

Abstract
Vulvar squamous cell carcinomas (VSCC), which constitute over 90% of vulvar malignancies in adults, are classifiable into 2 subgroups that are mostly clinicopathologically distinct, a classification that is fundamentally based whether or not the tumors are HPV-mediated. In this review, we aim to summarize the recent advances in the understanding of molecular events in the pathogenesis of VSCC, including common and targetable mutations, copy number alterations, epigenetics, noncoding RNAs, and tumor immune microenvironment, which may provide insight into the future management of the disease. These events show substantial differences between the 2 subgroups, although significant areas of overlap exist. Recurrent, driver mutations appear to be substantially more prevalent in HPV(−) VSCC. TP53 mutations are the most common somatic mutations in VSCC overall, and are notably predominant in the HPV(−) VSCC, where 30–88% show a mutation. TP53 mutations are associated with worse patient outcomes, and co-mutations between TP53 and either HRAS, PIK3CA or CDKN2A appear to define subsets with even worse outcomes. A wide variety of other somatic mutations have been identified, including a subset with different mutational frequencies between HPV(+) and HPV(−) VSCC. CDKN2A mutations are common, and have been identified in 21 to 55% of HPV(−) VSCC, and in 2 to 25% of HPV(+) VSCC. Hypermethylation of CDKN2A is the most frequently reported epigenetic alteration in VSCC and the expression of some microRNAs may be associated with patient outcomes. The PTEN/PI3K/AKT/mTOR pathway is commonly altered in HPV(+) VSCC, and is accordingly potentially targetable. HPV-positivity/p16 block expression by immunohistochemistry has been found to be an independent prognostic marker for improved survival in VSCC, and may have some predictive value in VSCC patients treated with definitive radiotherapy. 22–39.3% and 68% of VSCC show EGFR amplification and protein overexpression respectively, although the prognostic and predictive value of an EGFR alteration requires additional study. Recurrent chromosomal gains in VSCCs have been found at 1q, 2q, 3q, 4p, 5p, 7p, 8p, 8q, and 12q, and there may be differential patterns of alterations depending on HPV-status. At least one-third of VSCC patients may potentially benefit from immune checkpoint inhibition therapy, based on a high frequency of PD-L1 expression or amplification, or a high tumor mutational burden. Additional studies are ultimately required to better understand the global landscape of genetic and epigenetic alterations in VSCC, and to identify and test potential targets for clinical application.
Keywords: CDKN2A, differentiated vulvar intraepithelial neoplasia (dVIN), high-grade squamous intraepithelial lesions (HSIL), molecular events, mutation, PI3K/AKT/mTOR, TP53, vulvar squamous cell carcinoma (VSCC)
Introduction
Vulvar cancers are rare, and over 90% of them are squamous cell carcinoma (VSCC). Approximately 6120 new cases of vulvar cancer will be diagnosed in the United States in 2020, with roughly 1,350 deaths, accounting for 4–5% of all gynecologic cancers [1]. While nearly all squamous cell carcinomas of the uterine cervix are caused by carcinogenic types of high-risk human papillomavirus (HPV), VSCCs may or may not be associated with high-risk HPV infection, which has resulted in a dualistic model of approaching VSCC that is fundamentally based on whether or not the tumor is HPV-associated [2, 3]. HPV-unrelated VSCCs typically occur in older women and are associated with differentiated vulvar intraepithelial neoplasia (dVIN) and/or chronic inflammatory skin disorders such as lichen sclerosus [4–7]. HPV-related vulvar cancers, which represent approximately 30% of all VSCCs, often originate from usual type high-grade squamous intraepithelial lesions (HSIL) and typically present in comparatively younger women [7–10]. Histologically, HPV-unrelated VSCCs tend to be keratinizing and well to moderately differentiated, whereas HPV-related VSCCs are predominantly of basaloid or warty morphology.
As a group, patients with HPV-unrelated VSCCs may have a less favorable prognosis than their counterparts with HPV-related VSCCs [11–13]. However, the clinical management and prognostication of VSCC patients has generally not been based on their distinctive etiopathogenetic basis, but rather on conventional clinicopathologic parameters, as detailed by elsewhere in this issue of the Journal. Tumor stage and lymph nodes status are considered to be the most important independent determinants of patient outcome [14–16], with a 5-year survival rate for patients with distant metastasis of less than 20%. Accordingly, there is a need for novel approaches to understand and treat this tumor. The potential application of molecule/pathway-based targeted therapies to VSCC largely relies on identification of genetic alterations that are amenable to such therapies in these tumors. In this review, we aim to summarize the recent advances in the understanding of molecular events in the pathogenesis of VSCC, including common and targetable mutations, copy number alterations, epigenetics, noncoding RNAs, and tumor immune microenvironment, which may provide insight into the future management of the disease.
1. TP53
As a caretaker tumor suppressor and a multifunctional transcriptional factor, p53, encoded by TP53 gene in human, has been regarded as “the guardian of the genome” given its critical role in controlling cell cycle progression and cell survival, conserving DNA integrity, and preventing mutations of the genome [17]. Disturbance of p53 function, through mutation, deletion, or perturbation in pathways signaling to p53, for example, MDM2 amplification, occurs in more than half of human cancers [18]. Mutations of TP53 gene will lead to genomic instability, aberrant regulation of DNA damage response and apoptosis, and eventually carcinogenesis. In fact, as the most frequently altered gene in human tumors, TP53 mutations usually exert a dominant-negative effect that leads to loss of tumor suppressor activity of wild-type p53. Some TP53 mutations result in the acquisition of new oncogenic driver activities [19].
HPV-unrelated VSCCs harbor a wide variety of somatic TP53 gene mutations that commonly occur at the earliest stages of the disease. TP53 gene mutations have been reported to be present in 30–80% of HPV-negative SCCs of the vulva. In a recent study, we detected TP53 somatic mutations in 15 (88%) of 17 HPV-unrelated tumors (Figure 1), a high frequency that was likely related to advanced depth of next-generation sequencing technology [3]. The majority of tumors showed a single TP53 gene mutation, although a small subset displayed multiple TP53 mutations that probably represented a manifestation tumor heterogeneity. Perhaps more importantly, the multiplicity of mutations may also manifest itself in tumor progression. In one analysis, although 36% of recurrent, HPV-negative VSCCs carried TP53 gene mutations that were identical to the primary tumors, a majority of cases (63%) harbored different, occasionally more complex TP53 gene mutations [12]. Additionally, some originally TP53 wild type tumors became TP53 mutant in recurrences, and some of those recurrences were at multi-year intervals after the original resections, which suggests ongoing oncogenesis [12]. The background chronic inflammatory dermatoses may promote ongoing genetic instability that may set the stage for additional oncogenic events that are clinically interpreted as recurrences [18]. Irrespective of the mechanism of action, TP53-mutant VSCC appear to be associated with comparatively shorter disease-free intervals than their TP53-wild type counterparts [18]. However, it remains to be clarified what role specific TP53 mutations play in influencing survival. One study demonstrated that 45 (76%) of 59 HPV-negative VSCCs contained one or two TP53 somatic mutations [20]. 18 different “hot spot” mutations were identified in 22 (49%) of 45 cases with 5 Arg273, 3 Arg282, 2 Arg278, 2 Arg248, 2 Tyr220, and 1 hot spot for the rest of cases. Likewise, somatic mutations of Arg282, Arg273, Arg248 were also described in our study [3]. It has been demonstrated that gain-of-function mutations in the TP53 gene are associated with an aggressive clinical course through their influence on cell proliferation, motility and invasion, angiogenesis, and overall metastatic potential [21, 22]. For example, mutations on the Arg248 and Arg282 positions, but not other hotspot mutations, have been found to display significant association with shorter patient survival. Experimental expression of TP53 mutant Arg282Trp in cancer cell lines significantly upregulated CYP3A4 mRNA and protein levels which have been linked to chemoresistance [23].
Figure 1.
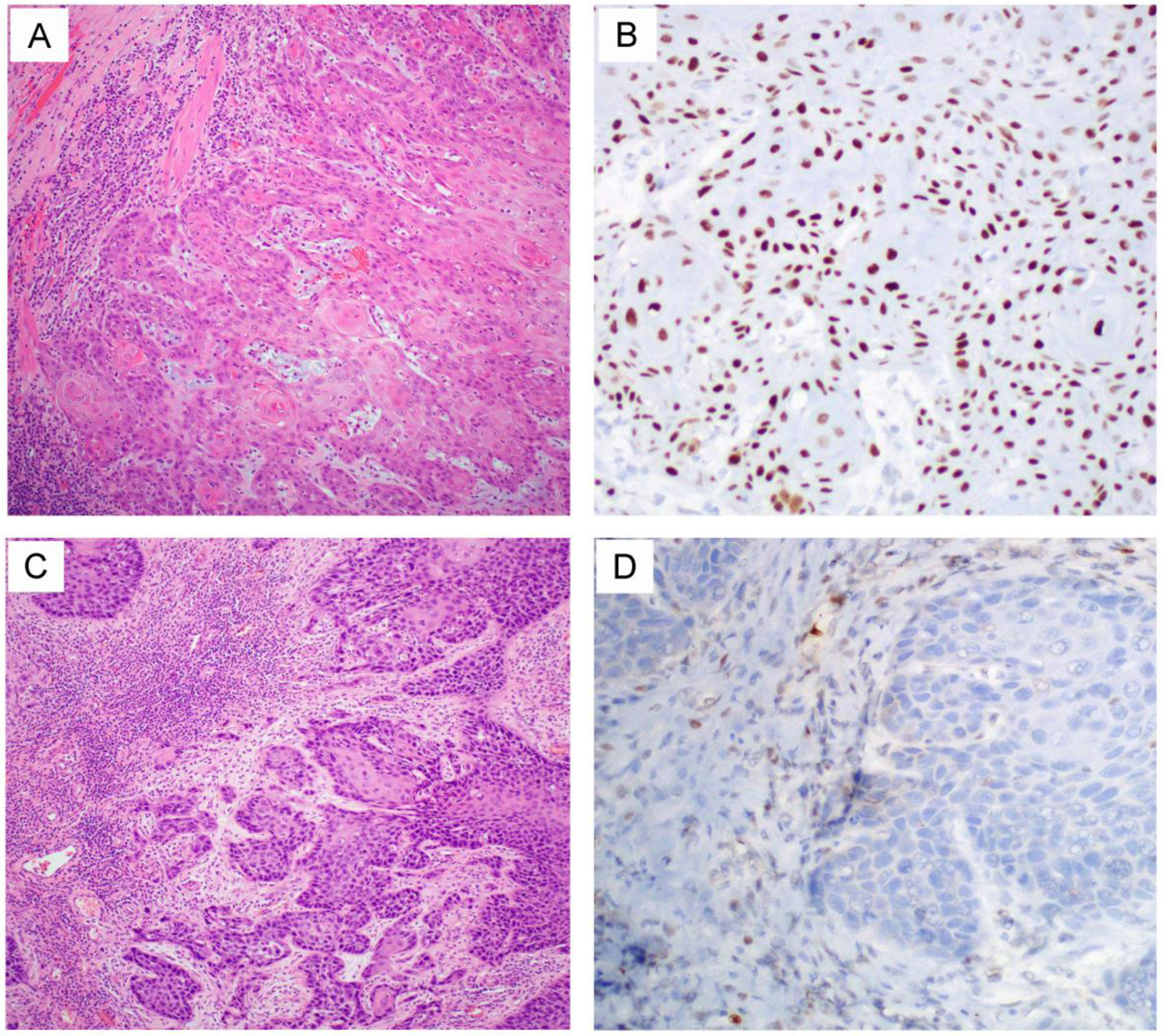
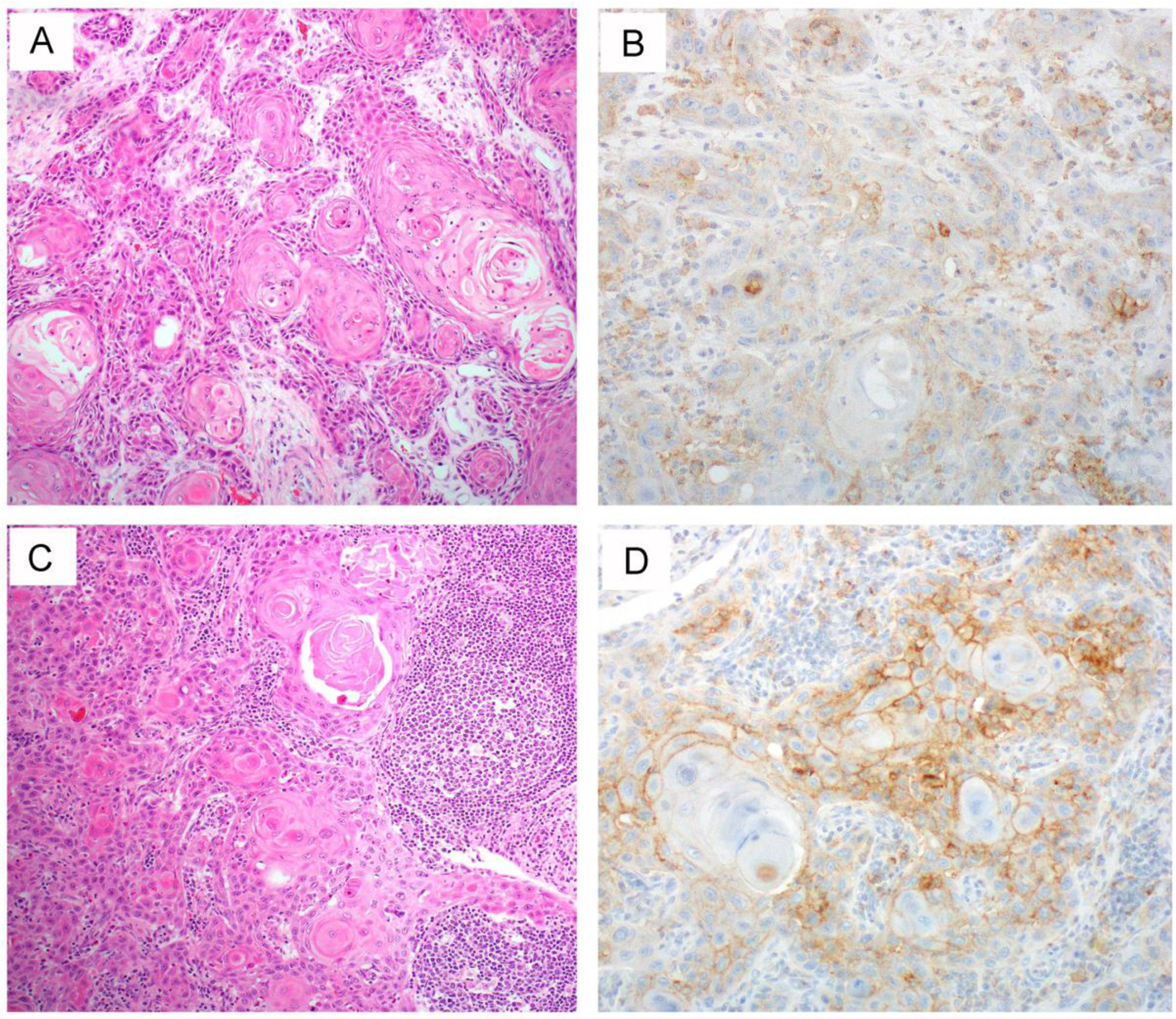
Two representative HPV-unrelated vulvar squamous cell carcinoma cases with TP53 mutation [3]. One tumor (A, H&E) harbors a TP53 p.Arg175His missense mutation. Consistent with this genotype, the tumor demonstrates aberrant/mutation-type p53 over-expression (B, strong and diffuse nuclear staining pattern). Another tumor (C, H&E) had a TP53 p.Gln165fs frameshift mutation that leads to synthesis of a truncated p53 protein. Thus, the tumor demonstrates aberrant/mutation-type complete loss of p53 expression (D) because the truncated protein cannot be recognized by the p53 antibody. A wild-type/normal pattern of p53 expression is seen in the stroma as an internal control.
