

Abstract
Approximately 7% of men worldwide suffer from infertility, with sperm abnormalities being the most common defect. Though genetic causes are thought to underlie a substantial fraction of idiopathic cases, the actual molecular bases are usually undetermined. Because the consequences of most genetic variants in populations are unknown, this complicates genetic diagnosis even after genome sequencing of patients. Some patients with ciliopathies, including primary ciliary dyskinesia and Bardet–Biedl syndrome, also suffer from infertility because cilia and sperm flagella share several characteristics. Here, we identified two deleterious alleles of RABL2A, a gene essential for normal function of cilia and flagella. Our in silico predictions and in vitro assays suggest that both alleles destabilize the protein. We constructed and analyzed mice homozygous for these two single-nucleotide polymorphisms, Rabl2L119F (rs80006029) and Rabl2V158F (rs200121688), and found that they exhibit ciliopathy-associated disorders including male infertility, early growth retardation, excessive weight gain in adulthood, heterotaxia, pre-axial polydactyly, neural tube defects and hydrocephalus. Our study provides a paradigm for triaging candidate infertility variants in the population for in vivo functional validation, using computational, in vitro and in vivo approaches.
Introduction
Infertility affects ~7% of the male population worldwide (1) and often manifests qualitative and/or quantitative defects in sperm parameters. Sperm tail (flagellum) defects can cause teratospermia (abnormal morphology), asthenospermia (reduced sperm motility), azoospermia and oligospermia (no and low sperm count, respectively), thus contributing to main categories of male infertility. Asthenozoospermia is often observed in patients suffering from primary ciliary dyskinesia (PCD), a group of autosomal recessive disorders caused by motile cilia dysfunction. Cilia are categorized into two types, motile cilia/flagella and non-motile cilia (also called primary cilia). The former is responsible for propelling cells or generating fluid flow over ciliated cells, whereas the latter functions as cellular antennae by sensing extracellular stimuli and transducing developmental signals. Defects in cilia result in a variety of congenital disorders, such as PCD, Bardet–Biedl syndrome (BBS), Joubert syndrome (JBTS) and Meckel–Grüber syndrome (MKS), which are collectively called the ciliopathies (2). PCD results in recurrent respiratory infections, male infertility, randomization of left–right (L/R) asymmetry and hydrocephalus. BBS is characterized by features including cognitive impairment, obesity, retinal degeneration, anosmia, neural tube defects (NTDs), cystic kidneys and polydactyly. MKS and JBTS are characterized by severe central nervous system (CNS) disorders.
Rab-like protein 2 (RABL2) is a small GTPase member of the Ras superfamily and is essential for proper function of flagella and cilia. In sperm tails, RABL2 localizes to the mid-piece region and can deliver a specific set of effector proteins, including key members of the glycolytic pathway, into the growing sperm tails by binding intraflagellar transport-B (IFT-B; a large of proteins) and guanosine triphosphate (GTP) (3). In cilia, RABL2 can be recruited by centrosomal protein 19 (CEP19) to the mother centriole, bind GTP and the IFT-B complex, triggering their entry into the cilium (4). The CEP19- RABL2-IFT pathway was proposed as a molecular mechanism for directing ciliary traffic, needed for cilium formation. A mouse model called ‘Mot’ was isolated in an ENU (N-ethyl-N-nitrosourea) mutagenesis screen, which carries a mutation (p.Asp73Gly) in Rabl2 (3). Homozygous mutant Rabl2Mot mice exhibited qualitatively normal spermatogenesis but were sterile due to the reduced sperm motility and oligospermia (3). Besides infertility, Mot mice also exhibited adult onset obesity and fatty livers, as well as impaired glucose and lipid metabolism (5). Knockout Rabl2 mice also display retinal degeneration and pre-axial polydactyly (4), which, together with the obesity and sperm defects, are reminiscent of symptoms observed in patients suffering from PCD and BBS. In humans, there are two highly similar paralogs, RABL2A and RABL2B. The extra copy arose after the split between humans and orangutans (6). A deletion allele (rs57719031) of RABL2A was identified as a potential risk factor for Australian infertility patients (7), though it was not reported whether these patients presented PCD- or BBS-associated symptoms.
It is important to characterize genetic causes of disease to aid in clinical diagnosis and possible treatment. Single-nucleotide polymorphisms (SNPs) are the most prevalent type of genetic variation in both coding and non-coding regions of the genome. Non-synonymous SNPs (nsSNPs; those which change an amino acid) can cause disease by affecting a protein’s structure or function, its interaction with other proteins, or ability to be modified at key residues (8). However, the effects of most SNPs are unknown and are referred to as variants of unknown significance (VUS) (9). Identifying those that contribute to disease is a major challenge in human genetics. To identify VUS that cause infertility in human populations, we have implemented an association- and linkage-free approach (10). This approach involves selecting nsSNPs in genes known to be required for gametogenesis and fertility, predicting their functional consequences, modeling the orthologous amino acid changes in mice using CRISPR/Cas9 genome editing and then assessing phenotypic consequences (10). To this end, several nsSNPs affecting infertility were identified (10–13).
Here, we present evidence that two nsSNP alleles of RABL2A, rs80006029 and rs200121688, are functionally deleterious. These variants disrupt protein stability and, when modeled in mice, cause PCD and BBS-associated disorders including male infertility, excessive weight, growth retardation, heterotaxia, pre-axial polydactyly, NTDs and hydrocephalus.
