

Abstract
Extracellular signal-regulated kinase (ERK), also known as classical mitogen-activated protein kinase, plays critical roles in cell regulation. ERK is activated through phosphorylation by a cascade of protein kinases including MEK. Various ligands activate the MEK/ERK pathway through receptor-dependent cell signaling. In cultured cells, many ligands such as growth factors, hormones, cytokines and vasoactive peptides elicit transient activation of MEK/ERK, often peaking at ~10 min after the cell treatment. Here, we describe a novel biological event, in which ligand-mediated cell signaling results in the dephosphorylation of MEK/ERK. Neuromedin N and neurotensin, peptides derived from the same precursor polypeptide, elicit cell signaling through the neurotensin receptors. In cultured human pulmonary artery smooth muscle cells (PASMCs), but not in human pulmonary artery endothelial cells (PAECs), we found that both neuromedin N and neurotensin promoted the dephosphorylation of ERK and MEK. Human PASMCs were found to express neurotensin receptor (NTR)-1, −2 and −3, while human PAECs only express NTR3. Neuromedin N-mediated dephosphorylation was suppressed by small chemical inhibitors of protein phosphatase 1/2A and peptidyl-prolyl isomerase. Transmission electron microscopy showed the formation of endocytic vesicles in response to neuromedin N treatment, and dephosphorylation did not occur when sorting nexin 9, a critical regulator of the endocytic vesicle formation, was knocked down. We conclude that neuromedin N and neurotensin elicit a unique dephosphorylation signaling in the MEK/ERK pathway that is regulated by endocytosis. Considering the pathophysiological importance of the MEK/ERK pathway, this discovery of the dephosphorylation mechanism should advance the field of cell signaling.
Keywords: Dephosphorylation, endocytosis, MAP kinase, neuromedin N, neurotensin
Introduction
Extracellular signal-regulated kinase (ERK), also known as classical mitogen-activated protein kinase (MAPK), plays critical roles in cell signaling events such as cellular proliferation, differentiation and neuronal plasticity [1]. ERK is activated through phosphorylation by a cascade of other kinases including MEK MAPK kinases. Various ligands activate the MEK/ERK pathway through receptor-dependent cell signaling [2]. In cultured cells, ligands such as growth factors, hormones, cytokines and vasoactive peptides elicit transient phosphorylation and activation of MEK and ERK, often peaking at ~10 min after cell treatment [3,4].
Neuromedin N and neurotensin are peptides derived from the same precursor polypeptide and interact with the neurotensin receptors [5]. Neurotensin as well as other neuropeptides have shown to activate MAPK in various cell types [6–8]. However, characteristics of MAPK activating pathways are highly dependent on cell and the receptor types [9]. Effects of these peptides in vascular smooth muscle cell MAPK systems are unknown. We initially hypothesized that, like many ligands including vasoactive peptides [3,10], neuromedin N and neurotensin may cause the activation of ERK. To test this, we treated cultured human pulmonary artery smooth muscle cells (PASMCs) with neuromedin N or neurotensin.
Surprisingly, we discovered that these peptides caused the dephosphorylation of ERK and MEK in these cells. To our knowledge, this is a unique ligand-mediated dephosphorylation-signaling event and is the first demonstration of ligand-mediated dephosphorylation of MAPKs. The present study reports this interesting and unique ligand-mediated dephosphorylation signaling process and further provides mechanistic insights that may involve endocytosis.
Materials and methods
Cell culture
Human PASMCs and human pulmonary artery endothelial cells (PAECs) were purchased from ScienCell Research Laboratories (Carlsbad, CA) and Cell Applications, Inc. (San Diego, CA) and were cultured in accordance with the manufacturers’ instructions in 5% CO2 at 37°C. Experimental results were confirmed in cells from multiple donors. Cells in passages 3–6 were used. Cells were grown in low fetal bovine serum (0.4%)-containing medium overnight before the treatment with ligands, neuromedin N (MedChem Express, Monmouth Junction NJ, USA) and neurotensin (Sigma-Aldrich, St. Louis, MO, USA).
siRNA experiments were performed using the Santa Cruz Biotechnology (Dallas, TX, USA) system in accordance with the manufacture’s instructions. Briefly, cells grown on 6-well plates were incubated with 1 μg siRNA and siRNA Transfection Reagent in siRNA Transfection Medium for 5 hours. Equal volume of growth medium containing 2 times the normal serum, growth supplements and antibiotics were added and cells were grown for 2 days. Cells were then maintained in 0.4% fetal bovine serum-containing medium overnight before the treatment.
Western blotting
Cell lysates and protein gel electrophoresis samples were prepared as previously described [11]. Equal protein amounts of samples were electrophoresed through a reducing SDS polyacrylamide gel and electroblotted onto a nitrocellulose membrane. Membranes were blocked and incubated with antibodies for pRaf-1 (S338; Catalog # 9427), pERK1/2 (T202/Y204; Catalog # 4370), pMEK1/2 (S217/221; Catalog # 9154), pStat3 (Y705; Catalog # 9145), phospho-threonine (Catalog # 9381), ERK1/2, MEK1/2 (Cell Signaling Technology, Danvers, MA, USA), neurotensin receptor (NTR)-1, NTR2, NTR3 and glutaraldehyde-3-phosphate-dehydrogenase (G3PDH) (Santa Cruz Biotechnology). Levels of proteins were detected by using horseradish peroxidase-linked secondary antibodies and an Enhanced Chemiluminescence System (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). Autoradiography was performed using UltraCruz Autoradiography Films (Santa Cruz Biotechnology). The developed films were scanned and optical densities of protein bands were quantified using NIH ImageJ.
