

Abstract
Growth hormone (GH) and its mediator, the insulin-like growth factor-1 (IGF-1) regulate somatic growth, metabolism and many aspects of aging. As such, actions of GH/IGF have been studied in many tissues and organs over decades. GH and IGF-1 are part of the hypothalamic/pituitary somatotrophic axis that consists of many other regulatory hormones, receptors, binding proteins, and proteases. In humans, GH/IGF actions peak during pubertal growth and regulate skeletal acquisition through stimulation of extracellular matrix production and increases in bone mineral density. During aging the activity of these hormones declines, a state called somatopause, which associates with deleterious effects on the musculoskeletal system. In this review, we will focus on GH/IGF-1 action in bone and cartilage. We will cover many studies that have utilized congenital ablation or overexpression of members of this axis, as well as cell-specific gene-targeting approaches used to unravel the nature of the GH/IGF-1 actions in the skeleton in vivo.
Keywords: growth hormone, insulin-like growth factor-1, bone, cartilage
Introduction:
The anabolic growth hormone (GH) / Insulin like growth factor-1 (IGF-1) axis has received a considerable amount of attention throughout the years due to its contribution to numerous physiological actions in many species. The mammalian GH, released from the anterior pituitary gland, is a major driver of somatic growth. It belongs to a family of hormones including prolactin (PRL), and the placental lactogens [1]. As is true for many hormones, GH is secreted in a pulsatile [2] and sexually dimorphic manner in both humans and animal models [3]. Pulsatile secretion of GH is regulated by two major hypothalamic factors: the GH releasing hormone (GHRH) that stimulates GH release, and the somatostatin release-inhibiting factor (SRIF, or somatostatin), which hampers GH secretion [4, 5]. Circulatory GH binds to the GH binding protein (GHBP), a truncated extracellular domain of the GH receptor (GHR), which increases the half-life of GH in the circulation [6]. In rodents, the GHBP is generated by alternative mRNA splicing [7], whereas in humans, it is formed as a result of a proteolytic cleavage of the extracellular domain of the GHR [8].
GH and IGF-1 act in an endocrine and an autocrine/paracrine manner. GH plays a central role in controlling circulatory IGF-1 levels (endocrine IGF-1) through its actions in liver, mainly during the postnatal period [9, 10]. The liver is the principal contributor to the endocrine pool of IGF-1 (75%), while other tissues, such as fat and muscle, contribute ~25% of IGF-1 in serum. While in circulation, IGF-1 is bound to IGF-binding proteins (IGFBPs) and the acid labile subunit (ALS), which prolong its half-life in serum and determine its bioavailability in tissues. Circulating IGF-1 provides negative feedback to the pituitary and regulates GH secretion [11]. During puberty, signals of the GH/IGF axis are upregulated in both humans and mice, and correlate with changes in bone formation markers [12]. Numerous in vivo studies of transgenic animal models and clinical human investigations have provided ample evidence for the importance of the endocrine and local actions of both GH and IGF-1 in skeletal growth and maintenance. Indeed, IGF-1 and IGF-2 are the most prominent growth factors stored in bone, and they are the most abundant growth factors released by osteoblast cells [13].
GH/IGF-1 axis and bone mass:
Bone mass refers to the amount of minerals—mostly calcium and phosphorus—that are contained in the collagen-rich matrix of the skeleton. Acquisition of bone mass is a complex process involving mesenchymal stromal cells (which give rise to osteoprogenitors), osteoblasts (bone-building cells), osteocytes (bone resident cells), and the hematopoietic originated cells [14], the osteoclasts (bone-resorbing cells). Peak bone mass (PBM), the greatest amount of bone an individual can attain, is achieved during postnatal growth and puberty, at which time also GH/IGF-1 peak in serum. It is estimated that bone mass increases about thirty-fold from birth to young adulthood, at which time it peaks [15]. Gains in bone mass correlate with longitudinal and radial growth in the skeleton. The skeleton contains two major compartments: cortical bone, which accounts for ~85% of skeletal tissue, and cancellous/trabecular bone, which accounts for the remaining 15% [16, 17]. There are considerable differences between these two compartments in terms of surface area and modeling/remodeling of bone. Additionally, cancellous bone is more metabolically active than cortical bone, and is largely affected by hormonal cues. Nonetheless, bone loss occurs across both compartments during aging.
Clinical studies have shown 40–50% increases in bone mineral density (BMD) during pubertal growth [18, 19], which were attributed mainly to the levels of expression and activity of sex steroid hormones and the GH/IGF axis. Accordingly, in humans, germline mutations leading to GH deficiency result in retarded growth, delayed sexual maturity, and consequently, reduced BMD [20]. Mutations in GHR in humans result in GH insensitivity syndrome (Laron’s syndrome) [21, 22]. Laron’s syndrome patients exhibit short stature and reduced serum IGF-1 [21]. They are born at a nearly normal size but show immediate postnatal growth decline with attenuated growth velocities [23]. Similarly, isolated GH deficiency (GHD) or multiple pituitary hormone deficiencies (MPHD) in humans have been associated with reduced BMD and low bone turnover, with a high risk of vertebral and nonvertebral fractures. This condition can be partially reversed by GH supplementation [24]. On the other hand, somatic alterations that lead to excess GH (such as pituitary adenomas) result in gigantism or acromegaly, and a robust skeleton with a high BMD [21, 25]. Notably, there are intimate interactions between the GH/IGF-1 and the sex steroid axes in multiple tissues [26]. Thus, impaired skeletal growth in states of GH/IGF-1 deficiency results also from concomitant impairment of the sex steroid axes [26].
Characterizations of the GH/IGF-1 axis in numerous animal models have unequivocally shown the importance of this axis in the regulation of skeletal acquisition (Figure 1). Mouse models with congenital inactivation of GH action are born with normal body weight but exhibit dwarfism and reduced skeletal acquisition during post-natal growth, and reach ~50–60% of normal adult body size [27–30]. Likewise, overexpression of a GH antagonist [31], which causes blunted activation of the GHR, result in a ~45% reduction in body size in mice. Overall, decreases in skeletal size in GH-deficient states is associated with significant decreases in bone size and BMD [28, 32] [33]. On the other hand, transgenic lines with overexpression of GHRH [34, 35], human (h) GH [36–42] or bovine (b) GH in mice, all show an excess of GH, increased body size, skeletal gigantism, and increased BMD [38, 43–49]. Accordingly, overexpression of hGHRH [48] resulted in systemic stimulation of endogenous GH and IGF-1 and initial increases in bone mass, but was later associated with increased bone resorption. Excessive GH action was also detected in the GHR inhibitor suppressor of cytokine signaling-2 null (SOCS2KO) mice. SOCS2KO mice were larger than controls and showed increased linear bone growth, radial bone growth, and trabecular bone volume [50, 51]. Unlike with GH, ubiquitous overexpression of IGF-1 in mice (which caused an inhibition of endogenous GH secretion) resulted in selective organomegaly of the spleen, pancreas, and brain. Radiographs of the tibia and radius of IGF-1 transgenic mice showed no differences in bone-length. However, detailed bone morphology was not reported in these cases [52]. On the other hand, hepatic IGF-1 transgenic (HIT) mice, which overexpress the rat-IGF-1 transgene exclusively in the liver (under the TTR promoter), show 2–3fold increases in serum IGF-1, exhibited 10% increases in skeletal size [53–56].
Figure 1: Mouse models of the GH/IGF axis.
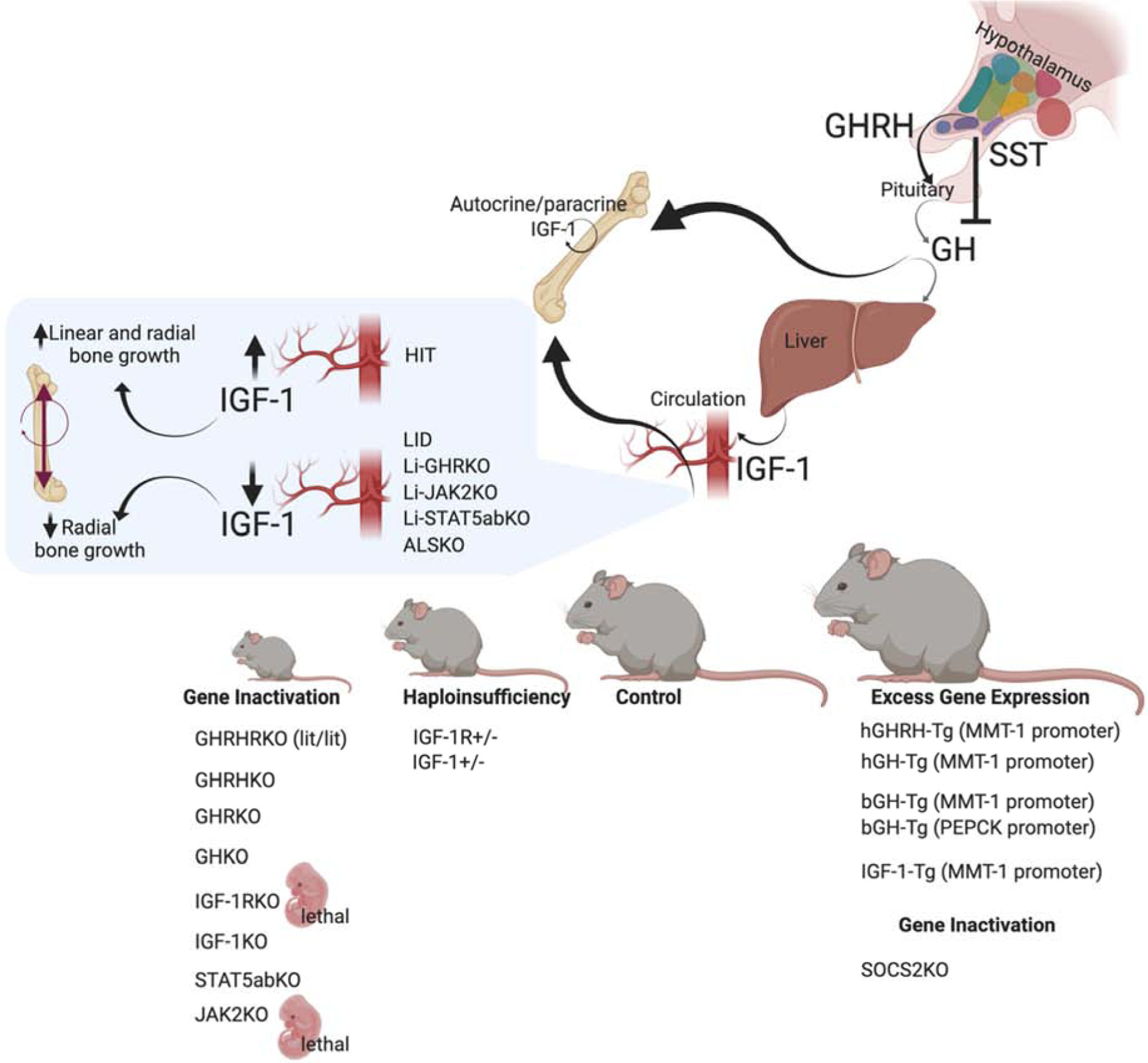
Several naturally occurring mutations in the GH/IGF-1 axis have been described over the years. Among them is the lit/lit mouse line, with a mutation in the GHRH-receptor which causes pituitary hypoplasia, reduced GH secretion, and dwarfism [220]. Similarly, targeted ablation of the GHRH gene causes GH deficiency, pituitary hypoplasia, and reductions in somatic growth [221]. GHRKO and the recently described GHKO mice [222] also show decreased linear bone growth and reduced whole-body BMD. Gene ablation of the IGF-1R was lethal [57], and caused severely retarded growth, with delayed ossification in all bones. Likewise, IGF-1KO mice showed severe dwarfism, associated with reduced BMD and decreased linear and radial bone growth [223]. In mice, inactivation of the downstream mediator of GH signaling, STAT5ab, also affected linear growth [224]. However, the details of skeletal morphology were not reported. Finally, in mice, knocking out JAK2, the GH mediator, was lethal [225], likely due to its central roles in cytokine signaling. Haploinsufficiency of IGF-1 or the IGR-1R associate with intermediate growth retardation and reduced linear and radial bone growth. Mouse models of excess GHRH [35, 226], hGH [36–42], and bGH [38, 43–49] show increased somatic growth with robust bones. Mice with excess IGF-1 resulted in selective organomegaly and as reported at the time showed minimal effects on skeletal growth [52, 227]. Lastly, SOCS2KO mice, which exhibit a “physiological” excess of GH action (as oppose to transgene driven GH expression), show increased linear and radial bone growth with increased trabecular bone volume [228].
Congenital IGF-1 or IGF-1R knockout mice were extensively studied in the 1990s. IGF-1RKO mice are born at 45% of normal size, show organ hypoplasia and delayed ossification, and die immediately after birth [57]. IGF-1R haploinsufficient mice (IGF-1R+/−) show significant decreases in IGF-1 gene expression in the liver—leading to reduced serum/endocrine IGF-1—as well as in the bone, testicles, and brain. Specifically, significant reductions in cortical bone thickness were shown [58]. On the other hand, ablation of IGF-1 in mice caused severely retarded growth, with adult skeletons reaching only 30% of normal size [57, 59] [60]. IGF-1 haploinsufficient mice (IGF-1+/−) showed intermediate levels of serum and tissue IGF-1 (as compared to the wildtype and IGF-1KO mice), which correlated with reduced body weight, body length, bone length, and BMD [61, 62]. Clearly, mouse models with germline ablation of IGF-1, IGF-1R, GH, or the GHR show compromised growth. Notably, in these models, both the endocrine and the autocrine/paracrine modes of IGF-1 were blocked.
Circulating IGF-1 is the biggest pool of IGF-1 in the body. However, using mouse models with germline mutations in IGF-1/IGF-1R did not allow researchers to dissect the specific roles of the serum IGF-1 pool in somatic growth. With the availability of the Cre/loxP technology, a few mouse models of selective reductions in liver-derived IGF-1 were generated. These models revealed the importance of the endocrine pool of IGF-1. Notably, however, most models with reduced serum IGF-1 levels show increases in pituitary secretion of GH, owing to the negative feedback loop between these two hormones. Thus, interpretation of skeletal data from these animal models should be done with caution. Liver-specific IGF-1-defficient (LID) mice were generated using the albumin promoter-derived cre (Alb-cre) [63, 64] and showed 75% reductions in serum IGF-1 levels. LID mice are born normal in size. Linear bone growth assessed at several ages was minimally reduced (~−5–6%). However, bone acquisition was compromised as evidenced by reduced BMD and slender bones (reductions in total cross-sectional area, cortical bone area, and cortical thickness). Alb-cre was also used to ablate the liver ghr gene (Liver-GHRKO mice) [65]. As seen with the LID mice, the Liver-GHRKO mice showed normal tibia and femur lengths despite >90% decreases in serum IGF-1 levels. However, they exhibited marked reductions in BMD and significantly reduced trabecular bone volume. Liver-specific signal transducer and activator of transcription 5ab (STAT5ab) inactivation [66], using the same cre line, did not show reductions in body weight or body length despite 50% reductions in serum IGF-1 [66]. Similarly, liver-specific JAK2KO mice [67, 68] showed significant decreases in total cross-sectional area, bone area, and cortical thickness (Yakar S, personal statement). Reduced serum IGF-1 levels were also achieved by gene ablation of the acid labile subunit (Igf1als). ALS knockout mice (ALSKO) [69] showed ~60% reductions in serum IGF-1 levels. They were born at normal weight, but showed ~15% reductions in length [70]. As seen with LID mice, ALSKO mice exhibited slender bones and reduced BMD.
