

Abstract
UCSF ChimeraX is the next‐generation interactive visualization program from the Resource for Biocomputing, Visualization, and Informatics (RBVI), following UCSF Chimera. ChimeraX brings (a) significant performance and graphics enhancements; (b) new implementations of Chimera's most highly used tools, many with further improvements; (c) several entirely new analysis features; (d) support for new areas such as virtual reality, light‐sheet microscopy, and medical imaging data; (e) major ease‐of‐use advances, including toolbars with icons to perform actions with a single click, basic “undo” capabilities, and more logical and consistent commands; and (f) an app store for researchers to contribute new tools. ChimeraX includes full user documentation and is free for noncommercial use, with downloads available for Windows, Linux, and macOS from https://www.rbvi.ucsf.edu/chimerax.
Keywords: cryoEM, density maps, interactive visualization and analysis, molecular graphics, molecular modeling, structural biology, virtual reality
1. INTRODUCTION
Over the past few decades, molecular graphics has moved from the purview of structural biologists into the realm of many life scientists. Originally focused on atomic‐scale data, molecular modeling now encompasses macromolecular assemblies and even larger‐scale data such as from optical microscopy. In recent years, the rapid increase in the size of data sets, the proliferation of web resources, and the rise of parallel computing have further affected modeling application development. 1
In early molecular graphics systems, the largest data sets that could be visualized interactively were single molecules, perhaps with density maps from X‐ray crystallography. Data size has grown rapidly as new experimental methods have been developed. Cryo‐electron microscopy (cryoEM) has produced density maps for larger molecules and assemblies. 2 , 3 As microscopy and imaging technologies advance, more and more data of varying scales (atomic, molecular, even cellular) have become available. Computational methods have also contributed to the growth. For example, molecular dynamics generates long trajectories, and integrative hybrid modeling 4 produces large ensembles of candidate structures of macromolecular assemblies.
The number of web resources, both databases and computational services, has increased steadily as well. 5 , 6 , 7 While the Worldwide Protein Data Bank 8 (wwPDB) is still the leading repository for experimentally solved atomic structures, other highly used resources include ModBase 9 for predicted atomic structures; EMDataBank 10 for electron microscopy density maps; and NCBI 11 , 12 and UniProt 13 for sequences, just to name a few.
Software engineering has also undergone significant changes. Concepts such as stable APIs as contracts, semantic versioning, 14 incremental updates, and an emphasis on extensibility have become common. Many software packages offer “plugins” that can extend the software, often in significant ways, and repositories o/f these extensions 15 , 16 that serve as avenues for sharing.
Finally, hardware improvement has changed in flavor in the past few years. 17 Previously, computing performance improved through faster individual processors. More recently, performance has improved by increasing the number of processors (CPUs) and taking advantage of the parallelism of graphics processors (GPUs). In addition, newer GPUs have enabled more advanced visualization techniques, including virtual reality. In the midst of all of these changes and opportunities, the need for molecular visualization and analysis has accelerated. During the first 6 months of the COVID‐19 pandemic, there were over 229 SARS‐CoV‐2 protein structures deposited in the public PDB, 18 with many more undoubtedly in the pipeline. These structures are helping to understand the potential mechanisms of treatments and vaccines. Tools that help with the modeling and analyses of these structures are necessary to shorten the time from experimental data collection to the availability of the structure. In addition, tools that help explain science to a broader audience are also important.
This is the context that has led us to develop ChimeraX, our next‐generation molecular visualization and analysis environment. ChimeraX builds on our experiences with UCSF Chimera, 19 but utilizes modern hardware and software approaches to meet the increasing demands of today's visualization requirements. ChimeraX 1.0 does not have all of the tools currently available in UCSF Chimera, but we have added many completely new capabilities and improved visualization, usability, and performance. With the release of ChimeraX version 1.0, the list of features has grown very long. We present an overview of ChimeraX's main features and advantages, followed by sections for researchers, educators, and developers.
2. OVERVIEW
ChimeraX allows interactively viewing very large data sets and analyzing different types of data together. Examples are atomic structures and cryo‐EM density maps of ribosomes and viral capsids, sequence alignments, multichannel and 4D light microscopy, and tomography at ever‐increasing scales, from whole cells to full‐body medical imaging. ChimeraX reads over 60 file formats, many for volume data (values on a grid: density maps, electrostatic potential, microscopy data), with the remainder for atomic structures, sequences, 3D objects, scripts or code, and composites of multiple data types such as ChimeraX session files and integrative hybrid models. 20 ChimeraX has a fast macromolecular crystallographic information file (mmCIF) reader and is able to write mmCIF files, in addition to several other formats. Data can be opened from local files and/or fetched directly from online repositories or web services, including the Research Collaboratory for Structure Bioinformatics (RCSB) Protein DataBank 18 (PDB), Electron Microscopy Data Bank 21 (EMDB), PubChem3D, 22 and UniProt. 13
The set of features for data analysis and modification includes superposition and fitting, morphing, map filtering and segmentation, coloring by lipophilicity and other properties, atomic interaction analysis, atomic structure building and modification, and many others described in more detail below.
ChimeraX also provides advanced graphics and lighting modes, such as interactive ambient occlusion and directional shadows. 23 A flat‐lighting mode with silhouettes (black outlines) can be used for a line‐drawing‐like appearance. Several lighting options are accessible via toolbar icons. ChimeraX molecular surface calculations are fast and robust. Protein helices can be shown as the traditional spiral ribbons or as curved tubes. 23 Display‐style presets for atomic structures are available from the menu, and numerous example images and the corresponding ChimeraX command scripts are presented in the feature highlights (https://www.rbvi.ucsf.edu/chimerax/features.html) and image gallery (https://www.rbvi.ucsf.edu/chimerax/gallery.html) sections of the ChimeraX website. ChimeraX is often used to make high‐quality figures and movies for publication. Movie content ranges from simple rotations to transitions between saved positions, morphing, and annotation with text and arrows. Interactive sessions may also be captured as movies. Capabilities for publication graphics are described in more detail below.
