

Abstract
Programmed cell death-1 (PD-1)/programmed cell death ligand-1 (PD-L1) blocking therapy has become a major pillar of cancer immunotherapy. Compared with antibodies targeting, small-molecule checkpoint inhibitors which have favorable pharmacokinetics are urgently needed. Here we identified berberine (BBR), a proven anti-inflammation drug, as a negative regulator of PD-L1 from a set of traditional Chinese medicine (TCM) chemical monomers. BBR enhanced the sensitivity of tumour cells to co-cultured T-cells by decreasing the level of PD-L1 in cancer cells. In addition, BBR exerted its antitumor effect in Lewis tumor xenograft mice through enhancing tumor-infiltrating T-cell immunity and attenuating the activation of immunosuppressive myeloid-derived suppressor cells (MDSCs) and regulatory T-cells (Tregs). BBR triggered PD-L1 degradation through ubiquitin (Ub)/proteasome-dependent pathway. Remarkably, BBR selectively bound to the glutamic acid 76 of constitutive photomorphogenic-9 signalosome 5 (CSN5) and inhibited PD-1/PD-L1 axis through its deubiquitination activity, resulting in ubiquitination and degradation of PD-L1. Our data reveals a previously unrecognized antitumor mechanism of BBR, suggesting BBR is small-molecule immune checkpoint inhibitor for cancer treatment.
Key words: PD-L1, Immune checkpoint blockade, COP9 signalosome 5, Berberine, PD-1/PD-L1 axis, T-cell immunity
Abbreviations: AMC, 7-amino-4-methylcoumarin; Baf, bafilomycin; BBR, berberine; CHX, cycloheximide; CQ, chloroquine; CSN5, COP9 signalosome 5; IB, immunoblotting; ICB, immune checkpoint blockade; IFN-γ, interferon-gamma; IHC, immunohistochemistry; MDSCs, myeloid-derived suppressor cells; NFAT, nuclear factor of activated T-cells; NSCLC, non-small cell lung cancer; PD-1, programmed cell death-1; PD-L1, programmed cell death ligand-1; qRT-PCR, quantitative real-time polymerase chain reaction; SPR, surface plasmon resonance; TCM, traditional Chinese medicine; TILs, tumor-infiltrating lymphocytes; TNF-α, tumor necrosis factor-α; Tregs, regulatory T-lymphocytes; Ub, ubiquitin
Graphical abstract
Berberine diminishes the expression of programmed cell death ligand-1 and promotes antitumor immunity via inhibiting the deubiquitination activity of COP9 signalosome 5 (CSN5) in non-small cell lung cancer.
1. Introduction
Immune escape is one of the major features of a variety of cancers, including non-small cell lung cancer (NSCLC)1,2. Cancer cells often bypass immune surveillance through suppressing interferon-gamma (IFN-γ) production, CD8+ T-cell proliferation, or oncolytic activity3. Recently, immune checkpoint blockade (ICB) therapy has dramatically reshaped the scenery of NSCLC treatment4. Agents that targeting immune checkpoints, particularly blockade of programmed cell death-1 (PD-1)/programmed cell death ligand-1 (PD-L1) axis, are recognized as the most efficient immunotherapies against NSCLC5. Despite these successes, responses to ICB usually remain restricted to a subpopulation of patients, and approximately 30% relapse6. Thus, identifying the optimal therapeutic approach that increases the efficacy of ICB is an attractive project deserving investigation.
PD-L1 is frequently high expressed in multiple cancers, including melanoma, lung, ovarian, pancreas, and colon cancer7,8. As a T-cell immune checkpoint molecule, PD-L1 can inactivate tumor-infiltrating lymphocytes (TILs) that express cell surface PD-1 (also known as CD279)9,10. The interaction between PD-L1 and PD-1 results in differentiation of naive CD4+ T-cells into regulatory T-lymphocytes (Tregs) thus inhibits the immune activation and effector response11. Disrupting the PD-1/PD-L1 axis and reactivating TILs is recognized as the promising target for cancer immunotherapy12. Considering the critical roles of PD-L1 in immunosuppression, understanding the regulatory mechanism of PD-L1 may develop combinatorial strategies for cancer treatment.
CSN5 (constitutive photomorphogenic-9 signalosome 5) is a evolutionarily conserved multifunctional protein involved in regulating DNA repair, modulating signal transduction, and controlling cell proliferation13. CSN5 acts as an oncogenic protein contributing to cell survival by functionally inactivating P27, P53, and cyclin E in cells14. The best understood function of CSN5 is regulation of Ub-mediated protein degradation through its deneddylation and deubiquitination activity, which is crucial for cancer progression15. For instance, CSN5 blocks the ubiquitination and degradation of survivin and snail thus enhances cancer cell invasion and migration16,17. Tumor necrosis factor-α (TNF-α)-induced CSN5 expression leads to PD-L1 stabilization in cancer cells to escape from immune surveillance18. Moreover, it has been reported that CSN5 upregulated in multiple human cancers and globally associated with poor prognosis19. Therefore, CSN5 is a promising therapeutic target for cancer treatment.
Berberine (BBR) is an isoquinoline quaternary alkaloid derived from Coptis chinensis which has been used as a therapeutic agent in the treatment of cancer, bacterial infections, diabetes, cardiovascular and inflammatory diseases20, 21, 22. BBR has been shown to have minimal cytotoxicity effects on healthy cells but has anti-proliferative effects on cancer cells (e.g., breast, liver, and colorectal cancer cells)23,24. Despite a number of studies have exploited the antitumor mechanism of BBR and suggest that BBR efficiently inhibits different cell signaling pathways, including NF-κB, JAK-STAT and MAPK/ERK signaling25, it is unclear whether BBR may functions as an immune modulator and mediates its antitumor efficacy. In the present study, we provide evidences that BBR suppressed the PD-L1 level in NSCLC cells and Lewis tumor xenografts mice. BBR enhances anti-NSCLC immunity through inhibiting the deubiquitination activity of CSN5 therefore triggers ubiquitination and degradation of PD-L1.
2. Materials and methods
2.1. Cell culture
NSCLC cell lines A549, H157, H358, H460, H1299, H1975, Lewis cells and Jurkat (E6-1) cells were purchased from Institute of Basic Medicine, Chinese Academy of Medical Sciences (Beijing, China). All cells were cultured in RPMI1640 medium (Gibco, Pittsburgh, PA, USA) except A549 cells cultured in DMEM/F-12 medium. The culture medium was supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 100 μg/mL streptomycin and 100 U/mL penicillin. Cells were incubated in a humidified atmosphere with 5% CO2 at 37 °C.
2.2. Mice
Eight-week-old female C57BL/6 mice and BALB/c-nu/nu nude mice were obtained from Beijing Vital River Laboratory Animal Technology (Beijing, China). All animal experiments were conducted in accordance with the Animal Ethics Committee of the Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences (Beijing, China).
2.3. Antibodies, plasmids and reagents
Antibodies are listed in Supporting Information Table S1. Traditional Chinese medicine (TCM) chemical monomers were purchased from Shanghai Standard Technology Co., Ltd. (Shanghai, China). CHX, MG132, bafilomycin (Baf), chloroquine (CQ), and Hoechst 33342 were purchased from Sigma (St. Louis, MO, USA). Human PD-1 Fc recombinant protein was purchased from R&D Systems (Minneapolis, MN, USA). The plasmids pcDNA3-HA-Ub (18712) obtained from Addgene (Watertown, MA, USA), pCMV3-PD-L1-Myc (HG10084-CM) and pCMV3-CSN5-Flag (HG17128-CF) plasmids were purchased from Sino Biological Inc. (Beijing, China).
