

Abstract
Most patients with chronic dry eye disease (DED) have episodic flares, which can be triggered by a variety of activities and environmental stresses. These flares are typically associated with rapid exacerbation of discomfort symptoms, followed by prolonged elevation of inflammation. In an acute flare, ocular surface inflammation begins with a nonspecific innate immune response, in some cases followed by a slower but more specific adaptive immune response. At the ocular surface, epithelial cells are central to the innate immune response, and we discuss their role in DED flares alongside the other core components. Epithelial cells and other cells of the innate response (neutrophils, monocytes, macrophages and dendritic cells) trigger flares in response to increased osmolarity, detected via pattern receptors on their cell surface. Ultimately, downstream signaling pathways activate innate and adaptive immune responses, with consequent inflammation and symptoms. In chronic DED, pathogenic T cells have infiltrated the ocular surface tissues. The established adaptive immune response is likely to lead to flare-ups at lower thresholds of stress, with inflammation maintained over a longer period. Increased understanding of the inflammatory cascades activated during a flare may guide management and improve outcomes.
Keywords: adaptive immunity, cornea, conjunctiva, dry eye syndromes, flare, innate immunity, pathology
1. Introduction
Dry eye disease (DED) is a common ocular condition, recognized as one of the most frequent reasons for patients to seek eye care (Bradley et al., 2019). DED characterized by a loss of homeostasis of the tear film, and inflammation of the ocular surface is fundamental to definition of the disease (Craig et al., 2017; Stern et al., 1998). The typical symptoms of DED — including irritation, discomfort, blurred or fluctuating vision — and signs, such as corneal epitheliopathy and loss of conjunctival goblet cells, are also closely associated with ocular surface inflammation.
The onset of DED may be initiated by desiccating and osmotic stress resulting from one or more of a variety of factors. Regardless of the specific initiating event, the disease is then perpetuated by a vicious circle of ocular surface inflammation, tear film instability, and hyperosmolarity (Bron et al., 2017; Ganesalingam et al., 2019; Pflugfelder and de Paiva, 2017; Stern et al., 2010). This results in an ongoing immune response, which is well documented in patients with DED. Numerous inflammatory cytokines associated with both the innate immune response as well as the adaptive T-cell response are elevated in the tear film of patients with DED and inflammatory markers are commonly used for diagnosis and severity grading of DED in clinical practice (Aragona et al., 2015; Lam et al., 2009; Lee et al., 2013). Some of the cytokines and chemokines associated with DED are known neurosensitizers that could lower the threshold required to trigger nerve impulses and potentiate eye discomfort (Watkins et al., 2007). Unfortunately, the cycle persists unless appropriate anti-inflammatory treatment is instituted.
As with other chronic inflammatory conditions, patients with DED generally have flares, typically with rapid exacerbation of symptoms. From the available studies of the natural history, it appears the majority of patients with DED have a chronic disease with an episodic rather than continuous symptom pattern (Lienert et al., 2016). Episodic symptoms can be triggered by a variety of activities and environmental stresses including dry or drafty environments, contact lens overwear, exacerbation of systemic autoimmune diseases, toxic medications and preservatives, allergens, as well as more predictably triggered by cataract and refractive surgery or experimental controlled adverse environments (Calonge et al., 2018). While it is thus widely agreed that the majority of patients with chronic DED experience flares, the pathophysiology is not well understood. Currently, literature on the exacerbation of signs and symptoms that might be termed a flare in DED is sparse. As there are few direct studies of flares, we have focused on the rapid onset of the innate immune response in DED as a model for acute flares, comparing this with the well-characterized adaptive immune responses associated with chronic disease.
2. Methods
Using the National Center for Biotechnology Information PubMed® database, the literature was searched for clinical studies in DED, with search terms “dry eye”, “inflammation”, and “flare”. The search was not restricted to controlled clinical trials because, by definition, flares are difficult to study in these circumstances. Studies of simulated adverse environments and patients undergoing surgery were included, as well as reported studies in animal models and human cell lines. In addition, the bibliographies of references identified by the searches were reviewed.
3. Innate and adaptive immune responses
For the purposes of discussion, we use the term flare to describe an orchestrated response, with a rapid onset followed by prolonged elevation of inflammation – for days, possibly even weeks – as opposed to an exacerbation of symptoms lasting a few hours following exposure to an adverse environment. These flares occur in patients with pre-existing chronic DED who may or may not already be on topical therapy.
In Figure 1, we show the concepts that we propose to underlie flares in DED, contrasting the key roles played by different components in acute flare (Fig. 1A) and flare in chronic DED (Fig. 1B). As shown in Fig. 1A, ocular surface inflammation in an acute flare begins with a rapid but nonspecific innate immune response, that can be followed by a slower but more specific adaptive immune response. At the ocular surface, epithelial cells are core components of the innate immune system. Corneal epithelial cells respond directly to hyperosmolar stress, producing matrix metalloproteinases (MMPs) as well as other mediators (Li et al., 2004). The MMPs are proteolytic enzymes that, in healthy eyes, have roles in physiological desquamation of the corneal epithelium, healing injuries, and remodeling the extracellular matrix. These enzymes can also initiate inflammatory cascades by cleaving pro-cytokines and other extracellular proteins (receptors, growth factors, and adhesion molecules), as well as establishing chemokine gradients. In DED, epithelial cells have also been noted to produce and release chemokines, tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1, IL-6, and IL-8, which amplify the immune response and attract inflammatory cells (Choi et al., 2012; Jones et al., 1994; Solomon et al., 2001). These mediators increase activation of sensory nerves, leading to the symptoms of discomfort associated with flare-ups. Furthermore, desiccating stress can upregulate innate inflammatory pathways in the epithelium, as discussed below.
Fig. 1. Proposed concepts and key components of the innate and immune responses over the time-course of a flare.
