

SUMMARY
Cellular functioning requires the orchestration of thousands of molecular interactions in time and space. Yet most molecules in a cell move by diffusion, which is sensitive to external factors like temperature. How cells sustain complex, diffusion-based systems across wide temperature ranges is unknown. Here, we uncover a mechanism by which budding yeast modulate viscosity in response to temperature and energy availability. This “viscoadaptation” uses regulated synthesis of glycogen and trehalose to vary the viscosity of the cytosol. Viscoadaptation functions as a stress response and a homeostatic mechanism, allowing cells to maintain invariant diffusion across a 20°C temperature range. Perturbations to viscoadaptation affect solubility and phase separation, suggesting that viscoadaptation may have implications for multiple biophysical processes in the cell. Conditions that lower ATP trigger viscoadaptation, linking energy availability to rate regulation of diffusion-controlled processes. Viscoadaptation reveals viscosity to be a tunable property for regulating diffusion-controlled processes in a changing environment.
Graphical Abstract
INTRODUCTION
Warm-blooded organisms thrive in a wide range of environments by maintaining nearly constant internal temperatures. Yet many organisms lacking homeothermic regulation also tolerate large and unpredictable fluctuations in temperature. The budding yeast S. cerevisiae, for example, can grow and divide at temperatures that span 30°C or more (Salvadó et al., 2011; Tai et al., 2007). Other organisms have adapted to more extreme conditions and can survive temperatures ranging from 80°C to 120°C (Frock and Kelly, 2012). Such versatility is remarkable given the numerous thermodynamic challenges that temperature variation poses for a cell. Many cellular processes, such as protein folding, are dictated by small energetic differences between states and are therefore highly sensitive to temperature (Dill, 1990).
In addition to intramolecular processes, temperature has a profound effect on intermolecular processes. In general, increasing the temperature of a liquid lowers its viscosity thereby increasing the diffusion rate of any constituent particles (Longsworth, 1954). As such, molecular interactions become more frequent. Varying the rate of molecular interactions is expected to have numerous consequences, including changing the rate of diffusion-controlled reactions, the kinetics of complex assembly, and the time scale of signal transduction (Johnson, 2018; Longsworth, 1954; Stojanovski et al., 2017). With so many diffusion-based cellular processes, a change in environmental temperature could be catastrophic to cellular function and yet many organisms routinely tolerate these changes. How the temperature-dependence of diffusion manifests in live cells with highly complex and dynamic interaction networks is largely unknown. Likewise, little is known about the cellular strategies that may exist to mitigate these challenges.
Seminal work in the 1970s revealed a homeostatic mechanism for controlling the fluidity of cell membranes in response to changing temperatures. Coined “homeoviscous adaptation”, cells respond to changes in temperature by adjusting the lipid content of their membranes to maintain their appropriate viscosity (Sinensky, 1974). In this way, the cell is able to control the rate of intermolecular interactions in the membrane in a temperature-independent way (Budin et al., 2018). No such mechanism had been identified for the cytosol. In this work, we compared the rate of a diffusion-controlled exogenous model reaction in vivo and in vitro as a function of temperature. In doing so, we discovered a form of cellular adaptation that renders the reaction rate invariant to temperature changes in vivo, despite having strong temperature dependence in vitro. Termed “viscoadaptation”, this response allows the cell to maintain a near-constant intracellular viscosity despite changes in temperature. As a result, diffusion rates and viscosity-controlled reaction rates are held invariant across at least 20°C. Viscoadaptation is both a homeostatic mechanism for growth at multiple temperatures as well as an acute response to environmental stressors.
RESULTS
Cells homeostatically regulate the rate of a diffusion-controlled reaction in response to temperature changes
To determine the effect of temperature on molecular interactions and diffusion-controlled reactions in live yeast, we used an exogenous biotinylation reaction in which the BirA enzyme from E. coli is co-expressed with its biotin-accepting substrate (the “Avi-tag”) fused to a “bait” protein (Chen et al., 2005; Jan et al., 2014)(Figure S1A). The BirA reaction is well-suited as a model, because it is temporally controlled by addition of biotin to the cell culture media, it can be performed in vitro and in vivo, and its progress is readily measured by a gel shift assay in which addition of streptavidin selectively causes the biotinylated species to run at a higher molecular weight (Figure S1A). We characterized the relationship between diffusion and the BirA biotinylation rate by measuring the reaction in solutions of varying viscosity due to sucrose content (0%, 20%, 40%, 60%) (Figure 1A). As a readout for diffusion we measured the mobility of GFP, a relatively compact protein, by Fluorescence Recovery After Photobleaching (FRAP), in which purified GFP was added to each solution and a circular region was photobleached. Fluorescence recovery in the bleached region was measured over time as an indicator of the diffusion rate of GFP, and thus the viscosity of the solution (Figure S1B). Recovery rates were quantified as the time for the bleached area to recover half of its initial intensity (“t-half”) or as diffusion coefficients of GFP (Figure S1B). t-half values increase with increasing viscosity whereas diffusion coefficients decrease with increasing viscosity. Example FRAP movies are provided via Mendeley. The BirA reaction rate scaled 1:1 with solution viscosity (reported as 1/t-half) for all concentrations of sucrose tested (Figure 1A). We thus concluded that the BirA reaction was diffusion-limited. Unless otherwise specified, reactions described occur between cytosolic BirA and GFP-Avi.
Figure 1. A viscosity-controlled model reaction is temperature-sensitive in cell lysates but not live cells.

(A) Biotinylation rate vs viscosity (1/t-half) for reactions carried out in 0%, 20%, 40%, or 60% sucrose solutions (●blue) or cell lysates (○orange). Reaction rates and t-half values were normalized to their respective values in H2O. Horizontal error bars are s.e.m. (n> 3 FRAP experiments). Vertical error bars are the standard error of the slope derived from the best fit line for multiple timecourses (n> 5 timecourses/condition) (B) Biotinylation rates in lysates. Cell growth temperature (left of arrow) and reaction temperature (right of arrow) are indicated in the figure key. Normalized to max labeling at 30°C (nonlinear regression, difference between slopes, p<.0001) (C) Diffusion coefficients for GFP in pure H20 (n=4), lysate from cells grown at 30°C (n=3), and live cells growing at 30°C (n=19) (unpaired t-test, H20 vs cell lysate p=.0037; cell lysate vs live cells p<.0001; H2O vs live cells p<.0001) (D) Biotinylation rates in live cells growing at 30°C (●blue) or 37°C (◼orange). Measurements are at the growth temperatures of the cells. Normalized to max labeling at 30°C (n=4 timecourses/temperature) (nonlinear regression, difference between slopes p=0.1) (E) Biotinylation rates in lysates from cells grown and measured at the indicated temperatures. Normalized to max labeling for 30°C→30°C (nonlinear regression difference between slopes, 30°C→30°C vs 30°C→37°C p>.0001; 30°C→30°C vs 37°C→30°C p>.0001; 30°C→37°C vs 37°C→30°C p>.0001) (F) Biotinylation rates measured at the same temperature of cell growth prior to lysis. Normalized to max labeling at 30°C→30°C (nonlinear regression, difference between slopes, 30°C→30°C vs 30°C→37°C p=.01) Error bars are s.e.m. Points without visible error bars have error within the size of the point. See also Figure S1.
We next tested the temperature sensitivity of the BirA reaction in vitro by performing the reaction in cell lysates at 30°C or 37°C (normal yeast growth temperatures) (Figure 1B). To make lysates, cells in liquid culture were filter-dried, frozen, and mechanically lysed. Subsequent centrifugation removed large structures such as cell wall debris, nuclei, and vacuoles that were not ruptured during lysis. The resulting lysates reflect the “average” cellular environment rather than the environment of a single compartment. FRAP measurements in the lysates estimated that they were approximately 10-fold less viscous than the cytosol of intact cells, with a viscosity similar to 40% sucrose (Figure 1C). This difference in viscosity is likely due to dilution by growth media that was incompletely removed during filtration and mixing of cellular compartments. We note that the diffusion coefficient we measured for GFP in live yeast is between the values observed in bacterial cytoplasm and mammalian cytoplasm (Mika and Poolman, 2011; Potma et al., 2001; Swaminathan et al., 1997).
To remove additional variables, each reaction contained equal amounts of exogenous BirA and biotin, and an excess of ATP. When we performed the reaction at 30°C or 37°C, the rate was 50% faster at 37°C, indicating that the BirA reaction is temperature-dependent in vitro (Figure 1B). However, when we measured the BirA reaction in live cells growing at 30°C or 37°C, we found no difference in rate (Figure 1D). Temperature insensitivity in vivo contrasted with the temperature dependence in vitro, suggesting that a change in the intracellular environment modifies the reaction rate at one or both temperatures. Since the reaction is exogenous, we predicted a broad cellular strategy of reaction regulation in response to temperature change.
The lack of temperature sensitivity in vivo suggested that either the viscosity of the cellular environment was not changing as expected with temperature or a separate cellular change was slowing the reaction at the higher temperature. We hypothesized that the viscosity of the cellular environment could be modulated by production of a viscosity-increasing molecule (“viscogen”) in cells growing at 37°C. If so, the reaction should be slower in lysates from cells grown at 37°C compared to those from cells grown at 30°C when the reactions are carried out at the same temperature. Consistent with this hypothesis, the reaction was 25% slower in the 37°C-derived lysate (Figure 1E). Furthermore, the measured viscosity of the lysate in each condition corresponded linearly to the observed reaction rate (Figure 1A).
The constant rate of the BirA reaction in live cells at 30°C and 37°C suggests that cells growing in steady state at different temperatures adapt homeostatically to maintain invariant reaction rates. If so, we predicted that the in vitro reaction rates would be equivalent for reactions performed at the growth temperatures of the cells prior to lysis (30°C or 37°C). Indeed, we found that changing the reaction temperature to match the cell culture growth temperature equalized the in vitro reaction rates (Figure 1F). These findings were not specific to GFP-Avi (Figure S1C). We conclude from these observations that cells maintain constant rates of viscosity-controlled reactions at different growth temperatures.
Cells regulate viscosity in response to temperature changes
We next tested whether cells regulate intracellular viscosity in response to temperature by measuring the mobility of GFP using FRAP in vivo and in vitro. Data are reported as diffusion coefficients or t-half values (raw FRAP values used for t-half determination and raw FRAP movies used for diffusion coefficient determination are provided via Mendeley). To determine the effect of temperature on diffusion in the absence of active cellular regulation, FRAP was performed on cell lysates at 22°C, 30°C, 37°C, and 42°C. The diffusion coefficient of GFP increased with temperature and was ~80% higher at 42°C relative to 22°C (Figure 2A). This result is consistent with the known effect of temperature on diffusion, though the magnitude of the effect is larger than would be predicted by the Stokes-Einstein equation (Miller Christina Cruickshank and Walker James, 1924). Deviations from the Stokes-Einstein prediction have previously been observed in cytosol (Sidell and Hazel, 1987).
Figure 2. Cells regulate intracellular viscosity in response to heat (“viscoadaptation”).

(A) Diffusion coefficients of GFP in lysates from cells grown at 30°C and measured at the indicated temperatures (n=4 lysates/temperature) (unpaired t-test, 22°C vs 42°C p=.0024; 22°C vs 37°C p=.0094; 22°C vs 30°C p=.083; 30°C vs 37°C p=.059; 30°C vs 42°C p=.0087; 37°C vs 42°C p=.432) (B) FRAP on live cells expressing eGFP, shifted from 22°C to the indicated temperature for >20 minutes before measurement. Striped bar represents cells measured within 5 minutes of temperature shift (n> 3 experiments/condition, >8 cells/experiment) (unpaired t-test, 22°C vs 42°C p<.0001; 22°C vs 40°C p<.0001; 22°C vs 37°C p<.0001; 22°C vs 30°C p=.0528; immediate 42°C vs delayed 42°C p=.0011; 22°C vs 42°C immediate p<.01) (C) Timecourse of FRAP measurements on live cells shifted from 22°C (T0) to 42°C (n>3 cells/timepoint). Points without visible error bars have error within the size of the point (nonlinear regression, slope of best fit line=.005) (D) Cells expressing eGFP were grown to log phase (minimum 3 hours) at the indicated temperatures. FRAP was performed at the same temperature as cell growth. (n> 3 replicates/temperature, >10 cells/replicate)(unpaired t-test, p>.05 for all pairwise comparisons) (E) Diffusion coefficients of purified GFP in lysates from cells grown to steady state at 22°C, 30°C, 37°C, or 40°C, determined by FRAP. All measurements were at 22°C (n> 3 lysates/condition) (unpaired t-test, 22°C vs 40°C p=.168; 22°C vs 37°C p=.046; 22°C vs 30°C p=.857; 30°C vs 37°C p=.0048; 30°C vs 40°C p=.075; 37°C vs 40°C p=.372) Error bars are s.e.m. See also Figure S2.
We next measured the diffusion of GFP in live cells. Cells at 22°C had an average t-half of 0.08 seconds and when transferred to 42°C showed an initial drop in t-half to 0.05 seconds, almost 40% lower and in agreement with the results in cell lysates (Figure 2B). However, when cells were given at least 20 minutes at the higher temperature before measurement, diffusion rates decreased with increasing temperature. In the most extreme case, the average t-half after adjustment to 42°C was 0.19s, almost 300% higher than the t-half in cells measured immediately after the temperature shift (Figure 2B). This result was not specific to GFP (Figure S2A). We conclude that cells respond to acute temperature increase by raising their viscosity, thereby slowing the diffusion of cytosolic proteins.
