

Abstract
The Genotype-Tissue Expression (GTEx) project was established to characterize genetic effects on the transcriptome across human tissues, and to link these regulatory mechanisms to trait and disease associations. Here, we present analyses of the v8 data, examining 15,201 RNA-sequencing samples from 49 tissues of 838 post-mortem donors. We comprehensively characterize genetic associations for gene expression and splicing in cis and trans, showing that regulatory associations are found for almost all genes, and describe the underlying molecular mechanisms and their contribution to allelic heterogeneity and pleiotropy of complex traits. Leveraging the large diversity of tissues, we provide insights into the tissue-specificity of genetic effects and show that cell type composition is a key factor in understanding gene regulatory mechanisms in human tissues.
Introduction
A pressing need in human genetics remains the characterization and interpretation of the function of the millions of genetic variants across the human genome. This is essential for identifying the molecular mechanisms of genetic risk for complex traits and diseases, which are mainly driven by non-coding loci with largely uncharacterized regulatory functions. To address this challenge, several projects have built comprehensive annotations of genome function across tissues and cell types (1, 2), and mapped the effects of regulatory variation across large numbers of individuals, primarily from whole blood and blood cell types (3–5). The Genotype-Tissue Expression (GTEx) project provides an essential intersection where variant function can be studied across a wide range of both tissues and individuals.
The GTEx project was launched in 2010 with the aim of building a catalog of genetic effects on gene expression across a large number of human tissues in order to elucidate the molecular mechanisms of genetic associations with complex diseases and traits, and improve our understanding of regulatory genetic variation (6). The project set out to collect biospecimens from ~50 tissues from up to ~1000 postmortem donors, and to create standards and protocols for optimizing postmortem tissue collection and donor recruitment (7, 8), biospecimen processing (7), and data sharing (www.gtexportal.org).
Following the GTEx pilot (9) and mid-stage results (10), we present a final analysis of the v8 data release from the GTEx Consortium. We provide a catalog of genetic regulatory variants affecting gene expression and splicing in cis and trans across 49 tissues, and describe patterns and mechanisms of tissue- and cell type specificity of genetic regulatory effects. Through integration of GTEx data with genome-wide association studies (GWAS), we characterize mechanisms of how genetic effects on the transcriptome mediate complex trait associations.
QTL discovery
The GTEx v8 data set after quality control (11) consists of 838 donors and 17,382 samples from 52 tissues and two cell lines. In the analysis of this study, we used 49 tissues or cell lines that had at least 70 individuals with both RNA sequence (RNA-seq) and genotype data from whole genome sequencing (WGS), comprising a total of 15,201 samples from 838 donors (Fig. 1A, and figs. S1 and S2). Of the 838 donors, 715 (85.3%) were European American, 103 (12.3%) African American, and 12 (1.4%) Asian American, with 16 (1.9%) reporting Hispanic or Latino ethnicity; 557 (66.4%) donors were male and 281 (33.5%) female (fig. S1). WGS was performed for each donor to a median depth of 32x, resulting in the detection of a total of 43,066,422 single nucleotide variants (SNVs) after QC and phasing (10,008,325 with MAF ≥ 0.01) and 3,459,870 small indels (762,535 with MAF ≥ 0.01) (fig. S3 and table S1, (11)). The mRNA of each of the tissue samples was sequenced to a median depth of 82.6 million reads, and alignment, quantification and quality control were performed as described in (11) (figs. S4, S5, and S6).
Figure 1. Sample and data types in the GTEx v8 study.
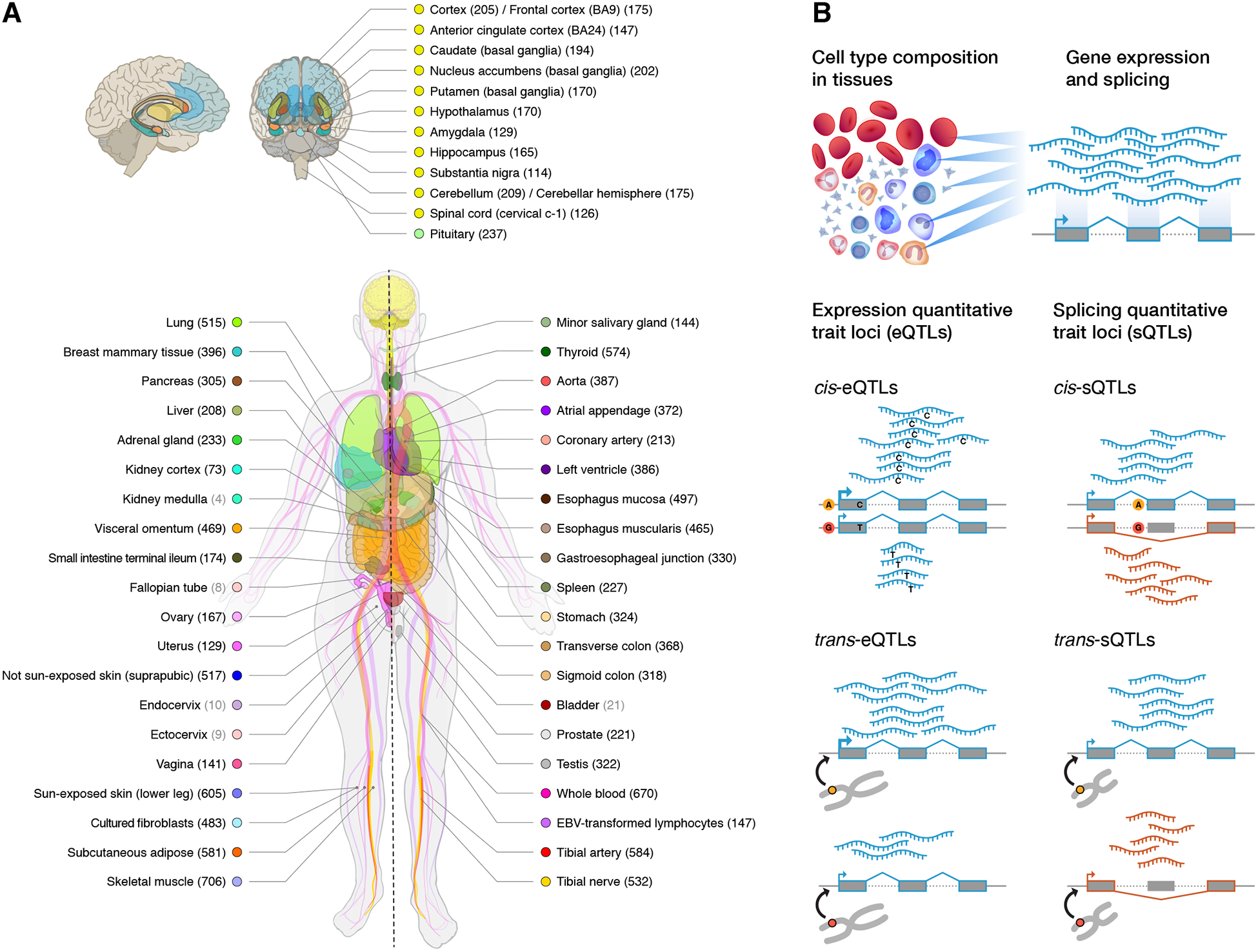
(A) Illustration of the 54 tissue types examined (including 11 distinct brain regions and 2 cell lines), with sample numbers from genotyped donors in parentheses and color coding indicated in the adjacent circles. Tissues with ≥70 samples were included in QTL analyses. (B) Illustration of the core data types used throughout the study. Gene expression and splicing were quantified from bulk RNA-seq of heterogenous tissue samples, and local and distal genetic effects (cis-QTLs and trans-QTLs, respectively) were quantified across individuals for each tissue.
The resulting data provide a broad survey of individual- and tissue- specific gene expression, enabling a comprehensive view of the impact of genetic variation on gene regulation (Fig. 1B). We mapped genetic loci that affect the expression (eQTL) or splicing (sQTL) of protein-coding and lincRNA genes, both in cis and trans. Genes with an eQTL or sQTL are called eGenes and sGenes, and significant variants eVariants and sVariants, respectively. Across all tissues, we discovered cis-eQTLs (5% FDR, per tissue (11) with 1% FDR results shown in fig. S7) for 18,262 protein coding and 5,006 lincRNA genes (23,268 total genes with a cis-eQTL, or cis-eGenes, corresponding to 94.7% of all protein coding and 67.3% of all detected lincRNA genes detected in at least one tissue), with a total of 4,278,636 genetic variants (43% of all variants with MAF ≥ 0.01) that were significant in at least one tissue (cis-eVariants) (Fig. 2A, figs. S7 and S8, and table S2). The discovered eQTLs had a high replication rate in external datasets (fig. S12 and S13). Cis-eQTLs for all long non-coding RNAs (lncRNAs), which include lincRNAs and other types, are characterized in a companion analysis (12). The genes lacking a cis-eQTL were enriched for those lacking expression in the tissues analyzed by GTEx, including genes involved in early development (fig. S9). While most of the discovered cis-eQTLs had small effect sizes measured as allelic fold change (aFC), across tissues an average of 22% of cis-eQTLs had an over 2-fold effect on gene expression (fig. S14). We mapped splicing QTLs (sQTLs) in cis with intron excision ratios from LeafCutter (11, 13), and discovered 12,828 (66.5%) protein coding and 1,600 (21.5%) lincRNA genes (14,424 total) with a cis-sQTL (5% FDR, per tissue) in at least one tissue (cis-sVariants) (Fig. 2A, table S2, with 1% FDR results shown in fig. S7). As expected (10), cis-QTL discovery was highly correlated with the sample size for each tissue (Spearman’s rho = 0.95 for cis-eQTLs, 0.92 for cis-sQTLs). The increased cis-eQTL discovery in larger tissues is primarily driven by additional power to discover small effects, with discovery of cis-eGenes with over two-fold effect saturating at ~1500 genes in tissues with >200 samples (fig. S14).
Figure 2. QTL discovery.
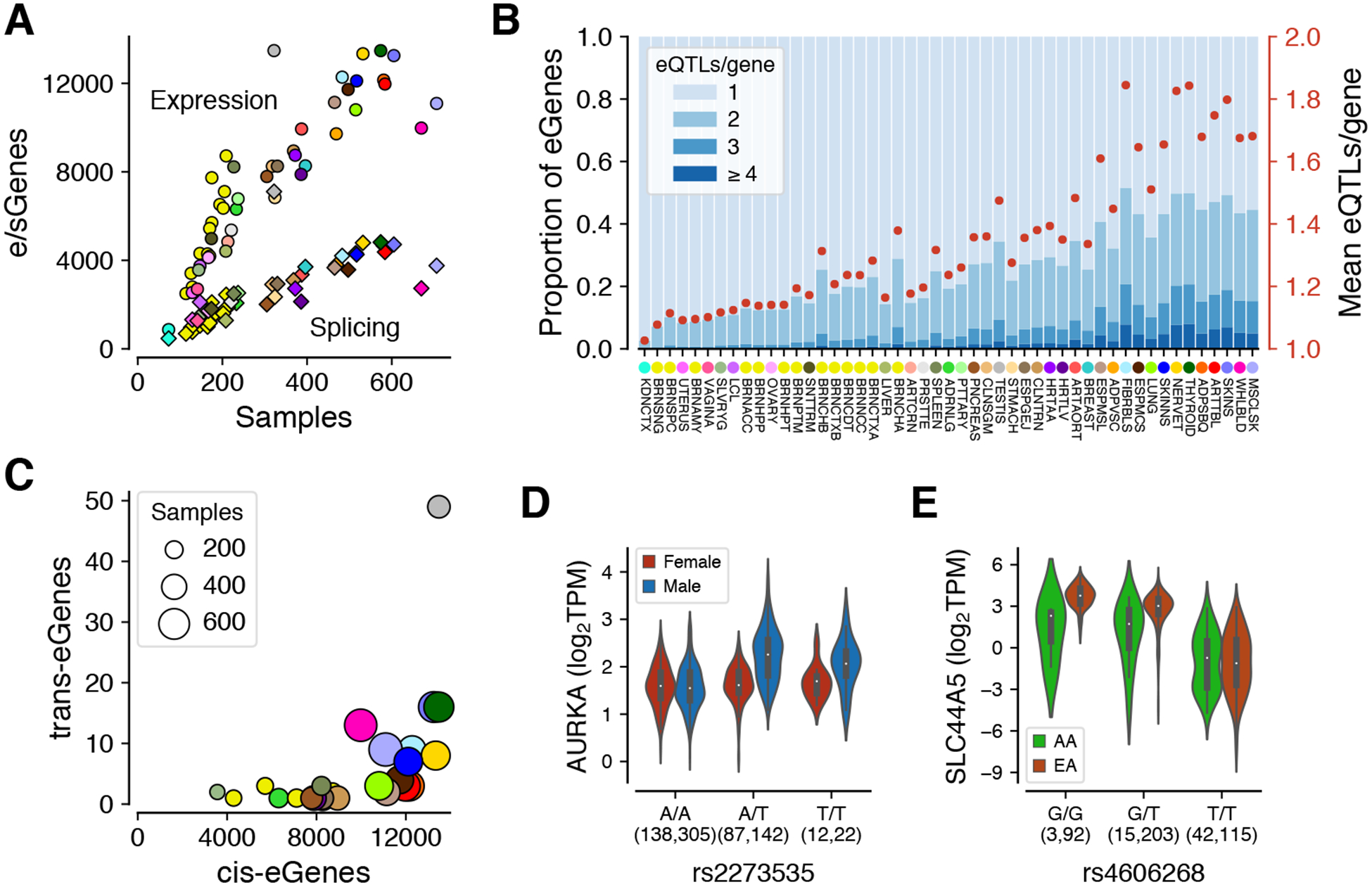
(A) The number of genes with a cis-eQTL (eGenes) or cis-sQTL (sGenes) per tissue, as a function of sample size. See Fig. 1A for the legend of tissue colors. (B) Allelic heterogeneity of cis-eQTLs depicted as proportion of eGenes with ≥1 independent cis-eQTLs (blue stacked bars; left y-axis) and as a mean number of cis-eQTLs per gene (red dots; right y-axis). The tissues are ordered by sample size. (C) The number of genes with a trans-eQTL as a function of the number of cis-eGenes. (D) Sex-biased cis-eQTL for AURKA in skeletal muscle, where rs2273535-T is associated with increased AURKA expression in males (p = 9.02×10−27) but not in females (p = 0.75). (E) Population-biased cis-eQTL for SLC44A5 in esophagus mucosa (allelic fold change = −2.85 and −4.82 and in African Americans (AA) and European Americans (EA), respectively; permutation p-value = 1.2×10−3).
