

Abstract
The sarcomere is the basic contractile unit of striated muscle and is a highly ordered protein complex with the actin and myosin filaments at its core. Assembling the sarcomere constituents into this organized structure in development, and with muscle growth as new sarcomeres are built, is a complex process coordinated by numerous factors. Once assembled, the sarcomere requires constant maintenance as its continuous contraction is accompanied by elevated mechanical, thermal, and oxidative stress, which predispose proteins to misfolding and toxic aggregation. To prevent protein misfolding and maintain sarcomere integrity, the sarcomere is monitored by an assortment of protein quality control (PQC) mechanisms. The need for effective PQC is heightened in cardiomyocytes as these cells are terminally differentiated and must survive for many years while preserving optimal mechanical output. To prevent toxic protein aggregation, molecular chaperones stabilize denatured sarcomere proteins and promote their refolding. However, when old and misfolded proteins cannot be salvaged by chaperones, they must be recycled via degradation pathways: the calpain and ubiquitin-proteasome systems, which operate under basal conditions, and the stress-responsive autophagy-lysosome pathway. Mutations to and deficiency of molecular chaperones and the associated factors charged with sarcomere maintenance commonly lead to sarcomere structural disarray and the progression of heart disease, highlighting the necessity of effective sarcomere PQC for maintaining cardiac function. This review focuses on the dynamic regulation of assembly and turnover at the sarcomere with an emphasis on the chaperones involved in these processes and describes the alterations to chaperones – through mutations and deficient expression – implicated in disease progression to heart failure.
Keywords: Sarcomere, Sarcomerogenesis, Protein quality control, Chaperone, Calpain, Ubiquitin, Proteasome, Autophagy
1. Introduction
The sarcomere is the fundamental molecular unit of contraction in striated muscle cells. It is a highly ordered structure containing myosin thick filaments and actin thin filaments, which directly engage to facilitate contraction. The myofilaments are accompanied by the contraction-tuning troponins (I, T, and C) and tropomyosin, which regulate the thin filament, as well as many associated signaling and adaptor proteins (Figure 1A). From its earliest histological description in the late 19th century [1], the composition and unique structural organization of the sarcomere have held a central focus in physiology. Long considered a rigid structure, with the advent of modern molecular biology techniques it has become clear in recent decades that assembly and turnover of sarcomere proteins is actually a highly dynamic process in constant flux [2–5]. The coordinated assembly of sarcomere proteins into organized paracrystalline structures in development (Figure 1B), and with muscle growth as new sarcomeres are added, is essential for heart function. Effective assembly alone, however, is insufficient, as the protein components of this contractile structure experience elevated stress and often denature, thus requiring regular turnover. Old and misfolded proteins must be swapped out for newly synthesized proteins without compromising mechanical function. Coordinating this task in cardiomyocytes, which contract persistently throughout the lifespan, is a challenging endeavor assisted by several protein degradation pathways and dozens of molecular chaperones. The need for effective sarcomeric protein quality control (PQC) increases under elevated stress conditions, such as occur over time with aging and the progression of heart disease with mechanical stress from increased afterload. Stress may also stem from mutations to sarcomeric proteins which promote misfolding and preclude adequate maintenance. Therefore, a mechanistic appreciation of the factors involved is warranted in order to characterize their functional significance for the sarcomere and manipulate their activity, toward the goal of targeting sarcomere PQC to forestall heart disease progression and restore cardiac function.
Figure 1. The cardiac sarcomere and assembly of the myofilaments.
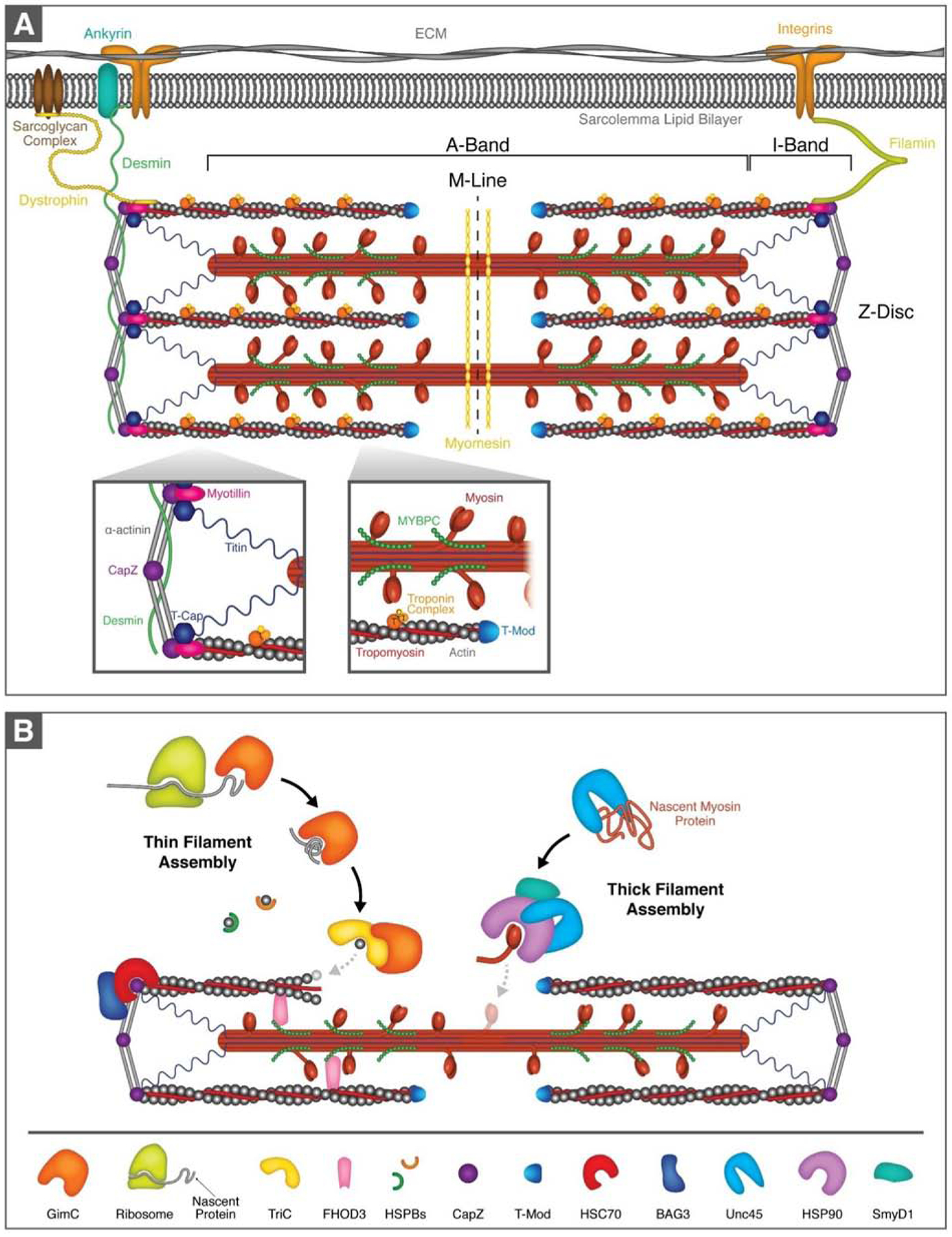
(A).Schematic diagram of the sarcomere and sarcomere-associated proteins. The sarcomere is the basic contractile unit in striated muscle with the thin actin and thick myosin myofilament proteins at its core. Sarcomeres are bounded at either end by α-actinin-rich Z-discs and are arranged sequentially to form myofibrils. (B). Simplified schematic of thin and thick filament assembly. GimC binds newly synthesized actin as it is translated by the ribosome and mediates early stages of folding. GimC passes now semi-folded actin to TriC, which assists in the assembly of the actin filament. Thin filament assembly is further supported by FHOD3, which stabilizes monomeric and filamentous actin and is crucial for actin filament nucleation and polymerization. The small heat shock proteins (heat shock protein B family, HSPBs) stabilize actin monomers and prevent protein aggregate formation. Actin capping proteins, CapZ and tropomodulin (T-Mod), bind to the ends of actin filaments and maintain stability. CapZ itself requires the BAG3/HSC70 chaperone complex to maintain its stability. Unc45 is the key chaperone for thick filament assembly and stability. It prevents myosin aggregation and assists with the folding of the myosin ATPase domain. Unc45 is a co-chaperone for HSP90, which participates in the early stages of myosin folding and myofibril assembly. The lysine methyltransferase SmyD1 is proposed to interact with the HSP90/Unc45/myosin complex.
Newly synthesized polypeptides require folding into their unique three-dimensional structures to achieve functional activity. Additionally, to maintain function and prevent cytotoxicity, proteins denatured due to stress or misfolding must be refolded or cleared from the cellular environment. Cardiomyocytes employ an assortment of molecular chaperones to ensure these vital tasks are accomplished. Chaperones promote proper protein folding, prevent protein misfolding through stabilizing interactions, and are required for the assembly of macromolecular complexes like the sarcomere [6]. They are essential at the dynamic sarcomere, where elevated stress conditions facilitate protein misfolding, and newly synthesized proteins must be rapidly incorporated. Unsurprisingly, mutations and deficiency of sarcomeric chaperones cause a failure of local PQC and aggregation of misfolded proteins, which can disrupt sarcomere structure, lead to muscle dysfunction, and ultimately heart failure.
The structure and composition of the sarcomere are highly conserved between skeletal and cardiac muscle, and from worms to mammals. As such, much of what we know about sarcomere PQC has been inferred from muscle studies in worms (C. elegans), flies (D. melanogaster), and fish (D. rerio) and will be included here. While there is reason to expect that these mechanisms translate to higher organisms, inferences should be made with care as critical differences may exist for species with longer lifespans and higher cardiac hemodynamic load. In this review, we summarize the current understanding of assembly and degradation processes at the cardiac sarcomere, discuss the chaperones and PQC pathways involved, and highlight emerging evidence for the dynamic remodeling processes that maintain sarcomere structure and function.
2. Sarcomere Assembly and Maintenance
2.1. Integrin-ECM interactions initiate myofibril assembly
Cardiomyocytes are efficiently organized into dozens of contractile substructures called myofibrils, which extend the length of the cell and are further organized into dozens of sarcomeres arranged sequentially. In developing myofibrils, mechanical tension communicated between myocyte cell surface proteins and the extracellular matrix (ECM) is required for sarcomere formation [7]. Integrins and their associated proteins are the first proteins to assemble with the regular periodicity characteristic of sarcomeres and mediate the membrane association of immature Z-disc structures, called Z-bodies [8–10]. These sites of high integrin concentration at the sarcolemma, termed protocostameres, recruit the Lim-domain binding protein ZASP/cypher which serves as a nucleation site for Z-body assembly [7,11,12]. The regional specificity of the integrin-rich protocostameres, which gives rise to the characteristic sarcomere periodicity, relies on their receptor counterparts in the ECM [13]. Genetic studies in invertebrates and a study of β1-integrin-KO mice support this and indicate myofibril assembly is integrin-dependent and requires specific ECM receptors [14–18]. Mutations to 18 proteins mediating myocyte-ECM interactions are linked to muscular dystrophy and cardiomyopathy through disruption of myocyte integrity [19–21].
2.2. Models of Sarcomerogenesis
The formation of sarcomeres (sarcomerogenesis) begins early in development as myofibrils form and continues in adult myocytes as they remodel with both physiological and pathological hypertrophy. However, the assembly mechanisms in hypertrophy remain poorly characterized due to limited approaches to assess mechanism in adult cardiomyocytes. Since it occurs in developing myocytes, more is known regarding sarcomere assembly compared with sarcomere turnover, as immature myocytes are more amenable to molecular biology approaches than adult myocytes, which rely heavily on maintenance. Even so, mechanisms of sarcomerogenesis remain controversial with multiple models proposed to explain this complex process.
2.2.1. The Titin Template Model
Titin is the largest known protein and extends from the sarcomere Z-disc to M-line (Figure 1A). The Titin Template Model proposes titin serves as a scaffold for sarcomerogenesis, establishing sarcomere length and mediating thick filament integration. It was initially proposed due to titin’s length of exactly one-half sarcomere and on evidence that titin associated with Z-bodies and assembled prior to myosin filaments [22,23]. Early evidence for the model was largely correlative, but support soon followed showing A-band titin assembly preceded myosin incorporation [24,25]. Further evidence came from a knockout study of titin, where myofibril assembly and thick filament formation were impaired [26]. Subsequent studies in developing myocytes used targeted deletion of C-terminal titin domains and found disrupted sarcomerogenesis [27–29]. The complexity of titin’s role is enhanced by the recent finding that absence of the Cronos/T2 titin isoform, which lacks C-terminal titin domains, also causes sarcomere disarray [30].
Despite the prevalence of “titin template” in myofilament nomenclature, in the decades since this model was proposed several lines of evidence have refuted its accuracy. For one, titin is not required for Z-body formation, as these α-actinin accumulations assemble prior to titin expression and titin I-band deletion does not impact Z-disc structure [31,32]. While titin may mediate thick filament integration, it is not required for assembly of the thick filament as myosin fibrils can form without titin [33]. It was also noted that invertebrate sarcomeres form despite their titin analogs only spanning the A-band region [34]. Thus, we speculate that titin is required for maintaining sarcomere spacing, but not for initial patterning of the myofibril.
