

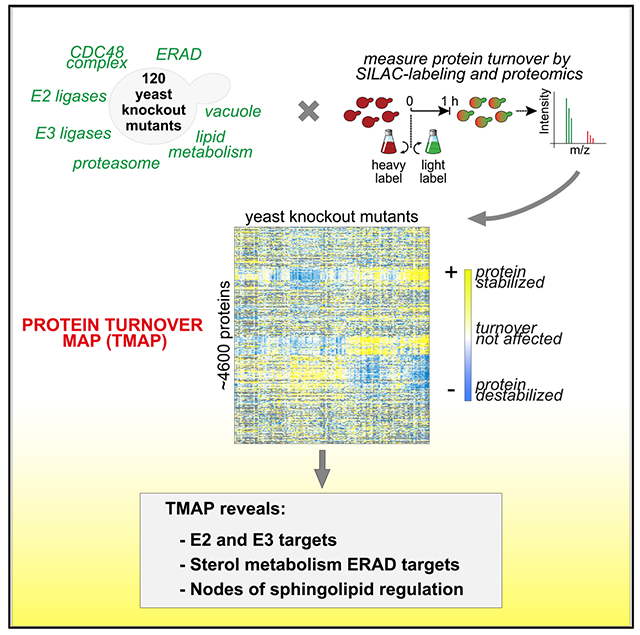
SUMMARY
Protein degradation is mediated by an expansive and complex network of protein modification and degradation enzymes. Matching degradation enzymes with their targets and determining globally which proteins are degraded by the proteasome or lysosome/vacuole have been a major challenge. Furthermore, an integrated view of protein degradation for cellular pathways has been lacking. Here, we present an analytical platform that combines systematic gene deletions with quantitative measures of protein turnover to deconvolve protein degradation pathways for Saccharomyces cerevisiae. The resulting turnover map (T-MAP) reveals target candidates of nearly all E2 and E3 ubiquitin ligases and identifies the primary degradation routes for most proteins. We further mined this T-MAP to identify new substrates of ER-associated degradation (ERAD) involved in sterol biosynthesis and to uncover regulatory nodes for sphingolipid biosynthesis. The T-MAP approach should be broadly applicable to the study of other cellular processes, including mammalian systems.
In Brief
How protein degradation is controlled is a central question in biology. Christiano et al. provide a global analysis of protein turnover looking at some 120 Saccharomyces cerevisiae mutants. The power of this T-MAP approach is illustrated by the analysis of specific pathways governing, for instance, lipid metabolism pathways.
Graphical Abstract
INTRODUCTION
The abundance and composition of specific proteins are key determinants of cell function. Degradation is crucial for controlling the abundance of many proteins, and is mediated by a large network of protein modification, sorting, and degradation machineries. Principally, proteins are degraded by either the proteasome (Arrigo et al., 1988) or acid proteases in the lysosomal compartment (the vacuole in yeast) (Gianetto and De Duve, 1955; Straus, 1954). Covalent modification with the small-protein-modifier ubiquitin (Ciehanover et al., 1978; Hershko et al., 1980) through the sequential actions of E1, E2, and E3 ligases is important for selecting substrates for either pathway. However, many mysteries remain about protein degradation, such as the routes of degradation taken by each protein individually, the identity of ubiquitin ligases that modify specific proteins, and how protein degradation pathways are integrated to regulate cell function. A major hurdle has been developing strategies to measure protein turnover globally, in a fashion similar to strategies that exist for measuring cellular mRNA expression levels (by RNA sequencing [RNA-seq] or microarrays) or protein synthesis (by ribosome footprinting).
With recent progress in mass spectrometry (MS)-based proteomics, unraveling these mysteries is now within reach. For example, global analyses of protein turnover in Saccharomyces cerevisiae and Schizosaccharomyces pombe revealed that proteins’ lifetimes in these organisms fall into three classes (Christiano et al., 2014; Eden et al., 2011). Most proteins in budding yeast (~85%) are long-lived (t1/2 > 5 h), and their amounts are determined chiefly by their rates of synthesis and dilution by cell growth and division. The remaining short- (t1/2 < 1.25 h) and medium-lived proteins (1.25 h < t1/2 < 5 h) are actively degraded at steady state, controlling their abundance. Turnover of this ~15% of the proteome is particularly important as this class is rich in regulated factors such as cell-cycle proteins, membrane transporters, or metabolic enzymes (Christiano et al., 2014). The ability to globally monitor turnover of native proteins (endogenous promoters and no tags) with minimal cellular perturbations (no drugs) opens the door to performing systematic turnover profiling studies in combination with genome perturbations to solve many long-standing questions pertaining to protein degradation. In particular, it is now possible to combine monitoring protein turnover rates across series of genetic deletions to elucidate biochemical networks.
RESULTS AND DISCUSSION
To comprehensively analyze protein degradation, we developed an approach to measure protein turnover in a large set of S. cerevisiae mutants deficient in specific components of the protein degradation machinery. We used state-of-the art proteomics, combined with pulse labeling of yeast with amino acids containing stable isotopes, to globally measure endogenous protein turnover in yeast strains with mutations of ~120 genes encoding components of the protein degradation machinery (Figures 1A and S1A). Our previous study showed 60 min after pulse labeling to be an optimal single time point for turnover analysis of most proteins (Christiano et al., 2014), and we used this time point for our measurements. Protein turnover rates derived from measurements after 1 h of chase showed good agreement with our previous time course measurements and with values from the literature (Figure 1B; Table S1). This strategy accurately captured changes for most proteins in a resultant turnover map (T-MAP), comprising 465,510 protein abundance and turnover measurements from 4,585 proteins derived from 86,502 peptides identified at a 1% false discovery rate (FDR) (Figure S1B). To ensure the accuracy and reproducibility of our measurements, we implemented a series of quality control analyses, such as independent biological duplicate measurements and testing for the absence of MS signals from peptides encoded by the deleted gene (see Supplementary Information; Figures S1C and S1D). While our protein abundance measurements are consistent with ribosome footprint data (Figure S1E; Ingolia et al., 2009), we did not find a correlation of protein abundance with our half-life measurements (Figure S1F). Analyzing the median of all of the measurements across mutants for each protein as a proxy of their turnover in a wild-type (WT) setting (see Supplementary Information; Figure S1C) revealed that each organelle contains short-, medium-, and long-lived proteins, suggesting that protein turnover is determined for proteins individually, rather than for a whole organelle (Figure S1G).
Figure 1. Overview of the Turnover Profiling Map (T-MAP) of Proteostasis Genes.
