

Summary
Perturbation biology is a powerful approach to modeling quantitative cellular behaviors and understanding detailed disease mechanisms. However, large-scale protein response resources of cancer cell lines to perturbations are not available, resulting in a critical knowledge gap. Here we generated and compiled perturbed expression profiles of ~210 clinically relevant proteins in >12,000 cancer cell line samples in response to ~170 drug compounds using reverse-phase protein arrays. We show that integrating perturbed protein response signals provides mechanistic insights into drug resistance, increases the predictive power for drug sensitivity, and helps identify effective drug combinations. We build a systematic map of “protein-drug” connectivity and develop a user-friendly data portal for community use. Our study provides a rich resource to investigate the behaviors of cancer cells and the dependencies of treatment responses, thereby enabling a broad range of biomedical applications.
Graphical Abstract
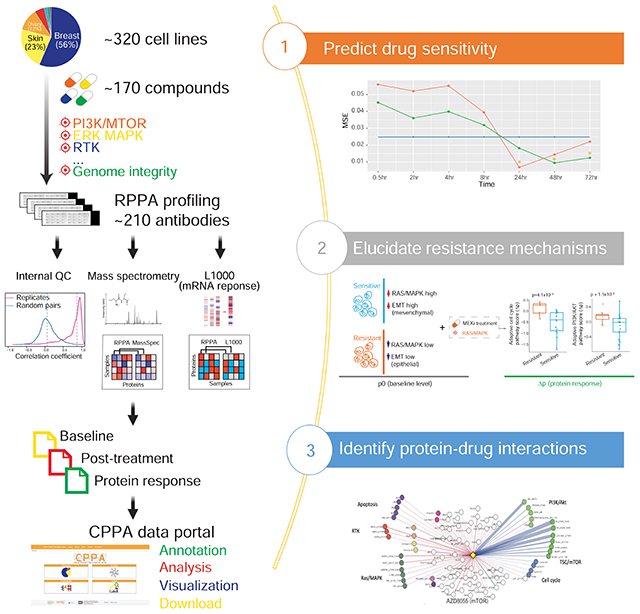
eTOC Blurb
Zhao et al. profile the protein responses of a large collection of cancer cell lines to drug perturbations using RPPA platform and build a systematic protein-drug connectivity map. The integration of perturbed protein responses provides better prediction to drug sensitivity and insights into drug resistance mechanisms and combination therapies.
Introduction
Cancer is a highly heterogeneous disease encompassing many tissue types and diverse oncogenic drivers, with treatment responses that are often variable in distinct tumor contexts. Over the last decade, extensive efforts have been made to characterize the tremendous heterogeneity of human cancers at the molecular level (Hutter and Zenklusen, 2018; Jiang et al., 2019). A real challenge in cancer research, however, is to obtain a systematic understanding of causality and mechanisms underlying the behaviors of cancer cells with the eventual goal of improving patient outcomes (Wise and Solit, 2019). To address this challenge, perturbation experiments provide a powerful approach in which cells are modulated by perturbagens, and downstream consequences are monitored (Korkut et al., 2015; Molinelli et al., 2013; Ng et al., 2018). The longitudinal data thus obtained provide considerably greater information content of both the basal biological network wiring and its associated changes under stress, thereby leading to a deeper understanding of mechanisms underlying cell survival under stress. Recently, large-scale compendia of the phenotypic and cellular effects of perturbed cancer cell lines have been established. For example, large-scale pharmacologic perturbation studies, cell viability measurements upon different drug treatments across many cell lines have been published (Barretina et al., 2012; Basu et al., 2013; Garnett et al., 2012; Iorio et al., 2016); several studies have built genome-wide “cancer dependency” maps across a large number of cell lines using loss-of-function siRNA, shRNA, or CRISPR-cas9 screens (McDonald et al., 2017; Tsherniak et al., 2017); a “connectivity map” of profiled mRNA responses of cancer cell lines to diverse perturbations using an efficient, robust RNA measurement platform, L1000 has been developed (Subramanian et al., 2017). These studies provide valuable resources for gaining a systems-level understanding of cancer mechanisms and phenotypes. However, similar large-scale resources for analysis and integration of protein responses of perturbed cancer cell lines have yet to be established. This knowledge gap is even more striking, considering that proteins comprise the basic functional units in biological processes and represent the major targets for cancer therapy.
To fill this gap, we generated and compiled a large compendium of perturbed protein expression profiles of cancer cell lines in response to a diverse array of clinically relevant drugs using reverse-phase protein arrays (RPPAs). RPPA is a quantitative antibody-based approach to assess protein markers in a large number of samples in a high-throughput, cost-effective, sensitive manner (Hennessy et al., 2010; Nishizuka et al., 2003; Tibes et al., 2006). This platform depends on antibodies for the detection of proteins, and currently, there is a limited but rapidly growing number of proteins for which high-quality antibodies exist that give an analyzable signal. We have applied this technology to quantify protein expression levels of large patient cohorts (e.g., The Cancer Genome Atlas) (Akbani et al., 2014) and cancer cell lines (e.g., MD Anderson Cell Line project and Cancer Cell Line Encyclopedia) (Ghandi et al., 2019; Li et al., 2017). The current antibody repertoire covers key oncogenic pathways such as PI3K/AKT, RAS/MAPK, Src/FAK, TGFβ/SMAD, JAK/STAT, DNA damage repair, Hippo, cell cycle, apoptosis, histone modification, and immune-oncology. Compared with proteome-wide mass spectrometry approaches, our RPPA-based approach has several advantages. First, although the number of protein markers in RPPA readout is much smaller (200-300), this highly select protein set is enriched in therapeutic targets and biomarkers, thereby greatly increasing the ability to generate clinically relevant hypotheses and make translational impacts. Statistically, this more focused assessment also substantially reduces multiple testing, a major challenge in identifying significant hits from unbiased proteomic searches (if no pre-filters are applied). Second, one RPPA slide can measure up to 1,000 samples simultaneously. Thus, the high-throughput and cost-effectiveness make RPPA a practical platform for assessing a large number of samples (e.g., >10,000), which is simply not feasible for alternative proteomic approaches. Third, protein-level responses, particularly changes in post-translational modifications, more likely reflect how cancer cells rewire their signaling pathways to adapt and survive a specific drug treatment, as most targeted therapies act by modulating protein phosphorylation and activity. The superior ability of RPPA to quantify some key post-translationally modified proteins has the potential to capture such adaptive responses and can provide stronger predictors of therapy response or resistance mechanisms (Mertins et al., 2014). Indeed, our recent studies have demonstrated the value of RPPA-based adaptive responses in the rational design of combination therapies (Fang et al., 2019; Iavarone et al., 2019; Korkut et al., 2015; Krepler et al., 2017; Krepler et al., 2016; Kwong et al., 2015; Molinelli et al., 2013; Muranen et al., 2012; Sun et al., 2017; Sun et al., 2018), with several of these translated to the clinic with patient benefit.
Results
A large, high-quality collection of perturbed RPPA profiles of cancer cell lines
To generate a high-quality resource of perturbed protein responses, we measured RPPA-based protein expression profiles of cancer cell lines in response to ~170 preclinical and clinical therapeutics (often across multiple time points), generated normalized RPPA data (including baseline level p0 and post-treatment level p1) and protein response to perturbation (Δp = p1 − p0) profiles using a standardized data processing pipeline, and made the data public through a user-friendly data portal (Figure 1A). We developed a multiple-stage, multiple platform quality control (QC) pipeline in which we first evaluated the reproducibility of both baseline and protein response profiles of our samples within the RPPA platform, and then validated the replicability of our data using independent platforms (i.e., protein, mass spectrometry; and mRNA, L1000) (STAR Methods, Figure 1B). By comparing protein response (Δp) correlations of replicate samples to those of random sample pairs, we demonstrated that replicate samples showed higher correlations across protein markers (mean R = 0.87) than random pairs (mean R = 0.058) (Figure 1C, D), indicating high reproducibility of our RPPA data. After excluding ~2.2% low confidence samples, the final compendium contained QC-passed RPPA profiles (~210 total and phosphorylated protein markers) of 15,492 samples (11,884 drug-treated samples and 3,608 control samples related to perturbation of 168 compounds in 319 cell lines) in total.
Figure 1. Summary of the perturbed RPPA profiling data in this study.