2.4. PD-L1/PD-1 blockade assay and T-cell-mediated tumor cell-killing assay
The PD-L1/PD-1 blockade assay Kit (Promega, Madison, WI, USA, J1250) was used to determine BBR-mediated functional changes in PD-1/PD-L1 interactions. The PD-L1-expressing H460 or H1975 cells (1 × 104) were pre-treatment with BBR (0–20 μmol/L) for 16 h. The following day, the drug-containing media were removed, and 1 × 104 Jurkat cells stably transfected with human PD-1 and nuclear factor of activated T-cells (NFAT)-luciferase reporter were added to each of the treated wells. Co-culture of these cells results in activation of NFAT-luciferase while PD-L1 to PD-1 interaction reduces the NFAT-luciferase. Six hours later, Bio-Glo reagent was added to each well, the plate was read with a LB942 multimode microplate reader (Berthold, Bad Wildbad, Germany) and the data were analyzed by ICE software.
Human T-cell-mediated tumor cell-killing assay was conducted by the xCELLigence system (Agilent, San Diego, CA, USA) as described previously26. Briefly, 50 μL of culture medium was placed in each well of the E-plate 16, followed by adding additional 50 μL medium containing of 1 × 103 H460 cells. Each treatment includes three replicates. After treated with the indicated conditions of BBR for 24 h, human T-cells (activated by anti-CD3 plus anti-CD28 co-stimulation) were added at the ratio of 1:5. Cell index values were measured by continuous impedance recordings every 15 min. The results were real-time analyzed by xCELLigence system (Agilent) with RTCA Software.
2.5. Membrane PD-L1 analysis and immunoblotting
After BBR treatment for the indicated time, cells were collected and incubated with PD-L1 extracellular domain-specific antibody (PE conjugate, Cell Signaling 71391) for 60 min at 4 °C. Cells were washed in incubation buffer and resuspended in 500 μL incubation buffer. The conjugate PE fluorescence was quantitatively analyzed by Cytoflex flow cytometer with CytExpert software (Beckman Coulter, Brea, CA, USA). Immunoblotting (IB) was performed as described previously26,27.
2.6. Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed as previously described26. The primers used are listed in the Supporting Information Table S2.
2.7. Animal experiments
C57BL/6 mice inoculated subcutaneously with 5 × 106 Lewis cells were intraperitoneally administered with 0, 2, 4, 8, 16 and 32 mg/kg of BBR when the average tumor volume reached approximately 50 mm3 (n = 5). During the 14-day treatment, 2 and 3 mice died in the 16 and 32 mg/kg group, respectively. No mice were died in other groups. Therefore, 4 and 8 mg/kg of BBR were used in our animal study as 2 mg/kg of BBR showed minor antitumor effect. Mice were then split to three groups randomly (n = 7) and intraperitoneally administered with 4 mg/kg BBR, 8 mg/kg BBR and PBS respectively when the average tumor volume reached approximately 50 mm3. The tumor volume and body weight of mice were measured every two days, and the tumor volumes were calculated by Eq. (1):
| (1) |
At the end, the mice were sacrificed, the major organs (lung, spleen, liver, and kidney) and blood samples were collected. The biochemistry indicators in blood samples were examined by automated biochemistry analyzer Synchron CX4 pro (Beckman Coulter, Brea, CA, USA). The harvested organs were fixed in 4% paraformaldehyde, stained with haematoxylin and eosin (H&E) and captured by microscope (Axio Vert.A1, Zeiss, Oberkochen, Germany). Immunohistochemistry was performed as described previously26.
2.8. Tumor-infiltrating lymphocyte isolation and T-cell profile analysis
Tumors tissues in mice treated with PBS or BBR (4 or 8 mg/kg) were collected and cut into small pieces, followed by enzyme digestion with type 4 collagenase (1 mg/mL, Sigma) and DNase 1 (0.1 mg/mL, Sigma) for 1 h at 37 °C. Suspension cells were blocked with anti-CD16/CD32 antibodies and stained with fixable viability dye for 15 min at 4 °C. Next, cells were incubation with surface marker antibodies CD45, CD3, CD8, CD69, CD25, CD137, CD11b, and Gr-1 for 30 min at 4 °C. After that, cells were fixed and permeabilized after the stimulation with Cell Stimulation Cocktail (eBioscience, San Diego, CA, USA) at incubator for 6 h, and labeled with anti-mouse IFN-γ or GzmB for 30 min at 4 °C. Cells were washed three times with cell staining buffer and quantitatively analyzed by Cytoflex flow cytometer with CytExpert software (Beckman Coulter).
2.9. CSN5 deubiquitinating activity assay
CSN5 deubiquitinating activity assay were conducted in a 96-well plate in a 100 μL reaction buffer with Ub–7-amino-4-methylcoumarin (AMC)-conjugated proteins (U-550; Boston Biochem, Cambridge, MA, USA). Purified CSN5 protein was incubated in Ub–AMC assay buffer (50 mmol/L Tris-HCl, pH 7.5, 1 mmol/L EDTA, 1 mg/mL ovalbumin, 5 mol/L MgCl2, 1 mmol/L DTT, 1 mmol/L ATP). Ub–AMC was added in assay buffer and incubated for 30 min. AMC fluorescence was measured with an excitation wavelength of 345 nm and an emission wavelength of 445 nm.
2.10. Small interfering RNA knockdown and transfection
Small interfering RNA (siRNA) targeting CSN5 (5′-CCAGACUAUUCCACUUAAU TT-3′) and control siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′) were purchased from GenePharma (Shanghai, China). For siRNA-mediated silencing, cells were transfected with 100 nmol/L of target siRNA and a control siRNA using Vigofect (Vigorous Biotechnology, Beijing, China) according to the manufacturer's recommendations. Forth-eight hours post-transfection, the protein expression was analyzed by immunoblotting.
2.11. Preparation of biotin-conjugated BBR (BBR–biotin)
To a stirred solution of 10-OH substituted protoberberine28 (1.0 g) in anhydrous CH3CN, K2CO3 (3 eq.) was added and heated to 70 °C. Then TsO-[(CH2)2O]2(CH2)2-NHBoc (1.5 eq.) was added and stirred for 24 h. The mixture was dissolved in CH3OH/HCl and stirred for 5–6 h, filtered and washed with ethanol to afford compound N8. Next, N8 (100 mg) was mixed with D-Biotin (1.1 eq.), EDCI (1.2 eq.) HOBT (1.2 eq.) and DIPEA (2.8 eq.) in 5 mL of DMF at room temperature for 12 h. The resulting mixture was evaporated and the residue was purified by flash chromatography using CH2Cl2/CH3OH to afford BBR–biotin as a yellow solid. The structure of BBR–biotin was elucidated by NMR and MS.