Previous studies have shown widespread allelic heterogeneity of gene expression in cis, i.e., multiple independent causal eQTLs per gene (4, 14, 15). We mapped independent cis-eQTLs and cis-sQTLs using stepwise regression, where the 5% FDR threshold for significance was defined by the single cis-QTL mapping (10). We observed widespread allelic heterogeneity, with up to 50% of eGenes having more than one independent cis-eQTL in the tissues with the largest sample sizes (Fig. 2B, and fig. S10). Our analysis captured a lower rate of allelic heterogeneity for cis-sQTLs, which can be a result of both underlying biology and lower power in cis-sQTL mapping (fig. S10). These results highlight gains in cis-eQTL mapping with increasing sample sizes even when the discovery of new eGenes in specific tissues starts to saturate.
Interchromosomal trans-eQTL mapping yielded 143 trans-eGenes (121 protein coding and 22 lincRNA at 5% FDR assessed at the gene level, separately for each gene type), after controlling for false positives due to read misalignment (11, 16) (table S13). The number of trans-eGenes discovered per tissue is correlated with sample size (Spearman’s rho = 0.68), and to the number of cis-eQTLs (rho = 0.77), with outlier tissues such as testis contributing disproportionately to both cis and trans (Fig. 2C). We identified a total of 49 trans-eGenes in testis, with 47 found in no other tissue even at FDR 50%. Over two-fold effect sizes on trans-eGene expression were observed for 19% of trans-eQTLs (fig. S14). Trans-sQTLs mapping yielded 29 trans-sGenes (5% FDR, per tissue), including a replication of a previously described trans-sQTL (3) and visual support of the association pattern in several loci (11) (fig. S11, table S14). These results suggest that while trans-sQTL mapping is challenging, we can discover robust genetic effects on splicing in trans.
We produced allelic expression (AE) data using two complementary approaches (11). In addition to the conventional AE data for each heterozygous genotype, we produced AE data by haplotype, integrating data from multiple heterozygous sites in the same gene, yielding 153 million gene-level measurements (≥8 reads) across all samples (17). Allelic expression reflects differential regulation of the two haplotypes in individuals that are heterozygous for a regulatory variant in cis; indeed, cis-eQTL effect size is strongly correlated with allelic expression (median rho = 0.82) (10). We hypothesized that cis-sQTLs could also partially contribute to allelic imbalance even if only for parts of transcripts. However, there is drastically less signal of increased allelic imbalance among individuals heterozygous for cis-sQTLs (median Spearman’s rho = −0.05) (fig S15). This indicates that allelic expression data primarily captures cis-eQTL effects, and that genetic splicing variation in cis is not strongly reflected in gene-level AE data.
Genetic regulatory effects across populations and sexes
Variability in human traits and diseases between sexes and population groups is likely to partially derive from differences in genetic effects (18–20). To study whether genetic regulatory variants manifest this, we analyzed variable cis-eQTL effects between males and females, as well as between individuals of European and African ancestry. Since external replication data sets are sparse, we developed an allelic expression approach for validation with an orthogonal data type from the same samples (17): allelic imbalance in individuals heterozygous for the cis-eQTL allows individual-level quantification of the cis-eQTL effect size (21), and can be correlated with the interaction terms used in cis-eQTL analysis to validate modifier effects of the cis-eQTL association (fig. S16).
To characterize sex-differentiated genetic effects on gene expression in GTEx tissues, we mapped sex-biased cis-eQTLs (sb-eQTLs). Analyzing the set of all conditionally independent cis-eQTLs, we identified eQTLs with significantly different effects between sexes by fitting a linear regression model and testing for a significant genotype-by-sex (G×S) interaction (11). Across the 44 GTEx tissues shared among sexes, we identified 369 sb-eQTLs (FDR ≤ 25%), characterized further in (22). Sex-biased eQTL discovery had a modest correlation with tissue sample size (Spearman’s rho = 0.39, p = 0.03), with most sb-eQTLs discovered in breast but also in muscle, skin and adipose tissues. In some cases, the cis-eQTL signal — identified with males and females combined — seems to be driven exclusively by one sex. For example, the cis-eQTL association of rs2273535 with the gene AURKA in skeletal muscle (cis-eQTL p = 6.92×1024) is correlated with sex (pG×S = 9.28×10−12, Storey qG×S = 1.07×10−7, AE validation p = 1.15×10−11) and present only in males (Fig. 2D, and fig. S17). AURKA is a member of the serine/threonine kinase family involved in mitotic chromosomal segregation that has been widely studied as a risk factor in several cancers (23–26) and has been recently shown to be involved in muscle differentiation (27).
We also characterized population-biased cis-eQTLs (pb-eQTLs), where a variant’s molecular effect on gene expression differs between individuals of European and African ancestry, controlling for differences in allele frequency, linkage disequilibrium (LD) and covariates (11). Analyzing 31 tissues with sample sizes >20 in both populations, we mapped genes with a different eQTL effect size measured by aFC. After applying stringent filters to remove differences potentially explained by LD or other artifacts (fig. S18A), we identified 178 pb-eQTLs for 141 eGenes (FDR ≤ 25%) that show a moderate degree of validation in allele-specific expression data (fig. S18C,D, table S10). While some of the pb-eQTL effects are tissue-specific, there are also effects that are shared across most tissues (fig. S18E). Fig. 2E shows an example of a pb-eQTL for the SLC44A5 gene involved in transport of sugars and amino acids, and expressed at different levels between epidermis of lighter and darker skin (reconstructed in vitro) (28, 29). In Europeans, the derived allele of rs4606268 decreases expression of the gene in esophagus mucosa (aFC = −4.82), but this effect is significantly lower in African Americans (aFC = −2.85, permutation p-value = 1.2×10−3, AE validation p = 0.002, fig. S18C).
Altogether, despite the relaxed FDR, we discovered only a few hundred sex- or population-biased cis-eQTLs out of tens of thousands of cis-eQTLs in GTEx. This indicates that there are few regulatory variants with major modifier effects, and that these associations continue to be challenging to identify without a much larger sample size. However, the discovered effects can provide insights in to sex- or population-specific regulatory effects on gene expression. Importantly, factors correlated with sex or population, e.g., cell type composition or environmental exposures, may contribute to sex- or population-biased cis-eQTLs. These effects are described in detail in (22).
Fine-mapping
A major challenge of all genetic association studies is to distinguish the causal variants from their LD proxies. We applied three different statistical fine-mapping methods — CaVEMaN (30), CAVIAR (31), and dap-g (32) — to infer likely causal variants of cis-eQTLs in each tissue (Fig. 3A) (11). For many cis-eQTLs the causal variant can be mapped with a high probability to a handful of candidates: the 90% credible set for each cis-eQTL consists of variants that include the causal variant with 90% probability; using dap-g, we identified a median of 6 variants in the 90% credible set for each cis-eQTL (fig. S19). Furthermore, 9.3% of the cis-eQTLs have a variant with a posterior probability > 0.8 according to dap-g, indicating a single likely causal variant for those cis-eQTLs. We defined a consensus set of 24,740 cis-eQTLs across all tissues (7,709 unique variants), for which the posterior probability was >0.8 across all three methods (fig. S20). Fine-mapped variants were significantly more highly enriched among experimentally validated causal variants from MPRA (33) and SuRE (34), compared to the lead eVariant across all eGenes (Fig. 3B). The highest enrichment was observed for the consensus set although with overlapping confidence intervals (Fig. 3B). This demonstrates how careful fine-mapping facilitates the identification of likely causal regulatory variants.
Figure 3. Fine mapping of cis-eQTLs.
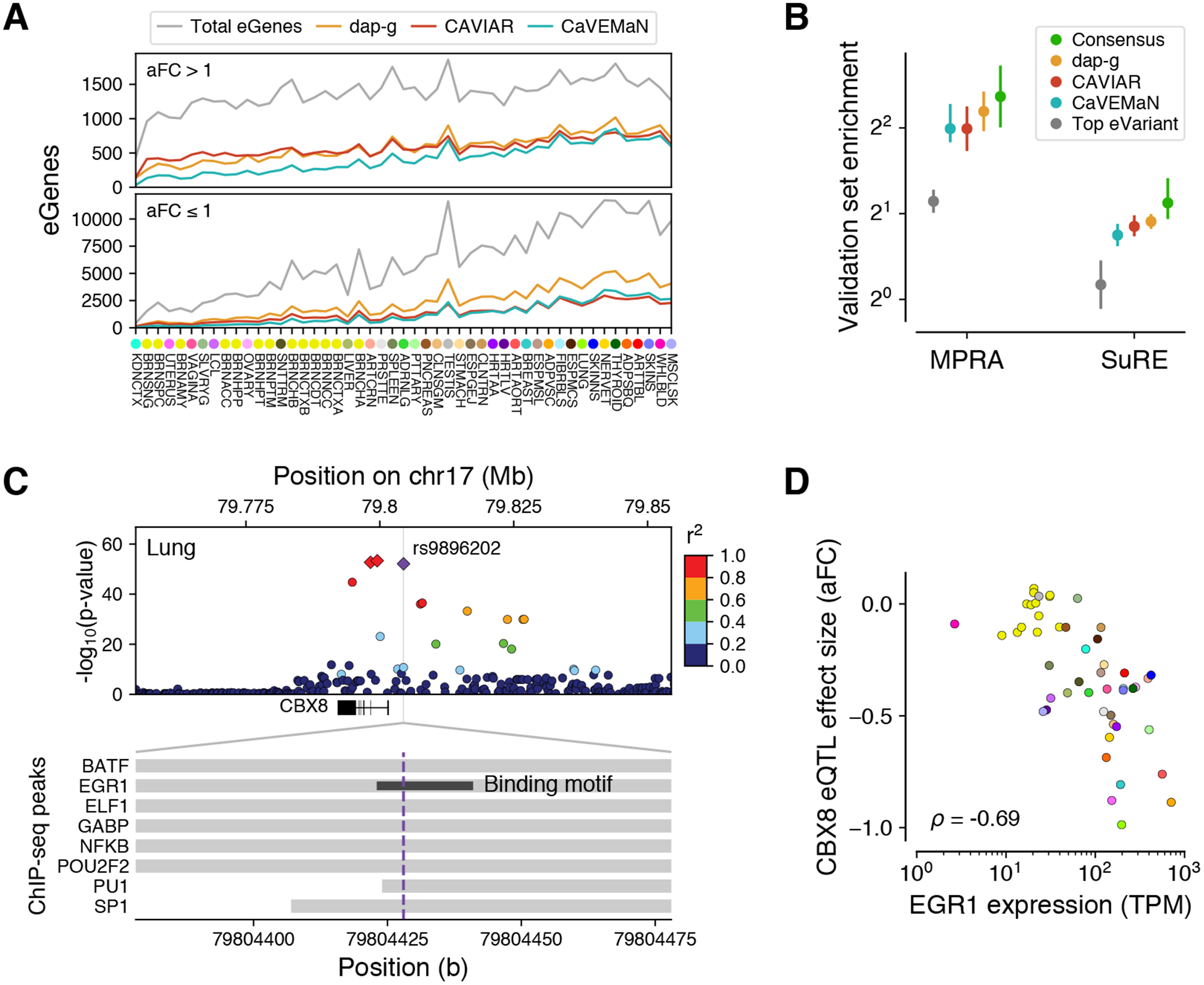
(A) Number of eGenes per tissue with variants fine-mapped with >0.5 posterior probability of causality, using three methods. The overall number of eGenes with at least one fine-mapped eVariant increases with sample size for all methods. However, this increase is in part driven by better statistical power to detect small effect size cis-eQTLs (aFC or allelic fold change ≤1 in log2 scale; see also fig. S14) with larger sample sizes, and the proportion of well fine-mapped eGenes with small effect sizes increases more modestly with sample size (bottom vs. top panels), indicating that such cis-eQTLs are generally more difficult to fine-map. (B) Enrichment of variants among experimentally validated regulatory variants, shown for the cis-eVariant with the best p-value (top eVariant), and those with posterior probability of causality >0.8 according to each of the three methods individually or all of them (consensus). Error bars: 95% CI. (C) The cis-eQTL signal for CBX8 is fine-mapped to a credible set of three variants (red and purple diamonds), of which rs9896202 (purple diamond) overlaps a large number of transcription factor binding sites in ENCODE ChIP-seq data and disrupts the binding motif of EGR1. (D) The potential role of EGR1 binding driving this cis-eQTL is further supported by correlation between EGR1 expression and the CBX8 cis-eQTL effect size across tissues.
Knowing the likely causal variant enables greater insights into the molecular mechanisms of individual eQTLs, including the mechanisms of their tissue-specific effects. Fig. 3C shows an example of an eQTL for the gene CBX8 that colocalizes with breast cancer risk and birth weight (posterior probability 0.68 for both in lung). One of the three variants in the confident set overlaps the binding site and disrupts the motif of the transcription factor EGR1 (1) (fig. S21). The role of EGR1 as an upstream driver of this eQTL is further supported by a cross-tissue correlation of the effect size of the eQTL and the expression level of EGR1 (Spearman’s rho = −0.69) (Fig. 3D).
