

Supplemental Digital Content is available in the text.
Keywords: exome, genetic counseling, human, population health, syncope
Background:
In population-based research exome sequencing, the path from variant discovery to return of results is not well established. Variants discovered by research exome sequencing have the potential to improve population health.
Methods:
Population-based exome sequencing and agnostic ExWAS were performed 5521 Amish individuals. Additional phenotyping and in vitro studies enabled reclassification of a KCNQ1 variant from variant of unknown significance to pathogenic. Results were returned to participants in a community setting.
Results:
A missense variant was identified in KCNQ1 (c.671C>T, p.T224M), a gene associated with long QT syndrome type 1, which can cause syncope and sudden cardiac death. The p.T224M variant, present in 1/45 Amish individuals is rare in the general population (1/248 566 in gnomAD) and was highly associated with QTc on electro-cardiogram (P=5.53E-24, β=20.2 ms/allele). Because of the potential importance of this variant to the health of the population, additional phenotyping was performed in 88 p.T224M carriers and 54 noncarriers. There was stronger clinical evidence of long QT syndrome in carriers (38.6% versus 5.5%, P=0.0006), greater history of syncope (32% versus 17%, P=0.020), and higher rate of sudden cardiac death in first degree relatives<age 30 (4.5% versus 0%, P=0.026). Expression of p.T224M KCNQ1 in Chinese hamster ovary cells showed near complete loss of protein function. Our clinical and functional data enabled reclassification of p.T224M from a variant of unknown significance to pathogenic. Of the 88 carriers, 93% met criteria for beta-blocker treatment and 5/88 (5.7%) were on medications that may further prolong QTc. Carriers were provided a Clinical Laboratory Improvement Amendments confirmed report, genetic counseling, and treatment recommendations. Follow-up care was coordinated with local physicians.
Conclusions:
This work provides a framework by which research exome sequencing can be rapidly translated in a culturally appropriate manner to directly benefit research participants and enable population precision health.
When exome sequencing (ES) is performed in a clinical setting, information that should be returned to the patient is generally clear.1,2 However, when ES is performed in research settings, as in population-based exome-wide association studies, the issue of return of results to study participants is far less settled. Research return of results for actionable findings is not currently obligatory, although increasingly recommended and being performed.3–5 Many ethical issues have been raised about research return of result,4,6 including the ethical obligations of investigators to return research results to study participants (whether or not this was anticipated in the Informed Consent), whether study participants should have the option to opt out of receiving actionable research findings, and implications to family members who may not be research participants.5,7,8 Furthermore, there are nontrivial practical and logistical issues and most research grants do not anticipate or provide funding for research return of result.9–11
Despite these issues, research return of result has the potential to positively impact the health of research participants, family members, and the population at large. These benefits may be magnified in founder populations in which pathogenic variants may occur at much higher frequency than in the general population due to genetic drift, the process whereby allele frequencies within a population change by chance over generations. In founder populations with high fecundity, genetic drift can lead to large changes in allele frequencies over a relatively short period of time.
As part of a large ongoing research program in the Old Order Amish (Amish) population from Lancaster County, Pennsylvania, we performed population-based ES of 5521 individuals. Exome-wide association studies identified a highly drifted variant in KCNQ1 (potassiium voltage-gated channel subfamily Q member; hg38.g.chr11:2571391[C>T]; c.671C>T; p.T224M; rs199472706), present in 1 in 45 Amish and significantly associated with increased electro-cardiogram (EKG)-derived QTc (QT corrected for heart rate) interval.
KCNQ1 encodes the potassium voltage-gated channel subfamily Q member 1. Pathogenic mutations in KCNQ1 cause long QT syndrome (LQTS) type 1 (LQT1, MIM No. 192500), an autosomal dominant disorder that increases the risk for syncope and sudden cardiac death (SCD).12–14 In individuals of European descent, the prevalence of LQTS is estimated to be ≈1:2500, with LQTS1 being the most common cause of this cardiac conduction disorder.15 KCNQ1 is included in the American College of Medical Genetics SF v2.02 list of medically actionable genes. Known pathogenic and likely pathogenic variants are to be reported to patients, when identified through clinical exome or genome sequencing. Beta-blockers are an effective therapeutic intervention in individuals with LQT1 as they may reduce the risk of syncope and sudden death by 70% to 90%.14
p.T224M KCNQ1 was reported previously in only 2 patients with LQTS15,16 and, therefore, was classified as a variant of unknown significance in ClinVar at the start of our study. Since the c.671C>T (p.T224M) variant was highly enriched in the Amish and had a large effect on QTc interval, we performed further clinical phenotyping and in vitro functional studies of this variant to determine its pathogenicity and better assess its possible health risk to the Amish community. We further describe the model we developed for reclassification of this variant to pathogenic and culturally appropriate return of results to individuals with the variant, including providing the option for disclosure of results, genetic counseling, recommendations for beta-blocker treatment, and cascade testing of first degree family members.
Methods
The authors declare that all supporting data are available within the article and in the Data Supplement. The study was approved by the Institutional Review Board of the University of Maryland School of Medicine. Participants signed written informed consent. Because of the sensitive nature of the data collected for this study, requests to access the data set from qualified researchers trained in human subject confidentiality protocols may be sent to The University of Maryland Program for Personalized and Genomic Medicine, Braxton Mitchell PhD at bmitchel@som.umaryland.edu. Additional information on methods can be found in the Data Supplement.
Results
Identification of the c.671C>T (p.T224M) Variant in KCNQ1
Population agnostic ES and exome-wide association studies were performed in 5521 Amish participants from the Amish Complex Disease Research Program (Table I in the Data Supplement). One of the strongest associations found was for EKG QTc interval with a missense variant in KCNQ1 (rs199472706; c.671C>T; p.T224M; P=5.53×10−24; Figure 1A). The p.T224M variant was associated with an average 20.2 ms higher QTc compared with the reference allele (Figure 1C). The p.T224M variant is highly drifted in the Amish, with a frequency of 0.011 (124 carriers among 5521 individuals); 1 in 45 Amish carry this variant in contrast to the general population in which there is only 1 carrier in 248 566 individuals overall and 1 carrier in 112 482 individuals of European descent from gnomAD (v2.1). As above, an additional p.T224M carrier was reported in the literature15 but not found in gnomAD. No p.T224M homozygotes were found, consistent with Hardy-Weinberg expectations (only 0.67 homozygotes would have been expected). The p.T224M variant was not associated at genome wide levels of significance with any other phenotypes present in our database, including cardiovascular risk markers (lipids, coronary calcification, blood pressure, body mass index), general chemistry and hematology studies, and DXA bone density measures. All p.T224M variant carriers were confirmed by Sanger sequencing in the University of Maryland Clinical Laboratory Improvement Amendments/College of American Pathologists-accredited Translational Genomics Laboratory.