Animal experiments
To promote pulmonary arterial hypertension, the SU5416/hypoxia model with pathologic features similar to those in humans was used [11]. Male Sprague-Dawley CD rats (Charles River Laboratories, Wilmington, MA, USA) were subcutaneously injected with 20-mg/kg-body weight SU5416 (MedChem Express), maintained in hypoxia for 3 weeks, then in normoxia for 5 weeks. Animals were subjected to sustained hypoxia in a chamber regulated by an OxyCycler Oxygen Profile Controller (BioSpherix, Redfield, NY, USA) that maintains 10% O2 with an influx of N2 gas. Georgetown University Animal Care and Use Committee approved all animal experiments. The investigation conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Histological measurements
Lung tissues from rats were immersed in buffered 10% formalin at room temperature, and were embedded in paraffin. Paraffin-embedded tissues were cut and mounted on glass slides. Tissue sections were subjected to immunohistochemistry using anti-neurotensin receptor 1 (Catalog # ab217134, Abcam, Cambridge, UK) and anti-neurotensin receptor 2 (Catalog # ab188910, Abcam) antibodies.
Transmission electron microscopy
Cells were fixed in the 2.5% glutaraldehyde/0.05 M cacodylate solution, post-fixed with 1% osmium tetroxide, and embedded in EmBed812. Ultrathin sections (70 nm) were post-stained with uranyl acetate and lead citrate and examined in the Talos F200X FEG transmission electron microscope (FEI Thermo Fisher Scientific, Hillsboro, OR, USA) at 80 KV located at the George Washington University Nanofabrication and Imaging Center. Digital electron micrographs were recorded with the TIA software (FEI).
Statistical analysis
Means and standard errors were calculated. Comparisons between two groups were analyzed by using a two-tailed Student’s t test and comparisons between three or more groups were analyzed by using one-way analysis of variance (ANOVA). P < 0.05 was considered to be significant.
Results
Neuromedin N and neurotensin trigger dephosphorylation of ERK and MEK in vascular smooth muscle cells
We initially tested the hypothesis that, like many other vasoactive peptides [3,10], neuromedin N and neurotensin activate ERK in pulmonary vascular smooth muscle cells. By using the antibody that detects phosphorylation of T202/Y204 residues of ERK that depicts the activation of this kinase, we surprisingly found that a 30-min treatment of human PASMCs with neuromedin N or neurotensin decreased the phosphorylation state of ERK (Fig. 1A). By contrast, neither neuromedin N nor neurotensin influenced ERK phosphorylation in human PAECs under conditions, in which these peptides triggered ERK dephosphorylation in PASMCs (Fig. 1B).
Fig. 1: Neurotensin (NT) and neuromedin N (NN) promote protein dephosphorylation of ERK MAP kinase in human PASMCs.
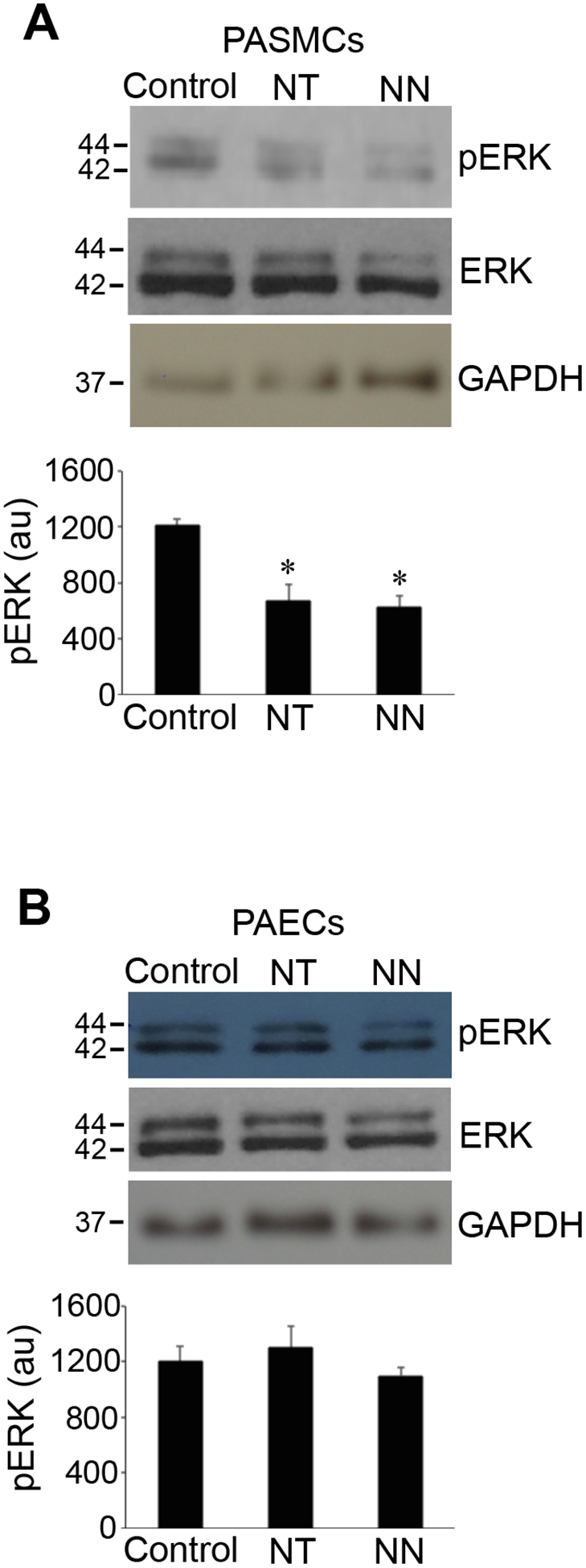
(A) Cultured human PASMCs or (B) human PAECs were treated with NT or NN (400 nM) for 30 min. Cell lysates were prepared and phosphorylated ERK (pERK) levels were monitored by Western blotting using the pERK1/2 (T202/Y204) antibody. Bar graphs represent means ± SEM of pERK levels determined by densitometry expressed in arbitrary unit (au). *Significantly different from the control value. (N=4 for PASMCs; N=3 for PAECs).