Functional mechanisms of QTL associations
Quantitative trait data from multiple molecular phenotypes, integrated with the regulatory annotation of the genome (table S3), offer a powerful way to understand the molecular mechanisms and phenotypic consequences of genetic regulatory effects. As expected, cis-eQTLs and cis-sQTLs are enriched in functional elements of the genome (Fig. 4A). While the strongest enrichments are driven by variant classes that lead to splicing changes or nonsense-mediated decay, these account for relatively few variants. Cis-sQTLs are enriched almost entirely in transcribed regions, while cis-eQTLs are enriched in transcriptional regulatory elements, as well. Previous studies (4, 35) have indicated that cis-eQTL and cis-sQTL effects on the same gene are typically driven by different genetic variants. This is corroborated by the GTEx v8 data, where the overlap of cis-eQTL credible sets of likely causal variants, from CAVIAR analysis, have only a 12% overlap with cis-sQTL credible sets (fig. S22). Functional enrichment of overlapping and non-overlapping cis-eQTLs and cis-sQTLs, using stringent LD filtering, showed that the patterns characteristic for each type — such as enrichment of cis-eQTL in enhancers and cis-sQTLs in splice sites — are even stronger for distinct loci (fig. S22).
Figure 4. Functional mechanisms of genetic regulatory effects.
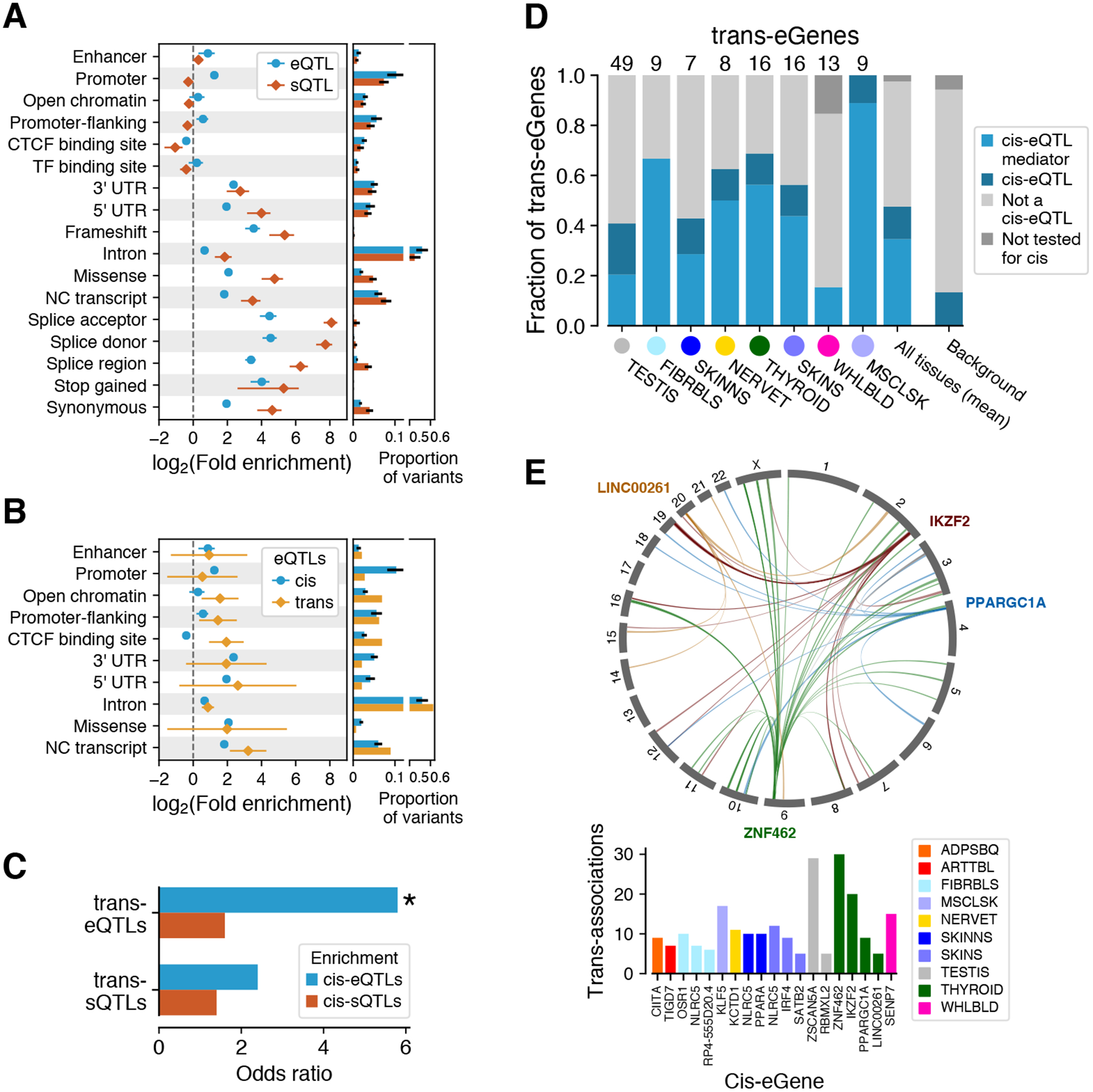
QTL enrichment in functional annotations for (A) cis-eQTLs and cis-sQTLs and for (B) trans-eQTLs. cis-QTL enrichment is shown as mean ± s.d. across tissues; trans-eQTL enrichment as 95% C.I. (C) Enrichment of lead trans-e/sVariants that have been tested for in cis-QTL effects being significant also cis-e/sVariants in the same tissue. * denotes significant enrichment, p < 10−21. (D) Proportion of trans-eQTLs that are significant cis-eQTLs or mediated by cis-eQTLs. (E) Trans associations of cis-mediating genes identified through colocalization (PP4 > 0.8 and nominal association with discovery trans-eVariant p < 10−5). Top: associations for four Thyroid cis-eQTLs (indicated by gene names); bottom: cis-mediating genes with ≥5 colocalizing trans-eQTLs.
We hypothesized that eVariants and their target eGenes in cis are more likely to be in the same topologically associated domains (TADs) that allow chromatin interactions between more distant regulatory regions and target gene promoters (36). To test this, we analyzed TAD data from ENCODE (1) and cis-eQTLs from matching GTEx tissues (table S3). Compared to matching random variant-gene pairs and controlling for distance from the transcription start site, cis-eVariant-eGene pairs were significantly enriched for being in the same TAD (median OR 4.55; all p<10−12) (fig. S23).
Trans-eQTLs are enriched in regulatory annotations that suggest both pre- and post-transcriptional mechanisms (Fig. 4B). Unlike cis-eQTLs, trans-eQTLs are enriched in CTCF binding sites, suggesting that disruption of CTCF binding may underlie distal genetic regulatory effects, potentially via its effect on interchromosomal chromatin interactions (36). Trans-eQTLs are also partially driven by cis-eQTLs (37, 38). Indeed, we observed a significant enrichment of lead trans-eVariants tested in cis being also cis-eVariants in the same tissue (5.9x; two-sided Fisher’s exact test p = 5.03×10−22, Fig. 4C). A lack of analogous enrichment suggests that cis-sQTLs are less important contributors to trans-eQTLs (p = 0.064), and trans-sVariants had no significant enrichment of either cis-eQTLs (p = 0.051) or cis-sQTLs (p = 0.53). A further demonstration of the important contribution of cis-eQTLs to trans-eQTLs is that, on the basis of mediation analysis, 77% of lead trans-eVariants that are also cis-eVariants (corresponding to 31.6% of all lead trans-eVariants) appear to act through the cis-eQTL (Fig. 4D, and fig. S24). Colocalization of cis-eQTLs and trans-eQTLs was widespread and often tissue-specific, with Fig. 4E showing cis-eQTLs with at least ten nominally significant colocalized trans-eQTLs each (PP4 > 0.8 and trans-eQTL p-value < 10−5), pinpointing how local effects on gene expression can potentially lead to downstream regulatory effects across the genome (fig. S25 and table S16). The many remaining trans-eQTLs that do coincide with a cis-eQTL may arise due to mechanisms including undetected cis effects in specific cell types or conditions, protein coding changes, effects on cell type heterogeneity, or more complex causality such as a variant that influences a trait with downstream consequences on gene expression.
Genetic regulatory effects mediate complex trait associations
In order to analyze the role of regulatory variants in genetic associations for human traits, we first asked whether variants in the GWAS catalog were enriched for significant QTLs, compared to all variants tested for QTLs (11). We observed a 1.46-fold enrichment for cis-eQTLs (63% vs 43%) and 1.86-fold enrichment for cis-sQTLs (37% vs 20%). The enrichment was even stronger, 6.97-fold (0.029% vs 0.0042%) for trans-eQTLs, consistent with other analyses (39) (Fig. 5A, fig. S26, tables S5 and S6). Cell type proportion may influence detection of trans-eQTLs in heterogeneous tissues, and may also be reflected in GWAS associations for blood cell count phenotypes and other complex traits. To minimize the possible impact of cell type heterogeneity on these enrichment statistics, we repeated these analyses among traits excluding blood cellularity traits. The resulting enrichments were 5.21-fold for trans-eQTLs, 1.43-fold for cis-eQTLs, and 1.81 for cis-sQTLs, largely preserving the patterns observed using the full set of GWAS traits.
Figure 5. Regulatory mechanisms of GWAS loci.
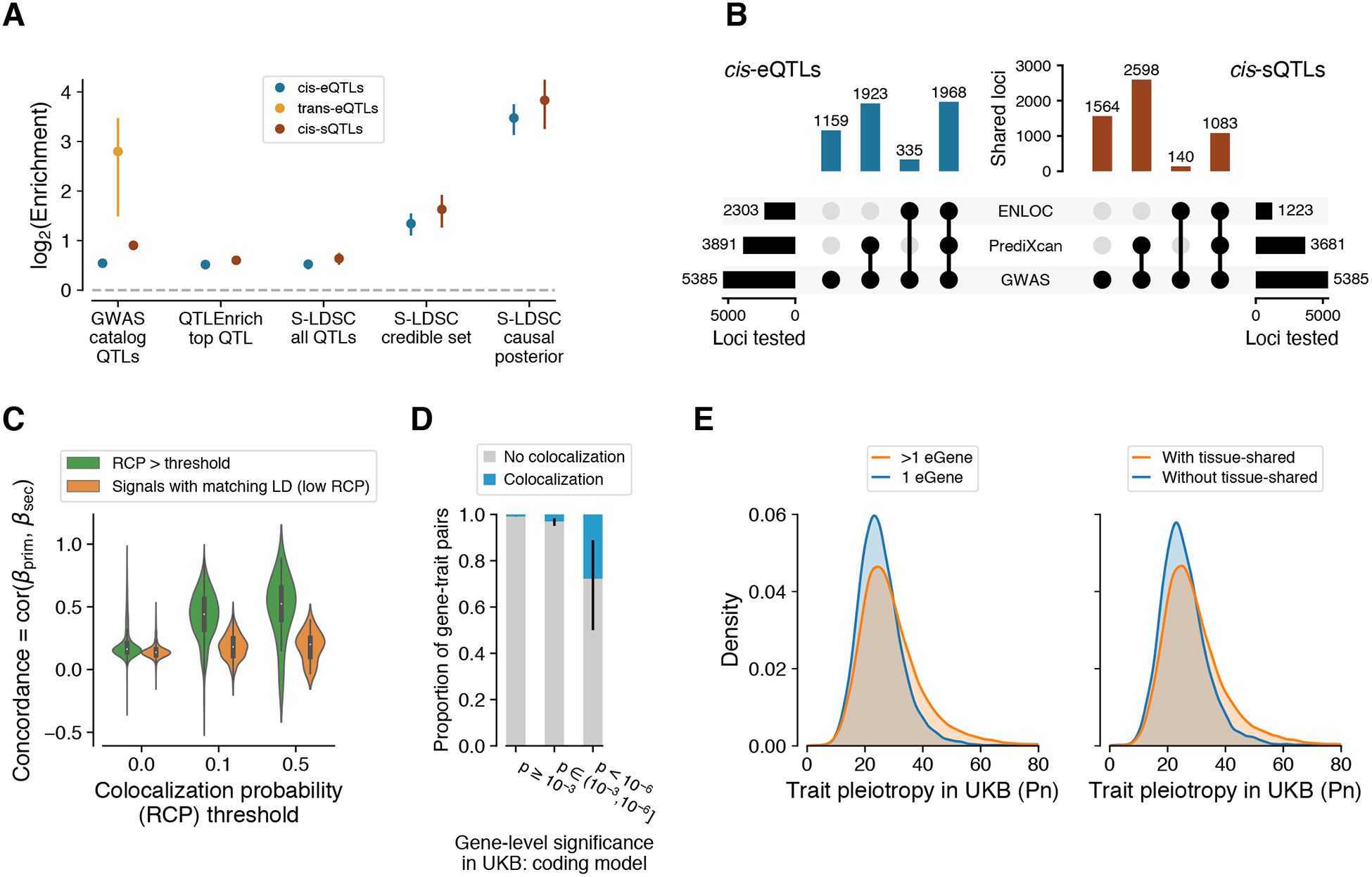
(A) GWAS enrichment of cis-eQTLs, cis-sQTLs, and trans-eQTLs measured with different approaches: enrichment calculated from GWAS summary statistics of the most significant cis-QTL per eGene/sGene with QTLEnrich and LD Score regression with all significant cis-QTLs (S-LDSC all cis-QTLs), simple QTL overlap enrichment with all GWAS catalog variants, and LD Score regression with fine-mapped cis-QTLs in the 95% credible set (S-LDSC credible set) and using posterior probability of causality as a continuous annotation (S-LDSC causal posterior). Enrichment is shown as mean and 95% CI. (B) Number of GWAS loci linked to e/sGenes through colocalization (ENLOC) and association (PrediXcan), aggregated across tissues. (C) Concordance of mediated effects among independent cis-eQTLs for the same gene, shown for different levels of regional colocalization probability (RCP (32)), which is used as a proxy for the gene’s causality. As the null, we show the concordance for LD matched genes without colocalization. (D) Proportion of colocalized cis-eQTLs with a matching phenotype for genes with different level of rare variant trait association in the UK Biobank (UKB). (E) Horizontal GWAS trait pleiotropy score distribution for cis-eQTLs that regulate multiple vs. a single gene (left), and for cis-eQTLs that are tissue-shared vs. specific.