Several nucleotide-based sequencing analyses of TP53 somatic mutation in the VSCCs have shown that the most frequent mutation types are C:G →T:A transitions, present in up to 57.9% of cases [24]. This mutation pattern is thought to result from hydrolytic deamination of 5-methylcytosine residues in these sites and usually occurs at first position of the Arg codon. Although commonly related to degenerative and chronic inflammatory diseases, this type of mutation is often induced by oxidative DNA damage in cancers. Mutations that occur in this setting would be consistent with current models of HPV-unrelated vulvar carcinogenesis, which is postulated to be a stepwise, multistage, multifactorial process that includes chronic inflammatory dermatoses such as lichen sclerosus, evolving to dVIN, and ultimately the invasive malignancy (Figure 2). Genome-wide analysis has provided genetic and epigenetic evidence for a clonal relationship between lichen sclerosus, dVIN, and HPV-unrelated VSCC. TP53 somatic mutation and CDKN2A promoter methylation have been observed in all three lesions. Additional genetic and epigenetic alterations (see below) acquired in the each step facilitate the oncogenic process (Figure 2).
Figure 2.
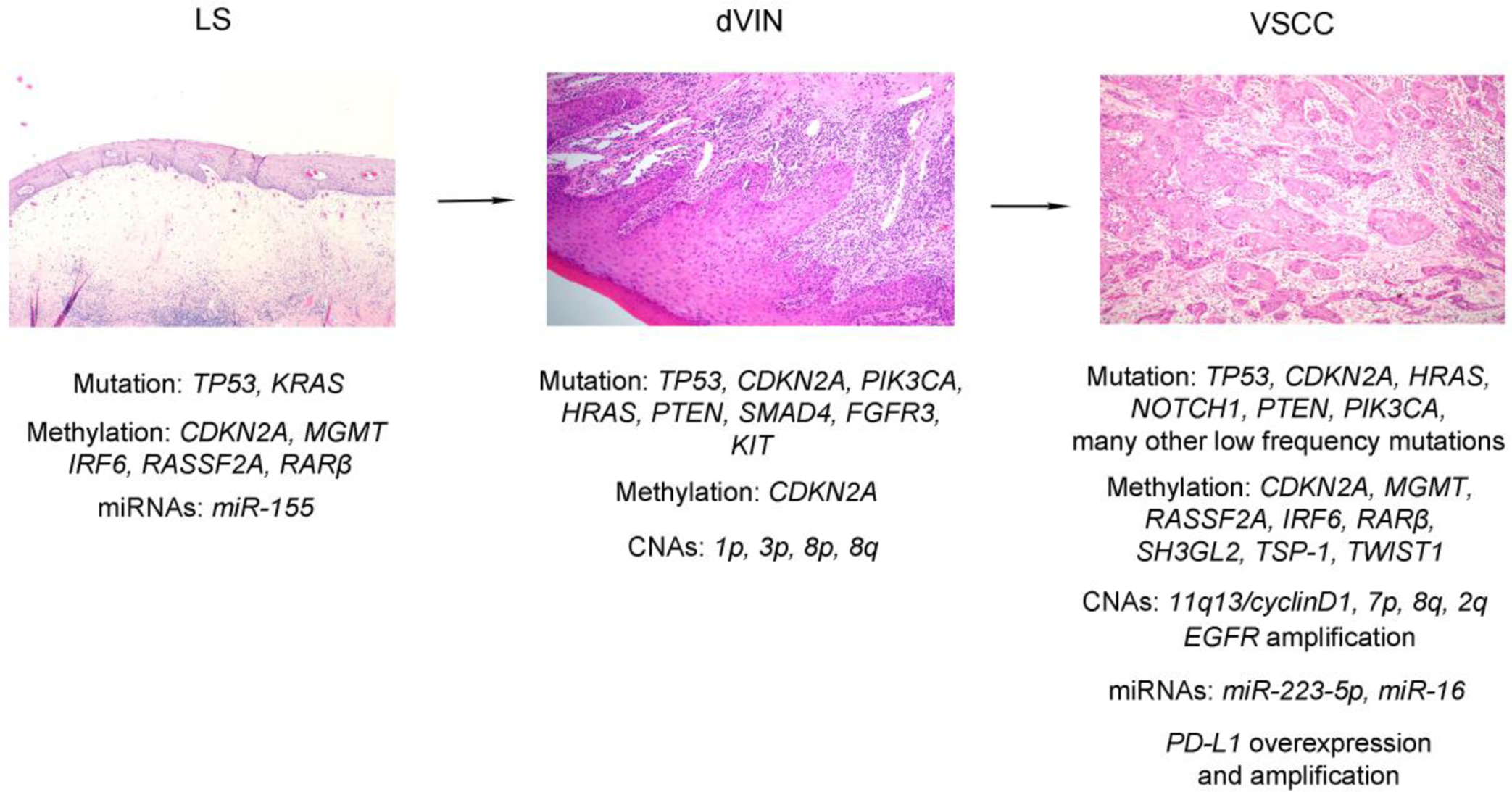
Overview of the multistage, stepwise evolution of HPV-unrelated vulvar squamous cell carcinoma (VSCC). The HPV-independent pathway often, but not always, starts with lichen sclerosus, which can develop into differentiated vulvar intraepithelial neoplasia (dVIN), which is thought to be the precursor of HPV-unrelated VSCC. Genome-wide analysis has provided genetic and epigenetic evidence for a clonal relationship between lichen sclerosus, dVIN, and HPV-unrelated VSCC. TP53 somatic mutation and CDKN2A promoter methylation have been observed in all three lesions. Additional genetic (for example, somatic mutations of CDKN2A, NOTCH1, and HRAS) and epigenetic alterations (for example, promoter methylation of MGMT, RASSF2A, RARβ and IRF6) acquired in the each step facilitate the oncogenic process.
With its central position to cellular machinery, TP53 likely plays a central role in this process that probably includes elevated levels of reactive oxygen species, oxidative stress-induced DNA damage, and chronically degenerative disease of the vulva. Indeed, TP53 mutations have been identified in every step of the process: For example, Rolfe et al. [25] found 77.8% of VSCCs, 46.7% of lichen sclerosus and 22.2% squamous hyperplasia to harbor at least one somatic mutation in TP53. Notably, in that study, the same mutation was identified in the VSCC and in the adjacent, non-dysplastic epidermis in 42% of cases [25]. Likewise, another study demonstrated that TP53 and KRAS somatic mutations occurred significantly more frequently in lichen sclerosus than in the adjacent and non-adjacent normal genital skin [26]. These findings indicate that TP53 and likely other mutations occur in the microenvironment of chronic inflammatory dermatoses in which most HPV-unrelated VSCC develop. The precise factors that drive the clonal selection process, and ultimately tumorigenesis, are unclear.
While TP53 mutations lead to loss of cell cycle arrest and uncontrolled cellular proliferation in HPV-unrelated VSCCs, cell cycle dysregulation is caused by the interaction of HPV viral oncoproteins with p53 in HPV-related VSCCs. Since the high-risk HPV viral oncoprotein E6 has the ability to neutralize the function of p53, the majority of high-risk HPV-related cancers would be expected to have a wild-type TP53 gene. Zieba et al. [5] found pathogenic mutations of TP53 to be present in 24% of HSIL cases, and other several studies have similarly reported a relatively low frequency of TP53 mutations in HPV-related VSCC and/or HSIL [3, 10, 24, 27, 28]. Not all studies have uniformly reported that TP53 mutations are more predominant in the HPV-unrelated pathway [29]. Ultimately, the role of TP53 mutation in the setting of high-risk HPV-mediated vulvar SCCs remain largely unknown; we speculate TP53 mutations are more likely to be involved in tumor progression or a passenger mutation, than being an initiating event in this subset of VSCCs.
Given the high frequency mutation of TP53 in HPV-unrelated VSCC, it has been proposed that depletion and destabilization of mutant p53 might serve as promising strategies for personalized therapy in these patients. However, mutated p53 is thought to be a challenging targetable biomarker. Nevertheless, some small molecules with a capacity for promoting reactivation of common mutant p53 proteins such as R175H and R273H have been identified. For example, APR-246 is a small-molecule that reactivates mutant and inactivated p53 protein by restoring p53 conformation and function, thereby inducing programmed cell death in cancer cells [30, 31]. This small molecule is currently in clinical trial, and may potentially benefit those VSCC patients with TP53 mutations.
2. CDKN2A (p16)
Since TP53 is generally regarded as a caretaker tumor suppressor gene that plays a critical role in conserving stability of genome by preventing mutational alterations, mutations of this gene will lead to genomic instability, aberrant regulation of DNA damage response and apoptosis, and eventually carcinogenesis. Not surprisingly, additional driving events, especially genetic alterations of gatekeeper oncogenes and other caretaker tumor suppressor genes, have been postulated to facilitate the progression of TP53 mutated precursor lesions to invasive SCC of the vulva. Somatic mutations in CDKN2A are probably the second most common genetic change in VSCCs. [5], but are absent in lichen sclerosus.
The CDKN2A gene is situated on chromosome 9p21, a region with high frequency of loss of heterozygosity (LOH) in different tumor types [32]. As a tumor suppressor gene, CDKN2A encodes the proteins p16INK4A and p14ARF that regulate cell cycle progression from G1 phase to S phase by promoting the activities of the retinoblastoma protein (RB) and p53 transcription factor, respectively. Normally, the cyclin D-dependent kinases, CDK4 and CDK6, stimulate the cells to continue through the cycle and divide. Binding of p16INK4a to CDK4 and CDK6 blocks their ability to stimulate cell cycle progression and thus, controls cell proliferation and division [33]. Unlike p16INK4A, p14ARF helps prevent tumor formation by protecting p53 from being broken down through a mechanism of antagonizing the function of the p53 negative regulator MDM2, a ubiquitin protein ligase that promotes p53 degradation [34].
Overall, CDKN2A mutations have been identified in 21 to 55% of HPV-negative SCC [3, 28, 35], and in 2 to 25% of HPV-positive SCC [29, 35], and, most studies conclude that CDKN2A mutations are also frequently seen in HPV-negative, rather than HPV-related VSCC patients [10, 27, 36–38].
CDKN2A mutations frequently coexist with TP53 mutation [29, 37, 39] and may have prognostic value. One study showed that the combination of mutations in both TP53 and CDKN2A correlated with a significantly worse prognosis in head and neck SCCs [40]. It has been postulated that the tumor cells tend to be more destabilized when both CDKN2A and TP53 are mutated, thus a conferring worse prognosis. Consistent with this hypothesis, Trietsch et al. [39] found that, when correcting for HPV infection, tumor size, and age, having both an HRAS and a TP53 mutation or both a CDKN2A and a TP53 mutation increased the hazard ratios to 5.1 (95% CI 1.504–17.062) and 4.1 (95% CI 1.2–14.074), respectively, for disease-specific death.
At the protein level, the loss of p16 expression is frequently seen in a variety of human tumors including HPV-unrelated VSCCs, mainly due to point or truncation mutations, gene deletion, or promoter hypermethylation (see below). In HPV-related VSCCs, the E7 oncoprotein binds and inactivates the tumor suppressor protein Rb, leading to p16 overexpression. Thus, a diffuse/block pattern of p16 immunohistochemical staining can serve as a surrogate marker for high-risk HPV infection (Figure 3). HPV infection leads to development of vulvar intraepithelial lesion 1 (VIN 1)/low-grade squamous intraepithelial lesion (LSIL). 20% of VIN 1/LSILs persist, of which half causes VIN 2–3/HSILs. A small subset of VIN 2–3/HSILs develops into HPV-related VSCC. The most common genetic and epigenetic alterations acquired in this oncogenic process is outlined in Figure 4.
Figure 3.