2.2.2. The Stitching Model
First described by Lu et al., in the Stitching Model elements of the A-band (myosin fibers, myomesin) and I-Z-I bands (α-actinin, actin fibers) assemble independently and are then “stitched” together by titin, which joins the two large subunits through interactions with myomesin and α-actinin [35,36]. This is supported by electron microscopy observations denoting independent sarcomere subunit assembly, where Z-discs with associated thin filaments formed in myosin-deficient muscle and A-band formation occurred in the absence of thin filament components [35,37,38]. Further work in Drosophila found that myosin filaments formed when various Z-disc components were deleted [39]. This study proposed that rather than sequential assembly of the sarcomere, multiple latent protein complexes are built simultaneously and then combined. Unfortunately, with its many moving pieces this model is challenging to assess using fluorescence-based approaches in live cells and is therefore largely based on observations in fixed preparations.
2.2.3. The Premyofibril Model
The Premyofibril Model is the prevailing model of sarcomere assembly. In this model, stress fiber-like structures composed of α-actinin, actin, and non-muscle myosin-II (NMM-2) assemble at the cell periphery [32]. These premyofibrils mature into nascent myofibrils, where NMM starts to be replaced by muscle myosin, and then into mature myofibrils containing only muscle myosin [32,40]. Prevention of NMM and muscle myosin co-polymerization in mature myofibrils is attributed to myosin binding protein C (MYBPC), which assembles only in the mature myofibril [32]. In support of the Premyofibril Model, deletion of NMM mRNA in myoblasts prevents actin stress fiber polymerization, and mice with germline KO of the NMM-2B isoform display significant cardiac structural defects and die in development [41–43]. The Premyofibril Model is not without contention, however, as sarcomerogenesis is unimpaired with cardiomyocyte-specific deletion of NMM-2B or NMM-2A [44,45]. Some suggest the two NMM isoforms serve overlapping functions allowing the cell to compensate if one is mutated or deficient, which is supported by studies in C. elegans [46–48]. Notably, NMM-2A expression increases in NMM-2B-deficient mice [41]. While such redundancy may explain the mouse single isoform KO results, whether NMM-2 isoforms have functional overlap in higher organisms or cardiomyocytes is not known.
A recent study of sarcomerogenesis in iPSC-cardiomyocytes suggested that aspects of each model are correct and proposed a unifying model of sarcomere assembly. This study found sarcomeres assemble directly from actin stress fiber templates and that assembly requires both NMM-2 isoforms and the actin nucleator FHOD3 [49].
2.3. Thin filament assembly and stability
As α-actinin is recruited to the maturing Z-disc by ZASP, it is organized into its lattice structure by nebulin-related anchoring protein (NRAP) [50]. With Z-discs in place, thin filament assembly proceeds with assistance from numerous chaperones (Figure 1B).
2.3.1. GimC & TRiC
As it is translated, nascent actin is bound by GimC (prefoldin), which promotes actin folding and prevents actin monomer aggregation [51]. GimC-bound actin is then targeted to TRiC (CCT), a general chaperone that localizes to the Z-disc [52]. TRiC is responsible for the final actin folding steps, and assists with actin polymerization [53–55]. Zebrafish with missense TRiC mutations develop normally but have impaired actin folding that precludes sarcomere assembly [52]. Mutations to TRiC are associated with heart failure in humans and knockout of TRiC in Drosophila resulted in disorganization of actin and myosin fibers [56,57]. However, whether this is due to impaired actin folding/incorporation into the thin filament is not known. TRiC has many different clients and has been proposed to interact with up to 10% of the proteome, including myosin [58].
2.3.2. FHOD3
Recent studies have identified a role in thin filament assembly for the formin family member FHOD3, which localizes to the sarcomere A-band [49]. Localization of FHOD3 to the A-band is dependent on MYBPC [59]. FHOD3 binds to both monomeric and filamentous actin and facilitates thin filament nucleation and polymerization; effects that are enhanced with GimC present [60]. As previously discussed, FHOD3 localizes with NMM in developing myofibrils and is required for de novo sarcomere assembly and organized A-band formation [49]. Notably, FHOD3 KO does not completely prevent myofibril formation, but impairs cardiac development and causes sarcomere disarray [61–63]. Mice expressing a mutant FHOD3 with disrupted actin binding die by embryonic day 12, however, expression of wild-type FHOD3 in these embryos rescued myofibril maturation by acting as an actin filament “reorganizer” at the center of the sarcomere [64]. A FHOD3 mutation causes dilated cardiomyopathy and FHOD3 expression decreases in human heart failure, likely one of many deficits contributing to the sarcomere disarray characteristic of the failing heart [65,66].
2.3.3. Regulating Actin Polymerization
Once the thin filament is formed, its length is dynamically regulated by two reciprocal factors, profilin and cofilin. The cofilin family of proteins are actin disassembly factors expressed in all eukaryotic cells [67]. Cofilin-2 is the isoform expressed in striated muscle and mediates thin filament turnover by promoting disassembly of actin at the pointed end [68,69]. Cofilin-2 binds to both ADP- and ATP-actin filaments, which it cleaves to provide actin monomers for new filament assembly [70–72]. Both cofilin-2 mutations and cofilin-2 deficiency are associated with cardiomyopathy [73–75]. Profilin catalyzes ADP to ATP exchange on monomeric actin and acts opposite to cofilin to enhance thin filament polymerization [76,77]. Profilin ultimately passes monomeric actin to FHOD3 for filament incorporation, though the site at which this occurs – Z-disc vs. A-band – is not clear [78,79].
Thin filament stability is supported by the actin capping proteins CapZ and tropomodulin. CapZ binds to the barbed end of the actin filament at the Z-disc and is critical for cardiac performance [80]. Inhibition of CapZ impairs myofibrillogenesis by disrupting actin filament assembly [81]. Tropomodulin binds to the pointed end of the actin filament and prevents polymerization. Tropomodulin knockout impairs sarcomere development and is embryonic lethal [82–84]. Its function is antagonized by Leiomodin, a structurally similar actin nucleating protein that is expressed later in the developing myofibril and maintains thin filament pointed end dynamics [85,86]. The stability of actin capping proteins is essential for sarcomere structural maintenance. This is emphasized by knockdown of the co-chaperone Bcl-2-associated athanogene 3 (BAG3) in neonatal ventricular myocytes, which caused rapid degradation of CapZ and sarcomere disarray [87].
2.3.4. The small heat shock proteins (HSPBs)
The HSPBs are ATP-independent chaperones and therefore do not have intrinsic folding activity. Instead, they operate as “holdases” for misfolded proteins to prevent protein aggregation and connect clients with the ATPase chaperones HSP70 and HSP90 [88]. Three HSPBs are implicated in actin stability: HSPB1 (HSP27), αB-crystallin (HSPB5), and HSPB7. Morpholino inhibition of HSPB1 translation in Xenopus embryo cardiac muscle impaired actin filament organization and caused myofibril assembly defects [89]. αB-crystallin also associates with actin and prevents formation of heat stress-induced actin aggregates [90–92]. HSPB7 binds to monomeric actin and inhibits actin polymerization, which likely prevents actin aggregation in vivo [93]. Cardiomyocyte-specific KO of HSPB7 resulted in abnormally long thin filaments with mislocalization of tropomodulin and was embryonic lethal [93]. The severe phenotypes in the KO studies suggest that the HSPBs serve non-overlapping roles where those still expressed are unable to compensate.
2.4. Thick filament assembly and stability
Contraction of the sarcomere is mediated by the molecular motor myosin, which ratchets along actin filaments by converting energy from ATP into mechanical work. Folding myosin polypeptides into their functional conformation, and assembly into the hexameric myosin complex found only at the sarcomere, requires chaperones (Figure 1B).
2.4.1. Unc45
Unc45 is the key myosin chaperone and is functionally conserved from worms to mammals. The association of Unc45 with myosin is mediated by the Unc45 c-terminal domain and prevents myosin aggregation [94,95]. Numerous studies show Unc45 is essential for myosin maturation, specifically for folding the myosin ATPase domain [96–104]. However, Unc45 expression must be tightly regulated as both knockdown and overexpression result in defective myofibril organization [105]. The association of Unc45 with myosin is enhanced with heat shock in vitro, suggesting denaturation-dependent binding [96,106]. A similar effect has been shown for αB-crystallin, which prevents heat shock-induced myosin aggregation [107]. A study in zebrafish found that physical cell stress triggers mobilization of Unc45 (and HSP90) to the sarcomere A-band from a reserve pool at the Z-disc that is regulated by differential affinity of Unc45 for denatured myosin [108]. The presence of a readily available myosin chaperone pool at the sarcomere facilitates a rapid response to muscle damage. Bi-allelic mutations in Unc45 are associated with congenital myopathy and Unc45 mutants impair sarcomerogenesis in zebrafish [109,110]. Notably, Unc45 mutations have not been found to cause cardiomyopathy. While Unc45 is fundamental for muscle function in lower organisms, it may be that other myosin chaperones are able compensate for impaired Unc45 activity in mammals.
2.4.2. HSP70 & HSP90
HSP70 and HSP90 are general chaperones each with a wide range of clients. Newly synthesized myosin is stabilized by forming a complex with HSP70 or HSP90, where the chaperones participate in early stages of myosin folding and myofibril assembly [111]. Less is known regarding HSP70-mediated stabilization of myosin and it likely plays a secondary role to HSP90. Both HSP90 mutations and deficiency are associated with significant sarcomere disarray [98,112,113]. While impaired myosin assembly contributes to the phenotype, careful interpretation is warranted as HSP90 is also involved in quality control of other sarcomere proteins, including titin [114–116]. The myosin folding activity of HSP70 and HSP90 are enhanced with Unc45, which interacts with HSP70/90 through its co-chaperone helix-turn-helix motif, though this association is dispensable for myosin folding in C. elegans [117,118]. A recent study in skeletal muscle indicated that another HSP70 co-chaperone, BAG3, also assists with myosin stabilization [119]. This suggests that, while Unc45/HSP90 is the primary myosin chaperone complex, these complexes can exist in at least one other chaperone/co-chaperone combination.
2.4.3. SmyD1
SmyD1 is a histone methyltransferase implicated in myofibril assembly. Though not a confirmed chaperone, SmyD1 localizes to the sarcomere M-line where it interacts with myosin and plays an essential role in thick filament integration [120–122]. A study in zebrafish found SmyD1 knockdown impaired myofibril maturation, disrupted heart contraction, and prevented swimming activity [123]. Knockdown of SmyD1 also inhibits myosin accumulation in the early sarcomere, causing instability and rapid myosin degradation. This effect could be due to disruption of the Unc45/HSP90/myosin complex, with which SmyD1 associates. In a probable attempt to compensate for myosin instability, Unc45 and HSP90 expression increase with SmyD1 deficiency [124]. These data suggest SmyD1 is a third member of the canonical myosin chaperone complex. SmyD1 levels are depressed in end-stage heart failure and a recently identified de novo SmyD1 mutation causes hypertrophic cardiomyopathy [125,126]. These studies add to the growing appreciation for the diversity of chaperone complexes that regulate thick filament assembly.
2.5. Maintaining titin and desmin stability
The factors maintaining titin stability are largely undefined but are known to include αB-crystallin. At physiological sarcomere lengths, αB-crystallin binds to the N2B region of titin in the I-band of the sarcomere and prevents ischemia/stretch-induced misfolding [127,128]. In overstretched myofibrils, αB-crystallin also binds to and stabilizes the I26/I27 Ig region of titin between the N2B domain and Z-disc titin [127]. This suggests increased affinity of αB-crystallin for the Ig region when it is mildly denatured. Higher stretching forces are required to denature titin when αB-crystallin is present and increased stiffness due to less distensible titin in failing ventricular tissue is reversed by introducing αB-crystallin [127,129]. Notably, a missense mutation in αB-crystallin (R157H) that causes familial dilated cardiomyopathy prevents association of αB-crystallin with titin [130]. The chaperones HSPB1 and HSP90 are also involved in titin maintenance and translocate to titin to prevent toxic aggregation in response to stress caused by genetic mutations to sarcomere proteins [115]. More research is needed to determine if these chaperones cooperate with αB-crystallin or act independently, and to identify additional chaperones involved in titin maintenance.
αB-crystallin is also the key chaperone for desmin, an intermediate filament protein involved in mechanotransduction that associates with the Z-disc, sarcolemma, and multiple organelles [131]. Nearly 70 desmin mutations and numerous αB-crystallin mutations have been linked to cardiomyopathy [132,133]. αB-crystallin binds to misfolded desmin and prevents aggregation [134]. The association of αB-crystallin with desmin in vitro increases with heat stress and pH changes, indicating stress/denaturation-dependent binding [91]. The R120G αB-crystallin mutation has received a lot of attention due to the severe phenotype it causes. This mutation decreases the dissociation constant for desmin by half and causes toxic protein aggregation, which leads to desmin-related cardiomyopathy [134–138]. Desmin- and αB-crystallin-containing protein aggregates are common features of cardiomyopathy and skeletal muscle myopathy tissue biopsies, even when mutations are not involved [139]. These data indicate that both disrupted stability and degradation of desmin can be pathogenic.
2.6. Maintenance of MYBPC
MYBPC is a flexible, thick-filament associated protein that regulates myosin crossbridge cycling [140]. Mutations to MYBPC are the leading cause of hypertrophic cardiomyopathy (HCM), accounting for up to 50% of cases [141]. Disease mechanisms hypothesized in HCM include: 1. Point mutations in sarcomere proteins that directly impair function (“poison peptide”), and 2. Protein haploinsufficiency caused by truncating mutations [142]. Unlike disease-associated mutations in other sarcomere proteins which are predominantly missense, truncations account for ~90% of MYBPC mutations [143]. Interestingly, studies of HCM biopsy samples from patients with MYBPC mutations failed to identify the truncated protein, suggesting that the protein either is not synthesized or is quickly degraded via PQC mechanisms [144]. In support of the latter, recent work by Glazier et al. found that HSC70 and HSP70 act as chaperones for both wild-type and mutant MYBPC where knockdown of HSC70 in cultured ventricular myocytes impaired their degradation [145]. However, wild-type and mutant MYBPC turnover was enhanced with a small molecule activator of HSP70/HSC70. In cases of missense MYBPC mutants, the role of Hsp70 in regulating MYBPC degradation may represent one mechanism in place to prevent the “poison peptide” effect. To date, HSP70/HSC70 is the only identified MYBPC chaperone, though future work may aim at determining the involvement of known HSP70 cochaperones in MYBPC turnover.