2.12. BBR–biotin pulldown assay and site-directed mutagenesis
BBR–biotin (100 μmol/L) was incubated with streptavidin–agarose beads in Hepes buffer (20 mmol/L Hepes, pH 7.4, 1 mmol/L dithiothreitol, 100 mmol/L NaCl, 0.05% NP-40) at room temperature for 2 h. BBR–biotin immobilized on streptavidin–agarose beads (10 μL) was then incubated with Flag–CSN5 protein (1.5 μg of each) with various concentrations of BBR, or with 293T-cell lysates expressed Flag–CSN5-wild type (WT), Flag–CSN5-E76A, M78A, H138A, Y143A, D151R at 25 °C for 2 h. The beads were then washed with Hepes buffer and bound proteins were subjected to IB with Flag antibody. Site-directed mutagenesis of CSN5 was performed with the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) using Flag–CSN5 as a template. The primers are listed in Supporting Information Table S2.
2.13. Surface plasmon resonance (SPR) analysis
SPR analysis was performed on a Biacore S200 instrument with CM5 sensor chip (BR-1005-30; GE Healthcare China Company, Beijing, China). Recombinant human CSN5 (TP300979; Origene, Rockville, MD, USA) was immobilized in parallel-flow channels of CM5 chip by using amine-coupling kit (BR-1000-50; GE Healthcare). BBR was dissolved in PBS-P (28-9950-84; GE Healthcare), series concentrations of BBR were injected into the flow system at the flow rate of 20 μL/min. The association time was 120 s and the dissociation time was 300 s. The binding kinetics was analyzed by Biaevaluation software 2.0.
2.14. Molecular modelling
The molecular docking model of BBR with 3D structure of CSN5 (PDB code: 5JOG) was carried out by Discovery Studio 4.5 and UCSF chimera 1.7. The regularized protein was used in determination of the important amino acids in the predicted binding pocket. Interactive docking for all the conformers of BBR to the selected active site was performed by LibDock after energy minimization using prepares ligand protocol. A score was assigned to the docked compound according to its binding mode onto the binding site.
2.15. Statistics
The data were presented as mean ± standard deviation (SD) and analyzed using GraphPad Prism 6 software (GraphPad Software, La Jolla, CA, USA). The unpaired, two-tailed Student's t-test and ANOVA with a Tukey's post hoc test were used to analyze the significance of differences between groups. P < 0.05 was considered statistically significant.
3. Results
3.1. BBR is a new negative regulator of PD-L1
Inspired by the bioactive substances of natural medicines in cancer treatment, we start our chemical screen by testing the ability of a panel of 55 TCM chemical monomers (Supporting Information Table S3) to alter PD-L1 protein expression in H460 cells. The compounds (10 μmol/L) were incubated with H460 cells for 24 h and PD-L1 level was determined by IB. Unexpectedly, we found that BBR (Fig. 1A) efficiently suppressed the constitutive PD-L1 expression in H460 cells (compound 8 in Fig. 1B). Although homoharringtonine, triptolide, and Paris I also reduced the constitutive PD-L1 expression (Supporting Information Fig. S1), a massive cell death was caused after treatment with the three compounds for 24 h (data not shown), therefore it was excluded for the further study. In addition, BBR did not induce apparent apoptosis and activation of cleaved caspase 9/3 as compared with staurosporine-treated control (Supporting Information Fig. S2A and S2B). At concentration up to 50 μmol/L, BBR also failed to affect cell viability of NSCLC cells as well as normal epithelial cells BEAS-2B (Fig. S2C). Therefore, 0–20 μmol/L BBR was used for the subsequent experiments in this study. Altogether, we identified BBR as a negative PD-L1 regulator with little cytotoxic effects on cancer cells.
Figure 1.

BBR decreases PD-L1 expression in NSCLC cells. (A) Chemical structure of BBR. (B) IB analysis of PD-L1 expression in H460 cells treatment with the indicated natural compounds (10 μmol/L) for 24 h. (C) and (D) H460, H1975, H358 and HCC827 cells were treated with 10 μmol/L BBR for the indicated times (C), or treated with different concentrations of BBR for 24 h (D), PD-L1 and CD47 expression levels were detected by IB. (E) A549 or H1299 cells were pre-treated with 10 μmol/L BBR for 2 h, followed by IFN-γ (5 ng/mL) stimulation for 24 h, PD-L1 and IDO1 expression levels were detected by IB. (F) H157 cells were treated with BBR (5 and 10 μmol/L) for 24 h, the plasma membrane PD-L1 was detected by flow cytometry. Statistic of PD-L1 mean fluorescence intensity (MFI) is shown in right (n = 3). Data shown are mean value of three independent experiments±standard error of mean (SEM). ∗∗P < 0.01, ∗∗∗P < 0.001 compared with DMSO group.
3.2. BBR effectively decreases PD-L1 expression in cancer cells
We sought to further validate the ability of BBR-mediated PD-L1 downregulation in NSCLC cells. We found that BBR induced a significant dose and time-responsive decrease of constitutive PD-L1 expression in H460, H1975, H358 and HCC827 cells (Fig. 1C and D). The expression of other immune inhibitory ligands, such as CD47 and B7–H3, were not eliminated when cells were treated with BBR (Fig. 1C and D and Supporting Information Fig. S3A). This data suggests that BBR selectively reduced the expression of PD-L1 in cancer cells. As PD-L1 in tumor cells can be upregulated by pro-inflammatory cytokines IFN-γ29, we then examined whether BBR affected inductive PD-L1 expression in IFN-γ-stimulated NSCLC cells26. BBR decreased IFN-γ-induced PD-L1 expression in both A549 and H1299 cells, while IFN-γ-induced IDO1 expression remained similar when cells were treated with BBR (Fig. 1E). Next, we investigated whether BBR regulates membranous PD-L1 expression. Indeed, flow cytometry assay revealed that BBR dramatically attenuated the cell surface PD-L1 in H157 (Fig. 1F) and H460 cells (Fig. S3B), indicating BBR could also decrease the PD-L1 functional expression on the cell membrane. Taken together, these results suggest that BBR decreased PD-L1 expression in NSCLC cells.
3.3. BBR recovers sensitivity of cancer cells to T-cell killing in vitro
Because PD-L1 expression on cancer cells binds its cognate receptor PD-1 on infiltrating T-cells and eliminates their antitumor activity30, we next tested whether BBR affected the ability of cancer cells bind to PD-1. H460 cells were incubated with recombinant PD-1 protein together with green fluorescence (Alexa 488)-conjugated human Fc antibody, the interaction between PD-L1 and PD-1 was measured by immunofluorescence microscope26. Decreased green fluorescence in BBR-treated H460 cells indicated that BBR attenuated the ability of cancer cells bind to PD-1 by reducing the PD-L1 level (Fig. 2A). To validate the functional changes in BBR-mediated PD-L1 downregulation in cancer cells, we co-cultured of PD-L1-expressing H460 or H1975 cells with PD-1-expressing NFAT-luciferase transfected Jurkat cells. PD-1/PD-L1 binding results in T-cell inactivation and the abrogation of luminescent signal, whereas blockade of PD-1/PD-L1 binding reactivates NFAT and results in luminescent signal31. BBR induced markedly transcriptional-mediated bioluminescent signal in a dose-dependent manner in both H460 and H1975 cells, indicating that BBR disrupted PD-L1's checkpoint activity thus promotes the activation of T-cells (Fig. 2B). To investigate the role of BBR in T-cell-mediated cancer cell killing, the cytotoxicity of activated human T-cells toward co-cultured H460 cells was measured by cell impedance assay26. The reduced cell index in BBR-treated H460 cells confirmed that BBR enhanced the cytotoxicity of human T-cells (Fig. 2C). The antitumor effect of BBR was further evaluated by determining the surviving tumor cells using crystal violet staining after co-culturing H460 cells and activated Jurkat cells. Treatment with BBR significantly reduced the surviving of H460 cells and induced caspase3/7 activity in H460 cells than those with control (Fig. 2D). All together, these results suggest that BBR increases the cytotoxicity of T-cells toward cancer cells by down-regulation of PD-L1 expression in cancer cells.