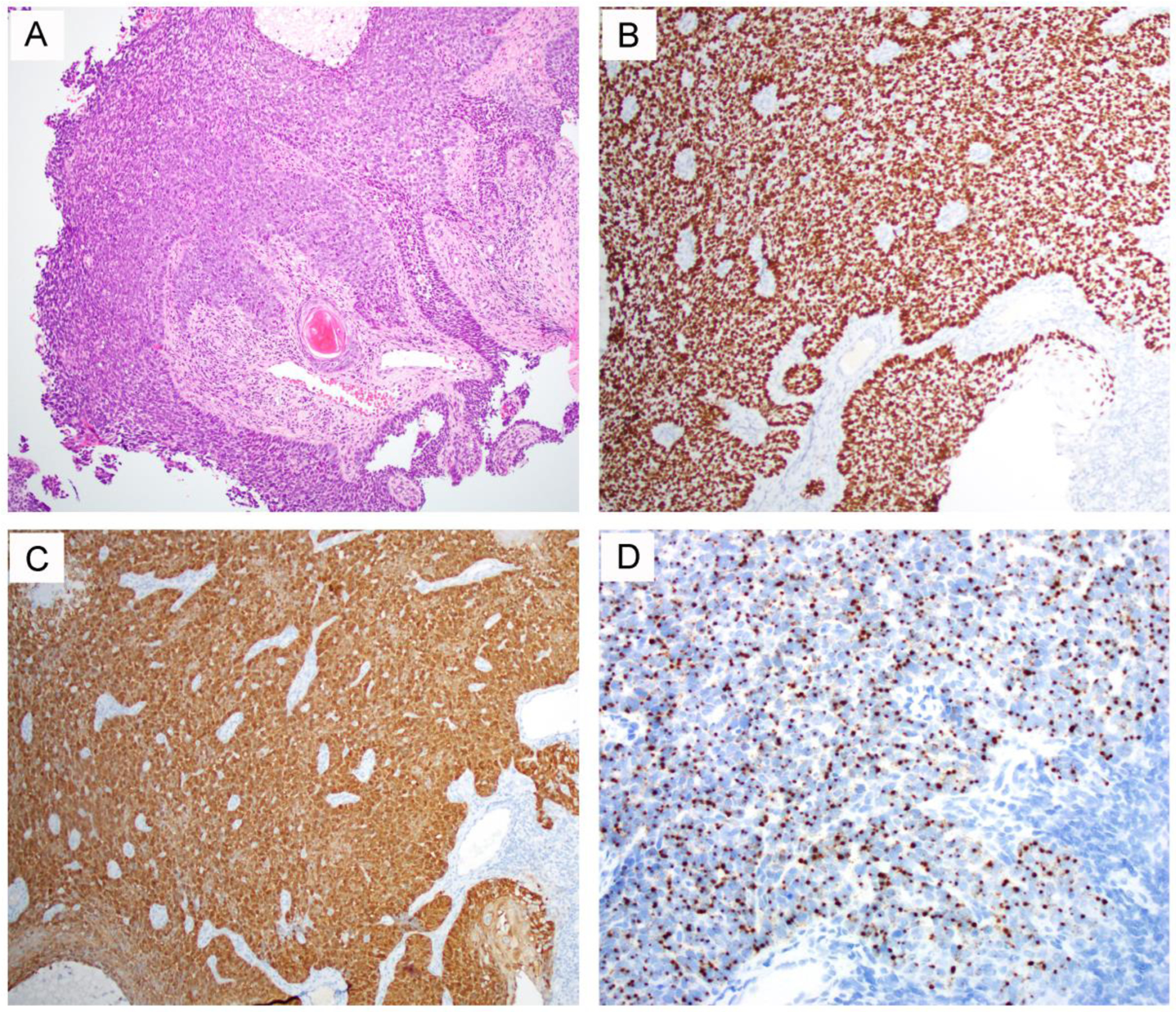
A representative HPV-related vulvar squamous cell carcinoma case with a wild-type TP53 [3]. The tumor displays a basaloid morphology with focal keratinization (A) and demonstrates strong and diffuse p63 (B) and p16 (C) expression with detected high-risk HPV by RNA in situ hybridization (D).
Figure 4.
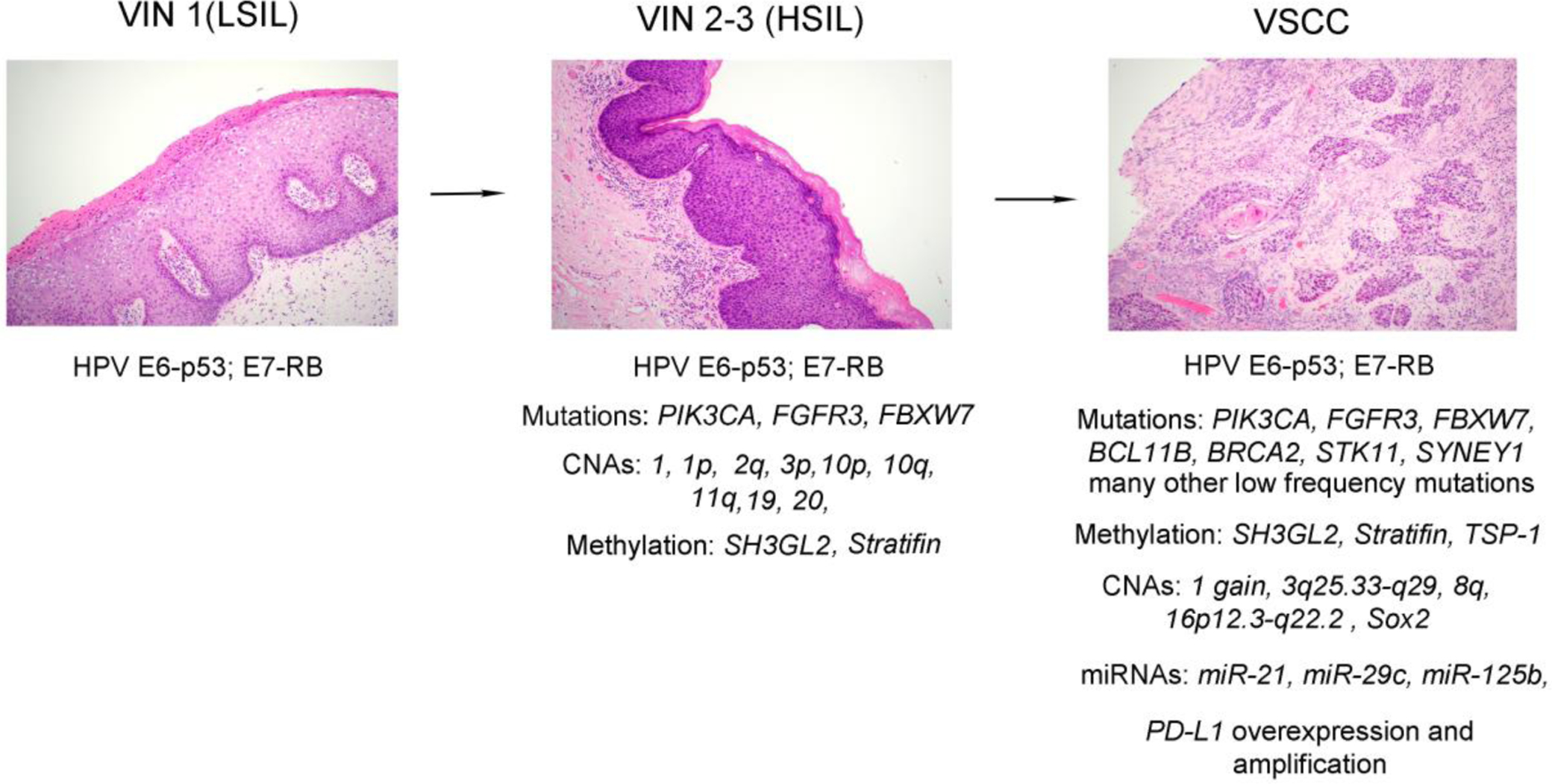
Overview of the multistage, stepwise evolution of HPV-related vulvar squamous cell carcinoma (VSCC). HPV infection leads to development of vulvar intraepithelial lesion 1 (VIN 1)/low-grade squamous intraepithelial lesion (LSIL). HPV E6 oncoprotein has the ability to neutralize the function of p53 and E7 oncoprotein binds and inactivates the tumor suppressor protein Rb. 20% of VIN 1/LSILs persist, of which half causes vulvar intraepithelial lesion 2–3 (VIN 2–3)/high-grade squamous intraepithelial lesions (HSILs). A small subset of VIN 2–3/HSILs develops into HPV-related VSCC. Additional genetic (for example, somatic mutations of PIK3CA, FGFR3, and FBXW7) and epigenetic alterations (for example, promoter methylation of SH3GL2 and Stratifin) acquired in the each step facilitate the oncogenic process.
A recent meta-analysis [13] that included 475 VSCC cases showed that p16 may be an independent prognostic marker for overall and disease specific survival after adjusting for potential confounders. Additionally, p16 overexpression may have some predictive value regarding response to radiotherapy, or at the very minimum, for predicting improved outcomes in VSCC patients treated with definitive radiotherapy [41–43].
3. PTEN/PI3K/AKT/mTOR pathway
Commonly disrupted across different cancer types, phosphatidylinositol-3-OH kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway regulates cell proliferation, differentiation, quiescence, apoptosis, longevity and oncogenesis [44]. Numerous studies have revealed that mTOR protein kinase plays a critical role in coordinating cell growth by sensing the level of nutrients in their environment [45]. Growth factors and nutrient inputs (glucose, amino acid, oxygen) send stimulatory signals to the small GTPase Ras (see below) and lipid kinase PI3K that continue to activate mTOR protein kinase. The disruption of crosstalk of the Ras, PI3K and mTOR signaling pathways drives cell growth in an uncontrolled manner and ultimately, drives tumorigenesis through the proteins that directly regulate global protein synthesis, cell cycle progression, metabolism, and survival [46].
The PI3K pathway has been implicated in cancer since its discovery and is thought to be among the most common mutations identified in all sporadic human cancers, including VSCCs [47]. It has been estimated that mutation in one of PI3K pathway component is present in up to 30% of all human cancers. Williams et al. estimate that 54% of HPV-positive VSCC have a potentially actionable alteration in the PTEN/PI3K/AKT/mTOR pathway [35].
3.1. PTEN
Phosphatase and Tensin Homolog deleted on Chromosome 10 (PTEN) is a dual phosphatase with both protein and lipid phosphatase activities that dephosphorylates phosphatidylinositol-3, 4, 5-phosphate (PIP3), a critical second messenger mediating the signal of growth factors and insulin [48]. As a negative regulator of PI3K signaling pathway, PTEN catalyzes removal of the D3 phosphate from PtdIns(3,4,5)P3 to limit and ultimately terminate PI3K signaling in cells. Loss of the PTEN lipid phosphatase appears to be the most common mechanism of activation of the PI3K pathway in human cancers [49]. Indeed, some investigators believe PTEN to be the second most commonly mutated tumor suppressor in humans, after TP53 [50].
In one study, Holway et al. [51] tested PTEN mutation in preneoplastic and neoplastic lesions obtained from 10 patients with VSCCs. They found five of eight VSCCs to harbor PTEN mutations that occurred in carcinoma in situ as well as squamous cell carcinoma. Importantly, PTEN mutations were identified in mucosal regions with mild or focal dysplasia (low-grade squamous intraepithelial lesion) in two patients. These results indicate that PTEN is frequently altered in VSCCs and can occur in early dysplastic changes in vulvar mucosa. Due to lack of HPV analysis in this study, the relationship between PTEN mutation and HPV infection cannot be assessed. Several studies have demonstrated that PTEN mutations can occur either in HPV-related or HPV-unrelated VSCCs, with an overall low mutational rate of less than 10% of cases [3, 27, 29, 37, 39, 52]. However, one large scale study found PTEN mutations to be substantially more common in HPV-positive VSCC (14% versus 2%, p<0.0001) [35]. Loss of PTEN expression, rather than somatic mutations, has also reported in these tumors [53, 54].
3.2. PIK3CA
As a known oncogene, PIK3CA is located at chromosome 3q26 which encodes the p110 catalytic subunit of phosphatidylinositol 3-kinase (PI3K). It has been demonstrated that PIK3CA plays a critical role in HPV-induced carcinogenesis, evidenced by the observation that activation of the PI3K/AKT/mTOR pathway through PIK3CA regulates various transformed phenotypes as well as growth and differentiation of HPV-immortalized cells [55]. In fact, PIK3CA is thought to be one of the most frequently mutated genes in HPV-related cancers including cervical squamous cell carcinoma, endocervical adenocarcinoma and HPV-positive head and neck tumors [56–58].
In our analysis of 12 HPV-related VSCCs, we identified PIK3CA hotspot mutations, p.Glu545Lys and p.Glu542Lys, in 25% of cases [3]. These findings are consistent with the mutational frequency reported by Williams et al. (31%) [35]. It is also broadly consistent previous results that that a subset of HPV-associated small cell neuroendocrine carcinomas (SCNECs) of the cervix harbor PIK3CA mutations [59]. Despite lacking a PIK3CA somatic mutation, one VSCC case, as discussed above, harbored PTEN (p.Gly129Arg) mutation that is speculated to cause excessive PIP3 at the plasma membrane, and further activate the PI3K/AKT/mTOR pathway that is similarly disrupted by PIK3CA mutation [60]. As such, the HPV-related VSCCs bearing these mutations, regardless of anatomic site, might be sensitive to mTOR or AKT inhibitor treatment, and is accordingly a potential target for therapy.
