

Abstract
Afatinib (AFA) is a potent aniline-quinazoline derivative, approved by the Food and Drug Administration (FDA) in 2013, as a first-line treatment for metastatic Non-Small Cell Lung Cancer (NSCLC). However, its clinical application is highly limited by its poor solubility, and consequently low bioavailability. We hypothesize that loading of AFA into biodegradable PLGA nanoparticles for localized inhalational drug delivery will be instrumental in improving therapeutic outcomes in NSCLC patients. Formulated AFA nanoparticles (AFA-NP) were evaluated for physicochemical properties (particle size: 180.2±15.6nm, zeta potential: −23.1±0.2mV, % entrapment efficiency: 34.4±2.3%), formulation stability, in-vitro aerosol deposition behavior and anti-cancer efficacy. Stability studies revealed the physicochemical stability of AFA-NP. Moreover, AFA-NP exhibited excellent inhalable properties (mass median aerodynamic diameter (MMAD): 4.7±0.1μm; fine particle fraction (FPF): 77.8±4.3%), indicating efficient particle deposition in deep lung regions. With respect to in-vitro drug release, AFA-NP showed sustained drug release with cumulative release of 56.8±6.4% after 48 hours. Cytotoxic studies revealed that encapsulation of AFA into PLGA nanoparticles significantly enhanced its cytotoxic potential in KRAS-mutated NSCLC cell lines (A549, H460). Cellular uptake studies revealed enhanced internalization of coumarin loaded nanoparticles compared to plain coumarin in A549. In addition, 3D tumor spheroid studies demonstrated superior efficacy of AFA-NP in tumor penetration and growth inhibition. To conclude, we have established in-vitro efficacy of afatinib-loaded PLGA nanoparticles as inhalable NSCLC therapy, which will be of great significance when designing preclinical and clinical studies.
Keywords: Non-small cell lung cancer, PLGA, afatinib, inhalation, 3D cell culture, KRAS mutation
Graphical Abstract
Introduction
Nanoparticulate systems have gained significant interest over past several years due to their high efficiency in drug delivery through providing a carrier system at nanoscale, which is very well demonstrated by approval of over 50 nanomedicines and nanotechnology based medical products [1–4]. Nanoparticulate systems can be classified into inorganic, lipid-based and polymeric nanoparticles. Polylactide co-glycoside (PLGA) is considered to be the most prominent polymer used for fabrication of polymeric nanoparticles; due to its excellent biocompatibility and well-known biodegradability. Its flexible properties allow it to be an excellent choice for targeting Non-small cell lung cancer (NSCLC) [5]. While understanding the importance of nanoparticulate systems in treating various prevalent diseases, higher attention has been placed on improving delivery and efficacy of drugs in treating tumors in difficult to access body parts, including lung cancer [6,7]. Being a predominant malignancy, lung cancer it is responsible for approximately 1.38 million annual mortalities worldwide, with non-small cell lung cancer (NSCLC) being responsible for approximately 85% cases [8,9]. Given that most of the approved NSCLC therapies suffer from off-target effects and acquired resistance, there is a dire need to emphasize on the delivery approaches to ensure efficient localized delivery of nanoparticulate systems to tumor tissue [10].
Pulmonary drug delivery has gained importance in recent times due to its ability to deliver therapeutics to target tissues in the lungs [11]. Local pulmonary inhalational delivery of anti-cancer drug loaded nanoparticulate system offers substantial benefits in lung cancer treatment as the most current systemic treatments are not very effective or lead to severe toxic effects [11,12]. It is worth adding that inhalation therapy is an advantageous approach to deliver nanomedicines to the lung due to its potential to increase local drug accumulation, prolong pulmonary residence time, protect drug from degradation while minimizing drug resistance, and reduction of therapeutic dose and off-target systemic toxicities [13,14]. In addition, inhalable therapy may also increase drug penetration into the tumoral mass by maintaining a significantly higher drug concentration at the lung tumor site [14]. Several studies have evaluated the accumulation of drug-loaded nanocarriers in the lungs after pulmonary delivery and have reported improved efficacy of inhalable carriers in NSCLC [15]. For instance, gemcitabine-loaded gelatin nanocarriers [16], paclitaxel-loaded nanocomposite microparticles [17], quinacrine loaded bovine serum albumin modified cationic nanoparticles [15], pirfenidone-loaded liposomes [18] and lactoferrin/chondroitin-functionalized monoolein nanocomposites [19] etc. have reported capability of inhaled nanocarriers to provide localized drug delivery in NSCLC treatment. In addition, Abdelaziz et al [6], Mangal et al [20], Ahmed et al [21] and Anderson et al [22] have also highlighted the significance of nanocarriers in pulmonary delivery along with details of further investigations being carried out and their respective advantages and disadvantages. These nanocarrier systems differ in their capability to encapsulate hydrophilic, hydrophobic drugs; flexibility for surface modifications and ability to endure nebulization forces [6]. Furthermore, encapsulation of chemotherapeutic drugs in inhalable nanocarriers and their pulmonary delivery has been shown to demonstrate enhanced tumor accumulation and anti-tumor activity, as reported in several studies [23,24]. For instance, Chishti et al reported the efficacy of docetaxel nanoparticles administered through pulmonary route in treating non-small cell lung cancer in preclinical in-vivo studies [24]. In another study, inhalable self-assembled albumin nanoparticles of doxorubicin were evaluated for their anti-tumor efficacy in xenograft models by Choi et al and the authors reported deep lung deposition of the inhaled albumin nanoparticles [23]. Other therapeutic applications of inhalational therapy include treatment and management of asthma and chronic obstructive pulmonary disease (COPD), pulmonary fibrosis, inflammation and cystic fibrosis [13,25]. Hence, inhalational therapy remains the mainstay of clinical treatment in the care of pulmonary diseases; and sought after for lung cancer therapeutics. Benefits of this kind of drug delivery route can be achieved through selection of proper drug and carrier system while considering the physicochemical properties of drug and the formulation characteristics [26].
Afatinib (AFA) is an aniline-quinazoline derivative, potent, irreversible small-molecule tyrosine kinase inhibitor (TKI), initially approved by the US Food and Drug Administration (FDA) in 2013 as first-line treatment of late stage metastatic NSCLC and EGFR (Epidermal growth factor receptor) mutation positive NSCLC (FDA approval in 2018) patients [27–33]. AFA irreversibly inhibits ErbB family of tyrosine kinases; including epidermal growth factor receptor (EGFR), HER2 and HER4; thus leading to decrease in tumor cell proliferation and vascularization [34]. AFA has been reported for its effect in impairment of tumorigenesis in NSCLC with KRAS mutations, known to drive acquired drug resistance [35]. While AFA is currently approved at a 40 mg once-a-day oral dose, it is a free base, practically insoluble drug (0.0128 mg/ml). Clinical outcomes include reduced risk of both lung cancer progression and therapy failure compared to other tyrosine kinase inhibitors [36]. AFA’s poor water solubility may contribute to its low bioavailability and poor penetration into tumor core. Moreover, AFA is shown to have very high volume of distribution (4,500 liters) indicating high tissue distribution and accumulation at non-cancerous sites [37]. AFA being a relatively new drug for NSCLC treatment; there have been major investigations for technologies to provide a safer way for its delivery with reduced side effects (e.g. diarrhea, skin rashes, paronychia) and underlying toxicity (e.g. gastrointestinal, dermatological etc.) along with better drug targeting [38]. Moreover, it is of utmost importance to develop inhalable delivery systems for efficient delivery of afatinib to the tumor site, so as to limit its off-target tissue accumulation and toxicity. Hence, it is hypothesized that utilizing a biodegradable nanoparticulate delivery system combined with inhalable delivery system provides an efficient carrier system to deliver afatinib to lung tumors; and present an innovative approach for NSCLC treatment, especially with KRAS mutations, while ensuring excellent deep lung deposition. In the present study, AFA loaded PLGA nanoparticles were formulated and characterized for their physicochemical properties, in-vitro aerosol performance and anti-cancer efficacy through in-vitro and ex-vivo studies in different KRAS-mutated NSCLC cell lines.
Materials and Methods
Chemicals and Reagents
Afatinib base (AFA) was purchased from LC Laboratories (Woburn, MA, USA). Acid-terminated Poly (lactic-co-glycolic) (PLGA 50:50; 7–17 kDa, Resomer 502H) and poly vinyl alcohol (PVA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). A549 and H460 NSCLC cell lines were obtained from ATCC (Manassas, VA, USA) and were maintained in RPMI-1640 medium (Corning, NY, USA) supplemented with 10% FBS (Atlanta Biologicals, R&D Systems, Minneapolis, MN, USA), sodium pyruvate, penicillin-streptomycin (Corning, NY, USA) at 5% CO2/37°C. Healthy non-cancerous porcine kidney proximal tubule cell line LLC-PK1 was obtained from Dr. Yong Yu (St. John’s university, NY, USA) and maintained in BioWhittaker® Medium 199 (Lonza Walkersville, Inc., MD, USA) with 5% FBS and 10% penicillin-streptomycin. 3- (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dichloromethane (DCM), dimethyl sulfoxide (DMSO), coumarin-6, crystal violet dye, 16% paraformaldehyde (PFA) solution, HPLC grade methanol, acetonitrile (ACN) and water were purchased from Fisher Scientific (Hampton, NH, USA). Molecular biology kits and supplies were purchased from other commercial vendors which are listed at appropriate places throughout the manuscript. All other solvents including UPLC grade chemicals and solvents, unless it is otherwise specified, were purchased from Fisher Scientific (Hampton, NH, USA).
UPLC Method Development for Afatinib (AFA)
A reverse-phase liquid chromatography method was developed for quantifying AFA using Waters series Acquity UPLC (Waters, Milford, MA, USA). The column used was Acquity UPLC BEH C18 column (50 × 2.1mm, 1.7μm, 130 Å). Mobile phase consisted of acetonitrile and phosphate buffer (adjusted to pH 7.55 using 10mM of potassium hydroxide) at 60:40 ratio. The flow rate was 0.25 ml/min and detection wavelength was set at 254nm. Retention time for afatinib was found to be 0.731min with total run time of 2min. Data were collected and analyzed using the Empower 3.0 software.
Preparation of AFA-loaded PLGA Nanoparticles
AFA nanoparticles were prepared following a procedure previously stated in literature [39,40]. Briefly, AFA (1mg) was first dissolved in 1ml of acetonitrile which was then added to organic solution of PLGA (Resomer RG 502H; 60 mg PLGA/3ml of dichloromethane) and subjected to ultra-probe sonication for 2min (10 seconds on-off cycle, 40% amplitude; QSonica Q500, Newtown, CT, USA). This primary suspension was further probe sonicated for 4min (10 seconds on-off cycle, 40% amplitude) in 1% w/v aqueous polyvinyl alcohol (PVA) stabilizer solution. Prepared nanoparticles were then stirred overnight at room temperature for removal of organic solvent and were subsequently washed to remove excess PVA and unentrapped drug. AFA-loaded nanoparticles were separated from the aqueous suspension medium by ultracentrifugation at 1,000 rcf at 4°C for 3min; and then washed twice with water through centrifugation at 22,000 rcf for 15min (4°C). Obtained nanoparticles were re-dispersed with 1ml of milliQ water.
Characterization of AFA-loaded NPs
NP size and polydispersity index (PDI) were measured using dynamic light scattering technique (DLS) on Zetasizer Nano (Malvern Instruments, Malvern, UK). For sizing, sample was prepared by diluting NP formulation in distilled water, followed by bath sonication for 2 minutes. Analysis was carried out at a scattering angle of 90° and at a temperature of T = 298.15 K. Z-average diameter and polydispersity index were calculated using Malvern software. To determine % encapsulation efficiency (%EE) and drug loading capacity (DLC), nanoparticles were lysed and amount of free Afatinib was determined using UPLC as discussed above. Briefly, 20μl DCM, 10μl DMSO and 1950 μl of acetonitrile: water (50:50) were added 20μl of AFA-NP. It was then centrifuged at 22,000 rcf for 45 min. Obtained supernatant was analyzed using UPLC system. The peak area of AFA was recorded and the concentration of free AFA in supernatant was calculated from a standard curve. The %EE and %DLC were calculated using Equations 1 and 2 respectively. All samples were measured in triplicate.
| (Eq. 1) |
| (Eq. 2) |
In-vitro Drug Release Study
In-vitro drug release profile from AFA-NPs was determined in phosphate buffered saline (PBS; pH 7.4) with 1% polysorbate 80 for a period of 48 hours. Briefly, 100 μl of nanoparticle formulation was dispersed in 1 mL of PBS with 1% polysorbate 80 (pH 7.4) in 2 mL microcentrifuge tubes. Tubes were incubated at 37°C and 120 rpm. At each specific time point (15, 30 min, 1, 2, 3, 4, 6, 12 and 48 hours), the amount of drug released was determined by analyzing the collected supernatants by UPLC after centrifuging the samples for 15 minutes at 22,000 rcf. Drug release testing was performed in triplicate and data obtained are represented as mean±SD. Kinetics and pattern of drug release from nanoparticle formulations was determined by fitting the release data to distinct models: first-order and Higuchi model as reported earlier [41].
Solid State Characterization Studies
Differential Scanning Calorimetry (DSC) Studies
The thermograms for AFA, AFA-NP, blank NP and physical mixture of AFA and blank NP were generated using a DSC 6000 (PerkinElmer, Inc.; Waltham, MA, USA); equipped with an intra-cooler accessory. Briefly, a fixed amount of accurately weighed sample (1–5 mg) was sealed in an aluminum pan, analyzed over a temperature range (30°C - 210°C) and compared to a sealed empty aluminum pan maintained as reference. The heating rate was maintained at 10°C/min under a nitrogen purge (flow rate: 50 mL/min).
