

Polyamine homeostasis determines T cell function.
Abstract
Robust and effective T cell–mediated immune responses require proper allocation of metabolic resources through metabolic pathways to sustain the energetically costly immune response. As an essential class of polycationic metabolites ubiquitously present in all living organisms, the polyamine pool is tightly regulated by biosynthesis and salvage pathway. We demonstrated that arginine is a major carbon donor and glutamine is a minor carbon donor for polyamine biosynthesis in T cells. Accordingly, the dependence of T cells can be partially relieved by replenishing the polyamine pool. In response to the blockage of biosynthesis, T cells can rapidly restore the polyamine pool through a compensatory increase in extracellular polyamine uptake, indicating a layer of metabolic plasticity. Simultaneously blocking synthesis and uptake depletes the intracellular polyamine pool, inhibits T cell proliferation, and suppresses T cell inflammation, indicating the potential therapeutic value of targeting the polyamine pool for managing inflammatory and autoimmune diseases.
INTRODUCTION
To cope with the capacity of pathogens for exponential growth, T cells have evolved mechanisms for rapidly adjusting their metabolism in response to T cell receptor (TCR) activation and additional signals indicating environmental change. A robust and effective metabolic reprogramming enables T cells to rapidly expand in number and differentiate to achieve large numbers of effector cells specialized in producing high levels of cytokines. Emerging evidence has shown that metabolic rewiring of the central carbon metabolism maximizes the acquisition and assimilation of energy and carbon sources and prepares T cells for growth, differentiation, immune regulation, and defense (1–5). Glycolysis, the pentose phosphate pathway (PPP), the Krebs cycle, and fatty acid oxidation (FAO) represent a core set of metabolic pathways that transform carbon and chemical energy from environmental nutrients to support the bioenergetic and biosynthesis needs of T cells. Beyond that, a myriad of peripheral metabolic pathways are integrated in complex metabolic networks and are tightly regulated to generate specialized metabolites, which are essential for maintaining homeostasis and immune functions of T cells.
Nonessential amino acids including arginine, glutamine, and proline have both anabolic and catabolic functions, providing building blocks for protein and nucleic acids, and connecting central carbon metabolism to a variety of specialized metabolic pathways, including polyamine biosynthesis (6–9). Polyamine is an essential class of polycationic metabolites ubiquitously present in all living organisms. In addition to de novo biosynthesis, other metabolic routes including polyamine catabolism, influx, and efflux act in concert to determine the size of the intracellular polyamine pool (10). Disrupting polyamine homeostasis can affect a plethora of cellular processes, including differentiation, transcription, translation, redox balance, and mitochondria quality control (10). Dysregulation of the level of polyamine and its amino acid precursors has been found to be associated with inflammation and autoimmune diseases (11, 12). We have previously reported that polyamines are one of the most up-regulated metabolite groups following T cell activation, and transcription factor MYC is responsible for its up-regulation (13). Emerging evidence has also shown that polyamine homeostasis is tightly regulated in cellular contexts other than T cells, which have critical roles in immune regulation and defense (14–18). As such, a better understanding of how polyamine homeostasis is regulated in immune cells will reveal the fundamental principles of the emerging connections between immune cell metabolic fitness and functional robustness. Further knowledge will also enable us to devise rational and practical approaches to treat inflammatory and autoimmune diseases.
Intrinsic T cell signaling cascades are instrumental in the control of T cell metabolic programming (5, 19–23). Numerous extrinsic environmental factors including oxygen and nutrient supplies also substantially influence T cell metabolic phenotypes and, thus, immune functions in vivo (24). Here, we report that the intracellular polyamine pool is tightly regulated by de novo biosynthesis and cellular uptake, through the import of extracellular polyamine. Heightened arginine and glutamine catabolism provide carbon sources to support polyamine de novo biosynthesis in vitro. Genetic and pharmacologic ablation of de novo biosynthesis of polyamine is sufficient to deplete the polyamine pool and suppress T cell proliferation in vitro. However, de novo biosynthesis is not essential for driving T cell proliferation in vivo, where T cells can salvage circulating polyamine to maintain intracellular polyamine pool. Simultaneously blocking polyamine synthesis and salvage inhibits T cell proliferation in vivo and confers protection against the pathogenic development of experimental autoimmune encephalomyelitis (EAE). Our findings implicate the potential therapeutic value of targeting polyamine metabolism in treating and managing inflammatory and autoimmune diseases.
RESULTS
Inhibition of ornithine decarboxylase reduces T cell proliferation and viability in vitro
We previously reported that an MYC-dependent noncanonical metabolic pathway links amino acid catabolism to the biosynthesis of polyamines during T cell activation (13). To investigate the role of polyamine metabolism in T cells, we used a genetic and a pharmacologic approach to ablate polyamine de novo biosynthesis. Because ornithine decarboxylase (ODC), the rate-limiting enzyme in polyamine de novo biosynthetic pathway, is essential for early embryo development, and ODC germline knockout is embryonically lethal (25), we obtained a mouse strain containing a reporter-tagged conditional allele of ODC (FRT-LacZ; ODCfl) generated by the European Mouse Mutant Archive (26). We first crossed this strain with the flippase (FLP) knock-in mouse strain, which removed the LacZ reporter allele and generated the strain containing the conditional allele (ODCfl). Then, we generated a T cell–specific ODC knockout strain (ODC cKO) by crossing the ODCfl strain with the CD4-Cre strain. The deletion of ODC was validated by quantitative polymerase chain reaction (qPCR) (fig. S1A), and the ablation of polyamine de novo synthesis was further validated by the accumulation of ornithine (substrate of ODC) and the depletion of putrescine, N-acetylputrescine, spermine, and spermidine (fig. S1B). ODC deletion did not affect the distribution of T cell subsets in the thymus, spleen, and lymph nodes (fig. S2, A and B). In addition, the percentage of naturally occurring interferon-γ (IFN-γ)–producing, interleukin-17 (IL-17)–producing, and FoxP3+ CD4 T cells is comparable in both wild-type (WT) and ODC cKO animals (fig. S2C). However, genetic deletion of ODC substantially delayed cell cycle progression from G0/G1 to the S phase after T cell activation and suppressed overall T cell proliferation in vitro (Fig. 1, A and B). Consistent with the impact of genetic deletion of ODC on T cells, difluoromethylornithine (DFMO), a potent inhibitor of ODC, inhibited activation-induced T cell cycle progression and proliferation in vitro (Fig. 1, C and D). Last, both genetic deletion of ODC and DFMO treatment caused moderately more cell death after activation in a time-dependent manner (Fig. 1, E and F). Together, our results indicate that polyamine homeostasis is critical for T cell proliferation and survival.
Fig. 1. Blockage of de novo polyamine biosynthesis suppresses T cell proliferation and reduces viability in vitro.
