

Abstract
The use of genomic approaches in toxicological studies has greatly increased our ability to define the molecular profiles of environmental chemicals associated with developmental neurotoxicity (DNT). Integration of these approaches with adverse outcome pathways (AOPs), a framework that translates environmental exposures to adverse developmental phenotypes, can potentially inform DNT testing strategies. Here, using retinoic acid (RA) as a case example, we demonstrate that the integration of toxicogenomic profiles into the AOP framework can be used to establish a paradigm for chemical testing. RA is a critical regulatory signaling molecule involved in multiple aspects of mammalian central nervous system (CNS) development, including hindbrain formation/patterning and neuronal differentiation, and imbalances in RA signaling pathways are linked with DNT. While the mechanisms remain unresolved, environmental chemicals can cause DNT by disrupting the RA signaling pathway. First, we reviewed literature evidence of RA and other retinoid exposures and DNT to define a provisional AOP related to imbalances in RA embryonic bioavailability and hindbrain development. Next, by integrating toxicogenomic datasets, we defined a relevant transcriptomic signature associated with RA-induced developmental neurotoxicity (RA-DNT) in human and rodent models that was tested against zebrafish model data, demonstrating potential for integration into an AOP framework. Finally, we demonstrated how these approaches may be systematically utilized to identify chemical hazards by testing the RA-DNT signature against azoles, a proposed class of compounds that alters RA-signaling. The provisional AOP from this study can be expanded in the future to better define DNT biomarkers relevant to RA signaling and toxicity.
Keywords: retinoids, retinoic acid, human, embryonic stem cells, whole embryo culture, zebrafish, neurogenesis, neurotoxicity, transcriptome, alternative model, in vitro, azoles, adverse outcome pathway, transcriptomics
Introduction
Retinoic acid (RA), also known as all-trans-retinoic acid, is required for mammalian development. Mammals are unable to synthesize RA de novo, deriving the compound through dietary intake of vitamin A or other retinoid precursors [1]. RA, a small lipophilic molecule, is synthesized in specific cells, released, and taken up by surrounding cells [2, 3]. Upon cellular entry via diffusion, RA transfers to the nucleus and acts as a transcriptional activating ligand by binding to nuclear receptors known as retinoic acid receptors (RARs) [4–6] that form heterodimer complexes with retinoid X receptors (RXRs) [7]. RA levels and ligand-activity is tightly regulated by the regional-and temporal-dependent expression of a combination of molecules including RAR/RXRs, co-factors, binding proteins, and metabolizing enzymes.
RA signaling is important for several aspects of central nervous system (CNS) development including 1) regional patterning and the 2) differentiation and maintenance of progenitors into neuronal populations [8–10]. Subsequently, disruption of RA signaling due to imbalances in RA bioavailability, in deficiency or in excess, can lead to developmental neurotoxicity (DNT) [11, 12]. While the effects of RA may be dependent on model and exposure paradigms utilized [13], CNS (and related axial) defects associated with perturbations in RA signaling include: anterior (exencephaly or anencephaly) and posterior (spina bifida) NTDs, hindbrain abnormalities, microcephaly, irregular somitogenesis, delayed or reduced caudal elongation, vascular damage, notochord defects, irregularities in the neural folds and other variants [14, 15]. Morphological alterations due to disrupted RA signaling are linked with abnormal changes on the cellular and molecular level (e.g., neuronal proliferation, differentiation, viability (apoptosis), and migration) [16, 17].
Classically, chemicals are evaluated for neurotoxicity [18, 19] and DNT [20–22] with standardized in vivo tests based on guidelines developed in the US and Europe (e.g., OECD TG 426). While informative and predictive in identifying potential developmental neurotoxicants [22], traditional in vivo assessments are costly, time-consuming, and suboptimal to rapidly screen the thousands of compounds being released in the environment [23]. In line with strategies to integrate the 3R principles (replacement, reduction, refinement) into toxicological testing and to appropriately model the complexities of the developing CNS, incorporating alternative methodologies as part of a DNT battery can increase screening-throughput, hasten validation efforts, and reduce reliance on traditional in vivo studies [24, 25]. Recent technological advancements in evaluating mRNA expression have enhanced our ability to determine global molecular perturbations that correlate with or precede toxicity in relation to chemical exposures (reviewed, [26]). The identification of molecular signatures linked with normal/abnormal neurodevelopmental processes [27] and/or toxic chemical exposures [28] may provide benchmarks that can be applied to determine common effects of developmental neurotoxicants across models. While toxicogenomic changes are highly dependent on several toxicological factors (e.g., dose, time, model, sex), the integration of like-datasets may lead to the discovery of common and consistent features that can be applied in hazard identification and screening approaches. As RA signaling is highly conserved among vertebrates and regulates various developmental processes in addition to CNS formation (e.g., skeletal [29], lung [30], heart [31], germ cell [32]), genomic approaches can define susceptible molecular players associated with toxicological interference, thereby facilitating and enhancing DNT hazard identification.
Adverse outcome pathways (AOPs) provide a framework to link environmental exposures to perturbations on molecular, cellular and functional levels that contribute to changes to DNT and disease [33]. AOPs can be implemented with in vitro, in silico, and in vivo toxicological testing strategies aimed at determining potential hazards. Recent work by Tonk et al. [34] constructed an AOP connecting perturbations in RA signaling and neural tube/axial patterning defects, with this framework also serving as a case study for a developmental toxicity ontology [35]. RA’s critical role across a diversity of developmental processes underscores the need to define key molecular interactions mediating organismal defects, as chemicals that directly or indirectly disrupt components of RA signaling—such as retinoids [14], azoles [36], or ethanol [37]–confer (neuro)developmental toxicity by disrupting RA homeostasis. Key examples of enzymes involved in RA metabolism were provided by Tonk et al. and their diminished function were linked with imbalances in RA and defects in the neural tube, axial patterning, and other malformations such as craniofacial and cardiac defects. As a proof-of-principle, the AOP framework was validated for hazard prediction utilizing pre-existing experimental data of flusilazole─ an antifungal agent and proposed RA-signaling disruptor─ in rat whole embryo culture (WEC), zebrafish (Zf) embryo, and embryonic stem cell (ESC) models.
Building on this approach, we constructed a provisional AOP framework connecting RA signaling and DNT, with a focus on CNS patterning and neuronal differentiation. In the following sections, we summarize existing literature linking RA signaling to CNS development and propose models and endpoints relevant for DNT assessment. Next, we integrated toxicogenomic datasets across mammalian model systems and performed a meta-analysis to identify common gene expression changes from excess RA exposure during early neurodevelopment, informing the design of our AOP framework. The resulting RA-DNT gene list was compared to a Zf embryo dataset to illustrate the applicability of these proposed gene targets in other model systems. Lastly, as a case study, we utilized the AOP and RA-DNT gene set to provide supporting evidence that azoles, a class of antifungals, disrupt RA signaling and cause DNT.
Molecules regulating RA signaling during early CNS development
Vitamin A is transformed into RA in two steps (Figure 1). First, retinol conversion to retinaldehyde is mediated by two classes of oxidizing enzymes (alcohol dehydrogenases and retinol dehydrogenases). In mouse, retinol dehydrogenase-10 (Rdh10) is necessary for conversion of retinol to retinaldehyde during early embryogenesis [38, 39]. Enzymes such as Short-chain Dehydrogenase/Reductase 3 (Dhrs3) facilitate the reverse conversion of retinaldehyde to retinol [40, 41]. In the second step, retinaldehyde is irreversibly oxidized to form RA by aldehyde dehydrogenases (ALDHs). Specifically, ALDH1A2, also known as retinaldehyde dehydrogenase 2 (Raldh2), is critical for RA synthesis during early CNS development [42]. The cytochrome p450 26 subfamily enzymes (Cyp26a1/Cyp26b1/Cyp26c1) are also necessary in regulating RA levels in the embryo by catalysing reactions converting RA to less active RA metabolites, thereby reducing bioavailability [43]. Although less defined, other enzymes, including other CYPs (Cyp1b1) [44, 45], may play secondary roles in RA metabolism during early CNS development.
Figure 1. Retinoic acid metabolism.
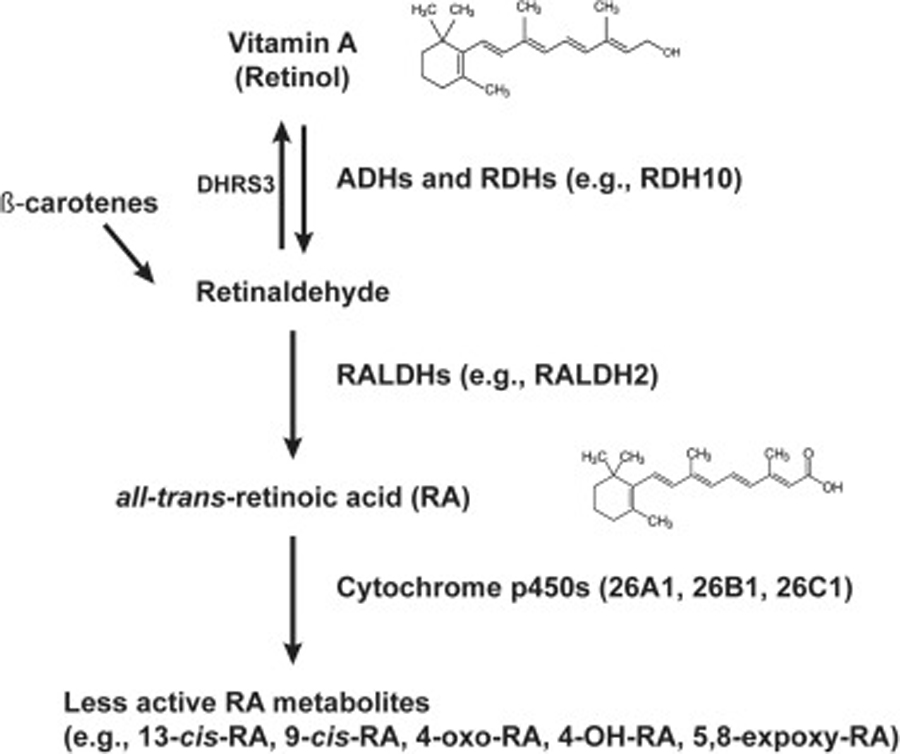
The availability of RA is tightly controlled in the mammalian CNS. Mammals are unable to synthesize RA de novo and require intake of vitamin A or other precursors (β-carotenes) from food sources. Vitamin A is converted to retinaldehyde by alcohol dehydrogenases (ADHs) and retinol dehydrogenases (RDHs). In mouse, RDH10 is necessary for conversion of retinol to retinaldehyde in the developing embryo [38]. Enzymes such as Short-chain Dehydrogenase/Reductase 3 (DHRS3) facilitates the reverse transformation of retinaldehyde to retinol [40, 68]. Retinaldehyde is further oxidized to form RA by aldehyde dehydrogenases (ALDHs or RALDHs) in an irreversible step. RALDH2 is critical for RA synthesis during early CNS development [42]. Cytochrome p450 26 subfamily enzymes regulate RA levels in the embryo and catalyze reactions to reduce RA bioavailability by converting RA to 4-OH-RA, 4-oxo RA, and other oxidized, less active metabolites [43]. These metabolites undergo glucuronidation which promote elimination pathways [180].
Cellular binding-proteins such as CRABP1 and CRABP2 facilitate RA transfer to the nucleus, though these proteins also play roles in RA cellular sequestration, metabolism, and function [46]. In the nucleus, retinoic acid receptors RARs and RXRs are activated by different forms of RA. The primary endogenous ligand for RARs is RA, while 9-cis-RA binds to both RARs and RXRs in vitro [47, 48]. After ligand binding, RARs form heterodimer complexes with RXRs and bind to motifs known as RA response elements (RAREs) to mediate gene transcription. Though the co-factors specific to RA-mediated CNS development remain undefined, transcriptional activation is influenced by binding of RAR/RXR heterodimer complexes, leading to the release of factors (e.g., SMRT [49], histone deacetylases) that restrict chromatin access and the recruitment of co-activators (e.g., CBP/p300 [50]) and co-repressors (e.g., LCoR [51, 52]). A suite of molecules that include RAR/RXRs, co-factors, binding proteins, and metabolizing enzymes are critical for controlling RA bioavailability and ligand-activity in the developing CNS.
RA signaling and CNS regional patterning
Neural induction is initiated by the release of signaling factors from notochord mesoderm to neighboring ectoderm cells overlying the notochord, giving rise to the neuroectoderm (as reviewed [53]). Along the dorsal region of the embryo, the neuroectoderm transforms to give rise to the neural plate. Thickening of neural plate tissue and elevation at the lateral edges of the neural tube results in neural folds which eventually merge at the dorsal midline and establish the neural tube, forming the backbone of the CNS.
