juhl H, Kahles A, Kane Christopher J., Kasaian K, Kerger M, Khurana E, Kim J, Klein Robert J., Kucherlapati R, Lacombe L, Ladanyi M, Lai Phillip H., Laird Peter W., Lander Eric S., Latour M, Lawrence Michael S., Lau K, LeBien T, Lee D, Lee S, Lehmann K-V, Leraas Kristen M., Leshchiner I, Leung R, Libertino John A., Lichtenberg Tara M., Lin P, Linehan WM, Ling S, Lippman Scott M., Liu J, Liu W, Lochovsky L, Loda M, Logothetis C, Lolla L, Longacre T, Lu Y, Luo J, Ma Y, Mahadeshwar Harshad S., Mallery D, Mariamidze A, Marra Marco A., Mayo M, McCall S, McKercher G, Meng S, Mes-Masson A-M, Merino Maria J., Meyerson M, Mieczkowski Piotr A., Mills Gordon B., Shaw Kenna R.M., Minner S, Moinzadeh A, Moore Richard A., Morris S, Morrison C, Mose Lisle E., Mungall Andrew J., Murray Bradley A., Myers Jerome B., Naresh R, Nelson J, Nelson Mark A., Nelson Peter S., Newton Y, Noble Michael S., Noushmehr H, Nykter M, Pantazi A, Parfenov M, Park Peter J., Parker Joel S., Paulauskis J, Penny R, Perou Charles M., Piché A, Pihl T, Pinto Peter A., Prandi D, Protopopov A, Ramirez Nilsa C., Rao A, Rathmell WK, Rätsch G, Ren X, Reuter Victor E., Reynolds Sheila M., Rhie Suhn K., Rieger-Christ K, Roach J, Robertson AG, Robinson B, Rubin Mark A., Saad F, Sadeghi S, Saksena G, Saller C, Salner A, Sanchez-Vega F, Sander C, Sandusky G, Sauter G, Sboner A, Scardino Peter T., Scarlata E, Schein Jacqueline E., Schlomm T, Schmidt Laura S., Schultz N, Schumacher Steven E., Seidman J, Neder L, Seth S, Sharp A, Shelton C, Shelton T, Shen H, Shen R, Sherman M, Sheth M, Shi Y, Shih J, Shmulevich I, Simko J, Simon R, Simons Janae V., Sipahimalani P, Skelly T, Sofia Heidi J., Soloway Matthew G., Song X, Sorcini A, Sougnez C, Stepa S, Stewart C, Stewart J, Stuart Joshua M., Sullivan Travis B., Sun C, Sun H, Tam A, Tan D, Tang J, Tarnuzzer R, Tarvin K, Taylor Barry S., Teebagy P, Tenggara I, Têtu B, Tewari A, Thiessen N, Thompson T, Thorne Leigh B., Tirapelli Daniela P., Tomlins Scott A., Trevisan Felipe A., Troncoso P, True Lawrence D., Tsourlakis Maria C., Tyekucheva S, Van Allen E, Van Den Berg David J., Veluvolu U, Verhaak R, Vocke Cathy D., Voet D, Wan Y, Wang Q, Wang W, Wang Z, Weinhold N, Weinstein John N., Weisenberger Daniel J., Wilkerson Matthew D., Wise L, Witte J, Wu C-C, Wu J, Wu Y, Xu Andrew W., Yadav Shalini S., Yang L, Yang L, Yau C, Ye H, Yena P, Zeng T, Zenklusen Jean C., Zhang H, Zhang J, Zhang J, Zhang W, Zhong Y, Zhu K, Zmuda E, The Molecular Taxonomy of Primary Prostate Cancer, Cell, 163 (2015) 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [340].Yamauchi Y, Furukawa K, Hamamura K, Furukawa K, Positive feedback loop between PI3K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: induction of the lipogenic pathway by a melanoma antigen, Cancer Research, 71 (2011) 4989–4997. [DOI] [PubMed] [Google Scholar]
- [341].Zadra G, Photopoulos C, Loda M, The fat side of prostate cancer, Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, 1831 (2013) 1518–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [342].Porstmann T, Santos C, Griffiths B, Cully M, Wu M, Leevers S, Griffiths J, Chung Y, Schultze A, SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth., Cell Metab., 8 (2008) 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Krycer JR, Sharpe LJ, Luu W, Brown AJ, The Akt–SREBP nexus: cell signaling meets lipid metabolism, Trends in Endocrinology & Metabolism, 21 (2010) 268–276. [DOI] [PubMed] [Google Scholar]
- [344].Shimano H, Sato R, SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology, Nature reviews. Endocrinology, 13 (2017) 710–730. [DOI] [PubMed] [Google Scholar]
- [345].Ricoult SJH, Yecies JL, Ben-Sahra I, Manning BD, Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP, Oncogene, 35 (2016) 1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [346].Lee G, Zheng Y, Cho S, Jang C, England C, Dempsey JM, Yu Y, Liu X, He L, Cavaliere PM, Chavez A, Zhang E, Isik M, Couvillon A, Dephoure NE, Blackwell TK, Yu JJ, Rabinowitz JD, Cantley LC, Blenis J, Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling, Cell, 171 (2017) 1545–1558.e1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [347].Lee Y-R, Pandolfi PP, PTEN Mouse Models of Cancer Initiation and Progression, Cold Spring Harb Perspect Med, 10 (2020) a037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [348].Zhou X, Yang X, Sun X, Xu X, Li X.a., Guo Y, Wang J, Li X, Yao L, Wang H, Shen L, Effect of PTEN loss on metabolic reprogramming in prostate cancer cells, Oncology letters, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [349].Bandyopadhyay S, Pai SK, Watabe M, Gross SC, Hirota S, Hosobe S, Tsukada T, Miura K, Saito K, Markwell SJ, Wang Y, Huggenvik J, Pauza ME, Iiizumi M, Watabe K, FAS expression inversely correlates with PTEN level in prostate cancer and a PI 3-kinase inhibitor synergizes with FAS siRNA to induce apoptosis, Oncogene, 24 (2005) 5389–5395. [DOI] [PubMed] [Google Scholar]
- [350].Robinson D, Van Allen Eliezer M., Wu Y-M, Schultz N, Lonigro Robert J., Mosquera J-M, Montgomery B, Taplin M-E, Pritchard Colin C., Attard G, Beltran H, Abida W, Bradley Robert K., Vinson J, Cao X, Vats P, Kunju Lakshmi P., Hussain M, Feng Felix Y., Tomlins Scott A., Cooney Kathleen A., Smith David C., Brennan C, Siddiqui J, Mehra R, Chen Y, Rathkopf Dana E., Morris Michael J., Solomon Stephen B., Durack Jeremy C., Reuter Victor E., Gopalan A, Gao J, Loda M, Lis Rosina T., Bowden M, Balk Stephen P., Gaviola G, Sougnez C, Gupta M, Yu Evan Y., Mostaghel Elahe A., Cheng Heather H., Mulcahy H, True Lawrence D., Plymate Stephen R., Dvinge H, Ferraldeschi R, Flohr P, Miranda S, Zafeiriou Z, Tunariu N, Mateo J, Perez-Lopez R, Demichelis F, Robinson Brian D., Schiffman M, Nanus David M., Tagawa Scott T., Sigaras A, Eng Kenneth W., Elemento O, Sboner A, Heath Elisabeth I., Scher Howard I., Pienta Kenneth J., Kantoff P, de Bono Johann S., Rubin Mark A., Nelson Peter S., Garraway Levi A., Sawyers Charles L., Chinnaiyan Arul M., Integrative Clinical Genomics of Advanced Prostate Cancer, Cell, 161 (2015) 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [351].Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL, Integrative Genomic Profiling of Human Prostate Cancer, Cancer cell, 18 (2010) 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [352].Lotan TL, Wei W, Ludkovski O, Morais CL, Guedes LB, Jamaspishvili T, Lopez K, Hawley ST, Feng Z, Fazli L, Hurtado-Coll A, McKenney JK, Simko J, Carroll PR, Gleave M, Lin DW, Nelson PS, Thompson IM, True LD, Brooks JD, Lance R, Troyer D, Squire JA, Analytic validation of a clinical-grade PTEN immunohistochemistry assay in prostate cancer by comparison with PTEN FISH, Mod Pathol, 29 (2016) 904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [353].Lotan TL, Gurel B, Sutcliffe S, Esopi D, Liu W, Xu J, Hicks JL, Park BH, Humphreys E, Partin AW, Han M, Netto GJ, Isaacs WB, De Marzo AM, PTEN Protein Loss by Immunostaining: Analytic Validation and Prognostic Indicator for a High Risk Surgical Cohort of Prostate Cancer Patients, Clinical Cancer Research, 17 (2011) 6563–6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [354].Lotan TL, Carvalho FLF, Peskoe SB, Hicks JL, Good J, Fedor HL, Humphreys E, Han M, Platz EA, Squire JA, De Marzo AM, Berman DM, PTEN loss is associated with upgrading of prostate cancer from biopsy to radical prostatectomy, Mod Pathol, 28 (2015) 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [355].Lotan TL, Heumann A, Rico SD, Hicks J, Lecksell K, Koop C, Sauter G, Schlomm T, Simon R, PTEN loss detection in prostate cancer: comparison of PTEN immunohistochemistry and PTEN FISH in a large retrospective prostatectomy cohort, Oncotarget, 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [356].Tosoian JJ, Almutairi F, Morais CL, Glavaris S, Hicks J, Sundi D, Humphreys E, Han M, De Marzo AM, Ross AE, Tomlins SA, Schaeffer EM, Trock BJ, Lotan TL, Prevalence and Prognostic Significance of PTEN Loss in African-American and European-American Men Undergoing Radical Prostatectomy, European Urology, 71 (2017) 697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [357].Jamaspishvili T, Berman DM, Ross AE, Scher HI, De Marzo AM, Squire JA, Lotan TL, Clinical implications of PTEN loss in prostate cancer, Nat Rev Urol, 15 (2018) 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [358].Chen M, Zhang J, Sampieri K, Clohessy J, Mendez L, Gonzalez-Billalabeita E, Liu X, Fung J, Katon J, Menon A, Webster K, Mg C, Palumbieiri M, Diolombi M, Breitkopf S, Teruya-Feldstein J, Signoretti S, Bronson R, Asara J, Castillo-Martin M, Cordon-Cardo C, Pandolfi P, An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer., Nat. Genetics, 50 (2018) 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [359].Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW, Negative Regulation of PKB/Akt-Dependent Cell Survival by the Tumor Suppressor PTEN, Cell, 95 (1998) 29–39. [DOI] [PubMed] [Google Scholar]
- [360].Sun H, Enomoto T, Shroyer KR, Ozaki K, Fujita M, Ueda Y, Nakashima R, Kuragaki C, Ueda G, Murata Y, Clonal Analysis and Mutations in the PTEN and the K-ras Genes in Endometrial Hyperplasia, Diagnostic Molecular Pathology, 11 (2002) 204–211. [DOI] [PubMed] [Google Scholar]
- [361].Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB, ATP citrate lyase is an important component of cell growth and transformation, Oncogene, 24 (2005) 6314–6322. [DOI] [PubMed] [Google Scholar]
- [362].Berwick DC, Hers I, Heesom KJ, Moule SK, Tavaré JM, The Identification of ATP-citrate Lyase as a Protein Kinase B (Akt) Substrate in Primary Adipocytes, Journal of Biological Chemistry, 277 (2002) 33895–33900. [DOI] [PubMed] [Google Scholar]
- [363].Jones NP, Schulze A, Targeting cancer metabolism – aiming at a tumour’s sweet-spot, Drug Discovery Today, 17 (2012) 232–241. [DOI] [PubMed] [Google Scholar]
- [364].Li X, Wu JB, Li Q, Shigemura K, Chung LWK, Huang W-C, SREBP-2 promotes stem cell-like properties and metastasis by transcriptional activation of c-Myc in prostate cancer, Oncotarget, 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [365].Wu Y, Chen K, Liu X, Huang L, Zhao D, Li L, Gao M, Pei D, Wang C, Liu X, Srebp-1 Interacts with c-Myc to Enhance Somatic Cell Reprogramming: Srebp-1 Enhances Reprogramming, Stem cells (Dayton, Ohio), 34 (2016) 83–92. [DOI] [PubMed] [Google Scholar]
- [366].Gouw AM, Margulis K, Liu NS, Raman SJ, Mancuso A, Toal GG, Tong L, Mosley A, Hsieh AL, Sullivan DK, Stine ZE, Altman BJ, Schulze A, Dang CV, Zare RN, Felsher DW, The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth, Cell Metabolism, 30 (2019) 556–572.e555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [367].Labbé DP, Zadra G, Yang M, Reyes JM, Lin CY, Cacciatore S, Ebot EM, Creech AL, Giunchi F, Fiorentino M, Elfandy H, Syamala S, Karoly ED, Alshalalfa M, Erho N, Ross A, Schaeffer EM, Gibb EA, Takhar M, Den RB, Lehrer J, Karnes RJ, Freedland SJ, Davicioni E, Spratt DE, Ellis L, Jaffe JD, DʼAmico AV, Kantoff PW, Bradner JE, Mucci LA, Chavarro JE, Loda M, Brown M, High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program, Nature communications, 10 (2019) 4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [368].Priolo C, Pyne S, Rose J, Regan ER, Zadra G, Photopoulos C, Cacciatore S, Schultz D, Scaglia N, McDunn J, De Marzo AM, Loda M, AKT1 and MYC Induce Distinctive Metabolic Fingerprints in Human Prostate Cancer, Cancer Research, 74 (2014) 7198–7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [369].Lee C.-k., Jeong S.-h., Jang C, Bae H, Kim YH, Park I, Kim SK, Koh GY, Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation, Science, 363 (2019) 644–649. [DOI] [PubMed] [Google Scholar]
- [370].Nguyen CDK, Yi C, YAP/TAZ Signaling and Resistance to Cancer Therapy, Trends in cancer, 5 (2019) 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [371].Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S, Rosato A, Piccolo S, Del Sal G, Metabolic control of YAP and TAZ by the mevalonate pathway, Nature Cell Biology, 16 (2014) 357–366. [DOI] [PubMed] [Google Scholar]
- [372].Parrales A, Iwakuma T, p53 as a Regulator of Lipid Metabolism in Cancer, International journal of molecular sciences, 17 (2016) 2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [373].Flöter J, Kaymak I, Schulze A, Regulation of Metabolic Activity by p53, Metabolites, 7 (2017) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [374].Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X, p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase, Nature Cell Biology, 13 (2011) 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [375].Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, Ide T, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Gotoda T, Nagai R, Kimura S, Ishibashi S, Osuga J-I, Yamada N, p53 Activation in adipocytes of obese mice, The Journal of Biological Chemistry, 278 (2003) 25395–25400. [DOI] [PubMed] [Google Scholar]
- [376].Rueda-Rincon N, Bloch K, Derua R, Vyas R, Harms A, Hankemeier T, Khan NA, Dehairs J, Bagadi M, Binda MM, Waelkens E, Marine JC, Swinnen JV, p53 attenuates AKT signaling by modulating membrane phospholipid composition, Oncotarget, 6 (2015) 21240–21254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [377].Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, Brinkman AB, Morganella S, Aure MR, Lingjærde OC, Langerød A, Ringnér M, Ahn S-M, Boyault S, Brock JE, Broeks A, Butler A, Desmedt C, Dirix L, Dronov S, Fatima A, Foekens JA, Gerstung M, Hooijer GKJ, Jang SJ, Jones DR, Kim H-Y, King TA, Krishnamurthy S, Lee HJ, Lee J-Y, Li Y, McLaren S, Menzies A, Mustonen V, O’Meara S, Pauporté I, Pivot X, Purdie CA, Raine K, Ramakrishnan K, Rodríguez-González FG, Romieu G, Sieuwerts AM, Simpson PT, Shepherd R, Stebbings L, Stefansson OA, Teague J, Tommasi S, Treilleux I, Van den Eynden GG, Vermeulen P, Vincent-Salomon A, Yates L, Caldas C, Veer L.v.t., Tutt A, Knappskog S, Tan BKT, Jonkers J, Borg Å, Ueno NT, Sotiriou C, Viari A, Futreal PA, Campbell PJ, Span PN, Van Laere S, Lakhani SR, Eyfjord JE, Thompson AM, Birney E, Stunnenberg HG, van de Vijver MJ, Martens JWM, Børresen-Dale A-L, Richardson AL, Kong G, Thomas G, Stratton MR, Landscape of somatic mutations in 560 breast cancer whole-genome sequences, Nature, 534 (2016) 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [378].The N Cancer Genome Atlas, Comprehensive molecular portraits of human breast tumours, Nature, 490 (2012) 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [379].Zhou G, Wang J, Zhao M, Xie T-X, Tanaka N, Sano D, Patel Ameeta A., Ward Alexandra M., Sandulache Vlad C., Jasser Samar A., Skinner Heath D., Fitzgerald Alison L., Osman Abdullah A., Wei Y, Xia X, Songyang Z, Mills Gordon B., Hung M-C, Caulin C, Liang J, Myers Jeffrey N., Gain-of-Function Mutant p53 Promotes Cell Growth and Cancer Cell Metabolism via Inhibition of AMPK Activation, Molecular cell, 54 (2014) 960–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [380].Loizou E, Banito A, Livshits G, Ho Y-J, Koche RP, Sánchez-Rivera FJ, Mayle A, Chen C-C, Kinalis S, Bagger FO, Kastenhuber ER, Durham BH, Lowe SW, A Gain-of-Function p53-Mutant Oncogene Promotes Cell Fate Plasticity and Myeloid Leukemia through the Pluripotency Factor FOXH1, Cancer Discovery, 9 (2019) 962–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [381].Li X, Wu JB, Chung LWK, Huang W-C, Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations, Oncotarget, 6 (2015) 41018–41032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [382].Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon S-H, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, Bissell MJ, Osborne TF, Tian B, Lowe SW, Silva JM, Børresen-Dale A-L, Levine AJ, Bargonetti J, Prives C, Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway, Cell, 148 (2012) 244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [383].Tamanoi F, Azizian M, Ashrafi M, Bathaie S, Mevalonate Pathway and Human Cancers, CMP, 10 (2017) 77–85. [DOI] [PubMed] [Google Scholar]
- [384].Lacroix M, Riscal R, Arena G, Linares LK, Le Cam L, Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer, Molecular Metabolism, (2019) S2212877819309214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [385].Liu J, Zhang C, Hu W, Feng Z, Tumor suppressor p53 and metabolism, Journal of Molecular Cell Biology, 11 (2019) 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [386].Humpton T, Vousden KH, Taking up the reins of power: metabolic functions of p53, Journal of Molecular Cell Biology, 11 (2019) 610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [387].Shahbandi A, Nguyen HD, Jackson JG, TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines, Trends in cancer, 6 (2020) 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [388].Hashimoto N, Nagano H, Tanaka T, The role of tumor suppressor p53 in metabolism and energy regulation, and its implication in cancer and lifestyle-related diseases, Endocr J, 66 (2019) 485–496. [DOI] [PubMed] [Google Scholar]
- [389].Shamma A, Takegami Y, Miki T, Kitajima S, Noda M, Obara T, Okamoto T, Takahashi C, Rb Regulates DNA Damage Response and Cellular Senescence through E2F-Dependent Suppression of N-Ras Isoprenylation, Cancer cell, 15 (2009) 255–269. [DOI] [PubMed] [Google Scholar]