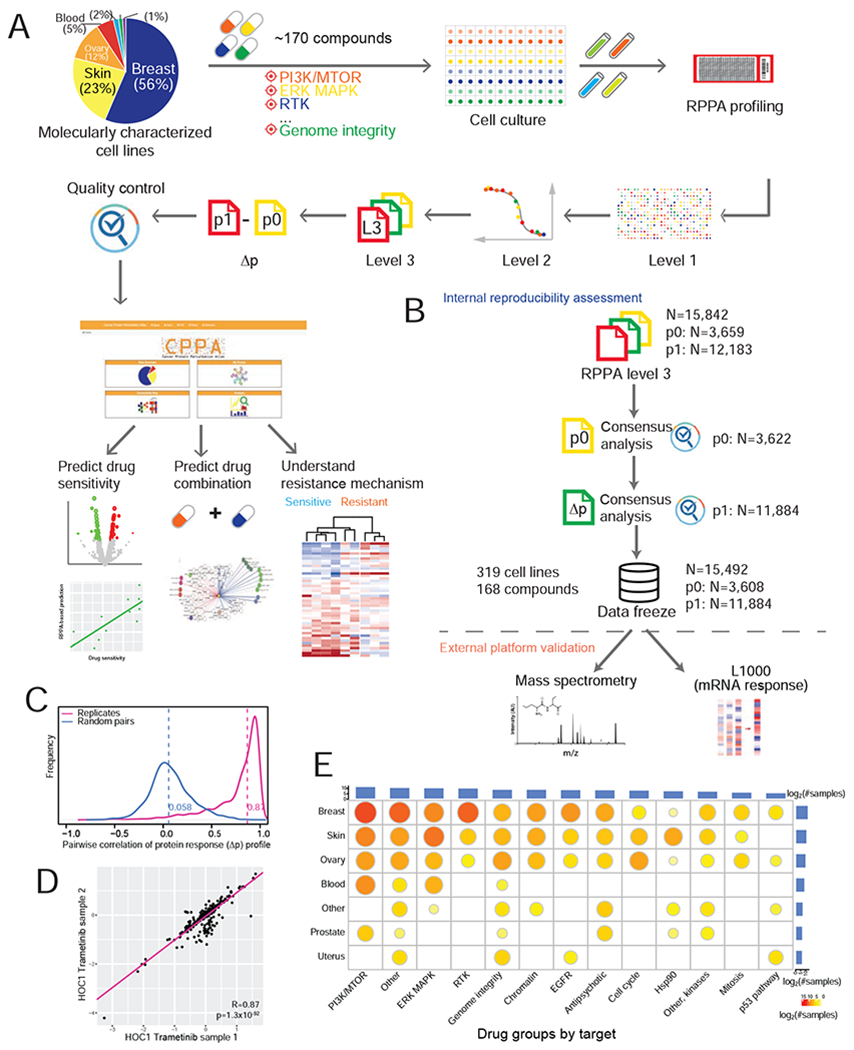
(A) Overview of the RPPA profiling experiments and data processing of cell line perturbations. The pie chart shows the lineage distribution of cancer cell lines profiled (n = 319). (B) The RPPA quality control pipeline, which contains within-platform assessment and external validation using independent platforms. (C) Reproducibility of perturbed RPPA data based on protein response (Δp) profiles of technical replicates (n = 2,753 pairs). (D) A representative scatter plot showing the correlation of Δp between two replicate samples across protein markers. (E) The distribution of drug-treated samples by cell lineages and drug groups. The bar plots show the numbers of samples profiled for each lineage or drug group, and the size of the circle is proportional to the number of samples profiled for each lineage-drug combination. See also Figure S1, Figure S6 and Table S1.
The cancer cell lines in our dataset come from several lineages, including breast, ovarian, uterus, skin, blood, and prostate; and the drug compounds target a broad range of cancer-related processes, including PI3K/mTOR signaling, ERK/MAPK signaling, RTK signaling, EGFR signaling, TP53 pathway, genome integrity, cell cycle, antipsychotic drugs, and chromatin remodeling (Figure 1E, Table S1). Due to time and cost constraints and the clinical relevance of different drugs, instead of profiling all possible perturbations across all cell line and drug combinations, we took a more pragmatic approach in which some cell lineages and drug groups were more frequently profiled but still represent an extensive survey of drug perturbations (Figure 1E). Our sample set is highly enriched in responses from a subset of common, well-characterized cancer cell lines that have rich molecular profiling and drug response data in public resources (Figure S1A). For example, >1,500 drug-treated samples were from MCF7, and >250 drug-treated samples were from BT20, SKBR3, MDA-MB-468, BT549, UACC812, BT474, SKOV3, and HCC1954 (Figure S1B). For drug treatment, ~86% of the samples were treated with monotherapy, and ~1,700 samples were treated with double or triple-drug combinations (Figure S1C). Among the drug compounds used, 23 compounds have >150 treated samples, with lapatinib (480 samples, HER2 inhibitor) and GSK690693 (454 samples, AKT inhibitor) being the top two drug treatments (Figure S1D). Importantly, for many of the therapeutic targets, we profiled multiple targeting agents, including those that target different members of the same pathway, to increase our ability to identify on-target activity.
Since high reproducibility in the same platform does not necessarily imply validity, to further confirm the quality of the RPPA data output, we sought to validate our protein response data using independent platforms (Figure 1B). First, we compared the baseline protein expression in our RPPA platform with baseline mass spectrometry data in a set of shared cell lines (Figure 2A). We found that the corresponding protein pairs between the two proteomic platforms showed substantially higher correlations across cell lines than random protein pairs (median correlation coefficient: 0.50 vs. 0.0, paired Student’s t-test, p = 7.8×10−11, Figure 2B). Indeed, the majority of protein pairs showed high correlations between the RPPA and mass spectrometry platforms. Second, since extensive data on protein changes in response to drug treatment are not available, we employed mRNA response data from the connectivity map (Subramanian et al., 2017). As this analysis is for different molecules (protein vs. RNA), and across different platforms (RPPA vs. L1000), we employed the Goodman-Kruskal’s gamma (γ) correlation to conduct a robust assessment. Based on the same cell lines perturbed by the same compounds (n = 46 unique cell line-drug perturbations), we converted the original continuous response scores into categorical response groups (i.e., upregulated, neutral, and downregulated) and compared the mRNA-protein response concordance by calculating mRNA-protein response association and sample-sample association (Figure 2C). We observed that the matched mRNA-protein responses from the same condition were highly associated with each other (median γ = 0.68), which is significantly higher than that from the randomly shuffled background distribution (paired Student’s t-test, p = 2.6×10−4, Figure 2D). Then, we tested whether the sample-sample association inferred from the RPPA-based protein responses were preserved in the L1000-based mRNA responses. Among the significant sample-sample associations identified by either platform (FDR < 0.01), the RPPA-based γ scores showed a strong, positive correlation with the L1000-based γ scores (Pearson’s correlation, R = 0.68, p = 2.7×10−6, Figure 2E). Further, categorized RPPA-based associations are highly consistent with L1000-based associations (Fisher’s exact test, p = 3×10−3). These external validations using cross-molecule, cross-platform, and cross-study comparisons strongly support the overall high quality of our protein response data.
Figure 2. Quality assessment of RPPA-based protein expression data using independent platforms.
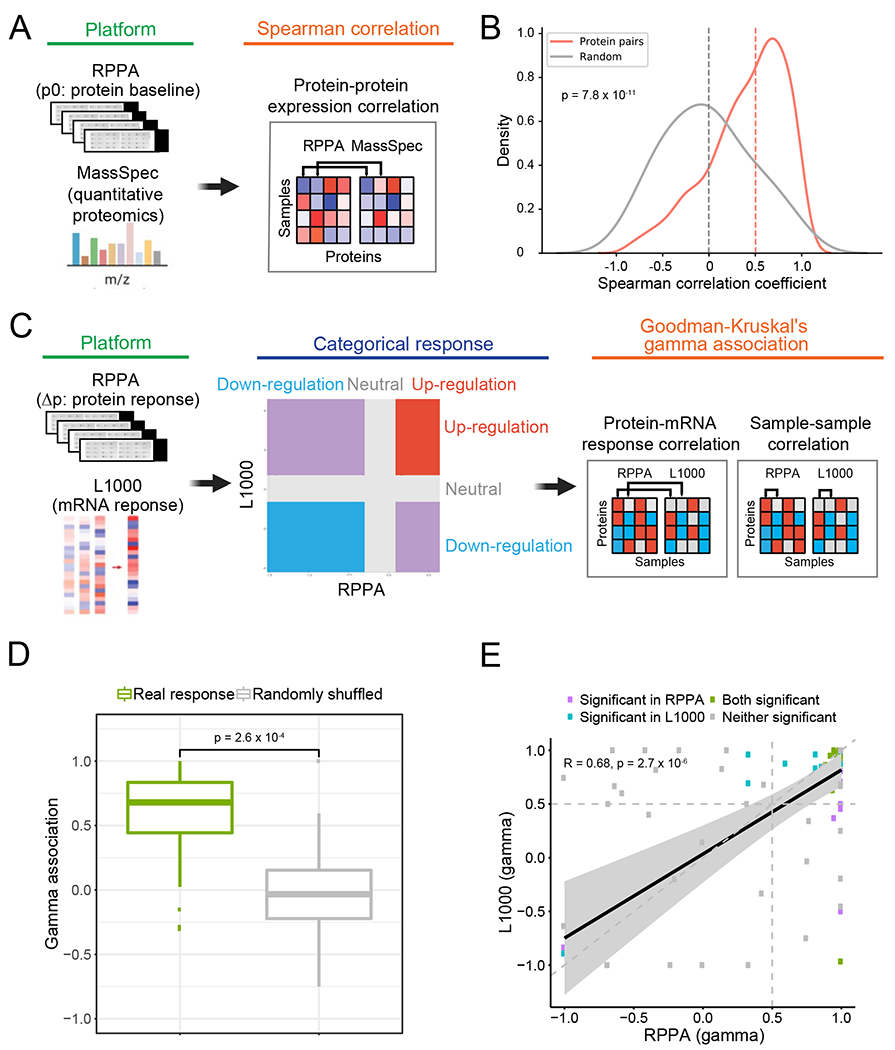
(A) Overview of the comparison between RPPA-based and mass spectrometry-based protein expression data. (B) The distribution of correlation coefficients of matched and random protein pairs between RPPA and mass spectrometry. The median values are marked by dash lines. (C) Overview of the comparison between RPPA-based protein response and L1000-based mRNA response (see STAR Methods for details). (D) Boxplots of protein-mRNA response associations between the RPPA and L1000 platforms using the same perturbations (i.e., the same cell line and the same compound, n = 46). The gamma associations from the real responses (green box) were compared to those from the randomly shuffled background distribution (grey box). The p-value is based on a paired Student’s t-test. The middle line in the box is the median, the bottom and top of the box are the first and third quartiles, and the whiskers extend to 1.5 IQR of the lower and upper quartiles, respectively. (E) Scatter plot showing the correlation of sample-sample gamma associations from the RPPA (x-axis) and L1000 (y-axis) platforms. Only significant data points (γ associations) with FDR < 0.01 in either platform are shown. Pearson’s correlation coefficient and p-value are shown.