Powder X-ray Diffraction (PXRD) Studies
X-ray diffraction spectroscopy was performed using XRD-6000 (Shimadzu, Kyoto, Japan). The diffractometry is composed of a graphite monochromator (copper-Kα1; radiation wavelength 1.5418 Å, 40 kV and 30 mA). Samples were uniformly spread on a micro sample glass holder and then analyzed (range of 5 – 80°; scanning speed of 2° (2θ) /minute).
Fourier Transform Infrared Spectroscopy (FT-IR)
FT-IR spectrum of AFA, Blank NP and AFA-NP were obtained where solvent (DMSO) was used for the background correction and 10μL of the formulation was used for the detection. Different peaks in the plotted IR spectrum were then interpreted to detect the presence of distinct AFA groups.
Morphological and Quantitative Analysis
AFA-NP formulation was imaged using transmission electron microscopy (TEM) utilizing our previously published protocols [41]. Briefly, the Formvar® carbon-coated copper grids 400-mesh (Electron Microscopy Sciences, Hatfield, PA, USA) were made hydrophilic using glow discharge plasma treatment for 60s. About 5 μL of diluted nanoparticles sample was then added to the grid and allowed to adhere for 1 minute. The samples were negatively stained by adding 5 μL of uranyl acetate (1%) and excess was removed by using Whatman filter paper after 30s. This process was repeated twice and the sample was finally allowed to air dry. The grids were then imaged using FEI Tecnai G2 Spirit TWIN TEM (Hillsboro, OR, USA) operated at 120 kV voltage. Quantitative analysis from obtained TEM images was performed using ImageJ software version 1.52v. The images were processed using Fast Fourier Transform (FFT) parameters and nanoparticles’ sizes were recorded after adjusting the threshold to reduce the noise from background. Results were obtained as average particle size of fifty (50) particles in terms of diameter measured in nanometer.
Atomic Force Microscopy Analysis (AFM):
The morphological characterization of AFA-NP was performed using AFM for a qualitative confirmation of their morphology and surface properties. Imaging was carried out using the cantilever in air using Bruker Multimode AFM 8 (Billerica, MA, USA). Briefly, nanoparticles were suspended in water, and were drop-casted on to freshly cleaved mica surface and imaged with Scanasyst-Air probe in peak force mode.
Stability studies
Stability of the formulated AFA-NP was evaluated while storing the samples at temperatures of 4, 25 and 37°C for four weeks as reported previously [42]. Samples were withdrawn after week 1, 2, 3 and 4; diluted with water (100-fold) and analyzed for particle size, PDI and zeta potential using Malvern Zeta Sizer. Entrapment efficiency was determined by lysing the samples as described earlier using UPLC. All experiments were performed in triplicate.
In-vitro Aerosol Performance Lung Deposition Test:
According to previously published studies, Next Generation Impactor™ (M170 NGI: MSP Corporation, Shoreview, MN, USA) was used to evaluate in-vitro lung deposition [15]. The NGI was equipped with a stainless-steel induction port (USP throat adaptor) attachment and specialized stainless steel NGI™ gravimetric insert cups (NGI Model 170, MSP Corporation, Shoreview, MN, USA). Prior to operation, NGI plates were placed in refrigerator (4°C; 3 h) for pre-conditioning NGI to minimize solvent evaporation during the process. To test in-vitro lung deposition, 2ml AFA-NP formulation was then placed into a PARI LC PLUS® nebulizer cup of Pari FAST-NEB compressor system. (Boehringer Ingelheim, Inc. Ridgefield, CT, USA) attached to a customized rubber mouth piece connected to the NGI™. To produce a flow rate (15 L/min; 4 min), a Copley HCP5 vacuum pump (Copley Scientific, Nottingham, UK) was used and flow rate was adjusted using a Copley DFM 2000 flow meter (Copley Scientific, Nottingham, UK). After the run, samples were collected from every stage (1–8) as well as from throat and induction port by using ACN: Water (75:25) for rinsing. Samples were centrifuged for 45 min at 21,000 rcf, and lysed supernatant was then analyzed using previously established UPLC method. Drug content and effective deposition was calculated for emitted dose deposited at each stage. To give an indication of aerosolization performance; fraction of the emitted dose deposited in the NGI stages with dae<5.39μm (Fine particle fraction; FPF %) was calculated. To determine the spread of aerodynamic particle size distribution, mass median aerodynamic diameter (MMAD, dae<5.00μm) and the geometric standard deviation (GSD) were calculated. Experiment was done in triplicate (n=3) and data are represented as mean ± SD.
In-vitro Cellular Uptake Studies:
A hydrophobic dye, coumarin-6 (C-6) has been very well established as a surrogate for demonstrating intracellular uptake of encapsulated drugs using nanocarriers. Efficacy of nanocarriers in traversing through the cellular lipid bilayers enables enhanced therapeutic efficacy of drugs [43,44]. Thus, to determine the impact of AFA encapsulation into biodegradable PLGA nanoparticles on its intracellular uptake, (C-6) was used in preparation of nanoparticles instead of AFA. Coumarin loaded nanoparticles were prepared using exactly the same method as mentioned in section 2.3. In-vitro cellular uptake studies were carried out according to previously published method [45]. Briefly, A549 cells were seeded in tissue culture treated 8-well chambered glass slides (Eppendorf, Hauppauge, NY, USA) at a seeding density of 10,000 cells per chamber, and then left overnight so that cells would adhere. Next day, cells were treated with either coumarin or coumarin-6 loaded nanoparticles at 1μg/mL concentration. At each time interval (1- and 3-hours), cells were washed twice with ice-cold PBS (pH 7.4) after which 4% paraformaldehyde (PFA) was added for 10min for fixation. Fixed cells were again washed twice with ice-cold PBS. Then, the chamber was removed and 20μL of Vectashield® hardset mountant (H1500, Vector Laboratories, Burlingame, CA) was placed on a glass slide dropwise followed by placing a cover glass and appropriate drying of slides. After hardening of mounting medium overnight at 4°C, the slides were imaged using EVOS-FL fluorescence microscope with 20X objective (Thermo Scientific, Waltham, MA, USA).
In-vitro Cytotoxicity Assay
In order to determine anti-proliferative activity of AFA-NP in comparison to plain AFA, an MTT assay was adopted as reported in literature [46]. In brief, immortalized NSCLC cells (A549 and H460) were seeded into tissue culture treated 96-well plates (Eppendorf, Hauppauge, NY, USA) at a seeding density of 2,500 cells/well, and were incubated overnight for adherence at 37°C/5% CO2. Next day, different concentrations of plain AFA and AFA-NP (0.098–25μM) were prepared in the culture media and were added to the adhered cells while having culture media without any treatment as control. Corresponding volumes of AFA-NPs were calculated based on the drug entrapment efficiency. After 48 hours of incubation, media was removed from each well followed by addition of MTT solution (1mg/ml in PBS pH 7.4) and 2-hour incubation so that formazan crystals would be formed. 100μL of dimethyl sulfoxide (DMSO) was added to each well after aspirating MTT solution from each well to dissolve the formed formazan crystals. Plates were then left on plate shaker for 30min, and absorbance was measured using a multi-modal plate-reader (Tecan Spark 10M; Tecan, Männedorf, Switzerland) at 570nm. Cytotoxicity studies were also performed following same protocol on healthy non-cancerous porcine kidney proximal tubule cell line LLC-PK1 to further evaluate the safety of drug-free nanoparticles using equivalent amounts of Blank NP at concentrations of 0.78 and 3.125μM with 48 hours of incubation period.
3D-Spheroid Cell Culture Study
Due to the complexity of tumor physiology, a 3D spheroid culture technique was proposed in order to mimic tumor environment giving a more realistic insight on drug activity. As previously reported, a 3D cell-based model was developed [15,46]. In brief, A549 cells were first seeded in Corning® ultralow attachment spheroid 96-well plates (Corning, NY, USA) at a density of 2.0×103 cells/well, and were incubated overnight at 37°C/5% CO2. Then, spheroids were grown for 3 days before treatment for obtaining a substantial tumoral core. The cells were subjected to two kinds of treatment regimens; either single dose or multiple dose. As for single-dose treatment, cells were given treatments (AFA or AFA-NP) only once and media was then replaced on respective days (day 1, 3, 6, 9,12 and 15). While in case of multiple dose study, cells were treated with either AFA or AFA-NP (1 and 3μM) on respective days (day 1, 3, 6, 9, 12 and 15). In both cases, only half of the media volume (100μL) was replaced with fresh media or treatments in order to prevent disturbance of the already developed spheroids or to eliminate chances spheroid aspiration hat may take place on complete removal of the media. Using inverted microscope (LAXCO, Mill Creek, WA, USA), spheroids were imaged at specified time intervals (n=5–6) at 10X magnification. Spheroid diameters were quantified using ImageJ software (Version 1.44) and spheroid volumes were calculated.
Live-Dead Cell Assay:
Live/Dead cell assay was performed using viability/cytotoxicity assay kit for Animal Live & Dead Cells (Biotium, Fremont, CA, USA), optimized for use in human cells. According to manufacturer’s protocol, live-dead cell study was performed on treated spheroids on day 15th of both single and multiple dosing in the therapeutic model. Briefly, after complete removal of media from the wells; 100 μL of staining solution (2μM calcein AM/4μM Ethidium homodimer III (EthD-III)) was added to treated spheroids; in order to give the green/red fluorescent staining for alive and dead cells respectively. Plate was then incubated at room temperature for 45 min in dark. Images were captured using (EVOS-FL, Thermo Fisher Scientific, Waltham, MA, USA) and ImageJ software was then used to quantify the mean red and green fluorescence intensity (RFP), signifying presence of dead and alive cells in core of spheroidal mass.
Data Analysis and Statistical Evaluation:
All data were addressed as mean±SD or SEM, with n=3 unless otherwise mentioned. Three trials of cytotoxicity studies were performed for each control or treatment group with n=6 for each trial. All data were evaluated by unpaired student’s t-test or one-way ANOVA followed by Tukey’s multiple comparisons test, using GraphPad Prism software (Version 6.0 for Windows, GraphPad Software, CA, USA). A p value of <0.05 was considered statistically significant and was presented in data figures as a single asterisk (*). However, some studies have demonstrated a smaller p-value of 0.01or less, which is included respectively.
Results and Discussion
UPLC Method Development for Afatinib (AFA)
For determination of AFA concentration, a precise and reproducible UPLC method was developed with a total run time of 2 min. The developed method exhibited excellent linearity (r2=1) for AFA concentration between 0.32–100 μg/ml (Equation. 3). Calibration curve for determination of AFA is shown in Fig. S1a. As shown in the UPLC chromatogram (Fig. S1b), a sharp peak with good resolution for AFA was eluted with retention time of 0.731min.
| (Eq. 3) |
Where Y = area under the curve; and X = concentration of afatinib (μg/ml)
Preparation and Characterization of AFA-NPs
As can be seen from the histogram presented in Fig. 1a, the formulated nanoparticles were found to be <200 nm in size with very monodispersed size distribution (represented by single size peak on the histogram). Mean particle size, polydispersity index and zeta potential of AFA-NPs were determined using dynamic light scattering (DLS) on Malvern Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK). For formulated AFA-NP, particle size was 180.2±15.6nm, polydispersity index was 0.112±0.004 and zeta potential was found to be −23.1±0.2mV. % Drug entrapment efficiency was determined to be 34.4±2.3%, along with actual % drug loading to be 0.58±0.05% as shown in Fig. 1b. Emulsification is considered to be one of the most significant steps in nanoparticle fabrication, which could lead to formation of large and polydispersed particle aggregates as a result of insufficient dispersion of phases. Therefore, a lot of investigation has been done on effects of experimental parameters including stabilizer species, stabilizer concentration and pH of aqueous phase, on particle size and drug entrapment efficiency [47,48]. It was previously reported that size of polymeric nanoparticles would strongly affect their rate of intracellular uptake inside the tumor microenvironment i.e. higher cellular uptake for smaller particle size [49,50]. Different pathways were suggested to be instrumental in cellular internalization of polymeric nanoparticles [44,45]. Sub 200nm sized nanoparticles most likely get internalized through receptor-mediated endocytosis. However bigger nanoparticles (>200 nm) are shown to get phagocytosed for gaining entry into the cells [51,52]. Carteria et al. studied the mechanism for internalization of rhodamine PLGA nanoparticles inside the cells using different body organ models and tracking their intracellular fate [53]. Analysis of images suggested that mechanism of cellular internalization of formulated PLGA nanoparticles to be through endocytosis.