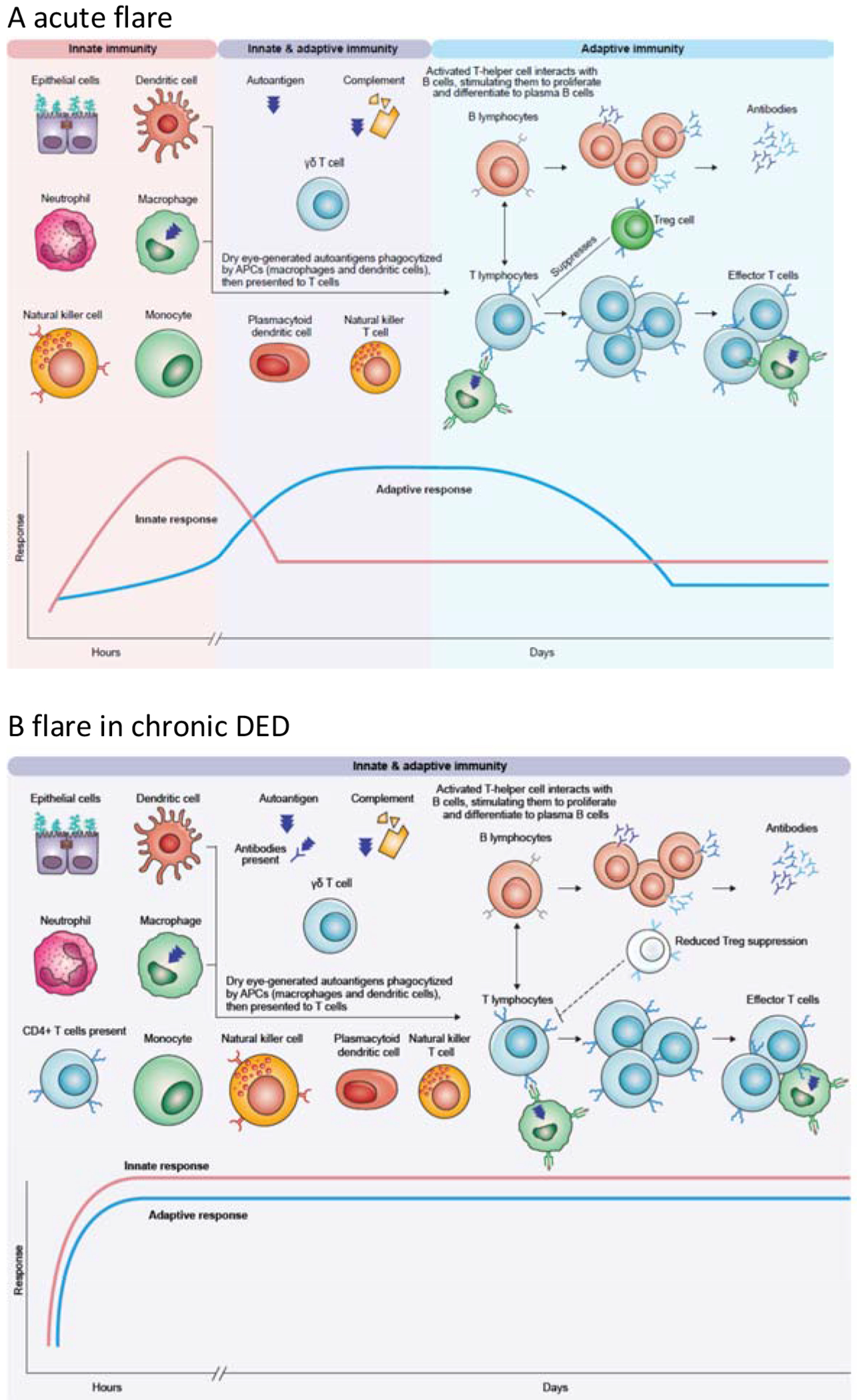
A. In an acute flare, the innate immune system generates a rapid response, which can activate the adaptive response. B. In chronic DED, components of the adaptive immune response are already activated and there are pathogenic T cells in the ocular surface, that leads to rapid increase in inflammation at lower thresholds. See text for details.
APC, antigen-presenting cell
Other key cells in the innate response are the phagocytic antigen-presenting cells (APCs); at the ocular surface, APCs include conventional dendritic cells, monocytes, and macrophages. In healthy eyes, immature APCs are resident in the corneal and conjunctival epithelium, and continuously sample antigens. As their name suggests, these cells present antigens to T cells, which coordinate the adaptive immune response. In dry eye flares, autoantigens are thought to be generated in response to desiccating stress by proteolytic cleavage or altered differentiation, although specific autoantigens are yet to be identified (Niederkorn et al., 2006). After phagocytizing the autoantigen, mature APCs migrate to the draining (cervical) lymph nodes and present the autoantigen to naïve T cells, which are essential for adaptive immune response in DED (Niederkorn et al., 2006; Stern and Pflugfelder, 2017). While antigen presentation is often perceived to be their main role, all these mononuclear phagocytes are also key innate immune cells and secrete proinflammatory cytokines (TNF-α, IL-6, and IL-12) and chemokines when activated. More recently, the role of plasmacytoid dendritic cells has also been recognized (Jamali et al., 2020; Stern and Pflugfelder, 2017). These cells secrete large amounts of interferon-alpha (IFN-α), as well as secreting multiple cytokines and chemokines, potentially playing a significant role in initiating and maintaining the autoimmune response (Jamali et al., 2020; Stern et al., 2013).
In acute inflammation, release of cytokines and MMPs stimulates neutrophils and natural killer (NK) cells to attack damaged cells. Neutrophils migrate to the ocular surface, and detect pathogens directly via recognition of pathogen-associated molecular patterns (PAMPs). Phagocytosis and fusion with the neutrophil granules not only destroys the pathogen but also generates reactive oxygen species (ROS), which can stimulate assembly of inflammasomes (as discussed below). On cell death, neutrophils can also generate neutrophil extracellular traps (NETs) by expelling DNA and other fibers; these NETs appear to form in response to hyperosmolarity, potentially triggering further inflammation (Estua-Acosta et al., 2019; Tibrewal et al., 2014). When activated, neutrophils also release proinflammatory cytokines and chemokines, modulating cells of both the innate and adaptive responses. Similarly, NK cells not only directly kill infected or damaged epithelial cells but also secrete large amounts of cytokines, notably IFN-γ, which activates other cells of the innate response, as well as T cells (Chen et al., 2011; Zhang et al., 2012).
NK T cells (NK cells that express conventional T-cell receptor) and gamma delta (γδ) T cells (T lymphocytes with a distinct T cell receptor) have features of both the innate and adaptive response; as lymphocytes without antigen specificity, they are considered innate cells with some similarities to effector lymphocytes. Furthermore, γδ T cells also secrete significant amounts of IL-17, which is known to diminish activity of regulatory T cells (Tregs) and stimulate MMP-3 and MMP-9 production (De Paiva et al., 2009).
The release of proinflammatory cytokines and action of APCs activates the adaptive immune system, resulting in activation of naive T cells. On activation, CD4+ T cells differentiate into different subsets of effector T cells, including the T-helper (Th) cells that are considered the drivers of adaptive responses in DED. After activation, Th1 and Th17 cells infiltrate the ocular surface, and secrete proinflammatory cytokines. Th1 cells secrete IFN-γ, which upregulates the production of chemokines, chemokine receptors, human leukocyte antigen (HLA), and cell adhesion molecules that facilitate migration of Th1 and Th17 cells to the ocular surface, as well as secondary stimulation of these cells on the ocular surface (Schaumburg et al., 2011). Notably for the long-term consequences, Th1-secreted IFN-γ inhibits secretion from goblet cells and promotes apoptosis of these cells (Garcia-Posadas et al., 2016). Th17 cells secrete IL-17, which recruits monocytes and neutrophils to the inflammation site, and further promotes epithelial damage by stimulating the production of proinflammatory cytokines and MMPs. Activated T cells also interact with B lymphocytes via cell-to-cell interaction and cytokine production, promoting expansion and differentiation to plasma B cells (Subbarayal et al., 2016). The role of B lymphocytes in DED is less well described than that of T cells, and while their key role is producing (auto)antibodies (Stern et al., 2012), they also secrete cytokines and serve as APCs. In healthy eyes, Tregs suppress T-cell activity to restore balance and prevent chronic inflammation (Siemasko et al., 2008), but Tregs are diminished in DED (Niederkorn et al., 2006).
