

Abstract
Rationale: Host inflammatory responses have been strongly associated with adverse outcomes in critically ill patients, but the biologic underpinnings of such heterogeneous responses have not been defined.
Objectives: We examined whether respiratory tract microbiome profiles are associated with host inflammation and clinical outcomes of acute respiratory failure.
Methods: We collected oral swabs, endotracheal aspirates (ETAs), and plasma samples from mechanically ventilated patients. We performed 16S ribosomal RNA gene sequencing to characterize upper and lower respiratory tract microbiota and classified patients into host-response subphenotypes on the basis of clinical variables and plasma biomarkers of innate immunity and inflammation. We derived diversity metrics and composition clusters with Dirichlet multinomial models and examined our data for associations with subphenotypes and clinical outcomes.
Measurements and Main Results: Oral and ETA microbial communities from 301 mechanically ventilated subjects had substantial heterogeneity in α and β diversity. Dirichlet multinomial models revealed a cluster with low α diversity and enrichment for pathogens (e.g., high Staphylococcus or Pseudomonadaceae relative abundance) in 35% of ETA samples, associated with a hyperinflammatory subphenotype, worse 30-day survival, and longer time to liberation from mechanical ventilation (adjusted P < 0.05), compared with patients with higher α diversity and relative abundance of typical oral microbiota. Patients with evidence of dysbiosis (low α diversity and low relative abundance of “protective” oral-origin commensal bacteria) in both oral and ETA samples (17%, combined dysbiosis) had significantly worse 30-day survival and longer time to liberation from mechanical ventilation than patients without dysbiosis (55%; adjusted P < 0.05).
Conclusions: Respiratory tract dysbiosis may represent an important, modifiable contributor to patient-level heterogeneity in systemic inflammatory responses and clinical outcomes.
Keywords: acute respiratory distress syndrome, endotypes, inflammation, bacterial infections, microbiota
At a Glance Commentary
Scientific Knowledge on the Subject
Subphenotypes of host inflammatory responses are strongly predictive of mortality in patients with acute respiratory failure, but their biologic underpinnings are not defined. Recent research has implicated the lung microbiome as a predictor of outcomes in critically ill patients, yet it remains unknown whether lung microbiota account for hyperinflammatory responses and related adverse outcomes.
What This Study Adds to the Field
In a cohort of 301 mechanically ventilated patients with acute respiratory failure, 16S rRNA gene sequencing in upper and lower respiratory tract specimens revealed heterogenous bacterial communities in terms of diversity and composition. Unsupervised clustering showed that a third of lower respiratory tract communities had low α diversity and enrichment for pathogens (e.g., Staphylococcus or Pseudomonadaceae), whereas the remainder of communities had an abundance of typical oral microbiota. Membership in the pathogen-enriched cluster was independently associated with the adverse hyperinflammatory subphenotype, worse survival, and longer time to liberation from mechanical ventilation. We also demonstrate the importance of upper respiratory tract communities, given that patients with dysbiosis (low α diversity and low abundance of “protective” oral bacteria) in both upper and lower respiratory tract communities had significantly worse outcomes. Our study highlights the respiratory tract microbiome as an important, modifiable contributor to patient-level heterogeneity in systemic inflammatory responses and to the evolution of critical illness.
The biologic heterogeneity of sepsis and acute respiratory distress syndrome (ARDS) has impaired our ability to discover broadly efficacious therapies (1, 2). To better understand such biologic heterogeneity, recent research with unsupervised classification methods has revealed distinct patient subgroups (subphenotypes), defined primarily by differential innate immune host responses (hyper- vs. hypoinflammatory), in patients both with and at risk for ARDS (3–6). Patients with the hyperinflammatory phenotype have been reproducibly found to have higher rates of organ dysfunction, worse patient-centered outcomes, and differential treatment responses (3–6). However, the biologic underpinnings of these differential host responses remain unknown.
The respiratory microbiome is emerging as an important contributor to host immune function and patient outcomes, in both acute and chronic lung diseases (7, 8). In health, the respiratory microbiome comprises a low biomass and highly diverse community of oral-origin bacteria, which have been reproducibly profiled by culture-independent sequencing studies (9, 10). The anatomic and pathophysiologic changes of the respiratory tract in the context of disease alter the ecologic conditions for the resident microbiota, with measurable changes in bacterial load, diversity, and composition (7). In acute respiratory failure, orotracheal intubation exposes the lower respiratory tract to increased movement of bacteria from the oropharyngeal space (owing to sedation, cough suppression, and constant opening of the glottis from the endotracheal tube, among other factors), whereas the nutrient-rich edema fluid flooding the injured alveolar compartment creates an environment conducive to microbial proliferation (11, 12). We thus hypothesized that perturbation of the lung microbiome of patients with acute respiratory failure (dysbiosis) is associated with systemic inflammatory responses and adverse clinical outcomes.
Given that the available evidence characterizing respiratory microbiota in mechanically ventilated patients is limited to small-scale studies (13–20), we sought to characterize the upper and lower respiratory tract microbiome in a larger cohort of critically ill mechanically ventilated patients and examine its relationship with host-response and patient-centered outcomes.
Methods
For details, see the Methods in the online supplement.
Clinical Cohort and Sample Collection
From March 2015 to December 2018, we prospectively enrolled a convenience sample of consecutive adult patients with acute respiratory failure, who were intubated and mechanically ventilated in the medical or cardiac ICU at the University of Pittsburgh Medical Center. Exclusion criteria included the inability to obtain informed consent, the presence of tracheostomy, or mechanical ventilation for more than 72 hours before enrollment. The study was approved by the University of Pittsburgh Institutional Review Board (protocol PRO10110387), and written informed consent was provided by all participants or their surrogates.
Upon enrollment, we collected noninvasive biospecimens for study of the upper and lower respiratory tract microbiota, with a posterior oropharyngeal swab (oral) and an endotracheal aspirate (ETA) collection, respectively (16). We also collected simultaneous blood samples for centrifugation, separation of plasma, and quantification of the host inflammatory response.
Laboratory Analyses
We extracted bacterial DNA directly from oral swabs and ETAs and amplified the V4 hypervariable region of the bacterial 16S ribosomal RNA (rRNA) gene for sequencing on the Illumina MiSeq platform (16). Simultaneously, we performed quantitative PCR (qPCR) of the V3–V4 region to obtain number of 16S rRNA gene copies per sample (surrogate for bacterial load) (21). For plasma biomarkers, we constructed a custom Luminex multianalyte panel (R&D Systems) targeting prognostic biomarkers for classification into hyper- versus hypoinflammatory subphenotypes (RAGE [receptor of advanced glycation end products], soluble TNFR1 [tumor necrosis factor receptor 1], IL-10, fractalkine, and angiopoietin-2), as previously described (6).
Clinical Classifications
A consensus committee reviewed clinical and radiographic data and performed retrospective classifications of the etiology and severity of acute respiratory failure without knowledge of microbiome sequencing or biomarker data. We retrospectively classified subjects as having ARDS per established criteria (22), being at risk for ARDS because of the presence of direct (pneumonia or aspiration) or indirect (e.g., extrapulmonary sepsis or acute pancreatitis) lung-injury risk factors (23) although lacking ARDS diagnostic criteria, having acute respiratory failure without risk factors for ARDS, or having acute-on-chronic respiratory failure. We recorded clinical microbiologic results of respiratory specimens obtained within 48 hours of research-sample acquisition and considered them as positive when pathogenic bacterial species were isolated by the clinical laboratory (semiquantitative reports). We recorded whether patients had received antibiotics in the 30 days before ICU admission and then modeled the systemic antibiotic exposure in the ICU (before microbiome sampling) with a published antibiotic-exposure score that took into account dosing duration, timing of administration, and specific antibiotic type (see Table E1 in the online supplement) (24). We followed patients prospectively for cumulative mortality and ventilator-free days (VFDs) at 30 days, as well as for time to liberation from mechanical ventilation and survival up to 30 days from intubation.
Data Processing and Statistical Analyses
From derived 16S sequences, we applied a custom pipeline for Operational Taxonomic Units classification (see online supplement) and performed analyses at the genus level (16, 25). We calculated descriptive statistics of baseline characteristics and performed nonparametric comparisons using R software (version 3.5.1; R Foundation for Statistical Computing). Biomarker values were log transformed. With logistic regression models combining biomarker (TNFR1, RAGE, fractalkine, IL-10, and angiopoietin-2) and clinical variables, patients were assigned to hyper- versus hypoinflammatory subphenotypes (6). Ecologic analyses of α diversity (Shannon index) and β diversity (Manhattan distances with permutational ANOVA [Permanova] at 1,000 permutations) were conducted using the R vegan package and visualized with principal-coordinates-analysis plots. To contextualize upper and lower respiratory microbial profiles from ICU patients against expected ecologic metrics of the corresponding microbiome in health, we used 16S sequencing data from a previous study that had analyzed oral washes and BAL and induced sputum specimens from healthy volunteers (26). To agnostically examine our samples for distinct clusters of microbial composition (“metacommunities”), we applied unsupervised Dirichlet multinomial models (DMMs) with Laplace approximations to define the optimal number of clusters in our data set (27).