Mechanistic insights into drug sensitivity by protein responses
To assess whether the protein response data can provide meaningful insights into phenotypic consequences of drug treatments, we focused on MCF7, a breast cancer cell line with the highest number of treated samples in our dataset, for an in-depth analysis. We first extracted RPPA data from >1,500 MCF7 samples treated by a variety of compounds. In total, these samples were treated by 19 compounds or combinations, 9 stimuli, and DMSO, at multiple time points, which covered major MCF7 drug targets including ER, PI3K/mTOR, AKT, MEK, and EGFR. To elucidate the signaling pathways underlying different drug responses, we further summarized the protein response (Δp) data in two dimensions by (i) grouping proteins into major cancer functional pathways (e.g., PI3K/AKT, RAS/MAPK, and TSC/mTOR), and (ii) categorizing compounds based on their target pathways (e.g., ER, PI3K/mTOR, and AKT). We next ranked the drug groups based on their median values of drug sensitivities in MCF7 using Cancer Therapeutics Response Portal v2 (CTRPv2) (Basu et al., 2013) and found that MCF7 was significantly more sensitive to two out of the eight drug groups (Figure 3A). The same pattern was confirmed using Genomics of Drug Sensitivity in Cancer 2 (GDSC2) (Garnett et al., 2012), another large-scale drug sensitivity data resource (Figure S2A). In particular, MCF7, an estrogen receptor (ER)-positive breast cancer cell line, showed the highest sensitivity to ER inhibitors, with PI3K/mTOR inhibitors being the second most effective group. We used the median of Δ pathway scores (Akbani et al., 2014) to represent the average pathway response to each drug group and found that, indeed, the two drug groups showed the most dramatic pathway responses (Figure 3B). Specifically, using pathway analysis as defined previously, ER inhibitors decreased TSC/mTOR and hormone_a, but increased EMT, core reactive, DNA damage, and hormone_b pathways; PI3K/mTOR compounds inhibited TSC/mTOR, PI3K/AKT, and cell cycle, but activated apoptosis and RTK signaling; other drug groups also specifically inhibited their target pathways, such as Abl/Src/c-Kit and MEK. We further compared the sensitive drug groups (ER and PI3K/mTOR) with others for each pathway and revealed several differentially altered response pathways (Figure 3C, 3D). Specifically, three pathways were inhibited in the sensitive groups, i.e., TSC/mTOR (t-test, p = 1.5×10−8), PI3K/AKT (p = 3.1×10−13), cell cycle (p = 8.1×10−8), and three pathways in the sensitive groups had significantly higher Δ pathway scores than those of other drugs, including apoptosis (p = 4.1×10−6), RTK ( p = 1.7×10−6), and RAS/MAPK (p = 7.4×10−6). These observations not only indicate that our RPPA-based protein response data successfully captured important phenotypic effects of drug treatments, but also demonstrate the ability to uncover molecular mechanisms underlying drug sensitivity.
Figure 3. Protein responses to various drug treatments in MCF7 cells.
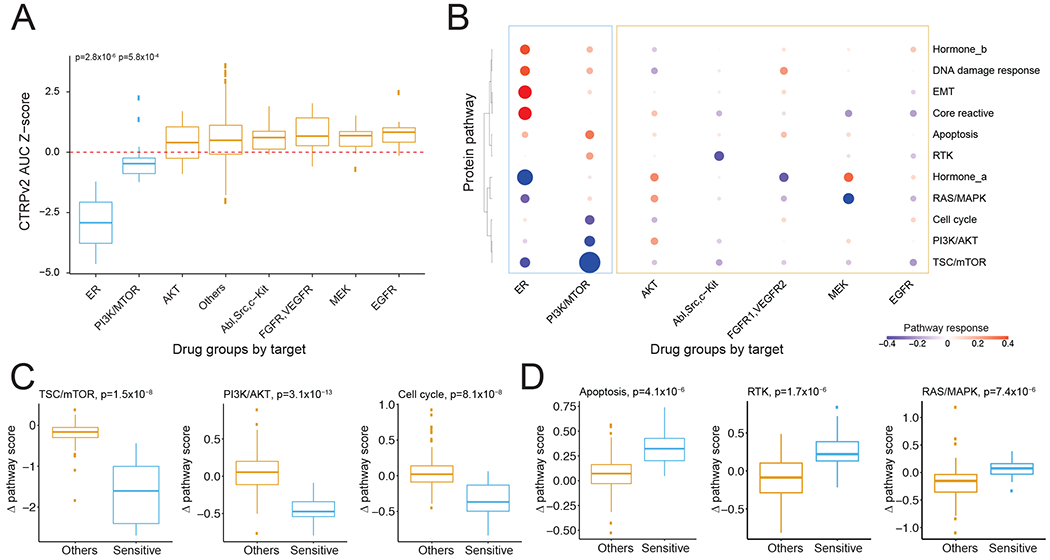
(A) A comparison of MCF7 drug sensitivity data between different drug groups using CTRPv2 data. (B) Heatmap showing the pathway responses among drug groups. The size of the circle is proportional to the effect size of the protein changes. (C, D) Boxplots showing significantly down- (C) and up-regulated pathways (D) in sensitive drug groups. The p-values were calculated by Student’s t-test. See also Figure S2.
To further demonstrate how protein response could help elucidate drug response mechanisms, we focused on the analysis of MEK inhibitors (MEKi) across different cell lines, using cobimetinib as an illustration example and considering both baseline (p0) and protein response levels (Δp) (Figure 4). Cell lines were divided into MEKi-resistant (OVCAR432: RAS pathway WT, OVCAR3: RAS pathway WT, and OAW28: MAP2K4 mutant) and MEKi-sensitive (OV90: BRAF mutant, CAOV3: RAS pathway WT, ES2: BRAF and MEK mutant, OVCAR5: KRAS mutant, JHOM1: RAS pathway WT, and OVCAR8: KRAS mutant) based on response to multiple MEK inhibitors in our and publicly available data (CTRPv2 and GDSC2). As expected, cell lines with aberrations in the RAS/MAPK pathway had a higher propensity to RAS/MAPK baseline pathway activity and sensitivity to MEKi, as indicated by low BIM and high EGFR, DUSP4, transglutaminase, pYB1, p90RSK, pMAPK, pMEK, and pJun (Pohl et al., 2005) (Figure 4A). There was also a suggestion that cell state and, particularly, decreased epithelial characteristics, or epithelial-mesenchymal transition (EMT) (low E-cadherin, beta-catenin, RAB25, ERalpha, GATA3, and high EPPK1, N-cadherin, AXL, PAI-1, and fibronectin) were associated with sensitivity to MEKi. The EMT characteristics were likely mediated, at least in part, by effects of the RAS/MAPK pathway activation noted above (Shao et al., 2014).
Figure 4. Differentially expressed protein markers between cobimetinib-sensitive and - resistant cell lines.

(A, B) Heatmaps showing baseline (A) and perturbed protein expressions (B) with significant differences between sensitive and resistant cell lines (FDR < 0.1). Each protein marker is annotated by whether it is a dual marker (i.e., significant both in p0 and Δp), BCL-2 family member, or belongs to a specific pathway. (C) Cartoon summary of baseline protein levels and adaptive protein responses to MEK inhibitors between the two cell groups. The difference of pathway scores between the two groups was assessed based on the Student’s t-test. See also Figure S3.
Sensitivity to cobimetinib was associated with evidence for a greater cobimetinib-induced decrease in RAS/MAPK pathway activity (decreased DUSP4, transglutaminase, FOXM1, p90RSK, pMAPK, pYB1, pS6, and pJun, and increased BIM), and decreased cell cycle progression (decreased pRB, cyclinB1 CDK1, PLK1, cdc25c, and Chk1, and increased p16, p21, and p27), likely as a consequence of RAS/MAPK signaling inhibition (Figure 4B). Further, there was a marked shift to an epithelial phenotype, as indicated by increased EMA, EPPK1, Claudin1, and beta-catenin (Figure 4B). Many of the associations with sensitivity to cobimetinib were identifiable in the pre-treatment samples, with the associations markedly accentuated and extended in cobimetinib-treated samples. The marked increase in BIM in response to MEKi has been identified previously and provides a biomarker for response to combined inhibition of MEKi and BCL2 family members (Cragg et al., 2008; Iavarone et al., 2019). We also performed a similar analysis using trametinib (Figure S3) and observed a marked overlap of potential biomarkers despite the analysis of different cell lines and different MEK inhibitors. We next calculated the pathway-level responses by aggregating the protein changes in the pathways (Akbani et al., 2014) and found that the adaptive pathway score changes associated with drug sensitivity include cell cycle inhibition in sensitive cell lines (t-test, p = 4.1 ×10−4, Figure 4C) and PI3K/AKT signaling activation in resistant cell lines (t-test, p = 0.015, Figure 4C). Together, the results suggest that (i) sensitivity to RAS/MAPK pathway inhibition is associated with baseline pathway activity and cell state, and (ii) acquired resistance to MEKi may come from the adaptive activation of PI3K/AKT pathways in resistant cells (Mirzoeva et al., 2009; Westin et al., 2019).