Results
Prioritizing potentially deleterious missense variants in RABL2A
We used Genome Aggregation Database (gnomAD), a database containing whole-exome sequencing data for >140 000 individuals (14), to identify potentially deleterious missense variants in RABL2A. We restricted the candidates to those with a minor allele frequency (MAF) of <2%, because infertility-causing variants are unlikely to persist at higher frequencies in populations. We also eliminated from consideration variants with a MAF of <0.02%, to focus efforts on alleles that are not exceedingly rare. Six missense variants fulfilled our criteria (Table 1). We further prioritized these six RABL2A mutations using SIFT (15) and PolyPhen-2 (16) and found that p.L119F, p.A94T and p.V158F scored as deleterious by both algorithms (Table 1). Because mutations predicted as being deleterious by variant classifiers do not always have phenotypic consequences (10,11,13), we further examined the biochemical impact of the variants. Structural analyses predicted that all three variants destabilize protein folding (Fig. 1A and Supplementary Material, Fig. S1). We therefore tested whether these amino acid changes impacted protein stability through western blot analysis of GFP-tagged versions of these proteins overexpressed in HEK293T cells. The levels of RABL2A protein containing the p.L119F and p.V158F variants were far lower than wild type (WT). The p.A94T variant did not cause a decrease in protein (Fig. 1B and C). We therefore selected p.L119F and p.V158F for in vivo validation in mice.
Table 1.
Selection of deleterious nsSNPs
| Variant | SNP ID | MAF | SIFT | PolyPhen-2 |
|---|---|---|---|---|
| p. Leu119Phe | rs80006029 | 0.80% | Deleterious | Probably damaging |
| p. Arg82Gln | rs145167719 | 0.42% | Tolerated | Possibly damaging |
| p. Glu217Lys | rs142229156 | 0.37% | Tolerated LC | Benign |
| p. Ala94Thr | rs145909437 | 0.13% | Deleterious | Probably damaging |
| p. Arg120Gln | rs564553154 | 0.035% | Tolerated | Possibly damaging |
| p. Val158Phe | rs200121688 | 0.023% | Deleterious | Probably damaging |
Bold text, predicted by both algorithms. LC, low confidence.
Figure 1.
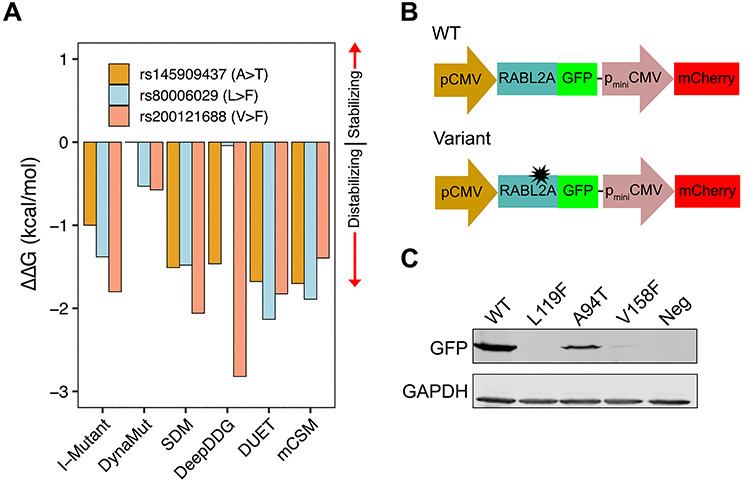
Experimental testing of nsSNPs. (A) Prediction of ΔΔG in p.Ala94Thr, p.Leu119Phe and p.Val158Phe variants by six protein stability prediction algorithms. For I-Mutant, ΔΔG (1 kcal/mol = 4184 J) < −0.5 indicates large decrease of stability, ΔΔG > 0.5 indicates large increase of stability and −0.5 ≤ ΔΔG ≤ 0.5 represents neutral stability. For DynaMut, SDM, DeepDDG, DUET and mCSM, positive ΔΔG indicates stabilization by the specified mutation and negative ΔΔG indicates destabilization. (B) Schematic of the pDEST-DUAL vector with RABL2A WT and variant ORFs used for protein stability assay. (C) Western blots for representative WT and variants detected using α-GFP. α-GAPDH was used as a loading control. Cells transfected with empty plasmid used as negative control.
Rabl2 variants cause male infertility in mice
To model the p.L119F and p.V158F alleles encoded by rs80006029 and rs200121688, respectively, equivalent point mutations were induced by CRISPR/Cas9-mediated genome editing in mouse Rabl2 (Fig. 2A). Founder mice with the correct mutation were backcrossed into strain FVB/NJ for two generations. A line with a 1 nt frameshift mutation in exon 5 was also established to serve as a presumptive null allele (Rabl2−/−) for comparison. As reported for Rabl2Mot (3) and null mice (4), Rabl2V158F/V158F (Rabl2VF/VF), Rabl2L119F/L119F (Rabl2LF/LF) and Rabl2−/− mice were all viable (Fig. 2B). Quantitative reverse transcription PCR (qRT-PCR) analysis indicated that testicular Rabl2 mRNA levels in the SNP alleles were comparable to WT but lower in the nulls (Fig. 2C).
Figure 2.
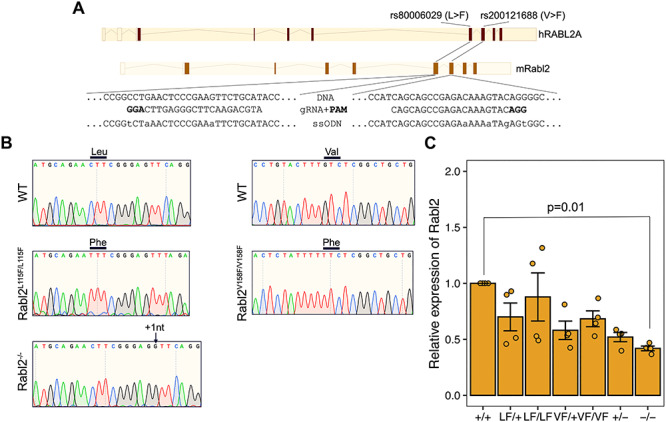
CRISPR/Cas9-mediated generation of Rabl2 mutant mice. (A) Diagram of CRISPR-Cas9 genome editing strategy to introduce the p.Leu119Phe and p.Val158Phe amino acid changes into mouse Rabl2. The human RABL2A p.Leu119Phe (rs80006029) and p.Val158Phe (rs200121688) variants are located in exons 5 and 6, respectively. (B) Representative Sanger sequencing chromatograms from WT, Rabl2L119F/L119F, Rabl2V158F/V158F and Rabl2−/− mice. The relevant codon bases are labeled above. Black arrow above the chromatogram indicates the insertion. (C) qRT-PCR analysis of Rabl2 expression in testis. Data in C are represented as the mean ± SEM and were analyzed using one-way analysis of variance (ANOVA) with Tukey’s post hoc test.