Associations of Microbial Profiles with Outcomes
We pursued complementary unsupervised and supervised analyses for examining associations between upper and lower respiratory tract microbial profiles and outcomes. In our main unsupervised approach, we examined associations between DMM clusters and outcomes (host-response subphenotypes, 30-day survival, and time to liberation from mechanical ventilation) in regression and Cox proportional hazard models, as appropriate (see online supplement). To delineate contributions of specific genera driving the associations observed at the DMM-cluster level, we performed relative-abundance transformations with the additive log ratio for the top 10 genera in each DMM cluster and examined our data for associations with outcomes, which were adjusted for covariates and multiple testing. In our supervised approach, we examined our data for differences in α and β diversity, bacterial load, and the relative abundance of individual genera for clinical outcome stratifications (i.e., survivors vs. nonsurvivors and VFD tertiles). In a final integrative analysis, we developed a simple index of dysbiosis on the basis of convergent predictive features identified in both unsupervised and supervised approaches, specifically α-diversity and relative-abundance combinations for individual genera. Finally, with multivariate adjusted models, we examined associations of clinical outcomes with the dysbiosis index in upper, lower, and combined upper–lower respiratory communities.
Results
Cohort Description
Three hundred one mechanically ventilated patients (median age, 59 yr; 52% men) contributed a total of 518 airway samples (262 oral swabs and 256 ETA samples) for analysis. Of the 301 subjects, ARDS was diagnosed in 24%, 84% were receiving systemic antibiotics at the time of sampling in the ICU, and 23% were classified into a hyperinflammatory subphenotype of host responses associated with worse outcomes (Table 1) (6). Comparison samples including 23 oral washes, 19 induced sputum samples, and 23 BAL samples from healthy volunteers and a total of 216 procedural control samples were used in our experimental pipelines.
Table 1.
Baseline Characteristics of Enrolled Patients
| Variable | All (n = 301) |
|---|---|
| Age, median (IQR), yr | 59.0 (46.6–67.1) |
| Males, n (%) | 156 (51.8) |
| BMI, median (IQR), kg/m2 | 29.8 (25.5–36.6) |
| Diabetes, n (%) | 110 (36.5) |
| COPD, n (%) | 76 (25.2) |
| Immunosuppression, n (%) | 68 (22.6) |
| ARDS, n (%) | 72 (23.9) |
| Pneumonia, n (%) | 120 (39.9) |
| Extrapulmonary sepsis, n (%) | 55 (18.3) |
| Aspiration, n (%) | 53 (17.6) |
| LIPS score, median (IQR) | 5.5 (4.0–7.0) |
| SOFA score, median (IQR)* | 6.0 (4.0–9.0) |
| PaO2/FiO2 ratio, median (IQR), mm Hg | 168.0 (117.0–205.0) |
| WBC, median (IQR), ×109 cells/L | 12.2 (8.9–17.0) |
| Plateau pressure, median (IQR), cm H2O | 20.0 (16.0–25.2) |
| Positive respiratory cultures†, n (%) | 73 (24.3) |
| Antibiotics before ICU admission, n (%) | 97 (32.4) |
| Antibiotics during ICU admission before sampling, n (%) | 254 (84.4) |
| Systemic steroids, n (%) | 110 (38.3) |
| Hyperinflammatory subphenotype, n (%) | 69 (22.9) |
Definition of abbreviations: ARDS = acute respiratory distress syndrome; BMI = body mass index; COPD = chronic obstructive pulmonary disease; IQR = interquartile range; LIPS = lung-injury prediction score; SOFA = Sequential Organ Failure Assessment; WBC = white blood cell count.
Data are presented as the median (with IQR) for continuous variables and as n (%) for categorical variables.
The SOFA score calculation does not include the neurologic component of the SOFA score because all patients were intubated and receiving sedative medications, impairing our ability to perform assessment of the Glasgow coma scale in a consistent and reproducible fashion.
Respiratory specimen cultures were defined as positive when pathogenic bacterial species were isolated by the clinical laboratory (semiquantitative reports). The denominator includes patients for whom respiratory specimen cultures were reported as negative (no growth or only normal respiratory flora detected, n = 152) as well as those for whom no cultures were obtained (n = 76).
Upper and Lower Respiratory Tract Communities Exhibit Substantial Heterogeneity
Clinical samples from ICU patients had a much higher number of 16S reads than procedural control samples (P < 0.001) (Figure E1). Microbial profiles from both oral swabs and ETAs exhibited substantial heterogeneity. The α diversity ranged from a Shannon index of zero (in collapsed, effectively monobacterial communities) to the range of the normal microbiome. By β-diversity comparisons, individual samples were distributed over wide spaces of principal-coordinates-analysis plots, indicating wide variation in microbial composition (Figures 1A and 1B and 2A and 2B for ETA samples and oral swabs, respectively). Compared with corresponding upper and lower respiratory tract samples from healthy volunteers, samples from critically ill subjects had significantly lower α-diversity (P < 0.001; Figure E2A) and β-diversity differences (P < 0.001; Figure E2B). In terms of bacterial load quantified by qPCR, oral swabs contained 16S rRNA gene copies at a median level that was 20-fold higher than that of the ETA samples (Figure E3), as expected for the higher bacterial biomass of the oropharyngeal microbiome. The ETA-sample bacterial load ranged from being effectively undetectable in a few samples to comprising a high biomass in the range of oral-swab communities. Thus, ecologic analyses revealed a pattern of respiratory communities that are widely variable among critically ill patients and significantly different from those of healthy individuals.
Figure 1.
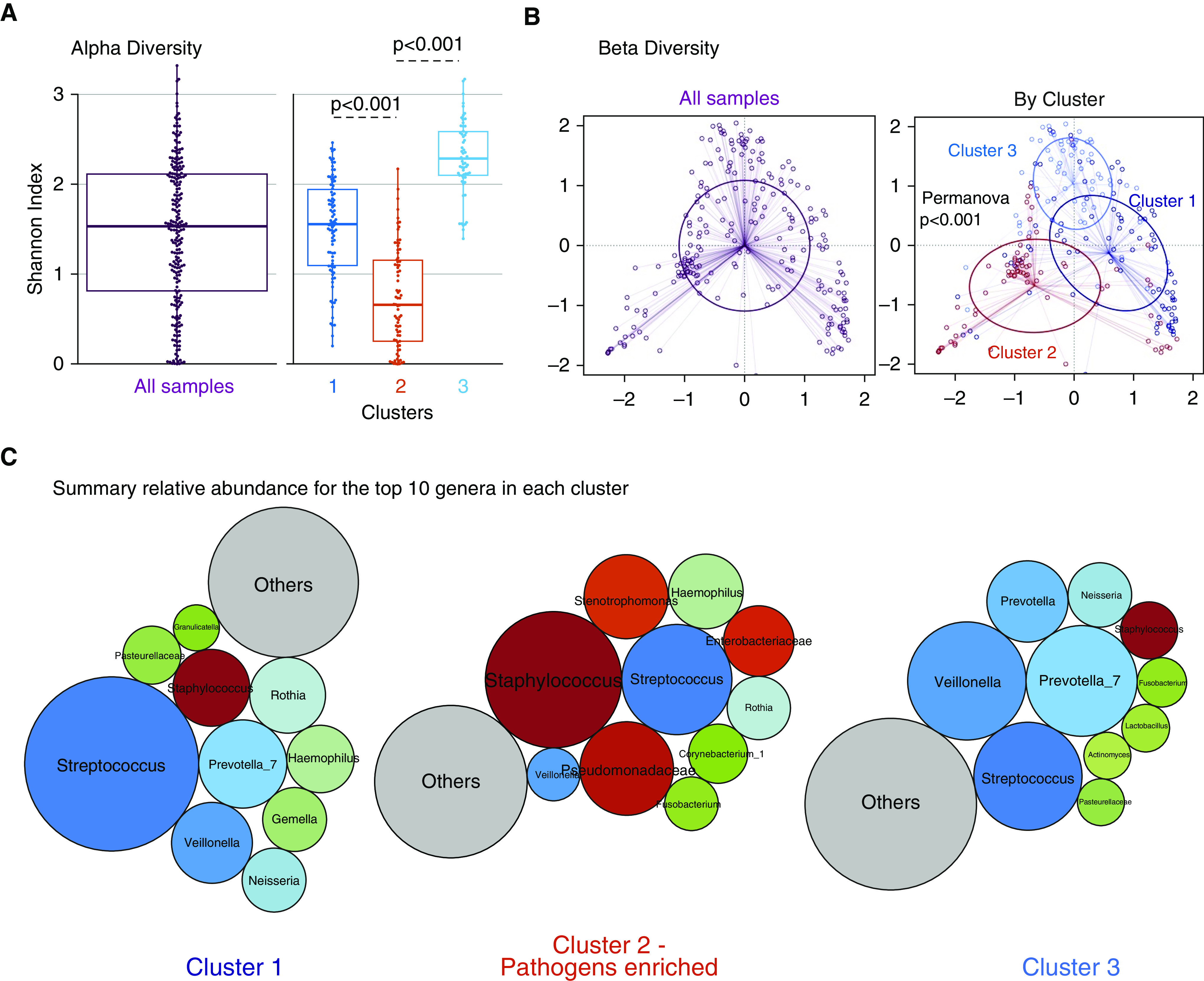
Dirichlet-multinomial-model clustering of endotracheal aspirate communities reveals a distinct cluster marked by pathogen abundance and low α diversity. (A) The α-diversity comparisons between clusters showed that cluster 2 had the lowest Shannon index, followed by cluster 1. (B) Principal coordinates analyses for β-diversity comparisons (Manhattan distances) with Permanova for all samples included and stratified by clusters. (C) Summary of the relative abundance for the top 10 genera in each cluster, visualized as bubble plots. The diameter of each circle corresponds to the mean relative abundance of each genus across all samples in the cluster. Streptococcus was the most abundant genus in cluster 1, and cluster 2 had high abundance for typical respiratory pathogens (shown in variations of red), such as Staphylococcus, Pseudomonadaceae, and Stenotrophomonas, whereas cluster 3 had high abundance of Prevotella_7, Veillonella, and Streptococcus genera. Genera beyond the top 10 genera demonstrated in these bubble plots were summarized to their overall relative abundance as a single bubble in gray and annotated as “Others.” Permanova = permutational ANOVA.