Increased predictive power for drug sensitivity by protein response
Our previous study demonstrated that RPPA-based baseline protein levels showed considerable predictive power for drug sensitivity in cancer cell lines (Li et al., 2017). Here we performed two complementary analyses to further assess the predictive power of protein responses for drug sensitivity. First, we evaluated the associations of perturbed RPPA data with drug sensitivity based on individual proteins. We integrated our RPPA data and drug sensitivity data available in GDSC2 (Iorio et al., 2016) and identified seven drugs whose sensitivity and protein expression data were available in at least five different cell lines. Then, for each drug, we defined three types of protein markers that may be informative about drug sensitivity: (i) p0: the baseline level of a protein shows a significant correlation with the sensitivity to the drug across cell lines (Pearson’s correlation, p < 0.05); (ii) Δp only: the protein response shows a significant correlation with drug sensitivity (Pearson’s correlation, p < 0.05); and (iii) Δp|p0: given p0, the protein response shows additional information content in predicting drug sensitivity. Across all the drugs, the numbers of Δp-informative (Δp only + Δp|p0) protein markers were significantly higher than those of p0-based markers (paired t-test, p = 1.38×10−3, n = 7 drugs, Figure 5A).
Figure 5. Comparison of the predictive power of protein markers for drug sensitivity.
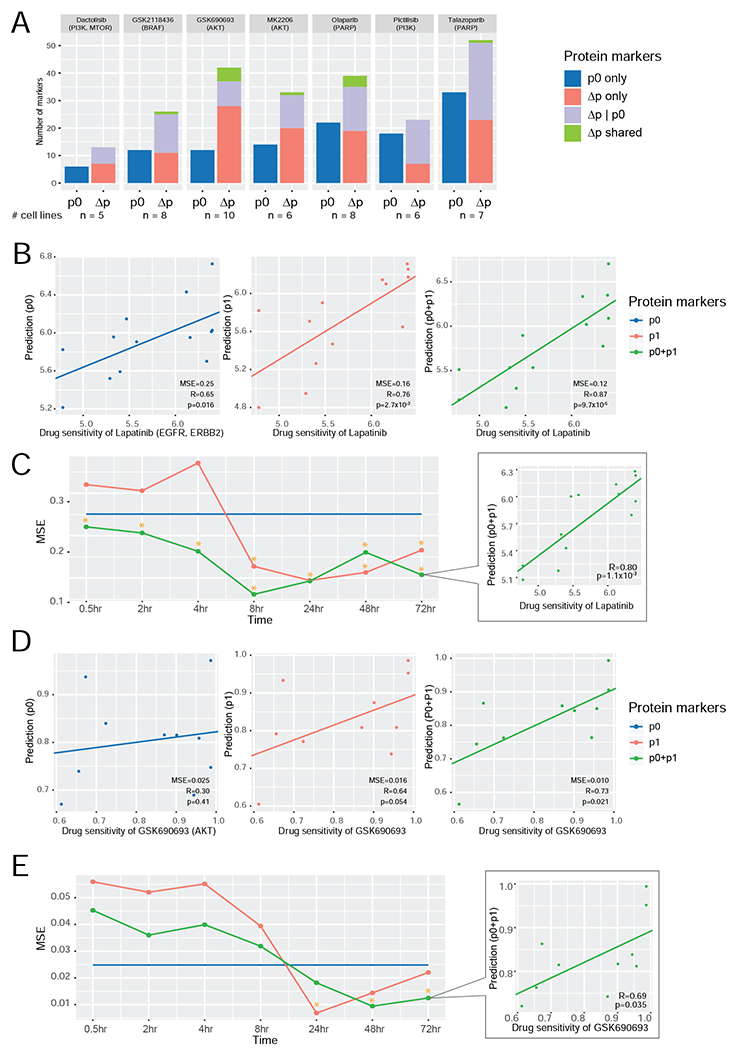
(A) A summary of predictive markers based on baseline level (p0) and protein response (Δp) using drug response data from GDSC2. Given a specific drug, three types of predictive markers were identified: (i) proteins whose p0 level is significantly correlated with drug sensitivity; (ii) proteins whose Δp level is significantly correlated with drug sensitivity; and (iii) proteins whose Δp level is significantly correlated with drug sensitivity, given the p0 contribution. Protein markers identified based on both Δp only and Δp|p0 are called Δp shared. The number of cell lines for each compound is shown at the bottom. (B, D) The scatter plots showing the correlations between the predicted and measured drug sensitivity values of lapatinib (B) and GSK690693 (D) based on the multivariate models using three sets of protein markers, respectively (left, p0; middle, p1; and right, p0 + p1). Measured sensitivity data of lapatinib (n = 13 cell lines) and GSK690693 (n = 10 cell lines) were from Daemen et al. (2013) and GDSC2, respectively. (C, E) The MSE curves of the three predictive models at different time points for lapatinib (C) and GSK690693 (E). The time points with significant correlations between the predicted and measured values are indicated with *. The scatter plot at the last time point is shown.
Second, we focused on two drugs, lapatinib, and GSK690693, which had RPPA protein and drug sensitivity data available in at least 10 cell lines ( Garnett et al., 2012; Daemen et al., 2013), in order to assess the overall performance of all RPPA protein markers for drug sensitivity prediction using a rigorous machine learning approach. For each drug, we had RPPA protein data available at seven post-treatment time points (Figure 5B–E). For comparison, we developed predictive models using elastic net based on three sets of protein markers: (i) p0: baseline level; (ii) p1: post-treatment level (averaged across different time points); and (iii) joint p0 and p1 profiles. Based on leave-one-out cross-validation, models based on joint p0 and p1 profiles had superior performance than models based on p0 or p1 only, and the predictions were significantly correlated with drug sensitivity (Figure 5B, D, Pearson’s correlation, lapatinib: p = 9.7×10−5, GSK690693: p = 0.021). Further, the p1-based models of both drugs showed better predictions than the p0-based models, and their associations with drug sensitivity were also significant (Pearson’s correlation, lapatinib: p = 2.7×10−3, GSK690693: p = 0.05). The p0-based lapatinib model was significant, but to a lesser extent (p = 0.016), and the GSK690693 model was not significant (p = 0.4). To obtain more insights into the model performance, we examined the predictive models based on the three sets of markers at each of the seven time points (Figure 5C, E). Overall, the p1 and p0+p1 joint models showed much better performance at later time points (e.g., >8hr) than earlier time points (e.g., ≤4hr). For both drugs, the prediction models based on the p0+p1 joint profiles at 48hr and 72hr had high predictive powers. The varied predictive performance at different time points suggests that during the initial stage after drug treatment, the protein responses reflect target inhibition and related collateral chaos, and it takes some time for cells to rewire signaling pathways to adapt to the treatment stress and, therefore, induced protein changes are more informative in predictive modeling. Collectively, the results in this section not only further support the high quality of the RPPA data, but also suggest that changes in protein levels on therapeutic challenge provide substantial additional information content beyond that provided by baseline protein levels for predicting treatment responses.
A systematic “protein-drug” connectivity map
To systematically evaluate the utility of our protein response data, we built a protein-drug connectivity map based on the RPPA data. In this map, each node represents a protein or a drug, protein-drug connections are based on whether the drug treatment caused a significant change of the protein, and drug-drug connections are based on whether the two drugs caused similar protein responses (Figure 6A). As expected, drugs for the same target are clustered together: for example, several MEK inhibitors and mTOR/PI3K inhibitors are highly connected, highlighting their similar downstream protein responses. This map also identifies intriguing connections: a PARP inhibitor showed both similar and opposite relationships with some drugs, suggesting potential additive or agonistic effects that could direct the development of rational drug combinations. Indeed, based on the assessment of functional proteomics changes as assessed by RPPA, we have validated the synergistic activity of PARP inhibitors and inhibition of PI3K pathway, MEK, ATR, and WEE1 inhibitors in preclinical and clinical studies (Fang et al., 2019; Shen et al., 2015; Sun et al., 2017; Sun et al., 2018).
Figure 6. A “drug-protein” connectivity map based on protein response signals.
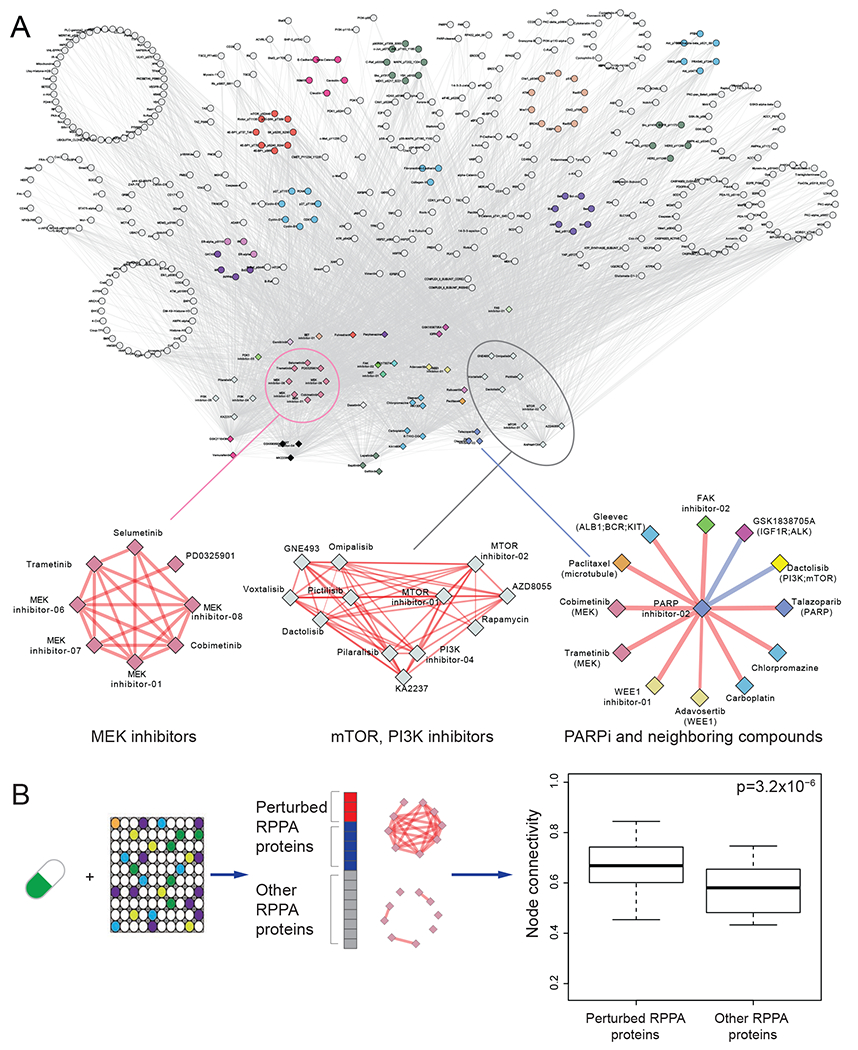
(A) A global view of the drug-protein connectivity map with highlighted examples of drug-drug correlation networks (i.e., MEK inhibitors, mTOR, PI3K inhibitors, and neighboring drugs of a PARP inhibitor). Red/blue edges represent positive/negative drug-drug correlations, respectively. Proteins were grouped and colored by their related functional pathways. Drugs were grouped and colored by their targeted genes or pathways. (B) Comparison of node connectivity between perturbed and neutral proteins in the protein interaction network. The p-value was computed based on a paired Wilcoxon test. The middle line in the box is the median, the bottom and top of the box are the first and third quartiles, and the whiskers extend to 1.5 IQR of the lower and upper quartiles, respectively. See also Figure S4 and S5.