Figure 1.
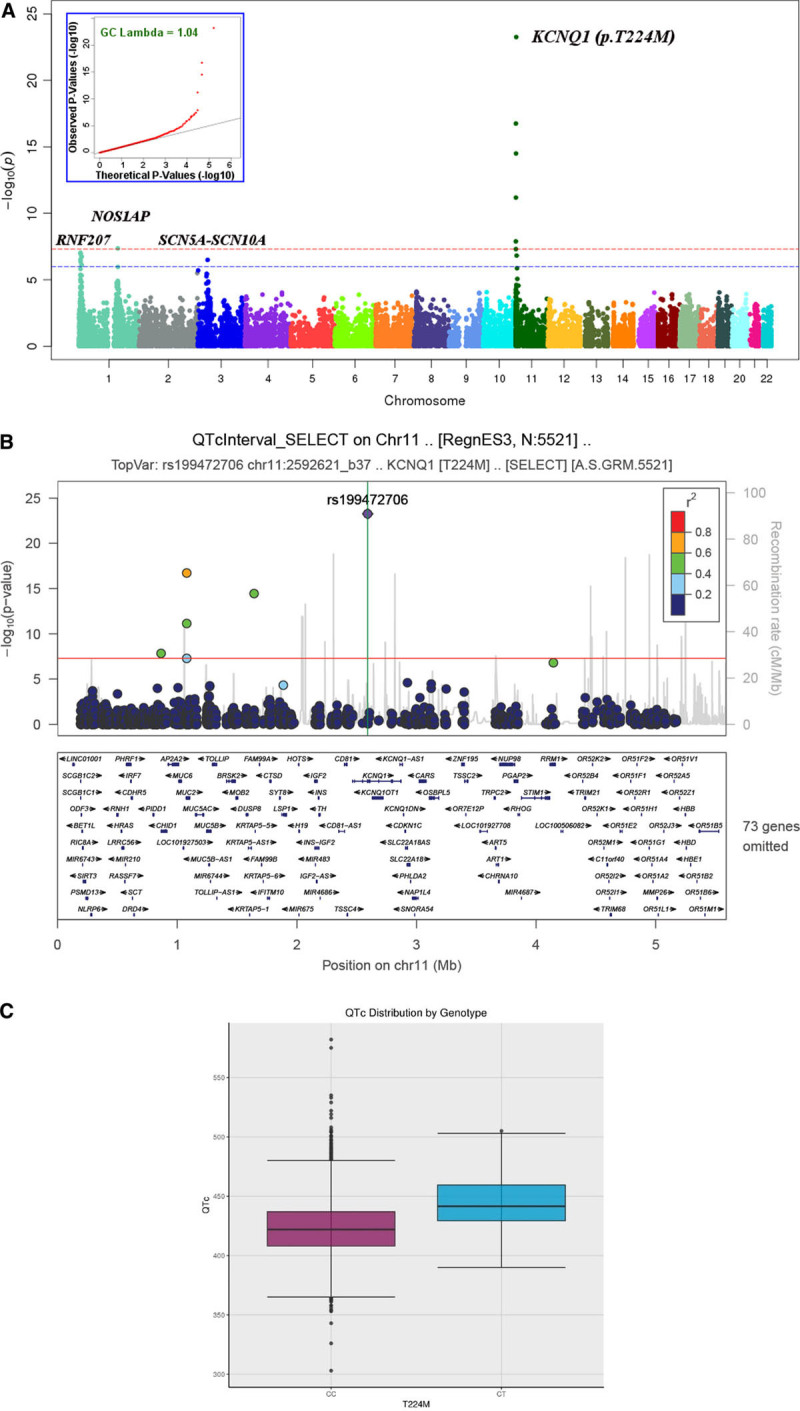
Exome-wide analysis of QTc in 5 521 Amish subjects.A, Manhattan plot, Q-Q plot (insert). The red dotted line represents the threshold for genomewide significance (P<5×10−8) and the blue dotted line represents a threshold of P <1×10−6. B, LocusZoom plot of KCNQ1 region on chromosome 11 showing all variants with P<5×10−8, for our study population. Peak association at p.T224M (rs199472706): Age and sex adjusted β=20.2 msec; P=5.53×10−24. The red dotted line represents the threshold for genomewide significance (P<5×10−8), and the linkage disequilibrium values are computed from the Amish. Recombination rate data are obtained from HapMap and may not necessarily pertain to the Amish. C, Boxplot comparing unadjusted mean QTc between KCNQ1 p.T224M carriers (CT) vs noncarriers (CC).
As indicated in Figure 1B, the KCNQ1 locus includes 5 additional variants showing evidence for association P<5×10−8, all of which are in moderate linkage disequilibrium (r2≥0.28) with rs199472706/p.T224M (r2: 0.41–0.61). Two of these 5 are predicted missense (1 in MUC2 and 1 in KRTAP5-4). Conditional analyses indicated that the association at this locus was likely due to a single variant since all of these associations were no longer statistically significant after accounting for p.T224M (all P>0.36; Table IIA in the Data Supplement).
Single nucleotide polymorphisms at 3 additional loci were identified in our analysis as being significantly or suggestively associated with QTc (Figure 1A). Notably, all 3 loci have been associated with QT interval previously in the QT Interval-International GWAS Consortium (Genome-Wide Association Study).17–19
Variable Expressivity of p.T224M KCNQ1 on QTc
To examine the impact of genetic variation at non-KCNQ1 loci on expressivity of QTc, we computed a polygenic risk score for QTc interval in all study subjects using summary results from 2 prior GWAS17,19 and estimated the correlation of QTc interval with polygenic risk score. In both KCNQ1 carriers and noncarriers, increasing polygenic risk score correlates with increasing QTc interval. The 39 single nucleotide polymorphisms contributing to the polygenic risk score are shown in Table III in the Data Supplement. Further analysis revealed no difference in the magnitude of the slopes between the 2 curves (P=0.36), and thus provided no evidence that the effects of KCNQ1 p.T224M differ between subjects at low and high polygenic risk for increased QTc (Figure I in the Data Supplement). We also tested for interactions between 4 single nucleotide polymorphisms previously reported to modify the effects of KCNQ1 Thr244Met on QTc20–22 and found no evidence for effect modification (Table IV in the Data Supplement).