Early in development, RA contributes to the antero-posterior and dorso-ventral patterning of the neural plate and neural tube via a tightly-regulated spatiotemporal concentration gradient [2, 54]. The specification of distinct regions in the CNS—forebrain, midbrain, hindbrain, and spinal cord— arises from the regulation of gene expression in each of these domains [55]. RA signaling influences transcription in these domains via “sources” and sinks,” represented by RALDHs in the posterior and CYP26 enzymes in the anterior ends of the embryo respectively. The bioavailability of RA is tightly controlled by the coordinated expression of these, and numerous other metabolic enzymes, throughout development [56]. During the presomite stages, Raldh2 and Rdh10 expression in the paraxial mesoderm produces RA in posterior trunk regions [57, 58]. The anterior neuroepithelium restricts diffusion of RA from these posterior regions via Cyp26a1 expression [43]. Meanwhile RA in the posterior regions activates FGF and WNT signaling to repress expansion of Cyp26a1 at the posterior regions [59]. Patterning of the CNS is regulated by this RA gradient.
The critical influence of RA on hindbrain patterning is well-established. During hindbrain formation, transient metameric units called rhombomeres develop (Figure 2). The hindbrain is composed of 7 rhombomeres and each segment generates a specific repertoire of genes that confers its distinct regional identity. RA regulates the formation of rhombomeres through interactions with RAREs on Hox genes. The function of Hox genes and their targets in establishing rhombomere identity has been thoroughly reviewed [60–62]. Briefly, RA regulates expression of Hox genes 1–4 [63] during hindbrain segmentation, with the highest concentrations of RA beginning at the caudal end and shifting throughout development [64]. In general, Hox expression varies within one or two-segment rhombomere boundary increments. Hoxa1 and Hoxb1 are induced early at the presomite stage and are critical for r4 patterning [65, 66]. Krox20 (Egr2), a zinc finger transcription factor, is subsequently activated and regulates the expression of Hoxa2 and Hoxb2 in r3 and r5 [67, 68]. Hoxa2 expression is required to maintain r2 identity [69], while the interactions of Hoxa2 and Hoxb2, in combination with Hoxa1 and Krox20, are necessary for r3 [70, 71]. Kreisler (Mafb) induces expression of Hoxa3 and Hoxb3, Hox genes important for the r5 and r6 segments [72, 73]. While no direct interaction between RA and Kreisler is known to us, indirect interactions could affect its expression. Krox20 and Kreisler synergize to regulate Hoxb3 expression [74], and Cdx1, which prevents hindbrain patterning in the spinal cord region [75], is activated by RA and represses Kreisler [76]. Hoxa4/Hoxb4/Hoxd4 maintain the r6/r7 boundaries respectively and all contain RAREs [77].
Figure 2. Retinoic acid and anterior-posterior axis formation.

During the formative stages of CNS development, retinoic acid (RA) is regionally restricted and induces posterior embryonic growth. CYP26A1 is exclusively expressed in anterior tissues leading to the breakdown of RA and inactivation of RAR/RXR-signaling. Growth factors (Fgf, Wnt) which promote a posterior phenotype inhibit CYP26A1 and promote RALDH2 biosynthesis of RA, contributing to a gradient of RA availability in the intermediate zone, enabling RAR/RXR activation of HOX genes and other molecules which promote expansion of the posterior domain. Modified from [10, 181].
RA signaling and neuronal differentiation
RA can differentiate cells towards various identities, including serotonergic, GABAergic, glutamatergic, and dopaminergic neurons. Transcriptional regulation of these different differentiation processes is context-dependent, based on factors such as RA levels [78] and positional cues [79]. One such example is the differentiation of cells from the ventral p3 hindbrain domain, which can become glutamatergic V3 spinal interneurons or serotonergic hindbrain neurons despite originating from the same progenitor pool. RA signaling influences the fate of these progenitors through dose-dependent attenuation of ASCL1 levels via the Notch pathway, with areas of low RA/high ASCL1 being permissive of serotoninergic differentiation and RA repression of ASCL1 inducing a glutamatergic V3 identity [80].
RA is critical for differentiation of neurons in the nigrostriatal system and cortical neurogenesis [81]. In a regional- and temporal-dependent manner, Raldh enzymes regulate RA bioavailability and, subsequently, differentiation. Raldh1 is expressed in neurons of the ventral midbrain early [82] and becomes restricted to the dorsal retina and dopaminergic neurons in the substantia nigra and ventral tegmentum [83] later in development. Raldh1 expression in these regions is transcriptionally regulated by Pitx3, as evidenced by loss of the enzyme in the meso-diencephalic dopaminergic neurons of Pitx3 knockout models and restoration with RA [84]. RA transcriptionally regulates dopaminergic differentiation via activation of the dopamine autoreceptor D2 at a RARE in its promoter region [85]. In contrast, Raldh3 expression shifts to the lateral ganglionic eminence (LGE) during development and extends to the piriform cortex and septum [86]. The LGE serves as a local site of RA during striatal neurogenesis, with production originating from these Raldh3 expressing cells [86, 87] and glia [88]. LGE cells and neuronal subpopulations migrating through the LGE require RA signaling for GABAergic differentiation [89]. RA production in the LGE is mediated by Gsh2 [90], with Gsh1 maintaining proliferation of LGE progenitors [91]. Gad67 upregulation is also a demonstrated effect of RA, though the mechanism is unclear as no RARE has been identified in the promoter region [92]. Maintaining the critical balance of RA during development extends into CNS maturation, as early regions of RA synthesis generally maintain their spatial distribution of RA later [93]. For example, in the postnatal rodent brain the highest levels of endogenous RA possess similar regional distinctions seen early in development (e.g, striatum) when measured with mass spectrometry [94, 95]. Thus altering RA homeostasis in sensitive regions or cellular populations may have lasting impact as RA is involved in the initial differentiation, and continued maintenance, of these neurons [96].
Genetic defects in RA signaling and adverse CNS outcomes
Due to the importance of RA bioavailability during hindbrain formation and anteroposterior patterning, proper RA signaling is necessary for development in these CNS regions. Genetic modulation of RA metabolic enzymes, receptors, and downstream signaling targets has provided substantial insight into the role of RA pathway members in hindbrain and AP development. Perturbations of various components of the RA signaling pathway has been linked to impaired patterning, neural tube closure, and CNS morphology (Supplemental Table 3). For example, Rdh10(−/−) embryos carrying a RARE-lacZ RA-reporter transgene display a loss of RARE-lacZ expression in the eye/forebrain and hindbrain regions, with defects in patterning and development in those areas [39]. Meanwhile, Raldh2(−/−) embryos generated insufficient levels of RA to activate a RAREhsplacZ transgene, subsequently resulting in hindbrain/axial defects [97]. Cyp26a1(−/−) embryos exhibit increased hindbrain defects, NTDs (exencephaly and spina bifida), and caudal truncation [43, 98]). Cyp26a1(−/−) models are sensitive to excessive concentrations of RA [99] and rescued by eliminating embryonic expression of Rarg [100].
Altered expression of RAR/RXR, transducers of RA-signaling, are similarly linked with abnormalities of the CNS. Observed phenotypes are related to the spatiotemporal patterns of the target molecule. For instance, Rarg is expressed prior to closure of the neural tube, while Rarb and Rara are expressed as the neural tube folds adjoin [101–103]. Rara(−/−):Rarg(−/−) double null mouse mutants [104] have significantly higher levels of exencephaly, while Rarg(−/−) are less susceptible to abnormalities in the neural tube [105, 106]. The genetic loss in expression of downstream target genes of RA, including Hoxa1 [107, 108], Hoxb1 [109, 110], Krox-20 (Egr2) [111] and Kreissler (Mafb) [112, 113] are also associated with abnormalities in hindbrain development.
Evidence of RA level imbalances in experimental models and developmental neurotoxicity
Various experimental models have a demonstrated concentration-dependent effect of RA on CNS development, with higher concentrations causing more overt impairments and teratogenicity (examples provided in Table 1). In animal models, including mouse [114], rat [115, 116], hamster [117], rabbit [118], and monkey [119], prenatal imbalances in retinoid levels —in excess or deficiency—results in DNT. In general, the most sensitive windows to retinoid-induced DNT appear when imbalances in RA signaling occurs during neurulation and early organogenesis [120–123], while later periods in development may be more vulnerable to deficiencies in brain weight and potentially postnatal survival [124]. Numerous studies in rodent models have exogenously manipulated RA signaling through diet or injection, leading to description of specific CNS malformations and structural variations. Sprague Dawley rats exposed on embryonic day 10 to RA (50–70 mg RA/kg maternal body weight, oral gavage) were significantly associated with multiple CNS deficiencies, including myelomeningocele, lumbosacral defects, exencephaly, and caudal regression syndrome [125]. Meanwhile, embryos of Vitamin A-deficient (VAD) pregnant rats (Sprague Dawley) display loss/disorganization of cranial nerves, loss of caudal hindbrain segmentation, expansion of rhombomeres R3–4, and otic vesicle abnormalities [126].
Table 1: Examples of concentration-dependent effects of RA in diverse models of CNS development and neurotoxicity.
We summarized a selection of studies detailing RA exposures in various models of DNT and their corresponding phenotypes based on increasing concentration.
| Model | Conc. | Effect | Reference |
|---|---|---|---|
| human ESC neural rosette | 0.002μM | dysregulated expression of RA-dependent signaling pathway members (e.g., Hox genes) | [135] |
| mouse ESC | 0.01μM | increased neural outgrowth and differentiation | [134] |
| human ESC neural rosette | 0.02μM | increased neural cell proliferation and differentiation; dysregulated expression of RA signaling pathways | [135] |
| zebrafish embryo | 0.33μM | dysregulated expression of RA metabolism enzymes, RA receptors, and downstream signaling molecules | [41] |
| rat maternal plasma (**Cmax; single dose RA, oral) | 1.2μM | low teratogencity | [116] |
| rat whole embryo culture | 1.7μM | moderate teratogenicity; dysregulated expression of RA metabolism enzymes and downstream signaling molecules | [128] |
| human ESC neural rosette | 2μM | inhibition of neural differentiation; dysregulated expression of RA signaling pathways | [135] |
| rat maternal plasma (**Cmax; single dose RA, ip) | 4.6μM | low-moderately teratogenic | [118] |
| rat maternal plasma (**Cmax; single dose RA, ip) | 6.3μM | moderately teratogenic | [118] |
| rat maternal plasma (**Cmax; single dose RA, ip) | 9.9μM | highly teratatogenic | [118] |
Abbreviations: concentration (Conc.); embryonic stem cells (ESC); concentration maximum (Cmax); intraperitoneal (ip).
= estimate from figure in manuscript.
Alternative models, including rat WEC, ESCs, and Zf embryo have also been utilized to evaluate retinoids in the context of DNT. The WEC model is a proposed tool to study the influence of both environmental and genetic factors on early embryonic development and has proven to be a valuable model to evaluate the teratogenic potency of retinoids. Morris and Steele demonstrated the use of the WEC model to profile the teratogenic effects of excess retinol and RA exposures during early organogenesis, validating in vivo observations that elevated exposures to retinoids during neurulation results in defects of the neural tube as well as malformations of the palate and limb [127]. Recent studies examining RA effects in WEC have incorporated transcriptomic approaches to identify global changes in gene expression which may underlie and precede morphological alterations. Luijten et al. utilized transcriptomic profiling to identify early responses to teratogenic levels of RA (0.5 μg/mL) in single cultured rat embryos initially exposed at 2–4 somites [16]. RA affected 260 genes after a 4h exposure duration, including genes involved in embryonic patterning and CNS development (e.g., Gbx2 and Otx2) as well as genes linked with RA metabolism (Cyp26a1, Cyp26b1, Dhrs3) and RA gene activation (Rarb). In a study by Robinson et al., RA effects on morphological and transcriptomic levels were compared between the rat postimplantation WEC model (0.5 μg/mL) and embryos in vivo undergoing neurulation (50 mg/kg) [128]. Consistent with Luitjen et al., perturbations were found in 845 genes and enriched for processes involved in CNS and embryonic development as well as neuronal differentiation; more than 50% of identified differentially expressed genes in either model overlapped. Thus, the WEC may be considered a useful alternative system to test retinoids and other environmental compounds in their effects on RA signaling in the context of early CNS development.