To track the adaptation of cells to heat shock over time, we collected a timecourse of FRAP measurements, beginning immediately after heat shock (22°C to 42°C) and extending to 40 minutes post heat shock (Figure 2C). Immediately after the temperature shift, we saw a drop in recovery time to 0.05 seconds, reflecting the effect of temperature increase on diffusion prior to cellular adaptation. However, t-half values then began to increase, reaching 0.17 seconds at 20 minutes and 0.24 seconds at 40 minutes. Again, the observed trends were not specific to GFP (Figure S2B).
Our observations suggest that acute heat shock increases cytosolic viscosity. Yet we observed a constant BirA reaction rate in cells growing at 30°C and 37°C (Figure 1D). We therefore investigated whether cytosolic diffusion is controlled homeostatically at longer time scales. To test this, we grew yeast to log-phase at 22°C, 30°C, 37°C, or 40°C (the highest permissible growth temperature) and performed FRAP measurements in a chamber matched to the growth temperature of the culture. Strikingly, the t-half for GFP was nearly constant at all temperatures, at a value of about 0.08 seconds (Figure 2D). This homeostatic control of viscosity in vivo corresponded to increased viscosity in cell lysates from cultures grown at higher temperatures when lysates were measured at 22°C (Figure 2E). Collectively, our results reveal that cells counteract the effect of temperature on protein diffusion by increasing their intracellular viscosity. We term this phenomenon “viscoadaptation”.
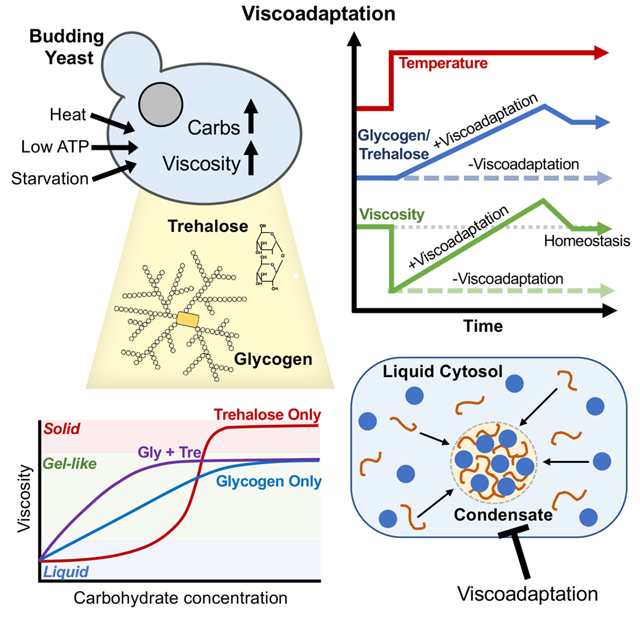
Accumulation of glycogen and trehalose in response to heat underlies viscoadaptation
We next investigated the mechanism underlying viscoadaptation. Neither the degree nor speed of viscoadaptation was significantly affected by pretreatment with the translation inhibitor cycloheximide, indicating that protein synthesis is not required for viscoadaptation in response to acute heat shock (Figure S3A). In addition to specialized proteins, temperature increase rapidly induces synthesis of two carbohydrates, glycogen and trehalose (Figure 3A,B,S3B) (Hottiger et al., 1987a, 1987b). Both glycogen and trehalose are assembled from glucose monomers and together can account for up to 20% of a cell’s dry mass in severe heat shock (Shi et al., 2010). Both glycogen and trehalose improve tolerance to heat stress (Gibney et al., 2013; Ruiz-Roig et al., 2010)(Quain and Tubb, 1983; Wilson et al., 2002). A glycogen molecule is composed of thousands of glucose monomers in an extensively branched spherical structure that can reach 40 nm in diameter in yeast (Prats et al., 2018). Glycogen acts as a viscogen in aqueous solutions (Kerly, 1930)(our observations below).
Figure 3. Heat-induced accumulation of the carbohydrates glycogen and trehalose increases cellular viscosity.

(A) Glycogen and trehalose are produced from glucose through conversion of glucose-1-p to glucose-6-p by phosphoglucomutase (PGM) which is inhibited by LiCl. (B) Quantification of glycogen (top) and trehalose (bottom) during 42°C heat shock. T0 is at 22°C. (n=3 timecourses each) (C) t-half values in the indicated strains at 22°C (leftmost, blue) or after >20 minutes at 42°C (red). Each dot represents a single cell. Corresponding iodine staining shown above (all iodine staining spots are 5 mm diameter) (unpaired t-test, WT 22°C vs WT 42°C p<.0001; WT 42°C vs ΔGlc3 42°C p=.4; WT 42°C vs ΔGsy2 42°C p=.924; WT 42°C vs ΔTps2 42°C p<.0001; WT 42°C vs ΔGΔGΔT 42°C p<.0001; WT 42°C vs WT 42°C +LiCl p<.0001) (D) Cells were treated with Validamycin A and DAB (10 ug/ml each) immediately prior to 42°C heat shock for 40 minutes. Each dot is a single cell (n=3 experiments)(unpaired t-test, vehicle 10 vs vehicle 90 p<.0001; +drug 10 vs +drug 90 p=.058; vehicle 10 vs +drug 10 p=.125; vehicle 90 vs +drug 90 p<.0001) (E) Cells expressing BirA and GFP-Avi were grown to steady state at 30°C (blue) or 37°C (red) with (▪) or without (●) 50 mM LiCl. Normalized to max labeling at 30°C -LiCl (nonlinear regression, difference between slopes, −LiCl 30°C vs +LiCl 30°C p=.48; −LiCl 30°C vs −LiCl 37°C p=.1; +LiCl 30°C vs +LiCl 37°C p<.0001; −LiCl 30°C vs +LiCl 37°C p<.0001) Error bars are s.e.m. Points without visible error bars have error within the size of the point. See also Figure S3.
Trehalose is a small disaccharide formed by a 1,1-glycosidic bond between two ɑ-glucose monomers (Jain and Roy, 2009). According to the “immobilization theory”, trehalose protects cells against desiccation by undergoing a liquid-to-glass phase transition that drastically reduces molecular mobility (Jain and Roy, 2009). Trehalose has also been proposed to act as a kosmotrope, substituting for water around biomolecules and causing the surrounding water molecules to become more ordered (Jain and Roy, 2009). Both proposed functions reduce the mobility of biomolecules (Jain and Roy, 2009).
Given the potential for glycogen and trehalose to influence the viscosity of solutions and their temperature-dependent production, we investigated how these two carbohydrates affect viscoadaptation. To do so, we generated yeast mutants deficient in the synthesis of one or both carbohydrates. Yeast lacking the trehalose synthesis enzyme Tps2 failed to increase viscosity in response to 42°C heat shock (Figure 3C), suggesting that trehalose production is necessary for viscoadaptation during temperature increase. We next examined diffusion after heat shock in strains lacking the glycogen synthase, Gsy2, or the glycogen branching enzyme, Glc3. To qualitatively and rapidly determine changes in glycogen in live yeast, we adapted an iodine vapor staining method in which darker staining indicates higher glycogen content (Figure S3C)(Demonte et al., 2014). As expected, iodine staining revealed that strains lacking Gsy2 or Glc3 failed to accumulate glycogen in response to heat shock at 42°C (Figure 3C). Unexpectedly, yeast deficient in glycogen synthesis demonstrated a wide range of diffusion phenotypes. Approximately 50% of cells failed to slow diffusion in response to heat (“non-responders”), but the remainder of cells responded normally or hyper-responded (“responders”)(Figure 3C). Glycogen and trehalose are made from the same precursor and disruption of one pathway can increase flux into the other (Seo et al., 2018)(Figure 3A). We therefore reasoned that the population of hyper-viscous glycogen deficient cells could result from increased flux through the trehalose synthesis pathway and resultant over-accumulation of trehalose in some cells. Consistent with this model, when production of both glycogen and trehalose was blocked by double knockout of GLC3 and TPS2, all cells failed to increase viscosity in response to heat shock (Figure 3C).
Genetic knockouts can have long term, indirect effects on cells. We therefore made use of several pharmacological tools to interfere with glycogen and trehalose production. Treatment with LiCl reduces levels of glycogen and trehalose accumulated in response to heat (Masuda et al., 2001)(Figure S3D) and prevented viscosity increase (Figure 3C). 1,4-imino-1,4-arabinitol (DAB), an inhibitor of glycogen phosphorylase (Walls et al., 2008) and Validamycin A, an inhibitor of trehalase (Müller et al., 1995), prevent the degradation of glycogen and trehalose after heat without affecting glycogen synthesis (Figure S3E). FRAP of cells that had been allowed to cool gradually from 42°C to room temperature over 90 minutes showed that untreated cells had decreased viscosity whereas drug-treated cells generally increased in viscosity (Figure 3D). This is predicted if glycogen and trehalose levels determine viscosity, and their levels remain high as temperature decreases. These pharmacological experiments suggest that glycogen and trehalose are necessary and sufficient to increase viscosity during heat shock in vivo.
Changes in cell size can increase molecular crowding with resultant effects on the mobility of large structures (Delarue et al., 2018; Joyner et al., 2016). Yet cell size was not correlated with viscoadaptation (Figure S3F) and thus crowding caused by changes in cell size does not explain viscoadaptation or its dependence on glycogen and trehalose production. Glucose, the precursor to glycogen and trehalose, also increases viscosity of solutions and thus its accumulation could contribute to viscoadaptation (Telis et al., 2007). Glucose levels were unaffected by steady state growth temperature but decreased by ~30% within 5 minutes of heat shock except when glycogen synthesis was impaired (Figure S3G). This is consistent with the conversion of glucose to glycogen and trehalose upon heat shock but not consistent with a direct role for glucose in controlling cytosolic viscosity.
To determine how glycogen and trehalose affect diffusion-controlled reaction rates, we sought to measure the rate of the BirA reaction in cells with compromised glycogen or trehalose production. However, we found that tps2Δ yeast could not grow in the low biotin media required for biotinylation assays. Glycogen deficient strains did not show a significant difference in reaction rate, presumably due to the heterogeneity of cellular viscosities (Figure S3H). Strikingly, LiCl treatment of cultures growing at 30°C or 37°C resulted in a disequalization of the BirA reaction rate with the reaction proceeding 50% faster at 37°C, in agreement with the results in cell lysates (Figure 3E). Treatment with an equivalent concentration of NaCl did not affect viscoadaptation, ruling out general effects of salt treatment (Figure S3I). These data suggest that accumulation of glycogen and trehalose is necessary and sufficient to increase intracellular viscosity in response to heat. Without ruling out a role for other factors, we propose that accumulation of glycogen and trehalose is the primary mechanism for viscoadaptation.
Viscoadaptation occurs in response to nutrient limitation
Our results thus far have demonstrated that regulation of glycogen and trehalose production by environmental temperature is closely linked to the diffusibility of cellular proteins. Cells upregulate glycogen and trehalose in response to cues in addition to temperature, including forms of nutrient deprivation (Lillie and Pringle, 1980; Thomsson et al., 2005). Accordingly, we observed glycogen accumulation in cells subjected to acute glucose deprivation, and both glycogen and trehalose accumulated in stationary phase cultures where nutrients were depleted over time (Figure 4A). We therefore tested whether glucose deprivation affects intracellular diffusion and consequently the rates of diffusion-controlled reactions. A switch from 2% to 0% glucose reduced the biotinylation rate of GFP-Avi by ~55% in live cells (Figure 4B). This observation held for other BirA-Protein/Avi-Protein pairs (Figure S4A). Additionally, the rate of biotinylation in vitro was ~65% lower in lysates from glucose starved cells relative to those from unstressed cells (Figure 4C). The similar magnitude of the rate discrepancy in vitro and in vivo (despite lysates being diluted relative to intact cells) suggests that increased viscosity may not be the only factor slowing biotinylation in glucose starved cells. To determine the contribution of viscoadaptation to the slowed BirA reaction rate, we compared biotinylation rates in live cells starved with or without LiCl. LiCl treatment decreased glycogen accumulation during starvation and increased the rate of biotinylation by ~50% relative to the untreated control (Figure 4D). This suggests that viscoadaptation contributes to the reduced rate of biotinylation in glucose starved cells and reveals that glycogen is sufficient for this effect.
Figure 4. Viscoadaptation occurs in response to nutrient limitation.

(A) Quantification of glycogen (left) and trehalose (right) for cells in 2% glucose, 0.1% glucose (30 minutes), and stationary phase for >24 hours (n=3 samples/condition). Below (bottom left) are iodine staining spots for the indicated conditions. (glycogen: unpaired t-test, log phase vs low glucose p=.004; log phase vs stationary phase p=.006) (trehalose: unpaired t-test, log phase vs low glucose p=.337; log phase vs stationary phase p=.005) (trehalose: unpaired t-test, log phase vs low glucose p=.337; log phase vs stationary phase p=.005) B) In vivo BirA labeling of GFP-Avi in 2% glucose (green) and 0% glucose (blue) (nonlinear regression, difference between slopes, p<.0001) (C) In vitro BirA labeling timecourse in lysates from cells grown in 2% glucose (green) and 0% glucose (blue) (nonlinear regression difference between slopes, p<.0001) (D) Fraction of GFP-Avi labeled by BirA after 30 minutes in 0% glucose with (checkered) and without (solid) 50mM LiCl. Normalized to the −LiCl condition (unpaired t-test, p=.01) (E) FRAP measurements on WT cells in the indicated growth conditions (n> 3 replicates, >9 cells/replicate) (unpaired t-test, 2% glucose vs low glucose p=.0006, 2% glucose vs low glucose +LiCl p>.05; 2% glucose vs stationary phase p=.0094; 2% glucose vs 0% glucose p>.05) (F) FRAP on the indicated strains in 2% glucose (leftmost) or 0.1% glucose for 30 minutes. Each dot represents one cell. (unpaired t-test, WT 2% glucose vs WT 0.1% glucose p<.0001; WT 0.1% glucose vs ΔGlc3 0.1% glucose p=.673; WT 0.1% glucose v. ΔGΔGΔT 0.1% glucose p=.006; WT 2% glucose vs ΔGΔGΔT 0.1% glucose p=.05) Error bars are s.e.m. See also Figure S4.