In a large series, Koncar et al. [53] analyzed molecular profiling of 743 TP53 wild-type, HPV-induced squamous cell carcinomas of anus, uterine cervix, oropharynx, and vulva. They found that the most commonly mutated gene was PIK3CA, with mutations most often affecting the helical domain of the protein and accompanied by concurrent lack of PTEN expression. The mutational rate of PIK3CA was 24.8% in anal SCC, 30.5% in cervical SCC, 22.3% in oropharyngeal SCC, and 10.2% in VSCC. The study concluded that the similar molecular profiles of the four cohorts indicate that treatment strategies by targeting PI3K pathway may be similarly efficacious across HPV-positive cancers. However, frequent PIK3CA mutations are reported in in HPV-unrelated VSCCs, which suggests that the oncogenesis of VSCC, at least for the subset of cases that either involve HPV infection or TP53 mutation, may converge towards a universal PI3K/AKT/mTOR pathway. Weberpals et al. [27] found that 27% of HPV-positive VSCCs contained PIK3CA mutation, whereas 19% of HPV-negative tumors harbored somatic mutations in this gene. In contrast, another study showed 7 (8%) of 89 HPV-negative VSCCs carried PIK3CA mutations, whereas none in HPV-positive tumors [39]. In a recent study, Tessier-Cloutier et al. [52] investigated genetic alteration in HPV-unrelated squamous epithelial lesions, including VSCC, verrucous carcinomas (VC), dVIN, differentiated exophytic vulvar intraepithelial lesions (DE-VIL), and vulvar acanthosis with altered differentiation (VAAD). They found that TP53 mutations occurred in 73% of VSCC, 85% of dVIN, and in 0% of VC, DE-VIL or VAAD. HRAS showed a mutational pattern comparable to PIK3CA, and both mutations predominated in lesions without a TP53 mutation (PIK3CA [50% VC, 33% DE-VIL, 100% VAAD, 40% VSCC]; HRAS [63% VC, 33% DE-VIL, 0% VAAD, 20% VSCC]). The absence of TP53 mutation in verrucous carcinoma, VAAD, and DE-VIL, and the frequent presence of PIK3CA and HRAS mutations, suggest a different carcinogenetic pathway than the typical keratinizing squamous cell carcinoma. Indeed, the occasionally observed temporal relationship between DE-VIL and verrucous carcinoma, as well as their shared mutational profile regarding HRAS and PIK3CA, has led some authors to conclude that DE-VIL is the precursor lesion for VC [61]. The presence of a PIK3CA mutation may also have prognostic significance, as patients whose VSCC have TP53 and PIK3CA co-mutations may have the worst clinical outcomes [52], which raises the possibility that there is a synergistic and/or additive effect between these two pathways.
3.3. AKT
AKT, also known as Protein kinase B/PKB, is a serine/threonine-specific protein kinase that plays a key role in cell proliferation, glucose metabolism, apoptosis, cell migration. In mammalian cells, there are three closely related Akt isoforms encoded by different genes: Akt1 (PKBα), Akt2 (PKBβ) and Akt3 (PKBγ) [62]. These isoforms are expressed in a tissue-specific manner but all are regulated by phosphoinositide-dependent kinase-1 (PDK1), a PH domain-containing kinase downstream of PI3K, which phosphorylates Akt isoforms [63].
Compared with PTEN and PIK3CA, somatic mutation of AKT appears to be less frequent in VSCCs. In the aforementioned study, which included 743 cases of HPV-unrelated SCCs of anus, uterine cervix, oropharynx, and vulva, the only gene found mutated within all four cohorts, aside from PIK3CA, was AKT1, although at a low rate that ranged from 0.8% to 5.5% [53]. Occasional somatic mutations of AKT1 in VSCCs have been similarly reported in other studies [29, 54]. Moreover, TSC2, a direct target of AKT1, has also been reported to harbor somatic mutations in VSCCs [10].
3.4. Pathway/molecule-based target therapy
Thus far, the PI3K/AKT/mTOR cascade is considered to be the most important targetable pathway in VSCC with involvement of two distinct etiologies. Theoretically, inhibition of PI3K pathway is a promising therapeutic strategy in the management of VSCC patients [64, 65]. For example, alpha-specific PI3K inhibitor alpelisib (BYL719) is an FDA-approved medication for treating patients with advanced hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, and PIK3CA-mutated (PIK3CAmut) breast cancer [66]. In vivo and in vitro studies have demonstrated that alpelisib can efficiently inhibit head and neck squamous cell carcinoma. A Phase 1b study of cetuximab and Alpelisib concurrent with intensity modulated radiation therapy in stage III-IVB head and neck squamous cell carcinoma is ongoing [67]. Accordingly, VSCC patients with PIK3CA mutation may potentially benefit from this targeted therapy.
As a downstream component of PI3K-Akt cascade, mTOR has been found to be widely expressed in the vast majority of VSCC samples by immunohistochemical staining [29]. An in vitro study showed that three mTOR inhibitors rapamycin, everolimus and AZD2014 can significantly inhibit proliferation of VSCC cell lines CAL-39 and SW-954 [29]. Some clinical trials investigating the role of different mTOR inhibitors indicate that targeting mTOR alone may lead to unsatisfactory outcomes in gynecological cancer. Ultimately, tumors such as VSCC with alternative survival pathways may require the inactivation of multiple individual pathways for successful treatment.
4. RAS-MAPK pathway
Similar to PI3K/AKT/mTOR pathway, the Ras-mitogen activated protein kinase (MAPK) pathway plays an essential role in the regulation of the cell cycle, differentiation, cell growth and senescence [68]. Ras is a family of related proteins that belong to small guanosine nucleotide-bound GTPases that involve cellular signal transduction [69]. When growth factors bind to receptor tyrosine kinases, extracellular matrix receptors, cytokine receptors, and G-protein-coupled receptors, Ras protein is activated and then sends activation signal to Raf, the first MAPK kinase of the pathway [70]. Raf phosphorylates and activates extracellular kinase 1 (MEK1) and MEK2, which further catalyze the activation of the effector extracellular signal regulated kinase 1 (ERK1) and ERK2 kinases. Once activated, ERK1/ERK2 translocate into the nucleus and exert broad regulatory functions. In an alternative pathway, RAS can activate PI3K/AKT/mTOR pathway by direct binding of RAS to PI3Kγ [71].
Ras family genes include HRAS, KRAS and NRAS, and are found to be mutated in 10–30% of cancers [72]. One study demonstrated that HRAS mutation is one of three most frequent mutations (TP53, CDKN2A, HRAS) in HPV-negative VSCCs [39]. Overall, the various studies have reported HRAS mutational frequencies of 4.5 to 7% in HPV-positive VSCC and 3–24% in HPV-negative VSCCs [3, 27, 29, 39]. Although mutations of KRAS, NRAS and BRAF have also been reported in the VSCCs, the frequency of these mutations is low compared with that of HRAS [39, 54]. Several studies have demonstrated that HRAS are commonly mutated in both HPV-positive and HPV-negative precursor lesions of VSCCs, but the role that any specific combination between HRAS and another effector may play in determining a particular malignant phenotype is unclear [28, 52]. Regarding potential therapeutic implications, in one cell line-based in vitro and in vivo study, constitutive activation of HRAS (Ras G12V mutation) was found to confer resistance to EGFR-targeted therapy in A431 human vulvar squamous carcinoma cells [73]. As a therapeutic strategy, direct targeting of RAS family proteins including HRAS has been unsuccessful, although searching for novel effective inhibitors still continues [74]. On the other hand, targeting RAS-MAPK pathway downstream effectors in some types of malignant neoplasms has shown promising results and may potentially have some clinical application in VSCC.
VSCC patients with somatic HRAS mutations have a significantly worse prognosis than patients lacking these changes, which could be partially due to epithelial-mesenchymal transition (EMT). While EMT is primarily driven by transcriptional repression of E-cadherin and other epithelial proteins, the MEK/ERK pathway is thought to be indirectly responsible for the process that may confer highly aggressive tumor behavior [75]. Theoretically, a dual blockade of upstream RAS by MEK inhibitors, and a more downstream inhibition of PI3K, may have better clinical results for the VSCC patients with HRAS mutations [76]. Similar to the coexistence of TP53 and CDKN2A or TP53 and PIK3CA mutations, having both an HRAS and a TP53 mutation may be associated with poorer clinical outcomes [39].
5. Receptor tyrosine kinase
Receptor tyrosine kinases (RTKs) are transmembrane protein receptors with tyrosine kinase activity that helps mediate cell-to-cell communication and control cell growth, metabolism, motility and migration, apoptosis, and differentiation [77]. All RTKs contain an extracellular domain with the ligand-binding site, single transmembrane region and cytosolic domain that contains a juxtamembrane regulatory region, a tyrosine kinase domain and a C-terminal tail. When signaling molecules (ligands) bind to extracellular region of RTKs, they cause the cytosolic regions of neighboring RTKs to cross-linked as dimers [78]. By cross-phosphorylation, i.e., one RTK in the dimer phosphorylates multiple tyrosine sites on the other RTK, or auto-phosphorylation, the tyrosine kinase activity is activated and initiates a series of enzymatic reactions, for example, activation of RAS-MAPK pathway and PI3K/AKT/mTOR pathway, that transmit the signal to the nucleus and alter patterns of protein transcription [79]. Dysregulation of RTK signaling by mutations, deletions, translocations and amplification and/or overexpression commonly occurs in human diseases, especially cancer [80]. In fact, it has been demonstrated that almost all types of human tumors including VSCC harbor genetic alterations in one or more RTKs.
The fibroblast growth factor receptors (FGFRs) are a gene family of RTKs with at least four designated members FGFR1, FGFR2, FGFR3, and FGFR4 in vertebrates, of which, FGFR3 Ser249Cys is commonly mutated genes in low-grade human urothelial cell carcinoma [81]. We and other have showed that FGFR3 p.Ser249Cys mutation was occasionally detected in HPV-related VSCC cases [3, 29]. Consistent with these findings, a recent study showed that 14% of HPV-positive tumors contained somatic mutations in this gene, raising this possibility of utilization of inhibitors of FGFR3 as a therapeutic strategy to treat a subset of vulvar SCC patients [27]. Although rare, somatic mutations in other RTK genes including FGFR2, JAK3, FLT3, ERBB4 have been reported in VSCCs [29, 54]. Over the past several decades, RTK inhibitors have proven to be effective anti-tumor agents in different cancers, and as such, may also have clinical application in VSCCs that carry RTK mutations or amplifications. HER2/neu amplification has been shown to be a rare event in VSCCs, with an amplification rate of 2% [54, 82]. In contrast, EGFR gene amplifications and/or overexpression has been identified more frequently. 22–39.3% of VSCC show EGFR amplification as assessed by fluorescence in situ hybridization (FISH) [54, 83–86] and EGFR aberrations appear to be restricted to the HPV-negative subset of VSCC. Up to 68% of VSCC show EGFR expression by immunohistochemistry [87]. The reported levels of correlation between EGFR protein expression and copy number increases have been somewhat incongruent across studies. Woelber et al. [98] identified a low positive correlation, whereas de Melo Maia et al. [88] showed that EGFR protein overexpression may be independent of gene amplification, suggestive of other molecular mechanisms that may regulate protein expression of EGFR in VSCCs. Dong et al found that 87.5% of EGFR amplified VSCC showed a 3+ staining level for EGFR by immunohistochemistry [85]. Growdon et al. [83] estimate that a 3+ staining for EGFR has 100% sensitivity, 79% specificity, and 39% positive predictive value to detect EGFR gene amplification.
It is unclear how well EGFR aberrations correlate with patient outcomes: Woelber et al. [98] found EGFR copy number increase to be related to unfavorable patient outcomes on multivariate analyses. Similarly, Growdon et al. [83] found EGFR amplification to be associated with decreased survival, even after adjusting for age at diagnosis, stage and lymph node metastasis. Johnson et al. found increased EGFR expression (assessed by IHC) to be associated with lymph node metastases and decreased survival [89]. Other authors have not found EGFR aberrations to retain their predictive value in multivariate models [85, 87]. Nonetheless, these findings support the potential utility of EGFR inhibitors as a therapeutic option in a subset of VSCCs. Indeed, in one Phase II trial evaluating the anti-EGFR medication erlotinib in VSCC, significant responses were observed, albeit of short durations [84].
6. Other miscellaneous mutations
While studies on somatic mutations other than those most frequently mutated genes used to be hampered by the absence of analytical tools, recently developed next-generation sequencing (NGS) technology allows for analysis of mutations and other genetic alterations, such as copy number change and translocations, in multiple genes. Distribution of 6 mutations including TP53, CDKN2A, HRAS, PIK3CA, FGFR3, described above, as well as NOTCH1 in HPV-unrelated and HPV-related VSCCs in 7 studies is outlined in Figure 5 [3, 10, 27–29, 36, 39].
Figure 5.