Figure 2.

BBR recovers the cytotoxicity of T-cells. (A) PD-L1/PD-1 binding assay in H157 cells treated with BBR (5 and 10 μmol/L, 24 h). The nuclei were stained with Hoechst. Scale bar, 200 μm. Bound PD-1 was calculated according to the intensity of green fluorescence (n = 3). ∗∗∗P < 0.001 compared with DMSO group. (B) PD-L1/PD-1 blockade assay performed with H460 and H1975 cells treated with 5, 10, and 20 μmol/L BBR for 16 h. Jurkat NFAT-luciferase reporter cells (10,000 cells/well) were added, and cells were co-cultured for 6 h. Data are presented as fold induction over untreated control (n = 3). ∗P < 0.05, ∗∗∗P < 0.001 compared with DMSO group. (C) Cell impedance assay analyzing human T-cell (activated by anti-CD3 plus anti-CD28 co-stimulation)-meditated tumor cell killing in H460 cells treated with BBR (5 or 10 μmol/L) for 24 h (n = 3). ∗∗∗P < 0.001 compared with control, ##P < 0.01, ###P < 0.001 compared with T-cell only group. (D) Jurkat cells were activated by 1 mg/mL phytohemagglutinin (PHA) plus 50 ng/mL phorbol 12-myristate 13-acetate (PMA) and co-cultured with H460 cells in 12-well plates for 2 days in the presence of BBR and the surviving tumor cells were visualized by crystal violet staining. Relative fold ratios of surviving cell intensity are shown (n = 3). ##P < 0.01 compared with H460 DMSO group; ∗∗P < 0.01, ∗∗∗P < 0.001 compared with H460+Jurkat DMSO group. Data shown are mean value of three independent experiments±standard error of mean (SEM).
3.4. BBR suppresses xenograft tumor growth in vivo
The antitumor effect of BBR was examined by comparing the tumor growth suppression of Lewis-tumor-bearing mice that were intraperitoneally administrated with vehicle or BBR (4 and 8 mg/kg) once a day for 18 days. BBR treatment displayed significant suppression in the growth of Lewis tumor xenografts with an inhibition rate of 46.8% and 75.8% at 4 and 8 mg/kg, respectively (Fig. 3A). This result was further demonstrated by the comparison of the tumor weights (Fig. 3B) and the ex vivo observation of the tumors (Fig. 3C). We thus confirm that BBR has intrinsic antitumor activity. Interestingly, in T-cell-deficient nude mice, the tumor-inhibiting effect of BBR against Lewis tumor was lost (Fig. 3D), suggesting that the antitumor effect of BBR is ascribed to its activating T-cell immunity. Furthermore, immunohistochemistry (IHC) assay show that the levels of PD-L1 and Ki67 (a marker of proliferation) were decreased, while the level of cleaved caspase 3 was increased in BBR-treated tumor xenografts mice (Fig. 3E), indicating BBR triggers an obvious apoptosis in tumor mice.
Figure 3.

BBR suppresses tumor xenograft growth in vivo. (A) C57BL/6 mice (n = 7) with subcutaneous Lewis tumor were i.p. treated with PBS or BBR (4 and 8 mg/kg) and the tumor growth was monitored. (B) Comparison of the weight of the tumors from the mice treatment with PBS or BBR. (C) Ex vivo observation of the tumors from the treated mice (A: PBS, B: BBR 4 mg/kg, C: BBR 8 mg/kg). (D) Nude mice (n = 6) bearing Leiws tumor were received daily i.p. injection of PBS or BBR for 2 weeks. Tumor growth was measured. (E) Representative IHC staining results for PD-L1, cleaved caspase 3 and Ki67. Scale bar, 200 μm. Quantification of IHC staining is shown (n = 3). (F) The body weight curves of the mice measured every two days. Data shown are mean value ± standard error of mean (SEM). ∗P < 0.05, ∗∗P < 0.01 and ∗∗∗P < 0.001 compared with PBS group.
During the treatment period, BBR did not induce significantly changes in mice body weight (Fig. 3F). BBR has no systemic toxicity on mice as demonstrated by the fact that BBR induced no change on the organ indexes, the serum biochemical indices and no significant changes on the major organs of mice (Supporting Information Figs. S4A–S4C).
3.5. BBR exerts its antitumor effect through activating tumor-infiltrating T-cell
To demonstrate BBR-mediated antitumor T-cell immunity, we performed TILs profile analysis. We found obvious elevations in the number of CD8+ T-cell and the ratio of CD8+ to CD4+ T-cell in BBR-treated mice (Fig. 4A and B), demonstrating that BBR increases intratumoral T-cell infiltration. Additionally, the populations of CD69+ CD137+ CD8+ T-cells in tumor tissues from BBR-treated mice were significantly increased compared to those treated with PBS (Fig. 4C), indicating BBR induces more effective T-cells activation in mice. IFN-γ plays a critical role in T-cell-mediated antitumor immunity32. BBR treatment boosted the level of IFN-γ producing by CD8+ T-cells (Fig. 4D), demonstrating BBR activates cytotoxic T-cell in TILs. Analysis the level of granzyme B, an indicator of cytotoxic T-cell activation, also supported the conclusion that the BBR increased the cytotoxic T-cell activity (Fig. 4E).
Figure 4.

BBR stimulates the activation of tumor-infiltrating T-cells. (A), (B) and (C) C57BL/6 mice (n = 5) with subcutaneous Lewis tumor were i.p. treated with PBS or BBR (4 and 8 mg/kg). The populations of CD8+ (A), CD8+/CD4+ (B) in CD3+ TILs, and CD69+CD137+ in CD3+CD8+ TILs (C) were analyzed by flow cytometry. (D) and (E) Representative flow cytometric plots and quantification of IFN-γ expression (D) and GzmB level (E) in CD3+CD8+ TILs from PBS or BBR-treated Lewis tumor was measured by flow cytometry. (F) and (G) Representative flow cytometric plots and quantification of MDSCs (F) number and Tregs (G) number in PBS or BBR-treated Lewis tumor. Data shown are mean value ± standard error of mean (SEM). ∗P < 0.05, ∗∗P < 0.01 and ∗∗∗P < 0.001 compared with PBS group.
