

Abstract
Patients with Hutchinson-Gilford progeria syndrome (HGPS) have low bone mass and an atypical skeletal geometry that manifests in a high risk of fractures. Using both in vitro and in vivo models of HGPS, we demonstrate that defects in the canonical WNT/β-catenin pathway, seemingly at the level of the efficiency of nuclear import of β-catenin, impair osteoblast differentiation and that restoring β-catenin activity rescues osteoblast differentiation and significantly improves bone mass. Specifically, we show that HGPS patient-derived iPSCs display defects in osteoblast differentiation, characterized by a decreased alkaline phosphatase activity and mineralizing capacity. We demonstrate that the canonical WNT/β-catenin pathway, a major signaling cascade involved in skeletal homeostasis, is impaired by progerin, causing a reduction in the active β-catenin in the nucleus and thus decreased transcriptional activity, and its reciprocal cytoplasmic accumulation. Blocking farnesylation of progerin restores active β-catenin accumulation in the nucleus, increasing signaling, and ameliorates the defective osteogenesis. Moreover, in vivo analysis of the Zmpste24−/− HGPS mouse model demonstrates that treatment with a sclerostin-neutralizing antibody (SclAb), which targets an antagonist of canonical WNT/β-catenin signaling pathway, fully rescues the low bone mass phenotype to wild-type levels. Together, this study reveals that the β-catenin signaling cascade is a therapeutic target for restoring defective skeletal microarchitecture in HGPS.
Keywords: Progeria, HGPS, Mesenchymal Stem Cell, WNT/β-Catenin Signaling, Osteogenic Differentiation and Aging
INTRODUCTION
Hutchinson-Gilford progeria syndrome (HGPS) is a rare genetic disorder of accelerated aging. The majority of HGPS cases are caused by a de novo single nucleotide mutation (C1824T, G608G) in the Lamin A gene (LMNA), which results in the activation of a cryptic splice donor site in exon 11. This point mutation leads to a deletion of 150 nucleotides in its mRNA and produces a permanently farnesylated mutant protein called progerin.(1)(2)(3) Progerin disturbs the function of proper nuclear structural scaffolding and attributes to defective progeroid phenotypes, which at the cellular level that are characterized by nuclear blebbing, nuclear pore clustering, disrupted peripheral heterochromatin, and misregulated gene expression.(4)(5) Patients with HGPS exhibit age-related conditions, including alopecia, growth retardation, lipodystrophy, wrinkling of skin, severe skeletal defects, and arterial diseases.(6)(7) Defects in cardiovascular function are the foremost cause of death in the patients, and can be managed with Lonafarnib, a farnesyltransferase inhibitor (FTI), improving cardiovascular function and extending the life span of patients.(8)(66) Statins and aminobisphosphonates (N-BPs) further inhibited the level of farnesylation of progerin and extended the lifespan in Zmpste24-deficient (Zmpste24−/−) mice, one of the HGPS mouse models.(9) These data confirm that farnesylation of progerin is key to its deleterious action, and that targeting farnesylation can be beneficial, but the molecular basis of its action is unclear.
In addition to cardiac defects, HGPS patients exhibit low bone mass with increased fracture risk, hypoplastic or aplastic clavicles, osteolysis in ribs, skeletal dysplasia, and delayed recovery of bone fractures.(10)(11)(12)(13)(14) However, the molecular basis leading to these profound skeletal changes are unknown, but likely involve defects in nuclear function caused by farnesylated progerin. A-type lamins have been implicated in the regulation of bone development. Lmna−/− mice exhibit low bone mass and a substantial reduction in osteoblast and osteoclast number.(15) Among HGPS mouse models,(16) Zmpste24−/− mice displays the most severe skeletal phenotype, which approximates that seen in HGPS patients.(17)(18)(19) Interestingly, a tissue-specific inducible transgenic mouse model that expresses progerin in osteoblasts not only exhibits defects in bone quality but and a significant reduction in the expression of Wnt-mediated Lef1,(20) suggesting impaired canonical β-catenin signaling. However, direct evidence of the involvement of WNT/β-catenin signaling to the skeletal phenotype in HGPS has not been established.
Canonical WNT/β-catenin signaling has major roles in stem cell proliferation, lineage allocation, and differentiation. In particular, β-catenin signaling is a major player in coordinating osteoblastogenesis, bone mass acquisition, and tissue remodeling.(21)(22)(23)(24)(25) β-catenin knockout mice have reduced femur length, diminished trabecular and cortical bone mass, and lower body weight than the control or heterozygous mice. Likewise, mice lacking β-catenin specifically in osteoblast- or osteocyte-lineage have progressively reduced bone mass in the axial and appendicular skeleton and exhibit increased bone resorption by osteoclasts.(23)(26)
A few pieces of evidence have suggested that HGPS might be associated with defective WNT/β-catenin signaling cascades.(20)(27)(28) Stem cells isolated from Zmpste24−/− mice exhibit alterations in the WNT signaling pathway.(27) Furthermore, this Zmpste24−/− mouse model shows a reduced level of active β-catenin in their epidermis and hair follicle cells.(27) These data imply that a global defect in β-catenin signaling may underpin the phenotypes of HGPS.
Here, we conducted in vitro and in vivo investigations of the osteoblast differentiation phenotypes in HGPS, and the potential roles of β-catenin signaling in this process. The results indicate that β-catenin signaling, osteoblast differentiation, and osteoblast function are impaired based on in vitro differentiation analysis using both HGPS patient-derived induced pluripotent stem cells (iPSCs) and human bone marrow derived mesenchymal stem cells (hBM-MSCs) overexpressing the mutant progerin. Furthermore, in vivo analysis shows that SclAb, which targets an antagonist of canonical WNT/β-catenin signaling pathway, restores active β-catenin and fully rescues low bone mass in the Zmpste24−/− mouse model.
MATERIALS AND METHODS
Cell Lines and in vitro Osteogenic Differentiation
Normal and two HGPS(I) and HGPS(II) fibroblast cell lines (HGADFN168, HGADFN164, HGADFN167) were obtained from the Progeria Research Foundation cell bank. These cell lines have been fully characterized, including the karyotypes.(39)(40)(41) Induced pluripotent stem cells (iPSCs) were generated by reprogramming with KLF4, SOX2, OCT4, and C-MYC of Yamanaka combinations, as we have described previously.(29) The iPSC derived osteoprogenitor cells were cultured by disassociating iPSC colonies that were treated with ROCK inhibitor Y-27632, and growing in MSCs derivation medium which contains alpha-MEM, 10%FBS, 100nM dexamethasone, and 50μM L-ascorbic acid.(29) These iPSC-derived progenitor cells were then maintained in alpha-MEM with 10% Hi-FBS, 2mM L-glutamine and non-essential amino acid. Our previous publication confirmed that these iPSC-progenitor cells could be induced into adipogenic cells, which was supported by the results from qPCR analysis, immunofluorescence, and lipid production using Oil Red O staining after 21days of culture.(29) Also, these MSC-like cells derived from iPSCs were characterized by labeling and sorting with positive (CD90) and negative (CD45) markers which are most representative surface markers for MSCs.(29) As controls, commercially prepared human bone-marrow derived -MSCs (hBM-MSCs) were purchased from Rooster Bio (#00022; Rooster Bio). The wild-type hBM-MSCs were infected with lentivirus to overexpress GFP-lamin A, GFP-progerin and GFP-SSIM progerin as previously described.(35)(67) Plasmids were sub-cloned into lentiviral vector pHR-SIN-CSGW d1NotI. After confirming the positive sequence, virus was generated by co-transfecting HEK293T cells with the lentiviral plasmids and the packaging vectors, CMV‐8.2ΔR, and pCMV‐VSVG using Fugene 6 (#E2692; Promega).(35)(67) hBM-MSCs were transduced with each type of lentiviruses for 48hrs, and GFP efficiency was checked under fluorescent microscope.
All osteoprogenitors were maintained in MSC complete medium, which includes alpha-MEM, L-glutamine, 10% FBS, MEM non-essential amino acids –NEAA (#11140; Gibco), Sodium bicarbonate at 37°C with 5% CO2. Osteogenic differentiation medium was composed of alpha-MEM, L-glutamine, 10% FBS, sodium bicarbonate, 100nM Dexamethasone (#D4902; Sigma-Aldrich), 100μM L-Ascorbic acid (#A8960; Sigma-Aldrich), and 10mM β-glycerophosphate (#G9422; Sigma-Aldrich).(30)(31)(32)
Alizarin Red S (ARS) and Von Kossa (VK) Staining
Alizarin Red S staining was performed as previously described.(33)(34) Quantification was performed by solubilizing the stained cell monolayer with 10% acetic acid, neutralized with 1% ammonium hydroxide, then heated at 85°C for 15 minutes to extract the dyes from the mineralized deposits. Lysates were centrifuged to remove the insoluble particles, and the extracted calcified minerals were quantified by colorimetric detection measuring absorbance at 405nm. For von Kossa staining, osteogenic differentiated iPSC-osteoprogenitor cells were fixed with 4% paraformaldehyde, then incubated with 1% silver nitrate solution (#204390; Sigma-Aldrich) under UV light for 1hour, then rinsed with distilled water several times.
Alkaline Phosphatase (AP) Histochemical Staining Assay
AP substrate solution was prepared by dissolving a single tablet of SIGMA FAST BCIP/NBT (#B5655; Sigma-Aldrich) in 10ml of distilled water. After the fixation of cells by 4% paraformaldehyde/PBS solution, the mineralized matrix was stained with AP substrate solution for 1 hour at room temperature in the dark, then rinsed with distilled water.
