

Abstract
Congenital hydrocephalus (CH) is caused by genetic mutations, but whether factors impacting human genetic mutations are disease-specific remains elusive. Given two factors associated with high mutation rates, we reviewed how many disease-susceptible genes match with (i) proximity to telomeres or (ii) high adenine and thymine (A + T) content in human CH as compared to other disorders of the central nervous system (CNS). We extracted genomic information using a genome data viewer. Importantly, 98 of 108 genes causing CH satisfied (i) or (ii), resulting in >90% matching rate. However, such a high accordance no longer sustained as we checked two factors in Alzheimer’s disease (AD) and/or familial Parkinson’s disease (fPD), resulting in 84% and 59% matching, respectively. A disease-specific matching of telomere proximity or high A + T content predicts causative genes of CH much better than neurodegenerative diseases and other CNS conditions, likely due to sufficient number of known causative genes (n = 108) and precise determination and classification of the genotype and phenotype. Our analysis suggests a need for identifying genetic basis of both factors before human clinical studies, to prioritize putative genes found in preclinical models into the likely (meeting at least one) and more likely candidate (meeting both), which predisposes human genes to mutations.
Keywords: Congenital hydrocephalus, Telomeres, A + T content, mutation, Chromosome, homologous recombination, familial Parkinson’s disease, Alzheimer’s disease
1. Introduction
Congenital hydrocephalus (CH) comprises one of the most common childhood brain disorders (Dewan et al., 2018) (~1 in 1000 worldwide births (Dewan et al., 2018; Isaacs et al., 2019)). CH, in which genetic variants are involved, may occur alone (non-syndromic) or as part of a syndrome with multiple anomalies (Zhang et al., 2006). If left untreated, hydrocephalus is fatal. The mainstay of treatments is surgical, through shunt insertion and a combination of endoscopic ventriculostomy and choroid plexus cauterization (Dewan and Naftel, 2017; Kulkarni et al., 2018; Vinchon et al., 2012). CH results in extremely poor neurological outcomes (McAllister 2nd, 2012) that are expensive to manage surgically (~$2 billion annually) with 29–15% failure rates (Lim et al., 2018; Ravindra et al., 2015; Wright et al., 2016).
Etiologically, CH is associated with developmental abnormalities such as genetic mutations linked to ciliopathies, neural tube defects, spina bifida, myelomeningocele, and exencephaly/anencephaly, while that with acquired form involves ischemia/hypoxia, hemorrhage, infection, trauma, and brain tumors. Collectively, all forms of hydrocephalus in children show hallmarks such as ventriculomegaly. The cause of this ventriculomegaly is claimed to be multi-factorial: excess CSF production, obstruction of CSF pathways, impaired CSF outflow, and/or imbalance of production and reabsorption at subarachnoid granulation (Ballabh, 2014; Bassan, 2009; Bateman, 2008; Bhattathiri et al., 2006; Garton et al., 2016; Kazan et al., 2005; Koschnitzky et al., 2018; Limbrick Jr. et al., 2010; Mbabazi-Kabachelor et al., 2019; McAllister 2nd, 2012; Murphy et al., 2002; Osterman et al., 2015; Perdaens et al., 2018; Radic et al., 2015; Rekate, 2007; Robinson, 2012; Romero et al., 2015; Shim et al., 2019; Shimada et al., 2019; Stoll et al., 2010; Zhang et al., 2006). Recent studies have identified that increases in the mutation rates of more than 100 distinct genes contribute directly to one or more of these causative factors, as well as the aforementioned developmental abnormalities, thus insinuating a strong correlation with the development of CH (Belal and Al Menabawy, 2017; de Paola et al., 2019; Fransen et al., 1995; Furey et al., 2018a; Furey et al., 2018b; Gable et al., 2019; Huh et al., 2009; Kousi and Katsanis, 2016; Rachel et al., 2015; Rowitch et al., 1999; Shim et al., 2019).
More specifically, a recent whole exome sequencing (WES) study suggests that CH results from impaired neurogenesis, leading to abnormal CSF drainage or obstruction of CSF flow (Furey et al., 2018b). One of the major neurodevelopmental pathways related with neurogenesis in CH is cilia/sonic hedgehog (SHH) signaling (Goetz and Anderson, 2010; Rohatgi et al., 2007). SHH is highly expressed in neural stem cells of the developing cortex, including the radial glia, intermediate progenitors, and outer radial astrocytes (Britto et al., 2002; Guemez-Gamboa et al., 2014; Han et al., 2008). The primary cilium, which is non-motile, is an essential organelle that mediates the SHH signaling. Using embryonic fibroblasts, it has been shown that when the ligand, SHH, binds to patched gene (PTCH) that blocks smoothened (SMO), SMO is released and translocates to cilia, which activates downstream signaling (Goetz and Anderson, 2010; Rohatgi et al., 2007). Mutations in a series of proteins that regulate the structure of cilia or SHH pathways have been implicated in CH as well. In a rat model of Meckel-Gruber Syndrome (MKS), mutated transmembrane protein 67 (TMEM67) resulted in CH with elongated primary cilia (Shim et al., 2019). The TMEM67 gene encodes for Meckelin, a protein that functions in centriole migration to the apical membrane and formation of the primary cilium (Abdelhamed et al., 2015). ZCCHC8, the zinc finger CCHC-type domain containing 8 protein, which is reported to interact with the TR component (a non-coding RNA) of telomerase, has been shown to cause CH as well (Gable et al., 2019). Interestingly, ZCCHC8−/− mice showed significant upregulations of transmembrane proteins (e.g. TMEM116) and cilia-regulating genes (e.g. ccdc33) (Gable et al., 2019).
However, despite these neuroanatomical and clinical findings, as well as attempts to treat CH surgically, there is no effective cure for this disease. Moreover, the process by which mutations in TMEM67 and ZCCHC8 have occurred in their chromosomes— affecting cilia and SHH pathways—is largely unknown. Thus, an attempt to elucidate the mechanism underlying these high mutation rates is vitally important, thereby providing valuable insight into the root cause for CH, as well as likely leading to a more accurate diagnosis. To this end, we tested how many causative genes of CH meet the following two criteria: i) proximity to a telomere and ii) high A + T content. These two factors have been associated with a high mutation rate—causing genetic diseases (Nusbaum et al., 2006) such as CH—as indicated previously. We further tested whether genes causing other genetic disorders of the CNS would meet (i) or (ii) at a rate comparable to CH, in order to determine if these criteria provide a clear indication of genes causing neurological disorders.
2. Materials and methods
2.1. Database and open access software
The literature survey was centered on CH and associated genes. We used PubMed database. During the literature survey, we also utilized the publicly open genome data viewer (https://www.ncbi.nlm.nih.gov/genome/gdv/), Pubmed database of nucleotide accession number (https://www.ncbi.nlm.nih.gov/nucleotide/) and GC content calculators to obtain A + T content ((www.endmemo.com/bio/gc.php) and/or (https://www.biologicscorp.com/tools/GCContent/)).
2.2. Data plot and statistical methods
Prism was used to plot a bar graph and box and violin plot of the data obtained during analysis with genome data viewer. Statistical analyses were performed using Prism (version 8, GraphPad Software Inc.). Normal distribution of the data was confirmed using Shapiro-Wilk normality test (α < 0.05). A two-sided unpaired t-test was used for comparison of two different groups, unless stated differently. Tukey’s multiple comparisons test following one-way analysis of variance was used for comparison of more than two groups. The difference between data sets was considered significant at P < 0.05; P values are identified in the figures and legends as *P < 0.05, **P < 0.01, ***P < 0.005.
3. Results
3.1. Most cases of CH belong to a type of neural tube defects
Our first question was how phenotypes of TMEM67 mutant rats (Shim et al., 2019), which were reported recently, might be linked to pediatric patients we studied previously (Shim et al., 2013). We reanalyzed the data (Shim et al., 2013) and sorted them based on the diagnosis. Interestingly, among seventy individual data points (31 controls & 35 children with hydrocephalus included; 4 babies with tumors excluded), more patients were diagnosed as non-hemorrhagic hydrocephalus as compared to PHH. Importantly, 40% of neonatal hydrocephalus at age ≤ 3 yr belongs to a broad classification of neural tube defects: spina bifida & hydrocephalus (23%), myelomeningocele & hydrocephalus (13%), and encephalocele & hydrocephalus (3%) (Fig. 1A). We then sorted the data by age (Fig. 1B). We found a significant number of patients belonged to ‘Not PHH’ such as CH, while ‘PHH’ occupied 26% of all patients. In the cohort of ‘age younger than 3 years’, encephalocele (Shim et al., 2019) was found in the ‘Not PHH’ group. This suggests a linkage to mutations in ciliopathy-related genes (Abdelhamed et al., 2015; Shim et al., 2019; Taulman et al., 2001). The aforementioned diagnosis by clinicians also highlighted the significance of CH associated with a form of spina bifida reported in Bardet Biedl syndrome (BBS) (Carter et al., 2012; Leitch et al., 2008; Smith et al., 2006) and our recent studies concerning TMEM67 defects (Shim et al., 2019; Smith et al., 2006). However, this diagnosis alone for pediatric patients was insufficient to uncover rationale of a high mutation rate in CH. Even in this single center study, we noticed that spina bifida (23%) was as frequent as PHH (26%) (Fig. 1A–B).
Fig. 1. Classification of pediatric hydrocephalus.
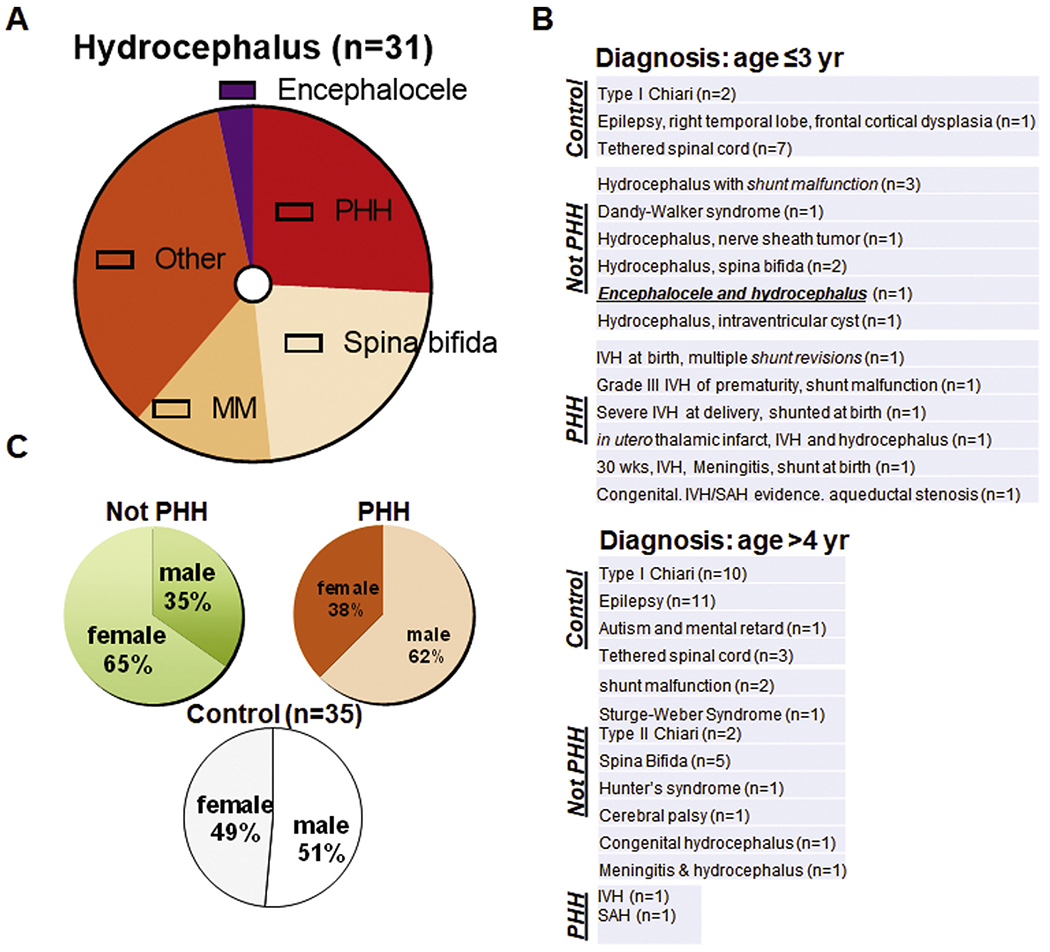
(A) Diagram summarizing sub-classification of hydrocephalus in children into post-hemorrhagic hydrocephalus (PHH) and not-PHH group involving diagnosis such as spina bifida (23%), myelomeningocele (MM; 13%), encephalocele (3%) and other conditions. Secondary causes such as hemorrhage leading to post-hemorrhagic hydrocephalus (PHH) accounted for fewer than one in three cases (26%). Collectively, 71% patients at age 0–26 years (n = 31) were diagnosed as hydrocephalus other than PHH (Not PHH). (B) Summary showing the gist of diagnosis for younger (upper) and older (lower) age group. (C) Sex was evenly distributed within control (n = 35) and hydrocephalus group (n = 31). However, when hydrocephalus group was sub-divided into two, female-male ratio became not even in Not PHH and PHH group (age 0–26 years). Single center study (Shim et al., 2013).
In all ages studied, male-female ratio was almost equal when sorted into control vs. hydrocephalus (Shim et al., 2013). As patients with hydrocephalus were further divided into PHH and ‘Not PHH’, however, there were more females in non-hemorrhagic hydrocephalus and fewer female patients in PHH (Fig. 1C). We then asked how abnormal neurogenesis was proposed as a cause of CH in recent literatures (Furey et al., 2018a; Rodriguez and Guerra, 2017). Previously (Shim et al., 2016), we reported that trajectories of doublecortin (DCX)-expressing young neurons in hydrocephalus were different in TMEM67−/− mutants as compared to WT during neonatal period (postnatal day 17, P17). A disorganized and widespread GFAP+ cells in the vicinity of radially and tangentially migrating neural progenitors in hydrocephalus is consistent with several previous studies by others (Doetsch and Alvarez-Buy 11a, 1996; Doetsch et al., 2002; Sawamoto et al., 2006; Shim et al., 2016) (Bothwell et al., 2019). In breeding animals, it has come to our attention that TMEM67−/− rats at P12 die in less than 10 days (~P21), even if many young neurons migrate postnatally in the presence of CH (Shim et al., 2019; Smith et al., 2006).
3.2. TMEM67 mutations detected in almost all exons of MKS and BBS patients with CH
It was not until recently that the spatial status of TMEM67 had been demonstrated in the ciliary zone close to basal body along with tectonicl, tectonic2, and Cc2d2a/MKS6, which collectively form a transition zone complex (Garcia-Gonzalo et al., 2011; Yang et al., 2015). In the DNA sequencing of five individuals with MKS, three types of mutations—namely, frameshift (exon 3 & exon6), splice site (exon 9 & exon 15), and missense mutation (exon 11)—were associated with neurological (encephalocele and Dandy Walker malformation), renal (polycystic kidney), and hepatic (fibrosis and ductal plate malformation) phenotypes (Smith et al., 2006). Using Wistar polycystic kidney (Wpk) rats, a missense mutation in exon 12 of TMEM67 has been shown, which results in a gene-dose dependent neurological and renal phenotype consistent with the aforementioned clinical report (Iannicelli et al., 2010). Specifically, hemorrhage in the dorsomedial subarachnoid space at birth, encephalocele-like protrusion, and late-onset hydrocephalus during adulthood were reported in TMEM67 heterozygous mutants, while the affected phenotype exhibited early-onset CH, polycystic kidney, and neonatal mortality before weaning age. This was observed with 100% penetrance of hydrocephalus in Wpk rats with TMEM67 homozygous mutant alleles (Shim et al., 2019). As pointed out recently, in comparison with twelve cases of MKS human fetuses in a wider ethnic background (United States, France, Italy, Algeria, and Morocco) than the prior report with limited ethnic identities (Oman and Pakistan) (Smith et al., 2006), there is no overt hepatic phenotype in the rat model of TMEM67 mutations (Iannicelli et al., 2010; Shim et al., 2019). As found in extra-renal organs, these TMEM67+/− rats developed late-onset hydrocephalus (Shim et al., 2019). Moreover, increasing evidence (Table S1)(Dempsey et al., 2017; Deveault et al., 2011; Doherty et al., 2010; Parisi, 2009) suggests that the correlation of genotype-phenotype is not simple and that TMEM67-involved MKS shares clinical features with Jourbert syndrome (JS), COACH syndrome, and BBS (Iannicelli et al., 2010; Leitch et al., 2008).
3.3. ZCCHC8 gene, whose KO mice develop CH, is in the vicinity of its telomere
The zinc finger CCHC-type domain containing 8 protein (ZCCHC8) is a component of the nuclear exosome targeting complex. Gable and colleagues recently showed that ZCCHC8 is required for the maturation of TR, a component of telomerase that is a specialized non-coding RNA (also known as TERC). They uncovered two distinct RNA dysregulation disease phenotypes caused by partial and complete loss of nuclear RNA exosome targeting. Genotypically, ZCCHC8+/− mice displayed lung disease, similar to human idiopathic pulmonary fibrosis related with defects in telomere length, while ZCCHC8−/− mice exhibited abnormal neurogenesis during embryonic development and CH in adulthood. The authors have claimed that hydrocephalus in ZCCHC8−/− mice is reminiscent of human mutation carriers with intellectual disability, although the presence of hydrocephalus in patients of that type has not been reported (Gable et al., 2019). Interestingly, transmembrane proteins (TMEMs) are upregulated in the RNA transcriptome analysis of ZCCHC8−/− mice, but whether or not they are related with cilia remains undetermined. Specifically, TMEM151a, TMEM151b, TMEM253, and TMEM116 were upregulated in their RNA-seq. Analysis. None of these four TMEMs have yet been shown to localize in the ciliaiy transition zone. Their interpretation of the mechanism by which CH develops in this model is built on three premises: 1) ZCCHC8 expression is low in the brain but detected in ependymal cells, 2) three TMEMs transcripts, one of which may be related with the ciliary transition zone, were upregulated, and 3) genes reported to regulate motile cilia, cede family proteins, were upregulated. Among TMEMs they uncovered, ‘the ciliary body’ connected to the dorsal iris of the eye, in which TMEM116 was highly expressed, could be considered a potential link to the cilia of the central nervous system (CNS) (Sousounis et al., 2013). Thus, clarification is warranted if, for example, TMEM116 upregulated in ZCCHC8−/− mice with CH is detected as one of transition zone complexes, similar to TMEM67, in conjunction with ‘ependymal’ ZCCHC8, which they briefly described (Gable et al., 2019).
3.4. An outline of genes causing CH
In the prior section, we compared two recently reported genes evoking CH. Similarly, here, we reviewed more than 100 genes causing CH and summarized the basic information (Table 1). Here, we found that 108 genes causing CH might be classified into four categories: 1) neurulation, 2) cilia, 3) syndromes, and 4) others. For more detailed classification, readers are recommended to visit the references (REF) listed in Table 1.
Table 1.