The ChimeraX graphics window and tool panels are integrated into a single, overall window (Figure 1). Individual panels can be shown, hidden, and repositioned, or detached entirely (undocked) to become floating panels. There is a Toolbar with icons across the top, a command line at the bottom, and a Log panel showing executed actions, results, and other messages. The icons perform commonly used actions with a single click, and the corresponding command is shown in the Log. Additional panels may be opened from the Tools menu, or appear as the result of opening a certain type of data or running a calculation. Panels may interact with the 3D display; for example, mousing over a bar in a histogram of H‐bond lengths highlights the corresponding H‐bonds in the structure. Which tools are shown upon startup, which are dockable, their approximate layout in the window when docked (top, bottom, left, right), and which icons appear in the Home tab of the Toolbar are customizable by the user.
FIGURE 1.
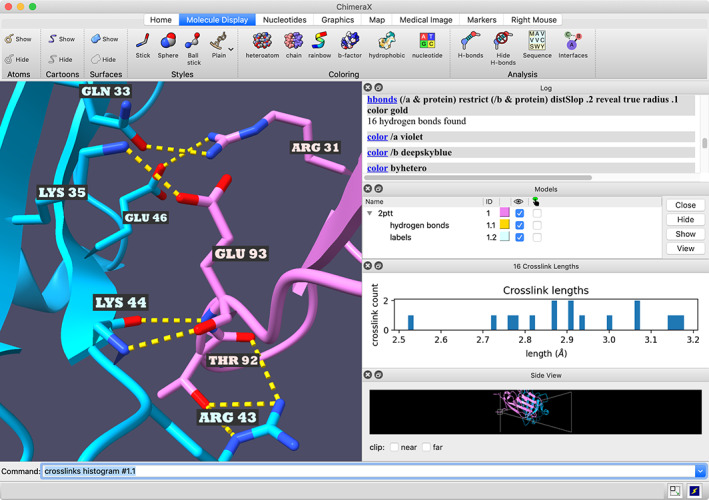
ChimeraX window showing interchain H‐bonds and length histogram (PDBid: 2ptt). The “crosslinks” command and resulting histogram were originally developed to analyze crosslinks, but may be used on any pseudobonds, including the H‐bonds shown here. For the corresponding ChimeraX command script, see: https://www.rbvi.ucsf.edu/chimerax/features.html#hbond‐histogram
Atomic structures, volume datasets, surfaces, and other objects in 3D are called “models” in ChimeraX. The hierarchy of open models is listed in the Model Panel, along with show/hide and selection checkboxes. Opening a 3D data file will generate one or more models, as will many calculations such as density‐map filtering or finding H‐bonds. Opening a volume dataset shows the Volume Viewer tool with a data histogram, a slider to adjust the contour level, and several other controls. Opening a sequence alignment shows it in a Sequence Viewer window and associates any similar‐sequence chains of the open atomic models with the sequences.
When all models are closed, a “rapid access” panel is shown in place of the graphics window. It contains thumbnail images for recent files that can be clicked to reopen the data.
Although the ChimeraX user interface appearance is different from Chimera, methods of interaction are similar: “mouse” modes, selection‐action, and command‐target.
2.1. Mouse modes
Functions that can be assigned to mouse buttons or their trackpad equivalents are referred to as mouse modes. They can also be assigned to VR hand‐controller buttons. Familiar modes include rotation, translation, zooming, and selection (more on this below), but in ChimeraX, dozens of other functions can be assigned, including moving selected models only, rotating bonds, adjusting clip planes, moving labels, making tape‐measure distance measurements, changing map contour level, cropping the volume box, and viewing different data planes or slabs. One of the Toolbar tabs is devoted to mouse modes, where clicking an icon assigns the corresponding mode to the right mouse button or its trackpad equivalent (trackpad + Alt on Windows, trackpad + command on Mac). The current right‐mouse assignment is shown with light green highlighting on the icon. Assignments to other buttons, alone and in combination with modifier keys, can be made with the mousemode command.
The Side View tool is another interface for intuitive clipping and zooming with the mouse (Figure 1).
2.2. Selection‐action
The Actions menu and toolbar action icons apply to the selected objects only (specific sets of atoms, residues, surfaces, models, etc.). If nothing is selected, however, such actions affect all applicable objects. By default, selected objects are indicated with bright green outlines in the graphics window. Objects can be selected in many ways, for example, with the Select menu, with the select command, using the “selection” mouse mode in the 3D graphics window, by dragging a box in a sequence that is associated with a structure chain, or by performing a calculation that can show results (such as clashing atoms) with selection.
The Select menu allows selecting by element, atom type, functional group, residue name, and chain, by convenient categorizations such as protein, nucleic acid, ligand, and solvent, and by distance zone from the current selection. The selection can be cleared by “selecting” with the mouse in an empty area of the graphics window, or by using the Select menu or command. The Actions menu includes showing, hiding, and adjusting the styles of atoms, cartoons, and molecular surfaces, labeling atoms and residues, basic coloring, and opening the Color Actions tool for a more extensive set of options. Whole models can be selected using checkboxes in the Model Panel.
In typed commands, the target objects can be listed in the command, or the word “sel” can indicate the current selection. The selection‐action approach is favored when it is easier to select the desired target(s) with one of the methods above than to name specific objects in a command.