The time course study demonstrated that neuromedin N time-dependently caused dephosphorylation of ERK at 30 and 60 min (Figs. 2A & 2B). Similar results were obtained when we monitored the phosphorylation of MEK, which phosphorylates ERK, suggesting that the effects of neuromedin N occurs upstream of MEK phosphorylation (Figs. 2A & 2B). By contrast, phosphorylation of Raf-1, which phosphorylates MEK was not influenced by neuromedin N (Figs. 2A & 2B). Neuromedin N-mediated dephosphorylation appears to occur specifically in the MEK/ERK pathway, as the phosphorylation of Stat3 (Figs. 2A & 2B) or the global threonine phosphorylation (Fig. 2C) were not affected by neuromedin N. Similarly to previous reports showing that neurotensin activates ERK in some cell lines, neuromedin N promoted phosphorylation of MEK and ERK in HeLa cells (Fig. 2D).
Fig. 2: Dephosphorylation of MEK MAP kinase kinase.
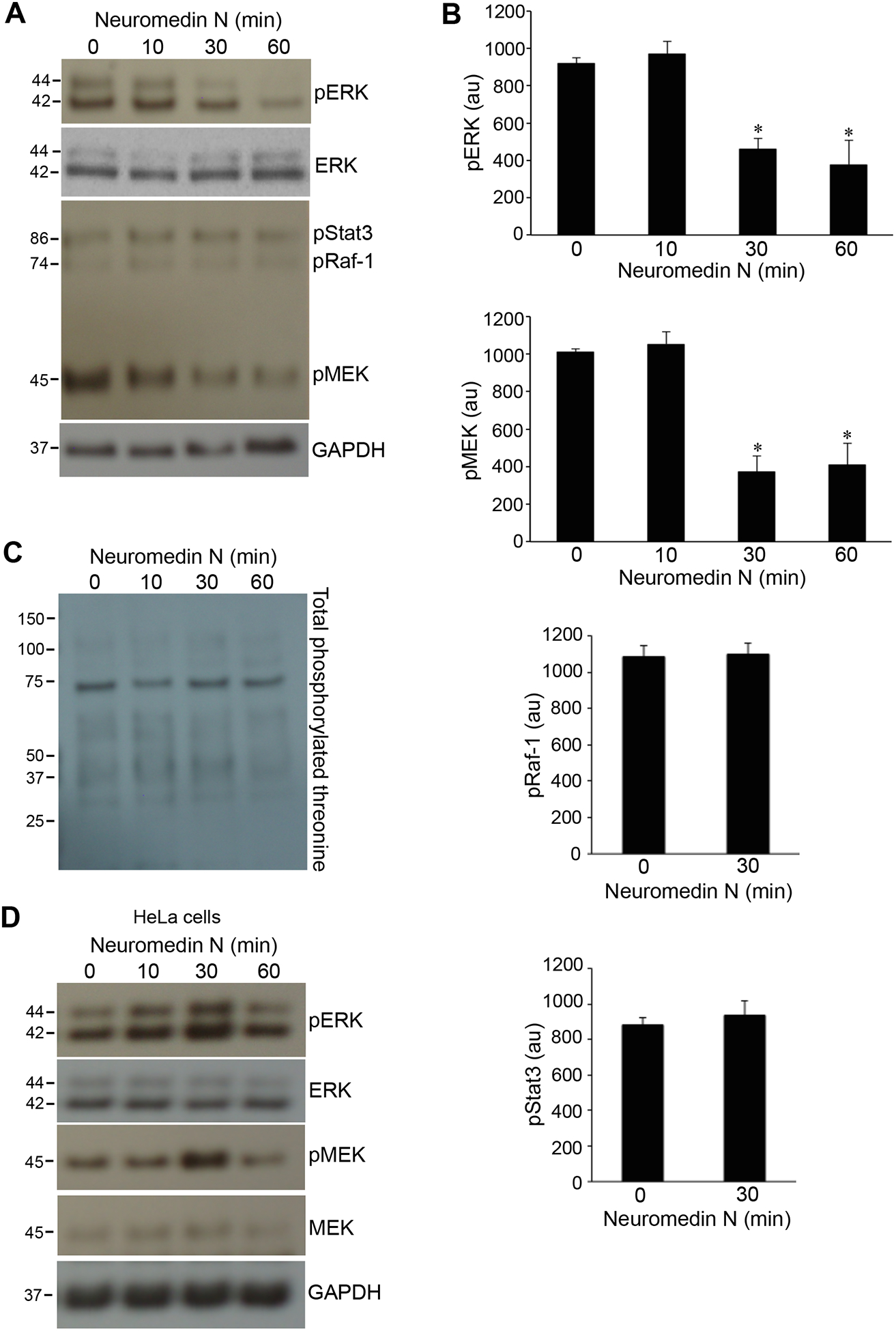
(A) Human PASMCs were treated with neuromedin N (400 nM) for various durations, cell lysates were prepared and phosphorylated forms of ERK (pERK), Stat3 (pStat3), Raf-1 (pRaf-1) and MEK (pMEK) were monitored by Western blotting using pERK1/2 (T202/Y204), pStat3 (Y705), pRaf-1 (S338) and pMEK1/2 (S217/221) antibodies. (B) Bar graphs represent means ± SEM of phospho-protein levels determined by densitometry expressed in arbitrary unit (au). * denotes significant difference from 0 min control at p < 0.01. N=6 (for pERK and pMEK); N=3 (for pRaf-1); N=5 (for pStat3). (C) Total threonine phosphorylation was monitored by Western blotting using the phosphorylated threonine antibody in the cell lysates of human PASMCs treated with neuromedin N (400 nM). (D) HeLa cells were treated with neuromedin N (400 nM) for various durations, cell lysates were prepared and pERK and pMEK were monitored by Western blotting.
Calyculin A, a small chemical inhibitor of protein phosphatases type 1 and/or 2B, promoted MEK phosphorylation and inhibited the neuromedin N-mediated dephosphorylation (Fig. 3). Juglone, a small chemical inhibitor of peptidyl-prolyl cis/trans isomerase, also exhibited similar effects as calyculin A (Fig. 3).
Fig. 3: Effects of calyculin A and juglone on neuromedin N-mediated MEK dephosphorylation.