An outline of genes causing CH.
| gene | class | protein | key functions | mutant phenotype | REF |
|---|---|---|---|---|---|
| AK7 | 2,4* | Adenylate Kinase 7 | catalyzes the reversible phosphorylation of adenine nucleotides, and plays a role in energy homeostasis of cell | Primary ciliary dyskinesia, spermatogenic failure 27, non-syndromic male infertility due to sperm motility disorder, and CH | (Lores et al., 2018) |
| AK8 | 4 | Adenylate Kinase 8 | catalyzes the reversible transfer of the terminal phosphate group between nucleoside triphosphates and monophosphates | Reticular Dysgenesis, Fetal Akinesia Deformation Sequence 1, and CH | (Vogel et al., 2010) |
| AKT3 | 3 | AKT Serine/Threonine Kinase 3 | Regulator of the PI3K-AKT-mTOR pathway (especially in the nervous system) and is essential for the development of the brain and other parts of the body | Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome, other disorders | (Riviere et al., 2012) |
| ALG13 | 3,4 | ALG13 UDP-N-Acetylglucosaminyltransferase Subunit | heterodimerizes with asparagine-linked glycosylation 14 homolog, helps to catalyze the second sugar addition of the highly conserved oligosaccharide precursor in endoplasmic reticulum N-linked glycosylation | Lennox-Gastaut syndrome, epileptic encephalopathy, and CH | (Epi et al., 2013) |
| AP1S2 | 3 | Adaptor Related Protein Complex 1 Subunit Sigma 2 | mediates the recruitment of clathrin to the membrane, as well as the recognition of sorting signals within the cytosolic tails of transmembrane receptors | Pettigrew syndrome, including choreoathetosis, Dandy-Walker malformation, seizures, iron/calcium deposition in the brain, and CH | (Cacciagli et al., 2014; Saillour et al., 2007) |
| ARHGAP31 | 3,4 | Rho GTPase Activating Protein 31 | Inactivates Cdc42 and Rac1 through stimulation of reaction to turn GTP into GDP | Adams-Oliver syndrome and CH | (Lehman et al., 1993) |
| gene | class | protein | key functions | mutant phenotype | REF |
| ARSB | 4* | Arylsulfatase B | Removes sulfate group from dermatan sulfate and chondroitin sulfate to aid in lysosome digestion | mucopolysaccharidosis type VI and CH | ( Valayannopoulos et al., 2010) |
| B3GALNT2 | 3,4 | Beta-1,3-N-Acetylgalactosaminyltransferase 2 | Involved in alpha-dystroglycan glycosylation | Walker-Warburg syndrome, cobblestone lissencephaly, eye malformations, profound mental disability, and CH | (Ducro et al., 2015) |
| B3GALTL | 3 | beta 3-glucosyltransferase | involved in a two-step glycosylation pathway to form a sugar structure made of fucose and glucose on a specific location of several different proteins | Peters plus syndrome (eye abnormalities, short stature, intellectual disability) and CH | (Schoner et al., 2013) |
| B3GNT1 | 4 | N-acetyllactosaminide beta-1,3-N-acetylglucosaminyltransferase | Essential for the synthesis of poly-N-acetyllactosamine, a determinant for the blood group i antigen | Congenital muscular dystrophy and CH | (Czeschik et al., 2013) |
| BRAF | 3,4 | B-Raf proto-oncogene, serine/threonine kinase | The gene is also referred to as proto-oncogene B-Raf and v-Raf murine sarcoma viral oncogene homolog B, while the protein is more formally known as serine/threonine-protein kinase B-Raf | a congenital anaplastic astrocytoma, a linear syringocystadenoma papilliferum, ocular abnormalities, and CH | (Watanabe et al., 2016) |
| CC2D2A | 2,3,4 | Coiled-Coil and C2 Domain Containing 2A | A coiled-coil and calcium binding protein that plays a critical role in cilia formation | Meckel syndrome type 6, Joubert syndrome type 9, and CH | (Bachmann-Gagescu et al., 2015) |
| CCDC85C | 1,4 | Coiled-Coil Domain Containing 85C | plays a role in cell-cell adhesion and epithelium development through its interaction with proteins of the beta-catenin family | Congenital disorder of glycosylation, Type lie, obstructive hydrocephalus, and CH | (Tanaka et al., 2015) |
| CCDC88C | 1 | coiled-coil domain containing 88C | interacts with the dishevelled protein and is a negative regulator of the Wnt signaling pathway | autosomal recessive, primary non-syndromic CH | (Ruggeri et al., 2018) |
| gene | class | protein | key functions | mutant phenotype | REF |
| CCL2 | 1,4* | chemokine (C–C motif) ligand 2 | Through signaling via the binding/ activation of CCR2, instigates a powerful chemotactic response and mobilization of calcium ions in the cell | Alopecia areata, neural tube defects, and CH | (Snowden et al., 2012) |
| CCND2 | 3 | G1/S-specific cyclin-D2 | Regulates G1-S transition step in the mitotic cycle and is regulated by the PI3K-AKT-mTOR pathway, influencing protein synthesis, cell proliferation and survival, and brain development | Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome | (Mirzaa et al., 2014) |
| CENPF | 3,4 | centromere protein F | Plays a role in chromosome segregation during mitotis by associating with the kinetochore in late G2 and maintaining this association through early anaphase | Stromme syndrome, intestinal atresia, ocular anomalies, renal and cardiac abnormalities, and CH | (Filges et al., 2016) |
| CEP83 | 2 | Centrosomal protein 83 | A centriolar protein involved in primary cilium assembly | Infantile nephronophthisis, intellectual disability, and CH | (Failler et al., 2014) |
| CFAP43 | 2 | cilia and flagella associated protein 43 | involved in sperm flagellum axoneme organization and function | Normal pressure hydrocephalus, spermatogenic failure 19, and CH | (Morimoto et al., 2019) |
| CFAP54 | 2 | Cilia- And Flagella-Associated Protein 54 | Required for assembly and function of cilia and flagella | Primary ciliary dyskinesia and CH | (McKenzie et al., 2015) |
| CRB2 | 1 | crumbs cell polarity complex component 2 | A member of a family of proteins that are components of the Crumbs cell polarity complex; important in early embryonic development | Cerebral ventriculomegaly and congenital nephrosis | (Slavotinek et al., 2015) |
| DAG1 | 3,4 | dystroglycan 1 | involved in laminin/basement membrane assembly, sarcolemmal stability, cell survival, peripheral nerve myelination, nodal structure, cell migration, and epithelial polarization | Walker-Warburg syndrome, limb-girdle muscular dystrophy, and CH | (Leibovitz et al., 2018) |
| gene | class | protein | key functions | mutant phenotype | REF |
| DNAAF1 | 2* | dynein axonemal assembly factor 1 | involved in the regulation of microtubule-based cilia and actin-based brush border microvilli | Primary ciliary dyskinesia and CH | (Ha et al., 2016) |
| DNAH5 | 2,3 | Dynein Axonemal Heavy Chain 5 | Produces the force within cilia necessary to cause movement | Primary ciliary dyskinesia heterotaxy syndrome, and CH | (Ibanez-Tallon et al., 2002 |
| DPCD | 2 | Deleted In Primary Ciliary Dyskinesia Homolog (Mouse) | involved in the generation and maintenance of ciliated cells | Primary ciliary dyskinesia and CH | (Vogel et al., 2010) |
| DYX1C1/DNAAF4 | 2,4 | Dynein axonemal assembly factor 4 | encodes a tetratricopeptide repeat domain-containing protein that interacts with estrogen receptors and heat shock proteins | involved in the generation and maintenance of ciliated cells | (Chandrasekar et al., 2013) |
| EML1 | 3 | echinoderm microtubule-associated protein-like 1 | Modulates the assembly and organization of the microtubule cytoskeleton, as well as likely the orientation of mitotic spindle and plane of cell division | Band heterotopia and CH | (Collin et al., 2020) |
| EN1 | 1 | homeobox protein engrailed-1; transcription factor | The human engrailed homologs 1 and 2 encode homeodomain-containing proteins and have been implicated in the control of pattern formation during development of the central nervous system | Ectopic expression leads to agenesis of subcommissural organ (SCO), ependymal differentiation defects in choroid plexus and CH | (Louvi and Wassef, 2000) |
| FBN1 | 3,4 | fibrillin 1 | Provides instructions for making fibrillin-1, which is transported out of cells into the extracellular matrix | Geleophysic dysplasia, aqueductal stenosis, and CH | (Porayette et al., 2014) |
| gene | class | protein | key functions | mutant phenotype | REF |
| FGFR2 | 3,4* | fibroblast growth factor receptor 2 | Promotes cell growth/division, maturation/ differentiation, bone development, angiogenesis, wound healing, and embryonic development | Apert syndrome, Beare-Stevenson cutis gyrate syndrome, Crouzon syndrome, Jackson-Weiss syndrome, Pfeiffer syndrome, breast cancer, cholangiocarcinoma, epidermal nevus, other cancers, and CH | (Wenger et al., 1993) |
| FGFR3 | 3,4 | fibroblast growth factor receptor 3 | Regulates bone growth through the limitation of ossification in long bones | Achondroplasia, Crouzon syndrome with acanthosis nigricans, epidermal nevus, hypochondroplasia, Muenke syndrome, SADDAN, multiple cancers, and CH | (Bodensteiner, 2019) |
| FKRP | 3,4 | fukutin-related protein | Like fukutin, adds a molecule called ribitol 5-phosphate to the chain of sugars attached to a protein called alpha (α)-dystroglycan | Walker-Warburg syndrome, limb-girdle muscular dystrophy, congenital muscular dystrophy type 1C, and CH | (Yis et al., 2007) |
| FKTN | 3,4 | Fukutin | adds a molecule called ribitol 5-phosphate to the chain of sugars attached to a protein called alpha (α)-dystroglycan | Fukuyama congenital muscular dystrophy, Walker-Warburg syndrome, limb-girdle muscular dystrophy, and CH | (Arora et al., 2019) |
| FLNA | 3,4 | Filamin A, alpha | participates in the anchoring of membrane proteins for the actin cytoskeleton | Periventricular heterotopia and CH | (Sheen et al., 2004) |
| FOXJ1 | 1,2 | Forkhead Box J1 | regulates the transcription of genes that control the production of motile cilia | Systemic lupus erythematosus, allergic rhinitis, and CH | (Wallmeier et al., 2019) |
| FUZ | 1 | Fuzzy Planar Cell Polarity Protein | involved in the creation of cilia and directional movement of cells | Neural tube defects and CH | (Kousi and Katsanis, 2016) |
| FZD3 | 4 | Frizzled Class Receptor 3 | encode seven-transmembrane domain proteins that are receptors for the wingless type MMTV integration site family of signaling proteins | Fallopian Tube Serous Adenocarcinoma and CH | ( Wang et al., 2014) |
| gene | class | protein | key functions | mutant phenotype | REF |
| GBA | 4* | Acid beta-glucocerebrosidase | Plays a role in lysosome digestion by breaking glucocerebroside into glucose and ceramide | Gaucher disease, Parkinson disease, dementia with Lewy bodies, and CH | (Cindik et al., 2010; Hruska et al., 2008) |
| GLI3 | 1,3 | GLI Family Zinc Finger 3 | One of the C2H2-type zinc finger proteins subclass of the Gli family; DNA-binding transcription factor, mediating SHH signaling | Greig cephalopolysyndactyly syndrome and CH | (Babbs et al., 2008; Balk and Biesecker, 2008) |
| GPSM2 | 3,4 | G-protein-signaling modulator 2 | Plays a significant role in mitotic spindle pole organization, as well as in the development of normal hearing | Nonsyndromic hearing loss, Chudley-McCullough syndrome, and CH | (Koenigstein et al., 2016) |
| HB-EGF | 4 | Heparin-binding EGF-like growth factor | Influences cell cycle progression, cell migration, and angiogenesis | Subarachnoid hemorrhage and CH | (Shim et al., 2016) |
| HRAS | 3,4 | GTPase HRas transforming protein p21 | Once bound to Guanosine triphosphate, H-Ras will activate a Raf kinase like c-Raf, the next step in the MAPK/ERK pathway | Costello syndrome, cerebellar tonsillar herniation, and CH | (Gripp et al., 2010) |
| HYDIN | 2 | axonemal central pair apparatus protein | Central pair-dynein adaptor | Missing central pair projection, ciliary motility defect, and CH | (Davy and Robinson, 2003) |
| HYLS1 | 2,3 | Hydrolethalus syndrome protein 1 | Incorporated into centrioles and required for the formation of cilia | Hydrolethalus syndrome, macrocephaly, midline cleft-lip, polydactyly, and CH | (Belengeanu et al., 2011) |
| IFT172 | 3,4 | intraflagellar transport 172 | encodes a subunit of the intraflagellar transport subcomplex IFT—B, which is necessary for ciliaryassembly and maintenance | Polydactyly, short-rib thoracic dysplasia, Joubert syndrome, and CH | (Friedland-Little et al., 2011) |
| ISLR2 | 4 | Immunoglobulin Superfamily Containing Leucine Rich Repeat 2 | Required for axon extension during neural development | Hernia, hiatus, chicken egg allergy, and CH | (Alazami et al., 2019) |
| gene | class | protein | key functions | mutant phenotype | REF |
| ISPD/crppa | 3,4* | Isoprenoid Synthase Domain-Containing Protein/CDP-L-Ribitol Pyrophosphorylase A | helps produce ribitol 5-phosphate, which is added to alpha (α)-dystroglycan | Walker-Warburg syndrome, limb-girdle muscular dystrophy, and CH | (Roscioli et al., 2012) |
| KIAA0586 | 2,3 | Homolog of TALPID3 | A centrosomal protein required for ciliogenesis and Polydactyly, abnormal dorsal | Polydactyly, abnormal dorsal patterning of the neural tube and CH | (Alby et al., 2015) |
| KIF7 | 2,3 | kinesin family member 7 | Provides instructions for making primary cilia | Growth retardation, infantile spasms, acrocallosal syndrome, and CH | (Tunovic et al., 2015) |
| L1CAM | 3,4 | L1 protein family cell adhesion molecule | Cell migration and adhesion | L1 syndrome or CRASH (corpus callosum hypoplasia, retardation, aphasia, spastic paraplegia, and hydrocephalus) | (Fransen et al., 1995) |
| LAMB1 | 4 | Laminin Subunit Beta 1 | involved in cell attachment, chemotaxis, and binding to the laminin receptor, as well as capable of inhibiting metastasis | Lissencephaly 5 (seizures and severely delayed psychomotor development) and CH | (Radmanesh et al., 2013) |
| LARGE | 3,4 | LARGE xylosyl- and glucuronyltransferase 1 | adds chains of sugar molecules composed of xylose and glucuronic acid to alpha (α)-dystroglycan | Walker-Warburg syndrome, congenital muscular dystrophy type 1D, and CH | (Kousi and Katsanis, 2016) |
| LRRC48 | 2,3 | Leucine-Rich Repeat-Containing Protein 48 | key regulator of ciliary/flagellar motility which maintains the alignment and integrity of the distal axoneme and regulates microtubule sliding in motile axonemes | Smith-Magenis syndrome, primary ciliary dyskinesia, and CH | (Ha et al., 2016) |
| MAF | 3,4 | MAF BZIP Transcription Factor | Regulates development of embryonic lens fiber cell, increased susceptibility of T-cells to apoptosis, and terminal differentiation of chondrocytes | Coloboma, multiple myeloma, Aymegripp syndrome, Cataract 21-multiple types, and CH | (Niceta et al., 2015) |
| gene | class | protein | key functions | mutant phenotype | REF |
| MBOAT7 | 4* | Membrane Bound O-Acyltransferase Domain Containing 7 | involved in the reacylation of phospholipids as part of the phospholipid remodeling pathway known as the Land cycle | Mental retardation (autosomal recessive 57), Autosomal Recessive Non-Syndromic Intellectual Disability, and CH | (Vogel et al., 2010) |
| MKS1 | 2,3 | Meckel Syndrome Type 1 | A part of the flagellar apparatus basal body proteome, required for cilium formation | Meckel syndrome, BBS, and CH | (Nabhan et al., 2015) |
| MKS4 | 2,3 | Meckel syndrome type 4 | a centrosomal protein that plays an important role in centrosome and cilia development | Suppression of ciliogenesis and hydrocephalus | (Rachel et al., 2015) |
| MPDZ | 4 | Multiple PDZ Domain Crumbs Cell Polarity Complex Component | Interacts with HTR2C receptor, causing potential clumping at cell surface | Hydrocephalus, congenital, 2, with or without brain or eye anomalies | (Yang et al., 2019) |
| MSX1 | 3 | Msh homeobox 1; transcription factor | a transcriptional repressor during embryogenesis | SCO agenesis; neuroependymal denudation and CH | (Bach et al., 2003) |
| MYH9 | 4 | Myosin Heavy Chain 9 | involved in cytokinesis, cell migration, polarization and adhesion, maintenance of cell shape, and signal transduction | Macrothrombocytopenia, inclusion bodies, nephropathy, and CH | (Belal and Al Menabawy, 2017) |
| NAPA | 4 | Alpha-soluble NSF attachment protein | involved in the intra-cellular trafficking and fusing of vesicles to target membranes in cells | Hydrocephalus with hop gait; male hyh mice with a strongly reduced fertility | (Batiz et al., 2009) |
| NF1 | 4 | Neurofibromatosis type 1 | A GTPase-activating protein that negatively regulates RAS/MAPK by accelerating the hydrolysis of Ras-bound GTP | Macrocephaly, vasculopathy, aqueduct stenosis, cancer, and CH | (Nix et al., 2020) |
| gene | class | protein | key functions | mutant phenotype | REF |
| NME5 | 2,4* | NME/NM23 Family Member 5 | Confers protection from cell death by Bax and alters the cellular levels of several antioxidant enzymes, including Gpx5 | Nemaline myopathy 5, primary ciliary dyskinesia, and CH | (Anderegg et al., 2019) |
| NME7 | 4 | NME/NM23 Family Member 7 | catalyzes phosphate transfer from nucleoside triphosphates to nucleoside diphosphates | Venous thromboembolism, dextrocardia with situs inversus, deafness, and CH | (Vogel et al., 2010) |
| NRAS | 3,4 | Neuroblastoma Proto-Oncogene, GTPase | The N-ras proto-oncogene is a member of the Ras gene family. It is mapped on chromosome 1, and it is activated in HL60, a promyelocytic leukemia line | Congenital melanocytic nevus syndrome and CH | (Recio et al., 2017; Uguen et al., 2015) |
| OFD1 | 1 | OFD1 Centriole and Centriolar Satellite Protein | Appears to play a role in the early development of the brain, limbs, and kidney | Joubert syndrome, polycystic kidney, and CH | ( Field et al., 2012) |
| PAC1/klf6 | 4 | pituitary adenylate cyclaseactivating polypeptide type I receptor | This gene encodes a member of the kinesin family of proteins. Members of this family are part of a multisubunit complex that functions as a microtubule motor in intracellular organelle transport. | Reduced neuronal proliferation, increased apoptosis in the developing cortex, reduced cerebral cortex, corpus callosu, and subcommissural organ with CH | (Lang et al., 2006; Picketts, 2006) |
| PAX6 | 1 | Paired box protein 6 | acts as a “master control” gene for the development of eyes and other sensory organs | SCO agenesis and CH | (Estivill-Torrus et al., 2001) |
| PI3K | 4 | Phosphoinositide-3-kinase | A group of genes that produce an enzyme involved in cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking | Cancers, other disorders | (Roy et al., 2019) |