While the predictive value of ESCs for DNT screening has yet to be fully defined [24], early investigations suggest that these model systems can be used to study a wide-range of risk factors on multiple aspects of neurogenesis [24, 129, 130], including retinoids and other environmental chemicals that cause neurotoxicity by perturbing RA-signaling pathways. Retinoids (RA or Vitamin A) are commonly added in ESC cultures to induce neural differentiation [15, 131] and can regulate patterning genes in a similar manner to in vivo [132, 133]. Utilizing a transcriptomic approach at four time points during mESC-embryoid body differentiation, Akanuma et al. (2012; [134]) demonstrated the stage-dependent sensitivity to RA at two concentrations (0.01 or 0.1μM). These analyses provided information regarding gene networks influenced by RA exposure at 0, 2, 8, and 36 days of mESC differentiation in parallel with cell morphological changes (increased growth and neuronal differentiation), and identified particular targets (e.g., Gfap, Gbx2) that may amplify RA-signaling and neuronal development.
The effects of RA have also been examined in various hESC models in relation to neurodevelopment and DNT in vitro. For example in a study using a hESC-based neural rosette model—a proposed screen to identify toxicants that disrupt early CNS morphogenesis and apical neuroepithelial organization of the neural tube—the concentration-dependent effects on cell morphology, function and global mRNA expression were evaluated [135]. Exposures to RA caused increased cytotoxicity (2μM), disorganized neural rosette morphology (≥0.2μM), and perturbed expression of molecules critical for CNS patterning (HOXA1, HOXA3, HOXB1, HOXB4) and neural differentiation (FOXA2, FOXC1, OTX2, PAX7)─ suspected target genes of RA in vivo [136, 137]─at concentrations as low as 0.02μM. As a model of early CNS development, ESC models may be used to assess cellular and molecular changes relevant to RA-induced DNT.
The zebrafish (Zf) embryo model has also been proposed as a predictive alternative screening system for developmental toxicology [138–141]. The relevance of RA regulation in Zf CNS development has been demonstrated through RARE visualization [142] and genetic knockdowns of metabolic enzymes which promote biosynthesis (rdh10 [143]; raldh2 [144]) or breakdown (cyp26a1 [145], dhrs3 [146]) of RA. Several investigations have examined the relationship between excess or deficiencies in RA and DNT using Zf. In a concentration-dependent manner, Holder and Hill demonstrated RA (0.001–1μM) to cause CNS and tail malformations [147]. Concentrations of 0.1–1μM RA produced loss of the mid-hindbrain border, rhombomere abnormalities and reduced neurons within the anterior lateral line and ganglia. Global transcriptomic analyses of Zf during early stages of embryogenesis demonstrate genes disrupted by RA (0.1μM) overlap with mammals, including cyp26a/b1, dhrs3a/b, aldh1a2 (raldh2), and hox genes [148]. In the same study, utilizing translational-blocking morpholinos, the authors demonstrated that the expression levels of a subset of genes (~27%; 85 genes) altered by RA are dependent on rar expression. These studies illustrate the ability of the Zf embryo model to be used to evaluate RA-induced DNT on morphological and molecular levels.
Material and Methods
Processing of transcriptomic datasets and identification of a RA-induced developmental neurotoxicity signature in mammalian models
To define a transcriptomic signature associated with RA and DNT, we searched for datasets in public online repositories maintained by the National Center for Biotechnology Information (Gene Expression Omnibus) and European Molecular Biology Laboratory-European Bioinformatics Institute (ArrayExpress) using search terms [“retinoic” or “retinoid”] and [“toxicity” or “neurotoxicity”]. Additional searches were completed ad hoc. In total, we identified twelve transcriptomic studies evaluating RA exposures in vertebrate model systems (Supplemental Table 1). First, we determined a common signature related to RA and DNT in mammalian models. We selected three datasets from two studies [E-MEXP-3577 and GSE33195] for meta-analysis. These studies examined the: 1) toxicological-response of RA in human embryonic stem cells (hESCs) differentiating towards a neural cell fate; or 2) time-dependent responses of a developmentally toxic concentration of RA in cultured embryos (WEC) or in embryos in vivo. As previously described [27], datasets were independently processed, i.e., log2 transformed, normalized via the Robust Multi-array Average (RMA) algorithm, and annotated. To identify genes commonly dysregulated by RA in association with DNT, a fixed effects linear model (ANOVA) was independently applied in each study to determine significance of RA across concentration or time. In the case of multiple probes per gene, the one with the lowest p-value, i.e., most significant changes with RA, was used for comparison purposes. Next, we determined genes commonly altered in all three studies by applying a cutoff of p<0.005, and incorporated a secondary filter that required genes to trend in the same direction in all 3 datasets with RA effects as determined by Pearson’s Correlation (across dose; E-MEXP-3577) or the computed average fold change in expression across time (GSE33195). This subset was termed the “RA-DNT” gene set (Supplemental Table 2). Datasets were merged using the Official Gene Symbol and R statistical package. Hierarchical clustering of fold change (FC) values for differentially expressed genes was determined using average linkage and Euclidean distance [149]. We performed enrichment analysis of RA-DNT genes utilizing Gene Set Enrichment Analysis [150] and a cutoff of q<1*10−10.
We examined the conservation of patterns of expression of RA-DNT genes, identified in mammalian model systems, in Zf embryos exposed to RA using the dataset GSE43755 (Supplemental Table 1). This transcriptomic study included profiles of RA-exposed (100nM at the 2 cell embryo stage; ~45 minutes post-fertilization) and control embryos at 3, 5, and 8h post exposure. In Zf embryos, exposures of 100nM RA have been shown to induce significant teratogenic effects, including malformations of the CNS. We processed, annotated, and merged the dataset with the RA-DNT gene set based on the OGS identifier. Genes identified as significantly altered due to RA exposure were defined with a p<0.05 (ANOVA). We performed a secondary filter to verify homology (≥ 60% similarity in protein sequence) between mammals and Zf via the National Center for Biotechnology Information Basic Local Alignment Search Tool (BLAST) [49].
We demonstrated the applicability of the RA-DNT gene set in two independent toxicogenomic assessments of azoles in cultured rat whole embryos (WEC) generated at the National Institute of Public Health and the Environment (GSE102082 [151] and [152]; Supplemental Table 1). The dataset GSE102082 [151] includes whole rat embryos exposed to a single concentration of either: difenoconazole, fenarimol, flusilazole, ketoconazole, miconazole, propiconazole, prothioconazole, tebuconazole, triadimefon, B595, B599 or B600, estimated to significantly induce developmental toxicity (reduce total morphological score (TMS) by 10%) after 4h exposure. The other dataset [152] utilized whole rat embryos exposed to either one of four concentrations of flusilazole or one of two concentrations of cyproconazole or triadimefon after 4h exposure. As described above, these datasets were normalized via Robust Multichip Average (RMA) and annotated. We identified RA-DNT genes significantly dysregulated by each compound (ANOVA; p<0.05). Within these gene subsets, we determined the number of differentially expressed RA-DNT genes that displayed similar directional changes, up or downregulated, as excessive RA exposure in the evaluated mammalian systems. Fold enrichment (observed vs. expected) and the significance of identifying the number of differentially expressed RA-DNT genes for each compound (Chi-Squared test with Yates’ continuity correction; https://www.graphpad.com/) was also calculated. In the case of multiple probes per gene, the one with the lowest p-value corresponding with all azoles tested in each study was used for comparison purposes.
An Adverse Outcome Pathway of RA-induced developmental neurotoxicity and the integration of a toxicogenomic signature of RA-DNT
We constructed a provisional RA-DNT AOP framework (as reviewed [153]) utilizing diverse literature sources (detailed above in the introduction). First, we assembled evidence supporting the role of RA in hindbrain formation/patterning and neuronal differentiation on the molecular, cellular, tissue, and organism level. Next, we connected a series of key events (KEs) to predict adverse outcome(s), with the first triggering KE being the molecular initiating event(s) (MIEs) that describe primary chemical-molecule interactions [33]. As the levels of RA and the activation of RA-dependent genes may be dependent on many factors—including spatiotemporal and concentration-dependent restrictions—multiple MIEs plausibly exist. Thus, we developed the RA-DNT AOP in a general manner to capture potential chemical-molecular interactions that contribute to the dysregulation of RA and RA-mediated transcription. Subsequent secondary KEs were categorized based on: 1) genes regulated by RA-RAR/RXR activation with evident roles in patterning and differentiation; 2) cellular regional identity and neuronal differentiation; 3) hindbrain (tissue) development; and 4) brain development and maturation on an organism level. Our defined transcriptomic signature associated with RA and DNT was incorporated into the AOP as a potential broad indicator of RA-dysregulation.
Results
Integration of toxicogenomic datasets to define a signature of RA-induced developmental neurotoxicity
We integrated genomic datasets related to RA exposures to identify a transcriptomic signature applicable across mammalian model systems, with the aim of defining predictive biomarkers of RA response for DNT assessments. We identified three datasets from two relevant studies: E-MEXP-3577 and GSE33195, examining the: 1) concentration-response of RA in hESCs differentiating towards neural rosettes or 2) time-dependent response of a developmentally toxic concentration of RA in cultured embryos (WEC) or in utero during neurulation/early organogenesis. Differentially expressed genes due to RA were identified via generalized linear models (ANOVA), and datasets were compared to identify common genes dysregulated across all three datasets. In total, we observed 131 differentially expressed genes (p<0.005; Figure 3A). Within this subset, 95 genes displayed common trends in regulation, of which 74 were upregulated, and 21 were downregulated. In general, the magnitude in RA-induced effects on expression were concentration- or time-dependent (Figure 3B). We performed enrichment analysis of the “RA-DNT” genes utilizing Gene Set Enrichment Analysis [150]. RA-DNT genes were significantly enriched for processes related to regulation of cell differentiation, embryo development, embryonic morphogenesis and regulation of gene expression (q<1*10−10; Figure 3C). Gene targets identified through this analysis (Figure 3D; Supplemental Table 2) included molecules involved in: RA metabolism (CYP26A1, CYP26B1, DHRS3), RA signaling (RARA, RARB), RA binding (CRABP2), patterning (HOXA1, HOXB2, MEIS1, MEIS2) and differentiation (MAFB, PBX1, SIX, SKIL). We propose that this gene set may be utilized to: 1) identify potential compounds that cause DNT via RA-mediated pathways, and 2) compare across mammalian and non-mammalian model systems for common/unique mechanisms of RA-induced DNT.
Figure 3. Characterization of common genes identified to be differentially expressed with RA exposure and associated with developmental neurotoxicity in mammalian models.

(A) We identified 131 genes to be commonly differentially expressed in association with DNT across in vitro, ex vivo and in vitro models (p<0.005, ANOVA). (B) Hierarchical clustering of the subset of 95 genes that displayed common trends in regulation, of which 74 were upregulated and 21 were downregulated with RA exposure in a concentration or time-dependent manner. This subset of genes was termed the RA-DNT gene set. (C) Identified enriched GO Biological Processes within the RA-DNT gene set (q<1*10−10). (D) RA-DNT genes with absolute average fold change ≥ 2 (log 2 scale) comparing RA vs. control per study. (E) We identified homologues for 64% (61 total genes) of the RA-DNT gene set. Within this subset, we identified 16 genes to be altered with RA in the Zf embryo model. All 16 genes trended in similar fashion to RA-exposed mammalian model systems. Referenced datasets: E-MEXP-3577 [135], GSE33195 [128], and GSE43755 [154].
Zebrafish case study
As a proof of principle, we examined for conservation of patterns of RA-DNT gene expression in the Zf embryo using the dataset GSE43755 [154], which included profiles of RA-exposed (100nM) and control embryos at 3, 5, and 8h post exposure, initially exposed at the 2 cell embryo stage. The concentration of RA (100nM) used in this study is significantly teratogenic (e.g., brain malformations) in the Zf [155, 156]. We identified homologues for 62 genes of the RA-DNT gene set. We observed an enrichment of differentially expressed RA-DNT genes (p<0.00001). In total, 16 genes were found to be altered with RA in the Zf embryo model, 15 of which were upregulated, 1 downregulated; and 100% of differentially expressed genes followed similar trends, i.e., up or downregulated, with observed responses in mammalian models (Figure 3E). The dysregulated genes included: crabp2a, cyp26a1, cyp26b1, dhrs3a, hoxb2a, lhx5 (downregulated), meis1b, meis2b, mmp11b, nrip1, raraa, rarab, skib, spsb4a, tiparp, and tshz1. Thus, our analysis led to the identification of relevant genes linked with mammalian RA-DNT that may be evaluated as biomarkers of RA exposure in Zf. These data support previous evidence suggesting significant conservation of members involved in the RA signaling pathway across vertebrates.
Integrating toxicogenomic signatures into the AOP framework
Based on available data in the literature and the RA-DNT gene set, we outlined a preliminary AOP of RA-induced DNT that describes the mechanistic links between excessive or deficiency in RA bioavailability and defects in hindbrain development (Figure 4).
Figure 4. A provisional adverse outcome pathway for RA and hindbrain development.

