

Abstract
Programmable nucleic acid nanoparticles (NANPs) with precisely controlled functional compositions can regulate the conditional activation of various biological pathways and responses in human cells. However, the intracellular delivery of NANPs alone is hindered by their susceptibility to nuclease activity and inefficient crossing of biological membranes. In this work, we optimized the internalization and therapeutic performance of several representative NANPs delivered with mesoporous silica nanoparticles (MSNPs) tailored for efficient electrostatic association with NANPs. We compared the immunostimulatory properties of different NA-MS-NP complexes formed with globular, planar, and fibrous NANPs and demonstrated the maximum immunostimulation for globular NANPs. As a proof of concept, we assessed the specific gene silencing by NA-MS-NP complexes functionalized with siRNA targeting green fluorescent protein expressed in triple-negative human breast cancer cells. We showed that the fibrous NANPs have the highest silencing efficiency when compared to globular or planar counterparts. Finally, we confirmed the multimodal ability of MSNPs to co-deliver a chemotherapy drug, doxorubicin, and NANPs targeting apoptosis regulator gene BCL2 in triple-negative breast cancer and melanoma cell lines. Overall, the combination of NANPs and MSNPs may become a new promising approach to efficiently treat cancer and other diseases via the simultaneous targeting of various pathways.
Keywords: nucleic acid nanoparticles (NANPs), mesoporous silica nanoparticles (MSNPs), small interfering RNA, combination therapy, triple-negative breast cancer, melanoma, doxorubicin
Graphical Abstract
1. INTRODUCTION
RNA interference (RNAi) triggered by exogenous RNA duplexes has gained prominence as a therapeutic and specific gene silencing mechanism. Recent advances have demonstrated its utility in treating complex diseases1,2 with two therapies (Onpattro3 and Givlaari4) already approved by the Food and Drug Administration (FDA). RNAi presents substantial opportunities as a therapeutic approach for intractable cancers by providing a massive number of targets that conventional strategies do not render.5 Although appealing as a therapeutic concept, the clinical positioning of RNAi therapies is hindered by their inefficient delivery and immunotoxic-related roadblocks. Nucleic acid nanoparticles (NANPs) present an alternative to traditional therapeutic nucleic acids, adding an additional layer of customizability.6 These complexes are composed entirely of nucleic acids (DNA, RNA, or their analogs) and utilize rational design along with the innate structures of nucleic acids to assume a plethora of complex three-dimensional structures.7 The limitless possibilities of these unique materials allow for highly tunable physicochemical properties, including size, charge, mass, thermodynamic and chemical stabilities, and multifunctionality of therapeutic nucleic acids,8–14 as well as the potential to regulate their immunostimulation.15–22 In addition to their roles as programmable scaffolds that coordinate therapeutic nucleic acids, NANPs have the inherent ability to be used as logic gates and biosensors, allowing for both therapeutic and diagnostic applications beyond current standards.23–26 Despite the promising advantages of NANP technology, there are several concerns that need to be tackled to drive their translation from benchtop to clinic such as susceptibility to enzymatic degradation, inability to cross biological membranes because of a negative charge, and the potential for triggering deleterious immune responses. Recently, our lab has demonstrated the use of polymeric and magnetic nanoparticles as carriers for NANPs’ delivery and compared the stability and efficacy of various NANP/delivery complexes.27,28 Both platforms were shown to efficiently protect and deliver NANPs, which was confirmed by the specific gene knockdown upon the release of siRNAs in various human cancer cell lines. While those systems are well-suited for the efficient delivery of NANPs, the additional possibility for embedding other therapeutic moieties into the core of the carrier would make this approach amenable to a much broader range of biomedical applications. Therefore, there is a critical need to develop robust platforms that not only deliver NANPs, but also synergize with other therapeutic agents such as conventional small-molecule drugs.
Mesoporous silica nanoparticles (MSNPs) are an attractive alternative as a delivery vehicle for various cancer-based therapies. Several advantages have been already demonstrated for MSNPs such as biocompatibility, large surface area, and multifunctionalization (e.g., therapeutic, imaging, and/or targeting agents).29–33 MSNPs are ideal carriers for siRNAs because of their efficient internalization by mammalian cells, ability to be modified to protect the siRNA cargo from enzymatic degradation, and can be engineered to escape from endosomes or lysosomes to release their cargo into the cytoplasm.26,34,35 Our group has previously developed silica-based nanoconstructs as a strategy for the efficient transport and delivery of siRNA.15,36,37 In this project, the MSNPs’ shell is composed of poly(ethylene glycol) (PEG)/polyethylenimine (PEI) to serve as the nonviral delivery vector for NANPs. Using a similar platform, we have recently shown the efficient transfection of siRNAs for silencing of tenascin C in hepatic cells.37 Additionally, we have tested the platform’s potential as a delivery vector for fibrous NANPs carrying siRNAs targeting green fluorescent protein (GFP) expressed in human breast cancer cells.15 Several advantages can be introduced by using MSNPs as a vector for NANPs such as modifying the immune response, improving targetability, and rendering multifunctional properties. In particular, the capability of MSNPs to carry a wide array of active pharmaceutical agents together with NANPs can be used to develop multimodal approaches. Nevertheless, the therapeutic performance and potential for the simultaneous delivery of NANPs with other therapeutics using MSNPs has not yet been demonstrated.
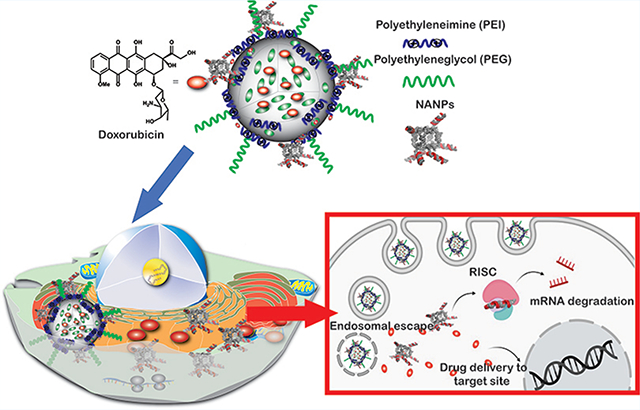
Here, we report on the synthesis, characterization, optimization, and in vitro application of a nanoplatform that combines two promising nanotechnologies: NANPs and MSNPs (NA-MS-NPs). The experimental scheme of the reported work is outlined in Figure 1. We carried out the detailed physicochemical characterization of all individual components as well as the resulting NA-MS-NP complexes. We then evaluated the relative immunostimulation, cellular uptake, and silencing capabilities of the NA-MS-NP system with different NANP geometries, including globular (cNANPs), planar (rNANPs), and fibrous (fNANPs) structures. Finally, we assessed the combinatorial effect of the NA-MS-NP platform loaded with doxorubicin (DOX) and targeting the antiapoptotic BCL2 gene in both triple-negative human breast cancer (MDA-MB-231) and human melanoma (A375) cell lines.
Figure 1.

Experimental work-flow reported in this project. (A) Fabrication of MSNPs and NANPs. The surfaces of MSNPs were functionalized in a multistep approach by grafting with phosphonate groups, followed by coating with PEI and PEG polymers. Three different shapes of NANP materials—globular (cNANPs), planar (rNANPs), and fibrous (fNANPs)—were synthesized via one-pot assembly protocols. (B) Different characterization techniques were used to confirm the fabrication of MSNPs, NANPs, and NA-MS-NPs. (C) In vitro experiments were carried out to validate cellular uptake, gene silencing, and assessment of cell growth and survival upon treatment with synthesized NA-MS-NPs.
2. EXPERIMENTAL SECTION
2.1. Synthesis of NANPs.
DNA strands were procured from Integrated DNA Technologies, Inc. Transcription was achieved by incubating DNA templates (containing T7 promoter regions) at 37 °C with homemade T7 RNA polymerase and transcription buffer (80 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES)–KOH, 2.5 mM spermidine, 50 mM dithiothreitol (DTT), and 25 mM MgCl2, and 5 mM each rNTP) for 4 h. The reaction was stopped with RQ1 DNase (Promega) for 30 min at 37 °C and then purified with denaturing gel electrophoresis (urea polyacrylamide gel electrophoresis (PAGE), 8 M urea, 8% acrylamide) by visualizing bands under UV, extracting gel slices, and eluting the samples into 300 mM NaCl, 89 mM tris-borate (pH 8.2), and 2 mM ethylenediaminetetraacetic acid (EDTA) overnight at 4 °C. On the following day, the samples were mixed with 2× volume of anhydrous ethanol and chilled to −20 °C for a minimum of 3 h. The samples were then spun at 14 000g for 30 min, followed by disposing off the supernatant. An additional washing step was carried out by adding 90% ethanol, centrifuging at 14 000g, and discarding the supernatant. Finally, the samples were processed by drying using a SpeedVac concentrator followed by resuspension in double-deionized endotoxin-free water. The concentrations were measured using a NanoDrop 2000.
The synthesis of all NANPs was completed using a “one-pot” assembly method.12 Cubes and rings (cNANPs and rNANPs) both consist of six scaffold ssRNAs with 3′-side dicer substrate (DS) antisense extensions and six complementary dicer substrate sense strands (Supporting Information (SI)). Fibers (fNANPs) consist of two ssRNAs with 3′-side dicer substrate antisense extensions and two complementary dicer substrate sense strands (SI). For cNANPs, the ssRNA strands were mixed in equimolar concentrations with 6 equiv of dicer substrate sense strands and heated to 95 °C for 2 min and then cooled down to 45 °C for another 2 min. Afterward, assembly buffer (final concentration 89 mM tris-borate, 2 mM MgCl2, 50 mM KCl) was added, and the solution was incubated at 45 °C for an additional 30 min. For rNANPs and fNANPs, the ssRNA strands were mixed in equimolar concentrations with either 6 (for rNANPs) or 2 (for fNANPs) equiv of dicer substrate sense strand. The mixture was heated to 95 °C for 2 min and then immediately cooled on ice (~4 °C) for another 2 min. Afterward, the assembly buffer was added and the solution was incubated at 30 °C for 30 min. To confirm the assembly of all structures, the NANPs were run in nondenaturing native-PAGE (8%, 37.5:1) in 89 mM tris-borate (pH 8.2), 2 mM MgCl2 buffer at 4 °C followed by ethidium bromide total staining and visualization on a ChemiDoc MP Imaging System.