| PIK3CA | 3,4 | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha | Produces a subunit of an enzyme important for cell proliferation and migration, protein production, intracellular materials transport, and cell survival | Klippel-Trenaunay syndrome, megalencephaly-capillary malformation syndrome, epidermal nevus, cancers, other disorders | (Riviere et al., 2012) |
| gene | class | protein | key functions | mutant phenotype | REF |
| PLOD2 | 3* | procollagen-lysine,2-oxoglutarate 5-dioxygenase 2 | a membrane-bound homodimeric enzyme that is localized to the cisternae of the rough endoplasmic reticulum. The enzyme catalyzes the hydroxylation of lysyl residues in collagen-like peptides. | Abnormal neurogenesis and prenatal communicating CH | (Furey et al., 2018a; Furey et al., 2018b) |
| POMGNT1 | 3,4 | protein O-linked mannose N-acetylglucosaminyltransferase 1 | encodes a type II transmembrane protein that participates in O-mannosyl glycosylation and is specific for alpha linked terminal mannose. involved in the presentation of the laminin-binding | Walker-Warburg syndrome, limb-girdle muscular dystrophy, muscle-eye-brain disease, and CH | (Vervoort et al., 2004) |
| POMK | 3,4 | protein O-mannose kinase | O-linked carbohydrate chain of alpha-dystroglycan (a-DG), which forms transmembrane linkages between the extracellular matrix and the exoskeleton | Walker-Warburg syndrome, heteropia, limb-girdle muscular dystrophy, and CH | (Preiksaitiene et al., 2020) |
| POMT1 | 4 | protein O-mannosyltransferase 1 | Along with POMT2, helps modify α-dystroglycan through glycosylation via the addition of mannose; this anchors the structural framework of the cytoskeleton to the ECM | Walker-Warburg syndrome, limb-girdle muscular dystrophy, muscle-eye-brain disease, and CH | (Currier et al., 2005) |
| POMT2 | 4 | protein O-mannosyltransferase 2 | Along with POMT2, helps modify α-dystroglycan through glycosylation via the addition of mannose; this anchors the structural framework of the cytoskeleton to the ECM | Walker-Warburg syndrome, limb-girdle muscular dystrophy, muscle-eye-brain disease, and CH | (van Reeuwijk et al., 2005) |
| PTCH1 | 1,3 | patched 1 | Receptor of Sonic Hedgehog in SHH pathway, which is essential in early cell growth and specialization | Gorlin syndrome, nonsyndromic holoprosencephaly, coloboma, cancers, and CH | (Furey et al., 2018a) |
| PTEN | 4 | phosphatase and tensin homolog | Provides instructions for making an enzyme that acts as a tumor suppressor | Enlargement of midbrain structures and CH | ( Ohtoshi, 2008) |
| PTPN11 | 3 | protein tyrosine phosphatase, non-receptor type 11 | helps regulate the RAS/MAPK signaling pathway | Noonan syndrome, cancers, and CH | (Zheng et al., 2018) |
| gene | class | protein | key functions | mutant phenotype | REF |
| RAF1 | 3,4* | Raf-1 proto-oncogene, serine/threonine kinase | part of the ERK1/2 pathway as a MAP kinase (MAP3K) that functions downstream of the Ras subfamily of membrane associated GTPases | Abnormal brain vasculature and CH | (Zarate et al., 2014) |
| RFX3 | 4 | Regulatory factor X3 | a transcriptional activator that can bind DNA as a monomer or as a heterodimer with other RFX family members. | neuroependymal defects and CH | (Baas et al., 2006) |
| RFX4 | 4 | Regulatory factor X4; transcription factor | a transcriptional repressor, which encodes transcription factors that contain a highly-conserved winged helix DNA binding domain | SCO agenesis; severe midline structure defects and CH | (Blackshear et al., 2003) |
| RPS6KA3 | 3,4 | Ribosomal protein S6 kinase alpha-3 | Involved in cellular proliferation, differentiation, and apoptosis and is important in pathways in the brain responsible for learning, the formation of long-term memories, and the survival of nerve cells | Coffin-Lowry syndrome, | (Kousi and Katsanis, 2016) |
| RSPH9 | 2 | radial spoke head component 9 | component of the axonemal radial spoke head | Primary ciliary dyskinesia and CH | (Zou et al., 2020) |
| RUVBL1 | 4 | RuvB Like AAA ATPase 1 | associates with several multisubunit transcriptional complexes and with protein complexes involved in both ATP-dependent remodeling and histone modification | Retinitis pigmentosa 50, hemangioma of spleen, and CH | (Dafinger et al., 2018) |
| SGSM3 | 4 | small G protein signaling modulator 3 | play a cooperative role in NF2-mediated growth suppression of cells | Protein-altering mutations found in human cases of CH | (Furey et al., 2018a) |
| SKI | 3,4 | Sloan-Kettering Institute (SKI) proto-oncogene | Controls TGF- β pathway by binding to certain SMAD proteins, thus interrupting pathway signaling | Shprintzen-Goldberg syndrome and CH | (O’Dougherty et al., 2019) |
| gene | class | protein | key functions | mutant phenotype | REF |
| SLC12A7 | 4* | Solute Carrier Family 12 Member 7 | Mediates electroneutral potassium-chloride cotransport when activated by cell swelling | Agenesis of corpus callosum with peripheral neuropathy, renal tubular acidosis, and CH | (Jin et al., 2019) |
| SMARCB1 | 3,4 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1 | regulates gene activity and expression by chromatin remodeling via the SWI/SNF protein complexes | Coffin-Siris syndrome, Rhabdoid tumor predisposition syndrome, Schwannomatosis, and CH | (Diets et al., 2019) |
| SMARCC1 | 1,4 | SWI/SNF complex subunit | The encoded protein is part of the large ATP-dependent chromatin remodeling complex SWI/SNF and contains a predicted leucine zipper motif typical of many transcription factors | Abnormal neurogenesis and prenatal communicating CH | (Furey et al., 2018a; Furey et al., 2018b) |
| SNX10 | 4 | Sorting Nexin 10 | a member of the sorting nexin family, involved in intracellular trafficking | Osteopetrosis, corpus callosum hypoplasia and CH | (Megarbane et al., 2013) |
| SNX27 | 4 | Sorting Nexin Family Member 27 | involved in endocytosis of plasma membrane receptors and protein trafficking | Epileptic encephalopathy, epilepsy, and CH | (Wang et al., 2016) |
| SOCS7 | 4 | Suppressor of cytokine signaling 7 | a target gene of microRNA-145, regulating interferon-b | A smaller body size and early neonatal death due to CH | (Krebs et al., 2004) |
| SPEF2 | 2,4 | Sperm Flagellar 2 | Plays a role in localization of the intraflagellar transport protein IFT20 to the manchette, and required for correct axoneme development in spermatozoa | Spermatogenic failure 43, primary ciliary dyskinesia, and CH | (Finn et al., 2014) |
| STK36 | 2 | Serine/Threonine Kinase 36 | positive regulator of the GLI zinc-finger transcription factors | Primary ciliary dyskinesia and CH | (Merchant et al., 2005) |
| gene | class | protein | key functions | mutant phenotype | REF |
| TBC1D7 | 3* | TBC1 domain family member 7 | Subunit of the TSC1-TSC2 complex and component of the TSC-TBC complex, regulates cell growth and differentiation, participates in the proper sensing of growth factors and glucose | Macrocephaly/megalencephaly syndrome (autosomal recessive) | (Lim and Crino, 2013) |
| TBX1 | 3,4 | T-Box Transcription Factor 1 | Aids formation of tissues and organs during embryonic development by binding to specific areas of DNA | 22q11.2 deletion syndrome, heart defects, cleft palate, hearing loss, and CH | (Kousi and Katsanis, 2016) |
| TGFBR1 | 3,4 | Transforming Growth Factor Beta Receptor 1 | Forms part of the TGF- β signaling pathway | Loeys-Dietz syndrome, familial thoracic aortic aneurism and dissection, prostate cancer, other cancers, and CH | (Yang et al., 2008) |
| TMEM216 | 2,3 | Transmembrane protein 216 | Part of the tectonic-like complex which is required for tissue-specific ciliogenesis and may regulate ciliary membrane composition | Meckel syndrome type 2, Joubert syndrome 2 also known as Cerebello-oculorenal syndrome 2, and CH | (Edvardson et al., 2010; Kousi and Katsanis, 2016) |
| TMEM67 | 2,3 | Meckelin or transmembrane protein 67 | centriole migration to the apical membrane and formation of the primary cilium | Meckel syndrome type 3 (MKS3), Joubert syndrome type 6 (JBTS6), and hydrocephalus | (Shim et al., 2019; Smith et al., 2006) |
| TRIM71 | 1,4 | Tripartite motif family protein 71 | One of group 1 members, which possesses a variety of C-terminal domains, and are represented in both vertebrate and invertebrates | Abnormal neurogenesis and prenatal communicating CH | (Furey et al., 2018a; Furey et al., 2018b) |
| ULK4 | 4 | unc-51 like kinase 4 | Plays a role in neuronal growth and endocytosis, specifically in neurite branching, neurite elongation and overall neuronal migration | Schizophrenia, bipolar disorder, autism, and CH | (Liu et al., 2016a) |
| VANGL1 | 1 | VANGL planar cell polarity protein 1 | Mediates intestinal trefoil factor induced wound healing in intestinal mucosa | Caudal regression sequence, neural tube defect | (Yamasaki and Kanemura, 2015) |
| gene | class | protein | key functions | mutant phenotype | REF |
| VANGL2 | 1* | VANGL planar cell polarity protein 2 | Involved in planar cell polarity (especially in the steriociliary bundles of the cochlea), neural plate development, and polarization and movement of myocardializing cells | Neural tube defect | (Bubenshchikova et al., 2012) |
| WASL/n-wasp | 3,4 | WASP Like Actin Nucleation Promoting Facto | interacts with several proteins involved in cytoskeletal organization, including cell division control protein 42 (CDC42) and the actin-related protein-2/3 (ARP2/3) complex | Wiskott-Aldrich Syndrome, Buruli Ulcer, and CH | (Jain et al., 2014) |
| WDR81 | 3,4 | WD repeat domain 81 | plays a role in endolysosomal trafficking | Cerebellar ataxia, severe mental disability, disequilibrium syndrome 2, and CH | (Cappuccio et al., 2017) |
| ZCCHC8 | 2,4 | zinc finger CCHC domain-containing protein 8 | an accessory factor to the nuclear RNA exosome complex | Pulmonary fibrosis and hydrocephalus | (Gable et al., 2019) |
| ZIC2 | 1 | Zic family member 2 | Provides instructions for making a protein that plays an important role in the development of the forebrain | Neural tube defect, holoprosencephaly and CH | (Yamasaki and Kanemura, 2015) |
| ZIC3 | 1,3 | Zinc finger protein ZIC 2 | A transcription factor in early stages of left-right axis formation | Dandy-Walker malformation, neural tube defects, and CH | Grinberg and Millen, 2005) |
class 1–4 (neurulation, cilia, syndrome, others).
While this classic summary of genotype-phenotype (Table 1) is appreciated, we additionally asked which fundamental characteristics of these genes are associated with mutations leading to CH. To find the answer to this question, we first examined the human ZCCHC8 gene using the genome data viewer. Interestingly, ZCCHC8 is located very close to its telomere (<50 Mbp) on chromosome 12. We also found that the human TMEM67 gene is located slightly farther than 50 Mbp from its telomere on human chromosome 8 (Table S2).
3.5. Mapping of select genes on human chromosomes: I. proximity to telomeres
During literature surveys of SHH signaling related with CH, we found that a gene located adjacent to a telomere in a chromosome interacts with genes located in the same chromosome and can also affect the expression of genes that are further apart. For example, when the ligand SHH (in chromosome 7) binds to its receptor, PTCH1 (in chromosome 9) that blocks SMO (in chromosome 7), SMO is released and translocated to cilia to activate the downstream signaling (Goetz and Anderson, 2010; Rohatgi et al., 2007). Here, both SHH (q36.3) and SMO (q32.1) are located at the long arm of chromosome 7. If two genes are in the same chromosome, their genetic linkage can alter recombination frequency. Therefore, we sought a way to identify which chromosome has which gene, pinpointing where each gene lies on its chromosome via genetic mapping.
Previously, the biological basis for the apparently high mutation rate in human chromosome 8, on which TMEM67 is located, has been described. A group of colleagues led by Eric Lander have proposed that three factors are associated with high mutation rates, highlighting i) proximity to telomeres, ii) high recombination rate, and iii) high A + T content (Chimpanzee and Analysis, 2005; Hellmann et al., 2005; Nusbaum et al., 2006). Strictly speaking, a high recombination rate (Chimpanzee and Analysis, 2005; Hellmann et al., 2005; Nusbaum et al., 2006) is obtained when exchanged nucleotide sequences of F2 offspring from homologous recombination of FI parent chromosomes are discerned. However, if we apply the rule of thumb of 1 centimorgan (cM) ~1 million base pair (Mbp) (Hastbacka et al., 1992), then measuring the distance between two genes will allow the recombination frequency (Ritter et al., 1990) or rate (Nusbaum et al., 2006) to be calculated. As a result, we examined two of the three factors mentioned above—focusing on the position of a gene and its distal end locus of each arm on a chromosome where the telomere lies—using the following premise:
If recombination frequency (Ritter et al., 1990) is less than ( ≤) 50 cM, genes are linked: (1)
if recombination frequency is higher than 50 cM, genes are not linked, (2)
where 1 centimorgan (cM) 1 million base pair (Mbp) (Hastbacka et al., 1992).
First, we retrieved each transcript using the genome data viewer and checked the position in the short (p) or long (q) arm of the chromosome relative to the telomere (the distal end of the p or q arm depends on where a gene is located) of select genes readily known in clinical and animal studies of CH (Table S2)(Botfield et al., 2013). We found that three causative genes of CH located at chromosome X (L1CAM) and chromosome 12 (ZCCHC8 and MKS4/cep290) met the first condition of ‘proximity to telomeres’ within 50 Mbp (Nusbaum et al., 2006). Interestingly, we found that three other genes related with CH are also located within this proximity (<50 Mbp) to their telomeres—namely, TMEM116, TRPV4, and decorin (DCN) (Bothwell et al., 2019; Gable et al., 2019). In addition, Orail, MYC, and IGF1, which mediate store-operated Ca2+ signaling (Miao et al., 2017), induction of pluripotency (Park et al., 2008), and ciliary signaling in embiyonic ventricular surface (Lehtinen et al., 2013), respectively, are located at less than 50 Mbp to telomeres of the q arm on either chromosome 8 or chromosome 12. Intriguingly, however, TMEM67 and BBS10—which are known to cause CH and BBS—are slightly farther than 50 Mbp from their telomeres (Forsythe and Beales, 2013; Forsythe et al., 2018; Shim et al., 2019; Smith et al., 2006). Inversely, GLI1, which is known to mediate SHH signaling (Swiderski et al., 2014), and alpha tubulin (TUBAB1) gene—a marker for tufts of cilia (Mirzadeh et al., 2008)—are clearly far from their telomeres. In comparison, WNT1 and LRRK2, both known to regulate PCP signaling (Bengoa-Vergniory and Kypta, 2015; Meng et al., 2015) and familial Parkinson’s disease (fPD) (Kluss et al., 2019), are much closer to the centromere (Fig. 2A) within chromosome 12 rather than the telomeres. Taken together, four genes known to cause CH or BBS (L1CAM, ZCCHC8, MKS4, and TMEM67) are noteworthy. However, an explanation other than ‘proximity to telomeres’ is sought to better correlate the genotype (mutations) with the reported phenotype in humans (CH or other syndromic diseases). Thus, we examined the % composition of guanine and cytosine (GC content) within all these gene transcripts to obtain the % composition of adenine and thymine (A + T content) (Nusbaum et al., 2006).
Fig. 2. Proximity to telomeres in genes known to cause CH.
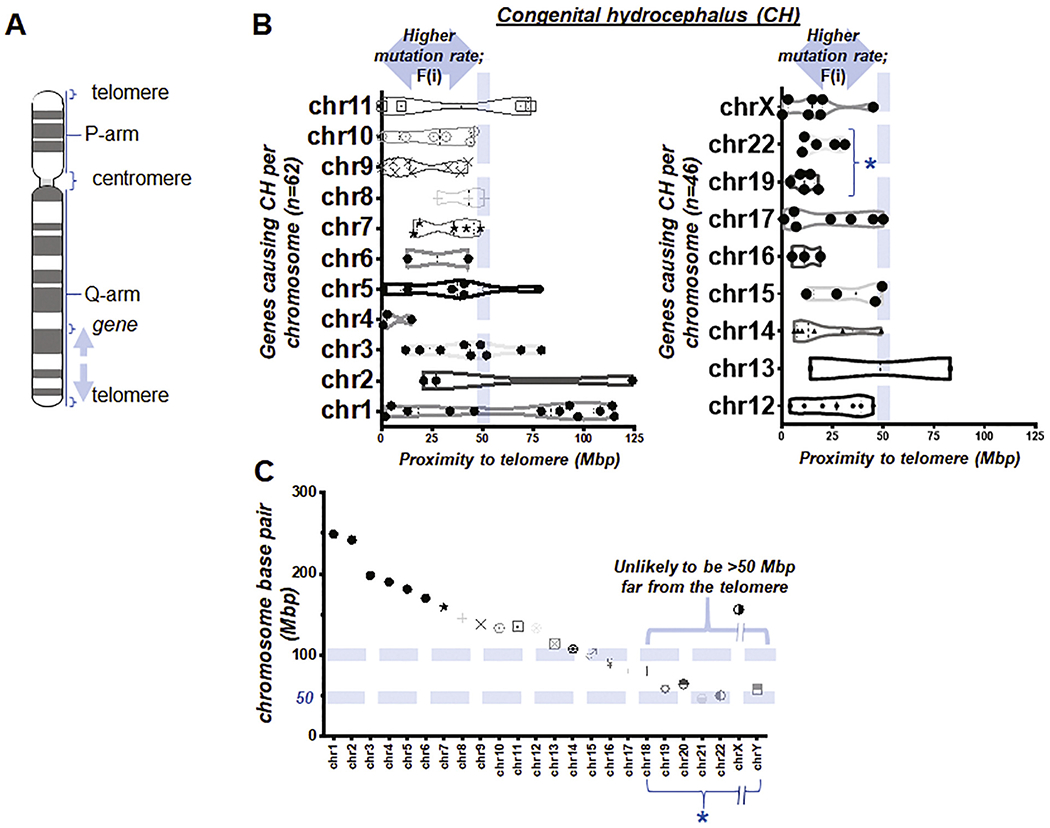
(A) Illustration simplified to indicate relative positions of telomeres, centromere, each arm, and a gene in a human chromosome (Nusbaum et al., 2006). Arrows indicate a distance between the gene and a telomere. (B) Box and violin plots showing the full distribution of the gene proximity to its telomere over chromosome (chr) 1 to 11 (left) and 12 to X (right). F(i) and * indicate ‘the first factor or proximity to telomeres’ and ‘the group of chromosomes unlikely to be >50 Mbp distant from the telomere due to their short physical size (Morton, 1991)’ (C) Length of all human chromosomes reported previously (Morton, 1991). * indicates the same in (B).
3.6. Mapping of 108 genes causing CH on human chromosomes: II. A + T content
The A + T content of 16 genes were obtained using a GC content calculator, after which we sorted the data by nucleotide size, in terms of full-length base pair number (bp). As is evident from Table S3, nucleotides longer than 6000 bp tended to show higher A + T content at >60%, which is higher than the threshold of 59% as proposed previously (Nusbaum et al., 2006). However, as the size of the nucleotide shortens, A + T content becomes less consistent across CH-related genes located on chromosome X, chromosome 8, and chromosome 12 (Table S3). As the sets of genes in Table S3 are compared to those of Table S2, despite having low A + T content (≤59%), two classic and relatively new causative genes of CH—L1CAM and ZCCHC8—demonstrated exemplary proximity (≤50Mbp) to their telomeres. In contrast, high A + T content (>59%) may have prevailed over insufficient proximity to telomeres in two marginally located genes, TMEM67 and BBS10. Although the select genes listed here may be a result of our unintended bias, TMEM67 and ZCCHC8, along with the aforementioned series of associated genes, have appeared to uncover that either proximity to telomeres or high A + T content might suffice to predict if human CH develops—based on CH found in mutant animals. To test this idea, we extended our analysis to 108 causative genes (4 in Table S2–S3 + 104 in Table S4) of CH, reported previously (Buysse et al., 2013; Belal and Al Menabawy, 2017; Chabas et al., 1995; de Paola et al., 2019; Fransen et al., 1995; Furey et al., 2018a; Furey et al., 2018b; Gable et al., 2019; Huh et al., 2009; Iliescu et al., 2011; Kibar et al., 2001; Konishi et al., 2020; Kousi and Katsanis, 2016; Liu et al., 2016b; Rachel et al., 2015; Rowitch et al., 1999; Shaheen et al., 2017; Shim et al., 2019; Sironen et al., 2011; Vogel et al., 2012; Wicker et al., 1991; Wild et al., 1997).