Under normal conditions during the initial stages of CNS development, RA is regionally restricted by enzymes that promote biosynthesis (Rdh10, Raldh2) or elimination (Cyp26a1, Cyp26c1) of RA. Binding proteins (e.g., Crabp1/2) facilitate RA transfer to the nucleus. RA binding of RAR/RXRs leads to recruitment of co-activators and co-repressors which mediate specificity of transcription. RA mediates expression of multiple gene and gene families involved in patterning and differentiation. On a localized level, changes in expression lead to cell specification (patterning) and cell differentiation, and underlie expansion of the hindbrain and maturation of the CNS. The hindbrain serves as the basis for the cerebellum, pons, and medulla. A prospective MIE that alters RA availability and/or activation of RAR/RXR can lead to KEs that change the expression of genes responsible for regulating cell regional identify and differentiation. These altered pathways underlie perturbations in hindbrain expansion and cell organization, which manifest as adverse outcomes brain development and structure.
The initial development and segmentation of the hindbrain occurs within the period of neurulation and early organogenesis, approximately between the 3rd and 4th weeks of human pregnancy, GD7.5–10 in mouse, and GD9.5–12 in rat. RA bioavailability is regionally controlled by a suite of enzymes. Rdh10 and Raldh2, expressed in the paraxial mesoderm, promote biosynthesis of RA and diffusion to proximal tissues. Cyp26a1 is highly expressed in the anterior region and strictly limits RA bioavailability in this region. Nuclear transfer of RA and activation of RAR/RXR leads to the recruitment of co-activators and co-repressors which mediate specificity of transcription. In a tightly regulated sequence, segments of the hindbrain are formed due to RA bioavailability and subsequent RA-induced gene expression. RA regulates the regional expression of several genes and signaling molecules that underlie hindbrain segmentation and regional identity, including Krox20(Egr2), MafB, and Hox family members [157] as well as target genes involved in the differentiation of cells within each region.
Alterations in the bioavailability of RA, in excess or deficiency, can result in severe malformations and/or variants in hindbrain development and posterior segmentation. These changes can occur on the molecular, cellular, and tissue level in the CNS, and depend on the timing/duration of RA exposure, dose, and factors such as genetic variants in RA metabolic enzymes. Excess RA results in hyper activation of RAR/RXR-mediated gene expression, whereas deficiency in RA, leads to lower activation of RAR/RXR-mediated gene expression. Subsequently, potential MIEs can occur through various means, such as 1) altering the availability of RA via enzymes that control the molecule’s spatiotemporal gradient or 2) interaction with RAR/RXR and/or its cofactors to alter RA-mediated gene expression. Regulation of RA-mediated target genes represents a key event (KE), as regional cellular identity (e.g., Hox family members, Krox20, MafB, Gbx2, Ncam1) and neuronal differentiation (e.g., Ngn2, Dbx1, Pax6) can be altered by aberrant expression of these genes. Perturbations in regional cell identity and differentiation underlies disruption of hindbrain expansion and disorganization on the tissue level. Postnatally, morphological defects may appear as irregularities in CNS morphology (abnormal segmentation), NTDs, and/or changes in brain patterning and size. Disruptions in the development of these regions can potentially manifest in, or increase the risk of, adverse neurobehavioral outcomes such as deficiencies in motor control or sleep on the organismal level. In the preliminary framework, we propose that alterations in either direction in specific gene/gene families may be biomarkers of changes in RA status.
Azole case study
Direct or indirect chemical-molecular interactions may lead to the disruption of RA levels and activation of RA-dependent gene expression, critical MIEs, which may result in subsequent perturbations on the molecular, cellular, tissue, and organism level. Here, utilizing the provisional AOP and the RA-DNT predictive signature, we classify evidence supporting the ability of azoles to cause DNT during early brain formation by altering RA signaling.
Azoles represent a diverse class of compounds (e.g., triazoles, imidazoles) which contain a five-membered ring comprised of at least one nitrogen atom and at least one other non-carbon atom. These compounds are widely-used as antifungal agents due to their ability to impair ergosterol formation, a necessary component of fungal cell-wall integrity, via inhibition of lanosterol 14α-demethylase (CYP51A1). In vertebrates, azoles are also suspected to cause DNT by altering RA signaling pathways by inhibiting CYP26 [36]. Metabolic disruption by select azoles leads to altered effects on the molecular, cellular, and organism level similar to observations as RA (at excess levels), including hindbrain segmentation and patterning defects as well as craniofacial and axial malformations [158, 159].
Evidence of azoles disrupting RA signaling has also been previously proposed on a global expression level [151, 152, 160]. Utilizing two transcriptomic datasets examining azoletoxicity in rat WEC (Supplemental Table 1), we interrogated the relationship between azole exposures and the expression of RA-DNT genes. In total, we examined the response of 13 independent azoles; two compounds (flusilazole, triadimefon) were evaluated in both datasets under different exposure designs. We identified enrichment of differentially expressed RA-DNT genes with 10 of the 13 azoles (p<0.01, Chi-Square). In both datasets, transcriptomic profiles of flusilazole or triadimefon exposed embryos were significantly enriched for differentially expressed RA-DNT genes (p<0.0001; fold enrichment ≈ 4X), and trends in dysregulation paralleled responses observed with excess RA exposure (≥96% agreement). Genes significantly altered by ≥5 compounds (p<0.05) included: DHRS3, CYP26A1, RARB, PRICKLE1, FNDC5, ZFP36L2, RARA, MAFB, RPS6KA2, WFIKKN1, HOXA1, MEIS1, GSE1, TSHZ1, RAB38, DHX32, SERPING1, DEPTOR, CRABP2, and MGAT4A. These analyses utilizing the independently derived RA-DNT gene set support the ability of azoles to alter expression of genes involved in RA-response and DNT, specific to hindbrain development and neural differentiation.
Discussion
In this study, we utilized existing literature and integrated relevant toxicogenomic datasets to construct a provisional RA-DNT AOP framework. Despite the central role of RA in embryonic growth and development, imbalances can be a human health risk; RA disruption during pregnancy leads to fetal CNS malformations and adverse neurodevelopmental outcomes [161]. Concentration-dependent effects of RA on various experimental models have been observed (Table 1), and as numerous xenobiotics classes may perturb RA signaling—including fungicides [36], styrenes [162], metals [163], and pesticides [164]—constructing an AOP framework could facilitate DNT evaluation of chemicals that disrupt the RA pathway. The use of alternative assays is a core tenet of AOP development and because RA signaling is conserved in vertebrates, we leveraged genomic approaches to create a RA-responsive gene set during CNS development, i.e. biomarkers of RA-DNT, that could refine and harmonize screening efforts. In the context of RA signaling, a multitude of enzymes regulate RA levels and their receptors. Accordingly, there potentially exists multiple MIEs, i.e., molecular interactions, that trigger global or local changes in RA bioavailability and signaling cascades. There is a vast literature detailing the components of the RA pathway and identifying the essential molecules for DNT assessment will be crucial. Our provisional AOP notes some of these potential key targets, informed by our review, and incorporates some examples from our toxicogenomic analysis that could inform comparisons across models.
We identified the RA-DNT gene set consisting of significantly responsive targets with shared directionality across hESC and rat embryos (ex vivo and in vivo) models. Proposed target genes included key enzymes involved in RA metabolism (CYP26A1, CYP26B1, DHRS3A), RARA and the transcriptional coregulator NRIP1, and the RA binding protein, CRABP2. Other identified RA-DNT genes control downstream KEs, including molecules critical in hindbrain patterning (HOXA1, HOXB2, TSHZ1, MEIS1, MEIS2) [165], cell migration (MMP11) [166], and neuron differentiation (LHX5) [167]. Uncharacterized RA-responsive genes, such as SPSB4 [168], SKIL, and TIPARP were also found in our analysis. Disruption of RA-target genes during this vulnerable time period in CNS development, such as those in the RA-DNT gene set, can lead to perturbations in regional cell identity and neuron differentiation during development, impacting hindbrain formation and expansion. Alterations in RA-sensitive brain structures can manifest as overt malformations, structural variations, and/or potentially, neurobehavioral deviations [122], such as respiration [169, 170] or motor activity [171]. By integrating like-transcriptomic datasets and identifying common dysregulated genes in mammalian models, we propose targets that can be introduced into the AOP framework to examine RA-DNT.
In an analysis of transcriptomic profiles of RA-exposed Zf embryos, we observed an enrichment of differentially expressed RA-DNT genes (Figure 3E), demonstrating the utility of the molecular signature in non-mammalian model systems. Common targets included well-described conserved elements of the RA-signaling pathway in vertebrates (e.g., cyp26a1, cyp26b1, crabp2a, dhrs3a, hoxb2a, nrip1, raraa, rarab). Divergence in RA responsive signatures between mammalian and Zf datasets may be due to several factors, such as differing RA exposure and developmental time-window, and additional comparisons with other Zf datasets is warranted to determine conservation of the RA-DNT signature. In addition, while many aspects of Zf development are comparable to mammals, the Zf may diverge in certain processes such as neural tube formation [288], and the need of RA for body axis extension [289]. The inclusion of hESCs in creating the RA-DNT gene set is also relevant. Though hESCs express and are capable of activating patterning genes, future protocols that emphasize optimizing culture cytoarchitecture will likely improve comparisons between the WEC and Zf embryo datasets [172]. These caveats also underscore the contextual nature of RA signaling, wherein effects are influenced by variables such as RA concentration and developmental time. Evaluating the responsiveness of multiple genes in the RA-DNT will be important, as the relative contribution of any single target can also vary by model system. For example CRABP2, while significantly responsive in our comparisons and essential for Zf hindbrain patterning [173], is dispensable in the mouse [174].
We applied the RA-DNT gene list to the case study of azoles to reinforce the value of toxicogenomic comparisons, as focusing on identifying common biomarker signatures within a biological pathway could hasten the identification of mechanisms of action by a chemical class. Our results, similar to reported findings which focused on specific RA and cholesterol metabolism genes [175, 176], revealed specific azoles, e.g., propiconazole, triadimefon, flusilazole, cyproconazole, to be more disruptive to RA-signaling at developmentally toxic levels (Table 2). By utilizing the RA-DNT gene set, we quantitatively described potency in terms of disruption of RA-signaling. Alterations in RA-DNT genes in either direction could be used as biomarkers and potentially, trends in expression may indicate excess vs. deficiency in RA levels. For example, in azoles that significantly disrupted the RA-DNT gene set, trends in dysregulation generally matched transcriptomic profiles associated with excess RA.
Table 2: Differentially expressed RA-DNT genes in cultured rat embryos exposed to triazole compounds.
We utilized two datasets (Study A: [152] and Study B: GSE102082 [151]) to assess the transcriptomic effects of triazole compounds in rat whole embryo culture (WEC) at concentration(s) associated with altered morphology, i.e., decline in total morphological score (TMS). Datasets were processed and analyzed for differentially expressed genes using a cutoff of p<0.05 (uncorrected). Within this subset, we determined the number of differentially expressed RA-DNT genes, the associated fold enrichment (FE) (i.e., ratio of observed vs. expected), statistical significance (Chi-Squared test with Yates’ continuity correction) in identifying with differentially expressed RA-DNT genes, and the agreement in trend with RA response in mammalian models.
| Study | Compound (# concentrations) | DE Genes | DE RA-DNT | FE | p-value | Trend w/RA |
|---|---|---|---|---|---|---|
| A | Flusilazole (4) | 728 | 21 | 4.4 | <0.0001 | 100% |
| Cyproconazole (2) | 828 | 22 | 4.0 | <0.0001 | 95% | |
| Triadimefon (2) | 869 | 25 | 4.0 | <0.0001 | 100% | |
| B | Propiconazole (1) | 920 | 31 | 5.1 | <0.0001 | 100% |
| Triadimefon (1) | 1056 | 31 | 4.4 | <0.0001 | 100% | |
| Flusilazole (1) | 1110 | 28 | 3.8 | <0.0001 | 96% | |
| Ketoconazole (1) | 1372 | 27 | 3.0 | <0.0001 | 89% | |
| B599 (1) | 1214 | 20 | 2.5 | <0.0001 | 95% | |
| Difenoconazole (1) | 747 | 14 | 2.8 | <0.0001 | 86% | |
| B600 (1) | 961 | 16 | 2.5 | 0.0002 | 100% | |
| Fenarimol (1) | 1471 | 20 | 2.1 | 0.0009 | 95% | |
| B595 (1) | 944 | 16 | 2.6 | 0.0054 | 100% | |
| Tebuconazole (1) | 969 | 11 | 1.7 | 0.0928 | 100% | |
| Miconazole (1) | 834 | 4 | 0.7 | 0.7254 | 100% | |
| Prothioconazole (1) | 1029 | 8 | 1.2 | 0.7806 | 63% |
The RA-DNT gene set and provisional AOP framework is not intended to be comprehensive or conclusive. There are significant challenges associated with developing AOPs informative for DNT, including the complexity and diversity of the brain, the multiple mechanisms that govern its development, and the potential of neurotoxicants to affect numerous molecular targets [177]. The RA signaling pathway is similarly complex and diverse, regulating the development of numerous organs [178]. Over 500 genes are associated with the RA signaling pathway [137] and deciphering transcriptional mechanisms and essential players is ongoing. While we have framed our AOP in the context of hindbrain development, the RA-DNT gene set and provisional AOP framework presented here provides only a general framework that necessitates additional, rigorous experimental input to be further developed. Within the AOP framework, alterations in the RA-DNT gene set potentially represents an early KE that signals general DNT risk. Although the cellular-, tissue-, and organismal- responses detailed in the provisional framework are DNT outcomes associated with disruptions in RA signaling, establishing the relative contributions of genomic alterations to these responses would significantly strengthen KE-relationships (KERs). Other MIEs outside of the presented AOP are also not accounted for; for instance, RA signaling can influence synaptic plasticity through non-genomic mechanisms [179], suggesting that RA and/or analogues can influence determinants of neural connectivity through other KEs. As knowledge of RA’s function expands, validation and incorporation of endpoints from nongenomic assays—e.g., cellular, functional, behavioral—that define sensitive target molecules or tissues would better elucidate these additional KEs and KERs. Alternative model systems, including those referenced above, will be relevant as they are adaptable to several DNT test methods used to evaluate KEs within the provisional RA-DNT framework. As AOPs are purposed to be deliberate simplifications of biological pathways, incorporating advances in mechanistic understanding will be necessary to both better evaluate chemicals in their ability to disrupt RA signaling pathways and to refine subsequent AOPs for RA-DNT.