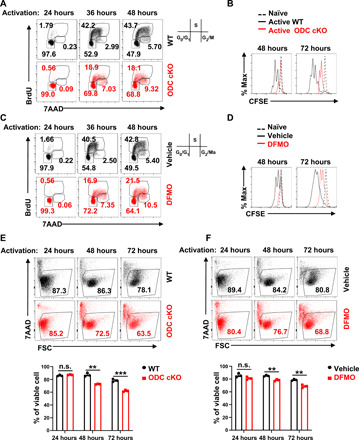
(A and C) Cell cycle of activated CD4+ T cells at 24, 36, and 48 hours with indicated genotypes or treatments was analyzed by using BrdU and 7AAD staining; the numbers indicate the percentage of cells in each cell cycle stage. Data represent three independent experiments. (B and D) Cell proliferation of naïve and activated CD4+ T cells at 48 and 72 hours with indicated genotypes or treatments was determined by CFSE dilution. Data represent six independent experiments. (E and F) Cell viability of activated CD4+ T cells at 24, 48, and 72 hours with indicated genotypes or treatments was assessed by 7AAD staining. Top panel is the representative dot plot, and bottom panel is the statistical analysis (means ± SEM). FSC, forward scatter. Data represent three independent experiments, **P < 0.01, ***P < 0.001 compared with WT, one-way analysis of variance (ANOVA). n.s., not significant.
ODC activity is dispensable for T cell proliferation and function in vivo
Controlling polyamine homeostasis is crucial for supporting cellular functions in all cell types, and both de novo biosynthesis and salvage by importing extracellular polyamine into the cell tightly regulate cellular polyamine pool size (10). Without exogenous polyamine supplements in cell culture media, we envisioned that T cells would solely depend on de novo biosynthesis to maintain the intracellular polyamine pool in vitro. However, circulating polyamines that are provided by dietary intake and by intestinal microbiota may be a source of exogenous polyamine for T cells in vivo (27). To assess the impact of ablating de novo biosynthesis on CD4 T cell proliferation in vivo, we used a well-established competitive homeostatic proliferation assay to determine the ratio and carboxyfluorescein succinimidyl ester (CFSE) dilution pattern of purified WT(Thy1.1+) or ODC cKO(Thy1.2+) CD4+ T cells in Rag1-deficient (Rag1−/−) mice, which lack endogenous T cells and permit spontaneous proliferation of adoptively transferred donor T cells. Unexpectedly, the ratio between WT and ODC cKO CD4+ T cells was similar before and after adoptive transfer. In addition, WT and ODC cKO CD4+ T cells display an overlapped CFSE dilution pattern, indicating that the loss of ODC did not affect T cell proliferation in vivo (Fig. 2A). Next, we sought to measure antigen-specific, TCR-dependent proliferation of WT or ODC cKO CD4+ T cells. We crossed Thy1.1 and CD4-Cre, ODCfl mice with OT-II transgenic mice to generate WT(Thy1.1+) and ODC cKO(Thy1.2+) donor OT-II strains in CD45.2+ background. We then adoptively transferred mixed and CFSE-labeled WT and ODC cKO CD4+ T cells into CD45.1+ mice that were immunized with chicken ovalbumin 323–339 peptide (OVA323–339) in complete Freund’s adjuvant (CFA). After 7 days, we measured the percentage ratio and CFSE dilution pattern of WT(Thy1.1+) and ODC cKO(Thy1.2+) CD4+ T cells in popliteal lymph node. Consistent with the homeostatic proliferation results, WT and ODC cKO OT-II–specific CD4+ T cells display a comparable antigen-specific proliferation (Fig. 2B). The expansion and balance between proinflammatory CD4+ T effector (Teff) cells determine the pathogenic development of EAE, a murine model of multiple sclerosis (MS), which is an inflammatory demyelinating disease of the central nervous system (CNS). We used this well-characterized system to further interrogate an in vivo CD4 T cell response in the absence of polyamine de novo biosynthesis. In line with our homeostatic and antigen-specific proliferation data, neither the genetic deletion of ODC in T cells nor the systemic delivery of DFMO changes the kinetics of pathogenic progression (Fig. 2, C and D). Together, our data indicate that polyamine salvage from circulation may be able to support T cell proliferation and effector function by compensating for the loss of de novo biosynthesis in the in vivo environment.
Fig. 2. De novo polyamine biosynthesis is dispensable for driving T cell proliferation and function in vivo.
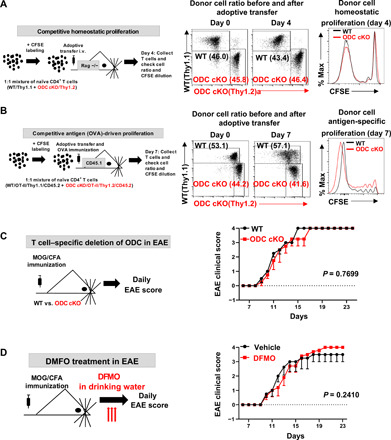
(A and B) An overview of in vivo competitive proliferation experimental procedure. The donor CD4 T cell ratios before and after adoptive transfer were evaluated by surface staining of isogenic markers, and cell proliferation was evaluated by CFSE dilution. Data represent three independent experiments. i.v., intravenous. (C and D) An overview of EAE experimental procedure. Clinical scores were evaluated daily (right). Data represent means ± SEM (n = 5) for each group; experiments were repeated three times.
Polyamine salvage compensates for the loss of biosynthesis activity in vitro
Next, we investigated the role of polyamine salvage (uptake) in regulating polyamine homeostasis in T cell. We measured polyamine uptake activity using radioactive-labeled putrescine (14C-putrescine) in naïve, active WT and ODC cKO T cells. Active T cells displayed higher polyamine uptake activity than naïve T cells (Fig. 3A). Ablation of ODC induces a compensatory increase in polyamine uptake (Fig. 3B). Next, we asked if polyamine uptake is sufficient to maintain polyamine homeostasis and support T cell proliferation in the absence of ODC activity. While genetic deletion or pharmacologic inhibition of ODC substantially delayed cell cycle progression from G0/G1 to the S phase and suppressed overall T cell proliferation, exogenous polyamine supplement could restore the cell cycle progression, proliferation, and viability in DFMO-treated and ODC cKO CD4+ T cells in vitro (Fig. 3, C and F, and fig. S3, A and B). We then sought to use a pharmacologic approach to block polyamine uptake and assessed the role of polyamine uptake in regulating polyamine homeostasis. AMXT 1501 (AMXT) is a novel lipophilic polyamine mimetic that potently blocks polyamine uptake in the low nanomolar concentration (28). The combination of AMXT and DFMO could effectively deplete the polyamine pool in tumor cells and suppress the growth of tumors (29). These promising preclinical studies led to a recently opened phase I clinical trial in solid tumors (NCT03077477). Similar to the genetic data (Fig. 3B), DFMO treatment induces a compensatory increase in polyamine uptake, which can be blocked by AMXT (Fig. 3G). In addition, AMXT could significantly suppresses exogenous polyamine-mediated cell proliferation and viability in ODC cKO, but not in WT CD4+ T cells (Fig. 3H and fig. S3C). Similarly, DFMO treatment suppresses human T cell viability and proliferation, which can be restored by polyamine supplement (fig. S4, A and B). AMXT treatment blocks exogenous polyamine-mediated cell proliferation and viability in human T cells (fig. S4, A and B). These results demonstrated that both human and mouse T cells require polyamines for supporting proliferation and survival. In addition, our results suggest that either polyamine biosynthesis or salvage is sufficient to maintain polyamine homeostasis in T cells. Supporting this idea, AMXT treatment alone failed to suppress T cell homeostatic proliferation or antigen-specific proliferation in vivo (fig. S5, A and B). These results, together with the results described above, suggest that the salvage pathway and the de novo biosynthesis pathway can compensate for the loss of each other, representing a layer of metabolic plasticity engaged by T cells to maintain polyamine homeostasis.