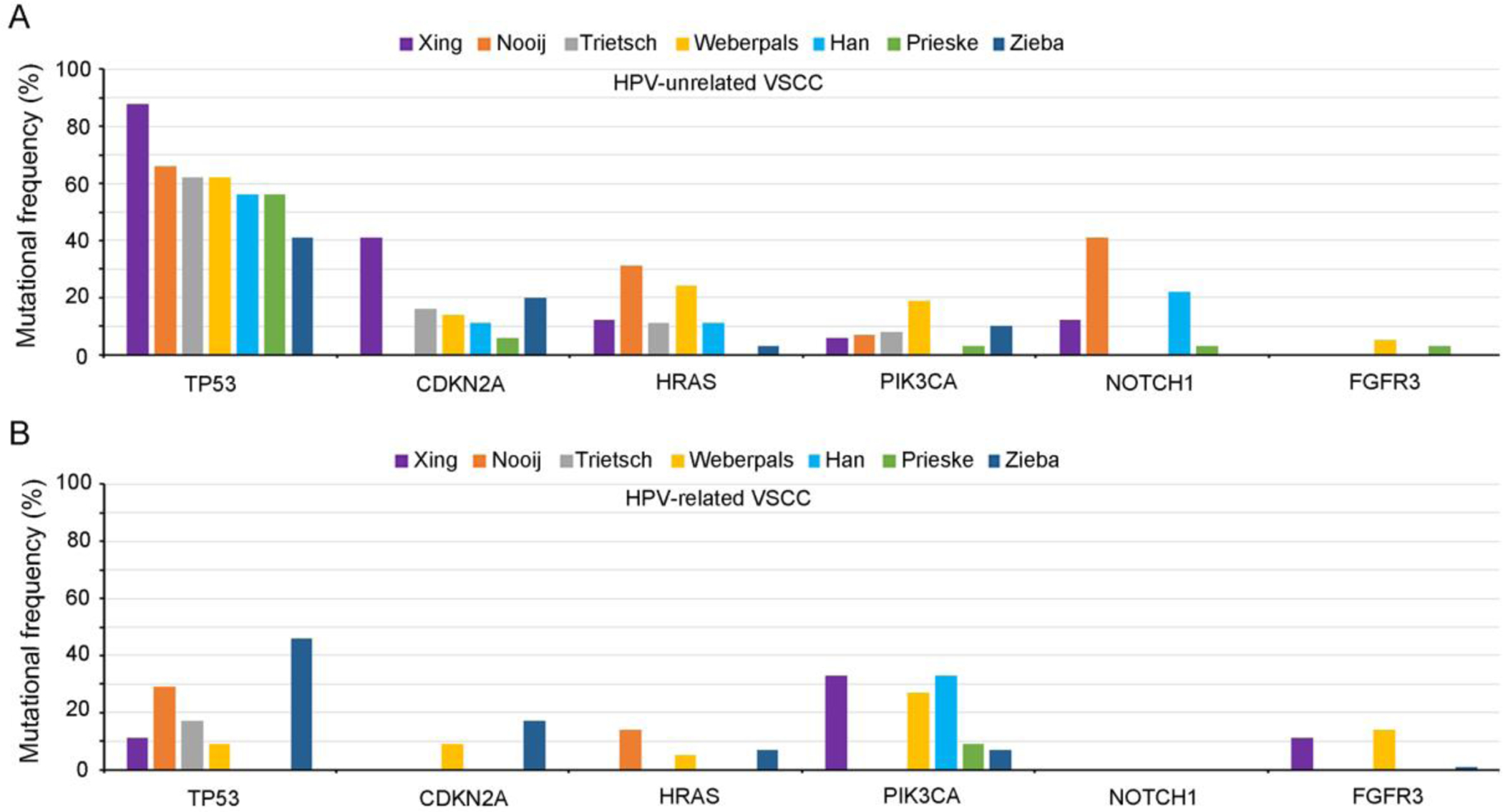
Distribution of 7 mutations including TP53, CDKN2A, HRAS, PIK3CA, NOTCH1, and FGFR3, in HPV-unrelated (A) and HPV-related (B) VSCCs. The graph presents the results of the following seven studies: Xing et al. [3], Nooij et al. [28], Trietsch et al. [39], Weberpals et al. [27], Han et al. [36], Prieske et al. [10], and Zieba et al. [29]. HPV-unrelated VSCCs display relatively high somatic mutational frequencies in TP53, CDKN2A, HRAS, and NOTCH1 genes, whereas HPV-related VSCCs harbored more mutations in PIK3CA and FGFR3 genes.
Like other pathways, NOTCH pathway plays an important role in cell growth, differentiation, and apoptosis. NOTCH1 gene has contextual functions with activities of both an oncogene and a tumor suppressor [90]. Similar to HRAS, which reportedly has a high mutational rate in HPV-negative tumors and their precursor lesions, NOTCH1 was found to be mutated in 20 (24%) of 82 all sequenced precursor lesions (3/22, 14% HPV-positive and 17/60, 28% HPV-negative) [28]. 29% (10 of 35) HPV-unrelated TP53 wild-type precursor lesions harbored NOTCH1 somatic mutations. Strikingly, up to 41% of HPV-negative vulvar cancer contained NOTCH1 mutations [28]. We have previously reported a NOTCH1 mutation in a small cell neuroendocrine carcinoma and 2 cases of HPV-unrelated VSCCs [3]. It has been well documented in the literature that one function of mutant NOTCH1 is to activate c-Myc and PI3K-AKT-mTOR1 signaling through transcriptional repression of PTEN and promoting growth factor receptor signaling to PI3K-AKT [91]. As such, we speculate that tumors, whether they are SCNECs or VSCCs, bearing these mutations might be sensitive to mTOR or AKT inhibitor treatment, thus providing a potential avenue of targeted therapy for these aggressive diseases.
F-box/WD repeat-containing protein 7 (FBXW7) gene encodes an F-box family protein that, as subunits of ubiquitin protein ligase complex called SCFs (SKP1-cullin-F-box), functions in phosphorylation-dependent ubiquitination [92]. FBXW7 is a less frequently mutated gene that has been reported in several studies on vulvar cancer, with reported mutation frequency of ≤10% in VSCCs [29, 36, 54]. Williams et al. reported that FBXW7 mutations are more common in HPV-positive VSCC (10% vs 3%, p=0.030) [35]. Interestingly, the main targets for degradation regulated by FBXW7 include NOTCH1, cyclin E, c-Jun, c-Myc and mTOR that are also commonly dysregulated/mutated in VSCCs.
An interesting potential target identified in our study of VSCCs is BRCA1-associated protein-1 (BAP1) that was mutated in 1 HPV-unrelated and 1 HPV-related VSCCs [3]. BAP1 is a nuclear deubiquitinating enzyme that regulates transcription, cell cycle, cell differentiation, DNA damage response and other cellular process [93]. It has been demonstrated that BAP1 can induce radioresistance in HPV-negative head and neck squamous cell cancer (HNSCC) cells and was associated with poor outcomes in patients with HNSCC [94]. Importantly, several studies showed that esophageal and laryngeal SCCs harbored recurrent somatic BAP1 mutations that may be associated with prognostic significance [94–96]. A role of BAP1 in the carcinogenic process, and its prognostic value, if any, in VSCCs remain unknown and warrants further investigation.
Although uncommon, BRCA2 somatic mutations have been described in VSCCs in several studies with an up to 10% of mutation rate [3, 36, 54]. The poly [ADP-ribose] polymerase (PARP) inhibitors have been successfully used for the treatment of germline BRCA-mutated ovarian cancer patients [97–99]. Importantly, patients with a somatic BRCA mutation also show benefit from treatment with PARP inhibitors [100, 101]. Identification of somatic BRCA mutations in patients with VSCC allows testing of PARP inhibitors to treat this aggressive tumor. Other somatic mutations, most with relatively low mutation frequency, that have been described in VSCCs include APC, ATM, BCL11B, BCORL1, CASC5, CASP8, CCND1, CDK12, CHD4, COL1A1, CREBBP, CTNNB1, CTRC1, DOCK2, EP300, ERC1, EXT2, EZH2, FANCA, FAT1, GNA11, GNAQ, GNAS, HOOK3, KAT6B, KDR, KMT2C, KMT2D, LRP1B, MACF1, MAP2, MET, MLLT6, MSH6, NACA, NBPF1, NCOA1, NCOAR2, NF1, NFE2L2, NOTCH2, NOTCH3, NRG1, NSD1, POLE, PPP2R1A, PMS2, PTPN11, PTPRB, PTPRK, RB1, RNF213, SMAD2, SMAD4, SMARCA4, SMARCB1, SPEN, STK11, SYNE1, SYNE2, TLX1, TRRAP, UBR5, ZFHX3, among many other genes [3, 10, 27–29, 36, 39, 52, 54]. A few of these show differential frequencies between HPV-negative and HPV-positive VSCC, including FAT1, pTERT, CCND1, and NOTCH1 which are reportedly more frequent in the HPV-negative group, and FBXW7, STK11, and EP300, reportedly more common in HPV-positive VSCC [35]. In a study that utilized whole exome sequencing of DNA isolated from 34 VSCC samples and matched normal tissue, Prieske and colleagues [10] detected total cohort 1848 cancer related mutations with median of 54.4 per sample. However, they found that while a multitude of cancer-related mutations was detected in various samples, only few mutations recur and/or affect concurrent signaling pathways.
7. Copy number alterations (CNAs) and selected other potentially targetable alterations
Copy number alterations (CNAs) are somatic changes in the chromosome that result in gain (duplication and/or amplification) or loss (deletion) in copies of sections of DNA. With a gain-of or loss-of-function, CNAs can promote tumor initiation and progression through changing gene expression levels at the affected genomic regions [102]. As one characteristic feature of many, if not all cancers, genomic instability can lead to accumulation of structural chromosome aberrancies, particularly CNAs. While somatic CNAs are nearly ubiquitous in cancer, different cancer types vary in their extent and location of CNAs, thus may have specific CNAs associated with cancer clinical characteristics and prognosis as well as the potential driver genes that are targetable [103]. Recurrent chromosomal gains in VSCCs have been found at 1q, 2q, 3q, 4p, 5p, 7p, 8p, 8q, and 12q [36, 104–108]. Han et al identified 18 recurrently altered CNA regions in HPV-negative VSCC, with gains on 7p and 8q and loss on 2q being the most recurrent CNAs. In that same analysis, 15 recurrently altered CNA regions were identified in HPV-positive VSCC, with gains at 3q25.33-q29 being the most common recurrent event. The authors also identified a recurrent region of kataegis at chromosome 16p12.3-q22.2 in a subset of HPV-positive VSCC [36]. Jee et al. [108] used CGH to analyze genetic alterations in 10 primary invasive VSCCs and they found frequent chromosomal losses of 4p13-pter, 3p and 5q as well as frequent chromosomal gains of 3q and 8p, a pattern of chromosomal imbalance similar to that in cervical cancers. Purdie et al. [109] used single-nucleotide polymorphism microarray analysis to perform the high-resolution investigation of genome-wide allelic imbalance in vulvar neoplasia (21 high-grade VIN cases and 6 VSCCs). The study showed that the most common recurrent aberrations were gains at 1p and 20, and the most frequent deletions observed at 2q, 3p and 10. Swarts et al. [110] used whole genome next generation shallow sequencing to analyze global CNAs in 41 HPV-related and 24 HPV-unrelated VSCCs including their precursor lesions. They found HPV-positive and -negative VSCC to display a partially overlapping pattern of recurrent CNA, including frequent gains of 3q and 8q. Chromosome 1 gain was detected in 81% of HPV-positive VIN and associated VSCCs and only in 21% of VIN cases without subsequent VSCCs. This study suggests that cancer risk in VIN seems to be reflected by the extent of CNA, in particular chromosome 1 gain in HPV-positive cases. In our study, a number of candidate driver genes were identified by clinically reportable threshold from the copy number variations [3]. From this group, 2 HPV-unrelated vulvar SCCs displayed BIRC3 amplification that was also reported in several human and mouse cancers [111–113]. BIRC3, as well as BIRC2, are members of the inhibitor of apoptosis family that inhibit apoptosis by interfering with the activation of caspases. In our study, while 1 tumor showed BIRC3 amplification, the other tumor demonstrated copy number gains including both BIRC2 and BIRC3 genes. In fact, these 2 genes are located in the chromosome 11q22 amplicon that is frequently seen in certain cancer types. One study showed 12.9% of cervical carcinomas had a high level amplification of BIRC2 that was significantly more frequent than CCND1 amplification (chromosome 11q13 amplicon) [114]. The tumor with BIRC3 amplification also showed copy number gains in other locations containing driver oncogenic genes such as MYC, MCL1, CDK4 and AEPX1, indicating genetic complexity. The patients whose tumors bear these genetic alterations may benefit from personalized therapy amenable to these targets.
Cyclin D1 (CCND1) has been found to be commonly amplified and overpressed in a variety of cancers, including VSCCs. Choschzick et al. [105] found that 32 (22.4%) of 143 VSCCs had CCND1 amplification, whereas detectable CCND1 expression was found in 83.2% of tumors. Increased levels of CCND1 expression, which is correlated with gene amplification, were associated with higher age, lymph node metastasis, and negative HPV status. Swarts et al. [110] reported that amplification of 11q13/cyclinD1 was exclusively found in HPV-negative lesions. SOX2, a SOX gene family transcription factor, was found to be correlated with basaloid phenotype and HPV-positive VSCCs. A study conducted by Gut et al. [106] showed 10 (20.8%) of 48 VSCCs had SOX2 amplification that was correlated with its overexpression. Amplification of the SOX2 locus was associated with high tumor grade and HPV positivity. This study provides further evidence for different molecular alterations in HPV-positive and HPV-negative VSCCs.
8. Epigenetic alterations – DNA methylation
An epigenetic alteration is a heritable change in gene expression that does not directly affect the sequence of the DNA. As a hallmark of cancer, epigenetic regulation plays a critical role in almost every aspect of tumor biology including cell growth and differentiation, cell cycle control, DNA repair, angiogenesis, migration, apoptosis and senescence [115]. DNA methylation, one of the most important areas of epigenetics, is probably the best studied epigenetic modification in mammals where methylation primarily occurs by the covalent modification of cytosine residues in CpG dinucleotides [116]. DNA methylation can lead to gene silencing. While overall genomic hypomethylation frequently occurs in cancers in comparison to control normal tissue, selective hypermethylation of promoters of tumor suppressor genes is considered as one of the most constant features of the cancer genome [117]. Indeed, CpG island hypermethylation has been described in almost every tumor type, including VSCCs.
It has been demonstrated that epigenetic inactivation of genes is a common event in VSCC and which can also be present in adjacent lesions, implying a possible role for these alterations in early carcinogenesis. Guerrero et al. [118] found increasing percentages of hypermethylation of genes from isolated lichen sclerosus to lichen sclerous associated with VSCC to pure VSCC. Interestingly, they found MGMT and RASSF2A hypermethylation exclusively in VSCC and lichen sclerosus associated with VSCC, but not in isolated pure lichen sclerosus. Rotondo et al. investigated the association of expression and promoter methylation of Retinoic Acid Receptor β (RARβ) gene [119] and Interferon Regulatory Factor 6 (IRF6) gene [120] with onset and progression of lichen sclerosus-associated VSCCs in two studies. In one study, the authors found that the RARβ promoter was hypermethylated in 90% of lichen sclerosus-associated VSCC, 55% of cancer-free lichen sclerosus, and 25% in the normal skin group [119]. Likewise, in another study, IRF6 promoter was found to be hypermethylated in 45% of lichen sclerosus specimens, 80% of VSCC specimens, 10% of cancer-free lichen sclerosus specimens, and not methylated in normal skin specimens [120]. These findings suggest that RARβ and IRF6 dysregulation may play a role in progression of lichen sclerosus-associated VSCCs, and their promoter methylation status may be used as a prognostic marker in clinical treatment of patients with lichen sclerosus-associated VSCCs.