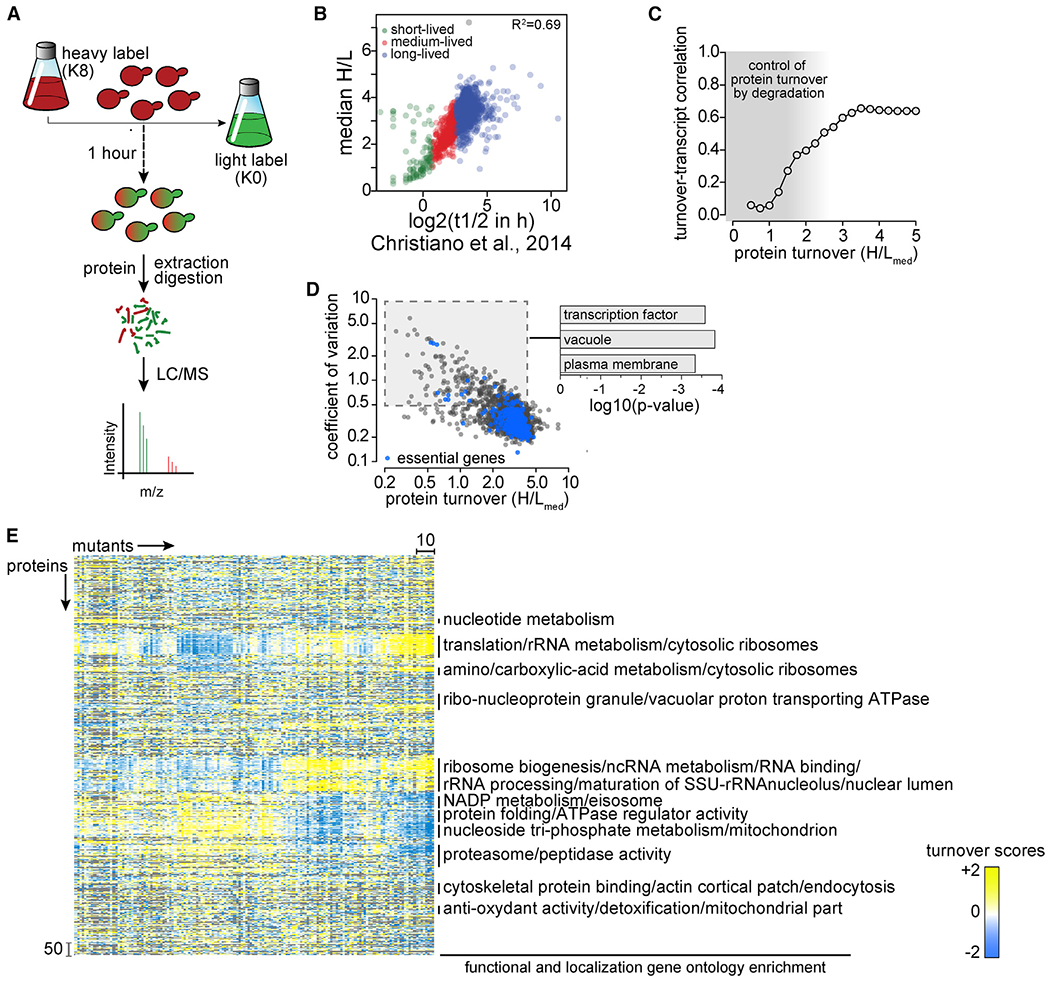
(A) Overview of single time point strategy for turnover profiling. Yeast cells were labeled for 3 days with heavy isotope lysine (K8). At t = 0, cells were washed with medium containing light isotope lysine (K0). After 1 h of chase, cells were harvested, and digested proteins analyzed by liquid chromatography-mass spectrometry (LC-MS) analysis to determine heavy to light (H:L) isotopic ratios for each detected protein, thereby enabling calculations of protein turnover.
(B) Comparison of median H:L ratios from a single (1-h) time point measurement in this study with multiple time point measurements in Christiano et al. (2014). Green, red, and blue puncta represent short-, medium-, and long-lived proteins, respectively.
(C) Distribution of Pearson correlation coefficients for pairwise turnover and expression correlations (transcript abundance; Kemmeren et al., 2014) as a function of protein turnover measurements in the present study. Gray shading indicates areas of weak correlation between mRNA levels and protein turnover, where protein abundance is controlled by protein degradation.
(D) Coefficients of variation of H:L ratios of each protein plotted as a function of the median of H:L ratios (H:Lmed). Essential proteins are in blue; other proteins are in black. p values indicate the enrichment of genes in the specific Gene Ontology categories.
(E) Hierarchical clustering of proteostasis mutants (left–right) and proteins (top–bottom). Enrichment for functional and localization categories in clusters is indicated on the right.
Whether changes in mRNA levels accurately predict changes in the proteome has been a long-standing question (Vogel and Marcotte, 2012). Integrating turnover data with measurements of mRNA transcript changes in a subset of our mutants (Kemmeren et al., 2014) showed that for long-lived proteins with slow degradation rates, transcriptional regulation accurately predicted protein turnover and abundance changes (Figure 1C). However, transcriptional changes in response to genome perturbation did not predict turnover for short-lived and medium-lived proteins. Instead, degradation had a major effect on these classes of proteins. This group of short- and medium-lived proteins contained proteins with the highest turnover variation due to genome perturbation (coefficient of variation ≥50%) and was enriched for transcription factors (p = 2.6 × 10−4), plasma membrane proteins (p = 1.5 × 10−4), and vacuolar proteins (p = 4.6 × 10−4), likely reflecting the importance of these proteins for maintaining cell homeostasis (Figure 1D).
To determine the effects of genetic perturbations on protein turnover, we developed a scoring function (robust T score) to measure stabilization and destabilization effects (Figure S1C). An overview of the T-MAP scores in hierarchical clusters of protein turnover profiles, detecting classes of co-regulated proteins, is shown in Figure 1E (see also Data S1). This analysis shows a similar response in the proteome turnover for numerous anabolic pathways, including several for protein synthesis (e.g., rRNA processing, ribosome biogenesis, translation) and metabolism (e.g., NADP metabolism, nucleotide metabolism). In addition to identi fying co-regulated proteins, the T-MAP provides comprehensive information on the turnover profiles of nearly all of the yeast proteins for each deletion mutant (Figure 1E; Data S1).
An important question for global protein turnover is which proteins are degraded by vacuolar versus proteasomal degradation pathways. We addressed this question by determining the effects of mutations in RPN4, a transcription factor that drives the expression of most proteasomal subunits (Mannhaupt et al., 1999; Xie and Varshavsky, 2001), and PEP4, the major vacuolar protease, on the turnover of endogenous proteins (Figures S2A–S2C). For the proteasomal degradation pathway, the analyses of proteins stabilized in rpn4Δ cells revealed 128 short- and medium-lived proteins (Table S1) that are enriched in cell-cycle-related proteins, kinases, ribosomal proteins, ubiquitination machinery, and some endoplasmic reticulum (ER) proteins (Figures 2A and 2B). In contrast, among the 106 short- and medium-lived proteins stabilized in pep4Δ cells were many proteins of the secretory and endolysosomal compartments (Figures 2A and S2C; Table S1). Unexpectedly, we also found soluble and membrane proteins from mitochondria that were stabilized in pep4Δ cells, suggesting the continuous degradation of specific mitochondrial proteins in the vacuole. Recently, a pathway was reported that removes specific mitochondrial membrane proteins in aged cells (Hughes et al., 2016); our data now suggest that a similar pathway may operate in yeast cells that are rapidly growing when mitophagy does not occur. We also identified 14 proteins that were stabilized in both vacuolar and proteasomal degradation mutants (Figures 2B and S2F), likely reflecting separate pathways for degrading distinct pools of protein. For example, Chs3 is present at the vacuolar membrane, where it is partially internalized and degraded (Arcones et al., 2016), and also at the ER, from where it may be delivered to the proteasome.