Figure 2.
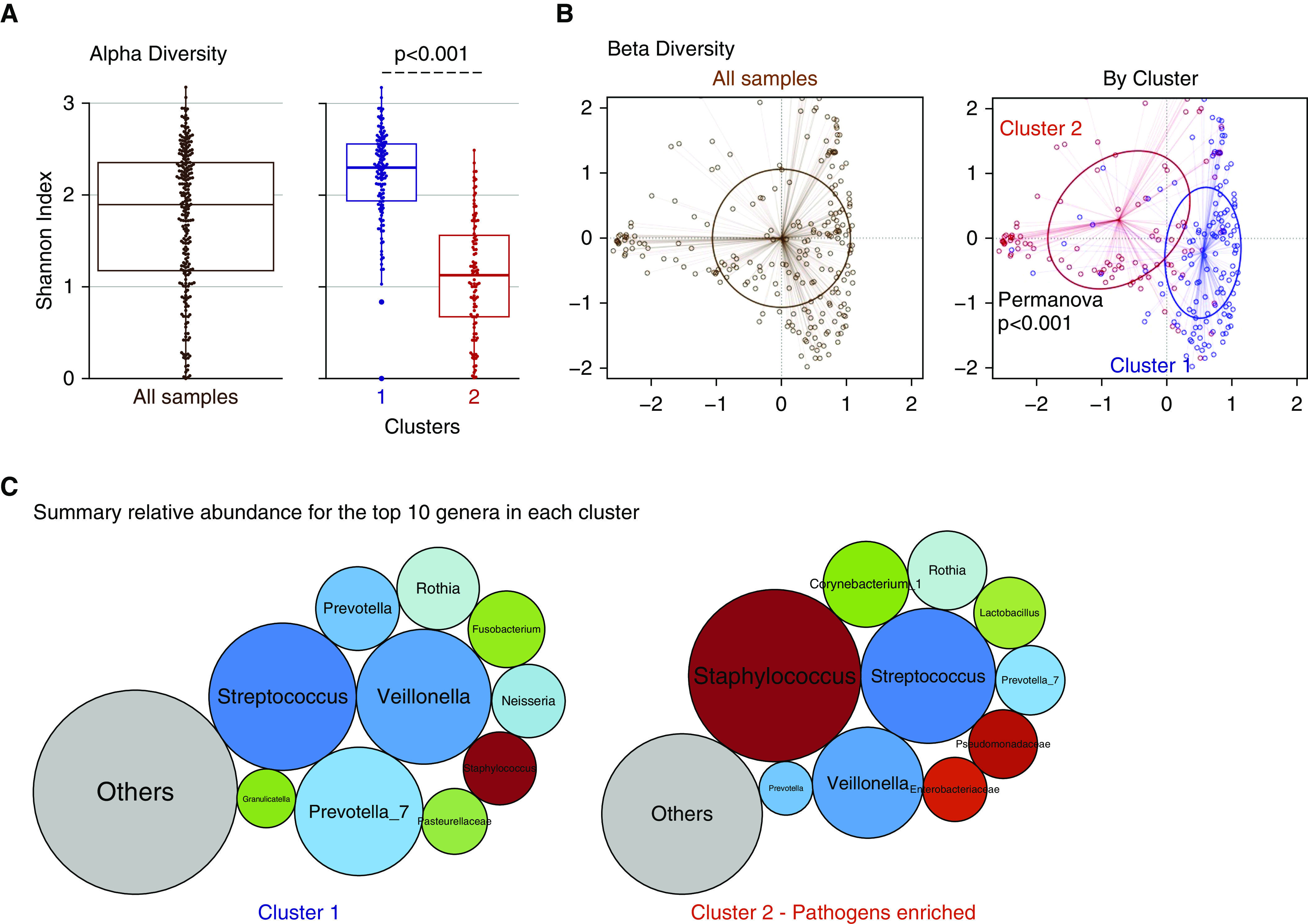
Dirichlet-multinomial-model clustering of oral-swab communities reveals a distinct cluster marked by pathogen abundance and low α diversity. (A) The α-diversity comparisons between clusters showed that cluster 2 had a significantly lower Shannon index. (B) Principal coordinates analyses for β-diversity comparisons (Manhattan distances) with Permanova for all samples included and stratified by cluster. (C) Summary of the relative abundance for the top 10 genera in each cluster, visualized as bubble plots. The diameter of each circle corresponds to relative abundance of each genus across all samples in the cluster. Streptococcus, Prevotella_7, and Veillonella were the most abundant genera in cluster 1, whereas Staphylococcus was the most abundant genus in cluster 2. Genera beyond the top 10 genera demonstrated in these bubble plots were summarized to their overall relative abundance as a single bubble in gray and annotated as “Others.” Permanova = permutational ANOVA.
Respiratory Communities Comprise Distinct Compositional Clusters
With demonstration of heterogeneous upper and lower respiratory communities in terms of bacterial load and α and β diversity, we sought to identify potential sources of microbial profile heterogeneity. First, we found no significant impact by time of sample acquisition (during our 72-h sampling period from intubation) on microbial profiles (Figure E4). Next, we derived unsupervised clusters of communities with the DMM approach. Laplace approximation of model fitting showed that three clusters offered the best fit in ETAs and that two clusters offered the best fit in oral swabs (Figure E5). In both sample types, we identified a distinct cluster (cluster 2) with lower α diversity (P < 0.001) and significantly different taxonomic composition (Permanova P < 0.001 for β-diversity differences) compared with the rest of cohort (Figures 1A and 1B and 2A and 2B). By summarizing the relative abundance for the top 10 genera across all samples, cluster 2 demonstrated a high abundance of typical respiratory pathogenic genera in ETAs (Staphylococcus, Stenotrophomonas, Enterobacteriaceae, and Pseudomonadaceae; Figure 1C) and oral swabs (Staphylococcus; Figure 2C). Clusters 1 and 3 in ETAs and cluster 1 in oral swabs had a high abundance of typical members of the respiratory microbiome (i.e., oral-origin bacteria, such as Streptococcus, Prevotella, and Veillonella). No significant differences in bacterial load by qPCR were observed in ETA clusters, whereas in oral swabs, cluster 2 had a lower bacterial load than cluster 1 (P < 0.001).
By baseline clinical characteristics, patients in ETA cluster 2 had a higher prevalence of chronic obstructive pulmonary disease (COPD), more often received a diagnosis of ARDS and extrapulmonary sepsis, had a higher incidence of positive respiratory cultures, and were more likely to have received systemic antibiotics before ICU admission (P < 0.05; Tables 2 and E3). For oral-swab clusters, patients in cluster 2 were older, had a higher prevalence of COPD and history of immunosuppression, and were also more likely to have received antibiotics before ICU admission (P < 0.05; Table E2).
Table 2.