Fig. 3. Ablation of de novo polyamine biosynthesis renders T cells dependent on polyamine uptake that can be blocked by AMXT 1501.
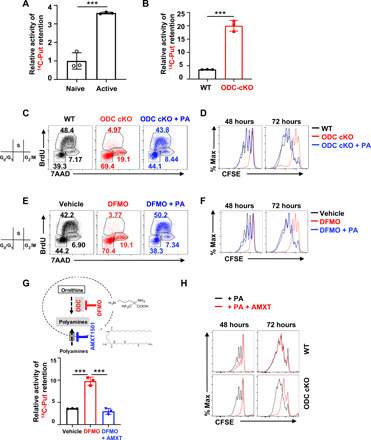
(A, B, and G) Putrescine uptake in the indicated experimental groups was determined by the cellular retention of 14C-labeled putrescine (means ± SEM). Data represent three independent experiments. ***P < 0.001, Student’s t test. (C and E) Cell cycle of 48-hour–activated CD4+ T cells with indicated genotypes or treatments was assessed by using BrdU and 7AAD staining. The numbers indicate the percentage of cells in each cell cycle stage. Data represent three independent experiments. (D, F, and H) Cell proliferation of activated CD4+ T cells at 48 and 72 hours with indicated genotypes or treatments was evaluated by CFSE dilution. Data represent three independent experiments.
Simultaneously blocking polyamine salvage and biosynthesis suppresses T cell proliferation in vivo
Given that either genetically ablating ODC or pharmacologically blocking polyamine uptake failed to affect T cell proliferation and function in vivo, we sought to assess the impacts of simultaneously blocking polyamine salvage and biosynthesis on T cells in vivo. We used competitive homeostatic proliferation and antigen-specific T cell proliferation assays to determine the proliferation of WT and ODC cKO donor CD4+ T cells in recipient mice, which were treated with vehicle or AMXT during the course of experiment. Donor ODC cKO CD4+ T cells recovered from AMXT-treated animals but not from vehicle-treated animals displayed reduced percentage and delayed proliferation compared with competitive WT CD4+ T cells (Fig. 4, A and B). We then sought to assess the impacts of simultaneously blocking polyamine salvage and biosynthesis on T cells in the EAE model. The genetic deletion of ODC in T cells failed to cause any notable changes in EAE pathogenic progression. Animals that were treated with AMXT displayed a delayed disease onset initially, but eventually proceeded with pathologic development and reached the end point. The combination of AMXT with genetic deletion of ODC in T cells conferred full protection against EAE pathogenic progression (Fig. 4C). ODC inhibitor DFMO is a Food and Drug Administration–approved medicine for hirsutism and African sleeping sickness and has been widely tested as a chemopreventive and chemotherapeutic agent against solid tumors (30, 31). Similar to the genetic data, DFMO alone failed to suppress EAE pathogenic progression (Fig. 4D). Although AMXT alone was sufficient to delay EAE onset moderately, it failed to protect animals from reaching the end point (Fig. 4D). We envision that the combination of DFMO and AMXT may be sufficient to deplete the T cell polyamine pool and consequently suppress T cell proliferation and effector function in vivo. Supporting this idea, the combination of AMXT and DFMO, but not single treatments, conferred full protection against EAE pathogenic progression (Fig. 4D). To evaluate the potential hematological toxicity of DFMO and AMXT combination treatment, we treated CD-1 mice with AMXT or AMXT plus DFMO at various dose levels for 28 days, and then evaluated a range of hematological parameters at days 29 and 43 (after 15-day recovery period). Our results showed that AMXT and DFMO combination treatment does not affect most of the hematological parameters (table S1). Inflammatory TH1, TH17, and FoxP3-expressing regulatory T cells (Tregs) are closely related to CD4 T cell subsets but with distinct functions. The balance between proinflammatory CD4+ Teff cells and Tregs determines the pathogenic development of EAE. Next, we examined polyamine’s role in CD4 Teff cell differentiation in vitro. Without exogenous polyamine supplemented in cell culture media, maintenance of the intracellular polyamine pool depends solely on ODC-mediated polyamine biosynthesis. Intracellular polyamine depletion resulting from ODC deficiency inhibited proinflammatory TH1 and TH17 cell differentiation while enhancing anti-inflammatory iTreg cell differentiation in vitro (fig. S6A). In EAE animals treated with DFMO in combination with AMXT, we observed a significant reduction in CD4+ T cell infiltration into the CNS and a reduction in IL17+ CD4+ T cell in CNS compared with the control group (fig. S6B). Together, our results indicate that the polyamine blocking approach, via ablation of salvage and biosynthesis pathways, suppresses T cell proliferation and inflammatory functions and may be a potential new therapy for treating inflammatory and autoimmune disease.
Fig. 4. Simultaneous blockage of polyamine uptake and de novo biosynthesis suppresses T cell proliferation and function in vivo.
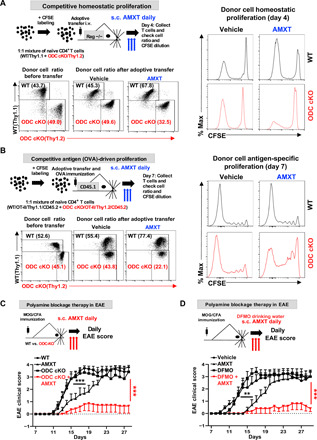
(A and B) Overview of in vivo competitive homeostatic and antigen-driven proliferation experimental procedure. The donor CD4+ T cell ratios before and after adoptive transfer were evaluated by surface staining of isogenic markers, and cell proliferation was evaluated by CFSE dilution. Data represent three independent experiments. s.c., subcutaneous. (C and D) Overview of the EAE experimental procedure. Clinical scores were evaluated daily. Data represent means ± SEM of n = 5 to 10 for each group. Experiments were repeated three times. **P < 0.01, ***P < 0.001, two-way analysis of variance (ANOVA).
Amino acid catabolism provides carbon sources for polyamine biosynthesis
Dietary intake and intestinal microbiota metabolism are major sources of circulating polyamine (27). We reasoned that an understanding of carbon sources that drive polyamine biosynthesis in T cells would enable the development of nutritional approaches for effectively controlling polyamine homeostasis in T cells. While arginine catabolism is integrated into the urea cycle to detoxify ammonia and provides the precursor for polyamine biosynthesis, we previously reported that glutamine-derived carbon could be funneled into polyamine biosynthesis through ornithine in T cells (13). In addition, proline can also provide carbon for polyamine biosynthesis in the placenta and in plants (32). To determine to what extent these three amino acids contribute to polyamine biosynthesis, we applied the stable isotope of carbon-13 (13C) labeling and mass spectrometry approach. We supplied 13C6-arginine, 13C5-glutamine, or 13C5-proline as metabolic tracers in T cell culture media and then followed 13C incorporation into individual metabolites (Fig. 5A). While not all selected metabolites were detected in our experiment because of technical limitation and a portion of proline was produced through de novo biosynthesis, our results clearly demonstrated that 13C5-proline only contributes a minimal amount of 13C5 isotopologues of ornithine and polyamine (Fig. 5B). In contrast, 13C6-arginine and 13C5-glutamine contribute 50 and 40% of 13C5-ornithine, respectively (Fig. 5B). 13C6-arginine and 13C5-glutamine contribute around 80 and 20% of 13C4 isotopologues of polyamine (putrescine and spermidine generated via decarboxylation of ornithine), respectively (Fig. 5B). Thus, we conclude that arginine is a major carbon donor, and glutamine is a minor carbon donor for supporting polyamine biosynthesis in T cells in vitro.