This approach does not leverage the full power of genome-wide GWAS and QTL association statistics, nor account for LD contamination, a situation wherein the causal variants for QTL and GWAS signals are distinct but LD between the two causal variants can suggest a false functional link (40). Hence, for subsequent analyses (below) we selected 87 Genome Wide Association Studies (GWAS) representing a broad array of binary and continuous complex traits that have summary results available in the public domain (11, 41), and cis-QTL statistics calculated from the European subset of GTEx donors to match the ancestry of GWAS studies (fig. S29). The analyses were performed for all pairwise combinations of 87 phenotypes and 49 tissues, and are summarized using an approach that accounts for similarity between tissues and variable standard errors of the QTL effect estimates, driven mainly by tissue sample size (fig. S27, and tables S4 and S11 (11)).
To analyze the mediating role of cis-regulation of gene expression on complex traits (35, 42), we used two complementary approaches, QTLEnrich (43) and stratified LD score regression (S-LDSC) (11, 44). To rule out the possibility that enrichment is driven by specific features of cis-QTLs such as allele frequency, distance to the transcription start site, or local level of LD (number of LD proxy variants; r2 ≥ 0.5), we used QTLEnrich. We found a 1.46-fold (SE=0.006) and 1.56-fold (SE=0.007) enrichment of trait associations among best cis-eQTLs and cis-sQTLs, respectively, adjusting for enrichment among matched null variants (Fig. 5A, table S7). The fact that these enrichment estimates differ little from those derived from the GWAS catalog overlap (above), even after accounting for the potential confounders, indicates how relatively robust these estimates are. Next, we used S-LDSC adjusting for functional annotations (44) to confirm the robustness of these results and to analyze how GWAS enrichment is affected by the causal e/sVariant being typically unknown (11). We computed the heritability enrichment of all cis-QTLs, fine-mapped cis-QTLs (in 95% credible set and posterior probability > 0.01 from dap-g), and fine-mapped cis-QTLs with maximum posterior inclusion probability as continuous annotation (MaxCPP) (45) (Fig. 5A). The largest increase in GWAS enrichment was for likely causal cis-QTL variants (11.1-fold (SE=1.2) for cis-eQTLs and 14.2-fold (SE=2.4) for and cis-sQTLs, for the continuous annotation), which is strong evidence of shared causal effects of cis-QTLs and GWAS, and for the importance of fine-mapping.
Joint enrichment analysis of cis-eQTLs and cis-sQTLs shows an independent contribution to complex trait variation from both (fig. S28, (11)), consistent with their limited overlap (fig. S22). The relative GWAS enrichments of cis-sQTLs and cis-eQTLs were similar (Fig. 5A; not significant for the robust QTLEnrich and LDSC analyses), but the larger number of cis-eQTLs discovered (Fig. 2) suggests a greater aggregated contribution of cis-eQTLs.
While these enrichment methods are powerful for genome-wide estimation of the QTL contribution to GWAS signals, they are not informative of regulatory mechanisms in individual loci. Thus, to provide functional interpretation of the 5,385 significant GWAS associations in 1,167 loci from approximately independent LD blocks (46) across the 87 complex traits, we performed colocalization with enloc (32) to quantify the probability that the cis-QTL and GWAS signals share the same causal variant. We also assessed the association between the genetically regulated component of expression or splicing and complex traits with PrediXcan (11, 41, 47). Both methods take multiple independent cis-QTLs into account, which is critical in large cis-eQTL studies with widespread allelic heterogeneity, such as GTEx. Of the 5,385 GWAS loci, 43% and 23% were colocalized with a cis-eQTL and cis-sQTL, respectively (Fig. 5B). A large proportion of colocalized genes coincide with significant PrediXcan trait associations with predicted expression or splicing (median of 86% and 88% across phenotypes respectively; figs. S30, S31, S32, S33, tables S8, S15), with the full resource available in (41). While colocalization does not prove a causal role of a QTL in any given locus nor a genome-wide proportion of GWAS loci driven by eQTLs, these results suggest target genes and their potential molecular changes for thousands of GWAS loci, sometimes including both cis and trans targets (fig. S34).
Having multiple independent cis-eQTLs for a large number of genes allowed us to test whether mediated effects of primary and secondary cis-eQTLs on phenotypes — the ratio of GWAS and cis-eQTL effect sizes — are concordant. To make sure that concordance is not driven by residual LD between primary and secondary signals, we used LD-matched cis-eGenes with low colocalization probability as controls (11, 41), and observed a significant increase in primary and secondary cis-eQTL concordance for colocalized genes (correlated t-test p-value < 10−30; Fig. 5C). Additionally, colocalization of a cis-eQTL increased the colocalization of an independent cis-sQTL in the same locus (OR = 4.27, Fisher’s exact test p < 10−16), and correspondingly colocalization of a cis-sQTL increased cis-eQTL colocalization (OR = 4.54, Fisher’s exact test p < 10−16; figs. S35 and S36). This indicates that multiple regulatory effects for the same gene often mediate the same complex trait associations. Furthermore, genes with suggestive rare variant trait associations in the UK Biobank (48) have a substantially increased proportion of colocalized eQTLs for the same trait (Fig. 5D, and fig. S37), showing concordant trait effects from rare coding and common regulatory variants (49). These genes, as well as those with multiple colocalizing cis-QTLs, represent bona fide disease genes with multiple independent lines of evidence.
The growing number of genome and phenome studies has revealed extensive pleiotropy, where the same variant or locus associates with multiple organismal phenotypes (50). We sought to analyze how this phenomenon can be driven by gene regulatory effects. First, we calculated the number of cis-eGenes of each fine-mapped and LD-pruned cis-eVariant per tissue at local false sign rate (LFSR) < 5%, with cross-tissue smoothing of effect sizes with mashr (11, 51). We observed that a median of 57% of variants were associated with more than one gene per tissue, typically co-occurring across tissues, indicating widespread regulatory pleiotropy. Using a binary classification of cis-eVariants with regulatory pleiotropy defined as those associated with more than one gene, we observed that they are more significantly associated with complex traits compared to matched cis-eVariants (fig. S38). This could be due to the fact that if a variant regulates multiple genes, there is a higher probability that at least one of them affects a GWAS phenotype. However, cis-eVariants with regulatory pleiotropy also have higher GWAS complex trait pleiotropy (50) than cis-eVariants with effects on a single gene (Fig. 5E). This observation suggests a mechanism for complex trait pleiotropy of genetic effects where the expression of multiple genes in cis, rather than a single eGene effect, translates into diverse downstream physiological effects. Furthermore, GWAS pleiotropy is higher for tissue-shared (41) than tissue-specific cis-eQTLs, indicating that regulatory effects affecting multiple tissues are more likely to translate to diverse physiological traits (Fig. 5E).
Tissue-specificity of genetic regulatory effects
The GTEx data provide an opportunity to study patterns and mechanisms of tissue-specificity of the transcriptome and its genetic regulation. Pairwise similarity of GTEx tissues was quantified from gene expression and splicing, as well as allelic expression, eQTLs in cis and trans, and cis-sQTLs (Fig. 6A, and fig. S41, (11)). These estimates show consistent patterns of tissue relatedness, indicating that the biological processes that drive transcriptome similarity also control tissue sharing of genetic effects (Fig. 6B). As seen in earlier versions of the GTEx data (9, 10), the brain regions form a separate cluster, and testis, LCLs, whole blood, and sometimes liver tend to be outliers, while most other organs have a notably high degree of similarity among each other. This indicates that blood is not an ideal proxy for most tissues, but that some other relatively accessible tissues, such as skin, may better capture molecular effects in other tissues.
Figure 6. Tissue-specificity of cis-QTLs.
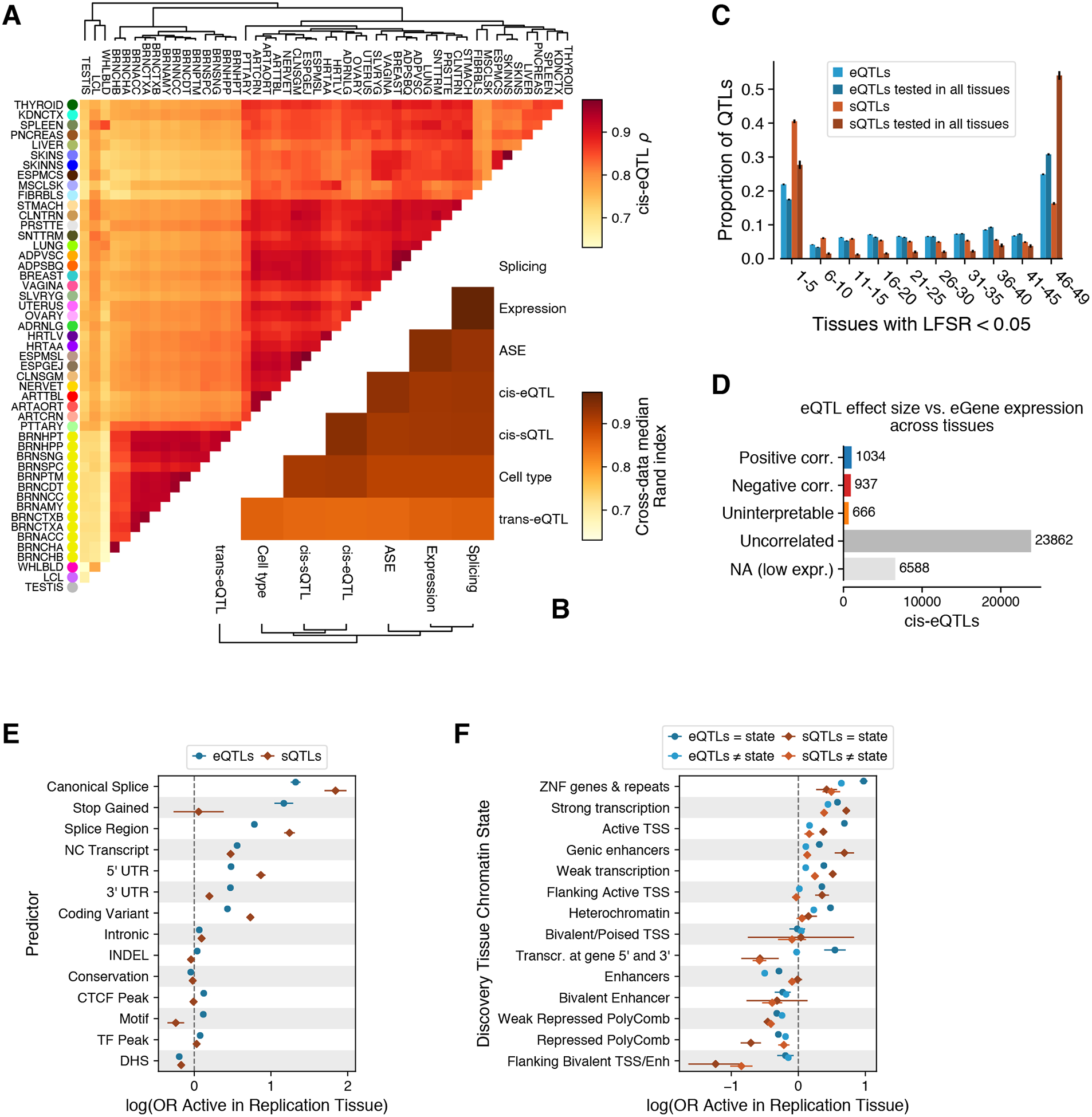
(A) Tissue clustering with pairwise Spearman correlation of cis-eQTL effect sizes. (B) Similarity of tissue clustering across core data types quantified using median pairwise Rand index calculated across tissues. (C) Tissue activity of cis expression and splicing QTLs, where an eQTL was considered active in a tissue if it had a mashr local false sign rate (LFSR, equivalent to FDR) of < 5%. This is shown for all cis-QTLs and only those that could be tested in all 49 tissues (red and blue). (D) Spearman correlation (corr.) between cis-eQTL effect size and eGene expression level across tissues. cis-eQTL counts are shown for those not tested due to low expression level, tested but without significant (FDR < 5%) correlation (uncorrelated), a significant correlation but effect sizes crossed zero which made the correlation direction unclear (uninterpretable), positively correlated, and negatively correlated. (E-F) The effect of genomic function on cis-QTL tissue sharing modeled using logistic regression with functional annotations (E) and chromatin state (F). CTCF Peak, Motif, TF Peak, and DHS indicate if the cis-QTL lies in a region annotated as having one of these features in any of the Ensembl Regulatory Build tissues. For chromatin states, model coefficients are shown for the discovery and replication tissues that have the same or different chromatin states.
The overall tissue specificity of QTLs ((11)) follows a U-shaped curve recapitulating previous GTEx analyses (9, 10), where genetic regulatory effects tend to be either highly tissue-specific or highly shared (Fig. 6C), with trans-eQTLs being more tissue-specific than cis-eQTLs (fig. S40). Cis-sQTLs appear to be significantly more tissue specific than cis-eQTLs when considering all mapped cis-QTLs, but this pattern is reversed when considering only those cis-QTLs where the gene or splicing event is quantified in all tissues (Fig. 6C, and fig. S39). This indicates that splicing measures are more tissue-specific than gene expression, but genetic effects on splicing tend to be more shared, consistent with pairwise tissue sharing patterns (fig. S41). This is important for understanding effects that disease-causing splicing variants may have across tissues, and for validation of splicing effects in cell lines that rarely are an exact match to cells in vivo. Next, we analyzed the sharing of allelic expression (AE) across multiple tissues of an individual, which is a metric of sharing of any heterozygous regulatory variant effects in that individual. Variation in AE has been useful for analysis of rare, potentially disease-causing variants (52). Using a clustering approach (11), we found that in 97.4% of the cases, AE across all tissues forms a single cluster. This suggests that in AE analysis, different tissues are often relatively good proxies for one another, provided that the gene of interest is expressed in the probed tissue. (fig. S42).