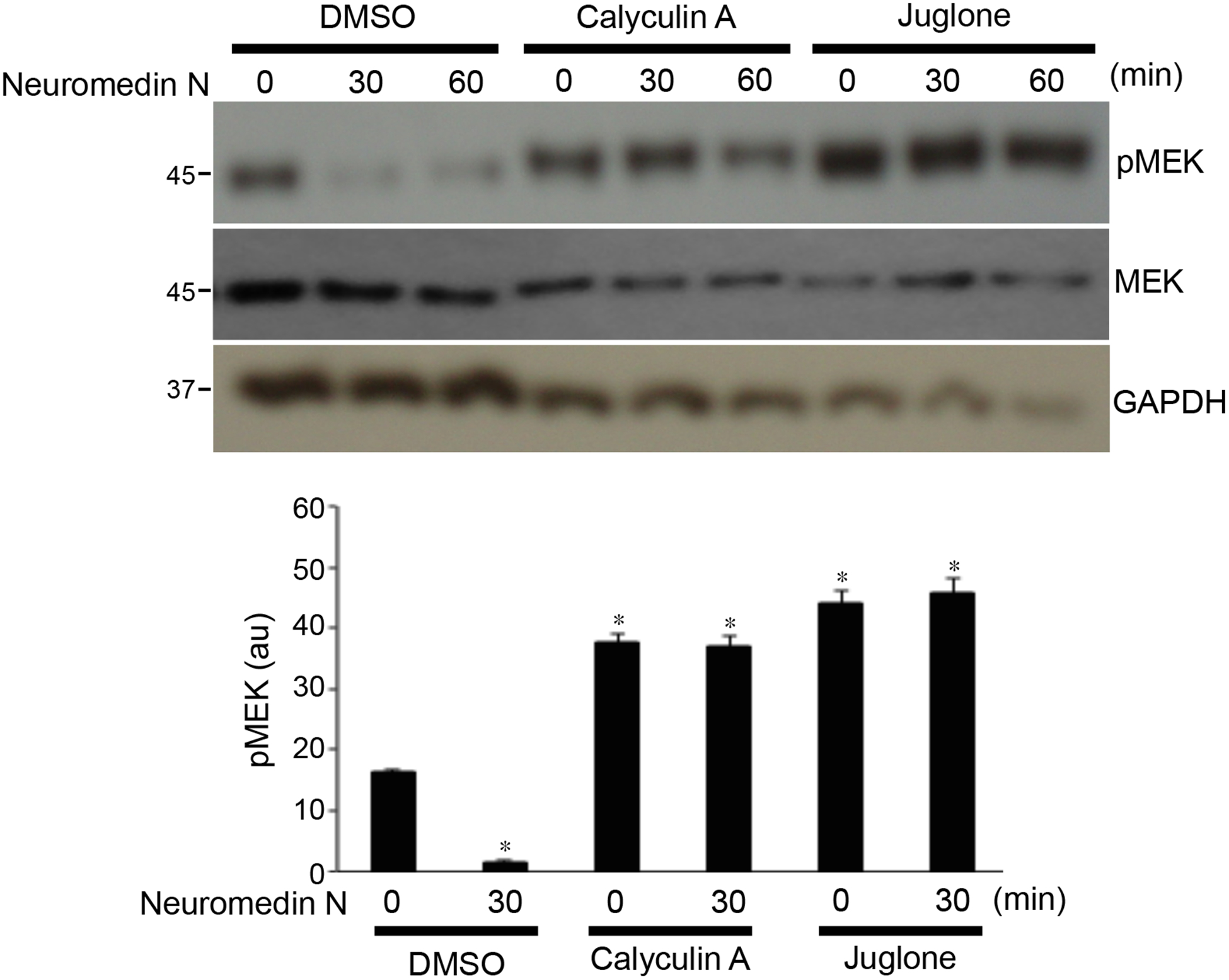
Human PASMCs were pre-treated with calyculin A (protein phosphatase type 1 and 2A inhibitor; 50 ng/ml) or juglone (peptidyl-prolyl cis/trans isomerase inhibitor; 20 μM) for 30 min, then treated with neuromedin N for 30 or 60 min. Phosphorylated MEK (pMEK) levels were monitored by Western blotting using the pMEK1/2 (S217/221) antibody. The bar graph represents means ± SEM of pMEK levels determined by densitometry expressed in arbitrary unit (au). * denotes significant difference from control at p < 0.01 (N=3).
Both neuromedin N and neurotensin are known to elicit signal transduction through binding to the neurotensin receptors [5]. Our experiments revealed that smooth muscle cells and endothelial cells exhibit different expression patterns of neurotensin receptor types. Human PASMCs were found to express NTR1, NTR2 and NTR3, while the predominant receptor in human PAECs was found to be NTR3 (Fig. 4A).
Fig. 4: Expression of neurotensin receptors (NTR) in pulmonary vascular cells.