Fig. 5. Arginine catabolism supports polyamine biosynthesis and T cell proliferation.
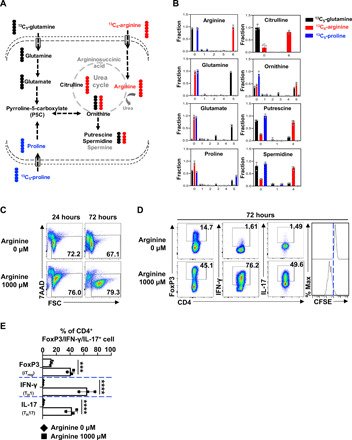
(A) Diagram of putative catabolic routes of 13C5-glutamine, 13C6-arginine, and 13C5-proline in T cells. (B) T cells were incubated in 13C5-glutamine or13C6-arginine, or 13C5-proline medium for 36 hours. Metabolites were extracted and analyzed using CE-TOFMS as described in Materials and Methods. Numbers in the x axis represent those of 13C atoms in given metabolites. Data represent means ± SEM of n = 3 for each group. (C) Cell viability of activated CD4+ T cell with indicated treatments was determined by 7AAD staining. Data represent three independent experiments. (D and E) CD4+ T cells were activated and polarized toward iTreg, TH1, and TH17 lineages with indicated treatment for 72 hours. The indicated proteins were quantified by intracellular staining. Cell proliferation was determined by CFSE staining. Data represent three independent experiments, ***P < 0.001, ****P < 0.0001, one-way ANOVA.
Arginine catabolism drives T cell proliferation partially through supporting polyamine biosynthesis
While arginine is generally considered a nonessential amino acid, T cells are arginine auxotrophic in vitro (18). Arginine is required to maintain CD4+ T cell viability, driving proliferative and proinflammatory lineage (TH17) differentiation (Fig. 5, C to E). While the amount of most amino acids is in the low micromolar (μM) range, the concentration of arginine is in the millimolar (mM) range in cell culture media. We envisioned that, in addition to a general requirement of arginine for protein synthesis, arginine might also support T cell proliferation and differentiation through polyamine biosynthesis. To test this idea, we titrated down the amount of arginine in cell culture media and found that 10 μM arginine was sufficient to support protein synthesis and the growth of cell mass at the early time point after T cell activation but failed to maintain cell viability and drive proliferation later (fig. S7, A and B). We envision that arginine supports protein synthesis and polyamine biosynthesis, both of which are required to drive T cell proliferation. Supporting this idea, we have shown that a low level of arginine (10 μM) in culture media is sufficient to support protein synthesis, but not proliferation (fig. S7A). Similar to what we found in arginine depletion condition, a low level of arginine (10 μM, with or without daily replenishment) in culture media failed to support T cell proliferation and an optimal TH17 differentiation (fig. S8, B and C). However, polyamine supplements can partially restore T cell proliferation and TH17 differentiation with a low level of arginine (10 μM, with or without daily replenishment) (fig. S8, B and C). In contrast, polyamine supplements failed to restore T cell proliferation and differentiation under arginine starvation condition (0 μM) (fig. S8A). Collectively, our data indicate that T cell activation engages a metabolic axis that connects arginine catabolism to polyamine de novo biosynthesis.
DISCUSSION
Fast-growing pathogens impose selective pressures on the metabolic fitness and metabolic plasticity of host immune cells, which are necessary for immune cells to maintain homeostasis while remaining ready to mount rapid responses under diverse metabolic and immune conditions (24, 33–36). A robust T cell–mediated adaptive immune response exhibits high and dynamic metabolic demands in T cells, which is accommodated through fine-tuned regulation on both the central carbon metabolic pathways and peripheral metabolic pathways including polyamine metabolism (4, 5, 9, 19). The standard formulation of cell culture media, which does not fully recapitulate the physiological metabolite composition in the plasma and tissue microenvironment, often leads to a substantial metabolic adaptation of growing cells in vitro. Metabolic phenotypes of cells growing in vivo may further differ from cells growing in vitro as a result of other environmental factors including oxygen level, cellular competition and cooperation, and the biophysical properties of the extracellular matrix (37–39). Similarly, T cell metabolism can respond and adapt to environmental nutrient levels (24, 40). Our studies revealed T cells’ capacity to engage in both de novo biosynthetic and salvage pathways to fine-tune polyamine homeostasis, which is required to maximize metabolic fitness and optimize CD4 Teff cell proliferation and differentiation. Such metabolic plasticity is likely to be crucial for T cells’ ability to elicit robust immune responses in different tissue contexts.
Glutamine and arginine are two nonessential amino acids that not only fulfill the general requirements for protein synthesis but also connect central carbon metabolism to a variety of biosynthetic pathways to produce specialized metabolites (41–44). Glutamine catabolism funnels the anaplerotic flux of carbon into the TCA cycle and also provides sources of nitrogen and carbon to support biosynthesis of nonessential amino acids, lipids, nucleotides, glutathione, and polyamines in T cells (13, 45, 46). Similarly, arginine is critical for maintaining T cell viability, driving proliferative and effector functions (14). We found that polyamine supplement could partially relieve T cell’s dependence on arginine, indicating that a key role of arginine catabolism in T cells is to support polyamine biosynthesis. Consistently, arginine serves as a major donor of carbon in polyamine biosynthesis, while glutamine only plays a minor role in funneling carbon into polyamine in the presence of arginine in vitro. These two amino acids contribute a comparable portion of carbon to ornithine—the precursor of polyamine. This finding is in line with previous studies showing that arginine-derived ornithine is not the only source for endogenous ornithine in most mammalian tissues (47). In addition, our findings may implicate ornithine as an important metabolic node representing a key branch point in both glutamine and arginine catabolic pathways. Ornithine can be committed toward the urea cycle, proline biosynthesis, or de novo synthesis of polyamine. The production of ornithine from two different metabolic precursors, glutamine and arginine, also enables fine-tuned coordination between the metabolic flux shunted toward polyamine synthesis and the metabolic flux shunted toward other specialized metabolites. Consistent with this idea, the overall high consumption rate of glutamine and arginine may provide a sensitive and precise regulation on intermediate metabolites that can be committed toward several metabolic branches, hence permitting rapid responses to meet the metabolic demands of cell growth and cytokine production.