- [390].Muranaka H, Hayashi A, Minami K, Kitajima S, Kohno S, Nishimoto Y, Nagatani N, Suzuki M, Kulathunga LAN, Sasaki N, Okada N, Matsuzaka T, Shimano H, Tada H, Takahashi C, A distinct function of the retinoblastoma protein in the control of lipid composition identified by lipidomic profiling, Oncogenesis, 6 (2017) e350–e350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [391].Flavin R, Zadra G, Loda M, Metabolic alterations and targeted therapies in prostate cancer, J. Pathol, 223 (2011) 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [392].Jeon SM, Chandel NS, Hay N, AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress, Nature, 485 (2012) 661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [393].Zadra G, Batista JL, Loda M, Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies, Molecular Cancer Research, 13 (2015) 1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [394].Jeon S-M, Hay N, The dark face of AMPK as an essential tumor promoter, Cellular Logistics, 2 (2012) 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [395].Luo Z, Zang M, Guo W, AMPK as a metabolic tumor suppressor: control of metabolism and cell growth, Future Oncology, 6 (2010) 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [396].Khan AS, Frigo DE, A spatiotemporal hypothesis for the regulation, role, and targeting of AMPK in prostate cancer, Nat Rev Urol, 14 (2017) 164–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [397].Scaglia N, Tyekucheva S, Zadra G, Photopoulos C, Loda M, De novo fatty acid synthesis at the mitotic exit is required to complete cellular division, Cell cycle (Georgetown, Tex.), 13 (2014) 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [398].Shackelford DB, Shaw RJ, The LKB1–AMPK pathway: metabolism and growth control in tumour suppression, Nature Reviews Cancer, 9 (2009) 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [399].DeRan M, Yang J, Shen C-H, Peters Eric C., Fitamant J, Chan P, Hsieh M, Zhu S, Asara John M., Zheng B, Bardeesy N, Liu J, Wu X, Energy Stress Regulates Hippo-YAP Signaling Involving AMPK-Mediated Regulation of Angiomotin-like 1 Protein, Cell reports, 9 (2014) 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [400].Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, Lazar AJ, Cherniack AD, Beroukhim R, Meyerson M, Caesar-Johnson SJ, Demchok JA, Felau I, Kasapi M, Ferguson ML, Hutter CM, Sofia HJ, Tarnuzzer R, Wang Z, Yang L, Zenklusen JC, Zhang J, Chudamani S, Liu J, Lolla L, Naresh R, Pihl T, Sun Q, Wan Y, Wu Y, Cho J, DeFreitas T, Frazer S, Gehlenborg N, Getz G, Heiman DI, Kim J, Lawrence MS, Lin P, Meier S, Noble MS, Saksena G, Voet D, Zhang H, Bernard B, Chambwe N, Dhankani V, Knijnenburg T, Kramer R, Leinonen K, Liu Y, Miller M, Reynolds S, Shmulevich I, Thorsson V, Zhang W, Akbani R, Broom BM, Hegde AM, Ju Z, Kanchi RS, Korkut A, Li J, Liang H, Ling S, Liu W, Lu Y, Mills GB, Ng K-S, Rao A, Ryan M, Wang J, Weinstein JN, Zhang J, Abeshouse A, Armenia J, Chakravarty D, Chatila WK, de Bruijn I, Gao J, Gross BE, Heins ZJ, Kundra R, La K, Ladanyi M, Luna A, Nissan MG, Ochoa A, Phillips SM, Reznik E, Sanchez-Vega F, Sander C, Schultz N, Sheridan R, Sumer SO, Sun Y, Taylor BS, Wang J, Zhang H, Anur P, Peto M, Spellman P, Benz C, Stuart JM, Wong CK, Yau C, Hayes DN, Parker JS, Wilkerson MD, Ally A, Balasundaram M, Bowlby R, Brooks D, Carlsen R, Chuah E, Dhalla N, Holt R, Jones SJM, Kasaian K, Lee D, Ma Y, Marra MA, Mayo M, Moore RA, Mungall AJ, Mungall K, Robertson AG, Sadeghi S, Schein JE, Sipahimalani P, Tam A, Thiessen N, Tse K, Wong T, Berger AC, Beroukhim R, Cherniack AD, Cibulskis C, Gabriel SB, Gao GF, Ha G, Meyerson M, Schumacher SE, Shih J, Kucherlapati MH, Kucherlapati RS, Baylin S, Cope L, Danilova L, Bootwalla MS, Lai PH, Maglinte DT, Van Den Berg DJ, Weisenberger DJ, Auman JT, Balu S, Bodenheimer T, Fan C, Hoadley KA, Hoyle AP, Jefferys SR, Jones CD, Meng S, Mieczkowski PA, Mose LE, Perou AH, Perou CM, Roach J, Shi Y, Simons JV, Skelly T, Soloway MG, Tan D, Veluvolu U, Fan H, Hinoue T, Laird PW, Shen H, Zhou W, Bellair M, Chang K, Covington K, Creighton CJ, Dinh H, Doddapaneni H, Donehower LA, Drummond J, Gibbs RA, Glenn R, Hale W, Han Y, Hu J, Korchina V, Lee S, Lewis L, Li W, Liu X, Morgan M, Morton D, Muzny D, Santibanez J, Sheth M, Shinbrot E, Wang L, Wang M, Wheeler DA, Xi L, Zhao F, Hess J, Appelbaum EL, Bailey M, Cordes MG, Ding L, Fronick CC, Fulton LA, Fulton RS, Kandoth C, Mardis ER, McLellan MD, Miller CA, Schmidt HK, Wilson RK, Crain D, Curley E, Gardner J, Lau K, Mallery D, Morris S, Paulauskis J, Penny R, Shelton C, Shelton T, Sherman M, Thompson E, Yena P, Bowen J, Gastier-Foster JM, Gerken M, Leraas KM, Lichtenberg TM, Ramirez NC, Wise L, Zmuda E, Corcoran N, Costello T, Hovens C, Carvalho AL, de Carvalho AC, Fregnani JH, Longatto-Filho A, Reis RM, Scapulatempo-Neto C, Silveira HCS, Vidal DO, Burnette A, Eschbacher J, Hermes B, Noss A, Singh R, Anderson ML, Castro PD, Ittmann M, Huntsman D, Kohl B, Le X, Thorp R, Andry C, Duffy ER, Lyadov V, Paklina O, Setdikova G, Shabunin A, Tavobilov M, McPherson C, Warnick R, Berkowitz R, Cramer D, Feltmate C, Horowitz N, Kibel A, Muto M, Raut CP, Malykh A, Barnholtz-Sloan JS, Barrett W, Devine K, Fulop J, Ostrom QT, Shimmel K, Wolinsky Y, Sloan AE, De Rose A, Giuliante F, Goodman M, Karlan BY, Hagedorn CH, Eckman J, Harr J, Myers J, Tucker K, Zach LA, Deyarmin B, Hu H, Kvecher L, Larson C, Mural RJ, Somiari S, Vicha A, Zelinka T, Bennett J, Iacocca M, Rabeno B, Swanson P, Latour M, Lacombe L, Têtu B, Bergeron A, McGraw M, Staugaitis SM, Chabot J, Hibshoosh H, Sepulveda A, Su T, Wang T, Potapova O, Voronina O, Desjardins L, Mariani O, Roman-Roman S, Sastre X, Stern M-H, Cheng F, Signoretti S, Berchuck A, Bigner D, Lipp E, Marks J, McCall S, McLendon R, Secord A, Sharp A, Behera M, Brat DJ, Chen A, Delman K, Force S, Khuri F, Magliocca K, Maithel S, Olson JJ, Owonikoko T, Pickens A, Ramalingam S, Shin DM, Sica G, Van Meir EG, Zhang H, Eijckenboom W, Gillis A, Korpershoek E, Looijenga L, Oosterhuis W, Stoop H, van Kessel KE, Zwarthoff EC, Calatozzolo C, Cuppini L, Cuzzubbo S, DiMeco F, Finocchiaro G, Mattei L, Perin A, Pollo B, Chen C, Houck J, Lohavanichbutr P, Hartmann A, Stoehr C, Stoehr R, Taubert H, Wach S, Wullich B, Kycler W, Murawa D, Wiznerowicz M, Chung K, Edenfield WJ, Martin J, Baudin E, Bubley G, Bueno R, De Rienzo A, Richards WG, Kalkanis S, Mikkelsen T, Noushmehr H, Scarpace L, Girard N, Aymerich M, Campo E, Giné E, Guillermo AL, Van Bang N, Hanh PT, Phu BD, Tang Y, Colman H, Evason K, Dottino PR, Martignetti JA, Gabra H, Juhl H, Akeredolu T, Stepa S, Hoon D, Ahn K, Kang KJ, Beuschlein F, Breggia A, Birrer M, Bell D, Borad M, Bryce AH, Castle E, Chandan V, Cheville J, Copland JA, Farnell M, Flotte T, Giama N, Ho T, Kendrick M, Kocher J-P, Kopp K, Moser C, Nagorney D, O’Brien D, O’Neill BP, Patel T, Petersen G, Que F, Rivera M, Roberts L, Smallridge R, Smyrk T, Stanton M, Thompson RH, Torbenson M, Yang JD, Zhang L, Brimo F, Ajani JA, Angulo Gonzalez AM, Behrens C, Bondaruk J, Broaddus R, Czerniak B, Esmaeli B, Fujimoto J, Gershenwald J, Guo C, Lazar AJ, Logothetis C, Meric-Bernstam F, Moran C, Ramondetta L, Rice D, Sood A, Tamboli P, Thompson T, Troncoso P, Tsao A, Wistuba I, Carter C, Haydu L, Hersey P, Jakrot V, Kakavand H, Kefford R, Lee K, Long G, Mann G, Quinn M, Saw R, Scolyer R, Shannon K, Spillane A, Stretch J, Synott M, Thompson J, Wilmott J, Al-Ahmadie H, Chan TA, Ghossein R, Gopalan A, Levine DA, Reuter V, Singer S, Singh B, Tien NV, Broudy T, Mirsaidi C, Nair P, Drwiega P, Miller J, Smith J, Zaren H, Park J-W, Hung NP, Kebebew E, Linehan WM, Metwalli AR, Pacak K, Pinto PA, Schiffman M, Schmidt LS, Vocke CD, Wentzensen N, Worrell R, Yang H, Moncrieff M, Goparaju C, Melamed J, Pass H, Botnariuc N, Caraman I, Cernat M, Chemencedji I, Clipca A, Doruc S, Gorincioi G, Mura S, Pirtac M, Stancul I, Tcaciuc D, Albert M, Alexopoulou I, Arnaout A, Bartlett J, Engel J, Gilbert S, Parfitt J, Sekhon H, Thomas G, Rassl DM, Rintoul RC, Bifulco C, Tamakawa R, Urba W, Hayward N, Timmers H, Antenucci A, Facciolo F, Grazi G, Marino M, Merola R, de Krijger R, Gimenez-Roqueplo A-P, Piché A, Chevalier S, McKercher G, Birsoy K, Barnett G, Brewer C, Farver C, Naska T, Pennell NA, Raymond D, Schilero C, Smolenski K, Williams F, Morrison C, Borgia JA, Liptay MJ, Pool M, Seder CW, Junker K, Omberg L, Dinkin M, Manikhas G, Alvaro D, Bragazzi MC, Cardinale V, Carpino G, Gaudio E, Chesla D, Cottingham S, Dubina M, Moiseenko F, Dhanasekaran R, Becker K-F, Janssen K-P, Slotta-Huspenina J, Abdel-Rahman MH, Aziz D, Bell S, Cebulla CM, Davis A, Duell R, Elder JB, Hilty J, Kumar B, Lang J, Lehman NL, Mandt R, Nguyen P, Pilarski R, Rai K, Schoenfield L, Senecal K, Wakely P, Hansen P, Lechan R, Powers J, Tischler A, Grizzle WE, Sexton KC, Kastl A, Henderson J, Porten S, Waldmann J, Fassnacht M, Asa SL, Schadendorf D, Couce M, Graefen M, Huland H, Sauter G, Schlomm T, Simon R, Tennstedt P, Olabode O, Nelson M, Bathe O, Carroll PR, Chan JM, Disaia P, Glenn P, Kelley RK, Landen CN, Phillips J, Prados M, Simko J, Smith-McCune K, VandenBerg S, Roggin K, Fehrenbach A, Kendler A, Sifri S, Steele R, Jimeno A, Carey F, Forgie I, Mannelli M, Carney M, Hernandez B, Campos B, Herold-Mende C, Jungk C, Unterberg A, von Deimling A, Bossler A, Galbraith J, Jacobus L, Knudson M, Knutson T, Ma D, Milhem M, Sigmund R, Godwin AK, Madan R, Rosenthal HG, Adebamowo C, Adebamowo SN, Boussioutas A, Beer D, Giordano T, Mes-Masson A-M, Saad F, Bocklage T, Landrum L, Mannel R, Moore K, Moxley K, Postier R, Walker J, Zuna R, Feldman M, Valdivieso F, Dhir R, Luketich J, Mora Pinero EM, Quintero-Aguilo M, Carlotti CG, Dos Santos JS, Kemp R, Sankarankuty A, Tirapelli D, Catto J, Agnew K, Swisher E, Creaney J, Robinson B, Shelley CS, Godwin EM, Kendall S, Shipman C, Bradford C, Carey T, Haddad A, Moyer J, Peterson L, Prince M, Rozek L, Wolf G, Bowman R, Fong KM, Yang I, Korst R, Rathmell WK, Fantacone-Campbell JL, Hooke JA, Kovatich AJ, Shriver CD, DiPersio J, Drake B, Govindan R, Heath S, Ley T, Van Tine B, Westervelt P, Rubin MA, Lee JI, Aredes ND, Mariamidze A, Genomic and Functional Approaches to Understanding Cancer Aneuploidy, Cancer cell, 33 (2018) 676–689.e673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [401].Stopsack KH, Whittaker CA, Gerke TA, Loda M, Kantoff PW, Mucci LA, Amon A, Aneuploidy drives lethal progression in prostate cancer, Proceedings of the National Academy of Sciences, 116 (2019) 11390–11395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [402].Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho Y-J, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao M-S, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M, The landscape of somatic copy-number alteration across human cancers, Nature, 463 (2010) 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [403].Cai Y, Crowther J, Pastor T, Abbasi Asbagh L, Baietti MF, De Troyer M, Vazquez I, Talebi A, Renzi F, Dehairs J, Swinnen JV, Sablina AA, Loss of Chromosome 8p Governs Tumor Progression and Drug Response by Altering Lipid Metabolism, Cancer cell, 29 (2016) 751–766. [DOI] [PubMed] [Google Scholar]
- [404].Bova GS, Carter BS, Bussemakers MJ, Emi M, Fujiwara Y, Kyprianou N, Jacobs SC, Robinson JC, Epstein JI, Walsh PC, et al. , Homozygous deletion and frequent allelic loss of chromosome 8p22 loci in human prostate cancer, Cancer Res, 53 (1993) 3869–3873. [PubMed] [Google Scholar]
- [405].Gallucci M, Merola R, Farsetti A, Orlandi G, Sentinelli S, De Carli P, Leonardo C, Carlini P, Guadagni F, Sperduti I, Cianciulli AM, Cytogenetic profiles as additional markers to pathological features in clinically localized prostate carcinoma, Cancer letters, 237 (2006) 76–82. [DOI] [PubMed] [Google Scholar]
- [406].Lu W, Takahashi H, Furusato B, Maekawa S, Ikegami M, Sudo A, Egawa S, Hano H, Allelotyping analysis at chromosome arm 8p of high-grade prostatic intraepithelial neoplasia and incidental, latent, and clinical prostate cancers, Genes, chromosomes & cancer, 45 (2006) 509–515. [DOI] [PubMed] [Google Scholar]
- [407].Kim JW, Cheng Y, Liu W, Li T, Yegnasubramanian S, Zheng SL, Xu J, Isaacs WB, Chang B-L, Genetic and epigenetic inactivation of LPL gene in human prostate cancer, International journal of cancer. Journal international du cancer, 124 (2009) 734–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [408].Carter SA, Foster NA, Scarpini CG, Chattopadhyay A, Pett MR, Roberts I, Coleman N, Lipoprotein lipase is frequently overexpressed or translocated in cervical squamous cell carcinoma and promotes invasiveness through the non-catalytic C terminus, British journal of cancer, 107 (2012) 739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [409].Rubin MA, Demichelis F, The Genomics of Prostate Cancer: emerging understanding with technologic advances, Mod Pathol, 31 (2018) 1–11. [DOI] [PubMed] [Google Scholar]
- [410].Parris TZ, Kovács A, Hajizadeh S, Nemes S, Semaan M, Levin M, Karlsson P, Helou K, Frequent MYC coamplification and DNA hypomethylation of multiple genes on 8q in 8p11-p12-amplified breast carcinomas, Oncogenesis, 3 (2014) e95–e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [411].Helms MW, Kemming D, Pospisil H, Vogt U, Buerger H, Korsching E, Liedtke C, Schlotter CM, Wang A, Chan SY, Brandt BH, Squalene epoxidase, located on chromosome 8q24.1, is upregulated in 8q+ breast cancer and indicates poor clinical outcome in stage I and II disease, British Journal of Cancer, 99 (2008) 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [412].Brown DN, Caffa I, Cirmena G, Piras D, Garuti A, Gallo M, Alberti S, Nencioni A, Ballestrero A, Zoppoli G, Squalene epoxidase is a bona fide oncogene by amplification with clinical relevance in breast cancer, Scientific Reports, 6 (2016) 19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [413].Sui Z, Zhou J, Cheng Z, Lu P, Squalene epoxidase (SQLE) promotes the growth and migration of the hepatocellular carcinoma cells, Tumor Biol, 36 (2015) 6173–6179. [DOI] [PubMed] [Google Scholar]
- [414].Chen J, Guccini I, Di Mitri D, Brina D, Revandkar A, Sarti M, Pasquini E, Alajati A, Pinton S, Losa M, Civenni G, Catapano CV, Sgrignani J, Cavalli A, D’Antuono R, Asara JM, Morandi A, Chiarugi P, Crotti S, Agostini M, Montopoli M, Masgras I, Rasola A, Garcia-Escudero R, Delaleu N, Rinaldi A, Bertoni F, Bono J.d, Carracedo A, Alimonti A, Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer, Nature genetics, 50 (2018) 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [415].Magnani L, Frigè G, Gadaleta RM, Corleone G, Fabris S, Kempe H, Verschure PJ, Barozzi I, Vircillo V, Hong S-P, Perone Y, Saini M, Trumpp A, Viale G, Neri A, Ali S, Colleoni MA, Pruneri G, Minucci S, Acquired CYP19A1 amplification is an early specific mechanism of aromatase inhibitor resistance in ERα metastatic breast cancer, Nature genetics, 49 (2017) 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [416].Nguyen VTM, Barozzi I, Faronato M, Lombardo Y, Steel JH, Patel N, Darbre P, Castellano L, Győrffy B, Woodley L, Meira A, Patten DK, Vircillo V, Periyasamy M, Ali S, Frige G, Minucci S, Coombes RC, Magnani L, Differential epigenetic reprogramming in response to specific endocrine therapies promotes cholesterol biosynthesis and cellular invasion, Nature communications, 6 (2015) 10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [417].Patten DK, Corleone G, Győrffy B, Perone Y, Slaven N, Barozzi I, Erdős E, Saiakhova A, Goddard K, Vingiani A, Shousha S, Pongor LS, Hadjiminas DJ, Schiavon G, Barry P, Palmieri C, Coombes RC, Scacheri P, Pruneri G, Magnani L, Enhancer mapping uncovers phenotypic heterogeneity and evolution in patients with luminal breast cancer, Nature medicine, 24 (2018) 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [418].Ellem SJ, Schmitt JF, Pedersen JS, Frydenberg M, Risbridger GP, Local Aromatase Expression in Human Prostate Is Altered in Malignancy, The Journal of Clinical Endocrinology & Metabolism, 89 (2004) 2434–2441. [DOI] [PubMed] [Google Scholar]
- [419].Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald Jeffrey G., Yuhanna Ivan S., Thompson B, Girard L, Mineo C, Brekken Rolf A., Umetani M, Euhus David M., Xie Y, Shaul Philip W., 27-Hydroxycholesterol Promotes Cell-Autonomous, ER-Positive Breast Cancer Growth, Cell reports, 5 (2013) 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [420].Baek AE, Yu Y-RA, He S, Wardell SE, Chang C-Y, Kwon S, Pillai RV, McDowell HB, Thompson JW, Dubois LG, Sullivan PM, Kemper JK, Gunn MD, McDonnell DP, Nelson ER, The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells, Nature communications, 8 (2017) 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [421].Shi S-Z, Lee E-J, Lin Y-J, Chen L, Zheng H-Y, He X-Q, Peng J-Y, Noonepalle SK, Shull AY, Pei FC, Deng L-B, Tian X-L, Deng K-Y, Shi H, Xin H-B, Recruitment of monocytes and epigenetic silencing of intratumoral CYP7B1 primarily contribute to the accumulation of 27-hydroxycholesterol in breast cancer, American journal of cancer research, 9 (2019) 2194–2208. [PMC free article] [PubMed] [Google Scholar]
- [422].Li X, Wang J, Wang L, Feng G, Li G, Yu M, Li Y, Liu C, Yuan X, Zang G, Li Z, Zhao L, Ouyang H, Quan Q, Wang G, Zhang C, Li O, Xiang J, Zhu J-K, Li W, Zhou Q, Zhang K, Impaired lipid metabolism by age-dependent DNA methylation alterations accelerates aging, Proceedings of the National Academy of Sciences, (2020) 201919403. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [423].Barfeld SJ, Itkonen HM, Urbanucci A, Mills IG, Androgen-regulated metabolism and biosynthesis in prostate cancer, Endocrine-related cancer, 21 (2014) T57–T66. [DOI] [PubMed] [Google Scholar]
- [424].Perone Y, Farrugia AJ, Rodríguez-Meira A, Győrffy B, Ion C, Uggetti A, Chronopoulos A, Marrazzo P, Faronato M, Shousha S, Davies C, Steel JH, Patel N, del Rio Hernandez A, Coombes C, Pruneri G, Lim A, Calvo F, Magnani L, SREBP1 drives Keratin-80-dependent cytoskeletal changes and invasive behavior in endocrine-resistant ERα breast cancer, Nature communications, 10 (2019) 2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [425].Rottiers V, Näär AM, MicroRNAs in metabolism and metabolic disorders, Nature Reviews Molecular Cell Biology, 13 (2012) 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [426].Aryal B, Singh AK, Rotllan N, Price N, Fernández-Hernando C, MicroRNAs and lipid metabolism, Current opinion in lipidology, 28 (2017) 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [427].Walker AK, Näär AM, SREBPs: regulators of cholesterol/lipids as therapeutic targets in metabolic disorders, cancers and viral diseases, Clinical Lipidology, 7 (2012) 27–36. [Google Scholar]
- [428].Kelsey I, Zbinden M, Byles V, Torrence M, Manning BD, mTORC1 suppresses PIM3 expression via miR-33 encoded by the SREBP loci, Scientific Reports, 7 (2017) 16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [429].Lin Y, Liu AY, Fan C, Zheng H, Li Y, Zhang C, Wu S, Yu D, Huang Z, Liu F, Luo Q, Yang CJ, Ouyang G, MicroRNA-33b Inhibits Breast Cancer Metastasis by Targeting HMGA2, SALL4 and Twist1, Scientific Reports, 5 (2015) 9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [430].Karatas OF, Ittmann M, MiR-33a and statins collaboratively reduce the proliferative capacity of prostate cancer cells, The European Research Journal, (2018). [Google Scholar]
- [431].Takwi AA, Li Y, Becker Buscaglia LE, Zhang J, Choudhury S, Park AK, Liu M, Young KH, Park WY, Martin RC, Li Y, A statin-regulated microRNA represses human c-Myc expression and function, EMBO molecular medicine, 4 (2012) 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [432].Pedroza-Torres A, Romero-Cordoba SL, Justo-Garrido M, Salido-Guadarrama I, Rodriguez-Bautista R, Montano S, Muniz-Mendoza R, Arriaga-Canon C, Fragoso-Ontiveros V, Alvarez-Gomez RM, Hernandez G, Herrera LA, MicroRNAs in Tumor Cell Metabolism: Roles and Therapeutic Opportunities, Front Oncol, 9 (2019) 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [433].Bai F, Yu Z, Gao X, Gong J, Fan L, Liu F, Simvastatin induces breast cancer cell death through oxidative stress up-regulating miR-140–5p, Aging, 11 (2019) 3198–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [434].Peng F, Wang JH, Fan WJ, Meng YT, Li MM, Li TT, Cui B, Wang HF, Zhao Y, An F, Guo T, Liu XF, Zhang L, Lv L, Lv DK, Xu LZ, Xie JJ, Lin WX, Lam EW, Xu J, Liu Q, Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia, Oncogene, 37 (2018) 1062–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [435].Li X, Chen YT, Josson S, Mukhopadhyay NK, Kim J, Freeman MR, Huang WC, MicroRNA-185 and 342 inhibit tumorigenicity and induce apoptosis through blockade of the SREBP metabolic pathway in prostate cancer cells, PloS one, 8 (2013) e70987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [436].Ru P, Hu P, Geng F, Mo X, Cheng C, Yoo JY, Cheng X, Wu X, Guo JY, Nakano I, Lefai E, Kaur B, Chakravarti A, Guo D, Feedback Loop Regulation of SCAP/SREBP-1 by miR-29 Modulates EGFR Signaling-Driven Glioblastoma Growth, Cell reports, 16 (2016) 1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [437].Xin M, Qiao Z, Li J, Liu J, Song S, Zhao X, Miao P, Tang T, Wang L, Liu W, Yang X, Dai K, Huang G, miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer, Oncotarget, 7 (2016) 44252–44265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [438].Krokker L, Nyiro G, Reiniger L, Darvasi O, Szucs N, Czirjak S, Toth M, Igaz P, Patocs A, Butz H, Differentially Expressed miRNAs Influence Metabolic Processes in Pituitary Oncocytoma, Neurochem Res, 44 (2019) 2360–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [439].Guo ST, Jiang CC, Wang GP, Li YP, Wang CY, Guo XY, Yang RH, Feng Y, Wang FH, Tseng HY, Thorne RF, Jin L, Zhang XD, MicroRNA-497 targets insulin-like growth factor 1 receptor and has a tumour suppressive role in human colorectal cancer, Oncogene, 32 (2013) 1910–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [440].Gharib E, Nasri Nasrabadi P, Reza Zali M, miR-497–5p mediates starvation-induced death in colon cancer cells by targeting acyl-CoA synthetase-5 and modulation of lipid metabolism, Journal of cellular physiology, (2020). [DOI] [PubMed] [Google Scholar]