To determine if these humanized alleles compromise reproduction, we tested fertility by mating and performed a series of histological and cytological analyses. Whereas female homozygotes had normal litter sizes (Fig. 3A), no males homozygous for any of the three alleles sired offspring when paired with WT females (Fig. 3A). Gross inspection of mutant males revealed phenotypically normal external genitalia, seminal vesicles and testicular descent. The testis weights from Rabl2VF/VF, Rabl2LF/LF and Rabl2−/− mice at 12 and 24 weeks were comparable to age-matched heterozygous and WT littermates (Supplementary Material, Fig. S2A). Histological examination of testis and epididymis sections revealed apparently normal architecture and organized distribution of germ cells, indicating completion of spermatogenesis (Supplementary Material, Fig. S2B and C). As expected, the concentrations of sperm from mutant cauda epididymides were similar to those from heterozygotes and WT mice (Supplementary Material, Fig. S2D). However, whereas mutant sperm had well-shaped heads and smoothly tapering flagella (Supplementary Material, Fig. S2E), sperm tail length was significantly shorter than those isolated from heterozygous and WT animals (Fig. 3B). This phenotype resembles that of Rabl2Mot mutant mice (3).
Figure 3.
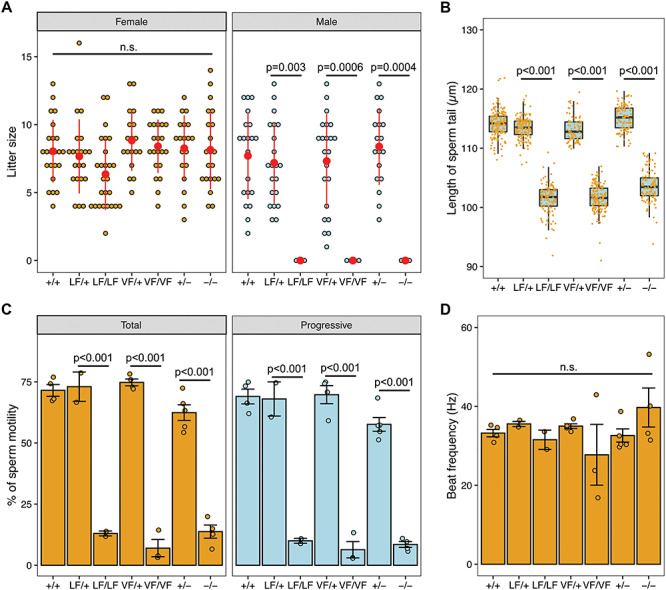
The Rabl2L119F/L119F and Rabl2V158F/V158F alteration cause male infertility. (A) Litter sizes from mating of Rabl2+/+ (+/+), Rabl2L119F/+ (LF/+), Rabl2L119F/L119F (LF/LF), Rabl2V158F/+ (VF/+), Rabl2V158F/V158F (VF/VF), Rabl2+/− (+/−) and Rabl2−/− (−/−) females and males to WT partners. (B) Boxplots showing the distribution of the length of sperm tails from +/+ (n = 174 from 4 mice), LF/+ (n = 158 from 3 mice), LF/LF (n = 120 from 3 mice), VF/+ (n = 83 from 2 mice), VF/VF (n = 160 from 3 mice), +/− (n = 120 from 3 mice) and −/− (n = 118 from 3 mice) mice. Black central line represents the median and boxes and whiskers represent the 25th and 75th and 2.5th and 97.5th percentiles. (C) Assessment of sperm total and progressive motilities of +/+, LF/+, LF/LF, VF/+, VF/VF, +/− and −/− mice. For each mouse, at least 100 sperms were analyzed. (D) Beat frequency (in hertz) of motile sperms from +/+, LF/+, LF/LF, VF/+, VF/VF, +/− and −/− mice. Data in A–D are represented as the mean ± SEM and were analyzed using one-way ANOVA with Tukey’s post hoc test. n.s. in A and D represents no significant difference.
Given that Rabl2Mot homozygotes are sterile due to defective sperm motility (3), we performed computer-assisted sperm analysis (CASA) on sperm recovered from the vas deferens of our mutants. Rabl2LF/LF, Rabl2VF/VF and Rabl2−/− sperm had significantly reduced motility and progressive movement compared to sperm from heterozygous and WT males (Fig. 3C). However, flagellar beat frequencies were not altered in the few mutant sperm that were motile (Fig. 3D), indicating that they were not defective in energy metabolism. Cumulatively, these data suggest that mutant male mice are sterile due to an inability of sperm to reach the oviducts and oocytes in the female genital tract.
Efferent duct motile cilia are necessary to propel immotile sperm from the testis to the epididymis. Loss of efferent duct motile cilia by genetic ablation of miR-34b/c and -449a/b/c causes sperm aggregation, agglutination and luminal obstruction. The resulting back pressure induces atrophy of the testis and ultimately male infertility (17). However, histology revealed no indication of efferent duct motile cilia abnormalities in Rabl2 mutants (Supplementary Material, Fig. S2F).
Rabl2 is important for normal growth, limb development and L/R patterning
Given that Rabl2 is expressed in many organs (18) and its ablation in mouse causes phenotypic hallmarks of PCD and BBS (3–5), we extended our observations to non-reproductive systems. Consistent with a previous study (5), Rabl2VF/VF, Rabl2LF/LF and Rabl2−/− mice became overweight with age (Fig. 4A and Supplementary Material, Fig. S3). However, we noticed that ~45% of mutant mice exhibited growth retardation during the lactation period (Fig. 4A and Supplementary Material, Fig. S3), despite having neonatal body weights similar to WT and heterozygous siblings. The growth retardation disappeared with age, approximately equalizing by 3 months after birth (Fig. 4A). Collectively, these data indicate that Rabl2 has distinct functions that impact mouse growth.