We next measured diffusion using FRAP in the same glucose starved condition and found that viscosity increased by ~50% (Figure S4B). Cells for BirA labeling were grown in low biotin media (0.5 nM compared to 8 nM) to prevent background biotinylation. To determine if viscoadaptation during glucose starvation occurs under less specialized conditions, we performed FRAP on cells in 0% glucose with biotin-replete media. In this case, 0% glucose did not cause increased viscosity (Figure S4B), but low glucose (0.1%) doubled the t-half relative to cells in 2% glucose (Figure 4E). The increased t-half in low glucose was reversed by LiCl treatment (Figure 4E). Accordingly, iodine staining demonstrated that 0% glucose caused accumulation of glycogen only when cells were grown in low biotin media (Figure 4SC). We tested whether differences in available glucose were impacting glycogen synthesis or cellular viscosity but found that glucose did not correlate with viscosity or explain the biotin-dependent discrepancy in glycogen production during starvation (Figure S4D). Thus, glycogen, but not glucose or trehalose, correlates with viscoadaptation in acute glucose starvation.
To test the requirement for glycogen and trehalose synthesis machinery in viscosity changes during starvation, we measured diffusion in strains transferred to 0.1% glucose with and without glycogen or trehalose synthesis enzymes. Strains lacking Glc3 or a combination of Glc3, Gsy2, and Tps2 showed impaired ability to increase viscosity in response to low glucose (Figure 4F). Occasional high viscosity cells were observed in the glc3Δ single knockout, which may be due to aberrant accumulation of trehalose as high-viscosity cells were not observed in the triple knockout (Figure 4F). Stationary phase, in which cell growth has stopped due to depletion of nutrients, also prompts accumulation of glycogen and trehalose (Huang et al., 1997)(Figure 4A). Measuring diffusion in these cells revealed a 300% increase in t-half relative to cells in logarithmic growth with 2% glucose (Figure 4E). Together, our findings suggest that viscoadaptation is induced by short and long-term starvation and that increased viscosity can be achieved through production of glycogen without a requirement for trehalose in the case of acute glucose starvation.
Glycogen and trehalose alter molecular movement in vitro
To better understand the potential for glycogen and trehalose to directly affect cellular viscosity during stress, we characterized viscosity, diffusion, and phase behavior in aqueous glycogen and trehalose solutions. Both glycogen and trehalose were sufficient to slow the diffusion of GFP with a combination of both carbohydrates showing the largest effect (Figure 5A). We next examined how diffusion in the same solutions was affected by temperature. Intriguingly, the diffusion rate was temperature sensitive in all solutions except 45% trehalose in which the t-half of GFP was unchanged between 22°C and 40°C (Figure 5A). Temperature-insensitive diffusion appears to be a unique property of trehalose solutions with potential implications for viscoadaptation. A 4-fold dilution of a glycogen and trehalose solution corresponded to a 42-fold reduction in viscosity (Figure 5B), consistent with LiCl treatment causing robust disequalization of the BirA reaction rate at 30°C and 37°C in vivo (Figure 3E) while only partially decreasing glycogen and trehalose accumulation (Figure S3D).
Figure 5. Glycogen and trehalose alter molecular movement in vitro.

(A) FRAP on purified GFP in aqueous solutions of glycogen and trehalose at 22°C and 40°C reported as t-half (n>3)(unpaired t-test, H2O 22°C vs H2O 40°C p=.0023; 45%Tre 22°C vs 45%Tre 40°C p=.636; 30%Gly 22°C vs 30%Gly 40°C p=.0013; Gly/Tre 22°C vs Gly/Tre 40°C p=.0286; H2O 22°C vs 45%Tre 22°C p=.005; H2O 22°C vs 30%Gly 22°C p<.0001; H2O 22°C vs Gly/Tre 22°C p<.0001) (B) FRAP on GFP in serial dilutions of glycogen and trehalose, measured at 22°C and reported as the diffusion coefficient of GFP (n=3)(unpaired t-test, 11.25/7.5 vs 45/30 p=.0001; 11.25/7.5 vs 22.5/15 p=.0011; 22.5/15 vs 45/30 p<.0001) C) Aqueous solutions of trehalose at the indicated concentrations in liquid droplets (top row) or after solidification(bottom row). Initial droplet diameter is 4 mm. (D) Summary of phase observations for glycogen and trehalose droplets of varying concentrations at 22°C, see Figure S5B for phase designation criteria. (E) Fluorescent images of purified GFP in glycogen and trehalose solutions. See also Figure S5.
Large complexes often move sub-diffusively (“anonymous diffusion”) due to steric hindrance in the crowded cellular environment (Luby-Phelps et al., 1986; Seksek et al., 1997). It is possible that glycogen and trehalose cause diffusion to become sub-diffusive for some molecules. Subdiffusive movement was only observed in the most concentrated aqueous solutions of glycogen and trehalose (30% and 45%, respectively) (Figure S5A), and in general the type of diffusion was independent of growth temperature (Figure S5A) and the ability to synthesize glycogen and trehalose (Figure S5A). We conclude that changes in mobility caused by glycogen and trehalose are more likely a consequence of increased viscosity than anomalous diffusion.
Trehalose undergoes an abrupt liquid-to-solid phase transition which dramatically restricts molecular movement and is adaptive in certain biological contexts (Buitink and Leprince, 2004). In contrast, viscoadaptation appears to be a tunable, graded response. We therefore tested whether the presence of glycogen could modify the propensity of trehalose to undergo phase transition in vitro. In the absence of glycogen, droplets of an aqueous trehalose solution underwent a concentration dependent liquid-to-solid phase transition (Figure 5C). Droplets containing only glycogen evaporated without undergoing phase transition, and droplets containing both glycogen and trehalose acquired an intermediate, gel-like state which remained malleable hours after the trehalose-only droplets had solidified (Figure 5D, S5B). These results indicate that glycogen can modify the phase behavior of trehalose, preventing abrupt phase transitions.
In comparison to trehalose, glycogen is poorly soluble in water, and it has been observed to aggregate into insoluble granules in healthy and diseased states (Kerly, 1930)(Palmucci et al., 1983; Rybicka, 1979). When performing FRAP on glycogen solutions, we observed that the distribution of GFP was irregular (Figure 5E), and GFP fluorescence was reduced relative to the equivalent GFP concentration in water. Addition of trehalose (45%) resulted in a more homogeneous GFP distribution and a partial restoration of fluorescence (Figure 5E). The fluorescence of exogenous GFP was also reduced in lysates from cells heat shocked at 42°C relative to lysates from unstressed cells (Figure S5C).
Together these data indicate that glycogen and trehalose impact the biophysical properties of solutions in distinct and complementary ways. Glycogen has a graded impact on viscosity with a larger effect per mass than trehalose but promotes loss of homogeneity of GFP fluorescence in solution. Trehalose has a comparatively low impact on the viscosity of solutions but can undergo an abrupt liquid-to-solid phase transition. Interactions between the two molecules reduce the tendency of glycogen to alter the fluorescence distribution of GFP and guard against abrupt phase transitions of trehalose. These complementary properties may make glycogen and trehalose effective in increasing viscosity while preserving the solubility of biomolecules.
Viscoadaptation affects phase separation and the solubility of biomolecules in vivo
Rapidly changing the composition and viscosity of the intracellular environment could affect myriad cellular processes, including phase separation and the solubility of biomolecules. Indeed, restricted molecular mobility caused by ribosome crowding has been shown to impact phase separation in an in vivo and in vitro synthetic system (Delarue et al., 2018). We investigated the impact of viscoadaptation on in vivo phase separation by examining the formation and dissolution of stress granules (SGs). SGs are condensates formed by liquid-liquid phase separation of endogenous nucleic acids and proteins during stress (Protter and Parker, 2016). We hypothesized that changing the biophysical properties of the cytosol in viscoadaptation could affect SG formation. To test this, we tracked SG formation after heat shock with and without LiCl using Pab1-GFP as an SG marker. LiCl treatment (which impairs viscoadaptation) resulted in a more rapid appearance of SGs, suggesting that viscoadaptation could impede SG formation (Figure 6A). Consistent with this, a condition which enhances viscoadaptation (low biotin vs normal biotin in 0% glucose) reduced the formation of SGs (Figure S6A). We tested whether viscoadaptation also affects the rate of SG dissolution by treating heat shocked cells with Validamycin A. Drug treatment hastened the disappearance of SGs relative to the untreated control, suggesting that higher trehalose after stress promotes disassembly of SGs (Figure S6B). These findings raise the possibility that viscoadaptation plays a previously unappreciated role in phase separation in cells.
Figure 6. Viscoadaptation affects phase separation and the solubility of biomolecules in vivo.

(A) Pab1-GFP in cells at 22°C (top) or after 40 minutes at 42°C (bottom) in the presence or absence of 50 mM LiCl (B) 10x brightfield images of the interior of dried lysate droplets produced from the strains and conditions indicated (C) (left) 10x phase contrast microscopy on lysate from cells heat shocked at 45°C for 30 minutes. Top and bottom images are different z-planes of the same region. (right) Diffusion was measured by microrheology in regions interior and exterior to the inclusions (n=3 exterior/3 interior measurements)(paired t-test, p<.0001) (D) Biotinylation of GFP-Avi in lysates from unstressed cells (blue) or cells heated at 45°C for 30 minutes (red). Normalized to max labeling in unstressed lysate (nonlinear regression, difference between slopes, p<.0001) (E) Lysates from the previous panel were diluted 1:50 in H20. Normalized to max labeling in the diluted unstressed lysate (n=3 timecourses/condition)(nonlinear regression, difference between slopes, p=.14) (F) The mobile fraction (MF) of GFP in WT and ΔReg1 cells classified as low (<0.8) or high (>0.8) in log phase and stationary phase cultures (unpaired t-test, WT log vs WT stationary phase p>.05; WT log vs ΔReg1 log p=.001; ΔReg1 log vs ΔReg1 stationary phase p=.0214)(n>3 replicates, >8 cells/replicate). Error bars are s.e.m. See also Figure S6.
Aberrant phase separation is associated with aggregate formation and loss of molecular solubility. While working with lysates from severely heat shocked cells (45°C for 30 minutes), we observed the formation of macroscopic ovoid inclusions (Figure 6B). Notably, these inclusions only appeared in lysates from heat shocked cells and their formation was prevented by loss of Tps2 (Figure 6B), implicating trehalose. Knockout of glycogen synthesis machinery did not prevent inclusion formation, but double knockout of TPS2 and GLC3 failed to form inclusions entirely (Figure 6B). To investigate the phase of the inclusions, we performed microrheology on regions interior and exterior to the structures. Interior regions showed a complete loss of mobility while exterior regions had much lower viscosities (Figure 6C). We mechanically probed the inclusions with magnetic beads to further determine their structure. Beads moved easily through regions without inclusions but became immobile within inclusions. We conclude that lysates from severely heat shocked cells separate into distinct liquid and solid components in a manner dependent on Tps2.
Because the phase of lysates from severely heat shocked cells is heterogenous, we predicted that the rate of BirA labeling in these lysates and their measured viscosities would not correspond. Consistent with this, biotinylation was 80% slower in lysates from severely heat shocked cells relative to lysates from unstressed cells (Figure 6D), yet the viscosities in liquid regions of lysates were not significantly different from those of unstressed lysates (Figure S6C). When lysates were diluted in water inclusions no longer formed (Figure S6D), and the BirA reaction rate became equivalent in all conditions (Figure 6E). We propose that strong viscoadaptation in severe heat shock results in unstable phase behavior in cell lysates.
Inclusion formation in lysates from severely heat shocked cells suggested that high levels of glycogen or trehalose could affect the solubility of biomolecules. To examine this possibility in vivo, we generated a GFP-expressing strain lacking the nutrient-sensing protein Reg1. reg1Δ cells have constitutively high glycogen and trehalose (Huang et al., 1996)(Figure S6E) and a growth defect which is suppressed by loss of glycogen synthesis machinery (Ruiz et al., 2011). We used FRAP to characterize the mobility of GFP in the reg1Δ strain and found the average t-half to be higher than in WT, consistent with glycogen and trehalose increasing cellular viscosity (Figure S6F). As a proxy for solubility, we determined the mobile fraction of GFP. The mobile fraction is a measure of the extent of fluorescence recovery after photobleaching rather than the rate of recovery (Figure S6G). If loss of GFP solubility causes its aggregation, this will be reflected in a decrease in the GFP mobile fraction. Consistent with this, 60% of reg1Δ cells in logarithmic growth had a mobile fraction below 0.8 compared to only 8% in the WT population (Figure 6F).
The increase in immobile GFP in the reg1Δ strain suggests that high levels of glycogen and trehalose could decrease protein solubility. However, similar accumulation during severe stress increases fitness (François and Parrou, 2001)(Wilson et al., 2010). Consistent with this, despite having higher absolute levels of glycogen and trehalose (Figure S6F), reg1Δ cells in stationary phase were less likely to have a mobile fraction below 0.8 than reg1Δ cells in log phase (14.5% and 60%, respectively)(Figure 6F) suggesting that the context of carbohydrate accumulation modifies its effect on protein solubility. We propose that accumulation of glycogen and trehalose in viscoadaptation has widespread effects on biophysical processes in the cell such as molecular solubility and phase separation and that “cellular context” (e.g. growth phase) can modulate these effects.