Supplementary Material
Highlights.
Review of retinoic acid (RA) signaling and developmental neurotoxicity (DNT)
Defined an adverse outcome pathway (AOP) related to imbalances in RA and DNT
By integrating diverse datasets, identified genes associated with RA exposure and DNT
This gene set, when applied to the zebrafish model, yielded shared targets of RA
Demonstrated application of RA-signature in AOP framework and chemical screening
Acknowledgements
The Organisation for Economic Co-operation and Development (OECD) Test Guidelines Programme Detailed Review Paper 178 (DRP178) provided an overview of in vitro and in vivo screening and testing methods and endpoints for evaluating endocrine disruptors. Due to the known interaction between RA signaling and endocrine pathways, within this review, molecular effect biomarkers of the retinoid system for tiered toxicity screening and monitoring studies were also summarized. As an extension of this project, a review of the Retinoid system was initiated (project 4.97b) within the OECD Endocrine Disruptors Testing and Assessment Advisory Group (EDTA AG). As a member of this working group, we reviewed literature ad hoc and summarized information related to retinoids and links to DNT to construct a review paper. In-part, language and content were modified from this report (written by Dr. Robinson and colleagues) to develop this study. The authors would like to thank Patience Browne (OECD), Thomas Knudsen (USEPA), and members of the OECD DRP Working Group for their tremendous feedback regarding content and structure of the OECD DRP Neurotoxicity Report, which provided the key elements of this manuscript.
Funding
This work was kindly supported by OECD and the National Institutes of Environmental Health Sciences (NIEHS) K99ES030401, K99ES023846, and R00ES023846.
Abbreviations:
- AOP
Adverse outcome pathway
- ADH
Alcohol dehydrogenase
- ALDH
Aldehyde dehydrogenase
- CNS
Central nervous system
- DNT
Developmental neurotoxicity
- ESC
Embryonic stem cell
- FC
Fold change
- hESC
Human embryonic stem cell
- NTD
Neural tube defect
- KE
Key event
- MIE
Molecular initiating event
- OECD
Organisation for Economic Co-operation and Development
- OGS
Official gene symbol
- RALDH
Retinaldehyde dehydrogenase
- RA
All-trans retinoic acid or retinoic acid
- RA-DNT
Retinoic acid-induced developmental neurotoxicity
- RAR
Retinoic acid receptor
- RARE
Retinoic acid response element
- RXR
Retinoid X receptor
- RDH
Retinol dehydrogenase
- RMA
Robust Multichip Average
- VAD
Vitamin A deficiency
- WEC
Whole embryo culture
- Zf
Zebrafish
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Competing Interests
The authors have no conflicts to declare.
Disclosure Statement:
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marceau G, Gallot D, Lemery D, and Sapin V, Metabolism of retinol during mammalian placental and embryonic development. Vitam Horm, 2007. 75: p. 97–115. [DOI] [PubMed] [Google Scholar]
- 2.Duester G, Retinoic acid synthesis and signaling during early organogenesis. Cell, 2008. 134(6): p. 921–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clagett-Dame M and Knutson D, Vitamin A in reproduction and development. Nutrients, 2011. 3(4): p. 385–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brand N, Petkovich M, Krust A, Chambon P, de The H, Marchio A, Tiollais P, and Dejean A, Identification of a second human retinoic acid receptor. Nature, 1988. 332(6167): p. 850–3. [DOI] [PubMed] [Google Scholar]
- 5.Krust A, Kastner P, Petkovich M, Zelent A, and Chambon P, A third human retinoic acid receptor, hRAR-gamma. Proc Natl Acad Sci U S A, 1989. 86(14): p. 5310–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petkovich M, Brand NJ, Krust A, and Chambon P, A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature, 1987. 330(6147): p. 444–50. [DOI] [PubMed] [Google Scholar]
- 7.Mangelsdorf DJ, Vitamin A receptors. Nutr Rev, 1994. 52(2 Pt 2): p. S32–44. [DOI] [PubMed] [Google Scholar]
- 8.Maden M, Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci, 2007. 8(10): p. 755–65. [DOI] [PubMed] [Google Scholar]
- 9.Maden M, Retinoid signalling in the development of the central nervous system. Nat Rev Neurosci, 2002. 3(11): p. 843–53. [DOI] [PubMed] [Google Scholar]
- 10.Cunningham TJ and Duester G, Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol, 2015. 16(2): p. 110–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCaffery PJ, Adams J, Maden M, and Rosa-Molinar E, Too much of a good thing: retinoic acid as an endogenous regulator of neural differentiation and exogenous teratogen. The European journal of neuroscience, 2003. 18(3): p. 457–72. [DOI] [PubMed] [Google Scholar]
- 12.Gutierrez-Mazariegos J, Theodosiou M, Campo-Paysaa F, and Schubert M, Vitamin A: a multifunctional tool for development. Seminars in cell & developmental biology, 2011. 22(6): p. 603–10. [DOI] [PubMed] [Google Scholar]
- 13.Louisse J, Gönen S, Rietjens IMCM, and Verwei M, Relative developmental toxicity potencies of retinoids in the embryonic stem cell test compared with their relative potencies in in vivo and two other in vitro assays for developmental toxicity. Toxicology letters, 2011. 203(1): p. 1–8. [DOI] [PubMed] [Google Scholar]
- 14.Collins MD and Mao GE, Teratology of retinoids. Annu Rev Pharmacol Toxicol, 1999. 39: p. 399–430. [DOI] [PubMed] [Google Scholar]
- 15.Maden M, Role and distribution of retinoic acid during CNS development. Int Rev Cytol, 2001. 209: p. 1–77. [DOI] [PubMed] [Google Scholar]
- 16.Luijten M, van Beelen VA, Verhoef A, Renkens MF, van Herwijnen MH, Westerman A, van Schooten FJ, Pennings JL, and Piersma AH, Transcriptomics analysis of retinoic acid embryotoxicity in rat postimplantation whole embryo culture. Reprod Toxicol, 2010. 30(2): p. 333–40. [DOI] [PubMed] [Google Scholar]
- 17.Melton KR, Iulianella A, and Trainor PA, Gene expression and regulation of hindbrain and spinal cord development. Frontiers in bioscience : a journal and virtual library, 2004. 9: p. 117–38. [DOI] [PubMed] [Google Scholar]
- 18.(OECD), O.f.E.C.-o.a.D. Neurotoxicity Study in Rodents. 1997. Paris, france. [Google Scholar]
- 19.Agency, U.S.E.P. Neurotoxicity Screening Battery. 1998. Washington, DC, USA. [Google Scholar]
- 20.Agency, U.S.E.P. Developmental Neurotoxicity Study. 1998. Washington, DC, USA. [Google Scholar]
- 21.Development, O.f.E.C.-o.a. Developmental Neurotoxicity Study. 2007. [Google Scholar]
- 22.Makris SL, Raffaele K, Allen S, Bowers WJ, Hass U, Alleva E, Calamandrei G, Sheets L, Amcoff P, Delrue N, and Crofton KM, A retrospective performance assessment of the developmental neurotoxicity study in support of OECD test guideline 426. Environmental health perspectives, 2009. 117(1): p. 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grandjean P and Landrigan PJ, Developmental neurotoxicity of industrial chemicals. Lancet, 2006. 368(9553): p. 2167–78. [DOI] [PubMed] [Google Scholar]
- 24.Bal-Price A, Hogberg HT, Crofton KM, Daneshian M, FitzGerald RE, Fritsche E, Heinonen T, Hougaard Bennekou S, Klima S, Piersma AH, Sachana M, Shafer TJ, Terron A, Monnet-Tschudi F, Viviani B, Waldmann T, Westerink RHS, Wilks MF, Witters H, Zurich MG, and Leist M, Recommendation on test readiness criteria for new approach methods in toxicology: Exemplified for developmental neurotoxicity. ALTEX, 2018. 35(3): p. 306–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crofton KM, Mundy WR, Lein PJ, Bal-Price A, Coecke S, Seiler AE, Knaut H, Buzanska L, and Goldberg A, Developmental neurotoxicity testing: recommendations for developing alternative methods for the screening and prioritization of chemicals. ALTEX, 2011. 28(1): p. 9–15. [PubMed] [Google Scholar]
- 26.Robinson JF and Piersma AH, Toxicogenomic approaches in developmental toxicology testing. Methods in molecular biology, 2013. 947: p. 451–73. [DOI] [PubMed] [Google Scholar]
- 27.Robinson JF, Gormley MJ, and Fisher SJ, A genomics-based framework for identifying biomarkers of human neurodevelopmental toxicity. Reprod Toxicol, 2016. 60: p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Dartel DA, Pennings JL, de la Fonteyne LJ, Brauers KJ, Claessen S, van Delft JH, Kleinjans JC, and Piersma AH, Evaluation of developmental toxicant identification using gene expression profiling in embryonic stem cell differentiation cultures. Toxicological sciences : an official journal of the Society of Toxicology, 2011. 119(1): p. 126–34. [DOI] [PubMed] [Google Scholar]
- 29.Zeller R, The temporal dynamics of vertebrate limb development, teratogenesis and evolution. Current opinion in genetics & development, 2010. 20(4): p. 384–90. [DOI] [PubMed] [Google Scholar]
- 30.Fernandes-Silva H, Araujo-Silva H, Correia-Pinto J, and Moura RS, Retinoic Acid: A Key Regulator of Lung Development. Biomolecules, 2020. 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sirbu IO, Chis AR, and Moise AR, Role of carotenoids and retinoids during heart development. Biochimica et biophysica acta. Molecular and cell biology of lipids, 2020: p. 158636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Endo T, Mikedis MM, Nicholls PK, Page DC, and de Rooij DG, Retinoic Acid and Germ Cell Development in the Ovary and Testis. Biomolecules, 2019. 9(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ankley GT and Edwards SW, The Adverse Outcome Pathway: A Multifaceted Framework Supporting 21(st) Century Toxicology. Current opinion in toxicology, 2018. 9: p. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tonk EC, Pennings JL, and Piersma AH, An adverse outcome pathway framework for neural tube and axial defects mediated by modulation of retinoic acid homeostasis. Reprod Toxicol, 2015. 55: p. 104–13. [DOI] [PubMed] [Google Scholar]
- 35.Baker N, Boobis A, Burgoon L, Carney E, Currie R, Fritsche E, Knudsen T, Laffont M, Piersma AH, Poole A, Schneider S, and Daston G, Building a developmental toxicity ontology. Birth Defects Res, 2018. 110(6): p. 502–518. [DOI] [PubMed] [Google Scholar]
- 36.Menegola E, Broccia ML, Di Renzo F, and Giavini E, Postulated pathogenic pathway in triazole fungicide induced dysmorphogenic effects. Reproductive toxicology, 2006. 22(2): p. 186–95. [DOI] [PubMed] [Google Scholar]
- 37.Deltour L, Ang HL, and Duester G, Ethanol inhibition of retinoic acid synthesis as a potential mechanism for fetal alcohol syndrome. FASEB J, 1996. 10(9): p. 1050–1057. [PubMed] [Google Scholar]
- 38.Sandell LL, Lynn ML, Inman KE, McDowell W, and Trainor PA, RDH10 oxidation of Vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PloS one, 2012. 7(2): p. e30698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chatzi C, Cunningham TJ, and Duester G, Investigation of retinoic acid function during embryonic brain development using retinaldehyde-rescued Rdh10 knockout mice. Developmental dynamics : an official publication of the American Association of Anatomists, 2013. 242(9): p. 1056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams MK, Belyaeva OV, Wu L, and Kedishvili NY, The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J Biol Chem, 2014. 289(21): p. 14868–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng L, Hernandez RE, Waxman JS, Yelon D, and Moens CB, Dhrs3a regulates retinoic acid biosynthesis through a feedback inhibition mechanism. Developmental biology, 2010. 338(1): p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niederreither K, Subbarayan V, Dolle P, and Chambon P, Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nature genetics, 1999. 21(4): p. 444–8. [DOI] [PubMed] [Google Scholar]