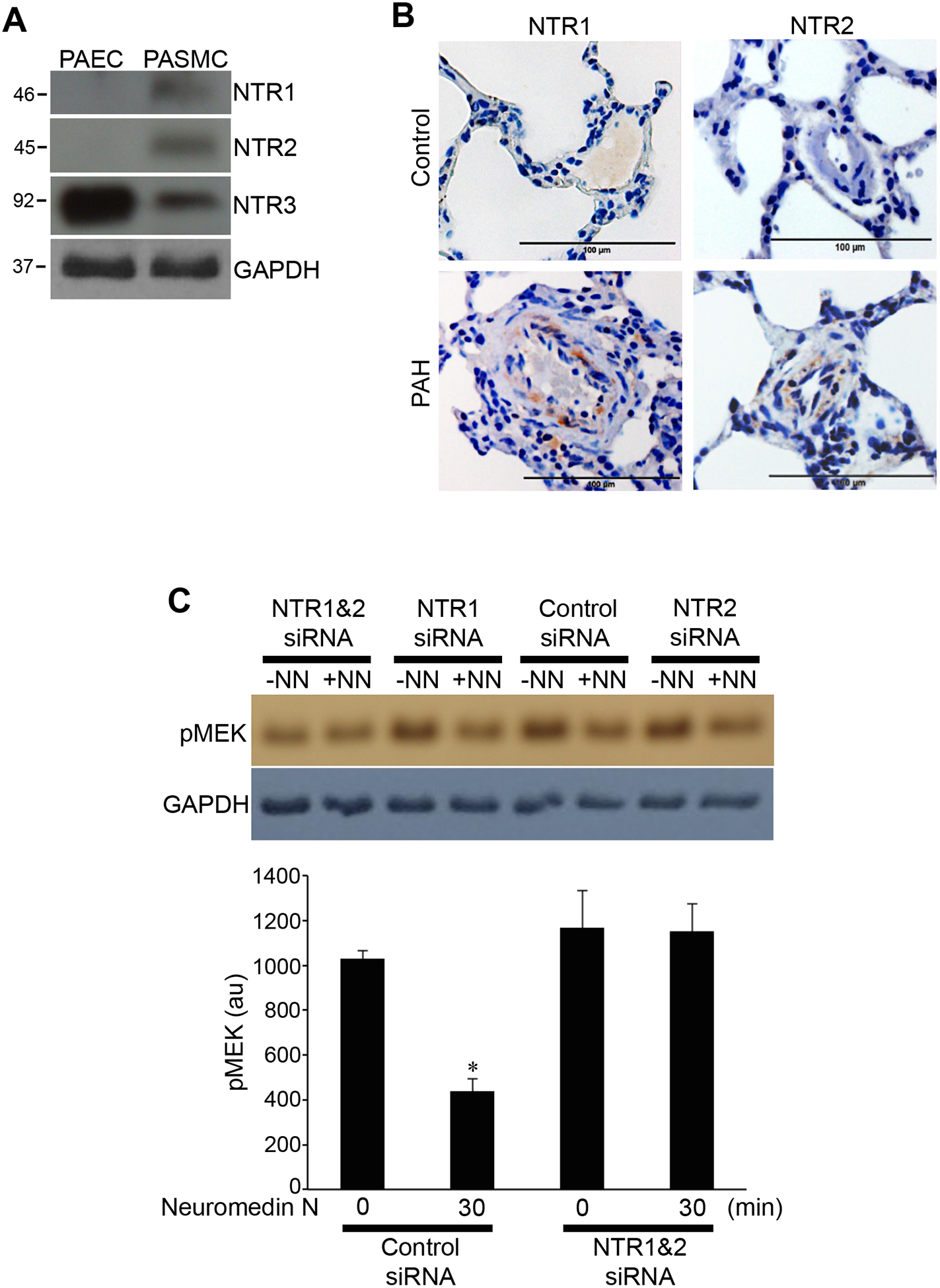
(A) Expression of NTR types 1, 2 and 3 (NTR1, 2 and 3) were monitored in cell lysates from untreated human PAECs and PASMCs by Western blotting. (B) Lung tissue sections from healthy control rats and rats with pulmonary arterial hypertension (PAH) were subjected to immunohistochemistry using antibodies for NTR1 and NTR2. Representative results of experiments with 5 rats per group are shown. (C) Human PASMCs were treated with control scrambled siRNA or siRNA to knockdown both NTR1 and NTR2. Three days later, cells were treated with neuromedin N (NN; 400 nM) for 30 min. Cell lysates were subjected to Western blotting using the pMEK1/2 (S217/221) antibody to monitor phosphorylated MEK (pMEK). The bar graph represents means ± SEM of pMEK levels determined by densitometry expressed in arbitrary unit (au). * denotes significant difference from untreated control at p < 0.05 (N=3).
Immunohistochemistry also detected the expression of NTRs (NTR1 and NTR2) in the medial smooth muscle layer of the pulmonary arteries in rats with pulmonary arterial hypertension, but not in control rats (Fig. 4B).
In cultured human PASMCs, knocking down both NTR1 and NTR2 by siRNA inhibited neuromedin N-induced MEK phosphorylation (Fig. 4C).
Neuromedin N-mediated protein dephosphorylation is mediated through endocytic vesicles
Transmission electron microscopy was used to visualize the cellular structure of human PASMCs that were untreated, treated with neuromedin N for 10 min and for 30 min. We found a dramatic increase in the number of endocytic vesicles near the plasma membrane of cells treated with neuromedin N when the dephosphorylation of MEK occurred. As shown in Fig. 5A, in untreated cells, endocytic vesicles were found with varied sizes ranging from 0.05 to 1.2 μm (arrows). Our analysis determined that the number of endocytic vesicles in untreated cell was 79 in a given field. The treatment of cells with neuromedin N for 10 min did not significantly alter these properties (arrow) with similar sizes and the number (107 vesicles). However, after 30 min treatment with neuromedin N, the dramatic increase of number of endocytic vesicles was observed, with most having uniform diameters of 0.08 −1.0 μm (arrows). The number of the vesicles also dramatically increased to 231 in cells treated with neuromedin N for 30 min. The increase in the endocytic vesicle formation did not occur in response to 10 or 30 min treatment of human PASMCs with platelet-derived growth factor that activates the phosphorylation of MEK and ERK (data not shown).
Fig. 5: Transmission electron microscopy studies of PASMCs treated with neuromedin N.