Overall, mouse models with reduced serum IGF-1 levels—such as LID, Liver-GHRKO, and ALSKO mice—show compromised cortical bone with marked reductions in total cross-sectional area, but only minor reductions in bone length. A genetic study comparing wild-type (two active IGF-1 alleles), IGF-1+/− (one IGF-1 active allele), IGF-1 null (two inactive IGF-1 alleles), and mice with liver IGF-1 production (LIP) has shown that endocrine IGF-1 plays significant roles in postnatal growth and contributes to approximately 30% of adult body size [71]. In a model expressing hepatic rat IGF-1 transgene (HIT), excess in endocrine IGF-1 levels were associated with increased radial bone growth with small increases in linear bone growth [55, 72, 73]. When HIT mice were crossed to an IGF-1 null background (IGF-1KO-HIT), where IGF-1 was produced only by the liver, mice grew to normal size and their bones were indistinguishable from controls both in morphology and mechanical properties [55, 72, 73], suggesting that serum IGF-1 can sustain growth in the absence of tissue IGF-1 production. The rescue of the IGF-1 null growth-phenotype was specific to IGF-1, as a similar strategy of crossing the IGF-1 null to IGF-2 transgenic (IGF-IKO-IGF-IITg) mice fail to rescue the growth retardation of the IGF-1 null mice [74]. Finally, when HIT mice were crossed to GHR null background (GHRKO-HIT) mice serum IGF-1 reached control levels, but was insufficient for rescuing the retarded growth of the GHR null mice [75]. This was due to compromised serum IGF-1 delivery system with reduced IGFBP-3 and the ALS proteins, as well as due to insufficient expression of IGF-1 in the bone tissue [75]. Overall, models with reduced/elevated serum GF-1 revealed that it regulates radial growth of the diaphysis in long bones and regulates overall skeletal BMD. Radial bone growth is an important determinant of bone strength.
GH/IGF-1 action in cells of the osseous system:
GH/IGF-1 effect on mesenchymal stem cells (MSCs)
The bone marrow (BM) stroma contains mesenchymal stem cells (MSCs), which are multipotent, adherent cells that can differentiate into osteoblasts, adipocytes, and chondroblasts [76]. However, MSCs are also found in adipose tissue [77, 78], the heart [77], trabecular bone [79], peripheral blood [80], and many other tissues. BM-MSCs comprise only 0.001% to 0.01% of the total cell number [81] and are usually identified by the expression of CD73, CD90, and CD105; they are also negative for the hematopoietic marker CD34 [82]. MSCs are distinct from the hematopoietic stem cell (HSC) lineage, which gives rise to osteoclasts (OCLs), the bone-resorbing cells. Factors that maintain the pluripotency of MSCs have not been fully discovered, but they include Sca-1 [83] and IL18 [84]. The molecular mechanisms that regulate MSCs’ commitment to becoming osteoblasts or chondrocytes are better understood and will be discussed below. However, it is worth noting that early-stage committed MSCs can also de-differentiate, depending on their local cellular microenvironment.
GH and IGF-1 stimulate the proliferation of MSCs and osteoprogenitors, but also promote their differentiation into osteoblasts (Figure 2). GH is also believed to be involved in MSCs’ commitment to becoming osteoblasts via its inhibitory effects on lipogenic genes. BM-MSCs— derived from human trabecular bone and cultured in lipogenic medium— provided with GH, showed inhibited lipid accumulation; decreased expression of C/EBPα, adiponectin, and acetylCoA carboxylase; and increased osteogenic factors like osterix and osteoprotegerin (OPG) [85]. These effects were associated with increased Wnt signaling, which is a “master switch” in MSCs commitment to becoming osteoblasts. Wnts play a widespread role in skeletogenesis including embryonic skeletal patterning, development and remodeling. Wnt10b was recognized as a regulator of fate choice towards OB via activation of the transcription factors runt-related transcription factor 2 (Runx2), distal-less homeobox 5 (Dlx5), and osterix [86, 87]. GH and IGF-1 are also known to increase the expression of bone morphogenetic proteins 2 and 4 (BMP2, and BMP4) [88, 89], which are involved in mesoderm induction, skeletal patterning, and limb morphogenesis [90–92]. BMP2 is currently being used to augment fracture repair.
Figure 2: The effects of GH and IGF-1 on cells of the osseous system.
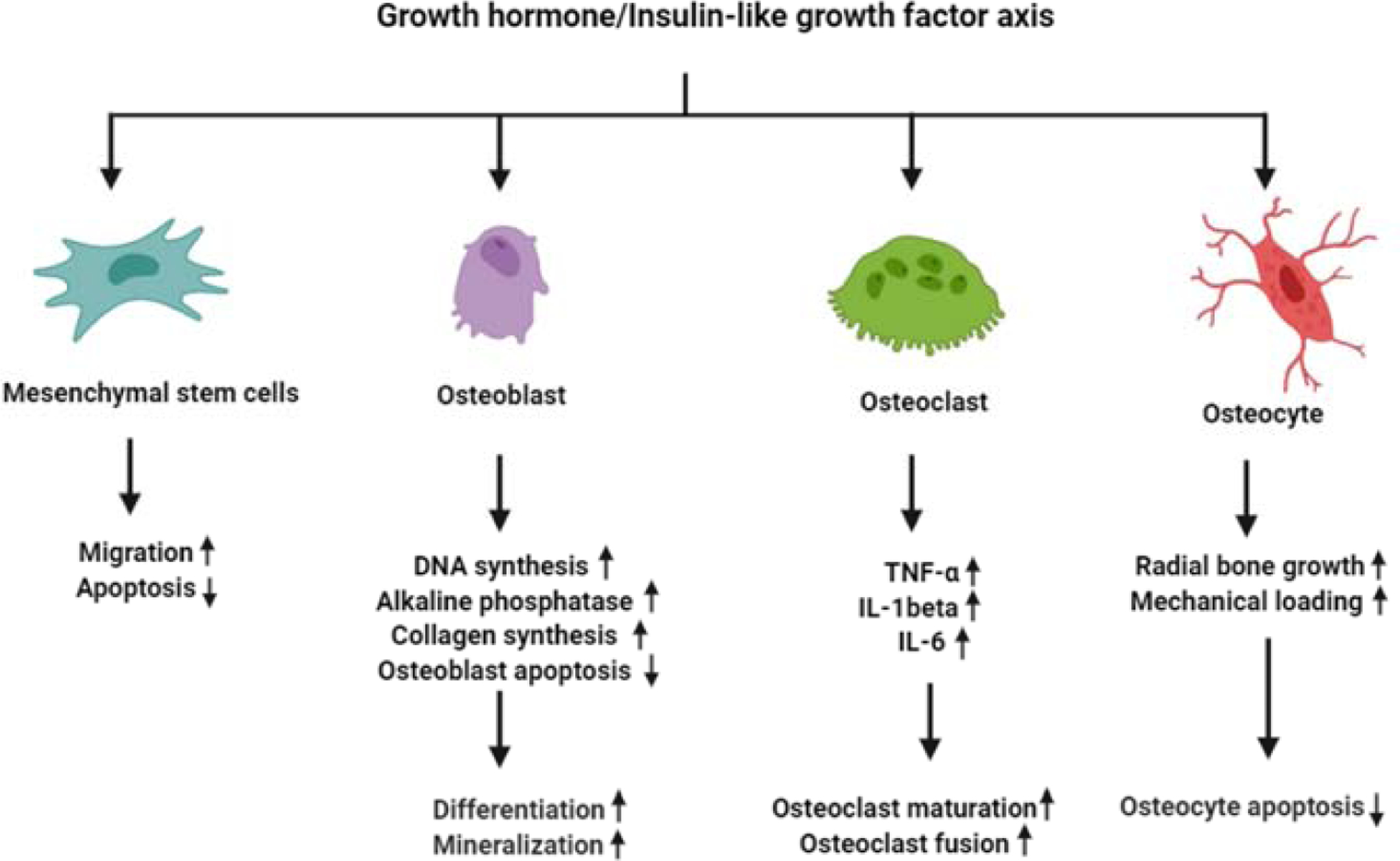
GH and IGF-1 stimulate the proliferation of MSCs and osteoprogenitors, and promote their differentiation into OB and chondrocytes. Activation of the IGF-1 and GH receptors in OB leads to increased OB proliferation, as evidenced by increased DNA synthesis. It also leads to OB cell differentiation, which is associated with increased collagen synthesis and elevated alkaline-phosphatase activity. Additionally, a mouse model of targeted inactivation of the IGF-1R in OB revealed the significant roles played by IGF-1 in matrix mineralization. Bone remodeling requires coupling of OB and OCL activity. Several mouse models have shown that IGF-1 also affects OCL maturation, fusion, and activity. The communication between OBs and OCLs is believed to be mediated by IGF-dependent expression of the membrane-bound ligand/receptor system ephrinB2/Eph4. Finally, GH IGF-1 receptors on osteocytes are important in regulation of radial bone growth and play significant roles in bone response to mechanical loading (not discussed in the current review).
GH and IGF-1 roles in the regulation of the osteoblast (OB) lineage:
Activation of the IGF-1R on MSCs triggers signaling cascades that lead to increased expression of the Runx2. Runx2, which is required for MSCs’ transition into osteoprogenitors, then activates osterix (osx), leading to the activation of osteoblastogenesis. In preosteoblasts, activation of the IGF-1R increases the expression of the proto-oncogene c-fos [93] and, via induction of osx, stimulates type I collagen synthesis, alkaline-phosphatase activity, and osteocalcin secretion [94, 95]. Additionally, IGF-1 activation of the mammalian target of rapamycin complex 2 (mTORC2, defined by Rictor)/AKT pathway activates Indian hedgehog (Ihh) and Gli2-mediated transcription [96], which are required for osteoblast differentiation.
In vivo, overexpression of human GH (hGH) [37, 97] or IGF-1 [98] under the osteocalcin promoter, specifically in OB, increased OB activity and enhanced collagen deposition. Similarly, overexpression of IGF-1, driven by the Col1a1(3.6) promoter (Col1a1(3.6)- IGF-1Tg) in late-stage differentiated osteoblasts caused increased bone remodeling, as evidenced by the thickening of calvarial bones and increases in femoral length and circumference [99, 100]. These observations were in agreements with data about transgenic mice expressing the PAPP-A protease, which increases IGF-1 bioavailability, under the Col1a1(2.3) promoter (Col1a1(2.3)-PAPP-ATg mice). Col1a1(2.3)-PAPP-ATg mice showed increased skeletal growth, which was associated with increased osteoid (unmineralized collagen) surface and bone formation rate [101]. By contrast, overexpression of IGFBP-4 [102], or IGFBP-5 [103] under the osteocalcin promoter in late-stage differentiated osteoblasts, significantly reduced IGF-1 bioavailability, resulting in decreased bone turnover and BMD. Accordingly, inactivation of the GH/IGF-1 axis in the OB lineage compromised bone matrix deposition and mineralization. Indeed, ablation of the IGF-1R in differentiated osteoblasts, using the osteocalcin promoter-derived Cre recombinase (OC-IGF-1RKO mice), resulted in significant reductions in trabecular bone volume and bone mineralization. This indicated that IGF-1 plays a role in skeletal mineralization [104, 105].
Finally, ablation of the IGF-1R early in the OB lineage, using the osx promoter-driven Cre (Osx-IGF-IRKO), resulted in impaired chondrogenesis, secondary ossification, trabecular bone formation, and compromised linear bone growth [106]. On the other hand, ablation of the GHR ([107]), IGF-1R ([108]), or IGF-1 [109] late in the OB lineage (osteocytes), using the DMP-1 promoter-driven Cre (DMP-GHRKO, DMP-IGF-IRKO, DMP-IGF-1KO, respectively), resulted in significant decreases in cortical bone area but only minimal (or negligible) effects on linear bone growth, suggesting that the GH/IGF-1 axis regulates radial bone growth partially via its actions on osteocytes.
GH and IGF-1 roles in the regulation of osteoclasts (OCL):
Clinical studies have shown that excess GH, and consequent elevations in serum IGF-1 (as seen in patients with acromegaly), were associated with increased bone turnover [110, 111]. Likewise, short-term GH treatment stimulated bone formation and bone resorption in postmenopausal women with osteopenia [112]. Ex-vivo experiments using fetal mouse metacarpal or metatarsal organ cultures showed that addition of IGF-1 increased resorption activity [113]. Furthermore, IGF-1 stimulated bone resorption in a dose-dependent manner even in fully differentiated, mature osteoclasts (OCLs) [114]. In contrast, IGF-1KO mice exhibit reduced OCL progenitors, inhibited fusion-induced multinucleation, and reduced OCL resorption activity [115]. Taken together, these studies indicate that GH/IGF-1 axis plays a role in OCLs formation and activity.
Bone resorption and formation are coupled and require communication between OCLs and OBs [116]. The communication between OBs and OCLs is facilitated by a membrane bound ligand, ephrin B2, and its receptor, EphB4. The EphB4 receptor belongs to a subfamily of receptor tyrosine kinases and can be activated by ephrin B2. Interactions between EphB4/ephrin B2 trigger bidirectional signaling such that the extracellular domains of ephrin B2 on one cell activate the tyrosine kinase domains of the EphB4 receptors on the adjacent cell. Further, the intracellular domains of ephrins engage the Src family kinases and other effector molecules. Members of the Eph/ephrin family are expressed on osteoprogenitor cells as well as on differentiated bone cells. As indicated above, expression of Eph receptors on OBs, and ephrins on OCLs and OBs, are implicated in coupling bone formation and bone resorption [117, 118]. Studies in vitro showed that OCL-derived IL-6 induces IGF1 and EphB2 expression in OCLs— and also induces EphB4 expression in OBs—leading to increased OB differentiation [119]. Igf1 gene ablation in OCLs using the TRACP-Cre (TRACP-IGF-1KO) did not affect femur length, body weight, cortical bone thickness or volume, or serum levels of IGF1 and IGFBP3. However, OCL of TRACP-IGF-1KO mice showed a marked decrease in EphB2 expression on OCLs and EphB4 expression on OBs [120]. IGF1 has been shown to be essential for OCL differentiation and to maintain OCL function [121, 122]. Accordingly, OCL formation, the number of nuclei per OCL, and the bone resorption capacity of OCLs in TRACP-IGF-1KO mice significantly decreased compared with controls, suggesting that OCL-derived IGF1 has autocrine effects on OCL formation and activity [120].