3. Sarcomeric Degradation
The regenerative capacity of cardiomyocytes is severely limited [146]. Sarcomere proteins, however, have half-lives on the order of weeks to days and require continuous turnover [147,148]. Sarcomeric protein turnover is thus essential to maintain function and promote cardiomyocyte longevity. In this section, we review the three mechanisms regulating sarcomere protein turnover (the calpains, the ubiquitin proteasome system, and the autophagy-lysosome system, Figure 2A) and discuss how their dysregulation in heart failure contributes to proteotoxicity and potentially sarcomere dysfunction (Figure 2B).
Figure 2. Sarcomeric protein degradation in the healthy and failing heart.
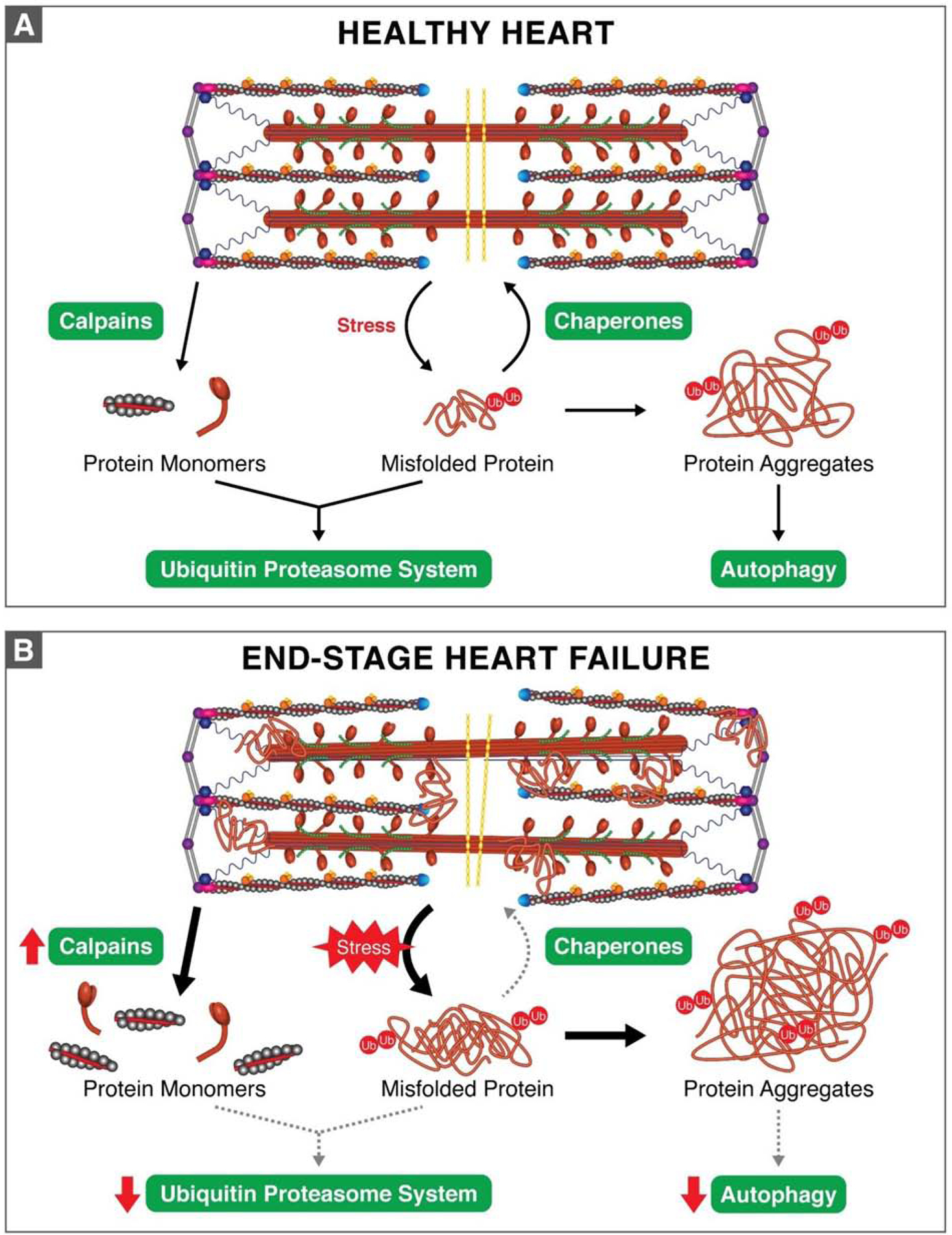
(A). Schematic representation of sarcomere PQC in the healthy heart. Three protein degradation pathways are in place for the sarcomere. The calpains proteolyze sarcomere proteins and release protein monomers to be degraded by the ubiquitin proteasome system (UPS). The UPS also mediates the degradation of individual misfolded proteins. Larger protein aggregates are removed by the autophagy/lysosome pathway. (B). Schematic representation of the changes to sarcomere PQC that occur in the end-stage failing heart. Proteins misfolded from mechanical (stemming from increased afterload), thermal, oxidative, and genetic stress aggregate in the end-stage failing heart. Aberrant protein misfolding as a result of these stressors is accompanied by elevated calpain activity, which produces excess proteolysis products. Compounding the issue of proteotoxicity, the activity of the systems designed to recycle these proteins, the UPS and autophagy, are downregulated in heart failure. Together, these factors culminate in toxic levels of protein aggregation, which – presumably – contribute to the sarcomere structural disarray and mechanical dysfunction commonly found in failing myocardium. Which specific proteins aggregate and to what extent remains to be fully elucidated.
3.1. The calpain system
Calpains are calcium-dependent cysteine proteases evolutionarily conserved from bacteria to mammals. The human genome contains at least 16 different calpain-encoding genes, three of which – calpain-1, calpain-2, and calpain-3 – are found in striated muscle [149,150]. Calpain-3 expression is skeletal muscle-specific [151]. Cardiomyocytes do not have a tissue-specific calpain and instead express modest amounts of calpains 1 and 2, each with many identified sarcomeric clients (Table 1) [152]. The susceptibility of sarcomere proteins to calpain proteolysis can be regulated by chaperones, which prevent calpain proteolysis by binding to cleavage sites. This was shown for HSPB1, which translocated to the Z-disc during oxidative stress and prevented calpain-mediated proteolysis of desmin [153]. HSPB1 binding can also prevent calpain cleavage of troponins I and T [154]. Calpain proteolysis of myofilament proteins is enhanced with sarcomere stretch [155].
Table 1.
Summary table of sarcomere and sarcomere-associated proteins with their proposed degradation mechanisms.
| Protein | Function | Proposed Degradation Mechanisms |
|---|---|---|
| Actin | Thin filament | TRIM32 [174] |
| α-Actinin | Actin cross-linking | Fbxl22 [186], TRIM32 [175] |
| Ankyrin | Cytoskeletal adaptor | Calpains [204,223] |
| Desmin | Cytoskeletal adaptor | Calpains [153,224], TRIM32 [175,176], Atrogin-1 [187], Cullin-5 [184,185] |
| Dystrophin | Cytoskeletal adaptor | Calpains [149,225] |
| FHL2 | Sarcomere/nucleus crosstalk | MuRF-3 [168] |
| Filamin A and C | Actin cross-linking | Calpains [226,227], Cullin-5 [184,185], Fbxl22 [186], MuRF-3 [168], CASA [196] |
| MYBPC | Thick filament regulation | Calpains [228,229], MuRF-1 [166] |
| β-MHC | Thick filament | MuRF1–3 [164,166], CHIP [178] |
| MLC-2 | Thick filament regulation | MuRF1–2 [164] |
| Myotilin | Z-disc integrity | MuRF1–2 [164], TRIM32 [175] |
| Nebulin/Nebulette | Thin filament stability | Calpains [230,231], MuRF1–2 [164] |
| NRAP | Thin filament stability | MuRF1–2 [164] |
| Telethonin/T-cap | Titin stability | Mdm2 [182], MuRF1–2 [164] |
| Titin | Strain-sensing, scaffold | Calpains [149,232], MuRF1–2 [164] |
| Tropomodulin/T-mod | Thin filament stability | Calpains [233] |
| Troponin C | Thin filament regulation | MuRF-1 [165] |
| Troponin I | Thin filament regulation | Calpains [208,211,234,235], MuRF1–2 [164,165], TRIM32 [175], c-Cbl [188] |
| Troponin T | Thin filament regulation | Calpains [208,234,235], MuRF1–2 [164], TRIM32 [175] |
| Tropomyosin | Thin filament regulation | Calpains [149,230], TRIM32 [175] |
Calpain activation is associated with an increase in protein ubiquitination and is thought to precede myofilament protein degradation by the ubiquitin-proteasome system (UPS), which requires proteins to be in monomeric form. In this model, calpains release the myofilament proteins from the sarcomere by proteolyzing several sarcomere components, allowing for their subsequent targeting to the UPS (Figure 2A) [156,157]. This observation was first made in the heart by Galvez et al. who observed that overexpression of calpain-1 resulted in increased ubiquitination and 26S proteasome activity while expression of the calpain inhibitor calpastatin decreased ubiquitination and caused cardiac dysfunction [158]. These findings suggest calpain proteolysis at the sarcomere is upstream of the UPS and is fundamental for cardiac function. However, the regulation of the UPS by calpains is incompletely understood and not all studies support the requirement for calpains upstream of the UPS [159]. More research is needed to characterize the calpain-UPS relationship at the sarcomere.
3.2. The Ubiquitin Proteasome System
UPS-mediated protein degradation is a concerted effort by multiple enzymes that culminates in a reaction catalyzed by an E3 ubiquitin ligase, which transfers ubiquitin to the protein of interest [160]. The ubiquitinated protein is subsequently degraded via the 26S proteasome, a large ATP-dependent protease complex with multi-catalytic activity that localizes to the sarcomere Z-disc [5]. Evidence from many studies indicates the UPS is fundamental for sarcomeric PQC and we focus our review on the E3 ligases found at the sarcomere.
3.2.1. The muscle RING finger E3 ligases (MuRFs)
The most well-characterized E3 ligases in striated muscle are the muscle RING finger (MuRF) proteins, which belong to the TRIM family of ubiquitin ligases. These localize to the sarcomere Z-disc and M-line and contribute to the proteasomal degradation of multiple sarcomere proteins [161–163]. MuRF-1 and MuRF-2 are functionally similar and associate with troponins I and T, NRAP, β-MHC, myosin light chain 2 (MLC2), telethonin (T-Cap), myotilin, titin, and nebulin [164–167]. MuRF-3 was the first MuRF to be discovered, however, it has only three identified sarcomere substrates: filamin C, β-MHC, and four-and-a-half LIM domain protein-2 (FHL2) (Table 1) [161,168,169]. Genetic mouse studies have contributed significantly to our understanding of the roles MuRFs serve in the myocardium. Absence of MuRF-2 or MuRF-3 caused protein aggregation and impaired cardiac function, while overexpression of MuRF-1 in a TAC heart failure model was detrimental [170–172]. Uniquely, when mice lacking MuRF-1 were challenged with TAC, they developed substantial hypertrophy which was not found in MuRF-2-deficient mice [173]. Together, these studies support that sarcomeric MuRFs share some redundancy and are fundamental for muscle structure and function but require tight regulation.
3.2.2. TRIM32
Another TRIM family member, TRIM32, is involved in the UPS-mediated degradation of thin filament-associated proteins. Work by Kuryashova et al. found TRIM32 binds to the myosin head/neck region and mediates the ubiquitination of sarcomeric actin [174]. More recently TRIM32 was shown to ubiquitinate the thin filament-associated proteins troponin T, troponin I, tropomyosin, myotilin, and α-actinin [175]. Studies of TRIM32 in muscle atrophy show that it is also an important mediator of desmin turnover, where TRIM32 knockdown prevented desmin degradation [175]. It seems TRIM32 is involved in an initial step of muscle atrophy requiring the disassembly of desmin filaments [176]. To date, most studies on TRIM32 have been performed in skeletal muscle, where TRIM32 mutations are associated with myofibrillar myopathy [174]. Despite high expression in the myocardium, very little is known regarding cardiac TRIM32. However, recent work with a mouse heart failure model indicated that TRIM32 has protective effects as TRIM32 overexpression prevented progression into heart failure in mice subjected to TAC [177]. More studies are needed to determine whether TRIM32 substrates identified in skeletal muscle are conserved in the heart.
3.2.3. Other E3 ligases at the sarcomere
Several other E3 ligases have been linked to sarcomere PQC, with more surely yet to be identified. One of these is carboxyl-terminus of HSP70-interacting protein (CHIP). CHIP localizes to the sarcomere Z-disc and A-band and assists with myosin turnover [178]. CHIP also mediates the degradation of the myosin chaperone Unc45 via the UPS [179]. Mice lacking CHIP have decreased survival and worsened myocardial injury following ischemia/reperfusion [180]. The E3 ligase murine double minute 2 (Mdm2) is canonically involved in the turnover of p53 [181]. However, in the heart it also ubiquitinates the titin-capping protein telethonin promoting its degradation [182]. Conditional knockout of Mdm2 in the heart causes spontaneous cardiac hypertrophy and significant cardiomyocyte cell death, though this is attributed to impaired degradation of a number of Mdm2 substrates [183]. Mdm2 gene expression decreases in heart failure [183]. Studies in recent years implicate several other E3 ligases in sarcomere PQC. These include Fbxl22 (α-actinin, filamin C), atrogin-1 (desmin), c-Cbl (troponin-I), and cullin-5/Asb2 (filamin A, filamin C, desmin) [184–188]. The number of E3 ligases involved in sarcomere protein turnover (Table 1) highlights the complexity of this process and suggests that proper turnover is achieved by engaging many E3 ligases with somewhat overlapping roles.