Western Blotting Analysis
Western blots were performed as previously described.(35) Subcellular fractions of normal and HGPS iPSC-osteoprogenitors were obtained by NE-PER Nuclear and Cytoplasmic Extraction Kit (#78835; Bio-Rad), according to manufacturer’s directions. For long bone extracts, tibiae were dissected from both genotypes and cleaned of soft tissue. Subsequently, the epiphyses were removed, the marrow cavity flushed with sterile saline, prior to lysis, as described.(34)(36) Primary antibodies used for immunoblotting analysis are as follows: rabbit anti-non-phosphorylated (Active) β-catenin protein Ser33/37/Thr41 (#4270; Cell Signaling Technology); mouse anti-lamin A/C (MAB3211; EMD Millipore); goat anti-lamin A/C N-18 (sc-6215; Santa Cruz Biotechnology); rabbit anti-RCC1 (#3589; Cell Signaling Technology); rabbit anti-S6 ribosomal protein (#2217; Cell Signaling Technology); monoclonal anti-β-Actin-peroxidase (#A3854; Sigma-Aldrich). Protein densitometry was analyzed by using Image lab software to eliminate the saturation of band and quantify normalized protein expression values.
TOP/FOP Flash Reporter Gene Assay
Normal and HGPS iPSC-derived osteoprogenitors were seeded at 10,000 cells/well into 24-well plate, and induced for differentiation for 1, 2 and 3 weeks by culturing the cells in the osteogenic differentiation medium (the culture medium was changed 3 times a week). After osteogenic differentiation for designated induction periods, the cells were transiently co-transfected with TOP- (#21–170; Upstate Biotechnology) or FOP- (#21–169; Upstate biotechnology) flash luciferase reporter vector and internal control Renilla plasmid, pRL-SV40 vector (#E2231; Promega) using Lipofectamine 2000 transfection reagent (#11668; Invitrogen) in Opti-MEM reduced serum medium (#31985; Gibco). TOP-flash reporter construct comprises three copies of β-catenin-responsive TCF/LEF sites driving expression of firefly luciferase. FOP-flash is a negative control with mutated copies of TCF/LEF binding sites. Dual-luciferase reporter assay kit (#E1910; Promega) was used for the detection of β-catenin activity. The TOP-flash reporter construct with wild-type TCF binding sites (0.5μg) or FOP-flash reporter construct with mutant TCF binding sites (0.5μg) was transfected with pRL-SV40 vector (0.1μg) for 48 hours at 37°C.(22) Passive lysis buffer provided by dual-luciferase reporter assay kit was used to lyse the cells. Firefly luciferase activity and Renilla Luciferase activity were in a luminometer (SoftMax Pro software).
HGPS Mouse Model and Isolation of bone cells
Zmpste24+/− mice, on a C57BL/6J background strain (stock number: 015958-UCD), were obtained from the Mutant Mouse Resource & Research Centers (MMRRC at Univ. of California, Davis). All animal studies were performed with approval by the Animal Care and Use Committee at the University of Maryland School of Medicine. Mice were group housed in micro-isolator cages, and food (standard rodent chow) and water were available ad libitum. Littermates from our breeding colony were used for all experiments.
In vivo SclAb Treatment and Bone Microarchitecture Assessment
All mouse work was conducted under the Zmpste24−/− mice and WT control littermate mice were treated with a sclerostin neutralizing antibody (SclAb, Novartis Pharma AG, 100mg/kg, 1x/week) or vehicle (Saline) for 4 weeks. The treatment was initiated at the age of 4-week-old, and intraperitoneal (IP) injected for once a week, until the mice reached the age of 8-week-old (4 weeks duration of the treatment). Then, the mice were euthanized and bone microarchitecture was evaluated by micro-computed tomography (microCT) analysis.
MicroCT
Femurs were dissected from 8-week old male Zmpste24+/+ and Zmpste24−/− mice and fixed in 4% PFA for two to four days, then transferred to 70% ethanol. Three-dimensional microCT was performed on the femurs of each genotype using SkyScan 1172 (Bruker, Kontich, Belgium), as described.(33)(36) The skeletal parameters assessed by microCT followed published nomenclature guidelines.(38) Bone morphology and microstructure were assessed at the mid-diaphysis for cortical parameters, including cortical thickness (Ct.Th) and mean polar moment of inertia (MMI). Trabecular parameters were assessed at the distal femoral metaphysis for trabecular parameters, including the trabecular bone volume fraction (BV/TV), trabecular bone thickness (Tb.Th), trabecular number (Tb.N) and trabecular separation (Tb.Sp). Analysis was completed on 8–9 animals for each genotype, treatment, and gender, respectively. Femurs were scanned with 2K resolution, 10-micron voxel size, 0.5 mm Al filter at 60kV and 167 μA. Trabecular bone was delineated manually in a region of interest 0.2 mm to 2.0 mm proximal to the distal femoral growth plate. For cortical bone parameters, transverse microCT scans were performed at the femoral diaphysis beginning at 56% of the femoral length (measured from the head of the femur) extending 0.6 mm distally.
Quantitative Real Time RT-PCR
Total RNA was isolated from tibia (flushed of marrow) using Tripure reagent (Roche, Indianapolis, IN, USA), reverse transcribed, and quantitative PCR carried out as described. (34)(37)(64) The data are simultaneously normalized to Gapdh, Rpl13, and Hprt using geNorm v3.5 software (Ghent University Hospital Ghent, Belgium), as described.(65) Primer sequences are available upon request.
Statistical Analysis
Results are presented as mean ±SD. Experiments were repeated at least three times, unless indicated otherwise. Data normality was assessed by GraphPad Prism 7 software by D’Agostino-Pearson omnibus normality test. For normally distributed data, samples were compared by an ANOVA for unpaired samples with a Holm-Sidak post hoc test, as appropriate, using GraphPad Prism 7 software. For nonparametric data, a two-tailed Mann-Whitney test or Kruskal-Wallis test was performed, as indicated. A P value of <0.05 was used as a threshold for statistical significance.
RESULTS
Osteoblastogenesis is Impaired in iPSCs Osteoprogenitors and hBM-MSCs with HGPS Mutation
The osteogenic differentiation potential of mesenchymal progenitor cells in HGPS was characterized in osteoprogenitors derived from iPSCs generated from normal and HGPS primary fibroblast cells.(29)(39) Early-stage osteoblasts, identified by alkaline phosphatase (AP) staining, were markedly reduced in both HGPS(I) and HGPS(II) iPSC-osteoprogenitors relative to the normal control line (Figure S1A,B). Similarly, HGPS iPSC-osteoprogenitors had reduced mineralizing capacity, with fewer calcified nodules than control cells, as shown by Alizarin Red S (ARS) staining (Figure 1A–C & Figure S1C) and Von Kossa staining (Figure S1D). In contrast to control iPSC-osteoprogenitors, HGPS iPSC-osteoprogenitors exhibit higher cell proliferative rate in comparison to normal osteoprogenitors. Nevertheless, despite this increase in cell number, the mineralization capacity in HGPS osteoprogenitors was reduced relative to normal iPSC-osteoprogenitors.
Figure 1: Osteoprogenitor cells with HGPS mutation undergo defective osteogenic differentiation.
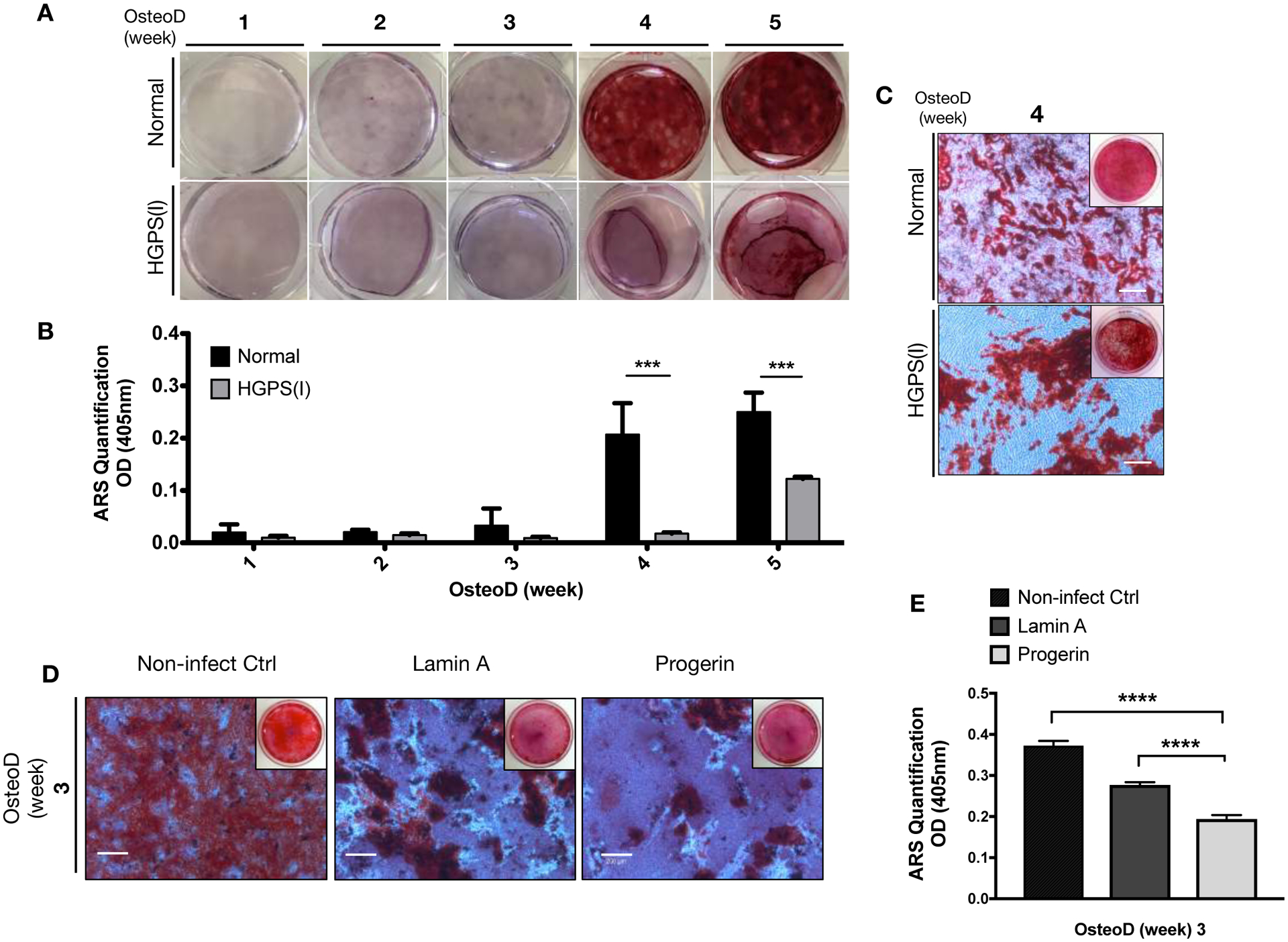
(A) Normal and HGPS iPSC-osteoprogenitors were induced for Osteogenic differentiation (OsteoD) for up to five weeks, and the calcium depositions in the cells were evaluated by Alizarin Red S (ARS) staining assay. Representative images are shown. (B) Quantification of ARS staining in normal and HGPS osteoprogenitors (n=3). (C) Representative whole plate views and magnified bright-field microscopy images of normal and HGPS osteoprogenitors that were differentiated for four weeks, and stained with ARS. Images are taken from biological replicate of (A). Scale bars, 200μm. (D) Representative whole plate views and magnified bright-field images of ARS stained hBM-MSCs with no lentiviruses (Control), or lentivirus-mediated overexpression of wild-type lamin A or progerin. Scale bars, 200μm. (E) Quantification of colorimetric detection of ARS stains (n=3) from samples in (D). Data are represented as mean ± SD. ***p < 0.001, ****p < 0.0001.