Figure 2. Turnover Effects of Lysosomal and Proteasomal Pathway Perturbations.
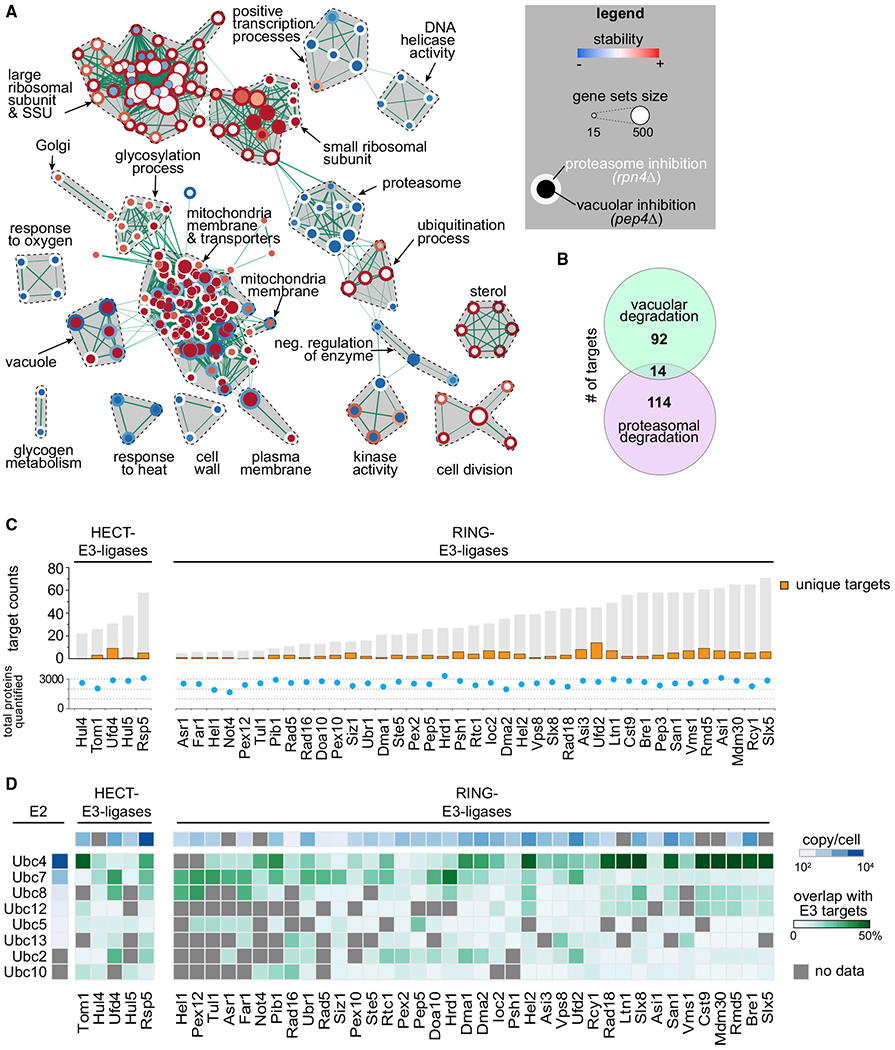
(A) Enrichment map of gene set enrichment analysis (GSEA) for proteins affected by inhibition of proteasomal and lysosomal degradation (p = 0.005, q = 0.1). Nodes represent gene sets that are enriched at the top or bottom of the ranking of differentially affected proteins (as determined by GSEA). Node size corresponds to the number of genes in the set. Edges indicate the overlap between gene sets, and the thickness indicates the size of the overlap. Red indicates increased and blue indicates decreased turnover. Node center corresponds to the effect of vacuolar degradation inhibition (pep4Δ cells), whereas node rims correspond to the effects of proteasomal degradation inhibition (rpn4Δ cells).
(B) Venn diagram representing the number of vacuolar (proteins stabilized in pep4Δ) and proteasomal (stabilized in rpn4Δ) targets.
(C) Number of targets of HECT and RING E3 ligases present in the T-MAP. Unique targets are indicated in orange. Shared targets are indicated in gray (shared between at least 2 E3 ligases). Total quantified proteins per mutant are indicated with blue dots.
(D) Target overlap between E3 and E2 enzymes (in percentage of E3 ligases total targets, green). E3 and E2 enzyme abundances are indicated (blue). Gray squares indicate no available data passing our quality criteria.
To validate some of these findings using an alternative approach, we examined the turnover of several proteins in pep4∆ cells (representing candidates over a wide range of T scores) by immunoblotting under conditions when new protein synthesis was blocked with cycloheximide (Figures S3A–S3C). The stabilization of proteins with high T scores (e.g., Fcy2, Cpr4) was confirmed by these analyses. However, the stabilization of proteins with lower T scores (e.g., Chs3, Pgk1) was not detectable in the immunoblotting assays. This possibly reflects the higher sensitivity of MS-based proteomics, compared with immunoblot analysis, to detect changes in protein abundance. In addition, inhibiting protein synthesis with cycloheximide may have independent effects on protein turnover.
Our analyses also revealed an unexpected coordination of the proteasomal and vacuolar protein degradation pathways. For instance, all proteasomal subunits were turned over faster in pep4Δ cells (Figures 2A and S2D). These changes in turnover were relatively small (T scores of ~−1) and were not detectable with immunoblotting in cycloheximide-chase experiments. We also found that vacuolar enzymes (e.g., Pep4, CPY/Prc1, Prb1, Cps1, Ape1, Ape3, Pho8) turned over faster in rpn4Δ cells (Figure S2E). How the degradation of the proteasome and the vacuolar proteases are coordinated when one component is blocked is unknown. Perhaps compromising vacuolar or proteasomal degradation slows cell anabolism (e.g., because components become limiting), and as a response, the alternative pathway is slowed.
A major goal for the field has been to identify the targets for specific ubiquitin ligases. Toward this goal, we systematically mutated the entire set of non-essential E3 and E2 ligases, expecting short-lived substrates to be stabilized in the absence of their cognate E2 and E3 enzymes. Consistently, we found that the vast majority of short- and medium-lived proteins are stabilized in at least one E3 ligase deletion strain (Figure 2C). By analyzing specific cases, we found that our data recapitulated prior findings on the requirements of substrates for particular E3 ligases, thus validating our dataset (Figure S2G; Table S1). In addition, we identified new degradation interactions for many E3 enzymes, with many proteins that apparently respond to the disruption of multiple E3 ligases, including 96 proteins (21%) that are affected by the deletion of ≥5 E3 ligases (Figure S2H). Conversely, mutations of some E3 ligases stabilized only a few targets (e.g., Hel1 has 6 identified targets), but other E3s target as many as ~70 proteins (e.g., Slx5) (Figure 2C). These findings now provide an opportunity to delineate direct enzyme-substrate relationships from more indirect effects of E3 ligase disruptions.