2.2. Complexation of NA-MS-NPs.
To enable the formation of NA-MS-NP complexes, 0.1 mg of MSNPs was dispersed in 100 μL of 1× assembly buffer (89 mM tris-borate (pH 8.2), 2 mM MgCl2, 50 mM KCl) followed by slow addition of either DNA/RNA duplexes or NANPs (cNANPs, rNANPs, or fNANPs) in solution. The specific volumes added for each material are shown in Table S1 of the SI. Additional 1× assembly buffer was added to the mixture to make a final volume of 200 μL. The final solution was mixed by pipetting several times followed by incubation for 30 min at room temperature. After that, NA-MS-NPs were separated from the dispersion by centrifugation at 8–10k rpm and redispersed in 100 μL of 1× assembly buffer. The optimal N/P mole ratio of 10 was used for all of the NANP–MSNP complexes fabricated in this work.
2.3. Characterization of NANPs, MSNPs, and NA-MS-NPs.
Atomic force microscopy (AFM) was used to investigate the morphology of the NANPs. Freshly cleaved mica was treated with 1-(2-aminopropyl)silatrane (APS) according to the established protocol.38,39 AFM was then performed as previously reported23 and described in depth in the Supporting Information.
The size and shape of the MSNPs were visualized using transmission electron microscopy (TEM) (JEM-1230 TEM) and quantified using ImageJ and previously reported.36 The size is reported as the average ± standard deviation (SD). Dynamic light scattering (DLS) was performed on a dilute dispersion (0.1 mg/mL) of nanoparticles in 1 mM phosphate-buffered saline (PBS) as previously reported.36 Thermogravimetric analysis (TGA) was performed using a Mettler Toledo small furnace thermogravimetric analyzer. The thermal degradation profiles were obtained for a heating rate of 1 °C/min between 25 and 800 °C followed by a 60 min hold at 800 °C. To measure the porous surface area and pore size, the nitrogen sorption isotherms were determined using a Quantachrome Instruments Nova series surface area and a pore size analyzer. The Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods were used to analyze the surface area and average pore diameter of the MSNPs, respectively.
To image the NA-MS-NPs by TEM, a drop of the NA-MS-NP solution (~5 μL) was deposited on a lacey carbon-coated copper grid. Before the sample was completely dried, a drop of the negative staining agent (Nano-W) was added, followed by a second drop a minute later. The sample was finally air-dried on the grid. Images were captured with a JEOL JEM 2100 LaB6 TEM. Particle size distributions were calculated measuring n = 50 nanoparticles using ImageJ and Origin software from several images taken at separate quartiles and various magnifications.
2.4. Nuclease Degradation Protection Studies.
To evaluate the ability of MSNPs to protect nucleic acids from enzymatic degradation, dsDNAs carrying both a fluorophore (Alexa Fluor 488-Al488) (5′) and a Quencher (Iowa Black) (3′) on subsequent strands were conjugated to MSNPs and treated with RQ1 DNase. MSNPs (0.33 mg/mL final) were mixed with the labeled dsDNA (0.8 μM final) in a solution of 30 μL total following the protocol described in Section 2.2. To this solution, 3 μL of RQ1 DNase was added and the samples were rapidly loaded into a CFX96 RT-thermocycler where the temperature was held at 37 °C. The relative fluorescence was measured every 30 s for 30 min using a Bio-Rad CFX96 real-time system. As a positive control, DNA duplexes without MSNs in the presence of RQ1 DNase were used.
2.5. Competitive Assay to Study the Release of NANPs from NA-MS-NPs.
NANPs and MSNPs were complexed for 30 min at room temperature for an N/P ratio of 10 following the method described in Section 2.2. After their attachment, aqueous heparin sulfate (Sigma) was added to the solution at a nucleic acid:heparin ratio of 1:6 w/w and incubated at 37 °C for 30 min. The samples were analyzed via an 8% native-PAGE as described above.
2.6. Immune Response by THP1-Dual Cells and HEK-Blue hTLR3 or 7 Cells.
THP1-Dual cells (InvivoGen) engineered to express secreted alkaline phosphatase (SEAP) upon nuclear factor κB (NF-κB) stimulation and luciferase upon interferon regulatory factor (IRF) stimulation were seeded at 40 000 cells per well in a 96-well plate. The cells were then immediately transfected with NA-MS-NPs for a final concentration of 50 nM NANPs, or cGAMP (for IRF activation) or R848 (for NF-κB activation), which were used as positive controls. To measure their relative immunostimulation, 24 h post transfection, 20 μL of the cell supernatant was mixed with 180 μL of prepared QUANTI-Blue solution (InvivoGen) and incubated for 75 min at 37 °C. The absorbance at 620 nm was then measured using a plate reader to quantify NF-κB stimulation. For IRF stimulation, 20 μL of the cell supernatant was mixed with 50 μL of prepared QUANTI-Luc solution and the luminescence was measured immediately using a plate reader.
HEK-Blue hTLR3 and 7 cells (InvivoGen) were used to assess contribution from specific nucleic acid receptors in detecting NA-MS-NPs. HEK-Blue hTLR3 or 7 cells were plated in a 96-well plate at a density of 40 000 cells per well and allowed to adhere overnight. The next day, the cells were treated with various NA-MS-NPs (50 nM NANPs) and incubated for 24 h at 37 °C and 5% CO2 atmosphere. Then, 20 μL of the cell supernatant was mixed with 180 μL of QUANTI-Blue solution and incubated for 75 min at 37 °C and 5% CO2 atmosphere. The absorbance at 620 nm was measured to quantify the activation of the respective TLRs. Positive controls such as poly I:C and R848 were used for TLR3 and 7, respectively.
2.7. Cellular Uptake of NA-MS-NPs.
For this study, fluorescein-labeled MSNPs (Fl-MSNPs) and Alexa546-labeled NANPs were used. Flow cytometry was utilized to study the uptake of the Alexa546-labeled NA-Fl-MS-NPs in the MDA-MB-231 cells. These cells were cultured at a density of 20 000 cells per well in a 24-well plate containing 500 μL of media and maintained for 24 h at 37 °C with 5% CO2 in a humidified incubator. The cells were then incubated with Alexa546-labeled NA-Fl-MS-NPs (0.5 mL) at a concentration of 30 μg/mL for a period of 24 h at 37 °C and 5% CO2 atmosphere. Afterward, the cells were washed with phosphate buffer, followed by detachment of the cells using 0.25% trypsin–EDTA. The cells were then gathered for analysis with the flow cytometer (BD LSR Fortessa cell analyzer) using green and red channels for fluorescein and Alexa546 fluorescence, respectively. The cells treated with free Alexa546-labeled NANPs were analyzed as control samples.
2.8. Cellular Uptake and Intracellular Localization of Alexa546-Labeled dsDNA Complexed With MSNPs.
A similar protocol as described in Section 2.7 was used to analyze the internalization of Alexa546-labeled dsDNA-Fl-MS-NPs by flow cytometry. To evaluate the temperature-dependent mechanism of uptake associated with Alexa546-labeled dsDNA and Fl-MSNPs in the MDA-MB-231 cells, the cells were cultured at a density of 20 000 cells per well in a 24-well plate containing 500 μL of media and maintained for 24 h at 37 °C with 5% CO2 in a humdified incubator. The cells were then treated with Alexa546-labeled dsDNA-Fl-MS-NPs (0.5 mL) at three different concentrations of MSNP (10, 20, or 30 μg/mL) and incubated for 4 h at 5% CO2 atmosphere at either 37 or 4 °C. Afterward, the cells were washed with phosphate buffer, followed by detachment of the cells using 0.25% trypsin–EDTA. The cells were then collected for analysis with the flow cytometer (BD LSR Fortessa cell analyzer) using green and red channels for fluorescein and Alexa546 fluorescence, respectively. Free nonlabeled and Alexa546-labeled dsDNAs were loaded using commercial Lipofectamine 2000 (L2 K) as controls.
For confocal laser scanning microscopy, the MDA-MB-231 cells at a density of 50 000 cells per well were seeded onto a coverslip placed in six-well plates and incubated at 37 °C with 5% CO2 atmosphere to promote adhesion. After incubation time of 24 h, the cells were treated with Alexa546-labeled dsDNA-Fl-MSNPs at a fixed MSNP concentration of 10 μg/mL in 2 mL of complete media for a period of 24 h. The cells were rinsed three times with cold PBS and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 15 min at 37 °C in a humified incubator. After an additional rinse with PBS, the coverslips were mounted onto the glass slides with mounting medium and images were acquired using an Olympus FluoView FV1000 confocal laser scanning microscope.
To determine intracellular localization, the MDA-MB-231 cells at a density of 5 × 106 cells per well were seeded onto a coverslip placed in six-well plates and maintained for 24 h at 37 °C with 5% CO2 atmosphere. IR700-labeled dsDNA-Fl-MSNPs (2 mL) were added at a fixed MSNP concentration of 10 μg/mL and incubated for another 6 h at 37 °C with 5% CO2 atmosphere. The cells were rinsed twice with PBS followed by incubation with either CellLight Early Endosomes-RFP or CellLight Late Endosomes-RFP (Molecular Probes, Invitrogen) for the labeling of early or late endosomes, respectively. First, the appropriate volume of the CellLight reagent was calculated for the number of cells using the following formula according to the manufacturer manual (https://www.thermofisher.com/order/catalog/product/C10589#/C10589)
where the number of cells is the estimated total number of cells at the time of labeling, PPC is the number of particles per cell (30 for this experiment), and 1 × 108 is the number of particles/mL of the reagent. Using the previous formula, it was determined that 90 μL of either CellLight Early Endosomes-RFP or CellLight late endosomes-RFP reagent was to be added to the cultured cells in growth media (2 mL) followed by gentle swirl for uniform mixing. The cells were incubated for an additional 16 h at 37 °C and 5% CO2 atmosphere. The microscopy images were obtained using an Olympus FluoView FV100 confocal laser scanning microscope.
