

Abstract
Ferredoxin reductase (FDXR) is a mitochondrial flavoprotein that initiates electron transport from NADPH to several cytochromes P450 via two electron carriers, ferredoxin 1 (FDX1) and FDX2. FDXR is the sole ferredoxin reductase in humans and plays a critical role in steroidogenesis and biosynthesis of heme and iron-sulfur clusters. However, much less is known about the role of FDXR in cancer. Here, we show that FDXR plays a role in tumorigenesis by modulating expression of the tumor suppressor p73. By using genetically modified mouse models, we recently showed that mice deficient in either Fdxr or Trp73 had a shorter lifespan and were prone to spontaneous tumors as compared with wild-type (WT) mice. Interestingly, compound Trp73+/−;Fdxr+/− mice lived longer and developed fewer tumors when compared with Fdxr+/− or Trp73+/− mice. Moreover, we found that cellular senescence was increased in Trp73+/− and Fdxr+/− mouse embryonic fibroblasts (MEFs), which was further increased in Trp73+/−;Fdxr+/− MEFs, as compared with that in WT MEFs. As FDXR is regulated by p73, we examined whether there was a feedback regulation between p73 and FDXR. Indeed, we found that Trp73 expression was decreased by loss of Fdxr in MEFs and that FDXR is required for p73 expression in multiple human cancer cell lines independent of p53. Mechanistically, we found that loss of FDXR, via FDX2, increased expression of iron-binding protein 2 (IRP2), which subsequently repressed TP73 mRNA stability. We also showed that TP73 transcript contained an iron response element in its 3′UTR, which was required for IRP2 to destabilize TP73 mRNA. Together, these data reveal a novel regulation of p73 by FDXR via IRP2 and that the FDXR-p73 axis plays a critical role in aging and tumor suppression.
Keywords: iron metabolism, iron response element, mRNA stability, FDX2, FDX1
Introduction
P73 belongs to the tumor suppressor p53 family, which also includes p53 and p63 [1]. These proteins are transcription factors and share a significant degree of sequence homology, especially in the DNA binding domain [2]. TP73 is expressed as multiple isoforms due to the usage of different promoters and alternative splicing [3]. At the N-terminus, TP73 is expressed as two major isoforms, TAp73 and ΔNp73. TAp73 isoforms are transcribed from the upstream promoter and contain an N-terminal activation domain with homology to that in p53, whereas the ΔNp73 isoforms are transcribed from the downstream promoter in intron 3 and are N-terminally truncated [4]. As a result, TAp73 isoforms can activate a series of p53 targets and, thus, are thought to function as tumor suppressors [4]. By contrast, ΔNp73 isoforms do not contain the transactivation domain and are thought to be inactive in transcription [4]. However, a unique ΔNp73 activation domain has been identified, which consists of the 13 distinct residues and the proline-rich domain [5]. At the C-terminus, p73 expresses at least seven isoforms due to alternative splicing. The function of these C-terminal isoforms is not fully understood.
p73 plays critical roles in tumor suppression, metabolism and neuronal development, as shown by several mouse models [6]. For example, mice deficient in all Trp73 isoforms exhibit abnormalities in neural development, such as hippocampal dysgenesis, hydrocephalus, loss of Cajal-Retzius neurons and defects in pheromone sensory pathways [7]. Recently, two studies showed that Trp73 is required for multiciliogenesis [8,9], which may explain the phenotypes observed in Trp73-KO mice. Like total Trp73-KO mice, mice deficient in ΔNp73 isoforms do not develop tumors but are prone to delayed onset of moderate neurodegeneration [10,11]. By contrast, mice deficient in TAp73 show an increased incidence of both spontaneous and carcinogen-induced tumors [12], as well as accelerated aging [13]. These different phenotypes observed in total or isoform-specific Trp73-KO mice clearly suggest that TAp73, but not ΔNp73, is a bona fide tumor suppressor. Notably, ΔNp73 has been found to be frequently upregulated in human cancers and is indicative of poor prognosis [14], suggesting that ΔNp73 has oncogenic potential. It is suggested that the ratio between TAp73 and ΔTAp73 isoforms is critical for tumorigenesis. Thus, understanding how p73 expression is controlled may help to develop p73-based therapeutic strategies for cancer management.
Ferredoxin reductase (FDXR, also known as adrenodoxin reductase) is a mitochondrial flavoprotein and functions as the first electron transfer protein of mitochondrial P450 systems such as P450scc [15,16]. FDXR 15,16]. FDXR receives two electrons from NADPH and transfers them one at a time to the electron transfer protein ferredoxin 1 (FDX1) and FDX2 [17], which then transfer them to cytochromes P450. FDXR is expressed in all tissues with the highest expression in tissues such as adrenal cortex, that specialize in steroid hormone synthesis [18]. Functionally, FDXR is suggested to be involved in various metabolic processes, including steroidogenesis, heme and iron-sulfur cluster biosynthesis. Notably, recent studies have shown that FDXR mutations are associated with mitochondrial disorders, probably due to its role in iron-sulfur cluster protein biosynthesis [19,20]. In addition, FDXR has also been found to be a sensitive and reliable biomarker of radiation exposure in vivo [21]. However, the role of FDXR in cancer is relatively under-studied. Previously, we and others showed that FDXR is a target of the p53 family and sensitizes cells to oxidative stress-induced apoptosis [22,23]. Recently, by a generating Fdxr-deficient mouse model, we showed that Fdxr+/− mice are prone to spontaneous tumors and liver abnormalities, presumably in part via reduced p53 expression [24]. As FDXR is a target of p53 family proteins, it is likely that FDXR may contribute to tumor suppression mediated by other p53 family proteins, such as p73. To test this, a cohort of wild-type(WT), Trp73+/−, Fdxr+/− and Trp73+/−; Fdxr+/− mice was generated and monitored for spontaneous tumor formation. Surprisingly, we found that compound Trp73+/−;Fdxr+/− mice showed better survival, as well as less tumor formation, when compared with Trp73+/− or Fdxr+/− mice. We also found that FDXR was required for p73 expression. Mechanistically, we found that loss of FDXR via FDX2 induced iron-binding protein 2 (IRP2) to destabilize TP73 mRNA. Together, our data reveal a novel crosstalk among FDXR, IRP2 and p73, which plays a critical role in iron homeostasis and tumor suppression.
Materials and methods
Mice
The use of animals and the study protocols were approved by the University of California at Davis Institutional Animal Care and Use Committee. Fdxr+/− mice were generated as described previously [24]. Trp73+/− mice were generated by the Mouse Biology Program at the University of California at Davis [25] (Davis, CA, USA). Irp2+/− mice were generated by the Mutant Mouse Resource and Research Center at the University of Missouri (Columbia, MO, USA). Trp53+/− mice were obtained from the Jackson Laboratory (Sacramento, CA, USA) as described previously [26]. Trp73+/−;Fdxr+/− mice were generated by intercrossing Trp73+/− mice with Fdxr+/− mice. To genotype the Trp73 WT allele we used the forward primer, 5′- CAC GAG CTT GGA AGG GAC TT -3′, and the reverse primer, 5′- TCA TTT AGG TGG CCT TTG AGG -3′. To genotype the Trp73 KO allele, we used the forward primer, 5′- CAT-GTC TGG ATC CGG AAT AAC TAA CT -3′ and the same reverse primer as for the WT allele.
Mouse embryonic fibroblast (MEF) isolation
MEFs were isolated from 12.5–13.5 days post-coitum (pc) mouse embryos, as described previously [26]. To generate WT, Trp73+/−, Fdxr+/− and Trp73+/−;Fdxr+/− MEFs, Trp73+/− mice were bred with Fdxr+/−mice for MEF isolation. To generate WT and Irp2−/− MEFs, Irp2+/− mice were bred with each other for MEF isolation. To generate Trp53−/− and Irp2−/−;Trp53−/− MEFs, Irp2+/− mice were bred with Trp53+/− mice to generate Irp2+/−;Trp53+/− mice, which were then bred with each other for MEF isolation. The MEFs were cultured in DMEM supplemented with 10% FBS (Hyclone Laboratories, Erie, PA, USA), 55 μM β-mercaptoethanol and 1× non-essential amino acids solution (Cellgro, Manassas, VA, USA).
Cell culture, cell line generation and reagents
HCT116, RKO, SW480, TP53−/− HCT116 and Mia-PaCa2 cells and their derivatives were cultured in DMEM supplemented with 10% FBS (Hyclone). FDXR+/−/− HCT116, FDX1-KO HCT116, FDX2+/− HCT116, IRP2-KO Mia-PaCa2 and IRP2-KO TP53−/− HCT116 cells were generated previously [24]. Doxorubicin and camptothecin were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Western blotting analyses
Western blotting was performed as described previously [27]. In brief, whole cell lysates were harvested in 2× SDS/sample buffer. Proteins were separated through a 7–13% SDS-polyacrylamide gel, transferred to a nitro-cellulose membrane, probed with the indicated antibodies, followed by detection using enhanced chemiluminescence.