Baseline Characteristics by Dirichlet-Multinomial-Model Clusters for Endotracheal Aspirates
| Variable | Cluster 1 (n = 90) | Cluster 2 (n = 78) | Cluster 3 (n = 62) | P Value |
|---|---|---|---|---|
| Age, median (IQR), yr | 59.1 (49.8–67.7) | 60.4 (47.6–68.4) | 56.5 (42.3–65.4) | 0.3 |
| Males, n (%) | 44 (48.9) | 43 (55.1) | 36 (58.1) | 0.5 |
| BMI, median (IQR), kg/m2 | 29.7 (25.2–36.2) | 27.8 (24.5–34.5) | 30.9 (26.3–36.6) | 0.2 |
| Diabetes, n (%) | 35 (38.9) | 29 (37.2) | 17 (27.4) | 0.31 |
| COPD, n (%) | 25 (27.8) | 27 (34.6) | 7 (11.3) | 0.01 |
| Immunosuppression, n (%) | 25 (27.8) | 17 (21.8) | 9 (14.5) | 0.15 |
| ARDS, n (%) | 12 (13.3) | 24 (30.8) | 11 (17.7) | 0.02 |
| Pneumonia, n (%) | 30 (33.3) | 34 (43.6) | 22 (35.5) | 0.37 |
| Sepsis, n (%) | 10 (11.1) | 22 (28.2) | 12 (19.4) | 0.02 |
| Aspiration, n (%) | 16 (17.8) | 10 (12.8) | 15 (24.2) | 0.22 |
| LIPS score, median (IQR) | 5.0 (3.5–6.5) | 5.5 (4.0–7.0) | 5.5 (4.0–6.5) | 0.22 |
| SOFA score, median (IQR)* | 6.0 (4.0–8.0) | 6.0 (5.0–9.0) | 6.0 (4.0–8.0) | 0.24 |
| PaO2/FiO2 ratio, median (IQR), mm Hg | 176.5 (137.0–207.2) | 158.0 (112.2–225.0) | 165.0 (118.5–205.0) | 0.44 |
| WBC, median (IQR), ×109/L | 11.3 (7.7–15.8) | 11.8 (8.7–17.4) | 12.8 (9.2–17.7) | 0.26 |
| Plateau pressure, median (IQR), cm H2O | 19.0 (15.0–24.0) | 20.0 (17.0–27.0) | 20.0 (16.0–27.0) | 0.28 |
| Positive respiratory cultures, n (%) | 20 (22.2) | 28 (35.9) | 10 (16.1) | 0.02 |
| Bacteremia, n (%) | 7 (8.0) | 13 (17.0) | 5 (8.0) | 0.12 |
| Systemic antibiotics before ICU admission, n (%) | 21 (23.3) | 42 (53.8) | 18 (29.0) | <0.01 |
| Antibiotics during ICU admission before sampling, n (%) | 73 (81.1) | 72 (90.0) | 54 (85.7) | 0.26 |
| Systemic steroids, n (%) | 41 (46.6) | 28 (40.6) | 13 (21.7) | 0.01 |
| Hyperinflammatory subphenotype, n (%) | 16 (17.8) | 23 (29.5) | 13 (21.0) | 0.18 |
For definition of abbreviations, see Table 1.
Data are presented as the median (with IQR) for continuous variables and as n (%) for categorical variables. P values for comparisons between clusters were obtained by using the Wilcoxon test for continuous variables and by using the Fisher’s exact test for categorical variables. Statistically significant P values (P < 0.05) are shown in bold.
The SOFA score calculation does not include the neurologic component of the SOFA score because all patients were intubated and receiving sedative medications, impairing our ability to perform assessment of the Glasgow coma scale in a consistent and reproducible fashion.
Therefore, DMMs uncovered compositionally and structurally distinct clusters of respiratory tract communities, which were significantly associated with key baseline clinical characteristics of critically ill patients. We then examined whether the three unsupervised DMM microbial community clusters were associated with clinical outcomes after adjustment for clinical-variable differences between clusters.
Unsupervised DMM Clusters Are Associated with Outcomes
Stratification of ETA microbiome profiles by DMM clusters revealed that patients belonging to cluster 2 (i.e., the pathogen-enriched cluster with low α diversity) had worse 30-day survival as well as longer time to liberation from mechanical ventilation than the other two clusters (Figures 3A and 3B and Table E4), effects that remained significant after adjustment for age, COPD, ARDS, extrapulmonary sepsis, antibiotics before ICU admission, and ICU antibiotic-exposure score. Similarly, oral-community cluster 2 membership was associated with worse 30-day survival and liberation outcomes, adjusted for age, COPD, immunosuppression, and antibiotic exposures (Figures 3C and 3D). Furthermore, patients belonging to cluster 2 in both the ETA and oral communities had worse outcomes than all other patient groups combined (Figures 3E and 3F). Inclusion of bacterial load by qPCR as a variable in these time-to-event models did not impact the observed effect sizes or statistical significance of DMM clusters.
Figure 3.
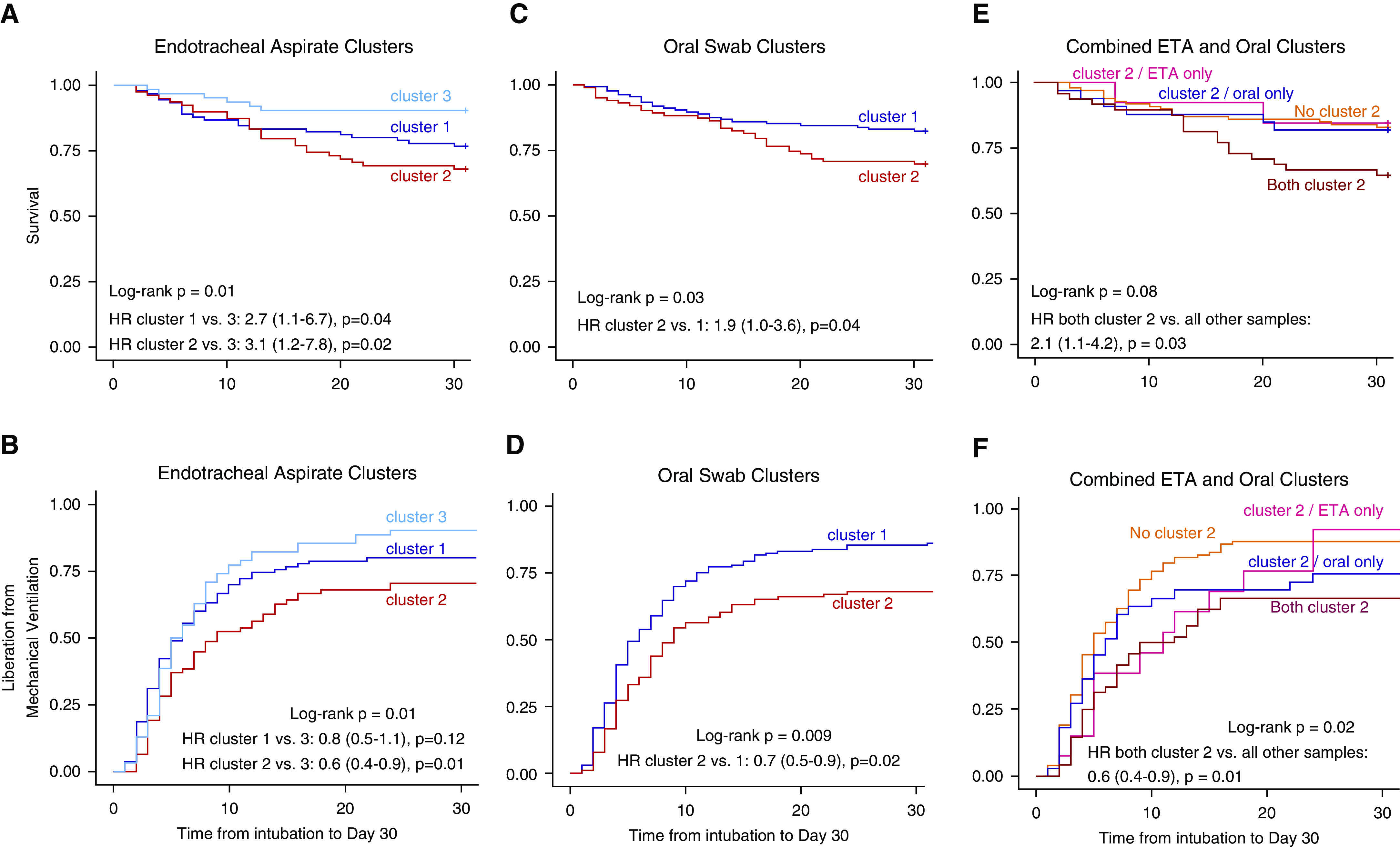
Patients with respiratory tract profiles belonging to the pathogen-enriched cluster 2 have worse 30-day survival and longer time to liberation from mechanical ventilation. (A and B) Kaplan-Meier curves for 30-day survival and time to liberation stratified by Dirichlet-multinomial-model (DMM) clusters for endotracheal aspirates (ETAs). P values were derived from the log-rank test, and hazard ratios (HRs) with corresponding 95% confidence intervals were derived from the Cox proportional hazard model adjusted for age, history of chronic obstructive pulmonary disease (COPD), diagnosis of acute respiratory distress syndrome, extrapulmonary sepsis, antibiotic administration before ICU admission, and antibiotic-exposure score in the ICU before sampling. (C and D) Kaplan-Meier curves for 30-day survival stratified by DMM clusters for oral swabs. HRs were adjusted for age, history of COPD, immunosuppression, antibiotic administration before ICU admission, and antibiotic-exposure score. (E and F) Kaplan-Meier curves for 30-day survival stratified by DMM clusters for ETA and oral-swab samples stratified in four categories: 1) both ETA and oral samples belonging to cluster 2 (n = 50), 2) cluster 2 in oral samples only (n = 34), 3) cluster 2 in ETA samples only (n = 13), and 4) neither oral nor ETA samples belonging to cluster 2 (n = 100). HRs were adjusted for age, history of COPD, diagnosis of acute respiratory distress syndrome, extrapulmonary sepsis, antibiotic administration before ICU admission, and antibiotic-exposure score.