In summary, it appears that GH and IGF-1 play significant roles in bone modeling during early development, as well as bone remodeling during growth and aging. In general, these two anabolic hormones tip the scale towards bone formation and mineral acquisition, but in excess, they can stimulate bone resorption and impair bone remodeling.
GH/IGF-1 axis and postnatal longitudinal bone growth- actions in chondrocytes of the growth plate:
Longitudinal bone growth is determined by the activity of chondrocytes in the growth plate. There, they undergo proliferation, differentiation, hypertrophy, and apoptosis. Growth plates are found at the ends of long bones, and in humans (but not in rodents) are fused after puberty, when growth ceases. Chondrocytes of the growth plate reside in three major zones: the germinal, proliferating, and hypertrophic zones (Figure 3). Chondrocytes in the germinal zone express parathyroid hormone-related protein (PTHrP) and a panel of markers for skeletal stem and progenitor cells [123]. Using an elegant approach of cell-lineage tracing, it was demonstrated that PTHrP-positive chondrocytes in the germinal zone give rise to columnar chondrocytes and undergo hypertrophy, and even give rise to osteoblasts and marrow stromal cells beneath the growth plate [123, 124]. Chondroprogenitors in the resting zone are gradually depleted. Using a multicolor-reporter system for clonal tracing it was demonstrated that longitudinal growth during the fetal and neonatal periods involves depletion of chondroprogenitors [125]. However, later in life, chondroprogenitors acquire the capacity for self-renewal. Regulation of the pool of self-renewing progenitors in the germinal zone involves activation of the hedgehog and mammalian target of rapamycin complex 1 (mTORC1) pathways [125]. Chondrocytes of the proliferating zone actively divide, are organized in columns, and secrete proteoglycans. The proliferating zone has a rich blood supply, allowing nutrients and hormonal signals to reach the cells in that zone. Finally, chondrocytes of the hypertrophic zone participate in mineralization of cartilage and, most importantly, determine bone length.
Figure 3: The growth-plate.
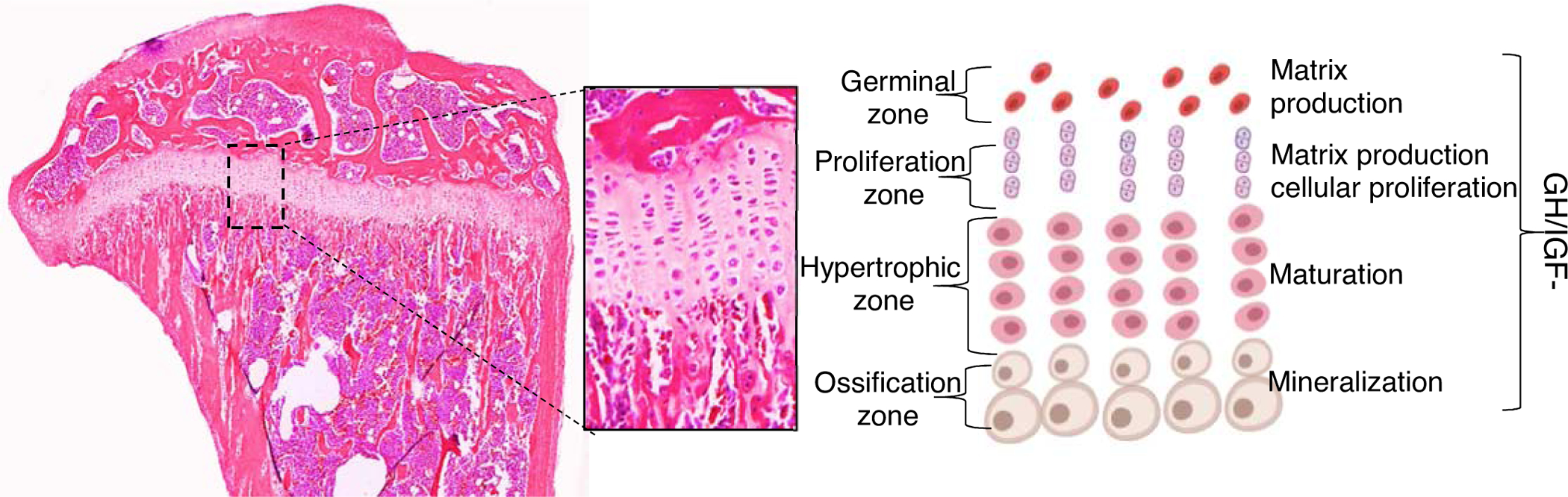
The effects of the GH/IGF axis on chondrocytes of the growth plate were extensively studied (and described in the current review). GH elicits direct and IGF-dependent effects on chondrocytes in all zones of the growth plate. However, IGF-1 actions were mostly detected at the proliferative zone, while those of GH were localized to the germinal zone.
Several clinical and experimental studies have demonstrated the essential roles of the GH/IGF axis in the growth plate. A study from 1940 showed increased linear bone growth in adult rats injected with extracts of the anterior pituitary [126]. A similar approach was taken using hypophysectomized (hypox) rats treated with GH, which also exhibited increased height of the growth plate and cartilage thickness [127]. Forty years later, a key experiment in which GH was injected locally into one hindlimb of hypox rats, while the other hindlimb served as a control, revealed local and direct roles of GH on chondrocytes in the growth plate [128]. Likewise, using hypox rats it was shown that chondrocytes in all zones of the growth plate responded to infusion of GH or recombinant human IGF-1 through osmotic pumps. This was evidenced not only by increased chondrocytes’ cell volume and matrix production but also by modulations in the cell cycle time [129]. GH increases chondrocyte proliferation in vivo and in vitro [130]. Supplementation of GH in culture medium without serum (avoiding an exogenous source of IGF-1) induced chondrocyte proliferation, suggesting direct effects of GH on chondrocytes [131]. Primary chondrocyte cultures, established from epiphyseal growth plates of the proximal tibia of hypox rats injected with GH (but not IGF-1) 24 hours before dissection, increased clonal expansion of chondrocytes in the growth plate [132]. Supplementation of IGF-1 to chondrocyte cultures primed with GH increased the size of chondrocyte colonies [132].
Using hypox rats, several studies established the concept that GH acts directly on chondrocytes to induce local production of IGF-1 and stimulate the proliferation and clonal expansion of chondrocytes [133, 134]. Local effects of GH were further evidenced in studies that showed that passive immunization against IGF-1 did not abolish the GH-induced growth of dwarf rats [135]. In humans, GHR is also expressed in the growth plate chondrocytes. Its expression is maturation-dependent in both intact tissue and primary cultures [136]. In vitro studies showed that chondrocytes from different zones of the growth plate responded differently to GH and IGF-1 stimulation [137]. Most affected cells were from the hypertrophic and proliferative zones, while resting zone cells were the least affected by GH and IGF-1 treatment. These distinct effects were attributed to zone- and age-specific expression of GHR, IGF-1, and IGF-1R in chondrocytes of the growth plate [138, 139].
The distinct and overlapping effects of GH and IGF-1 on growth plate chondrocytes were shown in vivo in a key experiment using GHR and IGF-1 knockout (GHRKO, IGF-1KO, respectively) mice [140]. IGF-1KO mice, which have high levels of GH, showed increases in the thickness of the germinal zone and significant reductions in the hypertrophic zone. However, chondrocyte proliferation and numbers in the proliferation zone were normal in IGF-1KO mice. In contrast, the germinal zone of the GHRKO mice was hypoplastic. Chondrocyte proliferation and number, as well as chondrocyte hypertrophy, were all reduced, suggesting that GH has direct and IGF-1-independent effects on chondrocytes of these zones. Furthermore, it was found that in the IGF-1KO, but not in the GHRKO mice, IGF-2 was upregulated, indicating that GH may exert its activities in growth plate partially through stimulation of IGF2 [140].
Ablation of the GHR mediator, STAT5ab in mice [33] [141], resulted in ~10% decreases in bone length in adult mice. These decreases were attributed to abnormalities in the growth plate detected as early at 2 weeks of age. In contrast, SOCS2KO mice, with enhanced GH action, show increased height of the proliferative and hypertrophic zones of the growth plate. Inhibition of excess GH production in the SOCS2 null mice, by means of crossing them with the lit/lit mice (which do not produce GH), normalized growth [27].
Chondrocyte specific IGF-1 knockout mice were generated by crossing the floxed IGF-1 mice with mice expressing the Cre recombinase transgene under the control of the procollagen type II alpha I promoter (Col2a1-IGF-1). Col2a1-IGF-1 mice showed decreased bone length and diameter, reduced growth plate chondrocytes proliferation, reduced height of the hypertrophic zone, and reduced BMD as compared to controls [142]. These morphological changes were associated with decreased expression of the parathyroid hormone related protein (PTHrP), parathyroid hormone receptor (PTH1R), Dlx-5, SRY-box containing gene-9 (Sox9), and IGFBP5 in the growth plate. A conditional deletion of the Igf1r in chondrocyte using the same Col2a1 promoter (Col2a1-IGF-1RKO mice) also showed growth arrest with reduced body length and shortened bones [143]. This was associated with minimized proliferation and hypertrophic differentiation, as well as increased apoptosis of chondrocytes and delayed endochondral ossification [143].
The severe phenotype of Col2a1-IGF1RKO mice resulted in intrauterine or early postnatal death, which made it impossible to understand the roles of IGF-1R specifically during postnatal growth. To overcome this difficulty, a tamoxifen (Tam)–inducible Col2a1-driven Cre recombinase (Col2a1Tam-cre) mice were used along with the Igf1r floxed mice. Col2a1Tam-IGF-1RKO mice that were depleted of Igf1r five days after birth showed retarded growth due to the delayed ossification, shortened bones, reduced cell proliferation and survival, and disorganized chondrocyte columns in the growth plate. This disorganization was accompanied with increased Pthrp and decreased Indian Hedgehog (Ihh) gene expressions [144]. These data were in line with the previous findings based on congenital Col2a1-IGF-1RKO mice, which showed partial disruption in the Ihh signaling pathway and severely affected linear bone growth [145]. Col2a1Tam-IGF-IRKO mice that were depleted of the IGF-1R at four weeks of age (the beginning of pubertal growth) also showed retarded growth. However, these mice responded to GH treatment as evidenced by increases in NF-kBp65 and BMP-2. They also showed increased linear bone growth and increased chondrocytes proliferation and differentiation, suggesting that GH actions on chondrocytes in the growth plate are at least partially IGF-1-independent [146].
As mentioned above, the chondro-osseous junction in the growth plate is rich with vasculature. Osteoclast activity at this site is important for vascular formation and invasion, which is followed by calcification of the cartilage. These events require chondrocytes to communicate with osteoclasts. The communication between these cells is facilitated by ephrin B2 and its receptor EphB4. Chondrocytes of the Col2a1Tam-IGF-1RKO mice show decreased expression of ephrin B2, EphB4, and the receptor activator of the NF-kB (RANK) ligand (RANKL), which is required for osteoclast activity [147]. In fact, the increase in trabecular bone volume, and concomitant decrease in osteoclast count in the metaphysis of the congenital IGF-1KO mice, may be explained by impaired chondrocyte-osteoclast interactions due to reduced EphB4/ephrin B2 expression. In summary, GH/IGF-1 regulate linear growth by binding to their cognate receptors on chondrocytes in the growth plate. Additionally, IGF-1 regulates the expression of other mediators, like EphB4/ephrin B2, that are essential for cellular communication in the growth plate.
Finally, we note that interpretation of the data coming from “cartilage-specific” knockouts is complicated by recent findings showing that growth plate chondrocytes can undergo trans-differentiation into osteoblast. The roles of GH/IGF-1 in that process are yet to be discovered. A recent study has demonstrated that the transcription factor Sox9 may play a role in chondrocytes’ trans-differentiation. Sox9 is expressed along the germinal and proliferating zones of the growth plate while its levels are naturally diminished in the hypertrophic zone. This physiological down regulation of Sox9 in hypetrophic chondrocytes was associated with increased expression of osteoblasts’ specific genes such as Col1a1, MMP13, MMP9, and Sp7, indicating that downregulation of Sox9 may induce trans-differentiation. In contrast, over expression of Sox9 across all chondrocytes of the growth plate inhibited the naturally enhanced expression of osteoblasts’ specific genes in hypertrophic chondrocytes and inhibited trans-differentiation [148]. Another example came from studies of endochondral ossification at the secondary ossification center of the femur in young mice. Using cell tracing approaches along with hypothyroid mouse model, the authors revealed the roles of thyroid hormone in chondrocytes to osteoblast trans-differentiation during the formation of the secondary ossification center [149]. Another study that followed the fate of hypertrophic chondrocytes used the Col10a1-Cre or the tamoxifen-induced aggrecan (Agc1)-CreERT2 along with EGFP, LacZ or Tomato expression [150]. Following labeling of chondrocytes, during prenatal and early postnatally, abundant labeled non-chondrocytic cells were present in the primary spongiosa, distributed along trabeculae surfaces, and later were present in the endosteum, and even embedded within the bone matrix. Overall, it is estimated that about sixty percent of all mature osteoblasts were originated from Col10a1-expressing hypertrophic chondrocytes in endochondral bones of one-month old mice. A similar process of chondrocyte to osteoblast transdifferentiation was involved during bone fracture healing in adult mice [150]. All together, these data support the notion that hypertrophic chondrocytes may act as a source of osteoprogenitors both during development and adulthood.
GH/IGF-1 actions in chondrocytes of the articular cartilage:
Cartilage is a connective tissue that supports and cushions for adjacent tissues. Articular cartilage, in particular, is a hyaline cartilage between 2 and 4 mm thick, found in diarthrodial joints (synovial joints). Articular cartilage serves as a friction-reducing and load-bearing cushion in synovial joints and is vital for mammalian skeletal movements. This cartilage is composed of a dense extracellular matrix (ECM) with a sparse distribution of highly specialized cells called chondrocytes. This tissue does not contain blood vessels, nerves, or lymphatics, and thus has a limited capacity for intrinsic healing and repair. The superficial zone of the articular cartilage, which has direct contact with the synovial fluid contains a relatively large number of flattened chondrocytes and articular cartilage progenitor cells. These cells play imperative roles in the protection and maintenance of the whole cartilage. Therefore, preservation and health of articular cartilage is essential for healthy joints, particularly during aging.