Patients with chronic DED will probably already have an established adaptive immune response (Brignole-Baudouin et al., 2017); consequently, inflammation will flare up more easily in response to adverse environments, and inflammation will also be maintained over a longer period (Fig. 1B). In mouse models of DED, anamnestic response has been observed for a prolonged period after return to a normal environment (Chen et al., 2014). This certainly seems to reflect the general experience in the clinic that patients with autoimmune DED, such as Sjögren’s syndrome, may have a heightened inflammatory response to dry and drafty environments. This would suggest that their threshold for a prolonged flare following an innate response is lowered, and the adaptive response is amplified and more rapidly activated than in people without chronic DED. This may be suppressed by immunomodulatory treatments, but to our knowledge it has not been studied. Indeed, given the difficulties of studying flares in practice, there is little solid evidence to demonstrate a reduced threshold, yet available studies do show patients with chronic DED have a greater increase in inflammatory markers compared to people without DED. A study comparing responses to a controlled adverse desiccating environment for 20 patients with mild-to-moderate DED versus 20 healthy volunteers showed increases in proinflammatory tear molecules, including IL-1 receptor antagonist (IL-1 RA), MMP-9, IL-6, and IL-8 (Fernandez et al., 2019). In a study of patients the day after cataract surgery, patients with DED had greater increases in inflammatory cytokines versus patients without DED, with significant increases in IL-1β, IL-6, IL-8, monocyte chemoattractant protein-1 (MCP-1), TNF-α, and IFN-γ (Park et al., 2016). In a study using a controlled environment that simulated an in-flight air cabin, IL-6 and MMP-9 were both significantly increased (Teson et al., 2013). Patients with DED also had rapid decreases in epidermal growth factor (EGF), an essential growth factor for maintaining homeostasis of the ocular surface epithelium. In addition, it appears that patients with chronic DED already have increased levels of immune markers that can initiate adaptive immune responses, such as HLA-DR, a key molecule required for antigen presentation to T-helper (CD4+) cells (Brignole-Baudouin et al., 2017). In controlled environment studies, rapid activation of APCs has also been indicated, when expression of HLA-DR was observed in patients with DED following exposure to a controlled adverse environment for 90 minutes (Moore et al., 2015).
4. Ocular surface changes
A flare results from a complex series of processes, which can be illustrated by comparing a healthy eye without inflammation, an acute flare, and a flare in chronic DED. For these three scenarios, Fig. 2 shows the staining likely to be observed, and compares the key elements of the cornea and conjunctiva for normal tissue and in acute flare or chronic dry eye. In healthy eyes (Fig. 2 left-hand panel), the ocular surface is protected and lubricated by a stable tear film that contains antimicrobial factors including defensins, immunoglobulin (Ig) A, lactoferrin, and lysozyme (produced by the lacrimal glands and the epithelial cells), to defend against bacteria. The epithelial cells of the ocular surface are tightly aligned, with intact epithelial tight junctions, and coated with a dense glycocalyx. Epithelial cells secrete soluble mucins, which help clear pathogens and allergens, as well as membrane-bound mucins, which are the main constituents of the glycocalyx and may have a role in sensing tear osmolarity (Mantelli and Argueso, 2008). Conjunctival epithelium is continuous with the corneal epithelium, and contains numerous goblet cells, which secrete mucins, notably the gel-forming mucin MUC5AC. The resulting gel is spread over the whole ocular surface, and has an integral role in maintaining tear stability. Goblet cells also produce cytokines, EGF, and retinoic acid, which all help to maintain immune tolerance, preventing activation of antigen-presenting cells (Alam et al., 2020). Resident immune cells are essential for defense against microbes, and NK cells, dendritic cells, macrophages, and γδ/CD4+/CD8+ T cells are all found in the conjunctival epithelium of normal eyes. Nerve endings located between the stratified corneal epithelial cells are shielded from environmental and inflammatory stimulation by epithelial tight junctions, extracellular matrix and glycocalyx, resulting in minimal nociceptor firing.
Fig. 2. Normal tissue and disruptions in acute flare or chronic dry eye.
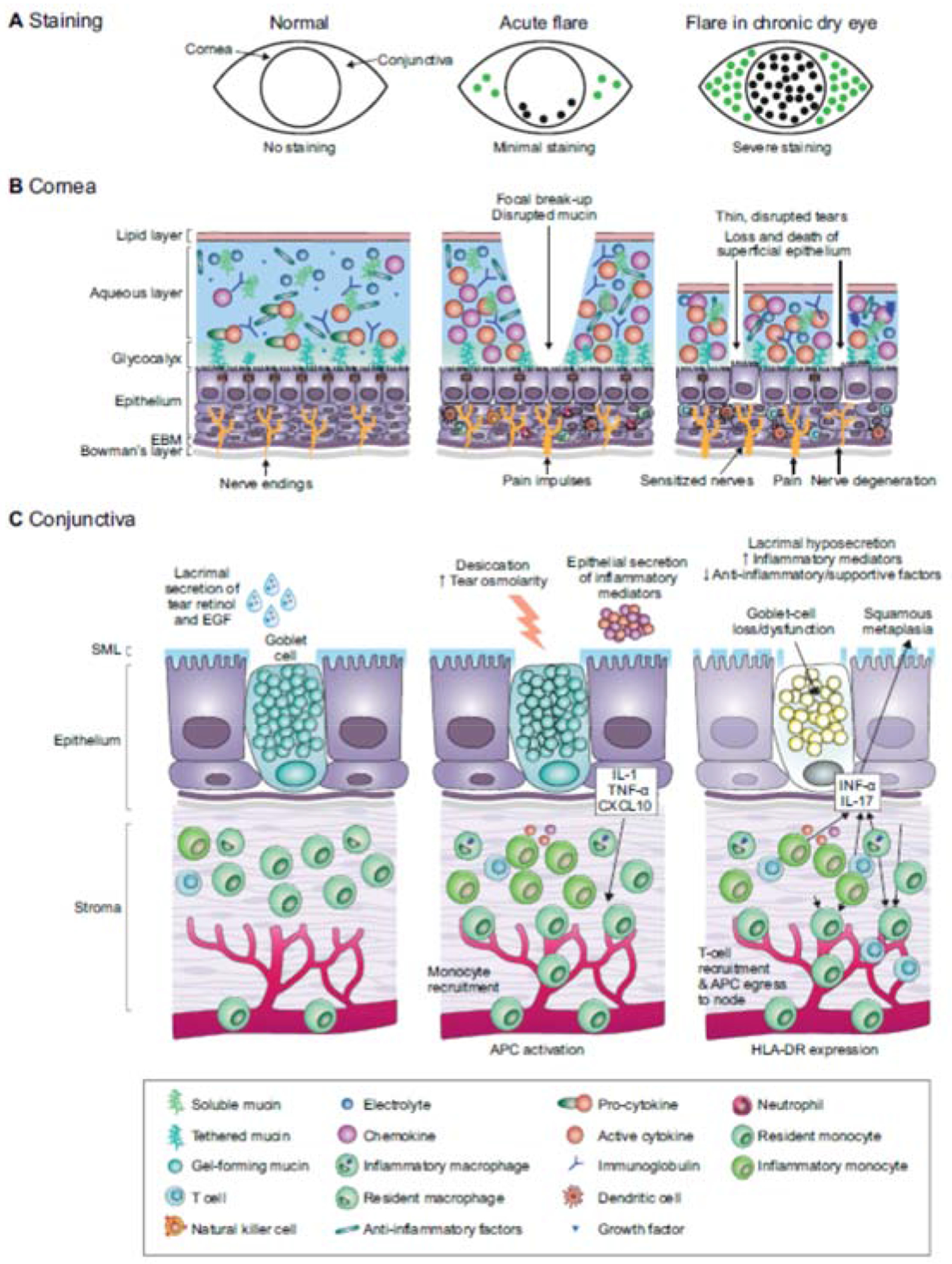
A. Patients with acute flares typically have minimal cornea and conjunctival dye staining, whereas there may be moderate-to-severe superficial punctate epitheliopathy and dye staining in chronic DED.