Fig. 1:
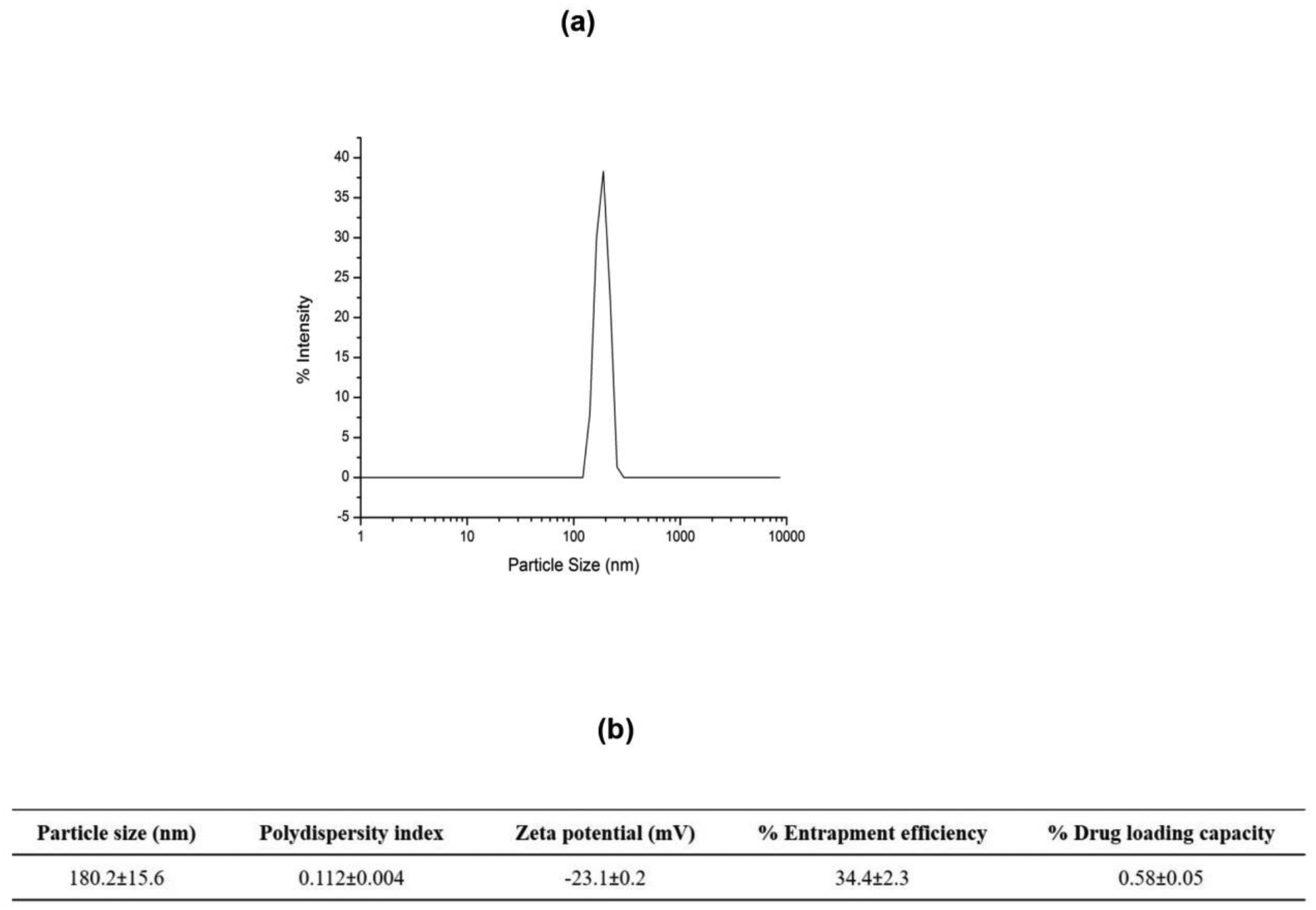
a. Particle size distribution histogram of afatinib loaded PLGA nanoparticles. b. Characterization of particle size, polydispersity index, zeta potential, % entrapment efficiency and % drug loading for afatinib loaded PLGA nanoparticle formulation. Data represent mean±SD (n=3).
In-vitro Drug Release from AFA-NPs
In order to determine drug release pattern, in-vitro drug release was measured in phosphate buffer saline (PBS) (pH 7.4, 37°C) mimicking environment of physiological fluid. A burst release of encapsulated AFA at 24.3±4.6% was observed within first 30 minutes followed by sustained cumulative drug release of 39.3±2.3% and 56.8±6.4% after 24 and 48 hours respectively (Fig. 2a). The drug release data were further analyzed for a perfect fit to either the first-order or Higuchi drug release models in order to predict best release kinetic and release patterns (Figs. 2b&c). In order to understand the release mechanism, release equations were determined from each model. The optimized equation was then chosen based on the value of correlation coefficient (r2). Release data were found to be the best fitting to first order kinetic model (r2 =0.86) (Fig. 2d). Several studies from different research groups have previously reported similar release patterns from drug-loaded PLGA nanoparticles [54–56]. In one such study, Yadav and Sawant [56] developed etoposide-loaded biodegradable nanoparticles, and reported that the release of etoposide from the PLGA NP followed first order kinetics. Matsuura et al [57] prepared PLGA nanoparticles loaded with itraconazole or miconazole, with drug release pattern best fitting the first order kinetics model predicted by higher correlation regression coefficient for first order compared to other release models. The burst release observed from AFA-NP may be due to rapid release of the drug adsorbed on or close to nanoparticles surface [58]. While at later stages, a decrease in drug release rate occurs, which is attributed to slow drug diffusion and gradual PLGA polymer degradation with subsequent release of entrapped drug from inside the polymeric core. Therefore, drug release from PLGA nanoparticles is assumed to be not dominated by a single mechanism but by a combination of mechanisms [59].
Fig. 2:
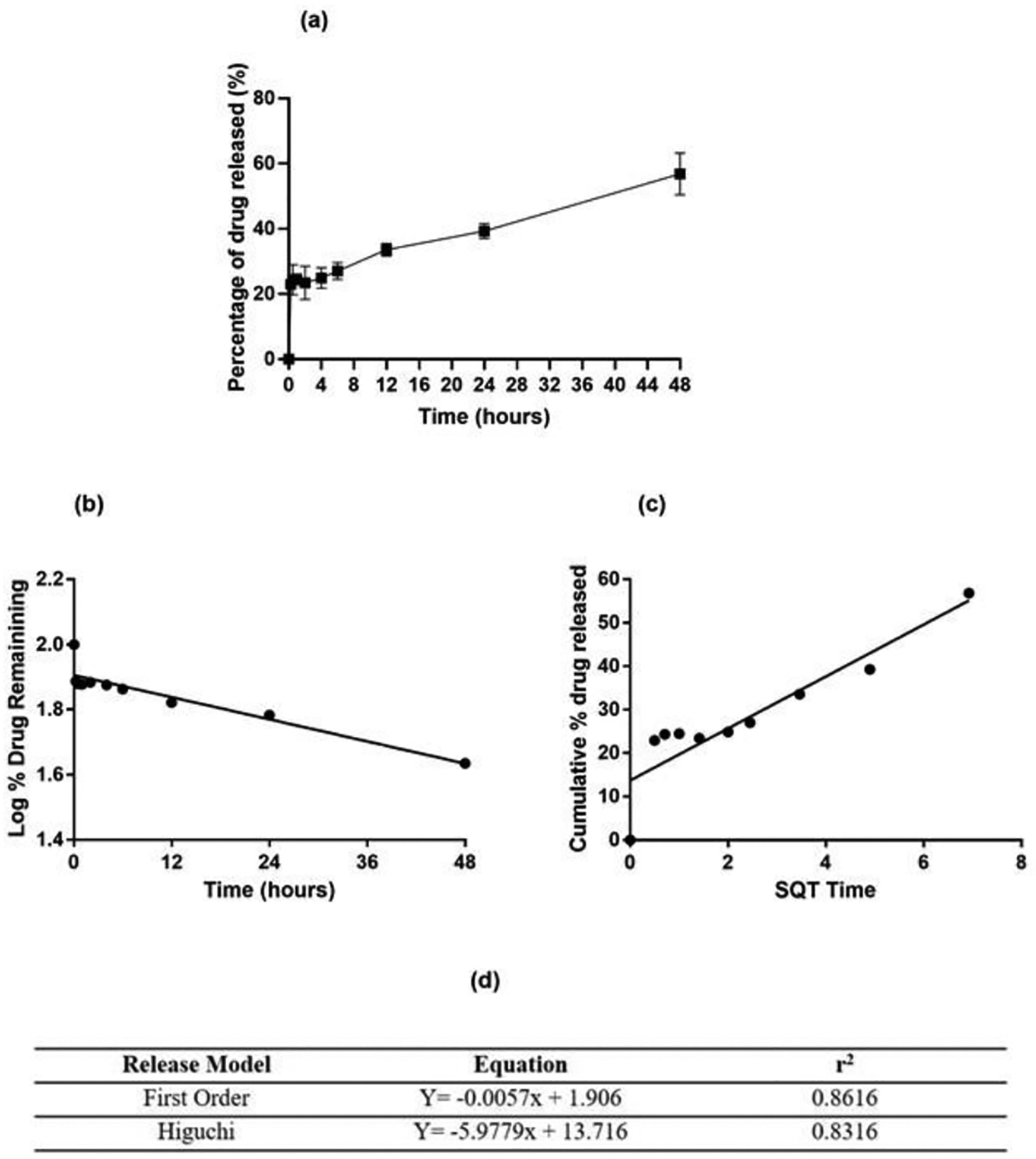
In-vitro drug release studies: a. % Cumulative in-vitro drug release profile of afatinib loaded PLGA nanoparticles, b. First order release kinetics and c. Higuchi release kinetics from afatinib loaded PLGA nanoparticles. Data represent mean±SD (n = 3). d. Release curve fitting details of AFA-NP using various models.
Solid State Characterization of AFA-NP
Differential Scanning Calorimetry (DSC)
DSC studies were performed to understand melting and crystallization behavior of AFA when present in different forms such as NP encapsulated AFA, physical mixture with blank NPs, as compared to control. As can be seen in Fig. 3a, thermogram of AFA showed sharp endothermic peaks at 95°C and 125°C due to its melting transition [60,61]. Absence of a sharp peak in AFA-NP indicated complete drug encapsulation in the nanoparticle formulation core and suggested a consistent result with that of XRD, discussed later.
Fig. 3:
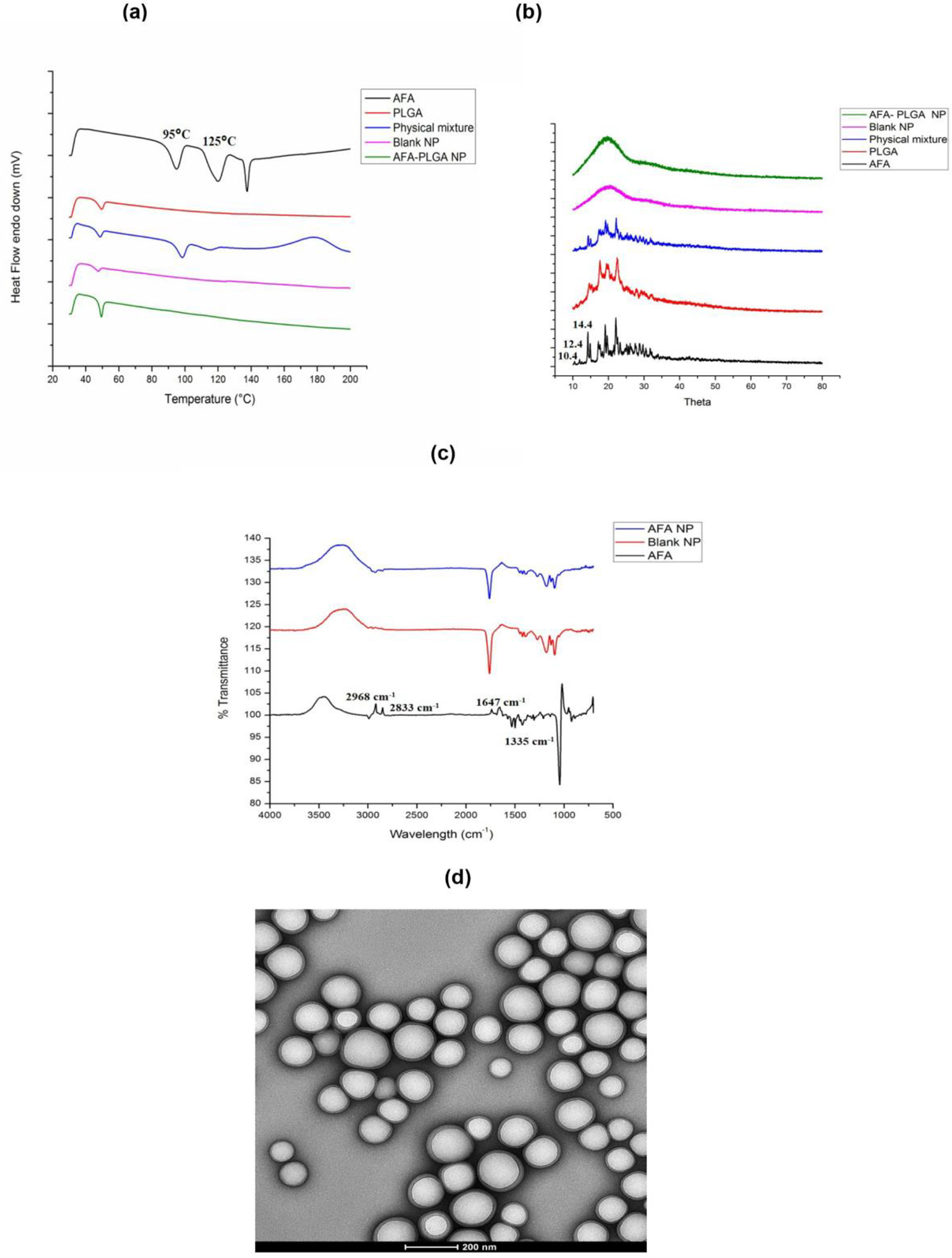
a. Thermograms of afatinib, PLGA, physical mixture of afatinib (AFA), blank NP and afatinib loaded PLGA nanoparticles (AFA-NP). b. XRD patterns of afatinib, PLGA, physical mixture of afatinib (AFA), blank NP and afatinib loaded PLGA nanoparticles (AFA-NP). c. FT-IR spectra of AFA, blank NP and AFA-NP. d. Transmission electron microscopy (TEM) images of AFA-NP reveal the morphology of nanoparticles. Magnification 42kX. The scale bar represents 200nm.