2.9. Specific Gene Silencing.
The MDA-MB-231/GFP cells expressing green fluorescent protein (GFP) (Biolabs, Inc.) for both flow cytometry and microscopy experiments were treated with NA-MS-NPs, where all NANPs were functionalized with dicer substrate (DS) RNAs against GFP. For analysis with flow cytometry, the MDA-MB-231/GFP cells were cultured in 12-well plates at a seeding density of 20 000 cells per well and maintained for 24 h at 37 °C in a 5% CO2 atmosphere. The cells were transfected with NA-MS-NPs with a NANP concentration of 50 nM (ratio N/P = 10) and left for 72 h at 37 °C (5% CO2) in a humidified incubator. Double-stranded (DS) RNAs targeting GFP were loaded to MSNPs (dsRNA-MS-NPs) and used as a control. After the incubation period, the cells were rinsed twice with PBS to remove any unbound particles. The cells were then detached from the culture plate by adding 100 μL of cell dissociation buffer per well (Gibco). The suspended cells were collected gently shaken before analysis. The GFP expression level was determined by fluorescence activated cell sorting (FACS) (BD Bioscience). A minimum of 15 000 events were collected per sample using Cell Quest software. The data is reported as percent GFP silencing.
For microscopy analysis, the MDA-MB-231/GFP cells were seeded at a density of 10 000 cells per well in a 24-well plate containing 500 μL of media and maintained for 24 h at 37 °C with 5% CO2 in a humidified incubator. The cells were then incubated with NA-MS-NPs at a NANP concentration of 50 nM (ratio N/P = 10) for 72 h at 37 °C in a 5% CO2 atmosphere. DS RNAs targeting GFP loaded to MSNPs (dsRNA-MS-NPs) were used as control. The cells were washed twice with PBS and further incubated for 24 h in a fresh culture medium. Finally, the samples were imaged to assess the GFP expression with an EVOS FL imaging system (inverted four-color imaging system).
2.10. Cytotoxicity of NA-MS-NPs.
Nonfunctional NANPs were assembled to evaluate the cytotoxicity of NA-MS-NPs in the absence of therapeutic DS RNA. The MDA-MB-231 cells were cultured in a 96-well plate at a density of 5000 cells per well and maintained for 24 h at 37 °C and 5% CO2 atmosphere. The cells were then treated with DS RNAs and DS RNA-functionalized fNA-, rNA-, cNA-MS-NPs or DOX-MSNPs at concentrations of 10, 20, 30, or 50 μg/mL and left for 48 h at 37 °C with 5% CO2 atmosphere in a humidified incubator. After the incubation period, the cell media was aspirated, and the cells were carefully rinsed twice with cold PBS followed by addition of 100 μL of fresh growth media per well. The cells were incubated for another 24 h at 37 °C and 5% CO2 atmosphere. Later, all cells were washed with PBS and replenished with 100 μL of fresh media and 20 μL of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) solution. Following 2.5 h of incubation at 37 °C and 5% CO2, the cytotoxicity was assessed by measuring the relative absorbance of the treatment groups with respect to the nonexposed cells at 490 nm using a microplate reader. Cytotoxicity analysis for the A375 cells was carried out using a similar protocol. For the A375 cells, an initial seeding density of 2000 cells per well and complete Dulbecco’s modified Eagle’s medium (DMEM) cell culture media were used.
2.11. Evaluation of Combined Therapy: Cell Viability, Apoptosis, and Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR).
To assess gene silencing efficiency in combination with chemotherapy, DOX-MSNPs were loaded with either DS RNAs designed to target BCL2 or DS RNA-functionalized fNANPs. The A375 cells were seeded into 96-well plates at a density of 2000 cells per well and maintained for 24 h at 37 °C and 5% CO2 atmosphere. DOX-MSNPs loaded with either dsDNA or nonfunctional fNANPs were employed as controls. The A375 cells were transfected with MSNPs at selected concentrations of 10, 20, 30, or 50 μg/mL. After an incubation for 48 h at 37 °C and 5% CO2, the cell media was discarded, the treated cells were rinsed twice with cold PBS, and 100 μL of fresh complete media was added to the wells. After incubating for 24 h at 37 °C and 5% CO2 atmosphere, all wells were washed with PBS and replenished with 100 μL of fresh growth media and 20 μL of MTS solution. Following 2.5 h of incubation at 37 °C and 5% CO2, the cytotoxicity was assessed using a plate reader by measuring the relative absorbance of the treatment groups with respect to the nonexposed cells. Combination therapy was also evaluated for the MDA-MB-231 cells using the same protocol, but with an initial seeding density of 5000 cells per well and RPMI cell culture media.
To evaluate the apoptosis of NA-DOX-MS-MP materials, the A375 or MDA-MB-231 cells were cultured in 24-well plates at a density of 2 × 104 or 4 × 104 cells per well, respectively. The cells were maintained for 24 h at 37 °C and 5% CO2 atmosphere in a humidified incubator. The cells were then treated with PEG-PEI-MSNPs, anti-BCL2-fNA-DOX-MS-NPs, anti-BCL2-RNA-DOX-MS-NPs, nonfunctionalized fNA-DOX-MS-NPs, nontherapeutic dsDNA-DOX-MS-NPs, or DOX-MS-NPs at a concentration of 10 μg/mL for 24 h. The cells were then rinsed once with PBS, detached using trypsin, and centrifuged. The pellet was gently mixed in 600 μL of 1× binding buffer (BD Pharmingen, 556547) and washed once. The cells were then evaluated using an annexin/propidium iodide (PI) solution as described in the SI.
The knockdown of BCL2 expression was evaluated by quantitative PCR. The A375 or MDA-MB-231 cells were grown in six-well plates at a seeding density of 5 × 104 or 1 × 105 cells per well, respectively. The cells were maintained for 24 h at 37 °C and 5% CO2 atmosphere. The cells were then treated with PEG-PEI-MSNPs, anti-BCL2-fNA-DOX-MS-NPs, anti-BCL2-RNA-DOX-MS-NPs, nonfunctionalized fNA-DOX-MS-NPs, nontherapeutic dsDNA-DOX-MS-NPs, or DOX-MS-NPs at a concentration of 5 μg/mL for 24 h. The cells were rinsed once with PBS, trypsinized, and the cell pellet was collected via centrifugation.
The isolation of RNA from the cells was carried out using Direct-zol RNA miniprep (Zymo Research). The cell pellet was mixed with 200 μL of TRI reagent (Zymo Research). The samples were centrifuged at 13 000 rcf for 30 s and the supernatant was transferred to a new tube. To this supernatant, 95–100% ethanol was added at 1:1 dilution, and the solution was briefly vortexed and centrifuged. Then, the ethanol–supernatant mix was transferred to a Zymo spin column, which was placed in the collection tube. The samples were centrifuged and the flow-through from the column was discarded. The columns were then treated with DNase I (Zymo Research) for 15 min at room temperature. Four hundred microlitres of the RNA wash buffer was added to the column, centrifuged, and the flow-through was discarded. Next, 400 μL of Direct-zol RNA PreWash was added to the column, centrifuged, and the flow-through was discarded. This washing step was repeated once again and the columns were transferred to a new tube. Once more, 700 μL of RNA wash buffer was run through the column. To elute the RNA, nuclease-free water was added to the column and centrifuged. The RNA samples were stored at −20 °C.
The cDNA synthesis was performed using an iScript cDNA synthesis kit (1708890, Bio-Rad). The extracted RNA was mixed with iScript reaction mix, iScript reverse transcriptase, and nuclease-free water as per the manufacturer’s protocol. The complete reaction mix was incubated in a thermal cycler programmed with the following protocol: 5 min priming at 25 °C, 20 min reverse transcription at 46 °C, and 1 min RT inactivation at 95 °C. The contents were held at 4 °C. The concentration of the cDNA product was measured using the Nanodrop 2000.
After the cDNA synthesis, RT-PCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) and a CFX96 real-time system (Bio-Rad). A 20 μL solution was prepared using 1× SsoAdvanced Universal SYBR Green Supermix, 500 nM primers, and 10 ng of cDNA. The solution was then subjected to the following protocol: an initial step of 95 °C for 30 s for polymerase activation and DNA denaturation, followed by 35× cycles of [95 °C for 15 s, 60 °C for 30 s, and a fluorescence reading]. The mRNA expression relative to the untreated cells was then quantified in the Bio-Rad CFX manager software using the ΔΔCq method with a GAPDH reference. A melt curve was performed following PCR to confirm the accuracy of amplification (data not shown).
2.12. Statistical Analysis.
All experimental results in this study are reported as mean ± standard deviation (SD) unless mentioned otherwise. For the analysis of nanoparticle size using TEM, 160 nanoparticles were evaluated using ImageJ. The hydrodynamic size, ζ-potential, and Kaiser’s test were carried out in triplicate or more. The amount of DOX loaded was analyzed in triplicate using different batches of nanoparticles. Immune response experiments were carried out in triplicate and statistical analysis was evaluated using a one-way analysis of variance (ANOVA), followed by a Tukey’s multiple comparison test. Cellular uptake using flow cytometry was measured with a minimum of 5000 gated cells and quantified in triplicates. The statistical analysis was performed with a one-way ANOVA using Tukey’s multiple comparison test. For the cell viability studies, GraphPad Prism was used to calculate the EC50 values (n = 3). Statistical analysis was done by one-way ANOVA using Tukey’s multiple comparison test. All of the statistical analysis was performed using GraphPad prism (v8.2.0) with a p-value <0.05 considered to be statistically significant.