In tumor microenvironment, myeloid-derived suppressor cells (MDSCs) and Tregs contribute to tumor immune escape by strongly inhibiting T-lymphocyte immunity33,34. Notably, activated MDSCs and Tregs express a large amount of PD-L1 that interacts with PD-1 on T-cells and eventually leads to their exhaustion35,36. Flow cytometric analysis revealed that the populations of activated MDSCs (CD11b+Gr-1+) and Tregs (CD4+CD25+Foxp3+) cells within the TILs were significantly reduced in BBR-treated tumors as compared with those in the control tumors (Fig. 4F and G). Furthermore, the levels of PD-L1 were reduced on BBR-treated tumor-infiltrating MDSCs and Tregs as compared with that in the PBS treated group (Support Information Fig. S5). Our results thus demonstrate BBR switches the immune microenvironment from immunosuppressive to immunoactivation. Overall, these data suggest that BBR can effectively stimulate the activation of tumor-infiltrating T-cells through down-regulation of PD-L1, resulting significant antitumor efficacy in Lewis-tumor-bearing mice.
3.6. Ub/proteasome pathway contributes to BBR-mediated PD-L1 degradation
Next, we sought to investigate the mechanisms through which BBR negatively regulates PD-L1 expression. Real-time PCR assay revealed that BBR did not significantly change the mRNA level of PD-L1 neither in H460, H1975, HCC827 and H157 cells, nor in IFN-γ-stimulated NSCLC cells (Fig. 5A and B), suggesting BBR down-regulation of PD-L1 expression is not a transcriptional event. This conclusion was further confirmed by half-life analysis of PD-L1. NSCLC cells were exposed to the protein translation inhibitor cycloheximide (CHX) with or without BBR. The CHX pulse-chase analysis show that the turnover rate of PD-L1 in BBR-treated cells was faster than that in untreated cells (Fig. 5C), demonstrating BBR regulated PD-L1 expression at the posttranslational level.
Figure 5.

BBR induces ubiquitin-dependent PD-L1 degradation. (A) and (B) Quantitative RT-PCR analysis of the mRNA level of PD-L1 in H460, H1975, HCC827 and H157 cells treated with BBR (10 μmol/L, 12 h) (A) or in A549 cells treated with BBR (10 μmol/L) and 5 ng/mL IFN-γ for 12 h (B). ∗P < 0.05 compared with DMSO group. (C) IB analysis of the PD-L1 expression in H460 cells treated with DMSO or BBR (10 μmol/L) for the indicated time points in the presence of CHX (25 μg/mL). Quantification of PD-L1 intensity is shown in right (n = 3). The abundance was normalized to GAPDH; each group was normalized as a percentage of that at 0 h. ∗P < 0.05 compared with DMSO group. (D) and (E) IB measuring the PD-L1 expression in H460 cells pre-treated with indicated concentration of MG132 (D), 200 nmol/L Baf or 100 μmol/L CQ (E), followed by 10 μmol/L BBR treatment for 24 h in the presence of CHX (25 μg/mL). (F) H1975 cells were transiently transfected with Myc-PD-L1 for 24 h, followed by Baf or CQ pretreatment for 1 h and BBR treatment for 24 h, PD-L1 level was analyzed by Myc antibody. (G) HEK293T-cells were transiently transfected with the indicated constructs. Ubiquitinated PD-L1 was immunoprecipitated (IP) and subjected to IB analysis with the ubiquitin antibody. Cells were treated with MG132 prior to ubiquitination analysis. Data shown are mean value of three independent experiments±standard error of mean (SEM). ∗P < 0.05 compared with DMSO group.
It has been reported that PD-L1 exerts both the proteasome- and lysosome-dependent degradation pathways37, 38, 39. To clarify which pathway involved in BBR-mediated PD-L1 down-regulation, H460 cells were co-treated BBR with proteasome inhibitor MG132 or lysosome inhibitor bafilomycin (Baf) or chloroquine (CQ). Co-incubation of MG132 alleviated BBR-mediated PD-L1 degradation (Fig. 5D and Fig. S5B), but Baf or CQ displayed no effect on the action of BBR (Fig. 5E). This finding suggests that BBR ubiquitinates PD-L1 to induce protein degradation. To further confirm this conclusion, we transfected Myc-PD-L1 into H460 cells and monitored its degradation. BBR-mediated Myc-PD-L1 degradation was completely restored by MG132 instead of Baf (Fig. 5F). Furthermore, we analyzed PD-L1 ubiquitination in the presence of BBR and found that BBR triggered PD-L1 ubiquitination (Fig. 5G). These findings consistently demonstrate that Ub/proteasome pathway contributes to BBR-mediated PD-L1 degradation.
3.7. BBR destabilizes PD-L1 by binding to and inhibition of CSN5 activity
Recent studies have demonstrated that PD-L1 is generally regulated by the Ub/proteasome pathway via E3 ligase cullin3, β-TrCP, C-terminus of HSP70 interacting protein (CHIP), and deubiquitinase CSN518,37,38,40. To clarify the possible contribution of Ub/proteasome regulator to BBR-mediated PD-L1 degradation, we tested whether BBR interacts with cullin3, β-TrCP, STUB1 or CSN5. To this end, we synthesized biotin-conjugated BBR probe (BBR–biotin) for pull-down assay. The streptavidin agarose beads precoupled with BBR–biotin were incubated with 293T-cell lysates, the precipitated proteins were subjected to IB to monitor the presence of β-TrCP, STUB1 or CSN5. Interestingly, CSN5 was specifically pulled down by BBR–biotin from 293T-cell lysates or Flag–CSN5 overexpressed 293T-cell lysates (Fig. 6A). In addition, BBR has no obvious effect on the mRNA and protein expressions of CSN5 (Supporting Information Fig. S6A). These data suggest that BBR targets CSN5 or a complex that contains CSN5.
Figure 6.

BBR directly binds to and inhibits CSN5 activity. (A) The 293T or 293T expressing Flag-CSN5 cell lysates were incubated with BBR-biotin at 4 °C overnight, the lysates were used for streptavidin–agarose pull-down assays, and the precipitates were resolved by IB for CSN5. (B) The recombinant Flag–CSN5 proteins were incubated with BBR–biotin for 1 h at 37 °C, followed by IB with biotin (upper band) or Flag (lower band). (C) The recombinant Flag–CSN5 protein was incubated with BBR–biotin in the absence or presence of indicated concentration of unlabelled BBR for 1.5 h at 37 °C, and the mixtures were IB for biotin or flag. (D) SPR analysis of the binding between BBR and CSN5. Recombinant human CSN5 protein was immobilized on an activated CM5 sensor chip, BBR was then flowed across the chip. (E) CSN5 activity in an in vitro deubiquitination assay. The activity was measured by 7-amino-4-methylcoumarin (AMC) released from the fluorogenic substrate, ubiquitin–AMC (n = 3). ∗∗P < 0.01 and ∗∗∗P < 0.001 compared with DMSO group. (F) IB analysis of the P53 and P27 levels in H460 cells treated with indicated concentration of BBR for 24 h. (G and H) H460 cells were transfected with siRNA control, siRNA targeting CSN5 for 24 h (G), or transfected with 2 μg empty vector, 2 μg Flag–CSN5 for 24 h (H), followed by BBR (10 μmol/L) treatment for 24 h, the PD-L1 expression level was determined by IB. Data shown are mean value of three independent experiments±standard error of mean (SEM).