The antibodies used in this study were: anti-TAp73 (A300-126A, 1:1000; Bethyl Laboratories, Montgomery, TX, USA) and anti-FDXR (sc-374 436, 1:2000; Santa Cruz Biotechnology, Dallas, TX, USA), anti-actin (sc-47 778, 1:5000; Santa Cruz Biotechnology), anti-IRP2 (sc-33 682, 1:2000; Santa Cruz Biotechnology), anti-HA (#901513, 1:2000; Covance, Princeton, NJ, USA), anti-FDX1 (ab108257, 1:1000; Abcam, Burlin-game, CA, USA), anti-vinculin (sc-73 614, 1:3000; Santa Cruz Biotechnology) and anti-FDX2 (sc-515 121, 1:1000; Santa Cruz Biotechnology).
RNA interference
Scrambled siRNA (5′- GGC CGA UUG UCA AAU AAU U- 3′), siRNAs against FDXR (5′-CAC CUU GAU CCA GCG GAC UUA -3′ and 5′- GCU CAG CAG CAU UGG GUA U -3′), siRNAs against FDX2 (5′- GCU GCA AUA AAU CGA UAA CAC -3′ and 5′- GCU GCC AGA UUG UCU GAC AC -3′) and siRNA against IRP2 (5′- GCA AAC AUG UGU CCG GAA U -3′) were purchased from Dharmacon (Chicago, IL, USA). RNAiMax from Life Technology (Carlsbad, CA, USA) was used for siRNA transfection according to the user manual. Cells were transfected with scrambled or target siRNA at 25 nM for 3 days.
Plasmids
pcDNA3 vectors for expressing EGFP, IRP2 or FDXR were generated previously [24]. To generate an EGFP expression vector containing various length of TP73 3′UTRs, PCR was carried out using a full-length TP73 3′UTR template to amplify fragment TP73 3′UTR-A, -B and -C. The PCR products were then cloned into the vector pcDNA3-HA-EGFP using NotI and XhoI. The primers for TP73 3′UTR-A were: forward primer, 5′- AAT GCG GCC GCG GGC CTC GCC TGG CTG CAG C -;3′, and reverse primer, 5′- ATC TCG AGC AGT GCT AGA GAA GGT GGC -3′. The primers for TP73 3′UTR-B were: forward primer, 5′- AAT GCG GCC GCG GCC ACC TTC TCT AGC ACT G -3′, and reverse primer, 5′- ATC TCG AGG GAC TCA GGG TGG ACA GGT A -3′. The primers for TP73 3′UTR-C were: forward primer, 5′- AAT GCG GCC GCA CCT GTC CAC CCT GAG TCC -3′, and reverse primer, 5′- ATC TCG AGA ATC CCC ACT GAA ACA CAG C -3′. To generate an EGFP expression vector containing a point mutation in IRE#1 of p73 3′UTR-C, two PCR rounds were used. The first round was to amplify two DNA fragments (#1 and #2) using pcDNA3-HA-EGFP-3′UTR-C as the template. Fragment #1 was amplified using forward P1 primer, 5′- AAT GCG GCC GCA CCT GTC CAC CCT GAG TCC -3′, and reverse P2 primer, 5′- CCC CTG CAC TTC TGG GAA AAC -3′. Fragment #2 was amplified using forward P3 primer, 5′- GTT TTC CCA GAA GTG CAG GGG -3′, and reverse P4 primer, 5′- ATC TCG AG A ATC CCC ACT GAA ACA CAG C -3′. The second round of PCR was performed by using both fragment #1 and #2 as templates with a forward P1 and a reverse P2 primer. The PCR products were then cloned into pcDNA3-HA-EGFP via NotI and XhoI. To generate an EGFP expression vector containing a point mutation in IRE#2 of p73 3′UTR-C, the same strategy was used except that the reverse P2 primer for fragment #1 was 5′- CGT TGG AGA CAC TTA CTG TTC -3′ and the forward P3 primer for fragment #2 was 5′- GAA CAG TAA GTG TCT CCA ACG -3′.
RNA isolation and RT-PCR
Total RNA was isolated using Trizol reagent as described according to the user manual (https://assets.thermofisher.com/TFS-Assets/LSG/manuals/trizol_reagent.pdf). cDNA was synthesized using reverse transcriptase (Promega, Madison, WI, USA) and used for PCR. The PCR program used for amplification was (1) 94 °C for 5 min, (2) 94 °C for 45 s, (3) 58 °C for 45 s, (4) 72 °C for 30 s and (5) 72 °C for 10 min. From steps 2 to 4, the cycle was repeated 22 times for ACTB, GAPDH, Gapdh and HPRT, 30 times for IRP2, FDXR, TFR, FDX1, FDX2, TP73, Cdkn2a, Cdkn2d and Cdkn1a. The sequence of primers used is listed in supplementary material, Table S5.
RNA-ChIP
RNA-ChIP was carried out as described previously [28] with some modifications. In brief, cell lysates were prepared with immunoprecipitation buffer (100 mM KCl, 5 mM MgCl2, 10 mM HEPES, 1 mM 1,4-dithiothreitol and 0.5% NP-40) and then incubated with 2 μg anti-IRP2 or an isotype control IgG at 4 °C over-night. The RNA-protein immunocomplexes were brought down using magnetic protein A/G beads (MCE, Monmouth Junction, NJ, USA), followed by RT-PCR analysis.
mRNA half-life assay
To measure the stability of TP73 mRNA, cells were treated at various times with 5, 6-dichloro-1-beta-d-ribofuranosylbenzimidazole (DRB), an inhibitor of transcription. The relative level of TP73 mRNA was quantified by UVP VisionWorks (Analytik Jena, Upland, CA, USA) and normalized using the level of ACTB mRNA. Values were plotted versus time and the half-life of TP73 mRNA calculated.
Senescence assay
Cells were seeded into a six-well plate and cultured for 3 days. A senescence assay was performed as described previously [26]. In brief, cells were fixed with 2% formaldehyde and 0.2% glutaraldehyde for 10–15 min at room temperature and then stained with fresh SA-β-galactosidase staining solution (1 mg/ml, 5-bromo-4-chloro-3-indolyl-beta-d-galactopyranoside, 40 mM citric acid/sodium phosphate [pH 6.0], 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl and 2 mM MgCl2) at 37 °C without CO2. The percentage of cells that were senescent was calculated.
Histological analysis
Mouse tissues were fixed in 10% neutral buffered formalin, processed and embedded in paraffin blocks. Tissues blocks were sectioned (6 μm) and stained with H&E.
Results
Loss of Trp73 extended life span and decreased tumor formation in Fdxr+/− mice
To determine whether FDXR plays a role in p73-mediated tumor suppression, we generated a cohort of WT, Trp73+/−, Fdxr+/− and Trp73+/−;Fdxr+/− mice and monitored these mice throughout their lifespan. To minimize the number of animals used, the data for Fdxr+/− and Trp73+/− mice were adapted from a previous study [24,25], whereas the data for WT mice were adapted from two previous studies [24,29]. All the mice were derived from the same C57BL/6 background, maintained in the same facility and were generated at similar times. The median lifespan of WT mice was 117 weeks, 82 weeks for Trp73+/− mice, 102 weeks for Fdxr+/− mice and 104 weeks for Trp73+/−;Fdxr+/− mice (Figure 1A and see supplementary material, Tables S1–S4). As expected, both Trp73+/− and Fdxr+/− mice had significantly shorter lifespans than WT mice, consistent with previous reports [24,30]. Surprisingly, we found that the lifespan of Trp73+/−;Fdxr+/− mice was significantly longer than that of Trp73+/− or slightly longer than that of Fdxr+/− mice but was not statistically different from that of WT mice (Figure 1A). Additionally, we found that the lifespan of Trp73+/− mice was significantly shorter than that of Fdxr+/− mice (p = 0.0077). Histological analyses were performed looking for pathological abnormalities of major organs from all these mice. We found that both Trp73+/− and Fdxr+/− mice were significantly more tumor prone, consistent with previous observations [24,25]. Twelve of 26 Trp73+/− mice compared with 26 of 29 Fdxr+/− mice developed tumors (Table 1 and see supplementary material, Tables S2,S3). However, only four of 20 Trp73+/−; Fdxr+/− mice developed tumors (Table 1 and see supplementary material, Table S4) and this was not significantly different from WT mice (p = 1.0 by Fisher’s exact test). Tumor penetrance was significantly lower in Trp73+/−;Fdxr+/− mice than that in Fdxr+/− mice (p = 0.0001 by Fisher’s exact test). Additionally, there was no difference in tumor penetrance between Trp73+/−;Fdxr+/− mice and Trp73+/− mice (p = 0.1173 by Fisher’s exact test).
Figure 1.