We next studied protein-protein relationships on the map. For any given drug treatment, we classified RPPA proteins into perturbed proteins and other proteins. We found that perturbed proteins were more likely to interact than other proteins, based on the STRING database (Szklarczyk et al., 2019) (t-test, p = 3.2 ×10−6), suggesting that proteins co-perturbed by a drug tend to be involved in the same biological processes and to interact as part of a signaling cascade (Figure 6B). This global assessment using prior protein interaction knowledge supports the utility of the approach to drive biological discoveries.
Using drug-centered protein neighborhoods, we initially focused on signaling through tyrosine kinases and their downstream networks: selumetinib (target: MEK) (Figure S4A), AZD8055 (target: mTOR) (Figure S4B), GSK1838705A (target: IGF1R/ALK) (Figure S4C), and sapatinib (target: EGFR/ERBB2) (Figure S4D), and demonstrated a marked overlap in protein networks in inhibitor-perturbed cells. Interestingly, the Hsp90 inhibitor (gamitrinib) protein neighborhood (Figure S5A) demonstrated similarities to that of the tyrosine kinase pathway inhibitors, potentially due to the role of Hsp90 in stabilizing multiple members of the tyrosine kinase signaling pathway. Indeed, the similarities in the protein networks argue that the major effects of Hsp90 are likely attributable to its effects on tyrosine kinase signaling pathways (Lee et al., 2017). In contrast, rabusertib (target: Chk1) (Figure S5B) and chlorpromazine (target: autophagy) (Figure S5C) demonstrated distinct protein neighborhoods consistent with markedly different mechanisms of action.
As described above, the MEKi protein neighborhood is strongly associated with signaling through the MAPK and mTOR pathways, cell cycle progression, and cell state. Based on extensive validation of the relationships between these pathways and RAS/MAPK signaling, the associations with multiple other proteins in the neighborhood map (Figure S4A) are likely valid. Given that the MAPK pathway is a key regulator of the TSC1/2 complex that is upstream of mTORC1 signaling, the protein neighborhood of mTOR inhibitor AZD8055 (Figure S4B) was indeed highly related to the selumetinib protein neighborhood. The most marked differences between the MEK and mTOR inhibitor protein neighborhoods were represented in the upper components of the PI3K and MAPK pathway that appeared relatively independent of each other. Interestingly, the IGF1R/ALK inhibitor, GSK1838705A, protein neighborhood encompassed components of both the MEK and mTOR protein neighborhoods, consistent with the IGF1R having input into both pathways. While the strong link to the PI3K pathway was expected, a link between the IGF1R and MAPK pathway has been suggested but less studied (Molina-Arcas et al., 2013) . The pan-EGFR family inhibitor, sapatinib, neighborhood reflects EGFR family receptors being the key regulators of the PI3K and MAPK pathways in epithelial cells (Akbani et al., 2014). The EGFR family has a stronger link than either mTOR or MEK inhibitors to the DNA damage repair pathway (i.e., 53BP1, Rad50, XRCC1, pChk1/2, and BRCA2), consistent with recent studies (Russo et al., 2019; Wang et al., 2013).
Prediction of drug combinations based on protein response
To further demonstrate the utility of protein response data, we developed an integrated analysis to predict drug combinations based on pathway-level protein responses (Figure 7A). Briefly, (i) we used a drug-centered connectivity map to infer upregulated and downregulated pathways; (ii) we performed a correlation analysis to confirm whether the pathways identified in step (i) correlated with drug resistance; and (iii) based on the pathway-drug matrix, we identified drugs that were affected by the resistance pathways identified in (ii). Through this strategy, we identified 150 drug combinations for 9 specific drugs, including mTOR/PI3K, EGFR, AKT, PARP, and FGFR inhibitors (Figure 7B and Table S2). We then evaluated our predictions by determining whether the proposed drug combinations have been reported in the literature or have been employed in clinical trials since each publication or clinical trial can be viewed as substantial evidence to support a specific prediction. On average, >50% of the predicted combinations had supporting evidence, and the validation rate ranged from 90% to 10% for each drug.
Figure 7. Prediction of drug combinations based on connectivity maps.
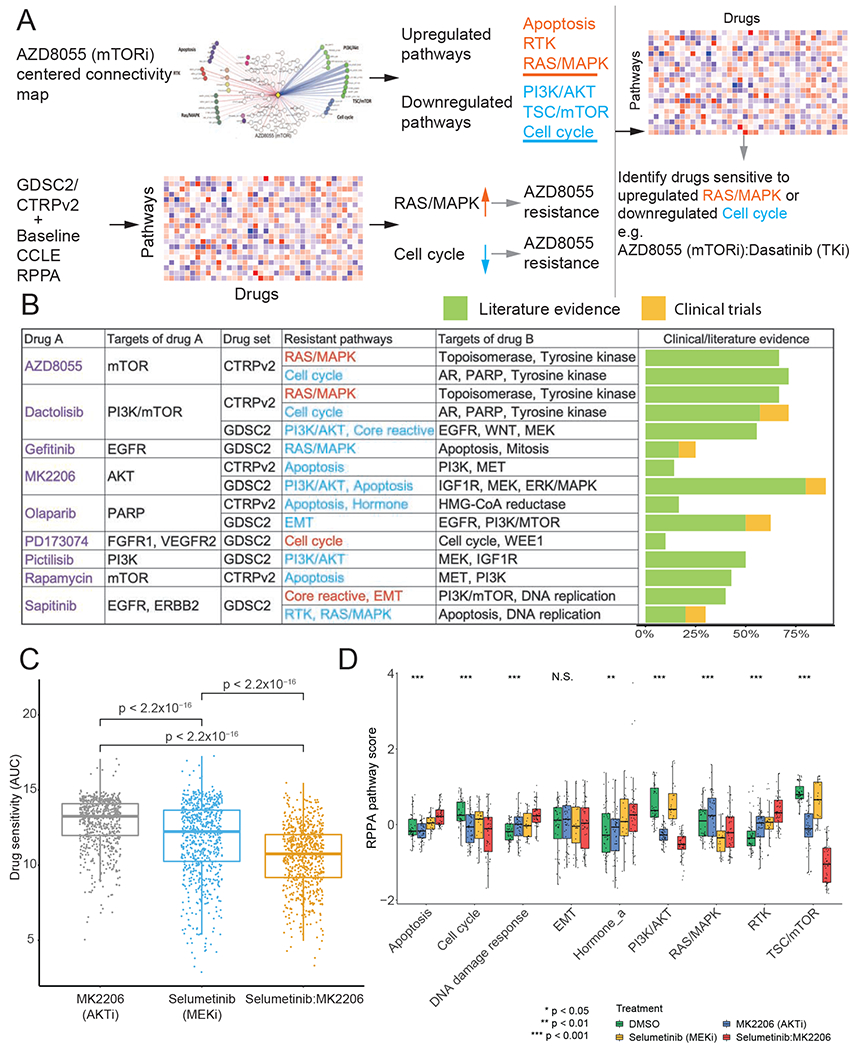
(A) The workflow of drug combination prediction. (B) Summary of predicted drug combinations and the corresponding literature/clinical evidence. (C) Boxplots showing CTRPv2 drug sensitivities of cell lines treated with MK2206 (AKT inhibitor), selumetinib (MEK inhibitor), and the combination (p-values were calculated by Wilcoxon tests; n = 706 cell lines for each treatment). (D) Protein pathway scores for samples treated with DMSO (n = 48 samples), MK2206 (n = 48 samples), selumetinib (n = 24 samples), and the combination (p-values were calculated by ANOVA tests; n = 48 samples). The middle line in the box is the median, the bottom and top of the box are the first and third quartiles, and the whiskers extend to 1.5 IQR of the lower and upper quartiles, respectively. See also Table S2.