We next computed the cross-tissue correlation of eQTL effect size and eGene expression level — often a proxy for gene functionality — and discovered that 1,971 cis-eQTLs (7.4%; FDR 5%) had a significant and robust correlation between eGene expression and cis-eQTL effect size across tissues (Fig 6D, and fig. S43). These correlated cis-eQTLs are split nearly evenly between negative (937) and positive (1,34) correlations. Thus, the tissues with the highest cis-eQTL effect sizes are equally likely to be among tissues with higher or lower expression levels for the gene. Trans-eQTLs show a different pattern, being typically observed in tissues with high expression of the trans-eGene relative to other tissues (fig. S43).
These observations raise the question of how to prioritize the relevant tissues for eQTLs in a disease context. To address this, we chose a subset of GWAS traits with a strong prior indication for the likely relevant tissue(s) (table S12). Analyzing colocalized cis-eQTLs for 1,778 GWAS loci (11), we discovered that the relevant tissues were significantly enriched in having high expression and effect sizes (paired Wilcoxon sign test p<1.5e−4), but the relatively weak signal indicates that pinpointing the likely relevant tissue GWAS loci is challenging (figs. S44, S45, table S9). This indicates that both effect sizes and gene expression levels are important for interpreting the tissue context where an eQTL may have downstream phenotypic effects.
The diverse patterns of QTL tissue-specificity raise the question of what molecular mechanisms underlie the ubiquitous regulatory effects of some genetic variants and the highly tissue-specific effects of others. To gain insight into this question, we modeled cis-eQTL and cis-sQTL tissue specificity using logistic regression as a function of the lead eVariant’s genomic and epigenomic context (11). Cis-QTLs where the top eVariant was in a transcribed region had overall higher sharing than those in classical transcriptional regulatory elements, indicating that genetic variants with post- or co-transcriptional expression or splicing effects have more ubiquitous effects (Fig. 6E). Canonical splice and stop gained variant effects had the highest probability of being shared across tissues, which may benefit disease-focused studies relying on likely gene-disrupting variants. We also considered whether varying regulatory activity between tissues contributed to tissue-specificity of genetic effects, and found that shared chromatin states between the discovery and query tissues were associated with increased probability of cis-eQTL sharing and vice-versa (Fig. 6F). cis-eQTLs and cis-sQTLs followed similar patterns. Since cis-sQTLs are more enriched in transcribed regions and likely arise via post-transcriptional mechanisms (Fig. 4A), this is likely to contribute to their higher overall degree of tissue-sharing (Fig. 6C). In comparison to cis-eQTLs, cis-sQTLs are more often located in regions where regulatory effects are shared.
These data indicate a possible means by which we can predict if a cis-eQTL observed in a GTEx tissue is active in another tissue of interest, using the variant’s annotation and properties in the discovery tissue (11). After incorporating additional features including cis-QTL effect size, distance to transcription start site, and eGene/sGene expression levels, we obtain reasonably good predictions of whether a cis-QTL is active in a query tissue (median AUC = 0.779 and 0.807, min = 0.703 and 0.721, max = 0.807 and 0.875 for cis-eQTLs and cis-sQTLs, respectively; fig. S46). This suggests that it is possible to extrapolate the GTEx cis-eQTL catalog to additional tissues and potentially developmental stages, where population-scale data for QTL analysis are particularly difficult to collect.
From tissues to cell types
The GTEx tissue samples consist of heterogeneous mixtures of multiple cell types. Hence, the RNA extracted and QTLs mapped from these samples reflect a composite of genetic effects that may vary across cell types and may mask cell type-specific mechanisms. To characterize the effect of cell type heterogeneity on analyses from bulk tissue, we used the xCell method (53) to estimate the enrichment of 64 reference cell types from the bulk expression profile of each sample (11). While these results need to be interpreted with caution given the scarcity of validation data (54), the resulting enrichment scores were generally biologically meaningful with, for example, myocytes enriched in heart left ventricle and skeletal muscle, hepatocytes enriched in liver, and various blood cell types enriched in whole blood, spleen, and lung, which harbors a large leukocyte population (fig. S47). Interestingly, the pairwise relatedness of GTEx tissues derived from their cell type composition is highly correlated with tissue-sharing of regulatory variants (cis-eQTL versus cell type composition Rand index = 0.92; Fig. 6B, and figs. S48 and S41), suggesting that similarity of regulatory variant activity between tissue pairs may often be due to the presence of similar cell types, and not necessarily shared regulatory networks within cells. This highlights the key role that characterizing cell type diversity will have for understanding not only tissue biology but genetic regulatory effects as well.
Enrichment of many cell types shows inter-individual variation within a given tissue, partially due to tissue sampling variation between individuals. This variation can be leveraged to identify cis-eQTLs and cis-sQTLs with cell type specificity, by including an interaction between genotype and cell type enrichment in the QTL model (11, 55). We applied this approach to seven tissue-cell type pairs with robustly quantified cell types in the tissue where each cell type was most enriched (Fig. 7A; an additional 36 pairs are described in (54)). The largest numbers of cell type interacting cis-eQTLs and cis-sQTLs (ieQTLs and isQTLs) were 1120 neutrophil ieQTLs and 169 isQTLs in whole blood and 1087 epithelial cell ieQTLs and 117 isQTLs in transverse colon (Fig. 7A). Of these ieQTLs, 76 and 229, respectively, involved an eGene for which no QTL was detected in bulk tissue. We validated these effects using published eQTLs from purified blood cell types (56), where neutrophil eQTLs had higher neutrophil ieQTL effect sizes than eQTLs from other blood cell types (fig. S49). For other cell types, external replication data was not available. Thus, we verified the robustness of the ieQTLs by the allelic expression validation approach that was used for sex- and population-biased cis-eQTL analyses: for ieQTL heterozygotes, we calculated the Spearman correlation between cell type enrichment and ieQTL effect size from AE data, and observed a high validation rate (54). It is important to note that ie/isQTLs should not be considered cell type-specific QTLs, because the enrichment of any cell type may be (anti-)correlated with other cell types (fig. S50). While full deconvolution of cis-eQTL effects driven by specific cell types remains a challenge for the future, ieQTLs and isQTLs can be interpreted as being enriched for cell type-specific effects.
Figure 7. Cell type interacting cis-eQTLs and cis-sQTLs.
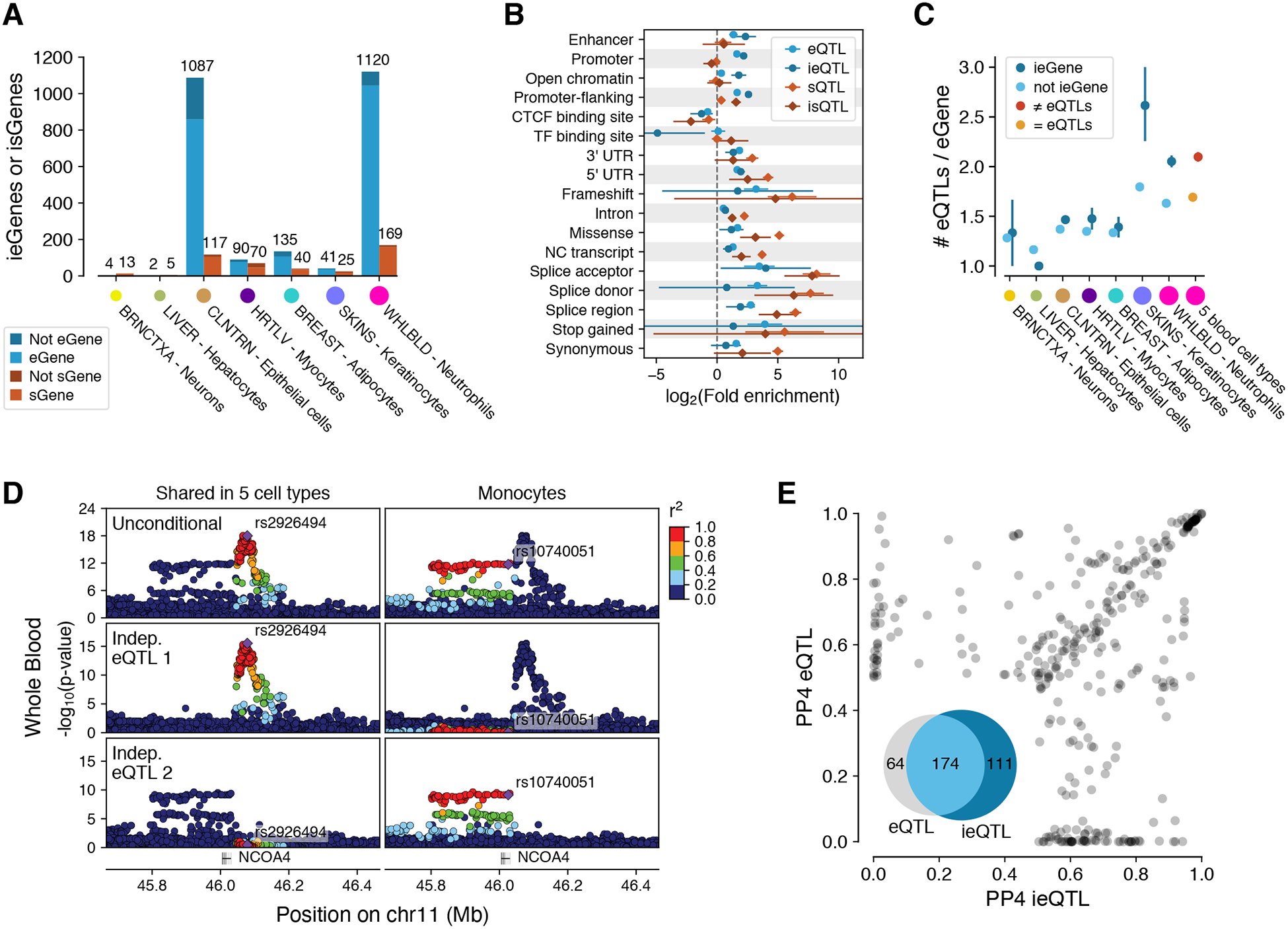
(A) Number of cell type interacting cis-eQTLs and cis-sQTLs (ieQTLs and isQTLs, respectively) discovered in seven tissue-cell type pairs, with shading indicating whether the ieGene or isGene was discovered by cis-eQTL/cis-sQTL analysis in bulk tissue. Colored dots are proportional to sample size. (B) Functional enrichment of neutrophil ieQTLs and isQTLs compared to cis-eQTLs and cis-sQTLs from whole blood. (C) Proportion of conditionally independent cis-eQTLs per eGene, for eGenes that do or do not have ieQTLs in GTEx, and for eGenes that have shared (= eQTLs) or non-shared (≠ eQTLs) cis-eQTL across five sorted blood cell types. (D) Whole blood cis-eQTL p-value landscape for NCOA4, for the standard analysis (top row, Unconditional) and for two independent cis-eQTLs (bottom rows). In a data set of 5 sorted cell types (56), analyses of all cell types yielded a lead eVariant, rs2926494 (left), which is in high LD with the first independent cis-eQTL but not the second. The lead variant in monocyte cis-eQTL analysis, rs10740051, is in high LD with the second conditional cis-eQTL, indicating that this cis-eQTL is active specifically in monocytes. Thus, the full GTEx whole blood cis-eQTL pattern and allelic heterogeneity is composed of cis-eQTLs that are active in different cell types. (E) COLOC posterior probability (PP4) of GWAS colocalization with whole blood ieQTLs and eQTLs of the same eGene. 349 gene-trait combinations across 132 genes and 36 GWAS traits showed evidence of colocalization (PP4 > 0.5) with an ieQTL and/or eQTL.
In most subsequent analyses to characterize the properties of ieQTLs and isQTLs, we focused on neutrophil ieQTLs, which are numerous and supported by external replication data. Functional enrichment analyses of these QTLs show that these largely follow the enrichment patterns observed for bulk tissue cis-QTLs (Fig. 7B). However, ieQTLs are more strongly enriched in promoter flanking regions and enhancers, which are known to be major drivers of cell type specific regulatory effects (2). Epithelial cell ieQTLs yielded similar patterns (fig. S51).
We hypothesized that the widespread allelic heterogeneity observed in the bulk tissue cis-eQTL data could be partially driven by an aggregate signal from cis-eQTLs that are each active in a different cell type present in the tissue. Indeed, the number of cis-eQTLs per gene is higher for ieGenes than for standard eGenes, especially in skin and blood (Fig. 7C). While differences in power could contribute to this pattern, it is corroborated by eGenes that have independent cis-eQTLs (r2 < 0.05) in five purified blood cell types (56) also showing an increased amount of allelic heterogeneity in GTEx whole blood (Fig. 7C and D). Thus, quantifying cell type specificity can provide mechanistic insights into the genetic architecture of gene expression, and may be leveraged to improve the resolution of complex patterns of allelic heterogeneity where we can distinguish effects manifesting in different cell types.
Next, we analyzed how cell type interacting cis-QTLs contribute to the interpretation of regulatory variants underlying complex disease risk. GWAS colocalization analysis of neutrophil ieQTLs (11) revealed multiple loci (111, ~32%) that colocalize only with ieQTLs and not with whole blood cis-eQTLs (Fig. 7E), even though 75% (42/56) of the corresponding eGenes have both cis-eQTLs and ieQTLs. Improved resolution into allelic heterogeneity appears to contribute to this. For example, the absence of colocalization between a platelet count GWAS signal and bulk tissue cis-eQTL for SPAG7 appears to be due to the whole blood signal being an aggregate of multiple independent signals (fig. S52). The neutrophil ieQTL analysis uncovers a specific signal that mirrors the GWAS association, suggesting that platelet counts are affected by SPAG7 expression only in specific cell type(s). Thus, in addition to previously undetected colocalizations pinpointing potential causal genes, ieQTL analysis has the potential to provide insights into cell type specific mechanisms of complex traits.