We then examined our data for associations between the unsupervised DMM clusters with the host-response subphenotypes defined by plasma biomarker levels and clinical variables. Cluster 2 membership in ETA samples was significantly associated with classification in the prognostically adverse hyperinflammatory subphenotype (odds ratio, 1.2 [95% confidence interval, 1.1–1.9]; P = 0.03, adjusted for age, COPD, and antibiotic exposures) (Table E5), but no significant association of cluster 2 membership was found for oral swabs only or in combined ETA and oral-swab analysis.
Overall, DMM clustering revealed significant associations with important patient-centered outcomes and host-response subphenotypes. We subsequently sought to identify the specific genera accounting for the observed associations.
Relative Abundance of Specific Genera Is Associated with Outcome
Despite the within-cluster structural and compositional similarity in terms of α and β diversity (Figures 1 and 2), each cluster included samples with important taxonomic dissimilarities. For example, cluster 2 included samples that were dominated by Staphylococcus genera but also included samples dominated by Pseudomonadaceae genera (Figures E6 and E7). We focused on the top 10 genera in each cluster for ETA samples or oral swabs and examined whether relative abundances (additive log ratio–transformed) of these common genera were associated with host-response subphenotypes or clinical outcomes.
In ETA samples, we examined a total of 18 unique genera and detected a dichotomous pattern of associations. A high relative abundance of typical pathogenic bacteria (i.e., Staphylococcus, Pseudomonadaceae, and Enterobacteriaceae) was associated with higher odds of classification into the hyperinflammatory subphenotype and fewer VFDs in the case of Staphylococcus relative abundance (adjusted P < 0.05; Figure 4A). Conversely, high relative abundance of typical members of the healthy lung microbiome (Prevotella_7, Streptococcus, or Haemophilus genera) had protective associations with lower mortality risk or more VFDs. In oral swabs, among 14 unique genera, we detected primarily protective associations of typical oral-origin bacteria (e.g., Prevotella_7, Streptococcus, Veillonella, Rothia, Granulicatella, and others), which were robust to multiple testing adjustments (Figure 4B). A similar pattern of results was seen for analyses restricted to samples without positive respiratory cultures (Figure E8). These genus-level analyses revealed that the significant associations of DMM cluster 2 with adverse clinical outcomes was accounted for by high relative abundance of specific pathogenic genera or, conversely, by low relative abundance of protective oral-origin bacteria.
Figure 4.
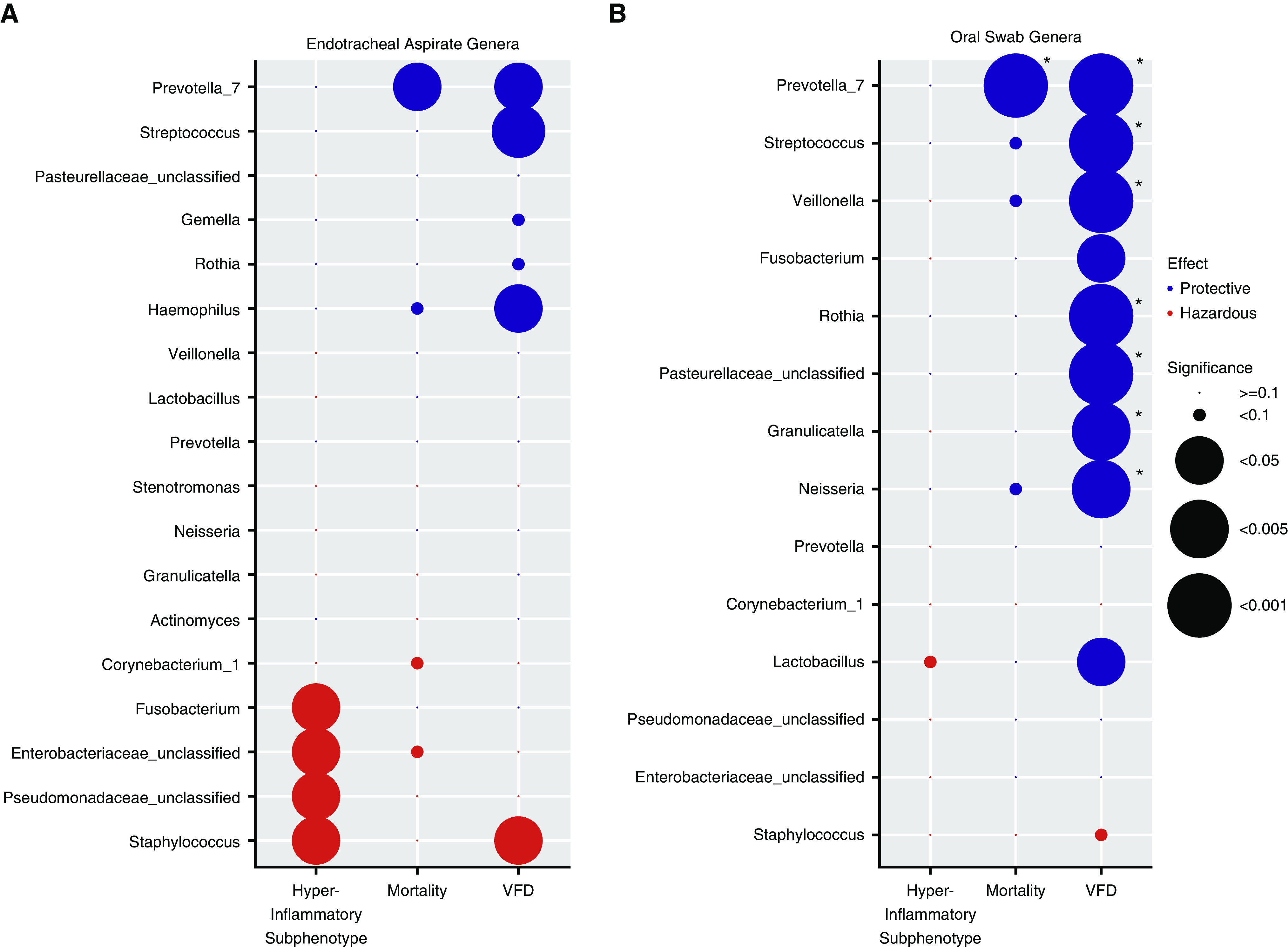
The relative abundance of individual genera is associated with clinical outcomes and host-response subphenotypes. (A) Endotracheal aspirate (ETA) genera. We examined data for associations between additive log ratio–transformed relative abundance for the top 10 genera in each cluster (total of 18 unique genera), which are shown on the y-axis with three outcome variables: hyperinflammatory subphenotype, 30-day mortality (logistic regression models), and ventilator-free days (VFDs; linear regression model). Models were adjusted for age, chronic obstructive pulmonary disease, and antibiotic exposures. In each column, the direction of the effect size of the coefficient and the statistical significance for each genus–outcome association are visually represented by color coding (with protective effects shown in blue and adverse effects shown in red) and the size of each circle, respectively. Typical pathogenic genera in ETA samples (Staphylococcus, Pseudomonadaceae, and Enterobacteriaceae) were associated with higher odds of having the hyperinflammatory subphenotype classification and fewer VFDs in the case of Staphylococcus genera, whereas typical members of the normal lung microbiome (e.g., Prevotella_7 and Streptococcus) were associated with improved outcomes. (B) Oral-swab genera. Among the 14 unique genera examined in oral swabs, a high relative abundance of typical members of the normal lung microbiome (e.g., Prevotella_7, Streptococcus, Veillonella, Rothia, etc.) was associated with improved outcomes (mainly more VFDs). Associations that remained significant after adjustment for multiple testing with the Benjamini-Hochberg method are highlighted with asterisks (*adjusted P < 0.05). In the case of Pseudomonadaceae_unclassified, Enterobacteriaceae_unclassified, and Pasterellaceae_unclassified, classification to specific genera within these families was not accomplished, and we thus used family-level descriptors for these genera.