In assessing the list of 108 causative genes of CH in Tables S2–S4, we summarized the survey of the distance between each gene locus and its telomere, defining it as the first factor or F(i) (Fig. 2B). As we went through each of these genes with the genome data viewer, it became obvious that if a causative gene of CH is located on chromosome 22, the 2nd shortest chromosome in physical length (Morton, 1991), “distance to a telomere” will almost always be within a 50 Mbp reach. This technically fulfills the first factor, F(i), indicating a higher mutation rate (Nusbaum et al., 2006), though it is less assertive than that indicated by longer chromosomes. By examining the known length of all chromosomes (Morton, 1991) and our data, we found that human chromosomes 18 to 22 had an absolute length less than 100 Mbp, meaning that their genes were unlikely to be >50 Mbp from the telomere, since the centromere is located between p- and q-arm (Fig. 2C), thereby easily meeting F (i). This implies that clinicians will have a different suggestion for patients with CH whose causative genes lie in chromosome 18, 19, 20, 21, 22, and Y—because any gene within these short chromosomes will likely have a range of distance from its telomere within 50 Mbp (Fig. 2A–C).
3.7. Causative genes of CH: III. Proximity to telomeres vs. A + T content
To further test our idea, we obtained the specific A + T (Fig. 3A) content of all of the aforementioned genes reported to cause CH. These 108 causative genes were found in almost all of the chromosomes except three (chromosome 18, 20, 21). Although a majority of these genes had less than 59% A + T content (dashed vertical lines in Fig. 3B), 32 of 108 genes with A + T content >59% (Fig. 3B; Table S2–S4). We defined this condition as the second factor or F(ii). When we counted the number of these factors to match with CH, >90% of genes causing CH satisfied the proximity to telomeres (n = 92/108) or high A + T content (n = 32/108). Furthermore, 20% of known genes met F(i) and F(ii) alike, while less than 10% (10 of 108 genes) met neither F(i) nor F(ii) (Fig. 3C). This raises the possibility that the disease-specific success rate of the previously proposed factors successfully projects CH, regardless of the particular locus on a certain chromosome (e.g. 8p) (Nusbaum et al., 2006).
Fig. 3. A + T content of genes and factors-CH matching rate.
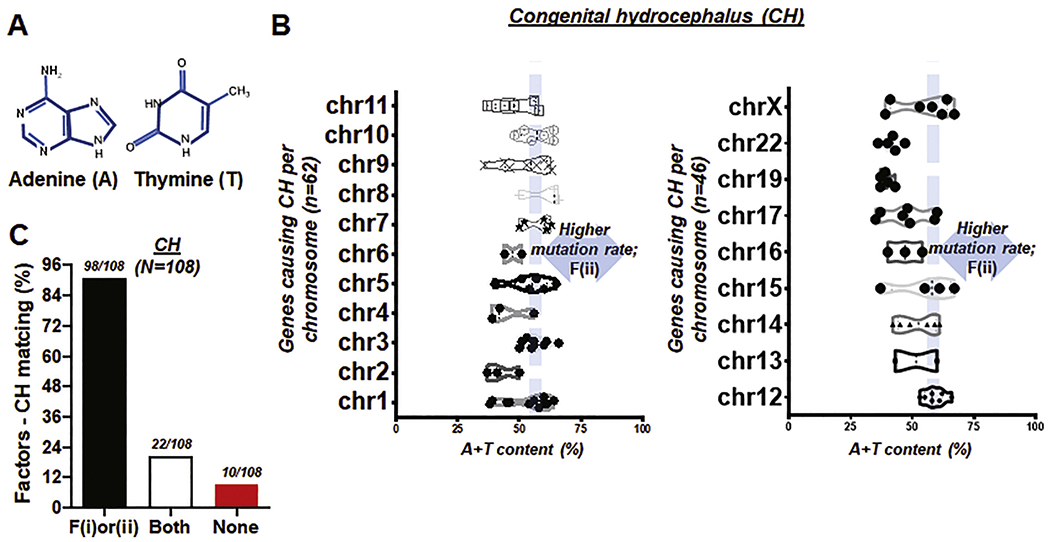
(A) Illustration showing two of four chemical bases comprising DNA, adenine (A) and thymine (T) whose combined content is associated with high mutation rate in human chromosomes (Nusbaum et al., 2006). (B) Box and violin plots showing the full distribution of A + T content over chromosome (chr) 1 to 11 (left) and 12 to X (right). F(ii) indicates ‘the second factor or A+T content’ (C) Bar graph demonstrating factors-CH matching rate. ‘F(i)or(ii)’ represents the genes causing CH satisfying either proximity to telomeres within 50 Mbp or A + T content higher than 59%; ‘Both’ represents the genes meeting two factors alike. Total number of genes, N = 108. Matching rate at >90%.
3.8. Genes causing CH uncovering the factor-nucleotide size relationship
During the survey of nucleotide base pairs of the 108 genes, data entries suggest that the full-length size of a gene, if it is larger than 6000 bp, likely leads to the said gene having high A + T content at >59%. The Pearson coefficient (r = 0.24) suggests that there is a correlation (P = 0.01) between the full-length size and A + T content in CH (Fig. 4A–B). We further organized factors (i) and (ii) with respect to the base pair number (bp) of each gene, by grouping into three categories with an approximately equal number per sub-group at 1–3000 bp (n = 40), 3001–6000 bp (n = 39), and 6001–15,000 bp (n = 29), respectively (total N = 108 causative genes of CH; Table S2, S3, and S4). Statistical analysis suggests that there is no significant overall difference in proximity to telomeres with respect to the gene full-length size. However, pair-wise comparisons using Tukey’s post-hoc test after one-way ANOVA suggest that there is a significant difference (P = 0.0075) in A + T content with respect to the full-length size (bp) in the shortest and the longest gene group. Genes with a full-length size longer than 6000 bp have a 41% chance (12/29) of harboring A + T content at >59%, compared to the two other groups with shorter sizes (20% and 24% chances for 1–3000 bp and ≥ 6001 bp group, respectively) (Fig. 4C–D).
Fig. 4. Proximity to telomere and A + T content over gene length (bp).
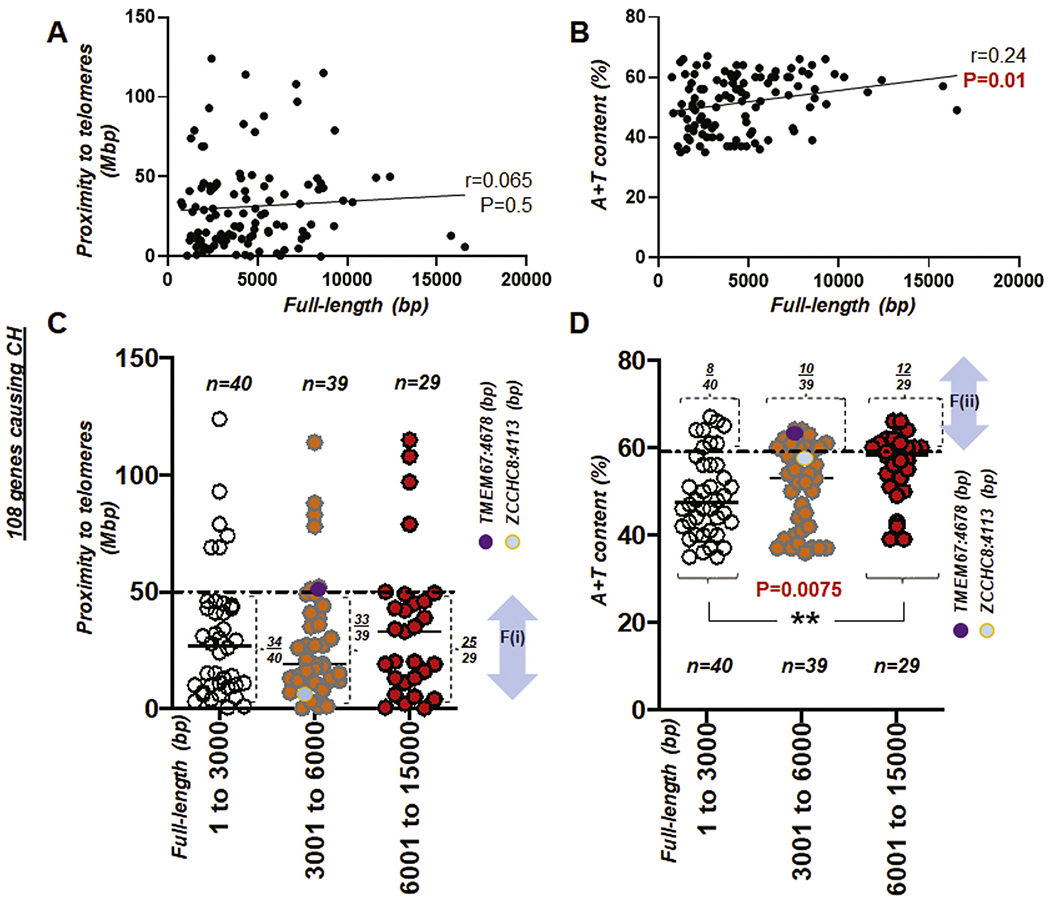
(A) Scattered plot showing the Pearson correlation between the full length (bp) of genes causing CH and proximity to telomeres. (B) Scattered plot showing the Pearson correlation between the full length (bp) of genes causing CH and A + T content. r: Pearson coefficient (A-B). (C) Scattered plot showing 108 genes causing CH with proximity to telomeres over full-length size of the gene (bp). A horizontal dotted line indicates 50 Mbp. (D) Scattered plot summarizing 108 genes causing CH with A + T content over full-length size of the gene (bp). A horizontal dotted line indicates A + T content at 59%. F(i) and F(ii) indicate the first and second factor, respectively. Note that circles in purple and light blue indicate TMEM67 and ZCCHC8 gene in (C) and (D). **, P < 0.01.
3.9. Genes associated with AD
We next checked the position of the p or q arm of the chromosome relative to the telomere of 70 genes associated with Alzheimer’s disease (AD) (Table S5)(Seshadri et al., 2010; Bird, 1993). In assessing these genes, we surveyed the distance between each gene locus and its telomere using the first factor, or F(i) (Fig. 5A). As we identified the 70 gene loci listed in Tables S5, it was evident that—in chromosomes of short physical length such as chromosome 19, 20, and 21—the “distance to a telomere” will almost always be within 50 cM, thereby fulfilling F(i). This is consistent with the previous result (Fig. 2B–C) above. Again, this confirms that clinicians can provide a convincing explanation for patients with AD whose susceptible genes lie on chromosome 19, 20, 21, 22 and Y—where mutations are highly likely due to the short chromosomal length (blue asterisk in Fig. 5A).
Fig. 5. Genes associated with Alzheimer’s disease (AD), satisfying either proximity to telomeres or high A + T content.
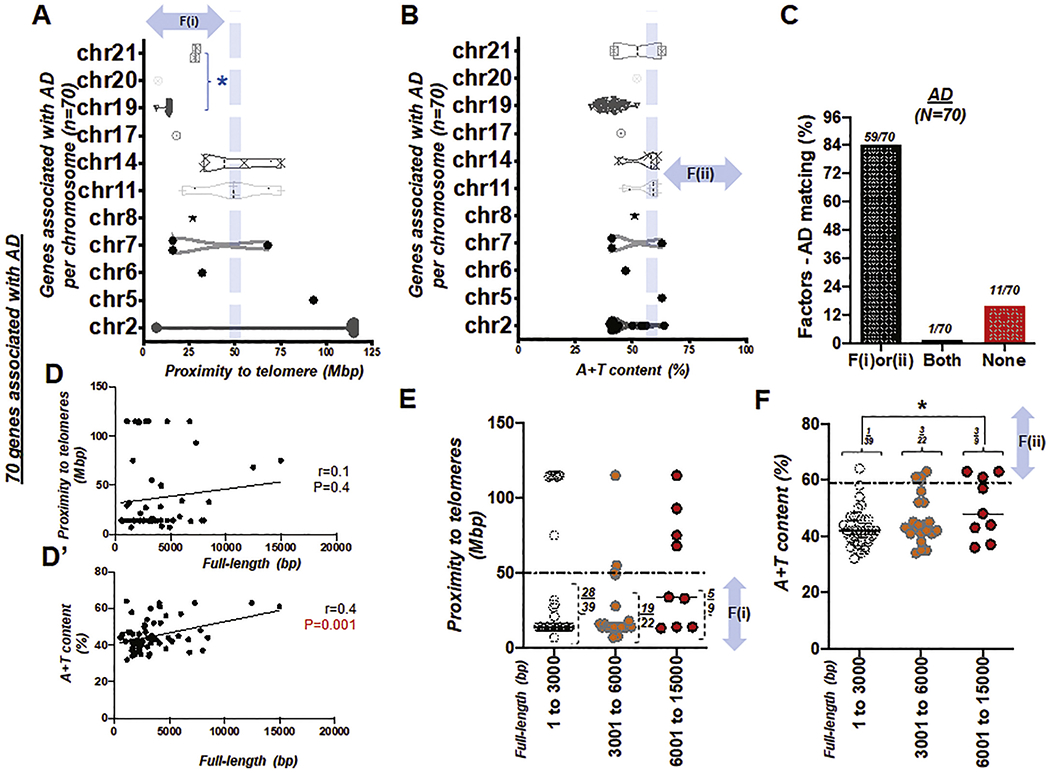
(A) Box and violin plots showing the full distribution of the gene proximity to its telomere over chromosome (chr) 1 to 21. An asterisk in blue, *, indicates ‘the group of chromosomes unlikely to be >50 Mbp distant from the telomere due to their short physical size (Morton, 1991)’ (B) Box and violin plots showing the full distribution of A + T content over chromosome (chr) 1 to 21. (C) Bar graph demonstrating factors-AD matching rate. Total number of genes, N = 70. Success (matching) rate at 84%. (D) Scattered plot showing the Pearson correlation between the full length (bp) of genes associated with AD and proximity to telomeres. (D’) Scattered plot showing the Pearson correlation between the full length (bp) of genes associated with AD and A + T content, r: Pearson coefficient (D-D’). (E) Scattered plot showing 70 genes of AD with proximity to telomeres over full-length size of the gene (bp). A horizontal dotted line indicates 50 Mbp. (F) Scattered plot summarizing 70 genes of AD with A + T content over full-length size of the gene (bp). A horizontal dotted line indicates A + T content at 59%. F(i) and F(ii) indicate the first and second factor, respectively. An asterisk in black, *, P < 0.05.
To further test our hypothesis, we obtained the A + T content (Fig. 5B) of genes reported to be susceptible to AD. The majority of genes susceptible to AD were distributed on chromosomes 2 and 19, forming a dense cluster. When we examined 70 genes of AD, 84% satisfied the proximity to telomeres (n = 53/70) or high A + T content (n = 8/70) conditions. Furthermore, 1 of these genes met both F(i) and F(ii), while less than 20% (11 of 70 genes) met neither F(i) nor F(ii) (Fig. 5C). This supports the idea that disease-specific matching of the previously proposed two factors can predict genetic disorders of the CNS—telomere proximity or A + T content might suffice to predict both diseases, although mismatch is higher for AD (84% matching) than CH (>90% matching).
We then organized the factors (i) and (ii) with respect to the base pair number (bp) of each gene. Consistent with the previous analysis (Fig. 4), we grouped the genes into three categories: 1–3000 bp (n = 39), 3001–6000 bp (n = 22), and 6001–15,000 bp (n = 9), respectively (total N = 70 genes susceptible to AD; Table S5; Fig. 5D–D’). Statistical analysis suggests that there is no significant difference in proximity to telomeres with respect to the gene full-length size. However, pair-wise comparisons using Tukey’s post-hoc test after one-way ANOVA suggest that there is a significant difference (P = 0.031) in A + T content with respect to the full-length size (bp) in the shortest and the longest gene group. Genes with a full-length size longer than 6000 bp have a 33% chance (3/9) of having >59% A + T content, compared to the two other groups with shorter sizes (3% and 14% chances for 1–3000 bp and ≥ 6001 bp group, respectively). (Fig. 5E–F).
3.10. Matching rate of two factors with other genetic diseases
We next analyzed the percentage of these two factors matching with familial Parkinson’s disease (fPD), since the series of causative genes was readily available: namely, PARK 1 to PARK 18. Using 17 genes of fPD identifiable via the genome data viewer, we found that less than 60% of genes associated with fPD satisfied either F(i) or F(ii) while no genes met F(i) and F(ii) at the same time. Inversely, we found that 41% of genes associated with fPD met neither proximity to telomeres nor high A + T content (Fig. 6A–C; Table S6)(Kumar et al., 2012; Le et al., 2003; Nichols et al., 2009). Statistical analysis suggests that there is no significant difference in proximity to telomeres with respect to the gene full-length size (Fig. 6D). Consistent with the previous assessment with CH, pair-wise comparisons using Tukey’s post-hoc test after one-way ANOVA indicate that A + T content of the fPD genes with molecular size higher than 6000 bp is significantly different than other groups (P < 0.01). However, fPD genes with full-length size longer than 6000 bp have only a 20% chance (1/5) of having A + T content at >59% while the two other groups of fPD genes showed a 0% likelihood (Fig. 6E).
Fig. 6. Genes causing fPD and other genetic diseases of the CNS.
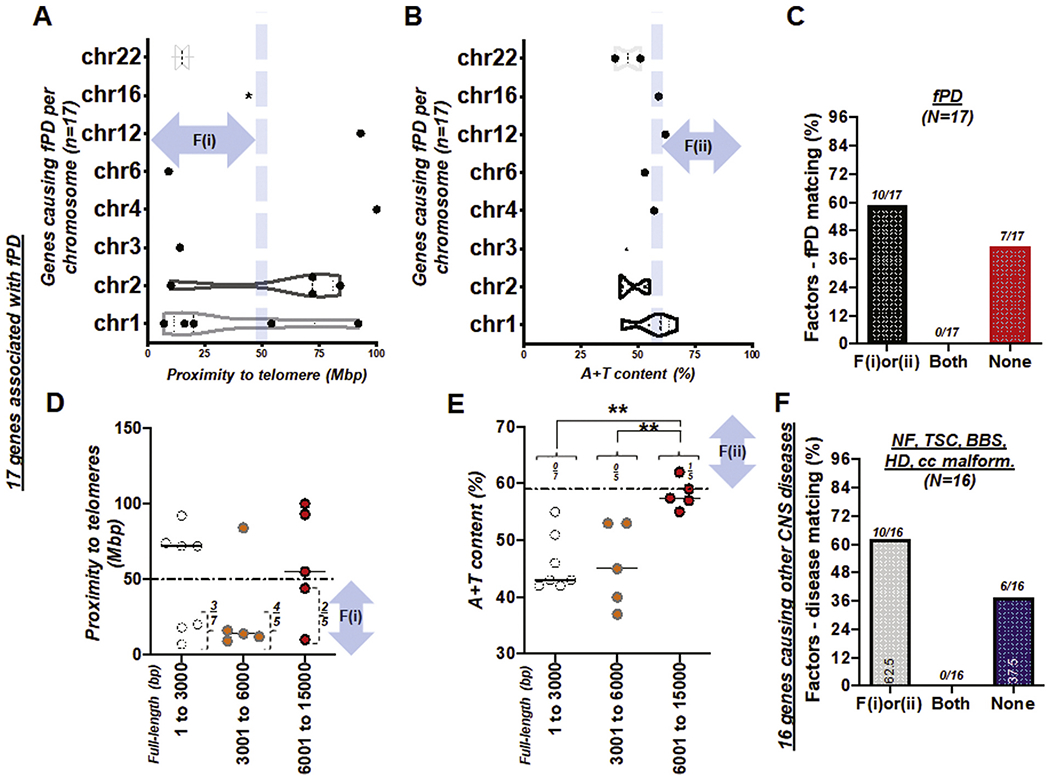
(A) Box and violin plots showing the full distribution of the gene proximity to its telomere over chromosome (chr) 1 to 22. (B) Box and violin plots showing the full distribution of A + T content over chromosome (chr) 1 to 22. fPD: familial Parkinson’s disease (A and B). (C) Bar graph demonstrating factors-fPD matching rate. Total number of genes, N = 17. Success (matching) rate at 59%. See also Table S6. (D) Scattered plot showing 17 genes causing fPD with proximity to telomeres over full-length size of the gene (bp). A horizontal dotted line indicates 50 Mbp. (E) Scattered plot summarizing 17 genes causing fPD with A + T content over full-length size of the gene (bp). A horizontal dotted line indicates A + T content at 59%. F(i) and F(ii) indicate the first and second factor, respectively. **, P < 0.01 (F) Bar graph demonstrating the factor-other diseases of the CNS matching rate. Total number of genes, N = 16. NF; neurofibromatosis (n = 2); TSC; tuberous sclerosis disease (n = 2); BBS (n = 5); HD (n = 1); CNS malformations involving agenesis of corpus callosum (cc; n = 6). Success (matching) rate at ~60%. See also Table S7.