Active T cells and other highly proliferative cells such as tumor cells share metabolic characteristics and are strictly dependent on the catabolism of glucose and glutamine through the central carbon metabolic pathways (48). Similarly, elevated levels of polyamine are associated with T cell activation and cell transformation (13, 49). Pharmacologic or genetic targeting of ODC, a transcriptional target of proto-oncogene MYC, could delay the development and progression of MYC-driven tumors in mice (50). While pharmacologic agents targeting polyamines such as the ODC inhibitor DFMO have generally low toxicity, DFMO yields only very marginal therapeutic benefits in patients with cancer as a single-agent therapy in clinical trials (51). Clearly, cancer cells are capable of importing polyamine from circulation to overcome the effect of DFMO. Supporting this idea, the simultaneous blockage of polyamine biosynthesis and uptake has generated promising results in tumor preclinical animal models (29). Mammalian cells can uptake polyamine through endocytosis and membrane transport system mediated by several solute carrier transporters (52). AMXT 1501 is a novel lipophilic polyamine mimetic that potently blocks polyamine uptake through competing with polyamine (28). The combination of AMXT 1501 and DFMO could effectively deplete the polyamine pool in tumor cells and suppress the growth of tumors in various animal models (29). These promising preclinical studies led to a recently opened phase I clinical trial in solid tumors (NCT03077477). Similarly, we have shown that ODC cKO CD4+ T cells proliferate and function normally in vivo. However, concurrent treatment of ODC cKO CD4+ T cells with AMXT 1501 abrogates their proliferation and inflammatory function in vivo. Moreover, the combination of AMXT 1501 and DFMO could confer full protection to mice against pathogenic development of EAE. Thus, polyamine blocking strategy via simultaneous blockade of polyamine biosynthesis and salvage may present a promising and novel therapy for treating inflammatory and autoimmune diseases.
MATERIALS AND METHODS
Mice
C57BL/6 (WT) mice, Rag1−/− mice (B6.129S7-Rag1tm1Mom/J), OT-II mice [B6.Cg-Tg(TcraTcrb)425Cbn/J], Thy1.1+ mice (B6.PL-Thy1a/CyJ), and CD45.1+ mice (B6.SJL-Ptprca Pepcb/BoyJ) were obtained from the Jackson laboratory (JAX, Bar Harbor, ME). Mice with one targeted allele of ODC on the C57BL/6 background (Odc1tm1a(EUCOMM)Hmgu/J) were generated by the Knockout Mouse Phenotyping Program (KOMP2) at the JAX. The mice were first crossed with a transgenic Flippase strain [B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/RainJ] to remove the LacZ reporter allele, and then crossed with the CD4-Cre strain to generate T cell–specific ODC knockout strain (ODC cKO). Gender- and age-matched mice (6 to 12 weeks old) were used for all the experiments. All mice were maintained in specific pathogen–free conditions at the Nationwide Children Hospital animal facility, and all the animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the Abigail Wexner Research Institute at Nationwide Children’s Hospital (protocol number AR13-00055).
Flow cytometry
For analysis of surface markers, cells were stained in phosphate-buffered saline (PBS) containing 2% (w/v) bovine serum albumin and the appropriate antibodies from BioLegend for 30 min at 4°C. For intracellular cytokine staining, cells were stimulated for 4 hours with Cell Stimulation Cocktails of phorbol 12-myristate 13-acetate (PMA), ionomycin, brefieldin A, and monensin (eBioscience). Cells were then washed with PBS, fixed, and permeabilized using FoxP3 Fixation/Permeabilization Kit according to the manufacturer’s instructions (eBioscience). Cells were then stained with anti–IFN-γ and anti–IL-17A antibodies (BioLegend); for intracellular FoxP3 staining, cells were fixed, permeabilized, and stained with anti–FoxP3 antibody (BioLegend). For protein content measurement, cells were fixed with 4% paraformaldehyde for 30 min at 4°C, permeabilized with FoxP3 permeabilization solution (eBioscience), and then stained with fluorescein isothiocyanate (FITC) (Thermo Fisher Scientific). For CFSE staining, freshly isolated T cells were incubated with 4 μM CFSE (Invitrogen) in PBS at room temperature for 10 min, and then washed three times with cell culture medium containing 10% fetal bovine serum (FBS). For cell viability analysis, cells were stained with 7AAD (BioLegend) for 5 min before running on a flow cytometer. All flow cytometry data were acquired on Novocyte (ACEA Biosciences) and were analyzed with FlowJo software (BD Biosciences).
Cell culture
Human peripheral blood mononuclear cells (PBMCs) were isolated from buffy coat (American Red Cross) using Ficoll/Hypaque density gradient centrifugation. Human CD4+ naïve T cells were directly enriched from PBMCs using human CD4 naïve T cell Isolation Kit (MojoSort, BioLegend) following the manufacturer’s instructions. Isolated human T cells (1 × 106 cells/ml) were stimulated with plate-bound anti-hCD3 and anti-hCD28 antibodies and cultured in RPMI-1640 medium (Gibco, Thermo Fisher Scientific) supplemented with 10% (v/v) heat-inactivated dialyzed FBS (DFBS), 2 mM l-glutamine, 1% sodium pyruvate (Sigma-Aldrich), penicillin (100 U/ml), and streptomycin (100 μg/ml), in the presence of hIL-2 (100 U/ml, Bio X Cell) at 37°C/5% CO2. Culture plates were precoated with anti-hCD3 (1 μg/ml) and anti–hCD28 antibodies (1 μg/ml) (Bio X Cell) overnight at 4°C. DFBS was made by dialyzing against 100 volumes of PBS (five changes in 3 days) using Slide-A-Lyzer G2 dialysis cassettes with cut-through MW size 2K (Thermo Fisher Scientific) at 4°C to remove any potential polyamines in the FBS (Atlanta Biologicals).
Total CD3+ T cells or naïve CD4+ T cells were isolated from mouse spleen and lymph nodes using murine CD3 or CD4 naïve T cell Isolation Kit (MojoSort, BioLegend) following the manufacturer’s instructions. Freshly isolated mouse T cells (1 × 106 cells/ml) were either maintained in culture media containing mouse IL-7 (5 ng/ml) or stimulated with mouse IL-2 (100 U/ml) as well as plate-bound anti-mCD3 and anti-mCD28 antibodies, and cultured with the medium described above with 0.05 mM 2-mercaptoethanol (Sigma-Aldrich). Culture plates were precoated with anti-mCD3 (2 μg/ml) (clone 145-2C11, Bio X Cell) and anti-mCD28 (2 μg/ml) (clone 37.51, Bio X Cell) antibodies overnight at 4°C. For CD4 T cell polarization, freshly isolated naïve CD4+ T cells (0.6 × 106/ml) were stimulated with cytokine and neutralizing antibody cocktail as well as plate-bound anti-mCD3 and anti-mCD28 antibodies. Specifically, culture plates were precoated with 5 μg/ml (for iTreg and TH1) or 10 μg/ml (for TH17) anti-mCD3 and anti-mCD28 antibodies overnight at 4°C. Cytokines and neutralizing antibody cocktails include mIL-2 (200 U/ml) and hTGF-β1 (5 ng/ml, PeproTech) for iTreg differentiation; mIL-6 (50 ng/ml, PeproTech), hTGF-β1 (2 ng/ml), anti–mIL-2 (8 μg/ml, Bio X Cell), anti–mIL-4 (8 μg/ml, Bio X Cell), and anti–mIFN-γ (8 μg/ml, Bio X Cell) for TH17 differentiation; and mIL-2 (200 U/ml), mIL-12 (5 ng/ml, PeproTech), and anti–mIL-4 (10 μg/ml) for TH1 differentiation. DFMO (2 mM, Carbosynth) and AMXT 1501 (1 μM, Aminex Therapeutics) were added into the cell culture medium in the indicated experiments. For polyamine supplementation experiment, polyamine mixture including putrescine, spermidine, and spermine (1 μM, Sigma-Aldrich) was added into culture medium along with aminoguanidine (0.2 mM, Sigma-Aldrich), which inhibits diamine oxidase in the FBS.