Discussion
The GTEx v8 data release represents a deep survey of both intra- and inter-individual transcriptome variation across a large number of tissues. With 838 donors and 15,253 samples — approximately twice the size of the v6 release used in the previous set of GTEx Consortium papers — we have created a comprehensive resource of genetic variants that influence gene expression and splicing in cis. This significantly expands and updates the GTEx catalog of sQTLs, doubles the number of eGenes per tissue, and saturates the discovery of eQTLs with over 2-fold effect sizes in ~40 tissues. The fine-mapping data of GTEx cis-eQTLs provides a set of thousands of likely causal functional variants. While trans-QTL discovery, as well as characterization of sex-specific and population-specific genetic effects, are still limited by sample size, analyses of the v8 data provide important insights into each. Cell type interacting cis-eQTLs and cis-sQTLs, mapped with computational estimates of cell type enrichment, constitute an important extension of the GTEx resource to effects of cell types within tissues. The strikingly similar tissue-sharing patterns across these data types suggests shared biology from cell type composition to transcriptome variation and genetic regulatory effects. Our results indicate that shared cell types between tissues may be a key factor behind tissue-sharing of genetic regulatory effects, which will constitute a key challenge to tackle in the future. Finally, GWAS colocalization with cis-eQTLs and cis-sQTLs provides rich opportunities for further functional follow-up and characterization of regulatory mechanisms of GWAS associations.
Given the very large number of cis-eQTLs, the extensive allelic heterogeneity – multiple independent regulatory variants affecting the same gene – is unsurprising. With well-powered cis-QTL mapping, it becomes possible and important to describe and disentangle these effects; the assumption of a single causal variant in a cis-eQTL locus no longer holds true for data sets of this scale. Similarly, we highlight cis-eQTL and cis-sQTL effects on the same gene, typically driven by distinct causal variants (4, 35). The joint complex trait contribution of independent cis-eQTLs and cis-sQTLs, and cis-eQTLs and rare coding variants for the same gene highlights how different genetic variants and functional perturbations can converge at the gene level to similar physiological effects. This orthogonal evidence pinpoints highly likely causal disease genes, and these associations could be leveraged to build allelic series, a powerful tool for estimating dosage-risk relationship for the purposes of drug development (57). Finally, we provide mechanistic insights into the cellular causes of allelic heterogeneity, showing the separate contributions from cis-eQTLs active in different cell types to the combined signal seen in a bulk tissue sample. With evidence that this increased cellular resolution improves colocalization in some loci, cell type specific analyses appear particularly promising for finer dissection of genetic association data.
Integration of GTEx QTL data and functional annotation of the genome provides powerful insights into the molecular mechanisms of transcriptional and post-transcriptional regulation that affect gene expression levels and splicing. A large proportion of cis-eQTL effects are driven by genetic perturbations in classical regulatory elements of promoters and enhancers. However, the magnitude of these enrichments is perhaps surprisingly modest, which likely reflects the fact that only a small fraction of variants in these large regions have true regulatory effects, leading to a lower resolution of annotating functional variants compared to the nucleotide-level annotation of, e.g., nonsense or canonical splice site variants. Context-specific genetic effects of tissue-specific and cell-type interacting cis-eQTLs are enriched in enhancers and related elements and their variable activity across tissues and cell types. While cis-eQTLs are enriched for a wide range of functional regions, the vast majority of cis-sQTL are located in transcribed regions, with likely co-/post-transcriptional regulatory effects. Interestingly, these appear to be less tissue-specific, which likely contributes to the higher tissue-sharing of cis-sQTLs than cis-eQTLs. The higher tissue-sharing of all co/post-transcriptional regulatory effects may facilitate interpretation of potentially disease-related functional effects of (rare) coding variants triggering nonsense-mediated decay or splicing changes, even when the disease-relevant tissues are not available.
Approximately a third of the observed trans-eQTLs are mediated by cis-eQTLs, demonstrating how local genetic regulatory effects can translate to effects at the level of cellular pathways. All types of QTLs that were studied are strong mediators of genetic associations to complex traits, with a higher relative enrichment for cis-sQTLs than cis-eQTLs, with trans-eQTLs having the highest enrichment of all (35). With large genome- and phenome-wide (GWAS/PheWAS) studies having uncovered extensive pleiotropy of complex trait associations, the GTEx data provide important insights into the molecular underpinnings of this observed pleiotropy: variants that affect the expression of multiple genes and multiple tissues have a higher degree of complex trait pleiotropy, indicating that some of the pleiotropy arises at the proximal regulatory level. Dissecting this complexity and pinpointing truly causal molecular effects that mediate specific phenotype associations will be a considerable challenge for the future.
This study of the GTEx v8 data has provided insights into genetic regulatory architecture and functional mechanisms. The catalog of QTLs and associated data sets of annotations, cell type enrichments, and GWAS summary statistics requires careful interpretation but provides insights into the biology of gene regulation and functional mechanisms of complex traits. We demonstrate how QTL data can be used to inform on multiple layers of GWAS interpretation: potential causal variants from fine-mapping, proximal regulatory mechanisms, target genes in cis, pathway effects in trans, in the context of multiple tissues and cell types. However, our understanding of genetic effects on cellular phenotypes is far from complete. We envision that further investigation into genetic regulatory effects in specific cell types, study of additional tissues and developmental time points not covered by GTEx, incorporation of a diverse set of molecular phenotypes, and continued investment in increasing sample sizes from diverse populations will continue to provide transformative scientific discoveries.
Supplementary Material
Acknowledgements
We thank the donors and their families for their generous gifts of organ donation for transplantation, and tissue donations for the GTEx research project; the Genomics Platform at the Broad Institute for data generation; J. Struewing for his support and leadership of the GTEx project; M. Khan and C. Stolte for the illustrations in Figure 1; and R. Do, D. Jordan, and M. Verbanck for providing GWAS pleiotropy scores.
Funding
This work was supported by the Common Fund of the Office of the Director, U.S. National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, NIA, NIAID, and NINDS through NIH contracts HHSN261200800001E (Leidos Prime contract with NCI: A.M.S., D.E.T., N.V.R., J.A.M., L.S., M.E.B., L.Q., T.K., D.B., K.R., A.U.), 10XS170 (NDRI: W.F.L., J.A.T., G.K., A.M., S.S., R.H., G.Wa., M.J., M.Wa., L.E.B., C.J., J.W., B.R., M.Hu., K.M., L.A.S., H.M.G., M.Mo., L.K.B.), 10XS171 (Roswell Park Cancer Institute: B.A.F., M.T.M., E.K., B.M.G., K.D.R., J.B.), 10X172 (Science Care Inc.), 12ST1039 (IDOX), 10ST1035 (Van Andel Institute: S.D.J., D.C.R., D.R.V.), HHSN268201000029C (Broad Institute: F.A., G.G., K.G.A., A.V.S., X.Li., E.T., S.G., A.G., S.A., K.H.H., D.T.N., K.H., S.R.M., J.L.N.), 5U41HG009494 (F.A., G.G., K.G.A.), and through NIH grants R01 DA006227-17 (Univ. of Miami Brain Bank: D.C.M., D.A.D.), Supplement to University of Miami grant DA006227 (D.C.M., D.A.D.), R01 MH090941 (Univ. of Geneva), R01 MH090951 and R01 MH090937 (Univ. of Chicago), R01 MH090936 (Univ. of North Carolina-Chapel Hill), R01MH101814 (M.M-A., V.W., S.B.M., R.G., E.T.D., D.G-M., A.V.), U01HG007593 (S.B.M.), R01MH101822 (C.D.B.), U01HG007598 (M.O., B.E.S.), U01MH104393 (A.P.F.), extension H002371 to 5U41HG002371 (W.J.K) as well as other funding sources: R01MH106842 (T.L., P.M., E.F., P.J.H.), R01HL142028 (T.L., Si.Ka., P.J.H.), R01GM122924 (T.L., S.E.C.), R01MH107666 (H.K.I.), P30DK020595 (H.K.I.), UM1HG008901 (T.L.), R01GM124486 (T.L.), R01HG010067 (Y.Pa.), R01HG002585 (G.Wa., M.St.), Gordon and Betty Moore Foundation GBMF 4559 (G.Wa., M.St.), 1K99HG009916-01 (S.E.C.), R01HG006855 (Se.Ka., R.E.H.), BIO2015-70777-P, Ministerio de Economia y Competitividad and FEDER funds (M.M-A., V.W., R.G., D.G-M.), la Caixa Foundation ID 100010434 under agreement LCF/BQ/SO15/52260001 (D.G-M.), NIH CTSA grant UL1TR002550-01 (P.M.), Marie-Skłodowska Curie fellowship H2020 Grant 706636 (S.K-H.), R35HG010718 (E.R.G.), FPU15/03635, Ministerio de Educación, Cultura y Deporte (M.M-A.), R01MH109905, 1R01HG010480 (A.Ba.), Searle Scholar Program (A.Ba.), R01HG008150 (S.B.M.), 5T32HG000044-22, NHGRI Institutional Training Grant in Genome Science (N.R.G.), EU IMI program (UE7-DIRECT-115317-1) (E.T.D., A.V.), FNS funded project RNA1 (31003A_149984) (E.T.D., A.V.), DK110919 (F.H.), F32HG009987 (F.H.), Massachusetts Lions Eye Research Fund Grant (A.R.H.).
GTEx Consortium*
Laboratory and Data Analysis Coordinating Center (LDACC): François Aguet1, Shankara Anand1, Kristin G Ardlie1, Stacey Gabriel1, Gad Getz1,30,31, Aaron Graubert1, Kane Hadley1, Robert E Handsaker33,34,35, Katherine H Huang1, Seva Kashin33,34,35, Xiao Li1, Daniel G MacArthur34,36, Samuel R Meier1, Jared L Nedzel1, Duyen T Nguyen1, Ayellet V Segrè1,17, Ellen Todres1
Analysis Working Group (funded by GTEx project grants):
François Aguet1, Shankara Anand1, Kristin G Ardlie1, Brunilda Balliu41, Alvaro N Barbeira2, Alexis Battle18,11, Rodrigo Bonazzola2, Andrew Brown3,4, Christopher D Brown24, Stephane E Castel5,6, Donald F Conrad42,43, Daniel J Cotter29, Nancy Cox16, Sayantan Das26, Olivia M de Goede29, Emmanouil T Dermitzakis3,27,28, Jonah Einson44,5, Barbara E Engelhardt7,8, Eleazar Eskin45, Tiffany Y Eulalio46, Nicole M Ferraro46, Elise D Flynn5,6, Laure Fresard12, Eric R Gamazon13,14,15,16, Diego Garrido-Martín22, Nicole R Gay29, Gad A Getz1,30,31, Michael J Gloudemans46, Aaron Graubert1, Roderic Guigó22,32, Kane Hadley1, Andrew R Hamel17,1, Robert E Handsaker33,34,35, Yuan He18, Paul J Hoffman5, Farhad Hormozdiari19,1, Lei Hou47,1, Katherine H Huang1, Hae Kyung Im2, Brian Jo7,8, Silva Kasela5,6, Seva Kashin33,34,35, Manolis Kellis47,1, Sarah Kim-Hellmuth5,6,9, Alan Kwong26, Tuuli Lappalainen5,6, Xiao Li1, Xin Li12, Yanyu Liang2, Daniel G MacArthur34,36, Serghei Mangul45,48, Samuel R Meier1, Pejman Mohammadi5,6,20,21, Stephen B Montgomery12,29, Manuel Muñoz-Aguirre22,23, Daniel C Nachun12, Jared L Nedzel1, Duyen T Nguyen1, Andrew B Nobel49, Meritxell Oliva2,10, YoSon Park24,25, Yongjin Park47,1, Princy Parsana11, Abhiram S Rao50, Ferran Reverter51, John M Rouhana17,1, Chiara Sabatti52, Ashis Saha11, Ayellet V Segrè1,17, Andrew D Skol2,53, Matthew Stephens37, Barbara E Stranger2,38, Benjamin J Strober18, Nicole A Teran12, Ellen Todres1, Ana Viñuela39,3,27,28, Gao Wang37, Xiaoquan Wen26, Fred Wright54, Valentin Wucher22, Yuxin Zou40
Analysis Working Group (not funded by GTEx project grants): Pedro G Ferreira55,56,57,58, Gen Li59, Marta Melé60, Esti Yeger-Lotem61,62
Leidos Biomedical - Project Management: Mary E Barcus63, Debra Bradbury63, Tanya Krubit63, Jeffrey A McLean63, Liqun Qi63, Karna Robinson63, Nancy V Roche63, Anna M Smith63, Leslie Sobin63, David E Tabor63, Anita Undale63
Biospecimen collection source sites: Jason Bridge64, Lori E Brigham65, Barbara A Foster66, Bryan M Gillard66, Richard Hasz67, Marcus Hunter68, Christopher Johns69, Mark Johnson70, Ellen Karasik66, Gene Kopen71, William F Leinweber71, Alisa McDonald71, Michael T Moser66, Kevin Myer68, Kimberley D Ramsey66, Brian Roe68, Saboor Shad71, Jeffrey A Thomas71,70, Gary Walters70, Michael Washington70, Joseph Wheeler69
Biospecimen core resource: Scott D Jewell72, Daniel C Rohrer72, Dana R Valley72
Brain bank repository: David A Davis73, Deborah C Mash73
Pathology: Mary E Barcus63, Philip A Branton74, Leslie Sobin63
ELSI study: Laura K Barker75, Heather M Gardiner75, Maghboeba Mosavel76, Laura A Siminoff75
Genome Browser Data Integration & Visualization: Paul Flicek77, Maximilian Haeussler78, Thomas Juettemann77, W James Kent78, Christopher M Lee78, Conner C Powell78, Kate R Rosenbloom78, Magali Ruffier77, Dan Sheppard77, Kieron Taylor77, Stephen J Trevanion77, Daniel R Zerbino77
eGTEx groups: Nathan S Abell29, Joshua Akey79, Lin Chen10, Kathryn Demanelis10, Jennifer A Doherty80, Andrew P Feinberg81, Kasper D Hansen82, Peter F Hickey83, Lei Hou47,1, Farzana Jasmine10, Lihua Jiang29, Rajinder Kaul84,85, Manolis Kellis47,1, Muhammad G Kibriya10, Jin Billy Li29, Qin Li29, Shin Lin86, Sandra E Linder29, Stephen B Montgomery12,29, Meritxell Oliva2,10, Yongjin Park47,1, Brandon L Pierce10, Lindsay F Rizzardi87, Andrew D Skol2,53, Kevin S Smith12, Michael Snyder29, John Stamatoyannopoulos84,88, Barbara E Stranger2,38, Hua Tang29, Meng Wang29
NIH program management: Philip A Branton74, Latarsha J Carithers74,89, Ping Guan74, Susan E Koester90, A. Roger Little91, Helen M Moore74, Concepcion R Nierras92, Abhi K Rao74, Jimmie B Vaught74, Simona Volpi93
Affiliations
1. The Broad Institute of MIT and Harvard, Cambridge, MA, USA
2. Section of Genetic Medicine, Department of Medicine, The University of Chicago, Chicago, IL, USA
3. Department of Genetic Medicine and Development, University of Geneva Medical School, Geneva, Switzerland
4. Population Health and Genomics, University of Dundee, Dundee, Scotland, UK
5. New York Genome Center, New York, NY, USA
6. Department of Systems Biology, Columbia University, New York, NY, USA
7. Department of Computer Science, Princeton University, Princeton, NJ, USA
8. Center for Statistics and Machine Learning, Princeton University, Princeton, NJ, USA
9. Statistical Genetics, Max Planck Institute of Psychiatry, Munich, Germany
10. Department of Public Health Sciences, The University of Chicago, Chicago, IL, USA
11. Department of Computer Science, Johns Hopkins University, Baltimore, MD, USA
12. Department of Pathology, Stanford University, Stanford, CA, USA
13. Data Science Institute, Vanderbilt University, Nashville, TN, USA
14. Clare Hall, University of Cambridge, Cambridge, UK
15. MRC Epidemiology Unit, University of Cambridge, Cambridge, UK
16. Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA
17. Ocular Genomics Institute, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, USA
18. Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD, USA
19. Department of Epidemiology, Harvard T.H. Chan School of Public Health, Boston, MA, USA
20. Scripps Research Translational Institute, La Jolla, CA, USA
21. Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA, USA
22. Centre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, Barcelona, Catalonia, Spain
23. Department of Statistics and Operations Research, Universitat Politècnica de Catalunya (UPC), Barcelona, Catalonia, Spain
24. Department of Genetics, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA
25. Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA
26. Department of Biostatistics, University of Michigan, Ann Arbor, MI, USA
27. Institute for Genetics and Genomics in Geneva (iGE3), University of Geneva, Geneva, Switzerland
28. Swiss Institute of Bioinformatics, Geneva, Switzerland
29. Department of Genetics, Stanford University, Stanford, CA, USA
30. Cancer Center and Department of Pathology, Massachusetts General Hospital, Boston, MA, USA
31. Harvard Medical School, Boston, MA, USA
32. Universitat Pompeu Fabra (UPF), Barcelona, Catalonia, Spain
33. Department of Genetics, Harvard Medical School, Boston, MA, USA
34. Program in Medical and Population Genetics, The Broad Institute of Massachusetts Institute of Technology and Harvard University, Cambridge, MA, USA
35. Stanley Center for Psychiatric Research, Broad Institute, Cambridge, MA, USA
36. Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA
37. Department of Human Genetics, University of Chicago, Chicago, IL, USA
38. Center for Genetic Medicine, Department of Pharmacology, Northwestern University, Feinberg School of Medicine, Chicago, IL, USA
39. Department of Twin Research and Genetic Epidemiology, King’s College London, London, UK
40. Department of Statistics, University of Chicago, Chicago, IL, USA
41. Department of Biomathematics, University of California, Los Angeles, Los Angeles, CA, USA
42. Department of Genetics, Washington University School of Medicine, St. Louis, Missouri, USA
43. Division of Genetics, Oregon National Primate Research Center, Oregon Health & Science University, Portland, OR, USA
44. Department of Biomedical Informatics, Columbia University, New York, NY, USA
45. Department of Computer Science, University of California, Los Angeles, Los Angeles, CA, USA
46. Program in Biomedical Informatics, Stanford University School of Medicine, Stanford, CA, USA
47. Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge, MA, USA
48. Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, Los Angeles, CA, USA
49. Department of Statistics and Operations Research and Department of Biostatistics, University of North Carolina, Chapel Hill, NC, USA