2.3. Command‐target
Objects to act upon can be specified directly in the command line, without going through a selection. The command‐line specification is quite powerful and may include any of the same types of information as in the Select menu, as well as residue number ranges, attribute values (e.g., atoms with a specific range of B‐values), and Boolean combinations of any of these. The syntax is similar to that in Chimera, but with several improvements, including better handling of chains and the ability to group with parentheses. Special symbols indicate model numbers (#), chain identifiers (/), residue names or numbers (:), atom names (@), distance zones (< and >), wild cards (? and *), negation (~), intersection (&), and union (|). For example, the command “label /A:his,lys & ligand :<4.8” would label histidine and lysine residues in chain A that are also within 4.8 Å of residues classified as ligand. Several built‐in command‐line specifiers are provided for convenience, such as sel for the current selection and hbonds for hydrogen‐bond pseudobonds.
Performing actions through the ChimeraX graphical interface (tool panels, menu, and toolbar icons) echoes the corresponding commands to the Log, making it easy for users to discover commands and to re‐execute them later. Commands can be saved to a plain text ChimeraX command file (.cxc), or such a file can be created directly in a text editor. Opening a command file executes its contents. Commands to execute upon startup can be specified in the user Settings (Preferences).
With commands or command scripts, users can create aliases (custom commands constructed from other commands), custom display‐style presets, named selections, and custom command‐line specifiers. Custom presets, named selections, and other custom specifiers can be accessed through the ChimeraX menu as well as the command line.
2.4. User conveniences
Undo is available for most changes in coloring, display, display style, position, and selection. The undo/redo stack can be navigated via the Edit menu or the commands undo and redo.
A bug reporter dialog can be opened from the Help menu or by clicking a button on the window that appears when an error occurs. Information about the user's platform (e.g., OS version and graphics driver), ChimeraX version, and (optionally) the entire contents of the Log are included in the report, and the user may also enter a description, attach a file, and include an e‐mail address if feedback is desired. Such reports automatically go into the bug‐tracking system. This mechanism makes it as easy as possible to report problems.
Even with complete documentation, users may need advice on what features are available or the details of applying them to a complicated task. A mailing list (chimerax‐users@cgl.ucsf.edu) is provided for users to ask questions about using ChimeraX and developing plugins. Suggestions and feature requests can also be sent. This list is monitored by the ChimeraX developers and responses are usually quick, typically within a work day, and thorough.
The ChimeraX Toolshed is a web repository for plugins, with mechanisms similar to the Cytoscape App Store 15 for developers to upload and update their bundles, and for users to install them in ChimeraX with minimal effort. Most notably, the interactive structure optimization by local direct exploration (ISOLDE) plugin to ChimeraX for interactive dynamics‐assisted structure building into cryo‐EM and crystallographic maps 24 (https://isolde.cimr.cam.ac.uk) has already been cited in over 30 publications, including two focused on proteins from severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). 25 , 26 The Toolshed (https://cxtoolshed.rbvi.ucsf.edu) can be opened from the ChimeraX Tools menu and is described in more detail in the Developers section.
2.5. Virtual reality
ChimeraX can be used with virtual‐reality (VR) headsets and hand controllers, 27 and it supports multi‐user sessions and recording augmented‐reality videos. Functions can be assigned to VR hand‐controller buttons using the Toolbar mouse‐mode icons. VR features are described in more detail in subsequent parts of the paper.
3. ANALYSIS AND PRESENTATION CAPABILITIES FOR RESEARCHERS
Many of the important analysis tools found in Chimera 19 have been ported to ChimeraX, often with enhancements and new capabilities. Multiple tools interact with one another to form a more powerful whole. For instance, the side‐chain rotamer tool provides a built‐in statistical probability score but also leverages hydrogen‐bond finding, clash detection, and density‐fitting to help identify the best rotamer in the context of the specific structure. ChimeraX's analysis capabilities can be grouped into three broad areas: structure, sequence, and volume.
3.1. Structure analysis
When an atomic structure is opened, it is displayed in a style designed to be the most informative given the structure's size. A small molecule is displayed as “sticks” colored by element type. Medium‐sized structures are displayed as cartoons with solvent hidden, but with ligands and nearby side chains shown in atomic detail. Large structures are shown as space‐filling spheres, with chains distinctly colored and shadowed to give a better sense of the structure's shape. In addition, information about the structure will be presented in the Log (if present in the input file), such as a description of the structure, experiment type, resolution, etc. As described in the Educators section, interactive tables of chain, residue, and assembly information shown in the Log facilitate rapid exploration and comprehension of the structure.
The mouse or trackpad can be used to rotate, translate, or zoom the structure, and hovering the mouse over the part of a structure will bring up a tooltip identifying the chain, residue, and atom under the cursor. Analysis features allow a user to:
Measure atomic distances and torsion angles
Find clashes or contacts between sets of atoms
Find hydrogen bonds or salt bridges
Show crosslink data on structures, as a histogram, or as a chain‐chain 2D plot
Display solvent‐excluded surfaces (SES); calculate areas of buried and exposed solvent‐accessible surfaces (SAS)
Color surfaces by lipophilicity or (externally computed) electrostatic potential
Plot 2D chain‐chain contacts based on buried surface areas
Superimpose structures using specified atoms or based on an automatically computed sequence alignment (matchmaker)
Color structures by B‐factor or other properties
Add and delete atoms for step‐by‐step building, rotate bonds manually
Generate a new model starting with a single atom, PubChem 28 entry, SMILES 29 string, or peptide sequence
Morph between similar (not necessarily identical) structures
View and evaluate molecular docking results (calculate H‐bonds, contacts, etc.)