Figure 4.
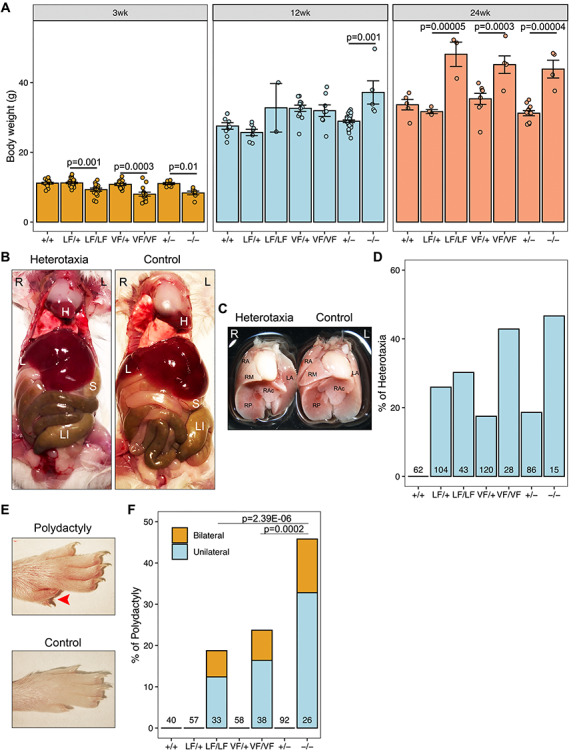
Rabl2 mutant mice have developmental defects. (A) Comparison of body weight of +/+, LF/+, LF/LF, VF/+, VF/VF, +/− and −/− males (see legend of Fig. 3 for genotype abbreviations) at 3, 12 and 24 weeks (wk) of age. (B) Examples of laterality defects in Rabl2 altered mice. Visceral dissection demonstrates the location of heart (H), liver (L), stomach (S) and large intestine (LI). (C) Relative location of heart and lung in heterotaxic and control mice. Ventral view of lung lobes was labeled. RA, right anterior; RM, right middle; RP, right posterior; RAc, right accessory and LA, left anterior lobe. (D) Quantification of the proportion of mice with heterotaxia. (E) Representative images of the hindlimb from Rabl2 mutant mouse with polydactyly. (F) Quantification of the proportion of mice with polydactyly. Data in A are represented as the mean ± SEM and were analyzed using one-way ANOVA with Tukey’s post hoc test. Data in F were analyzed using chi-square test.
Inspection of the internal viscera of Rabl2 mutant mice revealed abnormalities in L/R axis specification. About 30–50% of homozygous mutants displayed heterotaxia, a partial inversion defect, showing internal organs slightly shifted to the right side (Fig. 4B and C). However, we did not observe mice with complete situs inversus, a mirror image of normal anatomy. Heterotaxia was also found in heterozygotes albeit at lower frequencies, but not in WT mice (Fig. 4D), suggesting that there may be a certain degree of Rabl2 haploinsufficiency for certain functions in certain tissues. These findings demonstrate that RABL2 deficiency perturbs mechanisms controlling L/R asymmetry.
Similar to published observations that 67% of Rabl2 KO mice had polydactyly (4), approximately 19% of Rabl2LF/LF, 24% of Rabl2VF/VF and 42% of Rabl2−/− mice exhibited pre-axial polydactyly of the hind feet (Fig. 4E and F). Approximately 40% of affected mice exhibited bilateral polydactyly (Fig. 4F). Compared to Rabl2−/−, the incidences of polydactyly in Rabl2VF/VF and Rabl2LF/LF mice were lower, suggesting that these alleles are hypomorphic for limb development.
Rabl2LF/LF and Rabl2−/− mice have distinct perinatal neural defects
Breeding data from intercrosses of heterozygotes revealed that instead of yielding the expected 25% Mendelian ratio of homozygotes, we found only 14% of Rabl2LF/LF and Rabl2−/− live pups at postnatal day 7–14 (Fig. 5A), suggesting partially penetrant embryonic lethality. In contrast, Rabl2VF/VF mice were present at near-Mendelian frequency (23%) (Fig. 5A). To identify the stage at which embryonic lethality might be occurring in the former two alleles, we examined embryos at E12.5 and E14.5 from heterozygous intercrosses. We observed embryos with hypoplastic and exencephalic heads (Fig. 5B–E), suggesting that Rabl2 plays a role in neural tube closure. Additionally, we observed an increased incidence of resorption sites at E16.5–E18.5 (Fig. 5F). We did not observe liveborn offspring with NTDs; such pups may have been cannibalized by the mothers immediately after birth.
Figure 5.