Cooperation between E2 and E3 enzymes appears to be loose, inasmuch as there is great overlap between any E2 and multiple E3 targets in our analyses (Figure 2D). The most abundant E2 enzyme, Ubc4, has more overlap with E3 enzymes than less abundant E2 enzymes, such as Ubc10 or Ubc13 (Figure 2D), suggesting that cooperation between E2 and E3 enzymes may be driven to some extent by mass action instead of specific protein-protein interactions; however, more direct analyses will be necessary to test this possibility.
Our analyses delineate the specificity for interactions in the ubiquitin proteasome system, allowing for further mechanistic dissection of the network and for defining the rules that govern access to specific E3 ligases. For instance, previous work showed that the Hrd1 E3 ligase complex (Figure 3A) targets misfolded substrates that contain lesions either in their membrane domain (ERAD-M) or on the ER luminal side (ERAD-L), and each of these pathways is thought to involve different subunits of the complex (Carvalho et al., 2006). However, as these concepts were developed using mostly artificial model substrates, it is unclear to which extent these pathways degrade endogenous substrates and how these are selected for degradation in the absence of folding lesions. To address these questions, we used our dataset to define the bona fide substrates of the Hrd1 complex. In addition to the known substrate Hmg2 (T score in cue1Δ, 2.0; hrd1Δ, 1.05), we found 3 previously reported proteins (Erg3, Erg5, and Erg25) (Christiano et al., 2014; Jaenicke et al., 2011) and 2 newly identified short-lived proteins (Yjr015w and Ysp2; Figures 3B and 3E) that were stabilized in several mutants of the Hrd1 complex and in cells expressing a catalytically inactive version of Hrd1 (C399S) (Figure 3C) (Bordallo et al., 1998).
Figure 3. Systematic Prediction of Targets for the HRD1 Branch of ERAD.
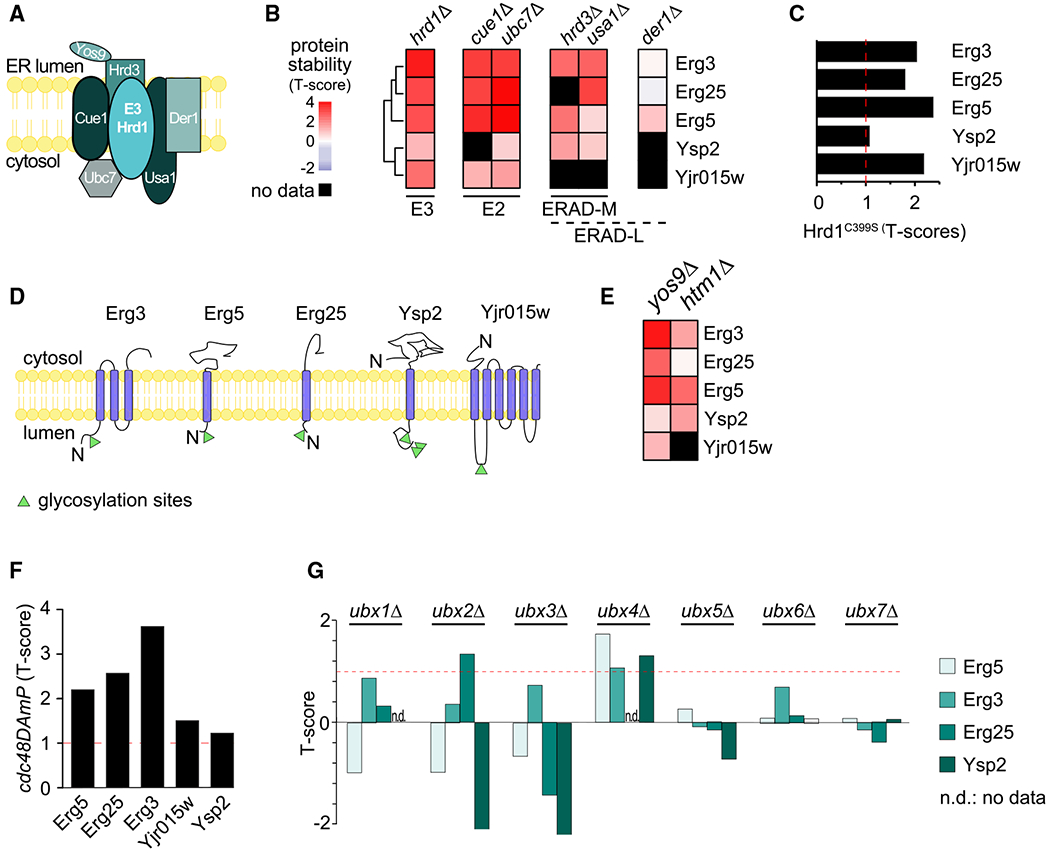
(A) Schematic of the Hrd1 branch of ERAD in yeast.
(B) Hierarchically clustered heatmap of the robust T scores of Hrd1 targets in response to deletion of the member of the Hrd1 complex.
(C) Stability of Hrd1 targets in cells expressing Hrd1C399S, a catalytically dead allele of HRD1.
(D) Localization and topology of bona fide substrates of the Hrd1 complex. Glycosylation sites are indicated as green triangles.
(E) Heatmap clustered and colored as in (B) of the robust T scores of Hrd1 targets in response to the deletion of YOS9 and HTM1.
(F) Stability of Hrd1 complex substrates in cells expressing cdc48DamP, a hypomorphic allele of CDC48.
(G) Stability of Erg5, Erg3, Erg25, and Ysp2 in cells deleted for individual UBX domain-containing substrate adaptors of Cdc48.
The T-MAP provides a powerful approach to determine the physiological function of a specific protein degradation pathway. For instance, each of the endogenous Hrd1 targets in the T-MAP is involved in sterol metabolism. Erg3, Erg5, and Erg25 are enzymes of ergosterol de novo synthesis (Arthington et al., 1991; Jaenicke et al., 2011; Kelly et al., 1995; Li and Kaplan, 1996), YJR015W encodes a protein with strong homology to human ABC transporters mediating the cellular efflux of sterols (HHPred; Figure S4A), and Ysp2/Ltc4 belongs to the recently discovered family of StART-domain proteins that localize at membrane contact sites and are responsible for the transport of sterols (Gatta et al., 2015; Murley et al., 2015). These findings expand our understanding of Hrd1-dependent ERAD, not only as a master regulator of sterol de novo synthesis but also sterol transport in cells. We further analyzed Erg25, as an example, and found that it interacts with Hrd1 complex members as well as the downstream machinery involved in its degradation (Figure S4B).