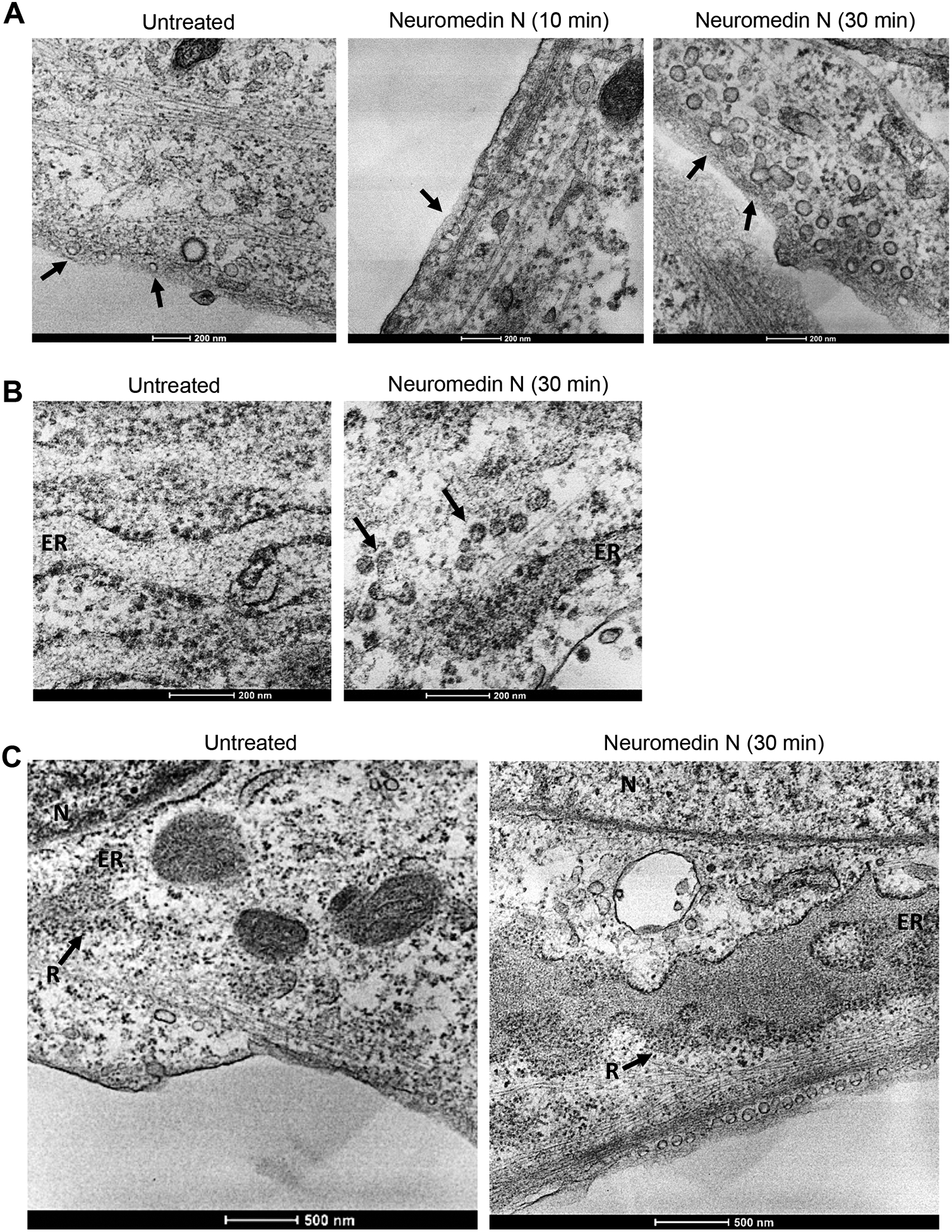
Human PASMCs were treated with neuromedin N (400 nM). Fix cells were subjected to transmission electron microscopy examinations. (A) Time course of 0, 10 and 30 min. Arrows indicate endocytic vesicles. (B) Transmission electron microscopy images showing neuromedin N-triggered endocytic vesicles (arrows) traveled to the endoplasmic reticulum (ER) cisternae at 30 min. Magnification x 26,000. (C) The transmission electron microscopy images of a larger field to indicate the locations of endocytic vesicles, the enlargement of ER-cisternae along with an increasing number of ribosomes of untreated cell and in the cell treated with neuromedin N for 30 min. N, nucleus. ER, endoplasmic reticulum. R, ribosomes. Magnification x 13,000.
Fig. 5B shows that endocytic vesicles occur near the endoplasmic reticulum (ER) in cells treated with neuromedin N for 30 min (arrows). The enlargement of ER-cisternae along with an increasing number of ribosomes (R) after 30 min treatment with neuromedin N is also evident (Fig. 5C). These results suggest that neuromedin N activates the endocytic pathway from the plasma membrane to the ER.
SNX9 is involved in endocytosis and intracellular endocytic vesicle trafficking [12,13]. To determine the role of endocytic vesicles in neuromedin N-mediated MEK dephosphorylation, we tested the effects of siRNA knockdown of SNX9. We found that SNX9 knockdown inhibited neuromedin N-mediated MEK dephosphorylation (Figs. 6A). By contrast, knocking down SNX1 or SNX5 had no effects (Fig. 6B). Transmission electron microscopy confirmed that neuromedin N-induced endocytic vesicle formation was inhibited by knocking down SNX9 (Fig. 6C).
Fig. 6: Effects of siRNA knockdown of sorting nexin 9 (SNX9).