To confirm a direct association between the HGPS mutation and impaired osteogenic differentiation, we conducted the same differentiation experiments in commercially available, well characterized hBM-MSCs. hBM-MSCs were overexpressing GFP-null, or GFP-lamin A, or GFP-progerin constructs (Figure S2). Consistent with the preceding results, ectopic overexpression of progerin in hBM-MSCs impaired osteogenic differentiation, reflected by fewer numbers of mineralized nodules (Figure 1D–E). Overall, these results support that the expression of progerin in osteoprogenitors disrupts osteoblast differentiation.
WNT/β-catenin Signaling is Impaired During the Osteogenic Differentiation of HGPS Osteoprogenitor Cells
Canonical β-catenin signaling is an essential regulator for the commitment of MSCs or progenitor cells toward osteoblast lineage.(42)(43)(44) Based on prior observations in HGPS mouse models,(20)(28) we hypothesize impaired β-catenin signaling affects HGPS osteogenic differentiation. In support of our hypothesis, we observed a large decrease in the protein level of non-phosphorylated (active) β-catenin in HGPS iPSC-derived osteoprogenitors after the osteogenic induction for two or three weeks (Figure 2A–B, Figure S3A–C).
Figure 2: WNT/β-catenin activity is diminished during the osteogenic differentiation of HGPS osteoprogenitors.
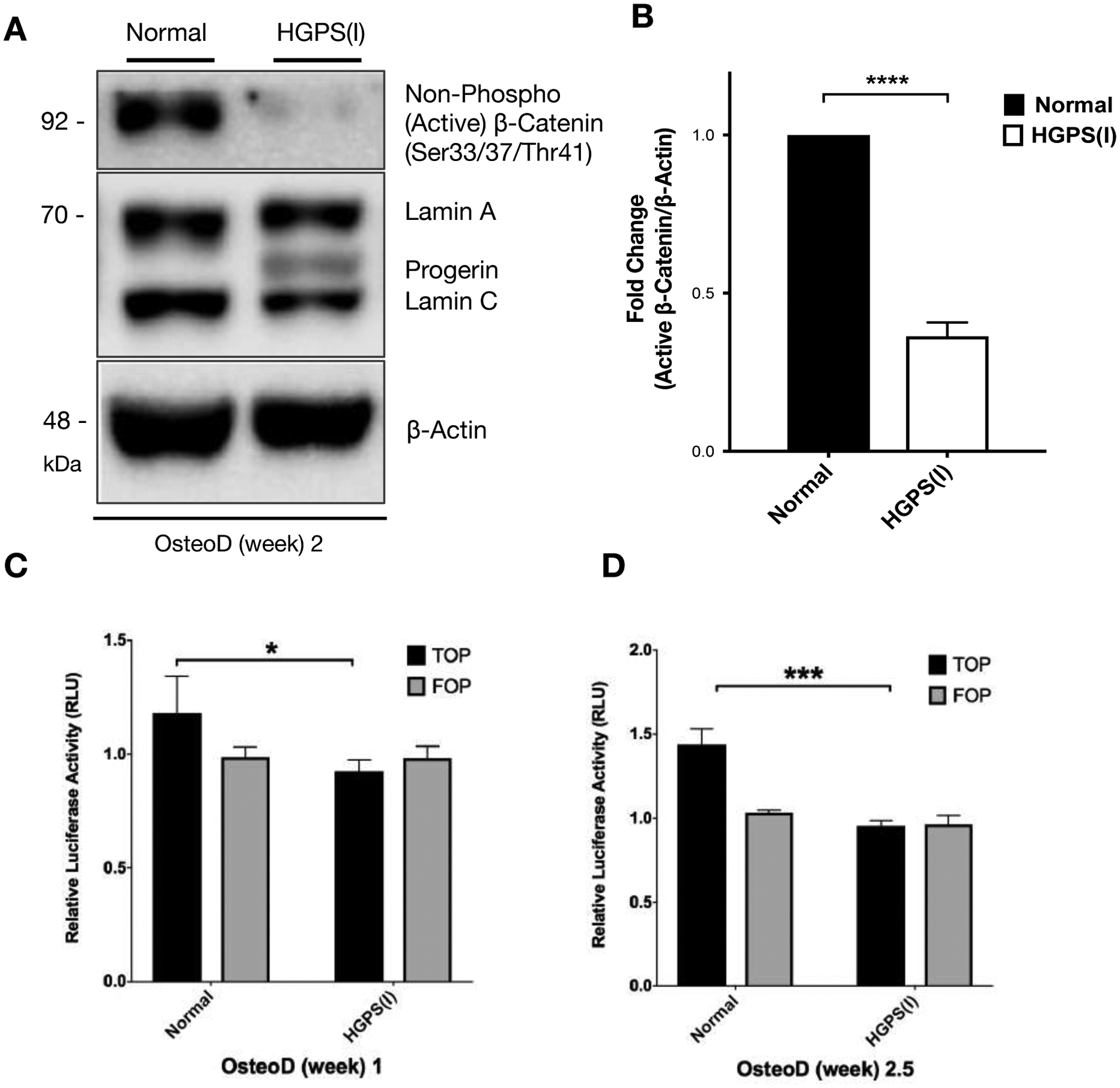
(A) A representative western blot showing detection of non-phosphorylated (active) β-catenin and lamin A/C proteins in normal and HGPS iPSC-osteoprogenitors that were induced for osteogenic differentiation for two weeks. β-actin protein was used as a loading control. (B) Quantification of fold change for western blot band densitometry of the relative non-phosphorylated (active) β-catenin protein levels normalized to its corresponding β-actin. (C-D) TOP and FOP flash luciferase reporter gene assay in normal and HGPS iPSC-osteoprogenitors during osteoblast differentiation (OsteoD) for 1 week (C) and 2.5 weeks (D). Relative luciferase activity (RLU) indicates a ratio of firefly/renilla luciferase activity. Data are represented as mean ± SD. *p < 0.05, ***p < 0.001.
Next, TOP/FOP flash luciferase reporter assay was employed to evaluate the β-catenin transcriptional activity in normal and HGPS osteoprogenitors during osteogenic induction. Consistent with the western blotting result of reduced active β-catenin protein in HGPS iPSC-derived osteoprogenitors (Figure S4), TOP-flash luciferase assay showed a significant decrease in β-catenin transcriptional activity in HGPS osteoprogenitors at osteogenic differentiation week 1 (Figure 2C) and week 2.5 (Figure 2D). In contrast, FOP-flash activity was similar between the normal and HGPS samples (Figure 2C–D).
To investigate whether defects in β-catenin signaling are responsible for the observed defects in HGPS osteoblast differentiation, the endogenous β-catenin in normal and HGPS osteoprogenitors was knocked down using siRNA, and the efficiency was assessed by western blot (Figure S5A). The knockdown β-catenin by siRNA in normal iPSC-osteoprogenitors resulted in decreased protein level of β-catenin to levels comparable to those seen in HGPS iPSC-osteoprogenitors (Figure S5A). Upon transient silencing of β-catenin in normal iPSC-osteoprogenitors followed by the induction of osteogenic differentiation, AP activity was markedly reduced, approaching the low levels seen in HGPS iPSC osteoprogenitors (Figure S5B). Given that HGPS iPSC-osteoprogenitors already express a very low level of β-catenin (Figure S5A), β-catenin knockdown in HGPS iPSC-osteoprogenitors displayed only mild reduction in AP activity (Figure S5C–D).
Farnesylated-Progerin is Required for Attenuated β-catenin Signaling
To examine how progerin suppresses β-catenin signaling during the osteogenic differentiation, we investigated the abnormal farnesylation of progerin, a key functional property of this mutant. Posttranslational processing of the LMNA gene involves farnesylation of the cysteine in the carboxyl-terminal CISM (CAAX motif) of prelamin A, followed by cleavage of the farnesyl group by Zmpste24.(44) Progerin permanently retains the farnesylation and is anchored to the nuclear membrane. SSIM-progerin carries a mutated form of CSIM (CAAX motif), which becomes non-farnesylable and rescues the nuclear blebbing phenotype by relocalizing progerin to the nucleoplasm.(46) Importantly, the abundance of active β-catenin protein in hBM-MSCs overexpressing SSIM-progerin was rescued to levels comparable to control hBM-MSCs overexpressing GFP-null or wild type lamin A (Figure 3A, B). In contrast, hBM-MSCs overexpressing GFP-progerin showed a reduced active β-catenin protein level compared to hBM-MSCs overexpressing control GFP-null vector or GFP-wild type lamin A. Consistent with the active β-catenin protein levels, the presence of the farnesylated-progerin, but not the non-farnesylable SSIM-progerin, was associated with impaired osteogenic differentiation, as indicated by AP activity (Figure 3C). In total, these results reveal that progerin farnesylation suppresses the active β-catenin, thereby inhibiting osteogenic differentiation.