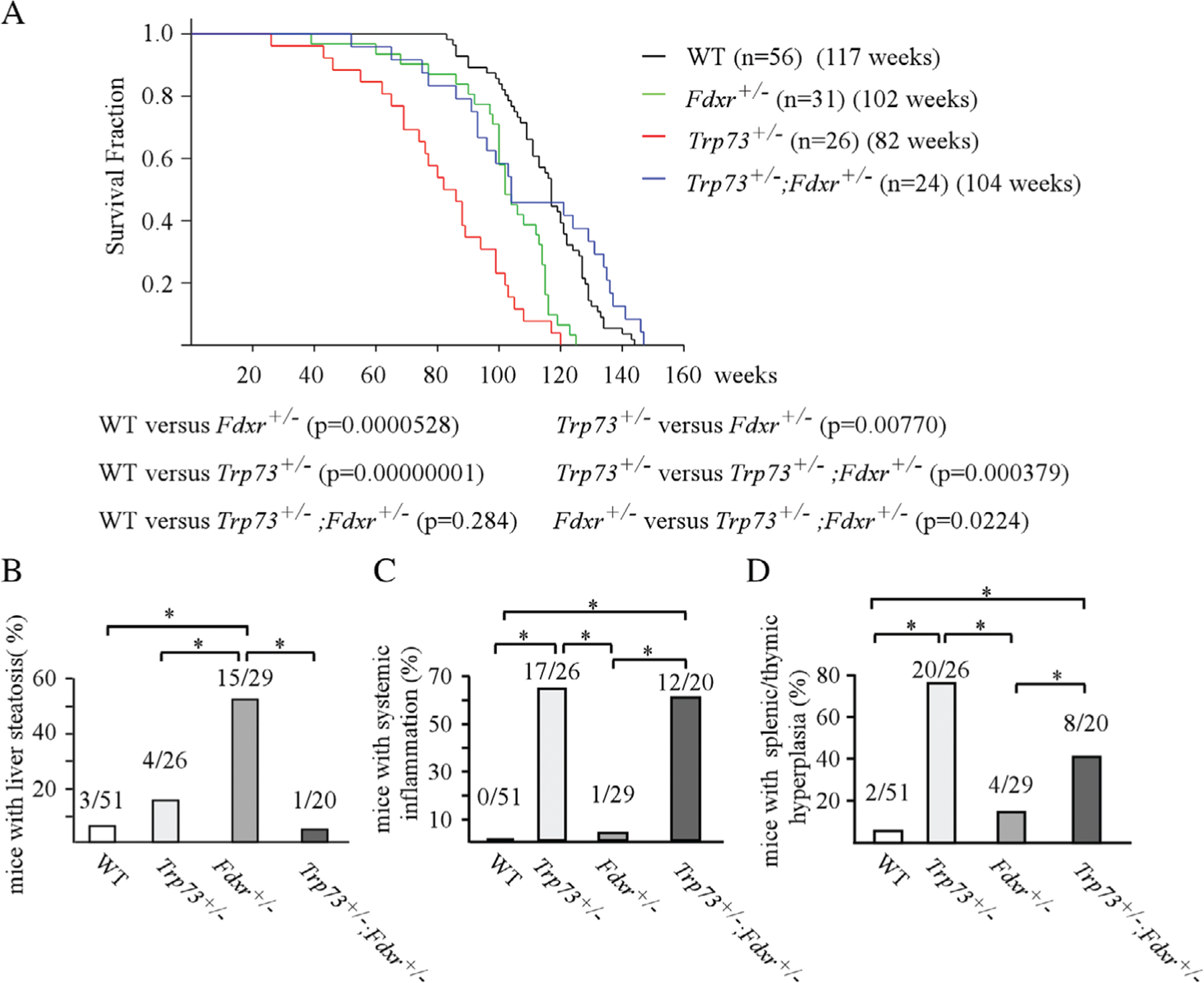
Loss of Trp73 extended life span and decreased tumor formation in Fdxr+/− mice. (A) Kaplan-Meier survival curve for WT (n = 56), Trp73+/− (n = 26), Fdxr+/− (n = 31) and Fdxr+/−;Trp73+/− (n = 24) mice. The median lifespan was 117 weeks for WT mice, 82 weeks for Trp73+/− mice, 102 weeks for Fdxr+/− mice and 104 weeks for Fdxr+/−;Trp73+/−. (B) The numbers and percentages of WT, Trp73+/−, Fdxr+/− and Fdxr+/−;Trp73+/− mice with liver steatosis. (C) The numbers and percentages of WT, Trp73+/−, Fdxr+/− and Fdxr+/−;Trp73+/− mice with systemic inflammation. (D) The numbers and percentages of WT, Trp73+/−, Fdxr+/− and Fdxr+/−;Trp73+/− mice with splenic or thymic hyperplasia.
Table 1.
Tumor spectrum and burden in WT, Trp73+/−, Fdxr+/− and Trp73+/−;Fdxr+/− mice
| Tumor | WT (n = 51)* | Trp73+/− (n = 26)* | Fdxr+/− (n = 29)* | Trp73+/−;Fdxr+/− (n = 20)* |
|---|---|---|---|---|
| DLBCL | 6 | 3 | 12 | 4 |
| T-LBL | 2 | 1 | 5 | 0 |
| Unclassified LM | 3 | 6 | 2 | 0 |
| Sarcoma | 1 | 1 | 7 | 0 |
| Adenoma | 0 | 1 | 2 | 0 |
| Hemangioma | 0 | 1 | 3 | 0 |
| Hepatoma | 0 | 1 | 0 | 0 |
| Lung adenocarcinoma | 0 | 0 | 4 | 0 |
| HCC | 0 | 0 | 2 | 0 |
| Plasmacytoma | 0 | 0 | 1 | 0 |
| Tumor penetrance | 11/51 | 12/26 | 26/29 | 4/20 |
DLBCL, diffuse large B-cell lymphoma; T-LBL, thymic lymphoblastic lymphoma; HCC, hepatocellular carcinoma; LM, lymphoma.
Dead/hydrocephalus mice were excluded.
In addition to tumors, these mice were also prone to other pathological abnormalities, including liver steatosis, systemic inflammation and splenic/thymic hyperplasia (see supplementary material, Tables S1–S4). We reported previously that loss of Fdxr led to liver steatosis in mice [24]. Interestingly, we found that the percentage of mice with liver steatosis was significantly decreased in Trp73+/−;Fdxr+/− mice (5%) as compared with that in Fdxr+/− mice (51.7%) (Figure 1B and see supplementary material, Tables S2, S4), suggesting that reduced expression of Trp73 ameliorates liver steatosis in Fdxr+/− mice. Moreover, Trp73+/− mice are known to be prone to systemic inflammation. Indeed, 17 of 26 mice developed inflammation in more than three organs (see supplementary material, Table S3). Similarly, 12 of 20 p73+/−;Fdxr+/− mice developed inflammation in more than three organs (see supplementary material, Table S4). The percentage of mice with chronic inflammation was significantly higher in both Trp73+/− and Trp73+/−;Fdxr+/− mice than that of WT or Fdxr+/− mice (Figure 1C). Furthermore, we observed that loss of Trp73 led to an enlarged spleen or thymus in mice, indicating splenic or thymic hyperplasia. H&E staining indicated that 20 of 26 Trp73+/− mice developed splenic or thymic hyperplasia (Figure 1D and see supplementary material, Table S3). Similarly, eight of 20 Trp73+/−; Fdxr+/− mice, but only four of 29 Fdxr+/− mice and two of 51 WT mice developed splenic or thymic hyperplasia (Figure 1D and see supplementary material, Tables S1, S2, S4). Statistical analyses indicated that the percentage of mice with splenic or thymic hyperplasia was significantly higher in Trp73+/− and Trp73+/−; Fdxr+/− mice than that in WT and Fdxr+/− mice. These data suggest that loss of Fdxr was unable to mitigate systemic inflammation and splenic/thymic hyperplasia in Trp73+/− mice.
Loss of Trp73 and Fdxr leads to enhanced senescence phenotypes
Cellular senescence has been considered as an important mechanism for tumor suppression although it can also promote tumor growth through senescence-associated secretory phenotype [31]. Thus, a cohort of WT, Trp73+/−, Fdxr+/− and Trp73+/−;Fdxr+/− MEFs were generated and cellular senescence assessed. We found that both Trp73+/− and Fdxr+/− MEFs were prone to senescence (Figure 2A). The percentage of SA-β-galactosidase-positive cells was 23.2% for p73+/− MEFs and 22.8% for Fdxr+/− MEFs, values which were significantly higher than for WT MEFs (2.8%) (Figure 2A, B). Notably, the percentage of SA-β-galactosidase-positive cells reached 43.5% in Trp73+/−;Fdxr+/− MEFs, which was significantly higher than in either Trp73+/− or Fdxr+/− MEFs (Figure 2A,B). To confirm this, the levels of senescence markers, Cdkn2a, Cdkn2d and Cdkn1a, were determined. As expected, the expression of Cdkn2a, Cdkn2d and Cdkn1a transcripts was increased in Trp73+/− or Fdxr+/− MEFs as compared with that in WT MEFs, which was then further increased in Trp73+/−;Fdxr+/− MEFs (Figure 2C). Notably, we also found that the expression of p73 transcripts was decreased in Fdxr+/− and Trp73+/−;Fdxr+/− when compared with that in WT and Trp73+/− MEFs, respectively (Figure 2C, p73 panel), suggesting that Fdxr regulates Trp73 expression.
Figure 2.
