

Abstract
Ultra-high field proton magnetic resonance spectroscopy (1HMRS) offers a unique opportunity to measure the concentration of neurometabolites implicated in psychosis (PSY). The extant 7T 1HMRS literature measuring glutamate-associated neurometabolites in the brain in PSY in vivo is small, but a comprehensive, quantitative summary of these data can offer insight and guidance to this emerging field. This meta-analysis examines proton spectroscopy (1HMRS) measures of glutamate (Glu), glutamine (Gln), glutamate+glutamine (Glx), gamma aminobutyric acid (GABA), and glutathione (GSH) across 255 individuals with PSY (121 first episode) and 293 healthy comparison participants (HC). While all five neurometabolites were lower in PSY as compared to HC, only Glu (Cohen’s d = −0.18) and GSH (Cohen’s d = −0.21) concentrations were significantly lower in PSY, whereas concentrations of Gln, Glx, and GABA did not significantly differ between groups. Notably, 1HMRS methodological choices and sample demographic characteristics did not impact study-specific effect sizes for PSY-related Glu or GSH differences. This review thus provides further evidence of neurometabolite dysfunction in first episode and chronic PSY, and thereby suggests that Glu and GSH abnormalities may additionally play a role in more incipient stages of the disorder: in clinical high risk stages. Additional 7T neurochemical imaging studies in larger, longitudinal, and unmedicated samples and in youth at risk for developing psychosis are needed. Such studies are critical for elucidating the neurodevelopmental and clinical time course of PSY-related neurometabolite alterations, and for assessing the potential for implicated metabolites to serve as druggable targets for decreasing PSY risk.
Keywords: meta-analysis, spectroscopy, 7T MRI, psychosis, glutamate
1.0. Introduction
Psychosis (PSY) is a complex brain disorder that typically presents in adolescence or early adulthood (Casey et al., 2014; Insel, 2010; Rapoport et al., 2012) that has a devastating impact on social, occupational, and daily functioning and large health costs (Wu et al., 2005; Zeidler et al., 2012). PSY is associated with progressive gray matter loss (Bora et al., 2011; Gur et al., 2000), reduced cortical neuropil density (Selemon and Goldman-Rakic, 1999), altered dopamine receptor availability (Laruelle et al., 2006; Poels et al., 2014), and abnormal excitatory and inhibitory neurotransmitter functioning (Coyle, 2006; Lewis and Moghaddam, 2006; Marsman et al., 2013; Moghaddam, 2004; Remington et al., 2011). Hence, neural circuit architecture and neurotransmission appear to be abnormal in PSY. Structural and neurochemical alterations have also been found, to a lesser extent, in youth at clinical high risk for PSY (CHR) (Allen et al., 2015; Egerton et al., 2014; Roalf et al., 2017, p. 201; Satterthwaite et al., 2016). CHR individuals exhibit brief or attenuated psychotic symptoms, which evolve into frank PSY in approximately 15–20% of individuals at one year (Fusar-Poli et al., 2016, 2012), 20–30% of individuals at two years (Fusar-Poli et al., 2016, 2012), and 35% of individuals at three to ten years following initial presentation to clinical services (Fusar-Poli et al., 2012; Nelson et al., 2013), with greatest transition risk occurring within the first two years (Fusar-Poli et al., 2016; Kempton et al., 2015; Nelson et al., 2013). There is evidence that the neuroetiological mechanisms associated with both PSY and CHR begin to emerge during early development, leading to the appearance of more apparent symptoms in the context of adolescent neuromaturation (Chung and Cannon, 2015; McGlashan and Hoffman, 2000). Mitigating PSY-related dysfunction while the brain is still developing may thus help to reduce the number of CHR individuals who convert to PSY, making identification of neural targets and corresponding interventions paramount.
There is a significant literature supporting the dopamine hypothesis of PSY, which was first proposed over 40 years ago (Howes and Kapur, 2009). Contemporary evidence furthermore indicates that dysregulation of the dopamine system in PSY and CHR may occur secondarily to deficits in glutamate (Glu) function (Poels et al., 2014). Interest in elucidating the role of Glu in the pathogenesis of PSY first emerged due to findings that phencyclidine (NMDA receptor antagonist), ketamine (NMDA receptor antagonist), and Δ−9-THC (transiently disturbs Glu and gamma aminobutyric acid (GABA)) have psychotomimetic properties, inducing both positive and negative symptoms similar to those seen in PSY (Bossong and Niesink, 2010, p. 20210; D’Souza et al., 2004; Frohlich and Van Horn, 2014; Kraguljac et al., 2017; Krystal et al., 1994; Tsai and Coyle, 2002).
Glu is the main excitatory neurotransmitter in the brain, present at approximately 80% of synapses, and it is additionally essential for cellular energy metabolism (Coyle, 2006; Curtis and Johnston, 1974; Erecińska et al., 1990; McEntee and Crook, 1993; Robbins and Murphy, 2006). Glu metabolism dynamically influences other neurochemicals, including glutamine (Gln), GABA, and glutathione (GSH). Glu released into the synapse and extrasynaptic space is taken up into astrocytes and converted to Gln, preventing excitotoxic cell death due to excess synaptic Glu. Gln is then converted back to Glu in both excitatory and inhibitory neuron presynaptic terminals. In inhibitory terminals, the predominant inhibitory neurotransmitter GABA is produced from Glu by the enzyme glutamate decarboxylase; GABA plays an integral role in maintaining the balance of excitation-inhibition throughout the brain. Finally, Glu is additionally incorporated into glutathione (GSH), an important antioxidant that protects the brain from oxidative stress. Alterations in Glu levels in the brain, such as those proposed to occur in PSY, may therefore co-occur with changes in GABA, Gln, and GSH, leading to multifaceted and detrimental neurobiological effects. Systematic investigation of this entire group of neurochemicals in prodromal and early PSY may therefore provide important and complimentary information.
Proton magnetic resonance spectroscopy (1HMRS) techniques can be used to quantify Glu, GABA, and related neurometabolite levels in vivo. While a number of studies have applied 1HMRS to study Glu-related metabolites in PSY (and some in CHR), results are somewhat heterogeneous (Merritt et al., 2016), and the vast majority of studies have been conducted at 3T. Recent improvements in MRI technology, particularly the use of ultra-high field (UHF, ≥ 7Tesla) magnets, has expanded and enhanced in vivo application of 1HMRS techniques. As reviewed in Godlewska et al. (Godlewska et al., 2017), 7T 1HMRS offers enhanced spectral resolution, thereby producing more specific and reliable metabolite measurements than can be resolved at lower magnetic fields (Godlewska et al., 2017; Terpstra et al., 2016). Moreover, 7T 1HMRS allows for increased metabolite sensitivity, improved image resolution, and approximately 1.6 times higher signal-to-noise ratio (SNR) compared to 3T (Godlewska et al., 2017). Critically, these improvements in specificity, sensitivity, and SNR at 7T are particularly significant for Glu, Gln, GABA, and GSH. For example, Glu, Gln, and GABA resonances can be more reliably distinguished at 7T, preventing the need for one combined measurement of Glu+Gln (i.e., “GLX”), and facilitating accurate measurement of GABA. Promisingly, studies harnessing UHF imaging to investigate PSY have begun to emerge over the past few years (Brandt et al., 2016, p. 20; Kumar et al., 2018; Marsman et al., 2014; Overbeek et al., 2019; Posporelis et al., 2018; Reid et al., 2018; Roalf et al., 2017; Rowland et al., 2016; Taylor et al., 2015; Thakkar et al., 2017; Wang et al., 2019). Here, we review these studies in order to provide insight into whether concentrations of the brain’s main excitatory and inhibitory neurotransmitters and associated neurometabolites are altered in PSY.
This review takes the form of a quantitative meta-analysis of existing 7T 1HMRS studies examining Glu-related neurometabolites in PSY. This is a relatively small, evolving, but important area of research. By consolidating early results across studies, this meta-analysis aims to provide a more comprehensive estimate of neurochemical differences between healthy individuals and those with PSY. We believe this will provide insight into the potential utility of UHF 1HMRS for understanding CHR and psychosis onset, while simultaneously shedding light on possible neurochemical targets for intervention.
2.0. Material and Methods
The report and extraction of relevant articles and accompanying data followed the Meta-Analysis of Observational Studies in Epidemiology (MOOSE) reporting standards (Supplemental Figure 1) (Stroup et al., 2000). The search strategy included an initial broad search in PubMed, MEDLINE, PsychINFO, and Google Scholar databases using the following search criteria: “psychosis/schizophrenia/clinical high risk for psychosis” AND “7T/UHF” AND “glutamate (Glu)/glutamine (Gln)/Glutamate+Glutamine (Glx)/GABA” OR “spectroscopy (HMRS)”, OR “brain”. The search was limited to articles published or in press prior to January 31, 2020 that were written in English and that enrolled human participants. A manual review of articles was additionally performed utilizing cross-references from identified original articles and reviews.