Use VR for enhanced perception of spatial relationships to aid in tasks such as evaluating docking poses or assessing structure fitting in volume data, individually or in a multi‐user session
3.2. Sequence analysis
Sequences from sequence‐alignment files or structure chains are displayed in a C/himeraX tool window. Single sequences and alignments are shown in a very similar fashion but with slight differences.
For sequence‐alignment files, the sequences can be laid out in a single strip with horizontal scrolling, or wrapped to the current window width. The alignment will display conservation and consensus headers above the sequences (Figure 2). Any chain(s) in a 3D structure with sufficient sequence similarity to alignment sequences (by default fewer than 10% mismatches) will be “associated” with the most similar sequence in the alignment, which means that selecting residues in the structure will show corresponding green boxes on the sequence, and dragging on the sequence will draw a box and select the corresponding structure residues. Associations can also be added or removed manually.
FIGURE 2.

Superimposed chains of four DNA‐binding proteins (PDBid: 1qvc, 1kaw, 1eyg, and 3ull), with names of associated sequences in the alignment highlighted with matching colors. The foreground helix was selected in the graphics window by dragging out the green/brown box on the sequence alignment. The missing structure is depicted by boxes on the alignment, outlined in black if missing from all chains, and in gray otherwise. The sequence window also shows a consensus sequence and sequence conservation histogram, with several choices for conservation scoring: the one in use in this figure is entropy‐based with independent counts as computed by the AL2CO program 30
For structure chains, the full sequence is displayed, including missing‐structure regions (if available from the input). All structure chains with the given sequence are associated with that sequence, and their secondary structure is shown on the sequence as colored boxes.
ChimeraX sequence capabilities include:
Fetch sequences from the UniProt database
Blast chain or other sequences against a PDB sequence database and display the hits as sequences or structures
Comparative modeling via an interface to Modeller, either installed locally or using an RBVI‐hosted web service, including support for modeling hetero‐multimeric complexes
More sequence tools are in development to provide a rich set similar to that in Chimera.
3.3. Volume analysis
Three‐dimensional image data from electron microscopy, light microscopy, x‐ray crystallography, and medical imaging can be visualized as surfaces, meshes, or colored voxels. Histograms of intensity values are shown in the Volume Viewer tool (Figure 4) where colors, threshold levels, and display styles are adjusted. Dozens of tools and commands allow an interactive analysis of volume data.
Many methods analyze cryoEM maps and atomic structures. Atomic models of components of a molecular assembly can be fitted taking into account map correlation, clashes, symmetry, allowing local rigid‐body optimization, or global searching (command fitmap). Surface and mesh can be displayed within a distance range of specified atoms to visually validate models (command volume zone) (Figure 3). Maps of assemblies can be colored to distinguish molecular components (command color zone). Maps can be smoothed, sharpened, dusted (hiding all small disconnected surface fragments). Distances, areas, enclosed volumes, and domain rotations and shifts can be measured (command measure). Symmetries such as icosahedral virus capsids, helical filaments, or rings can be quantified from maps and applied to asymmetric unit atomic models (command sym). Maps can be segmented using a clustering watershed method called Segger 31 to separate molecular components of an assembly.
FIGURE 3.
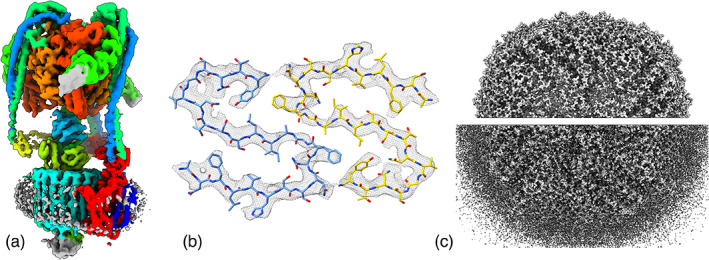
Visualizing electron microscopy maps. (a) Color molecular components from a fit atomic model, command color zone, mammalian V‐ATPase (EMDB 21317). (b) Density near atoms, command volume zone, islet amyloid fibril (EMDB 10669). (c) Hide small density blobs, command surface dust, apoferritin at 1.2 Å resolution 32
Several capabilities support 3D light microscopy analysis and electron tomography visualized as stacks of semi‐transparent color images (Figure 4). Multiple color channels can be blended and brightness curves for each channel adjusted with immediate graphics updates. Single‐plane slicing and slab slices tilted in any direction can be interactively adjusted for viewing in vivo structures that are not aligned with the image axes. Markers and paths can be hand‐placed and oriented boxes extracted using mouse modes. Scanning and jumping through time series with data subsampling allows fast exploration.
FIGURE 4.
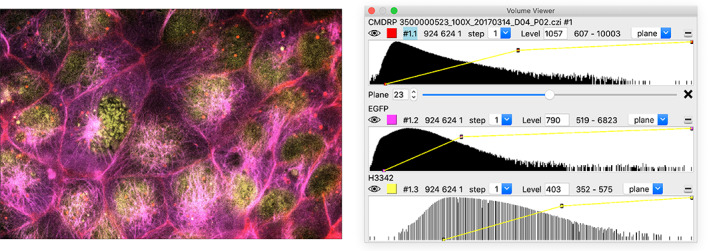
Multichannel 3D light microscopy. Histograms of each channel are shown in the Volume Viewer panel (right) where color, brightness curves, and display style can be adjusted
3.4. Segmentation display
Segmentations allow coloring and quantifying substructures. They are useful with electron tomography of cells or cryoEM maps of molecular assemblies where positions of the components are uncertain. Segmentations assign an integer value at each grid point of a volume indicating which region contains the grid point. Surfaces can be shown for each region (command segmentation surfaces) or the original grayscale image can be colored (Figure 5).
FIGURE 5.