Consistent with fPD, we found a similar trend of moderate success—an overall 62.5% matching rate—in other CNS genetic conditions including neurofibromatosis (NF), tuberous sclerosis (TSC), BBS, Huntington’s disease (HD) and malformations with agenesis of corpus callosum (cc) (Fig. 6F; Table S7)(Ferner and Gutmann, 2013; Hjortshoj et al., 2008; Ivanovski et al., 2018; Khalifa et al., 2015; Liu et al., 2019; Ly and Blakeley, 2019; Meczekalski et al., 2013; Nishimura et al., 2004; Rius et al., 2018; Sathasivam et al., 2013; Sheffield et al., 2018; Siegler et al., 2018; Tsai et al., 2012; Tyburczy et al., 2015; Uytingco et al., 2019; Zhang et al., 2011). Both tests of fPD and other CNS disease at ~60% matching rate were contrasted with the >90% rate generated from the 108 genes of CH (Fig. 3C vs. Fig. 6C&F). A mechanistic study elucidating disease-specific alterations of high mutation rates associated with proximity to telomeres and A + T content—as well as why CH demonstrates >90% accuracy while other genetic conditions of the CNS remain at ~60%—is warranted.
4. Discussion
We checked the two factors of telomere proximity and DNA composition focusing on AT-rich sequences in CH, fPD, AD, and a group of other genetic diseases—including neurofibromatosis (NF), tuberous sclerosis (TSC), Bardet-Biedl syndrome (BBS), and corpus callosum malformation (Fig. 1–6E and Fig. 6F). Chromosomal DNAs adjacent to telomeres are to some extent vulnerable to DNA damage or mutagenesis (Nusbaum et al., 2006). In this study, we considered this as being within 50 Mb from the chromosome ends (Nusbaum et al., 2006; Ritter et al., 1990). The threshold of 59% in A + T content is a genome-wide average reported in humans (Nusbaum et al., 2006). Telomeres, essentially characterized by a unique chromatin environment (Blasco, 2007), are composed of 3–20 kb of AT-rich repeats flanked by subtelomeric regions extending up to 300 kb into the chromosome (Misteli, 2014). In addition, tandem repeats of short GT-rich sequences are characteristic of almost all eukaryotic telomeres (McKnight and Shippen, 2004). Despite this repeated sequence and the number of repeats varying between species, human telomeres range in size from 2 to 50 kb and consist of 300–8000 precise repeats of the sequence CCCTAA/TTAGGG. Moreover, a common feature of all telomeres is the orientation of the G-rich strand. In all cases, this strand comprises the 3′-end of the chromosome, while its terminal part is single-stranded, generating a G-tail (Blackburn et al., 2006). It is also noted that genes in close proximity to telomeres are silenced due to the repressive nature of specific telomere-binding proteins such as heterochromatin (Misteli, 2014). The degree of repression declines with distance from the telomere, limited to ~100 kb (Kulkarni et al., 2010). Taken together, telomeres are the specialized mechanisms protecting chromosomes, since cells do not tolerate the presence of unprotected chromosome ends (McClintock, 1941)(O’Connor, 2008).
Our detailed analysis using the aforementioned two criteria associated with high mutation rate is primarily dedicated to CH (Fig. 2–4), AD (Fig. 5), and fPD (Fig. 6) because these diseases have multiple genes involved, rather than a small number of genes reported. We found that a genetic disease can have multiple genes with several types of mutations: i) point mutation, ii) indels, iii) transpositions, iv) inversions, v) nondisjunctions, and vi) changes in the ploidy level (Loewe and Hill, 2010). For instance, Down Syndrome is the most common genetic disorder by prevalence (1 in 1000 live births worldwide). But it is not single or multiple genes with type i (point mutation) through type v (nondisjunctions) mutations, but the presence of a third copy of chromosome 21 that causes this syndrome. In this situation (type vi—the ploidy level mutations) (Loewe and Hill, 2010), our analysis is inappropriate.
Previously, we characterized three different models of hydrocephalus starting from human CSF assays (Shim et al., 2016; Shim et al., 2013; Shim et al., 2019). Hence, our genomic analysis was centered on factors associated with high mutation rates (Nusbaum et al., 2006) in hydrocephalus resulting from genetic mutations. One of the critical physiological parameters determining the diagnosis of hydrocephalus is intracranial pressure (ICP) (Eide et al., 2010), which is associated with a rapid increase in head circumference or macrocephaly (Robinson et al., 2018), the most obvious sign of hydrocephalus in infants. Elevated ICP, a primary symptom manifested as macrocephaly, distinguishes neonatal hydrocephalus from similar types of the brain conditions with ventriculomegaly such as periventricular leukomalacia found in cerebral palsy (Albright et al., 2005; Limbrick Jr. and Park, 2006). However, a rise in ICP in hydrocephalus should not be confused with idiopathic intracranial hypertension, or pseudotumor cerebri (Friedman and Jacobson, 2002). Interestingly, unlike the SLC12A7 gene as we surveyed in CH (Table 1), a splicing mutation in SLC12A3 gene is linked to idiopathic intracranial hypertension (Godefroid et al., 2006). This suggests that there might be a novel population of undiscovered genes causing intracranial hypertension in the absence of hydrocephalus.
Our projection of genes causing CH highlights distinct biological processes over time: neurogenesis and neurodegeneration. CH is found in children as an early onset, mostly. However, some forms of genetically driven hydrocephalus are late-onset and found in young adults as well. Thus, we thought that hydrocephalus from genetic mutations is not always as a result of birth defects, because a set of patients with NPH (usually found in elderly persons) is known to have hydrocephalus arising from genetic defects (Morimoto et al., 2019). As such, other than age, we have sought what other critical factors can better explain CH. Since CH is primarily a CNS disease related with ‘abnormal neurogenesis’ or ‘generation’ as recently proposed (Furey et al., 2018a), we enthralled the idea that a CNS disease associated with ‘degeneration’ could potentially provide a valuable comparison. Thus, familial PD (fPD) was chosen, since there are more than 15 genes so far defined as PARK family genes.
In our investigation of these familial genes for fPD, we analyzed the dataset and found that it might be more meaningful to compare CH and “PD” with another type of degenerative disorder to investigate where each disease stands from a low-to-high matching rate standpoint with respect to the two factors utilized in this study. Hence, we have additionally surveyed Alzheimer’s Disease (AD). Surprisingly, AD was much more similar to CH than fPD was when comparing the matching rate using (i) proximity to telomeres and (ii) high A + T content—even though both AD and fPD are degenerative diseases of the CNS. Our interpretation is that genetic mutations that are highly associated with these two factors are sufficient to drive development of CH and AD in a similar manner. However, these factors might not be sufficient to explain the mutations detected in fPD, suggesting the involvement of ‘bigenic mechanisms’—such as environmental and genetic factors (Warner and Schapira, 2003). However, this observed discrepancy in fPD as compared to CH and/or AD does not exclude the possibility of acquired mutations (Veeriah et al., 2010) such as somatic mitochondrial DNA mutation (Coxhead et al., 2016).
Other than the two criteria tested in this study, linking a mutation to the disease through specific mediators might deepen our scientific understanding of the subject. For example, a more thorough evaluation of the phenotype revealing ‘hemorrhage’ in CH (Emmert et al., 2019; Shim et al., 2016; Shim et al., 2019) may shed light on mechanisms underlying PHH. Similarly, finding a phenotype of ‘age-related loss or toxin-induced injury in neural connectivity (Luk et al., 2012; Henderson et al., 2019)’ along the nigrostriatal pathway might better explain how genetic and environmental factors alike can contribute to the pathogenesis of fPD.
5. Conclusions
Per the criteria of <50 Mbp proximity to telomeres and A + T content at >59%, the TMEM67 gene is associated with mutations causing CH due primarily to a high A + T content of 64% with a marginal proximity to telomeres. In contrast, the ZCCHC8 gene is associated with mutations causing CH due primarily to proximity to telomeres.
Our results using the first factor suggests that causative genes located on chromosomes 18 to 22 more likely achieve a proximity to telomeres (<50 Mbp) due to short chromosomal length.
Factor-nucleotide size relationships suggest that the full-length size of a gene causing CH, if it is longer than 6000 bp, likely shows high A + T content at >59%.
- If genes causing genetic diseases with mutations do not satisfy the two criteria or the factors according to the previous suggestions (Nusbaum et al., 2006):
- Causative mutations may be found in a region that is less likely to be mutated or
- Off-target effects (mutations) might dominate in an animal study or
- During evolution, genes causing CH in animals have not survived in humans.
Supplementary Material
Acknowledgements
This work was supported by the state of West Virginia startup fund to faculty members at Marshall University. We wish to acknowledge the Marshall University Genomics Core for providing access to shared instrumentation. The Genomics Core is supported by funding from the WV-INBRE grant (NIH P20GM103434), the COBRE ACCORD grant (P20GM121299) and the West Virginia Clinical and Translational Science Institute (WV-CTSI) grant (2U54GM104942). We thank Dr. Jerome Gilbert for the Undergraduate Creative Discovery and Research Award by the Office of the President at Marshall University. We thank for the Faculty Research and Travel award by the West Virginia Space Grant Consortium (WVSGC) sponsored by NASA. We thank Paul Gross at the Hydrocephalus Association to connect J.W.S. with Dr. Bonnie Blazer-Yost at Indiana University Purdue University Indianapolis (IUPUI) through Wpk rat studies. We also thank Dr. Joseph Madsen at Boston Children’s Hospital for comments on this manuscript.
Footnotes
Conflict of interest
The authors declare no competing financial interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.expneurol.2020.113523.
References
- Abdelhamed ZA, Natarajan S, Wheway G, Inglehearn CF, Toomes C, Johnson CA, Jagger DJ, 2015. The Meckel-Gruber syndrome protein TMEM67 controls basal body positioning and epithelial branching morphogenesis in mice via the non-canonical Wnt pathway. Dis. Model. Mech 8, 527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alazami AM, Maddirevula S, Seidahmed MZ, Albhlal LA, Alkuraya FS, 2019. A novel ISLR2-linked autosomal recessive syndrome of congenital hydrocephalus, arthrogryposis and abdominal distension. Hum. Genet 138, 105–107. [DOI] [PubMed] [Google Scholar]
- Albright AL, Ferson S, Carlos S, 2005. Occult hydrocephalus in children with cerebral palsy. Neurosurgery 56, 93–96 (discussion 96-97). [DOI] [PubMed] [Google Scholar]
- Alby C, Piquand K, Huber C, Megarbane A, Ichkou A, Legendre M, Pelluard F, Encha-Ravazi F, Abi-Tayeh G, Bessieres B, El Chehadeh-Djebbar S, Laurent N, Faivre L, Sztriha L, Zombor M, Szabo H, Failler M, Garfa-Traore M, Bole C, Nitschke P, Nizon M, Elkhartoufi N, Clerget-Darpoux F, Munnich A, Lyonnet S, Vekemans M, Saunier S, Cormier-Daire V, Attie-Bitach T, Thomas S, 2015. Mutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome. Am J Hum Genet 97, 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderegg L, Im Hof Gut M, Hetzel U, Howerth EW, Leuthard F, Kyostila K, Lohi H, Pettitt L, Mellersh C, Minor KM, Mickelson JR, Batcher K, Bannasch D, Jagannathan V, Leeb T, 2019. NME5 frameshift variant in Alaskan Malamutes with primary ciliary dyskinesia. PLoS Genet 15, e1008378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora V, Bijarnia-Mahay S, Kulshreshtra S, Singh K, Puri RD, Verma IC, 2019. Prenatal presentation of a rare genetic disorder: a clinical, autopsy and molecular correlation. Autops Case Rep 9, e2019124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas D, Meiniel A, Benadiba C, Bonnafe E, Meiniel O, Reith W, Durand B, 2006. A deficiency in RFX3 causes hydrocephalus associated with abnormal differentiation of ependymal cells. Eur J Neurosci 24, 1020–1030. [DOI] [PubMed] [Google Scholar]
- Babbs C, Furniss D, Morriss-Kay GM, Wilkie AO, 2008. Polydactyly in the mouse mutant Doublefoot involves altered Gli3 processing and is caused by a large deletion in cis to Indian hedgehog. Mech Dev 125, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach A, Lallemand Y, Nicola MA, Ramos C, Mathis L, Maufras M, Robert B, 2003. Msxl is required for dorsal diencephalon patterning. Development 130, 4025–4036. [DOI] [PubMed] [Google Scholar]
- Bachmann-Gagescu R, Dempsey JC, Phelps IG, O’Roak BJ, Knutzen DM, Rue TC, Ishak GE, Isabella CR, Gorden N, Adkins J, Boyle EA, de Lacy N, O’Day D, Alswaid A, Ramadevi AR, Lingappa L, Lourenco C, Martorell L, Garcia-Cazorla A, Ozyurek H, Haliloglu G, Tuysuz B, Topcu M, University of Washington Center for Mendelian, G., Chance P, Parisi MA, Glass IA, Shendure J, Doherty D, 2015. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet 52, 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabh P, 2014. Pathogenesis and prevention of intraventricular hemorrhage. Clin. Perinatol 41, 47–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassan H, 2009. Intracranial hemorrhage in the preterm infant: understanding it, preventing it. Clin Perinatol 36, 737–762. [DOI] [PubMed] [Google Scholar]
- Bateman GA, 2008. The pathophysiology of idiopathic normal pressure hydrocephalus: cerebral ischemia or altered venous hemodynamics? AJNR Am. J. Neuroradiol 29, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batiz LF, De Bias GA, Michaut MA, Ramirez AR, Rodriguez F, Ratto MH, Oliver C, Tomes CN, Rodriguez EM, Mayorga LS, 2009. Sperm from hyh mice carrying a point mutation in alphaSNAP have a defect in acrosome reaction. PLoS One 4, e4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belal TA, Al Menabawy NM, 2017. Case 4: hydrocephalus, macrothrombocytopenia, inclusion bodies, and nephropathy in a 9-year-old boy. Pediatr. Rev 38, 335–336. [DOI] [PubMed] [Google Scholar]
- Belengeanu V, Viskari H, Tallila J, Lahtela J, Farcas S, Andreescu N, Stoian M, Bohiltea CL, Fryns JP, 2011. Lethal evolution of a newborn with consistent features of hydrolethalus syndrome–Romanian patient. Genet Couns 22, 293–304. [PubMed] [Google Scholar]
- Bengoa-Vergniory N, Kypta RM, 2015. Canonical and noncanonical Wnt signaling in neural stem/progenitor cells. Cell. Mol. Life Sci 72, 4157–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattathiri PS, Gregson B, Prasad KS, Mendelow AD, Investigators S, 2006. Intraventricular hemorrhage and hydrocephalus after spontaneous intracerebral hemorrhage: results from the STICH trial. Acta Neurochir Suppl 96, 65–68. [DOI] [PubMed] [Google Scholar]
- Bird TD, 1993. Alzheimer disease overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (Eds.), GeneReviews((R)). Seattle (WA). [Google Scholar]
- Blackburn EH, Greider CW, Szostak JW, 2006. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med 12, 1133–1138. [DOI] [PubMed] [Google Scholar]
- Blackshear PJ, Graves JP, Stumpo DJ, Cobos I, Rubenstein JL, Zeldin DC, 2003. Graded phenotypic response to partial and complete deficiency of a brain-specific transcript variant of the winged helix transcription factor RFX4. Development 130, 4539–4552. [DOI] [PubMed] [Google Scholar]
- Blasco MA, 2007. The epigenetic regulation of mammalian telomeres. Nat Rev Genet 8, 299–309. [DOI] [PubMed] [Google Scholar]
- Bodensteiner JB, 2019. Neurological Manifestations of Achondroplasia. Curr Neurol Neurosci Rep 19, 105. [DOI] [PubMed] [Google Scholar]
- Botfield H, Gonzalez AM, Abdullah O, Skjolding AD, Berry M, McAllister JP 2nd, Logan A, 2013. Decorin prevents the development of juvenile communicating hydrocephalus. Brain 136, 2842–2858. [DOI] [PubMed] [Google Scholar]
- Bothwell SW, Janigro D, Patabendige A, 2019. Cerebrospinal fluid dynamics and intracranial pressure elevation in neurological diseases. Fluids Barriers CNS 16, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britto J, Tannahill D, Keynes R, 2002. A critical role for sonic hedgehog signaling in the early expansion of the developing brain. Nat. Neurosci 5, 103–110. [DOI] [PubMed] [Google Scholar]
- Bubenshchikova E, Ichimura K, Fukuyo Y, Powell R, Hsu C, Morrical SO, Sedor JR, Sakai T, Obara T, 2012. Wtip and Vangl2 are required for mitotic spindle orientation and cloaca morphogenesis. Biol Open 1, 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buysse K, Riemersma M, Powell G, van Reeuwijk J, Chitayat D, Roscioli T, Kamsteeg EJ, van den Elzen C, van Beusekom E, Blaser S, Babul-Hirji R, Halliday W, Wright GJ, Stemple DL, Lin YY, Lefeber DJ, van Bokhoven H, 2013. Missense mutations in beta-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome. Hum. Mol. Genet 22, 1746–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacciagli P, Desvignes JP, Girard N, Delepine M, Zelenika D, Lathrop M, Levy N, Ledbetter DH, Dobyns WB, Villard L, 2014. AP1S2 is mutated in X-linked Dandy-Walker malformation with intellectual disability, basal ganglia disease and seizures (Pettigrew syndrome). Eur J Hum Genet 22, 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappuccio G, Pinelli M, Torella A, Vitiello G, D’Amico A, Alagia M, Del Giudice E, Nigro V, TudpBrunetti-Pierri N, 2017. An extremely severe phenotype attributed to WDR81 nonsense mutations. Ann Neurol 82, 650–651. [DOI] [PubMed] [Google Scholar]