3. RESULTS
3.1. Synthesis and Characterization of MSNPs.
Our group and others have demonstrated the enormous potential of MSNPs for delivering a wide range of therapeutic and imaging agents.37,40–42 The vast and dynamic biomedical applicability of this MSNPs’ platform capitalizes on their high surface area, diverse and facile surface chemistry, good biocompatibility, and well-defined pore structure. To promote the delivery of nucleic acid-based materials, we have previously reported on a stepwise synthetic approach involving surface modification of MSNPs with polyethylenimine (PEI) and poly(ethylene glycol) (PEG) polymers (Figure 1).15,37 Herein, we used this platform for the delivery of NANPs. The physicochemical properties of MSNPs were fully characterized and are demonstrated in the Supporting Information (Figure S1 and Table S2). As expected, the as-made MSNPs were spherical in shape with a diameter of 41 ± 3 nm (n = 50) according to TEM, a hydrodynamic diameter of 73 ± 1 nm (n = 5), and a highly negative surface charge of −50 ± 4 mV (n = 5). The MSNPs presented a high surface area of 638 m2/g and a pore diameter of 2.2 nm as determined using nitrogen sorption isotherms. After functionalization with the PEI and PEG polymers, a dramatic shift in the ζ-potential value from negative (−50 ± 4 mV) to positive (+14 ± 1 mV) values was observed. In addition, an increase in the hydrodynamic diameter to 118 ± 10 nm (n = 5) was also determined. Both thermogravimetric analysis and Kaiser’s test confirmed the presence of the polymers with a weight loss of 20.3 ± 1.5 and 28.1 ± 0.9 wt % and the presence of primary amine groups with amounts of 1861 ± 545 and 1207 ± 327 nmol/mg after modification with PEI and PEG, respectively (Table S2).
To prepare the DOX-loaded PEG-PEI-MSNPs, the DOX molecules were loaded into nonfunctionalized MSNPs under acidic conditions, which enhances the electrostatic interaction between nonfunctionalized MSNPs and DOX molecules. The nonfunctionalized MSNPs’ surface is negatively charged, while DOX is positively charged due to the primary amine group in its structure.43 This approach resulted in a high DOX loading of 16.7 ± 1.6 wt % (n = 3) for the final DOX-loaded PEG-PEI-MSNPs, which is in close agreement to previous reports for the MSNP materials.44 Unless stated otherwise, PEG-PEI-MSNPs and DOX-loaded PEG-PEI-MSNPs are referred as MSNPs and DOX-MSNPs, respectively, in the rest of the manuscript for simplicity.
3.2. Optimization of Nucleic Acid Binding to MSNPs, pH-Dependent Release, and Enzymatic Stability.
All initial optimization experiments in solution were carried out using Alexa488-labeled DNAs instead of NANPs. The binding between nucleic acid materials and MSNPs relies on the electrostatic interaction between the negatively charged phosphate groups (P) of nucleic acids and positive amine groups (N) on the MSNPs.15,45 First, we established the optimal N/P ratio for our system as visualized in gel electrophoresis by varying the N/P ratio from 1 to 40 (Figure 2A). DNA duplexes fluorescently labeled with Alexa488 (DNA-Alexa488), which did not bind with MSNPs, migrated faster through the gel, while the mobility of the duplexes electrostatically complexed with MSNPs was limited. We found that for the N/P ratios larger than 10, there is complete binding between the DNA-Alexa488 and MSNPs as indicated by the lack of the free DNA-Alexa488 band. This result was further corroborated by the lack of the fluorescent signal in the supernatant after centrifugation of the DNA-Alexa488-MSNP material (Figure 2B). Previous reports have shown that N/P ratios >8 for this MSNP platform provide optimal electrostatic interactions.45 Therefore, we chose to use an N/P ratio of 10 for this work since it shows strong binding and maximum loading of nucleic acids.
Figure 2.

Nucleic acid interactions with MSNPs. (A) Gel electrophoresis shows the binding effect of DNA-Alexa488 to MSNP at various N/P ratios. The presence of the green band indicates the decrease in electrostatic complexation of DNA-Alexa488 to the MSNPs at lower N/P ratios. (B) Strong binding at N/P = 10 was corroborated by analyzing the DNA duplexes remaining in the supernatant solution after binding with MSNs. (C) Treatment of DNA duplexes with quenching pair Alexa488 and Iowa Black with and without MSNP conjugation confirms the protection from nuclease activity.
We also investigated the MSNPs’ ability to protect nucleic acids from enzymatic degradation using fluorescently quenched duplexes assembled with DNA strands labeled with an Alexa488 fluorophore at the 5′-side of one strand and a complementary strand with an Iowa Black quencher on the 3′-side. These fluorescently quenched DNA duplexes were treated with RQ1 DNase. If there is no enzymatic protection, the degradation of the duplexes and further spatial separation of the fluorophore and quencher would lead to activation of the fluorescence signal; however, in the absence of degradation, the Iowa Black completely quenched the fluorescence of Alexa488 owing to their close proximity.27 The DNA duplexes were complexed with MSNPs and treated with RQ1 DNase (Figure 2C). MSNPs successfully protected the DNA duplexes from DNase degradation, as evidenced by the insignificant increase in fluorescence. In comparison, a control experiment with only DNA duplexes treated with DNase showed a dramatic increase in the fluorescence signal.
Our data demonstrated that MSNPs can efficiently complex with and protect nucleic acids from nuclease degradations. Nevertheless, it is also important to show that the nucleic material is released once it reaches the desired site inside the cells. We hypothesized that the release of nucleic acids from MSNPs can be triggered by an acidic pH such as is found in endosomes or lysosomes. To examine this hypothesis, we performed the release of DNA-Alexa488 loaded to MSNPs in buffer solutions at pH 5.2 and 7.4. There is a remarkable release of about 60% of DNA-Alexa488 compared with the sample at pH 7.4 (Figure S2). This pH-triggered release can be explained by the disruption of the interaction of the phosphate groups in the backbone of the RNA or DNA and the amine groups in the PEI polymer in the presence of protons at an acidic pH.46
3.3. Formation of NA-MS-NPs and NANPs’ Integrity Studies upon Their Release from NA-MS-NPs.
Three representative NANPs (globular cNANPs, planar rNANPs, and fibrous fNANPs) were all synthesized via one-pot assembly under the same buffer conditions. cNANPs are formed via intermolecular Watson–Crick base pairing,14 while rNANP and fNANP designs both are assembled via the initial intramolecular formation of Watson–Crick base pairings that facilitate magnesium-dependent intermolecular kissing loop interactions (Figure 1A, right panel).47 Each structure has the capacity to carry several DS RNAs against a specific target gene such as GFP or BCL2. The assembly of the different NANPs was achieved by mixing equimolar amounts of the constituent strands and undergoing a process of heating and cooling, outlined in detail in the methods section. To assess the formation of the NANPs, AFM and gel electrophoresis experiments were carried out, demonstrating their morphologies and monodispersity (Figure 3A). AFM micrographs clearly showed the formation of each assembled NANP, while the single band observed via native-PAGE demonstrated their monodisperse assemblies.
Figure 3.

Physical characterization of NANPs, MSNPs, and NA-MS-NPs. (A) Atomic force microscopy images and electromobility shift assays of GFP-functionalized NANPs demonstrate uniformity and morphology. (B) Transmission electron microscopy images demonstrate size, shape, and distribution of NA-MS-NPs. (C) Complexation and release of NANPs from MSNPs demonstrated by competitive binding with heparin. NANPs are released and stay intact post complexation with MSNPs.
The formation of the NA-MS-NPs relies on the electrostatic interaction between the negative charge associated to the phosphate backbone of NANPs and the positively charged surface of MSNPs. Based on our optimization experiments described above, we maintained the N/P ratio of 10 for the assembly of NA-MS-NPs. TEM was used to visualize the NA-MS-NPs after staining the material with a solution of organo-tungstate. Tungstate-based compounds are commonly used for negative stain electron microscopy (EM), which is an accessible and convenient approach. The negative staining strategy works by using heavy metal compounds embedded in a thin layer of biological macromolecules like proteins or nucleic acids to enable high contrast images of their morphologies.48,49 The TEM images for the NA-MS-NPs depicted a denser surface from the negative staining as an indication of the presence of NANPs on the surface of the MSNPs (Figure 3B). However, we were not able to distinguish the different morphology or shapes of the NANPs, which can be attributed to the drawbacks of negative stain EM.
To determine the ability of MSNPs to deliver intact NANPs, a heparin competition assay was carried out to disrupt the electrostatic interactions between NANPs and MSNPs.27 The highly negative charge of heparin outcompetes the NANPs’ binding with MSNPs, resulting in the release of the NANPs. Gel electrophoresis was used to evaluate the release of the NANPs (Figure 3C). The native-PAGE image clearly shows that the bands, corresponding to either cNANPs or rNANPs after their release from MSNPs in the presence heparin, travel a similar distance as the original NANPs. This is a clear demonstration that MSNPs can effectively carry and release NANPs without affecting their morphology. Due to the size of fNANPs and their inability to enter the gel, they were not evaluated using this technique.
3.4. Immunostimulation by NA-MS-NPs In Vitro.
Recently, we discovered that when NANPs are used with a delivery agent, the dimensionality, size, and composition of NANPs dictate their immunostimulatory properties.15,18,19,23,27,50 Therefore, to further characterize the immunostimulatory effects of NA-MS-NPs, we used human monocytic cells, THP1-Dual (InvivoGen), engineered to express secreted alkaline phosphatase (SEAP) and luciferase in response to NF-κB and IRF stimulation, respectively. We also utilized HEK-Blue hTLR cells to address the contributions from specific receptors. These model systems present a simple and straightforward way to measure immune signaling from nucleic acids. Poly I:C and R848, which are known inducers of immune response, were used as positive controls in these studies. We observed differential immune stimulation for MSNPs carrying different NANPs containing DS RNAs against GFP. Specifically, MSNPs modified with cNANPs elicited the greatest response in the THP1-Dual cells in the IRF pathway by exhibiting the highest production of interferons, which was consistent with the previous results.18,27 Both TLR3 and 7 are responsible for RNA detection in the endosomal compartments, with TLR3 recognizing dsRNA and TLR7 responsible for ssRNA detection.51 Our data show that all of the NA-MS-NPs generated a response in the HEK-Blue hTLR3 cells; however, MSNPs by themselves also initiated an immune response, which was not predicted with this cell line (Figure 4A). Upon contacting the manufacturer, it was determined that the HEK-Blue hTLR3 cells express a background level of endogenous TLR5, which can be activated by PEI.52,53 Furthermore, the NF-κB stimulation in the THP1-Dual cells may also be attributed to the presence of PEI on the nanoparticles (Figure 4D). cNA-MS-NPs were able to activate TLR7, while the other NA-MS-NPs were not (Figure 4B). Additionally, these cNA-MS-NPs were demonstrated to provoke an interferon response (Figure 4C). This particular performance of cNANPs has already been observed for polymeric delivery agents.33 These results demonstrate that not only is determining the immune response of NANPs themselves important, but also that NANPs associated with a carrier can have a major impact in the overall immune response of the nanocomplex. Interestingly, this concept also adds an additional layer of customizability for the therapeutic activity of NA-MS-NPs wherein the NANPs can be used as either a traditional therapeutic (RNAi, aptamer, antisense, etc.) or as an adjuvant and the MSNPs can also be customized to produce an additive immune response.