50. Department of Bioengineering, Stanford University, Stanford, CA, USA
51. Department of Genetics, Microbiology and Statistics, University of Barcelona, Barcelona. Spain.
52. Departments of Biomedical Data Science and Statistics, Stanford University, Stanford, CA, USA
53. Department of Pathology and Laboratory Medicine, Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL, USA
54. Bioinformatics Research Center and Departments of Statistics and Biological Sciences, North Carolina State University, Raleigh, NC, USA
55. Department of Computer Sciences, Faculty of Sciences, University of Porto, Porto, Portugal
56. Instituto de Investigação e Inovação em Saúde, University of Porto, Porto, Portugal
57. Institute of Molecular Pathology and Immunology, University of Porto, Porto, Portugal
58. Laboratory of Artificial Intelligence and Decision Support, Institute for Systems and Computer Engineering, Technology and Science, Porto, Portugal
59. Columbia University Mailman School of Public Health, New York, NY, USA
60. Life Sciences Department, Barcelona Supercomputing Center, Barcelona, Spain
61. Department of Clinical Biochemistry and Pharmacology, Ben-Gurion University of the Negev, Beer-Sheva, Israel
62. National Institute for Biotechnology in the Negev, Beer-Sheva, Israel
63. Leidos Biomedical, Rockville, MD, USA
64. UNYTS, Buffalo, NY, USA
65. Washington Regional Transplant Community, Annandale, VA, USA
66. Therapeutics, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
67. Gift of Life Donor Program, Philadelphia, PA, USA
68. LifeGift, Houston, TX, USA
69. Center for Organ Recovery and Education, Pittsburgh, PA, USA
70. LifeNet Health, Virginia Beach, VA. USA
71. National Disease Research Interchange, Philadelphia, PA, USA
72. Van Andel Research Institute, Grand Rapids, MI, USA
73. Department of Neurology, University of Miami Miller School of Medicine, Miami, FL, USA
74. Biorepositories and Biospecimen Research Branch, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Bethesda, MD, USA
75. Temple University, Philadelphia, PA, USA
76. Virginia Commonwealth University, Richmond, VA, USA
77. European Molecular Biology Laboratory, European Bioinformatics Institute, Hinxton, United Kingdom
78. Genomics Institute, University of California Santa Cruz, Santa Cruz, CA, USA
79. Carl Icahn Laboratory, Princeton University, Princeton, NJ, USA
80. Department of Population Health Sciences, The University of Utah, Salt Lake City, Utah, USA
81. Departments of Medicine, Biomedical Engineering, and Mental Health, Johns Hopkins University, Baltimore, MD, USA
82. Department of Biostatistics, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, USA
83. Department of Medical Biology, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia
84. Altius Institute for Biomedical Sciences, Seattle, WA, USA
85. Division of Genetics, University of Washington, Seattle, WA, University of Washington, Seattle, WA, USA
86. Department of Cardiology, University of Washington, Seattle, WA, USA
87. HudsonAlpha Institute for Biotechnology, Huntsville, AL, USA
88. Genome Sciences, University of Washington, Seattle, WA, USA
89. National Institute of Dental and Craniofacial Research, Bethesda, MD, USA
90. Division of Neuroscience and Basic Behavioral Science, National Institute of Mental Health, National Institutes of Health, Bethesda, MD, USA
91. National Institute on Drug Abuse, Bethesda, MD, USA
92. Office of Strategic Coordination, Division of Program Coordination, Planning and Strategic Initiatives, Office of the Director, National Institutes of Health, Rockville, MD, USA
93. Division of Genomic Medicine, National Human Genome Research Institute, Bethesda, MD, USA
Footnotes
Authors
# First author
* Alphabetical order
Lead Analysts*
François Aguet1#, Alvaro N Barbeira2, Rodrigo Bonazzola2, Andrew Brown3,4, Stephane E Castel5,6, Brian Jo7,8, Silva Kasela5,6, Sarah Kim-Hellmuth5,6,9, Yanyu Liang2, Meritxell Oliva2,10, Princy Parsana11
Analysts*
Elise D Flynn5,6, Laure Fresard12, Eric R Gamazon13,14,15,16, Andrew R Hamel17,1, Yuan He18, Farhad Hormozdiari19,1, Pejman Mohammadi5,6,20,21, Manuel Muñoz-Aguirre22,23, YoSon Park24,25, Ashis Saha11, Ayellet V Segrè1,17, Benjamin J Strober18, Xiaoquan Wen26, Valentin Wucher22
Manuscript Working Group*
François Aguet1, Kristin G Ardlie1, Alvaro N Barbeira2, Alexis Battle18,11, Rodrigo Bonazzola2, Andrew Brown3,4, Christopher D Brown24, Stephane E Castel5,6, Nancy Cox16, Sayantan Das26, Emmanouil T Dermitzakis3,27,28, Barbara E Engelhardt7,8, Elise D Flynn5,6, Laure Fresard12, Eric R Gamazon13,14,15,16, Diego Garrido-Martín22, Nicole R Gay29, Gad A Getz1,30,31, Roderic Guigó22,32, Andrew R Hamel17,1, Robert E Handsaker33,33,35, Yuan He18, Paul J Hoffman5, Farhad Hormozdiari19,1, Hae Kyung Im2, Brian Jo7,8, Silva Kasela5,6, Seva Kashin33,34,35, Sarah Kim-Hellmuth5,6,9, Alan Kwong26, Tuuli Lappalainen5,6, Xiao Li1, Yanyu Liang2, Daniel G MacArthur34,36, Pejman Mohammadi5,6,20,21, Stephen B Montgomery12,29, Manuel Muñoz-Aguirre22,23, Meritxell Oliva2,10, YoSon Park24,25, Princy Parsana11, John M Rouhana17,1, Ashis Saha11, Ayellet V Segrè1,17, Matthew Stephens37, Barbara E Stranger2,38, Benjamin J Strober18, Ellen Todres1, Ana Viñuela39,3,27,28, Gao Wang37, Xiaoquan Wen26, Valentin Wucher22, Yuxin Zou40
Analysis Team Leaders*
François Aguet1, Alexis Battle18,11, Andrew Brown3,4, Stephane E Castel5,6, Barbara E Engelhardt7,8, Farhad Hormozdiari19,1, Hae Kyung Im2, Sarah Kim-Hellmuth5,6,9, Meritxell Oliva2,10, Barbara E Stranger2,38, Xiaoquan Wen26
Senior Leadership*
Kristin G Ardlie1, Alexis Battle18,11, Christopher D Brown24, Nancy Cox16, Emmanouil T Dermitzakis3,27,28, Barbara E Engelhardt7,8, Gad A Getz1,30,31, Roderic Guigó22,33, Hae Kyung Im2, Tuuli Lappalainen5,6, Stephen B Montgomery12,29, Barbara E Stranger2,38
Manuscript Writing Group
François Aguet1, Hae Kyung Im2, Alexis Battle18,11, Kristin G Ardlie1, Tuuli Lappalainen5,6
Corresponding Authors
François Aguet1, Kristin G Ardlie1, Tuuli Lappalainen5,6
Competing interests
F.A. is an inventor on a patent application related to TensorQTL; S.E.C. is a co-founder, chief technology officer and stock owner at Variant Bio; E.R.G. is on the Editorial Board of Circulation Research, and does consulting for the City of Hope / Beckman Research Institut; E.T.D. is chairman and member of the board of Hybridstat LTD.; B.E.E. is on the scientific advisory boards of Celsius Therapeutics and Freenome; G.G. receives research funds from IBM and Pharmacyclics, and is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig, MSMuTect, MSMutSig, POLYSOLVER and TensorQTL. G.G. is a founder, consultant and holds privately held equity in Scorpion Therapeutics; S.B.M. is on the scientific advisory board of MyOme; D.G.M. is a co-founder with equity in Goldfinch Bio, and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Merck, Pfizer, and Sanofi-Genzyme; H.K.I. has received speaker honoraria from GSK and AbbVie.; T.L. is a scientific advisory board member of Variant Bio with equity and Goldfinch Bio. P.F. is member of the scientific advisory boards of Fabric Genomics, Inc., and Eagle Genomes, Ltd. P.G.F. is a partner of Bioinf2Bio.
Data and Materials Availability
All GTEx protected data are available via dbGaP (accession phs000424.v8). Access to the raw sequence data is now provided through the AnVIL platform (https://gtexportal.org/home/protectedDataAccess). Public-access data, including QTL summary statistics and expression levels, are available on the GTEx Portal, as downloadable files and through multiple data visualizations and browsable tables (www.gtexportal.org), as well as in the UCSC and Ensembl browsers. All components of the single tissue cis-QTL pipeline are available at https://github.com/broadinstitute/gtex-pipeline (https://doi.org/10.5281/zenodo.3727189), and analysis scripts are available at https://github.com/broadinstitute/gtex-v8 (https://doi.org/10.5281/zenodo.3930961). Residual GTEx biospecimens have been banked, and are available as a resource for further studies (access can be requested on the GTEx Portal, at https://www.gtexportal.org/home/samplesPage).
Supplementary Content
Supplementary Material, including methods, figures S1–S52 and tables S1–S9
Supplementary Tables S10–S16
References
- [1].ENCODE Project Consortium, An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Roadmap Epigenomics Consortium, et al. , Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Battle A, et al. , Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Research 24, 14–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lappalainen T, et al. , Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bonder MJ, et al. , Disease variants alter transcription factor levels and methylation of their binding sites. Nature Genetics 49, 131–138 (2017). [DOI] [PubMed] [Google Scholar]
- [6].GTEx Consortium, The Genotype-Tissue Expression (GTEx) project. Nature Genetics 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Carithers LJ, et al. , A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation and biobanking 13, 311–319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Siminoff LA, Wilson-Genderson M, Gardiner HM, Mosavel M, Barker KL, Consent to a Postmortem Tissue Procurement Study: Distinguishing Family Decision Makers’ Knowledge of the Genotype-Tissue Expression Project. Biopreservation and biobanking 16, 200–206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].GTEx Consortium, The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].GTEx Consortium, Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].See supplementary materials.