Figure 3: Wild-type hMSCs overexpressing a non-farnesylable form of progerin attain stabilized active β-catenin activity during the osteogenic differentiation.
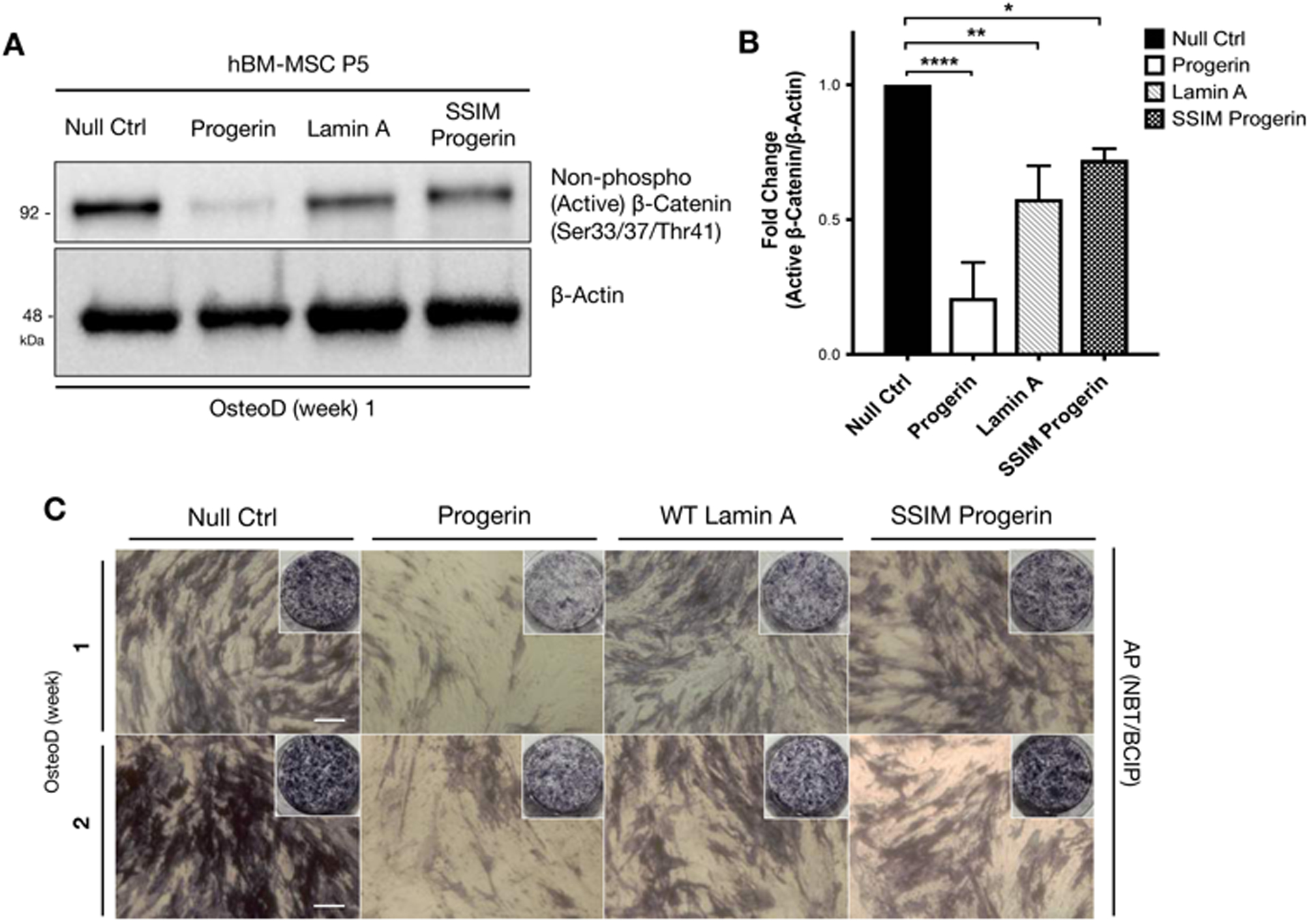
(A) Representative Western blot detecting non-phosphorylated (active) β-catenin in hBM-MSCs induced with lentiviruses expressing GFP, GFP-progerin, GFP-lamin A, or GFP-SSIM-progerin. β-actin protein was used as a loading control. The samples were collected at one week in osteogenic differentiation. (B) Quantification of western blot by analyzing the fold changes of non-phosphorylated (active) β-catenin/β-actin densitometry. Western blots were conducted at least three biological replicates (n>3) on separate gels. (C) Whole plate views and bright-field images of alkaline phosphatase (AP) staining in hBM-MSCs overexpressing GFP-null, GFP-progerin, GFP-lamin A and GFP-SSIM (non-farnesylable) progerin, then induced for osteogenic differentiation for one or two weeks.
Farnesylated Progerin Causes Abnormal Accumulation of Active β-catenin in the Cytosol of HGPS Osteoprogenitors During Osteogenic Differentiation
Nuclear translocation of β-catenin is an essential process to provide stabilized β-catenin levels in the nucleus to activate gene expression. Given that the farnesylated progerin anchors to the nuclear membrane and disrupts the normal nuclear lamina assembly, the abnormal nuclear scaffolding may impact the organization of nuclear pore complexes (NPCs) and the RanGTP gradient across the nuclear membrane.(39)(47) Since β-catenin has no nuclear localization signal (NLS), its import is mediated by the interactions with NPCs.(48) Accordingly, we hypothesized that the HGPS-associated changes in β-catenin signaling may be caused by inefficient transport of β-catenin from the cytosol to the nucleus, impairing the transcriptional signaling necessary to drive osteogenic differentiation.
Nuclear import of β-catenin was examined by the subcellular distribution of the non-phosphorylated (active) form of β-catenin during osteogenic differentiation. Subcellular fractionations were taken weekly during the four-week osteogenic differentiation of both normal and HGPS osteoprogenitors. The normal cells exhibited an increase in the ratio of nuclear to cytoplasmic active β-catenin protein, reaching a peak at 4 weeks, towards the late stage of osteogenic differentiation (Figure 4A–B). In contrast, HGPS cells showed marked reduction in the ratio of nuclear to cytoplasmic active β-catenin protein, reaching a nadir by 4 weeks, towards the later stage of osteogenic differentiation (Figure 4C–D). These ratios suggest that at a time that normal cells are accumulating active β-catenin in the nucleus, active β-catenin is selectively portioned in the cytoplasm of HGPS cells. This failure to accumulate in the nucleus of HGPS cells occurs despite increasing cytoplasmic levels active β-catenin in the cytoplasmic fraction over this same time course. Thus, these results suggest that inefficient nuclear import of active β-catenin leads to its accumulation in the cytosol of HGPS osteoprogenitors during the later stage of osteogenic differentiation.
Figure 4: Progerin causes abnormal accumulation of active β-catenin in the cytosol of HGPS osteoprogenitors during osteogenic differentiation.
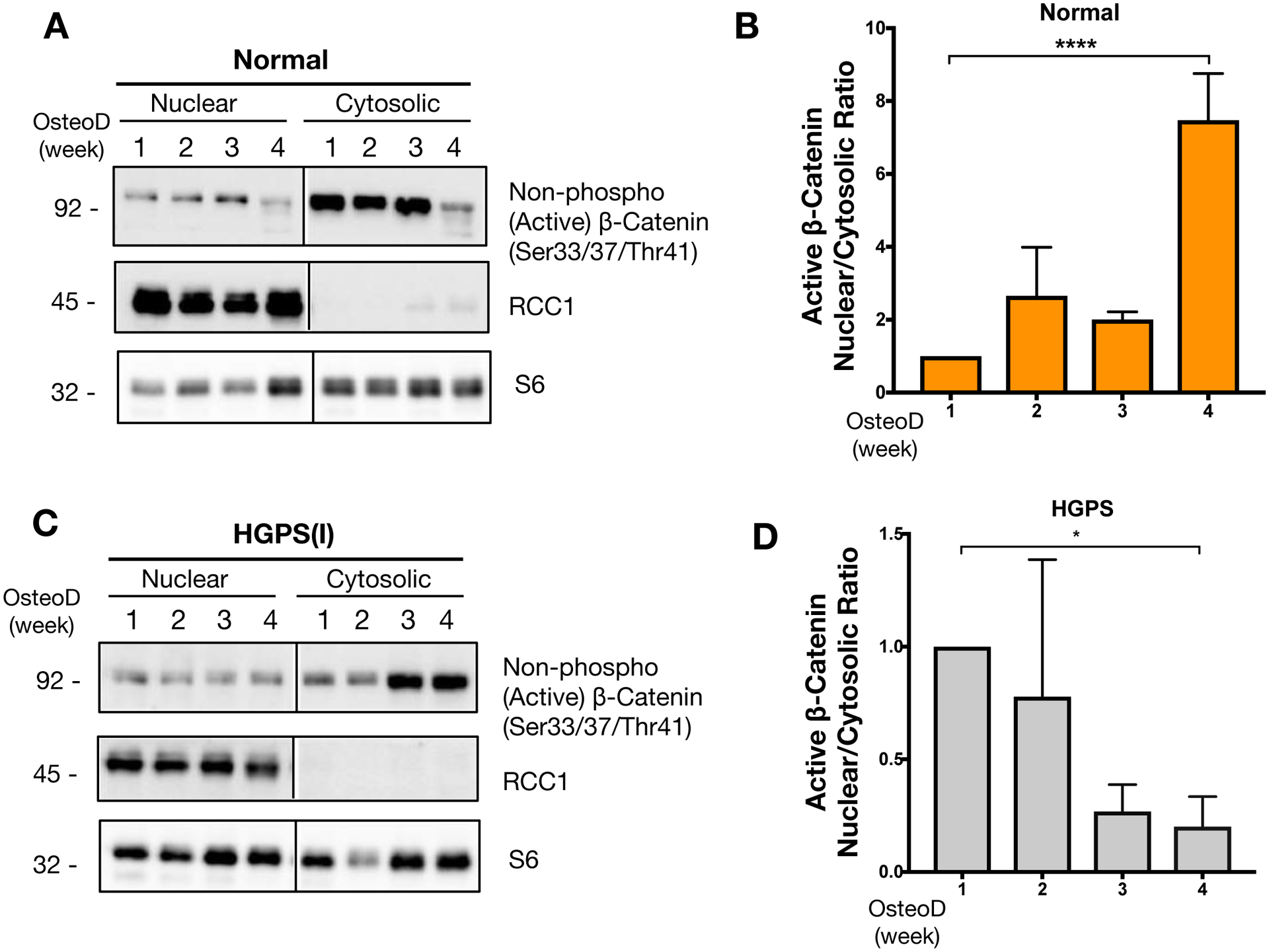
Representative image from western blotting analysis for active β-catenin protein in subcellular fractionations of (A) normal iPSC-osteoprogenitors or (C) HGPS iPSC-osteoprogenitors. The fractionated nuclear and cytosolic lysates were obtained weekly during osteogenic differentiation from week 1 to week 4. RCC1 represents the nuclear marker, and S6 was used to detect the overall loadings of samples. (B&D) Quantification for band intensities of nuclear to cytosolic β-catenin protein ratios at each osteogenic differentiation induction time points (week 1, 2, 3 and 4) for (B) normal iPSC-osteoprogenitors or (D) HGPS iPSC-osteoprogenitors. Western blots were conducted at least three biological replicates (n=3) on distinct blots. *p < 0.05, ****p < 0.0001.