Cell cycle analysis
T cell cycle analysis was performed using Phase-Flow Alexa Fluor 647 BrdU Kit (BioLegend) per the manufacturer’s instructions. Briefly, T cells were pulsed with BrdU (5-bromo-2′-deoxyuridine) (10 μg/ml) for 1 hour followed by surface staining, fixation, and permeabilization. BrdU incorporated into the DNA during the S phase was recognized by intracellular BrdU antibody staining, and total DNA content was used to differentiate the G1 and G2 stages by 7AAD labeling.
Quantitative polymerase chain reaction
Total RNA was isolated using the RNeasy Mini Kit (Qiagen) and was reverse transcribed using random hexamers and M-MLV Reverse Transcriptase (Invitrogen). SYBR Green–based reverse transcription qPCR (RT-qPCR) was performed using the Bio-Rad CFX96 Real-Time PCR Detection System. The relative gene expression was determined by the comparative CT method, also referred to as the 2−ΔΔCT method. The data were presented as the fold change in gene expression normalized to an internal reference gene (β2-microglobulin) and relative to the control (the first sample in the group). Fold change = 2−ΔΔCT = [(CTgene of interest − CTinternal reference)] sample A − [(CTgene of interest − CTinternal reference)] sample B. Samples for each experimental condition were run in triplicated PCRs. ODC1 primer sequences (forward: GCATGCATCTGCTTGATATTGG, reverse: GTCCAGAGCTGGGTTGATTAC) were obtained from PrimerBank.
Putrescine uptake assay
T cells (2 × 106) were suspended in 200 μl of PBS containing permeant putrescine (100 μM) and [1,4-14C]-putrescine dihydrochloride (0.2 μCi, ARC 0245) and then incubated at 37°C for 10 min (within the established linear phase of uptake). The uptake was stopped by loading all the transport mixture onto a discontinuous gradient of bromododecane and perchloric acid/sucrose and then centrifuged at 14, 000g for 90 s. The discontinuous gradient was prepared by overlaying 1-bromododecane (800 μl, Sigma-Aldrich) above 20% perchloric acid (100 μl, Sigma-Aldrich)/8% sucrose solution in a 1.5-ml microfuge tube. The samples were snap frozen in an ethanol-dry ice bath. The bottom part of the microfuge tubes containing T cell lysate in perchloric acid–sucrose was cut by a microfuge cutter. The cell lysates were diluted with 300 μl of 0.5% SDS–1% Triton X-100, and then transferred into scintillation vials with 10 ml of scintillation cocktail. The radioactivity was quantitated by liquid scintillation spectrometry.
Adoptive cell transfer and in vivo proliferation
For homeostatic proliferation assay, naïve CD4+ T cells isolated from WT(Thy1.1+) and ODC cKO(Thy1.2+) donor mice were labeled with CFSE as described above, and then were mixed at 1:1 ratio. Approximately 1 × 107 mixed cells in 150 μl of PBS were injected via the retro-orbital venous sinus into gender-matched Rag1−/− mice, which were euthanized 4 days later. Lymph nodes were collected, and cell proliferation was assessed by flow cytometry analysis.
For OVA antigen–driven proliferation assay, naïve CD4+ T cells isolated from OT-II–WT(Thy1.1+) and OT-II–ODC cKO(Thy1.2+) donor mice were labeled with CFSE as described above, and then were mixed at 1:1 ratio. Approximately 1 × 107 mixed cells in 150 μl of PBS were injected via the retro-orbital venous sinus into gender-matched CD45.1+ mice. One day after cell injection, host mice were immunized subcutaneously in the hock area of both legs with 50 μl OVA323–339 peptide (1 mg/ml, InvivoGen) emulsified with CFA (InvivoGen). Seven days after immunization, lymph organs of host mice were collected for cell surface staining and flow cytometry analysis.
EAE induction and assessment
Mice were immunized subcutaneously with 100 μg of myelin oligodendrocyte glycoprotein (MOG)35–55 peptide emulsified in CFA [mixture of IFA (incomplete Freund’s adjuvant; Difco) and Mycobacterium tuberculosis (Difco)]. Mice were intraperitoneally injected with 200 ng of pertussis toxin (PTX; List Biological Laboratories) on the day of immunization and 2 days later. For the DFMO-treated group, mice were fed with 1% DFMO in drinking water, which was replenished every 5 days throughout the experiment. For the AMXT 1501–treated group, mice received AMXT 1501 (3 mg/kg) subcutaneously daily throughout the experiment. All mice were observed daily for clinical signs and scored as described below in Table 1.
Table 1. EAE clinical score assessment sheet.
| Score 1 | Limp tail. When the mouse is picked up by the tail, the whole tail drapes over your finger instead of being erect. |
| Score 2 | Limp tail and weakness of hind legs. When mouse is picked up by tail, legs are not spread apart but held closer together. When the mouse is observed when walking, it has a clearly apparent wobbly walk. |
| Score 3 | Limp tail and complete paralysis of hind legs (most common). OR limp tail with paralysis of one front and one hind leg. |
| Score 4 | Limp tail, complete hind leg and partial front leg paralysis. Mouse is minimally moving around the cage but appears alert and feeding. Usually, euthanasia is recommended |
In some experiments, mice were euthanized when control mice reached the onset of symptom or when control mice reached terminal end stage as indicated. The CNS (brain and spinal cord), spleen, and peripheral lymph nodes were collected, and then single-cell suspension of each organ was prepared. To isolate mononuclear cells from the CNS, cell suspension was centrifuged on a 30%/70% Percoll gradient at 500g for 30 min. Isolated cells were activated for 4 hours using cell stimulation cocktail, and surface staining and intracellular staining were performed before flow cytometric analysis as described above.
Capillary electrophoresis–triple quadrupole/time-of-flight mass spectrometry (CE-QqQ/TOFMS) analysis
CD3+ T cells (around 1.5 × 107 cells per sample) isolated from WT or ODC cKO mice were activated for 36 hours and then used for the extraction of intracellular metabolites. The cells were collected by centrifugation (300g at 4°C for 5 min) and washed twice with 5% mannitol solution (10 ml first and then 2 ml), then treated with 800 μl of methanol and vortexed for 30 s to inactivate enzymes. Next, the cell extract was treated with 550 μl of Milli-Q water containing internal standards (H3304-1002, Human Metabolome Technologies Inc., Tsuruoka, Japan) and vortexed for 30 s. The extract was obtained and centrifuged at 2300g and 4°C for 5 min, and then 700 μl of upper aqueous layer was centrifugally filtered through a Millipore 5-kDa cutoff filter at 9100g and 4°C for 180 min to remove proteins. The filtrate was centrifugally concentrated and resuspended in 50 μl of Milli-Q water for CE-MS analysis. Cationic compounds were measured in the positive mode of CE-TOFMS, and anionic compounds were measured in the positive and negative modes of CE-MS/MS. Peaks detected by CE-TOFMS and CE-MS/MS were extracted using an automatic integration software (MasterHands, Keio University, Tsuruoka, Japan, and MassHunter Quantitative Analysis B.04.00, Agilent Technologies, Santa Clara, CA, USA, respectively) to obtain peak information including m/z (mass/charge ratio), migration time (MT), and peak area. The peaks were annotated with putative metabolites from the Human Metabolome Technologies (HMT) metabolite database based on their MTs in CE and m/z values determined by TOFMS. The tolerance range for the peak annotation was configured at ±0.5 min for MT and ± 10 ppm for m/z. In addition, concentrations of metabolites were calculated by normalizing the peak area of each metabolite with respect to the area of the internal standard and by using standard curves, which were obtained by three-point calibrations.