Studies included in this meta-analysis contained the following data: 1) experimental 1HMRS measurement of Glu, Gln, Glx (i.e. Glu+Gln), GABA, or GSH in PSY; 2) corresponding 1HMRS measurement in a healthy comparison (HC) group of unrelated participants with no history of major medical, neurological, or psychiatric conditions, and 3) statistical information for calculating effect sizes. DRR reviewed potential publications for inclusion based on the aforementioned criteria. Consensus of both authors was needed to exclude an article. We identified nine (n = 9) publications (Table 1) for inclusion that reported group comparisons (PSY v. HC) of Glu, Gln, Glx, GABA, and/or GSH 1HMRS measurements. One additional article was considered (Overbeek et al., 2019) but ultimately excluded given that the data overlapped almost entirely with an included manuscript (Reid et al., 2018). Across all nine included publications, there was a combined total of 61 effects (k). Examination of all 61 effects revealed that 59 effects utilized absolute neurometabolite concentrations, while 2 effects from Marsman et al. (2014) utilized creatine-normalized ratios (i.e. GABA/Creatine ratio). To ensure data type homogeneity in our meta-analysis, we conducted analyses using absolute concentration effects only (k = 59). DRR initially extracted relevant data from publications, and VJS quality checked all extracted data for accuracy. Variables extracted for meta-analytic analysis included 1HMRS concentration values and standard deviations, neurometabolite measured, 1HMRS sequence type, 1HMRS voxel size, brain region of interest (ROI), mean age of the PSY and HC samples, and sample sex distribution. Data used in this meta-analysis are available in the Supplemental Materials.
Table 1. Summary of Patient Characteristics, Sample Demographics, and1HMRS Acquisition Parameters for Studies Included in the Present Meta-Analysis.
HC = healthy comparison; PSY = psychosis. PSY Type Abbreviations: FEP-SZ = first episode schizophrenia; SAD = schizoaffective disorder; SZ = schizophrenia. Sex Abbreviations: %F = percent female; Region of Interest (ROI) Abbreviations: ACC = anterior cingulate cortex; B = bilateral; CSO = centrum semiovale; DLPFC = dorsolateral prefrontal cortex; L = left hemisphere; mPFC = medial prefrontal cortex; OCC = occipital cortex; OFR = orbitofrontal cortex. 1HMRS Sequence Abbreviations: MEGA = Mescher-Garwood; sLASER = semi-localized by adiabatic selective refocusing; STEAM = stimulated echo acquisition mode; Neurometabolite Abbreviations: GABA = gamma aminobutyric acid; Gln = glutamine; Glu = glutamate; Glx = glutamate+glutamine; GSH = glutathione.
| STUDY INFORMATION | PATIENT CHARACTERISTICS | SAMPLE DEMOGRAPHICS | 1HMRS ACQUISITION PARAMETERS | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study | Sample Size Total (HC/PSY) | PSY Type | Patients on Antipsychotics (%) | Class of Antipsychotics Reported | Mean Age HC | Mean Age PSY | Sex (%F) HC | Sex (%F) PSY | ROIs | 1HMRS Sequence | Echo Time (ms) | Neurometabolites Measured | ROI Size (mm3) |
| Brandt 2016 | 48 (24/24) | SZ, SAD | 100 | Typical and atypical | 36.6 | 37.5 | 17 | 21 | B ACC | STEAM | 14 | Glu, Gln, GABA, GSH | 7200 |
| Kumar 2018 | 73 (45/28) | SZ, SAD | Not reported, “most patients” | Not reported | 27.9 | 27.2 | 36 | 29 | B ACC L Insula B OCC |
STEAM | 17 | Glu, Gln, GSH | 9000 8640 8800 |
| Marsman 2014 | 40 (23/17) | SZ | 100 | Atypical | 27.7 | 27.6 | 30 | 24 | B mPFC B OCC |
sLASER | 28 | Glu | 8000f |
| Posporelis 2017 | 40 (20/20) | FEP-SZa | 80 | Typical and atypical | 23.1 | 24.3 | 35 | 35 | B ACC | STEAM | 15 | Glu | 27000 |
| Reid 2019 | 42 (21/21) | FEP-SZb | 95 | Atypical | 23.5 | 23.2 | 24 | 24 | B ACC | STEAM | 5 | Glu, Gln, GABA, GSH | 5400 |
| Rowland 2016 | 56 (29/27) | SZ, SAD | 83 | Typical and atypical | 29.7 | 34.4 | 52 | 37 | B ACC | STEAM | 14 | Glu, Gln, GABA, GSH | 12000 |
| Taylor 2015 | 32 (16/16) | SZ | 88 | Atypical | 23.9 | 22.7 | 31 | 19 | B ACC | STEAM | 10 | Glu, Gln, Glx | 8000 |
| Thakkar 2017 | 45 (24/21) | SZ, SAD | 100 | Not reported | 33.9 | 36.4 | 33 | 29 | B OCC B Striatum |
sLASER MEGA-sLASERc |
36d 74e |
Glu, Gln, GABA, Glx | 24000 |
| Wang 2019 | 172 (91/81) | FEP-SZa | Not reported | Not reported | 23.3 | 22.3 | 54 | 30 | B ACC L CSO L DLPFC L OFR B Thalamus |
STEAM | 14 | Glu, Gln, GABA, GSH | 12000 12000 10000 8000 9000 |
duration of illness ≤ 24 months;
duration of illness ≤ 24 months in 95% of patients;
used for GABA acquisition;
echo time used for sLASER sequence;
echo time used for MEGA-sLASER GABA sequence;
ROI size used for sLASER sequence (Glu) in both ROIs.
Additional notes: Clozapine is considered an atypical antipsychotic in this Table. ROI size is respective to the ROI listed, unless otherwise noted. Bilateral ROIs include both midline-crossing single voxel ROIs and two independent voxels placed in left and right hemisphere ROIs, depending on the study.
2.1. Statistical Analyses
An initial meta-analysis was conducted that included all 9 studies and all 5 neurometabolites to determine an overall effect, followed by meta-regression and analyses of publication bias. All analyses were carried out in Comprehensive Meta-Analysis version 2.21.064 (Biostat, 2005) with standard random-effects models. Neurometabolite concentration differences between PSY and HC participants were standardized using Cohen’s d effect sizes (calculated as the difference between the two raw mean scores divided by the pooled standard deviation). Confidence intervals (CI) were additionally calculated for each effect size. Studies were weighted according to their inverse variance estimates when calculating effect sizes to control for study sample size. Effect sizes can be classified as small (d = ±0.2), medium (d = ±0.5), or large (d ≥ ±0.8) (Cohen, 1988). Here, a negative effect size denotes lower neurometabolite level in PSY relative to HC. Homogeneity of effect sizes across studies was assessed using the Cochran Q-statistic (Hedges and Olkin, 1985).
2.1.1. Moderator Variables and Meta-regression
Meta-regression was used to investigate potential moderating variables, given that substantial heterogeneity was observed in the initial meta-analytic result. The following moderator variables were assessed via meta-regression: (1) neurometabolite studied (Glu, Gln, Glx, GABA, GSH), (2) 1HMRS protocol, (3) brain ROI, (4) 1HMRS acquisition voxel size (mm3), (5) echo time, (6) age, and (7) sex.
2.1.2. Analysis of Publication Bias
Meta-analytic results are susceptible to influence from publication bias (i.e. bias that arises when studies with large effects are overrepresented in the literature compared to similar studies with small or null effects). Publication bias was evaluated using complimentary methods: (1) funnel plot: an asymmetric scatterplot of effect size vs. study precision is indicative of publication bias; (2) adjusted rank-correlation (i.e. Spearman) test according to the methods of Begg and Mazumdar (Begg and Mazumdar, 1994), Egger et al. (Egger et al., 1997), and Duval and Tweedie (Duval and Tweedie, 2000): a high correlation between the effect size and corresponding sampling variances is indicative of publication bias; and (3) a post-hoc trim-and-fill method including calculation of a fail-safe N (Duval and Tweedie, 2000). The trim-and-fill method is a funnel plot-based approach that “trims” outlier studies from the plot and “fills” in potentially missing studies, allowing for estimation of a publication bias-adjusted meta-analytic effect size. This method controls for the possibility that certain studies are outliers, while additionally considering potentially unpublished data (i.e. “file drawer” studies).
3.0. Results
3.1. Summary of Extant Literature
Nine 7T studies comparing neurometabolite concentrations between HC and PSY groups were included in this meta-analysis. Study details including participant demographics, patient characteristics, and 1HMRS acquisition parameters are presented by study in Table 1. Data from 293 HC and 255 PSY participants (including 121 first episode patients within 24 months of PSY onset) were combined for the overall meta-analysis; this analysis does not contain data from CHR individuals due to lack of availability. The vast majority of PSY participants included in this meta-analysis were being treated with antipsychotic medications (both typical and atypical), with reported antipsychotic treatment rates ranging from 80–100% across studies.
The average age of HC and PSY individuals was 27.7 and 28.4 years, respectively. Most available studies were relatively small (< 30 individuals per group), however the most recently published study had a robust (n = 172) sample size (Wang et al., 2019). 1HMRS data was most frequently collected from the anterior cingulate cortex (ACC; 7/9 studies); no other ROI was reported in more than three studies. Average ROI size for 1HMRS acquisition was 11,269 mm3. Stimulated echo acquisition mode (STEAM) protocols with a short echo time (< 20 ms) were most commonly used to acquire data (7/9 studies). Glu concentration was reported in all studies, Gln in 7/9, GABA in 5/9, Glx in 2/9, and GSH in 5/9.