To test whether these endogenous substrates of the Hrd1 complex access the ERAD-M or the ERAD-L pathway, we analyzed the requirement of their degradation for Der1, which was implicated specifically in the degradation of ERAD-L model substrates, such as CPY* (Carvalho et al., 2006). We did not find striking effects of DER1 deletion on endogenous short-lived ERAD substrates (Figure 3B). We also did not detect any luminal proteins that were significantly affected by the deletion of ERAD genes under standard growing conditions. Consistently, we found no proteins with significantly altered turnover in cells expressing a version of Hrd1 (Hrd1KRK) that does not allow domain in vitro retrotranslocation of model ERAD-L substrates (data not shown; Baldridge and Rapoport, 2016). Instead, each endogenous Hrd1 substrate that we identified contained transmembrane domains, a common feature of ERAD-M (Figure 3D). Also, generally their degradation required Yos9, a lectin component of the HRD1 complex, suggesting that their degradation depends on glycosylation (Figure 3E). This model is supported by the stabilization of Erg3, Erg5, and Ysp2 in htm1Δ cells (Figure 3E), lacking the α-1,2-specific exo-mannosidase that trims a mannose residue from Man8GlcNac2 (Man8) glycans to form Man7GlcNac2 (Man7), a signal recognized by Yos9. Furthermore, database analysis showed that all endogenous Hrd1 substrates are glycosylated in the luminal portion of the proteins where interactions with Yos9 can occur (Figure 3D; Zielinska et al., 2012). These data show that these endogenous substrates of the Hrd1 complex are degraded by the ERAD-M pathway, likely being recognized through glycosylation.
To further dissect the Hrd1-dependent ERAD pathway, we analyzed the requirements for downstream effectors in targeting endogenous substrates for degradation. As expected, the turnover of Hrd1 targets required the activity of the AAA-ATPase Cdc48 involved in protein extraction from the membrane (Figure 3F) and required proteasomal activity (Figure S4C). Cdc48 is thought to be recruited to substrates by 1 of the 7 Cdc48-binding UBX domain-containing proteins (Ubx1–7). Surprisingly, different Hrd1 targets required different UBX proteins. Ubx2 is thought to be the canonical UBX protein involved in ERAD. However, it was only required for the degradation of Erg25, whereas Ubx4 was the predominant factor involved in the degradation of Erg3, Erg5, and Ysp2 (Figure 3G). Although the mechanistic basis for these differences is unknown, these findings illustrate the power of the T-MAP approach to discover specific degradation pathway requirements.
This T-MAP also allowed for the identification of potentially important regulatory proteins. For example, we found that key proteins involved in phospholipid (Tgl5 and Plb3) and sphingolipid (Tsc10, Orm2, Aur1, Sur1, Csg2, and Ipt1) metabolism are short or medium lived. (Figure 4A). Surprisingly, analyses of the T-MAP revealed that these proteins are targeted by different degradation pathways localized in different organelles. Sur1 and Csg2 are proteins in a complex involved in the synthesis of complex sphingolipids (Uemura et al., 2003). Both were stabilized when vacuolar degradation or the endosomal sorting complexes required for transport (ESCRT) machinery (delivering cargoes to vacuole) was impaired (Figure 4B; data not shown). In contrast, the regulatory protein Orm2, thought to be localized primarily in the ER (Breslow et al., 2010), is targeted by the Tul1 E3 ligase complex to degradation by the proteasome. The Tul1 complex shares similarities with the Hrd1 complex but is primarily localized in membranes of the Golgi apparatus and of the endolysosomal system (Yang et al., 2018) (Figures 4B and 4C). Finally, proteasomal degradation of Tsc10, an essential ER and lipid droplet enzyme (Currie et al., 2014) catalyzing the reduction of keto-dihydrosphingosine to dihydrosphingosine (Beeler et al., 1998), required ubiquitin (Ub) adaptors of ERAD (i.e., stabilized in ubc7Δ, cue1Δ, or ubc4Δ), Cdc48, and Asi1, one of the E3 ligases of the ASI branch of ERAD in yeast (Figures 4B, 4D, and 4E). In agreement with this, Tsc10 degradation is lower in asi1Δ and asi3Δ and, to a lesser extent, in doa10Δ mutants, than with WT cells, when new protein synthesis was blocked with cycloheximide (Figures 4F and S3D). A functional link between the Asi1/3 machinery and sphingolipid metabolism is further supported by published genetic interaction data, with the strongest interactions reported for ASI1 and ASI3 with sphingosine kinase (LCB5), sphingosine lyase (DPL1), and a component of serine-palmitoylcoenzyme A (CoA) transferase (TSC3) (Costanzo et al., 2016) (Figure 4G). Other proteins involved in sphingolipid and phospholipid metabolism (e.g., Sur2, involved in sphingolipid synthesis, and Lcb2 of the SPT complex) could not be assigned to a specific degradation route since they were stabilized in vacuolar (pep4Δ) and ERAD (hrd1Δ) mutants (Figure 4H). Overall, these results identify new potential nodes of regulation by degradation control for key enzymes in the sphingolipid metabolism.
Figure 4. Spatial Control of Degradation for Key Enzymes in the De Novo Sphingolipid Biosynthesis Pathway.
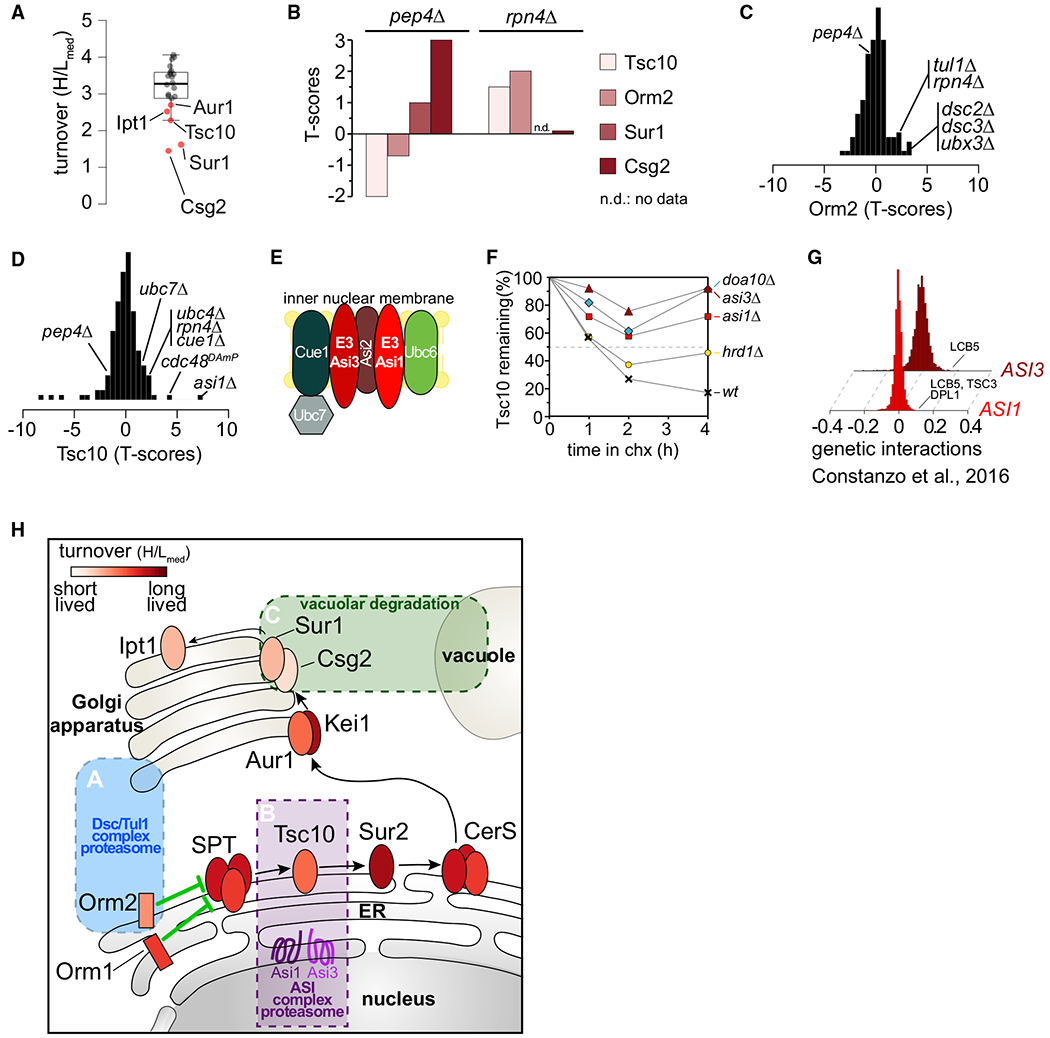
(A) Boxplot showing the turnover (H/Lmed) distribution of enzymes in the de novo sphingolipid biosynthesis pathway. Red dots indicate significantly deviating sphingolipid metabolism enzymes.