Visualizing segmentations. Milled block‐face electron microscopy of mouse barrel cortex showing neurons, dendritic spines, and synapses. 33 (a) Segmentation surfaces of 96 neurons (distinct colors). (b) EM map with colored axons (green), spine heads (blue), dendrites (red). (c) The surface of one neuron (yellow) with dendritic spines (red)
3.5. Virtual reality image and structure analysis
Models displayed by ChimeraX can be explored using virtual‐reality headsets that connect to a computer using the SteamVR system (e.g., Oculus Rift S, Valve Index, or Vive Pro headsets). ChimeraX user interface panels can be shown and used in VR. Some limitations are (a) using the keyboard to type parameters is difficult, (b) scenes with significantly slower than 90 fps rendering must be simplified, and (c) multitasking with a web browser or other applications is not convenient.
Three uses where VR offers significant advantages are (a) studying bound ligands, (b) building atomic models in cryoEM maps, and (c) placing molecular complexes in EM tomography (Figure 6). These tasks benefit from stereoscopic depth perception and hand manipulations with 6‐degree‐of‐freedom VR wands.
FIGURE 6.

Placing molecular complexes in EM tomography (EMDB 10780) of the Chlamydomonas thylakoid membrane using virtual reality: 34 photosystem I (green), photosystem II (blue), cytochrome b6f (orange), ATP synthase (magenta), ribosome (yellow)
3.6. Refining and validating atomic models in cryoEM and X‐ray maps using molecular dynamics
In addition to ChimeraX's built‐in analysis capabilities, its extensive third‐party developer support facilitates the development of sophisticated plugins, such as ISOLDE. 24 ISOLDE is an adaptation of the concept of molecular dynamics flexible fitting 35 (MDFF) to the task of interactively building and remodeling atomic models into experimental maps. While designed primarily for the experimental structural biology community, it is also hoped that it will prove useful to the downstream user community to evaluate and/or improve the quality of models prior to use.
Typical scenes from ISOLDE are pictured in Figure 7. ISOLDE aims to make modeling errors both immediately apparent (via real‐time markup of common geometric issues and ‐ where applicable ‐ recalculation of crystallographic maps) and straightforwardly correctable via interactive MD, without the need to worry about the details of the MD engine or forcefield. Atoms may be rearranged by direct tugging with the mouse or a VR pointer, or by the push‐button addition and removal of position, distance, or torsion restraints. More complex restraints (e.g., to restrain the conformation to a high‐resolution reference model) are accessible via the ChimeraX command line.
FIGURE 7.
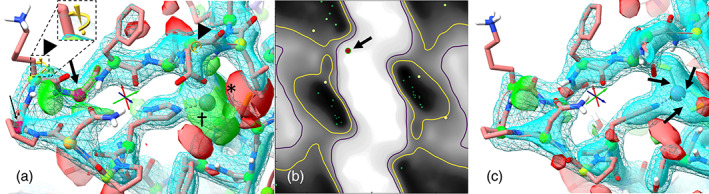
Typical scenes from an ISOLDE session, rebuilding a problematic loop in an x‐ray crystallographic model (PDBid: 7bq7). (a) Model as loaded from the wwPDB. Maps are calculated from the experimental structure factors and are updated on the fly in response to coordinate changes. Cyan wireframe/surface: electron density map at two different contours; green/red surface: difference map at +/− 3σ. Cryo‐EM sessions differ only in that the provided map(s) do not change over time. Live markup includes questionable rotamers (inset; marker grows and shades from yellow to red with decreasing probability) and backbone Ramachandran status (colored balls overlaid on alpha carbon atoms, shading from green through yellow to red with decreasing probability). This scene contains two Ramachandran outliers (arrows), two marginal rotamers (arrowheads), and a cysteine sidechain badly out of density as shown by the difference map (*/† show current and expected positions respectively). (b) ISOLDE's interactive Ramachandran plot, showing the glycine outlier selected in (a). This plot updates in real‐time. Hovering over a point identifies it, and clicking displays the corresponding residue in the main view. (c) Corrected model. Interactive distance restraints were applied to maintain the correct zinc‐sulfur distances in the Cys3HisZn coordination site at right (arrows)
3.7. Presentation images and videos
Images and videos in journal publications and talks help communicate findings on the structure and function of molecular complexes. Display styles such as nucleotides as slabs, 36 alpha‐helices as tubes, 23 molecular surfaces, and lower‐resolution Gaussian surfaces are used to depict the best level of detail to convey the message of the image (Figure 8). Text labels, arrows, colors, and transparency distinguish and focus attention on the different components of the molecular system. Advanced graphics capabilities such as ambient shadows, directional lights, silhouette edges, clipping, and fading distant objects maximize the three‐dimensional perception. Two‐dimensional images give a limited view of 3D structures and short videos that rotate, disassemble and morph molecular complexes can give a more informative view than static images. These capabilities are optimized to make images and videos render immediately on typical laptop computers and allow quickly composing scenes even with graphics‐intensive ambient shadows.
FIGURE 8.