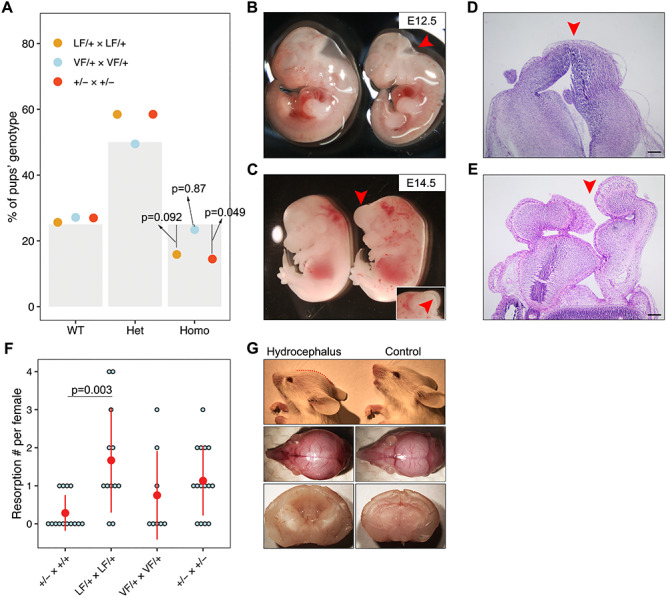
Rabl2 mutant mice have distinct neural defects. (A) Proportion of WT, heterozygous and homologous offspring obtained from het × het intercrosses. The grey bars represent the expected ratios, 25% for WT and homologous and 50% for heterozygous. The dots represent the observed ratios. n = 195 offspring obtained from LF/+ intercrosses, n = 188 offspring obtained from VF/+ intercrosses and n = 200 offspring obtained from +/− intercrosses. (B) General observation images of hypoplastic head phenotype (labeled by red arrowhead) at E12.5 in LF/LF embryo (right panel) as compared with WT control (left panel). (C) Exencephaly (labeled by red arrowhead) in LF/LF (LF = Rabl2L119F) embryo as compared to WT control (left panel) at E14.5. The insertion represents the other side showing the opened head. (D) Histology of head for control mouse. (E) Cranial histology of mouse with NTD. Red arrowheads in D and E indicate closed and opened head, respectively. (F) Plot of the number of embryo resorptions per female at E16.5–E18.5 from a heterozygous breeding pair. (G) Representative lateral images of 2-week-old LF/LF mouse with a domed cranial vault (red dashed line) compared with WT mouse (upper panel). The middle panel shows the top view of the whole skulls from the same mice. Sagittal sections of brains (lower panel) from a LF/LF and WT mouse revealing dilatation of the lateral and third ventricles in the mutant brain. Data in A were analyzed using chi-square test. Data in F are represented as the mean ± SEM and were analyzed using one-way ANOVA with Tukey’s post hoc test. Scale bars in D and E represent 200 μm.
Finally, we also observed dead or dying pups during the lactation period, partially explaining the lower frequency of homozygous mice observed at the time of genotyping (postnatal days 7–14). We occasionally found some Rabl2 mutant animals with a mildly dome-shaped cranial vault, typical of progressive hydrocephalus (Fig. 5G). Consistent with observations in another PCD mouse model (19), hydrocephalic mice always had a smaller body size (Fig. 4A). Collectively, these data suggest that Rabl2 plays a critical role in the neural tube and cranial development.
Discussion
A major challenge in medical genetics is to interpret VUS in terms of biological and clinical significance. Here, we identified two pathogenic variants in RABL2A, rs80006029 and rs200121688, by our integrated computational and experimental approaches. Both variants are predicted to be deleterious as per the consensus results of SIFT and PolyPhen-2 algorithms. Indeed, in silico analysis and in vitro protein stability assays found that both alleles destabilized the tertiary structure of RABL2A. These results provided the basis for building humanized mouse models—a non-trivial commitment—for purposes of exploring the pathogenesis of these variants in vivo. Consistent with the heterogeneous disorders caused by Rabl2 mutants reported previously (3,5,20), both of the humanized mice we generated exhibited PCD- and BBS-like features.
Rabl2 encodes an evolutionarily conserved member of the Rab-like Ras GTPase superfamily. The human paralogs RABL2A and RABL2B encode nearly identical protein sequences and are expressed in a wide range of tissues, raising the question as to why, or if, both copies are essential. There are three outcomes in the evolution of duplicated genes: (i) one copy becomes silenced by degenerative mutations; (ii) one copy acquires a novel, beneficial function and become preserved by natural selection, with the other copy retaining the original function and (iii) both copies become partially compromised by mutation accumulation to the point at which their total capacity is reduced to the level of the single-copy ancestral gene (21). There are four conserved functional domains within RABL2 proteins: two phosphate-Mg2+ binding motifs (G1 and G3), one effector region (G2) and one guanine base-binding motif (G4) (18). The effector region determines the specificity of the RAB molecules. In mouse, RABL2 mediates the delivery of effector proteins, including key members of the glycolytic pathway, into the growing sperm tail (3). In humans, the effector sequences that differ between RABL2A (DGKTILVDF) and RABL2B (DGRTILVDF) may confer slightly different functions (18). It remains to be addressed whether human RABL2A and RABL2B have identical or distinct functions during development, spermiogenesis and/or adult physiology.
We modeled the Rabl2 variants in mice for the primary purpose of determining if they affect fertility. Homozygous null and Rabl2Mot male mice are infertile (3,4), and a deletion allele in human RABL2A gene was identified as a risk factor for infertility (7). Here, we found that Rabl2LF/LF and Rabl2VF/VF mice were infertile and displayed sperm phenotypes consistent with those described in Rabl2Mot/Mot mice (3). Electron microscopy analysis of Mot sperm revealed ultrastructurally normal flagella (3). Consistent with this, our CASA data showed that the flagellar beat frequencies in motile Rabl2LF/LF and Rabl2VF/VF sperm were comparable to WT and heterozygous controls. The sperm tail truncation is consistent with a role for RABL2 in flagellar assembly, as demonstrated previously (3). Given that motile cilia and sperm flagella share the same highly conserved 9 + 2 microtubule doublet axoneme, and the efferent duct motile cilia are necessary for agitating sperm to prevent aggregation and clogging during the transition from the rete testis to epididymis (22), we examined efferent ductules. However, no defect such as sperm aggregation or agglutination was found in homozygous mutants, even though Rabl2 is expressed in efferent duct motile cilia (https://www.mousephenotype.org/). It is therefore possible that the efferent duct motile cilia have a mild defect in our Rabl2 mutant mouse models, but that any impairment in sperm agitation is insufficient to impede the dominant effect of testicular hydrostatic pressure during the sperm transition.
This study is a part of a larger scale project to identify human infertility alleles segregating in human populations. However, since Rabl2 disruption contributes to heterogeneous multisystem disorders such as PCD and BBS (3,4) that are caused by cilia defects, we explored the effects of our mutant alleles on non-reproductive systems. Consistent with previous observations (5), we found that Rabl2 mutants become overweight with advancing age (24 weeks old). Strikingly, ~45% exhibited marked growth retardation prior to weaning (3 weeks old) before reaching (or in some cases surpassing) the weight of controls by 12 weeks. There are several possible causes of the early growth retardation. One is hydrocephalus, as observed previously in PCD mouse models (19). Other potential causes to be investigated are impaired suckling (22) or defective release of growth hormone (23). The transition to becoming obese in later adulthood suggests a distinct function of RABL2. BBS patients have a high incidence of obesity and diabetes (24). A recent study found that the elimination of pancreatic β-cell primary cilia diminishes insulin levels and glucose homeostasis, leading to diabetes (25).