- [12].de Goede OM, et al. , Long non-coding RNA gene regulation and trait associations across human tissues. bioRxiv (2019). [Google Scholar]
- [13].Li YI, et al. , Annotation-free quantification of RNA splicing using LeafCutter. Nature Genetics 50, 151–158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jansen R, et al. , Conditional eQTL analysis reveals allelic heterogeneity of gene expression. Human molecular genetics 26, 1444–1451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hormozdiari F, et al. , Widespread Allelic Heterogeneity in Complex Traits. American Journal of Human Genetics 100, 789–802 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Saha A, Battle A, False positives in trans-eQTL and co-expression analyses arising from RNA-sequencing alignment errors. F1000Research 7, 1860–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Castel SE, Aguet F, Mohammadi P, Ardlie KG, Lappalainen T, A vast resource of allelic expression data spanning human tissues. bioRxiv (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Khramtsova EA, Davis LK, Stranger BE, The role of sex in the genomics of human complex traits. Nature Reviews Genetics 20, 173–190 (2019). [DOI] [PubMed] [Google Scholar]
- [19].Stranger BE, et al. , Patterns of cis regulatory variation in diverse human populations. PLoS Genetics 8, e1002639 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Raj T, et al. , Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344, 519–523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mohammadi P, Castel SE, Brown AA, Lappalainen T, Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Research 27, 1872–1884 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Oliva M, et al. , The role of sex in the human transcriptome. bioRxiv (2019). [Google Scholar]
- [23].Sun T, et al. , Functional Phe31Ile polymorphism in Aurora A and risk of breast carcinoma. Carcinogenesis 25, 2225–2230 (2004). [DOI] [PubMed] [Google Scholar]
- [24].Ewart-Toland A, et al. , Aurora-A/STK15 T+91A is a general low penetrance cancer susceptibility gene: a meta-analysis of multiple cancer types. Carcinogenesis 26, 1368–1373 (2005). [DOI] [PubMed] [Google Scholar]
- [25].Ruan Y, et al. , Genetic polymorphisms in AURKA and BRCA1 are associated with breast cancer susceptibility in a Chinese Han population. The Journal of Pathology 225, 535–543 (2011). [DOI] [PubMed] [Google Scholar]
- [26].Koh HM , et al. , Aurora Kinase A Is a Prognostic Marker in Colorectal Adenocarcinoma. Journal of pathology and translational medicine 51, 32–39 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dhanasekaran K, et al. , Unraveling the role of aurora A beyond centrosomes and spindle assembly: implications in muscle differentiation. The FASEB Journal 33, 219–230 (2019). [DOI] [PubMed] [Google Scholar]
- [28].Girardeau-Hubert S, et al. , Reconstructed Skin Models Revealed Unexpected Differences in Epidermal African and Caucasian Skin. Scientific Reports 9, 7456 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yin L, et al. , Epidermal gene expression and ethnic pigmentation variations among individuals of Asian, European and African ancestry. Experimental dermatology 23, 731–735 (2014). [DOI] [PubMed] [Google Scholar]
- [30].Brown AA, et al. , Predicting causal variants affecting expression by using whole-genome sequencing and RNA-seq from multiple human tissues. Nature Genetics 49, 1747–1751 (2017). [DOI] [PubMed] [Google Scholar]
- [31].Hormozdiari F, Kostem E, Kang EY, Pasaniuc B, Eskin E, Identifying causal variants at loci with multiple signals of association. Genetics 198, 497–508 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wen X, Pique-Regi R, Luca F, Integrating molecular QTL data into genome-wide genetic association analysis: Probabilistic assessment of enrichment and colocalization. PLoS Genetics 13, e1006646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tewhey R, et al. , Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165, 1519–1529 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].van Arensbergen J, et al. , Systematic identification of human SNPs affecting regulatory element activity. bioRxiv (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li YI, et al. , RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Delaneau O, et al. , Chromatin three-dimensional interactions mediate genetic effects on gene expression. Science 364 (2019). [DOI] [PubMed] [Google Scholar]
- [37].Small KS, et al. , Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nature Genetics 43, 561–564 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yang F, Wang J, GTEx Consortium, Pierce BL, Chen LS, Identifying cis-mediators for trans-eQTLs across many human tissues using genomic mediation analysis. Genome Research 27, 1859–1871 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Westra H-J, et al. , Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature Genetics 45, 1238–1243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu B, Gloudemans MJ, Rao AS, Ingelsson E, Montgomery SB, Abundant associations with gene expression complicate GWAS follow-up. Nature Genetics 51, 768–769 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Barbeira AN, et al. , Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. bioRxiv 42, 814350 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nicolae DL, et al. , Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genetics 6, e1000888 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gamazon ER, et al. , Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nature Genetics 50, 956–967 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Finucane HK, et al. , Partitioning heritability by functional annotation using genome-wide association summary statistics. Nature Genetics 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hormozdiari F, et al. , Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nature Genetics 50, 1041–1047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Berisa T, Pickrell JK, Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics 32, 283–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gamazon ER, et al. , A gene-based association method for mapping traits using reference transcriptome data. Nature Genetics 47, 1091–1098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cirulli ET, et al. , Genome-wide rare variant analysis for thousands of phenotypes in 54,000 exomes. bioRxiv 442, 199–22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ferraro NM, et al. , Diverse transcriptomic signatures across human tissues identify functional rare genetic variation. bioRxiv (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jordan DM, Verbanck M, Do R, HOPS: a quantitative score reveals pervasive horizontal pleiotropy in human genetic variation is driven by extreme polygenicity of human traits and diseases. Genome Biology 20, 222–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Urbut SM, Wang G, Carbonetto P, Stephens M, Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nature Genetics 51, 187–195 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mohammadi P, et al. , Genetic regulatory variation in populations informs transcriptome analysis in rare disease. Science 366, 351–356 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Aran D, Hu Z, Butte AJ, xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biology 18, 220 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kim-Hellmuth S, et al. , Cell type specific genetic regulation of gene expression across human tissues. bioRxiv 7, 1860 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhernakova DV, et al. , Identification of context-dependent expression quantitative trait loci in whole blood. Nature Genetics 49, 139–145 (2017). [DOI] [PubMed] [Google Scholar]
- [56].Peters JE, et al. , Insight into Genotype-Phenotype Associations through eQTL Mapping in Multiple Cell Types in Health and Immune-Mediated Disease. PLoS Genetics 12, e1005908 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Plenge RM, Scolnick EM, Altshuler D, Validating therapeutic targets through human genetics. Nature 12, 581–594 (2013). [DOI] [PubMed] [Google Scholar]
- [58].Fisher S, et al. , A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biology 12, R1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tukiainen T, et al. , Landscape of X chromosome inactivation across human tissues. Nature 550, 244–248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kim D, Langmead B, Salzberg SL, HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Handsaker RE, et al. , Large multiallelic copy number variations in humans. Nature Genetics 47, 296–303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Delaneau O, Zagury J-F, Marchini J, Improved whole-chromosome phasing for disease and population genetic studies. Nature Methods 10, 5–6 (2013). [DOI] [PubMed] [Google Scholar]
- [63].Dobin A, et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].van de Geijn B, McVicker G, Gilad Y, Pritchard JK, WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nature Methods 12, 1061–1063 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wright FA, et al. , Heritability and genomics of gene expression in peripheral blood. Nature Genetics 46, 430–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].DeLuca DS, et al. , RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28, 1530–1532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Robinson MD, Oshlack A, A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Stegle O, Parts L, Durbin R, Winn J, A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Computational Biology 6, e1000770 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Gay NR, et al. , Impact of admixture and ancestry on eQTL analysis and GWAS colocalization in GTEx. bioRxiv 95, 1.22.1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O, Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics 32, 1479–1485 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Storey JD, Tibshirani R, Statistical significance for genomewide studies. PNAS 100, 9440–9445 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Taylor-Weiner A, et al. , Scaling computational genomics to millions of individuals with GPUs. Genome Biology 20, 228–5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Giambartolomei C, et al. , Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genetics 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Buil A, et al. , Gene-gene and gene-environment interactions detected by transcriptome sequence analysis in twins. Nature Genetics 47, 88–91 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Võsa U, et al. , Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv 100, 228 (2018). [Google Scholar]
- [76].Castel SE, Levy-Moonshine A, Mohammadi P, Banks E, Lappalainen T, Tools and best practices for data processing in allelic expression analysis. Genome Biology 16, 195 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Panousis NI, Gutierrez-Arcelus M, Dermitzakis ET, Lappalainen T, Allelic mapping bias in RNA-sequencing is not a major confounder in eQTL studies. Genome Biology 15, 467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Castel SE, Mohammadi P, Chung WK, Shen Y, Lappalainen T, Rare variant phasing and haplotypic expression from RNA sequencing with phASER. Nature Communications 7, 12817 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Anders S, Huber W, Differential expression analysis for sequence count data. Genome Biology 11, R106–12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR, The ensembl regulatory build. Genome Biology 16, 56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wen X, Molecular QTL discovery incorporating genomic annotations using Bayesian false discovery rate control. The Annals of Applied Statistics 10, 1619–1638 (2016). [Google Scholar]
- [82].Michailidou K, et al. , Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Tang B, et al. , CBX8 exhibits oncogenic properties and serves as a prognostic factor in hepatocellular carcinoma. Cell death & disease 10, 52–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Chung C-Y, et al. , Cbx8 Acts Non-canonically with Wdr5 to Promote Mammary Tumorigenesis. Cell Reports 16, 472–486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zhang CZ, et al. , CBX8 Exhibits Oncogenic Activity via AKT/β-Catenin Activation in Hepatocellular Carcinoma. Cancer Research 78, 51–63 (2018). [DOI] [PubMed] [Google Scholar]
- [86].Buniello A, et al. , The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Research 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Bycroft C, et al. , The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Astle WJ, et al. , The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 167, 1415–1429.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Nikpay M, et al. , A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nature Genetics 47, 1121–1130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Hammerschlag AR, et al. , Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nature Genetics 49, 1584–1592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Paternoster L, et al. , Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nature Genetics 47, 1449–1456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Horikoshi M, et al. , Genome-wide associations for birth weight and correlations with adult disease. Nature 538, 248–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Hibar DP, et al. , Common genetic variants influence human subcortical brain structures. Nature 520, 224–229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zheng H-F, et al. , Whole-genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature 526, 112–117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wood AR, et al. , Defining the role of common variation in the genomic and biological architecture of adult human height. Nature Genetics 46, 1173–1186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Liu JZ, et al. , Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nature Genetics 47, 979–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lambert JC, et al. , Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature Genetics 45, 1452–1458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bentham J, et al. , Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nature Genetics 47, 1457–1464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Jones SE, et al. , Genome-Wide Association Analyses in 128,266 Individuals Identifies New Morningness and Sleep Duration Loci. PLoS Genetics 12, e1006125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Dupuis J, et al. , New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nature Genetics 42, 105–116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kettunen J, et al. , Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nature Communications 7, 11122–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Martin J, et al. , A Genetic Investigation of Sex Bias in the Prevalence of Attention-Deficit/Hyperactivity Disorder. Biological psychiatry 83, 1044–1053 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Schizophrenia Working Group of the Psychiatric Genomics Consortium, Biological insights from 108 schizophrenia- associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Okada Y, et al. , Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Okbay A, et al. , Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nature Genetics 48, 624–633 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Okbay A, et al. , Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Guo Q, et al. , Identification of novel genetic markers of breast cancer survival. Journal of the National Cancer Institute 107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Lee D, Bigdeli TB, Riley BP, Fanous AH, Bacanu S-A, DIST: direct imputation of summary statistics for unmeasured SNPs. Bioinformatics 29, 2925–2927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Pasaniuc B, et al. , Fast and accurate imputation of summary statistics enhances evidence of functional enrichment. Bioinformatics 30, 2906–2914 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Wen X, Lee Y, Luca F, Pique-Regi R, Efficient Integrative Multi-SNP Association Analysis via Deterministic Approximation of Posteriors. American Journal of Human Genetics 98, 1114–1129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Gazal S, et al. , Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nature Genetics 49, 1421–1427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Barbeira AN, et al. , Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nature Communications 9, 1825–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Barbeira AN, et al. , Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genetics 15, e1007889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Friedman J, Hastie T, Tibshirani R, Regularization Paths for Generalized Linear Models via Coordinate Descent. Journal of statistical software 33, 1–22 (2010). [PMC free article] [PubMed] [Google Scholar]
- [115].International HapMap 3 Consortium, et al. , Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Wang C, et al. , Deletion of mstna and mstnb impairs the immune system and affects growth performance in zebrafish. Fish & shellfish immunology 72, 572–580 (2018). [DOI] [PubMed] [Google Scholar]
- [117].Wan YY, GATA3: a master of many trades in immune regulation. Trends in immunology 35, 233–242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Wang W, Stephens M, Empirical Bayes Matrix Factorization. arXiv.org (2018). [PMC free article] [PubMed] [Google Scholar]
- [119].Sul JH, Han B, Ye C, Choi T, Eskin E, Effectively identifying eQTLs from multiple tissues by combining mixed model and meta-analytic approaches. PLoS Genetics 9, e1003491 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Zhou X, Stephens M, Genome-wide efficient mixed-model analysis for association studies. Nature Genetics 44, 821–824 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Han B, Eskin E, Interpreting Meta-Analyses of Genome-Wide Association Studies. PLoS Genetics 8, e1002555–11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Ongen H, et al. , Estimating the causal tissues for complex traits and diseases. Nature Genetics 49, 1676–1683 (2017). [DOI] [PubMed] [Google Scholar]
- [123].Majumdar A, et al. , Leveraging eQTLs to identify individual-level tissue of interest for a complex trait. bioRxiv (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Huang Y-F, Gulko B, Siepel A, Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nature Genetics 49, 618–624 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Davis JR, et al. , An Efficient Multiple-Testing Adjustment for eQTL Studies that Accounts for Linkage Disequilibrium between Variants. American Journal of Human Genetics 98, 216–224 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Malone J, et al. , Modeling sample variables with an Experimental Factor Ontology. Bioinformatics 26, 1112–1118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Köhler S, et al. , The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Research 42, D966–74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.