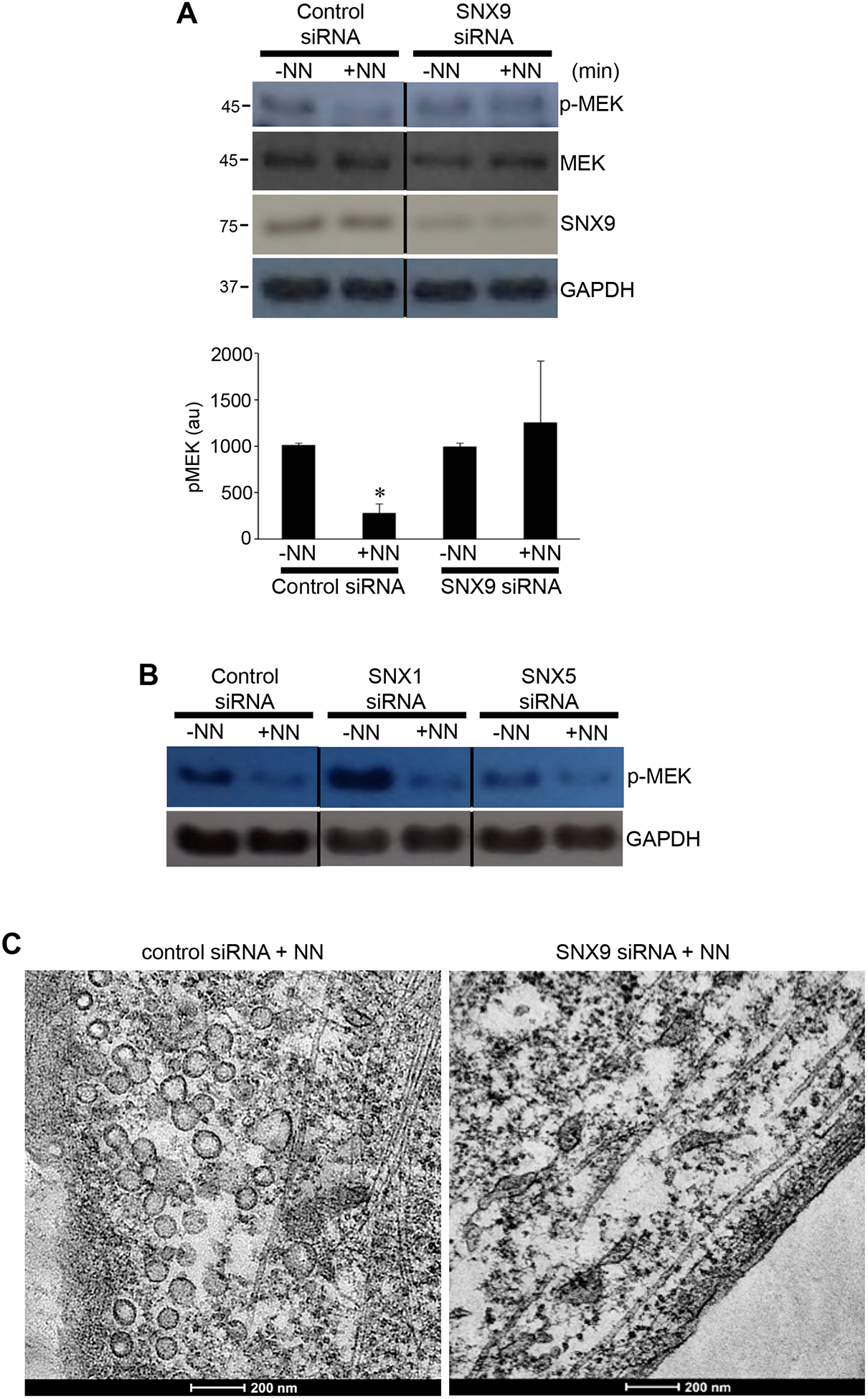
(A) Human PASMCs were treated with control scrambled siRNA or SNX9 siRNA. Three days later, cells were treated with neuromedin N (NN; 400 nM) for 30 min. Cell lysates were subjected to Western blotting using the pMEK1/2 (S217/221) antibody to monitor phosphorylated MEK (pMEK) as well as to assess the extent of SNX9 knockdown using the SNX9 antibody. The bar graph represents means ± SEM of pMEK levels determined by densitometry expressed in arbitrary unit (au). * denotes significant difference from untreated control at p < 0.05 (N=3). (B) Human PASMCs were treated with control scrambled siRNA, SNX1 siRNA or SNX5 siRNA. Three days later, cells were treated with NN. Cell lysates were subjected to Western blotting to monitor pMEK. (C) Human PASMCs were treated with SNX9 siRNA. Three days later, cells were treated with NN. Fix cells were subjected to transmission electron microscopy examinations.
Discussion
The major finding of this study is that the treatment of human vascular smooth muscle cells with neurotensin receptor agonists elicits cell signaling, resulting in dephosphorylation of MEK and ERK. To our knowledge, this is the first demonstration of the existence of ligand-mediated dephosphorylation signaling involving MAP kinase pathways. Mechanistically, we also demonstrated a novel cell-signaling event, in which the formation of endocytic vesicles mediates this dephosphorylation signaling.
In this study, we used human PASMCs as a model of vascular smooth muscle cells. The growth of PASMCs by various ligands serves as a critical event for the pathogenesis of pulmonary hypertension [14]. The MEK/ERK signaling pathway has also been attributed as a major mechanism of cell growth in this pathological condition as well as many diseases involving cell growth [15]. Thus, the inhibitory signaling process that is triggered by the natural neurotensin receptor agonists as presented in this study may have clinical significance. It is interesting to note that the signaling mechanism described in this study is specific to smooth muscle cells but does not occur in endothelial cells or HeLa cells. This cell type specificity may be defined by differential expression patterns of neurotensin receptors.
Protein phosphorylation is defined by the balance between phosphorylation by protein kinases and dephosphorylation by protein phosphatases. ERK MAP kinase is activated by the phosphorylation of T202 and Y204 residues by MEK MAP kinase kinase [16,17]. MEK is activated by the phosphorylation of two serine residues at 217 and 221 positions by Raf-1 MAP kinase kinase kinase [18]. Dephosphorylation of ERK by neurotensin and neuromedin N as shown in this study is likely a consequence of inhibited MEK activity due to the dephosphorylation of MEK. Raf-1 is an effector recruited by GTP-bound Ras for the activation of MEK/ERK pathway [19]. From our data, it is yet unclear if the signaling mechanism that is inhibited in response to neurotensin/neuromedin N is the phosphorylation of MEK or upstream events. Our experiments using calyculin A indicate the possible role of protein phosphatase type 1 and 2A in the mechanism of neurotensin/neuromedin N-mediated dephosphorylation signaling. Our study also presents results possibly linking the role of peptidyl-prolyl isomerase in the regulation of this dephosphorylation signaling event since juglone, that is capable of inhibiting this isomerase that defines the cis/trans state of proline residues [20], mimicked the action of calyculin A by inhibiting the dephosphorylation by neurotensin and neuromedin N. It has been reported that the protein phosphatase 2A activator is a peptidyl-prolyl isomerase [21]. Small chemical inhibitors can, however, exert non-specific effects, thus future studies are needed to more precisely define the protein phosphatase systems involved in this signaling mechanism.
In an attempt to define the mechanism of neuromedin N/neurotensin-mediated protein dephosphorylation, we employed transmission electron microscopy to study how cellular structures may change in response to the neuromedin N treatment of PASMCs. These experiments led us to fascinating observations that neuromedin N promotes the formation of endocytic vesicles from the plasma membrane and their possible transport routes include the caveolae-caveosome-ER pathway [22–24]. siRNA knockdown of a major regulator of endocytic vesicle formation, SNX9, demonstrated that endocytosis indeed regulates MEK dephosphorylation triggered by neuromedin N.