B. Focal tear breakup and osmotic stress activate corneal epithelium and immune cells to produce innate inflammatory mediators (center). In chronic DED, cornea barrier disruption and epithelial cell loss is accompanied by infiltration of myeloid and T cells.
C. In an acute flare, inflammatory mediators are produced by stressed conjunctival epithelial cells, including goblet cells that recruit inflammatory cells (center). In chronic DED, goblet cells are lost or dysfunctional, leading to reduced gel-forming mucin secretion that destabilizes the tear film and reduced production of anti-inflammatory factors by the goblet cells (right).
APC, antigen-presenting cell; EBM, epithelial basement membrane; EGF, epidermal growth factor; IL, interleukin; INF-α, interferon alpha; SML, secretory mucus layer; TNF-α, tumor necrosis factor alpha
In an acute flare (Fig. 2 center panel), little damage to the epithelium would be expected, as indicated by the low-grade of staining typically observed. The bulk aqueous/mucin layer of the tear film has not thinned as in chronic DED, but desiccating stress leads to increase in osmolarity, and breakup is observed in the precorneal tear film. These changes stimulate epithelial and immune cells to release cytokines, chemokines, and MMPs; the so-called cytokine storm. Release of cytokines and chemokines leads to activation of the innate immune response, which can sensitize nociceptors and cause pain, as well as activation of APCs, which mature and migrate to the lymph node, stimulating the adaptive immune response by presenting autoantigen to naive T lymphocytes. Differentiated effector CD4+ T cells can migrate to the ocular surface, in turn secreting cytokines that can cause epithelial disease and amplify the innate response, driving the ongoing flare.
In established DED (Fig. 2 right-hand panel), the chronically elevated levels of MMP-9 lead to degradation of the extracellular matrix and promote epithelial cell loss and barrier disruption. Eventually, tear hyperosmolarity leads to reduced epithelial cell volume, cell death, and desquamation. Dye staining in these eyes shows extensive damage to the epithelial cells of the cornea and conjunctiva, reflecting disruption of epithelial tight junctions or a compromised glycocalyx in the cornea, or cell membrane damage in the conjunctiva (Uchino, 2018). In the conjunctival epithelium, long-term stimulation with the proinflammatory cytokines IL-1 and IFN-γ causes squamous metaplasia and goblet cell secretory dysfunction and loss (Coursey et al., 2016; De Paiva et al., 2007; Garcia-Posadas et al., 2016; Solomon et al., 2001). As the amount of mucus secreted by goblet cells decreases, the lipid layer thins, further destabilizing the tear film.
In chronic DED, inflammation includes infiltration of the conjunctiva, cornea, and lacrimal glands by differentiated (antigen-specific) T cells, which secrete proinflammatory cytokines, and levels of tear cytokines are perpetually elevated (Chen et al., 2014). The proportion of Treg cells, which would normally suppress antigen-specific T cells, is reduced, and the secreted cytokines continue to re-trigger the innate immune system. The number of dendritic cells is increased; furthermore, a higher proportion are mature, as determined by expression of the antigen-presenting molecule HLA-DR and costimulatory molecules. The immune response to environmental factors that cause flares may be modulated in patients on immunomodulatory therapy. Cyclosporine inhibits T cell factors, while lifitegrast suppresses formation of the immune synapse between APCs and T cells, but neither suppress acute stress induced production of innate inflammatory mediators.
Damage to the epithelium exposes free nerve endings, but intraepithelial sensory nerve morphology and function is also altered (Stepp et al., 2018a). Changes are also seen in the density and structure of nerves, such as nerve sprouting and thickened stromal nerves (Benitez del Castillo et al., 2004; Simsek et al., 2019; Stepp et al., 2018b; Tuisku et al., 2008). Patients with chronic DED experience symptoms rapidly in response to triggers, presumably because the adaptive response is not required for nerve sensitization: innate signals during flare-up alter sensory nerves (nociceptors) and provoke rapid-onset of pain and DED symptoms. Inflammatory cytokines sensitize nociceptors, essentially making the nerves more sensitive by lowering their stimulation thresholds and increasing firing. Inflammatory mediators may trigger spontaneous, stimulus-independent pain impulses by nociceptors.
The adaptive response in chronic DED may also cause nerve alteration. Corneal nerve damage from chronic DED may cause spontaneous nerve firing, but can also reduce release of neuropeptides from nerve termini. This impact is unclear, as some neuropeptides can enhance proinflammatory cytokine production. Deregulation, inflammation, or loss of corneal nerves or nerve function may result in the loss of immune privilege and increased inflammation (Hamrah et al., 2016; Paunicka et al., 2015).
5. Priming of inflammation pathways
A flare begins when epithelial and resident immune cells of the ocular surface encounter a danger signal, such as refractive surgeries, or an environmental trigger. Desiccation of the ocular surface has been demonstrated to be a potent initiator of innate immune response (Corrales et al., 2006; Luo et al., 2005). Other triggers might be high osmolarity, various pollutants or allergens — or pathogens — but regardless of the specific event, they will trigger a flare via an inflammation pathway.
Fig. 3 shows how signal pathways prime inflammatory cascades in innate immune cells of the ocular surface. Signals are detected by cell surface receptors, which detect a variety of stimuli such as PAMPs, danger-associated molecular patterns (DAMPs), or ROS. This signal sets in motion a pathway of downstream effects, which activate nuclear factor-κB (NFκB) or other transcription factors (Guzman et al., 2016; Luo et al., 2005; Luo et al., 2004; Pelegrino et al., 2012). NFκB controls transcription of many genes involved in the inflammatory response, including proinflammatory cytokines. These pathways promote maturation of APCs, initiating the inflammatory cascade. In chronic DED, increase in pathways of antigen presentation has the potential to create a hyperactive antigen-presentation environment where adaptive immunity is rapidly ramped up (Schaumburg et al., 2011).
Fig. 3. Signal pathways priming inflammatory cascades.