- [441].De Santi C, Melaiu O, Bonotti A, Cascione L, Di Leva G, Foddis R, Cristaudo A, Lucchi M, Mora M, Truini A, Tironi A, Murer B, Boldorini R, Cipollini M, Gemignani F, Gasparini P, Mutti L, Landi S, Deregulation of miRNAs in malignant pleural mesothelioma is associated with prognosis and suggests an alteration of cell metabolism, Sci Rep, 7 (2017) 3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [442].Huang CM, Huang CS, Hsu TN, Huang MS, Fong IH, Lee WH, Liu SC, Disruption of Cancer Metabolic SREBP1/miR-142–5p Suppresses Epithelial-Mesenchymal Transition and Stemness in Esophageal Carcinoma, Cells, 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [443].Song H, Xu Y, Xu T, Fan R, Jiang T, Cao M, Shi L, Song J, CircPIP5K1A activates KRT80 and PI3K/AKT pathway to promote gastric cancer development through sponging miR-671–5p, Biomedicine & Pharmacotherapy, 126 (2020) 109941. [DOI] [PubMed] [Google Scholar]
- [444].Boguslawska J, Poplawski P, Alseekh S, Koblowska M, Iwanicka-Nowicka R, Rybicka B, Kedzierska H, Gluchowska K, Hanusek K, Tanski Z, Fernie AR, Piekielko-Witkowska A, MicroRNA-Mediated Metabolic Reprograming in Renal Cancer, Cancers (Basel), 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [445].Gholami M, Larijani B, Zahedi Z, Mahmoudian F, Bahrami S, Omran SP, Saadatian Z, Hasani-Ranjbar S, Taslimi R, Bastami M, Amoli MM, Inflammation related miRNAs as an important player between obesity and cancers, J Diabetes Metab Disord, 18 (2019) 675–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [446].Bengoechea-Alonso MT, Ericsson J, A Phosphorylation Cascade Controls the Degradation of Active SREBP1, Journal of Biological Chemistry, 284 (2009) 5885–5895. [DOI] [PubMed] [Google Scholar]
- [447].Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J, Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCFFbw7, Cell Metabolism, 1 (2005) 379–391. [DOI] [PubMed] [Google Scholar]
- [448].Bengoechea-Alonso MT, Punga T, Ericsson J, Hyperphosphorylation regulates the activity of SREBP1 during mitosis, Proceedings of the National Academy of Sciences, 102 (2005) 11681–11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [449].Punga T, Bengoechea-Alonso MT, Ericsson J, Phosphorylation and Ubiquitination of the Transcription Factor Sterol Regulatory Element-binding Protein-1 in Response to DNA Binding, Journal of Biological Chemistry, 281 (2006) 25278–25286. [DOI] [PubMed] [Google Scholar]
- [450].Bengoechea-Alonso MT, Ericsson J, The phosphorylation-dependent regulation of nuclear SREBP1 during mitosis links lipid metabolism and cell growth, Cell cycle (Georgetown, Tex.), 15 (2016) 2753–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [451].Hirano Y, Murata S, Tanaka K, Shimizu M, Sato R, Sterol Regulatory Element-binding Proteins Are Negatively Regulated through SUMO-1 Modification Independent of the Ubiquitin/26 S Proteasome Pathway, Journal of Biological Chemistry, 278 (2003) 16809–16819. [DOI] [PubMed] [Google Scholar]
- [452].Hirano Y, Yoshida M, Shimizu M, Sato R, Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway, The Journal of Biological Chemistry, 276 (2001) 36431–36437. [DOI] [PubMed] [Google Scholar]
- [453].Kuan Y-C, Hashidume T, Shibata T, Uchida K, Shimizu M, Inoue J, Sato R, Heat Shock Protein 90 Modulates Lipid Homeostasis by Regulating the Stability and Function of Sterol Regulatory Element-binding Protein (SREBP) and SREBP Cleavage-activating Protein, Journal of Biological Chemistry, 292 (2017) 3016–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [454].Gong Y, Lee JN, Lee PCW, Goldstein JL, Brown MS, Ye J, Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake, Cell Metabolism, 3 (2006) 15–24. [DOI] [PubMed] [Google Scholar]
- [455].Nakakuki M, Kawano H, Notsu T, Imada K, Mizuguchi K, Shimano H, A novel processing system of sterol regulatory element-binding protein-1c regulated by polyunsaturated fatty acid, Journal of biochemistry, 155 (2014) 301–313. [DOI] [PubMed] [Google Scholar]
- [456].Takeuchi Y, Yahagi N, Izumida Y, Nishi M, Kubota M, Teraoka Y, Yamamoto T, Matsuzaka T, Nakagawa Y, Sekiya M, Iizuka Y, Ohashi K, Osuga J, Gotoda T, Ishibashi S, Itaka K, Kataoka K, Nagai R, Yamada N, Kadowaki T, Shimano H, Polyunsaturated fatty acids selectively suppress sterol regulatory element-binding protein-1 through proteolytic processing and autoloop regulatory circuit, J Biol Chem, 285 (2010) 11681–11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [457].Yahagi N, Shimano H, Hasty AH, Amemiya-Kudo M, Okazaki H, Tamura Y, Iizuka Y, Shionoiri F, Ohashi K, Osuga J, Harada K, Gotoda T, Nagai R, Ishibashi S, Yamada N, A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids, J Biol Chem, 274 (1999) 35840–35844. [DOI] [PubMed] [Google Scholar]
- [458].Dong Q, Giorgianni F, Deng X, Beranova-Giorgianni S, Bridges D, Park EA, Raghow R, Elam MB, Phosphorylation of sterol regulatory element binding protein-1a by protein kinase A (PKA) regulates transcriptional activity, Biochem Biophys Res Commun, 449 (2014) 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [459].Lu M, Shyy JY, Sterol regulatory element-binding protein 1 is negatively modulated by PKA phosphorylation, Am J Physiol Cell Physiol, 290 (2006) C1477–1486. [DOI] [PubMed] [Google Scholar]
- [460].Yamamoto T, Shimano H, Inoue N, Nakagawa Y, Matsuzaka T, Takahashi A, Yahagi N, Sone H, Suzuki H, Toyoshima H, Yamada N, Protein kinase A suppresses sterol regulatory element-binding protein-1C expression via phosphorylation of liver X receptor in the liver, J Biol Chem, 282 (2007) 11687–11695. [DOI] [PubMed] [Google Scholar]
- [461].Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JYJ, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M, AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice, Cell Metabolism, 13 (2011) 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [462].Rios Garcia M, Steinbauer B, Srivastava K, Singhal M, Mattijssen F, Maida A, Christian S, Hess-Stumpp H, Augustin HG, Muller-Decker K, Nawroth PP, Herzig S, Berriel Diaz M, Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence, Cell Metab, 26 (2017) 842–855 e845. [DOI] [PubMed] [Google Scholar]
- [463].Luo J, Hong Y, Lu Y, Qiu S, Chaganty BK, Zhang L, Wang X, Li Q, Fan Z, Acetyl-CoA carboxylase rewires cancer metabolism to allow cancer cells to survive inhibition of the Warburg effect by cetuximab, Cancer letters, 384 (2017) 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [464].Gil G, Faust JR, Chin DJ, Goldstein JL, Brown MS, Membrane-bound domain of HMG CoA reductase is required for sterol-enhanced degradation of the enzyme, Cell, 41 (1985) 249–258. [DOI] [PubMed] [Google Scholar]
- [465].Hampton RY, Gardner RG, Rine J, Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein, Mol Biol Cell, 7 (1996) 2029–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [466].Graner E, Tang D, Rossi S, Baron A, Migita T, Weinstein LJ, Lechpammer M, Huesken D, Zimmermann J, Signoretti S, Loda M, The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer, Cancer cell, 5 (2004) 253–261. [DOI] [PubMed] [Google Scholar]
- [467].Priolo C, Tang D, Brahamandan M, Benassi B, Sicinska E, Ogino S, Farsetti A, Porrello A, Finn S, Zimmermann J, Febbo P, Loda M, The isopeptidase USP2a protects human prostate cancer from apoptosis, Cancer Res, 66 (2006) 8625–8632. [DOI] [PubMed] [Google Scholar]
- [468].Lin HP, Cheng ZL, He RY, Song L, Tian MX, Zhou LS, Groh BS, Liu WR, Ji MB, Ding C, Shi YH, Guan KL, Ye D, Xiong Y, Destabilization of Fatty Acid Synthase by Acetylation Inhibits De Novo Lipogenesis and Tumor Cell Growth, Cancer Res, 76 (2016) 6924–6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [469].Zaidi N, Swinnen JV, Smans K, ATP-citrate lyase: a key player in cancer metabolism, Cancer Res, 72 (2012) 3709–3714. [DOI] [PubMed] [Google Scholar]
- [470].Icard P, Wu Z, Fournel L, Coquerel A, Lincet H, Alifano M, ATP citrate lyase: A central metabolic enzyme in cancer, Cancer letters, 471 (2020) 125–134. [DOI] [PubMed] [Google Scholar]
- [471].Potapova IA, El-Maghrabi MR, Doronin SV, Benjamin WB, Phosphorylation of recombinant human ATP:citrate lyase by cAMP-dependent protein kinase abolishes homotropic allosteric regulation of the enzyme by citrate and increases the enzyme activity. Allosteric activation of ATP:citrate lyase by phosphorylated sugars, Biochemistry, 39 (2000) 1169–1179. [DOI] [PubMed] [Google Scholar]
- [472].Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y, ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer, Cancer Res, 68 (2008) 8547–8554. [DOI] [PubMed] [Google Scholar]
- [473].Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, Xiong Y, Lei QY, Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth, Molecular cell, 51 (2013) 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [474].Zhang C, Liu J, Huang G, Zhao Y, Yue X, Wu H, Li J, Zhu J, Shen Z, Haffty BG, Hu W, Feng Z, Cullin3-KLHL25 ubiquitin ligase targets ACLY for degradation to inhibit lipid synthesis and tumor progression, Genes & development, 30 (2016) 1956–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [475].Han C, Lu X, Nagrath D, Regulation of protein metabolism in cancer, Molecular & cellular oncology, 5 (2018) e1285384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [476].Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, Bon H, Zecchini V, Smith D-M, DeNicola GM, Mathews N, Osborne M, Hadfield J, MacArthur S, Adryan B, Lyons SK, Brindle KM, Griffiths J, Gleave ME, Rennie PS, Neal DE, Mills IG, The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis: AR coordinates anabolic program in prostate cancer, The EMBO Journal, 30 (2011) 2719–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [477].Butler LM, Centenera MM, Swinnen JV, Androgen control of lipid metabolism in prostate cancer: novel insights and future applications, Endocr Relat Cancer, 23 (2016) R219–227. [DOI] [PubMed] [Google Scholar]
- [478].Heemers HV, Verhoeven G, Swinnen JV, Androgen Activation of the Sterol Regulatory Element-Binding Protein Pathway: Current Insights, Molecular Endocrinology, 20 (2006) 2265–2277. [DOI] [PubMed] [Google Scholar]
- [479].Swinnen JV, Van Veldhoven PP, Esquenet M, Heyns W, Verhoeven G, Androgens markedly stimulate the accumulation of neutral lipids in the human prostatic adenocarcinoma cell line LNCaP, Endocrinology, 137 (1996) 4468–4474. [DOI] [PubMed] [Google Scholar]
- [480].Heemers H, Maes B, Foufelle F, Heyns W, Verhoeven G, Swinnen JV, Androgens stimulate lipogenic gene expression in prostate cancer cells by activation of the sterol regulatory element-binding protein cleavage activating protein/sterol regulatory element-binding protein pathway, Molecular Endocrinology, 15 (2001) 1817–1828. [DOI] [PubMed] [Google Scholar]
- [481].Swinnen JV, Heemers H, van de Sande T, de Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Androgens, lipogenesis and prostate cancer, The Journal of steroid biochemistry and molecular biology, 92 (2004) 273–279. [DOI] [PubMed] [Google Scholar]
- [482].Sharma NL, Massie CE, Ramos-Montoya A, Scott HE, Stark R, Warren AY, Mills IG, Neal DE, The androgen receptor induces a distinct transcriptional program in castration resistant prostate cancer in man, European Urology Supplements, 11 (2012) e942–e942a. [DOI] [PubMed] [Google Scholar]
- [483].Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, Cheng JX, Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness, Cell Metab, 19 (2014) 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [484].Mitra R, Chao O, Urasaki Y, Goodman OB, Le TT, Detection of lipid-rich prostate circulating tumour cells with coherent anti-Stokes Raman scattering microscopy, BMC Cancer, 12 (2012) 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [485].Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A, Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy, Cell, 168 (2017) 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [486].Huang WC, Li X, Liu J, Lin J, Chung LWK, Activation of Androgen Receptor, Lipogenesis, and Oxidative Stress Converged by SREBP-1 Is Responsible for Regulating Growth and Progression of Prostate Cancer Cells, Molecular Cancer Research, 10 (2012) 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [487].Yang T, Goldstein JL, Brown MS, Overexpression of Membrane Domain of SCAP Prevents Sterols from Inhibiting SCAP·SREBP Exit from Endoplasmic Reticulum, Journal of Biological Chemistry, 275 (2000) 29881–29886. [DOI] [PubMed] [Google Scholar]
- [488].Williams KJ, Argus JP, Zhu Y, Wilks MQ, Marbois BN, York AG, Kidani Y, Pourzia AL, Akhavan D, Lisiero DN, Komisopoulou E, Henkin AH, Soto H, Chamberlain BT, Vergnes L, Jung ME, Torres JZ, Liau LM, Christofk HR, Prins RM, Mischel PS, Reue K, Graeber TG, Bensinger SJ, An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity, Cancer Res, 73 (2013) 2850–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [489].Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, Nelson CC, Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence, Cancer Res, 64 (2004) 2212–2221. [DOI] [PubMed] [Google Scholar]
- [490].Mostaghel EA, Steroid hormone synthetic pathways in prostate cancer, Transl Androl Urol, 2 (2013) 212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [491].Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC, Androgen Levels Increase by Intratumoral De novo Steroidogenesis during Progression of Castration-Resistant Prostate Cancer, Cancer Research, 68 (2008) 6407–6415. [DOI] [PubMed] [Google Scholar]
- [492].Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS, Maintenance of Intratumoral Androgens in Metastatic Prostate Cancer: A Mechanism for Castration-Resistant Tumor Growth, Cancer Research, 68 (2008) 4447–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [493].Bennett MK, Lopez JM, Sanchez HB, Osborne TF, Sterol Regulation of Fatty Acid Synthase Promoter., J. Biol. Chem, 270 (1995) 25578–25583. [DOI] [PubMed] [Google Scholar]
- [494].Filippatos TD, Liberopoulos EN, Pavlidis N, Elisaf MS, Mikhailidis DP, Effects of hormonal treatment on lipids in patients with cancer, Cancer Treatment Reviews, 35 (2009) 175–184. [DOI] [PubMed] [Google Scholar]
- [495].Osborne CK, Schiff R, Estrogen-Receptor Biology: Continuing Progress and Therapeutic Implications, Journal of Clinical Oncology, 23 (2005) 1616–1622. [DOI] [PubMed] [Google Scholar]
- [496].Zhang Y, Klein K, Sugathan A, Nassery N, Dombkowski A, Zanger UM, Waxman DJ, Transcriptional Profiling of Human Liver Identifies Sex-Biased Genes Associated with Polygenic Dyslipidemia and Coronary Artery Disease, PloS one, 6 (2011) e23506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [497].Gao H, Fält S, Sandelin A, Gustafsson J-Å, Dahlman-Wright K, Genome-Wide Identification of Estrogen Receptor α-Binding Sites in Mouse Liver, Molecular Endocrinology, 22 (2008) 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [498].Han S.-i., Komatsu Y, Murayama A, Steffensen KR, Nakagawa Y, Nakajima Y, Suzuki M, Oie S, Parini P, Vedin L-L, Kishimoto H, Shimano H, Gustafsson J-Å, Yanagisawa J, Estrogen receptor ligands ameliorate fatty liver through a nonclassical estrogen receptor/Liver X receptor pathway in mice: Hepatology, Vol. XX, No. X, 2013 Han et al, Hepatology (Baltimore, Md.), 59 (2014) 1791–1802. [DOI] [PubMed] [Google Scholar]
- [499].Morita Y, Sakaguchi T, Ikegami K, Goto-Inoue N, Hayasaka T, Hang VT, Tanaka H, Harada T, Shibasaki Y, Suzuki A, Fukumoto K, Inaba K, Murakami M, Setou M, Konno H, Lysophosphatidylcholine acyltransferase 1 altered phospholipid composition and regulated hepatoma progression, Journal of hepatology, 59 (2013) 292–299. [DOI] [PubMed] [Google Scholar]
- [500].Du Y, Wang Q, Zhang X, Wang X, Qin C, Sheng Z, Yin H, Jiang C, Li J, Xu T, Lysophosphatidylcholine acyltransferase 1 upregulation and concomitant phospholipid alterations in clear cell renal cell carcinoma, Journal of Experimental & Clinical Cancer Research, 36 (2017) 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [501].Zhang J, Li Q, Wu Y, Wang D, Xu L, Zhang Y, Wang S, Wang T, Liu F, Zaky MY, Hou S, Liu S, Zou K, Lei H, Zou L, Zhang Y, Liu H, Cholesterol content in cell membrane maintains surface levels of ErbB2 and confers a therapeutic vulnerability in ErbB2-positive breast cancer, Cell Commun Signal, 17 (2019) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [502].Li YC, Park MJ, Ye S-K, Kim C-W, Kim Y-N, Elevated Levels of Cholesterol-Rich Lipid Rafts in Cancer Cells Are Correlated with Apoptosis Sensitivity Induced by Cholesterol-Depleting Agents, The American journal of pathology, 168 (2006) 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [503].Walther TC, Farese RV, Lipid Droplets and Cellular Lipid Metabolism, Annual review of biochemistry, 81 (2012) 687–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [504].Antalis CJ, Uchida A, Buhman KK, Siddiqui RA, Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification, Clinical & experimental metastasis, 28 (2011) 733–741. [DOI] [PubMed] [Google Scholar]
- [505].Li J, Gu D, Lee SSY, Song B, Bandyopadhyay S, Chen S, Konieczny SF, Ratliff TL, Liu X, Xie J, Cheng JX, Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer, Oncogene, 35 (2016) 6378–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [506].Geng F, Cheng X, Wu X, Yoo JY, Cheng C, Guo JY, Mo X, Ru P, Hurwitz B, Kim SH, Otero J, Puduvalli V, Lefai E, Ma J, Nakano I, Horbinski C, Kaur B, Chakravarti A, Guo D, Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis, Clinical cancer research : an official journal of the American Association for Cancer Research, 22 (2016) 5337–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [507].C. Chinese Human Proteome Project, Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, Xing B, Sun W, Ren L, Hu B, Li C, Zhang L, Qin G, Zhang M, Chen N, Zhang M, Huang Y, Zhou J, Zhao Y, Liu M, Zhu X, Qiu Y, Sun Y, Huang C, Yan M, Wang M, Liu W, Tian F, Xu H, Zhou J, Wu Z, Shi T, Zhu W, Qin J, Xie L, Fan J, Qian X, He F, Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma, Nature, 567 (2019) 257–261. [DOI] [PubMed] [Google Scholar]
- [508].Wang J, Tan M, Ge J, Zhang P, Zhong J, Tao L, Wang Q, Tong X, Qiu J, Lysosomal acid lipase promotes cholesterol ester metabolism and drives clear cell renal cell carcinoma progression, Cell proliferation, 51 (2018) e12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [509].Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, Puchowicz M, Serra D, Herrero L, Rini BI, Campbell S, Welford SM, HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism, Nat Commun, 8 (2017) 1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [510].Bensaad K, Favaro E, Lewis Caroline A., Peck B, Lord S, Collins Jennifer M., Pinnick Katherine E., Wigfield S, Buffa Francesca M., Li J-L, Zhang Q, Wakelam Michael J.O., Karpe F, Schulze A, Harris Adrian L., Fatty Acid Uptake and Lipid Storage Induced by HIF-1α Contribute to Cell Growth and Survival after Hypoxia-Reoxygenation, Cell reports, 9 (2014) 349–365. [DOI] [PubMed] [Google Scholar]