3.2. Overall Meta-analysis
The overall meta-analysis investigating differences between HC and PSY participants across all five neurometabolites (Glu, Gln, Glx, GABA, and GSH) revealed an overall significant but small meta-analytic effect size (k = 59, d = −0.13, 95% CI = −0.21 < δ < −0.06; z = 3.64, p < 0.001), indicating that metabolite levels were lower in PSY patients. However, an analysis of effect size homogeneity indicated that study-specific effect sizes were significantly heterogeneous (QB[58] = 84.4, p = 0.013).
3.3. Moderator Analyses
Given that variability in study-specific effect sizes across all neurometabolites differed more than would be expected from sampling error alone, a moderator analysis was performed that accounted for neurometabolite type. Effects sizes by study and neurometabolite are shown in Figure 1. Effect sizes for each neurometabolite were homogeneous in direction, as all five neurometabolites were lower in PSY as compared to HC (QB[4] = 5.01, p = 0.29). Only Glu (d = −0.18, 95% CI = −0.32 < δ < −0.04, k = 18; Z = 2.59, p = 0.010) and GSH (d = −0.21, 95% CI = −0.36 < δ < −0.06, k = 11; Z = 2.75, p = 0.006), however, were significantly lower in PSY (HC > PSY, Figure 2), i.e., GABA, Glx, and Gln concentrations did not significantly different between groups. Effects sizes for all neurometabolites are shown in Supplemental Table 1. Given these results, additional moderator analyses were performed for both Glu and GSH.
Figure 1.
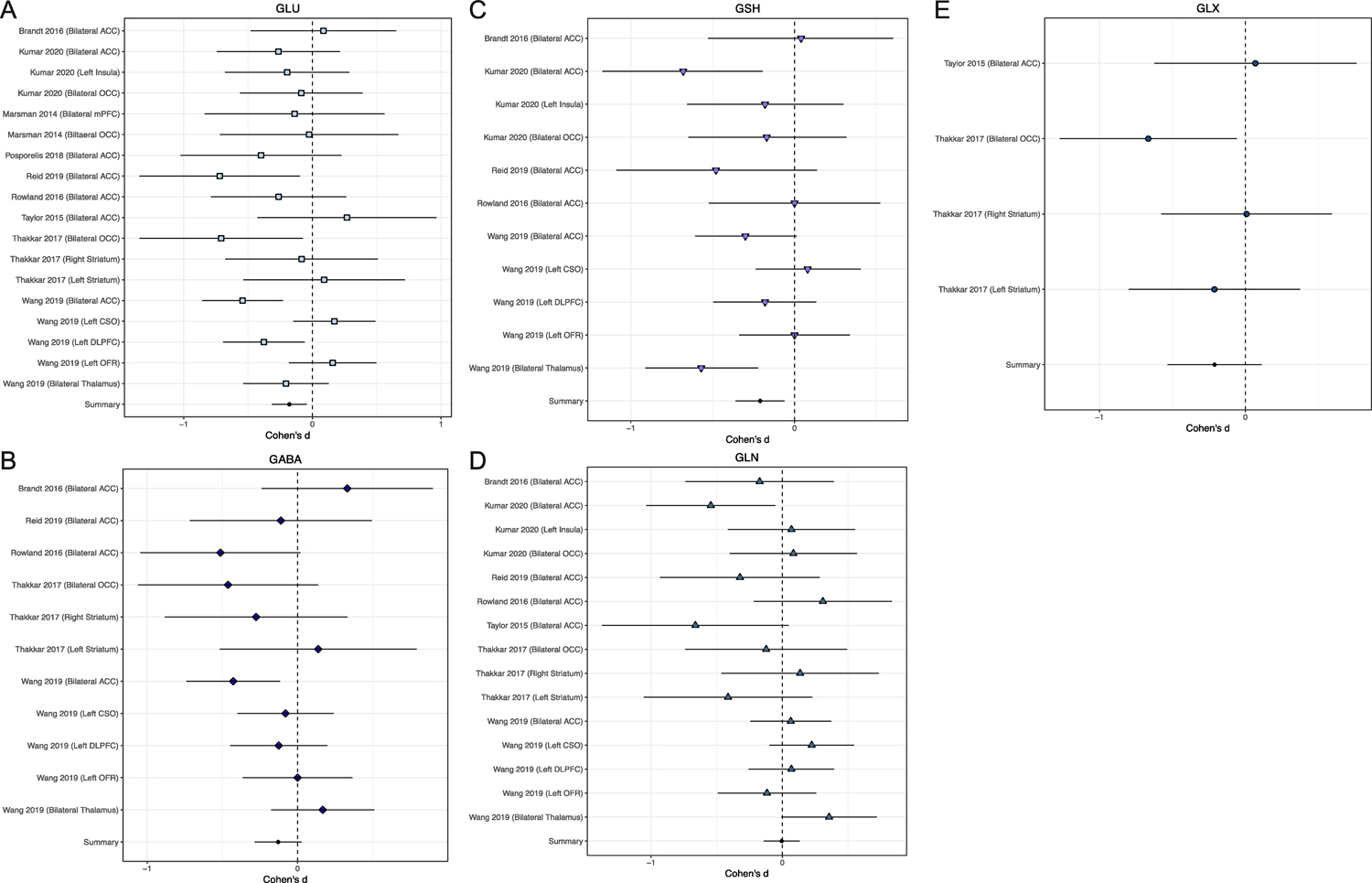
Study specific effect sizes (Cohen’s D +/− 95% CI) for each neurometabolite: A. Glutamate (Glu), B. Gamma aminobutyric acid (GABA), C. Glutathione (GSH), D. Glutamine (Gln) and E. Glutamate+Glutamine (Glx). Each forest plot displays the study and ROI in which the neurometabolite was measured. A summary effect size is provided for each neurometabolite. Negative effect sizes indicate lower concentration in PSY as compared to HC.
Figure 2.
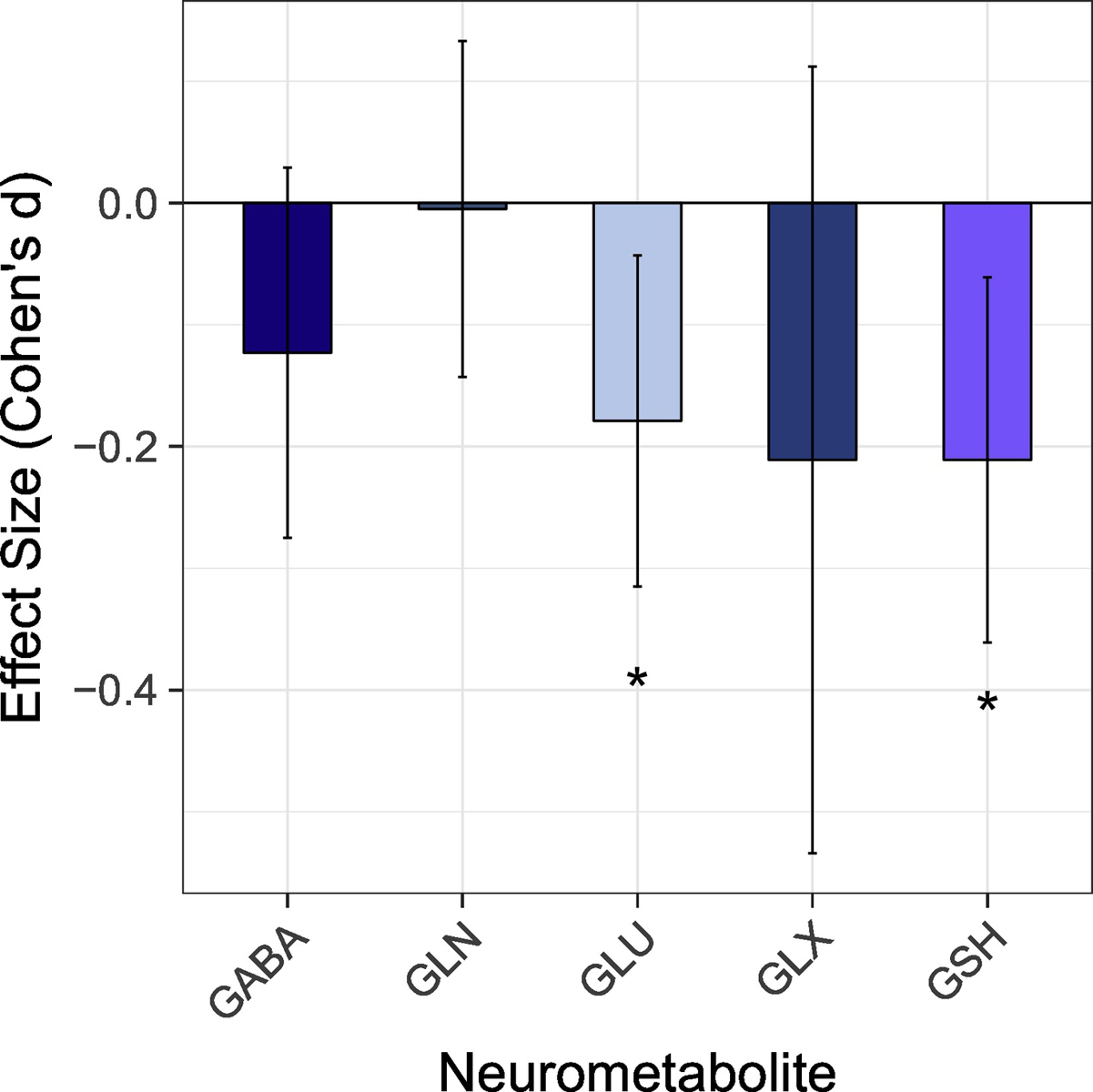
The meta-analytic derived average effect size (+/ 95% CI) for each neurometabolite. Gamma aminobutyric acid (GABA); Glutamine (Gln); Glutamate (Glu); Glutamate+Glutamine (Glx); Glutathione (GSH).