- Carter CS, Vogel TW, Zhang Q, Seo S, Swiderski RE, Moninger TO, Cassell MD, Thedens DR, Keppler-Noreuil KM, Nopoulos P, Nishimura DY, Searby CC, Bugge K, Sheffield VC, 2012. Abnormal development of NG2+PDGFR-alpha+ neural progenitor cells leads to neonatal hydrocephalus in a ciliopathy mouse model. Nat. Med 18, 1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabas A, Cormand B, Grinberg D, Burguera JM, Balcells S, Merino JL, Mate I, Sobrino JA, Gonzalez-Duarte R, Vilageliu L, 1995. Unusual expression of Gaucher’s disease: cardiovascular calcifications in three sibs homozygous for the D409H mutation. J. Med. Genet 32, 740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimpanzee S, Analysis C, 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87. [DOI] [PubMed] [Google Scholar]
- Cindik N, Ozcay F, Suren D, Akkoyun I, Gokdemir M, Varan B, Alehan F, Ozbek N, Tokel K, 2010. Gaucher disease with communicating hydrocephalus and cardiac involvement. Clin Cardiol 33, E26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin GB, Won J, Krebs MP, Hicks WJ, Charette JR, Naggert JK, Nishina PM, 2020. Disruption in murine Emil perturbs retinal lamination during early development. Sci Rep 10, 5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coxhead J, Kurzawa-Akanbi M, Hussain R, Pyle A, Chinnery P, Hudson G, 2016. Somatic mtDNA variation is an important component of Parkinson’s disease. Neurobiol aging 38, 217 e211–217 e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeschik JC, Hehr U, Hartmann B, Ludecke HJ, Rosenbaum T, Schweiger B, Wieczorek D, 2013. 160 kb deletion in ISPD unmasking a recessive mutation in a patient with Walker-Warburg syndrome. Eur J Med Genet 56, 689–694. [DOI] [PubMed] [Google Scholar]
- Dafinger C, Rinschen MM, Borgal L, Ehrenberg C, Basten SG, Franke M, Hohne M, Rauh M, Gobel H, Bloch W, Wunderlich FT, Peters DJM, Tasche D, Mishra T, Habbig S, Dotsch J, Muller RU, Bruning JC, Persigehl T, Giles RH, Benzing T, Schermer B, Liebau MC, 2018. Targeted deletion of the AAA-ATPase Ruvbl1 in mice disrupts ciliary integrity and causes renal disease and hydrocephalus. Exp. Mol. Med 50, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy BE, Robinson ML, 2003. Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum Mol Genet 12, 1163–1170. [DOI] [PubMed] [Google Scholar]
- de Paola M, Miro MP, Ratto M, Batiz LF, Michaut MA, 2019. Pleiotropic effects of alpha-SNAP M105I mutation on oocyte biology: ultrastructural and cellular changes that adversely affect female fertility in mice. Sci. Rep 9, 17374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey JC, Phelps IG, Bachmann-Gagescu R, Glass IA, Tully HM, Doherty D, 2017. Mortality in Joubert syndrome. Am. J. Med. Genet. A 173, 1237–1242. [DOI] [PubMed] [Google Scholar]
- Deveault C, Billingsley G, Duncan JL, Bin J, Theal R, Vincent A, Fieggen KJ, Gerth C, Noordeh N, Traboulsi EI, Fishman GA, Chitayat D, Knueppel T, Millan JM, Munier FL, Kennedy D, Jacobson SG, Innes AM, Mitchell GA, Boycott K, Heon E, 2011. BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum. Mutat 32, 610–619. [DOI] [PubMed] [Google Scholar]
- Dewan MC, Naftel RP, 2017. The global rise of endoscopic third Ventriculostomy with choroid plexus cauterization in pediatric hydrocephalus. Pediatr. Neurosurg 52, 401–408. [DOI] [PubMed] [Google Scholar]
- Dewan MC, Rattani A, Mekary R, Glancz LJ, Yunusa I, Baticulon RE, Fieggen G, Wellons JC, Park KB, Warf BC, 2018. Global hydrocephalus epidemiology and incidence: systematic review and meta-analysis. J. Neurosurg 1–15. [DOI] [PubMed] [Google Scholar]
- Diets IJ, Prescott T, Champaigne NL, Mancini GMS, Krossnes B, Fric R, Kocsis K, Jongmans MCJ, Kleefstra T, 2019. A recurrent de novo missense pathogenic variant in SMARCB1 causes severe intellectual disability and choroid plexus hyperplasia with resultant hydrocephalus. Genet Med 21, 572–579. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Alvarez-Buylla A, 1996. Network of tangential pathways for neuronal migration in adult mammalian brain. Proc. Natl. Acad. Sci. U. S. A 93, 14895–14900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A, 2002. EGF converts transit-amplifying neurogenic precursors in the adult brain into multi potent stem cells. Neuron 36, 1021–1034. [DOI] [PubMed] [Google Scholar]
- Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, Clericuzio C, Demir H, Dorschner M, van Essen AJ, Gahl WA, Gentile M, Gorden NT, Hikida A, Knutzen D, Ozyurek H, Phelps I, Rosenthal P, Verloes A, Weigand H, Chance PF, Dobyns WB, Glass IA, 2010. Mutations in 3 genes (MKS3, CC2D2A and RPGR1P1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J. Med. Genet 47, 8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducro BJ, Schurink A, Bastiaansen JW, Boegheim IJ, van Steenbeek FG, Vos-Loohuis M, Nijman IJ, Monroe GR, Hellinga I, Dibbits BW, Back W, Leegwater PA, 2015. A nonsense mutation in B3GALNT2 is concordant with hydrocephalus in Friesian horses. BMC Genomics 16, 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Shaag A, Zenvirt S, Erlich Y, Hannon GJ, Shanske AL, Gomori JM, Ekstein J, Elpeleg O, 2010. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am. J. Hum. Genet 86, 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide PK, Rapoport BI, Gormley WB, Madsen JR, 2010. A dynamic nonlinear relationship between the static and pulsatile components of intracranial pressure in patients with subarachnoid hemorrhage. J. Neurosurg 112, 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmert AS, Iwasawa E, Shula C, Schultz P, Lindquist D, Dunn RS, Fugate EM, Hu YC, Mangano FT, Goto J, 2019. Impaired neural differentiation and glymphatic CSF flow in the Ccdc39 rat model of neonatal hydrocephalus: genetic interaction with L1cam. Dis Model Mech 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi KC, Epilepsy Phenome/Genome, P., Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli Nieh S, O’Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin Thio L, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR, 2013. De novo mutations in epileptic encephalopathies. Nature 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estivill-Torrus G, Vitalis T, Fernandez-Llebrez P, Price DJ, 2001. The transcription factor Pax6 is required for development of the diencephalic dorsal midline secretory radial glia that form the subcommissural organ. Mech Dev 109, 215–224. [DOI] [PubMed] [Google Scholar]
- Failler M, Gee HY, Krug P, Joo K, Halbritter J, Belkacem L, Filhol E, Porath JD, Braun DA, Schueler M, Frigo A, Alibeu O, Masson C, Brochard K, Hurault de Ligny B, Novo R, Pietrement C, Kayserili H, Salomon R, Gubler MC, Otto EA, Antignac C, Kim J, Benmerah A, Hildebrandt F, Saunier S, 2014. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am J Hum Genet 94, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferner RE, Gutmann DH, 2013. Neurofibromatosis type 1 (NF1): diagnosis and management. Handb. Clin. Neurol 115, 939–955. [DOI] [PubMed] [Google Scholar]
- Filges I, Bruder E, Brandal K, Meier S, Undlien DE, Waage TR, Hoesli I, Schubach M, de Beer T, Sheng Y, Hoeller S, Schulzke S, Rosby O, Miny P, Tercanli S, Oppedal T, Meyer P, Selmer KK, Stromme P, 2016. Stromme Syndrome Is a Ciliary Disorder Caused by Mutations in CENPF. Hum Mutat 37, 711. [DOI] [PubMed] [Google Scholar]
- Finn R, Evans CC, Lee L, 2014. Strain-dependent brain defects in mouse models of primary ciliary dyskinesia with mutations in Pcdp1 and Spef2. Neuroscience 277, 552–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe E, Beales PL, 2013. Bardet-Biedl syndrome. Eur. J. Hum. Genet 21, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe E, Kenny J, Bacchelli C, Beales PL, 2018. Managing Bardet-Biedl syndrome-now and in the future. Front. Pediatr 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen E, Lemmon V, Van Camp G, Vits L, Coucke P, Willems PJ, 1995. CRASH syndrome: clinical spectrum of corpus callosum hypoplasia, retardation, adducted thumbs, spastic paraparesis and hydrocephalus due to mutations in one single gene, L1. Eur. J. Hum. Genet 3, 273–284. [DOI] [PubMed] [Google Scholar]
- Friedman DI, Jacobson DM, 2002. Diagnostic criteria for idiopathic intracranial hypertension. Neurology 59, 1492–1495. [DOI] [PubMed] [Google Scholar]
- Furey CG, Choi J, Jin SC, Zeng X, Timberlake AT, Nelson-Williams C, Mansuri MS, Lu Q, Duran D, Panchagnula S, Allocco A, Karimy JK, Khanna A, Gaillard JR, DeSpenza T, Antwi P, Loring E, Butler WE, Smith ER, Warf BC, Strahle JM, Limbrick DD, Storm PB, Heuer G, Jackson EM, Iskandar BJ, Johnston JM, Tikhonova I, Castaldi C, Lopez-Giraldez F, Bjornson RD, Knight JR, Bilguvar K, Mane S, Alper SL, Haider S, Guclu B, Bayri Y, Sahin Y, Apuzzo MLJ, Duncan CC, DiLuna ML, Gunel M, Lifton RP, Kahle KT, 2018a. De novo mutation in genes regulating neural stem cell fate in human congenital hydrocephalus. Neuron 99 (302–314), e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey CG, Zeng X, Dong W, Jin SC, Choi J, Timberlake AT, Dunbar AM, Allocco AA, Gunel M, Lifton RP, Kahle KT, 2018b. Human genetics and molecular mechanisms of congenital hydrocephalus. World Neurosurg 119, 441–443. [DOI] [PubMed] [Google Scholar]
- Gable DL, Gaysinskaya V, Atik CC, Talbot CC Jr., Kang B, Stanley SE, Pugh EW, Amat-Codina N, Schenk KM, Arcasoy MO, Brayton C, Florea L, Armanios M, 2019. ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev. 33, 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, Garcia-Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF, 2011. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet 43, 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garton T, Keep RF, Wilkinson DA, Strahle JM, Hua Y, Garton HJ, Xi G, 2016. Intraventricular hemorrhage: the role of blood components in secondary injury and hydrocephalus. Transl. Stroke Res 7, 447–451. [DOI] [PubMed] [Google Scholar]
- Godefroid N, Riveira-Munoz E, Saint-Martin C, Nassogne MC, Dahan K, Devuyst O, 2006. A novel splicing mutation in SLC12A3 associated with Gitelman syndrome and idiopathic intracranial hypertension. Am. J. Kidney Dis 48, e73–e79. [DOI] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV, 2010. The primary cilium: a signalling Centre during vertebrate development. Nat Rev Genet 11, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guemez-Gamboa A, Coufal NG, Gleeson JG, 2014. Primary cilia in the developing and mature brain. Neuron 82, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S, Lindsay AM, Timms AE, Beier DR, 2016. Mutations in Dnaaf1 and Lrrc48 cause hydrocephalus, laterality defects, and sinusitis in mice. G3 (Bethesda) 6, 2479–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A, 2008. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat. Neurosci 11, 277–284. [DOI] [PubMed] [Google Scholar]
- Hastbacka J, de la Chapelle A, Kaitila I, Sistonen P, Weaver A, Lander E, 1992. Linkage disequilibrium mapping in isolated founder populations: diastrophic dysplasia in Finland. Nat. Genet 2, 204–211. [DOI] [PubMed] [Google Scholar]
- Hellmann I, Prufer K, Ji H, Zody MC, Paabo S, Ptak SE, 2005. Why do human diversity levels vary at a megabase scale? Genome Res. 15, 1222–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson MX, Cornblath EJ, Darwich A, Zhang B, Brown H, Gathagan RJ, Sandler RM, Bassett DS, Trojanowski JQ, Lee VMY, 2019. Spread of alpha-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat Neurosci 22, 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjortshoj TD, Gronskov K, Philp AR, Nishimura DY, Adeyemo A, Rotimi CN, Sheffield VC, Rosenberg T, Brondum-Nielsen K, 2008. Novel mutations in BBSS highlight the importance of this gene in non-Caucasian Bardet-Biedl syndrome patients. Am. J. Med. Genet. A 146A, 517–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruska KS, LaMarca ME, Scott CR, Sidransky E, 2008. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 29, 567–583. [DOI] [PubMed] [Google Scholar]
- Huh MS, Todd MA, Picketts DJ, 2009. SCO-ping out the mechanisms underlying the etiology of hydrocephalus. Physiology (Bethesda) 24, 117–126. [DOI] [PubMed] [Google Scholar]
- Iannicelli M, Brancati F, Mougou-Zerelli S, Mazzotta A, Thomas S, Elkhartoufi N, Travaglini L, Gomes C, Ardissino GL, Bertini E, Boltshauser E, Castorina P, D’Arrigo S, Fischetto R, Leroy B, Loget P, Bonniere M, Starck L, Tantau J, Gentilin B, Majore S, Swistun D, Flori E, Lalatta F, Pantaleoni C, Penzien J, Grammatico P, International JSG, Dallapiccola B, Gleeson JG, Attie-Bitach T, Valente EM, 2010. Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum. Mutat 31, E1319–E1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez-Tallon I, Gorokhova S, Heintz N, 2002. Loss of function of axonemal dynein Mdnah5 causes primary ciliary dyskinesia and hydrocephalus. Hum Mol Genet 11, 715–721. [DOI] [PubMed] [Google Scholar]
- Iliescu A, Gravel M, Horth C, Kibar Z, Gros P, 2011. Loss of membrane targeting of Vangl proteins causes neural tube defects. Biochemistry 50, 795–804. [DOI] [PubMed] [Google Scholar]
- Isaacs AM, Riva-Cambrin J, Yavin D, Hockley A, Pringsheim TM, Jette N, Lethebe BC, Lowerison M, Dronyk J, Hamilton MG, 2019. Correction: age-specific global epidemiology of hydrocephalus: systematic review, metanalysis and global birth surveillance. PLoS One 14, e0210851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovski I, Djuric O, Caraffi SG, Santodirocco D, Pollazzon M, Rosato S, Cordelli DM, Abdalla E, Accorsi P, Adam MP, Ajmone PF, Badura-Stronka M, Baldo C, Baldi M, Bayat A, Bigoni S, Bonvicini F, Breckpot J, Callewaert B, Cocchi G, Cuturilo G, De Brasi D, Devriendt K, Dinulos MB, Hjortshoj TD, Epifanio R, Faravelli F, Fiumara A, Formisano D, Giordano L, Grasso M, Gronborg S, Iodice A, Iughetti L, Kuburovic V, Kutkowska-Kazmierczak A, Lacombe D, Lo Rizzo C, Luchetti A, Malbora B, Mammi I, Mari F, Montorsi G, Moutton S, Moller RS, Muschke P, Nielsen JEK, Obersztyn E, Pantaleoni C, Pellicciari A, Pisanti MA, Prpic I, Poch-Olive ML, Raviglione F, Renieri A, Ricci E, Rivieri F, Santen GW, Savasta S, Scarano G, Schanze I, Selicorni A, Silengo M, Smigiel R, Spaccini L, Sorge G, Szczaluba K, Tarani L, Tone LG, Toutain A, Trimouille A, Valera ET, Vergano SS, Zanotta N, Zenker M, Conidi A, Zollino M, Rauch A, Zweier C, Garavelli L, 2018. Phenotype and genotype of 87 patients with Mowat-Wilson syndrome and recommendations for care. Genet Med 20, 965–975. [DOI] [PubMed] [Google Scholar]
- Jain N, Lim LW, Tan WT, George B, Makeyev E, Thanabalu T, 2014. Conditional N-WASP knockout in mouse brain implicates actin cytoskeleton regulation in hydrocephalus pathology. Exp Neurol 254, 29–40. [DOI] [PubMed] [Google Scholar]
- Jin SC, Furey CG, Zeng X, Allocco A, Nelson-Williams C, Dong W, Karimy JK, Wang K, Ma S, Delpire E, Kahle KT, 2019. SLC12A ion transporter mutations in sporadic and familial human congenital hydrocephalus. Mol Genet Genomic Med 7, e892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazan S, Gura A, Ucar T, Korkmaz E, Ongun H, Akyuz M, 2005. Hydrocephalus after intraventricular hemorrhage in preterm and low-birth weight infants: analysis of associated risk factors for ventriculoperitoneal shunting. Surg Neurol 64 (Suppl. 2), S77–S81 discussion S81. [DOI] [PubMed] [Google Scholar]
- Khalifa O, Al-Sahlawi Z, Imtiaz F, Ramzan K, Allam R, Al-Mostafa A, Abdel-Fattah M, Abuharb G, Nester M, Verloes A, Al-Zaidan H, 2015. Variable expression pattern in Donnai-Barrow syndrome: report of two novel LRP2 mutations and review of the literature. Eur J Med Genet 58, 293–299. [DOI] [PubMed] [Google Scholar]
- Kibar Z, Vogan KJ, Groulx N, Justice MJ, Underhill DA, Gros P, 2001. Ltap, a mammalian homolog of Drosophila strabismus/van Gogh, is altered in the mouse neural tube mutant loop-tail. Nat. Genet 28, 251–255. [DOI] [PubMed] [Google Scholar]
- Kluss JH, Mamais A, Cookson MR, 2019. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans 47, 651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigstein K, Gramsch C, Kolodziej M, Neubauer BA, Weber A, Lechner S, Hahn A, 2016. Chudley-McCullough Syndrome: Variable Clinical Picture in Twins with a Novel GPSM2 Mutation. Neuropediatrics 47, 197–201. [DOI] [PubMed] [Google Scholar]
- Konishi S, Tanaka N, Mashimo T, Yamamoto T, Sakuma T, Kaneko T, Tanaka M, Izawa T, Yamate J, Kuwamura M, 2020. Pathological characteristics of Ccdc85c knockout rats: a rat model of genetic hydrocephalus. Exp. Anim 69, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschnitzky JE, Keep RF, Limbrick DD Jr., McAllister JP 2nd, Morris JA, Strahle J, Yung YC, 2018. Opportunities in posthemorrhagic hydrocephalus research: outcomes of the hydrocephalus association Posthemorrhagic hydrocephalus workshop. Fluids Barriers CNS 15, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousi M, Katsanis N, 2016. The genetic basis of hydrocephalus. Annu. Rev. Neurosci 39, 409–435. [DOI] [PubMed] [Google Scholar]
- Krebs DL, Metcalf D, Merson TD, Voss AK, Thomas T, Zhang JG, Rakar S, O’Bryan M,K, Willson TA, Viney EM, Mielke LA, Nicola NA, Hilton DJ, Alexander WS, 2004. Development of hydrocephalus in mice lacking SOCS7. Proc. Natl. Acad. Sci. U. S. A 101, 15446–15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A, Zschenker O, Reynolds G, Miller D, Murnane JP, 2010. Effect of telomere proximity on telomere position effect, chromosome healing, and sensitivity to DNA double-strand breaks in a human tumor cell line. Mol. Cell. Biol 30, 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni AV, Riva-Cambrin J, Rozzelle CJ, Naftel RP, Alvey JS, Reeder RW, Holubkov R, Browd SR, Cochrane DD, Limbrick DD, Simon TD, Tamber M, Wellons JC, Whitehead WE, Kestle JRW, 2018. Endoscopic third ventriculostomy and choroid plexus cauterization in infant hydrocephalus: a prospective study by the hydrocephalus clinical research network. J Neurosurg Pediatr 21, 214–223. [DOI] [PubMed] [Google Scholar]
- Kumar KR, Lohmann K, Klein C, 2012. Genetics of Parkinson disease and other movement disorders. Curr. Opin. Neurol 25, 466–474. [DOI] [PubMed] [Google Scholar]