Figure 4.

Immunostimulatory properties of MSNPs carrying GFP-functionalized NANPs treated in (A) HEK-Blue hTLR3, (B) HEK-Blue hTLR7, (C) THP1-Dual IRF pathway, and (D) THP1-Dual NF-κB pathway cells demonstrate the cellular pathways, which are activated by different NANPs. Statistics: one-way ANOVA using Tukey’s multiple comparison test was performed between different groups to determine the statistical difference. *p ≤ 0.05.
3.5. Cellular Uptake and Colocalization Studies for dsDNA-Loaded MSNPs.
Our group and others have previously demonstrated the ability of MSNPs to deliver nucleic acid materials.15,37,54 Nevertheless, in this work for the first time, we have shown the delivery of NANPs of different shapes using MSNPs. To carry out these experiments, NANPs and MSNPs were labeled with Alexa546 and fluorescein (Fl), respectively. Flow cytometry was used to assess the efficiency of internalization of the Alexa546-labeled-NA-Fl-MS-NPs in the MDA-MB-231 cancer cells. The flow cytometry data show comparable internalization of all NA-Fl-MS-NPs regardless of the NANPs’ morphology (Figure 5A). These results show that MSNPs can efficiently deliver NANPs regardless of the shape. In previous reports for the case of the polymeric nanoparticles, it is shown that the shape is an important factor for their internalization.55 However, it appears that this is not the case for the NA-MS-NPs, most likely owing to the strong electrostatically driven interaction between both nanoparticles. Control experiments of NANPs in the absence of any delivery vector show no internalization of the nanoparticles (Figure S3A).
Figure 5.

MDA-MB-231 cells uptake Alexa546-labeled NANPs or dsDNA loaded to Fl-MSNPs after inoculation for 24 h. (A) Mean fluorescence intensity associated to NANPs (gray) and Fl-MSNPs (black) obtained from flow cytometry experiments. (B) Mean fluorescence intensity associated to dsDNA (gray) and Fl-MSNPs (black) at different concentrations of the nanoparticle complex obtained from flow cytometry experiments. (C) Confocal micrographs of the MDA-MB-231 cells inoculated with Alexa546-labeled dsDNA-loaded Fl-MSNPs (10 μg/mL) for 24 h. The cell nuclei are observed in the blue channel after staining with Hoechst 33342 (C1). The fluorescence in the fluorescein isothiocyanate (FITC) (green) channel (C2) indicates the localization of Fl-MSNPs. The fluorescence in the tetramethylrhodamine (TRITC) (red) channel shows the presence of Alexa546-labeled dsDNA (C4). The merged micrographs (C4, C5) show the colocalization and localization of Alexa546-labeled dsDNA-loaded Fl-MSNs inside the MDA-MD-231 cells. The insets (C6, C7) clearly demonstrate the release of Alexa546-labeled dsDNA from Fl-MSNPs. Scale bar = 40 μm. Statistics: a one-way ANOVA was performed between different groups to determine the statistical difference. ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05.
We conducted a deeper investigation of the cellular uptake and colocalization of MSNPs in the MDA-MB-231 cells. For cost efficiency, these experiments were carried out using Alexa546-labeled dsDNA rather than NANPs. Similar to the results found for NA-MS-NPs, the data showed that both the Alexa546-labeled dsDNA and Fl-MSNPs are efficiently internalized by the MDA-MB-231 cells (Figure 5B). Control experiments with Alexa546-labeled dsDNA in the absence of Fl-MSNPs showed no internalization of DNA (Figure S3B). These data were further confirmed by confocal microscopy. Confocal images show the presence of Fl-MSNPs (green) and Alexa546-labeled dsDNA (red) inside the cells. It is clear after merging the green and red channels with the differential interference contrast (DIC) that most of the Alexa546-labeled dsDNA-loaded Fl-MSNs (yellow) have been internalized by the cells (Figure 5C1–C5). This is convincing evidence that the MSNPs successfully carried the dsDNA across the cell membrane. In addition, we detected spots in the micrographs where the green and red fluorescence do not completely overlap, which can be indicative that dsDNA has been released from the MSNPs (Figure 5C6,C7). As described above, we hypothesize that the main mechanism to account for the release of dsDNA is the displacement of dsDNA driven by acidic pH in organelles associated to the endolysosomal pathway.56
MSNPs have already been shown to be internalized through the endocytic mechanisms and to be trafficked by the endolysosomal pathway.57 A temperature-dependent experiment was carried out to assess whether the internalization of Alexa546-labeled dsDNA-loaded Fl-MSNPs follows an energy-dependent or passive mechanism. Figure S4 shows that the internalization of Alexa546-labeled dsDNA-loaded Fl-MSNPs is reduced under low temperature (4 °C) when compared to physiological temperature (37 °C), thus confirming that these complexes are indeed internalized by the cells through endocytic pathways.58
To determine the colocalization of dsDNA duplexes loaded to MSNPs inside the cells, early endosomes, late endosomes, or lysosomes were stained using CellLight Early Endosomes-RFP, CellLight Late Endosomes-RFP, or LysoTracker, respectively. For this experiment, to avoid any overlap with the organelle markers, DNA duplexes were labeled with IR700 fluorophore (IR700-labeled dsDNA). Confocal micrographs showed that Fl-MSNPs are co-localized with early endosomes, late endosomes, or lysosomes (Figure 6A–C). However, major localization is observed for lysosomes (Figure 6C). In several instances, the nanoparticles were not co-localized with any of the organelles as an indication of endolysosomal escape, most likely due to the so-called “proton sponge effect” associated to the PEI polymers.56 Moreover, similar to what was shown above, the release of IR700-labeled DNA duplexes is also corroborated in these experiments. Overall, these results demonstrate that the nanoconstructs are efficiently endocytosed by the MDA-MB-231 cells, transported through the endolysosomal pathway, escape from endosomes/lysosomes most likely owing to the “proton sponge effect”, and deliver DNA duplexes in the cytoplasm.
Figure 6.

Confocal micrographs of the MDA-MB-231 cells inoculated with NIR700-labeled dsDNA-loaded Fl-MSNPs (10 μg/mL) for 6 h. The fluorescence in the FITC (green) channel indicates the localization of Fl-MSNPs. The fluorescence in the TRITC (red) channel shows the labeling of organelles: CellLight Early Endosomes-RFP (A), CellLight Late Endosomes-RFP (B), or LysoTracker (C). The presence of IR700-labeled dsDNA is indicated in purple. The merged micrographs show the colocalization and localization of IR700-labeled dsDNA-loaded Fl-MSNPs inside the MDA-MB-231 cells. The insets clearly demonstrate the escape from endosomes/lysosomes and the release of IR700-labeled dsDNA from Fl-MSNPs. Scale bar = 40 and 20 μm (insets).
3.6. Specific Gene Silencing by NA-MS-NPs.
We have previously established that NANPs functionalized with DS RNA against GFP silence its expression when transfected into GFP-expressing human cell lines using magnetic nanoparticles or polymeric micelles.27,28 In this work, we assessed the NA-MS-NPs’ silencing efficacy using the MDA-MB-231 cell line modified to overexpress GFP. NANPs were functionalized with RNA duplexes against GFP. In these experiments, NA-MS-NPs depicted silencing-dependent efficacy based on the shape of NANPs (Figure 7A). The fNANPs and DS RNAs showed the higher knockdown efficiencies against GFP with 54 and 68% silencing, respectively, when compared to other NA-MS-NPs (one-way ANOVA, p < 0.05). In the case of cNANPs and rNANPs, silencing efficiencies of 40 and 33% were determined. Previous reports using polymeric micelles or magnetic nanoparticles as vectors did not show differences in GFP silencing efficiency depending on NANPs’ dimensionality.27,28 Considering that, based on our results, all NA-MS-NPs internalize into the cells with a similar efficiency and all NANPs carry an equal number of DS RNAs, the difference in silencing found in this study is most likely due to differences in electrostatic binding between NANPs and MSNPs, which can influence the intracellular release of NANPs. We hypothesize that cNANPs and rNANPs are likely to have stronger bindings to MSNPs than fNANPs or DS RNAs. A stronger electrostatic binding will result in fewer released NANPs, which will impact their processing by dicer and final knockdown of the target protein.
Figure 7.

(A) Percent GFP expression post treatment with DS RNA and MSNPs obtained using flow cytometry. (B) Fluorescence microscopy imaging for GFP gene silencing in the MDA-MB-231 cells/GFP. The controls cells show bright field and nontreated. The cells were treated with anti-GFP NA-MS-NPs. Scale bar = 400 μm. Statistics: one-way ANOVA was performed between different groups to determine the statistical difference. ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05.
Furthermore, fluorescence microscopy was utilized to validate the data obtained by flow cytometric analysis. As depicted by the negative control experiments, a significantly higher number of the MDA-MB-231/GFP cells expressing green fluorescent protein was silenced after transfection with NA-MS-NPs (Figure 7B). The fluorescence micrographs evidently illustrate a significant decrease in the expression of GFP after transfecting the MDA-MB-231/GFP cells with NA-MS-NPs. Based on our experimental results, fNANPs were the most efficient for gene silencing and also showed a reduced immunostimulatory effect.