- 43.Abu-Abed S, Dolle P, Metzger D, Beckett B, Chambon P, and Petkovich M, The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev, 2001. 15(2): p. 226–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross AC and Zolfaghari R, Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annual review of nutrition, 2011. 31: p. 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chambers D, Wilson L, Maden M, and Lumsden A, RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development, 2007. 134(7): p. 1369–83. [DOI] [PubMed] [Google Scholar]
- 46.Napoli JL, Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacology & therapeutics, 2017. 173: p. 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Oro AE, Kakizuka A, and Evans RM, Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes & development, 1992. 6(3): p. 329–44. [DOI] [PubMed] [Google Scholar]
- 48.Allenby G, Janocha R, Kazmer S, Speck J, Grippo JF, and Levin AA, Binding of 9-cis-retinoic acid and all-trans-retinoic acid to retinoic acid receptors alpha, beta, and gamma. Retinoic acid receptor gamma binds all-trans-retinoic acid preferentially over 9-cis-retinoic acid. The Journal of biological chemistry, 1994. 269(24): p. 16689–95. [PubMed] [Google Scholar]
- 49.Jepsen K, Solum D, Zhou T, McEvilly RJ, Kim HJ, Glass CK, Hermanson O, and Rosenfeld MG, SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature, 2007. 450(7168): p. 415–9. [DOI] [PubMed] [Google Scholar]
- 50.Lee S, Lee B, Lee JW, and Lee SK, Retinoid signaling and neurogenin2 function are coupled for the specification of spinal motor neurons through a chromatin modifier CBP. Neuron, 2009. 62(5): p. 641–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandes I, Bastien Y, Wai T, Nygard K, Lin R, Cormier O, Lee HS, Eng F, Bertos NR, Pelletier N, Mader S, Han VK, Yang XJ, and White JH, Ligand-dependent nuclear receptor corepressor LCoR functions by histone deacetylase-dependent and - independent mechanisms. Molecular cell, 2003. 11(1): p. 139–50. [DOI] [PubMed] [Google Scholar]
- 52.Koide T, Downes M, Chandraratna RA, Blumberg B, and Umesono K, Active repression of RAR signaling is required for head formation. Genes Dev, 2001. 15(16): p. 2111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nikolopoulou E, Galea GL, Rolo A, Greene ND, and Copp AJ, Neural tube closure: cellular, molecular and biomechanical mechanisms. Development, 2017. 144(4): p. 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niederreither K, Fraulob V, Garnier JM, Chambon P, and Dolle P, Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mechanisms of Development, 2002. 110(1–2): p. 165–171. [DOI] [PubMed] [Google Scholar]
- 55.Rhinn M and Brand M, The midbrain--hindbrain boundary organizer. Current opinion in neurobiology, 2001. 11(1): p. 34–42. [DOI] [PubMed] [Google Scholar]
- 56.Rhinn M and Dolle P, Retinoic acid signalling during development. Development, 2012. 139(5): p. 843–58. [DOI] [PubMed] [Google Scholar]
- 57.Molotkova N, Molotkov A, Sirbu IO, and Duester G, Requirement of mesodermal retinoic acid generated by Raldh2 for posterior neural transformation. Mech Dev, 2005. 122(2): p. 145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sandell LL, Sanderson BW, Moiseyev G, Johnson T, Mushegian A, Young K, Rey J-P, Ma J.-x., Staehling-Hampton K, and Trainor PA, RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev, 2007. 21(9): p. 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kudoh T, Wilson SW, and Dawid IB, Distinct roles for Fgf, Wnt and retinoic acid in posteriorizing the neural ectoderm. Development, 2002. 129(18): p. 4335–46. [DOI] [PubMed] [Google Scholar]
- 60.Alexander T, Nolte C, and Krumlauf R, Hox genes and segmentation of the hindbrain and axial skeleton. Annu Rev Cell Dev Biol, 2009. 25: p. 431–56. [DOI] [PubMed] [Google Scholar]
- 61.Svingen T and Tonissen KF, Hox transcription factors and their elusive mammalian gene targets. Heredity, 2006. 97(2): p. 88–96. [DOI] [PubMed] [Google Scholar]
- 62.Gavalas A and Krumlauf R, Retinoid signalling and hindbrain patterning. Curr Opin Genet Dev, 2000. 10(4): p. 380–6. [DOI] [PubMed] [Google Scholar]
- 63.Hunt P, Gulisano M, Cook M, Sham MH, Faiella A, Wilkinson D, Boncinelli E, and Krumlauf R, A distinct Hox code for the branchial region of the vertebrate head. Nature, 1991. 353(6347): p. 861–4. [DOI] [PubMed] [Google Scholar]
- 64.Sirbu IO, Gresh L, Barra J, and Duester G, Shifting boundaries of retinoic acid activity control hindbrain segmental gene expression. Development, 2005. 132(11): p. 2611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Studer M, Gavalas A, Marshall H, Ariza-McNaughton L, Rijli FM, Chambon P, and Krumlauf R, Genetic interactions between Hoxa1 and Hoxb1 reveal new roles in regulation of early hindbrain patterning. Development (Cambridge, England), 1998. 125(6): p. 1025–1036. [DOI] [PubMed] [Google Scholar]
- 66.Barrow JR, Stadler HS, and Capecchi MR, Roles of Hoxa1 and Hoxa2 in patterning the early hindbrain of the mouse. Development (Cambridge, England), 2000. 127(5): p. 933–944. [DOI] [PubMed] [Google Scholar]
- 67.Nonchev S, Vesque C, Maconochie M, Seitanidou T, Ariza-McNaughton L, Frain M, Marshall H, Sham MH, Krumlauf R, and Charnay P, Segmental expression of Hoxa-2 in the hindbrain is directly regulated by Krox-20. Development, 1996. 122(2): p. 543–54. [DOI] [PubMed] [Google Scholar]
- 68.Sham MH, Vesque C, Nonchev S, Marshall H, Frain M, Gupta RD, Whiting J, Wilkinson D, Charnay P, and Krumlauf R, The zinc finger gene Krox20 regulates HoxB2 (Hox2.8) during hindbrain segmentation. Cell, 1993. 72(2): p. 183–96. [DOI] [PubMed] [Google Scholar]
- 69.Gavalas A, Davenne M, Lumsden A, Chambon P, and Rijli FM, Role of Hoxa-2 in axon pathfinding and rostral hindbrain patterning. Development (Cambridge, England), 1997. 124(19): p. 3693–3702. [DOI] [PubMed] [Google Scholar]
- 70.Davenne M, Maconochie MK, Neun R, Pattyn A, Chambon P, Krumlauf R, and Rijli FM, Hoxa2 and Hoxb2 control dorsoventral patterns of neuronal development in the rostral hindbrain. Neuron, 1999. 22(4): p. 677–91. [DOI] [PubMed] [Google Scholar]
- 71.Helmbacher F, Pujades C, Desmarquet C, Frain M, Rijli FM, Chambon P, and Charnay P, Hoxa1 and Krox-20 synergize to control the development of rhombomere 3. Development, 1998. 125(23): p. 4739–48. [DOI] [PubMed] [Google Scholar]
- 72.Manzanares M, Cordes S, Kwan CT, Sham MH, Barsh GS, and Krumlauf R, Segmental regulation of Hoxb-3 by kreisler. Nature, 1997. 387(6629): p. 191–5. [DOI] [PubMed] [Google Scholar]
- 73.Manzanares M, Cordes S, Ariza-McNaughton L, Sadl V, Maruthainar K, Barsh G, and Krumlauf R, Conserved and distinct roles of kreisler in regulation of the paralogous Hoxa3 and Hoxb3 genes. Development, 1999. 126(4): p. 759–69. [DOI] [PubMed] [Google Scholar]
- 74.Manzanares M, Nardelli J, Gilardi-Hebenstreit P, Marshall H, Giudicelli F, Martinez-Pastor MT, Krumlauf R, and Charnay P, Krox20 and kreisler co-operate in the transcriptional control of segmental expression of Hoxb3 in the developing hindbrain. EMBO J, 2002. 21(3): p. 365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Skromne I, Thorsen D, Hale M, Prince VE, and Ho RK, Repression of the hindbrain developmental program by Cdx factors is required for the specification of the vertebrate spinal cord. Development, 2007. 134(11): p. 2147–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sturgeon K, Kaneko T, Biemann M, Gauthier A, Chawengsaksophak K, and Cordes SP, Cdx1 refines positional identity of the vertebrate hindbrain by directly repressing Mafb expression. Development, 2011. 138(1): p. 65–74. [DOI] [PubMed] [Google Scholar]
- 77.Niederreither K, Vermot J, Schuhbaur B, Chambon P, and Dolle P, Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development, 2000. 127(1): p. 75–85. [DOI] [PubMed] [Google Scholar]
- 78.Okada Y, Shimazaki T, Sobue G, and Okano H, Retinoic-acid-concentration-dependent acquisition of neural cell identity during in vitro differentiation of mouse embryonic stem cells. Developmental biology, 2004. 275(1): p. 124–42. [DOI] [PubMed] [Google Scholar]
- 79.Carcagno AL, Di Bella DJ, Goulding M, Guillemot F, and Lanuza GM, Neurogenin3 restricts serotonergic neuron differentiation to the hindbrain. Journal of Neuroscience, 2014. 34(46): p. 15223–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jacob J, Kong J, Moore S, Milton C, Sasai N, Gonzalez-Quevedo R, Terriente J, Imayoshi I, Kageyama R, Wilkinson DG, Novitch BG, and Briscoe J, Retinoid acid specifies neuronal identity through graded expression of Ascl1. Current biology : CB, 2013. 23(5): p. 412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA, Folias AE, Choe Y, May SR, Kume T, Napoli JL, Peterson AS, and Pleasure SJ, Retinoic acid from the meninges regulates cortical neuron generation. Cell, 2009. 139(3): p. 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wallen A, Zetterstrom RH, Solomin L, Arvidsson M, Olson L, and Perlmann T, Fate of mesencephalic AHD2-expressing dopamine progenitor cells in NURR1 mutant mice. Exp Cell Res, 1999. 253(2): p. 737–46. [DOI] [PubMed] [Google Scholar]
- 83.McCaffery P and Drager UC, High levels of a retinoic acid-generating dehydrogenase in the meso-telencephalic dopamine system. Proc Natl Acad Sci U S A, 1994. 91(16): p. 7772–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jacobs FM, Smits SM, Noorlander CW, von Oerthel L, van der Linden AJ, Burbach JP, and Smidt MP, Retinoic acid counteracts developmental defects in the substantia nigra caused by Pitx3 deficiency. Development, 2007. 134(14): p. 2673–84. [DOI] [PubMed] [Google Scholar]
- 85.Samad TA, Krezel W, Chambon P, and Borrelli E, Regulation of dopaminergic pathways by retinoids: activation of the D2 receptor promoter by members of the retinoic acid receptor-retinoid X receptor family. Proc Natl Acad Sci U S A, 1997. 94(26): p. 14349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li H, Wagner E, McCaffery P, Smith D, Andreadis A, and Drager UC, A retinoic acid synthesizing enzyme in ventral retina and telencephalon of the embryonic mouse. Mech Dev, 2000. 95(1–2): p. 283–9. [DOI] [PubMed] [Google Scholar]
- 87.Waclaw RR, Wang B, and Campbell K, The homeobox gene Gsh2 is required for retinoid production in the embryonic mouse telencephalon. Development (Cambridge, England), 2004. 131(16): p. 4013–4020. [DOI] [PubMed] [Google Scholar]
- 88.Toresson H, Mata de Urquiza A, Fagerstrom C, Perlmann T, and Campbell K, Retinoids are produced by glia in the lateral ganglionic eminence and regulate striatal neuron differentiation. Development, 1999. 126(6): p. 1317–26. [DOI] [PubMed] [Google Scholar]
- 89.Chatzi C, Brade T, and Duester G, Retinoic acid functions as a key GABAergic differentiation signal in the basal ganglia. PLoS biology, 2011. 9(4): p. e1000609-e1000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Waclaw RR, Wang B, and Campbell K, The homeobox gene <em>Gsh2</em> is required for retinoid production in the embryonic mouse telencephalon . Development, 2004. 131(16): p. 4013. [DOI] [PubMed] [Google Scholar]