3.3. The Autophagy-Lysosome Pathway
Proteins that cannot be refolded by chaperones or processed by the UPS may be degraded via macroautophagy, referred to hereafter as ‘autophagy’ (Figure 2A). In autophagy, membrane-enclosed vesicles (autophagosomes) are formed around a portion of the cytosol containing damaged organelles, protein aggregates, and/or other toxic cellular components [189]. Like the UPS, selectivity in autophagy is mediated by ubiquitin. Proteins carrying a K63-polyubiquitin tag, or aggregated proteins with K48-polyubiquitination, are recognized by adaptors P62/SQSTM1 and NBR1 which facilitate their association with proteins on the autophagosome membrane, or HDAC6 which first targets them to the aggresome [190,191]. Autophagosomes recycle their cargo by fusing with the lysosome, thus exposing autophagosome contents to lysosomal hydrolases. While it is evident from several studies that autophagy is required for sarcomere structural maintenance, relatively little is known regarding the specific involvement of autophagy in sarcomere PQC.
Impaired autophagy is associated with sarcomere structural abnormalities. Autophagy related protein 5 (ATG5) is essential for the formation of the autophagosome membrane and its knockout completely ablates autophagy [192]. One of the first studies to implicate autophagy in sarcomere PQC was by Nakai et al. using cardiomyocyte specific ATG5 knockout [193]. Baseline cardiac function was unaffected in ATG5-null mice. However, after one-week of pressure overload, ATG5 KO caused significant left ventricular dysfunction, chamber dilation, and sarcomere structural disarray [193]. Sarcomere disarray was also identified with ATG7 KO in skeletal muscle [194]. These studies indicate the importance of autophagy for the sarcomere, but do not specifically assess autophagy at the sarcomere level as autophagosome impairment affects global myocyte autophagy.
Chaperone-assisted selective autophagy (CASA), an autophagy pathway discovered in skeletal muscle, is thus far the only autophagy pathway that has been explicitly shown to operate at the sarcomere (Figure 3A). In CASA, the co-chaperone BAG3 operates as a scaffold for HSPB8 and HSC70/HSP70. The complex binds to misfolded filamin C, preventing its aggregation. Filamin C is then ubiquitinated by the E3 ubiquitin ligase CHIP, which docks on the carboxyl terminus of HSP70. This promotes client identification by the ubiquitin receptor P62/SQSTM1, which with synaptopodin-2 (SYNPO2) facilitates the association and engulfment of the complex by the autophagosome [195]. CASA was first described in Drosophila, where the complex was shown to localize to the sarcomere Z-disc and mediate the degradation of mechanically-misfolded filamin C [196]. While it has not been shown directly, it is possible that multiple HSPBs are interchangeable in the CASA complex (Figure 3B). This is supported in that multiple HSPBs are established BAG3 binding partners (HSPB1, HSPB6, HSPB8, αB-crystallin), some of which have also been implicated in filamin C maintenance [197–199]. Numerous BAG3 mutations and BAG3 deficiency have been linked to dilated cardiomyopathy in humans [200,201]. Developing work from our group has identified the CASA complex at the sarcomere in cardiomyocytes and found it helps maintain sarcomere proteostasis [202]. Future research is needed to determine the extent to which CASA operates in cardiomyocytes and whether it has additional sarcomeric clients beyond filamin C.
Figure 3. Chaperone-assisted selective autophagy at the sarcomere.
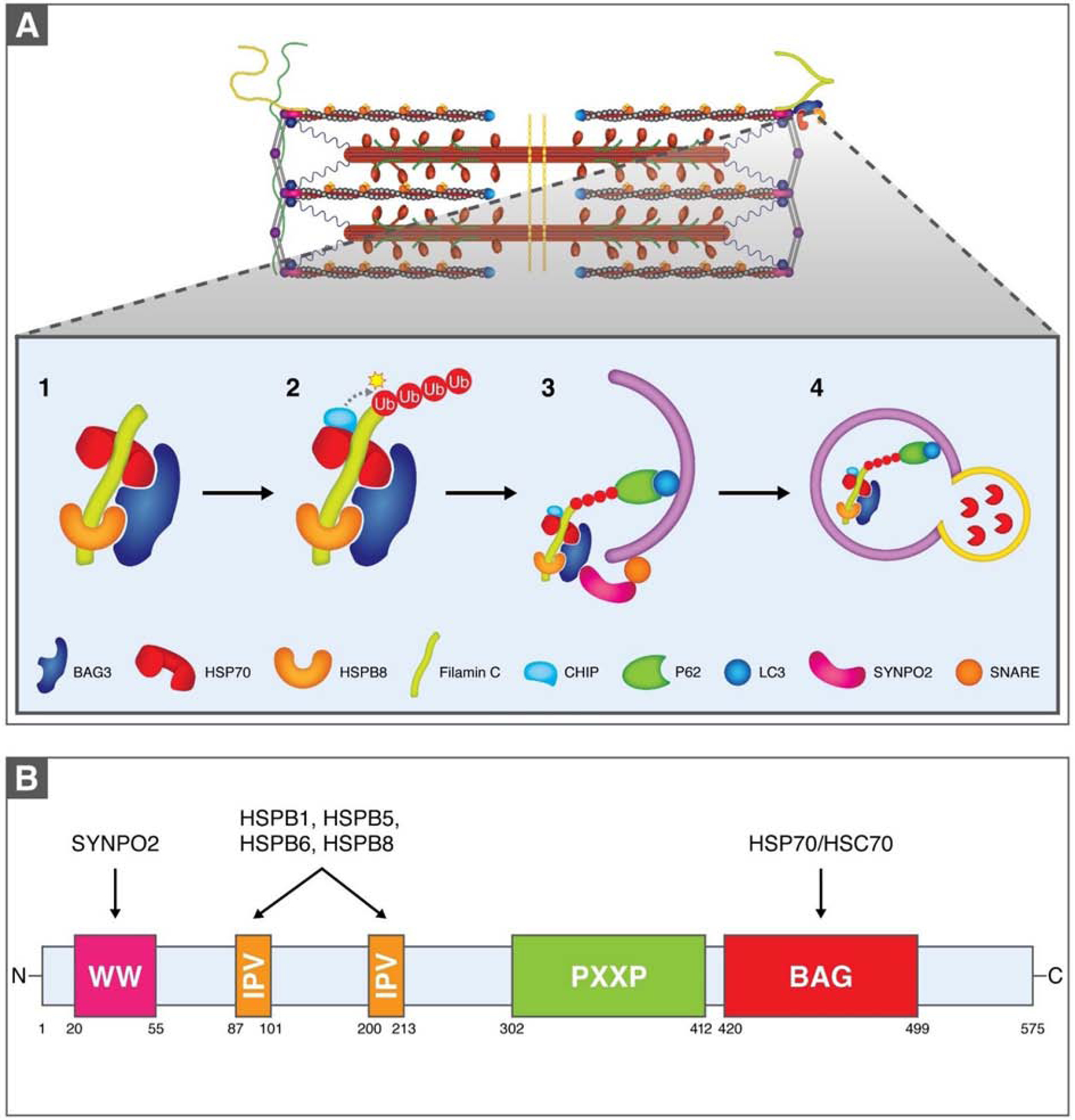
(A). The co-chaperone BAG3 operates as a scaffold for HSP70 and HSPB8, which bind to misfolded filamin C. The E3 ligase CHIP then ubiquitinates filamin C, allowing it to be recognized by the ubiquitin receptor P62, which facilitates the association of the CASA complex with LC3 on the autophagosome membrane. Through BAG3 this complex also associates with SYNPO-2, which mediates interaction with a SNARE protein on the autophagosome membrane that assists with autophagosome formation. Autophagosome contents are recycled via autophagosome-lysosome fusion. (B). BAG3 domains enable numerous binding activities. SYNPO2, which assists in autophagosome formation, binds to the N-terminal WW domain. Several HSPBs bind to two IPV motifs and the BAG domain associates with the ATPase domain of HSP70/HSC70.
3.4. Sarcomere Turnover in End-Stage Heart Failure
In end-stage heart failure, there is a dysregulation of PQC mechanisms resulting in old/misfolded protein aggregation that may cause sarcomere structural and functional impairment (Figure 2B). Calpain activity increases in both ischemic and dilated cardiomyopathy, causing an elevation in clients destined for degradation by the UPS [150,201–210]. However, in the end-stage of heart failure there is diminished UPS activity and thus the clients are not degraded rapidly enough [213]. In the healthy heart, protein aggregates not removed by these first two lines of defense can be removed by autophagy. Autophagy operates at low basal levels but is upregulated in response to proteasome-insufficiency, elevated mechanical strain – such as occurs with increased afterload –, and other stress conditions including those associated with genetic mutations to sarcomere proteins [214]. However, autophagy activity is dysregulated in heart failure [215]. Together, this dysregulation of sarcomeric PQC mechanisms in the progression of heart disease results in aberrant protein aggregation that may result in impaired contractile function and ultimately cell death.
4. Clinical Implications
Manipulating sarcomere PQC separately from global cardiomyocyte PQC is not feasible given our current understanding. However, as we gain mechanistic insight into the signaling pathways that mobilize chaperones from the cytosolic pool to the sarcomere, small molecule approaches targeting sarcomere PQC may become a reality. For now, manipulating global cardiomyocyte PQC to affect changes at the sarcomere appears the best option. Boosting HSP expression by exercise, caloric restriction, or small molecule therapeutics has proven cardioprotective in animal models of heart failure [216,217]. These therapies mostly target heat shock transcription factor-1 (HSF1), the master regulator of HSP expression [218]. The expected benefits of increasing HSP expression for the sarcomere extend from ensuring effective assembly and protein stability, to maintaining adequate protein degradation. Autophagy inducing drugs also show promise in restoring cardiac function in animal heart failure models, though their impact on the sarcomere is poorly characterized [189]. For cases of chaperone deficiency, which frequently occur in end-stage heart failure, we speculate AAV-based gene therapy may be a promising approach to restore proteostasis. Gene therapy has shown promise in restoring endoplasmic reticulum (ER) proteostasis, a central node of global cardiomyocyte PQC [219,220]. Moreover, a recent preprint of a study by our group highlights promise for gene therapy targeting sarcomere chaperones, where AAV9-BAG3 delivery in a mouse heart failure model rescued myofilament contractile function and restored sarcomere proteostasis [202]. However, gene therapy is complicated in larger species by difficulties with delivering ample amount of virus.
5. Summary & Future Directions
Protein quality control (PQC) at the sarcomere is fundamental for maintaining cardiac function. In this review, we describe the proteins involved in sarcomere assembly and stability, along with processes controlling protein degradation therein. The exact mechanism of sarcomere formation (sarcomerogenesis), while controversial, is thought to involve the formation of immature stress fiber complexes that are a blueprint for the mature sarcomere. Sarcomerogenesis is supported by a variety of chaperone and co-chaperone proteins, which stabilize sarcomere proteins and prevent their aggregation. While the lifetime of a cardiomyocyte extends for many years, that of the individual proteins is limited to days/weeks. Thus, new proteins must constantly be incorporated into the sarcomere in place of old members without compromising mechanical function. Protein turnover is accomplished via three different systems (the calpain system, the ubiquitin-proteasome system, and the autophagy-lysosome pathway), which assist in removal of old proteins to maintain sarcomere proteostasis.
The studies summarized in this review showcase the staggering complexity of sarcomere PQC and highlight dozens of factors essential for sarcomere assembly and maintenance. However, it is important to note that some of these studies came from skeletal myocytes. Unlike cardiomyocytes, skeletal myocytes do not persistently contract and are slowly turned over with age, and thus may not be faced with the same PQC requirements. Regardless, it is increasingly evident that we still do not fully understand many aspects of sarcomere PQC, which leads to key unanswered questions. Foremost among these are questions regarding sarcomere assembly and maintenance in the terminally differentiated adult cardiomyocytes: 1. What are the stimuli and mechanisms for de novo sarcomere assembly in adult myocytes? 2. Does the addition of new sarcomeres in eutrophy and exercise-induced hypertrophy differ from pathological hypertrophy? 3. How do mutations to sarcomere proteins, such as contribute to the “poison peptide” effect, impact their maintenance by chaperones? 4. Are there functionally distinct PQC mechanisms for the sarcomere? Much like mitochondria-specific autophagy (mitophagy), is there a sarcomere-specific autophagy (“sarcophagy”)? Answering these questions with current experimental systems/techniques is challenging. Adult cardiac and skeletal myocytes are not amenable to long-term culture, contracting cells present difficulties for live-cell imaging, and in vitro hypertrophy models require treatment paradigms of multiple days.