Heterotaxia is a birth defect involving randomization of L/R body axis, commonly observed in PCD patients. The leftward fluid flow generated by embryonic nodal cilia guides the asymmetric morphogenesis of developing organs and, if disrupted, impacts organ lateralization (26). Interestingly, we found that not only are Rabl2 homozygous mutants prone to heterotaxia but also heterozygotes, indicating that proper RABL2 levels in embryonic nodal cilia are important for establishing proper L/R asymmetry during gastrulation. This, and the commonality of spermiogenesis defects in PCD patients/mouse mutants, is consistent with the suggestion that motility functions of cilia (such as that involved in L/R asymmetry) are tightly regulated, and deficiencies in function or quantity are more likely to cause defects related to these functions (27). Consistent with this, hydrocephalus is rarer than heterotaxia in Rabl2 mutants.
Given the abundant expression of Rabl2 in the brain (18), it is likely most if not all the developmental phenotypes of Rabl2 mutants are a consequence of ciliary defects. The perinatal and juvenile phenotypes such as decreased weight may have a neurological basis, for example, by affecting suckling behavior. NTDs are occasionally present in BBS patients (28), and the severe ciliopathies, MKS and JBTS (29), commonly display CNS defects. Primary cilia are important for several pathways involved in neural tube patterning. Cerebrospinal fluid (CSF) flow is created by ependymal cells and is crucial for neurodevelopment and homeostasis of the ventricular system of the brain. The hydrocephalus in Rabl2 mutant mice may result from an accumulation of CSF due to the decreased ependymal cilia motility. The severity of hydrocephalus in PCD mouse models is affected by inbred strain background, with C57BL/6J mice being particularly severe (30). We used a mixed genetic background in these studies (FVB/NJ and B6(Cg)-Tyrc-2J/J), possibly explaining the rare incidence of hydrocephalus in our mutants.
In summary, we identified two variants in RABL2A causing male infertility and ciliopathy by functional interrogation in humanized mouse models. These genetic variants provide insight into the etiology of human asthenospermia, PCD and BBS, and can inform clinical management and genetic counseling of patients presenting relevant phenotypes and who are identified with these variants.
Materials and Methods
Computational identification of deleterious nsSNPs
The possible pathogenic functional effects of nsSNPs on human RABL2A were analyzed using SIFT (15) and PolyPhen-2 (16). The frequency of variants is as reported in gnomAD version 3.0 (gnomeAD.broadinstitute.org). To predict the functional consequences of the mutations, we modeled p.Ala94Thr, p.Leu119Phe and p.Val158Phe variants into the human RABL2A 3D structure (SWISS-MODEL accession code Q9UBK7) and used the following web servers: I-Mutant (31), DynaMut (32), SDM (33), DeepDDG (34), DUST (35) and mCSM (36). PyMol software was used to visualize the RABL2A 3D structure and protein destabilization.
Constructing vectors for DUAL-FLUO screen and western blot
Vector construction and western blot were performed as previously described (37). Briefly, the WT and mutant RABL2A open-reading frames were inserted into pDEST-DUAL vector by Gateway LR reactions and then the vectors were transfected into HEK293T cells. Anti-GFP (1:1000, SCBT, sc-9996) and anti-GAPDH (13 000, Proteintech, 60004-1-Ig) were used for immunoblotting analyses.
Production of CRISPR/Cas9-edited mice
All animal usage was approved by Cornell University’s Institutional Animal Care and Use Committee, under protocol 2004-0038 to J.C.S.
Rabl2 mutant mice were generated using CRISPR/Cas9-mediated homologous recombination in zygotes, as described previously (10). Design and selection of sgRNAs took into account the parameters of (i) on-target ranking, (ii) minimal predicted off-target sites and (iii) the distance to the target site (38). The sgRNAs were produced using MEGAshortscript™ T7 Transcription Kit (Ambion; AM1354). sgRNAs and ssODNs are listed in Supplementary Material, Table S1. Briefly, the sgRNA, ssODN and Cas9 mRNA (25 ng/μl, TriLink) were co-injected into zygotes (F1 hybrids between strains FVB/NJ and B6(Cg)-Tyrc-2J/J) and then transferred into the oviducts of pseudopregnant females. Founders carrying at least one copy of the desired alteration were identified and backcrossed into FVB/NJ. Initial phenotyping was done after one backcross generation and additional phenotyping was done with mice backcrossed two or more generations.
Genotyping
Mice were genotyped by PCR followed by Sanger sequencing. The PCR primer sequences are listed in Supplementary Material,Table S1. PCR was performed using EconoTaq and associated PCR reagents (Lucigen) with 4 μl of crude DNA lysate created as described previously (39) from ear punch biopsy specimens of 7- to 14-day-old mice. Annealing temperature was 56°C for Rabl2LF/LF and Rabl2−/− and 60°C for Rabl2VF/VF.
qRT-PCR
Total RNA was extracted from testis by TRIzol (ThermoFisher), and the reverse transcription was performed using a qScript™ cDNA SuperMix (Quantabio, 95048), according to the manufacturer’s instructions. For qRT-PCR, the specificity of PCRs was verified by a single peak according to melt curves. The primer sequences are listed in Supplementary Material, Table S1. qRT-PCR was performed with a C1000 Touch™ Thermal Cycler (Bio-Rad) amplification system using RT2 SYBR® Green qPCR Mastermixes (Qiagen). The relative levels of transcripts were calculated using the 2−ΔΔCT method and the Rabl2 expression levels were normalized to Gapdh.