Stimulation of β-catenin Signaling Cascade Rescues the Skeletal Phenotype in a Mouse Model of HGPS
The skeletal phenotypes of human HGPS patients are best recapitulated in one of the HGPS murine models, Zmpste24−/−, which exhibits similar severe osteopenia as that in HGPS patients.(7)(49)(50)(51) Consistent with our in vitro data, western blotting revealed that active β-catenin in the primary calvaria cells (Figure 5A–B) and the whole tibia extracts (Figure 5C–D) was significantly lower in samples of Zmpste24−/− mice than those of wild type control mice. This decrease β-catenin in occurred independent of any changes in Wnt signaling components (Figure S6), suggesting that the defect in β-catenin signaling occurred intracellularly, perhaps at the level of nuclear import. As expected, the cell autonomous nature of this defect was confirmed in primary calvarial osteoblasts from Zmpste24−/− mice, which exhibited the accumulation of farnesyl-prelamin A protein, the loss of mature lamin A, and reduction of active β-catenin protein (Figure 5A, B).
Figure 5: Zmpste24−/− osteoblasts show reduced active β-catenin, which can be rescued with sclerostin-neutralizing antibodies.
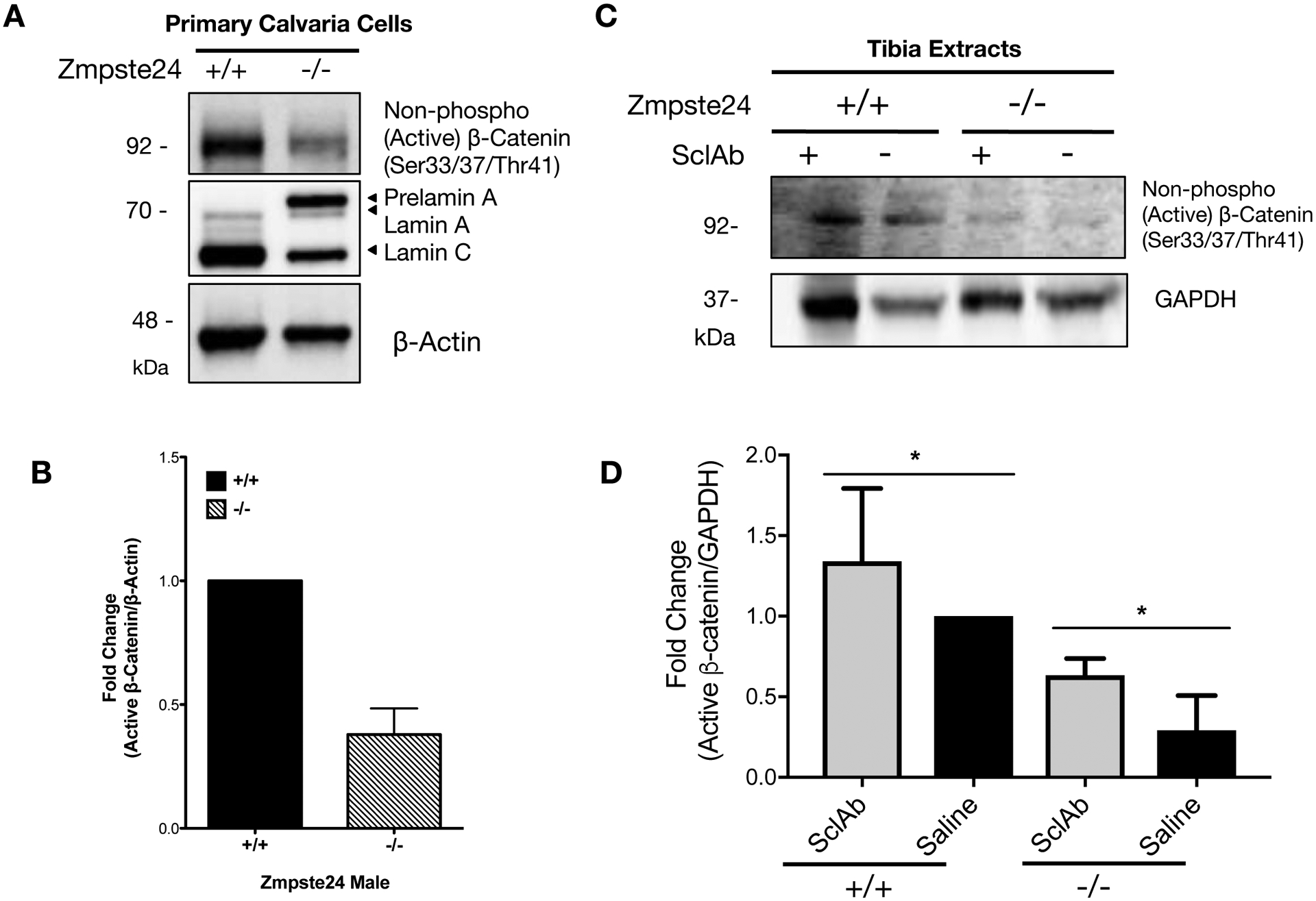
(A) Western blots active β-catenin protein, prelamin A, lamin A/C in primary calvaria cells isolated from a heterozygous (Zmpste24+/−), a wild-type (Zmpste24+/+), and two knockout (Zmpste24−/−) mice. β-actin protein was used as a loading control. (B) Quantification of fold changes of relative western blot band intensity of active β-catenin protein against β-actin protein band. Western blots were conducted at least three biological replicates (n>3) on separate gels. (C) Western blotting analysis to detect active β-catenin protein in tibia extracts from wild-type and Zmpste24−/− mice treated with vehicle (Saline) or 100mg/kg SclAb for 4 weeks during the age of week 4 to week 8. GAPDH was used a loading control. (D) Quantification of relative western blot band intensity from Figure 5C, indicating active β-catenin protein levels normalized to corresponding band intensities of GAPDH (n=4). Data are represented as mean ± SD. *p < 0.05.
Based on these data, we proposed that stimulating the pool of active β-catenin in the cell might compensate for the inefficiency in β-catenin nuclear import, restoring nuclear function, osteoblast differentiation, and ameliorating the skeletal phenotypes of HGPS. To activate the pathway in bone-specific aspect, we targeted the sclerostin, a bone-specific inhibitor of Wnt/β-catenin signaling, using sclerostin function blocking antibodies (SclAb), which enhance β-catenin signaling and osteoblast function.(52)(53) Consistent with the work of others,(50)(51) both male and female Zmspte24−/− mice have markedly reduced bone mass relative to littermate controls, including a >2-fold decrease in trabecular bone volume fraction (Figure 6A) and cortical thinning (Figure S7). Likewise, qPCR analysis of the long bones of these mice demonstrated defects in the osteoblast lineage, including down regulation of the mRNA encoding the key lineage driving transcription factors, Runx2 and Osterix, as well as the mRNA abundance of the differentiation markers, bone sialoprotein, alkaline phosphatase and osteocalcin. Further, the β-catenin responsive genes, RANKL, Cx43/Gja1 and Axin2 were affected as expected by Zmpste24 gene deletion (Figure S8).
Figure 6: Sclerostin neutralizing antibody treatment alleviates the defective bone quality of Zmpste24−/− mouse model.