- [511].Schulze A, Harris AL, How cancer metabolism is tuned for proliferation and vulnerable to disruption, Nature, 491 (2012) 364–373. [DOI] [PubMed] [Google Scholar]
- [512].Kuhajda F, Jenner K, Wood F, Hennigar R, Jacobs L, Dick J, Pasternak G, Fatty acid synthesis: a potential selective target for antineoplastic therapy., PNAS, 91 (1994) 6279–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [513].Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV, RNA interference-mediated silencing of the acetyl-CoA-carboxylase-alpha gene induces growth inhibition and apoptosis of prostate cancer cells, Cancer Research, 65 (2005) 6719–6725. [DOI] [PubMed] [Google Scholar]
- [514].Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV, Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells, Cancer Research, 67 (2007) 8180–8187. [DOI] [PubMed] [Google Scholar]
- [515].Scaglia N, Tyekucheva S, Zadra G, Photopoulos C, Loda M, De novo fatty acid synthesis at the mitotic exit is required to complete cellular division, Cell Cycle, 13 (2014) 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [516].Ackerman D, Hypoxia SC, lipids, and cancer: surviving the harsh tumor microenvironment, Trends in Cell Biology, 24 (2014) 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [517].Minville-Walz M, Pierre AS, Pichon L, Bellenger S, Fevre C, Bellenger J, Tessier C, Narce M, Rialland M, Inhibition of stearoyl-CoA desaturase 1 expression induces CHOP-dependent cell death in human cancer cells, PloS one, 5 (2010) e14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [518].Gouw AM, Eberlin LS, Margulis K, Sullivan DK, Toal GG, Tong L, Zare RN, Felsher DW, Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma, Proceedings of the National Academy of Sciences of the United States of America, 114 (2017) 4300–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [519].Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR, Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis, Proc Natl Acad Sci U S A, 113 (2016) E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [520].Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA, Chinnaiyan AM, Delineation of prognostic biomarkers in prostate cancer, Nature, 412 (2001) 822–826. [DOI] [PubMed] [Google Scholar]
- [521].Kumar-Sinha C, Shah RB, Laxman B, Tomlins SA, Harwood J, Schmitz W, Conzelmann E, Sanda MG, Wei JT, Rubin MA, Chinnaiyan AM, Elevated α-Methylacyl-CoA Racemase Enzymatic Activity in Prostate Cancer, The American journal of pathology, 164 (2004) 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [522].Zha S, Ferdinandusse S, Denis S, Wanders RJ, Ewing CM, Luo J, De Marzo AM, Isaacs WB, Alpha-methylacyl-CoA racemase as an androgen-independent growth modifier in prostate cancer, Cancer Res, 63 (2003) 7365–7376. [PubMed] [Google Scholar]
- [523].Selkala EM, Kuusisto SM, Salonurmi T, Savolainen MJ, Jauhiainen M, Pirila PL, Kvist AP, Conzelmann E, Schmitz W, Alexson SE, Kotti TJ, Hiltunen JK, Autio KJ, Metabolic adaptation allows Amacr-deficient mice to remain symptom-free despite low levels of mature bile acids, Biochim Biophys Acta, 1831 (2013) 1335–1343. [DOI] [PubMed] [Google Scholar]
- [524].Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC, The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth, Nature, 452 (2008) 230–233. [DOI] [PubMed] [Google Scholar]
- [525].Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, Sasaki AT, Anastasiou D, Mullarky E, Vokes NI, Sasaki M, Beroukhim R, Stephanopoulos G, Ligon AH, Meyerson M, Richardson AL, Chin L, Wagner G, Asara JM, Brugge JS, Cantley LC, Vander Heiden MG, Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis, Nature genetics, 43 (2011) 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [526].Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, Cantley LC, Evidence for an alternative glycolytic pathway in rapidly proliferating cells, Science, 329 (2010) 1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [527].Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, Shan C, Dai Q, Zhang L, Xie J, Gu TL, Jin P, Aleckovic M, LeRoy G, Kang Y, Sudderth JA, DeBerardinis RJ, Luan CH, Chen GZ, Muller S, Shin DM, Owonikoko TK, Lonial S, Arellano ML, Khoury HJ, Khuri FR, Lee BH, Ye K, Boggon TJ, Kang S, He C, Chen J, Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth, Cancer cell, 22 (2012) 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [528].Madak-Erdogan Z, Band S, Zhao Y, Smith B, Kulkoyloglu-Cotul E, Zuo Q, Casiano A, Wrobel K, Rossi G, Smith R, Kim S, Katznellenbogen J, Johnson M, Patel M, Marino N, Storniolo A, Flaws J, Free fatty acids rewire cancer metabolism in obesity-associated breast cancer via estrogen receptor and mTOR signaling., Cancer Res., 79 (2019) 2494–2510. [DOI] [PubMed] [Google Scholar]
- [529].Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, Nieman KM, Pascual G, Benitah SA, Montag A, Yamada SD, Abumrad NA, Lengyel E, Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis, Oncogene, 37 (2018) 2285–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [530].Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, Kim IS, Haldeman P, Mondal C, Yong-Gonzales V, Abu-Akeel M, Merghoub T, Jones DR, Zhu XG, Arora A, Ariyan CE, Birsoy K, Wolchok JD, Panageas KS, Hollmann T, Bravo-Cordero JJ, White RM, Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins, Cancer discovery, 8 (2018) 1006–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [531].Henderson F, Johnston HR, Badrock AP, Jones EA, Forster D, Nagaraju RT, Evangelou C, Kamarashev J, Green M, Fairclough M, Ramirez IB, He S, Snaar-Jagalska BE, Hollywood K, Dunn WB, Spaink HP, Smith MP, Lorigan P, Claude E, Williams KJ, McMahon AW, Hurlstone A, Enhanced Fatty Acid Scavenging and Glycerophospholipid Metabolism Accompany Melanocyte Neoplasia Progression in Zebrafish, Cancer Res, 79 (2019) 2136–2151. [DOI] [PubMed] [Google Scholar]
- [532].Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, Mogushi K, Shikami M, Ruvolo V, Ishizawa J, Hail N Jr., Kazuno S, Igarashi M, Matsushita H, Yamanaka Y, Arai H, Nagaoka I, Miida T, Hayashizaki Y, Konopleva M, Andreeff M, Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells, Cancer Res, 77 (2017) 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [533].Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS, Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment, Nature, 461 (2009) 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [534].Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VC, Laurent G, Adams AC, Sundvall M, Song SJ, Ito K, Finley LS, Egia A, Libermann T, Gerhart-Hines Z, Puigserver P, Haigis MC, Maratos-Flier E, Richardson AL, Schafer ZT, Pandolfi PP, A metabolic prosurvival role for PML in breast cancer, The Journal of clinical investigation, 122 (2012) 3088–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [535].Corbet C, Bastien E, Santiago de Jesus JP, Dierge E, Martherus R, Vander Linden C, Doix B, Degavre C, Guilbaud C, Petit L, Michiels C, Dessy C, Larondelle Y, Feron O, TGFbeta2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells, Nat Commun, 11 (2020) 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [536].Duman C, Yaqubi K, Hoffmann A, Acikgoz AA, Korshunov A, Bendszus M, Herold-Mende C, Liu HK, Alfonso J, Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation, Cell Metab, 30 (2019) 274–289 e275. [DOI] [PubMed] [Google Scholar]
- [537].Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, Zhang C, Lifshitz V, Song J, Yuan Y, Somlo G, Jandial R, Ann D, Hanash S, Jove R, Yu H, JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance, Cell Metab, 27 (2018) 1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [538].Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, Bosc C, Sugita M, Stuani L, Fraisse M, Scotland S, Larrue C, Boutzen H, Feliu V, Nicolau-Travers ML, Cassant-Sourdy S, Broin N, David M, Serhan N, Sarry A, Tavitian S, Kaoma T, Vallar L, Iacovoni J, Linares LK, Montersino C, Castellano R, Griessinger E, Collette Y, Duchamp O, Barreira Y, Hirsch P, Palama T, Gales L, Delhommeau F, Garmy-Susini BH, Portais JC, Vergez F, Selak M, Danet-Desnoyers G, Carroll M, Recher C, Sarry JE, Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism, Cancer discovery, 7 (2017) 716–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [539].Schoors S, Bruning U, Missiaen R, Queiroz KCS, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia J, Heggermont W, Goddé L, Vinckier S, Van Veldhoven PP, Eelen G, Schoonjans L, Gerhardt H, Dewerchin M, Baes M, De Bock K, Ghesquière B, Lunt SY, Fendt S-M, Carmeliet P, Fatty acid carbon is essential for dNTP synthesis in endothelial cells, Nature, 520 (2015) 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [540].Spector AA, Yorek MA, Membrane lipid composition and cellular function, J Lipid Res, 26 (1985) 1015–1035. [PubMed] [Google Scholar]
- [541].Dawaliby R, Trubbia C, Delporte C, Noyon C, Ruysschaert JM, Van Antwerpen P, Govaerts C, Phosphatidylethanolamine Is a Key Regulator of Membrane Fluidity in Eukaryotic Cells, J Biol Chem, 291 (2016) 3658–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [542].Cooper RA, Influence of increased membrane cholesterol on membrane fluidity and cell function in human red blood cells, J Supramol Struct, 8 (1978) 413–430. [DOI] [PubMed] [Google Scholar]
- [543].Taraboletti G, Perin L, Bottazzi B, Mantovani A, Giavazzi R, Salmona M, Membrane fluidity affects tumor-cell motility, invasion and lung-colonizing potential, International journal of cancer, 44 (1989) 707–713. [DOI] [PubMed] [Google Scholar]
- [544].Sok M, Sentjurc M, Schara M, Stare J, Rott T, Cell membrane fluidity and prognosis of lung cancer, Ann Thorac Surg, 73 (2002) 1567–1571. [DOI] [PubMed] [Google Scholar]
- [545].Sezgin E, Levental I, Mayor S, Eggeling C, The mystery of membrane organization: composition, regulation and roles of lipid rafts, Nature reviews. Molecular cell biology, 18 (2017) 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [546].Pike LJ, Growth factor receptors, lipid rafts and caveolae: an evolving story, Biochim Biophys Acta, 1746 (2005) 260–273. [DOI] [PubMed] [Google Scholar]
- [547].Casaletto JB, McClatchey AI, Spatial regulation of receptor tyrosine kinases in development and cancer, Nat Rev Cancer, 12 (2012) 387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [548].Swinnen JV, Van Veldhoven PP, Timmermans L, De Schrijver E, Brusselmans K, Vanderhoydonc F, Van de Sande T, Heemers H, Heyns W, Verhoeven G, Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains, Biochem Biophys Res Commun, 302 (2003) 898–903. [DOI] [PubMed] [Google Scholar]
- [549].Pike LJ, Lipid rafts: bringing order to chaos, J Lipid Res, 44 (2003) 655–667. [DOI] [PubMed] [Google Scholar]
- [550].Berndt N, Hamilton AD, Sebti SM, Targeting protein prenylation for cancer therapy, Nat Rev Cancer, 11 (2011) 775–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [551].Ogretmen B, Sphingolipid metabolism in cancer signalling and therapy, Nat Rev Cancer, 18 (2018) 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [552].McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA, Grimsrud PA, Hirschey MD, Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation, Cell reports, 17 (2016) 1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [553].Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB, ATP-citrate lyase links cellular metabolism to histone acetylation, Science, 324 (2009) 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [554].Kim J, Kim J, Bae JS, ROS homeostasis and metabolism: a critical liaison for cancer therapy, Exp Mol Med, 48 (2016) e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [555].Panieri E, Santoro MM, ROS homeostasis and metabolism: a dangerous liason in cancer cells, Cell death & disease, 7 (2016) e2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [556].Sabharwal SS, Schumacker PT, Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel?, Nat Rev Cancer, 14 (2014) 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [557].Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayir H, Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis, Nat Chem Biol, 13 (2017) 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [558].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR, Ferroptosis: an iron-dependent form of nonapoptotic cell death, Cell, 149 (2012) 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [559].Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, McCormick F, McManus MT, Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition, Nature, 551 (2017) 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [560].Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, Viswanathan SR, Chattopadhyay S, Tamayo P, Yang WS, Rees MG, Chen S, Boskovic ZV, Javaid S, Huang C, Wu X, Tseng Y-Y, Roider EM, Gao D, Cleary JM, Wolpin BM, Mesirov JP, Haber DA, Engelman JA, Boehm JS, Kotz JD, Hon CS, Chen Y, Hahn WC, Levesque MP, Doench JG, Berens ME, Shamji AF, Clemons PA, Stockwell BR, Schreiber SL, Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway, Nature, 547 (2017) 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [561].Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trumbach D, Mao G, Qu F, Bayir H, Fullekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JP, Conrad M, ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition, Nat Chem Biol, 13 (2017) 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [562].Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng YY, Deasy R, Kost-Alimova M, Dancik V, Leshchiner ES, Viswanathan VS, Signoretti S, Choueiri TK, Boehm JS, Wagner BK, Doench JG, Clish CB, Clemons PA, Schreiber SL, A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis, Nat Commun, 10 (2019) 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [563].Panzilius E, Holstein F, Dehairs J, Planque M, von Toerne C, Koenig A-C, Doll S, Bannier-Hélaouët M, Ganz HM, Hauck SM, Talebi A, Swinnen JV, Fendt S-M, Angeli JPF, Conrad M, Scheel CH, Cell density-dependent ferroptosis in breast cancer is induced by accumulation of polyunsaturated fatty acid-enriched triacylglycerides, bioRxiv, (2019) 417949. [Google Scholar]
- [564].Nassar ZD, Mah CY, Dehairs J, Burvenich IJG, Irani S, Centenera MM, Shrestha RK, Moldovan M, Don AS, Scott AM, Horvath LG, Lynn DJ, Selth LA, Hoy AJ, Swinnen JV, Butler LM, DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis, bioRxiv, (2019) 865626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [565].Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M, Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells, Biochim Biophys Acta, 1807 (2011) 726–734. [DOI] [PubMed] [Google Scholar]
- [566].Golan T, Parikh R, Jacob E, Vaknine H, Zemser-Werner V, Hershkovitz D, Malcov H, Leibou S, Reichman H, Sheinboim D, Percik R, Amar S, Brenner R, Greenberger S, Kung A, Khaled M, Levy C, Adipocytes sensitize melanoma cells to environmental TGF-beta cues by repressing the expression of miR-211, Science signaling, 12 (2019). [DOI] [PubMed] [Google Scholar]
- [567].Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, Polak K, Tondera D, Gounarides J, Yin H, Zhou F, Green MR, Chen L, Monti S, Marto JA, Shipp MA, Danial NN, Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma, Cancer cell, 22 (2012) 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [568].van Weverwijk A, Koundouros N, Iravani M, Ashenden M, Gao Q, Poulogiannis G, Jungwirth U, Isacke CM, Metabolic adaptability in metastatic breast cancer by AKR1B10-dependent balancing of glycolysis and fatty acid oxidation, Nat Commun, 10 (2019) 2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [569].Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, Saunders B, Howell M, Downward J, Felsher DW, Peck B, Schulze A, The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma, Oncogene, 37 (2018) 5435–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [570].Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, Bassik MC, Nomura DK, Dixon SJ, Olzmann JA, The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis, Nature, 575 (2019) 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [571].Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Grocin AG, Xavier da Silva TN, Panzilius E, Scheel CH, Mourao A, Buday K, Sato M, Wanninger J, Vignane T, Mohana V, Rehberg M, Flatley A, Schepers A, Kurz A, White D, Sauer M, Sattler M, Tate EW, Schmitz W, Schulze A, O’Donnell V, Proneth B, Popowicz GM, Pratt DA, Angeli JPF, Conrad M, FSP1 is a glutathione-independent ferroptosis suppressor, Nature, 575 (2019) 693–698. [DOI] [PubMed] [Google Scholar]
- [572].Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, Yucel B, Fiore D, Tavora B, Freinkman E, Chan SH, Lewis C, Min W, Inghirami G, Sabatini DM, Birsoy K, Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death, Nature, 567 (2019) 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [573].Wymann MP, Schneiter R, Lipid signalling in disease, Nature reviews. Molecular cell biology, 9 (2008) 162–176. [DOI] [PubMed] [Google Scholar]
- [574].Vegliante R, Di Leo L, Ciccarone F, Ciriolo MR, Hints on ATGL implications in cancer: beyond bioenergetic clues, Cell death & disease, 9 (2018) 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [575].Ou J, Miao H, Ma Y, Guo F, Deng J, Wei X, Zhou J, Xie G, Shi H, Xue B, Liang H, Yu L, Loss of abhd5 promotes colorectal tumor development and progression by inducing aerobic glycolysis and epithelial-mesenchymal transition, Cell reports, 9 (2014) 1798–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [576].Zagani R, El-Assaad W, Gamache I, Teodoro JG, Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells, Oncotarget, 6 (2015) 28282–28295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [577].Wang YY, Attane C, Milhas D, Dirat B, Dauvillier S, Guerard A, Gilhodes J, Lazar I, Alet N, Laurent V, Le Gonidec S, Biard D, Herve C, Bost F, Ren GS, Bono F, Escourrou G, Prentki M, Nieto L, Valet P, Muller C, Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells, JCI Insight, 2 (2017) e87489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [578].Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R, Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome, Cell Metab, 3 (2006) 309–319. [DOI] [PubMed] [Google Scholar]
- [579].Nomura DK, Long JZ, Niessen S, Hoover HS, Ng S-W, Cravatt BF, Monoacylglycerol Lipase Regulates a Fatty Acid Network that Promotes Cancer Pathogenesis, Cell, 140 (2010) 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [580].Xiang W, Shi R, Kang X, Zhang X, Chen P, Zhang L, Hou A, Wang R, Zhao Y, Zhao K, Liu Y, Ma Y, Luo H, Shang S, Zhang J, He F, Yu S, Gan L, Shi C, Li Y, Yang W, Liang H, Miao H, Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression, Nat Commun, 9 (2018) 2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [581].Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF, Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis, Cell, 140 (2010) 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [582].Sun H, Jiang L, Luo X, Jin W, He Q, An J, Lui K, Shi J, Rong R, Su W, Lucchesi C, Liu Y, Sheikh MS, Huang Y, Potential tumor-suppressive role of monoglyceride lipase in human colorectal cancer, Oncogene, 32 (2013) 234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [583].Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, Tumanov S, Allen-Petersen BL, Link J, Kendsersky ND, Vringer E, Schug M, Novo D, Hwang RF, Evans RM, Nixon C, Dorrell C, Morton JP, Norman JC, Sears RC, Kamphorst JJ, Sherman MH, A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression, Cancer discovery, 9 (2019) 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [584].Thery C, Zitvogel L, Amigorena S, Exosomes: composition, biogenesis and function, Nat Rev Immunol, 2 (2002) 569–579. [DOI] [PubMed] [Google Scholar]