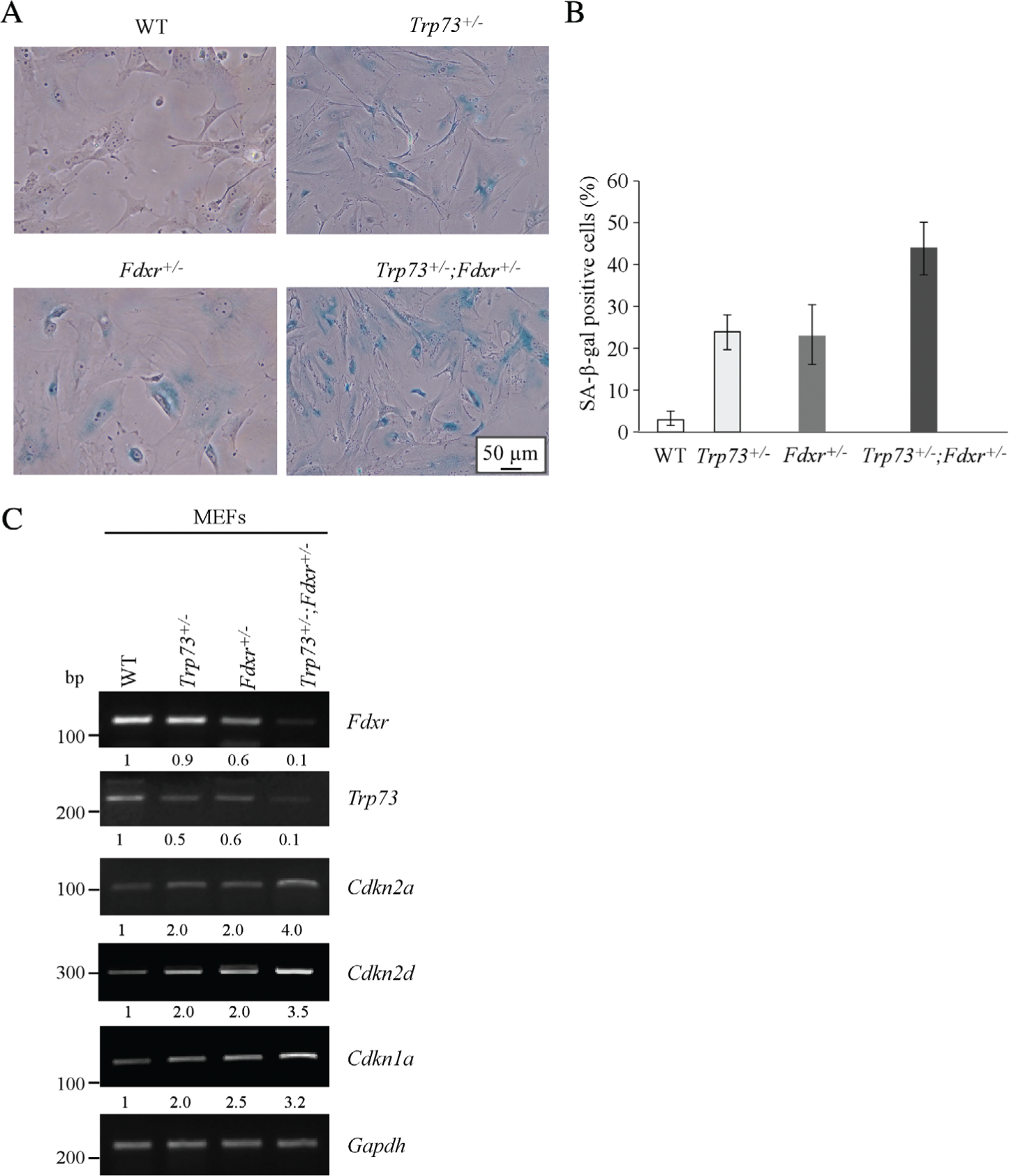
Loss of Trp73 and Fdxr leads to enhanced senescence phenotypes. (A) SA-β-galactosidase staining was performed with primary WT, Trp73+/−, Fdxr+/− and Trp73+/−; Fdxr+/− MEFs at passage 5. (B) Quantification of the percentage of SA-β-galactosidase-positive cells as shown in (A). (C) The levels of Fdxr, Trp73, Cdkn2a, Cdkn2d, Cdkn1a and Gapdh transcripts were examined in WT, Trp73+/−, Fdxr+/− and Trp73+/−;Fdxr+/− MEFs by RT-PCR.
FDXR is required for p73 expression
The expression of Trp73 transcripts was reduced by loss of Fdxr in MEFs (Figure 2C), thus it was important to determine whether FDXR regulates p73 expression in human cells. To test this, HCT116 cells were transfected with a scrambled or FDXR siRNA for 3 days, followed with or without doxorubicin treatment for 12 h. We found that the levels of FDXR proteins were decreased by siRNAs against FDXR (Figure 3A, FDXR panel, compare lanes 2, 3 and 5, 6 with lanes 1 and 4, respectively). Interestingly, we found that the levels of p73 proteins were decreased upon knockdown of FDXR regardless of doxorubicin treatment (Figure 3A, p73 panels, compare lanes 2, 3 and 5, 6 with lanes 1 and 4, respectively). To verify this, RKO cells were transfected with a scrambled or FDXR siRNA. We found that the levels of p73 proteins were decreased by FDXR knockdown in RKO cells treated with or without doxorubicin (Figure 3B, p73 panel, compare lanes 2, 3 and 5, 6 with lanes 1 and 4, respectively). Previously, we have shown that FDXR is required for p53 expression [24]. As p53 is known to regulate p73 expression, it is critical to confirm that FDXR regulates p73 expression independent of p53. To address this, mutant p53-containing SW480 cells were used and we found that the level of p73 protein was decreased by knockdown of FDXR (Figure 3C). Next, to confirm the above observations, FDXR+/−/− HCT116 cells, in which two of three FDXR alleles normally present in HCT116 cells were deleted by CRISPR/Cas9 technology [24], were used to examine the level of p73 protein together with isogenic control HCT116 cells. We found that the levels of p73 proteins were reduced in FDXR+/−/− HCT116cells as compared with that in isogenic control HCT116 cells under non-stress or stress-induced conditions (Figure 3D, p73 panel, compare lanes 1, 3 and 5 with 2, 4 and 6, respectively).
Figure 3.
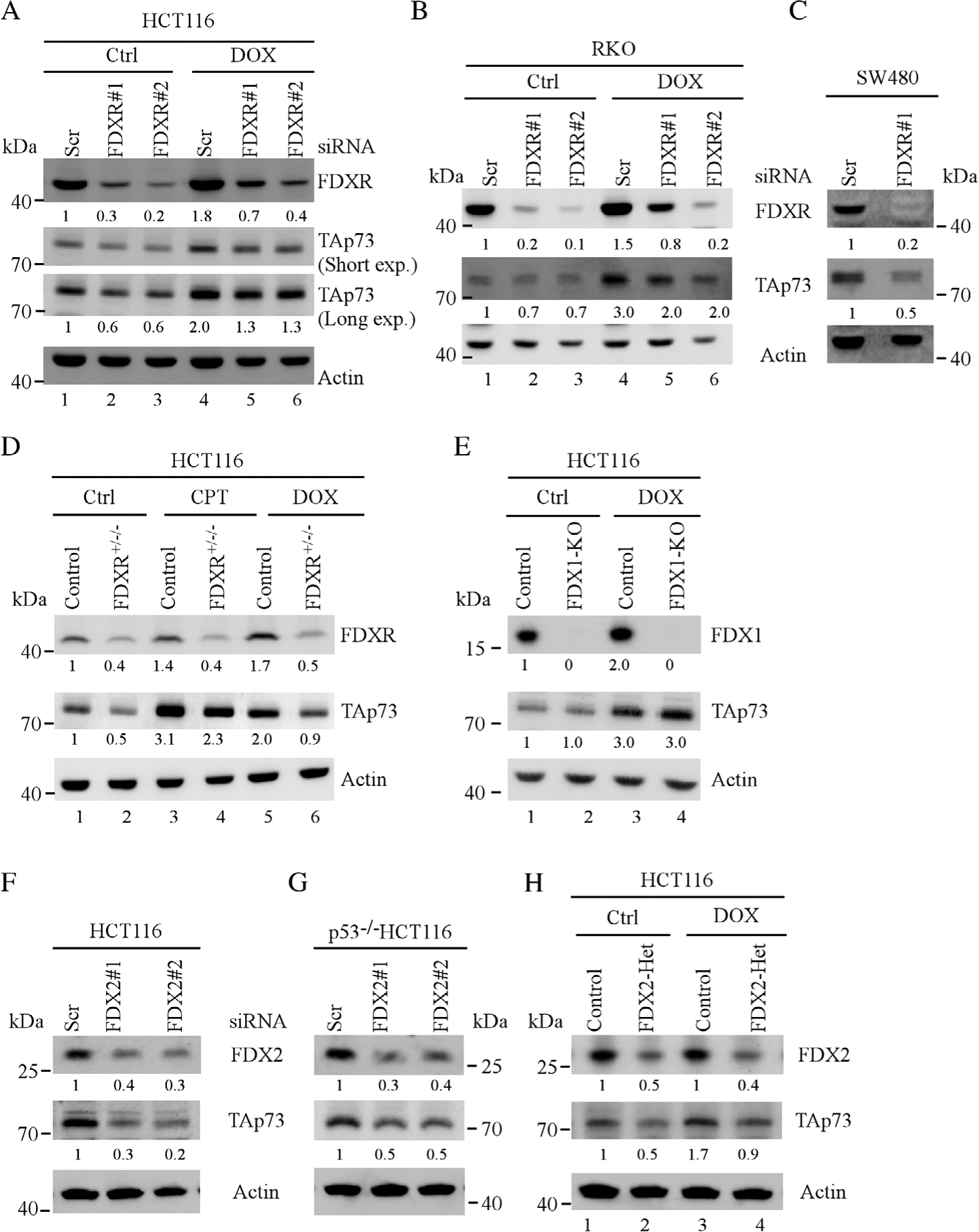
FDXR is required for p73 expression. (A) HCT116 and (B) RKO cells were transfected with scrambled or FDXR siRNA for 3 days, followed with or without doxorubicin treatment for 18 h. The levels of FDXR, TAp73 and β-actin proteins were examined by western blotting. (C) SW480 cells were transfected with a scrambled or FDXR siRNA for 3 days and the levels of FDXR, TAp73 and actin were determined by western blotting. (D) Isogenic control and FDXR+/−/− HCT116 cells were mock-treated or treated with camptothecin or doxorubicin for 18 h, and the levels of FDXR, TAp73 and β-actin proteins were determined by western blotting. (E) The levels of FDX1, TAp73 and β-actin were measured in isogenic control and FDX1-KO HCT116 cells. (F,G) HCT116 and TP53−/− HCT116 cells were transiently transfected with a scrambled or FDX2 siRNAs for 3 days, and the levels of FDX2, TAp73 and β-actin proteins were determined by western blotting. (H) The levels of FDX2, TAp73 and β-actin were measured in isogenic control and FDX2+/− HCT116 cells.