Conclusions
While further work is needed to elucidate precise mechanisms of this novel ligand-mediated protein dephosphorylation process, the present study provided evidence for a new cell signaling pathway, in which neuromedin N/neurotensin signaling activating SNX9-dependent endocytosis that, in turn, promotes dephosphorylation of MEK1/2. The reduction in the protein phosphorylation state by this ligand/receptor system may have pathophysiological and clinical significance, considering the crucial roles the MEK/ERK signaling pathway plays in the biological system.
Acknowledgments
This work was performed in part at the George Washington University Nanofabrication and Imaging Center (GWNIC).
Sources of Funding
This work was supported by the National Institutes of Health [R01HL72844] to YJS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- [1].Thomas G, Huganir RL, MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci 5 (2004) 173–183. [DOI] [PubMed] [Google Scholar]
- [2].Chung E, Kondo M, Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol. Res 49 (2011) 248–268. [DOI] [PubMed] [Google Scholar]
- [3].Arsenijevic T, Gregoire F, Chiadak J, Courtequisse E, Bolaky N, Perret J, Delporte C, Pituitary adenylate cyclase activating peptide (PACAP) participates in adipogenesis by activating ERK signaling pathway. PLoS ONE. 8 (2013) e72607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pratsinis H, Kletsas D, PDGF, bFGF and IGF-I stimulate the proliferation of intervertebral disc cells in vitro via the activation of the ERK and Akt signaling pathways. Eur. Spine J 16 (2007) 1858–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vincent JP, Mazella J, Kitabgi P, Neurotensin and neurotensin receptors. Trends Pharmacol. Sci 20 (1999) 302–309. [DOI] [PubMed] [Google Scholar]
- [6].Mannon PJ, Raymond JR, The neuropeptide Y/peptide YY Y1 receptor is coupled to MAP kinase via PKC and Ras in CHO cells. Biochem. Biophys. Res. Commun 246 (1998) 91–94. [DOI] [PubMed] [Google Scholar]
- [7].Poinot-Chazel C, Portier M, Bouaboula M, Vita N, Pecceu F, Gully D, Monroe JG, Maffrand JP, Le Fur G, Casellas P, Activation of mitogen-activated protein kinase couples neurotensin receptor stimulation to induction of the primary response gene Krox-24. Biochem. J 320 (1996) 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Seufferlein T, Rozengurt E, Galanin, neurotensin, and phorbol esters rapidly stimulate activation of mitogen-activated protein kinase in small cell lung cancer cells. Cancer Res. 56 (1996) 5758–5764 [PubMed] [Google Scholar]
- [9].Liebmann C, Regulation of MAP kinase activity by peptide receptor signalling pathway: paradigms of multiplicity. Cell. Signal 13 (2001) 777–785. [DOI] [PubMed] [Google Scholar]
- [10].Chen Q, Edvinsson L, Xu CB, Role of ERK/MAPK in endothelin receptor signaling in human aortic smooth muscle cells. BMC Cell Biology 10 (2009) 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ibrahim YF, Wong CM, Pavlickova L, Liu L, Trasar L, Bansal G, Suzuki YJ, Mechanism of the susceptibility of remodeled pulmonary vessels to drug-induced cell killing. J. Am. Heart Assoc 3 (2014) e000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lundmark R, Carlsson SR, Sorting nexin 9 participates in clathrin-mediated endocytosis through interactions with the core components. J. Biol. Chem 278 (2003) 46772–46781. [DOI] [PubMed] [Google Scholar]
- [13].Bendris N, Schmid SL, Endocytosis, metastasis and beyond: Multiple facets of SNX9. Trends Cell Biol. 27 (2017) 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol 54 (2009) S20–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Roberts RE, The extracellular signal-regulated kinase (ERK) pathway: a potential therapeutic target in hypertension. J. Exp. Pharmacol 4 (2012) 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Murphy LO, Blenis J, MAPK signal specificity: the right place at the right time. Trends Biochem. Sci 31 (2006) 268–275. [DOI] [PubMed] [Google Scholar]
- [17].Rubinfeld H, Seger R, The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol. 31 (2005) 151–174. [DOI] [PubMed] [Google Scholar]
- [18].Alessi DR, Saito Y, Campbell DG, Cohen P, Sithanandam G, Rapp U, Ashworth A, Marshall CJ, Cowley S, Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 13 (1994) 1610–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Avruch J, Zhang XF, Kyriakis JM, Raf meets Ras: completing the framework of a signal transduction pathway. Trends Biochem. Sci 19 (1994) 279–283. [DOI] [PubMed] [Google Scholar]
- [20].Chao SH, Greenleaf AL, Price DH, Juglone, an inhibitor of the peptidyl-prolyl isomerase Pin1, also directly blocks transcription. Nucleic Acids Res. 29 (2001) 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jordens J, Janssens V, Longin S, Stevens I, Martens E, Bultynck G, Engelborghs Y, Lescrinier E, Waelkens E, Goris J, Van Hoof C, The protein phosphatase 2A phosphatase activator is a novel peptidyl-prolyl cis/trans-isomerase. J. Biol. Chem 281 (2006) 6349–6357. [DOI] [PubMed] [Google Scholar]
- [22].Kiss AL, Botos E. Endocytosis via caveolae: alternative pathway with distinct cellular compartments to avoid lysosomal degradation? J. Cell Mol. Med 13 (2009) 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Norkin LC, Kuksin D. The caveolae-mediated sv40 entry pathway bypasses the golgi complex en route to the endoplasmic reticulum. Virol. J 2 (2005) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xu S, Olenyuk BZ, Okamoto CT, Hamm-Alvarez SF. Targeting receptor-mediated endocytotic pathways with nanoparticles: rationale and advances. Adv. Drug Deliv. Rev 65 (2013) 121–138. [DOI] [PMC free article] [PubMed] [Google Scholar]