To examine whether BBR directly binds to CSN5, we generated recombinant CSN5 protein by transfecting Flag–CSN5 plasmid into 293T-cells and purified it by anti-Flag beads. The purified Flag–CSN5 protein was incubated with DMSO or increasing concentrations of BBR–biotin, and BBR–biotin coupled with Flag–CSN5 were determined by IB with streptavidin antibody. As shown in Fig. 6B, BBR–biotin was captured by CSN5 in a concentration-dependent manner. In addition, CSN5-captured BBR–biotin could be competed away by excess unlabelled BBR (Fig. 6C). The direct interaction between BBR and CSN5 was further confirmed by SPR assay using a biacore platform. The determined equilibrium dissociation constant (KD) evaluating the BBR binding to the CSN5 protein is about 16.25 μmol/L (Fig. 6D). These results demonstrate that there is an interaction between BBR and CSN5.
Since CSN5 is a deubiquitination enzyme for PD-L118, we next examined whether BBR inhibits the deubiquitination activity of CSN5. In vitro deubiquitination assay revealed that BBR suppressed CSN5 hydrolyzing Ub from Ub–7-amido-4-methylcoumarin (AMC), a fluorogenic substrate for Ub hydrolases (Fig. 6E). As CSN5 interacts directly with P2741 and P5342 and induces their proteasome-dependent degradation in cancer cells, we further examined the effect of BBR on these two proteins. Indeed, BBR treatment induced an increase of P27 and P53 (Fig. 6F). Moreover, BBR-mediated PD-L1 ubiquitination and degradation was lost when CSN5 was silenced by siRNA, while overexpression of CSN5 reversed the action of BBR (Fig. 6G and H, and Supporting Information Figs. S6B and S6C). Taken together, these results suggest that BBR destabilization of PD-L1 by directly binding to and inhibition of CSN5 activity.
3.8. Glu76 of CSN5 is critical for binding to BBR
To further confirm BBR directly binds to and inactivates CSN5, a molecular docking model of BBR with crystal structure of CSN5 (PDB ID: 5JOG) was established. According to the computational study, BBR could dock to the JAMM Zn2+-metalloprotease motif (Fig. 7A) that provides the catalytic center to the CSN543. The score of binding affinity between BBR and CSN5 is 83. BBR forms favorable interactions with residues including Glu76, Met78, His138, Tyr143, and Asp151 (Fig. 7B). Next, we individually mutated these five residues of CSN5 into alanine to examine which residue is critical for binding of BBR. We found that E76A mutant dramatically reduced the ability of CSN5 to interact with BBR (Fig. 7C), suggesting Glu76 is critical for CSN5 binding of BBR. Moreover, PD-L1 ubiquitination assay revealed that BBR interrupted WT-CSN5, but not E76A-CSN5- mediated PD-L1 ubiquitination (Fig. 7D). Collectively, these results suggest that BBR inhibits CSN5 activity by directly binds to CSN5 at Glu76.
Figure 7.

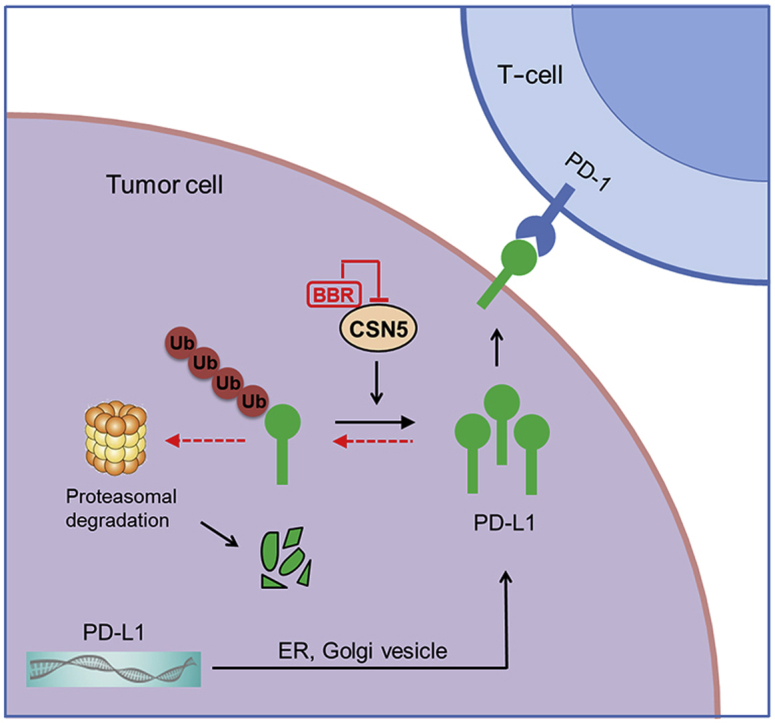
Glu76 of CSN5 is critical for binding to BBR. (A) Molecular docking model carried out by Discovery Studio 4.5 revealed that BBR binds to the JAMA domain of CSN5. (B) BBR forms an ionic bond with the backbone of Glu76 and interacts favorably with several residues including Met78, His138, Tyr143, and Asp151. (C) 293T-cells were transfected with WT-CSN5 and CSN5-mutant plasmids for 48 h, cell lysates were incubated with BBR–biotin at 37 °C for 2 h, followed by pull-down with streptavidin–agarose; the precipitates were then immunoblotted by flag antibody. (D) 293T-cells were co-transfected with Myc–PD-L1, HA–Ub and Flag–CSN5 WT or Flag–CSN5 E76A, followed by BBR treatment for 8 h. The ubiquitination of PD-L1 were analyzed by IB. (E) Proposed model of BBR-mediated PD-L1 degradation. BBR specific binds to and subsequently inactivates CSN5, which led to degradation of PD-L1 and activation of tumor-infiltrating T-cells.
4. Discussion
Compared with antibody-based PD-1/PD-L1 inhibitors, small-molecule inhibitors that disrupt PD-L1-mediated tumor tolerance are highly desirable. BBR, an old anti-inflammation drug, has been shown to inhibit the growth of various tumor cells in a number of preclinical models44. Here, we show that BBR exerts its antitumor activity through its immune-regulating function. PD-L1 was down-regulated by BBR in human NSCLC cells and in Lewis xenograft mice. In addition, BBR enhanced the sensitively of NSCLC cells to T-cells, increased the number effective CD8+ T-cells and decreased number of MSDCs and Tregs cells in Lewis xenograft mice, thus activating the immune microenvironment in tumor.
We provide evidence that BBR arouses T-cell immunity for its antitumor effect. First, the antitumor effect of BBR disappears in T-cell-deficient mice, and second, BBR induces more effective T-cells activation and boosts the production of IFN-γ and granzyme B by tumor-infiltrating CD8+ T-cells, demonstrating that BBR could activate cytotoxic T-cell in tumor microenvironment. Reactivating T-cells in tumor microenvironment is a promising therapeutic strategy to improve antitumor immunity. Currently, many antibody-based ICB target PD-1/PD-L1 axis to reactivate T-cell immunity. However, our present study provides an alternative strategy of reactivating T-cells. Being a small chemical compound, BBR possesses inherent advantages in terms of higher tissue and tumor penetration and better oral bioavailability, thus BBR is easily distributed in tumor microenvironment to target T-cells. Moreover, BBR is a extensively used drug with a confirmed safety record as it has been shown to have minimal effects on healthy cells23. Therefore, our findings identify BBR as a new tumor immunotherapeutic agent.