No additional moderator analyses were significant, indicating that meta-analytical effect sizes for Glu and GSH were not significantly influenced by 1HMRS protocol (e.g. STEAM versus sLASER), brain ROI, 1HMRS acquisition voxel size, echo time, sample sex, or sample age. Given minimal variability in the percentage of patients prescribed antipsychotics across studies as well as missing medication data (Table 1), we were unable to examine whether neurometabolite effect sizes differed as a function of antipsychotic treatment rate.
3.4. Publication Bias
Investigations were undertaken to gauge publication bias. The resulting funnel plots for Glu and GSH were symmetric (Supplemental Figures 2 and 3) and Begg (Glu p = 0.64; GSH p = 0.93) and Egger (Glu p = 0.99; GSH p = 0.71) tests of potential bias were non-significant. Trim-and-fill analyses identified no putative outlier effects for Glu or GSH. Calculation of a fail-safe N suggested that to negate the total meta-analytic effect observed across all five neurometabolites, 128 null studies would need to be incorporated into the primary analysis. Furthermore, to negative observed effects for Glu and GSH, 28 and 22 null studies would be needed, respectively. These results indicate that the current results are representative of the extant 7T literature in PSY.
4.0. Discussion
Results from the present 7T 1HMRS meta-analysis indicate that concentrations of brain Glu and GSH are significantly lower in patients with psychosis. Most, albeit not all, examined studies reported at least a trend towards lower brain Glu and GSH in PSY, thus the resulting meta-analytically derived summary effect sizes observed were small but significant. Meta-analytic summary effect sizes for GABA and Glx additionally indicated that levels may be lower in PSY as compared to HC, however summary effects for these metabolites were not significant. Assessments of publication bias indicated that the extant data is likely representative of the field (e.g., a large number of null studies would be needed to negate observed effects), though to date there are only nine independent 1HMRS studies of PSY at 7T. Interestingly, 1HMRS protocol, 1HMRS acquisition voxel size, protocol echo time, brain region studied, sample sex composition, and sample age were not found to systematically affect study-specific effect sizes, though the number of studies available may have been too small to detect subtle moderating effects. Altogether, this meta-analysis supports that 7T 1HMRS can offer promising insight into specific neurochemical disruptions in PSY, and further implicates Glu and GSH in the disorder. Nevertheless, additional data acquired from larger, younger, and unmedicated longitudinal samples is needed to elucidate the developmental time course of neurochemical alterations, the interplay between neurochemical alterations and changes in brain structure and function, and the relationship between such alterations and clinical and cognitive phenotypes that predict transition to PSY.
4.1. The Significance of Lower Brain Glu and GSH in Psychosis at 7T
The present meta-analytic findings add to a growing body of research implicating Glu dysfunction in PSY. Convergent evidence from animal models (Balla et al., 2001; Duncan et al., 2004; Javitt, 2004; Jentsch et al., 1997; Mohn et al., 1999), post-mortem tissue (Beneyto et al., 2007; Goff and Coyle, 2001; Hu et al., 2015), genetics studies (Harrison and Weinberger, 2005; Moghaddam, 2003; Rubio et al., 2013; Stefansson et al., 2002), peripheral markers (Palomino et al., 2007), and in vivo spectroscopy (Allen et al., 2015; Baiano et al., 2007) suggests that PSY involves significant abnormalities in the Glu system. Yet notably, the current 7T 1HMRS meta-analytic findings appear to contradict those reported in a large 3T 1HMRS meta-analysis of PSY, in which glutamate-related metabolites (Gln and Glx) were found to be, on average, higher in patients compared to controls (Merritt et al., 2016). Careful consideration of region-specific Glu effects within Merritt et al. (2016) reveals, however, that when Glu was analyzed and reported alone, levels indeed tended to be lower in individuals with PSY. Specifically, region-specific meta-analytic summary effect sizes indicated that Glu levels were lower in PSY patients than HC in the medial prefrontal cortex, frontal white matter, the medial temporal lobe, and the thalamus (though higher in PSY in the basal ganglia) (Merritt et al., 2016). This 7T meta-analysis thus corroborates these findings, as 72% (13/18, Figure 1) of the 7T Glu effects revealed lower Glu in PSY patients across a number of brain regions.
Lower levels of brain Glu may be indicative of a lower density of glutamatergic synapses (Olney and Farber, 1995), reduced excitatory neurotransmission, and/or disrupted glutamate metabolism in PSY. Though the mechanisms underlying putative glutamate dysfunction in PSY remain unknown, one model posits that a loss of inhibitory control due to NMDA-mediated interneuron dysfunction leads to elevated glutamate levels initially, glutamate-induced excitotoxicity, and an eventual loss of glutamatergic connections (Moghaddam and Javitt, 2012). If, as supported by the present meta-analysis, Glu levels are reduced in individuals diagnosed with PSY, this would suggest that disorder pathomechanisms (such as initially increased Glu) begin prior to when PSY is diagnosed.
GSH, the major intracellular antioxidant in the brain, plays a critical role in maintaining neuronal health. Lower levels of GSH have been found in the periphery in PSY (Raffa et al., 2009), as well as in the brain in post-mortem studies (Gawryluk et al., 2011; Yao et al., 2006). Further, a recent 3T meta-analysis assessing in vivo brain GSH in over 500 patients found lower GSH concentrations (effect size = −0.26) within the ACC in PSY (Das et al., 2019). The magnitude of the GSH meta-analytic summary effect size was similar in this 7T meta-analysis (effect size = −0.21) across fewer individuals, corroborating the 3T result and suggesting sensitivity improvements at UHF. GSH, which is synthesized from glutamate, cysteine, and glycine, detoxifies reactive oxygen species and potentiates the NMDA receptor response to glutamate (Do et al., 2009). Interestingly, animal studies have demonstrated that oxidative stress and NMDA receptor hypofunction are reciprocally linked, suggesting that the observed Glu and GSH reductions may be inter-dependent, though only one study in this review reported a significant positive relationship between Glu and GSH (Kumar et al., 2018).
4.2. Interactions Between Antipsychotic Medications and Glutamate Neurometabolites
The effects of antipsychotic psychotropics on neurometabolite levels remain poorly understood. This represents a significant limitation of the extant 1HMRS data to date. While changes in Glu metabolites appear related to the course of psychosis (Merritt et al., 2016), the presence and magnitude of these changes likely depend on stage of illness and pharmacological treatment status. A comprehensive meta-analysis of pharmacological treatment-associated changes in 1HMRS signals found that, across a number of psychotropic classes (antipsychotics and others), pharmacotherapy was linked to a significant decrease in frontal Glx levels in both first episode and chronic PSY patients (Kubota et al., 2020), with a more pronounced decline in first episode (effect size = −0.57) as compared to chronic PSY (effect size = −0.33). This meta-analytic pattern of medication-related neurometabolite reductions has also been observed in individual studies of unmedicated PSY patients and CHR individuals who transitioned to PSY. More specifically, antipsychotic treatment in these groups has been shown to result in lower levels of both Glu and GABA in the brain (de la Fuente-Sandoval et al., 2013b; Fuente-Sandoval et al., 2018), although these findings could be confounded by factors such as illness severity. Altogether, the literature offers evidence that reductions in Glu-related neurometabolites in PSY may be driven, influenced, and/or confounded by treatment with antipsychotics, however the nature and timing of treatment-metabolite relationships have not been fully elucidated, and analyses related to specific antipsychotic medication types are not available. Ultimately, more comprehensive data is needed to disambiguate endogenous disease-related alterations from treatment-induced effects (including effects during initial phases of treatment and after long-term use). This disambiguation will facilitate more direct interpretations of 1HMRS findings in PSY such as those reviewed in this meta-analysis.
4.3. Challenges and Complementary Approaches to UHF Neurochemical Imaging
1HMRS is a tremendously valuable tool for the in vivo study of brain neurochemicals. However, there are inherent methodological limitations, many which are more prominent at 3T. A primary limitation is the sensitivity of 1HMRS, given that the concentration of neurochemicals of interest is very low compared to the concentration of water in the brain. This can make neurometabolite detection difficult, necessitating long acquisitions and many averages to achieve sufficient SNR. An additional limitation comes from spectral overlap of signals from nuclei in different molecules, for example overlap of Glu, Gln, and GABA resonances. Ultra-high field 7T 1HMRS offers both improved sensitivity and specificity for quantifying brain neurochemistry (Choi et al., 2010), especially when shorter TEs are used, which may contribute to the significant effects we report in this meta-analysis across only a few studies. In fact, a direct comparison of brain metabolite quantification at 3T and 7T found that precision was higher for 12 brain metabolites using the 7T spectra (Mekle et al., 2009), despite a reduction in scan time by a factor of two. For Glu specifically, this increase in precision at 7T involves a 50% improvement in signal variability, and increased reliability for measuring coupled metabolites (e.g. Gln and Glu) (Terpstra et al., 2016). Continued implementation of 7T 1HMRS at diverse developmental and disease stages will thus likely result in a refined understanding of neurochemical alterations in attenuated psychosis syndrome and PSY.