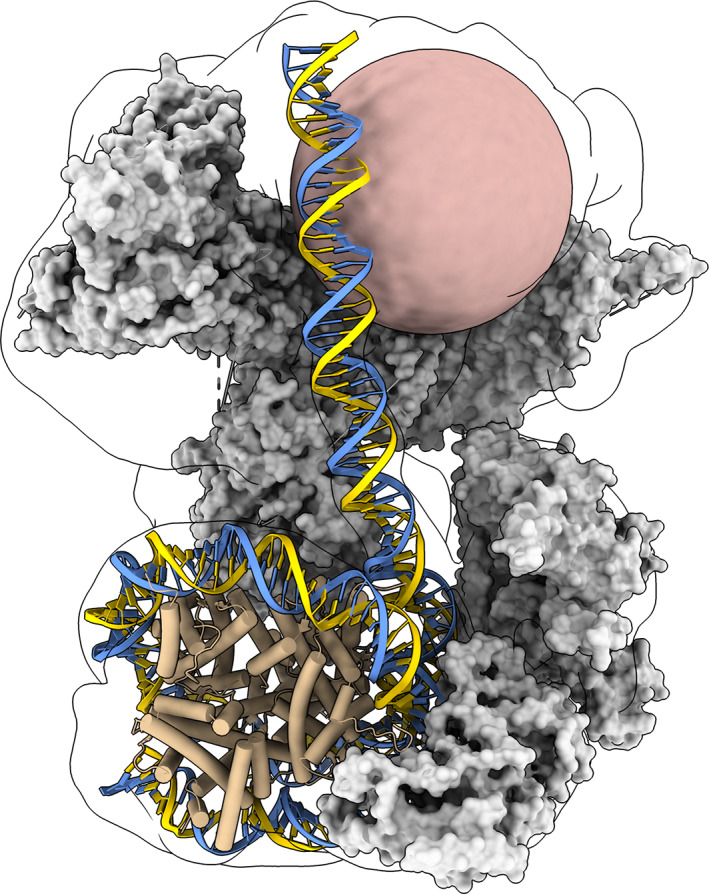
Display styles. SWI/SNF chromatin remodeling complex (PDBid: 6tda) illustrating various structure display styles. Molecular surfaces with ambient shadows for SWI/SNF proteins (gray). DNA nucleotide slabs (blue, gold). Alpha helix tubes for nucleosome histone proteins (tan). Unmodelled DNA interaction module as a sphere (pink). cryoEM density (15 Å resolution) as a transparent surface with silhouette edge
3.8. Making images and movies
ChimeraX has many features that enhance images. Ambient shadows cast light from all directions to provide good depth perception in space‐filling scenes, and silhouette edges on a white background help distinguish foreground and background objects. Saving a session for each image allows modifications later. A simple movie that rotates a scene 360° can be made by pressing the spin‐movie toolbar icon. A morph between conformations of a molecular assembly displays a slider, play button, and record button that creates a video on the user's desktop (Video S1). Online tutorials show how to make more elaborate videos with command scripts.
3.9. Augmented‐reality video capture
Augmented‐reality (AR) videos combine real‐world video of a presenter with computer‐generated 3‐dimensional objects and can be created with a VR headset and depth‐sensing camera (command realsense). Story‐telling with spoken narration and hand manipulation of molecular models can effectively communicate science research (Figure 9).
FIGURE 9.

Augmented‐reality video capture. Computer‐generated structures are blended with live video from a depth‐sensing camera to communicate science results. The presenter sees the structures and live video but not the room. Coronavirus cell entry example (YouTube https://youtu.be/dKNbRRRFhqY)
4. CHIMERAX FOR EDUCATORS AND STUDENTS
Several characteristics of a tool can make it attractive for use in learning environments. First, is the utility of the tool itself. Learning to use any computational tool takes time and effort, so it should provide a benefit in directly explaining the subject or in helping learners explore the subject on their own. Second, is the tool's usability. Usability includes the ease of learning, the materials available to learn, the discoverability of features, and the ease of use of the tool. Finally, the tool should grow with the learner, enabling increasingly advanced explorations and study of the subject. UCSF Chimera has been commonly used in educational settings, and ChimeraX has several features that we think will build on Chimera's use and offer new ways to enhance learning from the perspective of both educators and students. In the previous sections, we have addressed much of ChimeraX's utility. This section focuses on discoverability and learning materials available with ChimeraX.
A complete User Guide is included with each download, and the current version is shown at the ChimeraX website. The Help Viewer tool in ChimeraX is a built‐in HTML browser that can be used to view any web page. Importantly, it supports special links to execute ChimeraX commands. A user can view a tutorial and simply click links rather than type the commands into the command line. The Log also makes use of this mechanism, in a few different ways: (a) users may choose whether previous commands in the Log should be execution links or links to the command help pages; (b) the chains and nonstandard residues in a structure are logged automatically when the structure is opened, and clicking their identifiers selects them in the 3D structure, making it easy to survey its components while clicking on their descriptions shows their sequence or their RCSB Ligand Summary page, respectively; (c) for structures read from mmCIF format, the assemblies defined in the file are listed and can be generated with a single click. For example, a homotrimeric ion channel or an icosahedral capsid could be generated from the asymmetric unit.
Several tutorials on the website (https://www.rbvi.ucsf.edu/chimerax/tutorials.html) include click‐to‐execute links. The execution tag format is simple enough that anyone familiar with basic HTML can create new tutorials with executable links. This allows educators to create custom tutorials that combine learning the course material with learning about ChimeraX and how to view and analyze structures.
Other features to improve discoverability and learnability include the toolbars, which provide hints at functionality to be explored, the Log showing the commands as they are executed and linking to command documentation. Another key feature is the ability to save the Log, including optional thumbnails of the ChimeraX view.
Finally, it is important to consider the audience and situation, whether teaching students how to use the tool or demonstrating the science to a broader audience. ChimeraX provides several tools to assist in both of these spaces, including support for 3D printing such as the struts command to strengthen a model for 3D printing. For distance learning and interaction, ChimeraX offers nascent capabilities for multi‐user VR with the meeting command, which allows multiple participants to interact in the same virtual space, and for augmented reality presentations (described above), which can be used for classroom demonstrations as well as public outreach (see https://www.youtube.com/watch?v=dKNbRRRFhqY&t=7s for an example).