Supervised Analyses Reveal α Diversity and Relative Abundance of Specific Genera as Predictors of Outcome
Independent of the unsupervised DMM clustering approach, we sought to identify predictive features of the microbial profiles with supervised stratifications of observed clinical outcomes. We stratified our patient cohort on the basis of our two primary outcomes (30-d mortality [survivors vs. nonsurvivors] and VFD tertiles) and examined our data for microbial community differences in terms of α diversity, bacterial load, β diversity, and the relative abundance of individual genera. Nonsurvivors exhibited lower α diversity (P < 0.05) and significant differences in β diversity (Permanova P < 0.001) in ETA samples, with enrichment for pathogenic genera in nonsurvivors and for Prevotella_7 in survivors (Figure E9). Similarly, higher relative abundance of Streptococcus was associated with more VFDs (Figure E10). Oral-swab analyses revealed an association between enrichment with protective genera and favorable outcomes (Figures E11 and E12). Thus, supervised analyses by observed clinical outcomes uncovered two main predictive features of the respiratory tract microbiome: α diversity and relative abundance of pathogenic versus oral-origin bacteria.
A Simple Dysbiosis Index in Upper and Lower Respiratory Communities Is Predictive of Outcome
With supervised and unsupervised methods converging on the same two predictive features (α diversity and relative abundance of specific bacteria), we sought to develop a simple predictive index from these features. We aimed to define “normal” or “nondysbiotic” communities as those with high α diversity and relative abundance of protective oral-origin bacteria. With receiver operating characteristic curves for the mortality outcome (Figure E13), we derived optimal thresholds of protective-bacteria relative abundance (≥30% for ETAs and ≥70% for oral swabs). From the α-diversity distribution in DMM clusters (Figures 1A and 2A), we identified that a Shannon threshold of ≥1.98 distinguished the prognostically favorable clusters 3 (in ETAs) and 1 (in oral swabs) from the rest of the cohort. We then broadly defined communities with dysbiosis as those not meeting the “protective” Shannon index and relative-abundance thresholds. The so-called dysbiosis index in ETA samples was significantly associated with the hyperinflammatory subphenotype (adjusted odds ratio, 1.2 [1.1–1.3]; P = 0.008) as well as with worse survival and ventilation-liberation outcomes (Figure 5).
Figure 5.
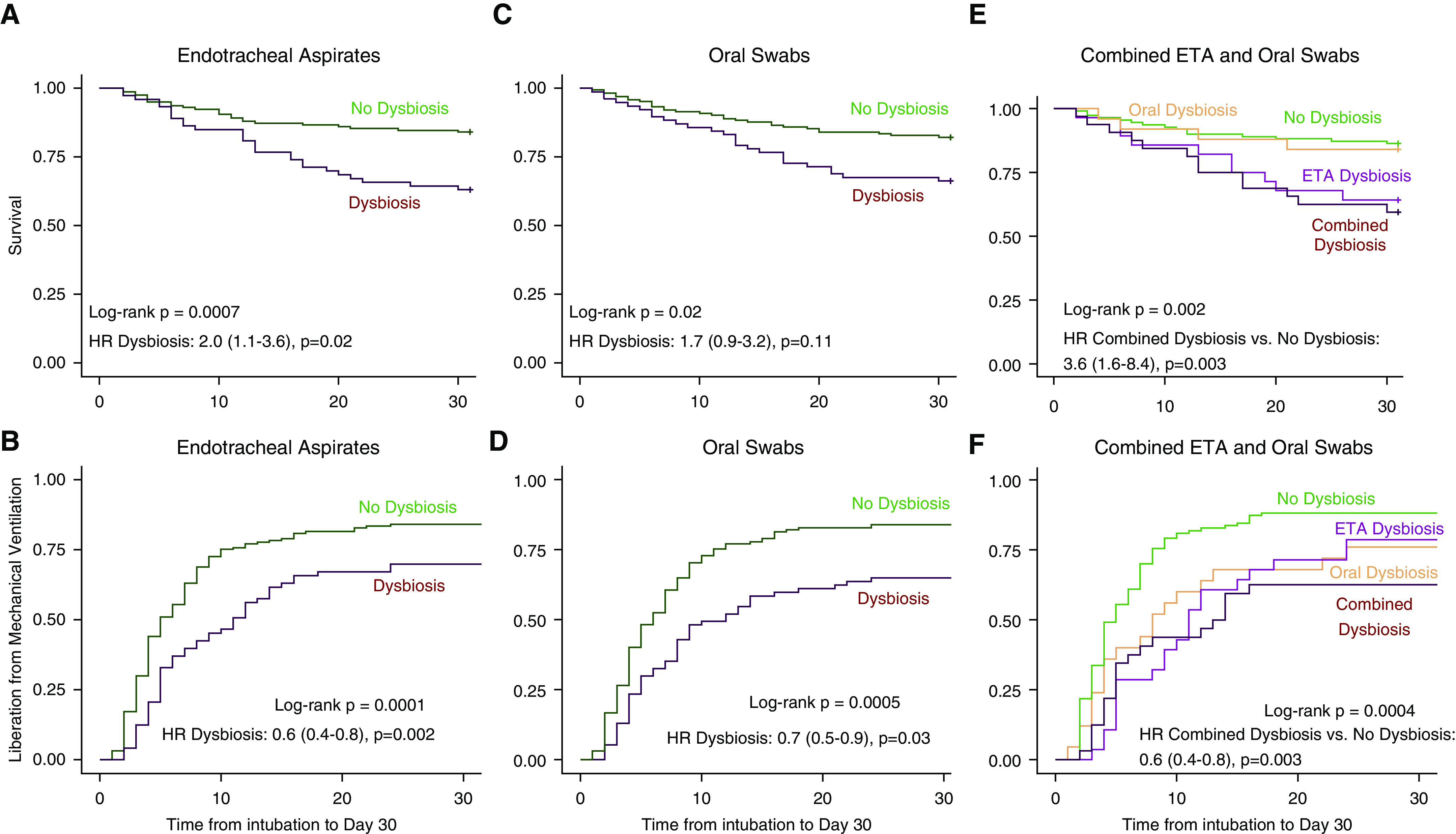
Patients with upper and lower respiratory tract dysbiosis have worse 30-day survival and longer time to liberation from mechanical ventilation. (A and B) Kaplan-Meier curves for 30-day survival and time to liberation stratified by the dysbiosis index (Shannon index ≥ 1.98 and protective-bacteria relative abundance ≥ 30%) for endotracheal aspirates (ETAs). P values were derived from the log-rank test, and hazard ratios (HRs) with corresponding 95% confidence intervals were derived from the Cox proportional hazard model adjusted for age, history of chronic obstructive pulmonary disease (COPD), diagnosis of acute respiratory distress syndrome, extrapulmonary sepsis, antibiotic administration before ICU admission, and antibiotic-exposure score in the ICU before sampling. (C and D) Kaplan-Meier curves for 30-day survival stratified by the dysbiosis index (Shannon index ≥ 1.98 and protective-bacteria relative abundance ≥ 70%) for oral swabs. HRs were adjusted for age, history of COPD, immunosuppression, antibiotic administration before ICU admission, and antibiotic-exposure score. (E and F) Kaplan-Meier curves for 30-day survival for ETA and oral-swab samples stratified in four categories: 1) both ETA and oral samples with dysbiosis (n = 34), 2) oral-swab samples with dysbiosis only (n = 28), 3) ETA samples with dysbiosis only (n = 28), and 4) neither oral nor ETA samples with dysbiosis (n = 110). HRs were adjusted for age, history of COPD, diagnosis of acute respiratory distress syndrome, extrapulmonary sepsis, antibiotic administration before ICU admission, and antibiotic-exposure score.
Discussion
In a large cohort of mechanically ventilated patients with acute respiratory failure, we demonstrate that respiratory tract dysbiosis is associated with systemic inflammatory responses and adverse clinical outcomes. Using culture-independent 16S rRNA gene sequencing in noninvasive samples from critically ill patients, we identified heterogeneous upper and lower respiratory tract communities in terms of bacterial load, α diversity, and composition. Respiratory community profiles from ICU patients not only systematically deviated from the representative communities of the healthy respiratory microbiome, but they also varied substantially among ICU patients. With an agnostic classification approach, we first identified clusters of respiratory communities that captured diversity and compositional differences and then discovered significant associations between cluster membership and outcomes. Importantly, DMMs revealed a cluster in lower respiratory tract communities with low α diversity and enrichment for pathogenic bacteria, which was then independently associated with a hyperinflammatory subphenotype of host responses and worse clinical outcomes. Microbe–outcome associations were apparent at the individual-genus level of relative abundance, with opposing effects between typical respiratory pathogens and oral-origin bacteria. Supervised analyses based on observed outcomes corroborated the predictive features of the unsupervised cluster analyses.