(B) Stability of Tsc10, Sur1, Csg2, and Orm2 under proteasomal and vacuolar degradation inhibition.
(C) Stability scores of Orm2 in the mutants present in T-MAP.
(D) Stability scores of Tsc10 in the mutants present in T-MAP.
(E) Schematic of the ASI branch of ERAD in yeast.
(F) Degradation of GFP-TSC10 after inhibition of protein synthesis by cycloheximide in wild-type (WT), asi1Δ, asi3Δ, doa10Δ, and hrd1Δ. The graph shows the quantification of the western blot shown in Figure S3D.
(G) ASI1 and ASI3 genetically interact with genes involved in sphingolipid metabolism (Costanzo et al., 2016). Positive SGA scores represent positive genetic interactions.
(H) Model for spatial control of degradation of key enzymes in the de novo sphingolipid biosynthesis pathway by the Tul1 complex (blue box), ASI1/3 complex (purple box), and the vacuole (green box). Ellipses represent sphingolipid metabolic enzymes and are color coded according to their half-lives.
In summary, we present an analytical platform that combines gene deletions with quantitative measures of protein turnover to deconvolve degradation pathways of S. cerevisiae. This particular T-MAP, focusing on protein degradation machineries, provides a rich, new resource for identifying and mechanistically dissecting the ubiquitin-mediated protein degradation system. It reveals endogenous target candidates of nearly all E2 and E3 ligases, as well as the scope of specific pathways, such as ERAD. The analysis of turnover of specific short-/medium-lived proteins reveals their degradation pathway and can be used to reveal previously unknown nodes of regulation in key cellular metabolisms as underscored by our analysis of sphingolipid metabolism (Figure 4H).
Similar to other large-scale analyses, the T-MAP approach has some limitations. Foremost, it cannot distinguish between direct enzyme-substrate relationships and indirect effects of protein deletion on the stability of targets. Also, the necessity to pick one time point for measurements complicates the analysis of proteins with much shorter or longer half-lives, and although MS-based proteomics now captures essentially all yeast proteins, low abundant proteins are less likely to yield robust data. Nonetheless, the T-MAP approach should be broadly applicable to the study of other cellular processes and systems. For example, extension to other gene sets could reveal target specificities of other components of the proteostasis network (e.g., protein folding, trafficking). Moreover, the extension of T-MAP analyses to other organisms, including mammals, provides a powerful new tool for investigating how protein turnover functions in normal physiology and disease. Finally, turnover profiling approaches will enable efforts in drug discovery to delineate the therapeutic potential of E3 ligase-modulating agents by allowing the rapid identification of targeted neo-substrates.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-GFP | Abcam | Cat# ab290; RRID: AB_303395 |
| Rabbit polyclonal anti-Rpn1 | Dan Finley | N/A |
| Rabbit polyclonal anti-Rpn5 | Dan Finley (Shi et al., 2016) | N/A |
| Rabbit polyclonal anti-Rpn8 | Dan Finley | N/A |
| Rabbit polyclonal anti-Rpn12 | Dan Finley | N/A |
| Mouse anti-IgG kappa binding protein-HRP | Santa Cruz Biotechnology | Cat# sc-516102; RRID: AB_2687626 |
| Mouse monoclonal anti-rabbit IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2357; RRID: AB_628497 |
| Mouse monoclonal anti-Pgk1 | Invitrogen | Cat# 459250; RRID: AB_2532235 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat# F1804; RRID AB_262044 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| heavy [13C6/15N2] L-lysine | Cambridge Isotope laboratories, Inc. | CNLM-291-H-PK |
| Drop-out Mix Minus Lysine w/o Yeast Nitrogen Base | US Biological | Cat# D9515B |
| Critical Commercial Assays | ||
| SuperSignal West Pico | SuperSignal West Pico Thermo Fisher Scientific Cat# 34580 | SuperSignal West Pico Thermo Fisher Scientific Cat# 34580 |
| SuperSignal West Dura | SuperSignal West Dura Thermo Fisher Scientific Cat# 34076 | SuperSignal West Dura Thermo Fisher Scientific Cat# 34076 |
| Deposited Data | ||
| Mass spectrometry proteomics data | Proteomics Identifications Database (PRIDE) repository | PRIDE: PXD008706 |
| Experimental Models: Yeast strains (Saccharomyces cerevisiae) | ||
| BY4743 | Dharmacon | N/A |
| BY4741 | Dharmacon | N/A |
| BY4741; pep4Δ::kanMX | Dharmacon | N/A |
| BY4741; CHS3-TEV-6xGly-3xFLAG::hphMX | This study | N/A |
| BY4741; pep4Δ::kanMX CHS3-TEV-6xGly-3xFLAG::hphMX | This study | N/A |
| BY4741; FCY2-TEV-6xGly-3xFLAG::hphMX | This study | N/A |
| BY4741; pep4Δ::kanMX FCY2-TEV-6xGly-3xFLAG::hphMX | This study | N/A |
| BY4741; CPR4-TEV-6xGly-3xFLAG::hphMX | This study | N/A |
| BY4741; pep4Δ::kanMX CPR4-TEV-6xGly-3xFLAG::hphMX | This study | N/A |
| Recombinant DNA | ||
| Plasmid: pTF272 (TEV-6xGly-3xFLAG) | Tim Formosa | Addgene #44083 |
| Software and Algorithms | ||
| MaxQuant (version 1.5.2.8) | (Tyanova et al., 2016) | https://www.maxquant.org/ |
| Perseus (version 1.5.1.7) | (Tyanova and Cox, 2018) | https://maxquant.net/perseus/ |
| TreeView | (Saldanha, 2004) | N/A |
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents generated in this study should be directed to and will be fulfilled by the Lead contact, Tobias C. Walther (twalther@hsph.harvard.edu).