GH has direct and IGF-mediated effects on articular chondrocytes. Articular chondrocytes from young rats exhibited dose-dependent effects of GH on DNA synthesis and IGF-1 expression and secretion to the conditioned media. Using an IGF-1 antibody in the same settings did not completely neutralize the effects of GH, suggesting that it acted independently of IGF-1 [151].
There are limited studies on the effects of GH on the development and homeostasis of healthy articular cartilage. However, numerous studies of models of damaged cartilage (as discussed below) revealed an association between GH levels and/or actions in articular cartilage. Nonetheless, GH actions particularly in articular cartilage are yet to be discovered.
GH/IGF-1 in osteoarthritis
Osteoarthritis (OA) is the most prevalent progressive and chronic degenerative joint disease affecting around 250 million people throughout the world [152]. Approximately 67 million Americans aged >65 years will suffer from OA by 2030 [153]. OA mostly affects the joints, and is a major contributor to musculoskeletal pain and disability.
OA is characterized by progressive deterioration of articular cartilage due to chondrocytes’ reduced or lost ability to produce ECM. This leads to changes in the entire synovial joint structure, including the synovium, meniscus, ligaments, adipose tissue, and subchondral bone [154]. During the early stages of OA, compensatory mechanisms induce ECM synthesis and chondrocyte proliferation [155]. Nonetheless, the newly synthesized, reparative matrix typically has abnormal molecular composition and altered structure [156]. This is evidenced by reduced aggrecan content, which is replaced by collagen [157]. Additionally, collagen synthesis shifts from type II collagen to type I collagen [158]. These structural and compositional changes in the ECM not only increase mechanical fault, but also activate catabolic pathways at the joint. Increased activities of NF-kB [159], interleukin 1β (IL-1β), tissue necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS) [160], IL-6 [161] IL-12 [162], IL-15 [163], IL-17 [164], IL-18 [165] and various chemokines, which all play significant roles in the progression of OA, are observed. Subsequently, chondrocytes increase the expression of matrix-degrading proteinases including matrix metalloproteinase (MMPs) −1, MMP-13 [166], and aggrecanases, among them, a disintegrin and metalloproteinase with a thrombospondin type 1 motif (ADAMTS)-4 [167], and ADAMTS-5 [168]). Once the catabolism in chondrocytes exceeds anabolism, cartilaginous tissue is completely lost. At later stages of OA, in an attempt to repair the damaged tissue, chondrocytes undergo aberrant cellular differentiation leading to the formation of osteophytes [169] or fibrosis [170]. As mentioned previously, the etiology of OA is multifactorial [171]. However, it is the failure of chondrocytes to balance the synthesis and degradation of ECM in cartilage that leads to OA.
The GH/IGF-1 axis is a vital component of both embryonic and postnatal skeletal/cartilage development, which has prompted researchers to investigate its relevance to OA. Several studies have investigated the possible association between the GH/IGF-1 axis and the severity of OA (Figure 4). A study of post-menopausal women with primary OA in the hip showed increased fasting serum GH as compared to age matched controls [172]. Similarly, symptomatic OA patients displayed increased levels of serum GH, but reduced IGF-1, when compared to asymptomatic OA or healthy individuals [173]. Further, a comparison of healthy and OA individuals controlled for demographic characteristics reported higher serum GH levels but lower IGF-1 levels among OA patients [174]. In patients with rheumatic disorders, (including rheumatoid arthritis, OA, gout, pseudogout, and diffuse idiopathic skeletal hyperostosis) GH levels in the synovial fluid increased significantly, while those of IGF-1 were markedly decreased. This indicated that elevated GH levels in the synovial fluid play roles in the pathogenesis of joint diseases [175]. Likewise, human OA chondrocytes, which express the GHR, were resistant to hGH treatment as evidenced by its lack of effects on cell viability, metabolic activity, collagen type 1 or 2, and even IGF-1 or 2 gene expression [176]. A study of symptomatic OA patients reported an association between polymorphism of the GH receptor (exon 3 deletion, d3-GHR) and OA, independent of age and BMI [177]. The product of this polymorphism in the GHR [178], called the d3-GHR isoform, augments GH signaling by nearly 30% more than the full-length isoform [179]. Interestingly, despite the increased expression of IGF-1 receptors (IGF-1R), chondrocytes of the OA cartilage were less responsive to IGF-1 [180]. Accordingly, increased levels of IGF-1 or IGF-2 in the fibrillated chondrocytes from OA cartilage did not activate the IGF-1R. On the other hand, increased production of IGF binding proteins (IGFBP) by OA chondrocytes, including IGFBP3 and IGFBP5, was associated with reduced IGF activity in the cartilage [181]. Taken together, OA chondrocytes show IGF-1 resistance, and IGFBPs play a vital role in the pathogenesis of OA, likely via sequestration of IGF-1. On the other hand, a modest association was reported between increased serum IGF-1 levels and bilateral knee OA or distal interphalangeal (DIP) joint disease in middle-aged women [182].
Figure 4: The effects of GH/IGF-1 axis on OA initiation and progression.
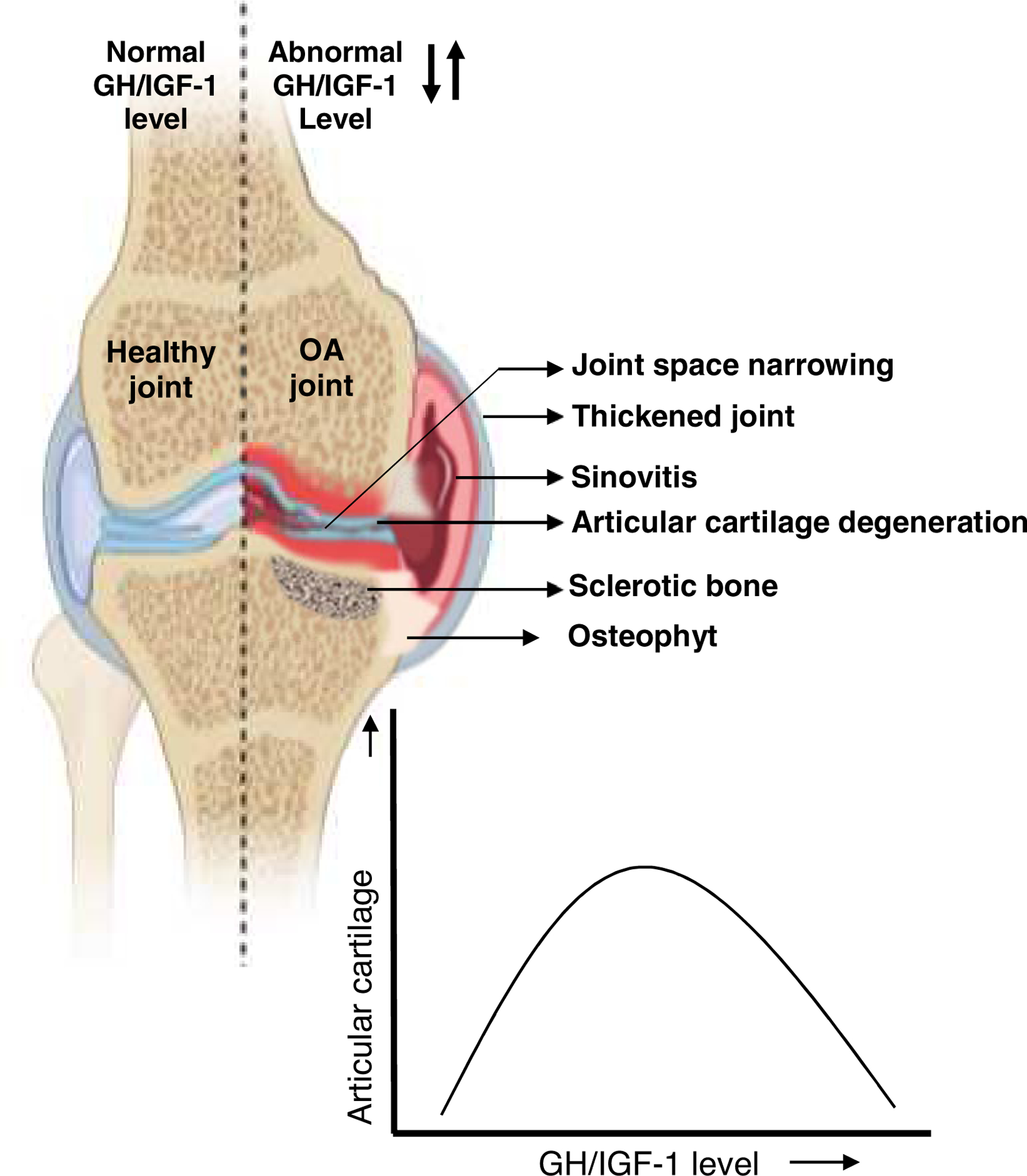
OA is characterized by a progressive degeneration of the articular cartilage, leading to changes in the entire synovial joint structure. These include narrowing of the joint space, sinovitis, deterioration of subchondral bone volume and structure, and formation of osteophytes. The roles of the GH/IGF-1 axis in OA are not fully understood. However, studies have shown a bell-shaped relationship between OA and GH/IGF signals, where too-low or too-high levels of these two hormones are associated with the joint disease.
Preclinical studies have also showed a relationship between the GH/IGF axis and OA. A study based on radiographic images revealed that increased hypophyseal GH production was associated with arthritis-like phenotypes in both intact or adrenal- and gonad-excised rats [183].
Injection of mice and rats with the malignant pituitary somatotroph cell line (mGH3), which produces excess GH, led to the development of osteoarthritis-like features. This was evidenced by reduced type II collagen and increased PCNA positive cells, indicating the proliferation of chondrocytes and osteoblasts, which then formed osteophytes. Compared to controls, GH-injected rats displayed increased IGF-1 and IGF-1R expression in the knee joint, synovial, perichondral, chondral, and osteophyte cells, suggesting that IGF-1 was involved in osteophyte formation via an autocrine and/or paracrine process [184].
During the progression of OA, abnormal remodeling of subchondral bone increases mineral density resulting in sclerosis and severe cartilage degradation [185]. Accumulation of free IGF-1 and prostaglandin E2 proteins in the subchondral bone of OA patients inhibited the expression of parathyroid hormone receptor (PTH-R) in osteoblasts. The PTH resistance of these OA osteoblasts is believed to impair PTH-dependent cyclic adenosine monophosphate (cAMP) signaling and to induce sclerosis [186]. OA synovial fluid was shown to contain elevated levels of phospholipids including phosphatidylcholine, lysophosphatidylcholine, and sphingomyelin, which mediate cartilage deterioration [187]. IGF-1 activation of fibroblast-like synoviocytes (FLS) induced the production of phospholipids and increased the severity of OA [188]. OA chondrocytes produce significantly more IGF-1 than healthy cells [189]. A recent study, it was reported that elevated local IGF-1 production increased the expression of pro-inflammatory cytokines and induced apoptosis and autophagy of chondrocytes. Inhibition of IGF-1 through miR-206 and the activation of the PI3K/AKT-mTOR signaling pathway in OA chondrocytes not only reduced the expression of pro-inflammatory cytokines but also reduced cell proliferation. Thus, IGF-1 signaling in OA chondrocytes was found to be largely regulated by the level of miR-206 [190].
A few studies have reported a negative association between GH/IGF-1 and OA. Chronic deficiency of GH and IGF-1 in dwarf rats adversely affected the integrity of the knee articular cartilage resulting in OA-like lesions. These observations were intriguing because age-associated OA in humans correlates with age-induced decrease in GH and IGF-1 levels [191]. Likewise, homozygous germline mutations in the GH-releasing hormone receptor (GHRHR) lead to GH deficiency [192]. In humans, when untreated with GH, this mutation leads to OA in the hips, with narrowed joint space and increased osteophyte formation [193]. Similarly, reduced IGF-1 levels in OA chondrocytes were accompanied by increased expression of the programmed cell death 5 (PDCD5) protein, suggesting that IGF-1 plays protective roles in chondrocytes [194]. Moreover, OA chondrocytes showed a reduced response to IGF-1. Endoplasmic reticulum (ER) stress resulting from the accumulation of misfolded and/or unfolded proteins in chondrocytes was associated with reduced IGF-1 signals. Growth arrest and DNA-damage inducible gene 153 (GADD153) and Tribbles (TRB)3, markers of ER stress, were elevated in OA chondrocytes compared to healthy cells. This study demonstrated that TRB3 inhibited the IGF-1 stimulation of Akt phosphorylation in chondrocytes, blocked proteoglycan synthesis, and reduced the likelihood of cell survival [195]. Interestingly, suppression of IGF-1 expression and activity in the fetus due to the prenatal nicotine exposure increased the susceptibility to develop OA in the adult offspring of rats. During strenuous running, prenatal nicotine-exposed offspring displayed cartilage destruction due to reduced AKT1/2 activation and decreased COL2a1, aggrecan, and SOX9 production by chondrocytes [196].
Finally, there were several studies showing a lack of association between IGF-1 and OA. Despite overall reduction in serum IGF-1 levels in subjects with OA as compared to healthy controls, no relationships were found after adjustment for age-related changes in IGF-1 levels [197, 198]. Likewise, the longitudinal Framingham osteoarthritis study, carried out for more than 20 years, also found a negative correlation between serum IGF-1 levels and age. Despite increases in OA incidence with age, however, an association between serum IGF-1 and the incidence or progression of OA in the knee was not established [199]. In mice, ablation of liver IGF-1 production, using the Mx1-cre/loxP system, resulted in significantly decreased serum IGF-1 levels. Surgical destabilization of the medial meniscus (DMM) of the knee did not intensify the cartilage damage in those mice, indicating that serum IGF-1 had no protective role in articular cartilage [200].
In summary, as with most hormones, there are U-shaped relationships between GH/IGF and OA, where too-low or too-high levels of these hormones lead to impaired cartilage integrity.
GH/IGF-1 in the treatment of OA
Several molecules have emerged as potential therapies, with hopes to alter the progression of OA. However, current approaches merely reduce pain and stiffness and improve joint function for a limited time. They cannot truly stop or reverse the degenerative process. In recent years, anti-catabolic therapeutic agents that target anabolic signaling pathways in cartilage and bone have been more closely studied as a possible treatment strategy.