Future work may aim at determining how local protein assembly and degradation are regulated at the sarcomere. How a cardiomyocyte ensures the delivery of new proteins to its hundreds of sarcomeres with both temporal and spatial accuracy is a puzzling question. Within a given myocyte, the mechanical strain on individual sarcomeres is non-uniform [221]. Higher mechanical strain increases the propensity of a protein to misfold and thus the rate of protein turnover at some sarcomeres must outpace that at others. In the same vein, the mobilization of protein degradation pathways to individual sarcomeres must therefore be locally regulated. Recent work by Lewis et al. made important observations regarding the spatiotemporal maintenance of the sarcomere [2]. They found ribosomes and sarcomere protein transcripts localize to the Z-disc, thus allowing for local translation of sarcomere proteins. They propose an excess synthesis model of sarcomere maintenance where an overabundance of sarcomere proteins are synthesized and serve as a readily available pool for incorporation into the complex [2]. Evidence also suggests that under increased stress, autophagy engages in local protein degradation at the sarcomere. In mechanically stressed myocytes, the autophagic ubiquitin receptor P62/SQSTM1 and autophagosome membrane protein LC3 localize to the sarcomere, suggesting sarcomere protein aggregates are engulfed locally by the autophagosome [195,222]. These studies shed some light on the mystery of regulating local PQC at the dynamic sarcomere and provide an excellent framework for future studies in the years to come.
Highlights.
Sarcomere assembly and maintenance are mediated by molecular chaperones
Mutations and deficiency of chaperones are linked to cardiomyopathy
Mechanical and oxidative stress at the sarcomere predispose to protein misfolding
Calpains, the UPS, and autophagy regulate sarcomeric protein turnover
Failure of PQC mechanisms occurs in the progression of heart disease
Acknowledgments
This work was supported by the National Heart Lung and Blood Institute/NIH (R01HL136737 to J.A.K.) and the American Heart Association (Predoctoral Fellowship 20PRE35170045 to T.G.M.). We thank Stefanie Papasoglu for assistance in creating the figures for this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
References
- [1].Rollett A, Examinations on the construction of striated muscle, Austrian Acad. Sci (1884). [Google Scholar]
- [2].Lewis YE, Moskovitz A, Mutlak M, Heineke J, Caspi LH, Kehat I, Localization of transcripts, translation, and degradation for spatiotemporal sarcomere maintenance, J. Mol. Cell. Cardiol 116 (2018) 16–28. [DOI] [PubMed] [Google Scholar]
- [3].Ono S, Dynamic regulation of sarcomeric actin filaments in striated muscle, Cytoskeleton. 67 (2010) 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ghosh SR, Hope IA, Determination of the mobility of novel and established Caenorhabditis elegans sarcomeric proteins in vivo, Eur. J. Cell Biol 89 (2010) 437–448. [DOI] [PubMed] [Google Scholar]
- [5].Rudolph F, Hüttemeister J, da Silva Lopes K, Jüttner R, Yu L, Bergmann N, Friedrich D, et al. , Resolving titin’s lifecycle and the spatial organization of protein turnover in mouse cardiomyocytes, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 25126–25136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Saibil H, Chaperone machines for protein folding, unfolding and disaggregation, Nat. Rev. Mol. Cell Biol 14 (2013) 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sparrow JC, Schöck F, The initial steps of myofibril assembly: Integrins pave the way, Nat. Rev. Mol. Cell Biol 10 (2009) 293–298. [DOI] [PubMed] [Google Scholar]
- [8].Kelly DE, Myofibrillogenesis and Z-band differentiation, Anat. Rec 163 (1969) 403–425. [DOI] [PubMed] [Google Scholar]
- [9].Wang J, Shaner N, Mittal B, Zhou Q, Chen J, Sanger JM, et al. , Dynamics of Z-band based proteins in developing skeletal muscle cells, Cell Motil. Cytoskeleton 61 (2005) 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].McDonald KA, Lakonishok M, Horwitz AF, α(v) and α 3 integrin subunits are associated with myofibrils during myofibrillogenesis, J. Cell Sci (1995). [DOI] [PubMed] [Google Scholar]
- [11].Katzemich A, Liao KA, Czerniecki S, Schöck F, Alp/Enigma Family Proteins Cooperate in Z-Disc Formation and Myofibril Assembly, PLoS Genet. 9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jani K, Schöck F, Zasp is required for the assembly of functional integrin adhesion sites, J. Cell Biol 179 (2007) 1583–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sanger JW, Wang J, Holloway B, Du A, Sanger JM, Myofibrillogenesis in skeletal muscle cells in zebrafish, Cell Motil. Cytoskeleton 66 (2009) 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Moerman DG, Williams BD, Sarcomere assembly in C elegans muscle., WormBook. (2006) 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bloor JW, Brown NH, Genetic analysis of the Drosophila α(PS2) integrin subunit reveals discrete adhesive, morphogenetic and sarcomeric functions, Genetics. 148 (1998) 1127–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Volk T, Fessler LI, Fessler JH, A role for integrin in the formation of sarcomeric cytoarchitecture, Cell. 63 (1990) 525–536. [DOI] [PubMed] [Google Scholar]
- [17].Pérez-Moreno JJ, Bischoff M, Martín-Bermudo MD, Estrada B, The conserved transmembrane proteoglycan Perdido/Kon-tiki is essential for myofibrillogenesis and sarcomeric structure in Drosophila, J. Cell Sci 127 (2014) 3162–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schwander M, Leu M, Stumm M, Dorchies OM, Ruegg UT, Schittny J, et al. , β1 integrins regulate myoblast fusion and sarcomere assembly, Dev. Cell 4 (2003) 673–685. [DOI] [PubMed] [Google Scholar]
- [19].Johnson K, Bertoli M, Phillips L, Töpf A, Van den Bergh P, Vissing J,et al. , Detection of variants in dystroglycanopathy-associated genes through the application of targeted whole-exome sequencing analysis to a large cohort of patients with unexplained limb-girdle muscle weakness, Skelet. Muscle 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bello L, Melacini P, Pezzani R, D’Amico A, Piva L, Leonardi E, et al. , Cardiomyopathy in patients with POMT1-related congenital and limb-girdle muscular dystrophy, Eur. J. Hum. Genet 20 (2012) 1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Di Costanzo S, Balasubramanian A, Pond HL, Rozkalne A, Pantaleoni C, Saredi S, et al. , POMK mutations disrupt muscle development leading to a spectrum of neuromuscular presentations, Hum. Mol. Genet 23 (2014) 5781–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Whiting A, Wardale J, Trinick J, Does titin regulate the length of muscle thick filaments?, J. Mol. Biol 205 (1989) 263–268. [DOI] [PubMed] [Google Scholar]
- [23].Tokuyasu KT, Immunocytochemical studies of cardiac myofibrillogenesis in early chick embryos. III. Generation of fasciae adherentes and costameres, J. Cell Biol 108 (1989) 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Van Der Loop FTL, Van Der Ven PFM, Fürst DO, Gautel M, Van Eys GJJM, Ramaekers FCS, Integration of titin into the sarcomeres of cultured differentiating human skeletal muscle cells, Eur. J. Cell Biol 69 (1996) 301–307. [PubMed] [Google Scholar]
- [25].Van Der Ven PFM, Fürst DO, Assembly of titin, myomesin and M-protein into the sarcomeric M band in differentiating human skeletal muscle cells in vitro, in: Cell Struct. Funct, 1997: pp. 163–171. [DOI] [PubMed] [Google Scholar]
- [26].Van Der Ven PFM, Bartsch JW, Gautel M, Jockusch H, Fürst DO, A functional knock-out of titin results in defective myofibril assembly, J. Cell Sci 113 (2000) 1405–1414. [DOI] [PubMed] [Google Scholar]
- [27].Miller G, Musa H, Gautel M, Peckham M, A targeted deletion of the C-terminal end of titin, including the titin kinase domain, impairs myofibrillogenesis, J. Cell Sci 116 (2003) 4811–4819. [DOI] [PubMed] [Google Scholar]
- [28].Person V, Kostin S, Suzuki K, Labeit S, Schaper J, Antisense oligonucleotide experiments elucidate the essential role of titin in sarcomerogenesis in adult rat cardiomyocytes in long-term culture, J. Cell Sci 113 (2000) 3851–3859. [DOI] [PubMed] [Google Scholar]
- [29].Musa H, Meek S, Gautel M, Peddie D, Smith AJH, Peckham M, Targeted homozygous deletion of M-band titin in cardiomyocytes prevents sarcomere formation, J. Cell Sci 119 (2006) 4322–4331. [DOI] [PubMed] [Google Scholar]
- [30].Zaunbrecher RJ, Abel AN, Beussman K, Leonard A, Von Frieling-Salewsky M, Fields PA, et al. , Cronos titin is expressed in human cardiomyocytes and necessary for normal sarcomere function, Circulation. 140 (2019) 1647–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Seeley M, Huang W, Chen Z, Wolff WO, Lin X, Xu X, Depletion of zebrafish titin reduces cardiac contractility by disrupting the assembly of Z-discs and A-bands, Circ. Res 100 (2007) 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rhee D, Sanger JM, Sanger JW, The premyofibril: Evidence for its role in myofibrillogenesis, Cell Motil. Cytoskeleton 28 (1994) 1–24. [DOI] [PubMed] [Google Scholar]
- [33].Du A, Sanger JM, Sanger JW, Cardiac myofibrillogenesis inside intact embryonic hearts, Dev. Biol (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Myhre JL, Pilgrim D, A titan but not necessarily a ruler: Assessing the role of titin during thick filament patterning and assembly, Anat. Rec 297 (2014) 1604–1614. [DOI] [PubMed] [Google Scholar]
- [35].Sanger JW, Kang S, Siebrands CC, Freeman N, Du A, Wang J, et al. , How to build a myofibril, in: J. Muscle Res. Cell Motil, 2005: pp. 343–354. [DOI] [PubMed] [Google Scholar]
- [36].Lu MH, DiLullo C, Schultheiss T, Holtzer S, Murray JM, Choi J, et al. , The vinculin/sarcomeric-α-actinin/α-actin nexus in cultured cardiac myocytes, J. Cell Biol 117 (1992) 1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Holtzer H, Hijikata T, Lin ZX, Zhang ZQ, Holtzer S, Protasi F, et al. , Independent assembly of 1.6 μm long bipolar MHC filaments and I-Z-I bodies, in: Cell Struct. Funct, 1997: pp. 83–93. [DOI] [PubMed] [Google Scholar]
- [38].Lin Z, Lu M-H, Schultheiss T, Choi J, Holtzer S, Dilullo C, et al. , Sequential appearance of muscle-specific proteins in myoblasts as a function of time after cell division: Evidence for a conserved myoblast differentiation program in skeletal muscle, Cell Motil. Cytoskeleton 29 (1994) 1–19. [DOI] [PubMed] [Google Scholar]
- [39].Rui Y, Bai J, Perrimon N, Sarcomere formation occurs by the assembly of multiple latent protein complexes, PLoS Genet. 6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dlugosz AA, Antin PB, Nachmias VT, Holtzer H, The relationship between stress fiber-like structures and nascent myofibrils in cultured cardiac myocytes, J. Cell Biol 99 (1984) 2268–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tullio AN, Accili D, Ferrans VJ, Yu ZX, Takeda K, Grinberg A, et al. , Nonmuscle myosin II-B is required for normal development of the mouse heart, Proc. Natl. Acad. Sci. U. S. A 94 (1997) 12407–12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Duan R, Gallagher PJ, Dependence of myoblast fusion on a cortical actin wall and nonmuscle myosin IIA, Dev. Biol 325 (2009) 374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Swailes NT, Colegrave M, Knight PJ, Peckham M, Non-muscle myosins 2A and 2B drive changes in cell morphology that occur as myoblasts align and fuse, J. Cell Sci 119 (2006) 3561–3570. [DOI] [PubMed] [Google Scholar]
- [44].Ma X, Takeda K, Singh A, Yu ZX, Zerfas P, Blount A, et al. , Conditional ablation of nonmuscle myosin II-B delineates heart defects in adult mice, Circ. Res 105 (2009) 1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Conti MA, Even-Ram S, Liu C, Yamada KM, Adelstein RS, Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice, J. Biol. Chem 279 (2004) 41263–41266. [DOI] [PubMed] [Google Scholar]
- [46].Hoppe PE, Waterston RH, A region of the myosin rod important for interaction with paramyosin in Caenorhabditis elegans striated muscle, Genetics. 156 (2000) 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hoppe PE, Waterston RH, Hydrophobicity variations along the surface of the coiled-coil rod may mediate striated muscle myosin assembly in Caenorhabditis elegans, J. Cell Biol 135 (1996) 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sanger JW, Wang J, Fan Y, White J, Sanger JM, Assembly and dynamics of myofibrils, J. Biomed. Biotechnol 2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fenix AM, Neininger AC, Taneja N, Hyde K, Visetsouk MR, Garde RJ, et al. , Muscle-specific stress fibers give rise to sarcomeres in cardiomyocytes, Elife. 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Manisastry SM, Zaal KJM, Horowits R, Myofibril assembly visualized by imaging N-RAP, alpha-actinin, and actin in living cardiomyocytes, Exp. Cell Res 315 (2009) 2126–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hansen WJ, Cowan NJ, Welch WJ, Prefoldin-nascent chain complexes in the folding of cytoskeletal proteins, J. Cell Biol 145 (2000) 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Berger J, Berger S, Li M, Jacoby AS, Arner A, Bavi N, et al. , In Vivo Function of the Chaperonin TRiC in α-Actin Folding during Sarcomere Assembly, Cell Rep. 22 (2018) 313–322. [DOI] [PubMed] [Google Scholar]