Materials Availability
Any unique reagents generated in this study are available upon request and will be handled according to the Harvard T. H. Chan School of Public Health policies regarding MTA and related matters.
Data and Code Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the Proteomics Identifications Database (PRIDE) repository with the dataset identifier PXD008706.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
To minimize compensatory mutations that might accumulate over time in the gene deletion strains, we freshly prepared all mutant strains. Haploid cells harboring the desired deletions were prepared from sporulation of the corresponding BY4743 heterozygous knockout diploid cells and selected to be isogenic to parental wild-type BY4742 genetic background (MATα his3Δ1, leu2Δ1, lys2Δ1, ura3Δ1). The deletion and the correct insertion of the kanamycin-resistance cassette were verified by two PCRs using primers described at the Saccharomyces Genome Deletion Project Web page (http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html). Missing haploid knock out strains, even after several rounds of sporulation and selection (28 strains), were directly purchased from Dharmacon and confirmed prior to further analysis. Tagging of genes with GFP (Janke et al., 2004) or TEV-6xGly-3xFLAG was carried out by insertion of homologs cassettes and verification by PCR and immunoblot.
METHOD DETAILS
Cell Culture and Pulsed SILAC Experiments
Yeast strains were grown in synthetic medium containing 6.7 g/l yeast nitrogen base, 2 g/l dropout mix containing all amino acids except lysine, and 2% glucose. For heavy pre-labeling, heavy [13C6/15N2] L-lysine was added to a final concentration of 30 mg/l. Cells were pre-cultured in 5 mL of medium containing heavy lysine overnight at 30°C and repeated twice. pSILAC experiments were performed by transferring exponentially growing heavy labeled cells, after three washes at 4°C with cold SILAC medium without lysine, to a fresh growth medium containing an excess of light lysine. Cells were harvested at 60 min and immediately prepared for MS analysis. At least two independent biological replicates for each strain were measured.
Cycloheximide pulse-chase
Cells were grown in yeast synthetic medium containing all amino acids including lysine and 2% glucose at 30°C till logarithmic growth phase (OD 0.6-1.0). Cycloheximide was added from a 12.5 mg/ml stock in H2O to a final concentration of 50 or 250 mg/ml. At each time point 10 OD units of cells were harvested and lysed as described in sample preparation and analyzed by SDS-PAGE and immunoblotting.
Dataset filtering
For each gene deletion turnover profiling dataset, we computed quality metrics including: (1) the number of proteins quantified with at least two ratio counts, (2) the presence of the aminoglycoside phosphotransferase enzyme encoded by the kanamycin cassette conferring resistance to G418, and (3) the absence of the protein encoded by the deleted gene. We excluded, from further analysis, mutant turnover profiling datasets with poor proteome coverage (≤2000 proteins identified).
Sample preparation
For each mutant, ~25 OD units of cells were harvested by centrifugation. Cells were lysed in 200 μl of buffer containing 50 mM Tris/HCl, pH 9.0, 5% SDS and 100 mM DTT for 30 min at 55°C. Lysates were cleared by centrifugation at 17,000 g for 10 min. Supernatants were diluted with buffer UA (8 M urea, 0.1 M Tris/HCl, pH 8.5) to a final concentration of 0.5% SDS. Proteins were digested with the endoproteinase LysC, following the protocol for filter-aided sample preparation (12). Briefly, protein extracts were loaded on a 30k centricon filter unit (Amicon) by centrifugation at 14,000 g. Samples were washed twice by addition of 200 μl of buffer UA and alkylated for 20 min in the dark with 5.5 mM iodoacetamide (IAA) in 200 μl of buffer UA. Samples were washed an additional four times by adding 200 μl of buffer UA and centrifugation. 60 μl of buffer UA containing 0.5 mg/ml LysC were added to the filter units and incubated at 37°C overnight. Peptides were recovered by centrifugation into afresh tube and additional elution with 200 μl of 0.5 M NaCl. Samples were acidified by adding trifluoroacidic acid (TFA) and cleared of precipitates by centrifugation at 17,000 g for 5 min. Peptide concentration was measured, and 5 μg of peptides were desalted following the protocol for StageTip purification (Rappsilber et al., 2007). Samples were eluted with 60 μL of buffer B (80% ACN, 0.1% formic acid in H2O) and reduced in a Vacufuge plus (Eppendorf) to a final volume of 3 μL. 2 μL of buffer A (0.1% formic acid in H2O) were added, and the resulting 5 μL were injected into the HPLC.
Chromatography and Mass Spectrometry
Reversed phase chromatography was performed on a Thermo Easy nLC 1000 system connected to a Q Exactive mass spectrometer (Thermo) through a nano-electrospray ion source. Peptides were separated on 50-cm columns (New Objective) with an inner diameter of 75 μm packed in house with 1.9 μm C18 resin (Dr. Maisch GmbH). Peptides were eluted from 50-cm columns with a linear gradient of acetonitrile from 5%−27% in 0.1% formic acid for 240 min at a constant flow rate of 250 nl/min. The column temperature was kept at 40°C by an oven (Sonation GmbH, Germany) with a Peltier element. Peptides eluted from the column were directly electrosprayed into the mass spectrometer. Mass spectra were acquired on the Q Exactive in a data-dependent-mode to automatically switch between full-scan MS and up to 10 data-dependent MS/MS scans. The maximum injection time for full scans was 20 ms with a target value of 3,000,000 at a resolution of 70,000 at m/z = 200. The 10 most intense multiple charged ions (z ≥ 2) from the survey scan were selected with an isolation width of 1.4Th and fragment with higher energy collision dissociation (HCD 14) with normalized collision energies of 25. Target values for MS/MS were set to 100,000 with a maximum injection time of 120 ms at a resolution of 17,500 at m/z = 200. To avoid repetitive sequencing, the dynamic exclusion of sequenced peptides was set to 45 s.
Raw MS Data Analysis
The resulting MS and MS/MS spectra were analyzed using MaxQuant (version 1.5.2.8) and its integrated ANDROMEDA search algorithms (Tyanova et al., 2016). Peak lists were searched against the UNIPROT databases for S. cerevisiae with common contaminants added. The search included carbamidomethylation of cysteines as fixed modification, and methionine oxidation and N-terminal acetylation as variable modifications. Maximum allowed mass deviation for MS peaks was set to 6 ppm and 20 ppm for MS/MS peaks. Maximum missed cleavages were 2. The false discovery rate was determined by searching a reverse database. Maximum false-discovery rates were 0.01 both on peptide and protein levels. Minimum required peptide length was six residues. Proteins with at least two peptides (one of them unique) were considered identified. The “match between runs” option was enabled with a time window of 1 min to match identification between replicates.
Protein Abundance Estimation
Absolute protein abundances as copies per cell were calculated by applying the “proteomic ruler” plugin in Perseus software (Tyanova and Cox, 2018) using the summed intensities of both heavy and light peptides for each protein groups.