Viscoadaptation is linked to low ATP levels
We reasoned that both heat and nutrient deprivation, two seemingly disparate stressors, may trigger viscoadaptation through a common signal. Both types of stress cause loss of intracellular ATP (Bermejo et al., 2010; Lambowitz et al., 1983; Patriarca and Maresca, 1990; Weitzel et al., 1987). We therefore investigated whether viscoadaptation could be triggered by pharmacological perturbations that reduce ATP levels. Treating cells with the metabolic poisons sodium azide, sodium arsenite, or a combination of Antimycin A, Oligomycin, and FCCP all lowered ATP and increased intracellular viscosity (Figure 7A,S7B). Furthermore, these viscosity changes were fully or partially prevented by treatment with LiCl, implicating viscoadaptation as a likely mechanism (Figure 7A).
Figure 7. Viscoadaptation occurs in cells with low ATP.

(A) FRAP measurements on WT cells untreated (UT) or treated with sodium azide, sodium arsenite, or a combination of oligomycin (O), antimycin A (A), and FCCP (F) +/− 50 mM LiCl (n>3 experiments, >10 cells/experiment)(unpaired t-test, UT vs Azide p=.0002, UT vs Arsenite p=.001, UT vs AOF p<.0001; Azide vs Azide +Li p=.054, Arsenite vs Arsenite +Li p=.025, AOF vs AOF +Li p=.016) Error bars are s.e.m. (B) WT cells expressing a FRET-based ATP nanosensor were switched from 2% to 0.1% glucose for 30–60 minutes. Each point is one cell. ATP levels for single cells were determined by the ratio of 528:488 fluorescence (see Figure S7A). Mobility was assessed by performing FRAP of the fluorescent sensor on the same cells. Orange dots represent cells treated with 50 mM LiCl during starvation (without LiCl R= −0.597 p<.0001; with LiCl R= −0.361 p=0.0063) (C) WT cells in 0% glucose SD media. Measurements occur between 30–60 minutes after starvation onset (R= −0.047, p=.38) (D) t-half values and ATP levels for unstressed WT cells in 2% glucose SD media. Cells were sampled to capture a range of ATP levels. (R= −0.555 p<.0001) (E) Cells expressing the ATP FRET sensor were heated over the course of an hour from 22°C to 42°C. ATP level and t-half was recorded for each cell. X-axis is time in minutes. Data is plotted as a rolling average of every 3 values and normalized on a scale of 0 to 1 to facilitate comparison. Dotted lines represent s.e.m. See also Figure S7.
To examine the relationship between viscosity and ATP in single cells, we used a genetically encoded FRET-based ATP sensor where a higher 528:488 nm emission ratio is indicative of higher ATP concentration (Bermejo et al., 2010; Imamura et al., 2009)(Figure S7A). By performing FRAP on the ATP sensor, we were able to measure ATP and diffusion in the same cells. Higher viscosity correlated with decreasing ATP content in conditions that prompt viscoadaptation (low glucose with regular biotin (Figure 7B) and 0% glucose with low biotin (Figure S7C)) but did not hold for cells in 0% glucose with regular biotin (Figure 7C), where viscoadaptation does not occur. Thus, ATP levels and intracellular diffusion are correlated in conditions where viscoadaptation takes place.
If low ATP triggers viscoadaptation, then impairing viscoadaptation should alter the relationship between ATP and viscosity. Treatment with LiCl reduced the strength of the correlation between viscosity and ATP in low glucose (r= −0.6 without LiCl and −0.36 with LiCl)(Figure 7B). We also performed FRAP on the ATP sensor in glycogen-deficient cells and observed a population of cells with low ATP and high mobility (“non-responders”) and a population with low ATP and very low mobility (“hyper-responders”)(Figure S7C). These populations mirror the variable viscosities observed in glycogen deficient strains (Figure 3C). Notably, hyper-responders had higher ATP than non-responders, suggesting that low viscosity may conserve energy. Prior work has demonstrated that ATP can act as a hydrotrope, keeping proteins soluble in cells and potentially affecting their mobility (Patel et al., 2017). The concurrence of low ATP and fast diffusion in the “non-responder” population of glycogen-deficient cells, however, indicates that low ATP does not directly cause low mobility (Figure S7C).
If low ATP is sufficient to prompt viscoadaptation, then stochastic drops in ATP in the absence of stress should also lead to increased viscosity. We measured diffusion in unstressed cells with a range of naturally occurring ATP levels. Once again lower ATP correlated with higher viscosity (r= −0.6, p<.0001) (Figure 7D), consistent with a model in which low ATP triggers viscoadaptation. This correlation could reflect direct stabilization of the “open” (low ATP) conformation of the sensor by high viscosity. We therefore created a condition predicted to have high ATP and high viscosity by shifting cells from growth at 37°C to measurement at 22°C, creating a temporary mismatch between glycogen and trehalose content and temperature. As predicted, cells measured after this temperature shift had both high ATP and high viscosity (Figure 7D). We conclude that the ATP sensor operates reliably in the relevant range of viscosities and that stochastic drops in ATP are linked to viscoadaptation.
If low ATP triggers viscoadaptation, ATP loss should precede viscosity change. We tracked ATP and t-half values for individual cells over 60 minutes of temperature increase (22°C to 40°C). ATP levels began to drop after 10 minutes (~33°C), while t-half values did not start increasing until 18–25 minutes (36–38°C), consistent with a causal relationship in which viscoadaptation occurs in response to ATP loss (Figure 7E). Collectively these findings link energy availability to control of cellular viscosity and suggest that low ATP may be a trigger for viscoadaptation.
Discussion:
This work reveals that cells broadly modulate intracellular diffusion through the regulated production of glycogen and trehalose (“viscoadaptation”). This viscosity regulation counters the effect of increased temperature on molecular diffusion and slows diffusion when ATP levels are low (starvation, mitochondrial damage, and stochastic drops in ATP). The homeostatic nature of viscosity control makes it unlikely that viscoadaptation is a chance byproduct of glycogen and trehalose accumulation. Furthermore, yeast grown at 37°C produce glycogen and trehalose yet fail to show classic signs of stress (e.g. slow growth) (Mühlhofer et al., 2019). Thus, the stress-protectant functions ascribed to glycogen and trehalose (energy reserve and molecular chaperone, respectively) may be unnecessary in this condition. We propose that cytosolic viscosity is a finely tunable cellular property and that viscosity regulation is an important function of glycogen and trehalose.
Additional mechanisms for regulating the mobility of biomolecules in cells have been described by others, including crowding-based mechanisms (Delarue et al., 2018; Joyner et al., 2016), the assembly of stress-induced condensates (Franzmann et al., 2018), and pH-induced protein polymerization (Munder et al., 2016). Some conditions that induce viscoadaptation may also induce these responses. However, viscoadaptation is phenomenologically and mechanistically distinct. Of these responses, only viscoadaptation affects mobility on the size scale of individual proteins or implicates glycogen and trehalose in the control of protein diffusion. In addition, viscoadaptation is unique in serving as both a homeostatic mechanism and an acute response to stress.
The discovery of viscoadaptation does not conflict with any of the proposed cellular functions of glycogen or trehalose but may inform previous observations in important ways. For example, viscoadaptation may be relevant to previously described phenomena including conversion of the bacterial cytoplasm into a “glassy” state with low metabolic activity (Parry et al., 2014) and changes in the viscosity of cancer cells which often store high levels of glycogen (Guyer and Claus, 1942; Rousset et al., 1981; Takahashi et al., 1999; Wu et al., 2019). Furthermore, the existence of viscoadaptation reveals that cytosolic viscosity may be an important but overlooked parameter in models of stress-induced phase separation. Many such models focus on changes in the biophysical properties of condensates while assuming the state of the greater cellular environment to be relatively constant (Alberti, 2017; Hyman et al., 2014; Mitrea and Kriwacki, 2016; Pelletier et al., 2012; Yoshizawa et al., 2020). Going forward it will be important to consider the potential impact of changing cytosolic viscosity on the formation and regulation of phase separated compartments.
Viscosity “buffering” in a changing environment may be adaptive by tuning the rates of diffusion-controlled molecular interactions, chemical reactions, signaling cascades, kinetic proofreading, and the assembly and disassembly of complexes. Furthermore, slowing cellular processes on a large scale could conserve energy when nutrients are scarce. Consistent with this, trehalose accumulation in dormant plant spores causes loss of molecular mobility with a large corresponding drop in metabolic activity (Buitink and Leprince, 2004). Similarly, viscoadaptation may be a strategy to reduce energy expenditure while maintaining critical cellular functions.
In heat, both glycogen and trehalose were required for viscoadaptation whereas only glycogen was required in acute glucose deprivation. Trehalose is produced exclusively by non-homeothermic organisms (Elbein et al., 2003), consistent with its role in buffering diffusion against temperature changes, whereas glycogen synthesis is conserved in humans (Thon et al., 1993). This raises the possibility that glycogen enables viscoadaptation in homeothermic organisms such as mammals. Future work may explore the manifestations of viscoadaptation across diverse life forms with unique evolutionary demands.
STAR Methods:
RESOURCE AVAILABILITY:
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Onn Brandman (onn@stanford.edu)
Materials Availability:
All unique reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability:
Raw data for this work are provided as a Mendeley dataset:
http://dx.doi.org/10.17632/n5cs6wjpjp.1
File contents are as follows:
Raw Data 1.zip: example FRAP movies
Raw Data 2.zip: FRAP raw data for t-half Determination using easyFRAP
Raw Data 3.zip: FRAP movies for diffusion coefficient determination using simFRAP
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains and growth conditions:
All yeast strains were in the BY4741 S288C background. Strains were grown at the indicated temperatures in synthetic complete or dropout media with 2% glucose according to the recipe provided by Hanscho et al. (Hanscho et al., 2012). For biotinylation experiments yeast were grown in low biotin SD complete media with 2% glucose (Sunrise Science -biotin YNB) supplemented with 0.5 nM d-biotin.
Deletion strains were constructed via transformation with PCR products containing antibiotic resistance cassettes (NATMX6, HYGMX6, or KANMX6). PCR products contained 40bp of homology to the 5’ and 3’ genomic regions immediately adjacent to the gene to be deleted. Cytosolic BirA was integrated at the LEU2 locus driven by a PGK1 promoter. BirA fusion proteins contain a 120 residue intrinsically disordered linker. Transformants were verified by genomic PCR.
Plasmids used in this study were cloned by the Gibson Assembly method using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs).
METHOD DETAILS
Cell Lysate Preparation:
For cell lysate preparation 1–2 L of log phase culture was vacuum filtered to remove media and cells were “flash frozen” by rapid submersion in liquid nitrogen. Cells in frozen pellets were then lysed by mechanical grinding into a fine powder with a Spex 6750 Freezer Mill at a rate of 10 for 1 minute. The powder was allowed to thaw with gentle shaking at 4°C, followed by centrifugation at 10,000 x g for 10 minutes at 4° C. Supernatant was removed and subjected to a second spin at 16,000 x g for 10 minutes at 4°C. Supernatant was once again removed and used directly in lysate experiments or stored at −80°C for later use.
Biotin Labeling Timecourses:
For in vivo biotinylation reactions, cells expressing BirA under the PGK1 promoter and an Avi-tagged substrate under a variable promoter were grown in low biotin media (0.5 nM biotin) to an OD of 0.4–0.6. The genomically integrated BirA strain and the BirA tagging plasmid were a gift from Calvin Jan at Calico. The BirA reaction was initiated by addition of biotin (SUPELCO #47868) to a final concentration of 1 μM. All samples were corrected for background labeling (labeling prior to the biotin pulse) by comparison to a no biotin-pulse control. Cells were collected by spinning down 1 ml of OD 0.4–0.6 culture, removing the media and resuspending the cell pellet in 12–15 ul 4x NuPage LDS Sample Buffer with 5% β-mercaptoethanol. Samples were boiled at 95°C for five minutes to ensure denaturation and used immediately in gel shift assays or stored at −20°C.
For in vitro biotinylation assays, lysates from cells expressing Avi-tagged substrates were prepared as described above. In vitro labeling experiments were conducted using the Avidity BirA biotin-protein ligase standard reaction kit (Avidity BirA500) according to manufacturer’s instructions. Each reaction used 13 uL of lysate with 0.5 uL mix A, 0.5 uL mix B, and 0.25 uL BirA enzyme. After collection of a no biotin control (time 0), reactions were initiated by addition of 0.2 uL of d-biotin, provided in the kit, and pipetted twice quickly to mix. Subsequent samples were collected at the indicated time points by mixing 2 uL of the reaction mixture with 6 ul of boiling 4x NuPage LDS Sample Buffer with 5% β-mercaptoethanol and boiling for 5 additional minutes. Samples were used immediately in gel shift assays or frozen at −20°C.
Gel Shift Assay:
Samples for quantification of biotinylation were allowed to cool for at least 10 minutes after boiling. 6 ul of boiled sample was combined with 1.5 ul streptavidin (Invitrogen #434302 at 10 ug/uL), vortexed briefly, and loaded into an Invitrogen SDS-PAGE 1.5 mm 4–12% Bis-Tris gel. Gels were run for 2 hours at 4°C and 110V in 1x MOPS running buffer. Transfer was performed onto nitrocellulose membrane using a semi-dry transfer apparatus (Bio-Rad Turbo Blot). Membranes were blocked for 30 minutes in 5% milk in TBST and then incubated with mouse anti-HA primary antibody (antiHA-12C5 Roche #11583816001) in 5% milk in TBST with gentle shaking overnight at 4°C. After primary antibody incubation, membranes were washed three times in TBST for a total of 15 minutes and incubated at room temperature with IRDye 800CW Goat anti-Mouse IgG Secondary Antibody (1:5000, Licor #926–32210) in 5% milk in TBST for 1 hour. Blots were imaged on a Licor gel imager and band intensities were quantified using ImageStudioLite. All ratiometric analyses are the intensity of the top band divided by the intensity of the bottom band plus the intensity of the top band in a given lane.