As previously noted, the p16/Rb/cyclin-D1 pathway is frequently altered in the VSCC and its precursor lesions, largely due to gene mutation, deletion, or amplification. Epigenetic regulation of p16 expression is likely a critical event in the development and progression of VSCCs [121, 122]. In one meta-analysis of the literature, there was a high variability in the reported frequencies of CDKN2A hypermethylation: up to 68% for VSCC, up to 72% for VIN, and up to 47% for lichen sclerosus [123].
Some studies have evaluated the correlation of methylation status in certain genes with clinicopathological parameters of VSCCs. Li et al. [124] reported that SH3GL2 gene methylation was detected in 28 (53.8%) of 52 cases of VSCC. The methylation status was closely related to tumor TNM stage and HPV infection status, but not related to age, tumor volume, tumor differentiation, lymph node metastasis and VIN grade. By the multivariate Cox proportional hazard ratio analysis, Guerrero et al. [118] showed that TSP-1 hypermethylation was an independent prognostic factor for disease-free survival independently of other significant factors as histological grade or lymph node involvement, indicating that TSP-1 hypermethylation is an unfavorable prognosis factor in VSCC. A study conducted by Oonk et al. [125] showed a high frequency of promoter methylation in TERT, p16, TEPI2, CADM1, MGMT, and TWIST1 in VSCC. A panel of three methylation markers (p16, TERT and TFPI2) reached a sensitivity of 67% and specificity of 100% for detection of metastatic lymph nodes. Although application of this panel for the detection lymph node metastases may be limited due to its suboptimal sensitivity, additional investigations of the possibility are warranted.
9. microRNAs and other non-coding RNAs
As key regulators of gene expression, microRNAs (miRNAs) are a class of small noncoding RNAs of ~22-nucleotide in length that post-transcriptionally repress gene expression by base-pairing to mRNAs [126]. It has been estimated that more than half of all mRNAs are targets of miRNAs and each miRNA is predicted to regulate from hundreds to thousands of targets [126]. In contrast, long noncoding RNAs (lncRNAs) are a class of RNA that comprises various RNA species longer than 200 nucleotides that are not translated into proteins. Both miRNAs and lncRNAs have been shown to regulate a broad range of biological processes, including proliferation, differentiation, and apoptosis [127]. miRNAs and lncRNAs may function as either oncogenes or tumor suppressors in different histopathological settings [128–130], and numerous studies have shown that these noncoding RNAs are not only involved in cancer initiation and development, but can also serve as potential biomarkers for human cancer diagnosis and management [131].
de Melo Maia et al. [132] characterized microRNA expression in VSCCs through an expression profile of 754 miRNAs: 25 microRNAs were differentially expressed between HPV-positive and HPV-negative groups whereas 79 were differentially expressed between the tumor and the normal samples. Downregulation of both miR-223–5p and miR-19-b1–5p were correlated with the presence of lymph node metastasis; downregulation of miR-100–3p and miR-19-b1–5p were correlated with presence of vascular invasion; overexpression of miR-519b and miR-133a were associated with advanced FIGO staging [132]. They further demonstrated an inverse correlation between miR-223–5p and p63 expressions in tumors from VSCC patients [133]. Another study revealed hsa-miR-93–5p and hsa-miR-425–5p as the most robust microRNAs in the plasma of VIN and VSCC patients, respectively [134].
Several studies have demonstrated that miRNAs can function as oncogenes in VSCCs. Yang et al. [135] found that the expression level of microRNA-4712–5p in VSCC tissues and the A431 vulvar cancer cell line was significantly increased. microRNA-4712–5p can reduce the expression of PTEN, further affecting its downstream p-AKT, p-GSK3β and cyclin D1 signaling pathways, promoting the proliferation and invasion of VSCC. Likewise, expression of miR-3147 was found to be markedly upregulated in VSCC tissues and its expression was associated with the depth of invasion [136]. The study showed that miR-3147 may participate in the process of epithelial-mesenchymal transition and reduce the expressions of downstream target genes in the transforming growth factor-β/Smad signaling pathway by directly binding to 3’ untranslated region of SMAD4 in A431 vulvar cancer cells. miRNAs that have an oncogenic functions in VSCC also include miR-590–5p, miR-182–5p and miR-183–5p [137].
However, microRNAs can also function as tumor suppressors in VSCCs. Agostini et al. [138] reported that HMGA2 was highly expressed in VSCCs whereas HMGA2-related miRNAs miR30c and let-7a were both downregulated. The latter, as a tumor suppressor miRNAs, is considered to be the cause of HMGA2 overexpression. A role of miRNAs in vulvar lichen sclerosus has also been described. Terlou et al. [139] found miR-155, a microRNA involved in regulation of the immune response, to be significantly upregulated in lichen sclerosus. Consistent with this finding, Ren et al. [140] demonstrated that an upregulated miR-155–5p facilitated cell proliferation, accelerated cell cycle progression and inhibited forkhead box O (FOXO) signaling pathway. Interestingly, Gao et al. [141] found urothelial cancer-associated 1 (UCA1) lncRNA functioned as a miR-103a sponge in VSCC cells. Their findings suggested exosomal UCA1 lncRNA derived from cancer-associated fibroblasts conferred VSCC cell resistance to cisplatin in vitro and in vivo at least partly through the miR-103a/WEE1 axis, highlighting a novel therapeutic method for improving the clinical benefits of cisplatin chemotherapy in VSCC patients. Other lncRNAs such as HOAIR, MALAT1, MEG3, NEAT1, MIR31HG and LINC00478 have been reported in VSCCs but their role in the tumorigenesis remains largely unknown [142].
10. The immune microenvironment in vulvar squamous cell carcinoma
It has been reported in the literature that vulvar cancer is an immunomodulatory tumor that harnesses the PD-1/PD-L1 pathway to induce tolerance. Our and other studies have documented that PD-L1 overexpression is readily detected in a substantial proportion of vulvar carcinomas [3, 143–145]. Sznurkowski et al. [146] showed that PD-L1 was expressed on VSCC (27/84, 32.1%) and peritumoral immune cells. PD-L1-positivity of cancer nests was correlated with higher infiltration of CD4+, CD8+, FOXP3+, CD68+ cells. High-risk HPV-status did not correlate with the PD-L1 status of VSCC and immune cells. PD-L1 positivity of peritumoral immune cells was found to be an independent favorable prognostic factor for overall survival. Similarly, Choschzick et al. [147] found that up to 72% of VSCCs expressed PD-L1, and of which, 27.3% showed moderate or strong PD-L1 expression that was independent of HPV status. Cocks et al. [148] reported nearly half of VSCCs (21 cases) were PD-L1 positive in tumor cells with over two-third of cases demonstrating PD-L1 positivity in immune cells. The overall risk for mortality was higher in tumors with higher PD-L1 and CD8 expression. Another study conducted by Hecking et al. [144] demonstrated that a minority of HPV-unrelated VSCCs expressed PD-L1 in the cell membrane and that its expression was an independent prognostic factor for poor outcome. In that analysis, PD-L1 expression was found to triple the risk of relapse. In contrast, Thangarajah et al. found PD-L1 expression to be independent from HPV status and to be unrelated to survival. The authors reported PD-L1 expression in tumor cells and peritumoral immune cells in 32.9% and 91.4% of VSCC patients respectively, with an observed correlation between PD-L1 expression in tumor cells and tumor stage [149]. In our study, PD-L1 expression was seen in 6 of 10 primary VSCCs and all 6 paired cases showed discordant PD-L1 expression in the primary and metastatic sites [3] (Figure 6); the latter finding highlights the importance of evaluating PD-L1 expression in different tumoral locations to avoid false negative results. There may be a role for anti- indoleamine dioxygenase-2,3 (IDO) therapy, in combination with anti PD-L1/PD-1 therapy, in a subset of VSCC patients, given that 13% of HPV-associated VSCC express this protein (135). Another checkpoint inhibitor that may potentially have efficacy in concert with anti PD-L1/PD-1 therapy is the checkpoint molecule, TIM3, which has been shown to expressed in the majority of PD-L1-positive VSCC [150]. A study by Williams et al. [35] reported that 39% of HPV-negative VSCC show at least one marker that may predict response to immunotherapy: PD-L1 high positive by IHC, a high tumor mutational burden, or PD-L1/PD-L2 amplification. The totality of findings suggest at least one-third of VSCC patients may potentially benefit from immune checkpoint inhibition therapy. One potential mechanism of resistance to immunotherapies in VSCC was evaluated by Dibbern et al. [151]. The authors showed that 11–22% of VSCC showed clonal or total loss of expression of class I major histocompatibility complex, including many PD-L1-positive cases, thereby potentially facilitating immune escape.
Figure 6.
Aberrant expression of PD-L1 in paired primary and metastatic VSCCs [3]. A representative primary vulvar SCC (A, H&E) shows PD-L1 expression with Combined Positive Score (CPS) approximately 50 (B); metastatic tumor (C, H&E) demonstrates increased PD-L1 expression with CPS of approximately 75 (D).
To determine the genetic basis of PD-L1 overexpression, Howitt and colleagues performed fluorescence in situ hybridization (FISH) using probes targeting CD274 (PD-L1) and PDCD1LG2 (PD-L2) on specimens from 48 cervical SCCs and 23 vulvar SCCs [143]. They observed cogain or coamplification of CD274 (PD-L1) and PDCD1LG2 (PD-L2) in up to 43% of VSCCs and median PD-L1 protein expression was highest among tumors with CD274 and PDCD1LG2 coamplification. This discovery provides a genetic basis (recurrent copy number gain) for PD-L1 expression in a subset of cervical and VSCCs and identifies a class of patients that are rational candidates for therapies targeting PD-1. As such, checkpoint inhibitor-based immunotherapy may have the potential to improve outcome in patients with VSCC and could complement conventional cancer treatment.
Conclusion
HPV-related and HPV-unrelated VSCCs are two mostly distinct clinicopathologic entities, each characterized by predominantly disparate genetic and epigenetic profiles, although there are substantial areas of overlap. Much remain unknown about the core molecular events in their pathogenesis. Although targeted therapies based on genetic and epigenetic alterations has shown clinical benefit in other gynecological malignancies, these approaches remain at an early stage in VSCC. Recent advances in cancer genetics and molecular oncology, especially the implementation of next-generation sequencing and other high throughput analyses in the genomic era, may ultimately facilitate a more precise evaluation of global landscape of genetic and epigenetic alterations in VSCCs. Additional studies are ultimately required to better understand the global landscape of genetic and epigenetic alterations in VSCC, and to identify and test potential targets for clinical application. Future management of VSCC patients may need to integrate molecule/pathway-based targeted therapies, immunotherapy, as well as traditional strategies, as the basis for a personalized/precision medicine approach to management [152–154].