- [53].Grantham J, Ruddock LW, Roobol A, Carden MJ, Eukaryotic chaperonin containing T-complex polypeptide 1 interacts with filamentous actin and reduces the initial rate of actin polymerization in vitro, Cell Stress Chaperones. 7 (2002) 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Siegers K, Waldmann T, Leroux MR, Grein K, Shevchenko A, Schiebel E, et al. , Compartmentation of protein folding in vivo: Sequestration of non-native polypeptide by the chaperonin-GimC system, EMBO J. 18 (1999) 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Vainberg IE, Lewis SA, Rommelaere H, Ampe C, Vandekerckhove J, Klein HL, Cowan NJ, Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin, Cell. 93 (1998) 863–873. [DOI] [PubMed] [Google Scholar]
- [56].Erdmann J, Stark K, Esslinger UB, Rumpf PM, Koesling D, DeWit C, et al. , Dysfunctional nitric oxide signalling increases risk of myocardial infarction, Nature. 504 (2013) 432–436. [DOI] [PubMed] [Google Scholar]
- [57].Melkani GC, Bhide S, Han A, Vyas J, Livelo C, Bodmer R, et al. , TRiC/CCT chaperonins are essential for maintaining myofibril organization, cardiac physiological rhythm, and lifespan, FEBS Lett. 591 (2017) 3447–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Srikakulam R, Winkelmann DA, Myosin II folding is mediated by a molecular chaperonin, J. Biol. Chem 274 (1999) 27265–27273. [DOI] [PubMed] [Google Scholar]
- [59].Matsuyama S, Kage Y, Fujimoto N, Ushijima T, Tsuruda T, Kitamura K, et al. , Interaction between cardiac myosin-binding protein C and formin Fhod3, Proc. Natl. Acad. Sci. U. S. A 115 (2018) E4386–E4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Paul AS, Pollard TD, Review of the mechanism of processive actin filament elongation by formins, Cell Motil. Cytoskeleton 66 (2009) 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kan-O M, Takeya R, Abe T, Kitajima N, Nishida M, Tominaga R, et al. , Mammalian formin Fhod3 plays an essential role in cardiogenesis by organizing myofibrillogenesis, Biol. Open 1 (2012) 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Iskratsch T, Lange S, Dwyer J, Kho AL, Dos Remedios C, Ehler E, Formin follows function: A muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance, J. Cell Biol 191 (2010) 1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Taniguchi K, Takeya R, Suetsugu S, Kan-o M, Narusawa M, Shiose A, et al. , Mammalian formin Fhod3 regulates actin assembly and sarcomere organization in striated muscles, J. Biol. Chem 284 (2009) 29873–29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Fujimoto N, Kan-O M, Ushijima T, Kage Y, Tominaga R, Sumimoto H, et al. , Transgenic expression of the formin protein fhod3 selectively in the embryonic heart: Role of actin-binding activity of fhod3 and its sarcomeric localization during myofibrillogenesis, PLoS One. 11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Arimura T, Takeya R, Ishikawa T, Yamano T, Matsuo A, Tatsumi T, et al. , Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor, Circ. J 77 (2013) 2990–2996. [DOI] [PubMed] [Google Scholar]
- [66].dos Remedios CG, Li A, Lal S, Non-sarcomeric causes of heart failure: a Sydney Heart Bank perspective, Biophys. Rev 10 (2018) 949–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bernstein BW, Bamburg JR, ADF/Cofilin: A functional node in cell biology, Trends Cell Biol. 20 (2010) 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ono S, Minami N, Abe H, Obinata T, Characterization of a novel cofilin isoform that is predominantly expressed in mammalian skeletal muscle, J. Biol. Chem 269 (1994) 15280–15286. [PubMed] [Google Scholar]
- [69].Vartiainen MK, Mustonen T, Mattila PK, Ojala PJ, Thesleff I, Partanen J, et al. , The three mouse actin-depolymerizing factor/cofilins evolved to fulfill cell-type-specific requirements for actin dynamics, Mol. Biol. Cell 13 (2002) 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kremneva E, Makkonen MH, Skwarek-Maruszewska A, Gateva G, Michelot A, Dominguez R, et al. , Cofilin-2 controls actin filament length in muscle sarcomeres, Dev. Cell 31 (2014) 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, et al. , Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: Implication in actin-based motility, J. Cell Biol 136 (1997) 1307–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Andrianantoandro E, Pollard TD, Mechanism of Actin Filament Turnover by Severing and Nucleation at Different Concentrations of ADF/Cofilin, Mol. Cell 24 (2006) 13–23. [DOI] [PubMed] [Google Scholar]
- [73].Agrawal PB, Greenleaf RS, Tomczak KK, Lehtokari VL, Wallgren-Pettersson C, Wallefeld W, et al. , Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2, Am. J. Hum. Genet 80 (2007) 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ockeloen CW, Gilhuis HJ, Pfundt R, Kamsteeg EJ, Agrawal PB, Beggs AH, et al. , Congenital myopathy caused by a novel missense mutation in the CFL2 gene, Neuromuscul. Disord 22 (2012) 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Subramanian K, Gianni D, Balla C, Assenza GE, Joshi M, Semigran MJ, et al. , Cofilin-2 phosphorylation and sequestration in myocardial aggregates: Novel pathogenetic mechanisms for idiopathic dilated cardiomyopathy, J. Am. Coll. Cardiol 65 (2015) 1199–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Witke W, The role of profilin complexes in cell motility and other cellular processes, Trends Cell Biol. 14 (2004) 461–469. [DOI] [PubMed] [Google Scholar]
- [77].Mockrin SC, Korn ED, Acanthamoeba Profilin Interacts with G-Actin to Increase the Rate of Exchange of Actin-Bound Adenosine 5’-Triphosphate, Biochemistry. 19 (1980) 5359–5362. [DOI] [PubMed] [Google Scholar]
- [78].Kan-o M, Takeya R, Taniguchi K, Tanoue Y, Tominaga R, Sumimoto H, Expression and subcellular localization of mammalian formin Fhod3 in the embryonic and adult heart, PLoS One. 7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Paul A, Pollard T, The Role of the FH1 Domain and Profilin in Formin-Mediated Actin-Filament Elongation and Nucleation, Curr. Biol 18 (2008) 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Pyle WG, Hart MC, Cooper JA, Sumandea MP, De Tombe PP, Solaro RJ, Actin capping protein: An essential element in protein kinase signaling to the myofilaments, Circ. Res 90 (2002) 1299–1306. [DOI] [PubMed] [Google Scholar]
- [81].Schafer DA, Hug C, Cooper JA, Inhibition of CapZ during myofibrillogenesis alters assembly of actin filaments, J. Cell Biol 128 (1995) 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ono Y, Schwach C, Antin PB, Gregorio CC, Disruption in the tropomodulin1 (Tmod1) gene compromises cardiomyocyte development in murine embryonic stem cells by arresting myofibril maturation, Dev. Biol 282 (2005) 336–348. [DOI] [PubMed] [Google Scholar]
- [83].McKeown CR, Nowak RB, Moyer J, Sussman MA, Fowler VM, Tropomodulin1 is required in the heart but not the yolk sac for mouse embryonic development, Circ. Res 103 (2008) 1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Fritz-Six KL, Cox PR, Fischer RS, Xu B, Gregorio CC, Zoghbi HY, Fowler VM, Aberrant myofibril assembly in tropomodulin1 null mice leads to aborted heart development and embryonic lethality, J. Cell Biol 163 (2003) 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Tsukada T, Pappas CT, Moroz N, Antin PB, Kostyukova AS, Gregorio CC, Leiomodin-2 is an antagonist of tropomodulin-1 at the pointed end of the thin filaments in cardiac muscle, J. Cell Sci 123 (2010) 3136–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Boczkowska M, Rebowski G, Kremneva E, Lappalainen P, Dominguez R, How Leiomodin and Tropomodulin use a common fold for different actin assembly functions, Nat. Commun 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Hishiya A, Kitazawa T, Takayama S, BAG3 and Hsc70 interact with actin capping protein CapZ to maintain myofibrillar integrity under mechanical stress, Circ. Res 107 (2010) 1220–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Bakthisaran R, Tangirala R, Rao CM, Small heat shock proteins: Role in cellular functions and pathology, Biochim. Biophys. Acta - Proteins Proteomics 1854 (2015) 291–319. [DOI] [PubMed] [Google Scholar]
- [89].Brown DD, Christine KS, Showell C, Conlon FL, Small heat shock protein Hsp27 is required for proper heart tube formation, Genesis. 45 (2007) 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Yin B, Tang S, Xu J, Sun J, Zhang X, Li Y, et al. , CRYAB protects cardiomyocytes against heat stress by preventing caspase-mediated apoptosis and reducing F-actin aggregation, Cell Stress Chaperones. 24 (2019) 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Bennardini F, Wrzosek A, Chiesi M, αB-crystallin in cardiac tissue: Association with actin and desmin filaments, Circ. Res 71 (1992) 288–294. [DOI] [PubMed] [Google Scholar]
- [92].Singh BN, Rao KS, Ramakrishna T, Rangaraj N, Rao CM, Association of αBCrystallin, a Small Heat Shock Protein, with Actin: Role in Modulating Actin Filament Dynamics in Vivo, J. Mol. Biol 366 (2007) 756–767. [DOI] [PubMed] [Google Scholar]
- [93].Wu T, Mu Y, Bogomolovas J, Fang X, Veevers J, Nowak RB, et al. , HSPB7 is indispensable for heart development by modulating actin filament assembly, Proc. Natl. Acad. Sci. U. S. A 114 (2017) 11956–11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Barral JM, Hutagalung AH, Brinker A, Hartl FU, Epstein HF, Role of the myosin assembly protein UNC-45 as a molecular chaperone for myosin, Science (80-.) (2002). [DOI] [PubMed] [Google Scholar]
- [95].Kachur T, Ao W, Berger J, Pilgrim D, Maternal UNC-45 is involved in cytokinesis and colocalizes with non-muscle myosin in the early Caenorhabditis elegans embryo, J. Cell Sci 117 (2004) 5313–5321. [DOI] [PubMed] [Google Scholar]
- [96].Barral JM, Hutagalung AH, Brinker A, Hartl FU, Epstein HF, Role of the myosin assembly protein UNC-45 as a molecular chaperone for myosin, Science (80-.) 295 (2002) 669–671. [DOI] [PubMed] [Google Scholar]
- [97].Barral JM, Bauer CC, Ortiz I, Epstein HF, Unc-45 mutations in Caenorhabditis elegans implicate a CRO1/She4p-like domain in myosin assembly, J. Cell Biol 143 (1998) 1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Etard C, Behra M, Fischer N, Hutcheson D, Geisler R, Strähle U, The UCS factor Steif/Unc-45b interacts with the heat shock protein Hsp90a during myofibrillogenesis, Dev. Biol 308 (2007) 133–143. [DOI] [PubMed] [Google Scholar]
- [99].Geach TJ, Zimmerman LB, Paralysis and delayed Z-disc formation in the Xenopus tropicalis unc45b mutant dicky ticker, BMC Dev. Biol 10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lee CF, Melkani GC, Yu Q, Suggs JA, Kronert WA, Suzuki Y, et al. , Drosophila UNC-45 accumulates in embryonic blastoderm and in muscles, and is essential for muscle myosin stability, J. Cell Sci 124 (2011) 699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Melkani GC, Bodmer R, Ocorr K, Bernstein SI, The UNC-45 chaperone is critical for establishing Myosin-Based myofibrillar organization and cardiac contractility in the Drosophila heart model, PLoS One. 6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Price MG, Landsverk ML, Barral JM, Epstein HF, Two mammalian UNC-45 isoforms are related to distinct cytoskeletal and muscle-specific functions, J. Cell Sci 115 (2002) 4013–4023. [DOI] [PubMed] [Google Scholar]
- [103].Wohlgemuth SL, Crawford BD, Pilgrim DB, The myosin co-chaperone UNC-45 is required for skeletal and cardiac muscle function in zebrafish, Dev. Biol 303 (2007) 483–492. [DOI] [PubMed] [Google Scholar]
- [104].Gazda L, Pokrzywa W, Hellerschmied D, Löwe T, Forné I, Mueller-Planitz F, et al. , The myosin chaperone UNC-45 is organized in tandem modules to support myofilament formation in C. elegans, Cell. 152 (2013) 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Bernick EP, Zhang PJ, Du S, Knockdown and overexpression of Unc-45b result in defective myofibril organization in skeletal muscles of zebrafish embryos, BMC Cell Biol. 11 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Melkani GC, Lee CF, Cammarato A, Bernstein SI, Drosophila UNC-45 prevents heat-induced aggregation of skeletal muscle myosin and facilitates refolding of citrate synthase, Biochem. Biophys. Res. Commun 396 (2010) 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Melkani GC, Cammarato A, Bernstein SI, αB-Crystallin Maintains Skeletal Muscle Myosin Enzymatic Activity and Prevents its Aggregation under Heat-shock Stress, J. Mol. Biol 358 (2006) 635–645. [DOI] [PubMed] [Google Scholar]
- [108].Etard C, Roostalu U, Strähle U, Shuttling of the chaperones Unc45b and Hsp90a between the A band and the Z line of the myofibril, J. Cell Biol 180 (2008) 1163–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Dafsari HS, Kocaturk NM, Daimagüler HS, Brunn A, Dötsch J, Weis J, et al. , Bi-allelic mutations in uncoordinated mutant number-45 myosin chaperone B are a cause for congenital myopathy, Acta Neuropathol. Commun 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Myhre JL, Hills JA, Jean F, Pilgrim DB, Unc45b is essential for early myofibrillogenesis and costamere formation in zebrafish, Dev. Biol (2014). [DOI] [PubMed] [Google Scholar]
- [111].Srikakulam R, Winkelmann DA, Chaperone-mediated folding and assembly of myosin in striated muscle, J. Cell Sci 117 (2004) 641–652. [DOI] [PubMed] [Google Scholar]