Powder X-ray Diffraction (PXRD)
Due to its crystalline nature, AFA showed distinct peaks at 2θ values of 5.2, 10.4, 12.4, 14.4 ±0.2 degrees two-theta in XRD spectra whereas there were no AFA peaks present in AFA-NP indicating encapsulation of drug inside the nanoparticles as shown in Fig. 3b. Results were found to be consistent with previous studies where afatinib free base have resulted in similar PXRD pattern [62]. Physical mixture of Blank NP and AFA also exhibited peaks inferring the crystalline nature of drug.
Fourier Transform Infrared Spectroscopy (FT-IR)
The FT-IR spectra of AFA, blank NP and AFA-NP are presented in Fig. 3c. The IR spectrum of AFA showed characteristic peaks at about 2,968 and 2,833cm−1 due to C-H stretching of CH3 group, 1,647 cm−1 due to C=O stretching of the amide group, and 1,335cm−1 due to C-N stretching of the aromatic amine group. A decrease and slight shift in the characteristic AFA peaks around 2,968 and 2,833cm−1 in case of AFA-NP with no peaks detected in the same region in case of blank NP, may be attributed to good entrapment of AFA inside the PLGA matrix of formulated nanoparticles [63].
Morphological and Quantitative Analysis
A representative TEM image presented in Fig. 3d indicated that the nanoparticles had uniform spherical shape with a smooth surface. No aggregation of nanoparticles was observed during TEM analysis which gives an indication of the uniform dispersion of nanoparticles in formulation. TEM images were also quantified for particle size which was found to be 118.7±24.0nm, and well in agreement with DLS size measurements shown in Fig. 1.
Atomic Force Microscopy Analysis (AFM)
Particle size of AFA-NP nanoparticles was determined by atomic force microscopy and the respective image is shown in Fig. S1c. Thus, morphology of AFA NP has been confirmed qualitatively using AFM. Results from the AFM correspond to the distribution acquired from DLS measurements and also narrow size distribution of AFA-NP is confirmed as seen in Fig. 3e.
Stability Studies
Stability is a major concern while dealing with nanosized drug delivery systems due to emulsion instability and particle aggregation [64]. Due to smaller particle size, there is a possibility of NP aggregation with AFA-NP formulation. Hence, studies were performed to determine stability of AFA nanoparticles at different storage conditions. As shown in Fig. S2, stability analysis data revealed that AFA-NP formulation was stable at 4° and 25°C, with no significant changes in particle size (Fig. S2a) or zeta potential (Fig. S2b) for up to 4 weeks’ storage. It can be postulated that negative zeta potential of AFA-NP prevents the particles from coalescing and aggregating [65]. There was no detrimental effect of temperature and storage time on entrapment efficiency either (Fig. S2c). Hence, the formulations were found to retain their physicochemical properties during their storage at different temperatures of 4° and 25°C over a period of 4 weeks. Formulations were also stored at 37°C to expose them for temperature excursions, and were found to exhibit increased particle size which could be due to coalescence. However, regular labeled storage conditions do not include 37°C. Ahmed et al studied the stability of a polyphenolic loaded PLGA nanoparticles at 4°C and attributed the good stability of the formulated nanoparticles to presence of a PVA coating barrier around the polymeric nanoparticles [66].
In-vitro Aerosol Performance and Lung Deposition
Pulmonary route of administration is well known for its ability to deliver therapeutics locally to site of action in deep lungs but is challenged by major obstacles like poor control over deposition rates and site of inhaled molecules for efficacious delivery at the same time [26]. NPs exhibiting optimal in-vitro aerosol lung deposition can be administered via inhalation, facilitating local delivery to the deep lungs, thus resulting in reduced exposure to other organs and reduced adverse events. Nebulizers can deliver the formulations as small droplets which will be deposited in the lung airways based on their aerodynamic properties like MMAD and FPF [67]. These aerodynamic properties of nebulized particles are used to reflect the in-vivo deposition profile in airways and alveolar part of deep lung regions. Deposition of inhaled particles in the respiratory airways depends on several parameters related to the physical properties of particles along with many physiological and anatomical factors of the respiratory system [68]. To assess the respirability of AFA nanoparticles; dose recovered from nebulizer cup and NGI stages, FPF (%), MMAD and GSD were calculated. Aerosol deposition pattern is presented graphically in Fig. 4a which refers to the % aerosol deposition at each stage of NGI, aiding in determination of aerosol dispersion performance. Relation between recovered cumulative mass and effective cut-off diameter is shown in Fig. 4b which represents the % cumulative particles deposited at each NGI stage Following aerosolization experiments, fine particle fraction (FPF) was found to be 77.8±4.3%, which suggests good aerosolization performance. In addition, MMAD and GSD values being 4.3±0.2μm, and 2.2±0.4 respectively as shown in Fig. 4c, suggest that majority of the emitted dose could be effectively delivered to targeted respirable region inside the lungs. Particle size, surface morphology and mass median aerodynamic diameter (MMAD) of inhaled therapeutics determine their fate and the site of deposition in deep lungs [69]. MMAD within the range of 2–5μm is essential for deep lung accumulation of particulate therapeutics, which is otherwise difficult to maintain due to inherent differences in particles’ physicochemical properties [70,71]. From the results obtained, the efficacy of formulated AFA-NPs for inhalable drug delivery of AFA could be established and it can be ensured that developed nanoparticles would be able to provide a promising strategy to overcome the physiological barriers involved in respiratory delivery of anticancer therapeutics.
Fig. 4:
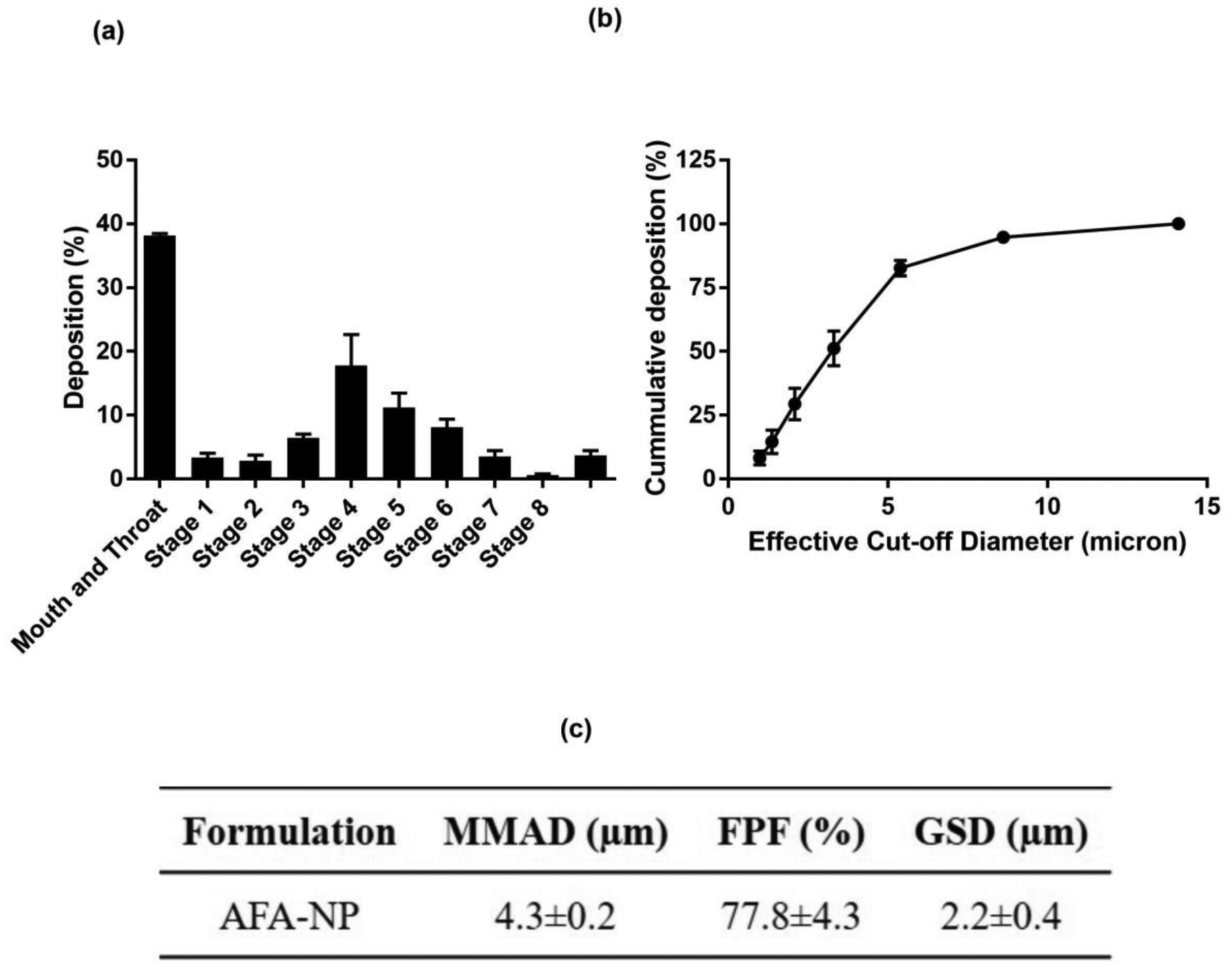
In-vitro deposition profile of AFA-NP. a. Aerosol dispersion performance as % deposited on each stage of the Next Generation Impactor™ (NGI™) for AFA-NP. For Q = 15 L/minute for 4min. The effective cutoff diameters (D50) for each impaction stage are as follows: stage 1 (14.1 μm), stage 2 (8.61 μm), stage 3 (5.39 μm), stage 4 (3.3 μm), stage 5 (2.08 μm), stage 6 (1.36 μm), and stage 7 (0.98 μm). (n=3, average ± SD). b. Cumulative % deposition plot representing cumulative % of particles deposited at each stage. c. Aerosolization properties of AFA-NP. Data represent mean±SD (n=3).
In-vitro Cellular Uptake in A549 NSCLC Cell Line
In-vitro cellular uptake studies were performed to evaluate efficiency of PLGA nanoparticles’ uptake into A549 cells (adenocarcinoma human alveolar basal epithelial cells). A549 cells were used for the cellular uptake studies to ensure the internalization of nanoparticles into these NSCLC cells for exhibiting their cytotoxic effect [72]. Based on recent studies, fluorescent dye (Coumarin-6) loaded polymeric nanoparticles were formulated and utilized in the current study to evaluate in-vitro cellular uptake [41]. It can be clearly seen from fluorescence images of cells incubated with Coumarin-6 loaded nanoparticles after 1 and 3 hours, that coumarin-loaded NPs showed significantly higher intracellular uptake as compared to plain coumarin (Fig. 5). It can be assumed that higher cellular uptake for drug-loaded NPs will ultimately lead to higher intracellular accumulation of afatinib, and thus sustained release of drug inside the cells, resulting in sustained and higher cytotoxicity as compared to plain afatinib. The proposed mechanism for internalization of negatively charged polymeric nanoparticles inside the cells could be through endocytosis [49]. When negatively charged particles get adsorbed on cell surface’s positive binding sites, endocytosis is known to be a favored pathway for nanoparticle internalization [73]. This observation comes in accordance with previously published studies reporting higher cellular uptake/internalization of drug loaded PLGA nanoparticles in A549 cells [45]. Tahara et al reported that A549 cellular uptake of PLGA NP increased when the particle size was controlled to be below the sub-micron region [74].