Next, we focused on the combination of selumetinib + MK-2206 for a detailed analysis and validation using the CTRPv2 sensitivity data of the two drugs and their combination. We compared the drug sensitivities of MK2206 (AKTi) and selumetinib (MEKi) with those of their combination and found that the drug sensitivity (area under the curve, AUC) of the individual drugs was significantly higher than their combination, indicating their synergistic effects (pairwise Wilcox sum rank test, p < 2.2 × 10−16, Figure 7C). To better understand the mechanisms underlying this synergistic effect, we further analyzed the protein response data for the individual drugs and their combination (Figure 7D). We observed three general patterns from the differential analysis: (i) both drugs had similar effects on the pathway which was accentuated by the combination (TSC/mTOR, DNA damage response, RTK, hormone signaling), (ii) one drug altered the pathway and this was accentuated by the combination (apoptosis, cell cycle, and PI3K/AKT), and (iii) each drug had the opposite effect (RAS/MAPK). The most significantly perturbed pathways were PI3K/AKT and the TSC/mTOR, which were both inhibited by the combination treatment, especially the TSC/mTOR pathway. This result suggests that the synergistic effect could be due to effective inhibition of PI3K/AKT and the TSC/mTOR pathway. Notably, the cell cycle pathway was also inhibited by the combination, which appeared to be mainly due to the effects of MK2206 (AKTi).
A user-friendly data portal for community use of protein responses
To facilitate the utilization of our protein response data, we provided unrestricted access to the data through a user-friendly portal, called “Cancer Perturbed Proteomics Atlas” for fluent data exploration and analysis, which can be accessed at https://bioinformatics.mdanderson.org/public-software/cppa. The data portal provides four interactive modules: “Data Summary,” “My Protein,” “Connectivity Map,” and “Analysis” (Figure S6). The “Data Summary” module provides detailed information about each sample (including cell line, compound, dose, time, and culture conditions). The datasets can be easily downloaded through a tree-view interface. “My protein” module provides annotation of RPPA protein markers, including the corresponding genes and antibody information. The “Connectivity Map” provides an interactive approach to exploring the map, through which protein-drug and drug-drug connectivity can be examined through different visual and layout styles. The “Analysis” module provides three common analyses through which users can explore protein responses associated with a drug/compound, including protein response (Δp) rank, volcano plots for the correlations between protein responses and drug sensitivity, and box plots for differential protein responses between sensitive and resistant cell lines. Collectively, this data portal enables researchers to explore, analyze, and visualize RPPA-based protein response data intuitively and efficiently.
Discussion
Here we present a large collection of protein responses (including total and post-translationally modified proteins) upon drug treatments (>12,000 treated samples) using RPPA, which is several magnitudes larger than previously published studies. We validated the quality of our datasets in several ways. First, we demonstrated the high reproducibility of replicate samples using the same platform. Second, we established a high consistency between RPPA measurements and independent platforms such as mass spectrometry and L1000. Third, the quality of our dataset is also supported by the meaningful patterns observed in a MCF7-focused analysis and a systematic “protein-drug” connectivity map, such as the clustering of similar drugs and higher node connectivity of perturbed proteins annotated in the STRING protein interaction database. Our study represents a unique, high-quality compendium of protein responses of cancer cell lines to a diversity of compound perturbations available for use by a broad community.
The utility of our protein response dataset is several-fold. First, our dataset provides a basis for understanding cause-effect relationships, which is complementary to correlation analyses and associations that can be obtained from patient cohorts. Based on these data, it will be possible to develop quantitative predictive models of how signaling networks function in intact cellular systems. Second, we show that while there is information content in biomarkers at baseline, the information content is markedly increased when baseline and response signals are combined.
This is predicted by systems biology and engineering precepts, wherein perturbed systems contain more information than static analysis. Biomarkers designed to select treatment using baseline data frequently have a limited power to predict benefit, and our results suggest that adaptive protein responses after initial treatment could be highly informative in terms of treatment response and clinical benefit. Further clinical investigations are warranted to assess the potential benefit gains using such a strategy. Third, since protein responses reflect how cancer cells critically rewire their signaling pathways to survive and adapt to the stress of specific drug treatment, these protein signals provide a strong basis for the rational design of combination therapies, as we have demonstrated previously (Iavarone et al., 2019; Krepler et al., 2017; Krepler et al., 2016; Kwong et al., 2015; Molinelli et al., 2013; Muranen et al., 2012; Sun et al., 2017; Sun et al., 2018).
We recognize some limitations of this study. First, compared with mass spectrometry-based protein level or mRNA level readout assays, the number of protein markers that can be effectively monitored by the RPPA technology is much smaller. However, the increased sensitivity (particularly for some key proteins and phosphoproteins), and cost considerations, make RPPA a practical platform for generating such a large resource. In capturing protein responses, RPPA and mass spectrometry are complementary because of their different scopes and focuses. Second, although many perturbed protein response profiles were generated, some cell lines and drug treatments (including different dosages) are still sparsely sampled. As a result, we could assess the ability of perturbed RPPA data to predict drug sensitivity only based on a small number of drugs. Further efforts are required to obtain more comprehensive sets. However, machine learning approaches may have the potential to fill some of these gaps. Finally, as with other high-throughput technologies, there can be technical measurement errors for individual samples, and interesting observations from our study should be followed by further in-depth investigations.
We have provided an interactive, user-friendly data portal through which biomedical researchers can explore, visualize, and intuitively analyze these data. With this bioinformatics tool, we expect an effective translation of the large-scale perturbed protein data into biological knowledge and clinical utility. Together with recent efforts that have systematically characterized phenotypic and molecular responses to drug treatment, our study provides a rich resource for the research community to investigate the behaviors of cancer cells and the dependencies of treatment responses.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Han Liang (hliang1@mdanderson.org).
Material Availability
This study did not generate new unique reagents.
Data and Code Availability
The RPPA data generated in this study can be found at the CPPA data portal: https://bioinformatics.mdanderson.org/public-software/cppa. The quantitative mass spectrometry data of CCLE were downloaded from Table S2 of Nusinow et al. (2020). The L1000 gene expression data were downloaded from Gene Expression Omnibus series GSE92742. The CTRPv2 drug sensitivity data were downloaded from CTRPv2 (https://portals.broadinstitute.org/ctrp/). The GDSC2 drug sensitivity data were downloaded from GDSC Release 6.1 (https://www.cancerrxgene.org). The drug sensitivity data of lapatinib were downloaded from Table S1 of Daemen et al. (2013). The protein-protein interaction network data were downloaded from STRING (https://string-db.org).
All software supporting the analysis in this study can be found in public repositories. SuperCurve is available at https://bioinformatics.mdanderson.org/public-software/supercurve/; Cytoscape is available at https://cytoscape.org; DataTables is available at https://datatables.net; and HighCharts is available at https://www.highcharts.com.
Experimental Model and Subject Details
Cell lines
We collected cancer cell lines through the MD Anderson Cancer Center (MDACC) CCSG-supported Cell Line Characterization Core Facility (Houston, TX, USA) and from several outside collaborations (see Table S1 for details). All cell lines prepared at MDACC were confirmed by short tandem repeat (STR) analysis in the core per institutional policy, and the outside collaborators also routinely confirmed cell lines by STR analysis.
Method Details
RPPA experiments
RPPA experiments were performed at the RPPA core facility at MDACC. Cell line samples were prepared, and antibodies were validated by comparison with immunoblotting, as previously described (Hennessy et al., 2010; Li et al., 2017). Briefly, lysates were manually serial-diluted in 5 two-fold dilutions with lysis buffer and printed on nitrocellulose-coated slides using an Aushon Biosystems 2470 arrayer. Slides were probed with validated primary antibodies, followed by detection with appropriate biotinylated secondary antibodies (Biotin conjugated-Goat anti-Rabbit IgG, Goat anti-Mouse IgG, or Rabbit anti-Goat IgG, from Vector Lab). The signal obtained was amplified using a Dako Cytomation–catalyzed system of avidin-biotin-peroxidase (from Agilent) binding to the secondary antibody and catalyzing a tyramide-biotin conjugation to form insoluble biotinylated phenols. Stained RPPA slides were first quantified using ArrayPro (Media Cybernetics) to generate signal intensities (level 1). Then, the SuperCurve software processed spots from all horizontal samples on the slide to determine the relative protein level for each sample (level 2). Standard parameters were used in this step, including nonparametric curve fitting via monotone increasing B-spline, and use of spatial adjustment for regional correction. Finally, protein measurements were corrected for loading using median polish (level 3). RPPA slide quality was assessed by a quality control classifier in the R package “SuperCurve” (Ju et al., 2015), which was trained in curated RPPA data sets using a generalized linear model and logistic function. Only slides with a quality score above 0.8 (range: 0-1) were retained for further analysis. In total, we generated RPPA data from 15,842 samples, including 12,183 treated cell line samples and 3,659 baseline samples (e.g., treated with DSMO).
Quality assessment within the RPPA platform
Internal quality assessment of RPPA data was first performed in baseline samples. We examined the consensus of the RPPA signals in baseline samples of the same cell line and excluded the samples for which average correlation coefficients with other baseline samples were < 0.5. Next, we performed a quality assessment of post-treatment samples. For each pair of post-treatment replicate samples, we generated protein response (Δp) profiles using the corresponding baseline samples that passed the consensus test and compared the Δp profile of the replicate samples (n = 2,753 pairs). Samples pairs with low Pearson correlation were removed from the analysis. In total, 11,884 post-treatment samples and 3,608 baseline samples passed the internal quality assessment. For each treated sample, we calculated Δp for each protein by deducting its protein level in the corresponding control sample. When replicates (technical or biological) were available, we used the average level across replicates. By combining replicates, we generated 1,916 unique baseline protein profiles and 7,941 unique protein response profiles of post-treatment samples. To generate the time-independent Δp profile for a specific treatment in a cell line, we merged the protein responses across different time points by taking the average.
Quality assessment using independent platforms
We obtained normalized CCLE protein expression data, generated through quantitative mass spectrometry, from a recent study (Nusinow et al., 2020). For each of the baseline RPPA sets that had an overlap with the proteomics dataset of ≥ 8 cell lines, we calculated the Spearman correlation coefficient between the normalized expressions generated by these two assays across the common cell lines, and in total, 169 unique proteins were included in the analysis. A background distribution of correlations was built by computing the Spearman correlation coefficient between each pair of different proteins across the same cell lines in the same sets.