Since GH/IGF-1 are known to modulate chondrocyte metabolism and exert effects on cartilage growth and maintenance, they are considered as possible candidates for OA treatment. The earliest report of GH application in OA treatment in a clinical setting was published in 1975. Human GH was directly injected into the knee joint of two women with moderate symmetrical OA at the age of 56 and 73 years. Unfortunately, clinical improvement was not observed even after 8 weeks of treatment [201]. In a porcine model of OA, GH had beneficial effects on damaged cartilage. It promoted healing of established articular cartilage defects by stimulating cell proliferation, new collagen synthesis, and preservation of the polysaccharide matrix [202]. Further, in a rabbit model of cartilage defect, GH increased mitosis of chondrocytes [203]. In aged rats with OA of the mandibular condyle cartilage—where irregularities and clefts along the articular cartilage could be observed—GH supplementation caused augmented cell proliferation and sulfated proteoglycan synthesis and mineralization [204]. Interestingly, intra-muscular administration of recombinant equine GH significantly elevated IGF-1 in the serum and synovial fluid of the horse. In exercising horses, elevations in IGF-1 were associated with reductions in glycosaminoglycan release in the synovium, without changes in aggrecan levels, and reduced progression of OA in the knee [205]. In a rat model of temporomandibular joint (TMJ) OA, local injection of recombinant human GH increased IGF-1 levels in the synovial fluid but not in serum. GH repaired both articular cartilage as well as subchondral bone in the damaged TMJs, resulting in improved OA scores without affecting systemic bone growth [206].
Besides GH, other factors were also reported to increase the concentration of IGF-1 in the knee joint of OA animal models. In a dog model of OA, following injection of the protease inhibitors pentosan polysulfate (intra-muscular) and PB-145 (intra-articular), IGFBP-3 and IGFBP-5-bound IGF-1 levels in the joint space were elevated [207]. Increased IGF-1 levels were associated with reduced collagenase levels and reduced eroded cartilage in the injured joints. Similarly, overexpression of IGF-1 in encapsulated chondrocytes or cartilage explants led to production of cartilage-like matrix. Production of IGF-1 in OA cartilage using the recombinant adeno-associated virus (rAAV) increased cell proliferation and proteoglycan and type II collagen synthesis. This was associated with reduced IGFBP-3 and IGFBP-4 expression, increased IGFBP-5 and IGF-1R expression, and activation of the MAPK/ERK-1/2 and PI3K/Akt pathways [208].
Because of the structural and functional complexity of articular cartilage, combination of GH or IGF-1 with other agents has been more effective in OA treatment. Intra-articular co-injection of HA and GH provided compelling evidence for the prevention and progression of OA in a rabbit model. This co-injection not only reduced the duration and severity of lameness, but also improved the macroscopic and histopathological scores of cartilage damage [209]. HA is a high molecular weight molecule found within the synovial fluid and cartilage forming the backbone of the large proteoglycan aggregates [210].
Pentosan polysulfate (PPS) is a heparin-like semi-synthetic polysaccharide ester derived from beechwood hemicellulose. PPS is considered a disease-modifying OA drug [211]. Intra-articular co-injection of PPS and IGF-1 to a dog model of OA inhibited ECM-degrading enzymes and stimulated both ECM synthesis and the anabolic activity of IGF-1 [212]. The OA-like lesions present in the mandibular condyle cartilage of aged animals were repaired when the condyle explants were cultured in the presence of TGF-beta1 along with IGF-1. TGF-beta1 and IGF-1 increased the height and area of toluidine blue staining and incorporation of sulfate, demonstrating that combination of these two growth factors improved proteoglycan synthesis, an important element of the repair process in OA [213].
Osteogenic protein-1 (OP-1, BMP7) is an anabolic growth factor that induces the synthesis of ECM in chondrocytes [214]. IGF-1 alone in the medium did not change the cellular activities of human OA chondrocytes in culture. However, addition of OP-1 increased cells’ sensitivity to IGF-1 and improved cell survival and proteoglycan synthesis, suggesting that the combined therapy is an effective strategy for OA treatment [215]. Transplantation of rabbit chondrocytes which exhibited co-overexpression of human IGF-1 and FGF-2 in a model of articular cartilage defect enhanced early repair events and protected against further degeneration of both cartilage and neighboring subchondral bone [216]. Combination of dexamethasone with IGF-1 was more effective than IGF-1 alone in preventing the degradation of IL1α cytokine-mediated cartilage degradation. Dexamethasone reduced the catabolic activities of IL-1α by suppressing the expression of ADAMTS3 and 5, MMP3 and 13, COX2, Furin, PACE2, and caspase-3. Meanwhile, IGF-1 activated the anabolic activities in cartilage chondrocytes via up-regulation of aggrecan and type II collagen [217].
For the effective treatment of any disease, the drug of choice must be delivered to the target cells efficiently. Fusion of the human IGF-1 gene with the heparin binding domain (HB-IGF-1) increased the targeted delivery and retention time of IGF-1 in cartilage, prolonged the stimulation of proteoglycan synthesis, and repaired the damaged cartilage in a rat model of OA [218]. Likewise, bio-conjugation of IGF-1 with PEGylated polyamidoamine (PAMAM) dendrimer nanocarriers improved the pharmacokinetics and efficacy of IGF-1. It increased the half-life of IGF-1 and successfully carried IGF-1 through the full thickness of the cartilage. In a rat model, increased bioactivity of IGF-1 rescued the cartilage from degeneration in OA induced by transection of the ACL and meniscectomy of the medial meniscus. Osteophyte formation and reduced inflammation in the synovium suggested that bio-conjugation protected both cartilage and bone [219].
In summary, the ability of both GH and IGF-1 to regulate anabolic and catabolic activities in articular cartilage makes them potent therapeutic agents/targets to treat OA. An appropriate delivery system to carry GH/IGF-1 to the complex structure of articular cartilage must also be considered to increase their action during cartilage repair.
Summary:
Cells of the osseous system express the GHR and the IGF-1R and secrete IGF-1 that acts in an autocrine/paracrine fashion. Activation of the GH/IGF-1 axis in osteogenic cells is essential for their differentiation and maturation. GH/IGF-1 stimulate proliferation and differentiation of chondrocytes in the growth plate, thus mediating linear bone growth. The roles of this axis in chondrocytes of the articular cartilage are less clear and likely involve a bell-shape relationship where too low or too high activities of GH/IGF-1 can be deleterious. GH/IGF-1 enhance OB differentiation, collagen secretion, and bone matrix mineralization were established in numerous studies. IGF-1 also affects OCL formation and activity and plays essential roles in bone remodeling by coupling OB-induced bone formation and OCL-mediated bone resorption through activation of the ephrinB2/Eph4 system. In osteocytes, the terminally differentiated OBs, IGF-1 is an established transducer of mechanical stimuli, promoting an anabolic response in bone.
Overall the GH/IGF axis plays central roles in the regulation of skeletal growth and mineral acquisition during development and growth. Human mutations of the GH/IGF-1 axis are rare. Inactivating mutations often present with retarded growth and skeletal abnormalities, while excess activation of this axis presents with abnormal increases in skeletal morphometric parameters and BMD. GH exhibits IGF-dependent and independent effects on the skeleton. It regulates the largest pool of endocrine IGF-1, serum, via its actions in liver. IGF-1, in turn, acts in an endocrine and autocrine/paracrine fashion. Its bioavailability in tissues, including bone, is controlled by IGFBPs and IGFBP-proteases. Reduced serum IGF-1 levels compromise not only linear growth, but also radial bone growth, which is an important determinant of bone mechanical properties. Notably, the GH/IGF-1 axis interacts with other hormones, such as sex steroids, the parathyroid hormone, insulin, and osteocalcin to regulate bone metabolism and whole-body metabolic homeostasis.
Highlights:
GH/IGF axis plays central roles in the regulation of skeletal growth and mineral acquisition during development and growth
Inactivation of GH or IGF-1 actions often present with retarded growth and skeletal abnormalities, while excess activation presents with abnormal increases in skeletal morphometric parameters and BMD.
Cells of the osseous system express the GHR and the IGF-1R and secrete IGF-1 that acts in an autocrine/paracrine fashion.
GH and IGF-1 stimulate proliferation and differentiation of chondrocytes in the growth plate, thus mediating linear bone growth.
IGF-1 enhances OB differentiation, collagen secretion, and bone matrix mineralization
IGF-1 affects OCL formation and activity and plays essential roles in bone remodeling by coupling OB-induced bone formation and OCL-mediated bone resorption through activation of the ephrinB2/Eph4 system.
IGF-1 transduces mechanical stimuli in osteocytes, the terminally differentiated OBs, and promotes bone anabolic response.
GH and IGF-1 interact with other hormones, such as sex steroids, parathyroid hormone, insulin, and osteocalcin to regulate bone metabolism and whole-body metabolic homeostasis.
Funding:
Financial support received from the National Institutes of Health, Grant R01AG056397 to SY.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Tuggle CK and Trenkle A, Control of growth hormone synthesis. Domest Anim Endocrinol, 1996. 13(1): p. 1–33. [DOI] [PubMed] [Google Scholar]
- 2.Gahete MD, et al. , Understanding the multifactorial control of growth hormone release by somatotropes: lessons from comparative endocrinology. Ann N Y Acad Sci, 2009. 1163: p. 137–53. [DOI] [PubMed] [Google Scholar]
- 3.Pincus SM, et al. , Females secrete growth hormone with more process irregularity than males in both humans and rats. Am J Physiol, 1996. 270(1 Pt 1): p. E107–15. [DOI] [PubMed] [Google Scholar]
- 4.Spiess J, Rivier J, and Vale W, Characterization of rat hypothalamic growth hormone-releasing factor. Nature, 1983. 303(5917): p. 532–5. [DOI] [PubMed] [Google Scholar]
- 5.Brazeau P, et al. , Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science, 1973. 179(4068): p. 77–9. [DOI] [PubMed] [Google Scholar]
- 6.Baumbach WR, Horner DL, and Logan JS, The growth hormone-binding protein in rat serum is an alternatively spliced form of the rat growth hormone receptor. Genes Dev, 1989. 3(8): p. 1199–205. [DOI] [PubMed] [Google Scholar]
- 7.Smith WC, Kuniyoshi J, and Talamantes F, Mouse serum growth hormone (GH) binding protein has GH receptor extracellular and substituted transmembrane domains. Mol Endocrinol, 1989. 3(6): p. 984–90. [DOI] [PubMed] [Google Scholar]
- 8.Trivedi B and Daughaday WH, Release of growth hormone binding protein from IM-9 lymphocytes by endopeptidase is dependent on sulfhydryl group inactivation. Endocrinology, 1988. 123(5): p. 2201–6. [DOI] [PubMed] [Google Scholar]
- 9.Bichell DP, Kikuchi K, and Rotwein P, Growth hormone rapidly activates insulin-like growth factor I gene transcription in vivo. Mol Endocrinol, 1992. 6(11): p. 1899–908. [DOI] [PubMed] [Google Scholar]
- 10.Mathews LS, Norstedt G, and Palmiter RD, Regulation of insulin-like growth factor I gene expression by growth hormone. Proc Natl Acad Sci U S A, 1986. 83(24): p. 9343–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamashita S, Ong J, and Melmed S, Regulation of human growth hormone gene expression by insulin-like growth factor I in transfected cells. J Biol Chem, 1987. 262(27): p. 13254–7. [PubMed] [Google Scholar]
- 12.Holmes SJ and Shalet SM, Role of growth hormone and sex steroids in achieving and maintaining normal bone mass. Horm Res, 1996. 45(1–2): p. 86–93. [DOI] [PubMed] [Google Scholar]
- 13.Rosen CJ and Donahue LR, Insulin-like growth factors and bone: the osteoporosis connection revisited. Proc Soc Exp Biol Med, 1998. 219(1): p. 1–7. [DOI] [PubMed] [Google Scholar]
- 14.Jacome-Galarza CE, et al. , Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature, 2019. 568(7753): p. 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trotter M and Hixon BB, Sequential changes in weight, density, and percentage ash weight of human skeletons from an early fetal period through old age. Anat Rec, 1974. 179(1): p. 1–18. [DOI] [PubMed] [Google Scholar]
- 16.Mora S, et al. , Age-related changes in cortical and cancellous vertebral bone density in girls: assessment with quantitative CT. AJR Am J Roentgenol, 1994. 162(2): p. 405–9. [DOI] [PubMed] [Google Scholar]
- 17.Riggs BL, et al. , Differential changes in bone mineral density of the appendicular and axial skeleton with aging: relationship to spinal osteoporosis. J Clin Invest, 1981. 67(2): p. 328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richman C, et al. , Postnatal and pubertal skeletal changes contribute predominantly to the differences in peak bone density between C3H/HeJ and C57BL/6J mice. J Bone Miner Res, 2001. 16(2): p. 386–97. [DOI] [PubMed] [Google Scholar]
- 19.Bonjour JP, et al. , Critical years and stages of puberty for spinal and femoral bone mass accumulation during adolescence. J Clin Endocrinol Metab, 1991. 73(3): p. 555–63. [DOI] [PubMed] [Google Scholar]
- 20.Boot AM, et al. , Changes in bone mineral density, body composition, and lipid metabolism during growth hormone (GH) treatment in children with GH deficiency. J Clin Endocrinol Metab, 1997. 82(8): p. 2423–8. [DOI] [PubMed] [Google Scholar]
- 21.Rosenfeld RG, Rosenbloom AL, and Guevara-Aguirre J, Growth hormone (GH) insensitivity due to primary GH receptor deficiency. Endocr Rev, 1994. 15(3): p. 369–90. [DOI] [PubMed] [Google Scholar]
- 22.Laron Z, Pertzelan A, and Mannheimer S, Genetic pituitary dwarfism with high serum concentation of growth hormone--a new inborn error of metabolism? Isr J Med Sci, 1966. 2(2): p. 152–5. [PubMed] [Google Scholar]
- 23.Rosenbloom AL, et al. , Growth hormone receptor deficiency in Ecuador. J Clin Endocrinol Metab, 1999. 84(12): p. 4436–43. [DOI] [PubMed] [Google Scholar]
- 24.Colao A, et al. , Bone loss is correlated to the severity of growth hormone deficiency in adult patients with hypopituitarism. J Clin Endocrinol Metab, 1999. 84(6): p. 1919–24. [DOI] [PubMed] [Google Scholar]