Fig. 5:
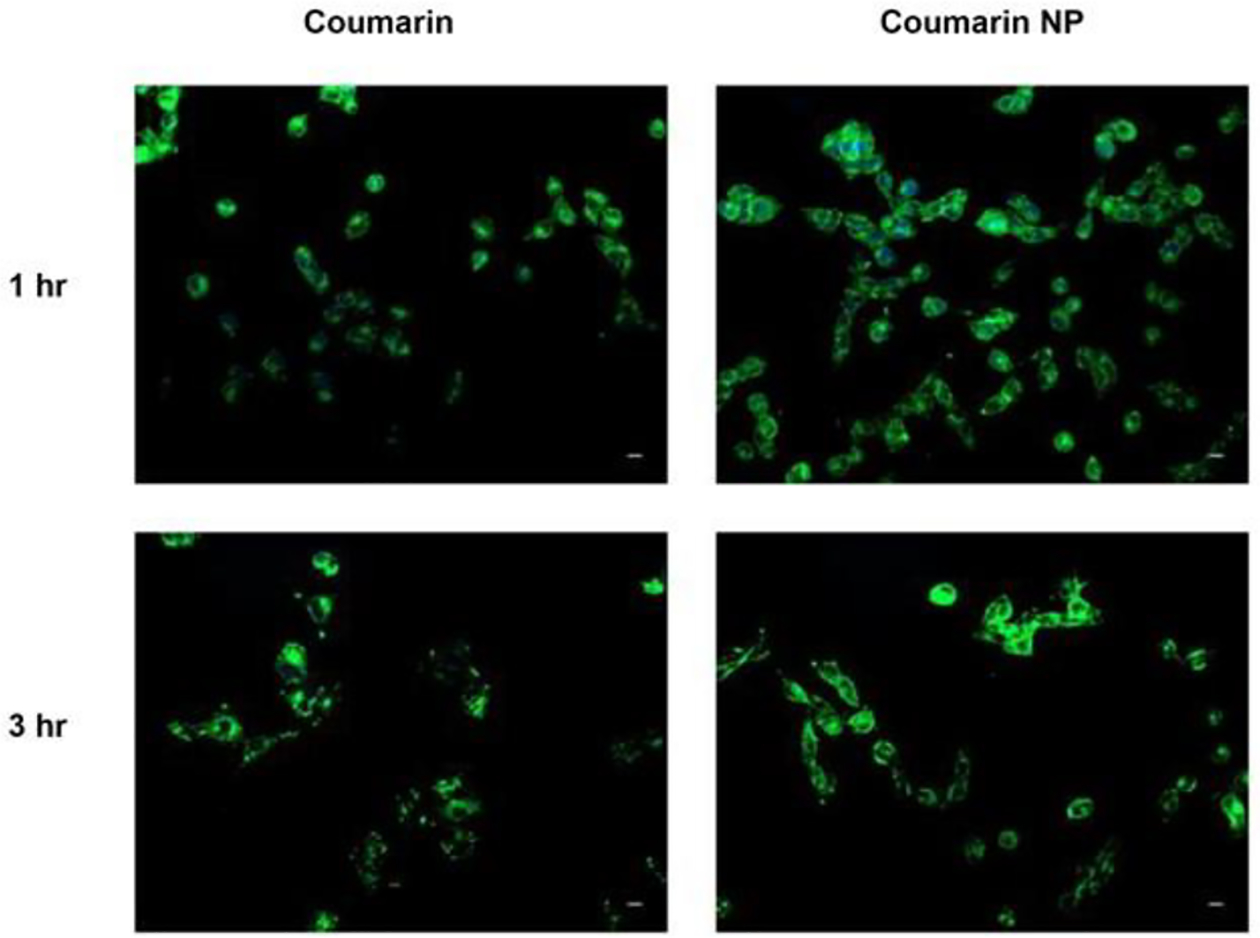
In-vitro cellular uptake of coumarin-6 loaded PLGA nanoparticles in A549 cells at two different time points; 1 and 3 hours. Coumarin-6 plain solution is used as control. Nuclei are stained blue (DAPI) and Coumarin NPs are green. Scale bar 100 μm. Representative images are presented from experiments run in triplicate (n=3).
In-vitro Cytotoxicity Studies
To evaluate improved therapeutic efficacy of AFA by encapsulation in PLGA nanoparticles against non-small cell lung cancer (NSCLC), in-vitro cellular cytotoxicity of prepared NPs in two different lung cancer cells (A549 and H460) was determined. Graphical plots of % cell viability for formulated AFA-NPs against plain AFA in two cell lines are shown in Fig. 6 and tabulated results for IC50 are represented in Table 1. IC50 was found to be 3.8±1.1 and 2.7±0.4μM for plain AFA; and 1.8±0.3 and 1.2±0.4μM for AFA-NP in A549 (Fig. 6a) and H460 (Fig. 6b) cell lines respectively with significantly improved cytotoxic effect of AFA-NP compared to AFA in A549 (p<0.01) (Table 1). Although A549 cell line is known to harbor wild type EGFR and HER2, activating K-ras G12s point mutation may affect activity of EGFR tyrosine kinase inhibitors (TKIs) [75,76]. On the contrary, H460 cells express wild-type of EGFR but carry KRAS mutation (KRASQ61H) [77,78]. As can be seen from both Figs. 6a and 6b, and also from Table 1, it was observed that IC50 value of AFA against A549 (Fig. 6a) and H460 (Fig. 6b) cells was lower in case of AFA-NPs compared to plain AFA which reflects improved anti-proliferative activity of AFA in two cell lines. This could be attributed to what has earlier been reported in literature that; although free drug enters the cells by passive diffusion, nanoparticles could be internalized by endocytosis [79,80]. This mechanism of internalization could also occur in tumor cells, resulting in higher intracellular AFA concentration with nanoparticles as compared to plain drug [79]. To establish safety of nanoparticle formulations to healthy tissues, blank (AFA free) PLGA nanoparticles were tested for toxicity in normal porcine kidney cells, LLC-PK1, derived from proximal tubules (Fig. 6c). As can be seen, blank nanoparticles without any AFA exhibited negligible cytotoxicity against LLC-PK1 cell line after 48 hours at AFA equivalent concentrations of 0.78 and 3.125μM with % cell viabilities of 98.8±13.6% and 87.3±9.7% respectively (Fig. 6c). These results also suggest that the prepared materials and nanoparticles were basically nontoxic, highly biocompatible and safe. These observations come in congruence with published data of cytotoxicity studies of PLGA loaded drugs on same studied cell lines [15,45]. Unal et al prepared paclitaxel loaded PLGA nanoparticles and studied their cytotoxicity on A549 cells, and demonstrated significantly improved cytotoxicity in comparison to plain drug solution [81]. Merlin et al prepared paclitaxel-PLGA nanoparticles and to study its anticancer efficacy in NCI-H460 cells in-vitro [82]. Results showed increased cytotoxicity of paclitaxel-PLGA nanoparticles compared to plain paclitaxel.
Fig. 6:
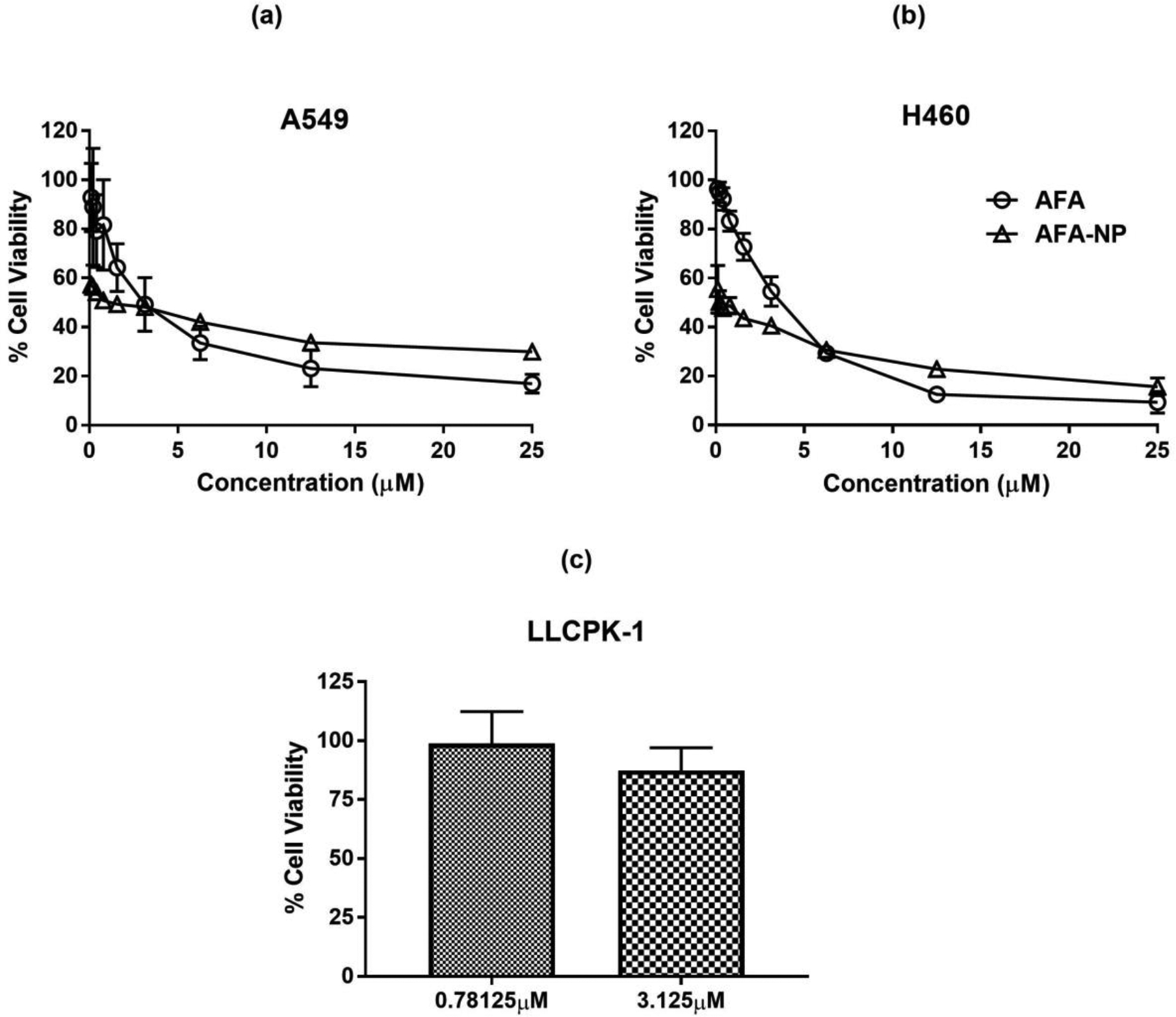
Cytotoxic effects on different NSCLC cell lines a. A549 and, b. H460; after treatments with AFA and AFA NP. AFA-NP was found to enhance cytotoxicity effects compared to AFA. Cells without treatment were considered as control (100%). Data represent mean±SD (n=6) of 3 individual experiments. c. Safety studies: Cytotoxicity studies on LLCPK-1 cell line after treatment with Blank NP. Data represent mean±SD (n=6).
Table 1:
Comparison of IC50 for AFA and AFA-NP in different cell lines. All data represent mean ± SD (n=6) of 3 individual experiments.
| Cell line | IC50 (μM) | |
|---|---|---|
| AFA | AFA-NP | |
| H460 | 3.8 ± 1.1 | 1.8 ± 0.3 |
| A549 | 2.7 ± 0.4 | 1.2 ± 0.4 |
3D-Spheroid Study for Tumor Penetrative Anticancer Efficacy
As described in Methods section, 3D-spheroid cell culture study (3D-SCC) was designed to better predict the physiological interaction of AFA-loaded NPs with tumoral mas. In case of solid tumors, the monolayer cell culture models do not accurately mimic tumor structure, resistance to drugs and drugs’ poor penetrability due to tumor microenvironment [83]. This could lead to inaccurate evaluation of actual performance of anticancer drugs within actual biological environment. These aspects are also responsible for inaccurate assessment of biological performance of nanotherapeutics [46]. Hence, the development of a 3D cell culture model has been thoroughly investigated to facilitate screening of various anticancer drugs. These cultures (tumor spheroids) combine the ability to mimic the 3D structure of malignant tissue accurately in addition to being relevant to cancer microenvironment to which the tumor cells are exposed (e.g., proliferation, survival, multi-drug resistance and gene expression heterogeneity) [84,85]. These models promote aggregation of cells which assists in constructing an in-vitro model with infrastructure of an in-vivo tumor [86]. As 3D in-vitro models allow creation of tumoral mass in three-dimensional space while mimicking the in-vivo model, they are able to recapitulate in-vivo biology. Moreover, these models promote aggregation of cells which assists in constructing an in-vitro model with infrastructure of in-vivo tumor. Although in-vivo experiments were not performed for the current study, few recent studies have compared in-vitro 3D spheroids with in-vivo efficacy, and have demonstrated to have similar outcomes in both the models [86,87]. Moreover, in-vitro tumor models serve as low-cost screening platforms in cancer research while in-vivo models present several challenges in quantitative mechanistic data extraction [88]. A 3D in-vitro model was developed successfully by using Corning ultra-low attachment spheroid plates as reported by Vaidya et al [89], in which cells are promoted to form a spheroidal mass under favorable incubation conditions. This in turn mimics the tumor mass physiology and gives better insight for drug’s cytotoxic effectiveness, compared to the cytotoxicity study, performed on 2D-cell monolayer.
For spheroid study, A549 cell line were chosen as these cells have strong intercellular communication, and could form spheroid masses from day 1 on culturing. After plating, the tumor spheroids were grown for 3 days before initiating the treatments. This study was performed in two separate treatment regimens; single and multiple. For each treatment regimen, two different concentrations (1 and 3μM) of both AFA and AFA-loaded NPs were used. Images were captured using inverted microscope (Laxco, Mill Creek, WA, USA) on respective days and were analyzed using the ImageJ software Version 1.44 (National Institute of Health, Bethesda, MD, USA). Spheroid images and a plot for spheroid volume comparisons are represented in Figs. 7a&7b for single dose; and Figs. S3a & S3b for multiple dose.