Cell Size Measurement:
Cytoplasmic area (excluding vacuoles) in yeast expressing GFP was measured using imageJ. Briefly, cell images were converted to binary masks based on GFP fluorescence distribution using automated thresholding. The resulting mask was designated as a selection and the area of the selection determined using the measure area tool. Images were taken at the largest cross-section of each cell, and cell buds were not included in size measurements.
Microrheology:
Microrheology experiments were performed on a Zeiss LSM 880 confocal microscope using an oil immersion 40x objective. Lysates were mixed with 0.5 um fluorescent beads (488 nm/512 nm). Chamber slides were prepared using double sided tape to delineate the chamber width (1 cm) and depth (100 um). Beads (Fluospheres, 2% solids) were added to lysates at a final dilution of 1:5,000 and ~20 ul of the bead-lysate mixture was loaded into the chamber. Open ends were sealed with tape to prevent evaporation and slides were allowed to rest 10 minutes before measurement to minimize transient effects of sample loading on bead movement. The focal plane was chosen to be halfway between the slide and coverslip (50 um from either surface of the chamber) and X and Y positions were centered in the chamber to avoid boundary effects. 500 images were collected over a total of 30 seconds at 5x zoom using a 488 laser at a power of 6%.
Particle tracking was done using Particle Tracker 2D/3D in the Imagej Mosaic plug-in. Particle detection settings were as follows: Radius 3, Cutoff .001, Per/Abs 0.15. Particle linking parameters were as follows: Link Range 2, Displacement 10, Dynamics: Brownian. Trajectories ≤ 20 frames were eliminated from the analysis to reduce noise. Diffusion coefficients extracted from multiple trajectories in the same frame were averaged. For analysis of anomalous diffusion the slope of the Moment Scaling Spectrum (sMSS) for each trajectory was recorded. The value of the sMSS is related to the degree and direction of departure from “perfect” diffusion in which the mean-squared displacement of a particle increases linearly with time (Arts et al., 2019; Oliveira et al., 2019).
Heat Shock:
For small cultures (<10 ml) heat shock experiments were performed by centrifugation to pellet cultures followed by removal of media and resuspension in media pre-heated to the appropriate temperature. Cultures were then placed in an incubator at the required temperature for the duration of the heat shock treatment.
For heat shock of large cultures (≥1 L) pre-existing media was removed by vacuum filtration of cells onto nitrocellulose membrane. The resulting cell paste was quickly scraped from the membrane and resuspended in preheated media. Flasks were then shaken in pre-warmed incubators for the duration of the heat shock treatment.
For FRAP experiments in heat shock, cells were placed on slides coated with Concavalin A (to prevent movement) and entered into a microscope chamber pre-warmed to the appropriate temperature. A given timecourse corresponds to a single slide being imaged repeatedly over time in the heated microscope chamber.
Fluorescence Recovery After Photobleaching (FRAP):
FRAP experiments were carried out on a Zeiss LSM 880 confocal microscope using a 40x oil immersion objective. For in vivo measurements, 100 images were taken at 11x zoom with one cycle of bleaching occurring after the second image. Images were separated by 52 milliseconds. Per cell, one bleach zone, one control zone within the cell, and one background zone outside the cell were specified (all circular). Bleach and control zones were chosen such that the circular ROI touched the cell wall at one point only, and regions were selected to be homogenous in their fluorescence intensity. Nuclei could often be discerned by increased fluorescence intensity and were avoided when choosing a bleach location. Within these parameters bleached regions varied in their location within the cytoplasm. We did not observe systematic differences in t-half based on the exact location of bleaching within the cytoplasm. In vivo FRAP experiments used for t-half calculations (see below) had ROIs of a constant size (600 nm diameter) for bleached and control spots. Background ROIs were sometimes larger. FRAP experiments analyzed for diffusion coefficients with the program simFRAP (see below) did not necessarily have bleached ROIs of constant size. simFRAP was used because determination of the diffusion coefficient with this method is independent of ROI size and geometry (Blumenthal et al., 2015). Except for experiments in which the mobile fraction was being compared, any measurement with a mobile fraction below 0.8 was not included in analyses. These were uncommon and potentially indicated that a region intersected a cellular compartment rather than being purely cytosolic. For timecourse experiments measurements taken plus or minus one minute of the indicated time point were binned.
For in vitro measurements, we used purified GFP (Chromtek, eGFP-250) at a concentration of .085 mg/mL (1 μL GFP into 10 μL lysate). The size of the bleached ROI was ~40 um. For lysate and solution measurements, between 200 and 1000 images were taken at 1x zoom, depending on the rate of recovery. Bleaching occurred after the second image using 100% laser power in the 405 nm, 456 nm, and 488 nm channels. Images were separated by 120 milliseconds. Bleach zones, control zones, and background zones were circular. All raw movies are at 1x speed.
Of note, for technical reasons the lysates used in Figure 2E were diluted approximately 5% relative to the lysates used in Figure 2A. Therefore, comparisons should be made within these data sets rather than between them.
FRAP Analysis:
The imageJ bioformats package was used to open .czi stacks. For t-half analysis intensity values for each region at each time point were extracted using the multi-measure tool set to “mean gray value”. Values were saved in excel spreadsheets in the format [column 1: time in seconds] : [column 2 : intensity of bleach ROI] : [column 3 : intensity of control ROI] : [column 4 : intensity of background ROI]. For in vitro experiments, the intensity of the background ROI was set to 0 for all time points since background did not apply as defined in this case. Spreadsheets were saved as either .xlsx or .czi files and were uploaded into easyFRAP, an open-source online FRAP analysis program (Koulouras et al., 2018), to calculate the mobile fraction, t-half, and R-squared values for each photobleaching experiment. Gap ratio and bleaching depth were used to determine the quality of the measurement. Images with a gap ratio below 0.6 or a bleaching depth below 0.2 were not included in the analyses. Example movies for FRAP experiments are provided via Mendeley in Raw Data 1 along with traces and raw data spreadsheets. The full set of raw data used for easyFRAP analysis is provided in Raw Data 2.
For determination of diffusion coefficients from FRAP experiments, we made use of the Fiji plug-in simFRAP according to developers’ instructions (Blumenthal et al., 2015). In the case of in vivo experiments the bleached cell, bleached region, and an unbleached reference cell were specified using the simFRAP interface. The inter-image time was 0.05 seconds and the pixel aspect ratio was 0.15. For in vitro experiments, region selection was modified slightly to accommodate the change in experiment type. The bleached region was in the center of the image and was specified as before, the unbleached reference cell was a circular region in a corner of the image, and the bleached cell was set as the circle of largest circumference that would fit in the square image and included the bleached region but excluded the unbleached reference region. The inter-image time was .12 seconds and the pixel aspect ratio was 1.66. Raw data movies for simFRAP analysis are provided via Mendeley in Raw Data 3.
Drug Treatments:
Cycloheximide (Calbiochem #239764) was used at a final concentration of 50 ug/ml. Sodium azide (Sigma #2002) and sodium arsenite (Sigma #7400) were used at final concentrations of 0.15 mM and 2 mM, respectively. Antimycin A (Sigma #A8674), oligomycin (Sigma #75351), and FCCP (Cayman Chemical #15218) were used in combination at 10 μM, 0.1 μM, and 50 nM, respectively. Cycloheximide was added 10 minutes prior to the start of the experiment; all other drugs were added 30 minutes prior to the start of the experiment.
Validamycin A (GoldBio, #V-170–1) and 1,4-dideoxy-1,4-imino-D-Arabinitol (DAB) (Cayman Chemical Company, #20939) were both used at 1 ug/mL. Validamycin was made fresh as a 1 mg/mL stock in H20 before each experiment, and DAB was used from a pre-made stock of 10 mg/mL in DMSO. DAB and Validamycin A were added to cultures immediately prior to transfer to slides and the initiation of heat shock. Heat shock was for 45 minutes in a microscope chamber pre-heated to 42°C after which slides were transferred to room temperature to recover. FRAP measurements or images of Pab1-GFP were collected after the indicated recovery period.
Iodine Staining:
Qualitative iodine staining experiments (see Figure S3D) were adapted from the method described by (Demonte et al., 2014). Briefly, cell spots for iodine staining were made by pipetting 1 ml of log phase (OD 0.4 - OD 0.6) culture onto nitrocellulose membrane using a round stencil as a guide (approx. 8 mm diameter). Media was pulled through the membrane by vacuum filtration leaving a layer of cells on the membrane. The membrane was then placed on a glass slide (to avoid direct contact between the iodine and the membrane) and onto a layer of iodine crystals in a closed chamber. Membranes were allowed to stain for 10 minutes in the chamber and then removed. Spot color generally began to decrease after removal from the iodine chamber so images were not taken until the color of all spots remained stable for 5 minutes (generally ~15 minutes after removal from the iodine chamber). Any spots to be compared were made on the same membrane to minimize variability in the staining conditions. Qualitative differences in glycogen content were determined by the color of each spot, with darker spots corresponding to cells with more glycogen. Spots were imaged using an iphone.
Quantitative Trehalose Measurements
Trehalose was measured using the Trehalose Assay Kit (K-TREH) from Megazyme. Manufacturer’s instructions were followed with the following exceptions: samples were collected in pre-weighed microcentrifuge tubes by pelleting 1 mL of OD 0.4–0.6 culture followed by washing in 1 mL of glucose-free SD media to remove background glucose signal. After removal of the wash, microcentrifuge tubes were quickly re-weighed and flash frozen in liquid nitrogen. Samples were used immediately or stored at −80°C until later use. To lyse the cells, 100 ul of boiling water was added directly to the frozen pellet with vigorous vortexing. Samples were boiled for 15 minutes with vortexing every 2 minutes then spun down in a table-top centrifuge for 3 minutes. The supernatant (roughly 100 uL) was transferred to new tubes. A second extraction of the pellet was performed using another 100 uL of boiling water with vortexing for 5 minutes. The second extraction was combined with the first extraction for a total of ~200 uL.
The trehalose measurements were performed according to the Megazyme microplate protocol with all reagent volumes cut in half and 2 uL of sample per well. Every sample was measured in technical triplicate, and each experiment was performed in at least biological triplicate. Absorbance readings were taken using a Tecan Plate Reader and all values were normalized to the wet weight of the cell pellet.
Quantitative Glycogen Measurements:
Cells for quantitative glycogen measurement were collected as for trehalose measurements. Glycogen extraction and measurement protocols were used from (Ewald et al., 2016). 125 ul of 0.25 M sodium carbonate was added to the frozen cell pellet, which was then vortexed vigorously and incubated at 85°C for 4 hours with gentle shaking. Next, 75 ul of 1 M acetic acid, 300 ul sodium acetate (200 mM, p.H. 5.2), and 2 ul amyloglucosidase (from Aspergillus Niger, Sigma, 1000 U/ml) were added to each sample. Samples were mixed by pipetting and incubated at 60°C overnight (~16 hours). Samples were centrifuged and the supernatants filtered using spin columns to remove cellular debris. Glucose released from glycogen digestion with amyloglucosidase was measured enzymatically using the Trehalose Assay Kit (Megazyme, K-TREH) without the trehalase.
Quantitative Glucose Measurements:
Glucose measurements were performed using the Sigma High Sensitivity Glucose Assay Kit (MAK181, Sigma-Aldrich), according to manufacturer’s instructions. In brief, yeast pellets were prepared as for glycogen and trehalose measurements. Cells were lysed in the same manner as for trehalose measurements and 2 uL of supernatant was used per measurement. All samples were measured in technical duplicates with a minimum of three biological replicates.
In Vitro Glycogen and Trehalose Phase Experiments:
Glycogen and trehalose solutions of the indicated concentrations were made in water using glycogen from oyster (Sigma #G8751) and trehalose dihydrate (EMD Millipore #625625). 40 uL droplets were made on parafilm at room temperature and left 8 hours, by which point trehalose-only droplets had solidified. Phase comparisons were done qualitatively by observing the response of the droplet to application of force with a pipette tip; droplets that dried out without retaining their 3-dimensional structure were considered not to have undergone phase transition. Droplets that distorted upon application of mechanical force without fracturing were categorized as gel-like, and droplets that fractured with application of mechanical force were categorized as solids (see Figure S5B for examples).
Glucose Starvation:
For glucose starvation experiments of small cultures (<10 mL), yeast were spun down in a tabletop centrifuge at 14,000 x g for 1 minute. Media was discarded and the cell pellet was washed once with glucose-free SD media, before being resuspended in the appropriate starvation media (either 0% glucose or 0.1% glucose). Cells were transferred to culture tubes and placed in a spinning wheel at 30°C for the duration of starvation.
For starvation of large cultures (1 L+), media exchange was performed using a vacuum filter flask as for heat shock of large cultures with the addition of one wash with 0.5 L of glucose-free media. The wash was performed after separation of cells from the previous growth media by flowing glucose-free media over cells adhered to the nitrocellulose membrane. After the wash cells were scraped from the membrane and resuspended in the appropriate starvation media. Flasks were shaken at 30°C for the duration of starvation.
ATP Measurements:
ATP levels in single cells were measured using a FRET based nanosensor expressed from a cen/ars plasmid, pDR-GW AT1.03YEMK, which was a gift from Wolf Frommer (Addgene plasmid # 28004; http://n2t.net/addgene:28004; RRID:Addgene_28004). Single-cell FRET measurements were recorded on a Zeiss LSM880 confocal microscope using a 40x oil immersion objective. Excitation was provided at 405 nm and emissions were detected at 460 nm - 510 nm (encompassing 485 nm) and 520 nm - 560 nm (encompassing 535 nm). Data is given as a ratio of 535:485 signal intensity. The detector gain was set at 750 for the 535 nm channel and 650 for the 485 nm channel, chosen such that the 535 nm:485 nm ratio was roughly 1:1 in unstressed cells. Images were then taken following perturbation to the initial state. Cells were grown in -URA dropout SD media.