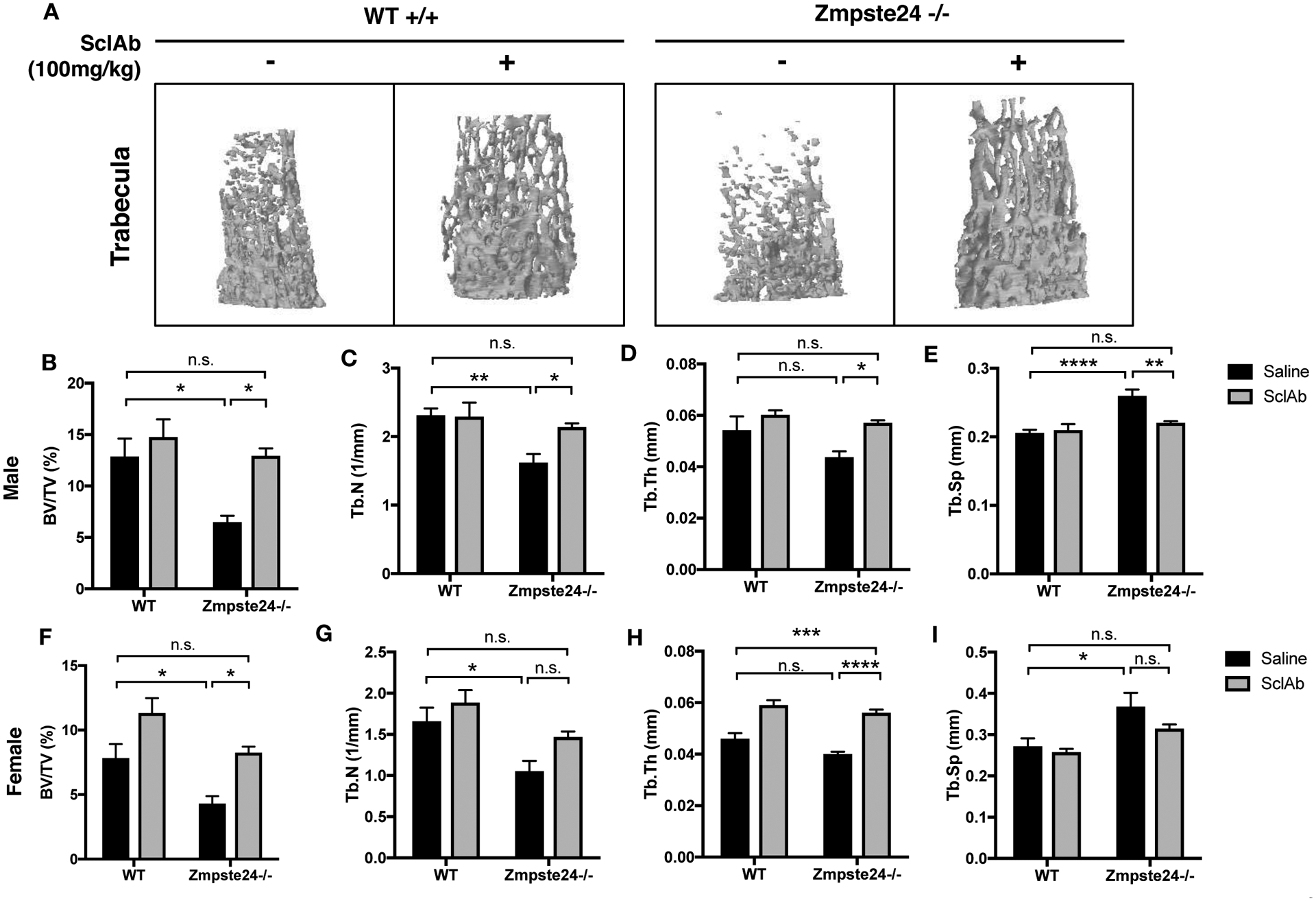
(A) Microcomputed tomography (μCT) analysis of femurs of normal and Zmpste24−/− male mice that were treated either by IP injection of 100mg/kg SclAb or saline control for 4 weeks, starting the treatment at the age of week 4. N=8–9 mice/treatment/group. (B-E) Bone quality assessment of SclAb -treated or non-treated normal or HGPS male mice elucidated by (B) Bone volume (BV/TV), (C) Trabecular number (Tb.N), (D) Trabecular thickness (Tb.Th), and (E) Trabecular spacing (Tb.Sp) was implemented in sclerostin-neutralizing antibody treated or non-treated male normal or HGPS mice. n.s. not significant, *p < 0.05, **p < 0.01, ****p < 0.0001. (F-I) Bone quality assessment of SclAb -treated or non-treated normal or HGPS female mice elucidated by (F) Bone volume, (G) Trabecular number, (H) Trabecular thickness, and (I) Trabecular spacing. n.s. not significant; *p < 0.05, ***p < 0.001, ****p < 0.0001.
SclAb were administered to wild type and Zmpste24−/− mice via IP injection for 4 weeks. Importantly, SclAb administration, not only enhanced the basal expression level of active β-catenin proteins in bone extracts from both wild type and Zmpste24−/− mice (Figure 5C–D), it also fully restored the trabecular and cortical bone parameters of Zmpste24−/− mice to age and sex matched WT control levels (Figure 6; Figure S7). The effects of SclAb on trabecular (and to a lesser extent cortical) microarchitecture were far more potent in Zmpste24−/− mice than in controls (Figure 6; Figure S7), strongly suggesting that defective β-catenin signaling is a fundamental driver of the skeletal phenotype in these mice. Likewise, markers of late osteoblast differentiation, including osterix, bone sialoprotein, and osteocalcin, were rescued by the antibody administration, as was the expression of β-catenin responsive genes, RANKL, Cx43/Gja1 and Axin2 (Figure S8). Together, these data indicate that targeting the β-catenin signaling cascade is a possible therapeutic target for restoring defective bone quality in HGPS patients, at least in part by restoring nuclear β-catenin function.
DISCUSSION
In the present study, we report that defects in β-catenin signaling, perhaps at the level of nuclear import, underpin delayed osteogenic differentiation in HGPS and that activating this pathway with sclerostin function blocking antibodies can restore bone mass in a mouse model of HGPS (Figure 7). We identified a diminished level of active β-catenin protein in the nucleus in two HGPS iPSC-derived osteoprogenitors lines, wild-type hBM-MSCs ectopically overexpressing progerin, and in osteoblasts and tissue extracts from Zmpste24−/− mice. This finding of decreased β-catenin in HGPS cells was associated with defects in osteoblast differentiation, lower matrix mineralization capacity, and a partitioning of active β-catenin in the cytoplasm of osteoblast during differentiation. We showed that the ability of progerin to inhibit the nuclear import and transcriptional activity of β-catenin required the farnesylation of progerin. Moreover, Zmpste24−/− mice, which express farnesylated pre-lamin A and phenotypically mimic HGPS, exhibit defects in β-catenin signaling and osteoblast differentiation that are consistent with the in vitro findings in iPSCs from HGPS patients. Further, we showed that targeting canonical WNT/β-catenin signaling with the sclerostin neutralizing antibodies fully restores trabecular bone mass and increases cortical bone parameters. In total, these data establish that defective β-catenin signaling, perhaps at the level of efficient nuclear import, is a fundamentally important driver of the skeletal phenotype of HGPS, and that therapeutically targeting β-catenin may be particularly effective at restoring skeletal function. Furthermore, given the fundamental nature of β-catenin signaling in stem cell renewal and lineage allocation, these findings may have broader implications, beyond skeletal biology, in the treatment of HGPS.
Figure 7: Schematic working model proposing the major hypothesis.
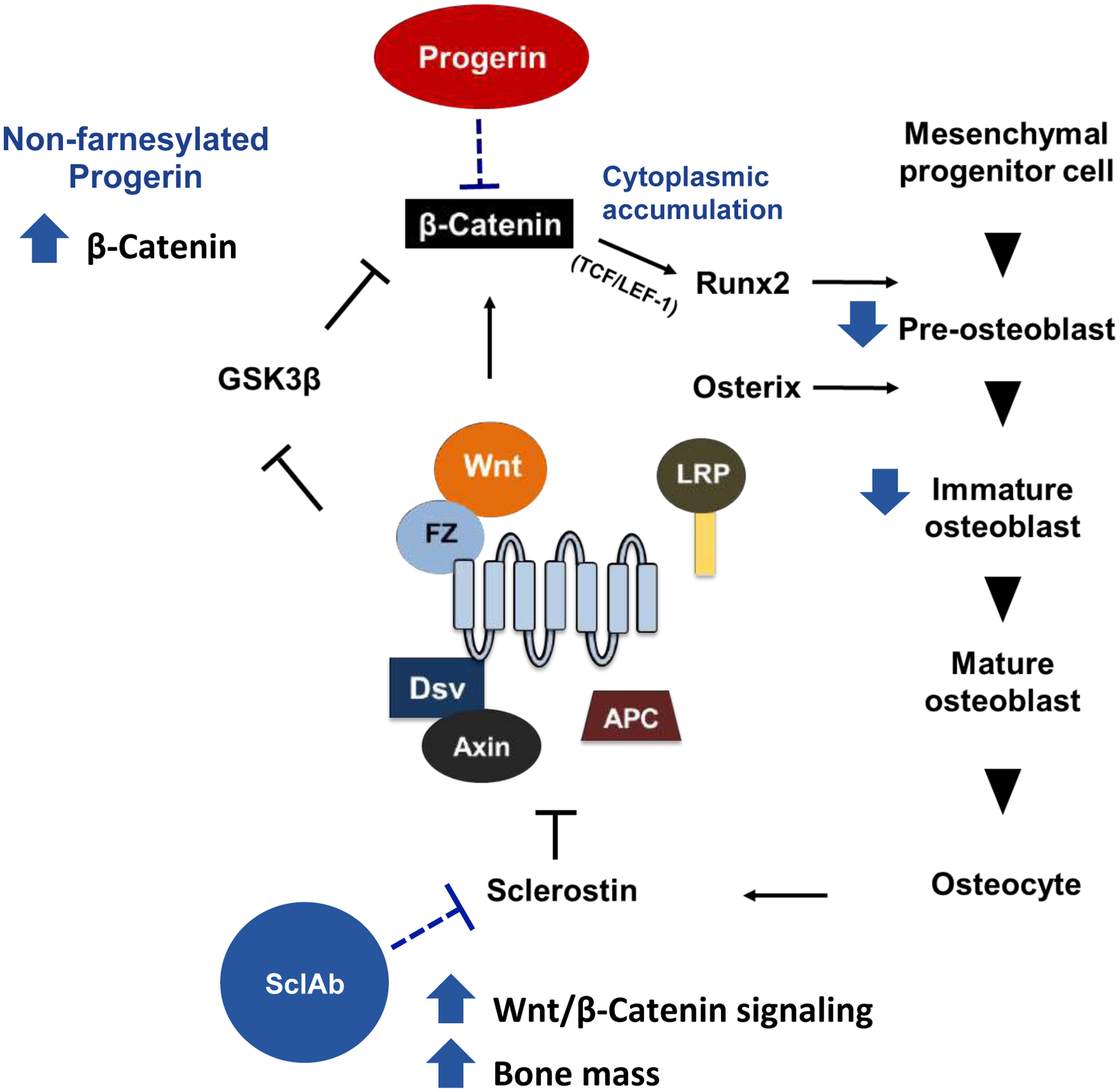
Progerin plays an inhibitory role in β-catenin signaling pathway and ultimately affects the downstream stages of osteogenesis.