Our findings highlight the respiratory microbiome as a previously understudied but potentially important contributor to patient-level heterogeneity in critical illness. Subphenotyping efforts for ARDS and sepsis by modeling clinical variables and blood biomarkers have identified subsets of patients with differential treatment responses and outcomes (6, 28, 29). Such associations have stimulated efforts to determine the biologic determinants of accentuated host inflammation (30). Our unsupervised clustering approach focused on the microbiome, by modeling the compositional heterogeneity of respiratory communities. For lung communities, we discovered a significant association between the pathogen-enriched cluster 2 and the adverse hyperinflammatory subphenotype (6). No significant association was found for the corresponding cluster 2 in oral communities. This site specificity of detectable host–microbiome associations suggests that in patients with acute respiratory failure, the lower respiratory tract may represent the active site of innate immune activation and inflammatory response to microbiota. However, the small effect size of the observed association of lung clusters with the hyperinflammatory subphenotype also points to multiple other sources of interindividual variability in inflammatory responses, unaccounted for by the study of respiratory bacteria (e.g., gut microbiota–host interactions, immunomodulatory medication effects, host genetic variation, etc.).
Our cross-sectional study design did not allow for establishing the directionality of the effects in the study of host–microbiome interactions, and it is possible that an inflammatory milieu in the alveolar space leads to secondary proliferation of pathogenic bacteria (12, 31, 32). Nonetheless, a causal role for respiratory pathogens stimulating host-response biomarkers (such as plasma TNFR1) is supported by temporality and biologic plausibility. With early (within 72 h of intubation) study of host–pathogen interactions in a cohort enriched for patients with direct lung-injury risk factors (pneumonia or aspiration), respiratory bacteria likely represent a proximal and primary insult. For example, the pathogenic genera associated with the hyperinflammatory subphenotype in our cohort (including Staphylococcus, Pseudomonadaceae, and Enterobacteriaceae) are known to potently stimulate Toll-like receptor signaling and systemic TNF-α levels (33).
Our analysis of patients without known cultivable pathogens in their respiratory tracts (i.e., excluding culture-positive patients) provided important insights. Routine microbiologic cultures have well-known limitations in detecting plausible respiratory pathogens in clinical samples (34), owing to high rates of antecedent antibiotics before ICU admission, among other reasons. DNA-based analyses cannot distinguish between viable or nonviable bacteria. Regardless of viability, pathogenic bacteria in the airways can present a variety of pathogen-associated molecular patterns in innate immune cells, such as LPS, flagellin, peptidoglycans, or nucleic acids, which can propagate the inflammatory cascade even in the absence of ongoing microbial proliferation (35). Although sensing of microbial viability through prokaryotic mRNA recognition (vita–pathogen-associated molecular pattern) is considered a key regulatory mechanism of innate immunity (36), the robust genus–outcome associations we observed even in subjects without viable respiratory pathogens in clinical biospecimens warrant further investigation to understand the mechanisms of host–microbiome interactions. Beyond mechanistic insights, identification of respiratory pathogen abundance by sequencing in culture-negative cases can offer critical diagnostic information for antibiotic decision-making and stewardship (16, 37, 38), if available within clinically actionable time frames.
Our unsupervised clustering approach with DMMs captured important interindividual variability and revealed major patterns of metacommunities in the respiratory tract. Previous studies in ICU patients have shown associations of nonspecific, global ecologic metrics (α diversity or bacterial load) or crude microbial composition at the phyla level with clinical variables or outcomes (13–19). More recently, higher bacterial load as well as enrichment for gut-associated bacteria (e.g., Lachnospiraceae and Enterobacteriaceae taxa) in mini-BAL samples were associated with fewer VFDs in a cohort of 91 mechanically ventilated patients (20). The larger sample size and the granular clinical data in our cohort allowed for robust cluster derivation and replicable associations with outcomes and host-response subphenotypes. The discovered lung and oral microbiota clusters were clinically meaningful and captured a range of α-diversity and microbial composition profiles. We established independent associations with outcomes for cluster 2 in both oral and lung communities, characterized by low α diversity and high relative abundance of typical pathogenic bacteria. Conversely, the clusters with high α diversity and composed of typical oral-origin bacteria represented symbiotic respiratory communities (9, 26). In a series of analyses, we showed that the DMM cluster effects could be traced to the relative abundance of individual genera in a dichotomous fashion: we detected hazardous associations for pathogenic bacteria and protective associations for oral-origin bacteria. Thus, although clustering approaches are not generalizable and transferrable in other cohorts, they offered us important insights into the ecology of respiratory microbiota in critical illness and key predictive features of microbial profiles.
On the basis of these predictive features of α diversity and relative abundance of specific genera, we developed a simple taxonomic index for detecting dysbiosis in respiratory communities. With the goal of generalizability in other patient populations, we recognize that dysbiotic communities can differ substantially from one another (e.g., because of dominance from different pathogenic bacteria in each case), whereas symbiotic communities are expected to be more similar to one another, characterized by high α diversity and typical oral-origin bacterial composition. We derived a simple dysbiosis index (defined as deviation from normal α diversity or oral-origin bacterial composition) and demonstrated significant associations with outcomes independent of clinical confounders. However, this index has not been independently validated, and different α-diversity or relative-abundance thresholds may offer better discrimination in other cohorts, experimental platforms, and analytic pipelines. Consequently, the dysbiosis index should be considered only as proof of concept for the ability of microbiome profiling to offer clinically relevant prediction and insights into host–microbiota interactions in critical illness.
Our study has limitations. It is a single-center study, and the generalizability of our findings in critically ill populations beyond our tertiary care ICU requires external validation. Internal cross-validation testing of clustering showed robust classifications. Our study is also limited by the available sample size. Despite being the largest study of next-generation sequencing in acute respiratory failure, results from analyses for patient subgroups and specific bacteria require cautious interpretation, as the effective sample size for such analyses is small. We were also only able to conduct cross-sectional analyses and thus cannot draw inferences about the longitudinal evolution of microbial communities and host outcomes, which should be studied in larger patient cohorts. Our results were also exclusively based on 16S rRNA gene sequencing, and, consequently, we could not reach species-level resolution or analyze viability and virulence factors. For lung-microbiota analyses, we only used ETA (and not BAL) samples, and we thus could not assess for regional variability of communities or study host–microbiome interactions directly in the alveolar space. We used noninvasive ETA samples for practical and ethical purposes (minimal risk exposure to participants), as well as because of evidence from clinical practice guidelines (39) and comparative studies (40) supporting the reliability of noninvasive samples. In addition, comparisons of microbial profiles from ICU patients with those from healthy control subjects should be considered exploratory, given that different types of samples were used for examination of upper and lower respiratory tract microbiota, which may provide systematically different profiles. However, the relevant finding from these comparisons remains the profound interpatient heterogeneity in microbial profiles revealed for critically ill patients.
In summary, our study underlines the role of respiratory tract dysbiosis in acute respiratory failure, linking sequencing-based microbiologic composition clusters and individual bacterial abundance with patient-level differences in systemic inflammation and outcomes. Our unsupervised clustering approach for respiratory microbial communities offers a new framework to model and understand biologic heterogeneity in critical illness, beyond isolated approaches on host responses or limited views of microbiota by culture-based techniques. Further study of the respiratory microbiome with culture-independent approaches will help delineate host–microbiome interactions in the intubated respiratory tract and define new therapeutic approaches for acute respiratory failure.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank all members of the research team of the Acute Lung Injury Registry and Biospecimen Repository at the University of Pittsburgh, the medical and nursing staff in the medical ICU at the University of Pittsburgh Medical Center, and all patients and their families for participating in this research project.
Footnotes
Supported by NIH grants K23 HL139987 (G.D.K.), U01 HL098962 (A.M.), P01 HL114453 (B.J.M.), R01 HL097376 (B.J.M.), K24 HL123342 (A.M.), U01 HL137159 (P.V.B.), R01 HL127349 (P.V.B.), F32 HL137258 (J.E.), F32 HL142172 (W.B.), and K23 GM122069 (F.S.).
Author Contributions: Conception and design: G.D.K., A.M., and B.J.M. Acquisition, analysis, or interpretation of data: G.D.K., H.Y., L.Y., S.Q., A.F., X.-H.W., K.F., J.E., W.B., F.S., K.L., B.M., P.V.B., A.M., and B.J.M. Clinical cohort phenotyping: G.D.K., J.E., W.B., F.S., A.M., and B.J.M. Drafting of work and/or revising for important intellectual content: G.D.K., H.Y., L.Y., J.E., W.B., F.S., K.L., B.M., P.V.B., A.M., and B.J.M. Final approval of version to be published and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: G.D.K., H.Y., L.Y., S.Q., A.F., X.-H.W., K.F., J.E., W.B., F.S., K.L., B.M., P.V.B., A.M., and B.J.M.