For the timecourse of viscosity and t-half (Figure 7E), cells expressing the ATP sensor started at room temperature (approx. 22°C) and were heated gradually in the microscope chamber to a final temperature of 40°C (approximately 40 minutes) at which they remained for the duration of the experiment. FRAP was performed on one cell every other minute over the course of 60 minutes. Three such timecourses were collected in total.
QUANTIFICATION AND STATISTICAL ANALYSIS
T-half values for FRAP experiments were calculated by first extracting intensity and time information for each ROI using ImageJ with the bioformats package (https://imagej.net/BioFormats). FRAP movies were processed manually or through an identical automated process by way of an ImageJ macro (developed internally). Information extracted from FRAP movies was then processed to produce t-half values using the open-source web-based platform easyFRAP (described in more detail above) (Koulouras et al., 2018). Spreadsheets, organized by figure panel, sample, and replicate, are provided in via Mendeley in Raw Data 2.
Diffusion coefficients were calculated directly from raw FRAP movies using the imagej plug-in simFRAP (Blumenthal et al., 2015). Movies are provided in Raw Data 3 and are organized by figure panel, sample, and replicate.
Information on statistical analysis and replicates, including how replicates were defined for individual experiments, is included in figure legends. Statistical analyses were performed using Graphpad Prism Version 8.4.
Supplementary Material
Figure S1 A viscosity-controlled model reaction is temperature dependent in cell lysates but not live cells, Related to Figure 1. (A) BirA biotinylates a 15-AA target sequence called the “Avi-tag”. Labeling rates are determined by a gel shift assay in which addition of streptavidin to cell lysates causes the biotinylated species to run at a higher molecular weight than its nonbiotinylated counterpart. All reactions use cytosolic BirA and GFP-Avi as “bait” (B) Schematic depicting the Fluorescence Recovery After Photobleaching (FRAP) technique. GFP was bleached and the rate of its subsequent recovery to the bleached region was measured to determine the viscosities of solutions, cell lysates, and live yeast cells. Hypothetical recovery curves for solutions of higher and lower viscosities are shown on the left where recovery time is being measured by t-half, the time to reach half-maximal fluorescence recovery. Recovery can also be reported as the diffusion coefficient of GFP (C) Labeling of RPL19b-Avi by BirA in lysates produced from cells grown at 30°C (●blue), 37°C (◼orange), or heat shocked for 30 minutes at 45°C (◆red). All reactions were performed at room temperature (nonlinear regression difference between slopes, 30°C vs 37°C p=.006; 30°C vs 45°C p<.0001; 37°C vs 45°C p<.0001) Error bars are s.e.m.
Figure S2 Cells regulate intracellular viscosity in response to heat (“viscoadaptation”), Related to Figure 2. (A) FRAP measurements on cells expressing mKate2 20 minutes after cells were shifted from 22°C to 42°C (n=3 cultures/temp, <10 cells/culture)(unpaired t-test, p=.0271) (B) FRAP timecourse on cells expressing mKate2. Cells were shifted from 22°C (T0) to 42°C (n=3 timecourses). Error bars are s.e.m. Points without visible error bars have error within the size of the point.
Figure S3 Heat-induced accumulation of the carbohydrates glycogen and trehalose increases cellular viscosity, Related to Figure 3. (A) Cells were treated with 50 ug/ml cycloheximide (CHX) or vehicle (EtOH) for 10 minutes prior to and during heat shock at 42°C. FRAP measurements were performed beginning at 20 minutes after temperature shift. (unpaired t-test, No CHX 22°C vs No CHX 42°C p<0.0001; No CHX 22°C vs +CHX 42°C p<.0001; No CHX 42°C vs +CHX 42°C, p=0.169) (B) Glycogen (left) and trehalose (right) levels in cells grown at the indicated steady state temperatures. Values are normalized to the minimum value at 30°C. (left) (n≥5 measurements/ temperature) (unpaired t-test, 22°C vs 30°C, p=.023; 22°C vs 37°C, p=.204; 30°C vs 37°C, p=.017; 30°C vs 40°C, p=.0007; 37°C vs 40°C, p=.262; 22°C vs 40°C, p=.018); (right)(n≥4 measurements/temperature) (unpaired t-test, 22°C vs 30°C, p=.1350, 22°C vs 37°C, p<.0001, 30°C vs 37°C, p=.0025, 30°C vs 40°C, p=.0008, 37°C vs 40°C, p=.0097, 22°C vs 40°C, p=.0006) (C) Schematic depicting the technique for iodine staining of glycogen in live cells. (D) (left) Glycogen content normalized to cell wet weight after 42°C heat shock with and without 50 mM LiCl. (n=2) (unpaired t-test, p=.07) (below, left) Iodine staining for glycogen content after heat shock with and without LiCl. (right) Trehalose content normalized to cell pellet wet weight after 42°C heat shock with and without 50 mM LiCl (n=3) (unpaired t-test, p<.05) (E) Iodine staining for glycogen levels in live cells unstressed or heated at 42°C with or without DAB. (F) Cell area was measured from a cross-section of the cell, excluding vacuoles. The included area was defined by the distribution of GFP. Heat shock was at 42°C for 20 minutes. (n≥2 experiments, ≥10 cells/experiment) (unpaired t-test, WT 22°C vs WT 42°C, p=.2165; ΔGΔT 22°C vs ΔGΔT 42°C, p=.267; WT 22°C vs ΔGΔT 22°C, p=.767; WT 42°C vs ΔGΔT 42°C, p=.412; WT 22°C vs ΔGΔT 42°C, p=.049) (G) (left) Glucose measured in strains grown in steady state at the indicated temperatures (n≥ 3 measurements per condition) (unpaired t-test, p≥.05 for all pairwise comparisons) (right) Glucose was measured in the indicated strains at the indicated times after shift from 30°C to 42°C (≥ 3 measurements per condition) (unpaired t-test, WT T0 vs T5, p=.032; WT T0 vs ΔGΔG T0, p=.006; ΔGΔG T0 vs T40 p=.014; WT T0 vs WT T20, p=.1; WT T0 vs WT T40, p=.065) (H) In vivo BirA labeling of GFP-Avi. Wildtype cells (●) and cells lacking Glc3 (◼) were grown at 30°C (blue) and 37°C (red) (n=4 timecourses for WT, n=2 timecourses for ΔGlc3) (nonlinear regression difference between slopes, WT 30°C vs WT 37°C p=.107; WT 30°C vs ΔGlc3 30°C p=.006; WT 37°C vs ΔGlc3 37°C p=.01; ΔGlc3 30°C vs ΔGlc3 37°C p=.001) (I) Cells were treated with no drug, 50 mM LiCl, or 50 mM NaCl for one hour prior to temperature shift. FRAP measurements began 20 minutes after temperature increase. (n≥ 2 experiments, ≥10 cells/experiment)(unpaired t-test, 22°C no drug vs 42°C no drug p<.0001, 22°C no drug vs 42°C + NaCl p=.0003, 42°C + LiCl vs 42°C + NaCl p=.018; 22°C no drug vs 42°C + LiCl, p=.22) Error bars are s.e.m.
Figure S4 Viscoadaptation occurs in response to nutrient limitation, Related to Figure 4. (A) Cytosolic BirA labeling of RPL19b-Avi (top left), RPL16-Avi (top right), RPS2-Avi (bottom left), and RPS20-Avi (bottom right) in 2% glucose (blue) and 0% glucose (orange). Cells were grown in low biotin media (0.5 nM) to prevent background biotinylation. RPS20-Avi labeling timecourse was shortened due to faster labeling of this substrate (n=3 replicates/strain) (nonlinear regression difference between slopes, top left p<.0001; top right p<.0001; bottom left p<.0001; bottom right p<.0001) (B) Cells were grown in 2% glucose with 8 nM biotin (regular biotin, RB) or 0.5 nM biotin (low biotin, LB) and transferred to 0% glucose as indicated. FRAP measurements start 30 minutes after removal of glucose. (n≥ 2 experiments, ≥ 15 cells/experiment) (unpaired t-test, 2% glucose RB vs 2% glucose LB, p≥.05; 2% glucose LB vs 0% glucose LB, p=.0006; 2% glucose RB vs 0% glucose LB, p≥.05) (C) Iodine staining on cells grown in media with the indicated biotin concentrations and 2% glucose or 0% glucose for 30 minutes (D) Intracellular glucose (normalized to cell pellet weight) in cells in 0% glucose for 30 minutes with normal SD media or low biotin SD media. (n≥ 3 measurements/condition)(unpaired t-test, 2% glucose RB vs 2% glucose LB p=.016; 2% glucose RB vs 0% glucose RB p=.004; 2% glucose LB vs 0% glucose LB p=.036; 0% glucose RB v. 0% glucose LB p=.009) Error bars are s.e.m.
Figure S5 Glycogen and trehalose alter molecular movement in vitro, Related to Figure 5. (A) Microrheology on fluorescent beads in the indicated aqueous solutions (left) and cell lysates (right). Plotted are the mean slopes of the Moment Scaling Spectrum (SMSS). SMSS values for individual particle trajectories were averaged to obtain each value (n≥58 trajectories) (left) (unpaired t-test, H20 vs 70% sucrose p=.54; H20 vs 30% gly p<.0001; H20 vs 45% tre p<.0001; H20 vs 30%gly+45%tre p<.0001) (right) (unpaired t-test, H20 vs 22°C lysate p=.026; H20 vs 42°C lysate p=.052; H20 vs 42°C lysate ΔGΔT p<.0001; 22°C lysate vs 42°C lysate p=.145; 42°C lysate vs 42°C lysate ΔGΔT p=.64) (B) Aqueous droplets (4 mm diameter) of glycogen and trehalose were monitored over time at room temperature. Phase properties of droplets were assessed when the trehalose-only droplets solidified, characterized by fracturing in response to mechanical force (left). Droplets that distorted upon mechanical force without fracturing were described as “gel-like” (middle). Droplets that evaporated without undergoing any phase transition were characterized as “liquids” (right) (C) 40x confocal fluorescence images of purified GFP (~0.1 mg/mL final concentration) in lysates from cells grown in the indicated conditions prior to lysis.
Figure S6 Viscoadaptation affects phase separation and the solubility of biomolecules in vivo, Related to Figure 6. (A) Cells expressing Pab1-GFP were starved of glucose in 8 nM biotin (solid, regular biotin, RB) or 0.5 nM biotin (checkered, low biotin, LB) media and the percentage of cells containing Pab1 foci was determined for 3 time windows after onset of glucose starvation (n=4 experiments)(unpaired t-test, RB vs LB 0–20 p=.655; RB vs LB 20–40 p=.041; RB vs LB 40–60 p=.181) (B) Pab1-GFP in cells that were heat shocked at 42°C for 40 minutes (top) and then given 1 hour for the temperature to cool to 24°C (bottom) with and without the trehalase inhibitor Validamycin A. Images are taken at 40x. (C) Microrheology was performed on H20, lysates from unstressed cells grown at 30°C, and lysate from cells heat shocked at 45°C for 30 minutes. Each dot is one sample. (unpaired t-test, H20 vs unstressed lysate p=.004; H20 vs 45°C HS lysate p=.017; unstressed lysate vs 45°C HS lysate p=.659) (D) 10x brightfield images of dried lysate droplets from cells heat shocked at 45°C for 30 minutes undiluted (left) or diluted 1:50 with H20 (right). (E) (left) Trehalose measurements in the same conditions/strains (unpaired t-test, log WT vs log ΔReg1 p=.004; stationary WT vs stationary ΔReg1 p<.0001) (right) Iodine staining for glycogen content in WT and ΔReg1 strains in logarithmic growth (unstressed) or stationary phase. (F) Average t-half for GFP in WT (solid) and ΔReg1 (checkered) cells in log phase growth at 30°C (n≥2 experiments, ≥8 cells/experiment)(unpaired t-test, p=.0173) Error bars are s.e.m. (G) (left) Schematic depicting estimation of the mobile fraction (MF) from FRAP recovery curves. (right) Example curve for an MF of 1 (top) and 0.7 (bottom) indicating 100% and 70% fluorescence recovery, respectively.