Previously it has been reported that downregulated activities of both Notch and Wnt signaling pathways and a possible role in impaired stem cell function in human HGPS osteoprogenitors and HGPS mouse models, including Δ9Lmna and tetop-LAG608G+ and K5tTA+.(28)(54)(55) Here, we demonstrated the sufficiency of enhancing β-catenin signaling to rescue of the skeletal phenotype of Zmpste24−/− mice. While numerous developmental signals are integrated into the regulatory cascade for osteoblast differentiation, including Hedgehog signaling, Notch signaling, bone morphogenetic protein (BMP) signaling and FGF signaling,(56) the canonical WNT/ β-catenin pathway has a key function in developmental processes involving lineage formation for organ-specific cells, especially vital to skeletal development.(44)(57)(58) Active β-catenin allows the osteoprogenitor cells to remain committed to the osteoblastic lineage, limiting the progenitor cell differentiation to adipocytes or chondrocytes, and drives the osteogenic differentiation program.(43)(59)(60) Future studies will be needed to see if aberrant β-catenin and lineage allocation issues underlie other tissue pathologies in HGPS.
Interestingly, two studies examined the osteoblast lineage in HGPS MSCs(61)(62) and reported an unpredicted enhancement in osteogenic differentiation, despite the low bone mass observed in HGPS patients and mouse models of HGPS. We also observed this phenotype transiently at the very early stage of osteogenic differentiation in both HGPS(I) and HGPS(II) iPSC-osteoprogenitors. However, when comparing HGPS and control cells at the same cell densities, we found that the HGPS cells failed to gain, but rather, lowered the mineralization capacity. These results were confirmed by assessing osteoblast lineage markers in the Zmpste24−/− mice. Furthermore, using ectopically expressed lamin A or progerin in hBM-MSCs to minimize the effects of different genetic backgrounds for the control versus HGPS iPSCs, we observed the same osteoblast differentiation defects as those in iPSC-derived HGPS osteoprogenitors.
Another finding is that farnesylation of progerin seems to disturb the cytosolic-nuclear distribution of active β-catenin in HGPS osteoprogenitor cells as they progress through osteogenic differentiation. While it is known that Ran-mediated nuclear transport is impaired in HGPS cells,(39)(47)(63) β-catenin has no nuclear localization signal (NLS), thus the nuclear import mechanism is independent of Ran gradient. Rather, β-catenin nuclear import is mediated by NPCs.(48) Progerin is a truncated form of lamin A that abnormally retains a farnesyl group, which anchors itself to the nuclear membrane. Our results suggest that farnesylation of progerin negatively affects the β-catenin nuclear entry and exacerbates the defects in osteogenesis. The MSCs with the mutation of CSIM sequence changed to SSIM in progerin, which prevents farnesylation and rescues nuclear blebbing,(46) restored β-catenin signaling, and osteoblast differentiation. While other points of disruption of β-catenin signaling may also exist in these models, the contribution of deficits in nuclear import of β-catenin are intriguing.
Regardless, activating β-catenin signaling fully restores defective bone formation in HGPS, and these results provide the mechanistic basis that defects in β-catenin signaling are underlying factors contributing to clinical features of bone in HGPS. The effects of SclAb on Zmpste24−/− HGPS mice were unambiguous. After the treatment with SclAb in Zmpste24−/− mice over a four-week period, the bone mass and the microarchitecture measures, such as trabecular bone volume, density, spacing, and cortical thickness, were fully restored to the levels of control mice (Figure 6; Figure S7). While the currently used clinical treatment with FTI slows down the rate of rapid bone turnover by blocking the high bone resorption process,(8) our study suggests a complementary therapeutic approach to synergistically restore bone formation by targeting β-catenin signaling in bone precursor cells in HGPS patients. Notably, FTIs may also partially restore β-catenin activity and osteoblast differentiation function in HGPS patient cells by blocking progerin farnesylation and preventing negative effects on Ran-GTPase system.
In normal cells, Ran is mostly localized in the nucleus, whereas HGPS-patient fibroblasts had more Ran in the cytoplasm.(47)(63) Disrupted Ran gradient by progerin was reported in the past, and Farnesyltransferase inhibitor (FTI) rescues disrupted Ran gradients in HGPS culture.(63) Moreover, other study demonstrated that progerin lowers the function of SUMOylation pathway which is involved in nuclear-cytosolic transport.(63) Since β-catenin nuclear transport is Ran machinery-independent, but its import is self-regulated by direct binding to NPC, we speculate that progerin provides a disrupted scaffold for organizing NPC, which consequently inhibits proper transport of active β-catenin in HGPS osteoprogenitors during the osteogenic differentiation.
The rationale for the intervention using SclAb is not to repair disrupted translocation activity by progerin, but rather to compensate for inefficient nuclear import of β-catenin in HGPS, by driving up the pool of active β-catenin. However, the skeletal impact may be more effective if β-catenin is directly targeted or targeted in conjunction with FTIs. We consider that combination therapies with FTIs and SclAb would potentially enhance bone formation aspect. Although abnormal skeletal development in HGPS is not fatal, a restoration of skeletal function could eminently improve the life quality of patients. Further, due to the role of β-catenin signaling in the lineage renewal and allocation of other stem cells populations, these findings in bone may be extrapolated to other aspects of HGPS. The complete rescue of skeletal microarchitecture by the activation of β-catenin is unexpected, novel, compelling and translational. As past studies have demonstrated defective β-catenin signaling in other cell types, it is likely not bone specific. While we were able to use a very bone specific model to target β-catenin signaling to fully rescue bone structure, it is highly likely that a more global strategy to rescue defective β-catenin signaling could be therapeutically beneficial to multiple organ systems in HGPS.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the grants from Maryland Stem Cell Research Fund; and University of Maryland, College Park - University of Maryland, Baltimore Research & Innovation Seed Program. We also acknowledge the support from CMNS Dean’s Matching Award for NIH Training Program in Cell and Molecular Biology (T32GM080201).
REFERENCES
- 1.Gordon LB, McCarten KM, Giobbie-Hurder A, Machan JT, Campbell SE, Berns SD, Kieran MW. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007; 120(4):824–33. [DOI] [PubMed] [Google Scholar]
- 2.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer MLN. Lamin A Truncation in Hutchinson Gilford Progeria. Science. 2003;300(5628):2055. [DOI] [PubMed] [Google Scholar]
- 4.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci 2004;101(24):8963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Csoka AB, English SB, Simkevich CP, Ginzinger DG, Butte AJ, Schatten GP, Rothman FG, Sedivy JM. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelarated atherosclerosis. Aging Cell. 2004;3(4):235–43. [DOI] [PubMed] [Google Scholar]
- 6.Gordon LB, McCarten KM, Giobbie-Hurder A, Machan JT, Campbell SE, Berns SD, Kieran MW. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007;120(4):824–33. [DOI] [PubMed] [Google Scholar]
- 7.Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith ACM, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, Reynolds JC, Gropman A, Yanovski JA, Gerhard-Herman M, Collins FS, Nabel EG, Cannon RO, Gahl WA, Introne WJ. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med 2008; 358(6):592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, Ploski C, Correia A, Quinn N, Ullrich NJ, Nazarian A, Liang MG, Huh SY, Schwartzman A, Kieran MW. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci 2012;109(41):16666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varela I, Pereira S, Ugalde AP, Navarro CL, Suárez MF, Cau P, Cadiñanos J, Osorio FG, Foray N, Cobo J, de Carlos F, Lévy N, Freije JMP, López-Otín C. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med 2008;14(7):767–72. [DOI] [PubMed] [Google Scholar]
- 10.De Paula Rodrigues GH, Tâmega IDE, Duque G, Spinola Dias Neto V. Severe bone changes in a case of Hutchinson-Gilford syndrome. Ann. Genet 2002;45(3):151–5. [DOI] [PubMed] [Google Scholar]
- 11.Gordon CM, Gordon LB, Snyder BD, Nazarian A, Quinn N, Huh S, Giobbie-Hurder A, Neuberg D, Cleveland R, Kleinman M, Miller DT, Kieran MW. Hutchinson-gilford progeria is a skeletal dysplasia. J. Bone Miner. Res 2011;26(7):1670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jansen T, Romiti R. Progeria infantum (Hutchinson-Gilford syndrome) associated with scleroderma-like lesions and acro-osteolysis: A case report and brief review of the literature. Pediatr. Dermatol 2000; 17(4):282–85. [DOI] [PubMed] [Google Scholar]
- 13.Hamer L, Kaplan F, Fallon M. The musculoskeletal manifestations of progeria. A literature review. Orthopedics. 1988; 11(5):763–9. [DOI] [PubMed] [Google Scholar]
- 14.Moen C Orthapedic aspects of progeria. J. bone Jt. Surg 1982;542–6. [PubMed] [Google Scholar]
- 15.Li W, Yeo LS, Vidal C, McCorquodale T, Herrmann M, Fatkin D, Duque G. Decreased bone formation and osteopenia in lamin A/C-deficient mice. PLoS One 2011;6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Kieckhaefer JE, Cao K. Mouse models of laminopathies. Aging Cell. 2013;12(1):2–10. [DOI] [PubMed] [Google Scholar]
- 17.Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RAD, Michaelis S, Griffey SM, Young SG. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. U. S. A 2002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rivas D, Li W, Akter R, Henderson JE, Duque G. Accelerated features of age-related bone loss in Zmpste24 metalloproteinase-deficient mice. Journals Gerontol. A Biol. Sci. Med. Sci 2009; 64A(10):1015–1024. [DOI] [PubMed] [Google Scholar]