- [112].Du Shao J, Li H, Bian Y, Zhong Y, Heat-shock protein 90α1 is required for organized myofibril assembly in skeletal muscles of zebrafish embryos, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Hawkins TA, Haramis AP, Etard C, Prodromou C, Vaughan CK, Ashworth R, et al. , The ATPase-dependent chaperoning activity of Hsp90a regulates thick filament formation and integration during skeletal muscle myofibrillogenesis, Development. 135 (2008) 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Codina M, Li J, Gutiérrez J, Kao JPY, Du SJ, Loss of Smyhc1 or Hsp90α1 function results in different effects on myofibril organization in skeletal muscles of zebrafish embryos, PLoS One. 5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Unger A, Beckendorf L, Böhme P, Kley R, von Frieling-Salewsky M, Lochmüller H, et al. , Translocation of molecular chaperones to the titin springs is common in skeletal myopathy patients and affects sarcomere function, Acta Neuropathol. Commun 5 (2017) 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Voelkel T, Andresen C, Unger A, Just S, Rottbauer W, Linke WA, Lysine methyltransferase Smyd2 regulates Hsp90-mediated protection of the sarcomeric titin springs and cardiac function, Biochim. Biophys. Acta - Mol. Cell Res 1833 (2013) 812–822. [DOI] [PubMed] [Google Scholar]
- [117].Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, et al. , Structure of TPR domain-peptide complexes: Critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine, Cell. 101 (2000) 199–210. [DOI] [PubMed] [Google Scholar]
- [118].Ni W, Hutagalung AH, Li S, Epstein HF, The myosin-binding UCS domain but not the Hsp90-binding TPR domain of the UNC-45 chaperone is essential for function in Caenorhabditis elegans, J. Cell Sci 124 (2011) 3164–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Hong J, Park JS, Lee H, Jeong J, Hyeon Yun H, Yun Kim H, et al. , Myosin heavy chain is stabilized by BCL-2 interacting cell death suppressor (BIS) in skeletal muscle, Exp. Mol. Med 48 (2016) e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Just S, Meder B, Berger IM, Etard C, Trano N, Patzel E, et al. , The myosin-interacting protein SMYD1 is essential for sarcomere organization, J. Cell Sci 124 (2011) 3127–3136. [DOI] [PubMed] [Google Scholar]
- [121].Li H, Xu J, Bian YH, Rotllant P, Shen T, Chu W, et al. , Smyd1b, a key regulator of sarcomere assembly, is localized on the M-Line of skeletal muscle fibers, PLoS One. 6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Cai M, Han L, Liu L, He F, Chu W, Zhang J, et al. , Defective sarcomere assembly in smyd1a and smyd1b zebrafish mutants, FASEB J. 33 (2019) 6209–6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Tan X, Rotllant J, Li H, DeDeyne P, Du SJ, SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 2713–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Li H, Zhong Y, Wang Z, Gao J, Xu J, Chu W, et al. , Smyd1b is required for skeletal and cardiac muscle function in zebrafish, Mol. Biol. Cell 24 (2013) 3511–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Fan LL, Ding DB, Huang H, Chen YQ, Jin JY, Xia K, et al. , A de novo mutation of SMYD1 (p.F272L) is responsible for hypertrophic cardiomyopathy in a Chinese patient, Clin. Chem. Lab. Med 57 (2019) 532–539. [DOI] [PubMed] [Google Scholar]
- [126].Borlak J, Thum T, Hallmarks of ion channel gene expression in end‐stage heart failure, FASEB J. 17 (2003) 1592–1608. [DOI] [PubMed] [Google Scholar]
- [127].Bullard B, Ferguson C, Minajeva A, Leake MC, Gautel M, Labeit D, et al. , Association of the Chaperone αβ-crystallin with Titin in Heart Muscle, J. Biol. Chem 279 (2004) 7917–7924. [DOI] [PubMed] [Google Scholar]
- [128].Golenhofen N, Arbeiter A, Koob R, Drenckhahn D, Ischemia-induced association of the stress protein αB-crystallin with I-band portion of cardiac titin, J. Mol. Cell. Cardiol 34 (2002) 309–319. [DOI] [PubMed] [Google Scholar]
- [129].Franssen C, Kole J, Musters R, Hamdani N, Paulus WJ, α-B crystallin reverses high diastolic stiffness of failing human cardiomyocytes, Circ. Hear. Fail 10 (2017). [DOI] [PubMed] [Google Scholar]
- [130].Inagaki N, Hayashi T, Arimura T, Koga Y, Takahashi M, Shibata H, et al. , αB-crystallin mutation in dilated cardiomyopathy, Biochem. Biophys. Res. Commun 342 (2006) 379–386. [DOI] [PubMed] [Google Scholar]
- [131].Raguz S, Hobbs C, Yagüe E, Ioannou PA, Walsh FS, Antoniou M, Muscle-specific locus control region activity associated with the human desmin gene, Dev. Biol 201 (1998) 26–42. [DOI] [PubMed] [Google Scholar]
- [132].Capetanaki Y, Papathanasiou S, Diokmetzidou A, Vatsellas G, Tsikitis M, Desmin related disease: A matter of cell survival failure, Curr. Opin. Cell Biol 32 (2015) 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Raju I, Abraham EC, Mutants of human αB-crystallin cause enhanced protein aggregation and apoptosis in mammalian cells: Influence of co-expression of HspB1, Biochem. Biophys. Res. Commun 430 (2013) 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Wang X, Klevitsky R, Huang W, Glasford J, Li F, Robbins J, αB-Crystallin Modulates Protein Aggregation of Abnormal Desmin, Circ. Res 93 (2003) 998–1005. [DOI] [PubMed] [Google Scholar]
- [135].Pan B, Lewno MT, Wu P, Wang X, Highly dynamic changes in the activity and regulation of macroautophagy in hearts subjected to increased proteotoxic stress, Front. Physiol 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Zobel ATC, Loranger A, Marceau N, Thériault JR, Lambert H, Landry J, Distinct chaperone mechanisms can delay the formation of aggresomes by the myopathy-causing R120G αB-crystallin mutant, Hum. Mol. Genet 12 (2003) 1609–1620. [DOI] [PubMed] [Google Scholar]
- [137].Vicart P, Caron A, Guicheney P, Li Z, Prévost MC, Faure A, et al. , A missense mutation in the αb-crystallin chaperone gene causes a desmin-related myopathy, Nat. Genet 20 (1998) 92–95. [DOI] [PubMed] [Google Scholar]
- [138].Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, Horwitz J, Mutation R120G in αB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function, Proc. Natl. Acad. Sci. U. S. A 96 (1999) 6137–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Singh S, Kadioglu H, Patel K, Carrier L, Agnetti G, Is Desmin Propensity to Aggregate Part of its Protective Function?, Cells. 9 (2020) 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Sadayappan S, de Tombe PP, Cardiac myosin binding protein-C: Redefining its structure and function, Biophys. Rev 4 (2012) 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Glazier AA, Thompson A, Day SM, Allelic imbalance and haploinsufficiency in MYBPC3-linked hypertrophic cardiomyopathy, Pflugers Arch. Eur. J. Physiol 471 (2019) 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Parbhudayal RY, Garra AR, Götte MJW, Michels M, Pei J, Harakalova M, et al. , Variable cardiac myosin binding protein-C expression in the myofilaments due to MYBPC3 mutations in hypertrophic cardiomyopathy, J. Mol. Cell. Cardiol 123 (2018) 59–63. [DOI] [PubMed] [Google Scholar]
- [143].Burke MA, Cook SA, Seidman JG, Seidman CE, Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy, J. Am. Coll. Cardiol 68 (2016) 2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, et al. , Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency, Circ. Res 105 (2009) 219–222. [DOI] [PubMed] [Google Scholar]
- [145].Glazier AA, Hafeez N, Mellacheruvu D, Basrur V, Nesvizhskii AI, Lee LM, et al. , HSC70 is a chaperone for wild-type and mutant cardiac myosin binding protein C, JCI Insight. 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Eschenhagen T, Bolli R, Braun T, Field LJ, Fleischmann BK, Frisén J, et al. , Cardiomyocyte regeneration: A consensus statement, Circulation. 136 (2017) 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Martin AF, Turnover of cardiac troponin subunits. Kinetic evidence for a precursor pool of troponin-I, J. Biol. Chem 256 (1981) 964–968. [PubMed] [Google Scholar]
- [148].Martin AF, Rabinowitz M, Blough R, Prior G, Zak R, Measurements of half life of rat cardiac myosin heavy chain with leucyl tRNA used as precursor pool, J. Biol. Chem 252 (1977) 3422–3429. [PubMed] [Google Scholar]
- [149].Pandurangan M, Hwang I, The role of calpain in skeletal muscle, Animal Cells Syst. (Seoul) 16 (2012) 431–437. [Google Scholar]
- [150].Ono Y, Sorimachi H, Calpains - An elaborate proteolytic system, Biochim. Biophys. Acta - Proteins Proteomics 1824 (2012) 224–236. [DOI] [PubMed] [Google Scholar]
- [151].Beckmann JS, Spencer M, Calpain 3, the “gatekeeper” of proper sarcomere assembly, turnover and maintenance, Neuromuscul. Disord 18 (2008) 913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Wang Y, Chen B, Huang CK, Guo A, Wu J, Zhang X, et al. , Targeting Calpain for Heart Failure Therapy: Implications From Multiple Murine Models, JACC Basic to Transl. Sci 3 (2018) 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Blunt BC, Creek AT, Henderson DAC, Hofmann PA, H2O2 activation of HSP25/27 protects desmin from calpain proteolysis in rat ventricular myocytes, Am. J. Physiol. - Hear. Circ. Physiol 293 (2007). [DOI] [PubMed] [Google Scholar]
- [154].Zhang D, Ke L, Mackovicova K, Der Want JJLV, Sibon OCM, Tanguay RM, et al. , Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation, J. Mol. Cell. Cardiol 51 (2011) 381–389. [DOI] [PubMed] [Google Scholar]
- [155].Weaver AD, Bowker BC, Gerrard DE, Sarcomere length influences mucalpain-mediated proteolysis of bovine myofibrils., J. Anim. Sci 87 (2009) 2096–2103. [DOI] [PubMed] [Google Scholar]
- [156].Hasselgren PO, Fischer JE, Muscle cachexia: Current concepts of intracellular mechanisms and molecular regulation, Ann. Surg 233 (2001) 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Neti G, Novak SM, Thompson VF, Goll DE, Properties of easily releasable myofilaments: Are they the first step in myofibrillar protein turnover?, Am. J. Physiol. - Cell Physiol 296 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Galvez AS, Diwan A, Odley AM, Hahn HS, Osinska H, Melendez JG, et al. , Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis, Circ. Res 100 (2007) 1071–1078. [DOI] [PubMed] [Google Scholar]
- [159].Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, et al. , Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions, J. Clin. Invest 113 (2004) 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Lecker SH, Goldberg AL, Mitch WE, Protein degradation by the ubiquitin-proteasome pathway in normal and disease states, J. Am. Soc. Nephrol 17 (2006) 1807–1819. [DOI] [PubMed] [Google Scholar]
- [161].Spencer JA, Eliazer S, Ilaria RL, Richardson JA, Olson EN, Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein, J. Cell Biol 150 (2000) 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, et al. , Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain, J. Mol. Biol 306 (2001) 717–726. [DOI] [PubMed] [Google Scholar]
- [163].McElhinny AS, Perry CN, Witt CC, Labeit S, Gregorio CC, Muscle-specific RING finger-2 (MURF-2) is important for microtubule, intermediate filament and sarcomeric M-line maintenance in striated muscle development, J. Cell Sci 117 (2004) 3175–3188. [DOI] [PubMed] [Google Scholar]
- [164].Witt SH, Granzier H, Witt CC, Labeit S, MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination, J. Mol. Biol 350 (2005) 713–722. [DOI] [PubMed] [Google Scholar]
- [165].Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C, Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I, Proc. Natl. Acad. Sci. U. S. A 101 (2004) 18135–18140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Chen SN, Czernuszewicz G, Tan Y, Lombardi R, Jin J, Willerson JT, Marian AJ, Human molecular genetic and functional studies identify TRIM63, encoding muscle RING finger protein 1, as a novel gene for human hypertrophic cardiomyopathy, Circ. Res 111 (2012) 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Witt CC, Witt SH, Lerche S, Labeit D, Back W, Labeit S, Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2, EMBO J. 27 (2008) 350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Fielitz J, Van Rooij E, Spencer JA, Shelton JM, Latif S, Van Der Nagel R, et al. , Loss of muscle-specific RING-finger 3 predisposes the heart to cardiac rupture after myocardial infarction, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 4377–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ, et al. , Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3, J. Clin. Invest 117 (2007) 2486–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Olivé M, Abdul-Hussein S, Oldfors A, González-Costello J, van der Ven PFM, Fürst DO, et al. , New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations, Hum. Mol. Genet 24 (2015) 3638–3650. [DOI] [PubMed] [Google Scholar]
- [171].Lodka D, Pahuja A, Geers-Knörr C, Scheibe RJ, Nowak M, Hamati J, et al. , Muscle RING-finger 2 and 3 maintain striated-muscle structure and function, J. Cachexia. Sarcopenia Muscle 7 (2016) 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Su M, Wang J, Kang L, Wang Y, Zou Y, Feng X, et al. , Rare variants in genes encoding MuRF1 and MuRF2 are modifiers of hypertrophic cardiomyopathy, Int. J. Mol. Sci 15 (2014) 9302–9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C, Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo, Circ. Res 100 (2007) 456–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Kudryashova E, Kudryashov D, Kramerova I, Spencer MJ, Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin, J. Mol. Biol 354 (2005) 413–424. [DOI] [PubMed] [Google Scholar]