- [585].Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, de Sousa M, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J, Lyden D, Tumour exosome integrins determine organotropic metastasis, Nature, 527 (2015) 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [586].Wong CH, Chen YC, Clinical significance of exosomes as potential biomarkers in cancer, World J Clin Cases, 7 (2019) 171–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [587].Faict S, Oudaert I, D’Auria L, Dehairs J, Maes K, Vlummens P, De Veirman K, De Bruyne E, Fostier K, Vande Broek I, Schots R, Vanderkerken K, Swinnen JV, Menu E, The Transfer of Sphingomyelinase Contributes to Drug Resistance in Multiple Myeloma, Cancers (Basel), 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [588].Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, Xia H, Zhou J, Li G, Li J, Li W, Wei S, Vatan L, Zhang H, Szeliga W, Gu W, Liu R, Lawrence TS, Lamb C, Tanno Y, Cieslik M, Stone E, Georgiou G, Chan TA, Chinnaiyan A, Zou W, CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy, Nature, 569 (2019) 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [589].Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, Ulas T, Papantonopoulou O, Van Eck M, Auphan-Anezin N, Bebien M, Verthuy C, Vu Manh TP, Turner M, Dalod M, Schultze JL, Lawrence T, Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression, Cell Metab, 29 (2019) 1376–1389 e1374. [DOI] [PubMed] [Google Scholar]
- [590].Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, Ricciotti E, DiRusso C, Murphy ME, Vonderheide RH, Lieberman PM, Mulligan C, Nam B, Hockstein N, Masters G, Guarino M, Lin C, Nefedova Y, Black P, Kagan VE, Gabrilovich DI, Fatty acid transport protein 2 reprograms neutrophils in cancer, Nature, 569 (2019) 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [591].Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, Schuchter LM, Xu W, Amaravadi R, Xiao M, Sadek N, Krepler C, Herlyn M, Freeman GJ, Rabinowitz JD, Ertl HCJ, Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy, Cancer cell, 32 (2017) 377–391 e379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [592].Harel M, Ortenberg R, Varanasi SK, Mangalhara KC, Mardamshina M, Markovits E, Baruch EN, Tripple V, Arama-Chayoth M, Greenberg E, Shenoy A, Ayasun R, Knafo N, Xu S, Anafi L, Yanovich-Arad G, Barnabas GD, Ashkenazi S, Besser MJ, Schachter J, Bosenberg M, Shadel GS, Barshack I, Kaech SM, Markel G, Geiger T, Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence, Cell, 179 (2019) 236–250 e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [593].Wang F, Beck-Garcia K, Zorzin C, Schamel WW, Davis MM, Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol, Nat Immunol, 17 (2016) 844–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [594].Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, Marbois BN, Komisopoulou E, Wilson EB, Osborne TF, Graeber TG, Reue K, Brooks DG, Bensinger SJ, Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity, Nat Immunol, 14 (2013) 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [595].Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, Barcinski M, Brekken RA, Huang X, Hutchins JT, Freimark B, Empig C, Mercer J, Schroit AJ, Schett G, Herrmann M, Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer, Cell death and differentiation, 23 (2016) 962–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [596].Kachler K, Bailer M, Heim L, Schumacher F, Reichel M, Holzinger CD, Trump S, Mittler S, Monti J, Trufa DI, Rieker RJ, Hartmann A, Sirbu H, Kleuser B, Kornhuber J, Finotto S, Enhanced Acid Sphingomyelinase Activity Drives Immune Evasion and Tumor Growth in Non-Small Cell Lung Carcinoma, Cancer Res, 77 (2017) 5963–5976. [DOI] [PubMed] [Google Scholar]
- [597].Perrotti F, Rosa C, Cicalini I, Sacchetta P, Del Boccio P, Genovesi D, Pieragostino D, Advances in Lipidomics for Cancer Biomarkers Discovery, Int J Mol Sci, 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [598].Bandu R, Mok HJ, Kim KP, Phospholipids as cancer biomarkers: Mass spectrometry-based analysis, Mass Spectrom Rev, 37 (2018) 107–138. [DOI] [PubMed] [Google Scholar]
- [599].Wang Y, Kuhajda FP, Sokoll LJ, Chan DW, Two-site ELISA for the quantitative determination of fatty acid synthase, Clinica chimica acta; international journal of clinical chemistry, 304 (2001) 107–115. [DOI] [PubMed] [Google Scholar]
- [600].Vatten LJ, Bjerve KS, Andersen A, Jellum E, Polyunsaturated fatty acids in serum phospholipids and risk of breast cancer: a case-control study from the Janus serum bank in Norway, Eur J Cancer, 29A (1993) 532–538. [DOI] [PubMed] [Google Scholar]
- [601].Lv W, Yang T, Identification of possible biomarkers for breast cancer from free fatty acid profiles determined by GC-MS and multivariate statistical analysis, Clin Biochem, 45 (2012) 127–133. [DOI] [PubMed] [Google Scholar]
- [602].Crotti S, Agnoletto E, Cancemi G, Di Marco V, Traldi P, Pucciarelli S, Nitti D, Agostini M, Altered plasma levels of decanoic acid in colorectal cancer as a new diagnostic biomarker, Anal Bioanal Chem, 408 (2016) 6321–6328. [DOI] [PubMed] [Google Scholar]
- [603].Dahm CC, Gorst-Rasmussen A, Crowe FL, Roswall N, Tjonneland A, Drogan D, Boeing H, Teucher B, Kaaks R, Adarakis G, Zylis D, Trichopoulou A, Fedirko V, Chajes V, Jenab M, Palli D, Pala V, Tumino R, Ricceri F, van Kranen H, Bueno-de-Mesquita HB, Quiros JR, Sanchez MJ, Lujan-Barroso L, Larranaga N, Chirlaque MD, Ardanaz E, Johansson M, Stattin P, Khaw KT, Wareham N, Wark PA, Norat T, Riboli E, Key TJ, Overvad K, Fatty acid patterns and risk of prostate cancer in a case-control study nested within the European Prospective Investigation into Cancer and Nutrition, Am J Clin Nutr, 96 (2012) 1354–1361. [DOI] [PubMed] [Google Scholar]
- [604].Alberg AJ, Armeson K, Pierce JS, Bielawski J, Bielawska A, Visvanathan K, Hill EG, Ogretmen B, Plasma sphingolipids and lung cancer: a population-based, nested case-control study, Cancer Epidemiol Biomarkers Prev, 22 (2013) 1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [605].Crowe FL, Allen NE, Appleby PN, Overvad K, Aardestrup IV, Johnsen NF, Tjonneland A, Linseisen J, Kaaks R, Boeing H, Kroger J, Trichopoulou A, Zavitsanou A, Trichopoulos D, Sacerdote C, Palli D, Tumino R, Agnoli C, Kiemeney LA, Bueno-de-Mesquita HB, Chirlaque MD, Ardanaz E, Larranaga N, Quiros JR, Sanchez MJ, Gonzalez CA, Stattin P, Hallmans G, Bingham S, Khaw KT, Rinaldi S, Slimani N, Jenab M, Riboli E, Key TJ, Fatty acid composition of plasma phospholipids and risk of prostate cancer in a case-control analysis nested within the European Prospective Investigation into Cancer and Nutrition, Am J Clin Nutr, 88 (2008) 1353–1363. [DOI] [PubMed] [Google Scholar]
- [606].Crowe FL, Appleby PN, Travis RC, Barnett M, Brasky TM, Bueno-de-Mesquita HB, Chajes V, Chavarro JE, Chirlaque MD, English DR, Gibson RA, Giles GG, Goodman GE, Henning SM, Kaaks R, King IB, Kolonel LN, Kristal AR, Neuhouser ML, Park SY, Severi G, Siddiq A, Stampfer MJ, Stattin P, Tangen CM, Tjonneland A, Trichopoulos D, Tumino R, Wilkens LR, Key TJ, Allen NE, Endogenous Hormones NB, Prostate Cancer Collaborative G, Circulating fatty acids and prostate cancer risk: individual participant meta-analysis of prospective studies, J Natl Cancer Inst, 106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [607].Schmidt JA, Fensom GK, Rinaldi S, Scalbert A, Appleby PN, Achaintre D, Gicquiau A, Gunter MJ, Ferrari P, Kaaks R, Kuhn T, Boeing H, Trichopoulou A, Karakatsani A, Peppa E, Palli D, Sieri S, Tumino R, Bueno-de-Mesquita B, Agudo A, Sanchez MJ, Chirlaque MD, Ardanaz E, Larranaga N, Perez-Cornago A, Assi N, Riboli E, Tsilidis KK, Key TJ, Travis RC, Patterns in metabolite profile are associated with risk of more aggressive prostate cancer: A prospective study of 3,057 matched case-control sets from EPIC, Int J Cancer, 146 (2020) 720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [608].Saito K, Ikeda M, Kojima Y, Hosoi H, Saito Y, Kondo S, Lipid profiling of pre-treatment plasma reveals biomarker candidates associated with response rates and hand-foot skin reactions in sorafenib-treated patients, Cancer Chemother Pharmacol, 82 (2018) 677–684. [DOI] [PubMed] [Google Scholar]
- [609].Li J, Xie H, Li A, Cheng J, Yang K, Wang J, Wang W, Zhang F, Li Z, Dhillon HS, Openkova MS, Zhou X, Li K, Hou Y, Distinct plasma lipids profiles of recurrent ovarian cancer by liquid chromatography-mass spectrometry, Oncotarget, 8 (2017) 46834–46845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [610].Murphy RA, Bureyko TF, Mourtzakis M, Chu QS, Clandinin MT, Reiman T, Mazurak VC, Aberrations in plasma phospholipid fatty acids in lung cancer patients, Lipids, 47 (2012) 363–369. [DOI] [PubMed] [Google Scholar]
- [611].Lin HM, Mahon KL, Weir JM, Mundra PA, Spielman C, Briscoe K, Gurney H, Mallesara G, Marx G, Stockler MR, Consortium PR, Parton RG, Hoy AJ, Daly RJ, Meikle PJ, Horvath LG, A distinct plasma lipid signature associated with poor prognosis in castration-resistant prostate cancer, Int J Cancer, 141 (2017) 2112–2120. [DOI] [PubMed] [Google Scholar]
- [612].Burla B, Arita M, Arita M, Bendt AK, Cazenave-Gassiot A, Dennis EA, Ekroos K, Han X, Ikeda K, Liebisch G, Lin MK, Loh TP, Meikle PJ, Oresic M, Quehenberger O, Shevchenko A, Torta F, Wakelam MJO, Wheelock CE, Wenk MR, MS-based lipidomics of human blood plasma: a community-initiated position paper to develop accepted guidelines, J Lipid Res, 59 (2018) 2001–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [613].Kim H, Min HK, Kong G, Moon MH, Quantitative analysis of phosphatidylcholines and phosphatidylethanolamines in urine of patients with breast cancer by nanoflow liquid chromatography/tandem mass spectrometry, Anal Bioanal Chem, 393 (2009) 1649–1656. [DOI] [PubMed] [Google Scholar]
- [614].Min HK, Lim S, Chung BC, Moon MH, Shotgun lipidomics for candidate biomarkers of urinary phospholipids in prostate cancer, Anal Bioanal Chem, 399 (2011) 823–830. [DOI] [PubMed] [Google Scholar]
- [615].Record M, Silvente-Poirot S, Poirot M, Wakelam MJO, Extracellular vesicles: lipids as key components of their biogenesis and functions, J Lipid Res, 59 (2018) 1316–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [616].Sorvina A, Bader CA, Caporale C, Carter EA, Johnson IRD, Parkinson-Lawrence EJ, Simpson PV, Wright PJ, Stagni S, Lay PA, Massi M, Brooks DA, Plush SE, Lipid profiles of prostate cancer cells, Oncotarget, 9 (2018) 35541–35552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [617].Burch TC, Isaac G, Booher CL, Rhim JS, Rainville P, Langridge J, Baker A, Nyalwidhe JO, Comparative Metabolomic and Lipidomic Analysis of Phenotype Stratified Prostate Cells, PLoS One, 10 (2015) e0134206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [618].Kim HY, Lee KM, Kim SH, Kwon YJ, Chun YJ, Choi HK, Comparative metabolic and lipidomic profiling of human breast cancer cells with different metastatic potentials, Oncotarget, 7 (2016) 67111–67128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [619].Kim HY, Lee H, Kim SH, Jin H, Bae J, Choi HK, Discovery of potential biomarkers in human melanoma cells with different metastatic potential by metabolic and lipidomic profiling, Sci Rep, 7 (2017) 8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [620].Roy J, Dibaeinia P, Fan TM, Sinha S, Das A, Global analysis of osteosarcoma lipidomes reveal altered lipid profiles in metastatic versus nonmetastatic cells, J Lipid Res, 60 (2019) 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [621].Jones DT, Valli A, Haider S, Zhang Q, Smethurst EA, Schug ZT, Peck B, Aboagye EO, Critchlow SE, Schulze A, Gottlieb E, Wakelam MJO, Harris AL, 3D Growth of Cancer Cells Elicits Sensitivity to Kinase Inhibitors but Not Lipid Metabolism Modifiers, Mol Cancer Ther, 18 (2019) 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [622].Wang Y, Hinz S, Uckermann O, Honscheid P, von Schonfels W, Burmeister G, Hendricks A, Ackerman JM, Baretton GB, Hampe J, Brosch M, Schafmayer C, Shevchenko A, Zeissig S, Shotgun lipidomics-based characterization of the landscape of lipid metabolism in colorectal cancer, Biochim Biophys Acta Mol Cell Biol Lipids, 1865 (2020) 158579. [DOI] [PubMed] [Google Scholar]
- [623].Mika A, Pakiet A, Czumaj A, Kaczynski Z, Liakh I, Kobiela J, Perdyan A, Adrych K, Makarewicz W, Sledzinski T, Decreased Triacylglycerol Content and Elevated Contents of Cell Membrane Lipids in Colorectal Cancer Tissue: A Lipidomic Study, J Clin Med, 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [624].Nagai K, Uranbileg B, Chen Z, Fujioka A, Yamazaki T, Matsumoto Y, Tsukamoto H, Ikeda H, Yatomi Y, Chiba H, Hui SP, Nakazawa T, Saito R, Koshiba S, Aoki J, Saigusa D, Tomioka Y, Identification of novel biomarkers of hepatocellular carcinoma by high-definition mass spectrometry: Ultrahigh-performance liquid chromatography quadrupole time-of-flight mass spectrometry and desorption electrospray ionization mass spectrometry imaging, Rapid Commun Mass Spectrom, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [625].Budhu A, Roessler S, Zhao X, Yu Z, Forgues M, Ji J, Karoly E, Qin LX, Ye QH, Jia HL, Fan J, Sun HC, Tang ZY, Wang XW, Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes, Gastroenterology, 144 (2013) 1066–1075 e1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [626].Hanel L, Kwiatkowski M, Heikaus L, Schluter H, Mass spectrometry-based intraoperative tumor diagnostics, Future Sci OA, 5 (2019) FSO373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [627].Patterson NH, Alabdulkarim B, Lazaris A, Thomas A, Marcinkiewicz MM, Gao ZH, Vermeulen PB, Chaurand P, Metrakos P, Assessment of pathological response to therapy using lipid mass spectrometry imaging, Sci Rep, 6 (2016) 36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [628].Batubara A, Carolan VA, Loadman PM, Sutton C, Shnyder SD, Clench MR, Thin-layer chromatography/matrix-assisted laser desorption/ionisation mass spectrometry and matrix-assisted laser desorption/ionisation mass spectrometry imaging for the analysis of phospholipids in LS174T colorectal adenocarcinoma xenografts treated with the vascular disrupting agent DMXAA, Rapid Commun Mass Spectrom, 29 (2015) 1288–1296. [DOI] [PubMed] [Google Scholar]
- [629].Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Arauzo-Bravo MJ, Kovacs WJ, Karalay O, Suter U, Machado RA, Roccio M, Lutolf MP, Semenkovich CF, Jessberger S, Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis, Nature, 493 (2013) 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [630].Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP, Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors, Science, 288 (2000) 2379–2381. [DOI] [PubMed] [Google Scholar]
- [631].Ventura R, Mordec K, Waszczuk J, Wang Z, Lai J, Fridlib M, Buckley D, Kemble G, Heuer TS, Inhibition of de novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression, EBioMedicine, 2 (2015) 808–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [632].Zaytseva YY, Rychahou PG, Le AT, Scott TL, Flight RM, Kim JT, Harris J, Liu J, Wang C, Morris AJ, Sivakumaran TA, Fan T, Moseley H, Gao T, Lee EY, Weiss HL, Heuer TS, Kemble G, Evers M, Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer, Oncotarget, 9 (2018) 24787–24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [633].Harriman G, Greenwood J, Bhat S, Huang X, Wang R, Paul D, Tong L, Saha AK, Westlin WF, Kapeller R, Harwood HJ Jr., Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats, Proceedings of the National Academy of Sciences of the United States of America, 113 (2016) E1796–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [634].Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, Gerken L, Greenwood J, Bhat S, Harriman G, Westlin WF, Harwood HJ Jr., Saghatelian A, Kapeller R, Metallo CM, Shaw RJ, Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models, Nature medicine, 22 (2016) 1108–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [635].Mallik R, Chowdhury TA, Metformin in cancer, Diabetes Res Clin Pract, 143 (2018) 409–419. [DOI] [PubMed] [Google Scholar]
- [636].Farooqi MAM, Malhotra N, Mukherjee SD, Sanger S, Dhesy-Thind SK, Ellis P, Leong DP, Statin therapy in the treatment of active cancer: A systematic review and meta-analysis of randomized controlled trials, PLoS One, 13 (2018) e0209486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [637].Emilsson L, Garcia-Albeniz X, Logan RW, Caniglia EC, Kalager M, Hernan MA, Examining Bias in Studies of Statin Treatment and Survival in Patients With Cancer, JAMA Oncol, 4 (2018) 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [638].Jeong GH, Lee KH, Kim JY, Eisenhut M, Kronbichler A, van der Vliet HJ, Hong SH, Shin JI, Gamerith G, Effect of Statin on Cancer Incidence: An Umbrella Systematic Review and Meta-Analysis, J Clin Med, 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [639].Jeong GH, Lee KH, Kim JY, Eisenhut M, Kronbichler A, van der Vliet HJ, Shin JI, Gamerith G, Statin and Cancer Mortality and Survival: An Umbrella Systematic Review and Meta-Analysis, J Clin Med, 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [640].Astanina K, Koch M, Jungst C, Zumbusch A, Kiemer AK, Lipid droplets as a novel cargo of tunnelling nanotubes in endothelial cells, Sci Rep, 5 (2015) 11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [641].Wang J, Li Y, CD36 tango in cancer: signaling pathways and functions, Theranostics, 9 (2019) 4893–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [642].Carbonetti G, Converso C, Clement T, Wang C, Trotman LC, Ojima I, Kaczocha M, Docetaxel/cabazitaxel and fatty acid binding protein 5 inhibitors produce synergistic inhibition of prostate cancer growth, Prostate, 80 (2020) 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [643].Rao E, Singh P, Zhai X, Li Y, Zhu G, Zhang Y, Hao J, Chi YI, Brown RE, Cleary MP, Li B, Inhibition of tumor growth by a newly-identified activator for epidermal fatty acid binding protein, Oncotarget, 6 (2015) 7815–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [644].Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, Shinohara T, Kawazoe Y, Sato S.-i., Asakura K, Choo H-YP, Sakai J, Wakil SJ, Uesugi M, A Small Molecule That Blocks Fat Synthesis By Inhibiting the Activation of SREBP, Chemistry & Biology, 16 (2009) 882–892. [DOI] [PubMed] [Google Scholar]
- [645].Shao W, Machamer CE, Espenshade PJ, Fatostatin blocks ER exit of SCAP but inhibits cell growth in a SCAP-independent manner, J Lipid Res, 57 (2016) 1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [646].Gholkar AA, Cheung K, Williams KJ, Lo YC, Hamideh SA, Nnebe C, Khuu C, Bensinger SJ, Torres JZ, Fatostatin Inhibits Cancer Cell Proliferation by Affecting Mitotic Microtubule Spindle Assembly and Cell Division, J Biol Chem, 291 (2016) 17001–17008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [647].Brovkovych V, Izhar Y, Danes JM, Dubrovskyi O, Sakallioglu IT, Morrow LM, Atilla-Gokcumen GE, Frasor J, Fatostatin induces pro- and anti-apoptotic lipid accumulation in breast cancer, Oncogenesis, 7 (2018) 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [648].Li X, Chen Y-T, Hu P, Huang W-C, Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling, Molecular Cancer Therapeutics, 13 (2014) 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [649].Siqingaowa, Sekar S, Gopalakrishnan V, Taghibiglou C, Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation, Biochem Biophys Res Commun, 488 (2017) 136–140. [DOI] [PubMed] [Google Scholar]