QUANTIFICATION AND STATISTICAL ANALYSIS
We developed custom scripts in R/Bioconductor for data analysis and plotting, which are available on request. Proteins were clustered hierarchically, based on a Pearson correlation of turnover robust score in Cluster, and visualized by TreeView (Saldanha, 2004). Statistical details of experiments can be found in figure legends or results section.
Supplementary Material
Highlights.
Systematic analysis of protein turnover in ~120 yeast mutants
Global protein turnover map (T-MAP) includes ~4,600 proteins
T-MAP identifies regulatory nodes of sterol and sphingolipid metabolism
T-MAP reveals candidate substrates for ERAD and alternative degradation routes
ACKNOWLEDGMENTS
We would like to thank Drs. Wade Harper and Dan Finley, as well as members of the Farese & Walther laboratory for critical discussion and comments on the manuscript. We thank Dan Finley, Tom Rapoport, and Ryan Baldridge for sharing reagents; Xiuling Guo for technical assistance; and Gary Howard for editorial help. This work was supported by a grant of the G. Harold and Leila Y. Mathers Foundation, NIH GM097194, and the Howard Hughes Medical Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108378.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arcones I, Sacristán C, and Roncero C (2016). Maintaining protein homeostasis: early and late endosomal dual recycling for the maintenance of intracellular pools of the plasma membrane protein Chs3. Mol. Biol. Cell 27, 4021–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigo AP, Tanaka K, Goldberg AL, and Welch WJ (1988). Identity of the 19S ‘prosome’ particle with the large multifunctional protease complex of mammalian cells (the proteasome). Nature 331, 192–194. [DOI] [PubMed] [Google Scholar]
- Arthington BA, Bennett LG, Skatrud PL, Guynn CJ, Barbuch RJ, Ulbright CE, and Bard M (1991). Cloning, disruption and sequence of the gene encoding yeast C-5 sterol desaturase. Gene 102, 39–44. [DOI] [PubMed] [Google Scholar]
- Baldridge RD, and Rapoport TA (2016). Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 166, 394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler T, Bacikova D, Gable K, Hopkins L, Johnson C, Slife H, and Dunn T (1998). The Saccharomyces cerevisiae TSC10/YBR265w gene encoding 3-ketosphinganine reductase is identified in a screen for temperature-sensitive suppressors of the Ca2+-sensitive csg2Delta mutant. J. Biol. Chem 273, 30688–30694. [DOI] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, and Wolf DH (1998). Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell 9, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, and Weissman JS (2010). Orm family proteins mediate sphingolipid homeostasis. Nature 463, 1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, and Rapoport TA (2006). Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373. [DOI] [PubMed] [Google Scholar]
- Christiano R, Nagaraj N, Fröhlich F, and Walther TC. (2014). Global proteome turnover analyses of the yeasts S. cerevisiae and S. pombe. Cell Rep. 9, 1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciehanover A, Hod Y, and Hershko A (1978). A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun 81, 1100–1105. [DOI] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. (2016). A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie E, Guo X, Christiano R, Chitraju C, Kory N, Harrison K, Haas J, Walther TC, and Farese RV Jr. (2014). High confidence proteomic analysis of yeast LDs identifies additional droplet proteins and reveals connections to dolichol synthesis and sterol acetylation. J. Lipid Res 55, 1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Geva-Zatorsky N, Issaeva I, Cohen A, Dekel E, Danon T, Cohen L, Mayo A, and Alon U (2011). Proteome half-life dynamics in living human cells. Science 331, 764–768. [DOI] [PubMed] [Google Scholar]
- Gatta AT, Wong LH, Sere YY, Calderon-Norena DM, Cockcroft S, Menon AK, and Levine TP (2015). A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. eLife 4, e07253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianetto R, and De Duve C (1955). Tissue fractionation studies. 4. Comparative study of the binding of acid phosphatase, beta-glucuronidase and cathepsin by rat-liver particles. Biochem. J 59, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A, Heller H, Haas AL, and Rose IA (1980). Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc. Natl. Acad. Sci. USA 77, 1783–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Hughes CE, Henderson KA, Yazvenko N, and Gottschling DE (2016). Selective sorting and destruction of mitochondrial membrane proteins in aged yeast. eLife 5, e13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenicke LA, Brendebach H, Selbach M, and Hirsch C (2011). Yos9p assists in the degradation of certain nonglycosylated proteins from the endoplasmic reticulum. Mol. Biol. Cell 22, 2937–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, and Knop M (2004). A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962. [DOI] [PubMed] [Google Scholar]
- Kelly SL, Lamb DC, Corran AJ, Baldwin BC, Parks LW, and Kelly DE (1995). Purification and reconstitution of activity of Saccharomyces cerevisiae P450 61, a sterol delta 22-desaturase. FEBS Lett. 377, 217–220. [DOI] [PubMed] [Google Scholar]
- Kemmeren P, Sameith K, van de Pasch LA, Benschop JJ, Lenstra TL, Margaritis T, O’Duibhir E, Apweiler E, van Wageningen S, Ko CW, et al. (2014). Large-scale genetic perturbations reveal regulatory networks and an abundance of gene-specific repressors. Cell 157, 740–752. [DOI] [PubMed] [Google Scholar]
- Li L, and Kaplan J (1996). Characterization of yeast methyl sterol oxidase (ERG25) and identification of a human homologue. J. Biol. Chem 271, 16927–16933. [DOI] [PubMed] [Google Scholar]
- Mannhaupt G, Schnall R, Karpov V, Vetter I, and Feldmann H (1999). Rpn4p acts as a transcription factor by binding to PACE, a nonamer boxfound upstream of 26S proteasomal and other genes in yeast. FEBS Lett. 450,27–34. [DOI] [PubMed] [Google Scholar]
- Murley A, Sarsam RD, Toulmay A, Yamada J, Prinz WA, and Nunnari J (2015). Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J. Cell Biol 209, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview-extensible visualization of microarray data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Shi Y, Chen X, Elsasser S, Stocks BB, Tian G, Lee BH, Shi Y, Zhang N, de Poot SA, Tuebing F, et al. (2016). Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 351,aad9421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus W (1954). Isolation and biochemical properties of droplets from the cells of rat kidney. J. Biol. Chem 207, 745–755. [PubMed] [Google Scholar]
- Tyanova S, and Cox J (2018). Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol 1711, 133–148. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, and Cox J (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- Uemura S, Kihara A, Inokuchi J, and Igarashi Y (2003). Csg1p and newly identified Csh1p function in mannosylinositol phosphorylceramide synthesis by interacting with Csg2p. J. Biol. Chem 278, 45049–5055. [DOI] [PubMed] [Google Scholar]
- Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet 13,227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, and Varshavsky A (2001). RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: a negative feedback circuit. Proc. Natl. Acad. Sci. USA 98, 3056–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Arines FM, Zhang W, and Li M (2018). Sorting of a multi-subunit ubiquitin ligase complex in the endolysosome system. eLife 7, e33116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinska DF, Gnad F, Schropp K, Wisniewski JR, and Mann M (2012). Mapping N-glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol. Cell 46, 542–548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the Proteomics Identifications Database (PRIDE) repository with the dataset identifier PXD008706.