We downloaded the level-5 data of L1000 phase 1 from the GEO database (GSE92742). For a fair comparison, we collected data from the same cell lines perturbed by the same compound. In total, 46 “perturbation-cell-line” IDs (60 samples) and 347 genes/proteins (total proteins but phosphoprotein if a total protein was not available) commonly shared by the two platforms were used in the subsequent analyses. For a perturbation-cell-line ID with multiple concentration and/or time points, we adopted the median value across all conditions as the representative response score. For each platform, we first converted the continuous response to a categorical response: upregulated, downregulated, or neutral. Random events were defined by the global median ± 35% quantile, calculated from the full matrix. Next, we excluded the random events and computed Goodman-Kruskal’s gamma (γ) to estimate sample associations across genes. We evaluated the concordance between RPPA and L1000 platforms through two analyses. (i) Protein-mRNA response associations: for each sample, a γ association between the two platforms was computed across genes/proteins when at least 12 genes showed up/downregulation. To generate the background distribution, we randomly shuffled protein labels and computed the response associations between the shuffled proteins and mRNAs (the seed used for randomization is “1234”). Then, a paired Student’s t-test was used to evaluate the statistical significance of the group difference between the real and matched randomly shuffled responses. (ii) Sample-sample associations: in our RPPA dataset, a perturbation-cell-line ID might have replicate samples. Here, we only retained the one with the best protein-mRNA response association from the previous analysis. Next, for samples that showed up/downregulation of > 3 genes, γ associations for every two such samples were computed within each platform (within the same batch). Then, Pearson’s correlation between the significant γ associations (FDR < 0.01 for each platform) was used to evaluate the consistency between the protein and mRNA responses.
Analysis of predictive protein markers of drug sensitivity
We collected drug sensitivity data from public resources including GDSC2, CTRPv2, and Daemen et al. (2013). For MCF7 analysis, the AUC Z-scores () were calculated based on AUCs for each drug across all screened cell lines. We calculated the median values of perturbed pathway scores (Δ pathway scores). The AUC difference between a specific drug group and other drugs was assessed by unpaired Student’s t-test, and the significant ones were identified with the cut-off of FDR < 0.01.
To identify the differential protein markers of drug sensitivity and resistance, cell lines were classified as sensitive or resistant to a specific drug based on the consensus call of CTRPv2, GDSC2, and in-house datasets. Baseline levels (p0) and protein response levels (Δp) with a significant difference between sensitive and resistant cell lines were identified by unpaired Student’s t-test. Pathway-level scores (Li et al., 2017) were similarly analyzed.
To assess the predictive power of perturbed RPPA data across cell lines, perturbation data of the same cell line treated with the same compound at different dosages or time points were averaged using mean values. For univariate analysis, seven compounds had both RPPA perturbation profiling and drug sensitivity data in ≥5 cell lines based on GDSC2 data (see Figure 5A for the sample size information of each drug). For each compound, the baseline levels (p0) and protein response levels (Δp) of each antibody were tested for associations with drug sensitivity (IC50 or AUC score) in univariate linear models. The joint markers (Δp|p0) were defined as the predictions of linear regression models, including both baseline and protein response for specific antibodies. Predictive markers were selected by Pearson correlation at a significance level of p = 0.05. We then developed a multivariate model to predict drug sensitivity using the leave-one-out approach and three sets of protein profiles: baseline level (p0), post-treatment level (p1), and joint profile (p0 and p1). For the machine-learning-based analysis, to provide sufficient statistical power for feature selection and cross validation, we examined compounds with both RPPA perturbation data and drug sensitivity data in ≥10 cell lines. Only lapatinib (drug data from Daemen et al., n = 13 cell lines.) and GSK690693 (drug data from GDSC2, n = 10 cell lines) were able to generate statistically meaningful models for assessment. In each round of cross-validation, one cell line was left out as the validation set. In the training set, candidate markers were first selected with a univariate correlation test at a significance level of p = 0.1, with a maximum size of 20. The model was then built by Elastic Net based on candidate markers in the training set and was applied to the validation set. In time-series experiments, we performed cross-validation for each time point using the three sets of protein profiles mentioned above. The performance was evaluated using the mean squared error (MSE) of prediction in the validation set.
Construction of a drug-protein connectivity map
The association of each drug-protein pair was assessed by testing the difference of protein expression between baseline (p0) and post-treatment level (p1) based on the paired t-test across cell lines. For each drug-drug pair, we used Goodman-Kruskal’s γ to calculate the associations, as described in the comparison between RPPA-based protein response and L1000-based mRNA response data. The significantly correlated drug-protein and drug-drug pairs (FDR < 0.1) were used to construct a global drug-protein connectivity map. In the connectivity map, proteins were grouped and colored by their related protein functional pathways, and drugs were grouped and colored by their targeted genes or pathways. For each drug, the network densities were calculated for the two subsets of RPPA proteins: (i) proteins significantly differentially expressed between p0 and p1 (perturbed RPPA proteins), and (ii) other RPPA proteins (neutral proteins). The network density D of a protein subset with size N was defined as a ratio of the number of protein-protein interactions (E) to the number of all possible protein pairs (), i.e., D = E/Emax. Protein-protein interaction information was obtained from the STRING database (Szklarczyk et al., 2019). A paired Wilcoxon test was performed to assess the difference of the network densities between the perturbed and other proteins of all the drugs. For Figure S4 and S5, the examples of drug-centered connectivity maps were generated separately with colored edges (red: upregulated in post-treatment; blue: downregulated in post-treatment). The edge widths were proportional to the differential expression between baseline (p0) and post-treatment levels (p1). All network views were generated by the Rcy3 library and Cytoscape (Otasek et al., 2019; Shannon et al., 2003).
Prediction and validation of drug combinations
Each drug-centered connectivity map was extracted from the full connectivity map (Figure 6). The perturbed protein markers were grouped into their associated protein pathways, e.g., apoptosis and RAS/MAPK. The baseline and perturbed pathway scores were calculated based on the weighted average of baseline (obtained from CCLE) and perturbed protein levels, respectively. The perturbed pathway scores were used to infer the pathway responses to each drug. The baseline pathway scores were used to perform correlation analysis between pathways and drug sensitivity data, which were further used to infer drug resistance pathways. Focusing on the resistance pathways, drugs were predicted as combination candidates when we found (i) positive correlations (FDR < 0.1) between AUC and downregulated pathways or (ii) negative correlations (FDR < 0.1) between AUC and upregulated pathways. The top 10 combinations for each drug were reported based on the rank of their correlation coefficients. The literature and clinical evidence were obtained from PubMed (https://pubmed.ncbi.nlm.nih.gov) and clinical trial database (https://clinicaltrials.gov). The sensitivity data (AUC) of MK-2206 (AKTi), selumetinib (MEKi), and their combination were obtained from CTRPv2.
Data portal development
All RPPA and drug sensitivity data accompanying the pre-calculated analytic results were stored in a CouchDB database. We generated all the analytic results in R. We implemented a user-friendly and interactive web interface in JavaScript. Specifically, tabular results were generated by DataTables, box and scatter plots were generated by HighCharts, and interactive network views were implemented by Cytoscape.js library.
Quantification and Statistical Analysis
Statistical analysis was performed using R (version 3.6.2). To assess the correlation between two continuous variables, Pearson or Spearman rank correlation test was used; to compare two groups within the RPPA platform, Wilcoxon test or Student’s t-test was used; to compare multiple groups with the RPPA platform, analysis of variance (ANOVA) was used; to compare the associations between RPPA and L1000 platforms, Goodman-Kruskal’s gamma (γ) test was used; for the machine-learning models for drug sensitivity prediction, elastic net regression was used. Detailed descriptions of statistical tests were provided in the Method Details section and in the respective figure legends.
Supplementary Material
Table S1. Detailed information of cell lines used in this study, related to Figure 1.
Table S2. Detailed information about the predicted drug combinations, related to Figure 7.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Perturbed RPPA data | CPPA data portal | https://bioinformatics.mdanderson.org/public-software/cppa |
| Quantitative mass spectrometry data of CCLE | Nusinow et al., 2020 | Table S2 |
| Connectivity map L1000 phase 1 gene expression assay data | Gene Expression Omnibus; Subramanian et al., 2017 | http://www.ncbi.nlm.nih.gov/geo/; GEO:GSE92742 |
| CTRPv2 drug sensitivity data | Cancer Therapeutics Response Portal v2 | https://portals.broadinstitute.org/ctrp/ |
| GDSC2 drug sensitivity data | Genomics of Drug Sensitivity in Cancer | https://www.cancerrxgene.org |
| Daemen et al. drug sensitivity data | Daemen et al., 2013 | Table S1 |
| Protein-protein interaction network data | STRING; Szklarczyk et al., 2019 | https://string-db.org |
| Experimental Models | ||
| Cell lines | This study | Table S1 |
| Software and Algorithms | ||
| SuperCurve | Ju et al., 2015 | https://bioinformatics.mdanderson.org/public-software/supercurve/ |
| Cytoscape | Otasek et al., 2019; Shannon et al., 2003 | https://cytoscape.org |
| DataTables | https://datatables.net | |
| HighCharts | https://www.highcharts.com | |
Highlights.