- 91.Toresson H and Campbell K, A role for Gsh1 in the developing striatum and olfactory bulb of Gsh2 mutant mice. Development, 2001. 128(23): p. 4769–80. [DOI] [PubMed] [Google Scholar]
- 92.Chatzi C, Cunningham TJ, and Duester G, Investigation of retinoic acid function during embryonic brain development using retinaldehyde-rescued Rdh10 knockout mice. Dev Dyn, 2013. 242(9): p. 1056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thompson Haskell G, Maynard TM, Shatzmiller RA, and Lamantia A-S, Retinoic acid signaling at sites of plasticity in the mature central nervous system. The Journal of comparative neurology, 2002. 452(3): p. 228–241. [DOI] [PubMed] [Google Scholar]
- 94.Kane MA, Chen N, Sparks S, and Napoli JL, Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochemical Journal, 2005. 388: p. 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kane MA, Folias AE, Wang C, and Napoli JL, Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Analytical Chemistry, 2008. 80(5): p. 1702–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zetterstrom RH, Lindqvist E, Mata de Urquiza A, Tomac A, Eriksson U, Perlmann T, and Olson L, Role of retinoids in the CNS: differential expression of retinoid binding proteins and receptors and evidence for presence of retinoic acid. European Journal of Neuroscience, 1999. 11(2): p. 407–16. [DOI] [PubMed] [Google Scholar]
- 97.Niederreither K, Subbarayan V, Dolle P, and Chambon P, Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet, 1999. 21(4): p. 444–8. [DOI] [PubMed] [Google Scholar]
- 98.Sakai Y, Meno C, Fujii H, Nishino J, Shiratori H, Saijoh Y, Rossant J, and Hamada H, The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev, 2001. 15(2): p. 213–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ribes V, Fraulob V, Petkovich M, and Dolle P, The oxidizing enzyme CYP26a1 tightly regulates the availability of retinoic acid in the gastrulating mouse embryo to ensure proper head development and vasculogenesis. Developmental dynamics : an official publication of the American Association of Anatomists, 2007. 236(3): p. 644–53. [DOI] [PubMed] [Google Scholar]
- 100.Abu-Abed S, Dolle P, Metzger D, Wood C, MacLean G, Chambon P, and Petkovich M, Developing with lethal RA levels: genetic ablation of Rarg can restore the viability of mice lacking Cyp26a1. Development, 2003. 130(7): p. 1449–59. [DOI] [PubMed] [Google Scholar]
- 101.Smith SM and Eichele G, Temporal and regional differences in the expression pattern of distinct retinoic acid receptor-beta transcripts in the chick embryo. Development, 1991. 111(1): p. 245–52. [DOI] [PubMed] [Google Scholar]
- 102.Chen WH, Morriss-Kay GM, and Copp AJ, Genesis and prevention of spinal neural tube defects in the curly tail mutant mouse: involvement of retinoic acid and its nuclear receptors RAR-beta and RAR-gamma. Development, 1995. 121(3): p. 681–91. [DOI] [PubMed] [Google Scholar]
- 103.Ruberte E, Dolle P, Chambon P, and Morriss-Kay G, Retinoic acid receptors and cellular retinoid binding proteins. II. Their differential pattern of transcription during early morphogenesis in mouse embryos. Development, 1991. 111(1): p. 45–60. [DOI] [PubMed] [Google Scholar]
- 104.Lohnes D, Mark M, Mendelsohn C, Dolle P, Dierich A, Gorry P, Gansmuller A, and Chambon P, Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development, 1994. 120(10): p. 2723–48. [DOI] [PubMed] [Google Scholar]
- 105.Lohnes D, Kastner P, Dierich A, Mark M, LeMeur M, and Chambon P, Function of retinoic acid receptor gamma in the mouse. Cell, 1993. 73(4): p. 643–58. [DOI] [PubMed] [Google Scholar]
- 106.Iulianella A and Lohnes D, Contribution of retinoic acid receptor gamma to retinoid-induced craniofacial and axial defects. Developmental dynamics : an official publication of the American Association of Anatomists, 1997. 209(1): p. 92–104. [DOI] [PubMed] [Google Scholar]
- 107.Mark M, Lufkin T, Vonesch JL, Ruberte E, Olivo JC, Dolle P, Gorry P, Lumsden A, and Chambon P, Two rhombomeres are altered in Hoxa-1 mutant mice. Development, 1993. 119(2): p. 319–38. [DOI] [PubMed] [Google Scholar]
- 108.Carpenter EM, Goddard JM, Chisaka O, Manley NR, and Capecchi MR, Loss of Hox-A1 (Hox-1.6) function results in the reorganization of the murine hindbrain. Development, 1993. 118(4): p. 1063–75. [DOI] [PubMed] [Google Scholar]
- 109.Goddard JM, Rossel M, Manley NR, and Capecchi MR, Mice with targeted disruption of Hoxb-1 fail to form the motor nucleus of the VIIth nerve. Development, 1996. 122(10): p. 3217–28. [DOI] [PubMed] [Google Scholar]
- 110.Studer M, Lumsden A, Ariza-McNaughton L, Bradley A, and Krumlauf R, Altered segmental identity and abnormal migration of motor neurons in mice lacking Hoxb-1. Nature, 1996. 384(6610): p. 630–4. [DOI] [PubMed] [Google Scholar]
- 111.Schneider-Maunoury S, Topilko P, Seitandou T, Levi G, Cohen-Tannoudji M, Pournin S, Babinet C, and Charnay P, Disruption of Krox-20 results in alteration of rhombomeres 3 and 5 in the developing hindbrain. Cell, 1993. 75(6): p. 1199–214. [DOI] [PubMed] [Google Scholar]
- 112.Cordes SP and Barsh GS, The mouse segmentation gene kr encodes a novel basic domain-leucine zipper transcription factor. Cell, 1994. 79(6): p. 1025–34. [DOI] [PubMed] [Google Scholar]
- 113.McKay IJ, Muchamore I, Krumlauf R, Maden M, Lumsden A, and Lewis J, The kreisler mouse: a hindbrain segmentation mutant that lacks two rhombomeres. Development, 1994. 120(8): p. 2199–211. [DOI] [PubMed] [Google Scholar]
- 114.Tibbles L and Wiley MJ, A comparative study of the effects of retinoic acid given during the critical period for inducing spina bifida in mice and hamsters. Teratology, 1988. 37(2): p. 113–25. [DOI] [PubMed] [Google Scholar]
- 115.Colakoglu N and Kukner A, Teratogenicity of retinoic acid and its effects on TGF-beta2 expression in the developing cerebral cortex of the rat. J Mol Histol, 2004. 35(8–9): p. 823–7. [DOI] [PubMed] [Google Scholar]
- 116.Collins MD, Tzimas G, Burgin H, Hummler H, and Nau H, Single versus multiple dose administration of all-trans-retinoic acid during organogenesis: differential metabolism and transplacental kinetics in rat and rabbit. Toxicology and applied pharmacology, 1995. 130(1): p. 9–18. [DOI] [PubMed] [Google Scholar]
- 117.Shenefelt RE, Morphogenesis of malformations in hamsters caused by retinoic acid: relation to dose and stage at treatment. Teratology, 1972. 5(1): p. 103–18. [DOI] [PubMed] [Google Scholar]
- 118.Tembe EA, Honeywell R, Buss NE, and Renwick AG, All-trans-retinoic acid in maternal plasma and teratogenicity in rats and rabbits. Toxicology and applied pharmacology, 1996. 141(2): p. 456–72. [DOI] [PubMed] [Google Scholar]
- 119.Hendrickx AG, Tzimas G, Korte R, and Hummler H, Retinoid teratogenicity in the macaque: verification of dosing regimen. J Med Primatol, 1998. 27(6): p. 310–8. [DOI] [PubMed] [Google Scholar]
- 120.Adams J, Structure-activity and dose-response relationships in the neural and behavioral teratogenesis of retinoids. Neurotoxicology and teratology, 1993. 15(3): p. 193–202. [DOI] [PubMed] [Google Scholar]
- 121.Cunningham ML, Mac Auley A, and Mirkes PE, From gastrulation to neurulation: transition in retinoic acid sensitivity identifies distinct stages of neural patterning in the rat. Developmental dynamics : an official publication of the American Association of Anatomists, 1994. 200(3): p. 227–41. [DOI] [PubMed] [Google Scholar]
- 122.Adams J, The neurobehavioral teratology of retinoids: a 50-year history. Birth Defects Res A Clin Mol Teratol, 2010. 88(10): p. 895–905. [DOI] [PubMed] [Google Scholar]
- 123.Holson RR, Gazzara RA, Ferguson SA, Ali SF, Laborde JB, and Adams J, Gestational retinoic acid exposure: a sensitive period for effects on neonatal mortality and cerebellar development. Neurotoxicology and teratology, 1997. 19(5): p. 335–346. [DOI] [PubMed] [Google Scholar]
- 124.Nolen GA, The effects of prenatal retinoic acid on the viability and behavior of the offspring. Neurobehavioral toxicology and teratology, 1986. 8(6): p. 643–654. [PubMed] [Google Scholar]
- 125.Danzer E, Schwarz U, Wehrli S, Radu A, Adzick NS, and Flake AW, Retinoic acid induced myelomeningocele in fetal rats: characterization by histopathological analysis and magnetic resonance imaging. Exp Neurol, 2005. 194(2): p. 467–75. [DOI] [PubMed] [Google Scholar]
- 126.White JC, Highland M, Kaiser M, and Clagett-Dame M, Vitamin A deficiency results in the dose-dependent acquisition of anterior character and shortening of the caudal hindbrain of the rat embryo. Developmental biology, 2000. 220(2): p. 263–84. [DOI] [PubMed] [Google Scholar]
- 127.Morriss GM and Steele CE, Comparison of the effects of retinol and retinoic acid on postimplantation rat embryos in vitro. Teratology, 1977. 15(1): p. 109–19. [DOI] [PubMed] [Google Scholar]
- 128.Robinson JF, Verhoef A, Pennings JL, Pronk TE, and Piersma AH, A comparison of gene expression responses in rat whole embryo culture and in vivo: time-dependent retinoic acid-induced teratogenic response. Toxicol Sci, 2012. 126(1): p. 242–54. [DOI] [PubMed] [Google Scholar]
- 129.Fritsche E, Barenys M, Klose J, Masjosthusmann S, Nimtz L, Schmuck M, Wuttke S, and Tigges J, Current Availability of Stem Cell-Based In Vitro Methods for Developmental Neurotoxicity (DNT) Testing. Toxicol Sci, 2018. 165(1): p. 21–30. [DOI] [PubMed] [Google Scholar]
- 130.Singh S, Srivastava A, Kumar V, Pandey A, Kumar D, Rajpurohit CS, Khanna VK, Yadav S, and Pant AB, Stem Cells in Neurotoxicology/Developmental Neurotoxicology: Current Scenario and Future Prospects. Mol Neurobiol, 2016. 53(10): p. 6938–6949. [DOI] [PubMed] [Google Scholar]
- 131.Janesick A, Wu SC, and Blumberg B, Retinoic acid signaling and neuronal differentiation. Cellular and molecular life sciences : CMLS, 2015. 72(8): p. 1559–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Merrill RA, Ahrens JM, Kaiser ME, Federhart KS, Poon VY, and Clagett-Dame M, All-trans retinoic acid-responsive genes identified in the human SH-SY5Y neuroblastoma cell line and their regulated expression in the nervous system of early embryos. Biological chemistry, 2004. 385(7): p. 605–614. [DOI] [PubMed] [Google Scholar]
- 133.Nadadhur AG, Leferink PS, Holmes D, Hinz L, Cornelissen-Steijger P, Gasparotto L, and Heine VM, Patterning factors during neural progenitor induction determine regional identity and differentiation potential in vitro. Stem Cell Res, 2018. 32: p. 25–34. [DOI] [PubMed] [Google Scholar]
- 134.Akanuma H, Qin XY, Nagano R, Win-Shwe TT, Imanishi S, Zaha H, Yoshinaga J, Fukuda T, Ohsako S, and Sone H, Identification of Stage-Specific Gene Expression Signatures in Response to Retinoic Acid during the Neural Differentiation of Mouse Embryonic Stem Cells. Front Genet, 2012. 3: p. 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Colleoni S, Galli C, Gaspar JA, Meganathan K, Jagtap S, Hescheler J, Sachinidis A, and Lazzari G, Development of a neural teratogenicity test based on human embryonic stem cells: response to retinoic acid exposure. Toxicol Sci, 2011. 124(2): p. 370–7. [DOI] [PubMed] [Google Scholar]