Fig. 7: 3D-Spheroid Study: Single dosing regimen:
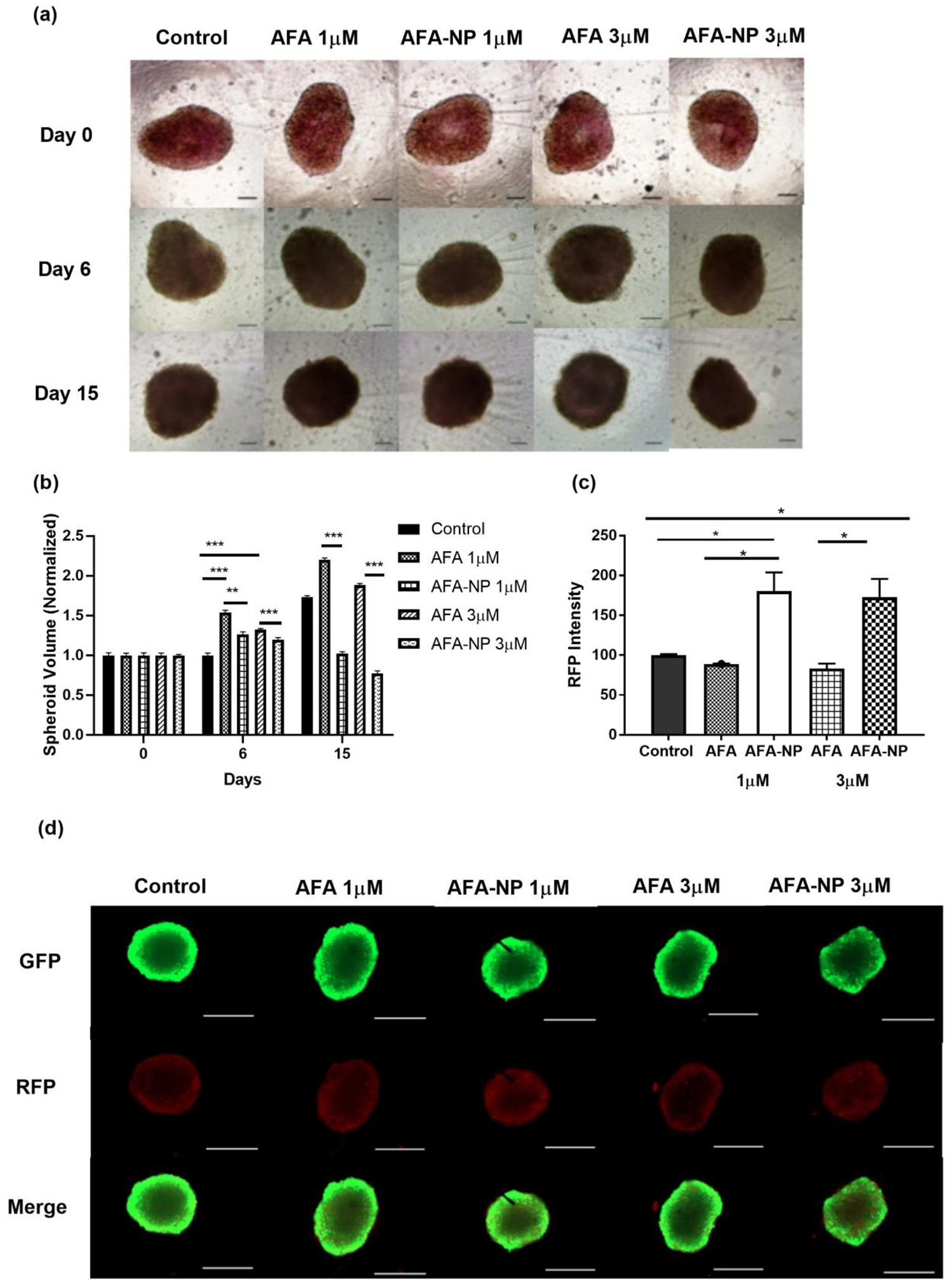
A549 cells were treated with control, AFA and AFA-NP (1, 3 μM). a. Images represent the spheroids on 0, 6 and 15 days of treatment. Scale bar for the images represents 400 μM. b. Effect of treatment on growth of tumor in 3D spheroids of A549 cells. % of spheroid volumes after 6th and 15th day of treatment were compared between the treatments. Data represent mean±SD (n = 6). Significance between the groups was analyzed by one-way ANOVA and Tukey’s multiple comparison test. c. The graph represents comparison of red fluorescence intensity (dead cells) of A549 spheroids through analyzing the fluorescent images by ImageJ software d. Images were captured using EVOS-FL fluorescence microscope. Regions showing green (GFP) indicate stained live cells and red (RFP) as stained dead cells. Merge images indicate overlay of GFP and RFP regions. Representative images of n=6 experiments. Scale bar represent 500 μM.
In the single dose study with a single treatment given on Day 1 following tumor mass establishment, the blank control spheroids were found to have a volume of 6.3±0.7mm3 on 15th Day as compared to a spheroid volume of 3.4±0.8 mm3 (1μM), and 2.7±1.1 mm3 (3μM) for AFA-loaded nanoparticles. In contrast, spheroids treated with plain afatinib had a volume of 6.3±0.8mm3 (1μM), and 6.2±0.8mm3 (3μM) respectively, thus underlining a more pronounced tumor reduction potential of AFA-loaded nanoparticles (Fig. 7a). Statistically speaking, a 1.21- (p<0.01), and 1.1- (p<0.01) fold reduction in spheroid volumes was observed for AFA-NP treated spheroids compared to AFA on day 6 at 1- and 3-μM concentrations respectively. Following prolonged exposure, a 2.14- (p<0.01), and 2.42- (p<0.01) fold reduction in tumor volume was observed for AFA-NP treated spheroids compared to plain AFA on day 15 at 1- and 3-μM concentrations respectively (Fig. 7b). Treatment with AFA-NP resulted in a significant difference in spheroid volumes as compared to AFA and control treated groups (control vs AFA-NP 1μM: p<0.0001; control vs AFA-NP 3μM: p<0.0001). Tumor growth suppression was continually observed until the end of the treatment.
In order to mimic physiological conditions inside the human body following multiple dosages, a multiple dose treatment was also performed (representative images shown in Fig. S3a). In this study on day 15th, average volume of untreated spheroids size was found to be 6.6±1.5 mm3, whereas volumes of AFA-NP treated spheroids was 5.3±4.3 mm3 and 4.0±1.3 mm3 for 1- and 3-μM respectively. Plain AFA treated spheroids were found to have volume of 6.1±1.4 mm3 and 6.5±1.1 mm3 for 1- and 3-μM respectively (Fig. S3b). AFA-NP treated spheroids exhibited a significant reduction in tumor volumes compared to both plain AFA treated and control spheroids on day 15 (AFA 1μM vs AFA-NP 1μM: p<0.0001; AFA 3μM vs AFA-NP 3μM: p<0.0001; Control vs AFA-NP 1μM: p<0.0001; Control vs AFA-NP 3μM: p<0.0001).
From obtained results, it can be clearly stated that AFA PLGA nanoparticles could significantly (p<0.05) decrease tumor progression following single dose treatment after 6 and 15 days of treatment with concentrations (1 and 3μM) compared to plain AFA. It was also observed that on multiple dose treatment; nanoparticle treatment could significantly (p<0.05) decrease tumor progression, especially after 6 days of treatment with AFA-NP at 1 and 3μM concentrations in tested A549 NSCLC cell line compared to plain drug. These data can also provide a strong evidence for capability of AFA-NPs to efficiently deliver the drug to spheroid’s/tumor’s core and their potential preclinical/clinical efficacy. To elaborate, diminished spheroid volumes on treatment with AFA-NP are indicative of nanoparticles’ ability to reach the solid core of the spheroid mass. This penetrability of nanoparticles is of advantage in reaching the tumor core, thus enabling nanoparticles to exhibit their superior tumor growth inhibition. Once AFA-NPs are internalized into the tumor cells of 3D spheroids, due to anti-cancer effects of AFA, increasing accumulation of non-proliferating cells in the central regions of the spheroids will take place. This progressive reduction in proliferating fraction of cells could be reflected by reduction of tumor volume [90]. Similarly, Gdowski et al developed a novel cabazitaxel bone targeted nanoparticle (NP) system for improved drug delivery to the bone tumor [91]. To evaluate its tumor inhibitory activity, 3D spheroid assay was used to compare cytotoxicity of nanoparticles to free drug. Guo et al formulated doxorubicin loaded polymeric nanoparticles which were evaluated and compared to plain doxorubicin on three-dimensional tumor spheroids, A549 cells [92]. They reported that when using plain drug, the tumor spheroids were observed to continue to grow in volume after 7 days to 128% of the original volume, while with doxorubicin loaded PLGA nanoparticles treatment, percentage decreased to almost 86% of its original volume.
Live-Dead Cell Assay
As the physiological and 3D tumors represent biological matrices, it is imperative for the drug to penetrate through tumor’s microenvironment, in order to demonstrate its full therapeutic potential. A live-dead cell assay helps us quantify the alive vs dead cells, not only on the tumor surface, but also in the core of the solid tumors [46]. As spheroid masses may consist of well-established outer intact periphery of living cells, it may also enclose a dead/necrotic cell core. Therefore, a superficial tumor volume or diameter measurement may not always assist in revealing the accurate effectiveness of the investigational therapy to kill tumor cells. Hence, this study was employed to confirm the ability of AFA nanoparticles to penetrate through solid mass of spheroids. Dead cell portion from treated spheroid mass was quantified through analyzing fluorescent images using ImageJ software. From the results, it was found that AFA-NP treated spheroids exhibit significantly increased presence of dead cells (red fluorescence) compared to live cells (green fluorescence) in case of both single (Fig. 7d), as well as multiple dosing (Fig. 8a) respectively. Red fluorescence intensity comparison on day 15 of single post treatment period revealed the presence of significantly higher number dead cells (RFP- AFA-NP 1μM: 180.4±23.5, AFA-NP 3μM: 172.6±23.5 in case of AFA-NP treated spheroids compared to both control (100±1.37) and plain AFA (AFA 1μM: 88.82±0.5, AFA 3μM: 83.18±6.2) treated groups as seen in Figs.7c & 7d (AFA 1μM vs AFA-NP 1μM: p<0.05; AFA 3μM vs AFA-NP 3μM: p<0.05; Control vs AFA-NP 1μM: p<0.05; Control vs AFA-NP 3μM: p<0.05). From the obtained results, it can be clearly stated that in case of nanoparticle treatment, there was significant (p<0.05) increase in RFP (red fluorescent protein) intensity in case of AFA-NP at both 1 and 3 μM concentration treatments compared to similar concentrations of plain AFA. This indicates the ability of AFA-NPs in reaching spheroid mass core leading to cell death. Therefore, it can be concluded that drug-loaded nanoparticles facilitate the entry of drug into deep tumor core, which would be inaccessible for plain drug, while the physicochemical properties of nanoformulations such as small particle size, charge or surface modifications are crucial in imparting good penetration behavior to them [93]. It has also been reported in the literature that smaller the nanoparticle size, more efficient will be their transport within the tumor [94]. However, in case of multiple dosing where spheroids were dosed at regular time periods, a distinct difference wasn’t observed in red fluorescence intensity after AFA and AFA-NP treatments as can be seen in Fig. 8b. As multiple dosing strategy involves repetitive dosing of respective treatments, it was assumed that higher concentrations of AFA provide cumulatively higher drug concentration of AFA, thus exhibiting similar effect to AFA-NP in tumor inhibition.
Fig. 8: Live-Dead Cell Assay-Multiple dosing regimen.
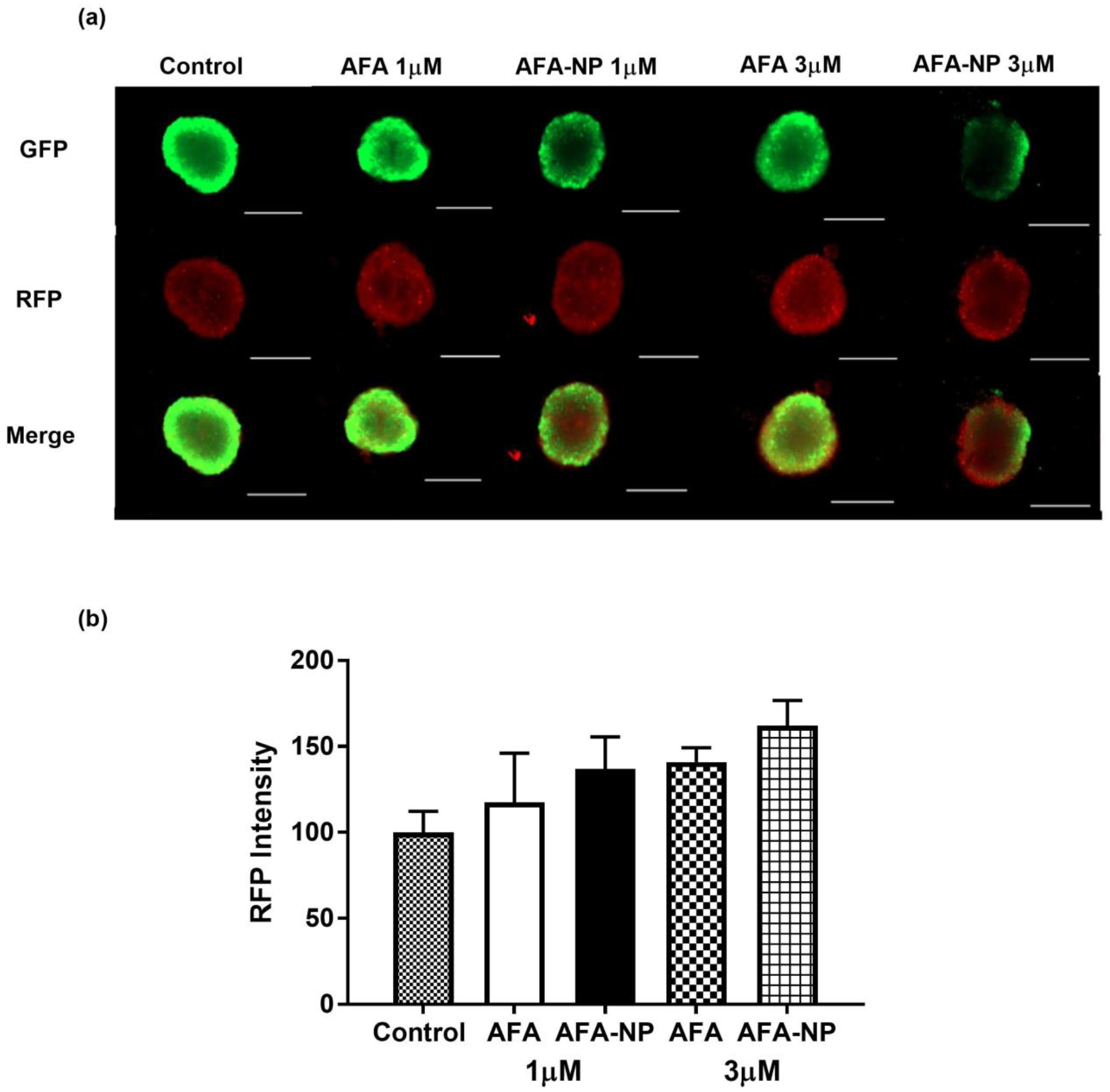
A549 spheroids treated with multiple dosing were stained using the viability/cytotoxicity assay kit to determine % of live and dead cells in respective spheroids. a. Images captured using EVOS-FL fluorescence microscope. b. The graph represents comparison of red fluorescence intensity (dead cells) of A549 spheroids treated with multiple dosing through analyzing the fluorescent images by ImageJ software. Data represent mean ± SEM (n = 3). Significance between the groups was analyzed by one-way ANOVA and Tukey’s multiple comparison test. Scale bar represent 500 μM.