5. EMPOWERING DEVELOPERS
5.1. Toolshed
One of the major goals of ChimeraX is to make it easy for external developers both to extend its functionality and to distribute their extensions. For users, extensions are easy to discover and install from the integrated, web‐based, ChimeraX Toolshed. For developers, the toolshed additionally collects download statistics and enables users to be notified of new releases.
In the toolshed parlance, extensions are called bundles. Bundles can include a mix of commands, graphical interfaces (tools), toolbar buttons, data formats, and more. The developer annotates the bundle so ChimeraX can integrate all components into the native interface. In addition, annotations are also displayed in Toolshed entries to help users discover details such as bundle commands and tools.
5.2. Documentation and versioning
The steps to create a new bundle are documented in detail, including tutorials and the application programming interfaces (API). ChimeraX itself is organized as bundles, with each bundle defining its own supported API. These APIs are semantically versioned where changes in the major version number indicate incompatible API changes. Each bundle declares the versions of other bundle APIs on which it depends. The semantic versioning approach guarantees that the bundle will continue to work as long as there is no major version change. This applies to all bundles, whether developed by the ChimeraX team or externally, and helps minimize “bit rot,” where a bundle that worked suddenly breaks after a software update. With developer tutorials, API reference documentation, examples, and semantic versioning, researchers can experiment with using ChimeraX as a development platform as well as a visualization and analysis tool. Developers can also share their work by publishing bundles on the ChimeraX Toolshed via the web interface at https://cxtoolshed.rbvi.ucsf.edu.
While bundles may be developed independently, they can also interact with and leverage other bundles' functionality in many ways. Bundles can invoke other bundles either by running a command or even directly executing code from another bundle. They can also monitor for events triggered by activity in other bundles. For example, both the graphical and sequence display tools monitor the “selected atoms changed” event and update their visual representations accordingly. Reusing already available functionality simplifies bundle development by reducing both code duplication and development effort.
5.3. mmCIF support
Another goal of ChimeraX is to handle large structures efficiently, such as the human immunodeficiency virus 1 (HIV‐1) capsid (PDBid: 3j3q, 2,440,800 atoms). Large structures from the wwPDB are only available as mmCIF files. We have developed the readcif C++ library that takes advantage of the “stylized” format of the wwPDB's mmCIF files to quickly extract data from mmCIF and CIF files. 37 The reader complies with the CIF 1.1 standard for data files and can be easily extended to handle CIF dictionaries. The readcif code is open source and available separately (https://github.com/RBVI/readcif) so non‐ChimeraX developers can use it.
5.4. Source availability
We are in the process of migrating the ChimeraX source code to GitHub to improve access for outside developers and encourage community contributions to the code through pull requests.
6. CONCLUSIONS AND FUTURE DIRECTIONS
ChimeraX 1.0 combines new features with improvements in performance, visualization, and usability. However, ChimeraX is far from complete and there are several areas that we are working on for future releases.
Currently, much of ChimeraX functionality is driven by commands. We will continue to develop equivalent graphical interfaces, enhance our menus, and provide more panels for quick access to features and useful information about structures and imaging data.
We will also be adding new analytical tools. For example, segmentation analysis, cryoEM model refinement, and building with VR are on the roadmap, and ISOLDE will be adding support for model building. We are also working on integrating some of the web services supported by UCSF Chimera 38 as well as integrating new ones to replace services that have been discontinued. Another area with much potential for development is 3D medical image analysis and handling of DICOM (Digital Imaging and Communications in Medicine) files, including reading and presenting the associated metadata.
The improved development environment and the availability of the Toolshed mean that other developers will be enhancing ChimeraX in ways that we have not envisioned. The Toolshed will also make improvements to built‐in tools immediately available to users regardless of the timing of the release cycle. We hope to see more packages like ISOLDE that add significant functionality to ChimeraX.
Finally, the development of specific features in ChimeraX will be responsive to the needs of users and collaborators, so please give it a try and suggest changes! We are open to new ideas.
AUTHOR CONTRIBUTIONS
Eric Pettersen: Conceptualization; software; writing‐original draft; writing‐review and editing. Thomas Goddard: Conceptualization; software; visualization; writing‐original draft; writing‐review and editing. Conrad Huang: Conceptualization; software; writing‐original draft; writing‐review and editing. Elaine Meng: Conceptualization; visualization; writing‐original draft; writing‐review and editing. Gregory Couch: Conceptualization; software; writing‐original draft. Tristan Croll: Software; writing‐original draft; writing‐review and editing. John Morris: Project administration; software; writing‐original draft; writing‐review and editing. Thomas Ferrin: Conceptualization; funding acquisition; supervision; writing‐review and editing.
Supporting information
Video S1 Morph movie of bacterial ATP synthase motor rotating through three conformations observed by cryoEM (PDBid: 6n2y, 6n2z, 6n30). Movie.
ACKNOWLEDGMENTS
We thank Michael Schmid at the Stanford‐SLAC cryoEM Facility for sharing his virtual reality tomogram extraction techniques. We are indebted to the many ChimeraX users who have contributed new feature suggestions and bug reports. This study was supported by NIH National Institute of General Medical Sciences, Grant R01‐GM129325; Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases; Wellcome Trust, grant 209407/Z/17/Z.
Pettersen EF, Goddard TD, Huang CC, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science. 2021;30:70–82. 10.1002/pro.3943
Eric F. Pettersen and Thomas D. Goddard contributed equally to this work.