Clinical Characteristics of p.T224M KCNQ1 Carriers
p.T224M KCNQ1 was classified as a variant of unknown significance in ClinVar. Because of the strong association of this variant with QTc and that it is one of the American College of Medical Genetics 59 actionable variants, we performed additional phenotyping of p.T224M carriers to further assess its pathogenicity and determine its importance to the health of the Amish community. We performed an EKG both supine and within 10 seconds of standing to improve the clinical diagnosis of LQTS,23,24 a full medical history, and a 3 generation family history. Of the 124 carriers identified, 88 consented to follow-up (Figure 2). Details of the follow-up study protocol are included in the Materials and Methods in the Data Supplement. Clinical characteristics are compared between p.T224M carriers and noncarriers in Table and Figure 3. Sex-specific data, stratified by carrier status, are shown in Table V in the Data Supplement. There were slightly more women than men in both carrier and noncarrier groups (Table). Mean and maximal QTc values supine and standing are shown in Table; maximal values in Figure 3. Mean and maximal QTc increased with standing, as expected in all groups, and both mean and maximal QTc were significantly higher in p.T224M carriers than in noncarriers both lying and standing (P<0.0001 for all comparisons; Table). In noncarriers, QTc was higher in females than males, as expected (Figure 3). However, in p.T224M carriers, QTc (both maximal and mean) between men and women was not significantly different (Figure 3, Table V in the Data Supplement). The maximal QTc was abnormal (>460 ms for women, >450 ms for men) in 83% of p.T224M carriers versus 18.5% of noncarriers (P<0.0001; odds ratio, 19.9 [95% CI, 8.41–47.15]; Table). The mean QTc was abnormal in 7% of noncarriers and 54% of carriers (P<0.0001; Table). No individuals had left bundle branch block or intraventricular conduction delay. Three individuals (1 carrier, 2 noncarriers) had high normal to elevated QRS duration (122, 122, and 120 ms) in a right bundle branch block pattern while supine. No carriers were deaf, nor were any of their children.
Figure 2.
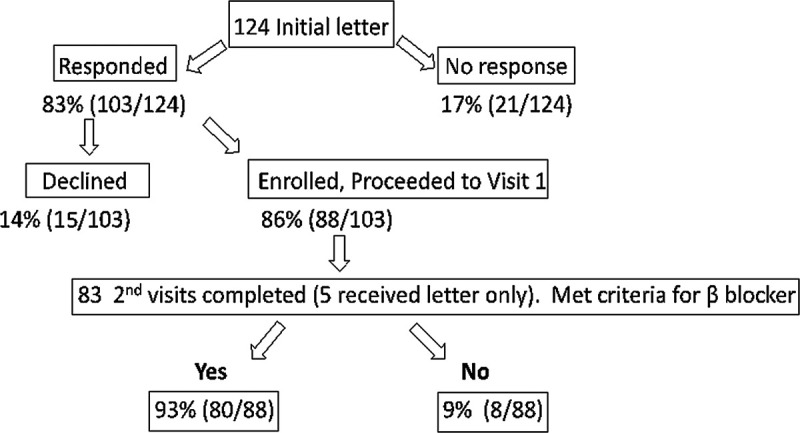
Recontact, clinical follow-up, and return of results for p.T224M KCNQ1 carriers. Of the124 carriers offered return of results, 88 (71% of those who received initial letter, 86% of those who responded) were enrolled. All 88 participants received their results with individualized clinical recommendations.
Figure 3.
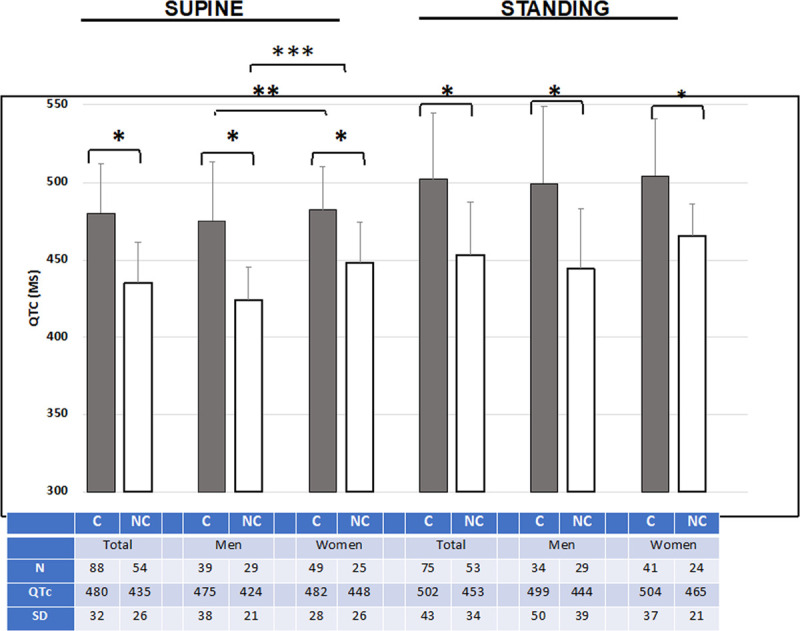
Higher maximal QTc in KCNQ1 p.T224M variant carriers (black bars) vs noncarriers (white bars). Group, QTc max±SD and N are shown below bars. C=p.T224M carrier; NC=noncarrier of p.T224M. TOTAL=men and women. The normal QTc for men is <450 ms; for women <460 ms. P values for comparisons: *<0.0001, **0.34, and ***0.0005.
Table.