As FDXR can transfer electrons to FDX1 and FDX2 to mediate its biological functions, we determined whether FDX1, FDX2 or both were involved in FDXR-mediated p73 expression. To test this, we assessed p73 expression in FDX1-KO HCT116 cells and isogenic control cells. We found that loss of FDX1 had very little effect on the level of p73 protein regardless of DNA damage (Figure 3E, p73 panel, compare lanes 1 and 3 with lanes 2 and 4, respectively), suggesting that FDX1 is not involved in FDXR-mediated p73 expression. Next, to determine whether FDX2 was involved in FDXR-mediated p73 expression, scrambled and FDX2 siRNAs were transiently transfected into HCT116 and TP53−/− HCT116 cells. We found that the levels of FDX2 proteins were decreased by both FDX2 siRNAs but not by scrambled siRNA (Figure 3F,G, FDX2 panel). Interestingly, the levels of p73 proteins were decreased upon knockdown of FDX2 in both HCT116 and TP53−/− HCT116 cells (Figure 3F,G, p73 panel). To verify that FDX2 regulates p73 expression, isogenic control and FDX2+/− HCT116 cells were treated with or without doxorubicin. We found that the levels of p73 proteins were decreased in FDX2+/− HCT116 cells as compared with that in isogenic control HCT116 cells regardless of doxorubicin treatment (Figure 3H, p73 panel, compare lanes 1 and 3 with 2 and 4, respectively). Together, these data suggest that FDXR signals through FDX2 to regulate p73 expression.
Loss of FDXR reduces p73 transcript abundance
To further understand the mechanism by which FDXR regulates p73 expression, RT-PCR analysis was performed to measure the relative expression of p73 transcripts in HCT116 transfected with scrambled or FDXR siRNAs. We found that upon knockdown of FDXR, the expression of TP73 transcripts was decreased (Figure 4A, p73 panel, compare lane 1 with 2 and 3). Similarly, the expression of TP73 transcripts was decreased by knockdown of FDXR in RKO cells treated with or without doxorubicin (Figure 4B, p73 panel, compare lanes 1 and 4 with lanes 2, 3 and 5, 6, respectively). Moreover, we showed that that TP73 transcript expression was reduced in FDXR+/−/− HCT116 cells as compared with isogenic control cells regardless of DNA damaging treatment (Figure 4C, TAp73 panel, compare lanes 1, 3 and 5 with 2, 4 and 6, respectively). To further confirm that Fdxr regulates Trp73 expression in mouse cells, we isolated two new sets of WT and Fdxr+/− MEF and found that the expression of Trp73 transcripts was reduced in Fdxr+/− MEFs as compared with that in WT MEFs (Figure 4D, p73 panel, compare lane 1 and 3 with 2 and 4, respectively), consistent with the data shown in Figure 2C. Furthermore, to determine whether FDX1 or FDX2 is required for regulating TP73 transcripts, the relative abundance of TP73 transcripts was measured in FDX1-KO and two FDX2-Het cell lines. We found that knockout of FDX1 had very little effect on the expression of TP73 transcripts (Figure 4E), consistent with the data that FDX1 is not required for p73 expression. Notably, the level of TP73 transcripts was decreased in FDX2-Het cells as compared with isogenic control cells (Figure 4F, TAp73 panel, compare lane 1 with lanes 2, 3). Taken together, these data indicate that FDXR regulates TP73 transcripts via FDX2.
Figure 4.
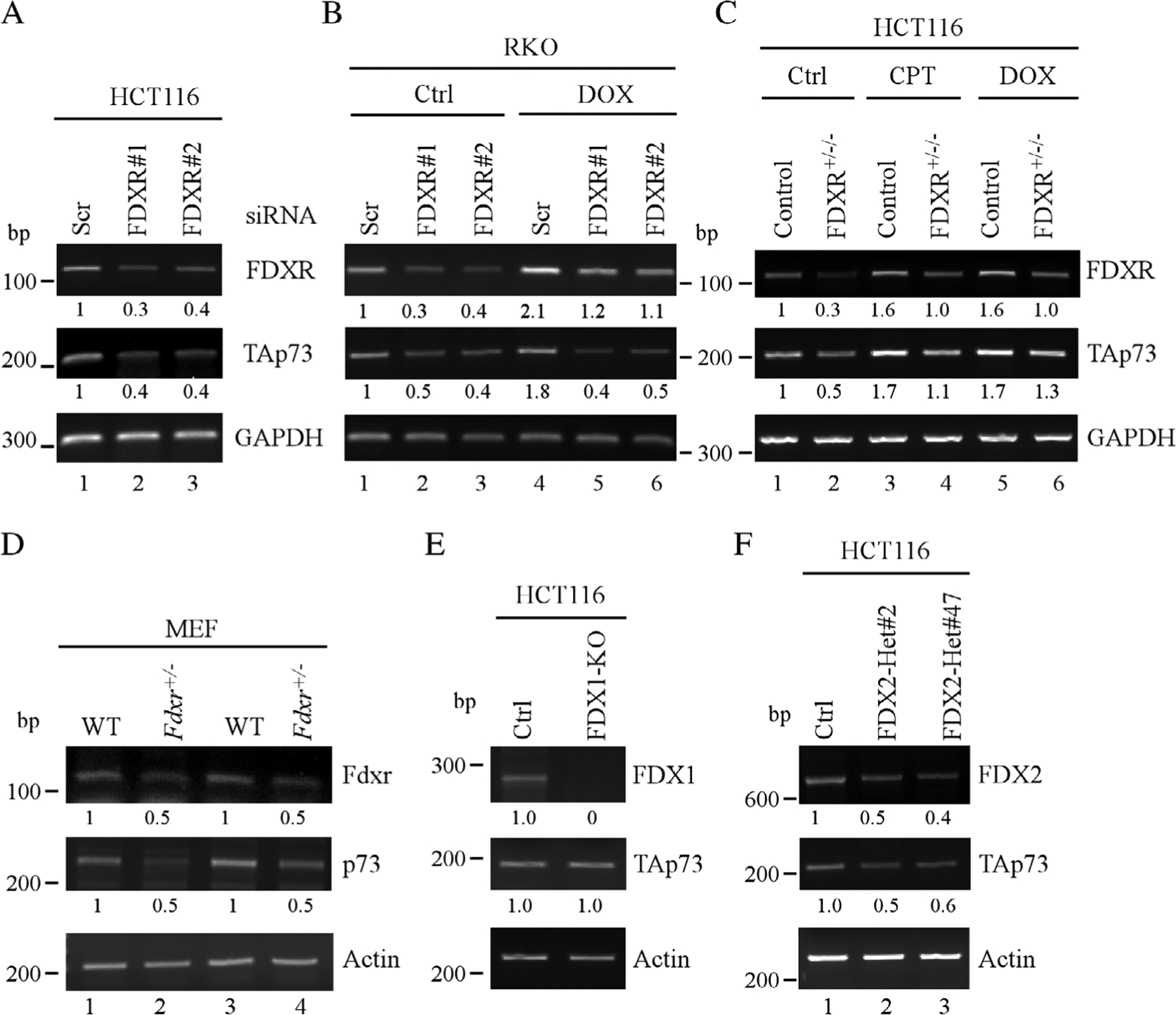
Loss of FDXR reduces p73 transcript abundance. (A) The levels of FDXR, TAp73 and ACTB transcripts were examined by RT-PCR analysis in HCT116 cells transiently transfected with scrambled or FDXR siRNAs. (B) RKO cells were transiently transfected with scrambled or FDXR siRNAs for 3 days, followed by mock-treatment or treatment with doxorubicin for 18 h. The levels of FDXR, TAp73 and ACTB transcripts were examined by RT-PCR analysis. (C) Isogenic control and FDXR+/−/− HCT116 cells were mock-treated or treated with camptothecin or doxorubicin for 18 h, and the levels of FDXR, TAp73 and GAPDH transcripts were assessed by RT-PCR. (D) The levels of Fdxr, Trp73 and Actb transcripts were measured in two sets of WT and Fdxr+/− MEF. (E) The levels of FDX1, TAp73 and ACTB transcripts were measured in isogenic control and FDX1-KO HCT116 cells. (F) The levels of FDX2, TAp73 and ACTB transcripts were measured in isogenic control and FDX2+/− HCT116 cells.