Grant support:
This study is supported by a Clinician Scientist Award at The Johns Hopkins University School of Medicine (D.X.) and partially supported by the Pilot Project Award by the Cervical Cancer SPORE program at Johns Hopkins (D.X.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
References
- 1.Siegel RL, Miller KD, and Jemal A, Cancer statistics, 2020. CA Cancer J Clin, 2020. 70(1): p. 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Kurman RJ, et al. , WHO Classification of Tumors of Female Reproductive Organs. Lyon: IARC; 2014. [Google Scholar]
- 3.Xing D, et al. , Recurrent genetic alterations and biomarker expression in primary and metastatic squamous cell carcinomas of the vulva. Hum Pathol, 2019. 92: p. 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pouwer AW, et al. , Clonal Relationship Between Lichen Sclerosus, Differentiated Vulvar Intra-epithelial Neoplasia and Non HPV-related Vulvar Squamous Cell Carcinoma. Cancer Genomics Proteomics, 2020. 17(2): p. 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zieba S, et al. , Somatic Mutation Profiling in Premalignant Lesions of Vulvar Squamous Cell Carcinoma. Int J Mol Sci, 2020. 21(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoang LN, et al. , Squamous precursor lesions of the vulva: current classification and diagnostic challenges. Pathology, 2016. 48(4): p. 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh N and Gilks CB, Vulval squamous cell carcinoma and its precursors. Histopathology, 2020. 76(1): p. 128–138. [DOI] [PubMed] [Google Scholar]
- 8.Eva LJ, et al. , Trends in HPV-dependent and HPV-independent vulvar cancers: The changing face of vulvar squamous cell carcinoma. Gynecol Oncol, 2020. 157(2): p. 450–455. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Zhang Y, and Zhang Z, Prevalence of human papillomavirus and its prognostic value in vulvar cancer: A systematic review and meta-analysis. PLoS One, 2018. 13(9): p. e0204162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prieske K, et al. , Genomic characterization of vulvar squamous cell carcinoma. Gynecol Oncol, 2020. [DOI] [PubMed] [Google Scholar]
- 11.Hinten F, et al. , Vulvar cancer: Two pathways with different localization and prognosis. Gynecol Oncol, 2018. 149(2): p. 310–317. [DOI] [PubMed] [Google Scholar]
- 12.Regauer S, Kashofer K, and Reich O, Time series analysis of TP53 gene mutations in recurrent HPV-negative vulvar squamous cell carcinoma. Mod Pathol, 2019. 32(3): p. 415–422. [DOI] [PubMed] [Google Scholar]
- 13.Sand FL, et al. , Response: Letter regarding “The prognostic value of p16 and p53 expression for survival after vulvar cancer: A systematic review and meta-analysis”. Gynecol Oncol Rep, 2019. 30: p. 100494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woelber L, et al. , Prognostic role of lymph node metastases in vulvar cancer and implications for adjuvant treatment. Int J Gynecol Cancer, 2012. 22(3): p. 503–8. [DOI] [PubMed] [Google Scholar]
- 15.Polterauer S, et al. , Lymph node ratio in inguinal lymphadenectomy for squamous cell vulvar cancer: Results from the AGO-CaRE-1 study. Gynecol Oncol, 2019. [DOI] [PubMed] [Google Scholar]
- 16.Burger MP, et al. , The importance of the groin node status for the survival of T1 and T2 vulval carcinoma patients. Gynecol Oncol, 1995. 57(3): p. 327–34. [DOI] [PubMed] [Google Scholar]
- 17.Brown CJ, et al. , Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer, 2009. 9(12): p. 862–73. [DOI] [PubMed] [Google Scholar]
- 18.Olivier M, Hollstein M, and Hainaut P, TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol, 2010. 2(1): p. a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amelio I and Melino G, Context is everything: extrinsic signalling and gain-of-function p53 mutants. Cell Death Discov, 2020. 6: p. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kashofer K and Regauer S, Analysis of full coding sequence of the TP53 gene in invasive vulvar cancers: Implications for therapy. Gynecol Oncol, 2017. 146(2): p. 314–318. [DOI] [PubMed] [Google Scholar]
- 21.Zhang C, et al. , Gain-of-function mutant p53 in cancer progression and therapy. J Mol Cell Biol, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oren M and Rotter V, Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol, 2010. 2(2): p. a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J, et al. , Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis, 2014. 5: p. e1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choschzick M, et al. , Role of TP53 mutations in vulvar carcinomas. Int J Gynecol Pathol, 2011. 30(5): p. 497–504. [DOI] [PubMed] [Google Scholar]
- 25.Rolfe KJ, et al. , TP53 mutations in vulval lichen sclerosus adjacent to squamous cell carcinoma of the vulva. Br J Cancer, 2003. 89(12): p. 2249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tapp RA, et al. , Single base instability is promoted in vulvar lichen sclerosus. J Invest Dermatol, 2007. 127(11): p. 2563–76. [DOI] [PubMed] [Google Scholar]
- 27.Weberpals JI, et al. , Vulvar Squamous Cell Carcinoma (VSCC) as Two Diseases: HPV Status Identifies Distinct Mutational Profiles Including Oncogenic Fibroblast Growth Factor Receptor 3. Clin Cancer Res, 2017. 23(15): p. 4501–4510. [DOI] [PubMed] [Google Scholar]
- 28.Nooij LS, et al. , Genomic Characterization of Vulvar (Pre)cancers Identifies Distinct Molecular Subtypes with Prognostic Significance. Clin Cancer Res, 2017. 23(22): p. 6781–6789. [DOI] [PubMed] [Google Scholar]
- 29.Zieba S, et al. , Somatic mutation profiling of vulvar cancer: Exploring therapeutic targets. Gynecol Oncol, 2018. 150(3): p. 552–561. [DOI] [PubMed] [Google Scholar]
- 30.Duffy MJ, et al. , Targeting p53 for the treatment of cancer. Semin Cancer Biol, 2020. [DOI] [PubMed] [Google Scholar]
- 31.Duffy MJ and Crown J, Drugging “undruggable” genes for cancer treatment: Are we making progress? Int J Cancer, 2020. [DOI] [PubMed] [Google Scholar]
- 32.Foulkes WD, et al. , The CDKN2A (p16) gene and human cancer. Mol Med, 1997. 3(1): p. 5–20. [PMC free article] [PubMed] [Google Scholar]
- 33.Ohtani N, et al. , The p16INK4a-RB pathway: molecular link between cellular senescence and tumor suppression. J Med Invest, 2004. 51(3–4): p. 146–53. [DOI] [PubMed] [Google Scholar]
- 34.Bates S, et al. , p14ARF links the tumour suppressors RB and p53. Nature, 1998. 395(6698): p. 124–5. [DOI] [PubMed] [Google Scholar]
- 35.Williams EA, W. A, Sharaf R, Montesion M, Shah N, Sokol ES, Pavlick DC, Danziger N, Killian JK, Lin DI, Miller VA, Ross JS, Tse JY. Elvin JA. , Vulvar squamous cell carcinoma: comprehensive genomic profiling of HPV(+) versus HPV(−) forms reveals a different set of potentially actionable biomarkers . SGO Annual meeting on women’s cancer. Toronto, Canada: March 2020., 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han MR, et al. , Mutational signatures and chromosome alteration profiles of squamous cell carcinomas of the vulva. Exp Mol Med, 2018. 50(2): p. e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clancy AA, Spaans JN, and Weberpals JI, The forgotten woman’s cancer: vulvar squamous cell carcinoma (VSCC) and a targeted approach to therapy. Ann Oncol, 2016. 27(9): p. 1696–705. [DOI] [PubMed] [Google Scholar]
- 38.Del Pino M, Rodriguez-Carunchio L, and Ordi J, Pathways of vulvar intraepithelial neoplasia and squamous cell carcinoma. Histopathology, 2013. 62(1): p. 161–75. [DOI] [PubMed] [Google Scholar]
- 39.Trietsch MD, et al. , CDKN2A(p16) and HRAS are frequently mutated in vulvar squamous cell carcinoma. Gynecol Oncol, 2014. 135(1): p. 149–55. [DOI] [PubMed] [Google Scholar]
- 40.Li Z, et al. , Cdkn2a suppresses metastasis in squamous cell carcinomas induced by the gain-of-function mutant p53(R172H). J Pathol, 2016. 240(2): p. 224–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Proctor L, et al. , Association of human papilloma virus status and response to radiotherapy in vulvar squamous cell carcinoma. Int J Gynecol Cancer, 2020. 30(1): p. 100–106. [DOI] [PubMed] [Google Scholar]
- 42.Horne ZD, et al. , Human papillomavirus infection mediates response and outcome of vulvar squamous cell carcinomas treated with radiation therapy. Gynecol Oncol, 2018. 151(1): p. 96–101. [DOI] [PubMed] [Google Scholar]
- 43.Lee LJ, et al. , Prognostic importance of human papillomavirus (HPV) and p16 positivity in squamous cell carcinoma of the vulva treated with radiotherapy. Gynecol Oncol, 2016. 142(2): p. 293–8. [DOI] [PubMed] [Google Scholar]
- 44.Dibble CC and Cantley LC, Regulation of mTORC1 by PI3K signaling. Trends Cell Biol, 2015. 25(9): p. 545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sabatini DM, Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc Natl Acad Sci U S A, 2017. 114(45): p. 11818–11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aoki M and Fujishita T, Oncogenic Roles of the PI3K/AKT/mTOR Axis. Curr Top Microbiol Immunol, 2017. 407: p. 153–189. [DOI] [PubMed] [Google Scholar]
- 47.Madsen RR, Vanhaesebroeck B, and Semple RK, Cancer-Associated PIK3CA Mutations in Overgrowth Disorders. Trends Mol Med, 2018. 24(10): p. 856–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sansal I and Sellers WR, The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol, 2004. 22(14): p. 2954–63. [DOI] [PubMed] [Google Scholar]
- 49.Li J, et al. , PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science, 1997. 275(5308): p. 1943–7. [DOI] [PubMed] [Google Scholar]
- 50.Yin Y and Shen WH, PTEN: a new guardian of the genome. Oncogene, 2008. 27(41): p. 5443–53. [DOI] [PubMed] [Google Scholar]
- 51.Holway AH, et al. , Somatic mutation of PTEN in vulvar cancer. Clin Cancer Res, 2000. 6(8): p. 3228–35. [PubMed] [Google Scholar]
- 52.Tessier-Cloutier B, et al. , Molecular characterization of invasive and in situ squamous neoplasia of the vulva and implications for morphologic diagnosis and outcome. Mod Pathol, 2020. [DOI] [PubMed] [Google Scholar]
- 53.Koncar RF, et al. , Comparative molecular profiling of HPV-induced squamous cell carcinomas. Cancer Med, 2017. 6(7): p. 1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palisoul ML, et al. , Identification of molecular targets in vulvar cancers. Gynecol Oncol, 2017. 146(2): p. 305–313. [DOI] [PubMed] [Google Scholar]
- 55.Henken FE, et al. , PIK3CA-mediated PI3-kinase signalling is essential for HPV-induced transformation in vitro. Molecular cancer, 2011. 10: p. 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ojesina AI, et al. , Landscape of genomic alterations in cervical carcinomas. Nature, 2014. 506(7488): p. 371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wright AA, et al. , Oncogenic mutations in cervical cancer: genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer, 2013. 119(21): p. 3776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, et al. , Subtypes of HPV-Positive Head and Neck Cancers Are Associated with HPV Characteristics, Copy Number Alterations, PIK3CA Mutation, and Pathway Signatures. Clinical cancer research : an official journal of the American Association for Cancer Research, 2016. 22(18): p. 4735–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xing D, et al. , Next-generation Sequencing Reveals Recurrent Somatic Mutations in Small Cell Neuroendocrine Carcinoma of the Uterine Cervix. Am J Surg Pathol, 2018. 42(6): p. 750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cantley LC and Neel BG, New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A, 1999. 96(8): p. 4240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Akbari A, et al. , Differentiated exophytic vulvar intraepithelial lesion: Clinicopathologic and molecular analysis documenting its relationship with verrucous carcinoma of the vulva. Mod Pathol, 2020. [DOI] [PubMed] [Google Scholar]
- 62.Watanabe M, et al. , Intrafractional gastric motion and interfractional stomach deformity during radiation therapy. Radiother Oncol, 2008. 87(3): p. 425–31. [DOI] [PubMed] [Google Scholar]
- 63.Nicholson KM and Anderson NG, The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal, 2002. 14(5): p. 381–95. [DOI] [PubMed] [Google Scholar]
- 64.Porta C, Paglino C, and Mosca A, Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol, 2014. 4: p. 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mirza-Aghazadeh-Attari M, et al. , Targeting PI3K/Akt/mTOR signaling pathway by polyphenols: Implication for cancer therapy. Life Sci, 2020. 255: p. 117481. [DOI] [PubMed] [Google Scholar]
- 66.Markham A, Alpelisib: First Global Approval. Drugs, 2019. 79(11): p. 1249–1253. [DOI] [PubMed] [Google Scholar]
- 67.Dunn LA, et al. , A Phase 1b Study of Cetuximab and BYL719 (Alpelisib) Concurrent with Intensity Modulated Radiation Therapy in Stage III-IVB Head and Neck Squamous Cell Carcinoma. Int J Radiat Oncol Biol Phys, 2020. 106(3): p. 564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCubrey JA, et al. , Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta, 2007. 1773(8): p. 1263–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nan X, et al. , Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc Natl Acad Sci U S A, 2015. 112(26): p. 7996–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Friday BB and Adjei AA, Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res, 2008. 14(2): p. 342–6. [DOI] [PubMed] [Google Scholar]
- 71.Suire S, Hawkins P, and Stephens L, Activation of phosphoinositide 3-kinase gamma by Ras. Curr Biol, 2002. 12(13): p. 1068–75. [DOI] [PubMed] [Google Scholar]
- 72.Prior IA, Hood FE, and Hartley JL, The Frequency of Ras Mutations in Cancer. Cancer Res, 2020. 80(14): p. 2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luwor RB, et al. , Constitutively active Harvey Ras confers resistance to epidermal growth factor receptor-targeted therapy with cetuximab and gefitinib. Cancer Lett, 2011. 306(1): p. 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Santarpia L, Lippman SM, and El-Naggar AK, Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets, 2012. 16(1): p. 103–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xing D and Orsulic S, Modeling resistance to pathway-targeted therapy in ovarian cancer. Cell Cycle, 2005. 4(8): p. 1004–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xing D and Orsulic S, A genetically defined mouse ovarian carcinoma model for the molecular characterization of pathway-targeted therapy and tumor resistance. Proc Natl Acad Sci U S A, 2005. 102(19): p. 6936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Du Z and Lovly CM, Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer, 2018. 17(1): p. 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ullrich A and Schlessinger J, Signal transduction by receptors with tyrosine kinase activity. Cell, 1990. 61(2): p. 203–12. [DOI] [PubMed] [Google Scholar]