Characteristics of KCNQ1 p.T224M Carriers Compared With Noncarriers
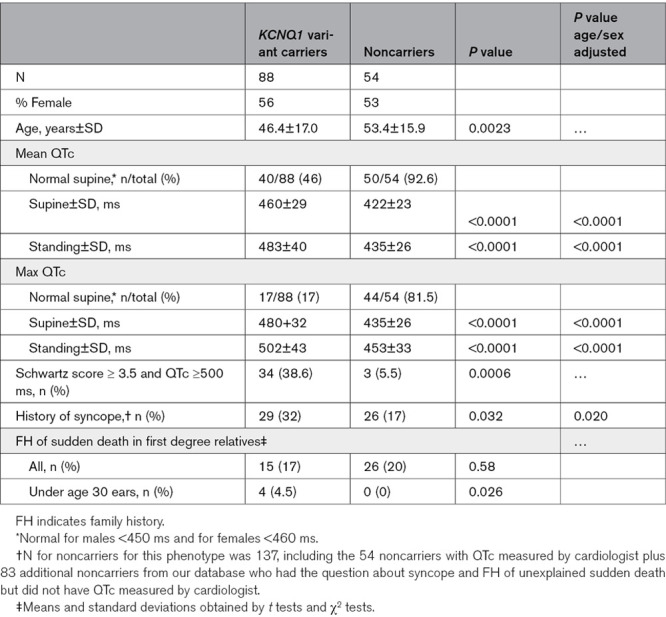
A self-reported personal history of syncope was higher in p.T224M carriers than noncarriers (32% versus 17%, P=0.020; Table). In 13/28 (46%) of p.T224M carriers who had a history of syncope, syncope occurred more than once, including 2 individuals with 10 or more syncopal episodes. Most syncope in noncarriers was vasovagal in character; in carriers, most did not have vasovagal characteristics. In the 2 carriers with ≥10 syncopal episodes, some of the episodes had vasovagal qualities. In 4 of the 18 female carriers with a history of syncope, syncope occurred within the first 24 hours postpartum. Most episodes of syncope in p.T224M carriers occurred in childhood or adolescence. The oldest age at first syncopal event was a male carrier at 60 years of age. This individual was on an antidepressant at the time of his syncopal episode that could have been a possible contributor but declined medical evaluation at the time of syncope to assess other causes.
A family history of SCD in first degree relatives under age 30 years was higher in p.T224M carriers than in noncarriers (4/88 [4.5%] versus 0/137 [0%], P=0.026), but was similar in relatives 30 years of age and over (15/88 [17%] versus 26/137 [20%], P=0.58; Table). The first degree relatives of p.T224M carriers with SCD under age 30 included 2 crib deaths, a 6 year old boy walking to school, and a 13 year old boy swimming (Figure 4). There were also 3 stillbirths in the families that included carriers of p.T224M. Stilbirths have recently been reported to be increased in LQT carriers.25 Combining first and second degree relatives of p.T224M carriers, 8% had a family history of sudden death under age 30 years, including 2 additional crib deaths in second degree relatives (Figure 4). Through pedigree review, we confirmed that each of these crib deaths was a distinct individual. Although genotyping for p.T224M was not available in the children who suffered sudden death or crib death, 4 of these children had a parent with the p.T224M variant, and the other parents were not genotyped. In p.T224M carriers, we found no association between the absolute value of the QTc and history of syncope, number of syncopal episodes or family history of SCD at any age. The p.T224M variant itself was associated with these outcomes.
Figure 4.
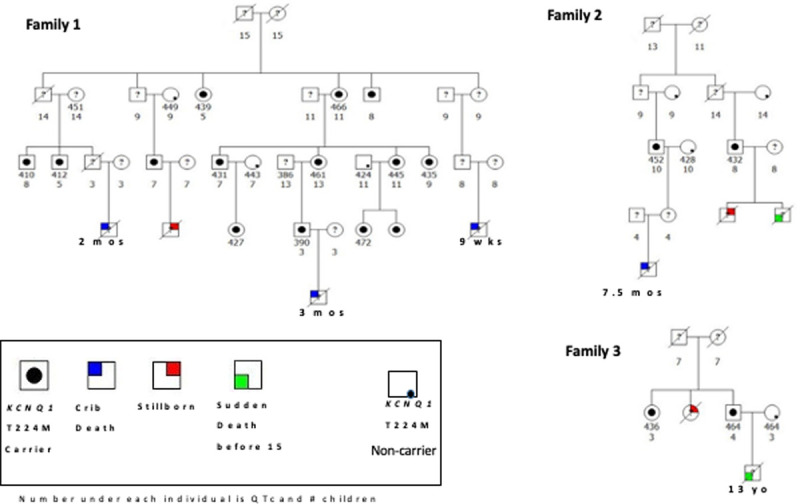
Family history of unexplained sudden deaths in children, crib deaths, and stillborns in the families of carriers of the KCNQ1 p.T224M variant. None of the children who died were genotyped for this variant, and none was known to be deaf.
The Schwartz score can be used to make a clinical diagnosis of LQTS, in the absence of a pathogenic genetic variant with criteria including QTc, T-wave changes, history of syncope with or without activity, and family history of SCD <30 years old. A Schwartz score of ≥3.5 is associated with a high probability of LQTS, 1.5 to 3.0 a moderate probability, and ≤1 a low probability. The Schwartz score was higher in p.T224M carriers than noncarriers (2.5±1.1 versus 0.70±1.0, P<0.0001). In the absence of a pathogenic variant, a QTc of ≥500 ms is also considered to be evidence of LQTS. Using the parameters QTc ≥500 ms and Schwartz score of ≥3.5 as clinical evidence of LQTS, 34/88 (38.6%) of p.T224M carriers versus 3/54 (5.5%) of noncarriers, had clinical evidence of LQTS (P=0.0006; Table).
In Vitro Functional Studies of KCNQ1 p.T224M
KCNQ1 encodes Kv7.1, a voltage-gated potassium channel that is present at the cell surface of cardiac cells. Kv7.1 associates with a function-modifying subunit encoded by KCNE1 to generate the slowly activating potassium current IKs that plays a key role in cardiac repolarization. As shown in Figure 5, in vitro expression of the p.T224M channel with KCNE1 in Chinese hamster ovary cells caused loss of IKs function compared with wild-type channels.26 The mutant T224M channel significantly reduced total activating and deactivating currents, with a marked positive shift in the voltage dependence of activation, by ≈26 mV (P<0.01).
Figure 5.