Data sharing statement: All deidentified sequencing data have been submitted to Sequence Read Archive database, with BioSample accession number SAMN13548552-13549028.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.201912-2441OC on July 27, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Matthay MA, McAuley DF, Ware LB. Clinical trials in acute respiratory distress syndrome: challenges and opportunities. Lancet Respir Med. 2017;5:524–534. doi: 10.1016/S2213-2600(17)30188-1. [DOI] [PubMed] [Google Scholar]
- 2.Meyer NJ, Calfee CS. Novel translational approaches to the search for precision therapies for acute respiratory distress syndrome. Lancet Respir Med. 2017;5:512–523. doi: 10.1016/S2213-2600(17)30187-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA NHLBI ARDS Network. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med. 2014;2:611–620. doi: 10.1016/S2213-2600(14)70097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bos LD, Schouten LR, van Vught LA, Wiewel MA, Ong DSY, Cremer O, et al. MARS consortium. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax. 2017;72:876–883. doi: 10.1136/thoraxjnl-2016-209719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, et al. ARDS Network. Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med. 2017;195:331–338. doi: 10.1164/rccm.201603-0645OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitsios GD, Yang L, Manatakis DV, Nouraie M, Evankovich J, Bain W, et al. Host-response subphenotypes offer prognostic enrichment in patients with or at risk for acute respiratory distress syndrome. Crit Care Med. 2019;47:1724–1734. doi: 10.1097/CCM.0000000000004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet. 2014;384:691–702. doi: 10.1016/S0140-6736(14)61136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang YJ, Charlson ES, Collman RG, Colombini-Hatch S, Martinez FD, Senior RM. The role of the lung microbiome in health and disease: a National Heart, Lung, and Blood Institute workshop report. Am J Respir Crit Care Med. 2013;187:1382–1387. doi: 10.1164/rccm.201303-0488WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Segal LN, Clemente JC, Tsay J-CJ, Koralov SB, Keller BC, Wu BG, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol. 2016;1:16031. doi: 10.1038/nmicrobiol.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carney SM, Clemente JC, Cox MJ, Dickson RP, Huang YJ, Kitsios GD, et al. Methods in lung microbiome research. Am J Respir Cell Mol Biol. 2020;62:283–299. doi: 10.1165/rcmb.2019-0273TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitsios GD, Morowitz MJ, Dickson RP, Huffnagle GB, McVerry BJ, Morris A. Dysbiosis in the intensive care unit: microbiome science coming to the bedside. J Crit Care. 2017;38:84–91. doi: 10.1016/j.jcrc.2016.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickson RP. The microbiome and critical illness. Lancet Respir Med. 2016;4:59–72. doi: 10.1016/S2213-2600(15)00427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zakharkina T, Martin-Loeches I, Matamoros S, Povoa P, Torres A, Kastelijn JB, et al. The dynamics of the pulmonary microbiome during mechanical ventilation in the intensive care unit and the association with occurrence of pneumonia. Thorax. 2017;72:803–810. doi: 10.1136/thoraxjnl-2016-209158. [DOI] [PubMed] [Google Scholar]
- 14.Kelly BJ, Imai I, Bittinger K, Laughlin A, Fuchs BD, Bushman FD, et al. Composition and dynamics of the respiratory tract microbiome in intubated patients. Microbiome. 2016;4:7. doi: 10.1186/s40168-016-0151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panzer AR, Lynch SV, Langelier C, Christie JD, McCauley K, Nelson M, et al. Lung microbiota is related to smoking status and to development of acute respiratory distress syndrome in critically ill trauma patients. Am J Respir Crit Care Med. 2018;197:621–631. doi: 10.1164/rccm.201702-0441OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitsios GD, Fitch A, Manatakis DV, Rapport SF, Li K, Qin S, et al. Respiratory microbiome profiling for etiologic diagnosis of pneumonia in mechanically ventilated patients. Front Microbiol. 2018;9:1413. doi: 10.3389/fmicb.2018.01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kyo M, Nishioka K, Nakaya T, Kida Y, Tanabe Y, Ohshimo S, et al. Unique patterns of lower respiratory tract microbiota are associated with inflammation and hospital mortality in acute respiratory distress syndrome. Respir Res. 2019;20:246. doi: 10.1186/s12931-019-1203-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamarche D, Johnstone J, Zytaruk N, Clarke F, Hand L, Loukov D, et al. PROSPECT Investigators; Canadian Critical Care Trials Group; Canadian Critical Care Translational Biology Group. Microbial dysbiosis and mortality during mechanical ventilation: a prospective observational study. Respir Res. 2018;19:245. doi: 10.1186/s12931-018-0950-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickson RP, Singer BH, Newstead MW, Falkowski NR, Erb-Downward JR, Standiford TJ, et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol. 2016;1:16113. doi: 10.1038/nmicrobiol.2016.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dickson RP, Schultz MJ, van der Poll T, Schouten LR, Falkowski NR, Luth JE, et al. Biomarker Analysis in Septic ICU Patients (BASIC) Consortium. Lung microbiota predict clinical outcomes in critically ill patients. Am J Respir Crit Care Med. 2020;201:555–563. doi: 10.1164/rccm.201907-1487OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu CM, Aziz M, Kachur S, Hsueh P-R, Huang Y-T, Keim P, et al. BactQuant: an enhanced broad-coverage bacterial quantitative real-time PCR assay. BMC Microbiol. 2012;12:56. doi: 10.1186/1471-2180-12-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. ARDS Definition Task Force. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 23.Neto AS, Barbas CSV, Simonis FD, Artigas-Raventós A, Canet J, Determann RM, et al. PRoVENT; PROVE Network investigators. Epidemiological characteristics, practice of ventilation, and clinical outcome in patients at risk of acute respiratory distress syndrome in intensive care units from 16 countries (PRoVENT): an international, multicentre, prospective study. Lancet Respir Med. 2016;4:882–893. doi: 10.1016/S2213-2600(16)30305-8. [DOI] [PubMed] [Google Scholar]
- 24.Zhao J, Murray S, Lipuma JJ. Modeling the impact of antibiotic exposure on human microbiota. Sci Rep. 2014;4:4345. doi: 10.1038/srep04345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fair K, Dunlap DG, Fitch A, Bogdanovich T, Methé B, Morris A, et al. Rectal swabs from critically ill patients provide discordant representations of the gut microbiome compared to stool samples. mSphere. 2019;4:e00358-19. doi: 10.1128/mSphere.00358-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, et al. Lung HIV Microbiome Project. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187:1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holmes I, Harris K, Quince C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS One. 2012;7:e30126. doi: 10.1371/journal.pone.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shankar-Hari M, Fan E, Ferguson ND. Acute respiratory distress syndrome (ARDS) phenotyping. Intensive Care Med. 2019;45:516–519. doi: 10.1007/s00134-018-5480-6. [DOI] [PubMed] [Google Scholar]
- 29.Seymour CW, Kennedy JN, Wang S, Chang CH, Elliott CF, Xu Z, et al. Derivation, validation, and potential treatment implications of novel clinical phenotypes for sepsis. JAMA. 2019;321:2003–2017. doi: 10.1001/jama.2019.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bos LDJ, Scicluna BP, Ong DSY, Cremer O, van der Poll T, Schultz MJ. Understanding heterogeneity in biologic phenotypes of acute respiratory distress syndrome by leukocyte expression profiles. Am J Respir Crit Care Med. 2019;200:42–50. doi: 10.1164/rccm.201809-1808OC. [DOI] [PubMed] [Google Scholar]
- 31.Poroyko V, Meng F, Meliton A, Afonyushkin T, Ulanov A, Semenyuk E, et al. Alterations of lung microbiota in a mouse model of LPS-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2015;309:L76–L83. doi: 10.1152/ajplung.00061.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickson RP, Erb-Downward JR, Prescott HC, Martinez FJ, Curtis JL, Lama VN, et al. Intraalveolar catecholamines and the human lung microbiome. Am J Respir Crit Care Med. 2015;192:257–259. doi: 10.1164/rccm.201502-0326LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 34.Kitsios GD. Translating lung microbiome profiles into the next-generation diagnostic gold standard for pneumonia: a clinical investigator’s perspective. mSystems. 2018;3:e00153-17. doi: 10.1128/mSystems.00153-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vance RE, Isberg RR, Portnoy DA. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. 2009;6:10–21. doi: 10.1016/j.chom.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, et al. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature. 2011;474:385–389. doi: 10.1038/nature10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langelier C, Kalantar KL, Moazed F, Wilson MR, Crawford ED, Deiss T, et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc Natl Acad Sci U S A. 2018;115:E12353–E12362. doi: 10.1073/pnas.1809700115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, Haidar G, Zia H, Nettles R, Qin S, Wang X, et al. Metagenomic identification of severe pneumonia pathogens in mechanically-ventilated patients: a feasibility and clinical validity study. Respir Res. 2019;20:265. doi: 10.1186/s12931-019-1218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalil AC, Metersky ML, Klompas M, Muscedere J, Sweeney DA, Palmer LB, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the infectious diseases society of America and the American thoracic society. Clin Infect Dis. 2016;63:e61–e111. doi: 10.1093/cid/ciw353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalantar KL, Moazed F, Christenson SC, Wilson J, Deiss T, Belzer A, et al. Metagenomic comparison of tracheal aspirate and mini-bronchial alveolar lavage for assessment of respiratory microbiota. Am J Physiol Lung Cell Mol Physiol. 2019;316:L578–L584. doi: 10.1152/ajplung.00476.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.