Figure S7 Viscoadaptation occurs in cells with low ATP, Related to Figure 7. (A) (left) Schematic of the FRET-based ATP sensor(Bermejo et al., 2010) (right) Example of log-phase cells in 2% glucose expressing the ATP sensor. Boxes highlight high and low ATP cells in the same culture (B) Cells expressing the ATP sensor after 30 minutes in the indicated conditions (C) t-half values and ATP levels plotted for single cells expressing the ATP sensor (left) WT cells in 0% glucose with low biotin (0.5 nM) (R = −0.589 p<.0001) (right) ΔGlc3ΔGsy2 cells in 0% glucose with low biotin. Boxes highlight responders (red) and non-responders (blue) (R= −0.132 p=0.135)
Highlights:
Cells tune their viscosity by synthesis of glycogen and trehalose (“viscoadaptation”)
Viscoadaptation enables constant diffusion and reaction rates across temperatures
Biophysical changes during viscoadaptation impact phase separation and solubility
Low ATP levels correlate with high cytosolic viscosity through viscoadaptation
Acknowledgements
We thank J. Ferrell, P. Geiduschek, P. Harbury, D. Herschlag, K.C. Huang and R. Das for invaluable guidance in the preparation of this manuscript. All members of the Brandman lab provided helpful discussions and continuous support throughout this project. This work was supported by NIGMS of the National Institutes of Health under award numbers T32GM007276 to L.P. and 5R01GM115968 to O.B. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References:
- Alberti S. (2017). Phase separation in biology. Curr. Biol 27, R1097–R1102. [DOI] [PubMed] [Google Scholar]
- Arts M, Smal I, Paul MW, Wyman C, and Meijering E. (2019). Particle Mobility Analysis Using Deep Learning and the Moment Scaling Spectrum. Sci. Rep 9, 17160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermejo C, Haerizadeh F, Takanaga H, Chermak D, and Frommer WB (2010). Dynamic analysis of cytosolic glucose and ATP levels in yeast using optical sensors. Biochem. J 432, 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal D, Goldstien L, Edidin M, and Gheber LA (2015). Universal Approach to FRAP Analysis of Arbitrary Bleaching Patterns. Sci. Rep 5, 11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budin I, de Rond T, Chen Y, Chan LJG, Petzold CJ, and Keasling JD (2018). Viscous control of cellular respiration by membrane lipid composition. Science 362, 1186–1189. [DOI] [PubMed] [Google Scholar]
- Buitink J, and Leprince O. (2004). Glass formation in plant anhydrobiotes: survival in the dry state. Cryobiology 48, 215–228. [DOI] [PubMed] [Google Scholar]
- Chen I, Howarth M, Lin W, and Ting AY (2005). Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2, 99–104. [DOI] [PubMed] [Google Scholar]
- Delarue M, Brittingham GP, Pfeffer S, Surovtsev IV, Pinglay S, Kennedy KJ, Schaffer M, Gutierrez JI, Sang D, Poterewicz G, et al. (2018). mTORC1 Controls Phase Separation and the Biophysical Properties of the Cytoplasm by Tuning Crowding. Cell 174, 338–349.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demonte AM, Asención Diez MD, Guerrero SA, Ballicora MA, and Iglesias AA (2014). Iodine staining of Escherichia coli expressing genes involved in the synthesis of bacterial glycogen. [Google Scholar]
- Dill KA (1990). Dominant forces in protein folding. Biochemistry 29, 7133–7155. [DOI] [PubMed] [Google Scholar]
- Elbein AD, Pan YT, Pastuszak I, and Carroll D. (2003). New insights on trehalose: a multifunctional molecule. Glycobiology 13, 17R – 27R. [DOI] [PubMed] [Google Scholar]
- Ewald JC, Kuehne A, Zamboni N, and Skotheim JM (2016). The Yeast Cyclin-Dependent Kinase Routes Carbon Fluxes to Fuel Cell Cycle Progression. Mol. Cell 62, 532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François J, and Parrou JL (2001). Reserve carbohydrates metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev 25, 125–145. [DOI] [PubMed] [Google Scholar]
- Franzmann TM, Jahnel M, Pozniakovsky A, Mahamid J, Holehouse AS, Nüske E, Richter D, Baumeister W, Grill SW, Pappu RV, et al. (2018). Phase separation of a yeast prion protein promotes cellular fitness. Science 359. [DOI] [PubMed] [Google Scholar]
- Frock AD, and Kelly RM (2012). Extreme Thermophiles: Moving beyond single-enzyme biocatalysis. Curr. Opin. Chem. Eng 1, 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney PA, Lu C, Caudy AA, Hess DC, and Botstein D. (2013). Yeast metabolic and signaling genes are required for heat-shock survival and have little overlap with the heat-induced genes. Proc. Natl. Acad. Sci. U. S. A 110, E4393–E4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyer MF, and Claus PE (1942). Increased Viscosity of Cells of Induced Tumors. Cancer Res. 2, 16–18. [Google Scholar]
- Hanscho M, Ruckerbauer DE, Chauhan N, Hofbauer HF, Krahulec S, Nidetzky B, Kohlwein SD, Zanghellini J, and Natter K. (2012). Nutritional requirements of the BY series of Saccharomyces cerevisiae strains for optimum growth. FEMS Yeast Res. 12, 796–808. [DOI] [PubMed] [Google Scholar]
- Hottiger T, Schmutz P, and Wiemken A. (1987a). Heat-induced accumulation and futile cycling of trehalose in Saccharomyces cerevisiae. J. Bacteriol 169, 5518–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger T, Boller T, and Wiemken A. (1987b). Rapid changes of heat and desiccation tolerance correlated with changes of trehalose content in Saccharomyces cerevisiae cells subjected to temperature shifts. FEBS Lett. 220, 113–115. [DOI] [PubMed] [Google Scholar]
- Huang D, Chun KT, Goebl MG, and Roach PJ (1996). Genetic interactions between REG1/HEX2 and GLC7, the gene encoding the protein phosphatase type 1 catalytic subunit in Saccharomyces cerevisiae. Genetics 143, 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Wilson WA, and Roach PJ (1997). Glucose-6-P control of glycogen synthase phosphorylation in yeast. J. Biol. Chem 272, 22495–22501. [DOI] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, and Jülicher F. (2014). Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol 30, 39–58. [DOI] [PubMed] [Google Scholar]
- Imamura H, Nhat KPH, Togawa H, Saito K, Iino R, Kato-Yamada Y, Nagai T, and Noji H. (2009). Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. U. S. A 106, 15651–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain NK, and Roy I. (2009). Effect of trehalose on protein structure. Protein Sci. 18, 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Williams CC, and Weissman JS (2014). Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 346, 1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ME (2018). Modeling the Self-Assembly of Protein Complexes through a Rigid-Body Rotational Reaction–Diffusion Algorithm. J. Phys. Chem. B 122, 11771–11783. [DOI] [PubMed] [Google Scholar]
- Joyner RP, Tang JH, Helenius J, Dultz E, Brune C, Holt LJ, Huet S, Müller DJ, and Weis K. (2016). A glucose-starvation response regulates the diffusion of macromolecules. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerly M. (1930). The solubility of glycogen. Biochem. J 24, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulouras G, Panagopoulos A, Rapsomaniki MA, Giakoumakis NN, Taraviras S, and Lygerou Z. (2018). EasyFRAP-web: a web-based tool for the analysis of fluorescence recovery after photobleaching data. Nucleic Acids Res. 46, W467–W472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambowitz AM, Kobayashi GS, Painter A, and Medoff G. (1983). Possible relationship of morphogenesis in pathogenic fungus, Histoplasma capsulatum, to heat shock response. Nature 303, 806–808. [DOI] [PubMed] [Google Scholar]
- Lillie SH, and Pringle JR (1980). Reserve carbohydrate metabolism in Saccharomyces cerevisiae: responses to nutrient limitation. J. Bacteriol 143, 1384–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longsworth LG (1954). Temperature Dependence of Diffusion in Aqueous Solutions. J. Phys. Chem 58, 770–773. [Google Scholar]
- Luby-Phelps K, Taylor DL, and Lanni F. (1986). Probing the structure of cytoplasm. J. Cell Biol 102, 2015–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda CA, Xavier MA, Mattos KA, Galina A, and Montero-Lomeli M. (2001). Phosphoglucomutase is an in vivo lithium target in yeast. J. Biol. Chem 276, 37794–37801. [DOI] [PubMed] [Google Scholar]
- Mika JT, and Poolman B. (2011). Macromolecule diffusion and confinement in prokaryotic cells. Curr. Opin. Biotechnol 22, 117–126. [DOI] [PubMed] [Google Scholar]
- Miller Christina Cruickshank, and Walker James (1924). The Stokes-Einstein law for diffusion in solution. Proceedings of the Royal Society of London. Series A, Containing Papers of a Mathematical and Physical Character 106, 724–749. [Google Scholar]
- Mitrea DM, and Kriwacki RW (2016). Phase separation in biology; functional organization of a higher order. Cell Commun. Signal 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlhofer M, Berchtold E, Stratil CG, Csaba G, Kunold E, Bach NC, Sieber SA, Haslbeck M, Zimmer R, and Buchner J. (2019). The Heat Shock Response in Yeast Maintains Protein Homeostasis by Chaperoning and Replenishing Proteins. Cell Rep. 29, 4593–4607.e8. [DOI] [PubMed] [Google Scholar]
- Müller J, Boller T, and Wiemken A. (1995). Effects of validamycin A, a potent trehalase inhibitor, and phytohormones on trehalose metabolism in roots and root nodules of soybean and cowpea. Planta 197, 362–368. [Google Scholar]
- Munder MC, Midtvedt D, Franzmann T, Nüske E, Otto O, Herbig M, Ulbricht E, Müller P, Taubenberger A, Maharana S, et al. (2016). A pH-driven transition of the cytoplasm from a fluid- to a solid-like state promotes entry into dormancy. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira FA, Ferreira RMS, Lapas LC, and Vainstein MH (2019). Anomalous Diffusion: A Basic Mechanism for the Evolution of Inhomogeneous Systems. Frontiers in Physics 7, 18. [Google Scholar]
- Palmucci L, Anzil AP, and Luh S. (1983). CRYSTALLINE AGGREGATES OF PROTEIN-GLYCOGEN COMPLEXES (ALIAS “VIRUS-LIKE PARTICLES”) IN SKELETAL MUSCLE: REPORT OF A CASE AND REVIEW OF THE LITERATURE. Neuropathol. Appl. Neurobiol 9, 61–71. [DOI] [PubMed] [Google Scholar]
- Parry BR, Surovtsev IV, Cabeen MT, O’Hern CS, Dufresne ER, and Jacobs-Wagner C. (2014). The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell 156, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Malinovska L, Saha S, Wang J, Alberti S, Krishnan Y, and Hyman AA (2017). ATP as a biological hydrotrope. Science 356, 753–756. [DOI] [PubMed] [Google Scholar]
- Patriarca EJ, and Maresca B. (1990). Acquired thermotolerance following heat shock protein synthesis prevents impairment of mitochondrial ATPase activity at elevated temperatures in Saccharomyces cerevisiae. Exp. Cell Res 190, 57–64. [DOI] [PubMed] [Google Scholar]
- Pelletier J, Bellot G, Gounon P, Lacas-Gervais S, Pouysségur J, and Mazure NM (2012). Glycogen Synthesis is Induced in Hypoxia by the Hypoxia-Inducible Factor and Promotes Cancer Cell Survival. Front. Oncol 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potma E, de Boeij WP, van Haastert PJ, and Wiersma DA (2001). Real-time visualization of intracellular hydrodynamics in single living cells. Proc. Natl. Acad. Sci. U. S. A 98, 1577–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prats C, Graham TE, and Shearer J. (2018). The dynamic life of the glycogen granule. J. Biol. Chem 293, 7089–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protter DSW, and Parker R. (2016). Principles and Properties of Stress Granules. Trends Cell Biol. 26, 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quain DE, and Tubb S. (1983). A rapid and simple method for the determination of glycogen in yeast. J. Inst. Brew 89, 38–40. [Google Scholar]
- Rousset M, Zweibaum A, and Fogh J. (1981). Presence of glycogen and growth-related variations in 58 cultured human tumor cell lines of various tissue origins. Cancer Res. 41, 1165–1170. [PubMed] [Google Scholar]
- Ruiz A, Xu X, and Carlson M. (2011). Roles of two protein phosphatases, Reg1-Glc7 and Sit4, and glycogen synthesis in regulation of SNF1 protein kinase. Proc. Natl. Acad. Sci. U. S. A 108, 6349–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Roig C, Viéitez C, Posas F, and De Nadal E. (2010). The Rpd3L HDAC complex is essential for the heat stress response in yeast. Mol. Microbiol 76, 1049–1062. [DOI] [PubMed] [Google Scholar]
- Rybicka K. (1979). Glycosomes (protein-glycogen complex) in the canine heart. Virchows Arch. B Cell Pathol. 30, 335–347. [DOI] [PubMed] [Google Scholar]
- Salvadó Z, Arroyo-López FN, Guillamón JM, Salazar G, Querol A, and Barrio E. (2011). Temperature adaptation markedly determines evolution within the genus Saccharomyces. Appl. Environ. Microbiol 77, 2292–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seksek O, Biwersi J, and Verkman AS (1997). Translational diffusion of macromolecule-sized solutes in cytoplasm and nucleus. J. Cell Biol 138, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y, Kingsley S, Walker G, Mondoux MA, and Tissenbaum HA (2018). Metabolic shift from glycogen to trehalose promotes lifespan and healthspan in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A 115, E2791–E2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Sutter BM, Ye X, and Tu BP (2010). Trehalose is a key determinant of the quiescent metabolic state that fuels cell cycle progression upon return to growth. Mol. Biol. Cell 21, 1982–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidell BD, and Hazel JR (1987). Temperature affects the diffusion of small molecules through cytosol of fish muscle. J. Exp. Biol 129, 191–203. [DOI] [PubMed] [Google Scholar]
- Sinensky M. (1974). Homeoviscous Adaptation—A Homeostatic Process that Regulates the Viscosity of Membrane Lipids in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 71, 522–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovski K, Ferrar T, Benisty H, Uschner F, Delgado J, Jimenez J, Solé C, de Nadal E, Klipp E, Posas F, et al. (2017). Interaction Dynamics Determine Signaling and Output Pathway Responses. Cell Rep. 19, 136–149. [DOI] [PubMed] [Google Scholar]
- Swaminathan R, Hoang CP, and Verkman AS (1997). Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J 72, 1900–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai SL, Daran-Lapujade P, Walsh MC, Pronk JT, and Daran J-M (2007). Acclimation of Saccharomyces cerevisiae to low temperature: a chemostat-based transcriptome analysis. Mol. Biol. Cell 18, 5100–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Satomi A, Yano K, Kawase H, Tanimizu T, Tuji Y, Murakami S, and Hirayama R. (1999). Estimation of glycogen levels in human colorectal cancer tissue: relationship with cell cycle and tumor outgrowth. J. Gastroenterol 34, 474–480. [DOI] [PubMed] [Google Scholar]
- Telis VRN, Telis-Romero J, Mazzotti HB, and Gabas AL (2007). Viscosity of Aqueous Carbohydrate Solutions at Different Temperatures and Concentrations. Int. J. Food Prop 10, 185–195. [Google Scholar]
- Thomsson E, Gustafsson L, and Larsson C. (2005). Starvation Response of Saccharomyces cerevisiae Grown in Anaerobic Nitrogen- or Carbon-Limited Chemostat Cultures. Applied and Environmental Microbiology 71, 3007–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thon VJ, Khalil M, and Cannon JF (1993). Isolation of human glycogen branching enzyme cDNAs by screening complementation in yeast. J. Biol. Chem 268, 7509–7513. [PubMed] [Google Scholar]
- Walls AB, Sickmann HM, Brown A, Bouman SD, Ransom B, Schousboe A, and Waagepetersen HS (2008). Characterization of 1,4-dideoxy-1,4-imino-d-arabinitol (DAB) as an inhibitor of brain glycogen shunt activity. J. Neurochem 105, 1462–1470. [DOI] [PubMed] [Google Scholar]
- Weitzel G, Pilatus U, and Rensing L. (1987). The cytoplasmic pH, ATP content and total protein synthesis rate during heat-shock protein inducing treatments in yeast. Exp. Cell Res 170, 64–79. [DOI] [PubMed] [Google Scholar]
- Wilson WA, Wang Z, and Roach PJ (2002). Systematic identification of the genes affecting glycogen storage in the yeast Saccharomyces cerevisiae: implication of the vacuole as a determinant of glycogen level. Mol. Cell. Proteomics 1, 232–242. [DOI] [PubMed] [Google Scholar]
- Wilson WA, Roach PJ, Montero M, Baroja-Fernández E, Muñoz FJ, Eydallin G, Viale AM, and Pozueta-Romero J. (2010). Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol. Rev 34, 952–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Shu W, Zeng C, Guo B, Shi J, Jing J, and Zhang X. (2019). A mitochondria targetable and viscosity sensitive fluorescent probe and its applications for distinguishing cancerous cells. Dyes Pigm. 168, 134–139. [Google Scholar]
- Yoshizawa T, Nozawa R-S, Jia TZ, Saio T, and Mori E. (2020). Biological phase separation: cell biology meets biophysics. Biophys. Rev 12, 519–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 A viscosity-controlled model reaction is temperature dependent in cell lysates but not live cells, Related to Figure 1. (A) BirA biotinylates a 15-AA target sequence called the “Avi-tag”. Labeling rates are determined by a gel shift assay in which addition of streptavidin to cell lysates causes the biotinylated species to run at a higher molecular weight than its nonbiotinylated counterpart. All reactions use cytosolic BirA and GFP-Avi as “bait” (B) Schematic depicting the Fluorescence Recovery After Photobleaching (FRAP) technique. GFP was bleached and the rate of its subsequent recovery to the bleached region was measured to determine the viscosities of solutions, cell lysates, and live yeast cells. Hypothetical recovery curves for solutions of higher and lower viscosities are shown on the left where recovery time is being measured by t-half, the time to reach half-maximal fluorescence recovery. Recovery can also be reported as the diffusion coefficient of GFP (C) Labeling of RPL19b-Avi by BirA in lysates produced from cells grown at 30°C (●blue), 37°C (◼orange), or heat shocked for 30 minutes at 45°C (◆red). All reactions were performed at room temperature (nonlinear regression difference between slopes, 30°C vs 37°C p=.006; 30°C vs 45°C p<.0001; 37°C vs 45°C p<.0001) Error bars are s.e.m.