Despite the advantages of 7T, there are still limitations of 1HMRS at UHF. Limitations include partial volume effects, susceptibility artifacts, persisting spectral overlap, and limited spatial coverage, given that most 1HMRS acquisitions are single-voxel based, with voxel sizes between 8–25 cm3 used in most 7T human studies (Table 1). Fortunately, a number of these limitations are overcome by Chemical Exchange Saturation Transfer (CEST) imaging techniques, which are complimentary approaches available only at 7T. By exploiting saturation exchange properties intrinsic to certain metabolites, CEST allows for metabolite detection with at least two orders of magnitude increased sensitivity as compared to 1HMRS (Cai et al., 2013, 2012). Specifically, CEST imaging uses water pool-dependent contrasts produced by endogenous compounds containing exchangeable protons (e.g. -OH, -NH2, -NH groups) (Henkelman et al., 2001; Kogan et al., 2013; Sherry and Woods, 2008; van Zijl and Yadav, 2011; Zhou and van Zijl, 2006), thus CEST is not as constrained by the concentration limitations of 1HMRS. Moreover, CEST is based on magnetization-transfer imaging sequences, and can therefore benefit from well-known fast acquisition strategies while both offering improved spatial coverage and resolution and facilitating multi-modal imaging analyses. To our knowledge, only glutamate CEST (GluCEST) has been applied to the study of PSY, revealing in vivo Glu hypofunction in youth on the PSY spectrum, including both PSY and CHR patients (Roalf et al., 2017). However CEST techniques capable of measuring creatine, myo-inositol (Haris et al., 2011), glutamine, and GABA (Lee et al., 2016) have been developed (Oh et al., 2017), and may provide new perspectives on PSY and PSY risk. Still, whether implementing 1HMRS or CEST techniques, multiple technical and practical challenges remain at 7T that must be considered, which have been comprehensively reviewed (Godlewska et al., 2017).
4.4. Implications for the Study of Clinical High Risk for Psychosis
There is an ample literature examining attenuated PSY and predictors of conversion from CHR to PSY (Lieberman et al., 2019). Indeed, individualized PSY risk calculators using data obtainable in clinical settings have demonstrated utility for predicting risk of conversion in CHR populations (Cannon et al., 2016; Carrión et al., 2016; Ciarleglio et al., 2019). Nonetheless, as supported by published risk calculators as well as by a recent meta-analysis of over 17,000 CHR individuals (Oliver et al., 2020), the most reliable non-biological predictors of PSY onset appear to be currently limited to more severe positive and negative psychotic symptoms and poorer neurocognitive and global functioning (Cannon et al., 2016; Oliver et al., 2020), i.e., attenuated symptoms of PSY itself. We believe that this underscores the need to identify sensitive, specific biological predictors that mediate the association between attenuated symptoms in CHR and transition to PSY, and that can thereby improve our understanding of what constitutes a high-risk state. This idea is supported by the fact that models predicting CHR outcomes are improved when neuroimaging and blood-derived measures are included in addition to clinical symptoms (Chan et al., 2015; Gschwandtner et al., 2009; Koutsouleris et al., 2018). We propose that 7T neurometabolite imaging represents one critical avenue for future investigation into mediators of CHR transition risk, given that these imaging techniques 1) produce quantifiable metrics that can be incorporated into risk calculators, and 2) inherently investigate druggable chemicals that could be targeted by early interventions. Yet, there is currently little available 7T 1HMRS neurometabolite data in CHR individuals, and CHR findings at 3T are even less consistent than those reported in PSY (Merritt et al., 2016; Treen et al., 2016; Wenneberg et al., 2019). Studies have found lower (Allen et al., 2015; Egerton et al., 2014; Keshavan et al., 2009; Uhl et al., 2011), higher (de la Fuente-Sandoval et al., 2013, 2011; Fusar-Poli et al., 2012; Stone et al., 2009; Tandon et al., 2013; Tibbo et al., 2004), and no difference (Natsubori et al., 2014; Purdon et al., 2008, p. 2; Yoo et al., 2009) in Glu (or Glu, Gln, Glx) concentrations between controls and CHR or genetic high risk individuals. It is unclear whether this is due to neurobiological factors (e.g., illness stage or disease heterogeneity), methodological factors (e.g. sensitivity, specificity and SNR limitations), or both. Intriguingly, a number of studies have found that Glu levels, particularly within the striatum, are associated with the transition to PSY (Allen et al., 2015; de la Fuente-Sandoval et al., 2013a; Egerton et al., 2014; Uhl et al., 2011). Additional studies are clearly needed that study Glu and Glu-related metabolites in CHR individuals.
Disruptions in Glu, GSH, and other neurochemicals are undoubtedly only one component of the complex, multi-faceted, and heterogeneous syndrome of PSY. Future efforts should aim to 1) uncover how brain neurochemistry interacts with structural and functional brain changes to increase risk of developing PSY, and to 2) integrate the study of neurochemicals with other genetic, biological, and clinical factors when evaluating Attenuated Psychosis Syndrome and CHR transition risk. As such, comprehensive, multimodal UHF neuroimaging approaches that include measures of neurochemistry are recommended. As 7T MRI becomes more common and is utilized within at-risk populations, the field should consider establishing UHF consortia with standardized acquisition and analysis guidelines, similar to the European Ultrahigh-Field Imaging Network for Neurodegenerative Diseases (EUFIND) (Düzel et al., 2019). This will be a vital step toward reducing methodological heterogeneity in 1HMRS acquisition protocols and improving our understanding of neurochemical disruptions in PSY and attenuated PSY syndrome.
Supplementary Material
Acknowledgements:
We thank Dr. Paul Moberg for his advice and assistance with implementation of the meta-analytic methods. His advice on this project was invaluable.
Funding Sources: This work was supported by the National Institute of Mental Health grants to R01MH119185 DRR & PJM, R01120174 (DRR) and the Life Span Brain Institute (LiBI)—a collaboration between the University of Pennsylvania School of Medicine and Children’s Hospital of Philadelphia. This work was also supported by a NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation (DRR). The funding sources were not directly involved in study design, collection, data analysis or interpretation, nor manuscript writing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
Valerie J. Sydnor, BA reports no potential conflicts of interest or financial disclosures related to this work.
David R. Roalf, PhD reports no potential conflicts of interest or financial disclosures related to this work.
Data Statement
Data for this meta-analysis was extracted from published literature. In addition, we provide a file containing the data we extracted for use in this meta-analysis in our Supplemental Material.