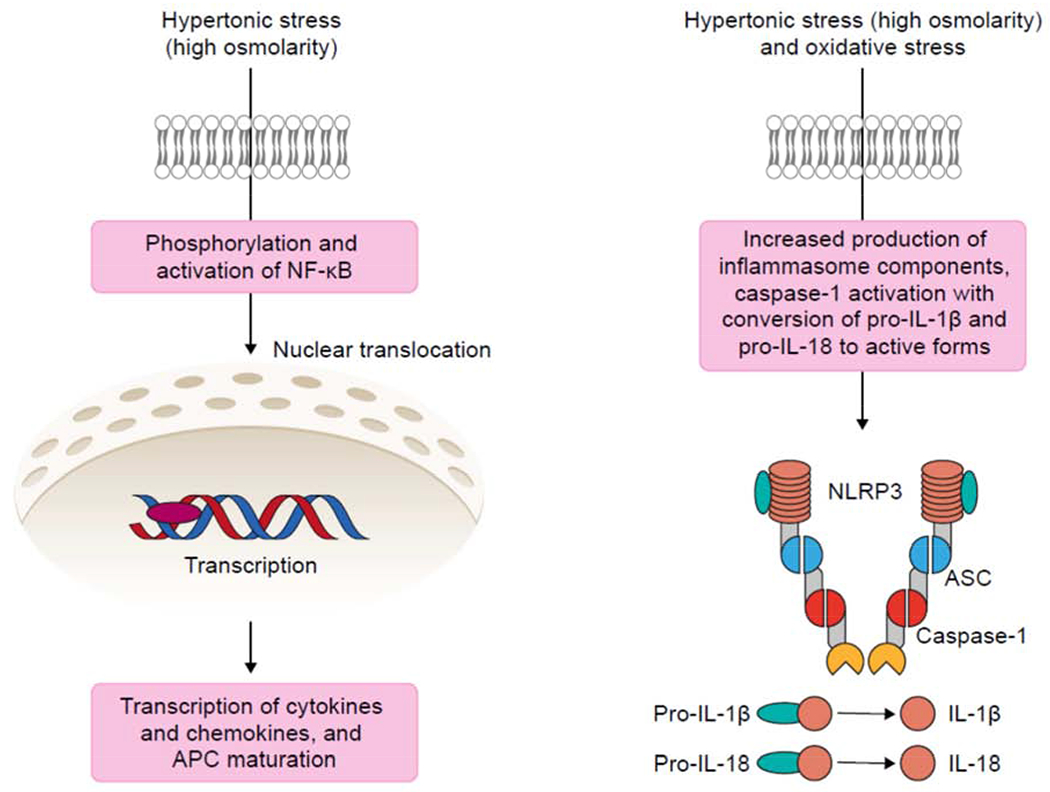
Innate immune cells (macrophages, dendritic cells, and neutrophils) and epithelial cells of the ocular surface detect hyperosmolarity via pattern receptors. Activation of signal transduction pathways, such as NFκB lead to production of proinflammatory cytokines and trigger innate and adaptive immune responses. Activation of the NLRP3 inflammasome results in IL-1B and IL-18 activation. See text for details.
APC, antigen-presenting cell; ASC, apoptosis speck-like protein; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3.
NF-κB is also known to regulate transcription of inflammasome components, in response to extracellular stimuli such as high osmolarity. Activation of inflammasome pathways is recognized as a key priming stage of epithelial inflammation (Chi et al., 2017). In essence, inflammasomes are multiprotein structures assembled in the cytoplasm of the epithelial cells, in response to extracellular signals (Fig. 3). Once assembled, the inflammasome activates caspase-1, which in turn activates proinflammatory cytokines IL-1β and IL-18.
Inflammasomes are molecular platforms, assembled from eight proteins – these vary with the specific inflammasome type, but the central protein is always a member of the protein family NLRP (NOD-, LRR- and pyrin domain-containing protein). Studies suggest NLRP3 is the key inflammasome protein in DED, via the ROS-NLRP3-IL-1β signaling axis (Chi et al., 2017; Niu et al., 2015; Zheng et al., 2014; Zheng et al., 2015), although NLRP6 inflammasomes have also been observed in models of DED (Chi et al., 2017).
On activation, the central protein (NLRP3 in the scenario shown in Fig. 3) recruits the adaptor apoptosis speck-like protein (ASC). ASC is activated, and cleaves procaspase to caspase-1. Active caspase-1 then cleaves the proinflammatory cytokines prointerleukin-1β and prointerleukin-18 to their active forms, IL-1β and IL-18. Inflammasome activation can lead to death of the cell via pyroptosis (cell death via rupture of plasma membrane) with subsequent release of intracellular components, releasing the active cytokines and initiating the immune response.
6. Anti-inflammatory therapies
Anti-inflammatory therapies have the potential to inhibit various points in the activation or maintenance of a flare. Of these agents, corticosteroids are probably the most familiar, yet the precise molecular mechanisms by which they modulate inflammation is not clear. The key mechanism is thought to be inhibiting binding of the transcription factor NF-κB to DNA, thus reducing transcription of cytokine genes (Lan et al., 2012). Given the central role of NF-κB in activation of inflammation, this proposed mechanism is supported by the broad-spectrum anti-inflammatory effects of corticosteroids. A corticosteroid can down-regulate both innate and adaptive immune response pathways, and inhibit signaling pathways and transcription of relevant cytokine and chemokines within hours. In mouse models of DED, corticosteroids suppress IL-1, IL-6, and MMP-9 (Bian et al., 2016), as well as activation of mitogen-activated protein (MAP) kinase stress signaling pathways (De Paiva et al., 2006b).
Other anti-inflammatory therapies used by clinicians for treating flares include autologous blood products (serum and plasma). By their nature, these include a variety of anti-inflammatory factors, such as vitamin A and transforming growth factor-β (TGF-β), as well as factors that can down-regulate inflammation by binding proinflammatory cytokines, in addition to promoting longer-term epithelial repair (Tsubota et al., 1999). Antibiotics with anti-inflammatory activity may also be used, namely doxycycline and azithromycin. Both of these antibiotics have been found to suppress production of MMP-9 and inflammatory cytokines, and doxycycline preserved corneal barrier function in the mouse desiccating stress model (De Paiva et al., 2006a; De Paiva et al., 2006b; Li et al., 2010).
Immunomodulatory therapies that focus on long-term inflammation in DED include cyclosporine and tacrolimus (calcineurin inhibitors) and lifitegrast (an antagonist of lymphocyte function-associated antigen-1 [LFA-1]). As with corticosteroids, these agents have been shown to improve DED signs and symptoms in clinical trials but — as expected — this is seen over a longer-term period of weeks to months (Chan and Prokopich, 2019; Moscovici et al., 2015; Tuan et al., 2020). Calcineurin inhibitors increase tear production and inhibit T cell activation, and cyclosporine treatment has been shown to modulate cytokines in mouse models (Daull et al., 2019). Lifitegrast also disrupts various key steps in T-cell-mediated inflammation, by blocking the interaction of intercellular adhesion molecule with LFA-1, which is critical in migration of dendritic cells to lymph nodes, naïve T-cell activation by dendritic cells, and T-cell transmigration into the ocular surface (Pflugfelder et al., 2017).