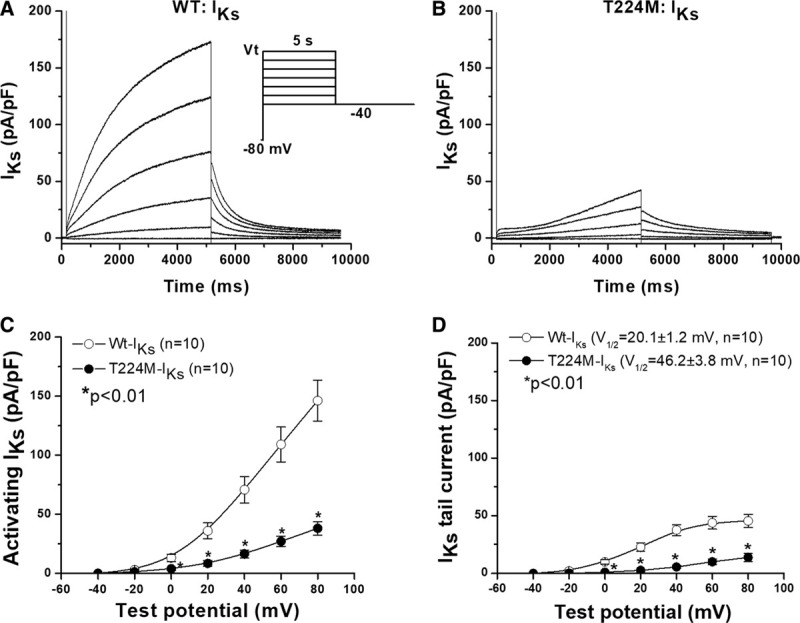
KCNQ1 T224M loss of IKs function.A and B, IKs recorded in Chinese hamster ovary (CHO) cells in which wild-type (WT) KCNQ1 or T224M were co-expressed with KCNE1 (the IKs accessory subunit). C and D, Summarize activating and deactivating IKs in the 2 groups of cells. The mutant T224M channel had significantly reduced total activating and deactivating currents, with a marked positive shift in the voltage dependence of activation, by ≈26 mV (P<0.01). Current densities were expressed in pA/pF after normalization of current amplitude to cell capacitance. The voltage clamp protocol is shown in the inset.
Reclassification of KCNQ1 p.T224M From a Variant of Unknown Significance to Pathogenic
The p.T224M variant was considered a variant of unknown significance at initial discovery in our cohort based on ClinVar classification and prior literature. Using the guidelines issued by the American College of Medical Genetics and Association for Molecular Pathology to reclassify p.T224M27 (Tables VI and VII in the Data Supplement), we determined that there was sufficient evidence to classify the variant as pathogenic. This upgraded classification was submitted to ClinVar to help inform future diagnoses of carriers of this variant elsewhere. Variants in KCNQ1 exon 4 listed in gnomAD and ClinVar are shown in Tables VIII and IX in the Data Supplement, which show the variation within the transmembrane region of the protein in exon 4, specifically within the S3 to S4 linker region. Variants within the region are classified by the sources as pathogenic, likely pathogenic, uncertain significance or not provided; no variation is classified by the sources as likely benign or benign.
Return of Results, Clinical Characterization, and Treatment Recommendations for KCNQ1 p.T224M Carriers
Based upon our reclassification of the p.T224M variant in KCNQ1 as pathogenic and the high prevalence of the variant in the Amish, we developed a plan to offer return of results to p.T224M carriers. The plan was reviewed with the University of Maryland Amish Research Clinic Advisory Committee, which includes Amish community leaders, and was approved by the University of Maryland Institutional Review Board. The plan is described in greater detail in the Materials and Methods in the Data Supplement.
As shown in Figures 2 and 83% (103/124) of p.T224M carriers who received a letter to inquire about their interest in obtaining additional information responded. Of these, 86% (88/103) expressed interest in obtaining additional information and 15/103 did not. Of those who declined, none provided a specific reason for declining participation. Home visit 1, which included repeat EKG (supine and standing), medical and family histories, and a blood draw for Clinical Laboratory Improvement Amendments confirmation of genotype was completed on all 88 participants who were enrolled in the study. Home visit 2, in which genetic results were disclosed and clinical recommendations were provided, was completed in 83 (5 participants did not call to schedule the second visit but received result letters by mail; see below). During home visit 2, many participants indicated their appreciation for the follow-up and information and expressed interest in cascade testing in their children. No participant asked to be withdrawn from the study.
Within one week of home visit 2, a final letter and doctor letter were mailed to p.T224M carriers. These letters reviewed in detail what was discussed at the visit, including genetic counseling and treatment recommendations. The doctor letter recommended using nadolol for all except women of child bearing age, in whom propranolol was recommended since nadolol is contraindicated in breastfeeding.28 Of 88 carriers, 80 (93%) qualified for beta-blocker treatment (Figure 2). Twenty-two (25%) of carriers were on prescription medications, including 5 (5.7%) who were on medications known to prolong QTc according to crediblemeds.com. In the doctor letter for these individuals, the recommendation was made to change to a medication not known to prolong QTc if possible, and a list of medications known to prolong QTc was included with recommendations to avoid these medications when possible.
Discussion
We describe the path from discovery of a variant in KCNQ1 in population research ES to its classification as pathogenic, clinical confirmation, and culturally sensitive return of results to Amish participants, including genetic counseling and treatment recommendations coordinated with local primary care providers, and opportunity for cascade testing of family members. The c.671C>T (p.T224M) variant in KCNQ1 was previously reported in only 2 individuals with LQTS15,16 and considered to be a variant of unknown significance. The variant is highly enriched in the Amish (carrier frequency 1/45), likely through a founder effect and genetic drift. Our phenotyping showed that p.T224M carriers had a 20.2 ms higher QTc, a higher rate of syncope, a higher number of SCD in first degree family members under age 30 years, and a higher Schwartz score than noncarriers. These data, in addition to in silico predictions (Table VI in the Data Supplement), location of the variant in an important functional domain with minimal benign variation,22 extremely low frequency in gnomAD, and in vitro functional studies showing variant significantly reduced activating and deactivating current densities and caused a markedly positive shift of voltage dependence of the channel activation by ≈26 mV compared with wild-type KCNQ1, enabled us to reclassify the c.671C>T (p.T224M) variant as pathogenic according to American College of Medical Genetics guidelines. To our knowledge, our work is the first electrophysiological evidence showing that p.T224M causes a loss of the KCNQ1 channel function.