- 25.Costa C, et al. , Transgenic rabbits overexpressing growth hormone develop acromegaly and diabetes mellitus. FASEB J, 1998. 12(14): p. 1455–60. [DOI] [PubMed] [Google Scholar]
- 26.Liu Z, Mohan S, and Yakar S, Does the GH/IGF-1 axis contribute to skeletal sexual dimorphism? Evidence from mouse studies. Growth Horm IGF Res, 2016. 27: p. 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenhalgh CJ, et al. , SOCS2 negatively regulates growth hormone action in vitro and in vivo. The Journal of clinical investigation, 2005. 115(2): p. 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasukawa Y, et al. , Evidence that sensitivity to growth hormone (GH) is growth period and tissue type dependent: studies in GH-deficient lit/lit mice. Endocrinology, 2003. 144(9): p. 3950–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohan S, et al. , Insulin-like growth factor regulates peak bone mineral density in mice by both growth hormone-dependent and -independent mechanisms. Endocrinology, 2003. 144(3): p. 929–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godfrey P, et al. , GHRH receptor of little mice contains a missense mutation in the extracellular domain that disrupts receptor function. Nature genetics, 1993. 4(3): p. 227–32. [DOI] [PubMed] [Google Scholar]
- 31.Stevenson AE, et al. , Does adiposity status influence femoral cortical strength in rodent models of growth hormone deficiency? American journal of physiology. Endocrinology and metabolism, 2009. 296(1): p. E147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sjogren K, et al. , Disproportional skeletal growth and markedly decreased bone mineral content in growth hormone receptor −/− mice. Biochemical and biophysical research communications, 2000. 267(2): p. 603–8. [DOI] [PubMed] [Google Scholar]
- 33.Sims NA, et al. , Bone homeostasis in growth hormone receptor-null mice is restored by IGF-I but independent of Stat5. The Journal of clinical investigation, 2000. 106(9): p. 1095–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayo KE, et al. , Dramatic pituitary hyperplasia in transgenic mice expressing a human growth hormone-releasing factor gene. Molecular endocrinology, 1988. 2(7): p. 606–12. [DOI] [PubMed] [Google Scholar]
- 35.Hammer RE, et al. , Expression of human growth hormone-releasing factor in transgenic mice results in increased somatic growth. Nature, 1985. 315(6018): p. 413–6. [DOI] [PubMed] [Google Scholar]
- 36.Steinke B, et al. , Human growth hormone transgene expression increases the biomechanical structural properties of mouse vertebrae. Spine, 1999. 24(1): p. 1–4. [DOI] [PubMed] [Google Scholar]
- 37.Tseng KF, et al. , Local expression of human growth hormone in bone results in impaired mechanical integrity in the skeletal tissue of transgenic mice. Journal of orthopaedic research : official publication of the Orthopaedic Research Society, 1996. 14(4): p. 598–604. [DOI] [PubMed] [Google Scholar]
- 38.Wanke R, et al. , The GH-transgenic mouse as an experimental model for growth research: clinical and pathological studies. Hormone research, 1992. 37 Suppl 3: p. 74–87. [DOI] [PubMed] [Google Scholar]
- 39.Wanke R, et al. , Accelerated growth and visceral lesions in transgenic mice expressing foreign genes of the growth hormone family: an overview. Pediatric nephrology, 1991. 5(4): p. 513–21. [DOI] [PubMed] [Google Scholar]
- 40.Wolf E, Rapp K, and Brem G, Expression of metallothionein-human growth hormone fusion genes in transgenic mice results in disproportionate skeletal gigantism. Growth, development, and aging : GDA, 1991. 55(2): p. 117–27. [PubMed] [Google Scholar]
- 41.Wolf E, et al. , Growth characteristics of metallothionein-human growth hormone transgenic mice as compared to mice selected for high eight-week body weight and unselected controls. II. Skeleton. Growth, development, and aging : GDA, 1991. 55(4): p. 237–48. [PubMed] [Google Scholar]
- 42.Wolf E, et al. , Growth characteristics of metallothionein-human growth hormone transgenic mice as compared to mice selected for high eight-week body weight and unselected controls. I. Body weight gain and external body dimensions. Growth, development, and aging : GDA, 1991. 55(4): p. 225–35. [PubMed] [Google Scholar]
- 43.Lim SV, et al. , Excessive growth hormone expression in male GH transgenic mice adversely alters bone architecture and mechanical strength. Endocrinology, 2015. 156(4): p. 1362–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmer AJ, et al. , Age-related changes in body composition of bovine growth hormone transgenic mice. Endocrinology, 2009. 150(3): p. 1353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eckstein F, et al. , Longitudinal in vivo effects of growth hormone overexpression on bone in transgenic mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 2004. 19(5): p. 802–10. [DOI] [PubMed] [Google Scholar]
- 46.Bartke A, et al. , Consequences of growth hormone (GH) overexpression and GH resistance. Neuropeptides, 2002. 36(2–3): p. 201–8. [DOI] [PubMed] [Google Scholar]
- 47.Eckstein F, et al. , Body composition, bone mass and microstructural analysis in GH-transgenic mice reveals that skeletal changes are specific to bone compartment and gender. Growth hormone & IGF research : official journal of the Growth Hormone Research Society and the International IGF Research Society, 2002. 12(2): p. 116–25. [DOI] [PubMed] [Google Scholar]
- 48.Tseng KF and Goldstein SA, Systemic over-secretion of growth hormone in transgenic mice results in a specific pattern of skeletal modeling and adaptation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 1998. 13(4): p. 706–15. [DOI] [PubMed] [Google Scholar]
- 49.Sandstedt J, et al. , Elevated levels of growth hormone increase bone mineral content in normal young mice, but not in ovariectomized mice. Endocrinology, 1996. 137(8): p. 3368–74. [DOI] [PubMed] [Google Scholar]
- 50.Dobie R, et al. , Direct stimulation of bone mass by increased GH signalling in the osteoblasts of Socs2−/− mice. J Endocrinol, 2014. 223(1): p. 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Metcalf D, et al. , Gigantism in mice lacking suppressor of cytokine signalling-2. Nature, 2000. 405(6790): p. 1069–73. [DOI] [PubMed] [Google Scholar]
- 52.Mathews LS, et al. , Growth enhancement of transgenic mice expressing human insulin-like growth factor I. Endocrinology, 1988. 123(6): p. 2827–33. [DOI] [PubMed] [Google Scholar]
- 53.Elis S, et al. , Elevated serum IGF-1 levels synergize PTH action on the skeleton only when the tissue IGF-1 axis is intact. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 2010. 25(9): p. 2051–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elis S, et al. , Elevated serum levels of IGF-1 are sufficient to establish normal body size and skeletal properties even in the absence of tissue IGF-1. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 2010. 25(6): p. 1257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elis S, et al. , Increased serum IGF-1 levels protect the musculoskeletal system but are associated with elevated oxidative stress markers and increased mortality independent of tissue igf1 gene expression. Aging cell, 2011. 10(3): p. 547–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu Y, et al. , Serum IGF-1 is insufficient to restore skeletal size in the total absence of the growth hormone receptor. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 2013. 28(7): p. 1575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu JP, et al. , Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell, 1993. 75(1): p. 59–72. [PubMed] [Google Scholar]
- 58.Castilla-Cortazar I, et al. , An experimental model of partial insulin-like growth factor-1 deficiency in mice. Journal of physiology and biochemistry, 2014. 70(1): p. 129–39. [DOI] [PubMed] [Google Scholar]
- 59.Powell-Braxton L, et al. , IGF-I is required for normal embryonic growth in mice. Genes & development, 1993. 7(12B): p. 2609–17. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, et al. , Insulin-like growth factor-I is essential for embryonic bone development. Endocrinology, 2006. 147(10): p. 4753–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He J, et al. , Postnatal growth and bone mass in mice with IGF-I haploinsufficiency. Bone, 2006. 38(6): p. 826–35. [DOI] [PubMed] [Google Scholar]
- 62.Mohan S and Baylink DJ, Impaired skeletal growth in mice with haploinsufficiency of IGF-I: genetic evidence that differences in IGF-I expression could contribute to peak bone mineral density differences. The Journal of endocrinology, 2005. 185(3): p. 415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yakar S, et al. , Serum IGF-1 determines skeletal strength by regulating subperiosteal expansion and trait interactions. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 2009. 24(8): p. 1481–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yakar S, et al. , Normal growth and development in the absence of hepatic insulin-like growth factor I. Proceedings of the National Academy of Sciences of the United States of America, 1999. 96(13): p. 7324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan Y, et al. , Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. The Journal of biological chemistry, 2009. 284(30): p. 19937–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cui Y, et al. , Loss of signal transducer and activator of transcription 5 leads to hepatosteatosis and impaired liver regeneration. Hepatology, 2007. 46(2): p. 504–13. [DOI] [PubMed] [Google Scholar]
- 67.Shi SY, et al. , Hepatocyte-specific deletion of Janus kinase 2 (JAK2) protects against diet-induced steatohepatitis and glucose intolerance. The Journal of biological chemistry, 2012. 287(13): p. 10277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nordstrom SM, et al. , Liver-derived IGF-I contributes to GH-dependent increases in lean mass and bone mineral density in mice with comparable levels of circulating GH. Molecular endocrinology, 2011. 25(7): p. 1223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ueki I, et al. , Inactivation of the acid labile subunit gene in mice results in mild retardation of postnatal growth despite profound disruptions in the circulating insulin-like growth factor system. Proceedings of the National Academy of Sciences of the United States of America, 2000. 97(12): p. 6868–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Courtland HW, et al. , Sex-specific regulation of body size and bone slenderness by the acid labile subunit. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 2010. 25(9): p. 2059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stratikopoulos E, et al. , The hormonal action of IGF1 in postnatal mouse growth. Proc Natl Acad Sci U S A, 2008. 105(49): p. 19378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elis S, et al. , Elevated serum IGF-1 levels synergize PTH action on the skeleton only when the tissue IGF-1 axis is intact. J Bone Miner Res, 2010. 25(9): p. 2051–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elis S, et al. , Elevated serum levels of IGF-1 are sufficient to establish normal body size and skeletal properties even in the absence of tissue IGF-1. J Bone Miner Res, 2010. 25(6): p. 1257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moerth C, et al. , Postnatally elevated levels of insulin-like growth factor (IGF)-II fail to rescue the dwarfism of IGF-I-deficient mice except kidney weight. Endocrinology, 2007. 148(1): p. 441–51. [DOI] [PubMed] [Google Scholar]
- 75.Wu Y, et al. , Serum IGF-1 is insufficient to restore skeletal size in the total absence of the growth hormone receptor. J Bone Miner Res, 2013. 28(7): p. 1575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pittenger MF, et al. , Multilineage potential of adult human mesenchymal stem cells. Science, 1999. 284(5411): p. 143–7. [DOI] [PubMed] [Google Scholar]
- 77.Correction to: Distinction Between Two Populations of Islet-1-Positive Cells in Hearts of Different Murine Strains, by Khattar P, Friedrich FW, Bonne G, Carrier L, Eschenhagen T, Evans SM, Schwartz K, Fiszman MY, and Vilquin J-T. Stem Cells Dev 2011;20(6):1043–1052 DOI: 10.1089/scd.2010.0374. Stem Cells Dev, 2018. 27(8): p. 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zuk PA, et al. , Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell, 2002. 13(12): p. 4279–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rompe JD, “Extracorporeal shock wave therapy for lateral epicondylitis--a double blind randomized controlled trial” by C.A. Speed et al., J Orthop Res 2002;20:895–8. J Orthop Res, 2003. 21(5): p. 958–9; author repy 961. [DOI] [PubMed] [Google Scholar]
- 80.Zvaifler NJ, et al. , Mesenchymal precursor cells in the blood of normal individuals. Arthritis Res, 2000. 2(6): p. 477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Suh MR, et al. , Human embryonic stem cells express a unique set of microRNAs. Dev Biol, 2004. 270(2): p. 488–98. [DOI] [PubMed] [Google Scholar]
- 82.Calloni R, et al. , Reviewing and updating the major molecular markers for stem cells. Stem Cells Dev, 2013. 22(9): p. 1455–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bonyadi M, et al. , Mesenchymal progenitor self-renewal deficiency leads to age-dependent osteoporosis in Sca-1/Ly-6A null mice. Proc Natl Acad Sci U S A, 2003. 100(10): p. 5840–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cornish J, et al. , Interleukin-18 is a novel mitogen of osteogenic and chondrogenic cells. Endocrinology, 2003. 144(4): p. 1194–201. [DOI] [PubMed] [Google Scholar]
- 85.Bolamperti S, et al. , GH prevents adipogenic differentiation of mesenchymal stromal stem cells derived from human trabecular bone via canonical Wnt signaling. Bone, 2018. 112: p. 136–144. [DOI] [PubMed] [Google Scholar]
- 86.Gaur T, et al. , Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem, 2005. 280(39): p. 33132–40. [DOI] [PubMed] [Google Scholar]
- 87.Bennett CN, et al. , Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A, 2005. 102(9): p. 3324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H, et al. , Growth hormone and insulin-like growth factor I induce bone morphogenetic proteins 2 and 4: a mediator role in bone and tooth formation? Endocrinology, 1998. 139(9): p. 3855–62. [DOI] [PubMed] [Google Scholar]
- 89.Wu S, et al. , Nuclear factor-kappaB (NF-kappaB) p65 interacts with Stat5b in growth plate chondrocytes and mediates the effects of growth hormone on chondrogenesis and on the expression of insulin-like growth factor-1 and bone morphogenetic protein-2. J Biol Chem, 2011. 286(28): p. 24726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Francis PH, et al. , Bone morphogenetic proteins and a signalling pathway that controls patterning in the developing chick limb. Development, 1994. 120(1): p. 209–18. [DOI] [PubMed] [Google Scholar]