3.7. Combination Therapy Using fNA-MS-NPs.
Despite all of the advantages of using NANPs for RNAi therapy, only few reports of the co-delivery of NANP/chemotherapeutic drugs have been published.3,22,26 Among different nanocarriers, MSNPs have shown remarkable features for the efficient co-delivery of siRNA and chemotherapeutic agents. This platform has been successfully administered as an effective gene delivery vector in different cancer models.59,60 As a proof of principle to demonstrate the therapeutic ability of the NA-MS-NPs for combined therapy, silencing of the antiapoptotic gene BCL2 in combination with doxorubicin (DOX) was evaluated. BCL2 is an attractive oncogene target because it activates the cellular antiapoptotic defense, which is one of the main mechanisms of cancer resistance.61 Inhibition of BCL2 enhances the sensitivity of the cancer cells to standard therapies,62 hence the importance of this gene as a promising therapeutic target in several human cancers. DOX, which is part of the family of anthracyclines drugs, mainly acts as a DNA intercalator that triggers apoptosis. Interestingly, recent investigations have shown that the combination of DOX with BCL2 inhibitors can be a promising treatment modality for TNBC and melanoma patients.63,64 Therefore, by combining NANPs that target the synthesis of BCL2 protein with DOX, we anticipate to have a major impact on cell survival.65–67
To evaluate the combination therapy of DOX and RNAi, inducers targeting BCL2 using the NA-MS-NPs, MDA-MB-231, and A375, which are triple-negative breast and skin cancer cell lines, were used. The MDA-MB-231 and A375 cells both overexpress BCL2.68,69 First, to rule out the possibility of any cytotoxicity associated to nonfunctionalized NANPs loaded to MSNPs, the viability of these cells in the presence of the nanoconstructs was tested using an MTS assay. The results showed a slight cytotoxicity due to the carrier at the tested concentrations, but no differences in growth inhibition for the nonfunctional NA-MS-NPs, indicating that the nonfunctionalized NANPs are not cytotoxic for either cell lines (Figure S5). In addition, we also evaluated any possible synergy between the nonfunctionalized NANPs and DOX-MSNPs. The cell viability results showed that the growth inhibition is only associated to the therapeutic effect of DOX, but does not dependent on the NANPs (Figure S6).
Our viability results demonstrated that there is no cytotoxic effect associated with the dimensionality of NANPs. Therefore, the fNANPs, which demonstrate a reduced immunostimulation effect and higher gene silencing as compared to other NANPs, were selected for the combined therapy. To test the combination therapy of BCL2 silencing and DOX in the A375 and MDA-MB-231 cell lines, fNANPs containing DS RNAs against BCL2 were engineered and complexed to DOX-MSNPs. As control experiments for this study, anti-BCL2 DS RNAs, nontherapeutic DNA duplexes, and nonfunctionalized fNANPs complexed with DOX-MSNPs were used. The viability results in both cell lines depict a concentration-dependent cytotoxic effect associated to the nanoparticles. It is observed that DOX is the main factor on the therapeutic outcome against both cell lines (Figure S7). Previous reports using the same combination have shown a similar trend.67,70 Nevertheless, in a closer look at specific concentrations, we found additive effects induced by anti-BCL2-fNA-DOX-MSNPs for both cell lines. In the case of the A375 cells at the concentration of 10 μg/mL, there is an evident cooperative effect between DOX and anti-BCL2-fNANPs, as shown in Figure 8A. The cytotoxic effect of the anti-BCL2-fNA-DOX-MS-NP platform was higher than DOX-MSNs, nontherapeutic DS DNA with DOX-MSNPs, or nonfunctionalized fNA-DOX-MS-NPs (one-way ANOVA, p < 0.01). To confirm this enhanced therapeutic effect, the number of apoptotic cells was analyzed using the annexin V assay (Figure 8B). A higher percentage of the apoptotic cells was observed for the anti-BCL2-fNA-DOX-MS-NPs as compared with the control groups (one-way ANOVA, p < 0.0001). We also studied the ability of this platform to silence the targeted mRNA expression associated to BCL2 with quantitative reverse transcriptase-polymerase chain reaction (RT-PCR). As shown in Figure 8C, the anti-BCL2-fNA-DOX-MS-NP platform suppressed the BCL2 mRNA level by half based on the control cells. This BCL2 mRNA suppression was better than nontherapeutic DS DNA with DOX-MSNPs or nonfunctionalized fNA-DOX-MS-NPs (one-way ANOVA, p < 0.01). Similarly, for the MDA-MB-231 cells, at the 50 μg/mL concentrations, a better cytotoxic effect for anti-BCL2-fNA-DOX-MS-NPs was observed as compared to DOX-MSNPs, nontherapeutic dsDNA-DOX-MSNPs, or nonfunctionalized fNA-DOX-MS-NPs (one-way ANOVA, p < 0.05) (Figure 8D). This platform also showed a higher number of the apoptotic cells than the control groups (Figure 8E). Additionally, the anti-BCL2-fNA-DOX-MS-NPs suppressed the BCL2 mRNA level by 25% compared to the control cells (Figure 8F). For both cell lines, the DS RNA targeting BCL2 loaded to DOX-MSNPs showed less cytotoxicity, apoptotic cells, and BCL2 mRNA suppression to anti-BCL2-fNA-DOX-MS-NPs, but it is not statistically significant.
Figure 8.

Cytotoxicity, apoptosis, and RT-PCR results of anti-BCL2-fNA-DOX-MS-NPs (gray/stripes), anti-BCL2-RNA-DOX-MS-NPs (light gray/stripes), nonfunctionalized fNA-DOX-MS-NPs (gray), nontherapeutic dsDNA-DOX-MS-NPs (light gray), and DOX-MS-NPs (dark gray) for the A375 cells (A–C) and for the MDA-MB-231 cells (D–F). Statistics: one-way ANOVA was performed between different groups to determine the statistical difference. ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05.
Our results demonstrated that the shape (either globular, planar, or fibrous) of NANPs does not have an impact on the cytotoxicity of bare nor DOX-loaded MSNPs. Nevertheless, when DS RNA or anti-BCL2-fNANPs are loaded to DOX-MSNPs, a clear additive effect with the chemotherapeutic drug is observed at specific concentrations.
4. CONCLUSIONS
We have evaluated and optimized the use of MSNPs as an efficient carrier for the delivery of NANPs. We demonstrated that the silencing efficacy and immunostimulatory activity are significantly impacted by the shape of NANPs. fNANPs demonstrate reduced immunostimulatory effects and greater gene silencing efficacy as compared to planar or globular NANPs. Nevertheless, the cytotoxicity of the NA-MS-NPs is not affected by the morphology of NANPs. fNANPs were used as a proof of principle to evaluate the combination of siRNA targeting BCL2 and the chemotherapeutic drug doxorubicin. An additive effect was determined for both A375 and MDA-MB-231 cell lines. The present results suggest that this novel platform has great potential for the combinatorial therapy of cancer.
Supplementary Material
ACKNOWLEDGMENTS
R.J. and H.V. thank the financial support from the Livingstone Endowment and Thomas Reynolds Graduate Research Award. J.D. and J.M. acknowledge summer support from NSF-REU and DOD-ASSURE. The research reported in this publication was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM120487 (to K.A.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The authors would also like to thank Drs. Alexander Lushnikov and Alexey Krasnoslobodtsev for performing AFM imaging of the nanoparticles at the Nanoimaging Core Facility at the University of Nebraska Medical Center and Lauren Lee (UNCC) for her assistance with the cell culture experiments.
Funding
NSF-REU DMR-1757619 and CBES award (UNC Charlotte); National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM120487.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c07106.
Extended materials section, synthesis of MSNPs and DOX-MSNPs, Kaiser’s test, cell culture, all sequences used in this work, and Supporting Figures S1–S7 and Tables S1 and S2 (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsami.0c07106
Contributor Information
Ridhima Juneja, Department of Chemistry, The University of North Carolina at Charlotte, Charlotte, North Carolina 28223, United States.
Hemapriyadarshini Vadarevu, Department of Chemistry and Nanoscale Science Program, The University of North Carolina at Charlotte, Charlotte, North Carolina 28223, United States.
Justin Halman, Department of Chemistry and Nanoscale Science Program, The University of North Carolina at Charlotte, Charlotte, North Carolina 28223, United States.
Mubin Tarannum, Department of Chemistry and Nanoscale Science Program, The University of North Carolina at Charlotte, Charlotte, North Carolina 28223, United States.
Lauren Rackley, Department of Chemistry, The University of North Carolina at Charlotte, Charlotte, North Carolina 28223, United States.
Jacob Dobbs, Department of Chemistry, The University of North Carolina at Charlotte, Charlotte, North Carolina 28223, United States.
Jose Marquez, Department of Chemistry, The University of North Carolina at Charlotte, Charlotte, North Carolina 28223, United States.
Morgan Chandler, Department of Chemistry and Nanoscale Science Program, The University of North Carolina at Charlotte, Charlotte, North Carolina 28223, United States.
Kirill Afonin, Department of Chemistry, Nanoscale Science Program, and The Center for Biomedical Engineering and Science, The University of North Carolina at Charlotte, Charlotte, North Carolina 28223, United States.
Juan L. Vivero-Escoto, Department of Chemistry, Nanoscale Science Program, and The Center for Biomedical Engineering and Science, The University of North Carolina at Charlotte, Charlotte, North Carolina 28223, United States.