- [650].Tang J, Li J, Qi W, Qiu W, Li P, Li B, Song B, Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques., Cell Metab, 13 (2011) 44–56. [DOI] [PubMed] [Google Scholar]
- [651].Li N, Zhou ZS, Shen Y, Xu J, Miao HH, Xiong Y, Xu F, Li BL, Luo J, Song BL, Inhibition of the sterol regulatory element-binding protein pathway suppresses hepatocellular carcinoma by repressing inflammation in mice, Hepatology (Baltimore, Md.), 65 (2017) 1936–1947. [DOI] [PubMed] [Google Scholar]
- [652].Guan M, Fousek K, Jiang C, Guo S, Synold T, Xi B, Shih CC, Chow WA, Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6, Clinical cancer research : an official journal of the American Association for Cancer Research, 17 (2011) 1796–1806. [DOI] [PubMed] [Google Scholar]
- [653].Guan M, Su L, Yuan YC, Li H, Chow WA, Nelfinavir and nelfinavir analogs block site-2 protease cleavage to inhibit castration-resistant prostate cancer, Sci Rep, 5 (2015) 9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [654].Nambiar DK, Deep G, Singh RP, Agarwal C, Agarwal R, Silibinin inhibits aberrant lipid metabolism, proliferation and emergence of androgen-independence in prostate cancer cells via primarily targeting the sterol response element binding protein 1, Oncotarget, 5 (2014) 10017–10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [655].Dankner R, Roth J, More recent, better designed studies have weakened links between antidiabetes medications and cancer risk, Diabet Med, 37 (2020) 194–202. [DOI] [PubMed] [Google Scholar]
- [656].Arrieta O, Barron F, Padilla MS, Aviles-Salas A, Ramirez-Tirado LA, Arguelles Jimenez MJ, Vergara E, Zatarain-Barron ZL, Hernandez-Pedro N, Cardona AF, Cruz-Rico G, Barrios-Bernal P, Yamamoto Ramos M, Rosell R, Effect of Metformin Plus Tyrosine Kinase Inhibitors Compared With Tyrosine Kinase Inhibitors Alone in Patients With Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: A Phase 2 Randomized Clinical Trial, JAMA Oncol, (2019) e192553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [657].Li L, Jiang L, Wang Y, Zhao Y, Zhang XJ, Wu G, Zhou X, Sun J, Bai J, Ren B, Tian K, Xu Z, Xiao HL, Zhou Q, Han R, Chen H, Wang H, Yang Z, Gao C, Cai S, He Y, Combination of Metformin and Gefitinib as First-Line Therapy for Nondiabetic Advanced NSCLC Patients with EGFR Mutations: A Randomized, Double-Blind Phase II Trial, Clin Cancer Res, 25 (2019) 6967–6975. [DOI] [PubMed] [Google Scholar]
- [658].Nanni O, Amadori D, De Censi A, Rocca A, Freschi A, Bologna A, Gianni L, Rosetti F, Amaducci L, Cavanna L, Foca F, Sarti S, Serra P, Valmorri L, Bruzzi P, Corradengo D, Gennari A, investigators M, Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial, Breast Cancer Res Treat, 174 (2019) 433–442. [DOI] [PubMed] [Google Scholar]
- [659].Zhao Y, Gong C, Wang Z, Zhang J, Wang L, Zhang S, Cao J, Tao Z, Li T, Wang B, Hu X, A randomized phase II study of aromatase inhibitors plus metformin in pre-treated postmenopausal patients with hormone receptor positive metastatic breast cancer, Oncotarget, 8 (2017) 84224–84236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [660].Qamri Z, Preet A, Nasser MW, Bass CE, Leone G, Barsky SH, Ganju RK, Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer, Mol Cancer Ther, 8 (2009) 3117–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [661].Caffarel MM, Andradas C, Mira E, Perez-Gomez E, Cerutti C, Moreno-Bueno G, Flores JM, Garcia-Real I, Palacios J, Manes S, Guzman M, Sanchez C, Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition, Mol Cancer, 9 (2010) 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [662].Blazquez C, Salazar M, Carracedo A, Lorente M, Egia A, Gonzalez-Feria L, Haro A, Velasco G, Guzman M, Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression, Cancer Res, 68 (2008) 1945–1952. [DOI] [PubMed] [Google Scholar]
- [663].Hasko J, Fazakas C, Molnar J, Nyul-Toth A, Herman H, Hermenean A, Wilhelm I, Persidsky Y, Krizbai IA, CB2 receptor activation inhibits melanoma cell transmigration through the blood-brain barrier, Int J Mol Sci, 15 (2014) 8063–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [664].Martinez-Martinez E, Martin-Ruiz A, Martin P, Calvo V, Provencio M, Garcia JM, CB2 cannabinoid receptor activation promotes colon cancer progression via AKT/GSK3beta signaling pathway, Oncotarget, 7 (2016) 68781–68791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [665].Khan MI, Sobocinska AA, Brodaczewska KK, Zielniok K, Gajewska M, Kieda C, Czarnecka AM, Szczylik C, Involvement of the CB2 cannabinoid receptor in cell growth inhibition and G0/G1 cell cycle arrest via the cannabinoid agonist WIN 55,212–2 in renal cell carcinoma, BMC Cancer, 18 (2018) 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [666].Lin C-Y, Gustafsson J-Å, Targeting liver X receptors in cancer therapeutics, Nature Reviews Cancer, 15 (2015) 216–224. [DOI] [PubMed] [Google Scholar]
- [667].Grommes C, Landreth GE, Heneka MT, Antineoplastic effects of peroxisome proliferatoractivated receptor γ agonists, The Lancet Oncology, 5 (2004) 419–429. [DOI] [PubMed] [Google Scholar]
- [668].Nagase T, Takahashi T, Sasaki T, Nagumo A, Shimamura K, Miyamoto Y, Kitazawa H, Kanesaka M, Yoshimoto R, Aragane K, Tokita S, Sato N, Synthesis and biological evaluation of a novel 3-sulfonyl-8-azabicyclo[3.2.1]octane class of long chain fatty acid elongase 6 (ELOVL6) inhibitors, J Med Chem, 52 (2009) 4111–4114. [DOI] [PubMed] [Google Scholar]
- [669].Sasaki T, Nagase T, Takahashi T, Nagumo A, Shimamura K, Miyamoto Y, Kitazawa H, Kanesaka M, Yoshimoto R, Aragane K, Tokita S, Sato N, Synthesis and evaluation of a novel 2-azabicyclo[2.2.2]octane class of long chain fatty acid elongase 6 (ELOVL6) inhibitors, Bioorg Med Chem, 17 (2009) 5639–5647. [DOI] [PubMed] [Google Scholar]
- [670].Shimamura K, Nagumo A, Miyamoto Y, Kitazawa H, Kanesaka M, Yoshimoto R, Aragane K, Morita N, Ohe T, Takahashi T, Nagase T, Sato N, Tokita S, Discovery and characterization of a novel potent, selective and orally active inhibitor for mammalian ELOVL6, Eur J Pharmacol, 630 (2010) 34–41. [DOI] [PubMed] [Google Scholar]
- [671].Shimamura K, Takahashi H, Kitazawa H, Miyamoto Y, Nagumo A, Tang C, Dean D, Nagase T, Sato N, Tokita S, Identification and characterization of a selective radioligand for ELOVL6, J Biochem, 146 (2009) 429–437. [DOI] [PubMed] [Google Scholar]
- [672].Takahashi T, Nagase T, Sasaki T, Nagumo A, Shimamura K, Miyamoto Y, Kitazawa H, Kanesaka M, Yoshimoto R, Aragane K, Tokita S, Sato N, Synthesis and evaluation of a novel indoledione class of long chain fatty acid elongase 6 (ELOVL6) inhibitors, J Med Chem, 52 (2009) 3142–3145. [DOI] [PubMed] [Google Scholar]
- [673].Schackmann MJ, Ofman R, Dijkstra IM, Wanders RJ, Kemp S, Enzymatic characterization of ELOVL1, a key enzyme in very long-chain fatty acid synthesis, Biochim Biophys Acta, 1851 (2015) 231–237. [DOI] [PubMed] [Google Scholar]
- [674].Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L, Cimic A, Sirintrapun SJ, Glode LM, Eckel RH, Cramer SD, Lipid catabolism via CPT1 as a therapeutic target for prostate cancer, Mol Cancer Ther, 13 (2014) 2361–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [675].Lin H, Patel S, Affleck VS, Wilson I, Turnbull DM, Joshi AR, Maxwell R, Stoll EA, Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells, Neuro Oncol, 19 (2017) 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [676].Cheng S, Wang G, Wang Y, Cai L, Qian K, Ju L, Liu X, Xiao Y, Wang X, Fatty acid oxidation inhibitor etomoxir suppresses tumor progression and induces cell cycle arrest via PPARgamma-mediated pathway in bladder cancer, Clin Sci (Lond), 133 (2019) 1745–1758. [DOI] [PubMed] [Google Scholar]
- [677].Itkonen HM, Brown M, Urbanucci A, Tredwell G, Ho Lau C, Barfeld S, Hart C, Guldvik IJ, Takhar M, Heemers HV, Erho N, Bloch K, Davicioni E, Derua R, Waelkens E, Mohler JL, Clarke N, Swinnen JV, Keun HC, Rekvig OP, Mills IG, Lipid degradation promotes prostate cancer cell survival, Oncotarget, 8 (2017) 38264–38275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [678].Ren XR, Wang J, Osada T, Mook RA Jr., Morse MA, Barak LS, Lyerly HK, Chen W, Perhexiline promotes HER3 ablation through receptor internalization and inhibits tumor growth, Breast Cancer Res, 17 (2015) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [679].Zhu J, Wu G, Song L, Cao L, Tan Z, Tang M, Li Z, Shi D, Zhang S, Li J, NKX2–8 deletion-induced reprogramming of fatty acid metabolism confers chemoresistance in epithelial ovarian cancer, EBioMedicine, 43 (2019) 238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [680].Tan Z, Xiao L, Tang M, Bai F, Li J, Li L, Shi F, Li N, Li Y, Du Q, Lu J, Weng X, Yi W, Zhang H, Fan J, Zhou J, Gao Q, Onuchic JN, Bode AM, Luo X, Cao Y, Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy, Theranostics, 8 (2018) 2329–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [681].Dheeraj A, Agarwal C, Schlaepfer IR, Raben D, Singh R, Agarwal R, Deep G, A novel approach to target hypoxic cancer cells via combining beta-oxidation inhibitor etomoxir with radiation, Hypoxia (Auckl), 6 (2018) 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [682].Flaig TW, Salzmann-Sullivan M, Su LJ, Zhang Z, Joshi M, Gijon MA, Kim J, Arcaroli JJ, Van Bokhoven A, Lucia MS, La Rosa FG, Schlaepfer IR, Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade, Oncotarget, 8 (2017) 56051–56065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [683].Yao CH, Liu GY, Wang R, Moon SH, Gross RW, Patti GJ, Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of beta-oxidation, PLoS Biol, 16 (2018) e2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [684].Hernandez-Alcoceba R, Fernandez F, Lacal JC, In vivo antitumor activity of choline kinase inhibitors: a novel target for anticancer drug discovery, Cancer Res, 59 (1999) 3112–3118. [PubMed] [Google Scholar]
- [685].Estevez-Braun A, Ravelo AG, Perez-Sacau E, Lacal JC, A new family of choline kinase inhibitors with antiproliferative and antitumor activity derived from natural products, Clin Transl Oncol, 17 (2015) 74–84. [DOI] [PubMed] [Google Scholar]
- [686].Mariotto E, Bortolozzi R, Volpin I, Carta D, Serafin V, Accordi B, Basso G, Navarro PL, Lopez-Cara LC, Viola G, EB-3D a novel choline kinase inhibitor induces deregulation of the AMPK-mTOR pathway and apoptosis in leukemia T-cells, Biochem Pharmacol, 155 (2018) 213–223. [DOI] [PubMed] [Google Scholar]
- [687].Mariotto E, Viola G, Ronca R, Persano L, Aveic S, Bhujwalla ZM, Mori N, Accordi B, Serafin V, Lopez-Cara LC, Bortolozzi R, Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer, Cancers (Basel), 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [688].Sola-Leyva A, Lopez-Cara LC, Rios-Marco P, Rios A, Marco C, Carrasco-Jimenez MP, Choline kinase inhibitors EB-3D and EB-3P interferes with lipid homeostasis in HepG2 cells, Sci Rep, 9 (2019) 5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [689].Xu T, Huang C, Qi XT, Yang XC, Zhang N, Cao J, Wang C, Zhu H, Yang B, He QJ, Shao XJ, Ying MD, 2-Bromopalmitate sensitizes osteosarcoma cells to adriamycin-induced apoptosis via the modulation of CHOP, Eur J Pharmacol, 844 (2019) 204–215. [DOI] [PubMed] [Google Scholar]
- [690].Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, Lu H, Fang C, Zhang Y, Liang L, Zhou X, Wang C, Xue Y, Cui Y, Xu J, Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours, Nat Biomed Eng, 3 (2019) 306–317. [DOI] [PubMed] [Google Scholar]
- [691].Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, Wei Y, Xia W, Hou J, Qiu Y, Hung MC, Palmitoylation stabilizes PD-L1 to promote breast tumor growth, Cell Res, 29 (2019) 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [692].Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, Erstad DJ, Fujiwara N, Leong V, Houde VP, Anagnostopoulos AE, Wang A, Broadfield LA, Ford RJ, Foster RA, Bates J, Sun H, Wang T, Liu H, Ray AS, Saha AK, Greenwood J, Bhat S, Harriman G, Miao W, Rocnik JL, Westlin WF, Muti P, Tsakiridis T, Harwood HJ Jr., Kapeller R, Hoshida Y, Tanabe KK, Steinberg GR, Fuchs BC, Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma, Cell Metab, 29 (2019) 174–182 e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [693].Vazquez-Martin A, Colomer R, Brunet J, Menendez JA, Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells, Int J Oncol, 31 (2007) 769–776. [PubMed] [Google Scholar]
- [694].Li J, Yan H, Zhao L, Jia W, Yang H, Liu L, Zhou X, Miao P, Sun X, Song S, Zhao X, Liu J, Huang G, Inhibition of SREBP increases gefitinib sensitivity in non-small cell lung cancer cells, Oncotarget, 7 (2016) 52392–52403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [695].Menendez JA, Lupu R, Fatty acid synthase regulates estrogen receptor-alpha signaling in breast cancer cells, Oncogenesis, 6 (2017) e299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [696].Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, Andreeff M, Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction, The Journal of Clinical Investigation, 120 (2010) 142–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [697].Aloia A, Mullhaupt D, Chabbert CD, Eberhart T, Fluckiger-Mangual S, Vukolic A, Eichhoff O, Irmisch A, Alexander LT, Scibona E, Frederick DT, Miao B, Tian T, Cheng C, Kwong LN, Wei Z, Sullivan RJ, Boland GM, Herlyn M, Flaherty KT, Zamboni N, Dummer R, Zhang G, Levesque MP, Krek W, Kovacs WJ, A Fatty Acid Oxidation-dependent Metabolic Shift Regulates the Adaptation of BRAF-mutated Melanoma to MAPK Inhibitors, Clinical cancer research : an official journal of the American Association for Cancer Research, 25 (2019) 6852–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [698].Alwarawrah Y, Hughes P, Loiselle D, Carlson DA, Darr DB, Jordan JL, Xiong J, Hunter LM, Dubois LG, Thompson JW, Kulkarni MM, Ratcliff AN, Kwiek JJ, Haystead TAJ, Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer, Cell Chemical Biology, 23 (2016) 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [699].Wu S, Naar AM, SREBP1-dependent de novo fatty acid synthesis gene expression is elevated in malignant melanoma and represents a cellular survival trait, Sci Rep, 9 (2019) 10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [700].Chuang HY, Lee YP, Lin WC, Lin YH, Hwang JJ, Fatty Acid Inhibition Sensitizes Androgen-Dependent and -Independent Prostate Cancer to Radiotherapy via FASN/NF-kappaB Pathway, Sci Rep, 9 (2019) 13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [701].Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, Dovas A, Higgins DM, Tan H, Zhang Y, Buonanno M, Wang TJC, Hei TK, Bruce JN, Canoll PD, Cheng SK, Stockwell BR, Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers, ACS chemical biology, 15 (2020) 469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [702].Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, Ajani JA, Xiao Q, Liao Z, Wang H, Gan B, The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression, Cell research, 30 (2020) 146–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [703].Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, Liao P, Zhou J, Zhang Q, Dow A, Saripalli AL, Kryczek I, Wei S, Szeliga W, Vatan L, Stone EM, Georgiou G, Cieslik M, Wahl DR, Morgan MA, Chinnaiyan AM, Lawrence TS, Zou W, Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11, Cancer discovery, 9 (2019) 1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [704].Xia S, Lin R, Jin L, Zhao L, Kang HB, Pan Y, Liu S, Qian G, Qian Z, Konstantakou E, Zhang B, Dong JT, Chung YR, Abdel-Wahab O, Merghoub T, Zhou L, Kudchadkar RR, Lawson DH, Khoury HJ, Khuri FR, Boise LH, Lonial S, Lee BH, Pollack BP, Arbiser JL, Fan J, Lei QY, Chen J, Prevention of Dietary-Fat-Fueled Ketogenesis Attenuates BRAF V600E Tumor Growth, Cell Metab, 25 (2017) 358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [705].Malvi P, Chaube B, Singh SV, Mohammad N, Pandey V, Vijayakumar MV, Radhakrishnan RM, Vanuopadath M, Nair SS, Nair BG, Bhat MK, Weight control interventions improve therapeutic efficacy of dacarbazine in melanoma by reversing obesity-induced drug resistance, Cancer & metabolism, 4 (2016) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [706].Martuscello RT, Vedam-Mai V, McCarthy DJ, Schmoll ME, Jundi MA, Louviere CD, Griffith BG, Skinner CL, Suslov O, Deleyrolle LP, Reynolds BA, A Supplemented High-Fat Low-Carbohydrate Diet for the Treatment of Glioblastoma, Clinical cancer research : an official journal of the American Association for Cancer Research, 22 (2016) 2482–2495. [DOI] [PubMed] [Google Scholar]
- [707].Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR, Bareja R, Ma Y, Harris EM, Sboner A, Beltran H, Rubin MA, Mukherjee S, Cantley LC, Suppression of insulin feedback enhances the efficacy of PI3K inhibitors, Nature, 560 (2018) 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [708].Mukherjee P, Augur ZM, Li M, Hill C, Greenwood B, Domin MA, Kondakci G, Narain NR, Kiebish MA, Bronson RT, Arismendi-Morillo G, Chinopoulos C, Seyfried TN, Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma, Commun Biol, 2 (2019) 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [709].Filik L, Ozyilkan O, Olive-oil consumption and cancer risk, European journal of clinical nutrition, 57 (2003) 191. [DOI] [PubMed] [Google Scholar]
- [710].Psaltopoulou T, Kosti RI, Haidopoulos D, Dimopoulos M, Panagiotakos DB, Olive oil intake is inversely related to cancer prevalence: a systematic review and a meta-analysis of 13,800 patients and 23,340 controls in 19 observational studies, Lipids in health and disease, 10 (2011) 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [711].Cardenas E, Ghosh R, Vitamin E: a dark horse at the crossroad of cancer management, Biochemical pharmacology, 86 (2013) 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [712].Yang P, Jiang Y, Fischer SM, Prostaglandin E3 metabolism and cancer, Cancer letters, 348 (2014) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [713].Huerta-Yepez S, Tirado-Rodriguez AB, Hankinson O, Role of diets rich in omega-3 and omega-6 in the development of cancer, Bol Med Hosp Infant Mex, 73 (2016) 446–456. [DOI] [PubMed] [Google Scholar]
- [714].Belury MA, Inhibition of carcinogenesis by conjugated linoleic acid: potential mechanisms of action, J Nutr, 132 (2002) 2995–2998. [DOI] [PubMed] [Google Scholar]
- [715].McGowan M, Eisenberg B, Lewis L, Froehlich H, Wells W, Eastman A, Kuemmerle N, Rosenkrantz K, Barth R, Schwartz G, Li Z, Tosteson T, Beaulieu B, Kinlaw W, A Proof of Principle Clinical Trial to Determine Whether Conjugated Linoleic Acid Modulates the Lipogenic Pathway in Human Breast Cancer Tissue, Breast Cancer Research and Treatment, 138 (2013) 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [716].Ruiz-Vela A, Aguilar-Gallardo C, Simon C, Building a framework for embryonic microenvironments and cancer stem cells, Stem Cell Rev Rep, 5 (2009) 319–327. [DOI] [PubMed] [Google Scholar]