Of critical clinical importance to the Amish population is to understand the absolute risk for SCD in p.T224M carriers. Unfortunately, we were unable to quantitatively estimate the risk of SCD in the Amish based on our data. The degree of lengthening of QTc in carriers was not associated with syncope or family history of SCD, so we presume all carriers of the p.T224M variant to be at increased risk, including the 46% of p.T224M carriers with a normal mean QTc. SCD in first degree family members of p.T224M carriers was more common than in noncarriers only among young relatives (<30 years). In addition, in carriers, most syncopal episodes occurred during childhood and adolescence. These data suggest that the highest risk of syncope and SCD of this variant may be in childhood. However, we have insufficient data to conclude that SCD risk attributable to p.T224M decreases or is modified with age. The similar rates of SCD in first degree relatives of carriers versus noncarriers age >30 years may reflect SCD risk increasing with age in the general population due to other causes, such as ischemic heart disease.
An unresolved issue in the field is what determines the variabile expressivity that is commonly seen LQT1. Our analyses indicated that polygenic background influences QTc interval in carriers as it does in noncarriers, although we found no evidence for a larger effect of p.T224M on QT interval in those with a stronger polygenic background. Nor did we observe any statistical evidence of modifying effects of common variants in other genes previously reported to modify the effects of other pathogenic KCNQ1 variants on QT interval. However, as in other studies, power to detect modifier effects in our study was limited.
Numerous medications are known to prolong QTc. In our cohort, 5 of 88 p.T224M carriers (5.7%) were on a medication known to prolong QT. This was brought to the attention of these individuals and their physicians, with recommendations for the physicians to change to a safer medication, if possible. Furthermore, all p.T224M carriers, even those with normal QTc, who could have only been identified through genetic testing, would be well-advised to avoid drugs that prolong QT interval. This is an example of the benefit of genomic information on medication safety in individuals carrying at risk genotypes, precision medicine.29
In addition, we found that 7% of noncarriers had a higher mean QTc (and 19.6% had a higher maximal QTc) than normal. However, only 3 noncarriers with greater than normal QTc had strong clinical evidence for LQTS (Schwartz score ≥3.5 or QTc ≥500 ms). For these 3 individuals, recommendations were made to consult with a cardiologist for further evaluation. Overlap of QTc interval between normal individuals and those with LQTS has been well described.30 Since the goal of this study was to compare carriers of the KCNQ1 p.T224M variant to noncarriers, a normal QTc was not required for inclusion in the noncarrier group. In noncarriers of p.T224M with a longer than normal QTc, none was on a medication known to prolong QTc, nor were there any obvious other causes to explain the longer than normal QTc in these individuals.
Our additional phenotyping revealed some other interesting findings, including that supine QTc was no different in p.T224M variant carrier women and men, whereas in noncarriers, the expected higher QTc in women was observed. The number of men and women with a history of syncope in our study was similar. Others have reported that women with a pathogenic KCNQ1 variant have a higher QTc than men.22,31
Founder pathogenic variants in KCNQ1 causing LQT1 have been described in other populations, including the Bitxsan First Nations population in Canada (c.613G>A, p.V205M), present in 1:12522 with a QTc effect size of 31 ms per allele. Founder pathogenic KCNQ1 variants have also been reported in the Finnish (c.1766G>A, p.G589D) present in 1:250 with an effect size of 50 ms32 and in the Swedish (c.1552C>T, p.A518X and c.332A>G, p.T111C), with effect sizes 50 and 30 ms, respectively.33 The founder variant in Afrikaners (c.641C>T, p.A341V) is associated with a SCD death rate of 14% before age 20 years.34 In the Saudi Arabian founder mutation (c.387-5T>A), there was a high incidence of homozygotes likely due to endogamy.35 The p.T224M variant in the Amish has a higher prevalence (1/45) than these other founder variants, with a smaller effect size on QTc (20.2 ms/allele) and possibly lower morbidity. To date, we have not found any p.T224M homozygotes but speculate that some of the stillbirths reported to us could have been homozygotes. In all examples of pathogenic founder variants in KCNQ1, the impact on survival or reproduction is likely minimal; otherwise, with time the variants would become progressively less prevalent. Recently, a multiexon duplication in RYR2 has been reported in 2 large Amish families with a high risk of sudden death.36 In our database of Pennsylvania Amish, we found 8 heterozygotes for this duplication and no homozygotes.
Currently, we have little information on how many KCNQ1 p.T224M carriers elected to be treated with a beta-blocker or how many family members will seek genetic testing. We know that currently, among the 88 participants, only 2 family members have undergone KCNQ1 genetic testing. Since the Amish do not own cars, we suspect that inconvenience and cost of transportation to a physician’s office or laboratory to obtain testing and the cost of testing may be major barriers to cascade testing in this population. We speculate that if this testing were offered at low cost or free of charge and without the need to travel, cascade testing for the p.T224M variant in the Amish might be more commonly done. A study is currently underway to attempt to answer these questions and to offer free in-home testing to offspring of probands with the p.T224M variant. Moreover, given its high prevalence in the Amish, adding this pathogenic variant to newborn screening in states with a significant Amish population
may be indicated and a more effective approach to identifying at risk individuals at the population level.
Limitations of this study include the absence of KCNQ1 genotyping and autopsy reports on family members who had SCD to confirm that they were carriers of the p.T224M variant. Second, we may have had fewer p.T224M carriers with a normal mean QTc if we had included stress testing in addition to the immediate standing EKG. In addition, we included 83 noncarriers who were not in the primary study population, to increase our power to assess for difference in family history of SD between carriers and noncarriers. Strengths of the study include that all carriers were seen by one physician, that family history in carriers were obtained by a medical geneticist, and that all EKGs were read by an electrophysiologist.
Through exome-wide association studies, we describe the identification of a highly drifted missense variant, p.T224M, in KCNQ1 in the Amish that is highly associated with QTc. Additional phenotyping and functional characterization led to reclassification of the c.671C>T (p.T224M) variant in KCNQ1 as pathogenic for LQT1. We implemented a culturally appropriate program for return of results including recommendations for cascade testing and treatment, coordinated with local health care providers. Furthermore, we have adopted a protocol for KCNQ1 genotyping and return of results for all ongoing studies at the Amish Research Clinic. This work provides an example of clinical implementation of an actionable genetic research result that has important health implications not only for research participants but also for community health in a founder population in which this pathogenic variant is common. We suggest this approach can be adapted for use in other genetic studies, particularly those whose protocol and consent did not anticipate return of medically actionable secondary findings.
Sources of Funding
This study was supported by National Institutes of Health grants U01 HL072515, U01 HL137181, R01 AG18728, R01 HL076768, U01 GM074518, R21 HG010412, R01 HL088119, GM115305, and American Heart Association grant 16MCPRP31350041. Partial support for this study, including exome sequencing was provided by the Regeneron Genetics Center, LLC.