- 136.Bally-Cuif L, Gulisano M, Broccoli V, and Boncinelli E, c-otx2 is expressed in two different phases of gastrulation and is sensitive to retinoic acid treatment in chick embryo. Mechanisms of Development, 1995. 49(1–2): p. 49–63. [DOI] [PubMed] [Google Scholar]
- 137.Balmer JE and Blomhoff R, Gene expression regulation by retinoic acid. Journal of lipid research, 2002. 43(11): p. 1773–1808. [DOI] [PubMed] [Google Scholar]
- 138.Ball JS, Stedman DB, Hillegass JM, Zhang CX, Panzica-Kelly J, Coburn A, Enright BP, Tornesi B, Amouzadeh HR, Hetheridge M, Gustafson AL, and Augustine-Rauch KA, Fishing for teratogens: a consortium effort for a harmonized zebrafish developmental toxicology assay. Toxicol Sci, 2014. 139(1): p. 210–9. [DOI] [PubMed] [Google Scholar]
- 139.Brannen KC, Panzica-Kelly JM, Danberry TL, and Augustine-Rauch KA, Development of a zebrafish embryo teratogenicity assay and quantitative prediction model. Birth defects research. Part B, Developmental and reproductive toxicology, 2010. 89(1): p. 66–77. [DOI] [PubMed] [Google Scholar]
- 140.Hermsen SA, van den Brandhof EJ, van der Ven LT, and Piersma AH, Relative embryotoxicity of two classes of chemicals in a modified zebrafish embryotoxicity test and comparison with their in vivo potencies. Toxicol In Vitro, 2011. 25(3): p. 745–53. [DOI] [PubMed] [Google Scholar]
- 141.Selderslaghs IW, Van Rompay AR, De Coen W, and Witters HE, Development of a screening assay to identify teratogenic and embryotoxic chemicals using the zebrafish embryo. Reprod Toxicol, 2009. 28(3): p. 308–20. [DOI] [PubMed] [Google Scholar]
- 142.Perz-Edwards A, Hardison NL, and Linney E, Retinoic acid-mediated gene expression in transgenic reporter zebrafish. Developmental Biology, 2001. 229(1): p. 89–101. [DOI] [PubMed] [Google Scholar]
- 143.D’Aniello E, Ravisankar P, and Waxman JS, Rdh10a Provides a Conserved Critical Step in the Synthesis of Retinoic Acid during Zebrafish Embryogenesis. PloS one, 2015. 10(9): p. e0138588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Retnoaji B, Akiyama R, Matta T, Bessho Y, and Matsui T, Retinoic acid controls proper head-to-trunk linkage in zebrafish by regulating an anteroposterior somitogenetic rate difference. Development, 2014. 141(1): p. 158–65. [DOI] [PubMed] [Google Scholar]
- 145.Emoto Y, Wada H, Okamoto H, Kudo A, and Imai Y, Retinoic acid-metabolizing enzyme Cyp26a1 is essential for determining territories of hindbrain and spinal cord in zebrafish. Developmental biology, 2005. 278(2): p. 415–27. [DOI] [PubMed] [Google Scholar]
- 146.Billings SE, Pierzchalski K, Butler Tjaden NE, Pang XY, Trainor PA, Kane MA, and Moise AR, The retinaldehyde reductase DHRS3 is essential for preventing the formation of excess retinoic acid during embryonic development. FASEB J, 2013. 27(12): p. 4877–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Holder N and Hill J, Retinoic acid modifies development of the midbrain-hindbrain border and affects cranial ganglion formation in zebrafish embryos. Development, 1991. 113(4): p. 1159–70. [DOI] [PubMed] [Google Scholar]
- 148.Samarut E, Gaudin C, Hughes S, Gillet B, de Bernard S, Jouve PE, Buffat L, Allot A, Lecompte O, Berekelya L, Rochette-Egly C, and Laudet V, Retinoic acid receptor subtype-specific transcriptotypes in the early zebrafish embryo. Molecular endocrinology, 2014. 28(2): p. 260–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, and Quackenbush J, TM4 microarray software suite. Methods Enzymol, 2006. 411: p. 134–93. [DOI] [PubMed] [Google Scholar]
- 150.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America, 2005. 102(43): p. 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dimopoulou M, Verhoef A, Pennings JLA, van Ravenzwaay B, Rietjens I, and Piersma AH, A transcriptomic approach for evaluating the relative potency and mechanism of action of azoles in the rat Whole Embryo Culture. Toxicology, 2017. 392: p. 96–105. [DOI] [PubMed] [Google Scholar]
- 152.Robinson JF, Tonk EC, Verhoef A, and Piersma AH, Triazole induced concentration-related gene signatures in rat whole embryo culture. Reproductive toxicology, 2012. 34(2): p. 275–83. [DOI] [PubMed] [Google Scholar]
- 153.Vinken M, The adverse outcome pathway concept: a pragmatic tool in toxicology. Toxicology, 2013. 312: p. 158–65. [DOI] [PubMed] [Google Scholar]
- 154.Weicksel SE, Xu J, and Sagerstrom CG, Dynamic nucleosome organization at hox promoters during zebrafish embryogenesis. PloS one, 2013. 8(5): p. e63175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Herrmann K, Teratogenic effects of retinoic acid and related substances on the early development of the zebrafish (Brachydanio rerio) as assessed by a novel scoring system. Toxicology in vitro : an international journal published in association with BIBRA, 1995. 9(3): p. 267–83. [DOI] [PubMed] [Google Scholar]
- 156.Selderslaghs IW, Van Rompay AR, De Coen W, and Witters HE, Development of a screening assay to identify teratogenic and embryotoxic chemicals using the zebrafish embryo. Reproductive toxicology, 2009. 28(3): p. 308–20. [DOI] [PubMed] [Google Scholar]
- 157.Tumpel S, Wiedemann LM, and Krumlauf R, Hox genes and segmentation of the vertebrate hindbrain. Current topics in developmental biology, 2009. 88: p. 103–37. [DOI] [PubMed] [Google Scholar]
- 158.Menegola E, Broccia ML, Di Renzo F, and Giavini E, Dysmorphogenic effects of some fungicides derived from the imidazole on rat embryos cultured in vitro. Reproductive toxicology, 2006. 21(1): p. 74–82. [DOI] [PubMed] [Google Scholar]
- 159.Menegola E, Broccia ML, Di Renzo F, Massa V, and Giavini E, Relationship between hindbrain segmentation, neural crest cell migration and branchial arch abnormalities in rat embryos exposed to fluconazole and retinoic acid in vitro. Reproductive toxicology, 2004. 18(1): p. 121–30. [DOI] [PubMed] [Google Scholar]
- 160.Dimopoulou M, Verhoef A, van Ravenzwaay B, Rietjens IM, and Piersma AH, Flusilazole induces spatio-temporal expression patterns of retinoic acid-, differentiation- and sterol biosynthesis-related genes in the rat Whole Embryo Culture. Reprod Toxicol, 2016. 64: p. 77–85. [DOI] [PubMed] [Google Scholar]
- 161.Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, Curry CJ, Fernhoff PM, Grix AW Jr., Lott IT, and et al. , Retinoic acid embryopathy. The New England journal of medicine, 1985. 313(14): p. 837–41. [DOI] [PubMed] [Google Scholar]
- 162.Kamata R, Shiraishi F, Nishikawa J, Yonemoto J, and Shiraishi H, Screening and detection of the in vitro agonistic activity of xenobiotics on the retinoic acid receptor. Toxicol In Vitro, 2008. 22(4): p. 1050–61. [DOI] [PubMed] [Google Scholar]
- 163.Cui Y and Freedman JH, Cadmium induces retinoic acid signaling by regulating retinoic acid metabolic gene expression. J Biol Chem, 2009. 284(37): p. 24925–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lemaire G, Balaguer P, Michel S, and Rahmani R, Activation of retinoic acid receptor-dependent transcription by organochlorine pesticides. Toxicology and applied pharmacology, 2005. 202(1): p. 38–49. [DOI] [PubMed] [Google Scholar]
- 165.Rezsohazy R, Saurin AJ, Maurel-Zaffran C, and Graba Y, Cellular and molecular insights into Hox protein action. Development, 2015. 142(7): p. 1212. [DOI] [PubMed] [Google Scholar]
- 166.Han J, Kim HJ, Schafer ST, Paquola A, Clemenson GD, Toda T, Oh J, Pankonin AR, Lee BS, Johnston ST, Sarkar A, Denli AM, and Gage FH, Functional Implications of miR-19 in the Migration of Newborn Neurons in the Adult Brain. Neuron, 2016. 91(1): p. 79–89. [DOI] [PubMed] [Google Scholar]
- 167.Zhao Y, Sheng HZ, Amini R, Grinberg A, Lee E, Huang S, Taira M, and Westphal H, Control of hippocampal morphogenesis and neuronal differentiation by the LIM homeobox gene Lhx5. Science, 1999. 284(5417): p. 1155–8. [DOI] [PubMed] [Google Scholar]
- 168.Savory JG, Edey C, Hess B, Mears AJ, and Lohnes D, Identification of novel retinoic acid target genes. Developmental Biology, 2014. 395(2): p. 199–208. [DOI] [PubMed] [Google Scholar]
- 169.Guimarães L, Domínguez-del-Toro E, Chatonnet F, Wrobel L, Pujades C, Monteiro LS, and Champagnat J, Exposure to retinoic acid at the onset of hindbrain segmentation induces episodic breathing in mice. European Journal of Neuroscience, 2007. 25(12): p. 3526–3536. [DOI] [PubMed] [Google Scholar]
- 170.Holson RR, Gazzara RA, Ferguson SA, and Adams J, A behavioral and neuroanatomical investigation of the lethality caused by gestational day 11–13 retinoic acid exposure. Neurotoxicology and teratology, 1997. 19(5): p. 347–53. [DOI] [PubMed] [Google Scholar]
- 171.Krezel W, Ghyselinck N, Samad TA, Dupé V, Kastner P, Borrelli E, and Chambon P, Impaired locomotion and dopamine signaling in retinoid receptor mutant mice. Science (New York, N.Y.), 1998. 279(5352): p. 863–867. [DOI] [PubMed] [Google Scholar]
- 172.Knight GT, Lundin BF, Iyer N, Ashton LM, Sethares WA, Willett RM, and Ashton RS, Engineering induction of singular neural rosette emergence within hPSC-derived tissues. eLife, 2018. 7: p. e37549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cai AQ, Radtke K, Linville A, Lander AD, Nie Q, and Schilling TF, Cellular retinoic acid-binding proteins are essential for hindbrain patterning and signal robustness in zebrafish. Development, 2012. 139(12): p. 2150–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lampron C, Rochette-Egly C, Gorry P, Dolle P, Mark M, Lufkin T, LeMeur M, and Chambon P, Mice deficient in cellular retinoic acid binding protein II (CRABPII) or in both CRABPI and CRABPII are essentially normal. Development, 1995. 121(2): p. 539–48. [DOI] [PubMed] [Google Scholar]
- 175.Dimopoulou M, Verhoef A, Pennings JLA, van Ravenzwaay B, Rietjens I, and Piersma AH, Embryotoxic and pharmacologic potency ranking of six azoles in the rat whole embryo culture by morphological and transcriptomic analysis. Toxicology and applied pharmacology, 2017. 322: p. 15–26. [DOI] [PubMed] [Google Scholar]
- 176.Robinson JF, Tonk EC, Verhoef A, and Piersma AH, Triazole induced concentration-related gene signatures in rat whole embryo culture. Reprod Toxicol, 2012. 34(2): p. 275–83. [DOI] [PubMed] [Google Scholar]
- 177.Bal-Price A, Lein PJ, Keil KP, Sethi S, Shafer T, Barenys M, Fritsche E, Sachana M, and Meek ME, Developing and applying the adverse outcome pathway concept for understanding and predicting neurotoxicity. Neurotoxicology, 2017. 59: p. 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ghyselinck NB and Duester G, Retinoic acid signaling pathways. Development, 2019. 146(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chen L, Lau AG, and Sarti F, Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology, 2014. 78: p. 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Samokyszyn VM, Gall WE, Zawada G, Freyaldenhoven MA, Chen G, Mackenzie PI, Tephly TR, and Radominska-Pandya A, 4-hydroxyretinoic acid, a novel substrate for human liver microsomal UDP-glucuronosyltransferase(s) and recombinant UGT2B7. J Biol Chem, 2000. 275(10): p. 6908–14. [DOI] [PubMed] [Google Scholar]
- 181.Bayha E, Jorgensen MC, Serup P, and Grapin-Botton A, Retinoic acid signaling organizes endodermal organ specification along the entire antero-posterior axis. PloS one, 2009. 4(6): p. e5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.