- [175].Cohen S, Zhai B, Gygi SP, Goldberg AL, Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy, J. Cell Biol 198 (2012) 575–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Volodin A, Kosti I, Goldberg AL, Cohen S, Myofibril breakdown during atrophy is a delayed response requiring the transcription factor PAX4 and desmin depolymerization, Proc. Natl. Acad. Sci. U. S. A 114 (2017) E1375–E1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Chen L, Huang J, Ji Y, Zhang X, Wang P, Deng K, et al. , Tripartite motif 32 prevents pathological cardiac hypertrophy, Clin. Sci 130 (2016) 813–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Nyamsuren O, Faggionato D, Loch W, Schulze E, Baumeister R, A mutation in CHN-1/CHIP suppresses muscle degeneration in Caenorhabditis elegans, Dev. Biol 312 (2007) 193–202. [DOI] [PubMed] [Google Scholar]
- [179].Janiesch PC, Kim J, Mouysset J, Barikbin R, Lochmüller H, Cassata G, et al. , The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy, Nat. Cell Biol 9 (2007) 379–390. [DOI] [PubMed] [Google Scholar]
- [180].Willis MS, Min JN, Wang S, Mcdonough H, Lockyer P, Wadosky KM, et al. , Carboxyl terminus of Hsp70-interacting protein (CHIP) is required to modulate cardiac hypertrophy and attenuate autophagy during exercise, Cell Biochem. Funct 31 (2013) 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Brooks CL, Gu W, p53 ubiquitination: Mdm2 and beyond, Mol. Cell 21 (2006) 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Tian LF, Li HY, Jin BF, Pan X, Man JH, Zhang PJ, et al. , MDM2 interacts with and downregulates a sarcomeric protein, TCAP, Biochem. Biophys. Res. Commun 345 (2006) 355–361. [DOI] [PubMed] [Google Scholar]
- [183].Hauck L, Stanley-Hasnain S, Fung A, Grothe D, Rao V, Mak TW, et al. , Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death, PLoS One. 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Métais A, Lamsoul I, Melet A, Uttenweiler-Joseph S, Poincloux R, Stefanovic S, et al. , Asb2α-filamin a axis is essential for actin cytoskeleton remodeling during heart development, Circ. Res 122 (2018) e34–e48. [DOI] [PubMed] [Google Scholar]
- [185].Thottakara T, Friedrich FW, Reischmann S, Braumann S, Schlossarek S, Krämer E, et al. , The E3 ubiquitin ligase Asb2β is downregulated in a mouse model of hypertrophic cardiomyopathy and targets desmin for proteasomal degradation, J. Mol. Cell. Cardiol 87 (2015) 214–224. [DOI] [PubMed] [Google Scholar]
- [186].Spaich S, Will RD, Just S, Spaich S, Kuhn C, Frank D, et al. , F-box and leucine-rich repeat protein 22 is a cardiac-enriched f-box protein that regulates sarcomeric protein turnover and is essential for maintenance of contractile function in vivo, Circ. Res 111 (2012) 1504–1516. [DOI] [PubMed] [Google Scholar]
- [187].Lokireddy S, Wijesoma IW, Sze SK, McFarlane C, Kambadur R, Sharma M, Identification of atrogin-1-targeted proteinsθδ during the myostatin-induced skeletal muscle wasting, Am. J. Physiol. - Cell Physiol 303 (2012). [DOI] [PubMed] [Google Scholar]
- [188].Rafiq K, Guo J, Vlasenko L, Guo X, Kolpakov MA, Sanjay A, et al. , c-Cbl ubiquitin ligase regulates focal adhesion protein turnover and myofibril degeneration induced by neutrophil protease cathepsin G, J. Biol. Chem 287 (2012) 5327–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Henning RH, Brundel BJJM, Proteostasis in cardiac health and disease, Nat. Rev. Cardiol 14 (2017) 637–653. [DOI] [PubMed] [Google Scholar]
- [190].Willis MS, Townley-Tilson WHD, Kang EY, Homeister JW, Patterson C, Sent to destroy: The ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease, Circ. Res 106 (2010) 463–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Johansen T, Lamark T, Selective autophagy mediated by autophagic adapter proteins, Autophagy. 7 (2011) 279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Ye X, Zhou XJ, Zhang H, Exploring the role of autophagy-related gene 5 (ATG5ATG5) yields important insights into autophagy in autoimmune/autoinflammatory diseases, Front. Immunol 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. , The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress, Nat. Med 13 (2007) 619–624. [DOI] [PubMed] [Google Scholar]
- [194].Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. , Autophagy Is Required to Maintain Muscle Mass, Cell Metab. 10 (2009) 507–515. [DOI] [PubMed] [Google Scholar]
- [195].Ulbricht A, Gehlert S, Leciejewski B, Schiffer T, Bloch W, Höhfeld J, Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle, Autophagy. 11 (2015) 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, et al. , Chaperone-Assisted Selective Autophagy Is Essential for Muscle Maintenance, Curr. Biol 20 (2010) 143–148. [DOI] [PubMed] [Google Scholar]
- [197].Collier MP, Reid Alderson T, De Villiers CP, Nicholls D, Gastall HY, Allison TM, et al. , HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C, Sci. Adv 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Wójtowicz I, Jabłońska J, Zmojdzian M, Taghli-Lamallem O, Renaud Y, Junion G, et al. , Drosophila small heat shock protein CryAB ensures structural integrity of developing muscles, and proper muscle and heart performance, Dev. 142 (2015) 994–1005. [DOI] [PubMed] [Google Scholar]
- [199].Stürner E, Behl C, The role of the multifunctional bag3 protein in cellular protein quality control and in disease, Front. Mol. Neurosci 10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Knezevic T, Myers VD, Gordon J, Tilley DG, Sharp TE, Wang J, et al. , BAG3: a new player in the heart failure paradigm, Heart Fail. Rev 20 (2015) 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Domínguez F, Cuenca S, Bilińska Z, Toro R, Villard E, Barriales-Villa R, et al. , Dilated Cardiomyopathy Due to BLC2-Associated Athanogene 3 (BAG3) Mutations, J. Am. Coll. Cardiol 72 (2018) 2471–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Martin TG, Myers VD, Dubey P, Dubey S, Perez E, Moravec CS, et al. , BAG3-dependent autophagy maintains sarcomere function in cardiomyocytes, BioRxiv. (2020). 10.1101/2020.04.10.022319. [DOI] [Google Scholar]
- [203].Yang D, Ma S, Tan Y, Li D, Tang B, Zhang X, et al. , Increased expression of calpain and elevated activity of calcineurin in the myocardium of patients with congestive heart failure, Int. J. Mol. Med 26 (2010) 159–164. [DOI] [PubMed] [Google Scholar]
- [204].Letavernier E, Zafrani L, Perez J, Letavernier B, Haymann JP, Baud L, The role of calpains in myocardial remodelling and heart failure, Cardiovasc. Res 96 (2012) 38–45. [DOI] [PubMed] [Google Scholar]
- [205].Arthur GD, Belcastro AN, A calcium stimulated cysteine protease involved in isoproterenol induced cardiac hypertrophy, Mol. Cell. Biochem 176 (1997) 241–248. [PubMed] [Google Scholar]
- [206].Sandmann S, Prenzel F, Shaw L, Schauer R, Unger T, Activity profile of calpains I and II in chronically infarcted rat myocardium - Influence of the calpain inhibitor CAL 9961, Br. J. Pharmacol 135 (2002) 1951–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Hernando V, Inserte J, Sartório CL, Parra VM, Poncelas-Nozal M, Garcia-Dorado D, Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion, J. Mol. Cell. Cardiol 49 (2010) 271–279. [DOI] [PubMed] [Google Scholar]
- [208].Ke L, Qi XY, Dijkhuis AJ, Chartier D, Nattel S, Henning RH, et al. , Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation, J. Mol. Cell. Cardiol 45 (2008) 685–693. [DOI] [PubMed] [Google Scholar]
- [209].McDonough JL, Arrell DK, Van Eyk JE, Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury, Circ. Res 84 (1999) 9–20. [DOI] [PubMed] [Google Scholar]
- [210].Van Eyk JE, Powers F, Law W, Larue C, Hodges RS, Solaro RJ, Breakdown and release of myofilament proteins during ischemia and ischemia/reperfusion in rat hearts identification of degradation products and effects on the pCa-force relation, Circ. Res 82 (1998) 261–271. [DOI] [PubMed] [Google Scholar]
- [211].Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marban E, Role of troponin I proteolysis in the pathogenesis of stunned myocardium, Circ. Res 80 (1997) 393–399. [PubMed] [Google Scholar]
- [212].Gao WD, Liu Y, Mellgren R, Marban E, Intrinsic Myofilament Alterations Underlying the Decreased Contractility of Stunned Myocardium: A Consequence of Ca2+-Dependent Proteolysis?, Circ. Res 78 (1996) 455–465. [DOI] [PubMed] [Google Scholar]
- [213].Powell SR, Herrmann J, Lerman A, Patterson C, Wang X, The ubiquitin-proteasome system and cardiovascular disease, in: Prog. Mol. Biol. Transl. Sci, 2012: pp. 295–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Wang C, Wang X, The interplay between autophagy and the ubiquitin-proteasome system in cardiac proteotoxicity, Biochim. Biophys. Acta - Mol. Basis Dis 1852 (2015) 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, et al. , Inhibition of autophagy in the heart induces age-related cardiomyopathy, Autophagy. 6 (2010) 600–606. [DOI] [PubMed] [Google Scholar]
- [216].Tarone G, Brancaccio M, Keep your heart in shape: Molecular chaperone networks for treating heart disease, Cardiovasc. Res 102 (2014) 346–361. [DOI] [PubMed] [Google Scholar]
- [217].Sheng Y, Lv S, Huang M, Lv Y, Yu J, Liu J, et al. , Opposing effects on cardiac function by calorie restriction in different-aged mice, Aging Cell. 16 (2017) 1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].De Thonel A, Le Mouël A, Mezger V, Transcriptional regulation of small HSP - HSF1 and beyond, Int. J. Biochem. Cell Biol (2012). [DOI] [PubMed] [Google Scholar]
- [219].Valenzuela V, Jackson KL, Sardi SP, Hetz C, Gene Therapy Strategies to Restore ER Proteostasis in Disease, Mol. Ther 26 (2018) 1404–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, et al. , ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart, Circ. Res 120 (2017) 862–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Moo EK, Herzog W, Single sarcomere contraction dynamics in a whole muscle, Sci. Rep 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, et al. , Cell biology: The kinase domain of titin controls muscle gene expression and protein turnover, Science (80-.) (2005). [DOI] [PubMed] [Google Scholar]
- [223].Kashef F, Li J, Wright P, Snyder J, Suliman F, Kilic A, Higgins RSD, et al. , Ankyrin-B protein in heart failure: Identification of a new component of metazoan cardioprotection, J. Biol. Chem 287 (2012) 30268–30281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Papp Z, Van Der Velden J, Stienen GJM, Calpain-I induced alterations in the cytoskeletal structure and impaired mechanical properties of single myocytes of rat heart, Cardiovasc. Res 45 (2000) 981–993. [DOI] [PubMed] [Google Scholar]
- [225].Cottin P, Poussard S, Mornet D, Brustis JJ, Mohammadpour M, Leger J, Ducastaing A, In vitro digestion of dystrophin by calcium-dependent proteases, calpains I and II, Biochimie. 74 (1992) 565–570. [DOI] [PubMed] [Google Scholar]
- [226].Potz BA, Sabe AA, Abid MR, Sellke FW, Calpains and coronary vascular disease, Circ. J 80 (2015) 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Reimann L, Wiese H, Leber Y, Schwable AN, Fricke AL, Rohland A, et al. , Myofibrillar Z-discs are a protein phosphorylation hot spot with protein kinase C (PKCα) modulating protein dynamics, Mol. Cell. Proteomics 16 (2017) 346–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Barefield DY, McNamara JW, Lynch TL, Kuster DWD, Govindan S, Haar L, et al. , Ablation of the calpain-targeted site in cardiac myosin binding protein-C is cardioprotective during ischemia-reperfusion injury, J. Mol. Cell. Cardiol 129 (2019) 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Previs MJ, Mun JY, Michalek AJ, Beck Previs S, Gulick J, Robbins J, et al. , Phosphorylation and calcium antagonistically tune myosin-binding protein C’s structure and function, Proc. Natl. Acad. Sci. U. S. A 113 (2016) 3239–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Huang J, Forsberg NE, Role of calpain in skeletal-muscle protein degradation, Proc. Natl. Acad. Sci. U. S. A 95 (1998) 12100–12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Moncman CL, Wang K, Targeted disruption of nebulette protein expression alters cardiac myofibril assembly and function, Exp. Cell Res 273 (2002) 204–218. [DOI] [PubMed] [Google Scholar]
- [232].Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, et al. , Anthracyclines Induce Calpain-dependent Titin Proteolysis and Necrosis in Cardiomyocytes, J. Biol. Chem 279 (2004) 8290–8299. [DOI] [PubMed] [Google Scholar]
- [233].Gokhin DS, Tierney MT, Sui Z, Sacco A, Fowler VM, Calpain-mediated proteolysis of tropomodulin isoforms leads to thin filament elongation in dystrophic skeletal muscle, Mol. Biol. Cell 25 (2014) 852–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Di Lisa F, De Tullio R, Salamino F, Barbato R, Melloni E, Siliprandi N, et al. , Specific degradation of troponin T and I by μ-calpain and its modulation by substrate phosphorylation, Biochem. J 308 (1995) 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Van Der Laarse A, Hypothesis: Troponin degradation is one of the factors responsible for deterioration of left ventricular function in heart failure, Cardiovasc. Res 56 (2002) 8–14. [DOI] [PubMed] [Google Scholar]