References
- Allen P, Chaddock CA, Egerton A, Howes OD, Barker G, Bonoldi I, Fusar-Poli P, Murray R, McGuire P, 2015. Functional outcome in people at high risk for psychosis predicted by thalamic glutamate levels and prefronto-striatal activation. Schizophrenia Bulletin 41, 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baiano M, David A, Versace A, Churchill R, Balestrieri M, Brambilla P, 2007. Anterior cingulate volumes in schizophrenia: a systematic review and a meta-analysis of MRI studies. Schizophrenia research 93, 1. [DOI] [PubMed] [Google Scholar]
- Balla A, Koneru R, Smiley J, Sershen H, Javitt DC, 2001. Continuous phencyclidine treatment induces schizophrenia-like hyperreactivity of striatal dopamine release. Neuropsychopharmacology 25, 157–164. [DOI] [PubMed] [Google Scholar]
- Begg CB, Mazumdar M, 1994. Operating characteristics of a rank correlation test for publication bias. Biometrics 50, 1088–1101. [PubMed] [Google Scholar]
- Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH, 2007. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 32, 1888–1902. [DOI] [PubMed] [Google Scholar]
- Bora E, Fornito A, Radua J, Walterfang M, Seal M, Wood SJ, Yücel M, Velakoulis D, Pantelis C, 2011. Neuroanatomical abnormalities in schizophrenia: a multimodal voxelwise meta-analysis and meta-regression analysis. Schizophrenia research 127, 46–57. [DOI] [PubMed] [Google Scholar]
- Bossong MG, Niesink RJ, 2010. Adolescent brain maturation, the endogenous cannabinoid system and the neurobiology of cannabis-induced schizophrenia. Progress in neurobiology 92, 370–385. [DOI] [PubMed] [Google Scholar]
- Brandt AS, Unschuld PG, Pradhan S, Lim IAL, Churchill G, Harris AD, Hua J, Barker PB, Ross CA, van Zijl PC, 2016. Age-related changes in anterior cingulate cortex glutamate in schizophrenia: a 1H MRS Study at 7 Tesla. Schizophrenia research 172, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai K, Haris M, Singh A, Kogan F, Greenberg JH, Hariharan H, Detre JA, Reddy R, 2012. Magnetic resonance imaging of glutamate. Nat. Med 18, 302–306. 10.1038/nm.2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai K, Singh A, Roalf DR, Nanga RPR, Haris M, Hariharan H, Gur R, Reddy R, 2013. Mapping glutamate in subcortical brain structures using high-resolution GluCEST MRI. NMR Biomed 26, 1278–1284. 10.1002/nbm.2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon TD, Yu C, Addington J, Bearden CE, Cadenhead KS, Cornblatt BA, Heinssen R, Jeffries CD, Mathalon DH, McGlashan TH, Perkins DO, Seidman LJ, Tsuang MT, Walker EF, Woods SW, Kattan MW, 2016. An Individualized Risk Calculator for Research in Prodromal Psychosis. AJP 173, 980–988. 10.1176/appi.ajp.2016.15070890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrión RE, Cornblatt BA, Burton CZ, Tso IF, Auther AM, Adelsheim S, Calkins R, Carter CS, Niendam T, Sale TG, Taylor SF, McFarlane WR, 2016. Personalized Prediction of Psychosis: External Validation of the NAPLS-2 Psychosis Risk Calculator With the EDIPPP Project. Am J Psychiatry 173, 989–996. 10.1176/appi.ajp.2016.15121565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey BJ, Oliveri ME, Insel T, 2014. A neurodevelopmental perspective on the research domain criteria (RDoC) framework. Biological psychiatry 76, 350–353. [DOI] [PubMed] [Google Scholar]
- Chan MK, Krebs M-O, Cox D, Guest PC, Yolken RH, Rahmoune H, Rothermundt M, Steiner J, Leweke FM, van Beveren NJM, Niebuhr DW, Weber NS, Cowan DN, Suarez-Pinilla P, Crespo-Facorro B, Mam-Lam-Fook C, Bourgin J, Wenstrup RJ, Kaldate RR, Cooper JD, Bahn S, 2015. Development of a blood-based molecular biomarker test for identification of schizophrenia before disease onset. Transl Psychiatry 5, e601 10.1038/tp.2015.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C, Dimitrov IE, Douglas D, Patel A, Kaiser LG, Amezcua CA, Maher EA, 2010. Improvement of resolution for brain coupled metabolites by optimized (1)H MRS at 7T. NMR in Biomedicine 23, 1044–52. 10.1002/nbm.1529 [doi] [DOI] [PubMed] [Google Scholar]
- Chung Y, Cannon TD, 2015. Brain imaging during the transition from psychosis prodrome to schizophrenia. The Journal of nervous and mental disease 203, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarleglio AJ, Brucato G, Masucci MD, Altschuler R, Colibazzi T, Corcoran CM, Crump FM, Horga G, Lehembre-Shiah E, Leong W, Schobel SA, Wall MM, Yang LH, Lieberman JA, Girgis RR, 2019. A predictive model for conversion to psychosis in clinical high-risk patients. Psychol Med 49, 1128–1137. 10.1017/S003329171800171X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J, 1988. Statistical Power Analysis for the Behavioral Sciences, 2nd ed. Routledge, New York, New York. [Google Scholar]
- Coyle JT, 2006. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cellular and molecular neurobiology 26, 363–382. [DOI] [PubMed] [Google Scholar]
- Curtis DR, Johnston GA, 1974. Amino acid transmitters in the mammalian central nervous system, in: Ergebnisse Der Physiologie Reviews of Physiology, Volume 69 Springer, pp. 97–188. [DOI] [PubMed] [Google Scholar]
- Das TK, Javadzadeh A, Dey A, Sabesan P, Théberge J, Radua J, Palaniyappan L, 2019. Antioxidant defense in schizophrenia and bipolar disorder: A meta-analysis of MRS studies of anterior cingulate glutathione. Progress in Neuro-Psychopharmacology and Biological Psychiatry 91, 94–102. [DOI] [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, León-Ortiz P, Azcárraga M, Favila R, Stephano S, Graff-Guerrero A, 2013a. Striatal glutamate and the conversion to psychosis: a prospective 1 H-MRS imaging study. The International Journal of Neuropsychopharmacology 16, 471–475. [DOI] [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, León-Ortiz P, Azcárraga M, Stephano S, Favila R, Díaz-Galvis L, Alvarado-Alanis P, Ramírez-Bermúdez J, Graff-Guerrero A, 2013b. Glutamate Levels in the Associative Striatum Before and After 4 Weeks of Antipsychotic Treatment in First-Episode Psychosis. JAMA Psychiatry 70, 1057–1066. 10.1001/jamapsychiatry.2013.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, Leon-Ortiz P, Favila R, Stephano S, Mamo D, Ramirez-Bermudez J, Graff-Guerrero A, 2011. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacology 36, 1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M, 2009. Redox dysregulation, neurodevelopment, and schizophrenia. Current opinion in neurobiology 19, 220–230. [DOI] [PubMed] [Google Scholar]
- D’Souza DC, Perry E, MacDougall L, Ammerman Y, Cooper T, Yu-te W, Braley G, Gueorguieva R, Krystal JH, 2004. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: implications for psychosis. Neuropsychopharmacology 29, 1558. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Moy SS, Perez A, Eddy DM, Zinzow WM, Lieberman JA, Snouwaert JN, Koller BH, 2004. Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behavioural brain research 153, 507–519. [DOI] [PubMed] [Google Scholar]
- Duval S, Tweedie R, 2000. Trim and fill: A simple funnel-plot-based method of testing and adjusting for publication bias in meta-analysis. Biometrics 56, 455–463. 10.1111/j.0006-341x.2000.00455.x [DOI] [PubMed] [Google Scholar]
- Düzel E, Acosta-Cabronero J, Berron D, Biessels GJ, Björkman-Burtscher I, Bottlaender M, Bowtell R, Buchem M. v, Cardenas-Blanco A, Boumezbeur F, 2019. European Ultrahigh-Field Imaging Network for Neurodegenerative Diseases (EUFIND). Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 11, 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerton A, Stone JM, Chaddock CA, Barker GJ, Bonoldi I, Howard RM, Merritt K, Allen P, Howes OD, Murray RM, McLean MA, Lythgoe DJ, O’Gorman RL, McGuire PK, 2014. Relationship between brain glutamate levels and clinical outcome in individuals at ultra high risk of psychosis. Neuropsychopharmacology 39, 2891–2899. 10.1038/npp.2014.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger M, Davey Smith G, Schneider M, Minder C, 1997. Bias in meta-analysis detected by a simple, graphical test. BMJ 315, 629–634. 10.1136/bmj.315.7109.629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erecińska M, Zaleska MM, Nelson D, Nissim I, Yudkoff M, 1990. Neuronal glutamine utilization: glutamine/glutamate homeostasis in synaptosomes. Journal of neurochemistry 54, 2057–2069. [DOI] [PubMed] [Google Scholar]
- Frohlich J, Van Horn JD, 2014. Reviewing the ketamine model for schizophrenia. Journal of psychopharmacology 28, 287–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuente-Sandoval C. de la, Reyes-Madrigal F, Mao X, León-Ortiz P, Rodríguez-Mayoral O, Jung-Cook H, Solís-Vivanco R, Graff-Guerrero A, Shungu DC, 2018. Prefrontal and Striatal Gamma-Aminobutyric Acid Levels and the Effect of Antipsychotic Treatment in First-Episode Psychosis Patients. Biological Psychiatry 83, 475–483. 10.1016/j.biopsych.2017.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusar-Poli P, Bonoldi I, Yung AR, Borgwardt S, Kempton MJ, Valmaggia L, Barale F, Caverzasi E, McGuire P, 2012. Predicting psychosis: meta-analysis of transition outcomes in individuals at high clinical risk. Archives of general psychiatry 69, 220. [DOI] [PubMed] [Google Scholar]