- 91.Kingsley DM, et al. , The mouse short ear skeletal morphogenesis locus is associated with defects in a bone morphogenetic member of the TGF beta superfamily. Cell, 1992. 71(3): p. 399–410. [DOI] [PubMed] [Google Scholar]
- 92.Dale L, et al. , Bone morphogenetic protein 4: a ventralizing factor in early Xenopus development. Development, 1992. 115(2): p. 573–85. [DOI] [PubMed] [Google Scholar]
- 93.Merriman HL, et al. , Insulin-like growth factor-I and insulin-like growth factor-II induce c-fos in mouse osteoblastic cells. Calcified tissue international, 1990. 46(4): p. 258–62. [DOI] [PubMed] [Google Scholar]
- 94.Mohan S, et al. , Characterization of the receptor for insulin-like growth factor II in bone cells. Journal of cellular physiology, 1989. 140(1): p. 169–76. [DOI] [PubMed] [Google Scholar]
- 95.Schmid C, Steiner T, and Froesch ER, Insulin-like growth factor I supports differentiation of cultured osteoblast-like cells. FEBS letters, 1984. 173(1): p. 48–52. [DOI] [PubMed] [Google Scholar]
- 96.Shi Y, et al. , Hedgehog signaling activates a positive feedback mechanism involving insulin-like growth factors to induce osteoblast differentiation. Proceedings of the National Academy of Sciences of the United States of America, 2015. 112(15): p. 4678–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baker AR, et al. , Osteoblast-specific expression of growth hormone stimulates bone growth in transgenic mice. Molecular and cellular biology, 1992. 12(12): p. 5541–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao G, et al. , Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology, 2000. 141(7): p. 2674–82. [DOI] [PubMed] [Google Scholar]
- 99.Brennan-Speranza TC, et al. , Selective osteoblast overexpression of IGF-I in mice prevents low protein-induced deterioration of bone strength and material level properties. Bone, 2011. 49(5): p. 1073–9. [DOI] [PubMed] [Google Scholar]
- 100.Jiang J, et al. , Transgenic mice with osteoblast-targeted insulin-like growth factor-I show increased bone remodeling. Bone, 2006. 39(3): p. 494–504. [DOI] [PubMed] [Google Scholar]
- 101.Qin X, et al. , Pregnancy-associated plasma protein-A increases osteoblast proliferation in vitro and bone formation in vivo. Endocrinology, 2006. 147(12): p. 5653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang M, et al. , Paracrine overexpression of IGFBP-4 in osteoblasts of transgenic mice decreases bone turnover and causes global growth retardation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 2003. 18(5): p. 836–43. [DOI] [PubMed] [Google Scholar]
- 103.Devlin RD, et al. , Transgenic mice overexpressing insulin-like growth factor binding protein-5 display transiently decreased osteoblastic function and osteopenia. Endocrinology, 2002. 143(10): p. 3955–62. [DOI] [PubMed] [Google Scholar]
- 104.Wang Y, et al. , IGF-I receptor is required for the anabolic actions of parathyroid hormone on bone. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 2007. 22(9): p. 1329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang M, et al. , Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. The Journal of biological chemistry, 2002. 277(46): p. 44005–12. [DOI] [PubMed] [Google Scholar]
- 106.Wang Y, et al. , IGF-I Signaling in Osterix-Expressing Cells Regulates Secondary Ossification Center Formation, Growth Plate Maturation, and Metaphyseal Formation During Postnatal Bone Development. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 2015. 30(12): p. 2239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu Z, et al. , DMP-1-mediated Ghr gene recombination compromises skeletal development and impairs skeletal response to intermittent PTH. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2016. 30(2): p. 635–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu Z, Mohan S, and Yakar S, Does the GH/IGF-1 axis contribute to skeletal sexual dimorphism? Evidence from mouse studies. Growth hormone & IGF research : official journal of the Growth Hormone Research Society and the International IGF Research Society, 2016. 27: p. 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sheng MH, et al. , Disruption of the insulin-like growth factor-1 gene in osteocytes impairs developmental bone growth in mice. Bone, 2013. 52(1): p. 133–44. [DOI] [PubMed] [Google Scholar]
- 110.Ezzat S, et al. , Biochemical assessment of bone formation and resorption in acromegaly. J Clin Endocrinol Metab, 1993. 76(6): p. 1452–7. [DOI] [PubMed] [Google Scholar]
- 111.Marazuela M, et al. , Serum bone Gla protein as a marker of bone turnover in acromegaly. Calcif Tissue Int, 1993. 52(6): p. 419–21. [DOI] [PubMed] [Google Scholar]
- 112.Brixen K, et al. , Short-term treatment with growth hormone stimulates osteoblastic and osteoclastic activity in osteopenic postmenopausal women: a dose response study. J Bone Miner Res, 1995. 10(12): p. 1865–74. [DOI] [PubMed] [Google Scholar]
- 113.Slootweg MC, et al. , Osteoclast formation together with interleukin-6 production in mouse long bones is increased by insulin-like growth factor-I. The Journal of endocrinology, 1992. 132(3): p. 433–8. [DOI] [PubMed] [Google Scholar]
- 114.Mochizuki H, et al. , Insulin-like growth factor-I supports formation and activation of osteoclasts. Endocrinology, 1992. 131(3): p. 1075–80. [DOI] [PubMed] [Google Scholar]
- 115.Wang Y, et al. , Role of IGF-I signaling in regulating osteoclastogenesis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 2006. 21(9): p. 1350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Matsuo K and Otaki N, Bone cell interactions through Eph/ephrin: bone modeling, remodeling and associated diseases. Cell Adh Migr, 2012. 6(2): p. 148–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Edwards CM and Mundy GR, Eph receptors and ephrin signaling pathways: a role in bone homeostasis. Int J Med Sci, 2008. 5(5): p. 263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao C, et al. , Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab, 2006. 4(2): p. 111–21. [DOI] [PubMed] [Google Scholar]
- 119.Teramachi J, et al. , Measles virus nucleocapsid protein increases osteoblast differentiation in Paget’s disease. J Clin Invest, 2016. 126(3): p. 1012–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Miyagawa K, et al. , Osteoclast-derived IGF1 is required for pagetic lesion formation in vivo. JCI Insight, 2020. 5(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bikle D, et al. , The skeletal structure of insulin-like growth factor I-deficient mice. J Bone Miner Res, 2001. 16(12): p. 2320–9. [DOI] [PubMed] [Google Scholar]
- 122.Fiorelli G, et al. , Characterization and function of the receptor for IGF-I in human preosteoclastic cells. Bone, 1996. 18(3): p. 269–76. [DOI] [PubMed] [Google Scholar]
- 123.Mizuhashi K, et al. , Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature, 2018. 563(7730): p. 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lui JC, Home for a rest: stem cell niche of the postnatal growth plate. J Endocrinol, 2020. 246(1): p. R1–R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Newton PT, et al. , A radical switch in clonality reveals a stem cell niche in the epiphyseal growth plate. Nature, 2019. 567(7747): p. 234–238. [DOI] [PubMed] [Google Scholar]
- 126.EVELYN SMITH ROSS FCM, THE INFLUENCE OF THE GROWTH PROMOTING HORMONE OF THE ANTERIOR LOBE OF THE PITUITARY UPON GROWTH ACTIVITY IN THE LONG BONES OF THE RAT. Endocrinology, 1940. 27(2): p. 329–339. [Google Scholar]
- 127.EVANS HERBERTM, S. ME, MARX WALTER, KIBRICK E, BIOASSAY OF THE PITUITARY GROWTH HORMONE. WIDTH OF THE PROXIMAL EPIPHYSIAL CARTILAE OF THE TIBIA IN HYPOPHYSECTOMI2ED RATS. Endocrinology, 1943. 32(1): p. 13–16. [Google Scholar]
- 128.Isaksson OG, Jansson JO, and Gause IA, Growth hormone stimulates longitudinal bone growth directly. Science, 1982. 216(4551): p. 1237–9. [DOI] [PubMed] [Google Scholar]
- 129.Hunziker EB, Wagner J, and Zapf J, Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo. J Clin Invest, 1994. 93(3): p. 1078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lindahl A, et al. , Growth hormone potentiates colony formation of epiphyseal chondrocytes in suspension culture. Endocrinology, 1986. 118(5): p. 1843–8. [DOI] [PubMed] [Google Scholar]
- 131.Madsen K, et al. , Growth hormone stimulates the proliferation of cultured chondrocytes from rabbit ear and rat rib growth cartilage. Nature, 1983. 304(5926): p. 545–7. [DOI] [PubMed] [Google Scholar]
- 132.Lindahl A, Isgaard J, and Isaksson OG, Growth hormone in vivo potentiates the stimulatory effect of insulin-like growth factor-1 in vitro on colony formation of epiphyseal chondrocytes isolated from hypophysectomized rats. Endocrinology, 1987. 121(3): p. 1070–5. [DOI] [PubMed] [Google Scholar]
- 133.Nilsson A, et al. , Regulation by GH of insulin-like growth factor-I mRNA expression in rat epiphyseal growth plate as studied with in-situ hybridization. J Endocrinol, 1990. 125(1): p. 67–74. [DOI] [PubMed] [Google Scholar]
- 134.Nilsson A, et al. , Regulation by growth hormone of number of chondrocytes containing IGF-I in rat growth plate. Science, 1986. 233(4763): p. 571–4. [DOI] [PubMed] [Google Scholar]
- 135.Spencer GS, Hodgkinson SC, and Bass JJ, Passive immunization against insulin-like growth factor-I does not inhibit growth hormone-stimulated growth of dwarf rats. Endocrinology, 1991. 128(4): p. 2103–9. [DOI] [PubMed] [Google Scholar]
- 136.Werther GA, et al. , Visual demonstration of growth hormone receptors on human growth plate chondrocytes. J Clin Endocrinol Metab, 1990. 70(6): p. 1725–31. [DOI] [PubMed] [Google Scholar]
- 137.Lindahl A, Nilsson A, and Isaksson OG, Effects of growth hormone and insulin-like growth factor-I on colony formation of rabbit epiphyseal chondrocytes at different stages of maturation. J Endocrinol, 1987. 115(2): p. 263–71. [DOI] [PubMed] [Google Scholar]
- 138.Cruickshank J, et al. , Spatial distribution of growth hormone receptor, insulin-like growth factor-I receptor and apoptotic chondrocytes during growth plate development. J Endocrinol, 2005. 184(3): p. 543–53. [DOI] [PubMed] [Google Scholar]
- 139.Oberbauer AM and Peng R, Growth hormone and IGF-I stimulate cell function in distinct zones of the rat epiphyseal growth plate. Connect Tissue Res, 1995. 31(3): p. 189–95. [DOI] [PubMed] [Google Scholar]
- 140.Wang J, et al. , Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol, 2004. 180(2): p. 247–55. [DOI] [PubMed] [Google Scholar]
- 141.Udy GB, et al. , Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proceedings of the National Academy of Sciences of the United States of America, 1997. 94(14): p. 7239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Govoni KE, et al. , Disruption of insulin-like growth factor-I expression in type IIalphaI collagen-expressing cells reduces bone length and width in mice. Physiol Genomics, 2007. 30(3): p. 354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Heilig J, Paulsson M, and Zaucke F, Insulin-like growth factor 1 receptor (IGF1R) signaling regulates osterix expression and cartilage matrix mineralization during endochondral ossification. Bone, 2016. 83: p. 48–57. [DOI] [PubMed] [Google Scholar]
- 144.Wang Y, et al. , IGF-1R signaling in chondrocytes modulates growth plate development by interacting with the PTHrP/Ihh pathway. J Bone Miner Res, 2011. 26(7): p. 1437–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Long F, et al. , Independent regulation of skeletal growth by Ihh and IGF signaling. Dev Biol, 2006. 298(1): p. 327–33. [DOI] [PubMed] [Google Scholar]
- 146.Wu S, Yang W, and De Luca F, Insulin-Like Growth Factor-Independent Effects of Growth Hormone on Growth Plate Chondrogenesis and Longitudinal Bone Growth. Endocrinology, 2015. 156(7): p. 2541–51. [DOI] [PubMed] [Google Scholar]
- 147.Wang Y, et al. , Ephrin B2/EphB4 mediates the actions of IGF-I signaling in regulating endochondral bone formation. J Bone Miner Res, 2014. 29(8): p. 1900–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lui JC, et al. , Persistent Sox9 expression in hypertrophic chondrocytes suppresses transdifferentiation into osteoblasts. Bone, 2019. 125: p. 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Aghajanian P, et al. , Epiphyseal bone formation occurs via thyroid hormone regulation of chondrocyte to osteoblast transdifferentiation. Sci Rep, 2017. 7(1): p. 10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhou X, et al. , Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet, 2014. 10(12): p. e1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tsukazaki T, et al. , Growth hormone directly and indirectly stimulates articular chondrocyte cell growth. Osteoarthritis Cartilage, 1994. 2(4): p. 259–67. [DOI] [PubMed] [Google Scholar]
- 152.Hunter DJ and Bierma-Zeinstra S, Osteoarthritis. Lancet, 2019. 393(10182): p. 1745–1759. [DOI] [PubMed] [Google Scholar]
- 153.Hootman JM and Helmick CG, Projections of US prevalence of arthritis and associated activity limitations. Arthritis Rheum, 2006. 54(1): p. 226–9. [DOI] [PubMed] [Google Scholar]
- 154.Mobasheri A, et al. , The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol, 2017. 13(5): p. 302–311. [DOI] [PubMed] [Google Scholar]
- 155.Man GS and Mologhianu G, Osteoarthritis pathogenesis - a complex process that involves the entire joint. J Med Life, 2014. 7(1): p. 37–41. [PMC free article] [PubMed] [Google Scholar]
- 156.Heinegard D and Saxne T, The role of the cartilage matrix in osteoarthritis. Nat Rev Rheumatol, 2011. 7(1): p. 50–6. [DOI] [PubMed] [Google Scholar]
- 157.Lark MW, et al. , Aggrecan degradation in human cartilage. Evidence for both matrix metalloproteinase and aggrecanase activity in normal, osteoarthritic, and rheumatoid joints. J Clin Invest, 1997. 100(1): p. 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lahm A, et al. , Changes in content and synthesis of collagen types and proteoglycans in osteoarthritis of the knee joint and comparison of quantitative analysis with Photoshop-based image analysis. Arch Orthop Trauma Surg, 2010. 130(4): p. 557–64. [DOI] [PubMed] [Google Scholar]
- 159.Bowles RD, et al. , In vivo luminescence imaging of NF-kappaB activity and serum cytokine levels predict pain sensitivities in a rodent model of osteoarthritis. Arthritis Rheumatol, 2014. 66(3): p. 637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Melchiorri C, et al. , Enhanced and coordinated in vivo expression of inflammatory cytokines and nitric oxide synthase by chondrocytes from patients with osteoarthritis. Arthritis Rheum, 1998. 41(12): p. 2165–74. [DOI] [PubMed] [Google Scholar]
- 161.Livshits G, et al. , Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: The Chingford Study. Arthritis Rheum, 2009. 60(7): p. 2037–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sakkas LI, et al. , Interleukin-12 is expressed by infiltrating macrophages and synovial lining cells in rheumatoid arthritis and osteoarthritis. Cell Immunol, 1998. 188(2): p. 105–10. [DOI] [PubMed] [Google Scholar]