Figure S2 Cells regulate intracellular viscosity in response to heat (“viscoadaptation”), Related to Figure 2. (A) FRAP measurements on cells expressing mKate2 20 minutes after cells were shifted from 22°C to 42°C (n=3 cultures/temp, <10 cells/culture)(unpaired t-test, p=.0271) (B) FRAP timecourse on cells expressing mKate2. Cells were shifted from 22°C (T0) to 42°C (n=3 timecourses). Error bars are s.e.m. Points without visible error bars have error within the size of the point.
Figure S3 Heat-induced accumulation of the carbohydrates glycogen and trehalose increases cellular viscosity, Related to Figure 3. (A) Cells were treated with 50 ug/ml cycloheximide (CHX) or vehicle (EtOH) for 10 minutes prior to and during heat shock at 42°C. FRAP measurements were performed beginning at 20 minutes after temperature shift. (unpaired t-test, No CHX 22°C vs No CHX 42°C p<0.0001; No CHX 22°C vs +CHX 42°C p<.0001; No CHX 42°C vs +CHX 42°C, p=0.169) (B) Glycogen (left) and trehalose (right) levels in cells grown at the indicated steady state temperatures. Values are normalized to the minimum value at 30°C. (left) (n≥5 measurements/ temperature) (unpaired t-test, 22°C vs 30°C, p=.023; 22°C vs 37°C, p=.204; 30°C vs 37°C, p=.017; 30°C vs 40°C, p=.0007; 37°C vs 40°C, p=.262; 22°C vs 40°C, p=.018); (right)(n≥4 measurements/temperature) (unpaired t-test, 22°C vs 30°C, p=.1350, 22°C vs 37°C, p<.0001, 30°C vs 37°C, p=.0025, 30°C vs 40°C, p=.0008, 37°C vs 40°C, p=.0097, 22°C vs 40°C, p=.0006) (C) Schematic depicting the technique for iodine staining of glycogen in live cells. (D) (left) Glycogen content normalized to cell wet weight after 42°C heat shock with and without 50 mM LiCl. (n=2) (unpaired t-test, p=.07) (below, left) Iodine staining for glycogen content after heat shock with and without LiCl. (right) Trehalose content normalized to cell pellet wet weight after 42°C heat shock with and without 50 mM LiCl (n=3) (unpaired t-test, p<.05) (E) Iodine staining for glycogen levels in live cells unstressed or heated at 42°C with or without DAB. (F) Cell area was measured from a cross-section of the cell, excluding vacuoles. The included area was defined by the distribution of GFP. Heat shock was at 42°C for 20 minutes. (n≥2 experiments, ≥10 cells/experiment) (unpaired t-test, WT 22°C vs WT 42°C, p=.2165; ΔGΔT 22°C vs ΔGΔT 42°C, p=.267; WT 22°C vs ΔGΔT 22°C, p=.767; WT 42°C vs ΔGΔT 42°C, p=.412; WT 22°C vs ΔGΔT 42°C, p=.049) (G) (left) Glucose measured in strains grown in steady state at the indicated temperatures (n≥ 3 measurements per condition) (unpaired t-test, p≥.05 for all pairwise comparisons) (right) Glucose was measured in the indicated strains at the indicated times after shift from 30°C to 42°C (≥ 3 measurements per condition) (unpaired t-test, WT T0 vs T5, p=.032; WT T0 vs ΔGΔG T0, p=.006; ΔGΔG T0 vs T40 p=.014; WT T0 vs WT T20, p=.1; WT T0 vs WT T40, p=.065) (H) In vivo BirA labeling of GFP-Avi. Wildtype cells (●) and cells lacking Glc3 (◼) were grown at 30°C (blue) and 37°C (red) (n=4 timecourses for WT, n=2 timecourses for ΔGlc3) (nonlinear regression difference between slopes, WT 30°C vs WT 37°C p=.107; WT 30°C vs ΔGlc3 30°C p=.006; WT 37°C vs ΔGlc3 37°C p=.01; ΔGlc3 30°C vs ΔGlc3 37°C p=.001) (I) Cells were treated with no drug, 50 mM LiCl, or 50 mM NaCl for one hour prior to temperature shift. FRAP measurements began 20 minutes after temperature increase. (n≥ 2 experiments, ≥10 cells/experiment)(unpaired t-test, 22°C no drug vs 42°C no drug p<.0001, 22°C no drug vs 42°C + NaCl p=.0003, 42°C + LiCl vs 42°C + NaCl p=.018; 22°C no drug vs 42°C + LiCl, p=.22) Error bars are s.e.m.
Figure S4 Viscoadaptation occurs in response to nutrient limitation, Related to Figure 4. (A) Cytosolic BirA labeling of RPL19b-Avi (top left), RPL16-Avi (top right), RPS2-Avi (bottom left), and RPS20-Avi (bottom right) in 2% glucose (blue) and 0% glucose (orange). Cells were grown in low biotin media (0.5 nM) to prevent background biotinylation. RPS20-Avi labeling timecourse was shortened due to faster labeling of this substrate (n=3 replicates/strain) (nonlinear regression difference between slopes, top left p<.0001; top right p<.0001; bottom left p<.0001; bottom right p<.0001) (B) Cells were grown in 2% glucose with 8 nM biotin (regular biotin, RB) or 0.5 nM biotin (low biotin, LB) and transferred to 0% glucose as indicated. FRAP measurements start 30 minutes after removal of glucose. (n≥ 2 experiments, ≥ 15 cells/experiment) (unpaired t-test, 2% glucose RB vs 2% glucose LB, p≥.05; 2% glucose LB vs 0% glucose LB, p=.0006; 2% glucose RB vs 0% glucose LB, p≥.05) (C) Iodine staining on cells grown in media with the indicated biotin concentrations and 2% glucose or 0% glucose for 30 minutes (D) Intracellular glucose (normalized to cell pellet weight) in cells in 0% glucose for 30 minutes with normal SD media or low biotin SD media. (n≥ 3 measurements/condition)(unpaired t-test, 2% glucose RB vs 2% glucose LB p=.016; 2% glucose RB vs 0% glucose RB p=.004; 2% glucose LB vs 0% glucose LB p=.036; 0% glucose RB v. 0% glucose LB p=.009) Error bars are s.e.m.
Figure S5 Glycogen and trehalose alter molecular movement in vitro, Related to Figure 5. (A) Microrheology on fluorescent beads in the indicated aqueous solutions (left) and cell lysates (right). Plotted are the mean slopes of the Moment Scaling Spectrum (SMSS). SMSS values for individual particle trajectories were averaged to obtain each value (n≥58 trajectories) (left) (unpaired t-test, H20 vs 70% sucrose p=.54; H20 vs 30% gly p<.0001; H20 vs 45% tre p<.0001; H20 vs 30%gly+45%tre p<.0001) (right) (unpaired t-test, H20 vs 22°C lysate p=.026; H20 vs 42°C lysate p=.052; H20 vs 42°C lysate ΔGΔT p<.0001; 22°C lysate vs 42°C lysate p=.145; 42°C lysate vs 42°C lysate ΔGΔT p=.64) (B) Aqueous droplets (4 mm diameter) of glycogen and trehalose were monitored over time at room temperature. Phase properties of droplets were assessed when the trehalose-only droplets solidified, characterized by fracturing in response to mechanical force (left). Droplets that distorted upon mechanical force without fracturing were described as “gel-like” (middle). Droplets that evaporated without undergoing any phase transition were characterized as “liquids” (right) (C) 40x confocal fluorescence images of purified GFP (~0.1 mg/mL final concentration) in lysates from cells grown in the indicated conditions prior to lysis.
Figure S6 Viscoadaptation affects phase separation and the solubility of biomolecules in vivo, Related to Figure 6. (A) Cells expressing Pab1-GFP were starved of glucose in 8 nM biotin (solid, regular biotin, RB) or 0.5 nM biotin (checkered, low biotin, LB) media and the percentage of cells containing Pab1 foci was determined for 3 time windows after onset of glucose starvation (n=4 experiments)(unpaired t-test, RB vs LB 0–20 p=.655; RB vs LB 20–40 p=.041; RB vs LB 40–60 p=.181) (B) Pab1-GFP in cells that were heat shocked at 42°C for 40 minutes (top) and then given 1 hour for the temperature to cool to 24°C (bottom) with and without the trehalase inhibitor Validamycin A. Images are taken at 40x. (C) Microrheology was performed on H20, lysates from unstressed cells grown at 30°C, and lysate from cells heat shocked at 45°C for 30 minutes. Each dot is one sample. (unpaired t-test, H20 vs unstressed lysate p=.004; H20 vs 45°C HS lysate p=.017; unstressed lysate vs 45°C HS lysate p=.659) (D) 10x brightfield images of dried lysate droplets from cells heat shocked at 45°C for 30 minutes undiluted (left) or diluted 1:50 with H20 (right). (E) (left) Trehalose measurements in the same conditions/strains (unpaired t-test, log WT vs log ΔReg1 p=.004; stationary WT vs stationary ΔReg1 p<.0001) (right) Iodine staining for glycogen content in WT and ΔReg1 strains in logarithmic growth (unstressed) or stationary phase. (F) Average t-half for GFP in WT (solid) and ΔReg1 (checkered) cells in log phase growth at 30°C (n≥2 experiments, ≥8 cells/experiment)(unpaired t-test, p=.0173) Error bars are s.e.m. (G) (left) Schematic depicting estimation of the mobile fraction (MF) from FRAP recovery curves. (right) Example curve for an MF of 1 (top) and 0.7 (bottom) indicating 100% and 70% fluorescence recovery, respectively.
Figure S7 Viscoadaptation occurs in cells with low ATP, Related to Figure 7. (A) (left) Schematic of the FRET-based ATP sensor(Bermejo et al., 2010) (right) Example of log-phase cells in 2% glucose expressing the ATP sensor. Boxes highlight high and low ATP cells in the same culture (B) Cells expressing the ATP sensor after 30 minutes in the indicated conditions (C) t-half values and ATP levels plotted for single cells expressing the ATP sensor (left) WT cells in 0% glucose with low biotin (0.5 nM) (R = −0.589 p<.0001) (right) ΔGlc3ΔGsy2 cells in 0% glucose with low biotin. Boxes highlight responders (red) and non-responders (blue) (R= −0.132 p=0.135)
Data Availability Statement
Raw data for this work are provided as a Mendeley dataset:
http://dx.doi.org/10.17632/n5cs6wjpjp.1
File contents are as follows:
Raw Data 1.zip: example FRAP movies
Raw Data 2.zip: FRAP raw data for t-half Determination using easyFRAP
Raw Data 3.zip: FRAP movies for diffusion coefficient determination using simFRAP