Hierarchical cluster analysis and principal components analysis were performed by our proprietary software PeakStat and SampleStat, respectively. Detected metabolites were plotted on metabolic pathway maps using VANTED (Visualization and Analysis of Networks containing Experimental Data) software.
CE-TOFMS analysis
CD3+ T cells (around 1.8 × 107 cells per sample) were activated for 36 hours in conditional media (RPMI-1640) containing 2 mM 13C5-glutamine, 1.15 mM 13C6-arginine, or 0.17 mM 13C5-proline, and then used for the extraction of intracellular metabolites as described above. Metabolome measurements were carried out through a facility service at Human Metabolome Technologies Inc. (Tsuruoka, Japan). CE-TOFMS measurement was carried out using an Agilent CE Capillary Electrophoresis System equipped with an Agilent 6210 Time of Flight mass spectrometer, Agilent 1100 isocratic HPLC pump, Agilent G1603A CE-MS adapter kit, and Agilent G1607A CE-ESI-MS sprayer kit (Agilent Technologies, Waldbronn, Germany). The systems were controlled by Agilent G2201AA ChemStation software version B.03.01 for CE (Agilent Technologies, Waldbronn, Germany). The metabolites were analyzed by using a fused silica capillary [50 μm internal diameter (i.d.)× 80 cm total length], with commercial electrophoresis buffer (solution ID: H3301-1001 for cation analysis and H3302-1021 for anion analysis, Human Metabolome Technologies) as the electrolyte. The sample was injected at a pressure of 50 mbar for 10 s (approximately 10 nl) in the cation analysis and 25 s (approximately 25 nl) in the anion analysis. The spectrometer was scanned from m/z 50 to 1000.
Statistical analysis
Statistical analysis was conducted using the GraphPad Prism software (GraphPad Software Inc.). P values were calculated with two-way analysis of variance (ANOVA) for the EAE experiments. Unpaired two-tailed Student’s t test, multiple comparisons of one- or two-way ANOVA, or Dunnett’s least significant difference (LSD) test was used to assess differences in other experiments. P values less than 0.05 were considered significant, with *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgments
We thank J. Sherman and H. Rodgers for critically reading and editing the manuscript. We thank Dr. Song Guo Zheng for insight and valuable discussions. Funding: This work was supported by 1R21CA227926-01A1 and 1UO1CA232488-01 from the National Institutes of Health (Cancer Moonshot program), 1R01AI114581 from the National Institutes of Health, V2014-001 from the V-Foundation, and 128436-RSG-15-180-01-LIB from the American Cancer Society (to R.W.). Author contributions: R. Wang conceptualized and supervised this work. R. Wu, X.C., and S.K. carried out the experiments. T.W., J.R.G., Y.Y., and L.L. were involved in data collection, analysis, and review. G.F. and M.R.B. provided conceptual input into the study development. M.R.B. provided experimental material and conceptual input into the study development. R. Wu, X.C., S.K., and R. Wang wrote the manuscript. All authors discussed the results and provided feedback on the manuscript. Competing interests: M.R.B., currently, is the president and CSO of Aminex Therapeutics Inc., which is a pharmaceutical start-up company that actively develops polyamine blocking agents. All other authors declare that they have no competing interest. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabc4275/DC1
REFERENCES AND NOTES
- 1.Finlay D., Cantrell D. A., Metabolism, migration and memory in cytotoxic T cells. Nat. Rev. Immunol. 11, 109–117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang R., Green D. R., Metabolic reprogramming and metabolic dependency in T cells. Immunol. Rev. 249, 14–26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weinberg S. E., Sena L. A., Chandel N. S., Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Neill L. A. J., Kishton R. J., Rathmell J., A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearce E. L., Poffenberger M. C., Chang C.-H., Jones R. G., Fueling immunity: Insights into metabolism and lymphocyte function. Science 342, 1242454 (2013).24115444 [Google Scholar]
- 6.Bertolo R. F., Burrin D. G., Comparative aspects of tissue glutamine and proline metabolism. J. Nutr. 138, 2032S–2039S (2008). [DOI] [PubMed] [Google Scholar]
- 7.Wu G., Borbolla A. G., Knabe D. A., The uptake of glutamine and release of arginine, citrulline and proline by the small intestine of developing pigs. J. Nutr. 124, 2437–2444 (1994). [DOI] [PubMed] [Google Scholar]
- 8.Majumdar R., Barchi B., Turlapati S. A., Gagne M., Minocha R., Long S., Minocha S. C., Glutamate, ornithine, arginine, proline, and polyamine metabolic interactions: The pathway is regulated at the post-transcriptional level. Front. Plant Sci. 7, 78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puleston D. J., Villa M., Pearce E. L., Ancillary activity: Beyond core metabolism in immune cells. Cell Metab. 26, 131–141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pegg A. E., Functions of polyamines in mammals. J. Biol. Chem. 291, 14904–14912 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bronte V., Zanovello P., Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 5, 641–654 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Miller-Fleming L., Olin-Sandoval V., Campbell K., Ralser M., Remaining mysteries of molecular biology: The role of polyamines in the cell. J. Mol. Biol. 427, 3389–3406 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Wang R., Dillon C. P., Shi L. Z., Milasta S., Carter R., Finkelstein D., McCormick L. L., Fitzgerald P., Chi H., Munger J., Green D. R., The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geiger R., Rieckmann J. C., Wolf T., Basso C., Feng Y., Fuhrer T., Kogadeeva M., Picotti P., Meissner F., Mann M., Zamboni N., Sallusto F., Lanzavecchia A., L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167, 829–842.e13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardbower D. M., Asim M., Luis P. B., Singh K., Barry D. P., Yang C., Steeves M. A., Cleveland J. L., Schneider C., Piazuelo M. B., Gobert A. P., Wilson K. T., Ornithine decarboxylase regulates M1 macrophage activation and mucosal inflammation via histone modifications. Proc. Natl. Acad. Sci. U.S.A. 114, E751–E760 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H., Alsaleh G., Feltham J., Sun Y., Napolitano G., Riffelmacher T., Charles P., Frau L., Hublitz P., Yu Z., Mohammed S., Ballabio A., Balabanov S., Mellor J., Simon A. K., Polyamines control eIF5A hypusination, TFEB translation, and autophagy to reverse B cell senescence. Mol. Cell 76, 110–125.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puleston D. J., Buck M. D., Klein Geltink R. I., Kyle R. L., Caputa G., O’Sullivan D., Cameron A. M., Castoldi A., Musa Y., Kabat A. M., Zhang Y., Flachsmann L. J., Field C. S., Patterson A. E., Scherer S., Alfei F., Baixauli F., Austin S. K., Kelly B., Matsushita M., Curtis J. D., Grzes K. M., Villa M., Corrado M., Sanin D. E., Qiu J., Pällman N., Paz K., Maccari M. E., Blazar B. R., Mittler G., Buescher J. M., Zehn D., Rospert S., Pearce E. J., Balabanov S., Pearce E. L., Polyamines and eIF5A hypusination modulate mitochondrial respiration and macrophage activation. Cell Metab. 30, 352–363.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray P. J., Amino acid auxotrophy as a system of immunological control nodes. Nat. Immunol. 