IRP2 is required for FDXR-regulated p73 expression
FDXR deficiency is known to alter iron homeostasis [24,32]. We showed previously that FDXR deficiency leads to enhanced expression of IRP2 [24], a RNA-binding protein that plays a critical role in cellular iron metabolism. Therefore, we examined whether IRP2 plays a role in FDXR-mediated p73 expression. To test this, isogenic control and IRP2-KO Mia-PaCa2 cells were transiently transfected with a control vector or vector expressing FDXR. We found that overexpression of FDXR was able to increase p73 expression in isogenic control Mia-PaCa2 cells, together with decreased expression of IRP2 (Figure 5A, p73 panel, compare lane 1 with 2). However, in the absence of IRP2, overexpression of FDXR had very little effect on TAp73 expression (Figure 5A, p73 panel, compare lane 3 with 4), suggesting that IRP2 is required for FDXR to regulate p73 expression. Moreover, we found that the levels of p73 proteins were increased by loss of IRP2 (Figure 5A, p73 panel, compare lane 1 with 3), suggesting that IRP2 represses p73 expression. To test this, HCT116 and Mia-PaCa2 cells were transiently transfected with scrambled or IRP2 siRNA and we found that the levels of p73 proteins were increased by knockdown of IRP2 (Figure 5B,C). To further verify that IRP2 regulates p73 expression, isogenic control and IRP2-KO HCT116 cells were used to examine p73 expression. We found that the levels of p73 proteins were increased by knockout of IRP2 (Figure 5D, TAp73 panel). Consistent with this, we found that loss of IRP2 resulted in increased levels of TAp73 proteins in TP53−/− HCT116 cells (Figure 5E) as well as in mutant p53-containingMia-PaCa2 cells (Figure 5F), suggesting that IRP2 represses TAp73 expression independent of p53. Moreover, to examine whether TP73 transcripts could be regulated by IRP2, their level was assessed in isogenic control, IRP2-KO HCT116 and Mia-PaCa2 cells. We found that the level of TP73 transcripts was increased by knockout of IRP2 (Figure 5G, H, TAp73 panel). Furthermore, to verify that the regulation of p73 by IRP2 is conserved in both human and mouse cells, a cohort of WT, Irp2−/−, Trp53−/− and Trp53−/−; Irp2−/− MEFs were generated and tested for Trp73 expression. We found that the expression of Trp73 transcripts was increased in Irp2−/− MEFs as compared with WT MEFs (Figure 5I, p73 panel). Similarly, loss of Irp2 led to increased levels of Trp73 transcripts in Trp53−/− MEFs (Figure 5J, p73 panel). Together, these data suggest that IRP2 represses p73 expression in both human and mouse cells independent of p53.
Figure 5.
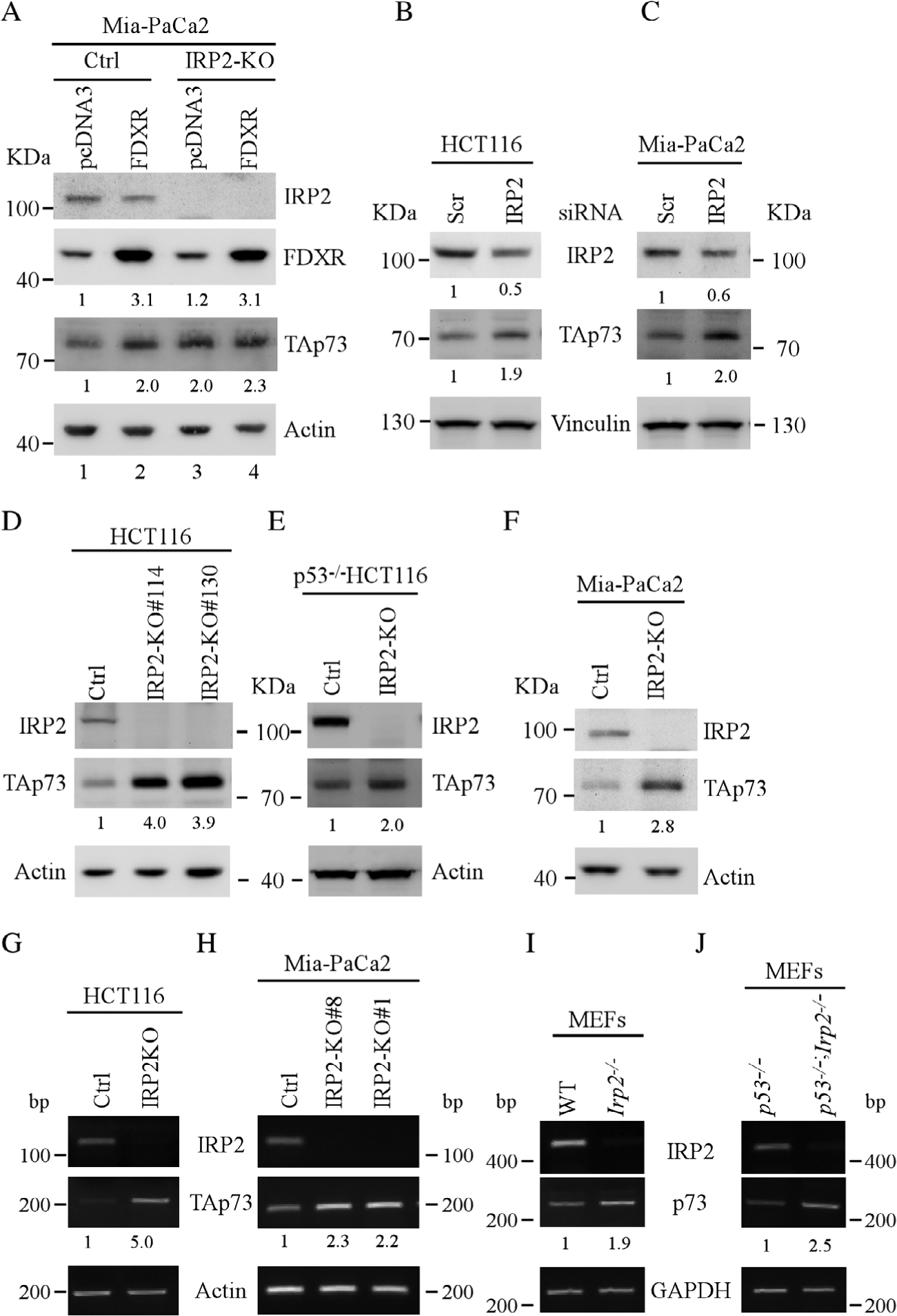
IRP2 is required for FDXR-regulated p73 expression. (A) Isogenic control and IRP2-KOMIA-PaCa2 cells were transiently transfected with control vector or FDXR expression vector for 24 h, and the levels of IRP2, FDXR, TAp73 and β-actin were measured by western blotting. (B,C) The levels of IRP2, TAp73 and vinculin were examined in HCT116 (B) or Mia-PaCa2(C) cells transfected with a scrambled or IRP2 siRNA for 3 days. (D–F) The levels of IRP2, TAp73 and actin proteins were examined in isogenic control and IRP2-KO HCT116 (D), TP53−/− HCT116 (E) or Mia-PaCa2(F) cells. (G,H) The levels of IRP2, TAp73 and ACTB transcripts were measured in isogenic control and IRP2-KO HCT116 (G) and Mia-PaCa2(H) cells. (I,J) The levels of Irp2, TAp73 and Actb transcripts were measured in WT and Irp2−/−(I) as well as in Trp53−/− and Irp2−/−;Trp53−/− MEFs (J).
IRP2 destabilizes TP73 mRNA via an iron response element (IRE) in the 3′UTR of TP73 mRNA
As shown above, FDXR deficiency leads to enhanced expression of IRP2, which subsequently represses p73 expression. Thus, to determine the mechanism by which IRP2 regulates p73 expression, an RNA immunoprecipitation assay was performed with isogenic control and IRP2-KO TP53−/− HCT116 and Mia-PaCa2 cells. We found that IRP2 was able to bind to the TP73 transcript in isogenic control, but not IRP2-KO p53−/− HCT116 or Mia-PaCa2 cells (Figure 6A,B, p73 panel, compare lane 3 with 6). Additionally, we found that IRP2 was able to bind to the transferrin receptor (TFR), a known target of IRP2 [33] (Figure 6A,B, TFR panel). By contrast, IRP2 was unable to bind to ACTB mRNA (Figure 6A,B, actin panel). Next, we examined whether IRP2 regulated TP73 mRNA stability. To test this, the half-life of TP73 transcripts was determined in both isogenic control and IRP2-KO Mia-PaCa2 cells. We found that the half-life of TP73 mRNA was significantly increased from 4.57 h in isogenic control cells to 7.26 h in IRP2-KO cells (Figure 6C,D), suggesting that IRP2 destabilizes TP73 mRNAs.
Figure 6.