Fertility testing
For fertility tests, 2-month-old mice were placed with age-matched FVB/NJ mates. After a period of at least 3 months without offspring, the analyzed individuals were considered infertile.
Histology and imaging
For the preparation of paraffin blocks, tissues were fixed overnight in Bouin’s solution or 4% paraformaldehyde at room temperature, washed in 70% ethanol and then dehydrated and embedded in paraffin. For histological analyses, paraffin sections at 6 μm thick were deparaffinized and then stained with hematoxylin and eosin (H&E). H&E slides were examined on an Olympus BX51 microscope using a 10× objective and Olympus cellSens standard software. Cropping, color and contrast adjustments were made with Adobe Photoshop CC 2019, using identical background adjustments for all images. Cauda epididymal sperm tail length was measured following staining with H&E. Forty tails per mouse were measured using Olympus cellSens software.
Sperm counts
Epididymal sperm counting was performed using a published method (40). One cauda epididymis per male was used for each data point.
Computer-assisted sperm analysis
Vas deferens were harvested from adult males and placed in 500 μl of in vitro fertilization (IVF) media (Research Vitro Fert K-RVFE-50; Cook Medical, Inc. , Bloomington, IN, USA). The semen was squeezed out by the needle and the sperm was allowed to swim out for 10 min at 37°C. A 150 μl drop of sperm suspension was placed into the same volume of pre-warmed IVF media (37°C). Then, the sperm were moved to a pre-warmed glass slide for motility assessment on an IVOS SpermAnalyzer (Hamilton-Thorne Research, Beverly, MA).
Statistics
All data were expressed as mean ± SEM. Statistical calculations were carried out using a Student’s t-test or one-way analysis of variance followed by the Tukey’s post hoc test or chi-squared test with Statistical Package for the Social Sciences (SPSS) software or within R. Graph generation was performed using R software.
Supplementary Material
Acknowledgements
The authors would like to thank R. Munroe and C. Abratte of Cornell’s transgenic facility for generating the edited mice.
Conflict of Interest statement. None declared.
Contributor Information
Xinbao Ding, Department of Biomedical Sciences, College of Veterinary Medicine, Cornell University, Ithaca, NY 14853, USA.
Robert Fragoza, Department of Biological Statistics and Computational Biology, Cornell University, Ithaca, NY 14853, USA; Weill Institute for Cell and Molecular Biology, Cornell University, Ithaca, NY 14853, USA.
Priti Singh, Department of Biomedical Sciences, College of Veterinary Medicine, Cornell University, Ithaca, NY 14853, USA.
Shu Zhang, Department of Biomedical Sciences, College of Veterinary Medicine, Cornell University, Ithaca, NY 14853, USA.
Haiyuan Yu, Department of Biological Statistics and Computational Biology, Cornell University, Ithaca, NY 14853, USA; Weill Institute for Cell and Molecular Biology, Cornell University, Ithaca, NY 14853, USA.
John C Schimenti, Department of Biomedical Sciences, College of Veterinary Medicine, Cornell University, Ithaca, NY 14853, USA.
Funding
National Institutes of Health (R01 HD082568 to J.C.S.); contract CO29155 from the NY State Stem Cell Program (NYSTEM). X.D. is supported by a postdoctoral fellowship from the Empire State Stem Cell Fund through New York State Department of Health contract no. C30293GG.
References
- 1. Agarwal A., Mulgund A., Hamada A. and Chyatte M.R. (2015) A unique view on male infertility around the globe. Reprod. Biol. Endocrinol., 13, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Waters A.M. and Beales P.L. (2011) Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol., 26, 1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lo J.C.Y., Jamsai D., O’Connor A.E., Borg C., Clark B.J., Whisstock J.C., Field M.C., Adams V., Ishikawa T., Aitken R.J. et al. (2012) RAB-like 2 has an essential role in male fertility, sperm intra-flagellar transport, and tail assembly. PLoS Genet., 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kanie T., Abbott K.L., Mooney N.A., Plowey E.D., Demeter J. and Jackson P.K. (2017) The CEP19-RABL2 GTPase complex binds IFT-B to initiate intraflagellar transport at the ciliary base. Dev. Cell, 42, 22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lo J.C.Y., O’Connor A.E., Andrews Z.B., Lo C., Tiganis T., Watt M.J. and O’Bryan M.K. (2016) RABL2 is required for hepatic fatty acid homeostasis and its dysfunction leads to steatosis and a diabetes-like state. Endocrinology, 157, 4732–4743. [DOI] [PubMed] [Google Scholar]
- 6. Martin C.L., Wong A., Gross A., Chung J., Fantes J.A. and Ledbetter D.H. (2002) The evolutionary origin of human subtelomeric homologies--or where the ends begin. Am. J. Hum. Genet., 70, 972–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jamsai D., Lo J.C.Y., McLachlan R.I. and O’Bryan M.K. (2014) Genetic variants in the RABL2A gene in fertile and oligoasthenospermic infertile men. Fertil. Steril., 102, 223–229. [DOI] [PubMed] [Google Scholar]
- 8. Yang Y., Peng X., Ying P., Tian J., Li J., Ke J., Zhu Y., Gong Y., Zou D., Yang N. et al. (2019) AWESOME: a database of SNPs that affect protein post-translational modifications. Nucleic Acids Res., 47, D874–D880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E. et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh P. and Schimenti J.C. (2015) The genetics of human infertility by functional interrogation of SNPs in mice. Proc. Natl. Acad. Sci. U. S. A., 112, 10431–10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tran T.N. and Schimenti J.C. (2018) A putative human infertility allele of the meiotic recombinase DMC1 does not affect fertility in mice. Hum. Mol. Genet., 27, 3911–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tran T.N. and Schimenti J.C. (2019) A segregating human allele of SPO11 modeled in mice disrupts timing and amounts of meiotic recombination, causing oligospermia and a decreased ovarian reserve†. Biol. Reprod., 101, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tran T.N., Martinez J. and Schimenti J.C. (2019) A predicted deleterious allele of the essential meiosis gene MND1, present in ~3% of East Asians, does not disrupt reproduction in mice. Mol. Hum. Reprod., 10, 668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P. et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumar P., Henikoff S. and Ng P.C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- 16. Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S. and Sunyaev S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yuan S., Liu Y., Peng H., Tang C., Hennig G.W., Wang Z., Wang L., Yu T., Klukovich R., Zhang Y. et al. (2019) Motile cilia of the male reproductive system require miR-34/miR-449 for development and function to generate luminal turbulence. Proc. Natl. Acad. Sci. U. S. A., 116, 3584–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wong A.C., Shkolny D., Dorman A., Willingham D., Roe B.A. and McDermid H.E. (1999) Two novel human RAB genes with near identical sequence each map to a telomere-associated region: the subtelomeric region of 22q13.3 and the ancestral telomere band 2q13. Genomics, 59, 326–334. [DOI] [PubMed] [Google Scholar]
- 19. Ibañez-Tallon I., Gorokhova S. and Heintz N. (2002) Loss of function of axonemal dynein Mdnah5 causes primary ciliary dyskinesia and hydrocephalus. Hum. Mol. Genet., 11, 715–721. [DOI] [PubMed] [Google Scholar]
- 20. Nishijima Y., Hagiya Y., Kubo T., Takei R., Katoh Y. and Nakayama K. (2017) RABL2 interacts with the intraflagellar transport-B complex and CEP19 and participates in ciliary assembly. Mol. Biol. Cell, 28, 1652–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lynch M. and Conery J.S. (2000) The evolutionary fate and consequences of duplicate genes. Science, 290, 1151–1155. [DOI] [PubMed] [Google Scholar]
- 22. Wu J., Bao J., Kim M., Yuan S., Tang C., Zheng H., Mastick G.S., Xu C. and Yan W. (2014) Two miRNA clusters, miR-34b/c and miR-449, are essential for normal brain development, motile ciliogenesis, and spermatogenesis. Proc. Natl. Acad. Sci. U. S. A., 111, E2851–E2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lucas-Herald A.K., Kinning E., Iida A., Wang Z., Miyake N., Ikegawa S., McNeilly J. and Ahmed S.F. (2015) A case of functional growth hormone deficiency and early growth retardation in a child with IFT172 mutations. J. Clin. Endocrinol. Metab., 100, 1221–1224. [DOI] [PubMed] [Google Scholar]
- 24. Mujahid S., Hunt K.F., Cheah Y.S., Forsythe E., Hazlehurst J.M., Sparks K., Mohammed S., Tomlinson J.W., Amiel S.A., Carroll P.V. et al. (2018) The endocrine and metabolic characteristics of a large Bardet–Biedl syndrome clinic population. J. Clin. Endocrinol. Metab., 103, 1834–1841. [DOI] [PubMed] [Google Scholar]
- 25. Hughes J.W., Cho J.H., Conway H.E., DiGruccio M.R., Ng X.W., Roseman H.F., Abreu D., Urano F. and Piston D.W. (2020) Primary cilia control glucose homeostasis via islet paracrine interactions. Proc. Natl. Acad. Sci. U. S. A., 117, 8912–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wagner M.K. and Yost H.J. (2000) Left-right development: the roles of nodal cilia. Curr. Biol., 10, R149–R151. [DOI] [PubMed] [Google Scholar]
- 27. Chiani F., Orsini T., Gambadoro A., Pasquini M., Putti S., Cirilli M., Ermakova O. and Tocchini-Valentini G.P. (2019) Functional loss of Ccdc151 leads to hydrocephalus in a mouse model of primary ciliary dyskinesia. Dis. Model. Mech., 12, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ross A.J., May-Simera H., Eichers E.R., Kai M., Hill J., Jagger D.J., Leitch C.C., Chapple J.P., Munro P.M., Fisher S. et al. (2005) Disruption of Bardet–Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet., 37, 1135–1140. [DOI] [PubMed] [Google Scholar]
- 29. Vogel T.W., Carter C.S., Abode-Iyamah K., Zhang Q. and Robinson S. (2012) The role of primary cilia in the pathophysiology of neural tube defects. Neurosurg. Focus., 33, E2–E9. [DOI] [PubMed] [Google Scholar]
- 30. McKenzie C.W., Preston C.C., Finn R., Eyster K.M., Faustino R.S. and Lee L. (2018) Strain-specific differences in brain gene expression in a hydrocephalic mouse model with motile cilia dysfunction. Sci. Rep., 8, 13370–13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Capriotti E., Fariselli P. and Casadio R. (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res., 33, W306–W310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rodrigues C.H., Pires D.E. and Ascher D.B. (2018) DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res., 46, W350–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pandurangan A.P., Ochoa-Montaño B., Ascher D.B. and Blundell T.L. (2017) SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res., 45, W229–W235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cao H., Wang J., He L., Qi Y. and Zhang J.Z. (2019) Deepddg: predicting the stability change of protein point mutations using neural networks. J. Chem. Inf. Model., 59, 1508–1514. [DOI] [PubMed] [Google Scholar]
- 35. Pires D.E.V., Ascher D.B. and Blundell T.L. (2014) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res., 42, W314–W319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pires D.E.V., Ascher D.B. and Blundell T.L. (2014) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics, 30, 335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fragoza R., Das J., Wierbowski S.D., Liang J., Tran T.N., Liang S., Beltran J.F., Rivera-Erick C.A., Ye K., Wang T.-Y. et al. (2019) Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun., 10, 4141–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh P., Schimenti J.C. and Bolcun-Filas E. (2015) A mouse geneticist’s practical guide to CRISPR applications. Genetics, 199, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Truett G.E., Heeger P., Mynatt R.L., Truett A.A., Walker J.A. and Warman M.L. (2000) Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). BioTechniques, 29, 52–54. [DOI] [PubMed] [Google Scholar]
- 40. Wang Y. (2003) Epididymal sperm count. Curr. Protoc. Toxicol., Chapter 16, Unit16.6.1–Unit16.6.5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.