Conclusion
In our present study, inhalable afatinib-loaded PLGA nanoparticles were successfully formulated using double emulsion-solvent evaporation method. Efficacy of proposed formulation in treatment of NSCLC was supported by many in-vitro studies, through which the AFA-loaded polymeric nanoparticles were compared to plain drug to order to comparatively assess its therapeutic efficacy. Our studies also confirmed physical and chemical stability of formulated nanoparticles in addition to their qualitative evaluation in intracellular uptake and efficacy in limiting tumor growth in NSCLC cancer cell lines. This study can provide a reliable platform for development of AFA-loaded polymeric nanoparticles for further preclinical and clinical studies. Developed PLGA nanoparticles of afatinib can exhibit higher residence time and accumulation in the lungs with improved pharmacokinetic profile. To the best of our knowledge, this study is one of a kind discussing delivery of afatinib in form of polymeric nanoparticles for deep lung/site-specific delivery by non-invasive inhalational route, thus overcoming limitations of marketed oral tablet. For future studies, surface modification of formulated afatinib polymeric nanoparticles could be assessed for better targeting and therapeutic efficacy.
Supplementary Material
Fig. S1: a. Standard curve for afatinib content performed by using UPLC method b. Representative chromatogram of afatinib obtained using Acquity UPLC BEH C18 column (50 × 2.1 mm, 1.7 μm particles); mobile phase: mixture of acetonitrile and phosphate buffer (pH 7.55) with the ratio of 60:40; flow rate: 0.25 ml/min, detection wavelength: 254 nm). Retention time was observed to be 0.731 min with total run time of 2 min. c. Atomic force microscopy (AFM) image for afatinib loaded nanoparticles.
Fig S2: Influence of storage temperature and length of storage on a. Particle size, b. Zeta potential and c. % Entrapment efficiency of AFA NP. Formulations were stored at 4, 25 and 37°C over a period of 4 weeks. Data represent mean±SD (n=3).
Fig S3: 3D Spheroid Study: Multiple dosing regimen a. Images represent the spheroids on 0, 6th and 15th days of treatment. Scale bar for the images represents 400 μM. b. Effect of treatment on growth of 3D spheroid tumors of A549 cells. % of spheroid volumes after 6th and 15th day of treatment were compared between the treatments. Data represent mean±SD (n = 6). Significance between the groups was analyzed by one-way ANOVA and Tukey’s multiple comparison test.
Acknowledgements:
The author(s) would like to acknowledge the Imaging Facility of CUNY Advanced Science Research Center for instrument use, scientific and technical assistance. The author(s) would also like to acknowledge Dr. Yong Yu (Associate Professor & Graduate Director, Biological Sciences, St. John’s University, NY, USA) for providing porcine kidney proximal tubule cell line (LLC-PK1).
Funding:
This project was funded with research funds to Vivek Gupta by College of Pharmacy and Health Sciences, St. John’s University, Queens, NY. Rasha S Elbatanony was supported by American Heart Association (AHA) (Grant #18AIREA33960072) to Vivek Gupta. Vineela Parvathaneni and Gautam Chauhan were supported by teaching assistantships from College of Pharmacy and Health Sciences, St. John’s University. Snehal K Shukla and Nishant S Kulkarni were supported with the research assistantship by National Institutes of Health (NIH) (Grant #1R15HL138606-01A1) to Vivek Gupta.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest and Financial Disclosures:
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References:
- 1.Anselmo AC, Mitragotri S. Nanoparticles in the clinic. Bioeng Transl Med. 2016;1:10–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martins JP, das Neves J, de la Fuente M, Celia C, Florindo H, Günday-Türeli N, et al. The solid progress of nanomedicine. Drug Deliv Transl Res. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farjadian F, Ghasemi A, Gohari O, Roointan A, Karimi M, Hamblin MR. Nanopharmaceuticals and nanomedicines currently on the market: challenges and opportunities. Nanomedicine (Lond). 2019;14:93–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leong HS, Butler KS, Brinker CJ, Azzawi M, Conlan S, Dufés C, et al. On the issue of transparency and reproducibility in nanomedicine. Nat Nanotechnol. 2019;14:629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.d’Angelo I, Quaglia F, Ungaro F. PLGA carriers for inhalation: where do we stand, where are we headed? Therapeutic Delivery. Future Science; 2015;6:1139–44. [DOI] [PubMed] [Google Scholar]
- 6.Abdelaziz HM, Gaber M, Abd-Elwakil MM, Mabrouk MT, Elgohary MM, Kamel NM, et al. Inhalable particulate drug delivery systems for lung cancer therapy: Nanoparticles, microparticles, nanocomposites and nanoaggregates. J Control Release. 2018;269:374–92. [DOI] [PubMed] [Google Scholar]
- 7.Din FU, Aman W, Ullah I, Qureshi OS, Mustapha O, Shafique S, et al. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int J Nanomedicine. 2017;12:7291–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–54. [DOI] [PubMed] [Google Scholar]
- 9.Ridge CA, McErlean AM, Ginsberg MS. Epidemiology of lung cancer. Semin Intervent Radiol. 2013;30:93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao J, Li H-R, Jin C, Jiang J-H, Ding J-Y. Strategies to overcome acquired resistance to EGFR TKI in the treatment of non-small cell lung cancer. Clin Transl Oncol. 2019;21:1287–301. [DOI] [PubMed] [Google Scholar]
- 11.Rosière R, Berghmans T, De Vuyst P, Amighi K, Wauthoz N. The Position of Inhaled Chemotherapy in the Care of Patients with Lung Tumors: Clinical Feasibility and Indications According to Recent Pharmaceutical Progresses. Cancers (Basel). 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh AP, Biswas A, Shukla A, Maiti P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduction and Targeted Therapy. 2019;4:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuzmov A, Minko T. Nanotechnology approaches for inhalation treatment of lung diseases. Journal of Controlled Release. 2015;219:500–18. [DOI] [PubMed] [Google Scholar]
- 14.Rosière R, Amighi K, Wauthoz N. Chapter 10 - Nanomedicine-Based Inhalation Treatments for Lung Cancer. In: Kesharwani P, editor. Nanotechnology-Based Targeted Drug Delivery Systems for Lung Cancer. Academic Press; 2019. p. 249–68. [Google Scholar]
- 15.Vaidya B, Kulkarni NS, Shukla SK, Parvathaneni V, Chauhan G, Damon JK, et al. Development of Inhalable Quinacrine Loaded Bovine Serum Albumin Modified Cationic Nanoparticles: Repurposing Quinacrine for Lung Cancer Therapeutics. International Journal of Pharmaceutics. 2020;118995. [DOI] [PubMed] [Google Scholar]
- 16.Youngren-Ortiz SR, Hill DB, Hoffmann PR, Morris KR, Barrett EG, Forest MG, et al. Development of Optimized, Inhalable, Gemcitabine-Loaded Gelatin Nanocarriers for Lung Cancer. J Aerosol Med Pulm Drug Deliv. 2017;30:299–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guzmán EAT, Sun Q, Meenach SA. Development and Evaluation of Paclitaxel-Loaded Aerosol Nanocomposite Microparticles and Their Efficacy Against Air-Grown Lung Cancer Tumor Spheroids. ACS Biomater Sci Eng. 2019;5:6570–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parvathaneni V, Kulkarni NS, Shukla SK, Farrales PT, Kunda NK, Muth A, et al. Systematic Development and Optimization of Inhalable Pirfenidone Liposomes for Non-Small Cell Lung Cancer Treatment. Pharmaceutics. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdelaziz HM, Elzoghby AO, Helmy MW, Abdelfattah E-ZA, Fang J-Y, Samaha MW, et al. Inhalable Lactoferrin/Chondroitin-Functionalized Monoolein Nanocomposites for Localized Lung Cancer Targeting. ACS Biomater Sci Eng. American Chemical Society; 2020;6:1030–42. [DOI] [PubMed] [Google Scholar]
- 20.Mangal S, Gao W, Li T, Zhou QT. Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: challenges and opportunities. Acta Pharmacol Sin. 2017;38:782–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmad J, Akhter S, Rizwanullah M, Amin S, Rahman M, Ahmad MZ, et al. Nanotechnology-based inhalation treatments for lung cancer: state of the art. Nanotechnol Sci Appl. 2015;8:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson CF, Grimmett ME, Domalewski CJ, Cui H. Inhalable nanotherapeutics to improve treatment efficacy for common lung diseases. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2020;12:e1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi SH, Byeon HJ, Choi JS, Thao L, Kim I, Lee ES, et al. Inhalable self-assembled albumin nanoparticles for treating drug-resistant lung cancer. J Control Release. 2015;197:199–207. [DOI] [PubMed] [Google Scholar]
- 24.Chishti N, Jagwani S, Dhamecha D, Jalalpure S, Dehghan MH. Preparation, Optimization, and In Vivo Evaluation of Nanoparticle-Based Formulation for Pulmonary Delivery of Anticancer Drug. Medicina (Kaunas). 2019;55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Velino C, Carella F, Adamiano A, Sanguinetti M, Vitali A, Catalucci D, et al. Nanomedicine Approaches for the Pulmonary Treatment of Cystic Fibrosis. Front Bioeng Biotechnol. 2019;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borghardt JM, Kloft C, Sharma A. Inhaled Therapy in Respiratory Disease: The Complex Interplay of Pulmonary Kinetic Processes. Canadian Respiratory Journal. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Y-L, Zhou C, Hu C-P, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. The lancet oncology. 2014;15:213–22. [DOI] [PubMed] [Google Scholar]
- 28.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. The lancet oncology. 2010;11:121–8. [DOI] [PubMed] [Google Scholar]
- 29.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–8. [DOI] [PubMed] [Google Scholar]
- 30.Mok TS, Wu Y-L, Thongprasert S, Yang C-H, Chu D-T, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. [DOI] [PubMed] [Google Scholar]
- 31.Zhou C, Wu Y-L, Chen G, Feng J, Liu X-Q, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. The lancet oncology. 2011;12:735–42. [DOI] [PubMed] [Google Scholar]
- 32.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. The lancet oncology. 2012;13:239–46. [DOI] [PubMed] [Google Scholar]
- 33.Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343:342–50. [DOI] [PubMed] [Google Scholar]
- 34.Subramaniam D, He AR, Hwang J, Deeken J, Pishvaian M, Hartley ML, et al. Irreversible multitargeted ErbB family inhibitors for therapy of lung and breast cancer. Curr Cancer Drug Targets. 2015;14:775–93. [DOI] [PubMed] [Google Scholar]
- 35.Moll HP, Pranz K, Musteanu M, Grabner B, Hruschka N, Mohrherr J, et al. Afatinib restrains K-RAS-driven lung tumorigenesis. Sci Transl Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Afatinib (Gilotrif) Improves Clinical Outcomes Compared With Gefitinib (Iressa) in Head-to-Head Lung Cancer Trial [Internet]. Managed Care magazine. 2016. [cited 2020 Apr 21]. Available from: https://www.managedcaremag.com/news/afatinib-gilotrif-improves-clinical-outcomes-compared-gefitinib-iressa-head-head-lung-cancer
- 37.Stopfer P, Marzin K, Narjes H, Gansser D, Shahidi M, Uttereuther-Fischer M, et al. Afatinib pharmacokinetics and metabolism after oral administration to healthy male volunteers. Cancer Chemother Pharmacol. 2012;69:1051–61. [DOI] [PubMed] [Google Scholar]
- 38.Edwards RL, Andan C, Lalla RV, Lacouture ME, O’Brien D, Sequist LV. Afatinib Therapy: Practical Management of Adverse Events With an Oral Agent for Non-Small Cell Lung Cancer Treatment. Clin J Oncol Nurs. 2018;22:542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel). 2011;3:1377–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasparini G, Kosvintsev SR, Stillwell MT, Holdich RG. Preparation and characterization of PLGA particles for subcutaneous controlled drug release by membrane emulsification. Colloids Surf B Biointerfaces. 2008;61:199–207. [DOI] [PubMed] [Google Scholar]
- 41.Shukla S, Kulkarni N, Chan A, Parvathaneni V, Farrales P, Muth A, et al. Metformin-Encapsulated Liposome Delivery System: An Effective Treatment Approach against Breast Cancer. Pharmaceutics. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta V, Gupta N, Shaik IH, Mehvar R, McMurtry IF, Oka M, et al. Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary preferential vasodilation in pulmonary arterial hypertension. J Control Release. 2013;167:189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mi Y, Wolfram J, Mu C, Liu X, Blanco E, Shen H, et al. Enzyme-responsive multistage vector for drug delivery to tumor tissue. Pharmacol Res. 2016;113:92–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foroozandeh P, Aziz AA. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res Lett. 2018;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vaidya B, Parvathaneni V, Kulkarni NS, Shukla SK, Damon JK, Sarode A, et al. Cyclodextrin modified erlotinib loaded PLGA nanoparticles for improved therapeutic efficacy against non-small cell lung cancer. Int J Biol Macromol. 2019;122:338–47. [DOI] [PubMed] [Google Scholar]