- [717].Ishay-Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R, Bantug G, Morini MF, Wang J, Hess C, Christofori G, Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis, Cancer cell, 35 (2019) 17–32 e16. [DOI] [PubMed] [Google Scholar]
- [718].Zaremberg V, Ganesan S, Mahadeo M, Lipids and Membrane Microdomains: The Glycerolipid and Alkylphosphocholine Class of Cancer Chemotherapeutic Drugs, Handb Exp Pharmacol, (2019). [DOI] [PubMed] [Google Scholar]
- [719].Escriba PV, Membrane-lipid therapy: A historical perspective of membrane-targeted therapies - From lipid bilayer structure to the pathophysiological regulation of cells, Biochim Biophys Acta Biomembr, 1859 (2017) 1493–1506. [DOI] [PubMed] [Google Scholar]
- [720].Vallabhapurapu SD, Blanco VM, Sulaiman MK, Vallabhapurapu SL, Chu Z, Franco RS, Qi X, Variation in human cancer cell external phosphatidylserine is regulated by flippase activity and intracellular calcium, Oncotarget, 6 (2015) 34375–34388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [721].Upadhyay R, Lipoproteins as drug delivery vehicles for cancer and tumor therapeutics, Stem Cell Research & Therapy, 4 (2018). [Google Scholar]
- [722].Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR, Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date, Pharm Res, 33 (2016) 2373–2387. [DOI] [PubMed] [Google Scholar]
- [723].Mulcahy LA, Pink RC, Carter DR, Routes and mechanisms of extracellular vesicle uptake, J Extracell Vesicles, 3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [724].Balachandran B, Yuana Y, Extracellular vesicles-based drug delivery system for cancer treatment, Cogent Medicine, 6 (2019) 1635806. [Google Scholar]
- [725].Chaudhary J, Bower J, Corbin IR, Lipoprotein Drug Delivery Vehicles for Cancer: Rationale and Reason, Int J Mol Sci, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [726].Tyurina YY, Tyurin VA, Anthonymuthu T, Amoscato AA, Sparvero LJ, Nesterova AM, Baynard ML, Sun W, He R, Khaitovich P, Vladimirov YA, Gabrilovich DI, Bayir H, Kagan VE, “Redox lipidomics technology: Looking for a needle in a haystack”, Chem Phys Lipids, 221 (2019) 93–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [727].Bowman AP, Bogie JFJ, Hendriks JJA, Haidar M, Belov M, Heeren RMA, Ellis SR, Evaluation of lipid coverage and high spatial resolution MALDI-imaging capabilities of oversampling combined with laser post-ionisation, Analytical and bioanalytical chemistry, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [728].Passarelli MK, Ewing AG, Winograd N, Single-cell lipidomics: characterizing and imaging lipids on the surface of individual Aplysia californica neurons with cluster secondary ion mass spectrometry, Anal Chem, 85 (2013) 2231–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [729].Wu H, Volponi JV, Oliver AE, Parikh AN, Simmons BA, Singh S, In vivo lipidomics using single-cell Raman spectroscopy, Proceedings of the National Academy of Sciences of the United States of America, 108 (2011) 3809–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- [1].Shan L, Chen YA, Davis L, Han G, Zhu W, Molina AD, Arango H, LaPolla JP, Hoffman MS, Sellers T, Kirby T, Nicosia SV, Sutphen R, Measurement of phospholipids may improve diagnostic accuracy in ovarian cancer, PLoS One, 7 (2012) e46846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang X, Zeng C, Lin J, Chen T, Zhao T, Jia Z, Xie X, Qiu Y, Su M, Jiang T, Zhou M, Zhao A, Jia W, Metabonomics approach to assessing the modulatory effects of St John’s wort, ginsenosides, and clomipramine in experimental depression, J Proteome Res, 11 (2012) 6223–6230. [DOI] [PubMed] [Google Scholar]
- [3].Qiu JF, Zhang KL, Zhang XJ, Hu YJ, Li P, Shang CZ, Wan JB, Abnormalities in Plasma Phospholipid Fatty Acid Profiles of Patients with Hepatocellular Carcinoma, Lipids, 50 (2015) 977–985. [DOI] [PubMed] [Google Scholar]
- [4].Guo Y, Wang X, Qiu L, Qin X, Liu H, Wang Y, Li F, Wang X, Chen G, Song G, Li F, Guo S, Li Z, Probing gender-specific lipid metabolites and diagnostic biomarkers for lung cancer using Fourier transform ion cyclotron resonance mass spectrometry, Clin Chim Acta, 414 (2012) 135–141. [DOI] [PubMed] [Google Scholar]
- [5].Yu Z, Chen H, Ai J, Zhu Y, Li Y, Borgia JA, Yang JS, Zhang J, Jiang B, Gu W, Deng Y, Global lipidomics identified plasma lipids as novel biomarkers for early detection of lung cancer, Oncotarget, 8 (2017) 107899–107906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Patel N, Vogel R, Chandra-Kuntal K, Glasgow W, Kelavkar U, A novel three serum phospholipid panel differentiates normal individuals from those with prostate cancer, PLoS One, 9 (2014) e88841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lv W, Yang T, Identification of possible biomarkers for breast cancer from free fatty acid profiles determined by GC-MS and multivariate statistical analysis, Clin Biochem, 45 (2012) 127–133. [DOI] [PubMed] [Google Scholar]
- [8].Crotti S, Agnoletto E, Cancemi G, Di Marco V, Traldi P, Pucciarelli S, Nitti D, Agostini M, Altered plasma levels of decanoic acid in colorectal cancer as a new diagnostic biomarker, Anal Bioanal Chem, 408 (2016) 6321–6328. [DOI] [PubMed] [Google Scholar]
- [9].Alberg AJ, Armeson K, Pierce JS, Bielawski J, Bielawska A, Visvanathan K, Hill EG, Ogretmen B, Plasma sphingolipids and lung cancer: a population-based, nested case-control study, Cancer Epidemiol Biomarkers Prev, 22 (2013) 1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schmidt JA, Fensom GK, Rinaldi S, Scalbert A, Appleby PN, Achaintre D, Gicquiau A, Gunter MJ, Ferrari P, Kaaks R, Kuhn T, Boeing H, Trichopoulou A, Karakatsani A, Peppa E, Palli D, Sieri S, Tumino R, Bueno-de-Mesquita B, Agudo A, Sanchez MJ, Chirlaque MD, Ardanaz E, Larranaga N, Perez-Cornago A, Assi N, Riboli E, Tsilidis KK, Key TJ, Travis RC, Patterns in metabolite profile are associated with risk of more aggressive prostate cancer: A prospective study of 3,057 matched case-control sets from EPIC, Int J Cancer, 146 (2020) 720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Saito K, Ikeda M, Kojima Y, Hosoi H, Saito Y, Kondo S, Lipid profiling of pre-treatment plasma reveals biomarker candidates associated with response rates and hand-foot skin reactions in sorafenib-treated patients, Cancer Chemother Pharmacol, 82 (2018) 677–684. [DOI] [PubMed] [Google Scholar]
- [12].Li J, Xie H, Li A, Cheng J, Yang K, Wang J, Wang W, Zhang F, Li Z, Dhillon HS, Openkova MS, Zhou X, Li K, Hou Y, Distinct plasma lipids profiles of recurrent ovarian cancer by liquid chromatography-mass spectrometry, Oncotarget, 8 (2017) 46834–46845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lin HM, Mahon KL, Weir JM, Mundra PA, Spielman C, Briscoe K, Gurney H, Mallesara G, Marx G, Stockler MR, Consortium PR, Parton RG, Hoy AJ, Daly RJ, Meikle PJ, Horvath LG, A distinct plasma lipid signature associated with poor prognosis in castration-resistant prostate cancer, Int J Cancer, 141 (2017) 2112–2120. [DOI] [PubMed] [Google Scholar]
- [14].Dill AL, Eberlin LS, Costa AB, Zheng C, Ifa DR, Cheng L, Masterson TA, Koch MO, Vitek O, Cooks RG, Multivariate statistical identification of human bladder carcinomas using ambient ionization imaging mass spectrometry, Chemistry, 17 (2011) 2897–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Eberlin LS, Norton I, Dill AL, Golby AJ, Ligon KL, Santagata S, Cooks RG, Agar NY, Classifying human brain tumors by lipid imaging with mass spectrometry, Cancer Res, 72 (2012) 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ide Y, Waki M, Hayasaka T, Nishio T, Morita Y, Tanaka H, Sasaki T, Koizumi K, Matsunuma R, Hosokawa Y, Ogura H, Shiiya N, Setou M, Human breast cancer tissues contain abundant phosphatidylcholine(36ratio1) with high stearoyl-CoA desaturase-1 expression, PLoS One, 8 (2013) e61204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cho YT, Su H, Chiang YY, Shiea J, Yuan SF, Hung WC, Yeh YT, Hou MF, Fine Needle Aspiration Combined With Matrix-assisted Laser Desorption Ionization Time-of-Flight/Mass Spectrometry to Characterize Lipid Biomarkers for Diagnosing Accuracy of Breast Cancer, Clin Breast Cancer, 17 (2017) 373–381 e371. [DOI] [PubMed] [Google Scholar]
- [18].Kawashima M, Tokiwa M, Nishimura T, Kawata Y, Sugimoto M, Kataoka TR, Sakurai T, Iwaisako K, Suzuki E, Hagiwara M, Harris AL, Toi M, High-resolution imaging mass spectrometry combined with transcriptomic analysis identified a link between fatty acid composition of phosphatidylinositols and the immune checkpoint pathway at the primary tumour site of breast cancer, Br J Cancer, 122 (2020) 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Santoro AL, Drummond RD, Silva IT, Ferreira SS, Juliano L, Vendramini PH, Lemos M, Eberlin MN, Andrade VP, In Situ DESI-MSI Lipidomic Profiles of Breast Cancer Molecular Subtypes and Precursor Lesions, Cancer Res, (2020). [DOI] [PubMed] [Google Scholar]
- [20].Porcari AM, Zhang J, Garza KY, Rodrigues-Peres RM, Lin JQ, Young JH, Tibshirani R, Nagi C, Paiva GR, Carter SA, Sarian LO, Eberlin MN, Eberlin LS, Multicenter Study Using Desorption-Electrospray-Ionization-Mass-Spectrometry Imaging for Breast-Cancer Diagnosis, Anal Chem, 90 (2018) 11324–11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Patterson NH, Alabdulkarim B, Lazaris A, Thomas A, Marcinkiewicz MM, Gao ZH, Vermeulen PB, Chaurand P, Metrakos P, Assessment of pathological response to therapy using lipid mass spectrometry imaging, Sci Rep, 6 (2016) 36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mirnezami R, Spagou K, Vorkas PA, Lewis MR, Kinross J, Want E, Shion H, Goldin RD, Darzi A, Takats Z, Holmes E, Cloarec O, Nicholson JK, Chemical mapping of the colorectal cancer microenvironment via MALDI imaging mass spectrometry (MALDI-MSI) reveals novel cancer-associated field effects, Mol Oncol, 8 (2014) 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Eberlin LS, Tibshirani RJ, Zhang J, Longacre TA, Berry GJ, Bingham DB, Norton JA, Zare RN, Poultsides GA, Molecular assessment of surgical-resection margins of gastric cancer by mass-spectrometric imaging, Proc Natl Acad Sci U S A, 111 (2014) 2436–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Muranishi Y, Sato T, Ito S, Satoh J, Yoshizawa A, Tamari S, Ueda Y, Yutaka Y, Menju T, Nakamura T, Date H, The Ratios of monounsaturated to saturated phosphatidylcholines in lung adenocarcinoma microenvironment analyzed by Liquid Chromatography-Mass spectrometry and imaging Mass spectrometry, Sci Rep, 9 (2019) 8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bednarczyk K, Gawin M, Chekan M, Kurczyk A, Mrukwa G, Pietrowska M, Polanska J, Widlak P, Discrimination of normal oral mucosa from oral cancer by mass spectrometry imaging of proteins and lipids, J Mol Histol, 50 (2019) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Doria ML, McKenzie JS, Mroz A, Phelps DL, Speller A, Rosini F, Strittmatter N, Golf O, Veselkov K, Brown R, Ghaem-Maghami S, Takats Z, Epithelial ovarian carcinoma diagnosis by desorption electrospray ionization mass spectrometry imaging, Sci Rep, 6 (2016) 39219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Clark AR, Calligaris D, Regan MS, Pomeranz Krummel D, Agar JN, Kallay L, MacDonald T, Schniederjan M, Santagata S, Pomeroy SL, Agar NYR, Sengupta S, Rapid discrimination of pediatric brain tumors by mass spectrometry imaging, J Neurooncol, 140 (2018) 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Randall EC, Zadra G, Chetta P, Lopez BGC, Syamala S, Basu SS, Agar JN, Loda M, Tempany CM, Fennessy FM, Agar NYR, Molecular Characterization of Prostate Cancer with Associated Gleason Score Using Mass Spectrometry Imaging, Mol Cancer Res, 17 (2019) 1155–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Goto T, Terada N, Inoue T, Nakayama K, Okada Y, Yoshikawa T, Miyazaki Y, Uegaki M, Sumiyoshi S, Kobayashi T, Kamba T, Yoshimura K, Ogawa O, The expression profile of phosphatidylinositol in high spatial resolution imaging mass spectrometry as a potential biomarker for prostate cancer, PLoS One, 9 (2014) e90242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Goto T, Terada N, Inoue T, Kobayashi T, Nakayama K, Okada Y, Yoshikawa T, Miyazaki Y, Uegaki M, Utsunomiya N, Makino Y, Sumiyoshi S, Yamasaki T, Kamba T, Ogawa O, Decreased expression of lysophosphatidylcholine (16:0/OH) in high resolution imaging mass spectrometry independently predicts biochemical recurrence after surgical treatment for prostate cancer, Prostate, 75 (2015) 1821–1830. [DOI] [PubMed] [Google Scholar]
- [31].Morse N, Jamaspishvili T, Simon D, Patel PG, Ren KYM, Wang J, Oleschuk R, Kaufmann M, Gooding RJ, Berman DM, Reliable identification of prostate cancer using mass spectrometry metabolomic imaging in needle core biopsies, Laboratory Investigation, (2019). [DOI] [PubMed] [Google Scholar]
- [32].Eberlin LS, Dill AL, Costa AB, Ifa DR, Cheng L, Masterson T, Koch M, Ratliff TL, Cooks RG, Cholesterol sulfate imaging in human prostate cancer tissue by desorption electrospray ionization mass spectrometry, Anal Chem, 82 (2010) 3430–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Marien E, Meister M, Muley T, Fieuws S, Bordel S, Derua R, Spraggins J, Van de Plas R, Dehairs J, Wouters J, Bagadi M, Dienemann H, Thomas M, Schnabel PA, Caprioli RM, Waelkens E, Swinnen JV, Non-small cell lung cancer is characterized by dramatic changes in phospholipid profiles, Int J Cancer, 137 (2015) 1539–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang J, Li SQ, Lin JQ, Yu W, Eberlin LS, Mass Spectrometry Imaging Enables Discrimination of Renal Oncocytoma from Renal Cell Cancer Subtypes and Normal Kidney Tissues, Cancer Res, 80 (2020) 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Margulis K, Chiou AS, Aasi SZ, Tibshirani RJ, Tang JY, Zare RN, Distinguishing malignant from benign microscopic skin lesions using desorption electrospray ionization mass spectrometry imaging, Proc Natl Acad Sci U S A, 115 (2018) 6347–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schcolnik-Cabrera A, Chavez-Blanco A, Dominguez-Gomez G, Taja-Chayeb L, Morales-Barcenas R, Trejo-Becerril C, Perez-Cardenas E, Gonzalez-Fierro A, Duenas-Gonzalez A, Orlistat as a FASN inhibitor and multitargeted agent for cancer therapy, Expert Opin Investig Drugs, 27 (2018) 475–489. [DOI] [PubMed] [Google Scholar]
- [37].Menendez JA, Vellon L, Lupu R, Antitumoral actions of the anti-obesity drug orlistat (XenicalTM) in breast cancer cells: blockade of cell cycle progression, promotion of apoptotic cell death and PEA3-mediated transcriptional repression of Her2/neu (erbB-2) oncogene, Ann Oncol, 16 (2005) 1253–1267. [DOI] [PubMed] [Google Scholar]
- [38].Liu H, Liu Y, Zhang JT, A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction, Mol Cancer Ther, 7 (2008) 263–270. [DOI] [PubMed] [Google Scholar]
- [39].Orita H, Coulter J, Lemmon C, Tully E, Vadlamudi A, Medghalchi SM, Kuhajda FP, Gabrielson E, Selective inhibition of fatty acid synthase for lung cancer treatment, Clin Cancer Res, 13 (2007) 7139–7145. [DOI] [PubMed] [Google Scholar]
- [40].Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, Syamala S, Bango C, Photopoulos C, Huang Y, Tyekucheva S, Bastos DC, Tchaicha J, Lawney B, Uo T, D’Anello L, Csibi A, Kalekar R, Larimer B, Ellis L, Butler LM, Morrissey C, McGovern K, Palombella VJ, Kutok JL, Mahmood U, Bosari S, Adams J, Peluso S, Dehm SM, Plymate SR, Loda M, Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer, Proc Natl Acad Sci U S A, 116 (2019) 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Heuer TS, Ventura R, Mordec K, Lai J, Fridlib M, Buckley D, Kemble G, FASN Inhibition and Taxane Treatment Combine to Enhance Anti-tumor Efficacy in Diverse Xenograft Tumor Models through Disruption of Tubulin Palmitoylation and Microtubule Organization and FASN Inhibition-Mediated Effects on Oncogenic Signaling and Gene Expression, EBioMedicine, 16 (2017) 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ventura R, Mordec K, Waszczuk J, Wang Z, Lai J, Fridlib M, Buckley D, Kemble G, Heuer TS, Inhibition of de novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression, EBioMedicine, 2 (2015) 808–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zaytseva YY, Rychahou PG, Le AT, Scott TL, Flight RM, Kim JT, Harris J, Liu J, Wang C, Morris AJ, Sivakumaran TA, Fan T, Moseley H, Gao T, Lee EY, Weiss HL, Heuer TS, Kemble G, Evers M, Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer, Oncotarget, 9 (2018) 24787–24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Alwarawrah Y, Hughes P, Loiselle D, Carlson DA, Darr DB, Jordan JL, Xiong J, Hunter LM, Dubois LG, Thompson JW, Kulkarni MM, Ratcliff AN, Kwiek JJ, Haystead TAJ, Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer, Cell Chemical Biology, 23 (2016) 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang C, Xu C, Sun M, Luo D, Liao DF, Cao D, Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis, Biochem Biophys Res Commun, 385 (2009) 302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Guseva NV, Rokhlin OW, Glover RA, Cohen MB, TOFA (5-tetradecyl-oxy-2-furoic acid) reduces fatty acid synthesis, inhibits expression of AR, neuropilin-1 and Mcl-1 and kills prostate cancer cells independent of p53 status, Cancer Biol Ther, 12 (2011) 80–85. [DOI] [PubMed] [Google Scholar]
- [47].Li S, Qiu L, Wu B, Shen H, Zhu J, Zhou L, Gu L, Di W, TOFA suppresses ovarian cancer cell growth in vitro and in vivo, Mol Med Rep, 8 (2013) 373–378. [DOI] [PubMed] [Google Scholar]
- [48].Jump DB, Torres-Gonzalez M, Olson LK, Soraphen A, an inhibitor of acetyl CoA carboxylase activity, interferes with fatty acid elongation, Biochemical pharmacology, 81 (2011) 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li EQ, Zhao W, Zhang C, Qin LZ, Liu SJ, Feng ZQ, Wen X, Chen CP, Synthesis and anticancer activity of ND-646 and its derivatives as acetyl-CoA carboxylase 1 inhibitors, Eur J Pharm Sci, 137 (2019) 105010. [DOI] [PubMed] [Google Scholar]
- [50].Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, Gerken L, Greenwood J, Bhat S, Harriman G, Westlin WF, Harwood HJ Jr., Saghatelian A, Kapeller R, Metallo CM, Shaw RJ, Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models, Nature medicine, 22 (2016) 1108–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Farooqi MAM, Malhotra N, Mukherjee SD, Sanger S, Dhesy-Thind SK, Ellis P, Leong DP, Statin therapy in the treatment of active cancer: A systematic review and meta-analysis of randomized controlled trials, PLoS One, 13 (2018) e0209486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Emilsson L, Garcia-Albeniz X, Logan RW, Caniglia EC, Kalager M, Hernan MA, Examining Bias in Studies of Statin Treatment and Survival in Patients With Cancer, JAMA Oncol, 4 (2018) 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jeong GH, Lee KH, Kim JY, Eisenhut M, Kronbichler A, van der Vliet HJ, Hong SH, Shin JI, Gamerith G, Effect of Statin on Cancer Incidence: An Umbrella Systematic Review and Meta-Analysis, J Clin Med, 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jeong GH, Lee KH, Kim JY, Eisenhut M, Kronbichler A, van der Vliet HJ, Shin JI, Gamerith G, Statin and Cancer Mortality and Survival: An Umbrella Systematic Review and Meta-Analysis, J Clin Med, 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Markovic SN, Suman VJ, Rao RA, Ingle JN, Kaur JS, Erickson LA, Pitot HC, Croghan GA, McWilliams RR, Merchan J, Kottschade LA, Nevala WK, Uhl CB, Allred J, Creagan ET, A phase II study of ABT-510 (thrombospondin-1 analog) for the treatment of metastatic melanoma, Am J Clin Oncol, 30 (2007) 303–309. [DOI] [PubMed] [Google Scholar]