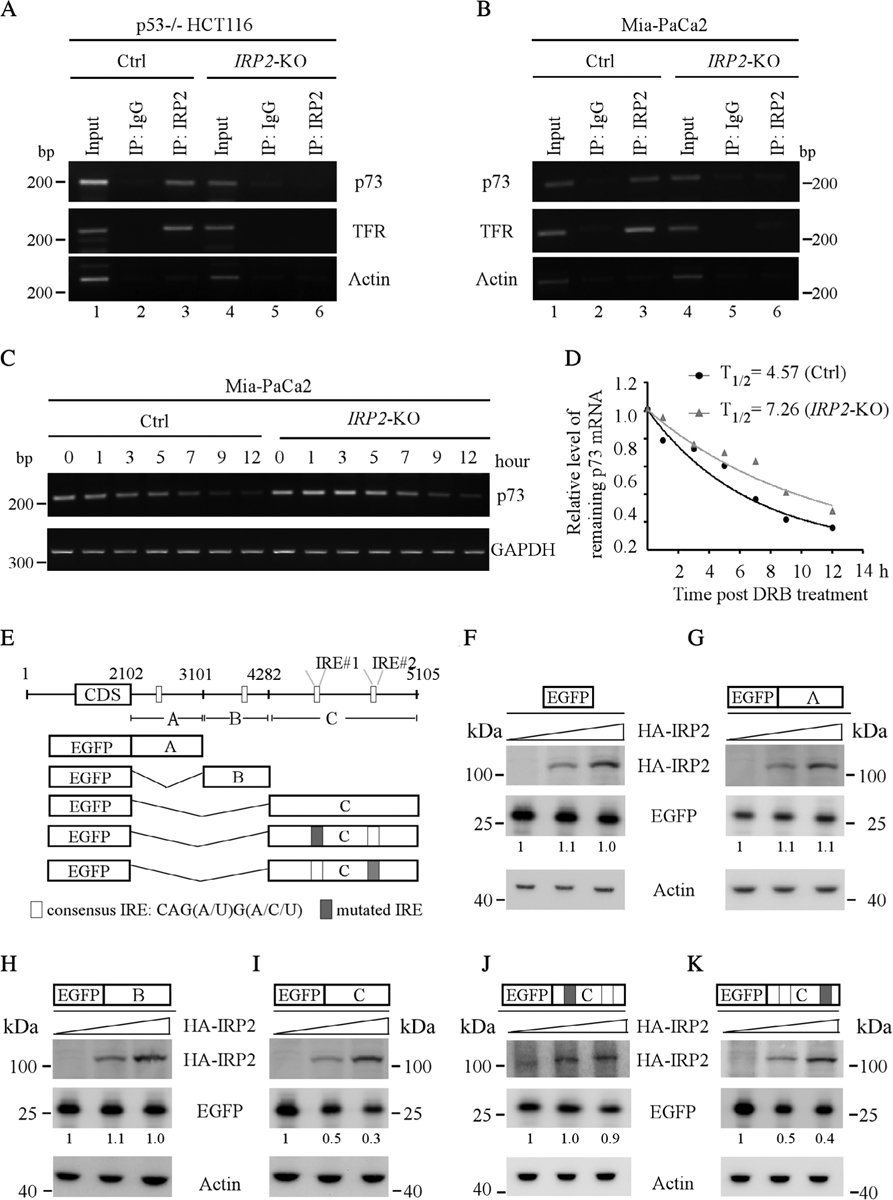
IRP2 destabilizes TP73 mRNA via an IRE in the 3′UTR of TP73 mRNA. (A,B) Isogenic control and (A) IRP2-KOTP53−/− HCT116 and (B) Mia-PaCa2 cell lysates were immunoprecipitated with an isotype control IgG or antibody against IRP2. Total RNAs were purified from immunocomplexes and subjected to RT-PCR to measure the level of p73, TFR and ACTB mRNAs associated with IRP2. (C) Isogenic control and IRP2-KO MiaPaCa2 were treated with or without DRB for various times, followed by RT-PCR to estimate the levels of TP73 and GAPDH transcripts. (D) The relative level of TAp73transcripts in (C) was normalized to GAPDH and the half-life of TAp73 transcript was calculated by plotting the remaining mRNA over time. (E) Schematic representation of TP73 transcript and the locations of 3′UTR fragments for the reporter assays. (F–K) Various amounts of IRP2 expression vector were transfected into IRP2-KO TP53−/− HCT116 cells together with a fixed amount of (F) EGFP or EGFP expression vectors that contain (G) p73 3′UTR-A, (H) p73 3′UTR-B, (I) p73 3′UTR-C, (J) p73 3′UTR-C-Mut#1 and (K) p73 3′UTR-C-Mut#2. The levels of HA-IRP2, EGFP and β-actin proteins were examined by western blotting.
As IRP2 is known to regulate its targets by binding to an IRE [34], we searched the TP73 mRNA and found several potential IREs in the 3′UTR (Figure 6E). To determine which IRE was required for IRP2 to repress p73 expression, several EGFP reporters containing various lengths of TP73 3′UTR (Figure 6E) were generated and then transfected into IRP2-KO HCT116 cells together with various amount of IRP2 expression vector. We found that overexpression of IRP2 had very little effect on the reporter that contained EGFP alone (Figure 6F). Similarly, ectopic expression of IRP2 did not alter EGFP expression when cotransfected with the reporter containing either the A or B fragment of the TP73 3′UTR (Figure 6G,H). By contrast, IRP2 was able to repress EGFP expression when fused with the C fragment in a dose-dependent manner (Figure 6I). Indeed, there are two potential IREs in the TP73 3′UTR C fragment (Figure 6E). A canonical IRE structure is composed of a 6-nt apical loop (5′- CAG WGH -3′; in which W is A or U and H is A, C or U) [35]. Additionally, the C1 and G5 in the loop forms a closing base pair, which is critical for RNA hairpin loop stability [36,37]. Thus, to examine which IRE was required for IRP2 to suppress TP73 expression, we generated two mutant reporters by substituting the ‘C’ nucleotide in the loop of each IRE with an ‘A’ nucleotide (Figure 6E). We found that IRP2 was unable to repress EGFP expression when cotransfected with a reporter in which the IRE#1 was mutated (Figure 6J). By contrast, EGFP expression was still repressed by IRP2 when the reporter contained a mutation in IRE#2 (Figure 6K). Taken together, these data support that the IRE#1 in the C fragment is required for IRP2 to destabilize the TP73 transcript.
Discussion
In this study, we found that FDXR is required for p73 expression. Mechanistically, we found that loss of FDXR signals through FDX2 and induces IRP2, which then destabilizes TP73 mRNA via an IRE in its 3′UTR. We also provide evidence that FDXR-mediated p73 expression is independent of p53. Considering that both FDXR and IRP2 are involved in regulating iron homeostasis, our data suggest that p73 plays a role in modulating iron metabolism. Indeed, p53 has been found to play a role in iron homeostasis and ferroptosis, an iron-mediated apoptosis [38,39]. Thus, it would be interesting to examine whether p73 is involved in similar pathways. Moreover, as deregulated iron metabolism underlies the pathogenesis of many human cancers [40], it would also be important to determine whether these regulations contribute to p73-mediated tumor suppression. In addition, as p73 is expressed as multiple isoforms with opposing functions, it will be important to dissect the function of these different isoforms and, subsequently, determine their roles in iron homeostasis. Furthermore, several recent studies showed that FDXR mutation was found in patients diagnosed with mitochondrial disorders [19,20,41]. Notably, patients with biallelic mutations of FDXR have neurodegeneration associated with inflammation, although the underlying mechanism is not clear [41]. As p73 deficiency is known to cause neuronal degeneration and inflammation [7], it is important to determine whether FDXR-mediated TP73 expression is involved in the pathogenies of the disease.
In our study we observed several interesting findings in compound Trp73+/−;Fdxr+/− mice when compared with Trp73+/− or Fdxr+/− mice. First, the median lifespan of Trp73+/−;Fdxr+/− mice was significantly longer (Figure 1A). Second, Trp73 deficiency alleviated tumor penetrance as well as liver steatosis observed in Fdxr+/− mice (Table 1, Figure 1C). Third, Fdxr deficiency was unable to attenuate pathological abnormalities observed in Trp73+/− mice, including chronic inflammation and splenic/thymic hyperplasia (Figure 1C,D). Taken together, these findings suggest that Fdxr and p73 function antagonistically in the pathways that mediate liver steatosis. It also suggests that FDXR may function upstream of p73 in regulating chronic inflammation or splenic/thymic hyperplasia. Moreover, although we showed that Trp73+/−;Fdxr+/− mice developed fewer tumors than Fdxr+/−, several lines of evidence may help explain it. First, we observed enhanced cellular senescence in Trp73+/−;Fdxr+/− when compared with Fdxr+/− MEFs (Figure 2). As cellular senescence is known to be a critical mechanism for tumor suppression, we postulate that the enhanced cellular senescence may contribute to the decreased tumor formation in Trp73+/−;Fdxr+/− mice. Second, like Trp73+/− mice, Trp73+/−;Fdxr+/− mice develop chronic inflammation in multiple organs (Figure 1C and see supplementary material, Tables S3, S4). It has been suggested that chronic inflammation serves as a double-edged sword in tumor promotion and suppression. Thus, it is likely that reduced p73 expression in Fdxr+/− mice alters the tumor microenvironment and, therefore, has a protective effect. Nevertheless, further studies are needed to elucidate the role of the FDXR-p73 loop in tumor suppression and aging.