- Lang B, Song B, Davidson W, MacKenzie A, Smith N, McCaig CD, Harmar AJ, Shen S, 2006. Expression of the human PAC1 receptor leads to dose-dependent hydrocephalus-related abnormalities in mice. J Clin Invest 116, 1924–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le WD, Xu P, Jankovic J, Jiang H, Appel SH, Smith RG, Vassilatis DK, 2003. Mutations in NR4A2 associated with familial Parkinson disease. Nat. Genet 33, 85–89. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Bjornsson CS, Dymecki SM, Gilbertson RJ, Holtzman DM, Monuki ES, 2013. The choroid plexus and cerebrospinal fluid: emerging roles in development, disease, and therapy. J. Neurosci 33, 17553–17559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibovitz Z, Mandel H, Falik-Zaccai TC, Ben Harouch S, Savitzki D, Krajden-Haratz K, Gindes L, Tamarkin M, Lev D, Dobyns WB, Lerman-Sagie T, 2018. Walker-Warburg syndrome and tectocerebellar dysraphia: A novel association caused by a homozygous DAG1 mutation. Eur J Paediatr Neurol 22, 525–531. [DOI] [PubMed] [Google Scholar]
- Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL, Badano JL, Katsanis N, 2008. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet 40, 443–448. [DOI] [PubMed] [Google Scholar]
- Lim KC, Crino PB, 2013. Focal malformations of cortical development: new vistas for molecular pathogenesis. Neuroscience 252, 262–276. [DOI] [PubMed] [Google Scholar]
- Lim J, Tang AR, Liles C, Hysong AA, Hale AT, Bonfield CM, Naftel RP, Wellons JC, Shannon CN, 2018. The cost of hydrocephalus: a cost-effectiveness model for evaluating surgical techniques. J Neurosurg Pediatr 23, 109–118. [DOI] [PubMed] [Google Scholar]
- Limbrick DD Jr., Park TS, 2006. Occult hydrocephalus in children with cerebral palsy. Neurosurgery 58 E590; author reply E590. [DOI] [PubMed] [Google Scholar]
- Limbrick DD Jr., Mathur A, Johnston JM, Munro R, Sagar J, Inder T, Park TS, Leonard JL, Smyth MD, 2010. Neurosurgical treatment of progressive posthemorrhagic ventricular dilation in preterm infants: a 10-year single-institution study. J Neurosurg Pediatr 6, 224–230. [DOI] [PubMed] [Google Scholar]
- Liu M, Guan Z, Shen Q, Lalor P, Fitzgerald U, O’Brien T, Dockery P, Shen S, 2016a. Ulk4 is essential for Ciliogenesis and CSF flow. J. Neurosci 36, 7589–7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zheng H, Li X, Wang S, Meyerson HJ, Yang W, Neel BG, Qu CK, 2016b. Gain-of-function mutations of Ptpn11 (Shp2) cause aberrant mitosis and increase susceptibility to DNA damage-induced malignancies. Proc. Natl. Acad. Sci. U. S. A 113, 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang L, Xu B, Yang Y, Shan D, Wu Q, 2019. X-linked dominant chondrodysplasia punctata with severe phenotype in a female fetus: a case report. Medicine (Baltimore) 98, el3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L, Hill WG, 2010. The population genetics of mutations: good, bad and indifferent. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci 365, 1153–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lores P, Coutton C, El Khouri E, Stouvenel L, Givelet M, Thomas L, Rode B, Schmitt A, Louis B, Sakheli Z, Chaudhry M, Fernandez-Gonzales A, Mitsialis A, Dacheux D, Wolf JP, Papon JF, Gacon G, Escudier E, Arnoult C, Bonhivers M, Savinov SN, Amselem S, Ray PF, Dulioust E, Toure A, 2018. Homozygous missense mutation L673P in adenylate kinase 7 (AK7) leads to primary male infertility and multiple morphological anomalies of the flagella but not to primary ciliary dyskinesia. Hum Mol Genet 27, 1196–1211. [DOI] [PubMed] [Google Scholar]
- Louvi A, Wassef M, 2000. Ectopic engrailed 1 expression in the dorsal midline causes cell death, abnormal differentiation of circumventricular organs and errors in axonal pathfinding. Development 127, 4061–4071. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM, 2012. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly KI, Blakeley JO, 2019. The diagnosis and Management of Neurofibromatosis Type 1. Med Clin North Am 103, 1035–1054. [DOI] [PubMed] [Google Scholar]
- Mbabazi-Kabachelor E, Shah M, Vaughan KA, Mugamba J, Ssenyonga P, Onen J, Nalule E, Kapur K, Warf BC, 2019. Infection risk for Bactiseal universal shunts versus Chhabra shunts in Ugandan infants: a randomized controlled trial. J Neurosurg Pediatr 23, 397–406. [DOI] [PubMed] [Google Scholar]
- McAllister JP 2nd, 2012. Pathophysiology of congenital and neonatal hydrocephalus. Semin. Fetal Neonatal Med 17, 285–294. [DOI] [PubMed] [Google Scholar]
- McClintock B, 1941. The stability of broken ends of chromosomes in Zea Mays. Genetics 26, 234–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie CW, Craige B, Kroeger TV, Finn R, Wyatt TA, Sisson JH, Pavlik JA, Strittmatter L, Hendricks GM, Witman GB, Lee L, 2015. CFAP54 is required for proper ciliary motility and assembly of the central pair apparatus in mice. Mol. Biol. Cell 26, 3140–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight TD, Shippen DE, 2004. Plant telomere biology. Plant Cell 16, 794–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meczekalski B, Podfigurna-Stopa A, Smolarczyk R, Katulski K, Genazzani AR, 2013. Kallmann syndrome in women: from genes to diagnosis and treatment. Gynecol. Endocrinol 29, 296–300. [DOI] [PubMed] [Google Scholar]
- Megarbane A, Pangrazio A, Villa A, Chouery E, Maarawi J, Sabbagh S, Lefranc G, Sobacchi C, 2013. Homozygous stop mutation in the SNX10 gene in a consanguineous Iraqi boy with osteopetrosis and corpus callosum hypoplasia. Eur J Med Genet 56, 32–35. [DOI] [PubMed] [Google Scholar]
- Meng H, Li F, Hu R, Yuan Y, Gong G, Hu S, Feng H, 2015. Deferoxamine alleviates chronic hydrocephalus after intraventricular hemorrhage through iron chelation and Wnt1/Wnt3a inhibition. Brain Res. 1602, 44–52. [DOI] [PubMed] [Google Scholar]
- Merchant M, Evangelista M, Luoh SM, Frantz GD, Chalasani S, Carano RA, van Hoy M, Ramirez J, Ogasawara AK, McFarland LM, Filvaroff EH, French DM, de Sauvage FJ, 2005. Loss of the serine/threonine kinase fused results in postnatal growth defects and lethality due to progressive hydrocephalus. Mol Cell Biol 25, 7054–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Bhushan J, Dani A, Vig M, 2017. Na(+) influx via Orail inhibits intracellular ATP-induced mTORC2 signaling to disrupt CD4 T cell gene expression and differentiation. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa G, Parry DA, Fry AE, Giamanco KA, Schwartzentruber J, Vanstone M, Logan CV, Roberts N, Johnson CA, Singh S, Kholmanskikh SS, Adams C, Hodge RD, Hevner RF, Bonthron DT, Braun KPJ, Faivre L, Riviere JB, St-Onge J, Gripp KW, Mancini GM, Pang K, Sweeney E, van Esch H, Verbeek N, Wieczorek D, Steinraths M, Majewski J, Consortium FC, Boycot KM, Pilz DT, Ross ME, Dobyns WB, Sheridan EG, 2014. De novo CCND2 mutations leading to stabilization of cydin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat Genet 46, 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadeh Z, Merkle FT, Soriano-Navarro M, Garcia-Verdugo JM, Alvarez-Buylla A, 2008. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell 3, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, 2014. The long reach of telomeres. Genes Dev. 28, 2445–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto Y, Yoshida S, Kinoshita A, Satoh C, Mishima H, Yamaguchi N, Matsuda K, Sakaguchi M, Tanaka T, Komohara Y, Imamura A, Ozawa H, Nakashima M, Kurotaki N, Kishino T, Yoshiura KI, Ono S, 2019. Nonsense mutation in CFAP43 causes normal-pressure hydrocephalus with ciliary abnormalities. Neurology 92, e2364–e2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton NE, 1991. Parameters of the human genome. Proc. Natl. Acad. Sci. U. S. A 88, 7474–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BP, Inder TE, Rooks V, Taylor GA, Anderson NJ, Mogridge N, Horwood LJ, Volpe JJ, 2002. Posthaemorrhagic ventricular dilatation in the premature infant: natural history and predictors of outcome. Arch. Dis. Child. Fetal Neonatal Ed 87, F37–F41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabhan MM, Abdelaziz H, Xu Y, El Sayed R, Santibanez-Koref M, Soliman NA, Sayer JA, 2015. Case Report: Whole-exome analysis of a child with polycystic kidney disease and ventriculomegaly. Genet Mol Res 14, 3618–3624. [DOI] [PubMed] [Google Scholar]
- Niceta M, Stellacci E, Gripp KW, Zampino G, Kousi M, Anselmi M, Traversa A, Ciolfi A, Stabley D, Bruselles A, Caputo V, Cecchetti S, Prudente S, Fiorenza MT, Boitani C, Philip N, Niyazov D, Leoni C, Nakane T, Keppler-Noreuil K, Braddock SR, Gillessen-Kaesbach G, Palleschi A, Campeau PM, Lee BH, Pouponnot C, Stella L, Bocchinfuso G, Katsanis N, Sol-Church K, Tartaglia M, 2015. Mutations Impairing GSK3-Mediated MAF Phosphorylation Cause Cataract, Deafness, Intellectual Disability, Seizures, and a Down Syndrome-like Facies. Am J Hum Genet 96, 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols WC, Pankratz N, Marek DK, Pauciulo MW, Elsaesser VE, Halter CA, Rudolph A, Wojcieszek J, Pfeiffer RF, Foroud T, Parkinson Study Group PI, 2009. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 72, 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, Andorf JL, Mykytyn K, Swiderski RE, Yang B, Carmi R, Stone EM, Sheffield VC, 2004. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl. Acad. Sci. U. S. A 101, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusbaum C, Mikkelsen TS, Zody MC, Asakawa S, Taudien S, Garber M, Kodira CD, Schueler MG, Shimizu A, Whittaker CA, Chang JL, Cuomo CA, Dewar K, FitzGerald MG, Yang X, Allen NR, Anderson S, Asakawa T, Blechschmidt K, Bloom T, Borowsky ML, Butler J, Cook A, Corum B, DeArellano K, DeCaprio D, Dooley KT, Dorris L 3rd, Engels R, Glockner G, Hafez N, Hagopian DS, Hall JL, Ishikawa SK, Jaffe DB, Kamat A, Kudoh J, Lehmann R, Lokitsang T, Macdonald P, Major JE, Matthews CD, Mauceli E, Menzel U, Mihalev AH, Minoshima S, Murayama Y, Naylor JW, Nicol R, Nguyen C, O’Leary SB, O’Neill K, Parker SC, Polley A, Raymond CK, Reichwald K, Rodriguez J, Sasaki T, Schilhabel M, Siddiqui R, Smith CL, Sneddon TP, Talamas JA, Tenzin P, Topham K, Venkataraman V, Wen G, Yamazaki S, Young SK, Zeng Q, Zimmer AR, Rosenthal A, Birren BW, Platzer M, Shimizu N, Lander ES, 2006. DNA sequence and analysis of human chromosome 8. Nature 439, 331–335. [DOI] [PubMed] [Google Scholar]
- O’Connor C, 2008. Telomeres of human chromosomes. Nature Education 1 (1), 166. [Google Scholar]
- O’Dougherty GR, Fulkerson DH, Kern M, Haidar K, Calhoun B, 2019. Complications of Insufficient Dura and Blood Loss During Surgical Intervention in Shprintzen-Goldberg Syndrome: A Case Report. Am J Case Rep 20, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtoshi A, 2008. Hydrocephalus caused by conditional ablation of the Pten or beta-catenin gene. Cerebrospinal Fluid Res 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterman MJ, Kochanek KD, MacDorman MF, Strobino DM, Guyer B, 2015. Annual summary of vital statistics: 2012-2013. Pediatrics 135, 1115–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA, 2009. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet 151C, 326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ, 2008. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146. [DOI] [PubMed] [Google Scholar]
- Perdaens O, Koerts G, Nassogne MC, 2018. Hydrocephalus in children under the age of five from diagnosis to short–/medium–/long-term progression: a retrospective review of 142 children. Acta Neurol. Belg 118, 97–103. [DOI] [PubMed] [Google Scholar]
- Picketts DJ, 2006. Neuropeptide signaling and hydrocephalus: SCO with the flow. J Clin Invest 116, 1828–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porayette P, Fruitman D, Lauzon JL, Le Goff C, Cormier-Daire V, Sanders SP, Pinto-Rojas A, Perez-Atayde AR, 2014. Novel mutations in geleophysic dysplasia type 1. Pediatr Dev Pathol 17, 209–216. [DOI] [PubMed] [Google Scholar]
- Preiksaitiene E, Voisin N, Gueneau L, Benusiene E, Krasovskaja N, Blazyte EM, Ambrozaityte L, Rancelis T, Reymond A, Kucinskas V, 2020. Pathogenic homozygous variant in POMK gene is the cause of prenatally detected severe ventriculomegaly in two Lithuanian families. Am J Med Genet A 182, 536–542. [DOI] [PubMed] [Google Scholar]
- Rachel RA, Yamamoto EA, Dewanjee MK, May-Simera HL, Sergeev YV, Hackett AN, Pohida K, Munasinghe J, Gotoh N, Wickstead B, Fariss RN, Dong L, Li T, Swaroop A, 2015. CEP290 alleles in mice disrupt tissue-specific cilia biogenesis and recapitulate features of syndromic ciliopathies. Hum. Mol. Genet 24, 3775–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radic JA, Vincer M, McNeely PD, 2015. Temporal trends of intraventricular hemorrhage of prematurity in Nova Scotia from 1993 to 2012. J Neurosurg Pediatr 15, 573–579. [DOI] [PubMed] [Google Scholar]
- Radmanesh F, Caglayan AO, Silhavy JL, Yilmaz C, Cantagrel V, Omar T, Rosti B, Kaymakcalan H, Gabriel S, Li M, Sestan N, Bilguvar K, Dobyns WB, Zaki MS, Gunel M, Gleeson JG, 2013. Mutations in LAMB1 cause cobblestone brain malformation without muscular or ocular abnormalities. Am J Hum Genet 92, 468–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindra VM, Kraus KL, Riva-Cambrin JK, Kestle JR, 2015. The need for cost-effective neurosurgical innovation–a global surgery initiative. World Neurosurg 84, 1458–1461. [DOI] [PubMed] [Google Scholar]
- Recio A, Sanchez-Moya AI, Felix V, Campos Y, 2017. Congenital Melanocytic Nevus Syndrome: A Case Series. Actas Dermosifiliogr 108, e57–e62. [DOI] [PubMed] [Google Scholar]
- Rekate HL, 2007. Comments on the article by D. Greitz “paradigm shift in hydrocephalus research in legacy of Dandy’s pioneering work: rationale for third ventriculostomy in communicating hydrocephalus”. Childs Nerv. Syst 23, 1227–1228 (author reply 1229-1231). [DOI] [PubMed] [Google Scholar]
- Ritter E, Gebhardt C, Salamini F, 1990. Estimation of recombination frequencies and construction of RFLP linkage maps in plants from crosses between heterozygous parents. Genetics 125, 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius R, Gonzalez-Del Angel A, Velazquez-Aragon JA, Cordero-Guzman LM, Munoz-Hernandez SE, Alcantara-Ortigoza MA, 2018. Identification of a novel SLC12A6 pathogenic variant associated with hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC) in a non-French-Canadian family. Neurol. India 66, 1162 1165. [DOI] [PubMed] [Google Scholar]
- Robinson S, 2012. Neonatal posthemorrhagic hydrocephalus from prematurity: pathophysiology and current treatment concepts. J Neurosurg Pediatr 9, 242–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S, Conteh FS, Oppong AY, Yellowhair TR, Newville JC, Demerdash ND, Shrock CL, Maxwell JR, Jett S, Northington FJ, Jantzie LL, 2018. Extended combined neonatal treatment with erythropoietin plus melatonin prevents Posthemorrhagic hydrocephalus of prematurity in rats. Front. Cell. Neurosci 12, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez EM, Guerra MM, 2017. Neural stem cells and fetal-onset hydrocephalus. Pediatr. Neurosurg 52, 446–461. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP, 2007. Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376. [DOI] [PubMed] [Google Scholar]
- Romero L, Ros B, Arraez MA, Rius F, Gonzalez L, Martin A, Carrasco A, Segura M, 2015. Analysis of risk factors for hydrocephalus development in newborn infants with germinal matrix hemorrhage. Minerva Pediatr. 67, 401–406. [PubMed] [Google Scholar]
- Roscioli T, Kamsteeg EJ, Buysse K, Maystadt I, van Reeuwijk J, van den Elzen C, van Beusekom E, Riemersma M, Pfundt R, Vissers LE, Schraders M, Altunoglu U, Buckley MF, Brunner HG, Grisart B, Zhou H, Veltman JA, Gilissen C, Mancini GM, Delree P, Willemsen MA, Ramadza DP, Chitayat D, Bennett C, Sheridan E, Peeters EA, Tan-Sindhunata GM, de Die-Smulders CE, Devriendt K, Kayserili H, El-Hashash OA, Stemple DL, Lefeber DJ, Lin YY, van Bokhoven H, 2012. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of alpha-dystroglycan. Nat Genet 44, 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch DH, Danielian PS, McMahon AP, Zee N, 1999. Cystic malformation of the posterior cerebellar vermis in transgenic mice that ectopically express Engrailed-1, a homeodomain transcription factor. Teratology 60, 22–28. [DOI] [PubMed] [Google Scholar]
- Ruggeri G, Timms AE, Cheng C, Weiss A, Kollros P, Chapman T, Tully H, Mirzaa GM, 2018. Bi-allelic mutations of CCDC88C are a rare cause of severe congenital hydrocephalus. Am J Med Genet A 176, 676–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saillour Y, Zanni G, Des Portes V, Heron D, Guibaud L, Iba-Zizen MT, Pedespan JL, Poirier K, Castelnau L, Julien C, Franconnet C, Bonthron D, Porteous ME, Chelly J, Bienvenu T, 2007. Mutations in the AP1S2 gene encoding the sigma 2 subunit of the adaptor protein 1 complex are associated with syndromic X-linked mental retardation with hydrocephalus and calcifications in basal ganglia. J Med Genet 44, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, Smith DL, Faull RL, Roos RA, Howland D, Detloff PJ, Housman DE, Bates GP, 2013. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U. S. A 110, 2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawamoto K, Wichterle H, Gonzalez-Perez O, Cholfin JA, Yamada M, Spassky N, Murcia NS, Garcia-Verdugo JM, Marin O, Rubenstein JL, Tessier-Lavigne M, Okano H, Alvarez-Buylla A, 2006. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science 311, 629–632. [DOI] [PubMed] [Google Scholar]
- Schoner K, Kohlhase J, Muller AM, Schramm T, Plassmann M, Schmitz R, Neesen J, Wieacker P, Rehder H, 2013. Hydrocephalus, agenesis of the corpus callosum, and cleft lip/palate represent frequent associations in fetuses with Peters’ plus syndrome and B3GALTL mutations. Fetal PPS phenotypes, expanded by Dandy Walker cyst and encephalocele. Prenat Diagn 33, 75–80. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, Debette S, Longstreth WT Jr., Janssens AC, Pankratz VS, Dartigues JF, Hollingworth P, Aspelund T, Hernandez I, Beiser A, Kuller LH, Koudstaal PJ, Dickson DW, Tzourio C, Abraham R, Antunez C, Du Y, Rotter JI, Aulchenko YS, Harris TB, Petersen RC, Berr C, Owen MJ, Lopez-Arrieta J, Varadarajan BN, Becker JT, Rivadeneira F, Nalls MA, Graff-Radford NR, Campion D, Auerbach S, Rice K, Hofman A, Jonsson PV, Schmidt H, Lathrop M, Mosley TH, Au R, Psaty BM, Uitterlinden AG, Farrer LA, Lumley T, Ruiz A, Williams J, Amouyel P, Younkin SG, Wolf PA, Launer LJ, Lopez OL, van Duijn CM, Breteler MM, Consortium C, Consortium G, Consortium E, 2010. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Sebai MA, Patel N, Ewida N, Kurdi W, Altweijri I, Sogaty S, Almardawi E, Seidahmed MZ, Alnemri A, Madirevula S, Ibrahim N, Abdulwahab F, Hashem M, Al-Sheddi T, Alomar R, Alobeid E, Sallout B, AlBaqawi B, AlAali W, Ajaji N, Lesmana H, Hopkin RJ, Dupuis L, Mendoza-Londono R, Al Rukban H, Yoon G, Faqeih E, Alkuraya FS, 2017. The genetic landscape of familial congenital hydrocephalus. Ann. Neurol 81, 890–897. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Basel-Vanagaite L, Goodman JR, Scheffer IE, Bodell A, Ganesh VS, Ravenscroft R, Hill RS, Cherry TJ, Shugart YY, Barkovich J, Straussberg R, Walsh CA, 2004. Etiological heterogeneity of familial periventricular heterotopia and hydrocephalus. Brain Dev 26, 326–334. [DOI] [PubMed] [Google Scholar]