- [56].Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, Liu XS, Lee YR, Fung J, Katon JM, Menon AV, Webster KA, Ng C, Palumbieri MD, Diolombi MS, Breitkopf SB, Teruya-Feldstein J, Signoretti S, Bronson RT, Asara JM, Castillo-Martin M, Cordon-Cardo C, Pandolfi PP, An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer, Nat Genet, 50 (2018) 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li X, Chen YT, Hu P, Huang WC, Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling, Mol Cancer Ther, 13 (2014) 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nguyen VTM, Barozzi I, Faronato M, Lombardo Y, Steel JH, Patel N, Darbre P, Castellano L, Győrffy B, Woodley L, Meira A, Patten DK, Vircillo V, Periyasamy M, Ali S, Frige G, Minucci S, Coombes RC, Magnani L, Differential epigenetic reprogramming in response to specific endocrine therapies promotes cholesterol biosynthesis and cellular invasion, Nature communications, 6 (2015) 10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Brovkovych V, Izhar Y, Danes JM, Dubrovskyi O, Sakallioglu IT, Morrow LM, Atilla-Gokcumen GE, Frasor J, Fatostatin induces pro- and anti-apoptotic lipid accumulation in breast cancer, Oncogenesis, 7 (2018) 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Li X, Chen Y-T, Hu P, Huang W-C, Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling, Molecular Cancer Therapeutics, 13 (2014) 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li X, Wu JB, Chung LWK, Huang W-C, Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations, Oncotarget, 6 (2015) 41018–41032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Siqingaowa S Sekar V Gopalakrishnan C Taghibiglou, Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation, Biochem Biophys Res Commun, 488 (2017) 136–140. [DOI] [PubMed] [Google Scholar]
- [63].Alakurtti S, Makela T, Koskimies S, Yli-Kauhaluoma J, Pharmacological properties of the ubiquitous natural product betulin, Eur J Pharm Sci, 29 (2006) 1–13. [DOI] [PubMed] [Google Scholar]
- [64].Krol SK, Kielbus M, Rivero-Muller A, Stepulak A, Comprehensive review on betulin as a potent anticancer agent, Biomed Res Int, 2015 (2015) 584189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Li N, Zhou ZS, Shen Y, Xu J, Miao HH, Xiong Y, Xu F, Li BL, Luo J, Song BL, Inhibition of the sterol regulatory element-binding protein pathway suppresses hepatocellular carcinoma by repressing inflammation in mice, Hepatology (Baltimore, Md.), 65 (2017) 1936–1947. [DOI] [PubMed] [Google Scholar]
- [66].Becker-Schiebe M, Mengs U, Schaefer M, Bulitta M, Hoffmann W, Topical use of a silymarin-based preparation to prevent radiodermatitis : results of a prospective study in breast cancer patients, Strahlenther Onkol, 187 (2011) 485–491. [DOI] [PubMed] [Google Scholar]
- [67].Ladas EJ, Kroll DJ, Oberlies NH, Cheng B, Ndao DH, Rheingold SR, Kelly KM, A randomized, controlled, double-blind, pilot study of milk thistle for the treatment of hepatotoxicity in childhood acute lymphoblastic leukemia (ALL), Cancer, 116 (2010) 506–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Elyasi S, Hosseini S, Niazi Moghadam MR, Aledavood SA, Karimi G, Effect of Oral Silymarin Administration on Prevention of Radiotherapy Induced Mucositis: A Randomized, Double-Blinded, Placebo-Controlled Clinical Trial, Phytother Res, 30 (2016) 1879–1885. [DOI] [PubMed] [Google Scholar]
- [69].Zadra G, Photopoulos C, Tyekucheva S, Heidari P, Weng QP, Fedele G, Liu H, Scaglia N, Priolo C, Sicinska E, Mahmood U, Signoretti S, Birnberg N, Loda M, A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis, EMBO molecular medicine, 6 (2014) 519–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Blazquez C, Salazar M, Carracedo A, Lorente M, Egia A, Gonzalez-Feria L, Haro A, Velasco G, Guzman M, Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression, Cancer Res, 68 (2008) 1945–1952. [DOI] [PubMed] [Google Scholar]
- [71].Qamri Z, Preet A, Nasser MW, Bass CE, Leone G, Barsky SH, Ganju RK, Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer, Mol Cancer Ther, 8 (2009) 3117–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Caffarel MM, Andradas C, Mira E, Perez-Gomez E, Cerutti C, Moreno-Bueno G, Flores JM, Garcia-Real I, Palacios J, Manes S, Guzman M, Sanchez C, Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition, Mol Cancer, 9 (2010) 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hasko J, Fazakas C, Molnar J, Nyul-Toth A, Herman H, Hermenean A, Wilhelm I, Persidsky Y, Krizbai IA, CB2 receptor activation inhibits melanoma cell transmigration through the blood-brain barrier, Int J Mol Sci, 15 (2014) 8063–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Martinez-Martinez E, Martin-Ruiz A, Martin P, Calvo V, Provencio M, Garcia JM, CB2 cannabinoid receptor activation promotes colon cancer progression via AKT/GSK3beta signaling pathway, Oncotarget, 7 (2016) 68781–68791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Khan MI, Sobocinska AA, Brodaczewska KK, Zielniok K, Gajewska M, Kieda C, Czarnecka AM, Szczylik C, Involvement of the CB2 cannabinoid receptor in cell growth inhibition and G0/G1 cell cycle arrest via the cannabinoid agonist WIN 55,212–2 in renal cell carcinoma, BMC Cancer, 18 (2018) 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Fu S, Xiang W, Chen J, Ma L, Chen L, Synthesis and biological evaluation of 1, 2, 4-oxadiazole derivatives as novel GPR119 agonists, Chem Biol Drug Des, 89 (2017) 815–819. [DOI] [PubMed] [Google Scholar]
- [77].Im JH, Kang KW, Kim SY, Kim YG, An YJ, Park S, Jeong BH, Choi SY, Lee JS, Kang KW, GPR119 agonist enhances gefitinib responsiveness through lactate-mediated inhibition of autophagy, J Exp Clin Cancer Res, 37 (2018) 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Arrieta O, Barron F, Padilla MS, Aviles-Salas A, Ramirez-Tirado LA, Arguelles Jimenez MJ, Vergara E, Zatarain-Barron ZL, Hernandez-Pedro N, Cardona AF, Cruz-Rico G, Barrios-Bernal P, Yamamoto Ramos M, Rosell R, Effect of Metformin Plus Tyrosine Kinase Inhibitors Compared With Tyrosine Kinase Inhibitors Alone in Patients With Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: A Phase 2 Randomized Clinical Trial, JAMA Oncol, (2019) e192553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Li L, Jiang L, Wang Y, Zhao Y, Zhang XJ, Wu G, Zhou X, Sun J, Bai J, Ren B, Tian K, Xu Z, Xiao HL, Zhou Q, Han R, Chen H, Wang H, Yang Z, Gao C, Cai S, He Y, Combination of Metformin and Gefitinib as First-Line Therapy for Nondiabetic Advanced NSCLC Patients with EGFR Mutations: A Randomized, Double-Blind Phase II Trial, Clin Cancer Res, 25 (2019) 6967–6975. [DOI] [PubMed] [Google Scholar]
- [80].Nanni O, Amadori D, De Censi A, Rocca A, Freschi A, Bologna A, Gianni L, Rosetti F, Amaducci L, Cavanna L, Foca F, Sarti S, Serra P, Valmorri L, Bruzzi P, Corradengo D, Gennari A, investigators M, Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial, Breast Cancer Res Treat, 174 (2019) 433–442. [DOI] [PubMed] [Google Scholar]
- [81].Zhao Y, Gong C, Wang Z, Zhang J, Wang L, Zhang S, Cao J, Tao Z, Li T, Wang B, Hu X, A randomized phase II study of aromatase inhibitors plus metformin in pre-treated postmenopausal patients with hormone receptor positive metastatic breast cancer, Oncotarget, 8 (2017) 84224–84236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Theodoropoulou S, Brodowska K, Kayama M, Morizane Y, Miller JW, Gragoudas ES, Vavvas DG, Aminoimidazole carboxamide ribonucleotide (AICAR) inhibits the growth of retinoblastoma in vivo by decreasing angiogenesis and inducing apoptosis, PLoS One, 8 (2013) e52852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Su CC, Hsieh KL, Liu PL, Yeh HC, Huang SP, Fang SH, Cheng WC, Huang KH, Chiu FY, Lin IL, Huang MY, Li CY, AICAR Induces Apoptosis and Inhibits Migration and Invasion in Prostate Cancer Cells Through an AMPK/mTOR-Dependent Pathway, Int J Mol Sci, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Katz A, Udata C, Ott E, Hickey L, Burczynski ME, Burghart P, Vesterqvist O, Meng X, Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants, J Clin Pharmacol, 49 (2009) 643–649. [DOI] [PubMed] [Google Scholar]
- [85].Schweiger M, Romauch M, Schreiber R, Grabner GF, Hutter S, Kotzbeck P, Benedikt P, Eichmann TO, Yamada S, Knittelfelder O, Diwoky C, Doler C, Mayer N, De Cecco W, Breinbauer R, Zimmermann R, Zechner R, Pharmacological inhibition of adipose triglyceride lipase corrects high-fat diet-induced insulin resistance and hepatosteatosis in mice, Nat Commun, 8 (2017) 14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF, Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects, Nat Chem Biol, 5 (2009) 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].von Roemeling CA, Marlow LA, Wei JJ, Cooper SJ, Caulfield TR, Wu K, Tan WW, Tun HW, Copland JA, Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma, Clin Cancer Res, 19 (2013) 2368–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Piao C, Cui X, Zhan B, Li J, Li Z, Li Z, Liu X, Bi J, Zhang Z, Kong C, Inhibition of stearoyl CoA desaturase-1 activity suppresses tumour progression and improves prognosis in human bladder cancer, J Cell Mol Med, 23 (2019) 2064–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].She K, Fang S, Du W, Fan X, He J, Pan H, Huang L, He P, Huang J, SCD1 is required for EGFR-targeting cancer therapy of lung cancer via re-activation of EGFR/PI3K/AKT signals, Cancer Cell Int, 19 (2019) 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Imamura K, Tomita N, Kawakita Y, Ito Y, Ono K, Nii N, Miyazaki T, Yonemori K, Tawada M, Sumi H, Satoh Y, Yamamoto Y, Miyahisa I, Sasaki M, Satomi Y, Hirayama M, Nishigaki R, Maezaki H, Discovery of Novel and Potent Stearoyl Coenzyme A Desaturase 1 (SCD1) Inhibitors as Anticancer Agents, Bioorg Med Chem, 25 (2017) 3768–3779. [DOI] [PubMed] [Google Scholar]
- [91].Nishizawa S, Sumi H, Satoh Y, Yamamoto Y, Kitazawa S, Honda K, Araki H, Kakoi K, Imamura K, Sasaki M, Miyahisa I, Satomi Y, Nishigaki R, Hirayama M, Aoyama K, Maezaki H, Hara T, In vitro and in vivo antitumor activities of T-3764518, a novel and orally available small molecule stearoyl-CoA desaturase 1 inhibitor, Eur J Pharmacol, 807 (2017) 21–31. [DOI] [PubMed] [Google Scholar]
- [92].von Roemeling CA, Caulfield TR, Marlow L, Bok I, Wen J, Miller JL, Hughes R, Hazlehurst L, Pinkerton AB, Radisky DC, Tun HW, Kim YSB, Lane AL, Copland JA, Accelerated bottom-up drug design platform enables the discovery of novel stearoyl-CoA desaturase 1 inhibitors for cancer therapy, Oncotarget, 9 (2018) 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Theodoropoulos PC, Gonzales SS, Winterton SE, Rodriguez-Navas C, McKnight JS, Morlock LK, Hanson JM, Cross B, Owen AE, Duan Y, Moreno JR, Lemoff A, Mirzaei H, Posner BA, Williams NS, Ready JM, Nijhawan D, Discovery of tumor-specific irreversible inhibitors of stearoyl CoA desaturase, Nat Chem Biol, 12 (2016) 218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Cheng S, Wang G, Wang Y, Cai L, Qian K, Ju L, Liu X, Xiao Y, Wang X, Fatty acid oxidation inhibitor etomoxir suppresses tumor progression and induces cell cycle arrest via PPARgamma-mediated pathway in bladder cancer, Clin Sci (Lond), 133 (2019) 1745–1758. [DOI] [PubMed] [Google Scholar]
- [95].Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L, Cimic A, Sirintrapun SJ, Glode LM, Eckel RH, Cramer SD, Lipid catabolism via CPT1 as a therapeutic target for prostate cancer, Mol Cancer Ther, 13 (2014) 2361–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lin H, Patel S, Affleck VS, Wilson I, Turnbull DM, Joshi AR, Maxwell R, Stoll EA, Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells, Neuro Oncol, 19 (2017) 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ren XR, Wang J, Osada T, Mook RA Jr., Morse MA, Barak LS, Lyerly HK, Chen W, Perhexiline promotes HER3 ablation through receptor internalization and inhibits tumor growth, Breast Cancer Res, 17 (2015) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Itkonen HM, Brown M, Urbanucci A, Tredwell G, Ho Lau C, Barfeld S, Hart C, Guldvik IJ, Takhar M, Heemers HV, Erho N, Bloch K, Davicioni E, Derua R, Waelkens E, Mohler JL, Clarke N, Swinnen JV, Keun HC, Rekvig OP, Mills IG, Lipid degradation promotes prostate cancer cell survival, Oncotarget, 8 (2017) 38264–38275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Zhu J, Wu G, Song L, Cao L, Tan Z, Tang M, Li Z, Shi D, Zhang S, Li J, NKX2–8 deletion-induced reprogramming of fatty acid metabolism confers chemoresistance in epithelial ovarian cancer, EBioMedicine, 43 (2019) 238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, Foa R, Nicolai R, Peluso G, Tafuri A, Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias, Blood, 126 (2015) 1925–1929. [DOI] [PubMed] [Google Scholar]
- [101].Pacilli A, Calienni M, Margarucci S, D’Apolito M, Petillo O, Rocchi L, Pasquinelli G, Nicolai R, Koverech A, Calvani M, Peluso G, Montanaro L, Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis, J Natl Cancer Inst, 105 (2013) 489–498. [DOI] [PubMed] [Google Scholar]
- [102].Henkels KM, Muppani NR, Gomez-Cambronero J, PLD-Specific Small-Molecule Inhibitors Decrease Tumor-Associated Macrophages and Neutrophils Infiltration in Breast Tumors and Lung and Liver Metastases, PLoS One, 11 (2016) e0166553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Rao E, Singh P, Zhai X, Li Y, Zhu G, Zhang Y, Hao J, Chi YI, Brown RE, Cleary MP, Li B, Inhibition of tumor growth by a newly-identified activator for epidermal fatty acid binding protein, Oncotarget, 6 (2015) 7815–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Carbonetti G, Converso C, Clement T, Wang C, Trotman LC, Ojima I, Kaczocha M, Docetaxel/cabazitaxel and fatty acid binding protein 5 inhibitors produce synergistic inhibition of prostate cancer growth, Prostate, 80 (2020) 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hernandez-Alcoceba R, Fernandez F, Lacal JC, In vivo antitumor activity of choline kinase inhibitors: a novel target for anticancer drug discovery, Cancer Res, 59 (1999) 3112–3118. [PubMed] [Google Scholar]
- [106].Estevez-Braun A, Ravelo AG, Perez-Sacau E, Lacal JC, A new family of choline kinase inhibitors with antiproliferative and antitumor activity derived from natural products, Clin Transl Oncol, 17 (2015) 74–84. [DOI] [PubMed] [Google Scholar]
- [107].Trousil S, Kaliszczak M, Schug Z, Nguyen QD, Tomasi G, Favicchio R, Brickute D, Fortt R, Twyman FJ, Carroll L, Kalusa A, Navaratnam N, Adejumo T, Carling D, Gottlieb E, Aboagye EO, The novel choline kinase inhibitor ICL-CCIC-0019 reprograms cellular metabolism and inhibits cancer cell growth, Oncotarget, 7 (2016) 37103–37120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Mariotto E, Bortolozzi R, Volpin I, Carta D, Serafin V, Accordi B, Basso G, Navarro PL, Lopez-Cara LC, Viola G, EB-3D a novel choline kinase inhibitor induces deregulation of the AMPK-mTOR pathway and apoptosis in leukemia T-cells, Biochem Pharmacol, 155 (2018) 213–223. [DOI] [PubMed] [Google Scholar]
- [109].Mariotto E, Viola G, Ronca R, Persano L, Aveic S, Bhujwalla ZM, Mori N, Accordi B, Serafin V, Lopez-Cara LC, Bortolozzi R, Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer, Cancers (Basel), 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Sola-Leyva A, Lopez-Cara LC, Rios-Marco P, Rios A, Marco C, Carrasco-Jimenez MP, Choline kinase inhibitors EB-3D and EB-3P interferes with lipid homeostasis in HepG2 cells, Sci Rep, 9 (2019) 5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Nghiemphu PL, Ebiana VA, Wen P, Gilbert M, Abrey LE, Lieberman F, DeAngelis LM, Robins HI, Yung WKA, Chang S, Drappatz J, Mehta MP, Levin VA, Aldape K, Dancey JE, Wright JJ, Prados M, Kuhn J, Cloughesy TF, Phase I study of sorafenib and tipifarnib for recurrent glioblastoma: NABTC 05–02, J Neurooncol, 136 (2018) 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Yam C, Murthy RK, Valero V, Szklaruk J, Shroff GS, Stalzer CJ, Buzdar AU, Murray JL, Yang W, Hortobagyi GN, Moulder SL, Arun B, A phase II study of tipifarnib and gemcitabine in metastatic breast cancer, Invest New Drugs, 36 (2018) 299–306. [DOI] [PubMed] [Google Scholar]
- [113].Ho AL, Chau N, Bauman J, Bible K, Chintakuntlawar A, Cabanillas ME, Wong DJ, Brana Garcia I, Brose MS, Boni V, Even C, Razaq M, Mishra V, Bracken K, Wages D, Scholz C, Gualberto A, Preliminary results from a phase II trial of tipifarnib in squamous cell carcinomas (SCCs) with HRAS mutations, Ann Oncol, 29 Suppl 8 (2018) viii373. [Google Scholar]
- [114].Castaneda C, Meadows KL, Truax R, Morse MA, Kaufmann SH, Petros WP, Zhu Y, Statkevich P, Cutler DL, Hurwitz HI, Phase I and pharmacokinetic study of lonafarnib, SCH 66336, using a 2-week on, 2-week off schedule in patients with advanced solid tumors, Cancer Chemother Pharmacol, 67 (2011) 455–463. [DOI] [PubMed] [Google Scholar]
- [115].Milojkovic Kerklaan B, Dieras V, Le Tourneau C, Mergui-Roelvink M, Huitema AD, Rosing H, Beijnen JH, Marreaud S, Govaerts AS, Piccart-Gebhart MJ, Schellens JH, Awada A, Phase I study of lonafarnib (SCH66336) in combination with trastuzumab plus paclitaxel in Her2/neu overexpressing breast cancer: EORTC study 16023, Cancer Chemother Pharmacol, 71 (2013) 53–62. [DOI] [PubMed] [Google Scholar]
- [116].Yust-Katz S, Liu D, Yuan Y, Liu V, Kang S, Groves M, Puduvalli V, Levin V, Conrad C, Colman H, Hsu S, Yung WK, Gilbert MR, Phase 1/1b study of lonafarnib and temozolomide in patients with recurrent or temozolomide refractory glioblastoma, Cancer, 119 (2013) 2747–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Eder JP Jr., Ryan DP, Appleman L, Zhu AX, Puchalski T, He X, Sonnichsen DS, Cooper M, Wright J, Clark JW, Supko JG, Phase I clinical trial of the farnesyltransferase inhibitor BMS-214662 administered as a weekly 24 h continuous intravenous infusion in patients with advanced solid tumors, Cancer Chemother Pharmacol, 58 (2006) 107–116. [DOI] [PubMed] [Google Scholar]
- [118].Bailey HH, Alberti DB, Thomas JP, Mulkerin DL, Binger KA, Gottardis MM, Martell RE, Wilding G, Phase I trial of weekly paclitaxel and BMS-214662 in patients with advanced solid tumors, Clin Cancer Res, 13 (2007) 3623–3629. [DOI] [PubMed] [Google Scholar]
- [119].Xu T, Huang C, Qi XT, Yang XC, Zhang N, Cao J, Wang C, Zhu H, Yang B, He QJ, Shao XJ, Ying MD, 2-Bromopalmitate sensitizes osteosarcoma cells to adriamycin-induced apoptosis via the modulation of CHOP, Eur J Pharmacol, 844 (2019) 204–215. [DOI] [PubMed] [Google Scholar]
- [120].Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, Lu H, Fang C, Zhang Y, Liang L, Zhou X, Wang C, Xue Y, Cui Y, Xu J, Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours, Nat Biomed Eng, 3 (2019) 306–317. [DOI] [PubMed] [Google Scholar]
- [121].Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, Wei Y, Xia W, Hou J, Qiu Y, Hung MC, Palmitoylation stabilizes PD-L1 to promote breast tumor growth, Cell Res, 29 (2019) 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Klutzny S, Lesche R, Keck M, Kaulfuss S, Schlicker A, Christian S, Sperl C, Neuhaus R, Mowat J, Steckel M, Riefke B, Prechtl S, Parczyk K, Steigemann P, Functional inhibition of acid sphingomyelinase by Fluphenazine triggers hypoxia-specific tumor cell death, Cell death & disease, 8 (2017) e2709. [DOI] [PMC free article] [PubMed] [Google Scholar]