17, 132–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang R., Green D. R., Metabolic checkpoints in activated T cells. Nat. Immunol. 13, 907–915 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Zeng H., Chi H., mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr. Opin. Immunol. 46, 103–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gnanaprakasam J. N. R., Sherman J. W., Wang R., MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 35, 63–70 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Powell J. D., Delgoffe G. M., The mammalian target of rapamycin: Linking T cell differentiation, function, and metabolism. Immunity 33, 301–311 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi L. Z., Wang R., Huang G., Vogel P., Neale G., Green D. R., Chi H., HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slack M., Wang T., Wang R., T cell metabolic reprogramming and plasticity. Mol. Immunol. 68, 507–512 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pendeville H., Carpino N., Marine J.-C., Takahashi Y., Muller M., Martial J. A., Cleveland J. L., The ornithine decarboxylase gene is essential for cell survival during early murine development. Mol. Cell. Biol. 21, 6549–6558 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skarnes W. C., Rosen B., West A. P., Koutsourakis M., Bushell W., Iyer V., Mujica A. O., Thomas M., Harrow J., Cox T., Jackson D., Severin J., Biggs P., Fu J., Nefedov M., de Jong P. J., Stewart A. F., Bradley A., A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larqué E., Sabater-Molina M., Zamora S., Biological significance of dietary polyamines. Nutrition 23, 87–95 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Burns M. R., Graminski G. F., Weeks R. S., Chen Y., O’Brien T. G., Lipophilic lysine-spermine conjugates are potent polyamine transport inhibitors for use in combination with a +polyamine biosynthesis inhibitor. J. Med. Chem. 52, 1983–1993 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gamble L. D., Purgato S., Murray J., Xiao L., Yu D. M. T., Hanssen K. M., Giorgi F. M., Carter D. R., Gifford A. J., Valli E., Milazzo G., Kamili A., Mayoh C., Liu B., Eden G., Sarraf S., Allan S., Giacomo S. D., Flemming C. L., Russell A. J., Cheung B. B., Oberthuer A., London W. B., Fischer M., Trahair T. N., Fletcher J. I., Marshall G. M., Ziegler D. S., Hogarty M. D., Burns M. R., Perini G., Norris M. D., Haber M., Inhibition of polyamine synthesis and uptake reduces tumor progression and prolongs survival in mouse models of neuroblastoma. Sci. Transl. Med. 11, eaau1099 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Kennedy P. G., Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 12, 186–194 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Vissing A. C., Taudorf E. H., Haak C. S., Philipsen P. A., Haedersdal M., Adjuvant eflornithine to maintain IPL-induced hair reduction in women with facial hirsutism: A randomized controlled trial. J. Eur. Acad. Dermatol. Venereol. 30, 314–319 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Wu G., Bazer F. W., Hu J., Johnson G. A., Spencer T. E., Polyamine synthesis from proline in the developing porcine placenta. Biol. Reprod. 72, 842–850 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Weis S., Carlos A. R., Moita M. R., Singh S., Blankenhaus B., Cardoso S., Larsen R., Rebelo S., Schäuble S., Barrio L. D., Mithieux G., Rajas F., Lindig S., Bauer M., Soares M. P., Metabolic adaptation establishes disease tolerance to sepsis. Cell 169, 1263–1275.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinberg E. D., Nutritional immunity Host’s attempt to withold iron from microbial invaders. JAMA 231, 39–41 (1975). [DOI] [PubMed] [Google Scholar]
- 35.Abu Kwaik Y., Bumann D., Microbial quest for food in vivo: ‘Nutritional virulence’ as an emerging paradigm. Cell. Microbiol. 15, 882–890 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Troha K., Ayres J. S., Metabolic adaptations to infections at the organismal level. Trends Immunol. 41, 113–125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sullivan M. R., Danai L. V., Lewis C. A., Chan S. H., Gui D. Y., Kunchok T., Dennstedt E. A., Vander Heiden M. G., Muir A., Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cantor J. R., The rise of physiologic media. Trends Cell Biol. 29, 854–861 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ackermann T., Tardito S., Cell culture medium formulation and its implications in cancer metabolism. Trends Cancer 5, 329–332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma E. H., Verway M. J., Johnson R. M., Roy D. G., Steadman M., Hayes S., Williams K. S., Sheldon R. D., Samborska B., Kosinski P. A., Kim H., Griss T., Faubert B., Condotta S. A., Krawczyk C. M., DeBerardinis R. J., Stewart K. M., Richer M. J., Chubukov V., Roddy T. P., Jones R. G., Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity 51, 856–870.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Kvamme E., Svenneby G., Effect of anaerobiosis and addition of keto acids on glutamine utilization by Ehrlich ascites-tumor cells. Biochim. Biophys. Acta 42, 187–188 (1960). [DOI] [PubMed] [Google Scholar]
- 42.DeBerardinis R. J., Lum J. J., Hatzivassiliou G., Thompson C. B., The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Altman B. J., Stine Z. E., Dang C. V., From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 619–634 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morris S. M., Jr., Arginine metabolism: Boundaries of our knowledge. J. Nutr. 137, 1602S–1609S (2007). [DOI] [PubMed] [Google Scholar]
- 45.Lian G., Gnanaprakasam J. R., Wang T., Wu R., Chen X., Liu L., Shen Y., Yang M., Yang J., Chen Y., Vasiliou V., Cassel T. A., Green D. R., Liu Y., Fan T. W., Wang R., Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T cell differentiation. eLife 7, e36158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klysz D., Tai X., Robert P. A., Craveiro M., Cretenet G., Oburoglu L., Mongellaz C., Floess S., Fritz V., Matias M. I., Yong C., Surh N., Marie J. C., Huehn J., Zimmermann V., Kinet S., Dardalhon V., Taylor N., Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 8, ra97 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Deignan J. L., Livesay J. C., Yoo P. K., Goodman S. I., O’Brien W. E., Iyer R. K., Cederbaum S. D., Grody W. W., Ornithine deficiency in the arginase double knockout mouse. Mol. Genet. Metab. 89, 87–96 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Wang T., Liu G., Wang R., The intercellular metabolic interplay between tumor and immune cells. Front. Immunol. 5, 358 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerner E. W., Meyskens F. L. Jr., Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 4, 781–792 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Bello-Fernandez C., Packham G., Cleveland J. L., The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. U.S.A. 90, 7804–7808 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casero R. A. Jr., Murray Stewart T., Pegg A. E., Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 18, 681–695 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abdulhussein A. A., Wallace H. M., Polyamines and membrane transporters. Amino Acids 46, 655–660 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabc4275/DC1