- 163.Scanzello CR, et al. , Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthritis Cartilage, 2009. 17(8): p. 1040–8. [DOI] [PubMed] [Google Scholar]
- 164.Wang Z, et al. , Interleukin-17 Can Induce Osteoarthritis in Rabbit Knee Joints Similar to Hulth’s Method. Biomed Res Int, 2017. 2017: p. 2091325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cunningham CC, et al. , Osteoarthritis-associated basic calcium phosphate crystals induce pro-inflammatory cytokines and damage-associated molecules via activation of Syk and PI3 kinase. Clin Immunol, 2012. 144(3): p. 228–36. [DOI] [PubMed] [Google Scholar]
- 166.Vincenti MP and Brinckerhoff CE, Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res, 2002. 4(3): p. 157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cheung KS, et al. , Expression of ADAMTS-4 by chondrocytes in the surface zone of human osteoarthritic cartilage is regulated by epigenetic DNA de-methylation. Rheumatol Int, 2009. 29(5): p. 525–34. [DOI] [PubMed] [Google Scholar]
- 168.Ji Q, et al. , The IL-1beta/AP-1/miR-30a/ADAMTS-5 axis regulates cartilage matrix degradation in human osteoarthritis. J Mol Med (Berl), 2016. 94(7): p. 771–85. [DOI] [PubMed] [Google Scholar]
- 169.Hashimoto S, et al. , Development and regulation of osteophyte formation during experimental osteoarthritis. Osteoarthritis Cartilage, 2002. 10(3): p. 180–7. [DOI] [PubMed] [Google Scholar]
- 170.Remst DF, Blaney Davidson EN, and van der Kraan PM, Unravelling osteoarthritis-related synovial fibrosis: a step closer to solving joint stiffness. Rheumatology (Oxford), 2015. 54(11): p. 1954–63. [DOI] [PubMed] [Google Scholar]
- 171.Xia B, et al. , Osteoarthritis pathogenesis: a review of molecular mechanisms. Calcif Tissue Int, 2014. 95(6): p. 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dequeker J, Burssens A, and Bouillon R, Dynamics of growth hormone secretion in patients with osteoporosis and in patients with osteoarthrosis. Horm Res, 1982. 16(6): p. 353–6. [DOI] [PubMed] [Google Scholar]
- 173.Denko CW and Boja B, Growth factors in asymptomatic osteoarthritis - insulin, insulin-like growth factor-1, growth hormone. InflammoPharmacology, 1993. 2(1): p. 71–76. [Google Scholar]
- 174.Denko CW, Boja B, and Moskowitz RW, Growth promoting peptides in osteoarthritis: insulin, insulin-like growth factor-1, growth hormone. J Rheumatol, 1990. 17(9): p. 1217–21. [PubMed] [Google Scholar]
- 175.Denko CW, Boja B, and Moskowitz RW, Growth factors, insulin-like growth factor-1 and growth hormone, in synovial fluid and serum of patients with rheumatic disorders. Osteoarthritis Cartilage, 1996. 4(4): p. 245–9. [DOI] [PubMed] [Google Scholar]
- 176.Gabusi E, et al. , Age-independent effects of hyaluronan amide derivative and growth hormone on human osteoarthritic chondrocytes. Connect Tissue Res, 2015. 56(6): p. 440–51. [DOI] [PubMed] [Google Scholar]
- 177.Claessen KM, et al. , Relationship between the functional exon 3 deleted growth hormone receptor polymorphism and symptomatic osteoarthritis in women. Ann Rheum Dis, 2014. 73(2): p. 433–6. [DOI] [PubMed] [Google Scholar]
- 178.Pantel J, et al. , Species-specific alternative splice mimicry at the growth hormone receptor locus revealed by the lineage of retroelements during primate evolution. J Biol Chem, 2000. 275(25): p. 18664–9. [DOI] [PubMed] [Google Scholar]
- 179.Dos Santos C, et al. , A common polymorphism of the growth hormone receptor is associated with increased responsiveness to growth hormone. Nat Genet, 2004. 36(7): p. 720–4. [DOI] [PubMed] [Google Scholar]
- 180.Dore S, et al. , Human osteoarthritic chondrocytes possess an increased number of insulin-like growth factor 1 binding sites but are unresponsive to its stimulation. Possible role of IGF-1-binding proteins. Arthritis Rheum, 1994. 37(2): p. 253–63. [DOI] [PubMed] [Google Scholar]
- 181.Olney RC, et al. , Chondrocytes from osteoarthritic cartilage have increased expression of insulin-like growth factor I (IGF-I) and IGF-binding protein-3 (IGFBP-3) and −5, but not IGF-II or IGFBP-4. J Clin Endocrinol Metab, 1996. 81(3): p. 1096–103. [DOI] [PubMed] [Google Scholar]
- 182.Lloyd ME, et al. , Relation between insulin-like growth factor-I concentrations, osteoarthritis, bone density, and fractures in the general population: the Chingford study. Ann Rheum Dis, 1996. 55(12): p. 870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Reinhardt WO and Li CH, Experimental production of arthritis in rats by hypophyseal growth hormone. Science, 1953. 117(3038): p. 295–7. [DOI] [PubMed] [Google Scholar]
- 184.Okazaki K, et al. , Expression of insulin-like growth factor I messenger ribonucleic acid in developing osteophytes in murine experimental osteoarthritis and in rats inoculated with growth hormone-secreting tumor. Endocrinology, 1999. 140(10): p. 4821–30. [DOI] [PubMed] [Google Scholar]
- 185.Li G, et al. , Subchondral bone in osteoarthritis: insight into risk factors and microstructural changes. Arthritis Res Ther, 2013. 15(6): p. 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hilal G, et al. , Endogenous prostaglandin E2 and insulin-like growth factor 1 can modulate the levels of parathyroid hormone receptor in human osteoarthritic osteoblasts. J Bone Miner Res, 2001. 16(4): p. 713–21. [DOI] [PubMed] [Google Scholar]
- 187.Kosinska MK, et al. , Articular Joint Lubricants during Osteoarthritis and Rheumatoid Arthritis Display Altered Levels and Molecular Species. PLoS One, 2015. 10(5): p. e0125192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sluzalska KD, et al. , Growth factors regulate phospholipid biosynthesis in human fibroblast-like synoviocytes obtained from osteoarthritic knees. Sci Rep, 2017. 7(1): p. 13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Dore S, et al. , Increased insulin-like growth factor 1 production by human osteoarthritic chondrocytes is not dependent on growth hormone action. Arthritis Rheum, 1995. 38(3): p. 413–9. [DOI] [PubMed] [Google Scholar]
- 190.Yu Q, et al. , microRNA-206 is required for osteoarthritis development through its effect on apoptosis and autophagy of articular chondrocytes via modulating the phosphoinositide 3-kinase/protein kinase B-mTOR pathway by targeting insulin-like growth factor-1. J Cell Biochem, 2019. 120(4): p. 5287–5303. [DOI] [PubMed] [Google Scholar]
- 191.Ekenstedt KJ, et al. , Effects of chronic growth hormone and insulin-like growth factor 1 deficiency on osteoarthritis severity in rat knee joints. Arthritis Rheum, 2006. 54(12): p. 3850–8. [DOI] [PubMed] [Google Scholar]
- 192.Salvatori R, et al. , Familial dwarfism due to a novel mutation of the growth hormone-releasing hormone receptor gene. J Clin Endocrinol Metab, 1999. 84(3): p. 917–23. [DOI] [PubMed] [Google Scholar]
- 193.Epitacio-Pereira CC, et al. , Isolated GH deficiency due to a GHRH receptor mutation causes hip joint problems and genu valgum, and reduces size but not density of trabecular and mixed bone. J Clin Endocrinol Metab, 2013. 98(11): p. E1710–5. [DOI] [PubMed] [Google Scholar]
- 194.Yi C, et al. , Down-regulation of programmed cell death 5 by insulin-like growth factor 1 in osteoarthritis chondrocytes. Int Orthop, 2013. 37(5): p. 937–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cravero JD, et al. , Increased expression of the Akt/PKB inhibitor TRB3 in osteoarthritic chondrocytes inhibits insulin-like growth factor 1-mediated cell survival and proteoglycan synthesis. Arthritis Rheum, 2009. 60(2): p. 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tie K, et al. , Intrauterine low-functional programming of IGF1 by prenatal nicotine exposure mediates the susceptibility to osteoarthritis in female adult rat offspring. FASEB J, 2016. 30(2): p. 785–97. [DOI] [PubMed] [Google Scholar]
- 197.Hochberg MC, et al. , Serum levels of insulin-like growth factor in subjects with osteoarthritis of the knee. Data from the Baltimore Longitudinal Study of Aging. Arthritis Rheum, 1994. 37(8): p. 1177–80. [DOI] [PubMed] [Google Scholar]
- 198.McAlindon TE, Teale JD, and Dieppe PA, Levels of insulin related growth factor 1 in osteoarthritis of the knee. Ann Rheum Dis, 1993. 52(3): p. 229–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Fraenkel L, et al. , Longitudinal analysis of the relationship between serum insulin-like growth factor-I and radiographic knee osteoarthritis. Osteoarthritis Cartilage, 1998. 6(5): p. 362–7. [DOI] [PubMed] [Google Scholar]
- 200.Tornqvist AE, et al. , Liver-derived IGF-I is not required for protection against osteoarthritis in male mice. Am J Physiol Endocrinol Metab, 2019. 317(6): p. E1150–E1157. [DOI] [PubMed] [Google Scholar]
- 201.Le Vay D, Letter: Intraarticular human growth in osteoarthritis. Lancet, 1975. 2(7936): p. 666–7. [DOI] [PubMed] [Google Scholar]
- 202.Chrisman OD, The effect of growth hormone on established cartilage lesions. A presidential address to the Association of Bone and Joint Surgeons, 1974. Clin Orthop Relat Res, 1975(107): p. 232–8. [PubMed] [Google Scholar]
- 203.Hendricson AS, Havdrup T, and Telhag H, The effect of growth hormone and thyroxine on adult joint cartilage. Clin Orthop Relat Res, 1982(162): p. 270–5. [PubMed] [Google Scholar]
- 204.Livne E, Laufer D, and Blumenfeld I, Comparison of in vitro response to growth hormone by chondrocytes from mandibular condyle cartilage of young and old mice. Calcif Tissue Int, 1997. 61(1): p. 62–7. [DOI] [PubMed] [Google Scholar]
- 205.Dart AJ, et al. , Recombinant equine growth hormone administration: effects on synovial fluid biomarkers and cartilage metabolism in horses. Equine Vet J, 2003. 35(3): p. 302–7. [DOI] [PubMed] [Google Scholar]
- 206.Ok SM, et al. , Local Injection of Growth Hormone for Temporomandibular Joint Osteoarthritis. Yonsei Med J, 2020. 61(4): p. 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Clemmons DR, et al. , Inhibition of insulin-like growth factor binding protein 5 proteolysis in articular cartilage and joint fluid results in enhanced concentrations of insulin-like growth factor 1 and is associated with improved osteoarthritis. Arthritis Rheum, 2002. 46(3): p. 694–703. [DOI] [PubMed] [Google Scholar]
- 208.Weimer A, et al. , Benefits of recombinant adeno-associated virus (rAAV)-mediated insulinlike growth factor I (IGF-I) overexpression for the long-term reconstruction of human osteoarthritic cartilage by modulation of the IGF-I axis. Mol Med, 2012. 18: p. 346–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kim SB, et al. , Additive effects of intra-articular injection of growth hormone and hyaluronic acid in rabbit model of collagenase-induced osteoarthritis. J Korean Med Sci, 2010. 25(5): p. 776–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Uebelhart D and Williams JM, Effects of hyaluronic acid on cartilage degradation. Curr Opin Rheumatol, 1999. 11(5): p. 427–35. [DOI] [PubMed] [Google Scholar]
- 211.Kumagai K, et al. , Sodium pentosan polysulfate resulted in cartilage improvement in knee osteoarthritis--an open clinical trial. BMC Clin Pharmacol, 2010. 10: p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Rogachefsky RA, et al. , Treatment of canine osteoarthritis with insulin-like growth factor-1 (IGF-1) and sodium pentosan polysulfate. Osteoarthritis Cartilage, 1993. 1(2): p. 105–14. [DOI] [PubMed] [Google Scholar]
- 213.Blumenfeld I, et al. , Enhancement of toluidine blue staining by transforming growth factor-beta, insulin-like growth factor and growth hormone in the temporomandibular joint of aged mice. Cells Tissues Organs, 2000. 167(2–3): p. 121–9. [DOI] [PubMed] [Google Scholar]
- 214.Flechtenmacher J, et al. , Recombinant human osteogenic protein 1 is a potent stimulator of the synthesis of cartilage proteoglycans and collagens by human articular chondrocytes. Arthritis Rheum, 1996. 39(11): p. 1896–904. [DOI] [PubMed] [Google Scholar]
- 215.Loeser RF, Pacione CA, and Chubinskaya S, The combination of insulin-like growth factor 1 and osteogenic protein 1 promotes increased survival of and matrix synthesis by normal and osteoarthritic human articular chondrocytes. Arthritis Rheum, 2003. 48(8): p. 2188–96. [DOI] [PubMed] [Google Scholar]
- 216.Orth P, et al. , Transplanted articular chondrocytes co-overexpressing IGF-I and FGF-2 stimulate cartilage repair in vivo. Knee Surg Sports Traumatol Arthrosc, 2011. 19(12): p. 2119–30. [DOI] [PubMed] [Google Scholar]
- 217.Li Y, et al. , Effects of insulin-like growth factor-1 and dexamethasone on cytokine-challenged cartilage: relevance to post-traumatic osteoarthritis. Osteoarthritis Cartilage, 2015. 23(2): p. 266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Loffredo FS, et al. , Targeted delivery to cartilage is critical for in vivo efficacy of insulin-like growth factor 1 in a rat model of osteoarthritis. Arthritis Rheumatol, 2014. 66(5): p. 1247–55. [DOI] [PubMed] [Google Scholar]
- 219.Geiger BC, et al. , Cartilage-penetrating nanocarriers improve delivery and efficacy of growth factor treatment of osteoarthritis. Sci Transl Med, 2018. 10(469). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lin SC, et al. , Molecular basis of the little mouse phenotype and implications for cell type-specific growth. Nature, 1993. 364(6434): p. 208–13. [DOI] [PubMed] [Google Scholar]
- 221.Alba M and Salvatori R, A mouse with targeted ablation of the growth hormone-releasing hormone gene: a new model of isolated growth hormone deficiency. Endocrinology, 2004. 145(9): p. 4134–43. [DOI] [PubMed] [Google Scholar]
- 222.List EO, et al. , The Effects of 20-kDa Human Placental GH in Male and Female GH-deficient Mice: An Improved Human GH? Endocrinology, 2020. 161(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Baker J, et al. , Role of insulin-like growth factors in embryonic and postnatal growth. Cell, 1993. 75(1): p. 73–82. [PubMed] [Google Scholar]
- 224.Udy GB, et al. , Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A, 1997. 94(14): p. 7239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Parganas E, et al. , Jak2 is essential for signaling through a variety of cytokine receptors. Cell, 1998. 93(3): p. 385–95. [DOI] [PubMed] [Google Scholar]
- 226.Mayo KE, et al. , Dramatic pituitary hyperplasia in transgenic mice expressing a human growth hormone-releasing factor gene. Mol Endocrinol, 1988. 2(7): p. 606–12. [DOI] [PubMed] [Google Scholar]
- 227.D’Ercole AJ, Expression of insulin-like growth factor-I in transgenic mice. Annals of the New York Academy of Sciences, 1993. 692: p. 149–60. [DOI] [PubMed] [Google Scholar]
- 228.Macrae VE, et al. , Increased bone mass, altered trabecular architecture and modified growth plate organization in the growing skeleton of SOCS2 deficient mice. J Cell Physiol, 2009. 218(2): p. 276–84. [DOI] [PubMed] [Google Scholar]