- Fusar-Poli P, Cappucciati M, Borgwardt S, Woods SW, Addington J, Nelson B, Nieman DH, Stahl DR, Rutigliano G, Riecher-Rössler A, Simon AE, Mizuno M, Lee TY, Kwon JS, Lam MML, Perez J, Keri S, Amminger P, Metzler S, Kawohl W, Rössler W, Lee J, Labad J, Ziermans T, An SK, Liu C-C, Woodberry KA, Braham A, Corcoran C, McGorry P, Yung AR, McGuire PK, 2016. Heterogeneity of Psychosis Risk Within Individuals at Clinical High Risk: A Meta-analytical Stratification. JAMA Psychiatry 73, 113–120. 10.1001/jamapsychiatry.2015.2324 [DOI] [PubMed] [Google Scholar]
- Gawryluk JW, Wang J-F, Andreazza AC, Shao L, Young LT, 2011. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. International Journal of Neuropsychopharmacology 14, 123–130. [DOI] [PubMed] [Google Scholar]
- Godlewska BR, Clare S, Cowen PJ, Emir UE, 2017. Ultra-high-field magnetic resonance spectroscopy in psychiatry. Frontiers in psychiatry 8, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Coyle JT, 2001. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 158, 1367–1377. 10.1176/appi.ajp.158.9.1367 [DOI] [PubMed] [Google Scholar]
- Gschwandtner U, Pflueger MO, Semenin V, Gaggiotti M, Riecher-Rössler A, Fuhr P, 2009. EEG: a helpful tool in the prediction of psychosis. Eur Arch Psychiatry Clin Neurosci 259, 257–262. 10.1007/s00406-008-0854-3 [DOI] [PubMed] [Google Scholar]
- Gur RE, Turetsky BI, Cowell PE, Finkelman C, Maany V, Grossman RI, Arnold SE, Bilker WB, Gur RC, 2000. Temporolimbic volume reductions in schizophrenia. Archives of general psychiatry 57, 769–75. https://doi.org/yoa9447b [pii] [DOI] [PubMed] [Google Scholar]
- Haris M, Cai K, Singh A, Hariharan H, Reddy R, 2011. In vivo Mapping of Brain Myo-Inositol. Neuroimage 54, 2079–2085. 10.1016/j.neuroimage.2010.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR, 2005. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 10, 40–68; image 5. 10.1038/sj.mp.4001558 [DOI] [PubMed] [Google Scholar]
- Hedges LV, Olkin I, 1985. Statistical Methods for Meta-Analysis. Harcourt Brace Jovanovich Publishers. [Google Scholar]
- Henkelman RM, Stanisz GJ, Graham SJ, 2001. Magnetization transfer in MRI: a review. NMR in Biomedicine 14, 57–64. [DOI] [PubMed] [Google Scholar]
- Howes OD, Kapur S, 2009. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophrenia Bulletin 35, 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, MacDonald ML, Elswick DE, Sweet RA, 2015. The glutamate hypothesis of schizophrenia: evidence from human brain tissue studies. Annals of the New York Academy of Sciences 1338, 38–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR, 2010. Rethinking schizophrenia. Nature 468, 187–193. 10.1038/nature09552 [DOI] [PubMed] [Google Scholar]
- Javitt DC, 2004. Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry 9, 984–997, 979. 10.1038/sj.mp.4001551 [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Redmond DE, Elsworth JD, Taylor JR, Youngren KD, Roth RH, 1997. Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science 277, 953–955. [DOI] [PubMed] [Google Scholar]
- Kempton MJ, Bonoldi I, Valmaggia L, McGuire P, Fusar-Poli P, 2015. Speed of Psychosis Progression in People at Ultra-High Clinical Risk: A Complementary Meta-analysis. JAMA Psychiatry 72, 622–623. 10.1001/jamapsychiatry.2015.0094 [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Dick RM, Diwadkar VA, Montrose DM, Prasad KM, Stanley JA, 2009. Striatal metabolic alterations in non-psychotic adolescent offspring at risk for schizophrenia: a 1 H spectroscopy study. Schizophrenia research 115, 88–93. [DOI] [PubMed] [Google Scholar]
- Kogan F, Singh A, Debrosse C, Haris M, Cai K, Nanga RP, Elliott M, Hariharan H, Reddy R, 2013. Imaging of glutamate in the spinal cord using GluCEST. NeuroImage 77, 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsouleris N, Kambeitz-Ilankovic L, Ruhrmann S, Rosen M, Ruef A, Dwyer DB, Paolini M, Chisholm K, Kambeitz J, Haidl T, Schmidt A, Gillam J, Schultze-Lutter F, Falkai P, Reiser M, Riecher-Rössler A, Upthegrove R, Hietala J, Salokangas RKR, Pantelis C, Meisenzahl E, Wood SJ, Beque D, Brambilla P, Borgwardt S, PRONIA Consortium, 2018. Prediction Models of Functional Outcomes for Individuals in the Clinical High-Risk State for Psychosis or With Recent-Onset Depression: A Multimodal, Multisite Machine Learning Analysis. JAMA Psychiatry 75, 1156–1172. 10.1001/jamapsychiatry.2018.2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraguljac NV, Frölich MA, Tran S, White DM, Nichols N, Barton-McArdle A, Reid MA, Bolding MS, Lahti AC, 2017. Ketamine modulates hippocampal neurochemistry and functional connectivity: a combined magnetic resonance spectroscopy and resting-state fMRI study in healthy volunteers. Molecular psychiatry 22, 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Charney DS, 1994. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Archives of general psychiatry 51, 199–214. [DOI] [PubMed] [Google Scholar]
- Kubota M, Moriguchi S, Takahata K, Nakajima S, Horita N, 2020. Treatment effects on neurometabolite levels in schizophrenia: A systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Schizophr. Res 10.1016/j.schres.2020.03.069 [DOI] [PubMed] [Google Scholar]
- Kumar J, Liddle EB, Fernandes CC, Palaniyappan L, Hall EL, Robson SE, Simmonite M, Fiesal J, Katshu MZ, Qureshi A, 2018. Glutathione and glutamate in schizophrenia: a 7T MRS study. Molecular psychiatry 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M, Kegeles LS, Abi-Dargham A, 2006. Glutamate, dopamine, and schizophrenia. Annals of the New York Academy of Sciences 1003, 138–158. [DOI] [PubMed] [Google Scholar]
- Lee J-S, Xia D, Jerschow A, Regatte RR, 2016. In vitro study of endogenous CEST agents at 3 T and 7 T. Contrast media & molecular imaging 11, 4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Moghaddam B, 2006. Cognitive Dysfunction in Schizophrenia: Convergence of {gamma}-Aminobutyric Acid and Glutamate Alterations. Archives of Neurology 63, 1372. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Small SA, Girgis RR, 2019. Early Detection and Preventive Intervention in Schizophrenia: From Fantasy to Reality. AJP 176, 794–810. 10.1176/appi.ajp.2019.19080865 [DOI] [PubMed] [Google Scholar]
- Marsman A, Mandl RC, Klomp DW, Bohlken MM, Boer VO, Andreychenko A, Cahn W, Kahn RS, Luijten PR, Pol HEH, 2014. GABA and glutamate in schizophrenia: A 7 T 1H-MRS study. NeuroImage: Clinical 6, 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman A, van den Heuvel MP, Klomp DWJ, Kahn RS, Luijten PR, Pol HEH, 2013. Glutamate in schizophrenia: a focused review and meta-analysis of 1H-MRS studies. Schizophrenia Bulletin 39, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEntee WJ, Crook TH, 1993. Glutamate: its role in learning, memory, and the aging brain. Psychopharmacology 111, 391–401. [DOI] [PubMed] [Google Scholar]
- McGlashan TH, Hoffman RE, 2000. Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Archives of general psychiatry 57, 637–648. [DOI] [PubMed] [Google Scholar]
- Mekle R, Mlynárik V, Gambarota G, Hergt M, Krueger G, Gruetter R, 2009. MR spectroscopy of the human brain with enhanced signal intensity at ultrashort echo times on a clinical platform at 3T and 7T. Magnetic Resonance in Medicine: An Official Journal of the International Society for Magnetic Resonance in Medicine 61, 1279–1285. [DOI] [PubMed] [Google Scholar]
- Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK, 2016. Nature of Glutamate Alterations in Schizophrenia: A Meta-analysis of Proton Magnetic Resonance Spectroscopy Studies. JAMA Psychiatry 73, 665–674. 10.1001/jamapsychiatry.2016.0442 [DOI] [PubMed] [Google Scholar]
- Moghaddam B, 2004. Targeting metabotropic glutamate receptors for treatment of the cognitive symptoms of schizophrenia. Psychopharmacology 174, 39–44. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, 2003. Bringing order to the glutamate chaos in schizophrenia. Neuron 40, 881–884. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Javitt D, 2012. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 37, 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH, 1999. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98, 427–436. [DOI] [PubMed] [Google Scholar]
- Natsubori T, Inoue H, Abe O, Takano Y, Iwashiro N, Aoki Y, Koike S, Yahata N, Katsura M, Gonoi W, 2014. Reduced frontal glutamate+ glutamine and N-acetylaspartate levels in patients with chronic schizophrenia but not in those at clinical high risk for psychosis or with first-episode schizophrenia. Schizophrenia Bulletin 40, 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson B, Yuen HP, Wood SJ, Lin A, Spiliotacopoulos D, Bruxner A, Broussard C, Simmons M, Foley DL, Brewer WJ, Francey SM, Amminger GP, Thompson A, McGorry PD, Yung AR, 2013. Long-term Follow-up of a Group at Ultra High Risk (“Prodromal”) for Psychosis: The PACE 400 Study. JAMA Psychiatry 70, 793–802. 10.1001/jamapsychiatry.2013.1270 [DOI] [PubMed] [Google Scholar]
- Oh J-H, Kim H-G, Woo D-C, Jeong H-K, Lee SY, Jahng G-H, 2017. Chemical-exchange-saturation-transfer magnetic resonance imaging to map gamma-aminobutyric acid, glutamate, myoinositol, glycine, and asparagine: Phantom experiments. Journal of the Korean Physical Society 70, 545–553. 10.3938/jkps.70.545 [DOI] [Google Scholar]
- Oliver D, Reilly TJ, Baccaredda Boy O, Petros N, Davies C, Borgwardt S, McGuire P, Fusar-Poli P, 2020. What causes the onset of psychosis in individuals at clinical high risk? A meta-analysis of risk and protective factors. Schizophrenia Bulletin 46, 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Farber NB, 1995. Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry 52, 998–1007. 10.1001/archpsyc.1995.03950240016004 [DOI] [PubMed] [Google Scholar]