Consistent with previous reports23,24, we found that BBR is unable to directly kill NSCLC cells at concentrations lower than 20 μmol/L. Therefore, it is likely that BBR inhibits tumor growth through modulating tumor microenvironment. Our in vivo animal data also suggested that BBR exerts its immune-regulating function mainly at 4 mg/kg, because 8 mg/kg of BBR may inhibit the mitochondrial energy metabolism and induce the decrease of the mice body weight. MDSCs and Tregs are the immunoregulatory cells that profoundly inhibits tumor-specific T-cell immune response via PD-1/PD-L1 interaction35,36. Our results show that BBR attenuates the activation of MDSCs and Tregs by reducing their PD-L1 expression in the tumor microenvironment, which is in line with previous reports that blockade of PD-L1 expression in MDSC and Tregs decreased their immunosuppressive phenotype35,45. Our study thus demonstrates that BBR eliminates the suppressive effect on T-cells and promotes antitumor T-cell immunity through down-regulation of PD-L1.
An important finding in this study is that BBR triggers PD-L1 Ub/proteasome-dependent degradation via inhibition of CSN5. It has been established that PD-L1 is generally regulated by the Ub/proteasome pathway46, suggesting that targeting of PD-L1 polyubiquitination is an alternative strategy to enhancing immune checkpoint therapy. CSN5 directly deubiquitinates and stabilizes PD-L1 in cancer cells to escape from immune surveillance18. In the present study, we found BBR specific binds to and inhibits the activity of CSN5, leading to ubiquitination and subsequent degradation of PD-L1. Disrupting the MPN domain of CSN5 affected CSN5-mediated PD-L1 stabilization and deubiquitination43. Aberrant upregulation of CSN5 has been observed in the progression of diverse types of human cancer and closely correlated with poor prognosis17,41. NSCLC patients with high CSN5 expression levels have poor 5-year overall survival rate of 43.9%, compared with 63.1% for patients with low CSN5 expression17,47. In this regard, inhibition of CSN5 activity by BBR reduces PD-L1-based immunosuppression and is likely to have a significant effect on cancer treatment. Except CSN5, we also found other DUBs, such as USP28, could stabilize PD-L1 expression in cancer cells. However, BBR failed to interact with them except CSN5 (unpublished data). In addition, although pull-down assay suggested that biotin-conjugated BBR did not directly bind with cullin3, β-TrCP, STUB1, we could not exclude the possibility that BBR may strength the E3 ligase activity to destabilize PD-L1. This issue needs further investigation.
Accumulating studies suggest that BBR suppresses the growth of different tumor cells through multiple mechanisms48. Our findings show that BBR inhibited CSN5 activity and reduced PD-L1-based immunosuppression. This suggests that functioning as an immune-modulator is one of BBR's antitumor mechanisms. Of note, CSN5 knockout mice may be needed to further validate the in vivo antitumor effect of BBR. However, it is impossible for us to compare the antitumor effect of BBR between WT and CSN5 knockout mice since ablation of CSN5 results in embryonic death14. Further study may address this issue by using mice with myeloid-specific deleting CSN5 or Glu76 of CSN5 knock in mice.
In summary, our study shows that BBR diminishes PD-L1 expression and promotes antitumor immunity via inhibiting the deubiquitination activity of CSN5 in NSCLC (Fig. 7E). BBR specific binds to the Glu76 of CSN5 and subsequently inactivates CSN5, which led to degradation of PD-L1 and activation of tumor-infiltrating T-cells. Therefore, our study reveals that BBR functions as an immune-modulator by facilitating antitumor T-cell immunity. Our findings provide a rationale for the potential application of BBR as small-molecule inhibitor that disrupts PD-L1-mediated immunosuppression.
Acknowledgments
We thank Dr. Hualong Yu (Beijing Dequan Xingye Commerce and Trade Co., Ltd., Beijing, China) for the valuable flow cytometer technical support. This study was supported by grants from National Natural Science Foundation of China (81973366, 81773782 and 81903695), CAMS Innovation Fund for Medical Sciences (2016-12M-1-011, China), Open Project of State Key Laboratory of Bioactive Substance and Function of Natural Medicines (GTZK201908, China), National Mega-project for Innovative Drugs (2019ZX09721-001, China) and Chinese Pharmaceutical Association-Yiling Pharmaceutical Innovation Fund for Biomedicine (GL-1-B04-20180366, China).
Footnotes
Peer review under the responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.06.014.
Contributor Information
Lu Liu, Email: liulu208@163.com.
Hongbin Deng, Email: hdeng@imb.pumc.edu.cn.
Author contributions
Hongbin Deng, Yang Liu and Lu Liu designed research and analyzed data. Yang Liu, Xiaojia Liu, Na Zhang, Mingxiao Yin and Jingwen Dong conducted experiments. Qingxuan Zeng and Danqing Song synthesized the BBR-biotin and did the molecular docking. Genxiang Mao provided the TCM chemical monomers. Hongbin Deng obtained the funding, supervised the whole study, wrote and revised the manuscript. All authors approved the final version of the manuscript.
Conflicts of interest
The authors have declared no conflicts of interest.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Dunn G.P., Old L.J., Schreiber R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Ettinger D.S., Akerley W., Borghaei H., Chang A.C., Cheney R.T., Chirieac L.R. Non-small cell lung cancer, version 2.2013. J Natl Compr Canc Netw. 2013;11:645–653. doi: 10.6004/jnccn.2013.0084. [DOI] [PubMed] [Google Scholar]
- 3.Wei S.C., Duffy C.R., Allison J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Canc Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 4.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Canc. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwai Y., Hamanishi J., Chamoto K., Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci. 2017;24:26. doi: 10.1186/s12929-017-0329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pitt J.M., Vetizou M., Daillere R., Roberti M.P., Yamazaki T., Routy B. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44:1255–1269. doi: 10.1016/j.immuni.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Fusi A., Festino L., Botti G., Masucci G., Melero I., Lorigan P. PD-L1 expression as a potential predictive biomarker. Lancet Oncol. 2015;16:1285–1287. doi: 10.1016/S1470-2045(15)00307-1. [DOI] [PubMed] [Google Scholar]
- 8.Chen X., Pan X., Zhang W., Guo H., Cheng S., He Q. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B. 2020;10:723–733. doi: 10.1016/j.apsb.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boussiotis V.A. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767–1778. doi: 10.1056/NEJMra1514296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma P., Allison J.P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun C., Mezzadra R., Schumacher T.N. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lipson E.J., Sharfman W.H., Drake C.G., Wollner I., Taube J.M., Anders R.A. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Canc Res. 2013;19:462–468. doi: 10.1158/1078-0432.CCR-12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hannss R., Dubiel W. COP9 signalosome function in the DDR. FEBS Lett. 2011;585:2845–2852. doi: 10.1016/j.febslet.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 14.Tomoda K., Yoneda-Kato N., Fukumoto A., Yamanaka S., Kato J.Y. Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J Biol Chem. 2004;279:43013–43018. doi: 10.1074/jbc.M406559200. [DOI] [PubMed] [Google Scholar]