Funding information National Institute of Allergy and Infectious Diseases; National Institute of General Medical Sciences, Grant/Award Number: R01‐GM129325; Wellcome Trust, Grant/Award Number: 209407/Z/17/Z
REFERENCES
- 1. O'Donoghue SI, Goodsell DS, Frangakis AS, et al. Visualization of macromolecular structures. Nat Methods. 2010;7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baker ML, Hryc CF, Zhang Q, et al. Validated near‐atomic resolution structure of bacteriophage epsilon15 derived from cryo‐EM and modeling. Proc Natl Acad Sci USA. 2013;110:12301–12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baker ML, Zhang J, Ludtke SJ, Chiu W. Cryo‐EM of macromolecular assemblies at near‐atomic resolution. Nat Protoc. 2010;5:1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sali A, Berman HM, Schwede T, et al. Outcome of the first wwPDB hybrid/integrative methods task force workshop. Structure. 2015;23:1156–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mcwilliam H, Valentin F, Goujon M, et al. Web services at the European Bioinformatics Institute‐2009. Nucleic Acids Res. 2009;37(Web Server):W6–W10. 10.1093/nar/gkp302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhagat J, Tanoh F, Nzuobontane E, et al. BioCatalogue: a universal catalogue of web services for the life sciences. Nucleic Acids Res. 2010;38(Web Server):W689–W694. 10.1093/nar/gkq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rose PW, Beran B, Bi C, et al. The RCSB protein data bank: redesigned web site and web services. Nucleic Acids Res. 2011;39(Database):D392–D401. 10.1093/nar/gkq1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berman H, Henrick K, Nakamura H. Announcing the worldwide protein data Bank. Nat Struct Biol. 2003;10:980. [DOI] [PubMed] [Google Scholar]
- 9. Pieper U, Webb BM, Barkan DT, et al. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2011;39:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lawson CL, Patwardhan A, Baker ML, et al. EMDataBank unified data resource for 3DEM. Nucleic Acids Res. 2016;44:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Resource Coordinators NCBI. Database resources of the National Center for biotechnology information. Nucleic Acids Res. 2016;44:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marchler‐Bauer A, Derbyshire MK, Gonzales NR, et al. CDD: NCBI's conserved domain database. Nucleic Acids Res. 2015;43:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. UniProt Consortium . UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Semantic Versioning Specification 2.0.0 . Available from: https://semver.org/spec/v2.0.0.html.
- 15. Lotia S, Montojo J, Dong Y, Bader GD, Pico AR. Cytoscape app store. Bioinformatics. 2013;29:1350–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eclipse Marketplace . Available from: https://marketplace.eclipse.org/.
- 17. Waldrop MM. The chips are down for Moore's law. Nature. 2016;530:144–147. [DOI] [PubMed] [Google Scholar]
- 18. Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 20. Vallat B, Webb B, Westbrook JD, Sali A, Berman HM. Development of a prototype system for archiving integrative/hybrid structure models of biological macromolecules. Structure. 2018;26:894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abbott S, Iudin A, Korir PK, Somasundharam S, Patwardhan A. EMDB web resources. Curr Protoc Bioinformatics. 2018;61:5.10.1–5.10.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bolton EE, Chen J, Kim S, et al. PubChem3D: A new resource for scientists. J Chem. 2011;3:32–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goddard TD, Huang CC, Meng EC, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Croll TI. ISOLDE: A physically realistic environment for model building into low‐resolution electron‐density maps. Acta Cryst. 2018;D74:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wrapp D, De Vlieger D, Corbett KS, et al. Structural basis for potent neutralization of betacoronaviruses by single‐domain camelid antibodies. Cell. 2020;181:1004–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goddard TD, Brilliant AA, Skillman TL, et al. Molecular visualization on the holodeck. J Mol Biol. 2018;430:3982–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Y, Bryant SH, Cheng T, et al. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2017;45:D955–D963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. OpenSMILES . Available from: http://opensmiles.org/opensmiles.html.
- 30. Pei J, Grishin NV. AL2CO: Calculation of positional conservation in a protein sequence alignment. Bioinformatics. 2001;17:700–712. [DOI] [PubMed] [Google Scholar]
- 31. Pintilie G, Chiu W. Comparison of Segger and other methods for segmentation and rigid‐body docking of molecular components in cryo‐EM density maps. Biopolymers. 2012;97:742–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakane T, Kotecha A, Sente A, McMullan G, Masiulis S, Brown, Patricia M. G. E. , Grigoras IT, Malinauskaite L, Malinauskas T, Miehling J, Yu L, Karia D, Pechnikova EV, de Jong E, Keizer J, Bischoff M, McCormack J, Tiemeijer P, Hardwick SW, Chirgadze DY, Murshudov G, Aricescu AR, Scheres SHW (2020). Single‐particle cryo‐EM at atomic resolution. bioRxiv, 110189. http://dx.doi.org/10.1101/2020.05.22.110189. [DOI] [PMC free article] [PubMed]
- 33. Motta A, Berning M, Boergens KM, et al. Dense connectomic reconstruction in layer 4 of the somatosensory cortex. Science. 2019;366:eaay3134. [DOI] [PubMed] [Google Scholar]
- 34. Wietrzynski W, Schaffer M, Tegunov D, Albert S, Kanazawa A, Plitzko JM, Baumeister W, Engel BD. Charting the native architecture of Chlamydomonas thylakoid membranes with single‐molecule precision. Elife. 2020;9:e53740 10.7554/eLife.53740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Couch GS, Hendrix DK, Ferrin TE. Nucleic acid visualization with UCSF chimera. Nucleic Acids Res. 2006;34:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. ChimeraX Fast mmCIF Guidelines . Available from: https://www.rbvi.ucsf.edu/chimerax/docs/devel/bundles/mmcif/src/mmcif_guidelines.html.
- 38. Huang CC, Meng EC, Morris JH, Pettersen EF, Ferrin TE. Enhancing UCSF chimera through web services. Nucleic Acids Res. 2014;42:478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1 Morph movie of bacterial ATP synthase motor rotating through three conformations observed by cryoEM (PDBid: 6n2y, 6n2z, 6n30). Movie.