- Overbeek G, Gawne TJ, Reid MA, Salibi N, Kraguljac NV, White DM, Lahti AC, 2019. Relationship Between Cortical Excitation and Inhibition and Task-Induced Activation and Deactivation: A Combined Magnetic Resonance Spectroscopy and Functional Magnetic Resonance Imaging Study at 7T in First-Episode Psychosis. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging 4, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomino A, González-Pinto A, Aldama A, González-Gómez C, Mosquera F, González-García G, Matute C, 2007. Decreased levels of plasma glutamate in patients with first-episode schizophrenia and bipolar disorder. Schizophrenia research 95, 174–178. [DOI] [PubMed] [Google Scholar]
- Poels EMP, Kegeles LS, Kantrowitz JT, Slifstein M, Javitt DC, Lieberman JA, Abi-Dargham A, Girgis RR, 2014. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Molecular psychiatry 19, 20–29. [DOI] [PubMed] [Google Scholar]
- Posporelis S, Coughlin JM, Marsman A, Pradhan S, Tanaka T, Wang H, Varvaris M, Ward R, Higgs C, Edwards JA, 2018. Decoupling of brain temperature and glutamate in recent onset of schizophrenia: a 7T proton magnetic resonance spectroscopy study. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging 3, 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdon SE, Valiakalayil A, Hanstock CC, Seres P, Tibbo P, 2008. Elevated 3T proton MRS glutamate levels associated with poor Continuous Performance Test (CPT-0X) scores and genetic risk for schizophrenia. Schizophrenia research 99, 218–24. https://doi.org/S0920-9964(07)00529-4 [pii] 10.1016/j.schres.2007.11.028 [doi] [DOI] [PubMed] [Google Scholar]
- Raffa M, Mechri A, Othman LB, Fendri C, Gaha L, Kerkeni A, 2009. Decreased glutathione levels and antioxidant enzyme activities in untreated and treated schizophrenic patients. Progress in Neuro-Psychopharmacology and Biological Psychiatry 33, 1178–1183. [DOI] [PubMed] [Google Scholar]
- Rapoport J, Giedd J, Gogtay N, 2012. Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry 17, 1228–1238. 10.1038/mp.2012.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MA, Salibi N, White DM, Gawne TJ, Denney TS, Lahti AC, 2018. 7T proton magnetic resonance spectroscopy of the anterior cingulate cortex in first-episode schizophrenia. Schizophrenia Bulletin 45, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remington G, Agid O, Foussias G, 2011. Schizophrenia as a disorder of too little dopamine: implications for symptoms and treatment. Expert Review of Neurotherapeutics 11, 589–607. 10.1586/ern.10.191 [doi] [DOI] [PubMed] [Google Scholar]
- Roalf DR, Nanga RPR, Rupert PE, Hariharan H, Quarmley M, Calkins ME, Dress E, Prabhakaran K, Elliott MA, Moberg PJ, Gur RC, Gur RE, Reddy R, Turetsky BI, 2017. Glutamate imaging (GluCEST) reveals lower brain GluCEST contrast in patients on the psychosis spectrum. Mol. Psychiatry 22, 1298–1305. 10.1038/mp.2016.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW, Murphy ER, 2006. Behavioural pharmacology: 40+ years of progress, with a focus on glutamate receptors and cognition. Trends in pharmacological sciences 27, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LM, Pradhan S, Korenic S, Wijtenburg SA, Hong LE, Edden RA, Barker PB, 2016. Elevated brain lactate in schizophrenia: a 7 T magnetic resonance spectroscopy study. Translational psychiatry 6, e967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio MD, Wood K, Haroutunian V, Meador-Woodruff JH, 2013. Dysfunction of the ubiquitin proteasome and ubiquitin-like systems in schizophrenia. Neuropsychopharmacology 38, 1910–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterthwaite TD, Wolf DH, Calkins ME, Vandekar SN, Erus G, Ruparel K, Roalf DR, Linn KA, Elliott MA, Moore TM, 2016. Structural brain abnormalities in youth with psychosis spectrum symptoms. JAMA psychiatry 73, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selemon LD, Goldman-Rakic PS, 1999. The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biological psychiatry 45, 17–25. [DOI] [PubMed] [Google Scholar]
- Sherry AD, Woods M, 2008. Chemical exchange saturation transfer contrast agents for magnetic resonance imaging. Annual review of biomedical engineering 10, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Petursson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, Gunnarsdottir S, Ivarsson O, 2002. Neuregulin 1 and susceptibility to schizophrenia. The American Journal of Human Genetics 71, 877–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JM, Day F, Tsagaraki H, Valli I, McLean MA, Lythgoe DJ, O’Gorman RL, Barker GJ, McGuire PK, 2009. Glutamate dysfunction in people with prodromal symptoms of psychosis: relationship to gray matter volume. Biological psychiatry 66, 533–539. [DOI] [PubMed] [Google Scholar]
- Stroup DF, Berlin JA, Morton SC, Olkin I, Williamson GD, Rennie D, Moher D, Becker BJ, Sipe TA, Thacker SB, 2000. Meta-analysis of observational studies in epidemiology: a proposal for reporting. Jama 283, 2008–2012. [DOI] [PubMed] [Google Scholar]
- Tandon N, Bolo NR, Sanghavi K, Mathew IT, Francis AN, Stanley JA, Keshavan MS, 2013. Brain metabolite alterations in young adults at familial high risk for schizophrenia using proton magnetic resonance spectroscopy. Schizophr. Res 148, 59–66. 10.1016/j.schres.2013.05.024 [DOI] [PubMed] [Google Scholar]
- Taylor R, Neufeld RWJ, Schaefer B, Densmore M, Rajakumar N, Osuch EA, Williamson PC, Théberge J, 2015. Functional magnetic resonance spectroscopy of glutamate in schizophrenia and major depressive disorder: anterior cingulate activity during a color-word Stroop task. npj Schizophrenia 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terpstra M, Cheong I, Lyu T, Deelchand DK, Emir UE, Bednařík P, Eberly LE, Öz G, 2016. Test-retest reproducibility of neurochemical profiles with short-echo, single-voxel MR spectroscopy at 3T and 7T. Magn Reson Med 76, 1083–1091. 10.1002/mrm.26022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar KN, Rösler L, Wijnen JP, Boer VO, Klomp DW, Cahn W, Kahn RS, Neggers SF, 2017. 7T proton magnetic resonance spectroscopy of gamma-aminobutyric acid, glutamate, and glutamine reveals altered concentrations in patients with schizophrenia and healthy siblings. Biological psychiatry 81, 525–535. [DOI] [PubMed] [Google Scholar]
- Tibbo P, Hanstock C, Valiakalayil A, Allen P, 2004. 3-T proton MRS investigation of glutamate and glutamine in adolescents at high genetic risk for schizophrenia. American Journal of Psychiatry 161, 1116–1118. [DOI] [PubMed] [Google Scholar]
- Treen D, Batlle S, Mollà L, Forcadell E, Chamorro J, Bulbena A, Perez V, 2016. Are there glutamate abnormalities in subjects at high risk mental state for psychosis? A review of the evidence. Schizophr. Res 171, 166–175. 10.1016/j.schres.2016.01.005 [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT, 2002. Glutamatergic mechanisms in schizophrenia. Annual review of pharmacology and toxicology 42, 165–179. [DOI] [PubMed] [Google Scholar]
- Uhl I, Mavrogiorgou P, Norra C, Forstreuter F, Scheel M, Witthaus H, Ozgurdal S, Gudlowski Y, Bohner G, Gallinat J, 2011. 1H-MR spectroscopy in ultra-high risk and first episode stages of schizophrenia. Journal of psychiatric research 45, 1135–1139. [DOI] [PubMed] [Google Scholar]
- van Zijl P, Yadav NN, 2011. Chemical exchange saturation transfer (CEST): what is in a name and what isn’t? Magnetic resonance in medicine 65, 927–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AM, Pradhan S, Coughlin JM, Trivedi A, DuBois SL, Crawford JL, Sedlak TW, Nucifora FC, Nestadt G, Nucifora LG, 2019. Assessing brain metabolism with 7-T proton magnetic resonance spectroscopy in patients with first-episode psychosis. JAMA psychiatry 76, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenneberg C, Glenthøj BY, Hjorthøj C, Zingenberg FJB, Glenthøj LB, Rostrup E, Broberg BV, Nordentoft M, 2019. Cerebral glutamate and GABA levels in high-risk of psychosis states: A focused review and meta-analysis of 1H-MRS studies. Schizophrenia research. [Google Scholar]
- Wu EQ, Birnbaum HG, Shi L, Ball DE, Kessler RC, Moulis M, Aggarwal J, 2005. The economic burden of schizophrenia in the United States in 2002. Journal of Clinical Psychiatry. [DOI] [PubMed] [Google Scholar]
- Yao JK, Leonard S, Reddy R, 2006. Altered glutathione redox state in schizophrenia. Disease markers 22, 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SY, Yeon S, Choi C-H, Kang D-H, Lee J-M, Shin NY, Jung WH, Choi J-S, Jang D-P, Kwon JS, 2009. Proton magnetic resonance spectroscopy in subjects with high genetic risk of schizophrenia: investigation of anterior cingulate, dorsolateral prefrontal cortex and thalamus. Schizophrenia research 111, 86–93. [DOI] [PubMed] [Google Scholar]
- Zeidler J, Slawik L, Fleischmann J, Greiner W, 2012. The costs of schizophrenia and predictors of hospitalisation from the statutory health insurance perspective. Health economics review 2, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, van Zijl PC, 2006. Chemical exchange saturation transfer imaging and spectroscopy. Progress in Nuclear Magnetic Resonance Spectroscopy 48, 109–136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.