- 15.Cope G.A., Suh G.S., Aravind L., Schwarz S.E., Zipursky S.L., Koonin E.V. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science. 2002;298:608–611. doi: 10.1126/science.1075901. [DOI] [PubMed] [Google Scholar]
- 16.Wu Y., Deng J., Rychahou P.G., Qiu S., Evers B.M., Zhou B.P. Stabilization of snail by NF-κB is required for inflammation-induced cell migration and invasion. Canc Cell. 2009;15:416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J., Li Y., Wang B., Ma Y., Chen P. CSN5/Jab1 facilitates non-small cell lung cancer cell growth through stabilizing survivin. Biochem Biophys Res Commun. 2018;500:132–138. doi: 10.1016/j.bbrc.2018.03.183. [DOI] [PubMed] [Google Scholar]
- 18.Lim S.O., Li C.W., Xia W., Cha J.H., Chan L.C., Wu Y. Deubiquitination and stabilization of PD-L1 by CSN5. Canc Cell. 2016;30:925–939. doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo Z., Wang Y., Zhao Y., Shu Y., Liu Z., Zhou H. The pivotal oncogenic role of Jab1/CSN5 and its therapeutic implications in human cancer. Gene. 2019;687:219–227. doi: 10.1016/j.gene.2018.11.061. [DOI] [PubMed] [Google Scholar]
- 20.Ni W.J., Ding H.H., Tang L.Q. Berberine as a promising anti-diabetic nephropathy drug: an analysis of its effects and mechanisms. Eur J Pharmacol. 2015;760:103–112. doi: 10.1016/j.ejphar.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 21.Feng X., Sureda A., Jafari S., Memariani Z., Tewari D., Annunziata G. Berberine in cardiovascular and metabolic diseases: from mechanisms to therapeutics. Theranostics. 2019;9:1923–1951. doi: 10.7150/thno.30787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H., Fan C., Lu H., Feng C., He P., Yang X. Protective role of berberine on ulcerative colitis through modulating enteric glial cells–intestinal epithelial cells–immune cells interactions. Acta Pharm Sin B. 2020;10:447–461. doi: 10.1016/j.apsb.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu B., Wang G., Yang J., Pan X., Yang Z., Zang L. Berberine inhibits human hepatoma cell invasion without cytotoxicity in healthy hepatocytes. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chidambara Murthy K.N., Jayaprakasha G.K., Patil B.S. The natural alkaloid berberine targets multiple pathways to induce cell death in cultured human colon cancer cells. Eur J Pharmacol. 2012;688:14–21. doi: 10.1016/j.ejphar.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Zhu T., Li L.L., Xiao G.F., Luo Q.Z., Liu Q.Z., Yao K.T. Berberine increases doxorubicin sensitivity by suppressing STAT3 in lung cancer. Am J Chin Med. 2015;43:1487–1502. doi: 10.1142/S0192415X15500846. [DOI] [PubMed] [Google Scholar]
- 26.Zhang N., Dou Y., Liu L., Zhang X., Liu X., Zeng Q. SA-49, a novel aloperine derivative, induces MITF-dependent lysosomal degradation of PD-L1. EBioMedicine. 2019;40:151–162. doi: 10.1016/j.ebiom.2019.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang N., Liu X., Liu L., Deng Z., Zeng Q., Pang W. Glycogen synthase kinase-3β inhibition promotes lysosome-dependent degradation of c-FLIPL in hepatocellular carcinoma. Cell Death Dis. 2018;9:230. doi: 10.1038/s41419-018-0309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y.X., Liu L., Zeng Q.X., Fan T.Y., Jiang J.D., Deng H.B. Synthesis and identification of novel berberine derivatives as potent inhibitors against TNF-α-induced NF-κB activation. Molecules. 2017;22:1257. doi: 10.3390/molecules22081257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zou W., Wolchok J.D., Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 31.Maher C.M., Thomas J.D., Haas D.A., Longen C.G., Oyer H.M., Tong J.Y. Small-molecule sigma1 modulator induces autophagic degradation of PD-L1. Mol Canc Res. 2018;16:243–255. doi: 10.1158/1541-7786.MCR-17-0166. [DOI] [PubMed] [Google Scholar]
- 32.Dunn G.P., Koebel C.M., Schreiber R.D. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 33.Nishikawa H., Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7. doi: 10.1016/j.coi.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Kumar V., Patel S., Tcyganov E., Gabrilovich D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noman M.Z., Desantis G., Janji B., Hasmim M., Karray S., Dessen P. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dieterich L.C., Ikenberg K., Cetintas T., Kapaklikaya K., Hutmacher C., Detmar M. Tumor-associated lymphatic vessels upregulate PD-L1 to inhibit T-cell activation. Front Immunol. 2017;8:66. doi: 10.3389/fimmu.2017.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J., Bu X., Wang H., Zhu Y., Geng Y., Nihira N.T. Cyclin D–CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–95. doi: 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burr M.L., Sparbier C.E., Chan Y.C., Williamson J.C., Woods K., Beavis P.A. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101–105. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y., Wang H., Yao H., Li C., Fang J.Y., Xu J. Regulation of PD-L1: emerging routes for targeting tumor immune evasion. Front Pharmacol. 2018;9:536. doi: 10.3389/fphar.2018.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C.W., Lim S.O., Xia W., Lee H.H., Chan L.C., Kuo C.W. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan Y., Zhang Q., Tian L., Wang X., Fan X., Zhang H. Jab1/CSN5 negatively regulates p27 and plays a role in the pathogenesis of nasopharyngeal carcinoma. Canc Res. 2012;72:1890–1900. doi: 10.1158/0008-5472.CAN-11-3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X.C., Chen J., Su C.H., Yang H.Y., Lee M.H. Roles for CSN5 in control of p53/MDM2 activities. J Cell Biochem. 2008;103:1219–1230. doi: 10.1002/jcb.21504. [DOI] [PubMed] [Google Scholar]
- 43.Lingaraju G.M., Bunker R.D., Cavadini S., Hess D., Hassiepen U., Renatus M. Crystal structure of the human COP9 signalosome. Nature. 2014;512:161–165. doi: 10.1038/nature13566. [DOI] [PubMed] [Google Scholar]
- 44.McCubrey J.A., Lertpiriyapong K., Steelman L.S., Abrams S.L., Yang L.V., Murata R.M. Effects of resveratrol, curcumin, berberine and other nutraceuticals on aging, cancer development, cancer stem cells and microRNAs. Aging (Albany NY) 2017;9:1477–1536. doi: 10.18632/aging.101250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujimura T., Ring S., Umansky V., Mahnke K., Enk A.H. Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J Invest Dermatol. 2012;132:1239–1246. doi: 10.1038/jid.2011.416. [DOI] [PubMed] [Google Scholar]
- 46.Hsu J.M., Li C.W., Lai Y.J., Hung M.C. Posttranslational modifications of PD-L1 and their applications in cancer therapy. Canc Res. 2018;78:6349–6353. doi: 10.1158/0008-5472.CAN-18-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osoegawa A., Yoshino I., Kometani T., Yamaguchi M., Kameyama T., Yohena T. Overexpression of Jun activation domain-binding protein 1 in nonsmall cell lung cancer and its significance in p27 expression and clinical features. Cancer. 2006;107:154–161. doi: 10.1002/cncr.21961. [DOI] [PubMed] [Google Scholar]
- 48.Hesari A., Ghasemi F., Cicero A.F.G., Mohajeri M., Rezaei O., Hayat S.M.G. Berberine: a potential adjunct for the treatment of gastrointestinal cancers?. J Cell Biochem. 2018;119:9655–9663. doi: 10.1002/jcb.27392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.