Disclosures
Drs Shuldiner, Van Hout, Gosalia, Gonzaga-Jauregu, and Economides are employees of Regeneron Pharmaceuticals, Inc and receive compensation for their employment. The other authors report no conflicts. Details are present in the Data Supplement. This scientific journal article was prepared or accomplished by Dr Jeng in her personal capacity. The opinions expressed in this article are the author’s own and do not reflect the view of the US Food and Drug Administration, the Department of Health and Human Services, or the United States government.
Supplementary Material
Nonstandard Abbreviation and Acronyms
- ES
- exome sequencing
- LQT1
- long QT syndrome type 1
- LQTS
- long QT syndrome
- SCD
- sudden cardiac death
The members of the author group can be found in the Data Supplement.
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCGEN.120.003133.
For Sources of Funding and Disclosures, see page 586.
Contributor Information
Vincent Y. See, Email: vsee@som.umaryland.edu.
Linda B.J. Jeng, Email: ljeng@som.umaryland.edu.
Kristin A. Maloney, Email: kmaloney1@som.umaryland.edu.
Megan Lynch, Email: mlynch@som.umaryland.edu.
Andrew M. Glazer, Email: andrew.m.glazer@vumc.org.
Tao Yang, Email: tao.yang@vumc.org.
Dan Roden, Email: Dan.Roden@VUMC.org.
Toni I. Pollin, Email: tpollin@medicine.umaryland.edu.
Melanie Daue, Email: mdaue@som.umaryland.edu.
Kathleen A. Ryan, Email: kryan@som.umaryland.edu.
Cristopher Van Hout, Email: cristopher.vanhout@regeneron.com.
Nehal Gosalia, Email: ngosalia@gmail.com.
Claudia Gonzaga-Jauregui, Email: clau.gonzagajauregui@regeneron.com.
Aris Economides, Email: aris@regeneron.com.
James A. Perry, Email: jperry@som.umaryland.edu.
Jeffrey O’Connell, Email: joconnel@som.umaryland.edu.
Amber Beitelshees, Email: abeitels@som.umaryland.edu.
Kathleen Palmer, Email: kpalmer@som.umaryland.edu.
Braxton D. Mitchell, Email: bmitchel@som.umaryland.edu.
Alan R Shuldiner, Email: ashuldin@medicine.umaryland.edu.
References
- 1.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, et al. ; American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical genetics and genomics. Genet Med. 2017;19:249–255. doi: 10.1038/gim.2016.190 [DOI] [PubMed] [Google Scholar]
- 3.Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, O’Dushlaine C, Van Hout CV, Staples J, Gonzaga-Jauregui C, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354:aaf6814. doi: 10.1126/science.aaf6814 [DOI] [PubMed] [Google Scholar]
- 4.Fabsitz RR, McGuire A, Sharp RR, Puggal M, Beskow LM, Biesecker LG, Bookman E, Burke W, Burchard EG, Church G, et al. Ethical and practical guidelines for reporting genetic research results to study participants:updated guidelines from a national heart, lung and blood institute working group. Circ Cardiovasc Genet. 2010;3:574–580. doi: 10.1161/CIRCGENETICS.110.958827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bombard Y, Brothers KB, Fitzgerald-Butt S, Garrison NA, Jamal L, James CA, Jarvik GP, McCormick JB, Nelson TN, Ormond KE, et al. The responsibility to recontact research participants after reinterpretation of genetic and genomic research results. Am J Hum Genet. 2019;104:578–595. doi: 10.1016/j.ajhg.2019.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kollek R, Petersen I. Disclosure of individual research results in clinico-genomic trials: challenges, classification and criteria for decision-making. J Med Ethics. 2011;37:271–275. doi: 10.1136/jme.2009.034041 [DOI] [PubMed] [Google Scholar]
- 7.Bui ET, Anderson NK, Kassem L, McMahon FJ. Do participants in genome sequencing studies of psychiatric disorders wish to be informed of their results? A survey study. PLoS One. 2014;9:e101111. doi: 10.1371/journal.pone.0101111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller FA, Hayeems RZ, Li L, Bytautas JP. One thing leads to another: the cascade of obligations when researchers report genetic research results to study participants. Eur J Hum Genet. 2012;20:837–843. doi: 10.1038/ejhg.2012.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fossey R, Kochan D, Winkler E, Pacyna JE, Olson J, Thibodeau S, Connolly JJ, Harr M, Behr MA, Prows CA, et al. Ethical considerations related to return of results from genomic medicine projects: the eMERGE network (phase III) experience. J Pers Med. 2018;8:2. doi: 10.3390/jpm8010002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kostick KM, Brannan C, Pereira S, Lázaro-Muñoz G. Psychiatric genetics researchers’ views on offering return of results to individual participants. Am J Med Genet B Neuropsychiatr Genet. 2019;180:589–600. doi: 10.1002/ajmg.b.32682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz MLB, McCormick CZ, Lazzeri AL, Lindbuchler DM, Hallquist MLG, Manickam K, Buchanan AH, Rahm AK, Giovanni MA, Frisbie L, et al. A model for genome-first care: returning secondary genomic findings to participants and their healthcare providers in a large research cohort. Am J Hum Genet. 2018;103:328–337. doi: 10.1016/j.ajhg.2018.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phillips J, Ichinose H. Clinical and pathologic studies in the hereditary syndrome of a long QT interval, syncopal spells and sudden death. Chest. 1970;58:236–243. doi: 10.1378/chest.58.3.236 [DOI] [PubMed] [Google Scholar]
- 13.Waddell-Smith KE, Skinner JR; members of the CSANZ Genetics Council Writing Group. Update on the diagnosis and management of familial long QT syndrome. Heart Lung Circ. 2016;25:769–776. doi: 10.1016/j.hlc.2016.01.020 [DOI] [PubMed] [Google Scholar]