A large collection of cancer cell line protein responses to drug perturbations
Perturbed protein responses greatly increase predictive power for drug sensitivity
Build a systematic map of protein-drug connectivity based on response profiles
Develop a user-friendly, interactive data portal for community use
Acknowledgments
This study was supported by the National Institutes of Health (U01CA168394, U24CA143883, U54HG008100, P50CA098258, and P50CA217685 to G.B.M., U24CA209851 and U01CA217842 to H.L. and G.B.M., P50CA221703 to H.L., U24CA210950 and U24CA210949 to R.A. and G.B.M., R50CA221675 to Y.L., UL1TR003167 to A.K., and Cancer Center Support Grant P30CA016672), a kind gift from the Sheldon and Miriam Adelson Medical Research Foundation, Susan G Komen (SAC110052), Ovarian Cancer Research Foundation (545152), and Breast Cancer Research Foundation (BCRF-18-110) to G.B.M., the Lorraine Dell Program in Bioinformatics for Personalization of Cancer Medicine to H.L., a DoD/CDMRP (W81XWH-16-1-0237) to R.A., Cancer Prevention and Research Institute of Texas (CPRIT) training fellowships (Grant No. RP170593 to M.M.C. and Grant No. RP160015 to S.V.K.), and a CPRIT grant (RP170640), an OCRA collaborative research development award, and an ICI Fund Award to A.K. This work was supported by the National Cancer Institute’s Office of Cancer Genomics Cancer Target Discovery and Development (CTD^2) initiative. The results published here are based in whole or in part upon data generated by CTD^2 Network (https://ocg.cancer.gov/programs/ctd2/data-portal) established by the National Cancer Institute’s Office of Cancer Genomics. We thank Kamalika Mojumdar for editorial assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
R.A. is a bioinformatics consultant for the University of Houston. G.B.M. is on the Scientific Advisory Board or a consultant for AstraZeneca, ImmunoMet, Lilly, Nuevolution, PDX Pharmaceuticals, Symphogen, and Tarveda, has stock options with Catena Pharmaceuticals, Immunomet, Signalchem, and Tarveda, and has licensed technologies to Myriad and Nanostring. H. L. is a shareholder and scientific advisor of Precision Scientific Ltd.
References
- Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J, et al. (2014). A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun 5, 3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 492, 290–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, et al. (2013). An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, and Scott CL (2008). Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest 118, 3651–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daemen A, Griffith OL, Heiser LM, Wang NJ, Enache OM, Sanborn Z, Pepin F, Durinck S, Korkola JE, Griffith M, et al. (2013). Modeling precision treatment of breast cancer. Genome Biol 14, R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, Ju Z, Vellano CP, Lu Y, Li Y, et al. (2019). Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell 35, 851–867 e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. (2012). Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Lu Y, Gonzalez-Angulo AM, Carey MS, Myhre S, Ju Z, Davies MA, Liu W, Coombes K, Meric-Bernstam F, et al. (2010). A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin Proteomics 6, 129–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter C, and Zenklusen JC (2018). The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 173, 283–285. [DOI] [PubMed] [Google Scholar]
- Iavarone C, Zervantonakis IK, Selfors LM, Palakurthi S, Liu JF, Drapkin R, Matulonis UA, Hallberg D, Velculescu VE, Leverson JD, et al. (2019). Combined MEK and BCL-2/XL Inhibition Is Effective in High-Grade Serous Ovarian Cancer Patient-Derived Xenograft Models and BIM Levels Are Predictive of Responsiveness. Mol Cancer Ther 18, 642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H, et al. (2016). A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, Xiao Y, Yu KD, Liu YR, Yu Y, et al. (2019). Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 35, 428–440 e425. [DOI] [PubMed] [Google Scholar]
- Korkut A, Wang W, Demir E, Aksoy BA, Jing X, Molinelli EJ, Babur O, Bemis DL, Onur Sumer S, Solit DB, et al. (2015). Perturbation biology nominates upstream-downstream drug combinations in RAF inhibitor resistant melanoma cells. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepler C, Sproesser K, Brafford P, Beqiri M, Garman B, Xiao M, Shannan B, Watters A, Perego M, Zhang G, et al. (2017). A Comprehensive Patient-Derived Xenograft Collection Representing the Heterogeneity of Melanoma. Cell Rep 21, 1953–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepler C, Xiao M, Sproesser K, Brafford PA, Shannan B, Beqiri M, Liu Q, Xu W, Garman B, Nathanson KL, et al. (2016). Personalized Preclinical Trials in BRAF Inhibitor-Resistant Patient-Derived Xenograft Models Identify Second-Line Combination Therapies. Clin Cancer Res 22, 1592–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong LN, Boland GM, Frederick DT, Helms TL, Akid AT, Miller JP, Jiang S, Cooper ZA, Song X, Seth S, et al. (2015). Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J Clin Invest 125, 1459–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Saini N, Parris AB, Zhao M, and Yang X (2017). Ganetespib induces G2/M cell cycle arrest and apoptosis in gastric cancer cells through targeting of receptor tyrosine kinase signaling. Int J Oncol 51, 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao W, Akbani R, Liu WB, Ju ZL, Ling SY, Vellano CP, Roebuck P, Yu QH, Eterovic AK, et al. (2017). Characterization of Human Cancer Cell Lines by Reverse-phase Protein Arrays. Cancer Cell 31, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ER 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, et al. (2017). Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 170, 577–592 e510. [DOI] [PubMed] [Google Scholar]
- Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, Clauser KR, Qiao JW, Gritsenko MA, Moore RJ, et al. (2014). Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics 13, 1690–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva OK, Das D, Heiser LM, Bhattacharya S, Siwak D, Gendelman R, Bayani N, Wang NJ, Neve RM, Guan Y, et al. (2009). Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res 69, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Arcas M, Hancock DC, Sheridan C, Kumar MS, and Downward J (2013). Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov 3, 548–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinelli EJ, Korkut A, Wang W, Miller ML, Gauthier NP, Jing X, Kaushik P, He Q, Mills G, Solit DB, et al. (2013). Perturbation biology: inferring signaling networks in cellular systems. PLoS Comput Biol 9, e1003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, and Brugge JS (2012). Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 21, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PK, Li J, Jeong KJ, Shao S, Chen H, Tsang YH, Sengupta S, Wang Z, Bhavana VH, Tran R, et al. (2018). Systematic Functional Annotation of Somatic Mutations in Cancer. Cancer Cell 33, 450–462 e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka S, Charboneau L, Young L, Major S, Reinhold WC, Waltham M, Kouros-Mehr H, Bussey KJ, Lee JK, Espina V, et al. (2003). Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc Natl Acad Sci U S A 100, 14229–14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER 3rd, Kalocsay M, Jane-Valbuena J, Gelfand E, Schweppe DK, Jedrychowski M, et al. (2020). Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402 e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otasek D, Morris JH, Boucas J, Pico AR, and Demchak B (2019). Cytoscape Automation: empowering workflow-based network analysis. Genome Biol 20, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl G, Ho CL, Kurman RJ, Bristow R, Wang TL, and Shih Ie M (2005). Inactivation of the mitogen-activated protein kinase pathway as a potential target-based therapy in ovarian serous tumors with KRAS or BRAF mutations. Cancer Res 65, 1994–2000. [DOI] [PubMed] [Google Scholar]
- Russo M, Crisafulli G, Sogari A, Reilly NM, Arena S, Lamba S, Bartolini A, Amodio V, Magri A, Novara L, et al. (2019). Adaptive mutability of colorectal cancers in response to targeted therapies. Science 366, 1473–1480. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, et al. (2014). KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, Kapoor P, Ju Z, Mo Q, Shih Ie M, et al. (2015). ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov 5, 752–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437–1452 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Fang Y, Yin J, Chen J, Ju Z, Zhang D, Chen X, Vellano CP, Jeong KJ, Ng PK, et al. (2017). Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, Vellano CP, Ju Z, Zhao W, Zhang D, et al. (2018). BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 33, 401–416 e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, and Kornblau SM (2006). Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther 5, 2512–2521. [DOI] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yuan JL, Zhang YT, Ma JJ, Xu P, Shi CH, Zhang W, Li YM, Fu Q, Zhu GF, et al. (2013). Inhibition of both EGFR and IGF1R sensitized prostate cancer cells to radiation by synergistic suppression of DNA homologous recombination repair. PLoS One 8, e68784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westin SN, Sill MW, Coleman RL, Waggoner S, Moore KN, Mathews CA, Martin LP, Modesitt SC, Lee S, Ju Z, et al. (2019). Safety lead-in of the MEK inhibitor trametinib in combination with GSK2141795, an AKT inhibitor, in patients with recurrent endometrial cancer: An NRG Oncology/GOG study. Gynecol Oncol 155, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise HC, and Solit DB (2019). Precision Oncology: Three Small Steps Forward. Cancer Cell 35, 825–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Detailed information of cell lines used in this study, related to Figure 1.
Table S2. Detailed information about the predicted drug combinations, related to Figure 7.
Data Availability Statement
The RPPA data generated in this study can be found at the CPPA data portal: https://bioinformatics.mdanderson.org/public-software/cppa. The quantitative mass spectrometry data of CCLE were downloaded from Table S2 of Nusinow et al. (2020). The L1000 gene expression data were downloaded from Gene Expression Omnibus series GSE92742. The CTRPv2 drug sensitivity data were downloaded from CTRPv2 (https://portals.broadinstitute.org/ctrp/). The GDSC2 drug sensitivity data were downloaded from GDSC Release 6.1 (https://www.cancerrxgene.org). The drug sensitivity data of lapatinib were downloaded from Table S1 of Daemen et al. (2013). The protein-protein interaction network data were downloaded from STRING (https://string-db.org).
All software supporting the analysis in this study can be found in public repositories. SuperCurve is available at https://bioinformatics.mdanderson.org/public-software/supercurve/; Cytoscape is available at https://cytoscape.org; DataTables is available at https://datatables.net; and HighCharts is available at https://www.highcharts.com.