- 46.Kulkarni NS, Parvathaneni V, Shukla SK, Barasa L, Perron JC, Yoganathan S, et al. Tyrosine kinase inhibitor conjugated quantum dots for non-small cell lung cancer (NSCLC) treatment. Eur J Pharm Sci. 2019;133:145–59. [DOI] [PubMed] [Google Scholar]
- 47.Kwon H-Y, Lee J-Y, Choi S-W, Jang Y, Kim J-H. Preparation of PLGA nanoparticles containing estrogen by emulsification-diffusion method. Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2001;182:123–30. [Google Scholar]
- 48.Quintanar-Guerrero D, Fessi H, Allémann E, Doelker E. Influence of stabilizing agents and preparative variables on the formation of poly(d,l-lactic acid) nanoparticles by an emulsification-diffusion technique. International Journal of Pharmaceutics. 1996;143:133–41. [Google Scholar]
- 49.Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC, et al. Cellular Uptake of Nanoparticles: Journey Inside the Cell. Chem Soc Rev. 2017;46:4218–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Toy R, Roy K. Engineering nanoparticles to overcome barriers to immunotherapy. Bioeng Transl Med. 2016;1:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Panariti A, Miserocchi G, Rivolta I. The effect of nanoparticle uptake on cellular behavior: disrupting or enabling functions? Nanotechnol Sci Appl. 2012;5:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377:159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cartiera MS, Johnson KM, Rajendran V, Caplan MJ, Saltzman WM. The uptake and intracellular fate of PLGA nanoparticles in epithelial cells. Biomaterials. 2009;30:2790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soltani S, Zakeri-Milani P, Barzegar-Jalali M, Jelvehgari M. Fabrication and in-vitro Evaluation of Ketotifen Fumarate-loaded PLGA Nanoparticles as a Sustained Delivery System. Iran J Pharm Res. 2017;16:22–34. [PMC free article] [PubMed] [Google Scholar]
- 55.Srikar G Study on influence of polymer and surfactant on in vitro performance of biodegradable aqueous-core nanocapsules of tenofovirdisoproxil fumarate by response surface methodology. Braz J Pharm Sci. 2019;55. [Google Scholar]
- 56.Yadav KS, Sawant KK. Formulation optimization of etoposide loaded PLGA nanoparticles by double factorial design and their evaluation. Curr Drug Deliv. 2010;7:51–64. [DOI] [PubMed] [Google Scholar]
- 57.Matsuura K, Kojima H, Haraguchi T, Yoshida M, Suzuki S, Suzuki T, et al. Preparation and Characterization of Itraconazole- or Miconazole-Loaded PLGA Microspheres. Chemical and Pharmaceutical Bulletin. 2019;67:106–11. [DOI] [PubMed] [Google Scholar]
- 58.Soni A, Dandagi P, Gadad A, Mastiholimath V. Simvastatin-loaded PLGA nanoparticles for improved oral bioavailability and sustained release: Effect of formulation variables. Asian Journal of Pharmaceutics. 2011;5:57. [Google Scholar]
- 59.Yang H, Li J, Patel SK, Palmer KE, Devlin B, Rohan LC. Design of Poly(lactic-co-glycolic Acid) (PLGA) Nanoparticles for Vaginal Co-Delivery of Griffithsin and Dapivirine and Their Synergistic Effect for HIV Prophylaxis. Pharmaceutics. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cabri W, Lahiri S, Mishra BB, Abul A, Panda N, Bhavanam PR, et al. Polymorphic forms of afatinib free base and afatinib dimaleate. Google Patents; 2018. [Google Scholar]
- 61.Khanna R, Kumar N, Sharma VK, Ankit G, Khattar D. Pharmaceutical compositions comprising afatinib. Google Patents; 2018. [Google Scholar]
- 62.Kumar P, Dhande P, Mazlee MTF, Yaman SM, Chandran NSNBMS, Makhtar MZB, et al. A controlled, efficient and robust process for the synthesis of an epidermal growth factor receptor inhibitor: Afatinib Dimaleate. 1. 2019;1:3–12. [Google Scholar]
- 63.Sun S-B, Liu P, Shao F-M, Miao Q-L. Formulation and evaluation of PLGA nanoparticles loaded capecitabine for prostate cancer. Int J Clin Exp Med. 2015;8:19670–81. [PMC free article] [PubMed] [Google Scholar]
- 64.Salome Amarachi C, Kenechukwu F, Attama A. Nanoemulsions - Advances in Formulation, Characterization and Applications in Drug Delivery. 2014;76–126. [Google Scholar]
- 65.Müller KH, Motskin M, Philpott AJ, Routh AF, Shanahan CM, Duer MJ, et al. The effect of particle agglomeration on the formation of a surface-connected compartment induced by hydroxyapatite nanoparticles in human monocyte-derived macrophages. Biomaterials. 2014;35:1074–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahmed R, Tariq M, Ahmad IS, Fouly H, Fakhar-i-Abbas, Hasan A, et al. Poly(lactic-co-glycolic acid) Nanoparticles Loaded with Callistemon citrinus Phenolics Exhibited Anticancer Properties against Three Breast Cancer Cell Lines. Journal of Food Quality. 2019. [Google Scholar]
- 67.Labiris NR, Dolovich MB. Pulmonary drug delivery. Part I: Physiological factors affecting therapeutic effectiveness of aerosolized medications. Br J Clin Pharmacol. 2003;56:588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moreno-Sastre M, Pastor M, Salomon CJ, Esquisabel A, Pedraz JL. Pulmonary drug delivery: a review on nanocarriers for antibacterial chemotherapy. J Antimicrob Chemother. 2015;70:2945–55. [DOI] [PubMed] [Google Scholar]
- 69.Carvalho TC, Peters JI, Williams RO. Influence of particle size on regional lung deposition--what evidence is there? Int J Pharm. 2011;406:1–10. [DOI] [PubMed] [Google Scholar]
- 70.Dabbagh A, Abu Kasim NH, Yeong CH, Wong TW, Abdul Rahman N. Critical Parameters for Particle-Based Pulmonary Delivery of Chemotherapeutics. Journal of Aerosol Medicine and Pulmonary Drug Delivery. 2017;31:139–54. [DOI] [PubMed] [Google Scholar]
- 71.Johal B, Howald M, Fischer M, Marshall J, Venthoye G. Fine Particle Profile of Fluticasone Propionate/Formoterol Fumarate Versus Other Combination Products: the DIFFUSE Study. Comb Prod Ther. 2013;3:39–51. [Google Scholar]
- 72.Tang J, Li J, Li G, Zhang H, Wang L, Li D, et al. Spermidine-mediated poly(lactic--glycolic acid nanoparticles containing fluorofenidone for the treatment of idiopathic pulmonary fibrosis. International Journal of Nanomedicine. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilhelm C, Billotey C, Roger J, Pons JN, Bacri J-C, Gazeau F. Intracellular uptake of anionic superparamagnetic nanoparticles as a function of their surface coating. Biomaterials. 2003;24:1001–11. [DOI] [PubMed] [Google Scholar]
- 74.Tahara K, Sakai T, Yamamoto H, Takeuchi H, Hirashima N, Kawashima Y. Improved cellular uptake of chitosan-modified PLGA nanospheres by A549 cells. Int J Pharm. 2009;382:198–204. [DOI] [PubMed] [Google Scholar]
- 75.Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–9. [DOI] [PubMed] [Google Scholar]
- 76.Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang S, Zheng X, Huang H, Wu K, Wang B, Chen X, et al. Afatinib increases sensitivity to radiation in non-small cell lung cancer cells with acquired EGFR T790M mutation. Oncotarget. 2015;6:5832–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xie C, Li Y, Li L-L, Fan X-X, Wang Y-W, Wei C-L, et al. Identification of a New Potent Inhibitor Targeting KRAS in Non-small Cell Lung Cancer Cells. Front Pharmacol. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao Y-Z, Sun C-Z, Lu C-T, Dai D-D, Lv H-F, Wu Y, et al. Characterization and anti-tumor activity of chemical conjugation of doxorubicin in polymeric micelles (DOX-P) in vitro. Cancer Lett. 2011;311:187–94. [DOI] [PubMed] [Google Scholar]
- 80.Oh JE, Nam YS, Lee KH, Park TG. Conjugation of drug to poly (D, L-lactic-co-glycolic acid) for controlled release from biodegradable microspheres. J Controlled Release. 1999;57:269–80. [DOI] [PubMed] [Google Scholar]
- 81.Unal R, Yao-Borengasser A, Varma V, Rasouli N, Labbate C, Kern PA, et al. Matrix metalloproteinase-9 is increased in obese subjects and decreases in response to pioglitazone. J Clin Endocrinol Metab. 2010;95:2993–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Merlin JPJ, Venkadesh B, Hussain R, Prasad NR, Shibli SMA, Raj AVM, et al. Paclitaxel loaded poly-d,l-lactide-co-glycolide nanoparticles: Enhanced anticancer effect in non-small cell lung carcinoma cell line. 2013.
- 83.Jo Y, Choi N, Kim K, Koo H-J, Choi J, Kim HN. Chemoresistance of Cancer Cells: Requirements of Tumor Microenvironment-mimicking In Vitro Models in Anti-Cancer Drug Development. Theranostics. 2018;8:5259–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Breslin S, O’Driscoll L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discov Today. 2013;18:240–9. [DOI] [PubMed] [Google Scholar]
- 85.Fennema E, Rivron N, Rouwkema J, van Blitterswijk C, de Boer J. Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol. 2013;31:108–15. [DOI] [PubMed] [Google Scholar]
- 86.Lazzari G, Couvreur P, Mura S. Multicellular tumor spheroids: a relevant 3D model for the in vitro preclinical investigation of polymer nanomedicines. Polym Chem. The Royal Society of Chemistry; 2017;8:4947–69. [Google Scholar]
- 87.Yang Y, Roy A, Zhao Y, Undzys E, Li S-D. Comparison of Tumor Penetration of Podophyllotoxin-Carboxymethylcellulose Conjugates with Various Chemical Compositions in Tumor Spheroid Culture and In Vivo Solid Tumor. Bioconjugate Chem. American Chemical Society; 2017;28:1505–18. [DOI] [PubMed] [Google Scholar]
- 88.Katt ME, Placone AL, Wong AD, Xu ZS, Searson PC. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front Bioeng Biotechnol. 2016;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vaidya B, Parvathaneni V, Kulkarni NS, Shukla SK, Damon JK, Sarode A, et al. Cyclodextrin modified erlotinib loaded PLGA nanoparticles for improved therapeutic efficacy against non-small cell lung cancer. Int J Biol Macromol. 2019;122:338–47. [DOI] [PubMed] [Google Scholar]
- 90.Galateanu B, Hudita A, Negrei C, Ion R-M, Costache M, Stan M, et al. Impact of multicellular tumor spheroids as an in vivo‑like tumor model on anticancer drug response. International Journal of Oncology. Spandidos Publications; 2016;48:2295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gdowski AS, Ranjan A, Sarker MR, Vishwanatha JK. Bone-targeted cabazitaxel nanoparticles for metastatic prostate cancer skeletal lesions and pain. Nanomedicine. 2017;12:2083–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.GUO YWANG L, LV P, ZHANG P. Transferrin-conjugated doxorubicin-loaded lipid-coated nanoparticles for the targeting and therapy of lung cancer. Oncol Lett. 2015;9:1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tchoryk A, Taresco V, Argent RH, Ashford M, Gellert PR, Stolnik S, et al. Penetration and Uptake of Nanoparticles in 3D Tumor Spheroids. Bioconjugate Chem. American Chemical Society; 2019;30:1371–84. [DOI] [PubMed] [Google Scholar]
- 94.Zhang Y-R, Lin R, Li H-J, He W, Du J-Z, Wang J. Strategies to improve tumor penetration of nanomedicines through nanoparticle design. WIREs Nanomedicine and Nanobiotechnology. 2019;11:e1519. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: a. Standard curve for afatinib content performed by using UPLC method b. Representative chromatogram of afatinib obtained using Acquity UPLC BEH C18 column (50 × 2.1 mm, 1.7 μm particles); mobile phase: mixture of acetonitrile and phosphate buffer (pH 7.55) with the ratio of 60:40; flow rate: 0.25 ml/min, detection wavelength: 254 nm). Retention time was observed to be 0.731 min with total run time of 2 min. c. Atomic force microscopy (AFM) image for afatinib loaded nanoparticles.
Fig S2: Influence of storage temperature and length of storage on a. Particle size, b. Zeta potential and c. % Entrapment efficiency of AFA NP. Formulations were stored at 4, 25 and 37°C over a period of 4 weeks. Data represent mean±SD (n=3).
Fig S3: 3D Spheroid Study: Multiple dosing regimen a. Images represent the spheroids on 0, 6th and 15th days of treatment. Scale bar for the images represents 400 μM. b. Effect of treatment on growth of 3D spheroid tumors of A549 cells. % of spheroid volumes after 6th and 15th day of treatment were compared between the treatments. Data represent mean±SD (n = 6). Significance between the groups was analyzed by one-way ANOVA and Tukey’s multiple comparison test.