7. Conclusions and future research
When we consider DED as a chronic autoimmune condition, it is not surprising that most patients experience flares of inflammation. In chronic DED, the innate immune system is primed to react to hypertonic and desiccating stress, triggering an orchestrated immune response with rapid onset of symptoms. Whether such flares influence progression is unknown and in fact, although DED is generally accepted to be a progressive disease, relatively little information is available on the natural history. Long-term studies of associations between flares and signs, symptoms, and progression of DED would therefore be valuable. Additional research is needed to better understand the pathogenesis and durations of acute flares and flares in chronic DED. Identification of biomarkers associated with these flares could lead to potential new therapeutic targets. Understanding of the inflammatory cascades activated during a flare may help to identify specific “flare targets” and guide management and we are optimistic that additional research will further improve outcomes in the future.
Highlights.
Episodic inflammatory flares occur in most patients with chronic dry eye disease.
Flares typically show rapid onset followed by prolonged exacerbation of symptoms.
Flares result from complex inflammatory cascades.
Increased understanding of flares may guide management and improve outcomes.
Author contributions and acknowledgments
All authors made substantial contributions to concepts discussed in the manuscript content, participated in drafting the article and revising it critically for important intellectual content, and approved of the final version. The authors thank Geraldine Thompson, of Engage Scientific Solutions, Horsham, UK, who provided medical writing assistance funded by Kala Pharmaceuticals, Inc.
Abbreviations
- ASC
apoptosis speck-like protein
- APC
antigen-presenting cell
- DED
dry eye disease
- EGF
epidermal growth factor
- HLA
human leukocyte antigen
- IFN
interferon
- IL
interleukin
- LFA-1
lymphocyte function-associated antigen-1
- MAP
mitogen-activated protein
- MMP
matrix metalloproteinase
- NETs
neutrophil extracellular traps
- NLRP3
NOD-, LRR- and pyrin domain-containing protein 3
- PAMP
pathogen-associated molecular pattern
- ROS
reactive oxygen species
- TNF
tumor necrosis factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
V.L.P., S.C.P., and M.E.S. are consultants for Kala Pharmaceuticals, Inc. The authors received no compensation related to the development of the manuscript.
References
- Alam J, de Paiva CS, Pflugfelder SC, 2020. Immune–Goblet cell interaction in the conjunctiva. Ocul. Surf 18, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragona P, Aguennouz M, Rania L, Postorino E, Sommario MS, Roszkowska AM, De Pasquale MG, Pisani A, Puzzolo D, 2015. Matrix metalloproteinase 9 and transglutaminase 2 expression at the ocular surface in patients with different forms of dry eye disease. Ophthalmology 122, 62–71. [DOI] [PubMed] [Google Scholar]
- Benitez del Castillo JM, Wasfy MA, Fernandez C, Garcia-Sanchez J, 2004. An in vivo confocal masked study on corneal epithelium and subbasal nerves in patients with dry eye. Invest. Ophthalmol. Vis. Sci 45, 3030–3035. [DOI] [PubMed] [Google Scholar]
- Bian F, Pelegrino FS, Henriksson JT, Pflugfelder SC, Volpe EA, Li DQ, de Paiva CS, 2016. Differential effects of dexamethasone and doxycycline on inflammation and MMP production in murine alkali-burned corneas associated with dry eye. Ocul. Surf 14, 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley JL, Ozer Stillman I, Pivneva I, Guerin A, Evans AM, Dana R, 2019. Dry eye disease ranking among common reasons for seeking eye care in a large US claims database. Clin. Ophthalmol 13, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignole-Baudouin F, Riancho L, Ismail D, Deniaud M, Amrane M, Baudouin C, 2017. Correlation between the inflammatory marker HLA-DR and signs and symptoms in moderate to severe dry eye disease. Invest. Ophthalmol. Vis. Sci 58, 2438–2448. [DOI] [PubMed] [Google Scholar]
- Bron AJ, de Paiva CS, Chauhan SK, Bonini S, Gabison EE, Jain S, Knop E, Markoulli M, Ogawa Y, Perez V, Uchino Y, Yokoi N, Zoukhri D, Sullivan DA, 2017. TFOS DEWS II pathophysiology report. Ocul. Surf 15, 438–510. [DOI] [PubMed] [Google Scholar]
- Calonge M, Labetoulle M, Messmer EM, Shah S, Akova YA, Boboridis KG, Merayo-Lloves J, Aragona P, Benitez-Del-Castillo J, Geerling G, Rolando M, Baudouin C, 2018. Controlled adverse environment chambers in dry eye research. Curr. Eye Res 43, 445–450. [DOI] [PubMed] [Google Scholar]
- Chan CC, Prokopich CL, 2019. Lifitegrast ophthalmic solution 5.0% for treatment of dry eye disease: overview of clinical trial program. J. Pharm. Pharm. Sci 22, 49–56. [DOI] [PubMed] [Google Scholar]
- Chen Y, Chauhan SK, Lee HS, Saban DR, Dana R, 2014. Chronic dry eye disease is principally mediated by effector memory Th17 cells. Mucosal Immunol. 7, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chauhan SK, Saban DR, Sadrai Z, Okanobo A, Dana R, 2011. Interferon-γ-secreting NK cells promote induction of dry eye disease. J. Leukoc. Biol 89, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi W, Hua X, Chen X, Bian F, Yuan X, Zhang L, Wang X, Chen D, Deng R, Li Z, Liu Y, de Paiva CS, Pflugfelder SC, Li DQ, 2017. Mitochondrial DNA oxidation induces imbalanced activity of NLRP3/NLRP6 inflammasomes by activation of caspase-8 and BRCC36 in dry eye. J. Autoimmun 80, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Li Z, Oh HJ, Im SK, Lee SH, Park SH, You IC, Yoon KC, 2012. Expression of CCR5 and its ligands CCL3, −4, and −5 in the tear film and ocular surface of patients with dry eye disease. Curr. Eye Res 37, 12–17. [DOI] [PubMed] [Google Scholar]
- Corrales RM, Stern ME, De Paiva CS, Welch J, Li DQ, Pflugfelder SC, 2006. Desiccating stress stimulates expression of matrix metalloproteinases by the corneal epithelium. Invest. Ophthalmol. Vis. Sci 47, 3293–3302. [DOI] [PubMed] [Google Scholar]
- Coursey TG, Tukler Henriksson J, Barbosa FL, de Paiva CS, Pflugfelder SC, 2016. Interferon-γ–induced unfolded protein response in conjunctival goblet cells as a cause of mucin deficiency in Sjögren syndrome. Am. J. Pathol 186, 1547–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig JP, Nichols KK, Akpek EK, Caffery B, Dua HS, Joo CK, Liu Z, Nelson JD, Nichols JJ, Tsubota K, Stapleton F, 2017. TFOS DEWS II definition and classification report. Ocul. Surf 15, 276–283. [DOI] [PubMed] [Google Scholar]