In our study, we found that IRP2 represses TP73 expression via destabilizing TP73 mRNA. We also demonstrated that an IRE in the TP73 3′UTR is required for IRP-mediated p73 expression. As IRP2 has been shown to promote tumor growth [42], IRP2-mediated suppression of p73, most likely TAp73, contributes to its oncogenic activities. However, as TAp73 and ΔNp73 share the same 3′UTR, it is likely that IRP2 may regulate ΔNp73 isoforms. Due to the opposing functions between TAp73 and ΔNp73, further studies are warranted to analyze the biological function of IRP2-mediated suppression of ΔNp73.
As shown by genome-wide association studies [43] and mouse models [44], IRP2 is found to be elevated in chronic obstructive pulmonary disease (COPD). IRP2 expression is also found to be altered in people with lung tumors [45]. Interestingly, loss of p73 is also associated with COPD owing to its role in regulating multiciliogenesis [8,9]. Additionally, mice deficient in TAp73 are prone to lung adenocarcinoma. Thus, further studies are needed to elucidate whether IRP2-mediated suppression of p73 is involved in COPD and in lung cancers.
Supplementary Material
Table S1. Survival, tumor spectrum steatosis, inflammation and other abnormalities in WT mice (n = 56)
Table S2. Survival, tumor spectrum steatosis, inflammation and other abnormalities in Fdxr+/− mice (n = 31)
Table S3. Survival, tumor spectrum steatosis, inflammation and other abnormalities in Trp73+/− mice (n = 28)
Table S4. Survival, tumor spectrum steatosis, inflammation and other abnormalities in Fdxr+/−;Trp73+/− mice (n = 24)
Table S5. The sequences of primers used in this study
Acknowledgements
This work was supported in part by National Institutes of Health R01 grants CA224433 and CA081237 to X. Chen and by the CCAH (Center for Companion Animal Health, UC Davis) grant 2018-18F to J. Zhang.
Footnotes
No conflicts of interest were declared.
References
- 1.Dotsch V, Bernassola F, Coutandin D, et al. p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol 2010; 2: a004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harms K, Nozell S, Chen X. The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci 2004; 61: 822–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res 2004; 2: 371–386. [PubMed] [Google Scholar]
- 4.Melino G, De Laurenzi V, Vousden KH. p73: friend or foe in tumorigenesis. Nat Rev Cancer 2002; 2: 605–615. [DOI] [PubMed] [Google Scholar]
- 5.Liu G, Nozell S, Xiao H, et al. DeltaNp73beta is active in transactivation and growth suppression. Mol Cell Biol 2004; 24: 487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rufini A, Agostini M, Grespi F, et al. p73 in cancer. Genes Cancer 2011; 2: 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang A, Walker N, Bronson R, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000; 404: 99–103. [DOI] [PubMed] [Google Scholar]
- 8.Nemajerova A, Kramer D, Siller SS, et al. TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes Dev 2016; 30: 1300–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marshall CB, Mays DJ, Beeler JS, et al. p73 is required for multiciliogenesis and regulates the Foxj1-associated gene network. Cell Rep 2016; 14: 2289–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tissir F, Ravni A, Achouri Y, et al. DeltaNp73 regulates neuronal survival in vivo. Proc Natl Acad Sci U S A 2009; 106: 16871–16876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilhelm MT, Rufini A, Wetzel MK, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev 2010; 24: 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomasini R, Tsuchihara K, Wilhelm M, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008; 22: 2677–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rufini A, Niklison-Chirou MV, Inoue S, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev 2012; 26: 2009–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez N, Pelaez A, Barderas R, et al. Clinical implications of the deregulated TP73 isoforms expression in cancer. Clin Transl Oncol 2018; 20: 827–836. [DOI] [PubMed] [Google Scholar]
- 15.Hanukoglu I Steroidogenic enzymes: structure, function, and role in regulation of steroid hormone biosynthesis. J Steroid Biochem Mol Biol 1992; 43: 779–804. [DOI] [PubMed] [Google Scholar]
- 16.Hanukoglu I, Gutfinger T, Haniu M, et al. Isolation of a cDNA for adrenodoxin reductase (ferredoxin-NADP+ reductase). Implications for mitochondrial cytochrome P-450 systems. Eur J Biochem 1987; 169: 449455. [DOI] [PubMed] [Google Scholar]
- 17.Lambeth JD, Kamin H. Adrenodoxin reductase. Properties of the complexes of reduced enzyme with NADP+ and NADPH. J Biol Chem 1976; 251: 4299–4306. [PubMed] [Google Scholar]
- 18.Hanukoglu I Conservation of the enzyme-coenzyme interfaces in FAD and NADP binding adrenodoxin reductase - a ubiquitous enzyme. J Mol Evol 2017; 85: 205–218. [DOI] [PubMed] [Google Scholar]
- 19.Paul A, Drecourt A, Petit F, et al. FDXR mutations cause sensorial neuropathies and expand the spectrum of mitochondrial Fe-S-synthesis diseases. Am J Hum Genet 2017; 101: 630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng Y, Shinde DN, Valencia CA, et al. Biallelic mutations in the ferredoxin reductase gene cause novel mitochondriopathy with optic atrophy. Hum Mol Genet 2017; 26: 4937–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Brien G, Cruz-Garcia L, Majewski M, et al. FDXR is a biomarker of radiation exposure in vivo. Sci Rep 2018; 8: 684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu G, Chen X. The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress-induced apoptosis. Oncogene 2002; 21: 7195–7204. [DOI] [PubMed] [Google Scholar]
- 23.Hwang PM, Bunz F, Yu J, et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med 2001; 7: 1111–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Qian Y, Zhang J, et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev 2017; 31: 1243–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Sun W, Kong X, et al. Mutant p53 antagonizes p63/-p73-mediated tumor suppression via Notch1. Proc Natl Acad Sci U S A 2019; 116: 24259–24267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Cho SJ, Shu L, et al. Translational repression of p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes Dev 2011; 25: 1528–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dohn M, Zhang S, Chen X. p63alpha and DeltaNp63alpha can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene 2001; 20: 3193–3205. [DOI] [PubMed] [Google Scholar]
- 28.Peritz T, Zeng F, Kannanayakal TJ, et al. Immunoprecipitation of mRNA-protein complexes. Nat Protoc 2006; 1: 577–580. [DOI] [PubMed] [Google Scholar]
- 29.Yang HJ, Zhang J, Yan W, et al. Ninjurin 1 has two opposing functions in tumorigenesis in a p53-dependent manner. Proc Natl Acad Sci U S A 2017; 114: 11500–11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flores ER, Sengupta S, Miller JB, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005; 7: 363–373. [DOI] [PubMed] [Google Scholar]
- 31.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192: 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Y, Ghosh M, Kovtunovych G, et al. Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron-sulfur cluster biogenesis. Biochim Biophys Acta 1823; 2012: 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erlitzki R, Long JC, Theil EC. Multiple, conserved iron-responsive elements in the 3′-untranslated region of transferrin receptor mRNA enhance binding of iron regulatory protein 2. J Biol Chem 2002; 277: 42579–42587. [DOI] [PubMed] [Google Scholar]
- 34.Pantopoulos K Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci 2004; 1012: 1–13. [DOI] [PubMed] [Google Scholar]
- 35.Kuhn LC. Iron regulatory proteins and their role in controlling iron metabolism. Metallomics 2015; 7: 232–243. [DOI] [PubMed] [Google Scholar]
- 36.Blose JM, Proctor DJ, Veeraraghavan N, et al. Contribution of the closing base pair to exceptional stability in RNA tetraloops: roles for molecular mimicry and electrostatic factors. J Am Chem Soc 2009; 131: 8474–8484. [DOI] [PubMed] [Google Scholar]
- 37.Henderson BR, Menotti E, Kuhn LC. Iron regulatory proteins 1 and 2 bind distinct sets of RNA target sequences. J Biol Chem 1996; 271: 4900–4908. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Chen X. p53 tumor suppressor and iron homeostasis. FEBS J 2019; 286: 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy ME. Ironing out how p53 regulates ferroptosis. Proc Natl Acad Sci U S A 2016; 113: 12350–12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manz DH, Blanchette NL, Paul BT, et al. Iron and cancer: recent insights. Ann N Y Acad Sci 2016; 1368: 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slone J, Peng Y, Chamberlin A, et al. Biallelic mutations in FDXR cause neurodegeneration associated with inflammation. J Hum Genet 2018; 63: 1211–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang W, Deng Z, Hatcher H, et al. IRP2 regulates breast tumor growth. Cancer Res 2014; 74: 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeMeo DL, Mariani T, Bhattacharya S, et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am J Hum Genet 2009; 85: 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cloonan SM, Glass K, Laucho-Contreras ME, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med 2016; 22: 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khiroya H, Moore JS, Ahmad N, et al. IRP2 as a potential modulator of cell proliferation, apoptosis and prognosis in nonsmall cell lung cancer. Eur Respir J 2017; 49: 1600711. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Survival, tumor spectrum steatosis, inflammation and other abnormalities in WT mice (n = 56)
Table S2. Survival, tumor spectrum steatosis, inflammation and other abnormalities in Fdxr+/− mice (n = 31)
Table S3. Survival, tumor spectrum steatosis, inflammation and other abnormalities in Trp73+/− mice (n = 28)
Table S4. Survival, tumor spectrum steatosis, inflammation and other abnormalities in Fdxr+/−;Trp73+/− mice (n = 24)
Table S5. The sequences of primers used in this study