- 14.Schwartz PJ, Ackerman MJ, George AL, Jr, Wilde AAM. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol. 2013;62:169–180. doi: 10.1016/j.jacc.2013.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AA, Ackerman MJ. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–1303. doi: 10.1016/j.hrthm.2009.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arking DE, Pulit SL, Crotti L, van der Harst P, Munroe PB, Koopmann TT, Sotoodehnia N, Rossin EJ, Morley M, Wang X, et al. ; CARe Consortium; COGENT Consortium; DCCT/EDIC; eMERGE Consortium; HRGEN Consortium. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet. 2014;46:826–836. doi: 10.1038/ng.3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bihlmeyer NA, Brody JA, Smith AV, Warren HR, Lin H, Isaacs A, Liu CT, Marten J, Radmanesh F, Hall LM, et al. ExomeChip-wide analysis of 95 626 individuals identifies 10 novel loci associated with QT and JT intervals. Circ Genom Precis Med. 2018;11:e001758. doi: 10.1161/CIRCGEN.117.001758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Setten J, Verweij N, Mbarek H, Niemeijer MN, Trompet S, Arking DE, Brody JA, Gandin I, Grarup N, Hall LM, et al. Genome-wide association meta-analysis of 30,000 samples identifies seven novel loci for quantitative ECG traits. Eur J Hum Genet. 2019;27:952–962. doi: 10.1038/s41431-018-0295-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Villiers CP, van der Merwe L, Crotti L, Goosen A, George AL, Jr, Schwartz PJ, Brink PA, Moolman-Smook JC, Corfield VA. AKAP9 is a genetic modifier of congenital long-QT syndrome type 1. Circ Cardiovasc Genet. 2014;7:599–606. doi: 10.1161/CIRCGENETICS.113.000580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Post W, Shen H, Damcott C, Arking DE, Kao WH, Sack PA, Ryan KA, Chakravarti A, Mitchell BD, Shuldiner AR. Associations between genetic variants in the NOS1AP (CAPON) gene and cardiac repolarization in the old order Amish. Hum Hered. 2007;64:214–219. doi: 10.1159/000103630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kapplinger JD, Erickson A, Asuri S, Tester DJ, McIntosh S, Kerr CR, Morrison J, Tang A, Sanatani S, Arbour L, et al. KCNQ1 p.L353L affects splicing and modifies the phenotype in a founder population with long QT syndrome type 1. J Med Genet. 2017;54:390–398. doi: 10.1136/jmedgenet-2016-104153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chorin E, Havakuk O, Adler A, Steinvil A, Rozovski U, van der Werf C, Postema PG, Topaz G, Wilde AA, Viskin S, et al. Diagnostic value of T-wave morphology changes during “QT stretching” in patients with long QT syndrome. Heart Rhythm. 2015;12:2263–2271. doi: 10.1016/j.hrthm.2015.06.040 [DOI] [PubMed] [Google Scholar]
- 24.Viskin S, Postema PG, Bhuiyan ZA, Rosso , Kalman JM, Vohra JK, Guevara-Valdivia ME, Marquez MF, Kogan E, Belhassen B, et al. The response of the QT interval to the brief tachycardia provoked by standing a bedside test for diagnosing long QT syndrome. J Am Coll Card. 2010;55:1955–1961. doi: 10.1016/j.jacc.2009.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cuneo BF, Kaizer AM, Clur SA, Swan H, Herberg U, Winbo A, Rydberg A, Haugaa K, Etheridge S, Ackerman MJ, et al. ; Fetal LQTS Consortium. Mothers with long QT syndrome are at increased risk for fetal death: findings from a multicenter international study. Am J Obstet Gynecol. 2020;222:263.e1–263.e11. doi: 10.1016/j.ajog.2019.09.004 [DOI] [PubMed] [Google Scholar]
- 26.Yang T, Chung SK, Zhang W, Mullins JG, McCulley CH, Crawford J, MacCormick J, Eddy CA, Shelling AN, French JK, et al. Biophysical properties of 9 KCNQ1 mutations associated with long-QT syndrome. Circ Arrhythm Electrophysiol. 2009;2:417–426. doi: 10.1161/CIRCEP.109.850149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davanzo R, Bua J, Paloni G, Facchina G. Breastfeeding and migraine drugs. Eur J Clin Pharmacol. 2014;70:1313–1324. doi: 10.1007/s00228-014-1748-0 [DOI] [PubMed] [Google Scholar]
- 29.Ginsburg GS, Phillips KA. Precision medicine: from science to value. Health Aff (Millwood). 2018;37:694–701. doi: 10.1377/hlthaff.2017.1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viskin S. The QT interval: too long, too short or just right. Heart Rhythm. 2009;6:711–715. doi: 10.1016/j.hrthm.2009.02.044 [DOI] [PubMed] [Google Scholar]
- 31.Winbo A, Stattin EL, Westin IM, Norberg A, Persson J, Jensen SM, Rydberg A. Sex is a moderator of the association between NOS1AP sequence variants and QTc in two long QT syndrome founder populations: a pedigree-based measured genotype association analysis. BMC Med Genet. 2017;18:74. doi: 10.1186/s12881-017-0435-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marjamaa A, Salomaa V, Newton-Cheh C, Porthan K, Reunanen A, Karanko H, Jula A, Lahermo P, Väänänen H, Toivonen L, et al. High prevalence of four long QT syndrome founder mutations in the finnish population. Ann Med. 2009;41:234–240. doi: 10.1080/07853890802668530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stattin EL, Boström IM, Winbo A, Cederquist K, Jonasson J, Jonsson BA, Diamant UB, Jensen SM, Rydberg A, Norberg A. Founder mutations characterise the mutation panorama in 200 Swedish index cases referred for Long QT syndrome genetic testing. BMC Cardiovasc Disord. 2012;12:95. doi: 10.1186/1471-2261-12-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, Heradien M, Geldenhuys G, Vanoli E, Bacchini S, et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation. 2005;112:2602–2610. doi: 10.1161/CIRCULATIONAHA.105.572453 [DOI] [PubMed] [Google Scholar]
- 35.Bdier AY, Al-Ghamdi S, Verma PK, Dagriri K, Alshehri B, Jiman OA, Ahmed SE, Wilde AAM, Bhuiyan ZA, Al-Aama JY. Autosomal recessive long QT syndrome, type 1 in eight families from Saudi Arabia. Mol Genet Genomic Med. 2017;5:592–601. doi: 10.1002/mgg3.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tester DJ, Bombei HM, Fitzgerald KK, Giudicessi JR, Pitel BA, Thorland EC, Russell BG, Hamrick SK, Kim CSJ, Haglund-Turnquist CM, et al. Identification of a novel homozygous multi- exon duplication in RYR2 among children with exertion-related unexplained sudden deaths in the Amish community. JAMA Cardiol. 2020;5:13–18. doi: 10.1001/jamacardio.2019.5400 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.