- Daull P, Barabino S, Feraille L, Kessal K, Docquier M, Parsadaniantz SM, Baudouin C, Garrigue JS, 2019. Modulation of inflammation-related genes in the cornea of a mouse model of dry eye upon treatment with cyclosporine eye drops. Curr. Eye Res 44, 476–485. [DOI] [PubMed] [Google Scholar]
- De Paiva CS, Chotikavanich S, Pangelinan SB, Pitcher JD 3rd, Fang B, Zheng X, Ma P, Farley WJ, Siemasko KF, Niederkorn JY, Stern ME, Li DQ, Pflugfelder SC, 2009. IL-17 disrupts corneal barrier following desiccating stress. Mucosal Immunol. 2, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paiva CS, Corrales RM, Villarreal AL, Farley W, Li DQ, Stern ME, Pflugfelder SC, 2006a. Apical corneal barrier disruption in experimental murine dry eye is abrogated by methylprednisolone and doxycycline. Invest. Ophthalmol. Vis. Sci 47, 2847–2856. [DOI] [PubMed] [Google Scholar]
- De Paiva CS, Corrales RM, Villarreal AL, Farley WJ, Li DQ, Stern ME, Pflugfelder SC, 2006b. Corticosteroid and doxycycline suppress MMP-9 and inflammatory cytokine expression, MAPK activation in the corneal epithelium in experimental dry eye. Exp. Eye Res 83, 526–535. [DOI] [PubMed] [Google Scholar]
- De Paiva CS, Villarreal AL, Corrales RM, Rahman HT, Chang VY, Farley WJ, Stern ME, Niederkorn JY, Li DQ, Pflugfelder SC, 2007. Dry eye-induced conjunctival epithelial squamous metaplasia is modulated by interferon-γ. Invest. Ophthalmol. Vis. Sci 48, 2553–2560. [DOI] [PubMed] [Google Scholar]
- Estua-Acosta GA, Zamora-Ortiz R, Buentello-Volante B, Garcia-Mejia M, Garfias Y, 2019. Neutrophil extracellular traps: current perspectives in the eye. Cells 8, 979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez I, Lopez-Miguel A, Enriquez-de-Salamanca A, Teson M, Stern ME, Gonzalez-Garcia MJ, Calonge M, 2019. Response profiles to a controlled adverse desiccating environment based on clinical and tear molecule changes. Ocul. Surf 17, 502–515. [DOI] [PubMed] [Google Scholar]
- Ganesalingam K, Ismail S, Sherwin T, Craig JP, 2019. Molecular evidence for the role of inflammation in dry eye disease. Clin. Exp. Optom 102, 446–454. [DOI] [PubMed] [Google Scholar]
- Garcia-Posadas L, Hodges RR, Li D, Shatos MA, Storr-Paulsen T, Diebold Y, Dartt DA, 2016. Interaction of IFN-γ with cholinergic agonists to modulate rat and human goblet cell function. Mucosal Immunol. 9, 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M, Keitelman I, Sabbione F, Trevani AS, Giordano MN, Galletti JG, 2016. Desiccating stress-induced disruption of ocular surface immune tolerance drives dry eye disease. Clin. Exp. Immunol 184, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamrah P, Seyed-Razavi Y, Yamaguchi T, 2016. Translational immunoimaging and neuroimaging demonstrate corneal neuroimmune crosstalk. Cornea 35 Suppl 1, S20–S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamali A, Kenyon B, Ortiz G, Abou-Slaybi A, Sendra VG, Harris DL, Hamrah P, 2020. Plasmacytoid dendritic cells in the eye. Prog. Retin. Eye Res, 100877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Monroy D, Ji Z, Atherton SS, Pflugfelder SC, 1994. Sjögren’s syndrome: cytokine and Epstein-Barr viral gene expression within the conjunctival epithelium. Invest. Ophthalmol. Vis. Sci 35, 3493–3504. [PubMed] [Google Scholar]
- Lam H, Bleiden L, de Paiva CS, Farley W, Stern ME, Pflugfelder SC, 2009. Tear cytokine profiles in dysfunctional tear syndrome. Am. J. Ophthalmol 147, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan W, Petznick A, Heryati S, Rifada M, Tong L, 2012. Nuclear factor-κB: central regulator in ocular surface inflammation and diseases. Ocul. Surf 10, 137–148. [DOI] [PubMed] [Google Scholar]
- Lee SY, Han SJ, Nam SM, Yoon SC, Ahn JM, Kim TI, Kim EK, Seo KY, 2013. Analysis of tear cytokines and clinical correlations in Sjögren syndrome dry eye patients and non-Sjögren syndrome dry eye patients. Am. J. Ophthalmol 156, 247–253. [DOI] [PubMed] [Google Scholar]
- Li DQ, Chen Z, Song XJ, Luo L, Pflugfelder SC, 2004. Stimulation of matrix metalloproteinases by hyperosmolarity via a JNK pathway in human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci 45, 4302–4311. [DOI] [PubMed] [Google Scholar]
- Li DQ, Zhou N, Zhang L, Ma P, Pflugfelder SC, 2010. Suppressive effects of azithromycin on zymosan-induced production of proinflammatory mediators by human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci 51, 5623–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienert JP, Tarko L, Uchino M, Christen WG, Schaumberg DA, 2016. Long-term natural history of dry eye disease from the patient’s perspective. Ophthalmology 123, 425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Li DQ, Corrales RM, Pflugfelder SC, 2005. Hyperosmolar saline is a proinflammatory stress on the mouse ocular surface. Eye Contact Lens 31, 186–193. [DOI] [PubMed] [Google Scholar]
- Luo L, Li DQ, Doshi A, Farley W, Corrales RM, Pflugfelder SC, 2004. Experimental dry eye stimulates production of inflammatory cytokines and MMP-9 and activates MAPK signaling pathways on the ocular surface. Invest. Ophthalmol. Vis. Sci 45, 4293–4301. [DOI] [PubMed] [Google Scholar]
- Mantelli F, Argueso P, 2008. Functions of ocular surface mucins in health and disease. Curr. Opin. Allergy Clin. Immunol 8, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore QL, De Paiva CS, Pflugfelder SC, 2015. Effects of dry eye therapies on environmentally induced ocular surface disease. Am. J. Ophthalmol 160, 135–142 e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscovici BK, Holzchuh R, Sakassegawa-Naves FE, Hoshino-Ruiz DR, Albers MB, Santo RM, Hida RY, 2015. Treatment of Sjogren’s syndrome dry eye using 0.03% tacrolimus eye drop: Prospective double-blind randomized study. Cont Lens Anterior Eye 38, 373–378. [DOI] [PubMed] [Google Scholar]
- Niederkorn JY, Stern ME, Pflugfelder SC, De Paiva CS, Corrales RM, Gao J, Siemasko K, 2006. Desiccating stress induces T cell-mediated Sjögren’s syndrome-like lacrimal keratoconjunctivitis. J. Immunol 176, 3950–3957. [DOI] [PubMed] [Google Scholar]