- Sheffield ID, McGee MA, Glenn SJ, Baek DY, Coleman JM, Dorius BK, Williams C, Rose BJ, Sanchez AE, Goodman MA, Daines JM, Eggett DL, Sheffield VC, Suli A, Kooyman DL, 2018. Osteoarthritis-like changes in Bardet-Biedl syndrome mutant ciliopathy mice (Bbs1(M390R/M390R)): evidence for a role of primary cilia in cartilage homeostasis and regulation of inflammation. Front. Physiol 9, 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim JW, Sandlund J, Han CH, Hameed MQ, Connors S, Klagsbrun M, Madsen JR, Irwin N, 2013. VEGF, which is elevated in the CSF of patients with hydrocephalus, causes ventriculomegaly and ependymal changes in rats. Exp. Neurol 247, 703–709. [DOI] [PubMed] [Google Scholar]
- Shim JW, Sandlund J, Hameed MQ, Blazer-Yost B, Zhou FC, Klagsbrun M, Madsen JR, 2016. Excess HB-EGF, which promotes VEGF signaling, leads to hydrocephalus. Sci. Rep 6, 26794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim JW, Territo PR, Simpson S, Watson JC, Jiang L, Riley AA, McCarthy B, Persohn S, Fulkerson D, Blazer-Yost BL, 2019. Hydrocephalus in a rat model of Meckel Gruber syndrome with a TMEM67 mutation. Sci. Rep 9, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada IS, Somatilaka BN, Hwang SH, Anderson AG, Shelton JM, Rajaram V, Konopka G, Mukhopadhyay S, 2019. Derepression of sonic hedgehog signaling upon Gprl61 deletion unravels forebrain and ventricular abnormalities. Dev. Biol 450, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegler L, Erber R, Burghaus S, Brodkorb T, Wachter D, Wilkinson N, Bolton J, Stringfellow H, Haller F, Beckmann MW, Hartmann A, Agaimy A, 2018. Fumarate hydratase (FH) deficiency in uterine leiomyomas: recognition by histological features versus blind immunoscreening. Virchows Arch. 472, 789–796. [DOI] [PubMed] [Google Scholar]
- Sironen A, Kotaja N, Mulhern H, Wyatt TA, Sisson JH, Pavlik JA, Miiluniemi M, Fleming MD, Lee L, 2011. Loss of SPEF2 function in mice results in spermatogenesis defects and primary ciliary dyskinesia. Biol. Reprod 85, 690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek A, Kaylor J, Pierce H, Cahr M, DeWard SJ, Schneidman-Duhovny D, Alsadah A, Salem F, Schmajuk G, Mehta L, 2015. CRB2 mutations produce a phenotype resembling congenital nephrosis, Finnish type, with cerebral ventriculomegaly and raised alpha-fetoprotein. Am J Hum Genet 96, 162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, Aligianis IA, Ward CJ, Pasha S, Punyashthiti R, Malik Sharif S, Batman PA, Bennett CP, Woods CG, McKeown C, Bucourt M, Miller CA, Cox P, Algazali L, Trembath RC, Torres VE, Attie-Bitach T, Kelly DA, Maher ER, Gattone VH 2nd, Harris PC, Johnson CA, 2006. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet 38, 191–196. [DOI] [PubMed] [Google Scholar]
- Snowden JN, Beaver M, Smeltzer MS, Kielian T, 2012. Biofilm-infected intracerebroventricular shunts elicit inflammation within the central nervous system. Infect Immun 80, 3206–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousounis K, Looso M, Maki N, Ivester CJ, Braun T, Tsonis PA, 2013. Transcriptome analysis of newt lens regeneration reveals distinct gradients in gene expression patterns. PLoS One 8, e61445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, Hale EC, Newman NS, Schibler K, Carlo WA, Kennedy KA, Poindexter BB, Finer NN, Ehrenkranz RA, Duara S, Sanchez PJ, O’Shea TM, Goldberg RN, Van Meurs KP, Faix RG, Phelps DL, Frantz ID 3rd, Watterberg KL, Saha S, Das A, Higgins RD, Eunice Kennedy Shriver National Institute of Child, H., Human Development Neonatal Research, N, 2010. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics 126, 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiderski RE, Nakano Y, Mullins RF, Seo S, Banfi B, 2014. A mutation in the mouse ttc26 gene leads to impaired hedgehog signaling. PLoS Genet. 10, e1004689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Izawa T, Takenaka S, Yamate J, Kuwamura M, 2015. Ccdc85C, a causative protein for hydrocephalus and subcortical heterotopia, is expressed in the systemic epithelia with proliferative activity in rats. Histol Histopathol 30, 823–832. [DOI] [PubMed] [Google Scholar]
- Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK, 2001. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol. Biol. Cell 12, 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M, 2012. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tscl mutant mice. Nature 488, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyburczy ME, Dies KA, Glass J, Camposano S, Chekaluk Y, Thorner AR, Lin L, Krueger D, Franz DN, Thiele EA, Sahin M, Kwiatkowski DJ, 2015. Mosaic and Intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet. 11, el005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uguen A, Laurent C, Samaison L, Boisselier B, Talagas M, Costa S, Aziza J, Mokhtari K, Le Marechal C, Marcorelles P, 2015. Severe hydrocephalus caused by diffuse leptomeningeal and neurocutaneous melanocytosis of antenatal onset: a clinical, pathologic, and molecular study of 2 cases. Hum Pathol 46, 1189–1196. [DOI] [PubMed] [Google Scholar]
- Uytingco CR, Williams CL, Xie C, Shively DT, Green WW, Ukhanov K, Zhang L, Nishimura DY, Sheffield VC, Martens JR, 2019. BBS4 is required for intraflagellar transport coordination and basal body number in mammalian olfactory cilia. J Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valayannopoulos V, Nicely H, Harmatz P, Turbeville S, 2010. Mucopolysaccharidosis VI. Orphanet J Rare Dis 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Reeuwijk J, Janssen M, van den Elzen C, Beltran-Valero de Bernabe D, Sabatelli P, Merlini L, Boon M, Scheffer H, Brockington M, Muntoni F, Huynen MA, Verrips A, Walsh CA, Barth PG, Brunner HG, van Bokhoven H, 2005. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet 42, 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, Holland EC, Paty PB, Mischel PS, Liau L, Cloughesy TF, Mellinghoff IK, Solit DB, Chan TA, 2010. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet 42, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinchon M, Rekate H, Kulkarni AV, 2012. Pediatric hydrocephalus outcomes: a review. Fluids Barriers CNS 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel P, Read R, Hansen GM, Freay LC, Zambrowicz BP, Sands AT, 2010. Situs inversus in Dpcd/Poll−/−, Nme7−/−. and Pkd1l1−/− mice. Vet Pathol 47, 120–131. [DOI] [PubMed] [Google Scholar]
- Vogel P, Read RW, Hansen GM, Payne BJ, Small D, Sands AT, Zambrowicz BP, 2012. Congenital hydrocephalus in genetically engineered mice. Vet. Pathol 49, 166–181. [DOI] [PubMed] [Google Scholar]
- Wallmeier J, Frank D, Shoemark A, Nothe-Menchen T, Cindric S, Olbrich H, Loges NT, Aprea I, Dougherty GW, Pennekamp P, Kaiser T, Mitchison HM, Hogg C, Carr SB, Zariwala MA, Ferkol T, Leigh MW, Davis SD, Atkinson J, Dutcher SK, Knowles MR, Thiele H, Altmuller J, Krenz H, Woste M, Brentrup A, Ahrens F, Vogelberg C, Morris-Rosendahl DJ, Omran H, 2019. De Novo Mutations in FOXJ1 Result in a Motile Ciliopathy with Hydrocephalus and Randomization of Left/Right Body Asymmetry. Am J Hum Genet 105, 1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Shangguan S, Chang S, Wang Z, Lu X, Wu L, Li R, Bao Y, Qiu Z, Niu B, Zhang T, 2014. Impaired methylation modifications of FZD3 alter chromatin accessibility and are involved in congenital hydrocephalus pathogenesis. Brain Res 1569, 48–56. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhou Y, Wang J, Tseng IC, Huang T, Zhao Y, Zheng Q, Gao Y, Luo H, Zhang X, Bu G, Hong W, Xu H, 2016. SNX27 deletion causes hydrocephalus by impairing ependymal cell differentiation and Ciliogenesis. J. Neurosci 36, 12586–12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner TT, Schapira AH, 2003. Genetic and environmental factors in the cause of Parkinson’s disease. Ann Neurol 53 (Suppl. 3). S16–23; discussion S23-15. [DOI] [PubMed] [Google Scholar]
- Wicker G, Prill V, Brooks D, Gibson G, Hopwood J, von Figura K, Peters C, 1991. Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). An intermediate clinical phenotype caused by substitution of valine for glycine at position 137 of arylsulfatase B. J. Biol. Chem 266, 21386–21391. [PubMed] [Google Scholar]
- Wild A, Kalff-Suske M, Vortkamp A, Bornholdt D, Konig R, Grzeschik KH, 1997. Point mutations in human GLI3 cause Greig syndrome. Hum. Mol. Genet 6, 1979–1984. [DOI] [PubMed] [Google Scholar]
- Wright Z, Larrew TW, Eskandari R, 2016. Pediatric hydrocephalus: current state of diagnosis and treatment. Pediatr. Rev 37, 478–490. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Kanemura Y, 2015. Molecular Biology of Pediatric Hydrocephalus and Hydrocephalus-related Diseases. Neurol Med Chir (Tokyo) 55, 640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LT, Li WY, Kaartinen V, 2008. Tissue-specific expression of Cre recombinase from the Tgfb3 locus. Genesis 46, 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Simonneau C, Kilker R, Oakley L, Byrne MD, Nichtova Z, Stefanescu I, Par deep-Kumar F, Tripathi S, Londin E, Saugier-Veber P, Willard B, Thakur M, Pickup S, Ishikawa H, Schroten H, Smeyne R, Horowitz A, 2019. Murine MPDZ-linked hydrocephalus is caused by hyperpermeability of the choroid plexus. EMBO Mol Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang TT, Su J, Wang WJ, Craige B, Witman GB, Tsou MF, Liao JC, 2015. Super resolution pattern recognition reveals the architectural map of the ciliary transition zone. Sci. Rep 5, 14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Williams MA, Rigamonti D, 2006. Genetics of human hydrocephalus. J. Neurol 253, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate YA, Lichty AW, Champion KJ, Clarkson LK, Holden KR, Matheus MG, 2014. Unique cerebrovascular anomalies in Noonan syndrome with RAF1 mutation. J Child Neurol 29, NP13–17. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Nishimura D, Seo S, Vogel T, Morgan DA, Searby C, Bugge K, Stone EM, Rahmouni K, Sheffield VC, 2011. Bardet-Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes. Proc. Natl. Acad. Sci. U. S. A 108, 20678–20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Yu WM, Waclaw RR, Kontaridis MI, Neel BG, Qu CK, 2018. Gain-of-function mutations in the gene encoding the tyrosine phosphatase SHP2 induce hydrocephalus in a catalytically dependent manner. Sci Signal 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Lv Y, Liu ZI, Xia P, Li H, Jiao J, 2020. Loss of Rsph9 causes neonatal hydrocephalus with abnormal development of motile cilia in mice. Sci. Rep 10, 12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere JB, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkei SM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA, Albrecht B, Armour CM, Armstrong L, Caluseriu O, Cytrynbaum C, Drolet BA, Innes AM, Lauzon JL, Lin AE, Mancini GM, Meschino WS, Reggin JD, Saggar AK, Lerman-Sagie T, Uyanik G, Weksberg R, Zirn B, Beaulieu CL, Finding of Rare Disease Genes Canada, C., Majewski J, Bulman DE, O’Driscoll M, Shendure J, Graham JM Jr., Boycott KM, Dobyns WB, 2012. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet 44, 934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman A, Wuyts W, Patel MS, 1993. Adams-Oliver Syndrome, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (Eds.), GeneReviews((R)), Seattle (WA) [Google Scholar]
- Lehtinen MK, Bjornsson CS, Dymecki SM, Gilbertson RJ, Holtzman DM, Monuki ES, 2013. The choroid plexus and cerebrospinal fluid: emerging roles in development, disease, and therapy. J Neurosci 33, 17553–17559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Shido K, Niihori T, Niizuma H, Katata Y, Iizuka C, Oba D, Moriya K, Saito-Nanjo Y, Onuma M, Rikiishi T, Sasahara Y, Watanabe M, Aiba S, Saito R, Sonoda Y, Tominaga T, Aoki Y, Kure S, 2016. Somatic BRAF c.1799T>A p.V600E Mosaicism syndrome characterized by a linear syringocystadenoma papilliferum, anaplastic astrocytoma, and ocular abnormalities. Am J Med Genet A 170A, 189–194. [DOI] [PubMed] [Google Scholar]
- Chandrasekar G, Vesterlund L, Hultenby K, Tapia-Paez I, Kere J, 2013. The zebrafish orthologue of the dyslexia candidate gene DYX1C1 is essential for cilia growth and function. PLoS One 8, e63123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimpanzee S, Analysis C, 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87. [DOI] [PubMed] [Google Scholar]
- Wenger TL, Hing AV, Evans KN, 1993. Apert Syndrome, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (Eds.), GeneReviews((R)), Seattle (WA). [Google Scholar]
- Wicker G, Prill V, Brooks D, Gibson G, Hopwood J, von Figura K, Peters C, 1991. [PubMed] [Google Scholar]
- Yis U, Uyanik G, Kurul S, Dirik E, Ozer E, Gross C, Hehr U, 2007. A case of Walker-Warburg syndrome resulting from a homozygous POMT1 mutation. Eur J Paediatr Neurol 11, 46–49. [DOI] [PubMed] [Google Scholar]
- Zhang J, Williams MA, Rigamonti D, 2006. Genetics of human hydrocephalus. J Neurol 253, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balk K, Biesecker LG, 2008. The clinical atlas of Greig cephalopolysyndactyly syndrome. Am J Med Genet A 146A, 548–557. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Hopkins E, Doyle D, Dobyns WB, 2010. High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am J Med Genet A 152A, 1161–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guemez-Gamboa A, Coufal NG, Gleeson JG, 2014. Primary cilia in the developing and mature brain. Neuron 82, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland-Little JM, Hoffmann AD, Ocbina PJ, Peterson MA, Bosnian JD, Chen Y, Cheng SY, Anderson KV, Moskowitz IP, 2011. A novel murine allele of Intraflagellar Transport Protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum Mol Genet 20, 3725–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DI, Jacobson DM, 2002. Diagnostic criteria for idiopathic intracranial hypertension. Neurology 59, 1492–1495. [DOI] [PubMed] [Google Scholar]
- Tunovic S, Baranano KW, Barkovich JA, Strober JB, Jamal L, Slavotinek AM, 2015. Novel KIF7 missense substitutions in two patients presenting with multiple malformations and features of acrocallosal syndrome. Am J Med Genet A 167A, 2767–2776. [DOI] [PubMed] [Google Scholar]
- Nix JS, Blakeley J, Rodriguez FJ, 2020. An update on the central nervous system manifestations of neurofibromatosis type 1. Acta Neuropathol 139, 625–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusbaum C, Mikkelsen TS, Zody MC, Asakawa S, Taudien S, Garber M, Kodira CD, Schueler MG, Shimizu A, Whittaker CA, Chang JL, Cuomo CA, Dewar K, FitzGerald MG, Yang X, Allen NR, Anderson S, Asakawa T, Blechschmidt K, Bloom T, Borowsky ML, Butler J, Cook A, Corum B, DeArellano K, DeCaprio D, Dooley KT, Dorris L 3rd, Engels R, Glockner G, Hafez N, Hagopian DS, Hall JL, Ishikawa SK, Jaffe DB, Kamat A, Kudoh J, Lehmann R, Lokitsang T, Macdonald P, Major JE, Matthews CD, Mauceli E, Menzel U, Mihalev AH, Minoshima S, Murayama Y, Naylor JW, Nicol R, Nguyen C, O’Leary SB, O’Neill K, Parker SC, Polley A, Raymond CK, Reichwald K, Rodriguez J, Sasaki T, Schilhabel M, Siddiqui R, Smith CL, Sneddon TP, Talamas JA, Tenzin P, Topham K, Venkataraman V, Wen G, Yamazaki S, Young SK, Zeng Q, Zimmer AR, Rosenthal A, Birren BW, Platzer M, Shimizu N, Lander ES, 2006. DNA sequence and analysis of human chromosome 8. Nature 439, 331–335. [DOI] [PubMed] [Google Scholar]
- Field M, Scheffer IE, Gill D, Wilson M, Christie L, Shaw M, Gardner A, Glubb G, Hobson L, Corbett M, Friend K, Willis-Owen S, Gecz J, 2012. Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet 20, 806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filges I, Bruder E, Brandal K, Meier S, Undlien DE, Waage TR, Hoesli I, Schubach M, de Beer T, Sheng Y, Hoeller S, Schulzke S, Rosby O, Miny P, Tercanli S, Oppedal T, Meyer P, Selmer KK, Stromme P, 2016. Stromme Syndrome Is a Ciliary Disorder Caused by Mutations in CENPF. Hum Mutat 37, 711. [DOI] [PubMed] [Google Scholar]
- Roy A, Murphy RM, Deng M, MacDonald JW, Bammler TK, Aldinger KA, Glass IA, Millen KJ, 2019. PI3K-Yap activity drives cortical gyrification and hydrocephalus in mice. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggeri G, Timms AE, Cheng C, Weiss A, Kollros P, Chapman T, Tully H, Mirzaa GM, 2018. Bi-allelic mutations of CCDC88C are a rare cause of severe congenital hydrocephalus. Am J Med Genet A 176, 676–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoort VS, Holden KR, Ukadike KC, Collins JS, Saul RA, Srivastava AK, 2004. POMGnTl gene alterations in a family with neurological abnormalities. Ann Neurol 56, 143–148. [DOI] [PubMed] [Google Scholar]
- Vinchon M, Rekate H, Kulkarni AV, 2012. Pediatric hydrocephalus outcomes: a review. Fluids Barriers CNS 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currier SC, Lee CK, Chang BS, Bodell AL, Pai GS, Job L, Lagae LG, Al-Gazali LI, Eyaid WM, Enns G, Dobyns WB, Walsh CA, 2005. Mutations in POMT1 are found in a minority of patients with Walker-Warburg syndrome. Am J Med Genet A 133A, 53–57. [DOI] [PubMed] [Google Scholar]
- Grinberg I, Millen KJ, 2005. The ZIC gene family in development and disease. Clin Genet 67, 290–296. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Hopkins E, Doyle D, Dobyns WB, 2010. High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am J Med Genet A 152A, 1161–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.