REFERENCES
- (1).Elbashir SM; Harborth J; Lendeckel W; Yalcin A; Weber K; Tuschl T Duplexes of 21-nucleotide RNAs Mediate RNA Interference in Cultured Mammalian Cells. Nature 2001, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- (2).Bumcrot D; Manoharan M; Koteliansky V; Sah DW RNAi therapeutics: A Potential New Class of Pharmaceutical Drugs. Nat. Chem. Biol. 2006, 2, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Adams D; Gonzalez-Duarte A; O’Riordan WD; Yang CC; Ueda M; Kristen AV; Tournev I; Schmidt HH; Coelho T; Berk JL; Lin KP; Vita G; Attarian S; Plante-Bordeneuve V; Mezei MM; Campistol JM; Buades J; Brannagan TH 3rd; Kim BJ; Oh J; Parman Y; Sekijima Y; Hawkins PN; Solomon SD; Polydefkis M; Dyck PJ; Gandhi PJ; Goyal S; Chen J; Strahs AL; Nochur SV; Sweetser MT; Garg PP; Vaishnaw AK; Gollob JA; Suhr OB Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [DOI] [PubMed] [Google Scholar]
- (4).Chan A; Liebow A; Yasuda M; Gan L; Racie T; Maier M; Kuchimanchi S; Foster D; Milstein S; Charisse K; Sehgal A; Manoharan M; Meyers R; Fitzgerald K; Simon A; Desnick RJ; Querbes W Preclinical Development of a Subcutaneous ALAS1 RNAi Therapeutic for Treatment of Hepatic Porphyrias Using Circulating RNA Quantification. Mol. Ther.—Nucleic Acids 2015, 4, No. e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mansoori B; Sandoghchian Shotorbani S; Baradaran B RNA interference and its Role in Cancer Therapy. Adv. Pharm. Bull. 2014, 4, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pecot CV; Calin GA; Coleman RL; Lopez-Berestein G; Sood AK RNA Interference in the Clinic: Challenges and Future Directions. Nat. Rev. Cancer 2011, 11, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Guo P The Emerging Field of RNA Nanotechnology. Nat. Nanotechnol. 2010, 5, 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Parlea L; Puri A; Kasprzak W; Bindewald E; Zakrevsky P; Satterwhite E; Joseph K; Afonin KA; Shapiro BA Cellular Delivery of RNA Nanoparticles. ACS Comb. Sci. 2016, 18, 527–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Afonin KA; Kasprzak W; Bindewald E; Puppala PS; Diehl AR; Hall KT; Kim TJ; Zimmermann MT; Jernigan RL; Jaeger L; Shapiro BA Computational and Experimental Characterization of RNA Cubic Nanoscaffolds. Methods 2014, 67, 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Afonin KA; Kireeva M; Grabow WW; Kashlev M; Jaeger L; Shapiro BA Co-transcriptional Assembly of Chemically Modified RNA Nanoparticles Functionalized with siRNAs. Nano Lett. 2012, 12, 5192–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kireeva ML; Afonin KA; Shapiro BA; Kashlev M In Cotranscriptional Production of Chemically Modified RNA Nanoparticles. RNA Nanostructures; Bindewald E; Shapiro B, Eds.; Methods in Molecular Biology; Humana Press: New York, NY, 2017; Vol. 1632, pp 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Afonin KA; Grabow WW; Walker FM; Bindewald E; Dobrovolskaia MA; Shapiro BA; Jaeger L Design and Self-assembly of siRNA-functionalized RNA Nanoparticles for use in Automated Nanomedicine . Nat. Protoc. 2011, 6, 2022–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Afonin KA; Kasprzak WK; Bindewald E; Kireeva M; Viard M; Kashlev M; Shapiro BA in silico Design and Enzymatic Synthesis of Functional RNA Nanoparticles. Acc. Chem. Res. 2014, 47, 1731–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Afonin KA; Bindewald E; Yaghoubian AJ; Voss N; Jacovetty E; Shapiro BA; Jaeger L in vitro Assembly of Cubic RNA-based Scaffolds Designed in silico. Nat. Nanotechnol. 2010, 5, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Rackley L; Stewart JM; Salotti J; Krokhotin A; Shah A; Halman J; Juneja R; Smollett J; Lee L; Roark B; Viard M; Tarannum M; Vivero-Escoto JL; Johnson P; Dobrovolskaia MA; Dokholyan NV; Franco E; Afonin KA RNA Fibers as Optimized Nanoscaffolds for siRNA Coordination and Reduced Immunological Recognition. Adv. Funct. Mater. 2018, 28, No. 1805959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ke W; Hong E; Saito RF; Rangel MC; Wang J; Viard M; Richardson M; Khisamutdinov EF; Panigaj M; Dokholyan NV; Chammas R; Dobrovolskaia MA; Afonin KA RNA-DNA Fibers and Polygons with Controlled Immunorecognition Activate RNAi, FRET and Transcriptional Regulation of NF-kappaB in Human Cells. Nucleic Acids Res. 2019, 47, 1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chandler M; Afonin KA Smart-Responsive Nucleic Acid Nanoparticles (NANPs) with the Potential to Modulate Immune Behavior. Nanomaterials 2019, 9, No. 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hong E; Halman JR; Shah AB; Khisamutdinov EF; Dobrovolskaia MA; Afonin KA Structure and Composition Define Immunorecognition of Nucleic Acid Nanoparticles. Nano Lett. 2018, 18, 4309–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hong E; Halman JR; Shah A; Cedrone E; Truong N; Afonin KA; Dobrovolskaia MA Toll-Like Receptor-Mediated Recognition of Nucleic Acid Nanoparticles (NANPs) in Human Primary Blood Cells. Molecules 2019, 24, No. 1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chandler M; Johnson MB; Panigaj M; Afonin KA Innate Immune Responses Triggered by Nucleic Acids Inspire the Design of Immunomodulatory Nucleic Acid Nanoparticles (NANPs). Curr. Opin. Biotechnol. 2020, 63, 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Khisamutdinov EF; Li H; Jasinski DL; Chen J; Fu J; Guo P Enhancing Immunomodulation on Innate Immunity by Shape Transition among RNA Triangle, Square and Pentagon Nanovehicles. Nucleic Acids Res. 2014, 42, 9996–10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Guo S; Xu C; Yin H; Hill J; Pi F; Guo P Tuning the Size, Shape and Structure of RNA Nanoparticles for Favorable Cancer Targeting and Immunostimulation. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2020, 12, No. e1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Halman JR; Satterwhite E; Roark B; Chandler M; Viard M; Ivanina A; Bindewald E; Kasprzak WK; Panigaj M; Bui MN; Lu JS; Miller J; Khisamutdinov EF; Shapiro BA; Dobrovolskaia MA; Afonin KA Functionally-interdependent Shape-switching Nanoparticles with Controllable Properties. Nucleic Acids Res. 2017, 45, 2210–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Afonin KA; Bindewald E; Kireeva M; Shapiro BA Computational and Experimental Studies of Reassociating RNA/DNA Hybrids Containing Split Functionalities. Methods Enzymol. 2015, 553, 313–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Roark BK; Tan LA; Ivanina A; Chandler M; Castaneda J; Kim HS; Jawahar S; Viard M; Talic S; Wustholz KL; Yingling YG; Jones M; Afonin KA Fluorescence Blinking as an Output Signal for Biosensing. ACS Sens 2016, 1, 1295–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Alibakhshi MA; Halman JR; Wilson J; Aksimentiev A; Afonin KA; Wanunu M Picomolar Fingerprinting of Nucleic Acid Nanoparticles Using Solid-State Nanopores. ACS Nano 2017, 11, 9701–9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Halman JR; Kim KT; Gwak SJ; Pace R; Johnson MB; Chandler MR; Rackley L; Viard M; Marriott I; Lee JS; Afonin KA A Cationic Amphiphilic Co-polymer as a Carrier of Nucleic Acid Nanoparticles (Nanps) for Controlled Gene Silencing, Immunostimulation, and Biodistribution. Nanomedicine 2020, 23, No. 102094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cruz-Acuña M; Halman JR; Afonin KA; Dobson J; Rinaldi C. Magnetic Nanoparticles Loaded with Functional RNA Nanoparticles. Nanoscale 2018, 10, 17761–17770. [DOI] [PubMed] [Google Scholar]
- (29).Vivero-Escoto JL; Slowing II; Trewyn BG; Lin VS Mesoporous Silica Nanoparticles for Intracellular Controlled Drug Delivery. Small 2010, 6, 1952–1967. [DOI] [PubMed] [Google Scholar]
- (30).Paris JL; Baeza A; Vallet-Regi M Overcoming the Stability, Toxicity, and Biodegradation Challenges of Tumor Stimuli-Responsive Inorganic Nanoparticles for Delivery of Cancer Therapeutics. Expert Opin. Drug Delivery 2019, 16, 1095–1112. [DOI] [PubMed] [Google Scholar]
- (31).Baeza A; Ruiz-Molina D; Vallet-Regi M Recent Advances in Porous Nanoparticles for Drug Delivery in Antitumoral Applications: Inorganic Nanoparticles and Nanoscale Metal-Organic Frameworks. Expert Opin. Drug Delivery 2017, 14, 783–796. [DOI] [PubMed] [Google Scholar]
- (32).Vivero-Escoto JL; Huxford-Phillips RC; Lin W Silica-based Nanoprobes for Biomedical Imaging and Theranostic Applications. Chem. Soc. Rev. 2012, 41, 2673–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Croissant JG; Fatieiev Y; Khashab NM Degradability and Clearance of Silicon, Organosilica, Silsesquioxane, Silica Mixed Oxide, and Mesoporous Silica Nanoparticles. Adv. Mater. 2017, 29, No. 1604634. [DOI] [PubMed] [Google Scholar]
- (34).Castillo RR; Baeza A; Vallet-Regi M Recent Applications of the Combination of Mesoporous Silica Nanoparticles with Nucleic Acids: Development of Bioresponsive Devices, Carriers and Sensors. Biomater. Sci 2017, 5, 353–377. [DOI] [PubMed] [Google Scholar]
- (35).Pinese C; Lin J; Milbreta U; Li M; Wang Y; Leong KW; Chew SY Sustained Delivery of siRNA/Mesoporous Silica Nanoparticle Complexes from Nanofiber Scaffolds for Long-Term Gene Silencing. Acta Biomater. 2018, 76, 164–177. [DOI] [PubMed] [Google Scholar]