- Niu L, Zhang S, Wu J, Chen L, Wang Y, 2015. Upregulation of NLRP3 inflammasome in the tears and ocular surface of dry eye patients. PLoS One 10, e0126277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Hwang HB, Kim HS, 2016. Observation of influence of cataract surgery on the ocular surface. PLoS One 11, e0152460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paunicka KJ, Mellon J, Robertson D, Petroll M, Brown JR, Niederkorn JY, 2015. Severing corneal nerves in one eye induces sympathetic loss of immune privilege and promotes rejection of future corneal allografts placed in either eye. Am. J. Transplant 15, 1490–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrino FS, Pflugfelder SC, De Paiva CS, 2012. Low humidity environmental challenge causes barrier disruption and cornification of the mouse corneal epithelium via a c-jun N-terminal kinase 2 (JNK2) pathway. Exp. Eye Res. 94, 150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflugfelder SC, de Paiva CS, 2017. The pathophysiology of dry eye disease: what we know and future directions for research. Ophthalmology 124, S4–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflugfelder SC, Stern M, Zhang S, Shojaei A, 2017. LFA-1/ICAM-1 interaction as a therapeutic target in dry eye disease. J. Ocul. Pharmacol. Ther 33, 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaumburg CS, Siemasko KF, De Paiva CS, Wheeler LA, Niederkorn JY, Pflugfelder SC, Stern ME, 2011. Ocular surface APCs are necessary for autoreactive T cell-mediated experimental autoimmune lacrimal keratoconjunctivitis. J. Immunol 187, 3653–3662. [DOI] [PubMed] [Google Scholar]
- Siemasko KF, Gao J, Calder VL, Hanna R, Calonge M, Pflugfelder SC, Niederkorn JY, Stern ME, 2008. In vitro expanded CD4+CD25+Foxp3+ regulatory T cells maintain a normal phenotype and suppress immune-mediated ocular surface inflammation. Invest. Ophthalmol. Vis. Sci 49, 5434–5440. [DOI] [PubMed] [Google Scholar]
- Simsek C, Kojima T, Nagata T, Dogru M, Tsubota K, 2019. Changes in murine subbasal corneal nerves after scopolamine-induced dry eye stress exposure. Invest. Ophthalmol. Vis. Sci 60, 615–623. [DOI] [PubMed] [Google Scholar]
- Solomon A, Dursun D, Liu Z, Xie Y, Macri A, Pflugfelder SC, 2001. Pro- and anti-inflammatory forms of interleukin-1 in the tear fluid and conjunctiva of patients with dry-eye disease. Invest. Ophthalmol. Vis. Sci 42, 2283–2292. [PubMed] [Google Scholar]
- Stepp MA, Pal-Ghosh S, Tadvalkar G, Williams A, Pflugfelder SC, de Paiva CS, 2018a. Reduced intraepithelial corneal nerve density and sensitivity accompany desiccating stress and aging in C57BL/6 mice. Exp. Eye Res 169, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepp MA, Pal-Ghosh S, Tadvalkar G, Williams AR, Pflugfelder SC, de Paiva CS, 2018b. Reduced corneal innervation in the CD25 null model of Sjogren syndrome. Int. J. Mol. Sci 19, 3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern ME, Beuerman RW, Fox RI, J. G,Mircheff AK, Pflugfelder SC, 1998. A unified theory of the role of the ocular surface in dry eye In: Sullivan DA, Dartt DA, Meneray MA (eds) Lacrimal gland, tear film, and dry eye syndromes 2. Advances in experimental medicine and biology, vol 438 Springer, Boston, MA: [DOI] [PubMed] [Google Scholar]
- Stern ME, Pflugfelder SC, 2017. What we have learned from animal models of dry eye. Int. Ophthalmol. Clin 57, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern ME, Schaumburg C, Gao J, Ratanapinta A, Calder V, Wheeler L, Niederkorn JY, Pflugfelder S, Beutler B, Theofilopoulos A, 2013. Plasmacytoid dendritic cells are the primary source of IFN-α during the immunopathogenesis of desiccating stress-induced dry eye disease. Invest. Ophthalmol. Vis. Sci. 54, 4323. [Google Scholar]
- Stern ME, Schaumburg CS, Dana R, Calonge M, Niederkorn JY, Pflugfelder SC, 2010. Autoimmunity at the ocular surface: pathogenesis and regulation. Mucosal Immunol. 3, 425–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern ME, Schaumburg CS, Siemasko KF, Gao J, Wheeler LA, Grupe DA, De Paiva CS, Calder VL, Calonge M, Niederkorn JY, Pflugfelder SC, 2012. Autoantibodies contribute to the immunopathogenesis of experimental dry eye disease. Invest. Ophthalmol. Vis. Sci 53, 2062–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarayal B, Chauhan SK, Di Zazzo A, Dana R, 2016. IL-17 augments B cell activation in ocular surface autoimmunity. J. Immunol 197, 3464–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teson M, Gonzalez-Garcia MJ, Lopez-Miguel A, Enriquez-de-Salamanca A, Martin-Montanez V, Benito MJ, Mateo ME, Stern ME, Calonge M, 2013. Influence of a controlled environment simulating an in-flight airplane cabin on dry eye disease. Invest. Ophthalmol. Vis. Sci 54, 2093–2099. [DOI] [PubMed] [Google Scholar]
- Tibrewal S, Ivanir Y, Sarkar J, Nayeb-Hashemi N, Bouchard CS, Kim E, Jain S, 2014. Hyperosmolar stress induces neutrophil extracellular trap formation: implications for dry eye disease. Invest. Ophthalmol. Vis. Sci 55, 7961–7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubota K, Goto E, Fujita H, Ono M, Inoue H, Saito I, Shimmura S, 1999. Treatment of dry eye by autologous serum application in Sjogren’s syndrome. Br. J. Ophthalmol 83, 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuan HI, Chi SC, Kang YN, 2020. An updated systematic review with meta-analysis of randomized trials on topical cyclosporin A for dry-eye disease. Drug Des. Devel. Ther 14, 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuisku IS, Konttinen YT, Konttinen LM, Tervo TM, 2008. Alterations in corneal sensitivity and nerve morphology in patients with primary Sjögren’s syndrome. Exp. Eye Res 86, 879–885. [DOI] [PubMed] [Google Scholar]
- Uchino Y, 2018. The ocular surface glycocalyx and its alteration in dry eye disease: a review. Invest. Ophthalmol. Vis. Sci 59, DES157–DES162. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF, 2007. Norman Cousins Lecture. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain. Behav. Immun 21, 131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Volpe EA, Gandhi NB, Schaumburg CS, Siemasko KF, Pangelinan SB, Kelly SD, Hayday AC, Li DQ, Stern ME, Niederkorn JY, Pflugfelder SC, De Paiva CS, 2012. NK cells promote Th-17 mediated corneal barrier disruption in dry eye. PLoS One 7, e36822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Ren Y, Reinach PS, She Y, Xiao B, Hua S, Qu J, Chen W, 2014. Reactive oxygen species activated NLRP3 inflammasomes prime environment-induced murine dry eye. Exp. Eye Res 125, 1–8. [DOI] [PubMed] [Google Scholar]
- Zheng Q, Ren Y, Reinach PS, Xiao B, Lu H, Zhu Y, Qu J, Chen W, 2015. Reactive oxygen species activated NLRP3 inflammasomes initiate inflammation in hyperosmolarity stressed human corneal epithelial cells and environment-induced dry eye patients. Exp. Eye Res 134, 133–140. [DOI] [PubMed] [Google Scholar]