- 19.Strandgren C, Nasser HA, McKenna T, Koskela A, Tuukkanen J, Ohlsson C, Rozell B, Eriksson M. Transgene silencing of the Hutchinson-Gilford progeria syndrome mutation results in a reversible bone phenotype, whereas resveratrol treatment does not show overall beneficial effects. FASEB J. 2015;29(8):3193–205. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt E, Nilsson O, Koskela A, Tuukkanen J, Ohlsson C, Rozell B, Eriksson M. Expression of the Hutchinson-Gilford progeria mutation during osteoblast development results in loss of osteocytes, irregular mineralization, and poor biomechanical properties. J. Biol. Chem 2012;287(40):33512–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glass DA, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 2005;8(5):751–64. [DOI] [PubMed] [Google Scholar]
- 22.Mbalaviele G, Sheikh S, Stains JP, Salazar VS, Cheng SL, Chen D, Civitelli R. beta-catenin and BMP-2 synergize to promote osteoblast differentiation and new bone formation. J. Cell. Biochem 2005;94(2):403–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, Deng L, Clemens TL, Williams BO. Essential role of beta-catenin in postnatal bone acquisition. J. Biol. Chem 2005;280(22):21162–8. [DOI] [PubMed] [Google Scholar]
- 24.Day TF, Guo X, Garrett-Beal L, Yang Y. mouse study: Wnt/β-Catenin Signaling in Mesenchymal Progenitors Controls Osteoblast and Chondrocyte Differentiation during Vertebrate Skeletogenesis. Dev. Cell 2005;8(5):739–50. [DOI] [PubMed] [Google Scholar]
- 25.Hu H Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2004;132(1):49–60. [DOI] [PubMed] [Google Scholar]
- 26.Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol 2010;30(12):3071–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Espada J, Varela I, Flores I, Ugalde AP, Cadiñanos J, Pendás AM, Stewart CL, Tryggvason K, Blasco MA, Freije JMP, López-Otín C. Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J. Cell Biol 2008;181(1):27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandez L, Roux KJ, Wong ESM, Mounkes LC, Mutalif R, Navasankari R, Rai B, Cool S, Jeong J-W, Wang H, Lee H-S, Kozlov S, Grunert M, Keeble T, Jones CM, Meta MD, Young SG, Daar IO, Burke B, Perantoni AO, Stewart CL. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev. Cell. Elsevier Inc 2010;19(3):413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiong ZM, LaDana C, Wu D, Cao K. An inhibitory role of progerin in the gene induction network of adipocyte differentiation from iPS cells. Aging. 2013;5(4):288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Almeida M Aging mechanisms in bone. Bonekey Rep. 2012; 1(102). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruedigam C, van Driel M, Koedam M, van de Peppel J, van der Eerden BCJ, Eijken M, van Leeuwen JPTM. Basic techniques in human mesenchymal stem cell cultures: Differentiation into osteogenic and adipogenic lineages, genetic perturbations, and phenotypic analyses. Curr. Protoc. Stem Cell Biol 2011;1H.3.1–1H.3.20. [DOI] [PubMed] [Google Scholar]
- 32.Langenbach F, Handschel J. Effects of dexamethasone, ascorbic acid and β-glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem Cell Res. Ther 2013;(4):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moorer MC, Hebert C, Tomlinson RE, Iyer SR, Chason M, Stains JP. Defective signaling, osteoblastogenesis, and bone remodeling in a mouse model of connexin43 C-terminal truncation. J. Cell Sci 2017;130(3):531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moorer MC, Buo AM, Garcia-Pelagio K, Stains JP, Bloch RJ. Deficiency of the intermediate filament synemin reduces bone mass in vivo. Am J Physiol Cell Physiol. 2016;311(6):839–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiong ZM, Choi JY, Wang K, Zhang H, Tariq Z, Wu D, Ko E, Ladana C, Sesaki H, Cao K. Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria. Aging Cell. 2016;15(2):279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buo AM, Tomlinson RE, Eidelman ER, Chason M, Stains JP. Connexin43 and Runx2 Interact to Affect Cortical Bone Geometry, Skeletal Development, and Osteoblast and Osteoclast Function. J. Bone Miner. Res 2017;32(8):1727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buo AM, Williams MS, Kerr JP, Stains JP. A cost-effective method to enhance adenoviral transduction of primary murine osteoblasts and bone marrow stromal cells. Bone Res. 2016;4:16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM. Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res 2013;28(1):2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H, Xiong Z-M, Cao K. HGPS defective nuclear import: Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1. Proc. Natl. Acad. Sci 2014;111(22):E2261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wheaton K, Campuzano D, Weili Ma, Michal Sheinis, Brandon Ho GWB, Benchimol S. crossm Progerin-Induced Replication Stress Facilitates Premature Senescence in. Mol. Cell. Biol 2017;37(14):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Atchison L, Zhang H, Cao K, Truskey GA. A Tissue Engineered Blood Vessel Model of Hutchinson-Gilford Progeria Syndrome Using Human iPSC-derived Smooth Muscle Cells. Sci. Rep 2017;7(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M. Osteocyte Wnt/ -Catenin Signaling Is Required for Normal Bone Homeostasis. Mol. Cell. Biol 2010;30(12):3071–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burgers TA, Williams BO. Regulation of Wnt/beta-catenin signaling within and from osteocytes. Bone. 2013; 54(2):244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krishnan V, Byrant HU, MacDougald OA. Regulation of bone mass by Wnt signaling. J. Clin. Invest 2006;116(5):1202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davies BSJ, Fong LG, Yang SH, Coffinier C, Young SG. The posttranslational processing of prelamin A and disease. Annu. Rev. Genomics Hum. Genet 2009;10:153–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci 2005;102(36):12879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snow CJ, Dar A, Dutta A, Kehlenbach RH, Paschal BM. Defective nuclear import of Tpr in Progeria reflects the Ran sensitivity of large cargo transport. J. Cell Biol 2013;201(4):541–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fagotto F et al. Nuclear localization signal-independent and importin/karyopherin-independent nuclear import of beta-catenin. Curr. Biol 1998;8(4):181–90. [DOI] [PubMed] [Google Scholar]
- 49.Hennekam RCM. Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. Part A 2006. p. 140A:2603–2624. [DOI] [PubMed] [Google Scholar]
- 50.Ibrahim MX, Sayin VI, Akula MK, Liu M, Fong LG, Young SG, Bergo MO. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science. 2013;340(6138):1330–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, Lopez-Otin C. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet 2002;31(1):94–9. [DOI] [PubMed] [Google Scholar]
- 52.Li X, Ominsky MS, Niu Q-T, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res 2008;23(6):860–9. [DOI] [PubMed] [Google Scholar]
- 53.Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Geng Z, Niu Q-T, Ke HZ, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C. Sclerostin Antibody Treatment Increases Bone Formation, Bone Mass, and Bone Strength in a Rat Model of Postmenopausal Osteoporosis. J. Bone Miner. Res 2009;24(4):578–88. [DOI] [PubMed] [Google Scholar]
- 54.Rosengardten Y, Mckenna T, Grochová D, Eriksson M. Stem cell depletion in Hutchinson-Gilford progeria syndrome. Aging Cell. 2011;10(6):1011–20. [DOI] [PubMed] [Google Scholar]
- 55.Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol 2008;10(4):452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N, Takada S. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002;(16):870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med 2013;19(2):179–92. [DOI] [PubMed] [Google Scholar]
- 58.Kim JH et al. Wnt signaling in bone formation and its therapeutic potential for bone diseases. Rev. Ther Adv Musculoskel Dis 2013;5(1):13–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PVN, Komm BS, Javed A, Van Wijnen AJ, Stein JL, Stein GS, Lian JB. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem 2005;280(39):33132–40. [DOI] [PubMed] [Google Scholar]
- 60.James AW. Review of Signaling Pathways Governing MSC Osteogenic and Adipogenic Differentiation. Scientifica (Cairo). 2013;Articl ID 684736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blondel S, Jaskowiak A, Egesipe A, Corf A Le, Cordette V, Martinat C, Laabi Y, Djabali K, Sandre-giovannoli A De, Levy N, Peschanski M, Nissan X, Val E Induced Pluripotent Stem Cells Reveal Functional Differences Between Drugs Currently Investigated in Patients With Hutchinson-Gilford Progeria Syndrome. Stem Cells Transl. Med 2014;3(4):510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lo Cicero A, Jaskowiak A-L, Egesipe A-L, Tournois J, Brinon B, Pitrez PR, Ferreira L, de Sandre-Giovannoli A, Levy N, Nissan X. A High Throughput Phenotypic Screening reveals compounds that counteract premature osteogenic differentiation of HGPS iPS-derived mesenchymal stem cells. Sci. Rep 2016;6:34798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kelley JB, Datta S, Snow CJ, Chatterjee M, Ni L, Spencer A, Yang C-SC-S, Cubeñas-Potts C, Matunis MJ, Paschal BM. The defective nuclear lamina in Hutchinson-gilford progeria syndrome disrupts the nucleocytoplasmic Ran gradient and inhibits nuclear localization of Ubc9. Mol. Cell. Biol 2011;31(16):3378–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gupta A, Anderson H, Buo AM, Moorer MC, Ren M, Stains JP. Communication of cAMP by connexin43 gap junctions regulates osteoblast signaling and gene expression. Cell Signal. 2016;28(8):1048–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Niger C, Luciotti MA, Buo AM, Hebert C, Ma V, Stains JP. The regulation of runt-related transcription factor 2 by fibroblast growth factor-2 and connexin43 requires the inositol polyphosphate/protein kinase Cδ cascade. J Bone Miner Res. 2013;(6):1468–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gordon LB, Shappell H, Massaro J, D’Agostino RB, Brazier J, Campbell SE, Kleinman ME, Kieran MW. Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA. 2018;319(16):1687–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu D, Yates PA, Zhang H, Cao K. Comparing lamin proteins post-translational relative stability using a 2A peptide-based system reveals elevated resistance of progerin to cellular degradation. Nucleus. 2016; 7(6): 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.