- (36).Juneja R; Lyles Z; Vadarevu H; Afonin KA; Vivero-Escoto JL Multimodal Polysilsesquioxane Nanoparticles for Combinatorial Therapy and Gene Delivery in Triple-Negative Breast Cancer. ACS Appl. Mater. Interfaces 2019, 11, 12308–12320. [DOI] [PubMed] [Google Scholar]
- (37).Vivero-Escoto JL; Vadarevu H; Juneja R; Schrum LW; Benbow JH Nanoparticle Mediated Silencing of Tenascin C in Hepatic Stellate Cells: Effect on Inflammatory Gene Expression and Cell Migration. J. Mater. Chem. B 2019, 7, 7396–7405. [DOI] [PubMed] [Google Scholar]
- (38).Shlyakhtenko LS; Gall AA; Lyubchenko YL Mica Functionalization for Imaging of DNA and Protein-DNA Complexes with Atomic Force Microscopy. Methods Mol. Biol. 2013, 931, 295–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Shlyakhtenko LS; Gall AA; Filonov A; Cerovac Z; Lushnikov A; Lyubchenko YL Silatrane-Based Surface Chemistry for Immobilization of DNA, Protein-DNA Complexes and Other Biological Materials. Ultramicroscopy 2003, 97, 279–287. [DOI] [PubMed] [Google Scholar]
- (40).Vivero-Escoto JL; Elnagheeb M Mesoporous Silica Nanoparticles Loaded with Cisplatin and Phthalocyanine for Combination Chemotherapy and Photodynamic Therapy in vitro. Nanomaterials 2015, 5, 2302–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Dreáu D; Moore LJ; Alvarez-Berrios MP; Tarannum M; Mukherjee P; Vivero-Escoto JL. Mucin-1-Antibody-Conjugated Mesoporous Silica Nanoparticles for Selective Breast Cancer Detection in a Mucin-1 Transgenic Murine Mouse Model. J. Biomed. Nanotechnol. 2016, 12, 2172–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Alvarez-Berríos MP; Vivero-Escoto JL in vitro Evaluation of Folic Acid-Conjugated Redox-Responsive Mesoporous Silica Nanoparticles for the Delivery of Cisplatin. Int. J. Nanomed. 2016, 11, 6251–6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shen J; He Q; Gao Y; Shi J; Li Y Mesoporous Silica Nanoparticles Loading Doxorubicin Reverse Multidrug Resistance: Performance and Mechanism. Nanoscale 2011, 3, 4314–4322. [DOI] [PubMed] [Google Scholar]
- (44).Shahabi S; Doscher S; Bollhorst T; Treccani L; Maas M; Dringen R; Rezwan K Enhancing Cellular Uptake and Doxorubicin Delivery of Mesoporous Silica Nanoparticles via Surface Functionalization: Effects of Serum. ACS Appl. Mater. Interfaces 2015, 7, 26880–26891. [DOI] [PubMed] [Google Scholar]
- (45).Meng H; Liong M; Xia T; Li Z; Ji Z; Zink JI; Nel AE Engineered Design of Mesoporous Silica Nanoparticles to Deliver Doxorubicin and P-glycoprotein siRNA to Overcome Drug Resistance in a Cancer Cell Line. ACS Nano 2010, 4, 4539–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Tatiparti K; Sau S; Kashaw SK; Iyer AK siRNA Delivery Strategies: A Comprehensive Review of Recent Developments. Nanomaterials 2017, 7, No. 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Grabow WW; Zakrevsky P; Afonin KA; Chworos A; Shapiro BA; Jaeger L Self-assembling RNA Nanorings based on RNAI/II Inverse Kissing Complexes. Nano Lett. 2011, 11, 878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Asadi J; Ferguson S; Raja H; Hacker C; Marius P; Ward R; Pliotas C; Naismith J; Lucocq J Enhanced Imaging of Lipid Rich Nanoparticles Embedded in Methylcellulose Films for Transmission Electron Microscopy Using Mixtures of Heavy Metals. Micron 2017, 99, 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).De Carlo S; Harris JR Negative Staining and Cryo-Negative Staining of Macromolecules and Viruses for TEM. Micron 2011, 42, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bui MN; Brittany Johnson M; Viard M; Satterwhite E; Martins AN; Li Z; Marriott I; Afonin KA; Khisamutdinov EF Versatile RNA Tetra-U Helix Linking Motif as a Toolkit for Nucleic Acid Nanotechnology. Nanomedicine 2017, 13, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kawasaki T; Kawai T Toll-like Receptor Signaling Pathways. Front. Immunol. 2014, 5, No. 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hu Z; Xing Y; Qian Y; Chen X; Tu J; Ren L; Wang K; Chen Z Anti-Radiation Damage Effect of Polyethylenimine as a Toll-like Receptor 5 Targeted Agonist. J. Radiat. Res. 2013, 54, 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Cubillos-Ruiz JR; Engle X; Scarlett UK; Martinez D; Barber A; Elgueta R; Wang L; Nesbeth Y; Durant Y; Gewirtz AT; Sentman CL; Kedl R; Conejo-Garcia JR Polyethylenimine-based siRNA Nanocomplexes Reprogram Tumor-associated Dendritic Cells via TLR5 to Elicit Therapeutic Antitumor Immunity. J. Clin. Invest. 2009, 119, 2231–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Cha W; Fan R; Miao Y; Zhou Y; Qin C; Shan X; Wan X; Li J Mesoporous Silica Nanoparticles as Carriers for Intracellular Delivery of Nucleic Acids and Subsequent Therapeutic Applications. Molecules 2017, 22, No. 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Wang J; Byrne JD; Napier ME; DeSimone JM More Effective Nanomedicines Through Particle Design. Small 2011, 7, 1919–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wang M; Li X; Ma Y; Gu H Endosomal Escape Kinetics of Mesoporous Silica-based System for Efficient siRNA Delivery. Int. J. Pharm. 2013, 448, 51–57. [DOI] [PubMed] [Google Scholar]
- (57).Walker WA; Tarannum M; Vivero-Escoto JL Cellular Endocytosis and Trafficking of Cholera Toxin B-Modified Mesoporous Silica Nanoparticles. J. Mater. Chem. B 2016, 4, 1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Bharti C; Nagaich U; Pal AK; Gulati N Mesoporous Silica Nanoparticles in Target Drug Delivery System: A review. Int. J. Pharm. Invest. 2015, 5, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Shen J; Zhang W; Qi R; Mao ZW; Shen H Engineering Functional Inorganic-Organic Hybrid Systems: Advances in siRNA therapeutics. Chem. Soc. Rev. 2018, 47, 1969–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Darvishi B; Farahmand L; Majidzadeh AK Stimuli-Responsive Mesoporous Silica NPs as Non-viral Dual siRNA/Chemotherapy Carriers for Triple Negative Breast Cancer. Mol. Ther.—Nucleic Acids 2017, 7, 164–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Pakunlu RI; Wang Y; Tsao W; Pozharov V; Cook TJ; Minko T Enhancement of the Efficacy of Chemotherapy for Lung Cancer by Simultaneous Suppression of Multidrug Resistance and Antiapoptotic Cellular Defense: Novel Multicomponent Delivery System. Cancer Res. 2004, 64, 6214–6224. [DOI] [PubMed] [Google Scholar]
- (62).Tabuchi Y; Matsuoka J; Gunduz M; Imada T; Ono R; Ito M; Motoki T; Yamatsuji T; Shirakawa Y; Takaoka M; Haisa M; Tanaka N; Kurebayashi J; Jordan VC; Naomoto Y Resistance to Paclitaxel Therapy is Related with Bcl-2 Expression through an Estrogen Receptor Mediated Pathway in Breast Cancer. Int. J. Oncol. 2009, 34, 313–319. [PubMed] [Google Scholar]
- (63).Inao T; Iida Y; Moritani T; Okimoto T; Tanino R; Kotani H; Harada M Bcl-2 Inhibition Sensitizes Triple-negative Human Breast Cancer Cells to Doxorubicin. Oncotarget 2018, 9, 25545–25556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wang Y; Sun HJ; Li RG; Wang XM; Cheng ZQ; Lou N Reprogramming Factors Induce Proliferation and Inhibit Apoptosis of Melanoma Cells by Changing the Expression of Particular Genes. Mol. Med. Rep. 2019, 19, 967–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Deng ZJ; Morton SW; Ben-Akiva E; Dreaden EC; Shopsowitz KE; Hammond PT Layer-by-layer Nanoparticles for Systemic Codelivery of an Anticancer Drug and siRNA for Potential Triple-negative Breast Cancer Treatment. ACS Nano 2013, 7, 9571–9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Chen AM; Zhang M; Wei D; Stueber D; Taratula O; Minko T; He H Co-delivery of Doxorubicin and Bcl-2 siRNA by Mesoporous Silica Nanoparticles Enhances the Efficacy of Chemotherapy in Multidrug-resistant Cancer Cells. Small 2009, 5, 2673–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Pan QS; Chen TT; Nie CP; Yi JT; Liu C; Hu YL; Chu X In Situ Synthesis of Ultrathin ZIF-8 Film-Coated MSNs for Codelivering Bcl 2 siRNA and Doxorubicin to Enhance Chemotherapeutic Efficacy in Drug-Resistant Cancer Cells. ACS Appl. Mater. Interfaces 2018, 10, 33070–33077. [DOI] [PubMed] [Google Scholar]
- (68).Tekedereli I; Alpay SN; Akar U; Yuca E; Ayugo-Rodriguez C; Han HD; Sood AK; Lopez-Berestein G; Ozpolat B. Therapeutic Silencing of Bcl-2 by Systemically Administered siRNA Nanotherapeutics Inhibits Tumor Growth by Autophagy and Apoptosis and Enhances the Efficacy of Chemotherapy in Orthotopic Xenograft Models of ER (−) and ER (+) Breast Cancer. Mol. Ther.—Nucleic Acids 2013, 2, No. e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Looi CY; Moharram B; Paydar M; Wong YL; Leong KH; Mohamad K; Arya A; Wong WF; Mustafa MR Induction of Apoptosis in Melanoma A375 Cells by a Chloroform Fraction of Centratherum anthelminticum (L.) Seeds involves NF-kappaB, p53 and Bcl-2-controlled Mitochondrial Signaling Pathways. BMC Complementary Altern. Med 2013, 13, No. 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Zhou X; Chen L; Nie W; Wang W; Qin M; Mo X; Wang H; He C Dual-Responsive Mesoporous Silica Nanoparticles Mediated Codelivery of Doxorubicin and Bcl-2 SiRNA for Targeted Treatment of Breast Cancer. J. Phys. Chem. C 2016, 120, 22375–22387. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.