- 79.Montor WR, Salas A, and Melo FHM, Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: the current arsenal of inhibitors. Mol Cancer, 2018. 17(1): p. 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gschwind A, Fischer OM, and Ullrich A, The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer, 2004. 4(5): p. 361–70. [DOI] [PubMed] [Google Scholar]
- 81.Iyer G and Milowsky MI, Fibroblast growth factor receptor-3 in urothelial tumorigenesis. Urol Oncol, 2013. 31(3): p. 303–11. [DOI] [PubMed] [Google Scholar]
- 82.Choschzick M, et al. , HER2 amplification in squamous cell carcinomas of the vulva. Histopathology, 2013. 62(6): p. 965–7. [DOI] [PubMed] [Google Scholar]
- 83.Growdon WB, et al. , Decreased survival in EGFR gene amplified vulvar carcinoma. Gynecol Oncol, 2008. 111(2): p. 289–97. [DOI] [PubMed] [Google Scholar]
- 84.Horowitz NS, et al. , Phase II trial of erlotinib in women with squamous cell carcinoma of the vulva. Gynecol Oncol, 2012. 127(1): p. 141–6. [DOI] [PubMed] [Google Scholar]
- 85.Dong F, et al. , Squamous Cell Carcinoma of the Vulva: A Subclassification of 97 Cases by Clinicopathologic, Immunohistochemical, and Molecular Features (p16, p53, and EGFR). Am J Surg Pathol, 2015. 39(8): p. 1045–53. [DOI] [PubMed] [Google Scholar]
- 86.Woelber L, et al. , EGFR gene copy number increase in vulvar carcinomas is linked with poor clinical outcome. J Clin Pathol, 2012. 65(2): p. 133–9. [DOI] [PubMed] [Google Scholar]
- 87.Oonk MH, et al. , EGFR expression is associated with groin node metastases in vulvar cancer, but does not improve their prediction. Gynecol Oncol, 2007. 104(1): p. 109–13. [DOI] [PubMed] [Google Scholar]
- 88.de Melo Maia B, et al. , EGFR expression in vulvar cancer: clinical implications and tumor heterogeneity. Hum Pathol, 2014. 45(5): p. 917–25. [DOI] [PubMed] [Google Scholar]
- 89.Johnson GA, et al. , Epidermal growth factor receptor in vulvar malignancies and its relationship to metastasis and patient survival. Gynecol Oncol, 1997. 65(3): p. 425–9. [DOI] [PubMed] [Google Scholar]
- 90.Lobry C, Oh P, and Aifantis I, Oncogenic and tumor suppressor functions of Notch in cancer: it’s NOTCH what you think. J Exp Med, 2011. 208(10): p. 1931–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hales EC, Taub JW, and Matherly LH, New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of gamma-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal, 2014. 26(1): p. 149–61. [DOI] [PubMed] [Google Scholar]
- 92.Akhoondi S, et al. , FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res, 2007. 67(19): p. 9006–12. [DOI] [PubMed] [Google Scholar]
- 93.Jensen DE and Rauscher FJ 3rd, BAP1, a candidate tumor suppressor protein that interacts with BRCA1. Ann N Y Acad Sci, 1999. 886: p. 191–4. [DOI] [PubMed] [Google Scholar]
- 94.Liu X, et al. , BAP1 Is a Novel Target in HPV-Negative Head and Neck Cancer. Clin Cancer Res, 2018. 24(3): p. 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang L, et al. , Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am J Hum Genet, 2015. 96(4): p. 597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mori T, et al. , Somatic alteration and depleted nuclear expression of BAP1 in human esophageal squamous cell carcinoma. Cancer Sci, 2015. 106(9): p. 1118–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Farmer H, et al. , Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 2005. 434(7035): p. 917–21. [DOI] [PubMed] [Google Scholar]
- 98.Pujade-Lauraine E, et al. , Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. The Lancet. Oncology, 2017. 18(9): p. 1274–1284. [DOI] [PubMed] [Google Scholar]
- 99.Matulonis UA, et al. , Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Annals of oncology : official journal of the European Society for Medical Oncology, 2016. 27(6): p. 1013–9. [DOI] [PubMed] [Google Scholar]
- 100.Ledermann JA, et al. , Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. The Lancet. Oncology, 2016. 17(11): p. 1579–1589. [DOI] [PubMed] [Google Scholar]
- 101.Dougherty BA, et al. , Biological and clinical evidence for somatic mutations in BRCA1 and BRCA2 as predictive markers for olaparib response in high-grade serous ovarian cancers in the maintenance setting. Oncotarget, 2017. 8(27): p. 43653–43661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zack TI, et al. , Pan-cancer patterns of somatic copy number alteration. Nat Genet, 2013. 45(10): p. 1134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Beroukhim R, et al. , The landscape of somatic copy-number alteration across human cancers. Nature, 2010. 463(7283): p. 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Allen DG, et al. , Genetic aberrations detected by comparative genomic hybridisation in vulvar cancers. Br J Cancer, 2002. 86(6): p. 924–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Choschzick M, et al. , Role of cyclin D1 amplification and expression in vulvar carcinomas. Hum Pathol, 2012. 43(9): p. 1386–93. [DOI] [PubMed] [Google Scholar]
- 106.Gut A, Moch H, and Choschzick M, SOX2 Gene Amplification and Overexpression is Linked to HPV-positive Vulvar Carcinomas. Int J Gynecol Pathol, 2018. 37(1): p. 68–73. [DOI] [PubMed] [Google Scholar]
- 107.Aulmann S, et al. , Gains of chromosome region 3q26 in intraepithelial neoplasia and invasive squamous cell carcinoma of the vulva are frequent and independent of HPV status. J Clin Pathol, 2008. 61(9): p. 1034–7. [DOI] [PubMed] [Google Scholar]
- 108.Jee KJ, et al. , Loss in 3p and 4p and gain of 3q are concomitant aberrations in squamous cell carcinoma of the vulva. Mod Pathol, 2001. 14(5): p. 377–81. [DOI] [PubMed] [Google Scholar]
- 109.Purdie KJ, et al. , High-resolution genomic profiling of human papillomavirus-associated vulval neoplasia. Br J Cancer, 2010. 102(6): p. 1044–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Swarts DRA, et al. , Molecular heterogeneity in human papillomavirus-dependent and - independent vulvar carcinogenesis. Cancer Med, 2018. 7(9): p. 4542–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bashyam MD, et al. , Array-based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia, 2005. 7(6): p. 556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ma O, et al. , MMP13, Birc2 (cIAP1), and Birc3 (cIAP2), amplified on chromosome 9, collaborate with p53 deficiency in mouse osteosarcoma progression. Cancer Res, 2009. 69(6): p. 2559–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cheng L, et al. , Rb inactivation accelerates neoplastic growth and substitutes for recurrent amplification of cIAP1, cIAP2 and Yap1 in sporadic mammary carcinoma associated with p53 deficiency. Oncogene, 2010. 29(42): p. 5700–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Choschzick M, et al. , BIRC2 amplification in squamous cell carcinomas of the uterine cervix. Virchows Arch, 2012. 461(2): p. 123–8. [DOI] [PubMed] [Google Scholar]
- 115.Sharma S, Kelly TK, and Jones PA, Epigenetics in cancer. Carcinogenesis, 2010. 31(1): p. 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Esteller M and Herman JG, Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J Pathol, 2002. 196(1): p. 1–7. [DOI] [PubMed] [Google Scholar]
- 117.Ehrlich M, DNA methylation in cancer: too much, but also too little. Oncogene, 2002. 21(35): p. 5400–13. [DOI] [PubMed] [Google Scholar]
- 118.Guerrero D, et al. , Differential hypermethylation of genes in vulvar cancer and lichen sclerosus coexisting or not with vulvar cancer. Int J Cancer, 2011. 128(12): p. 2853–64. [DOI] [PubMed] [Google Scholar]
- 119.Rotondo JC, et al. , Association of Retinoic Acid Receptor beta Gene With Onset and Progression of Lichen Sclerosus-Associated Vulvar Squamous Cell Carcinoma. JAMA Dermatol, 2018. 154(7): p. 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rotondo JC, et al. , Hypermethylation-Induced Inactivation of the IRF6 Gene as a Possible Early Event in Progression of Vulvar Squamous Cell Carcinoma Associated With Lichen Sclerosus. JAMA Dermatol, 2016. 152(8): p. 928–33. [DOI] [PubMed] [Google Scholar]
- 121.Aide S, et al. , Promoter hypermethylation of death-associated protein kinase and p16 genes in vulvar lichen sclerosus. J Low Genit Tract Dis, 2012. 16(2): p. 133–9. [DOI] [PubMed] [Google Scholar]
- 122.Lerma E, et al. , Alterations of the p16/Rb/cyclin-D1 pathway in vulvar carcinoma, vulvar intraepithelial neoplasia, and lichen sclerosus. Hum Pathol, 2002. 33(11): p. 1120–5. [DOI] [PubMed] [Google Scholar]
- 123.Trietsch MD, et al. , Genetic and epigenetic changes in vulvar squamous cell carcinoma and its precursor lesions: a review of the current literature. Gynecol Oncol, 2015. 136(1): p. 143–57. [DOI] [PubMed] [Google Scholar]
- 124.Li B, et al. , Aberrant promoter methylation of SH3GL2 gene in vulvar squamous cell carcinoma correlates with clinicopathological characteristics and HPV infection status. Int J Clin Exp Pathol, 2015. 8(11): p. 15442–7. [PMC free article] [PubMed] [Google Scholar]
- 125.Oonk MH, et al. , Identification of inguinofemoral lymph node metastases by methylation markers in vulvar cancer. Gynecol Oncol, 2012. 125(2): p. 352–7. [DOI] [PubMed] [Google Scholar]
- 126.Gurtan AM and Sharp PA, The role of miRNAs in regulating gene expression networks. J Mol Biol, 2013. 425(19): p. 3582–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yao RW, Wang Y, and Chen LL, Cellular functions of long noncoding RNAs. Nat Cell Biol, 2019. 21(5): p. 542–551. [DOI] [PubMed] [Google Scholar]
- 128.Gailhouste L and Ochiya T, Cancer-related microRNAs and their role as tumor suppressors and oncogenes in hepatocellular carcinoma. Histol Histopathol, 2013. 28(4): p. 437–51. [DOI] [PubMed] [Google Scholar]
- 129.Nohata N, et al. , MicroRNAs function as tumor suppressors or oncogenes: aberrant expression of microRNAs in head and neck squamous cell carcinoma. Auris Nasus Larynx, 2013. 40(2): p. 143–9. [DOI] [PubMed] [Google Scholar]
- 130.Voorhoeve PM, MicroRNAs: Oncogenes, tumor suppressors or master regulators of cancer heterogeneity? Biochim Biophys Acta, 2010. 1805(1): p. 72–86. [DOI] [PubMed] [Google Scholar]
- 131.Peng Y and Croce CM, The role of MicroRNAs in human cancer. Signal Transduct Target Ther, 2016. 1: p. 15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.de Melo Maia B, et al. , microRNA portraits in human vulvar carcinoma. Cancer Prev Res (Phila), 2013. 6(11): p. 1231–41. [DOI] [PubMed] [Google Scholar]
- 133.de Melo Maia B, et al. , MiR-223–5p works as an oncomiR in vulvar carcinoma by TP63 suppression. Oncotarget, 2016. 7(31): p. 49217–49231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zalewski K, et al. , Normalizers for microRNA quantification in plasma of patients with vulvar intraepithelial neoplasia lesions and vulvar carcinoma. Tumour Biol, 2017. 39(11): p. 1010428317717140. [DOI] [PubMed] [Google Scholar]
- 135.Yang S, et al. , MicroRNA47125p promotes proliferation of the vulvar squamous cell carcinoma cell line A431 by targeting PTEN through the AKT/cyclin D1 signaling pathways. Oncol Rep, 2019. 42(5): p. 1689–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang XH and Guo F, miR3147 serves as an oncomiR in vulvar squamous cell cancer via Smad4 suppression. Mol Med Rep, 2018. 17(5): p. 6397–6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yang X and Wu X, miRNA expression profile of vulvar squamous cell carcinoma and identification of the oncogenic role of miR-590–5p. Oncol Rep, 2016. 35(1): p. 398–408. [DOI] [PubMed] [Google Scholar]
- 138.Agostini A, et al. , Expressions of miR-30c and let-7a are inversely correlated with HMGA2 expression in squamous cell carcinoma of the vulva. Oncotarget, 2016. 7(51): p. 85058–85062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Terlou A, et al. , An autoimmune phenotype in vulvar lichen sclerosus and lichen planus: a Th1 response and high levels of microRNA-155. J Invest Dermatol, 2012. 132(3 Pt 1): p. 658–66. [DOI] [PubMed] [Google Scholar]
- 140.Ren L, et al. , MiR-155–5p promotes fibroblast cell proliferation and inhibits FOXO signaling pathway in vulvar lichen sclerosis by targeting FOXO3 and CDKN1B. Gene, 2018. 653: p. 43–50. [DOI] [PubMed] [Google Scholar]
- 141.Gao Q, et al. , Exosomal lncRNA UCA1 from cancer-associated fibroblasts enhances chemoresistance in vulvar squamous cell carcinoma cells. J Obstet Gynaecol Res, 2020. [DOI] [PubMed] [Google Scholar]
- 142.Ni S, Zhao X, and Ouyang L, Long non-coding RNA expression profile in vulvar squamous cell carcinoma and its clinical significance. Oncol Rep, 2016. 36(5): p. 2571–2578. [DOI] [PubMed] [Google Scholar]
- 143.Howitt BE, et al. , Genetic Basis for PD-L1 Expression in Squamous Cell Carcinomas of the Cervix and Vulva. JAMA Oncol, 2016. 2(4): p. 518–22. [DOI] [PubMed] [Google Scholar]
- 144.Hecking T, et al. , Tumoral PD-L1 expression defines a subgroup of poor-prognosis vulvar carcinomas with non-viral etiology. Oncotarget, 2017. 8(54): p. 92890–92903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chinn Z, Stoler MH, and Mills AM, PD-L1 and IDO expression in cervical and vulvar invasive and intraepithelial squamous neoplasias: implications for combination immunotherapy. Histopathology, 2019. 74(2): p. 256–268. [DOI] [PubMed] [Google Scholar]
- 146.Sznurkowski JJ, et al. , PD-L1 expression on immune cells is a favorable prognostic factor for vulvar squamous cell carcinoma patients. Oncotarget, 2017. 8(52): p. 89903–89912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Choschzick M, Gut A, and Fink D, PD-L1 receptor expression in vulvar carcinomas is HPV-independent. Virchows Arch, 2018. 473(4): p. 513–516. [DOI] [PubMed] [Google Scholar]
- 148.Cocks M, et al. , Immune checkpoint status and tumor microenvironment in vulvar squamous cell carcinoma. Virchows Arch, 2020. 477(1): p. 93–102. [DOI] [PubMed] [Google Scholar]
- 149.Thangarajah F, et al. , Clinical impact of PD-L1 and PD-1 expression in squamous cell cancer of the vulva. J Cancer Res Clin Oncol, 2019. 145(6): p. 1651–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Curley J, et al. , Looking past PD-L1: expression of immune checkpoint TIM-3 and its ligand galectin-9 in cervical and vulvar squamous neoplasia. Mod Pathol, 2020. 33(6): p. 1182–1192. [DOI] [PubMed] [Google Scholar]
- 151.Dibbern ME, et al. , Loss of MHC Class I Expression in HPV-associated Cervical and Vulvar Neoplasia: A Potential Mechanism of Resistance to Checkpoint Inhibition. Am J Surg Pathol, 2020. 44(9): p. 1184–1191. [DOI] [PubMed] [Google Scholar]
- 152.Woelber L, Jaeger A, and Prieske K, New treatment standards for vulvar cancer 2020. Curr Opin Obstet Gynecol, 2020. 32(1): p. 9–14. [DOI] [PubMed] [Google Scholar]
- 153.Zieba S, et al. , Genes, pathways and vulvar carcinoma - New insights from next-generation sequencing studies. Gynecol Oncol, 2020. 158(2): p. 498–506. [DOI] [PubMed] [Google Scholar]
- 154.Giulia M, et al. , Molecular pathways in vulvar squamous cell carcinoma: implications for target therapeutic strategies. J Cancer Res Clin Oncol, 2020. 146(7): p. 1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]